
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 22 August 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.956340
This article is part of the Research TopicGlobal Excellence in Inflammatory Diseases: Latin America 2021View all 6 articles
 Andrea V. Loureiro1†
Andrea V. Loureiro1† Lauro I. Moura-Neto1†
Lauro I. Moura-Neto1† Conceição S. Martins1Pedro I. M. Silva1Matheus B.S. Lopes1
Conceição S. Martins1Pedro I. M. Silva1Matheus B.S. Lopes1 Renata F. C. Leitão1
Renata F. C. Leitão1 Juliana M. Coelho-Aguiar2
Juliana M. Coelho-Aguiar2 Vivaldo Moura-Neto2Cirle A. Warren3
Vivaldo Moura-Neto2Cirle A. Warren3 Deiziane V.S. Costa3*‡
Deiziane V.S. Costa3*‡ Gerly A. C. Brito1,4*‡
Gerly A. C. Brito1,4*‡Clostridioides difficile (C. difficile) produces toxins A (TcdA) and B (TcdB), both associated with intestinal damage and diarrhea. Pannexin-1 (Panx1) channels allows the passage of messenger molecules, such as adenosine triphosphate (ATP), which in turn activate the P2X7 receptors (P2X7R) that regulate inflammation and cell death in inflammatory bowel diseases. The aim of this study was to verify the effect of C. difficile infection (CDI) in the expression of Panx1 and P2X7R in intestinal tissues of mice, as well as their role in cell death and IL-6 expression induced by TcdA and TcdB in enteric glial cells (EGCs). Male C57BL/6 mice (8 weeks of age) were infected with C. difficile VPI10463, and the control group received only vehicle per gavage. After three days post-infection (p.i.), cecum and colon samples were collected to evaluate the expression of Panx1 by immunohistochemistry. In vitro, EGCs (PK060399egfr) were challenged with TcdA or TcdB, in the presence or absence of the Panx1 inhibitor (10Panx trifluoroacetate) or P2X7R antagonist (A438079), and Panx1 and P2X7R expression, caspase-3/7 activity and phosphatidylserine binding to annexin-V, as well as IL-6 expression were assessed. CDI increased the levels of Panx1 in cecum and colon of mice compared to the control group. Panx1 inhibitor decreased caspase-3/7 activity and phosphatidylserine-annexin-V binding, but not IL-6 gene expression in TcdA and TcdB-challenged EGCs. P2X7 receptor antagonist accentually reduced caspase-3/7 activity, phosphatidylserine-annexin-V binding, and IL-6 gene expression in TcdA and TcdB-challenged EGCs. In conclusion, Panx1 is increased during CDI and plays an important role in the effects of C. difficile toxins in EGCs, participating in cell death induced by both toxins by promoting caspase-3/7 activation via P2X7R, which is also involved in IL-6 expression induced by both toxins.
Clostridioides difficile (C. difficile) is a gram-positive, spore-forming, toxin-producing, anaerobic bacillus and is one of the main causes of antibiotic use-associated nosocomial diarrhea (1, 2) exhibiting an incidence of 8.3 C. difficile infection (CDI) cases per 10000 patient-day, costing approximately US $ 4 billion per year in the US (3). The main virulence factors of C. difficile are toxin A (TcdA), toxin B (TcdB) and binary toxin (CDT) (4, 5). The intestinal disease caused by CDI can range from mild diarrhea to fulminant disease (2).
The enteric nervous system (ENS) is composed by enteric neurons and enteric glial cells (EGCs) that together control the intestinal reflex (peristalsis), secretion and inflammatory response (6). Enteric glial cells (EGCs), as an important cellular component of ENS, play a key role in regulating intestinal homeostasis, inflammatory response due its ability in interacting with other intestinal cells, as well as in responding to bacteria stimuli (7–11). Complete ablation of EGCs promoted fulminant colitis in mice (12), showing the potential role of these cells in regulating key functions in the gut. It is known that TcdB induces EGCs death via reactive oxygen species (ROS)/nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)/c-Jun N-terminal kinase (JNK) in a caspase-dependent manner but mitochondria-independent pathway, as well as senescence (13–15). In vitro, co-culture of ileum mucosal and submucosal layer challenged with TcdB presented higher IL-8 secretion than the culture of each layer alone (16). The fact that EGCs can be found on these two layers (6), suggest a potential role of these cells in the inflammatory response to C. difficile toxin.
Pannexin-1 (Panx1) is a well-characterized membrane protein that participates in cell-extracellular environment communication (17–19) and is part of an important mechanism for release of ATP (adenosine triphosphate), an endogenous P2X7 receptor (P2X7R) agonist, to the extracellular environment. P2X7R is involved in many cellular functions, such as metabolism, proliferation, migration, neurotransmitters release and cytokine synthesis (20–24). In the gut, P2X7R can be found in epithelial cells, neurons, macrophages, T cells and EGCs (25–27). Activation of P2X7R begins with the release of ATP through Panx1 channels, which are activated via Ca2+ signaling (6, 28). In high levels, ATP via P2X7 activation, promotes tissue damage by stimulating cell death (29). In a colitis murine model, activation of P2X7R in EGCs resulted in inflammatory response and cell death (28). However, it is still unknown the role of Panx1 and P2X7R in EGCs death and inflammatory response induced by C. difficile toxins.
Given that TcdA and TcdB induces cell death (13, 14) and upregulation of pro-inflammatory mediators (such as IL-6) in EGCs (30), we investigated whether Panx1/P2X7R signaling is involved in these deleterious effects.
Male C57BL/6 mice (Jackson Laboratory, Farmington, US, 8 weeks old) were housed in temperature-controlled rooms under 12 h light-dark cycles. The animals received water and food ad libitum. All surgical procedures and treatments performed with C57BL/6 mice were conducted in accordance with the Guidelines for Institutional and Animal Care and Use of the University of Virginia, Charlottesville, US. The protocol has been approved by the committee on the Ethics of Animal Experiments of the University of Virginia (Protocol number: 4096).
Our murine CDI model is broadly used due to the ability to mimic severe diarrhea presented by humans and was performed as previously described (31–35). C57BL/6 mice (n=6 for each group) received an antibiotic treatment (0.035 mg per mL gentamicin, 850 U per mL colistin, 0.215 mg per mL metronidazole, and 0.045 mg per mL vancomycin) in the drinking water for 3 days. After 1 day off antibiotics, an intraperitoneal injection of clindamycin (32 mg per kg) was given 1 day before C. difficile challenge. Then, 105 CFU (in 100 μL of Chopped meat broth, a pre-reduced medium) of the vegetative C. difficile strain VPI10463 (ATCC 43255, tcdA+tcdB+cdtB-) was given by oral gavage (Figure 1A). The inoculum was prepared as previously described (30). Control mice received chopped meat broth (100 μL). Mouse weights and the development of disease symptoms were monitored daily. The animals were euthanized three days after infection using ketamine and xylazine (180 and 15 mg/kg, i.p.).
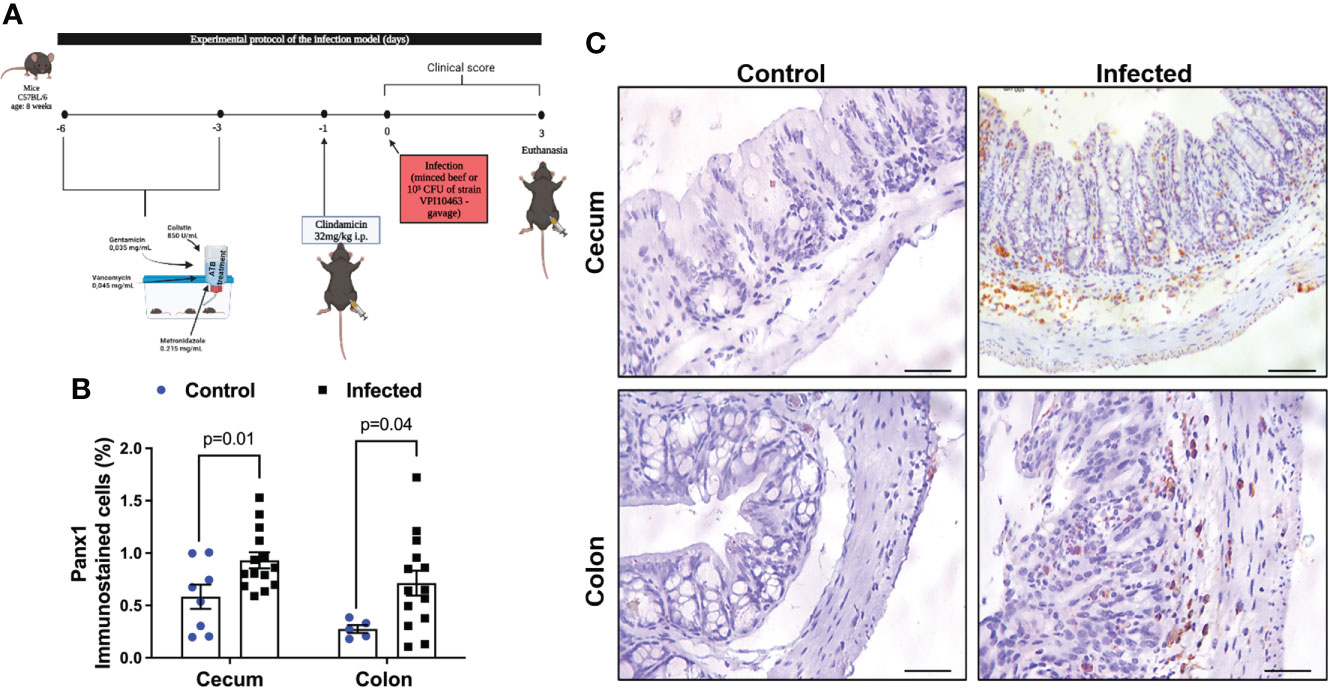
Figure 1 CDI increases Panx1 expression in mouse colon and cecum samples. (A) Experimental protocol of the infection model. (B) CDI increases Panx1 immunostaining in the cecum and colon of mice. The data are the mean ± SEM of the percentage of the immunopositive area for Panx1 in the cecum and colon of mice submitted to CDI and uninfected in relation to the total area; t test of Student; the value of p is represented in the graph. (C) immunohistochemical imaging for Panx1 in mouse colon and cecum tissues with CDI (Bars=100 μm).
Sections (4 µm thick) were prepared from paraffin-embedded mouse colon and cecum tissues. After deparaffinization, antigens were recovered by incubating the slides in citrate buffer (pH 9.0) for 20 min in PT link tank (DAKO). Endogenous peroxidase was blocked with 3% H2O2 for 30 min to reduce nonspecific binding. The sections were then incubated with an Panx-1 antibody (R&D system) for 3 hours. The sections were then incubated for 30 min with polymer (K4061, Dako). The antibody binding sites were visualized by incubating the samples with diaminobenzidine–H2O2 (DAB, Dako) solution. Sections incubated with antibody diluent without a primary antibody were used as negative controls. Immunohistochemical images were captured using a light microscope coupled to a camera with a LAZ 3.5 acquisition system (LEICA DM1000, Germany). To quantify the positive immunostaining area for Panx-1, the Adobe Photoshop 8.0 program was used to obtain the total tissue and the positive area. The percentage of immunopositive area was calculated by dividing the number of DAB-positive pixels (immunostaining-positive pixels) by the number of pixels per total tissue image and multiplying the result by 100, as previously described (36).
The immortalized rat enteric glial cell (EGC) line PK060399egfr (ATCC CRL-2690, VA, United States), which has been shown to exhibit similar morphology and functional properties to primary enteric glial cells (37), was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) and supplemented with 10% fetal bovine serum, 1% antibiotics (100 μg/mL penicillin and 100 μg/mL streptomycin, Gibco) and 1 mM sodium pyruvate (Gibco) at 37°C in a humidified incubator under 5% CO2 for no more than 16 passages. For all experiments, EGCs were released using 0.05% trypsin-EDTA for 5 min. Cells were incubated with 10 and 50 µM 10Panx trifluoroacetate (Panx1 inhibitor, Sigma-Aldrich SML2152) or 300 µM A438079 (P2X7R antagonist, Tocris 2972) 1 h before incubation with TcdA (50 ng/mL) or TcdB (1 ng/mL). All drug concentrations used were based on MTT assay results (Figure S1). Purified TcdA and TcdB (TechLab, VA, United States) produced by C. difficile strain VPI10463 were used in this study.
EGCs line (5x103 cells/well) were seeded in 96-well plates and treated with 10Panx trifluoroacetate (10, 100 and 300µM) and A438079 (1, 3, 10, 30, 100 and 300μM) for 18h. Then, the cells were incubated with thiazolyl blue tetrazolium bromide (MTT, 0.5 mg/mL reconstituted in supplemented DMEM, Sigma-Aldrich, M2128) for 2 h at 37°C in a humidified incubator under 5% CO2. After removal of the MTT solution, 150 µL of dimethylsulfoxide was added to each well. The plates were then shaken for 2 min at room temperature, and the absorbance of the reaction at 570 nm was measured using an ELISA reader.
EGCs line (6x105 cells/well) were seeded in 6-well plates and treated with TcdA or TcdB and pharmacologic modulators. After incubation, total RNA was extracted with a RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) using QIAcube (Qiagen). RNA was quantified with a Qubit 3.0 fluorometer (Life Technologies) using a Qubit RNA BR Assay Kit (Invitrogen, Q10211). After DNA contamination was removed by RNA treatment with DNase I (Invitrogen, 18068-015), a total of 600 ng of RNA was then reverse transcribed using an iScript cDNA Synthesis Kit (Bio-Rad, 1708891) according to the manufacturer’s protocol. qPCR amplification of Panx-1, IL-6 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in cell samples was performed in a CFX Connect system (Bio-Rad) with the following conditions: 95°C for 30 s, 40 cycles of 95°C for 5 s and 60°C for 30s, and melt curve analysis from 65-95°C in 0.5°C increments for 2 s each. All PCRs were performed with iTaq Universal SYBR Green Supermix (Bio-Rad, 172-5124). The primer sets are listed in Supplementary Material.
EGCs line (4x104 cells/well) plated on 8-chamber glass tissue culture slides in a polystyrene vessel and treated with TcdA or TcdB for 18 h were fixed in 4% PFA solution (Alfa Aesar) in PBS for 30 min at room temperature and permeabilized with 0.5% Triton X-100 (Sigma-Aldrich) and 3% bovine serum albumin (BSA, Sigma) in PBS for 10 min at 4°C. After blocking with 5% normal bovine serum albumin in PBS for 40 min at room temperature, the cells were incubated with anti-Panx-1 antibody (1:100, Invitrogen, 488100), anti-P2X7 antibody (1:50, Millipore, AB5246), anti-IL-6 (1:20, R&D system, AF506) or phosphorylated NFκB (1:100, Thermo Scientific, PA537718) overnight at 4°C. After three washes with washing buffer (0.01% Tween 20 in PBS), the cells were incubated for 2h with secondary antibody conjugated with Alexa Fluor 488 or 594 (1:400, Invitrogen, A21206/abcam, ab150129/abcam, ab150064) at room temperature, washed with PBS and mounted with ProLong Gold antifade reagent containing DAPI (Thermo Scientific, P36931). The samples were visualized by fluorescence microscopy (Zeiss).
Caspase 3/7 activity was measured by using a Caspase-Glo assay kit (Promega, G8091) following the manufacturer’s instructions. EGCs (104 cells/well) seeded in a white tissue culture-treated 96-well plates (Falcon, solid white bottom) were treated with TcdA or TcdB alone or in the presence with 10Panx (50µM) or A438079 (300µM) for 18 h. Then, the plates containing the cells were removed from the incubator for 30 min. A volume of 100 µL of Caspase-Glo reagent was added to each well, and the wells were mixed with a plate shaker at 500 rpm for 30 s. The plates were incubated for 2 h at room temperature. Then, the luminescence of each sample was acquired in a plate-reading luminometer (Promega) to obtain the relative luminescent units (RLUs) subtracted by the background.
Apoptosis was evaluated with a live cell real-time assay (RealTime-Glo annexin V apoptosis assay, Promega, JA1000), following the manufacturer’s instructions. EGCs (104 cells/well) were treated with TcdA or TcdB alone or in the presence of 10Panx (50 µM) or A438079 (300 µM) 1h prior to toxin challenge. Then, 200 µL of 2x detection reagent was added to each well, and the cells were incubated at 37°C in a humidified incubator under 5% CO2. The luminescence was recorded using a luminometer (NanoLuc technology ready, Promega), and the intrinsic reagent luminescence (no-cell, no-compound background control) was subtracted from the luminescence signals in the sample wells to obtain the relative luminescent units (RLUs).
Level of extracellular ATP was measured with a live cell real-time assay (RealTime-Glo extracellular ATP assay, Promega, GA5010), following the manufacturer’s instructions. EGCs (104 cells/well) were treated with TcdA or TcdB alone or in the presence of 10Panx (50 µM) or A438079 (300 µM) 1h prior to toxin challenge. Then, 50 µL of 4x RealTime-Glo extracellular ATP assay reagent was added to each well, and the cells were incubated at 37°C in a humidified incubator under 5% CO2. The luminescence was recorded using a luminometer (NanoLuc technology ready, Promega), and the intrinsic reagent luminescence (no-cell, no-compound background control) was subtracted from the luminescence signals in the sample wells to obtain the relative luminescent units (RLUs) and the data was normalized by the control cells to obtain the fold change.
EGC lines (6×105 cells/well) were seeded in six-well plates and treated with TcdA or TcdB in the presence or absence of P2X7R antagonist (A438079) 1h prior to toxin challenge. After incubation, the supernatant was removed and the cells were lysed using RIPA lysis buffer (Thermo Fisher Scientific, containing EDTA and phosphatase-free protease inhibitor), centrifuged (17 min, 4°C, 13000 rpm) and the supernatant was collected. Protein concentrations were determined through the bicinchoninic acid assay according to the manufacturer’s protocol (Thermo Fisher Scientific). 40 µg of protein (previously prepared with Laemmil sample buffer and β-mercaptoethanol) were denatured at 95°C for 5 min, separated on 10% BIS-Tris gel and transferred to PVDF membranes for 2 h. After blocking with 5% blocking solution (BioRad) at room temperature for 1 h, the membranes were incubated overnight with the primary antibodies (anti-β-actin 1:500, Millipore EP1123Y and cleaved caspase-3 1:1000, Sigma-Aldrich PC679) at 4°C and secondary antibodies (anti-mouse 1:500 and anti-rabbit 1:500) for 1 h and 30 min. Membranes were washed in Tris-buffered saline containing 0.05% Tween 20 (TSB-T) and incubated with Enhanced Chemiluminescence – ECL (Biorad 1705060). The chemiluminescence signal was detected using a ChemiDoc system (BioRad). Densitometric quantification of bands was performed using ImageLab software (BioRad).
Analyses were performed using GraphPad software 9.0 (San Diego, CA, USA). The data are presented as the mean ± standard error of the mean (SEM). Student’s t test or one- or two-way analysis of variance (ANOVA) followed by the Tukey test was used to compare means. P< 0.05 was considered to indicate significance.
Cecum and colon are the intestinal segments more affected by CDI in mice and in human (38–40). To investigate if levels of Panx1 in the intestine was affected by CDI, we performed an immunohistochemistry analysis. Using a CDI pre-clinical model (Figure 1A), we found increased positive immunostaining for Panx1 in the cecum (p = 0.01, Figure 1B) and in the colon (p = 0.04, Figure 1B) of infected mice compared to the control group (Figure 1B). Cecum and colon samples from mice with CDI showed notable increased immunostaining for Panx1 in the lamina propria and submucosa layer compared to the control group, together with epithelial cell disruption, inflammatory cell infiltrate, and submucosal edema (Figure 1C).
Next, we evaluated whether C. difficile toxins induce alteration on the gene expression of Panx1 in EGCs. We found that TcdA and TcdB upregulated the gene expression of Panx1 in EGCs at 18h incubation compared to the control cells (p = 0.03 TcdA; p<0.0001 TcdB, Figure 2A). However, TcdB, but not TcdA, reduced the gene expression of Panx1 at 12h incubation compared to the control group (p = 0.01, Figure 2A). The immunofluorescence data also showed increased positive immunostaining for Panx1 in EGCs incubated with TcdA and TcdB at 18h (Figures 2B, C). Due to the higher deleterious effects of C. difficile toxins occurred on the later time point, we focused on 18h incubation time, as shown by others (13, 14, 30).
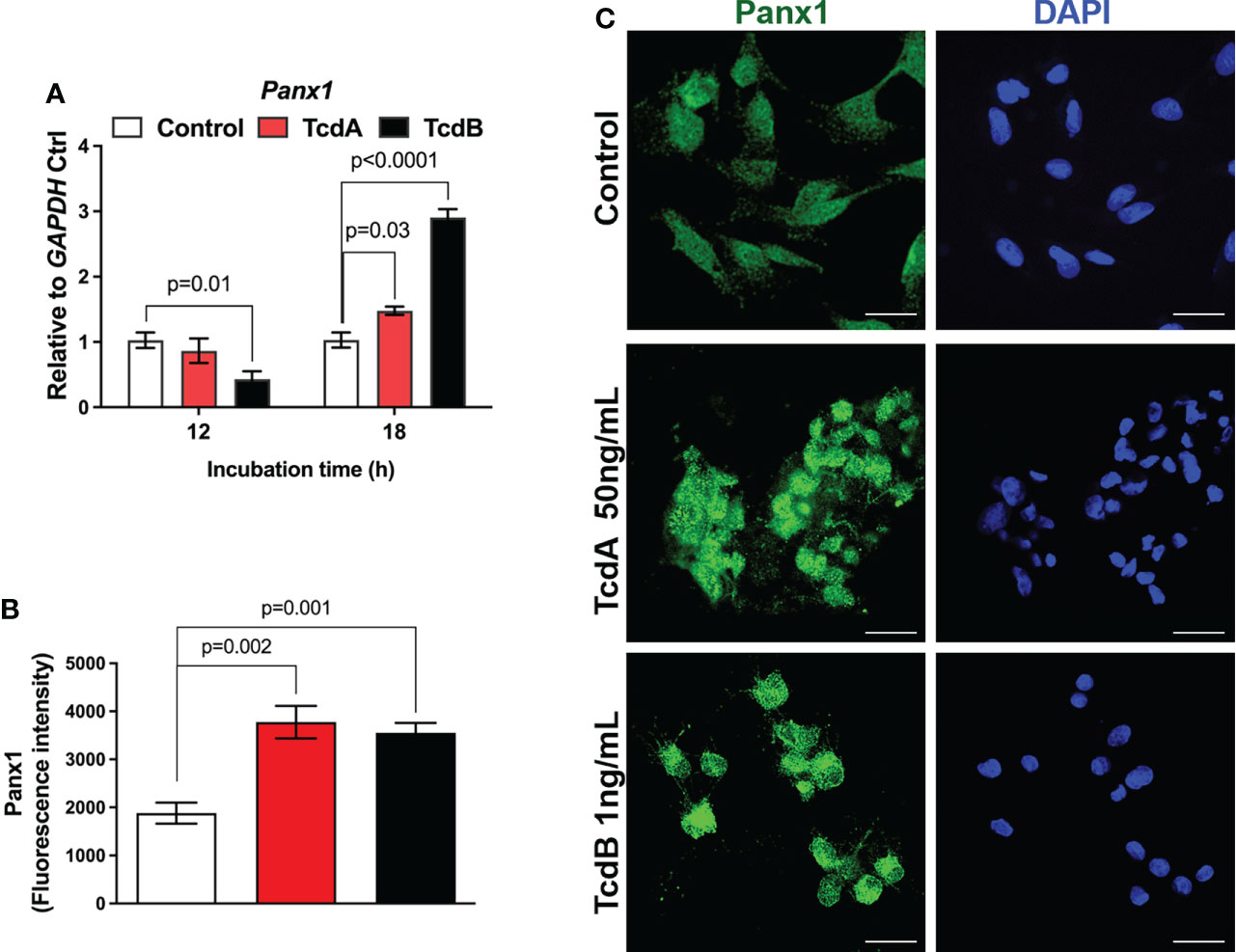
Figure 2 TcdA and TcdB alter Panx1 gene expression in EGCs in vitro. (A) Gene expression of Panx1 were evaluated by qPCR in EGC incubated with DMEM only (control), TcdA (50 ng/mL) and TcdB (1 ng/mL). The data are the mean ± SEM. p value is represented in the graph; the one-way ANOVA test followed by the Tukey test was used. (B) Fluorescence intensity of Panx1 immunostaining in EGCs measured by ImageJ software. The data are the mean ± SEM. One-way ANOVA followed by the Tukey test was used. p value is represented in the graph. (C) Representative photomicrographs of Panx1 (green) immunostaining and DAPI (blue) nuclear staining in EGCs exposed to TcdA and TcdB after 18 h of incubation.
To assess whether Panx1 participate on cell death and IL-6 expression induced by C. difficile toxins in EGCs, we used a pharmacological approach (10Panx) to inhibit Panx1 before challenge EGCs with TcdA or TcdB. Inhibition of Panx1 (50 µM 10Panx) decreased phosphatidylserine-annexin V binding (p = 0.01 TcdA; p = 0.001 TcdB, Figure 3A) and the levels of caspase 3/7 activity (p=0.02 TcdA; p = 0.002 TcdB, Figure 3B), markers of cell death, induced by TcdA and TcdB in EGCs. The Panx1 inhibitor (50 µM 10Panx) did not prevent TcdA and TcdB-induced IL-6 upregulation in EGCs (Figure 3C).
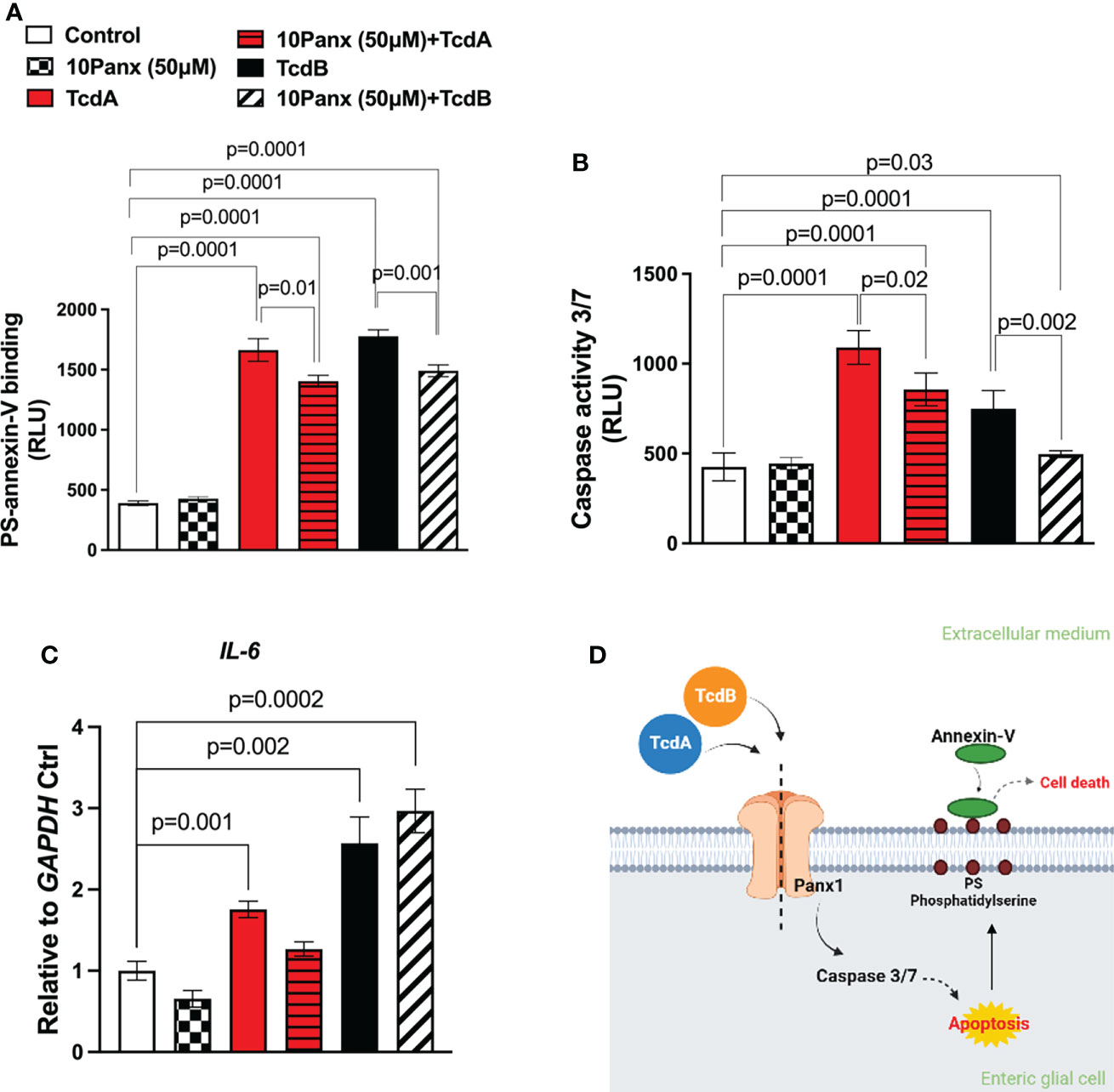
Figure 3 Panx1 inhibitor on caspase 3/7 activity and annexin V phosphatidylserine binding in EGCs challenged by TcdA and TcdB in vitro. (A) Cell death was analyzed by RealTime-Glo annexin V apoptosis assay in EGCs incubated for 18 h with TcdA, TcdB and 10Panx trifluoroacetate (10Panx; 50µM), a Panx1 antagonist, one hour prior to toxin challenge. For statistical analysis, the one-way ANOVA test was used followed by the Tukey test; the p value is represented in the graph. (B) The activity of caspase 3/7 was analyzed by luminescence assay in EGCs incubated for 18 h with TcdA, TcdB and 10Panx (50µM), one hour prior to toxin challenge. (C) IL-6 gene expression was evaluated by qPCR in EGCs challenged by TcdA and TcdB for 18h, previously incubated or not with Panx1 inhibitor (10Panx, 50 μM). The data are the mean ± SEM. For statistical analysis, the one-way ANOVA test was used followed by the Tukey test; the p value is represented in the graph. (D) Proposed model of the role of Panx1 signaling in TcdA and TcdB-induced cell death in EGCs. TcdA and TcdB activate Panx1 in EGCs. Its activation results in the activation of caspase 3/7 and phosphatidylserine expose related to cell death.
Taken together, these data demonstrated that Panx1 is involved in TcdA and TcdB-induced EGC death but not IL-6 expression (Figure 3D).
Once activated, Panx1 releases ATP, which in turn promotes P2X7R activation (41, 42). As shown in our Supplementary Data, inhibition of Panx1, in fact, decreased the levels of extracellular ATP in EGCs challenged by C. difficile toxins (Figure S2A). Next, we investigated if C. difficile toxins affected the expression of P2X7R. Our qPCR data showed that TcdA (p = 0.005) and TcdB (p<0.0001) upregulated P2X7R at 12h, which persisted at 18h incubation (p<0.0001) compared to control cells (Figure 4A). Confirming these findings, increased fluorescence intensity for P2X7R was found in EGCs challenged with TcdA and TcdB (p = 0.04 TcdA; p = 0.0004 TcdB, Figures 4B, C).
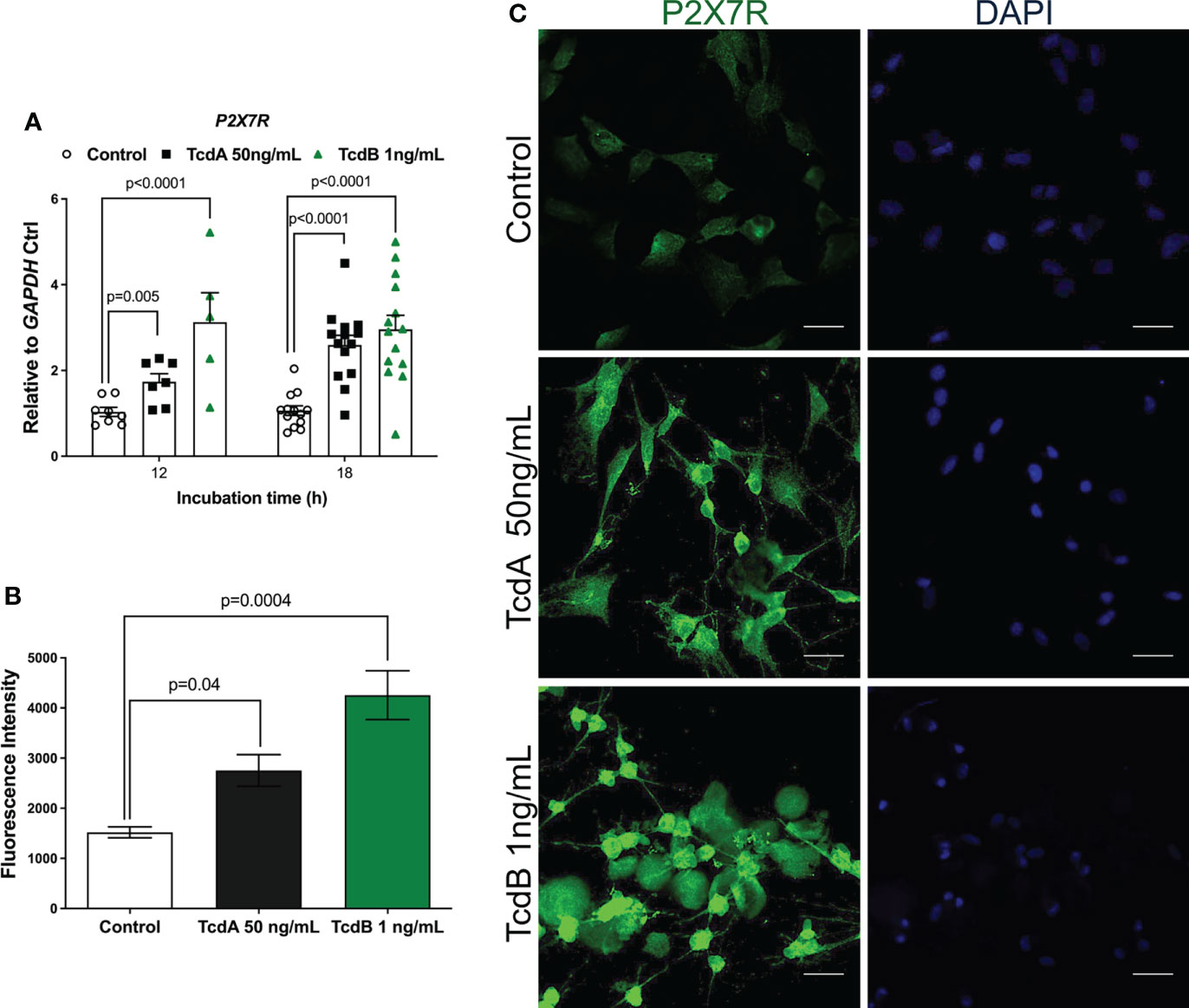
Figure 4 TcdA and TcdB upregulate P2X7R expression in EGCs. (A) Gene expression of P2X7R were evaluated by qPCR in EGCs incubated with DMEM only (control), TcdA (50 ng/mL) and TcdB (1 ng/mL). The data are the mean ± SEM. Two-way ANOVA followed by the Tukey test was used. (B) Fluorescence intensity of P2X7R immunostaining in EGCs measured by ImageJ software. The data are the mean ± SEM. One-way ANOVA followed by the Tukey test was used. p value is represented in the graph (C) Representative photomicrographs of P2X7R (green) immunostaining and DAPI (blue) nuclear staining in EGCs exposed to TcdA and TcdB after 18 h of incubation.
To determine whether P2X7R participated on the deleterious effects induced by TcdA and TcdB in EGCs, we inhibited this receptor using a selective antagonist (300 µM A438079) before challenging EGCs with the toxins. In fact, A438079 did not decrease the levels of extracellular ATP in EGCs challenged by C. difficile toxins (Figure S2B). Blockage of P2X7R markedly decreased the caspase 3/7 activity (p<0.0001, Figure 5A), as well as the levels of protein expression of cleaved caspase-3 (Figures 5B–D), in EGCs challenged by TcdA (p = 0.003) and TcdB (p<0.05). A438079 (P2X7R antagonist) also prevented cell death analyzed by phosphatidylserine-annexin V binding (p<0.0001, Figure 5E).
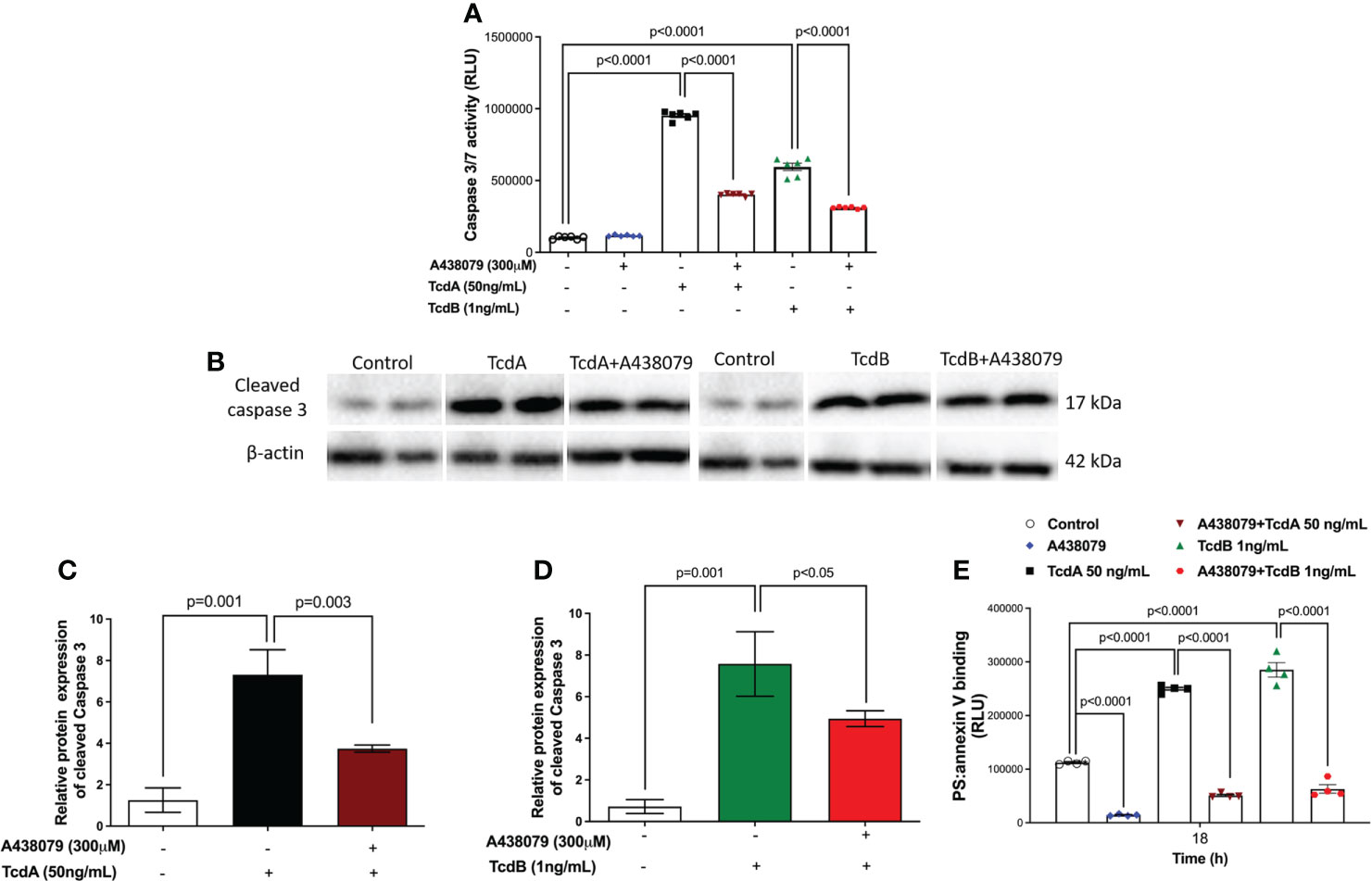
Figure 5 A438079, a P2X7R antagonist, prevents TcdA- and TcdB-induced EGCs death. (A) Activity of caspase 3/7 analyzed by luminescence assay in EGCs incubated for 18 h with TcdA, TcdB by qPCR. A438079, a P2X7R antagonist, was added to EGCs one hour prior to toxin challenge. (B) Representative Western Blot images showing the prevention of TcdA and TcdB induced cleavage of caspase 3 protein expression by A438079, a P2X7R antagonist. β-actin was used as a loading control. The WB images for the control group are the same for both toxins. (C, D) Graphs represents cleaved relative protein expression of caspase 3. The data are the mean ± SEM. One-way ANOVA followed by the Tukey test was used. (E) A438079 (P2X7R antagonist) prevented cell death analyzed by RealTime-Glo annexin V apoptosis assay in EGCs. The data are the mean ± SEM. Two-way ANOVA followed by Tukey test was used. p value is represented in the graph.
When we evaluated whether P2X7R participates on upregulation of IL-6 induced by TcdA and TcdB on EGCs, we found that the inhibition of this receptor decreased IL-6 gene expression induced by both toxins in the enteric glia (p<0.03 TcdA, p<0.007 TcdB, Figure 6A). Our protein analysis of IL-6 also showed similar findings (p<0.0001 TcdA, p=0.0003 TcdB, Figures S3, S4).
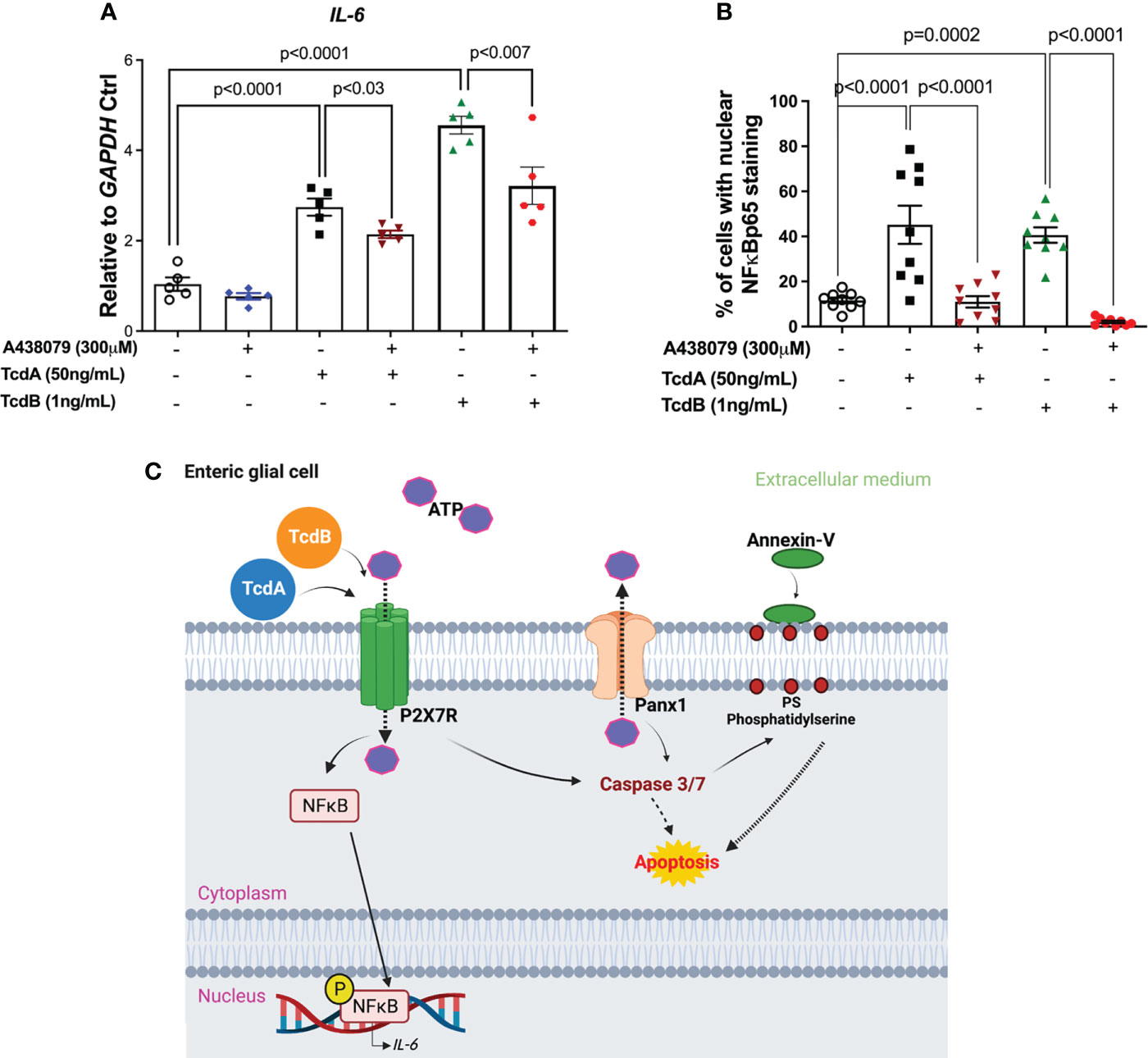
Figure 6 A438079, a P2X7R antagonist, inhibits TcdA- and TcdB-induced IL-6 upregulation in EGCs. (A) IL-6 mRNA expression in EGCs incubated for 18h with TcdA, TcdB and A438079 (300µM) 1h prior to toxin challenge, as analyzed by qPCR. The data are the mean ± SEM.; one-way ANOVA followed by the Tukey test was used. (B) Percentage of cells with positive nuclear phosphorylated NFκBp65 staining. The data are the mean ± SEM. p value is represented in the graph. (C) Proposed model of the role of Panx1/P2X7R signaling in TcdA- and TcdB-induced IL-6 expression and in EGC death. TcdA and TcdB activate Panx1 in EGCs, promoting the release of ATP, which in turn activates P2X7R. Its activation results on nuclear translocation of NFκB and IL-6 expression, as well as caspase 3/7 activation that promotes cell death.
Finally, when we analyzed whether P2X7R blockage decreased the cells with nuclear phosphorylated NFκB, we found that A438079 reduced the percentage of EGCs with positive nuclear NFκBp65 staining induced by TcdA and TcdB (p<0.0001, Figures 6B, S4).
Thus, these data together indicated that P2X7R is involved in EGCs death, as well as on upregulation of IL-6 induced by both C. difficile toxins.
In the present study, high expression of Panx1 was found in colon and cecum samples from animals infected with C. difficile. The cecum, in fact, is the region of the intestine most affected by CDI (30, 38–40), with extensive rupture of the intestinal epithelium, hemorrhage, edema and recruitment of inflammatory cells. Increased Panx1 has been reported in several conditions, such as cerebral ischemia (43, 44), in metastatic cell lines of patients with breast cancer (45), in Crohn’s disease and in ulcerative colitis (9, 46).
We detected an increase in Panx1 protein levels on intestinal mucosa and submucosa layer. Given that EGCs is one of the ENS component that can be found on these two layers (6), we hypothesized that EGCs expressing Panx1 could have a role in CDI pathogenesis. Enteric glia is an intriguing population of cells that act modulating intestinal physiological processes such as motility, secretion, and maintenance of barrier function (9, 47). The importance of EGC on CDI has been demonstrated previously. Von Boyen et al. (48) showed that Glial fibrillary acidic protein (GFAP), an enteric glial marker is increased in colonic tissue from patients with CDI. Study from our group reported that S100B expression is increased in colonic biopsies and on fecal samples from patients with CDI, and in colon tissues from C. difficile-infected mice (30). We also demonstrated that S100B signaling is involved in EGC inflammatory response and that its inhibition attenuates the intestinal injury and diarrhea caused by C. difficile toxins (30).
The Panx1 channel establishes a communication between the cytosol and extracellular environment (17, 49). Furthermore, it is essential for the release of signaling molecules such as ATP (50–52). By detecting cellular danger signals, such as ATP, EGCs can be activated releasing other mediators that induce neuronal death and inflammatory response (47).
Here, the activation of Panx1 led to EGCs death induced by TcdA and TcdB. In an experimental colitis model, Panx1 was associated with enteric neurons death via caspase activation (9, 46) as also shown here. Cleaved caspase 3/7 target multiple cytoskeletal proteins, including key components of actin filaments, intermediate filaments, and microtubules (53). Both Caspase 3 and 7 are considered executor caspases, which are activated by initiator caspases arising from intrinsic or extrinsic apoptosis (54). It has been demonstrated that TcdA induces apoptosis in human epithelial cell line (T84) through intrinsic and extrinsic pathway via activation of initiator caspases 8 and 9 followed by activation of executor caspases 3 and 6, and bid cleavage (55). TcdB-promoted cell death has also been shown to occur via caspase 3 activation (13, 15, 56). The catalytic activity of executor caspases promotes cell death as they are responsible for a variety of morphological and biochemical changes that favor this process, such as DNA fragmentation, phosphatidylserine exposure and formation of apoptotic bodies (54, 57–59). In the present work, we demonstrated that Panx1 is involved in caspase 3/7 activation and in phosphatidylserine (PS) exposure. Caspase-3-mediated phosphatidylserine (PS) exposure through the activation of proteins related to its externalization, such as scrambling phospholipids, or inactivation of factors that promote its internalization, such as phospholipid flipases (60–62). Unlike caspase 3, which is the main caspase activated in the apoptosis process, caspase 7 is responsible for disintegrating cells from their extracellular component and their adhesion to other cells, as demonstrated in an in vitro study using embryogenic fibroblasts (63). Therefore, our data agree with previous demonstration that the opening of Panx1 channels can lead to cell death via release of ATP, which results in activation of purinergic receptors and increased intracellular calcium flow that stimulate the activation of initiator and effector caspases culminating in the apoptotic process (44, 49, 51, 54, 64).
Accordingly, we identified that both TcdA and TcdB stimulated the expression of P2X7R in EGCs. P2X7R has been extensively investigated due to its upregulation in a variety of diseases, such as Crohn’s disease, ulcerative colitis, systemic lupus erythematosus, depression and Alzheimer’s disease (65–69).
Interestingly, the P2X7R antagonist (A438079) decreased the EGCs death, as demonstrated here by reduced PS and annexin V binding and caspase 3/7 activity, showing the potential role of this receptor in promotes EGCs death induced by C. difficile toxins. The involvement of P2X7R in cell death has also been shown in other cells such as podocytes (70), macrophages (71), cerebellar astrocytes (72) and in neurons (73, 74). Mitogen-activated protein kinase (MAPKs) such as extracellular signal-regulated kinase (ERK) 1/2 and JNK have also been associated with cell death mediated by this receptor. In primary cortical neurons, P2X7R agonists mediate nuclear condensation and DNA fragmentation by expressing a functioning P2X7R and activating ERK1/2 and JNK1 in a manner dependent on caspase 8/9/3 activation (74). This could be one of the mechanisms of caspase 3 activation in EGCs challenged with TcdA and TcdB. In addition, a previous study showed that TcdB-induced EGCs apoptosis via NADPH oxidase/ROS/JNK/caspase-3 (15). Unlike this study that identified the intracellular signaling pathway involved in TcdB-induced EGCs death, here we showed a membrane receptor, P2X7R, involved in this process not only induced by TcdB, but also by TcdA.
However, further investigations are needed to verify the involvement of this receptor in CDI clinical outcomes, having in mind that drugs that block membrane receptors may be potentially more tolerable with fewer side effects than drugs that block components of the intracellular pathways, since they may participate in other processes important to the physiological functions of the cell.
Here we also show that Panx1 did not participate in IL-6 upregulation induced by TcdA and TcdB. IL-6 can be synthesized by a variety of cells, including EGCs (30, 37, 75). IL-6 is a pleiotropic cytokine considered a predictor of CDI severity (76). The effects of IL-6 vary between promoting cell survival and pro-inflammatory effect (77–79). Consistent with our data, it was shown that Panx1 is not required for inflammasome activation, but participates in cell apoptosis (80, 81)
In contrast, P2X7R has been shown to regulate the expression and secretion of several cytokines and inflammatory mediators, including IL-2, IL-4, IL-13, IL-18, TNF-α, NO and IL-6 (82–86). Here, the P2X7R antagonist reduced the expression of IL-6 induced by TcdA and TcdB. These data suggest the important role of this receptor in regulating the expression of this cytokine. In agreement with our findings, P2X7R has indeed been implicated in the regulation of IL-6 in various cell types including fibroblasts (87), skeletal muscle cells (88), macrophages (89) and microglia (90).
Our study is the first to show the involvement of Panx1/P2X7R signaling in cell death of EGCs challenged by TcdA and TcdB. TcdA and TcdB activate Panx1, releasing ATP, which in turn activates P2X7R leading to activation of caspase-3/7, culminating in exposure of PS in the plasma membrane, which characterizes EGC in process of cell death (Figure 6C). Further, activation of P2X7R promotes IL-6 expression induced by C. difficile toxins. Further research on exploring Panx1/P2X7R signaling using pre-clinical model of CDI are needed to better understating the participation of these molecules in the pathogenesis of this disease.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Committee on the Ethics of Animal Experiments of the University of Virginia (Protocol number: 4096).
AL, LM-N, DC, and GB wrote sections of the manuscript. AL and LM-N performed all the experiments and analyzed the data. CM, PS, ML, RL, JC-A, VM-N, and CW helped in the acquisition of data. AL, DC, and GB contributed to the organization of the manuscript, reviewed and approved the final version. All authors had full access, revised and approved the manuscript for publication.
This study was funded by PRONEX CNPq/FUNCAP grant number PR2-0101-00060.01.00/15.
The authors would like to thank Flávia A. Silva and Rosemayre Freire (analytics central/Federal University of Ceara) for their technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.956340/full#supplementary-material
1. Abt MC, McKenney PT, Pamer EG. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol (2016) 14(10):609–20. doi: 10.1038/nrmicro.2016.108
2. Leffler DA, Lamont JT. Clostridium difficile infection. New Engl J Med (2015) 372(16):1539–48. doi: 10.1056/NEJMra1403772
3. Marra AR, Perencevich EN, Nelson RE, Samore M, Khader K, Chiang HY, et al. Incidence and outcomes associated with clostridium difficile infections. JAMA Network Open (2020) 3(1):e1917597. doi: 10.1001/jamanetworkopen.2021.41089
4. Rustandi RR, Hamm M. Development of an ADP-ribosylation assay for residual toxicity in c. difficile binary toxin CDTa using automated capillary western blot. J Pharm Biomed Analysis (2020) 182:113125. doi: 10.1016/j.jpba.2020.113125
5. del Prete R, Ronga L, Addati G, Magrone R, Abbasciano A, Decimo M, et al. Clostridium difficile. a review on an emerging infection. Clin Ter (2019) 170(1):e41–7. doi: 10.7417/CT.2019.2106
6. Seguella L, Gulbransen BD. Enteric glial biology, intercellular signalling and roles in gastrointestinal disease. Nat Rev Gastroenterol Hepatol (2021) 18(8):571–87. doi: 10.1038/s41575-021-00423-7
7. Yu YB, Li YQ. Enteric glial cells and their role in the intestinal epithelial barrier. World J Gastroenterol (2014) 20(32):11273–80. doi: 10.3748/wjg.v20.i32.11273
8. Gulbransen BD. Colloquium neuroglia in biology and medicine: From physiology to disease. In: Gulbransen B, editor. Enteric glia, 2nd ed. TX, United States: Morgan & Claypool Life Sciences (2014). p. 1–70.
9. Gulbransen BD, Sharkey KA. Novel functional roles for enteric glia in the gastrointestinal tract. Nat Rev Gastroenterol Hepatol (2012) 9(11):625–32. doi: 10.1038/nrgastro.2012.138
10. Cirillo C. S100B protein in the gut: The evidence for enteroglial-sustained intestinal inflammation. World J Gastroenterol (2011) 17(10):1261. doi: 10.3748/wjg.v17.i10.1261
11. Bassotti G. The role of glial cells and apoptosis of enteric neurones in the neuropathology of intractable slow transit constipation. Gut (2006) 55(1):41–6. doi: 10.1136/gut.2005.073197
12. Bush TG, Savidge TC, Freeman TC, Cox HJ, Campbell EA, Mucke L, et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell (1998) 93(2):189–201. doi: 10.1016/S0092-8674(00)81571-8
13. Fettucciari K, Ponsini P, Gioè D, Macchioni L, Palumbo C, Antonelli E, et al. Enteric glial cells are susceptible to clostridium difficile toxin b. Cell Mol Life Sci (2017) 74(8):1527–51. doi: 10.1007/s00018-016-2426-4
14. Fettucciari K, Macchioni L, Davidescu M, Scarpelli P, Palumbo C, Corazzi L, et al. Clostridium difficile toxin b induces senescence in enteric glial cells: A potential new mechanism of clostridium difficile pathogenesis. Biochim Biophys Acta (BBA) - Mol Cell Res (2018) 1865(12):1945–58. doi: 10.1016/j.bbamcr.2018.10.007
15. Macchioni L, Davidescu M, Fettucciari K, Petricciuolo M, Gatticchi L, Gioè D, et al. Enteric glial cells counteract clostridium difficile toxin b through a NADPH oxidase/ROS/JNK/caspase-3 axis, without involving mitochondrial pathways. Sci Rep (2017) 7(1):45569. doi: 10.1038/srep45569
16. Tixier E, Lalanne F, Just I, Galmiche JP, Neunlist M. Human mucosa/submucosa interactions during intestinal inflammation: involvement of the enteric nervous system in interleukin-8 secretion. Cell Microbiol (2005) 7(12):1798–810. doi: 10.1111/j.1462-5822.2005.00596.x
17. Jalaleddine N, El-Hajjar L, Dakik H, Shaito A, Saliba J, Safi R, et al. Pannexin1 is associated with enhanced epithelial-To-Mesenchymal transition in human patient breast cancer tissues and in breast cancer cell lines. Cancers (Basel) (2019) 11(12)1–22. doi: 10.3390/cancers11121967
18. Makarenkova HP, Shah SB, Shestopalov VI. The two faces of pannexins: new roles in inflammation and repair. J Inflammation Res (2018) 11:273–88. doi: 10.2147/JIR.S128401
19. Penuela S, Gehi R, Laird DW. The biochemistry and function of pannexin channels. Biochim Biophys Acta (2013) 1828(1):15–22. doi: 10.1016/j.bbamem.2012.01.017
20. di Virgilio F, Schmalzing G, Markwardt F. The elusive P2X7 macropore. Trends Cell Biol (2018) 28(5):392–404. doi: 10.1016/j.tcb.2018.01.005
21. Arguin G, Bourzac JF, Placet M, Molle CM, Paquette M, Beaudoin JF, et al. The loss of P2X7 receptor expression leads to increase intestinal glucose transit and hepatic steatosis. Sci Rep (2017) 7(1):12917. doi: 10.1038/s41598-017-13300-8
22. di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity (2017) 47(1):15–31. doi: 10.1016/j.immuni.2017.06.020
23. Miras-Portugal MT, Sebastián-Serrano Á, de Diego García L, Díaz-Hernández M. Neuronal P2X7 receptor: Involvement in neuronal physiology and pathology. J Neurosci (2017) 37(30):7063–72. doi: 10.1523/JNEUROSCI.3104-16.2017
24. Giannuzzo A, Pedersen SF, Novak I. The P2X7 receptor regulates cell survival, migration and invasion of pancreatic ductal adenocarcinoma cells. Mol Cancer (2015) 14(1):203. doi: 10.1186/s12943-015-0472-4
25. da Silva MV, Marosti AR, Mendes CE, Palombit K, Castelucci P. Submucosal neurons and enteric glial cells expressing the P2X7 receptor in rat experimental colitis. Acta Histochem (2017) 119(5):481–94. doi: 10.1016/j.acthis.2017.05.001
26. McClain JL, Fried DE, Gulbransen BD. Agonist-evoked Ca2+ signaling in enteric glia drives neural programs that regulate intestinal motility in mice. Cell Mol Gastroenterol Hepatol (2015) 1(6):631–45. doi: 10.1016/j.jcmgh.2015.08.004
27. Li CKF, Bowers K, Pathamakanthan S, Gray T, Lawson M, Fagura M, et al. Expression and functions of purinergic receptor P2X7 in colonic macrophages and T lymphocytes from normal and inflammatory bowel disease mucosa. Gastroenterology (2001) 120(5):A522. doi: 10.3389/fimmu.2020.01882
28. Brown IAM, McClain JL, Watson RE, Patel BA, Gulbransen BD. Enteric glia mediate neuron death in colitis through purinergic pathways that require connexin-43 and nitric oxide. Cell Mol Gastroenterol Hepatol (2016) 2(1):77–91. doi: 10.1016/j.jcmgh.2015.08.007
29. Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol (2006) 176(7):3877–83. doi: 10.4049/jimmunol.176.7.3877
30. Costa DVS, Moura-Neto V, Bolick DT, Guerrant RL, Fawad JA, Shin JH, et al. S100B inhibition attenuates intestinal damage and diarrhea severity during clostridioides difficile infection by modulating inflammatory response. Front Cell Infect Microbiol (2021) 11:11. doi: 10.3389/fcimb.2021.739874
31. Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, et al. A mouse model of clostridium difficile–associated disease. Gastroenterology (2008) 135(6):1984–92. doi: 10.1053/j.gastro.2008.09.002
32. Moore JH, Pinheiro CCD, Zaenker EI, Bolick DT, Kolling GL, van Opstal E, et al. Defined nutrient diets alter susceptibility to clostridium difficile associated disease in a murine model. PloS One (2015) 10(7):e0131829. doi: 10.1371/journal.pone.0131829
33. Shin JH, Gao Y, Moore JH, Bolick DT, Kolling GL, Wu M, et al. Innate immune response and outcome of clostridium difficile infection are dependent on fecal bacterial composition in the aged host. J Infect Dis (2018) 217(2):188–97. doi: 10.1093/infdis/jix414
34. Broecker F, Wegner E, Seco BMS, Kaplonek P, Bräutigam M, Ensser A, et al. Synthetic oligosaccharide-based vaccines protect mice from clostridioides difficile infections. ACS Chem Biol (2019) 14(12):2720–8. doi: 10.1021/acschembio.9b00642
35. Schmidt CJ, Wenndorf K, Ebbers M, Volzke J, Müller M, Strübing J, et al. Infection with clostridioides difficile attenuated collagen-induced arthritis in mice and involved mesenteric treg and Th2 polarization. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.571049
36. Costa DVS, Bon-Frauches AC, Silva AMHP, Lima-Júnior RCP, Martins CS, Leitão RFC, et al. 5-fluorouracil induces enteric neuron death and glial activation during intestinal mucositis via a S100B-RAGE-NFκB-Dependent pathway. Sci Rep (2019) 9(1):665. doi: 10.1038/s41598-018-36878-z
37. Ruhl A, Trotter J, Stremmel W. Isolation of enteric glia and establishment of transformed enteroglial cell lines from the myenteric plexus of adult rat. Neurogastroenterol Motility (2001) 13(1):95–106. doi: 10.1046/j.1365-2982.2001.00246.x
38. Chen YS, Chen IB, Pham G, Shao TY, Bangar H, Way SS, et al. IL-17-producing γδ T cells protect against clostridium difficile infection. J Clin Invest (2020) 130(5):2377–90. doi: 10.1172/JCI127242
39. Cheng S, Zhu L, Faden HS. Interactions of bile acids and the gut microbiota: learning from the differences in clostridium difficile infection between children and adults. Physiol Genomics (2019) 51(6):218–23. doi: 10.1152/physiolgenomics.00034.2019
40. Jenior ML, Leslie JL, Young VB, Schloss PD. Clostridium difficile alters the structure and metabolism of distinct cecal microbiomes during initial infection to promote sustained colonization. mSphere (2018) 3(3):1–15. doi: 10.1128/mSphere.00261-18
41. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity (2015) 43(5):923–32. doi: 10.1016/j.immuni.2015.10.009
42. Aguirre A, Shoji KF, Sáez JC, Henríquez M, Quest AFG. FasL-triggered death of jurkat cells requires caspase 8-induced, ATP-dependent cross-talk between fas and the purinergic receptor P2X(7). J Cell Physiol (2013) 228(2):485–93. doi: 10.1002/jcp.24159
43. Bargiotas P, Krenz A, Hormuzdi SG, Ridder DA, Herb A, Barakat W, et al. Pannexins in ischemia-induced neurodegeneration. Proc Natl Acad Sci (2011) 108(51):20772–7. doi: 10.1073/pnas.1018262108
44. Cisneros-Mejorado A, Pérez-Samartín A, Gottlieb M, Matute C. ATP signaling in brain: Release, excitotoxicity and potential therapeutic targets. Cell Mol Neurobiol (2015) 35(1):1–6. doi: 10.1007/s10571-014-0092-3
45. Furlow PW, Zhang S, Soong TD, Halberg N, Goodarzi H, Mangrum C, et al. Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat Cell Biol (2015) 17(7):943–52. doi: 10.1038/ncb3194
46. Diezmos EF, Sandow SL, Markus I, Shevy Perera D, Lubowski DZ, King DW, et al. Expression and localization of pannexin-1 hemichannels in human colon in health and disease. Neurogastroenterol Motility (2013) 25(6):e395–405. doi: 10.1111/nmo.12130
47. Gulbransen BD, Christofi FL. Are we close to targeting enteric glia in gastrointestinal diseases and motility disorders? Gastroenterology (2018) 155(2):245–51. doi: 10.1053/j.gastro.2018.06.050
48. von Boyen GB, Schulte N, Pflüger C, Spaniol U, Hartmann C, Steinkamp M. Distribution of enteric glia and GDNF during gut inflammation. BMC Gastroenterol (2011) 11(1):3. doi: 10.1186/1471-230X-11-3
49. Crespo Yanguas S, Willebrords J, Johnstone SR, Maes M, Decrock E, de Bock M, et al. Pannexin1 as mediator of inflammation and cell death. Biochim Biophys Acta (BBA) - Mol Cell Res (2017) 1864(1):51–61. doi: 10.1016/j.bbamcr.2016.10.006
50. Bennett MVL, Garré JM, Orellana JA, Bukauskas FF, Nedergaard M, Giaume C, et al. Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res (2012) 1487:3–15. doi: 10.1016/j.brainres.2012.08.042
51. Kameritsch P, Pogoda K. The role of connexin 43 and pannexin 1 during acute inflammation. Front Physiol (2020) 11:594097. doi: 10.3389/fphys.2020.594097
52. Wong J, Chopra J, Chiang LLW, Liu T, Ho J, Wu WKK, et al. The role of connexins in gastrointestinal diseases. J Mol Biol (2019) 431(4):643–52. doi: 10.1016/j.jmb.2019.01.007
53. Wen LT, Caldwell CC, Knowles AF. Poly(ADP-ribose) polymerase activation and changes in bax protein expression associated with extracellular ATP-mediated apoptosis in human embryonic kidney 293-P2X 7 cells. Mol Pharmacol (2003) 63(3):706–13. doi: 10.1124/mol.63.3.706
54. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
55. Brito GAC, Fujji J, Carneiro-Filho BA, Lima AAM, Obrig T, Guerrant RL. Mechanism of clostridium difficile toxin a–induced apoptosis in T84 cells. J Infect Dis (2002) 186(10):1438–47. doi: 10.1086/344729
56. Qa’Dan M, Ramsey M, Daniel J, Spyres LM, Safiejko-Mroczka B, Ortiz-Leduc W, et al. Clostridium difficile toxin b activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell Microbiol (2002) 4(7):425–34. doi: 10.1046/j.1462-5822.2002.00201.x
57. Nagata S. DNA DEGRADATION IN DEVELOPMENT AND PROGRAMMED CELL DEATH. Annu Rev Immunol (2005) 23(1):853–75. doi: 10.1146/annurev.immunol.23.021704.115811
58. Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol (2001) 3(4):339–45. doi: 10.1038/35070009
59. Sebbagh M, Renvoizé C, Hamelin J, Riché N, Bertoglio J, Bréard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol (2001) 3(4):346–52. doi: 10.1038/35070019
60. Segawa K, Kurata S, Nagata S. Human type IV p-type ATPases that work as plasma membrane phospholipid flippases and their regulation by caspase and calcium. J Biol Chem (2016) 291(2):762–72. doi: 10.1074/jbc.M115.690727
61. Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science (1979) 344(6188):1164–8. doi: 10.1126/science.1252809
62. Yabas M, Coupland LA, Cromer D, Winterberg M, Teoh NC, D’Rozario J, et al. Mice deficient in the putative phospholipid flippase ATP11C exhibit altered erythrocyte shape, anemia, and reduced erythrocyte life span*. J Biol Chem (2014) 289(28):19531–7. doi: 10.1074/jbc.C114.570267
63. Brentnall M, Rodriguez-Menocal L, de Guevara RL, Cepero E, Boise LH. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol (2013) 14(1):32. doi: 10.1186/1471-2121-14-32
64. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature (2010) 467(7317):863–7. doi: 10.1038/nature09413
65. Ribeiro DE, Roncalho AL, Glaser T, Ulrich H, Wegener G, Joca S. P2X7 receptor signaling in stress and depression. Int J Mol Sci (2019) 20(11):2778. doi: 10.3390/ijms20112778
66. Faliti CE, Gualtierotti R, Rottoli E, Gerosa M, Perruzza L, Romagnani A, et al. P2X7 receptor restrains pathogenic tfh cell generation in systemic lupus erythematosus. J Exp Med (2019) 216(2):317–36. doi: 10.1084/jem.20171976
67. Figliuolo VR, Savio LEB, Safya H, Nanini H, Bernardazzi C, Abalo A, et al. P2X7 receptor promotes intestinal inflammation in chemically induced colitis and triggers death of mucosal regulatory T cells. Biochim Biophys Acta (BBA) - Mol Basis Dis (2017) 1863(6):1183–94. doi: 10.1016/j.bbadis.2017.03.004
68. Neves AR, Castelo-Branco MTL, Figliuolo VR, Bernardazzi C, Buongusto F, Yoshimoto A, et al. Overexpression of ATP-activated P2X7 receptors in the intestinal mucosa is implicated in the pathogenesis of crohn’s disease. Inflamm Bowel Dis (2014) 20(3):444–57. doi: 10.1097/01.MIB.0000441201.10454.06
69. McLarnon JG, Ryu JK, Walker DG, Choi HB. Upregulated expression of purinergic P2X 7 receptor in Alzheimer disease and amyloid-β peptide-treated microglia and in peptide-injected rat hippocampus. J Neuropathol Exp Neurol (2006) 65(11):1090–7. doi: 10.1097/01.jnen.0000240470.97295.d3
70. Zhu Y, Li Q, Xun W, Chen Y, Zhang C, Sun S. Blocking P2X7 receptor ameliorates oxidized LDL-mediated podocyte apoptosis. Mol Biol Rep (2019) 46(4):3809–16. doi: 10.1007/s11033-019-04823-6
71. Hanley PJ, Kronlage M, Kirschning C, del Rey A, di Virgilio F, Leipziger J, et al. Transient P2X7 receptor activation triggers macrophage death independent of toll-like receptors 2 and 4, caspase-1, and pannexin-1 proteins. J Biol Chem (2012) 287(13):10650–63. doi: 10.1074/jbc.M111.332676
72. Salas E, Carrasquero LMG, Olivos-Oré LA, Bustillo D, Artalejo AR, Miras-Portugal MT, et al. Purinergic P2X7 receptors mediate cell death in mouse cerebellar astrocytes in culture. J Pharmacol Exp Ther (2013) 347(3):802–15. doi: 10.1124/jpet.113.209452
73. Chen S, Ma Q, Krafft PR, Chen Y, Tang J, Zhang J, et al. P2X7 receptor antagonism inhibits p38 mitogen-activated protein kinase activation and ameliorates neuronal apoptosis after subarachnoid hemorrhage in rats. Crit Care Med (2013) 41(12):e466–74. doi: 10.1097/CCM.0b013e31829a8246
74. Kong Q, Wang M, Liao Z, Camden JM, Yu S, Simonyi A, et al. P2X7 nucleotide receptors mediate caspase-8/9/3-dependent apoptosis in rat primary cortical neurons. Purinergic Signalling (2005) 1(4):337–47. doi: 10.1007/s11302-005-7145-5
75. Puzan M, Hosic S, Ghio C, Koppes A. Enteric nervous system regulation of intestinal stem cell differentiation and epithelial monolayer function. Sci Rep (2018) 8(1):6313. doi: 10.1038/s41598-018-24768-3
76. Yu H, Chen K, Sun Y, Carter M, Garey KW, Savidge TC, et al. Cytokines are markers of the clostridium difficile-induced inflammatory response and predict disease severity. Clin Vaccine Immunol (2017) 24(8):1–11. doi: 10.1128/CVI.00037-17
77. Codeluppi S, Fernandez-Zafra T, Sandor K, Kjell J, Liu Q, Abrams M, et al. Interleukin-6 secretion by astrocytes is dynamically regulated by PI3K-mTOR-Calcium signaling. PloS One (2014) 9(3):e92649. doi: 10.1371/journal.pone.0092649
78. Brasier AR. The nuclear factor- b-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res (2010) 86(2):211–8. doi: 10.1093/cvr/cvq076
79. Murakami M, Kamimura D, Hirano T. Pleiotropy and specificity: Insights from the interleukin 6 family of cytokines. Immunity (2019) 50(4):812–31. doi: 10.1016/j.immuni.2019.03.027
80. Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, Dubyak GR. Active caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by lanthanides. J Immunol (2016) 197(4):1353–67. doi: 10.4049/jimmunol.1600699
81. Qu Y, Misaghi S, Newton K, Gilmour LL, Louie S, Cupp JE, et al. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol (2011) 186(11):6553–61. doi: 10.4049/jimmunol.1100478
82. Brough D, le Feuvre RA, Wheeler RD, Solovyova N, Hilfiker S, Rothwell NJ, et al. Ca 2+ stores and Ca 2+ entry differentially contribute to the release of IL-1β and IL-1α from murine macrophages. J Immunol (2003) 170(6):3029–36. doi: 10.4049/jimmunol.170.6.3029
83. Verkhratsky A, Toescu EC. Endoplasmic reticulum Ca 2+ homeostasis and neuronal death. J Cell Mol Med (2003) 7(4):351–61. doi: 10.1111/j.1582-4934.2003.tb00238.x
84. Loomis WH, Namiki S, Ostrom RS, Insel PA, Junger WG. Hypertonic stress increases T cell interleukin-2 expression through a mechanism that involves ATP release, P2 receptor, and p38 MAPK activation. J Biol Chem (2003) 278(7):4590–6. doi: 10.1074/jbc.M207868200
85. Mehta VB, Hart J, Wewers MD. ATP-stimulated release of interleukin (IL)-1β and IL-18 requires priming by lipopolysaccharide and is independent of caspase-1 cleavage. J Biol Chem (2001) 276(6):3820–6. doi: 10.1074/jbc.M006814200
86. Hu Y, Fisette PL, Denlinger LC, Guadarrama AG, Sommer JA, Proctor RA, et al. Purinergic receptor modulation of lipopolysaccharide signaling and inducible nitric-oxide synthase expression in RAW 264.7 macrophages. J Biol Chem (1998) 273(42):27170–5. doi: 10.1074/jbc.273.42.27170
87. Inoue Y, Tanaka N, Tanaka Y, Inoue S, Morita K, Zhuang M, et al. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J Virol (2007) 81(16):8722–9. doi: 10.1128/JVI.00253-07
88. Bustamante M, Fernández-Verdejo R, Jaimovich E, Buvinic S. Electrical stimulation induces IL-6 in skeletal muscle through extracellular ATP by activating Ca 2+ signals and an IL-6 autocrine loop. Am J Physiology-Endocrinol Metab (2014) 306(8):E869–82. doi: 10.1152/ajpendo.00450.2013
89. Hanley PJ, Musset B, Renigunta V, Limberg SH, Dalpke AH, Sus R, et al. Extracellular ATP induces oscillations of intracellular Ca 2+ and membrane potential and promotes transcription of IL-6 in macrophages. Proc Natl Acad Sci (2004) 101(25):9479–84. doi: 10.1073/pnas.0400733101
Keywords: Clostridioides difficile, Clostridioides difficile infection, Pannexin-1, P2X7R, Enteric glia
Citation: Loureiro AV, Moura-Neto LI, Martins CS, Silva PIM, Lopes MBS, Leitão RFC, Coelho-Aguiar JM, Moura-Neto V, Warren CA, Costa DVS and Brito GAC (2022) Role of Pannexin-1-P2X7R signaling on cell death and pro-inflammatory mediator expression induced by Clostridioides difficile toxins in enteric glia. Front. Immunol. 13:956340. doi: 10.3389/fimmu.2022.956340
Received: 30 May 2022; Accepted: 01 August 2022;
Published: 22 August 2022.
Edited by:
Thomas Burris, Washington University in St. Louis, United StatesReviewed by:
Mindy Engevik, Medical University of South Carolina, United StatesCopyright © 2022 Loureiro, Moura-Neto, Martins, Silva, Lopes, Leitão, Coelho-Aguiar, Moura-Neto, Warren, Costa and Brito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerly A. C. Brito, Z2VybHlicml0b0Bob3RtYWlsLmNvbQ==; Deiziane V.S. Costa, ZGVpemlhbmUyMDA5QGdtYWlsLmNvbQ==
†These authors share first co-authorship
‡These authors share last co-authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.