 Michele K. Anderson
Michele K. Anderson- 1Department of Biological Sciences, Sunnybrook Research Institute, Toronto, ON, Canada
- 2Department of Immunology, University of Toronto, Toronto, ON, Canada
Shifting levels of E proteins and Id factors are pivotal in T cell commitment and differentiation, both in the thymus and in the periphery. Id2 and Id3 are two different factors that prevent E proteins from binding to their target gene cis-regulatory sequences and inducing gene expression. Although they use the same mechanism to suppress E protein activity, Id2 and Id3 play very different roles in T cell development and CD4 T cell differentiation. Id2 imposes an irreversible choice in early T cell precursors between innate and adaptive lineages, which can be thought of as a railway switch that directs T cells down one path or another. By contrast, Id3 acts in a transient fashion downstream of extracellular signals such as T cell receptor (TCR) signaling. TCR-dependent Id3 upregulation results in the dislodging of E proteins from their target sites while chromatin remodeling occurs. After the cessation of Id3 expression, E proteins can reassemble in the context of a new genomic landscape and molecular context that allows induction of different E protein target genes. To describe this mode of action, we have developed the “Clutch” model of differentiation. In this model, Id3 upregulation in response to TCR signaling acts as a clutch that stops E protein activity (“clutch in”) long enough to allow shifting of the genomic landscape into a different “gear”, resulting in accessibility to different E protein target genes once Id3 decreases (“clutch out”) and E proteins can form new complexes on the DNA. While TCR signal strength and cytokine signaling play a role in both peripheral and thymic lineage decisions, the remodeling of chromatin and E protein target genes appears to be more heavily influenced by the cytokine milieu in the periphery, whereas the outcome of Id3 activity during T cell development in the thymus appears to depend more on the TCR signal strength. Thus, while the Clutch model applies to both CD4 T cell differentiation and T cell developmental transitions within the thymus, changes in chromatin accessibility are modulated by biased inputs in these different environments. New emerging technologies should enable a better understanding of the molecular events that happen during these transitions, and how they fit into the gene regulatory networks that drive T cell development and differentiation.
Introduction
Conventional T cells acquire their functional properties in two main phases. The first occurs in the thymus, as T cells transit through successive stages that install the gene expression programs that will run at steady state. The second phase of differentiation occurs in the periphery after exposure to signals that occur during an immune response. These signals activate accessible but latent sub-routines that are kept in check prior to the initiation of the immune response. Both processes depend on the activity of E protein transcription factors and their antagonists, the Id factors. One of the most intriguing aspects of E proteins is their context-dependent use in many different T cell lineages, and the propensity of T cell receptor (TCR) signaling and Id3 activity, in collaboration with other extracellular signals, to create those contexts. While TCR signaling is required for peripheral CD4 T cell differentiation, the specific functional pathways accessed in the periphery are very sensitive to the cytokine milieu. By contrast, the progression of T cell precursors into different pathways in the thymus appears to be driven more by TCR signal strength. In both cases, TCR-dependent upregulation of Id3 is important for allowing changes in changes in chromatin remodeling and gene expression that are needed to restrict E protein activity to the appropriate targets.
T helper cell differentiation and function
Conventional CD4 T cells emerge from the thymus as “naïve” cells ready for activation. The functional T helper cell differentiation pathways they take upon antigen encounter depends on the types of inflammatory molecules produced during the innate immune response (1) (Figure 1A). Each T helper cell subset is dependent on a specific “master regulator” transcription factor that directly induces the effector genes of each program (2). The Th17 lineage, characterized by secretion of IL-17A, IL-17F, and IL-22, is triggered by the innate response to bacteria and fungi. RORγt (Rorc) is the Th17 master regulator. Viruses and other intracellular pathogens induce differentiation into the T-bet (Tbx21) dependent Th1 pathway, leading to IL-2, TFNα, and IFNγ production. Helminth infection induces the Th2 fate, leading to secretion IL-4, IL-5, and IL-13, under the control of GATA3 (3).
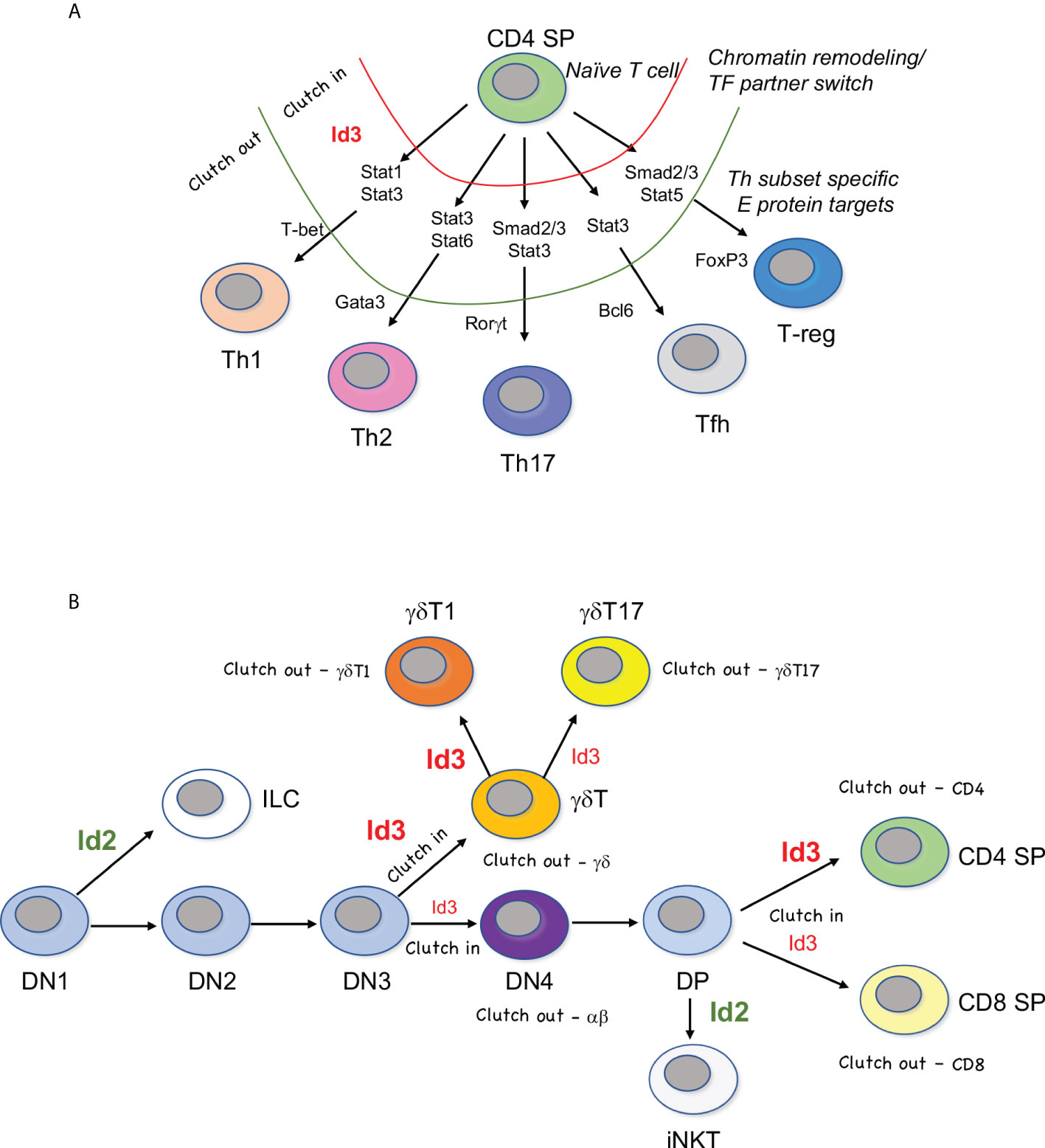
Figure 1 “Clutch” model of Id3-E protein mediated fate choice. (A) E proteins regulate a core CD4 T cell program in naïve T cells. Upregulation of Id3 causes a transient inhibition of E protein activity (red, clutch in), during which time initiating transcription factors provide access to Th subset-specific genes. Once remodeling is finished, Id3 activity ceases and E protein activity resumes (green, clutch out). E proteins can then induce master regulators and effector genes specific to each Th lineage. (B) Waves of graded Id3 (red) induction in response to TCR signaling pauses a subset E protein activity (clutch in) followed by reassembly of E proteins at successive stages of T cell development (clutch out). Levels of Id3 dictate lineage choice, but E proteins are often engaged in both choices downstream of lineage commitment. Id2 (green) is responsible for differentiation away from the adaptive T cell lineage and into the innate lymphoid cell lineage (ILC) or invariant natural killer T (iNKT) cell lineage. DN=double negative CD4-CD8-, DP=double positive CD4+CD8+, γδT1 = IFNγ-producing γδ T cells, γδT17 = IL-17 producing γδ T cells.
Other Th subsets generated in the periphery include Bcl6-driven T-follicular helper cells (Tfh) (4), specialized for B cell help in the germinal center, and induced T-reg cells, which, like thymic-derived T-regs, depend on FoxP3 (5). In addition to playing unique roles in immunity, Th subsets also have pathogenic impacts when dysregulated (6). In general, Th1 and Th17 cells contribute to autoimmune pathology, Th2 cells are largely responsible for allergic reactions, and T-regs inhibit anti-cancer immunity (7, 8). Most Th subsets retain plasticity after activation, and some can transdifferentiate from one type to another (2). Additional Th subsets continue to be identified, including Th22, Th9, Tfh13, and Tr1 cells, suggesting that the networks controlling these effector functions are dynamic, and represent more of a physiological state than a committed fate, rendering them open to manipulation during an immune response (9–11).
Transcriptional control of Th differentiation
Differentiation of naïve CD4 T cells into the Th subsets is coordinated by several sets of signal-dependent transcription factors (12). Triggering of the αβ TCR and co-stimulatory receptors leads to activation of NFkB, NFAT, and IRF transcription family members, as well as upregulation of AP1 transcription factor family members such as BATF and Jun (13, 14). Cytokine receptor signaling leads to the activation of different sets of transcription factors, most notably members of the STAT and SMAD families (15, 16). BATF, IRF4, and the cytokine-responsive factors recruit chromatin remodeling enzymes that provide access to genes of specific Th subsets, while restricting access to genes of alternative Th subsets (13, 17). After chromatin remodeling, the master regulators are induced, providing the final key needed for functional activation during the immune response.
E proteins and Id proteins are involved in regulation of the naïve CD4 T cell state, and in the differentiation of Th2, Th17, and T-reg cells (18–20). In general, E protein activity is regulated post-translationally by Id proteins, which sequester them in inactive dimers. The requirement for E proteins for Th17 differentiation has been especially well studied. A comprehensive study conducted by the Strober group in 2013 showed that mice carrying a conditional double HEB/E2A deletion on a CD4-Cre background had a profound defect in Th17 development in vitro, and compromised immune function in vivo, using both autoimmunity and infection models (21). This study also showed that HEB and E2A can directly bind and activate the Rorc locus, but only in the context of Th17 cells, not in naïve CD4 T cells. Studies of Id3-deficient mice suggest that E proteins restrain the Th2 and Tfh lineages and promote the Th9 lineage, whereas Th1 cells appear to require Id proteins and to be E protein independent (21–24). Interestingly, T-regs require both Id3 and E2A in a sequential manner. TGFβ induces transient expression of Id3, which is needed to prevent repression of the FoxP3 promoter (25). This repression is not mediated directly by E proteins, but rather results from E protein-mediated upregulation of GATA3. Subsequently, E2A activity is required to directly activate the FoxP3 promoter. However, if E2A levels are too high, FoxP3 expression becomes unstable in T-regs, emphasizing the importance of transcription factors levels in maintaining stable outcomes (26).
The Clutch model of E protein/Id3 activity in T cell transitional states
The theme of transient Id3 expression followed by shifting E protein target gene activation suggests what we term a “Clutch” model of Th differentiation (Figure 1). In this model, Id3-mediated pausing of E protein activity would act like the clutch of a car, withholding access to the engine (E protein activity) until the appropriate gear (chromatin context) is engaged, and then allowing the engine to move the car (Th differentiation) forward in a controlled fashion (Figure 1A). E proteins bind to many effector genes in Th subsets. Therefore, it is likely that restriction of E protein binding to the “right” set of mediators within each lineage is essential for linking environmental input to functional output in Th subsets. This is clearly a strong paradigm for peripheral T cell differentiation (27). The Clutch model also applies to T cell development in the thymus, but with a twist, as described below (Figure 1B). Moreover, the role of Id2 in thymic T cell development exhibits stark differences from Id3 during thymic development and does not conform to the Clutch model.
Id2 regulates the innate/adaptive fate choice in early T cell precursors
The earliest T cell progenitors (ETPs) to enter the thymus are not yet committed to the T-cell lineage and have alternative fates available to them depending on their access to microenvironmental signals. One of the key molecular switches that must be flipped to gain access to the T cell pathway is to increase E protein activity. This occurs in at least two different ways. The first is upregulation of E proteins at the mRNA level, and the second is the downregulation of Id2 (28). Id2 is a critical mediator of the innate/adaptive lineage split (18, 29). ETPs express “legacy genes”, thus termed because they are expressed in hematopoietic stem cells (30). ETP legacy genes include Id2, the Ets protein PU.1, and the Class II bHLH factor SCL. All three of these factors can act in opposition to T-lineage commitment: PU.1 drives expression of myeloid and B cell genes (31), SCL can re-direct E proteins to stem cell gene loci and away from T cell gene loci (32), and Id2 interferes with E protein activity. E protein activity is essential for the expression of Rag recombinase genes, which are necessary for the generation of TCRs and thus T cells (33). Unlike Id3, Id2 does not appear to be under the influence of transient signals during thymocyte development but rather is subject to degradation in a cell cycle-dependent manner (34, 35). Downregulation of PU.1 and upregulation of Bcl11b in early T cell development results in the cessation of Id2 mRNA expression, which allows upregulation of T-lineage E protein target genes (36, 37). Conversely, Id2 expression is maintained in mature innate cells including ILCs, NK cells, and myeloid cells, and appears to support the maintenance of lineage fidelity.
Notch signaling shifts the E protein-Id2 balance to allow T cell development
As ETPs enter the thymus, they are exposed to Delta-like (Dll) ligands of Notch receptors, resulting in strong Notch signaling. Notch signaling is indispensable for T cell specification and lineage commitment, acting upstream of an elegant cascade of transcription factors that inhibits alternative fates and induces T-cell genes (38). While Notch regulates a wide swath of important target genes, one of the most important roles plays in T-lineage commitment is by shifting the balance between Id and E protein activity in ETPs, in three complimentary ways. First, Notch redirects PU.1 away from Id2 and towards more T-lineage friendly genes (39). Secondly, Notch upregulates the E protein HEBAlt, increasing the overall E protein availability (40). Thirdly, Notch directly upregulates Bcl11b, which downregulates Id2 at the transcriptional level (37). This delivers a one-two-three punch that directs cells permanently away from Id2-dependent ILCs and into the T-cell lineage. Thus, Id2 does not acts as a way station for changing gene availability to E proteins, but instead is more akin to a railroad switch that directs cells down one pathway or another (Figure 2).
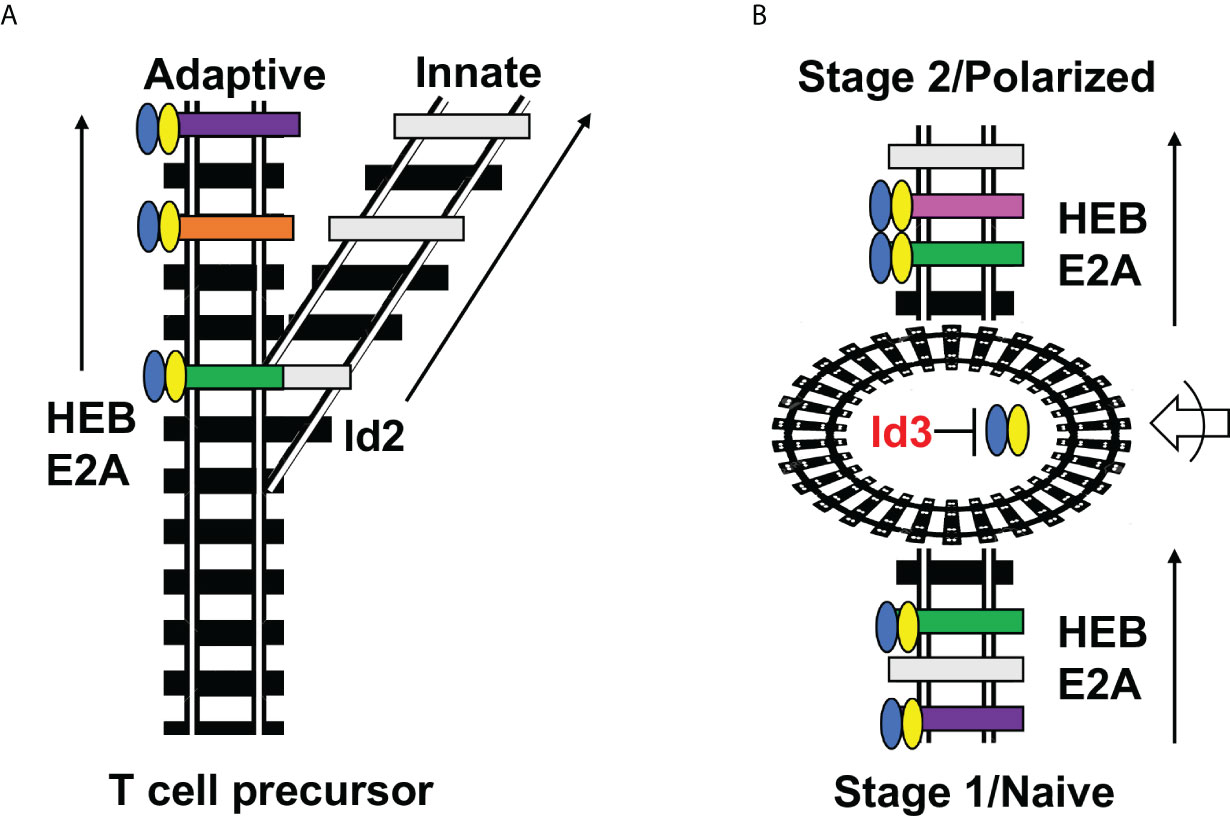
Figure 2 Railroad analogy of Id2 versus Id3 outcomes. (A) Id2 acts as a switch that diverts T cell precursors away from the adaptive fate and towards the innate fate by permanently silencing E protein activity and E protein target gene expression. (B) Id3 serves as a way station (roundhouse) that allows changes in accessibility of E protein target genes while E proteins are inactive, followed by E protein engagement with different E protein target genes at the next stage of development. This occurs in both CD4 T cell differentiation (Naïve/Polarized) and multiple stages of thymic differentiation (Stage 1/Stage 2). Blue-Yellow ovals = HEB/E2A. Black railroad ties = completely inaccessible genes; gray railroad ties = accessible genes lacking the proper combination of transcription factors for induction; colored railroad ties = E protein target genes bound by HEB/E2A and undergoing active transcription. Roundabout = Id3-mediated pause in E protein activity during which changes in accessibility of E protein target sites occurs. Arrow with half circle = extracellular signaling inputs that direct which genes undergo changes in chromatin accessibility.
TCR signal strength determines lineage outcomes during the intrathymic T cell lineage choices
Once cells have been switched onto the T-lineage track, they progress towards the first “checkpoint” of T cell development. There are two main checkpoints that occur during T cell development, so called because they serve as testing of the cells for functional TCR rearrangement and function (Figure 1B). During the first checkpoint, the TCRβ chain pairs with the pre-Tα chain to form a pre-TCR. The only requirement for the pre-TCR to allow “passage” through the checkpoint is for it to complex with CD3 chains and translocate to the cell membrane long enough to invoke a weak set of signaling cascades (41). Alternatively, the cell can rearrange and express a TCR composed of TCRγ and TCRδ chains. In this situation, the γδ TCR/CD3 complex is stably expressed on the surface, transmitting a stronger signal than that transduced by the pre-TCR, which directs cells away from the αβ T cell fate and into the γδ T cell fate (42, 43). After commitment to the αβ T cell lineage, cells expressing αβ TCRs are subjected to second “checkpoint” which vets these TCRs for their ability to bind to MHC/peptide and assesses the affinity of the interaction. As with the first checkpoint, this signal also serves as a lineage branchpoint, with cells experiencing lower and briefer TCR signaling adopting the CD8 fate, and cells experiencing longer and stronger TCR signaling progressing into the CD4 T cell lineage (44). This paradigm also applies to committed γδ T cells that progress along the IFNγ-producing γδT1 fate or the γδT17 fate (45) (Figure 1B). Engagement of strong γδ TCR ligands in conjunction with co-stimulatory molecules results in strong TCR signaling and the γδT1 developmental outcome, whereas a less strong TCR signal leads to the γδT17 fate (46–48). All these lineage choices are intimately associated with the balance between Id3 and E proteins (49, 50).
Translation of TCR signal strength into Id3 activity modulates E protein target gene accessibility
As in peripheral CD4 T cells, TCR signaling in early precursors leads to upregulation of Id3, and a pause in E protein activity allows chromatin remodeling and shifting of E protein target availability. There may also be a role for TCR signal strength during T helper cell differentiation, particularly in combination with cytokine signaling (51). However, there is a clear hierarchy of TCR signal strength that is induced at each checkpoint in thymic T cell development (52). During T cell development, TCR signaling may shift the balance between Id3 and E proteins to different degrees, allowing retention of E protein occupancy on some sites but not others. E2A and HEB are direct regulators of most of the genes needed for assembly of the TCR genes and formation of the pre-TCR (53, 54). Id3 is also induced in response to αβTCR signaling at the DP stage, and is necessary to overcome the gatekeeper function of E proteins at the DP to SP transition (55, 56). However, past this checkpoint, E proteins are required for the generation of CD4 SP cells (57). E proteins also regulate genes in γδ-T committed cells that dictate functional programming, including Tcf7 (58). An elegant study by Hosoya and colleagues shed considerable light on the chromatin remodeling events that occur during αβ T cell development using ATAC-seq, which detects open chromatin and predicts the presence of transcriptional complexes (59). This study showed that the loci for both γδ-lineage and αβ-lineage genes were accessible in DN thymocytes. However, as cells transitioned from the DN to the DP stage and then to the CD4 and CD8 stages, cis-regulatory elements with predicted binding by the key γδ-lineage factor Sox13 showed a dramatic loss of accessibility. Likewise, predicted HEB sites shifted in accessibility according to the stage of αβ T cell development, consistent with Id3-facilitated chromatin remodeling at these transitions. This is doubtless just the beginning of this new phase of our journey towards a deeper understanding of T cell developmental transitions, and it will be exciting to learn how E protein genomic site occupancy changes after they are dislodged and then reassembled on different loci at progressive stages of T cell development and differentiation.
Limitations of the Clutch model of Id3-facilitated shifts in E protein targets
Like E proteins, Id3 is used widely in different contexts outside of T cell development (60, 61). Clearly, the Clutch model does not apply in all situations, but rather appears to be restricted to certain types of cells and developmental transitions. Moreover, an examination of E2A occupancy at the DN3 to DN4 transition revealed both overlapping and unique sites of E2A occupancy in both subsets, indicating that E2A was only dislodged from a subset of sites during the transition, while others were maintained (19). Release of E proteins from specific sites likely depends on both the Id3/E protein ratio and the availability of E protein binding partners. For instance, the downregulation of Notch1 in response to pre-TCR signaling would be predicted to increase the disengagement of E proteins from sites that require both Notch factors and E proteins, but not from other sites that maintain the core T-lineage program. Importantly, E proteins themselves are important mediators of chromatin remodeling, interacting directly with both positive and negative regulators of chromatin configuration such as p300, CHD4, LSD1, and PRC2 (62–66). It is important to note that chromatin remodeling in this context does not indicate simply a shift between “open” and “closed” configurations, but also includes the transition from “poised” to “active” states (67). This may be mediated in part by fresh access to new binding partners that become available after the transition. Furthermore, the plasticity of CD4 T cell subsets suggests that lineage-specifying E protein sites remain accessible during and after CD4 T cell differentiation (68). A comprehensive understanding of global E protein occupancy changes that occur during these processes awaits further studies. Likewise, the relative contributions of E2A versus HEB to these processes are not well understood.
Discussion
While it is well understood that Id proteins inhibit E protein activity and interfere with the expression of E protein target genes, much less is known about how E protein targets shift during the developmental transitions that occur during Id3 expression, and the molecular events that underpin them. Here, the Clutch model is presented as a conceptual scaffold that will provoke questions and undergo modifications and stratification as new data is obtained revealing E protein chromatin occupancy before, during, and after T cell stages transitions, and identifying stage-specific E protein partners. Due to technical limitations, earlier studies largely relied on in vitro models of T cell development or differentiation such as OP9-DL co-culture derived T cell precursors or in vitro polarization of naïve peripheral T cells (69, 70). While these studies have provided a wealth of information into the global events that orchestrate T cell development, they cannot completely replicate the complex thymic niches that shift over time as cells migrate through different niches in the thymus, nor can they fully provide the complex medley of signals that transpire during a coordinated immune response. The advent of single cell RNA-seq, and multiomic approaches such as scRNA-seq/ATAC-seq and CITE-seq that allows that require fewer input cells are now providing unprecedented access to ex vivo precursors and products that arise during T cell development. Moreover, computational methods such as pseudotime modeling and RNA velocity are further advancing our understanding of transient states of development (71). Importantly, there is a fourth dimension that is rarely considered in these snapshot approaches: time. Single cell live imaging has revealed that Id3 transcription is “bursty”, occurring in only a small number of cells within a population at any one time, in the B cell lineage (72). It remains to be seen whether this is true in T cell precursors, and whether TCR signaling can synchronize cells into uniformly high Id3 expressers. Alternatively, burstiness may contribute to the gradation of Id3 that mediates intrathymic T cell fate choices. By contrast, Id2 acts as a permanent switch into the innate lineage choice. This distinction highlighting the unique nature of Id3 in regulating fate choices by facilitating E protein target changes as T cells journey through development in the thymus or differentiate in the periphery during an immune response.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
The ideas presented in this work were made possible by support from NIH (1P01AI102853-06), CIHR (201610PJT) and NSERC (RGPIN 05333-14).
Acknowledgments
The ideas presented in this Mini Review have been strongly influenced by my conversations with colleagues too numerous to present here, and I would like to apologize to any that were not cited due to space limitations. I am particularly indebted to Juan Carlos Zúñiga-Pflücker, David Wiest, Yuan Zhuang, and Cornelis Murre for thoughtful and lively discussions and for their ongoing contributions to the field.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Martinez-Sosa P, Mendoza L. The regulatory network that controls the differentiation of T lymphocytes. Biosystems (2013) 113(2):96–103. doi: 10.1016/j.biosystems.2013.05.007
2. Hirahara K, Poholek A, Vahedi G, Laurence A, Kanno Y, Milner JD, et al. Mechanisms underlying helper T-cell plasticity: implications for immune-mediated disease. J Allergy Clin Immunol (2013) 131(5):1276–87. doi: 10.1016/j.jaci.2013.03.015
3. Butcher MJ, Zhu J. Recent advances in understanding the Th1/Th2 effector choice. Fac Rev (2021) 10:30. doi: 10.12703/r/10-30
4. Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A, et al. BCL6 orchestrates tfh cell differentiation via multiple distinct mechanisms. J Exp Med (2015) 212(4):539–53. doi: 10.1084/jem.20141380
5. Georgiev P, Charbonnier LM, Chatila TA. Regulatory T cells: the many faces of Foxp3. J Clin Immunol (2019) 39(7):623–40. doi: 10.1007/s10875-019-00684-7
6. Nakayama T, Hirahara K, Onodera A, Endo Y, Hosokawa H, Shinoda K, et al. Th2 cells in health and disease. Annu Rev Immunol (2017) 35:53–84. doi: 10.1146/annurev-immunol-051116-052350
7. Dobrzanski MJ. Expanding roles for CD4 T cells and their subpopulations in tumor immunity and therapy. Front Oncol (2013) 3:63. doi: 10.3389/fonc.2013.00063
8. Sarkar T, Dhar S, Sa G. Tumor-infiltrating T-regulatory cells adapt to altered metabolism to promote tumor-immune escape. Curr Res Immunol (2021) 2:132–41. doi: 10.1016/j.crimmu.2021.08.002
9. Plank MW, Kaiko GE, Maltby S, Weaver J, Tay HL, Shen W, et al. Th22 cells form a distinct Th lineage from Th17 cells In vitro with unique transcriptional properties and tbet-dependent Th1 plasticity. J Immunol (2017) 198(5):2182–90. doi: 10.4049/jimmunol.1601480
10. Schmitt E, Klein M, Bopp T. Th9 cells, new players in adaptive immunity. Trends Immunol (2014) 35(2):61–8. doi: 10.1016/j.it.2013.10.004
11. Gowthaman U, Chen JS, Zhang B, Flynn WF, Lu Y, Song W, et al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science (2019) 365(6456):1–22. doi: 10.1126/science.aaw6433
12. Rogers D, Sood A, Wang H, van Beek JJP, Rademaker TJ, Artusa P, et al. Pre-existing chromatin accessibility and gene expression differences among naive CD4(+) T cells influence effector potential. Cell Rep (2021) 37(9):110064. doi: 10.1016/j.celrep.2021.110064
13. Iwata A, Durai V, Tussiwand R, Briseno CG, Wu X, Grajales-Reyes GE, et al. Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat Immunol (2017) 18(5):563–72. doi: 10.1038/ni.3714
14. Carr TM, Wheaton JD, Houtz GM, Ciofani M. JunB promotes Th17 cell identity and restrains alternative CD4(+) T-cell programs during inflammation. Nat Commun (2017) 8(1):301. doi: 10.1038/s41467-017-00380-3
15. Yoo SA, Kim M, Kang MC, Kong JS, Kim KM, Lee S, et al. Placental growth factor regulates the generation of TH17 cells to link angiogenesis with autoimmunity. Nat Immunol (2019) 20:1348–59. doi: 10.1038/s41590-019-0456-4
16. Malhotra N, Robertson E, Kang J. SMAD2 is essential for TGF beta-mediated Th17 cell generation. J Biol Chem (2010) 285(38):29044–8. doi: 10.1074/jbc.C110.156745
17. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity (2009) 30(1):155–67. doi: 10.1016/j.immuni.2008.12.009
18. Zook EC, Li ZY, Xu Y, de Pooter RF, Verykokakis M, Beaulieu A, et al. Transcription factor ID2 prevents e proteins from enforcing a naive T lymphocyte gene program during NK cell development. Sci Immunol (2018) 3(22):1–17. doi: 10.1126/sciimmunol.aao2139
19. Miyazaki M, Rivera RR, Miyazaki K, Lin YC, Agata Y, Murre C. The opposing roles of the transcription factor E2A and its antagonist Id3 that orchestrate and enforce the naive fate of T cells. Nat Immunol (2011) 12(10):992–1001. doi: 10.1038/ni.2086
20. Zhou H, Wang L, Xu Q, Liu Q, Liu H, Qiu W, et al. ID3 may protect mice from antiGBM glomerulonephritis by regulating the differentiation of Th17 and treg cells. Mol Med Rep (2017) 16(6):9086–94. doi: 10.3892/mmr.2017.7724
21. Zhang J, Kalkum M, Yamamura S, Chait BT, Roeder RG. E protein silencing by the leukemogenic AML1-ETO fusion protein. Science (2004) 305(5688):1286–9. doi: 10.1126/science.1097937
22. Nakatsukasa H, Zhang D, Maruyama T, Chen H, Cui K, Ishikawa M, et al. The DNA-binding inhibitor Id3 regulates IL-9 production in CD4(+) T cells. Nat Immunol (2015) 16(10):1077–84. doi: 10.1038/ni.3252
23. Miyazaki M, Miyazaki K, Chen S, Chandra V, Wagatsuma K, Agata Y, et al. The e-id protein axis modulates the activities of the PI3K-AKT-mTORC1-Hif1a and c-myc/p19Arf pathways to suppress innate variant TFH cell development, thymocyte expansion, and lymphomagenesis. Genes Dev (2015) 29(4):409–25. doi: 10.1101/gad.255331.114
24. Han X, Liu H, Huang H, Liu X, Jia B, Gao GF, et al. ID2 and ID3 are indispensable for Th1 cell differentiation during influenza virus infection in mice. Eur J Immunol (2019) 49(3):476–89. doi: 10.1002/eji.201847822
25. Maruyama T, Li J, Vaque JP, Konkel JE, Wang W, Zhang B, et al. Control of the differentiation of regulatory T cells and T(H)17 cells by the DNA-binding inhibitor Id3. Nat Immunol (2011) 12(1):86–95. doi: 10.1038/ni.1965
26. Rauch KS, Hils M, Lupar E, Minguet S, Sigvardsson M, Rottenberg ME, et al. Id3 maintains Foxp3 expression in regulatory T cells by controlling a transcriptional network of E47, spi-b, and SOCS3. Cell Rep (2016) 17(11):2827–36. doi: 10.1016/j.celrep.2016.11.045
27. Jiang Q, Wang J, Jiang H, Li W, Sun Y, Shan Y, et al. Competitive binding of transcription factors underlies flexibility of T peripheral helper cells and T follicular helper cells in SLE. Rheumatol (Oxford) (2022) 1–11. doi: 10.1093/rheumatology/keac112
28. Rothenberg EV, Moore JE, Yui MA. Launching the T-cell-lineage developmental programme. Nat Rev Immunol (2008) 8(1):9–21. doi: 10.1038/nri2232
29. Miyazaki M, Miyazaki K, Chen K, Jin Y, Turner J, Moore AJ, et al. The e-id protein axis specifies adaptive lymphoid cell identity and suppresses thymic innate lymphoid cell development. Immunity (2017) 46(5):818–34 e4. doi: 10.1016/j.immuni.2017.04.022
30. David-Fung ES, Butler R, Buzi G, Yui MA, Diamond RA, Anderson MK, et al. Transcription factor expression dynamics of early T-lymphocyte specification and commitment. Dev Biol (2009) 325(2):444–67. doi: 10.1016/j.ydbio.2008.10.021
31. Anderson MK, Hernandez-Hoyos G, Diamond RA, Rothenberg EV. Precise developmental regulation of ets family transcription factors during specification and commitment to the T cell lineage. Development (1999) 126(14):3131–48. doi: 10.1242/dev.126.14.3131
32. Gerby B, Tremblay CS, Tremblay M, Rojas-Sutterlin S, Herblot S, Hebert J, et al. SCL, LMO1 and Notch1 reprogram thymocytes into self-renewing cells. PloS Genet (2014) 10(12):e1004768. doi: 10.1371/journal.pgen.1004768
33. Miyazaki K, Miyazaki M. The interplay between chromatin architecture and lineage-specific transcription factors and the regulation of rag gene expression. Front Immunol (2021) 12:659761. doi: 10.3389/fimmu.2021.659761
34. Lasorella A, Stegmuller J, Guardavaccaro D, Liu G, Carro MS, Rothschild G, et al. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature (2006) 442(7101):471–4. doi: 10.1038/nature04895
35. Sullivan JM, Havrda MC, Kettenbach AN, Paolella BR, Zhang Z, Gerber SA, et al. Phosphorylation regulates Id2 degradation and mediates the proliferation of neural precursor cells. Stem Cells (2016) 34(5):1321–31. doi: 10.1002/stem.2291
36. Anderson MK, Weiss AH, Hernandez-Hoyos G, Dionne CJ, Rothenberg EV. Constitutive expression of PU.1 in fetal hematopoietic progenitors blocks T cell development at the pro-T cell stage. Immunity (2002) 16(2):285–96. doi: 10.1016/s1074-7613(02)00277-7
37. Hosokawa H, Romero-Wolf M, Yui MA, Ungerback J, Quiloan MLG, Matsumoto M, et al. Bcl11b sets pro-T cell fate by site-specific cofactor recruitment and by repressing Id2 and Zbtb16. Nat Immunol (2018) 19(12):1427–40. doi: 10.1038/s41590-018-0238-4
38. Rothenberg EV. Dynamic control of the T-cell specification gene regulatory network. Curr Opin Syst Biol (2019) 18:62–76. doi: 10.1016/j.coisb.2019.10.012
39. Del Real MM, Rothenberg EV. Architecture of a lymphomyeloid developmental switch controlled by PU.1, notch and Gata3. Development (2013) 140(6):1207–19. doi: 10.1242/dev.88559
40. Wang D, Claus CL, Vaccarelli G, Braunstein M, Schmitt TM, Zuniga-Pflucker JC, et al. The basic helix-loop-helix transcription factor HEBAlt is expressed in pro-T cells and enhances the generation of T cell precursors. J Immunol (2006) 177(1):109–19. doi: 10.4049/jimmunol.177.1.109
41. Dutta A, Zhao B, Love PE. New insights into TCR beta-selection. Trends Immunol (2021) 42(8):735–50. doi: 10.1016/j.it.2021.06.005
42. Haks MC, Lefebvre JM, Lauritsen JP, Carleton M, Rhodes M, Miyazaki T, et al. Attenuation of gammadeltaTCR signaling efficiently diverts thymocytes to the alphabeta lineage. Immunity (2005) 22(5):595–606. doi: 10.1016/j.immuni.2005.04.003
43. Hayes SM, Li L, Love PE. TCR signal strength influences alphabeta/gammadelta lineage fate. Immunity (2005) 22(5):583–93. doi: 10.1016/j.immuni.2005.03.014
44. Karimi MM, Guo Y, Cui X, Pallikonda HA, Horkova V, Wang YF, et al. The order and logic of CD4 versus CD8 lineage choice and differentiation in mouse thymus. Nat Commun (2021) 12(1):99. doi: 10.1038/s41467-020-20306-w
45. Zarin P, In TS, Chen EL, Singh J, Wong GW, Mohtashami M, et al. Integration of T-cell receptor, notch and cytokine signals programs mouse gammadelta T-cell effector differentiation. Immunol Cell Biol (2018) 96(9):994–1007. doi: 10.1111/imcb.12164
46. Sumaria N, Grandjean CL, Silva-Santos B, Pennington DJ. Strong TCRgammadelta signaling prohibits thymic development of IL-17A-Secreting gammadelta T cells. Cell Rep (2017) 19(12):2469–76. doi: 10.1016/j.celrep.2017.05.071
47. Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, et al. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat Immunol (2009) 10(4):427–36. doi: 10.1038/ni.1717
48. Chen ELY, Lee CR, Thompson PK, Wiest DL, Anderson MK, Zuniga-Pflucker JC. Ontogenic timing, T cell receptor signal strength, and notch signaling direct gammadelta T cell functional differentiation in vivo. Cell Rep (2021) 35(10):109227. doi: 10.1016/jcelrep.2021.109227
49. Jones ME, Zhuang Y. Stage-specific functions of e-proteins at the beta-selection and T-cell receptor checkpoints during thymocyte development. Immunol Res (2011) 49(1-3):202–15. doi: 10.1007/s12026-010-8182-x
50. Anderson MK, Selvaratnam JS. Interaction between gammadeltaTCR signaling and the e protein-id axis in gammadelta T cell development. Immunol Rev (2020) 298(1):181–97. doi: 10.1111/imr.12924
51. Bhattacharyya ND, Feng CG. Regulation of T helper cell fate by TCR signal strength. Front Immunol (2020) 11:624. doi: 10.3389/fimmu.2020.00624
52. Zarin P, Chen EL, In TS, Anderson MK, Zuniga-Pflucker JC. Gamma delta T-cell differentiation and effector function programming, TCR signal strength, when and how much? Cell Immunol (2015) 296(1):70–5. doi: 10.1016/j.cellimm.2015.03.007
53. Miyazaki K, Watanabe H, Yoshikawa G, Chen K, Hidaka R, Aitani Y, et al. The transcription factor E2A activates multiple enhancers that drive rag expression in developing T and b cells. Sci Immunol (2020) 5(51):1–15. doi: 10.1126/sciimmunol.abb1455
54. Schwartz R, Engel I, Fallahi-Sichani M, Petrie HT, Murre C. Gene expression patterns define novel roles for E47 in cell cycle progression, cytokine-mediated signaling, and T lineage development. Proc Natl Acad Sci U S A (2006) 103(26):9976–81. doi: 10.1073/pnas.0603728103
55. Bain G, Quong MW, Soloff RS, Hedrick SM, Murre C. Thymocyte maturation is regulated by the activity of the helix-loop-helix protein, E47. J Exp Med (1999) 190(11):1605–16. doi: 10.1084/jem.190.11.1605
56. Jones ME, Zhuang Y. Acquisition of a functional T cell receptor during T lymphocyte development is enforced by HEB and E2A transcription factors. Immunity (2007) 27(6):860–70. doi: 10.1016/j.immuni.2007.10.014
57. Jones-Mason ME, Zhao X, Kappes D, Lasorella A, Iavarone A, Zhuang Y. E protein transcription factors are required for the development of CD4(+) lineage T cells. Immunity (2012) 36(3):348–61. doi: 10.1016/j.immuni.2012.02.010
58. Fahl SP, Contreras AV, Verma A, Qiu X, Harly C, Radtke F, et al. The e protein-TCF1 axis controls gammadelta T cell development and effector fate. Cell Rep (2021) 34(5):108716. doi: 10.1016/j.celrep.2021.108716
59. Hosoya T, D'Oliveira Albanus R, Hensley J, Myers G, Kyono Y, Kitzman J, et al. Global dynamics of stage-specific transcription factor binding during thymocyte development. Sci Rep (2018) 8(1):5605. doi: 10.1038/s41598-018-23774-9
60. Hai Y, Sun M, Niu M, Yuan Q, Guo Y, Li Z, et al. BMP4 promotes human sertoli cell proliferation via Smad1/5 and ID2/3 pathway and its abnormality is associated with azoospermia. Discovery Med (2015) 19(105):311–25.
61. Micheli L, Ceccarelli M, Farioli-Vecchioli S, Tirone F. Control of the normal and pathological development of neural stem and progenitor cells by the PC3/Tis21/Btg2 and Btg1 genes. J Cell Physiol (2015) 230(12):2881–90. doi: 10.1002/jcp.25038
62. Hyndman BD, Thompson P, Denis CM, Chitayat S, Bayly R, Smith SP, et al. Mapping acetylation sites in E2A identifies a conserved lysine residue in activation domain 1 that promotes CBP/p300 recruitment and transcriptional activation. Biochim Biophys Acta (2012) 1819(5):375–81. doi: 10.1016/j.bbagrm.2011.11.013
63. Williams CJ, Naito T, Arco PG, Seavitt JR, Cashman SM, De Souza B, et al. The chromatin remodeler mi-2beta is required for CD4 expression and T cell development. Immunity (2004) 20(6):719–33. doi: 10.1016/j.immuni.2004.05.005
64. Teachenor R, Beck K, Wright LY, Shen Z, Briggs SP, Murre C. Biochemical and phosphoproteomic analysis of the helix-loop-helix protein E47. Mol Cell Biol (2012) 32(9):1671–82. doi: 10.1128/MCB.06452-11
65. Yoon SJ, Foley JW, Baker JC. HEB associates with PRC2 and SMAD2/3 to regulate developmental fates. Nat Commun (2015) 6:6546. doi: 10.1038/ncomms7546
66. Yi S, Huang X, Zhou S, Zhou Y, Anderson MK, Zuniga-Pflucker JC, et al. E2A regulates neural ectoderm fate specification in human embryonic stem cells. Development (2020) 147(23):1–14. doi: 10.1242/dev.190298
67. Dutta A, Venkataganesh H, Love PE. Epigenetic regulation of T cell development. Int Rev Immunol (2021) 1–9. doi: 10.1080/08830185.2021.2022661
68. Lu KT, Kanno Y, Cannons JL, Handon R, Bible P, Elkahloun AG, et al. Functional and epigenetic studies reveal multistep differentiation and plasticity of in vitro-generated and in vivo-derived follicular T helper cells. Immunity (2011) 35(4):622–32. doi: 10.1016/j.immuni.2011.07.015
69. Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell (2012) 149(2):467–82. doi: 10.1016/j.cell.2012.01.056
70. Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, et al. A validated regulatory network for Th17 cell specification. Cell (2012) 151(2):289–303. doi: 10.1016/j.cell.2012.09.016
71. Zhou W, Yui MA, Williams BA, Yun J, Wold BJ, Cai L, et al. Single-cell analysis reveals regulatory gene expression dynamics leading to lineage commitment in early T cell development. Cell Syst (2019) 9(4):321–37.e9. doi: 10.1016/j.cels.2019.09.008
Keywords: thymus, T-cell development, transcription factor, chromatin, Id proteins, E proteins
Citation: Anderson MK (2022) Shifting gears: Id3 enables recruitment of E proteins to new targets during T cell development and differentiation. Front. Immunol. 13:956156. doi: 10.3389/fimmu.2022.956156
Received: 29 May 2022; Accepted: 08 July 2022;
Published: 02 August 2022.
Edited by:
Yi Ding, National Cancer Institute (NIH), United StatesReviewed by:
Ellen V. Rothenberg, California Institute of Technology, United StatesZhongmei Zhang, National Cancer Institute (NIH), United States
Copyright © 2022 Anderson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michele K. Anderson, bWFuZGVyc29Ac3JpLnV0b3JvbnRvLmNh