
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 September 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.955654
This article is part of the Research TopicPlatelets and Megakaryocytes dysfunctions in Infectious DiseasesView all 8 articles
 Mengqi Xiang1
Mengqi Xiang1 Xiaoming Wu1Haijiao Jing1Langjiao Liu1Chunxu Wang1Yufeng Wang1Valerie A. Novakovic2
Xiaoming Wu1Haijiao Jing1Langjiao Liu1Chunxu Wang1Yufeng Wang1Valerie A. Novakovic2 Jialan Shi1,2,3*
Jialan Shi1,2,3*Patients with COVID-19 often have hypoxemia, impaired lung function, and abnormal imaging manifestations in acute and convalescent stages. Alveolar inflammation, pulmonary vasculitis, and thromboembolism synergistically damage the blood-air barrier, resulting in increased pulmonary permeability and gas exchange disorders. The incidence of low platelet counts correlates with disease severity. Platelets are also involved in the impairment of pulmonary microcirculation leading to abnormal lung function at different phases of COVID-19. Activated platelets lose the ability to protect the integrity of blood vessel walls, increasing the permeability of pulmonary microvasculature. High levels of platelet activation markers are observed in both mild and severe cases, short and long term. Therefore, the risk of thrombotic events may always be present. Vascular endothelial injury, immune cells, inflammatory mediators, and hypoxia participate in the high reactivity and aggregation of platelets in various ways. Microvesicles, phosphatidylserine (PS), platelets, and coagulation factors are closely related. The release of various cell-derived microvesicles can be detected in COVID-19 patients. In addition to providing a phospholipid surface for the synthesis of intrinsic factor Xase complex and prothrombinase complex, exposed PS also promotes the decryption of tissue factor (TF) which then promotes coagulant activity by complexing with factor VIIa to activate factor X. The treatment of COVID-19 hypercoagulability and thrombosis still focuses on early intervention. Antiplatelet therapy plays a role in relieving the disease, inhibiting the formation of the hypercoagulable state, reducing thrombotic events and mortality, and improving sequelae. PS can be another potential target for the inhibition of hypercoagulable states.
Many COVID-19 patients, in both acute and convalescent stages, have symptoms of shortness of breath and dyspnea, accompanied by hypoxemia, impaired lung function (the diffusing capacity of the lung for carbon monoxide (DLCO) <80%), and abnormal imaging manifestations (interstitial infiltration, ground-glass opacity (GGO) and fibrosis) (1–5). Pulmonary diffusing dysfunction is ultimately attributed to damage to the blood-air barrier, including abnormalities in alveolar ventilation and pulmonary blood perfusion. Alveolar inflammation, pulmonary vasculitis, and thrombosis/embolism all have detrimental effects on pulmonary alveoli and microcirculation. Under the migration of macrophages that exist in the alveolar interstitium, pulmonary inflammation first appears on the alveolar side of the blood-air barrier. Further infection of type II alveolar epithelial cells by a large number of viruses leads to various immune cells, inflammatory mediators, and cytokines interacting to initiate inflammatory cascades, forming a cytokine storm and allowing more extensive infiltration of the alveoli (6, 7). In severe cases, the immune system is compromised, and the population of lymphocytes decreases or even collapses. The immune response can spread to vascular endothelial cells, allowing SARS-CoV-2 to directly invade angiotensin-converting enzyme 2 (ACE2) receptor-expressing vascular endothelial cells. Local vasoconstriction occurs after pulmonary vasculitis. The exposed collagen at the injury site promotes platelet activation, initiates the coagulation cascade, and eventually forms microthrombi or large thrombi in situ, resulting in increased intravascular pressure (8, 9). The mechanism by which platelets lead to impaired lung function has not been elucidated in detail. The function of platelets is complex. Platelet count and activity may be abnormal during both the acute and convalescent phases of COVID-19. The influence of virus-induced microenvironmental changes which induce platelets into a procoagulant phenotype appear to exist during many stages of COVID-19. COVID-19 patients have been shown to exhibit high levels of platelet-derived microvesicles (pEVs), similar to patients with acute respiratory distress syndrome (ARDS) and H1N1. Therefore, this review focuses on the effect of platelets on pulmonary microcirculation (including vascular endothelial injury and thrombosis) and explores the capability of endothelial injury, inflammation, and hypoxia to promote the formation of procoagulant platelets throughout COVID-19 disease progression. Additionally, we emphasize the role of phosphatidylserine (PS)-positive microvesicles (MVs) and discuss the treatment for COVID-19 hypercoagulability and thrombosis.
In SARS-CoV-2 infection, platelet consumption induced by pulmonary or extrapulmonary thrombosis/embolism and platelet destruction by immune-associated complement and antibodies are directly responsible for thrombocytopenia (9, 10). However, in the early stage of COVID-19, platelet consumption results from the formation of microthrombi, which then stimulates the release of new platelets by megakaryocytes from the bone marrow in a system of feedback compensation, resulting in a slight or no reduction in platelet count. In a retrospective, observational study conducted by Barett et al., high immature platelet fraction and increased mean platelet volume were found in patients with COVID-19, indicating an accelerated rate of platelet renewal (11). In other comparative studies, similar increased immature platelet count was shown in patients with COVID-19 (12, 13). For severe and critically ill patients, the consumption of platelets and coagulation factors is massively increased due to platelet activation by cytokine storm, causing severe endothelial damage leading to enhanced contact between platelets and endothelial cells, white blood cells and erythrocytes, which promotes extensive formation of thrombi. When the bone marrow cannot mobilize and release sufficient platelets, exhaustion of platelet levels occurs (14). Thus, low platelet count has been shown to be associated with the severity of COVID-19 (15). The results of a retrospective cohort study showed that 36.2% of COVID-19 patients had low platelet counts (< 150*109/L) on admission. With increasing disease severity, the incidence of low platelet counts increased. And platelet levels were lower in critically ill patients than in severe patients (16). Chen et al. studied low platelet counts that occurred 14 days after symptom onset (known as delayed-phase thrombocytopenia) and showed that 11.8% of patients developed transient delayed-phase thrombocytopenia (17). This could be a combination of the immune system and impaired megakaryocyte growth. Townsend et al. evaluated coagulation markers four months after SARS-CoV-2 infection, and more than 90% of convalescent patients had platelet counts in the normal range (18). Interestingly, a rare complication of adenovirus-based vaccination against SARS-CoV-2, vaccine-induced immune thrombotic thrombocytopenia (VITT), also leads to low platelet counts (19–23). Additionally, in a self-controlled case series study, low platelet counts and an increased risk of thrombotic events were observed following initial vaccination with ChAdOx1 nCoV-19 and BNT162b2 mRNA vaccines (24). The specific pathogenesis in these phenomena is not fully understood, although initial research into VITT suggests that it is mediated through anti-platelet factor 4 antibodies that activate platelets leading to thrombosis and consumptive coagulopathy (25). It may also be related to variant proteins produced in human cells in response to the mRNA carried by the vector (26).
During inflammation, platelets not only coordinate the chemotaxis of leukocytes but also maintain the integrity of the endothelial barrier at the injury site in what is termed “inflammation-related hemostasis”. In fact, evidence has shown that platelets protect the integrity of microcirculation. For example, in mice with acute skin inflammation, bleeding events occurred early and persisted in the platelet-depleted group, while only a few ecchymoses occurred in the normal platelet group (27). Another study found that mice deficient in the platelet-derived angiogenesis protein 1 (ANGPT1) gene had increased microsphere leaks at the site of leukocyte extravasation, with similar results using an anti-glycoprotein Ibα antibody. The authors propose that platelet ANGPT1 interacts with endothelial Tie-2 to prevent passive plasma leakage (28). The permeability of pulmonary microcirculation is of particular concern. The blood-air barrier, where the lungs communicate with the outside world, is highly susceptible to inflammation when pathogens invade, and studies have shown that megakaryocytes exist in large quantities in the pulmonary circulation where they are able to dynamically release platelets in the lungs (29–31). Histological and experimental examination of patients with inflammatory lung disease revealed an increased number of peripheral blood megakaryocytes (32). Postmortem reports of non-survivors of COVID-19 also showed significantly elevated megakaryocyte levels, which may be related to the promotion of thrombopoietin production and upregulation of JAK-STAT signaling by the high inflammatory response (33). At the same time, shortened megakaryopoiesis and retention of megakaryocytes in the lung result in no concomitant increase in platelet count (34). In studies of isolated rabbit and sheep lungs, an artificially low platelet state increased pulmonary microvascular permeability, leading to pulmonary edema and fluid accumulation (35, 36). Pulmonary capillary leakage and low platelet counts are often observed together in critically ill ARDS patients (37, 38). Human platelets have also been observed to reduce vascular endothelial permeability in vitro (39). In another ARDS study, the use of the platelet C-type lectin-like 2 (CLEC-2) ligand (podoplanin) protected alveolar-capillary integrity (40). Therefore, the protective effect of platelets on the vascular endothelium may be underappreciated in acute lung infection. Prior to the occurrence of pulmonary capillary leakage, platelets, together with endothelial cells, can protect the vascular wall structure of pulmonary microcirculation in the early immune response to avoid the leakage of blood components into the interstitium and alveolar cavity and to ensure the normal lung function.
In severe and critical COVID-19 patients, the incidence of severe pulmonary inflammation is high, often accompanied by the pathological manifestations of diffuse alveolar injury including the leakage of plasma components into the alveolar cavity (41). At this time, the viral load increases sharply, and abundant immune cells are recruited and activated to release inflammatory mediators and cytokines, which damage alveolar epithelial cells and vascular endothelial cells. Although cytokine storm is an important factor in causing severe pneumonia and pulmonary dysfunction (6, 7, 42), the weakening of vascular endothelial protection caused by the reduction of platelets also contributes. The integrity of the blood vessel wall cannot be maintained, and the permeability of the pulmonary microcirculation is further increased by the combined effects of viruses, inflammation, and increased pulmonary vascular pressure. With the gradual accumulation of fluid, the effective volume of the alveolar space, the tidal volume and vital capacity decrease, worsening dyspnea. Airflow leads to the evaporation of water in the alveolar cavity, causing residual plasma proteins and necrotic alveolar epithelial debris to form a hyaline membrane (43). Even if sufficient oxygen can be ensured, the pulmonary condition cannot be reversed due to the destruction of the lung structure and the reduction of the effective volume of the alveolar space (43, 44).
Physiological hemostasis is a repair process involving platelets, coagulation cascade, and a variety of factors that form platelet-fibrin complexes locally at the damaged blood vessel site (45). Thrombosis is a disordered pathological process (46, 47). Platelets bind to von Willebrand factor (vWF), which is attached to subcutaneous collagen fibers via glycoprotein (GP) Ib/IX/V complexes. Platelet granules are then released. Further, adenosine diphosphate (ADP), thromboxane A2 (TXA2), 5-hydroxytryptamine, and platelet factor IV are released from the granules. Platelets aggregate due to the action of GP IIb/IIIa, Ca2+, and fibrinogen. Under the synergistic effect of thrombin, ADP, TXA2, and intrinsic and extrinsic coagulation pathways, platelets aggregate inside the growing thrombus, which locally seals off damaged blood vessels. The risks of thrombosis and embolism are important to consider in pulmonary syndrome. They affect the ventilation/perfusion ratio and cause hypoxia in serious cases. From the acute phase to the recovery phase, platelet activity should be continuously monitored in COVID-19 patients. A single-center cross-sectional study showed that the platelet activation marker soluble CD40 ligand (sCD40L) was significantly higher in intensive care unit (ICU) patients than in non-COVID-19 and non-hospitalized controls (48). Barrett et al. found that in hospitalized patients with COVID-19, platelet activation markers (such as thromboxane B2 (TXB2), p-selectin, and sCD40L) were associated with increased mortality, with plasma TXB2 increasing the risk of thrombosis (Figure 1A) (49). Alterations in platelet phenotype can also occur in patients with mild symptoms and may even persist into the recovery phase. McCafferty et al. compared the phenotype of platelets in SARS-CoV-2 positive group and SARS-CoV-2 exposed individuals and found an increase in both PAC1-positive and CD62P-positive platelets (50). The data confirm platelet activation and suggest that patients with mild disease may be at risk for thrombosis during both the acute and convalescent periods. These studies indicate that the risk of thrombotic events may always exist in COVID-19 patients whether with mild or severe disease in the short or long term. The presence of SARS-CoV-2 particles and viral proteins in a significant fraction of platelets has been detected in severe COVID-19 patients, and virus-platelet interactions amplify platelet activation. As part of the defense mechanisms against SARS-CoV-2, platelets recognize intracellular viral components to degrade them via autophagosomes and autophagolysosomes (51). Platelets containing viruses may also originate from bone marrow and lung megakaryocytes infected by SARS-CoV-2 and become infectious through being captured by lung macrophages (34). SARS-CoV-2 appears to have the ability to impact mitochondria in platelets and lead to thrombotic or bleeding events depending on the extent of mitochondrial damage (52).
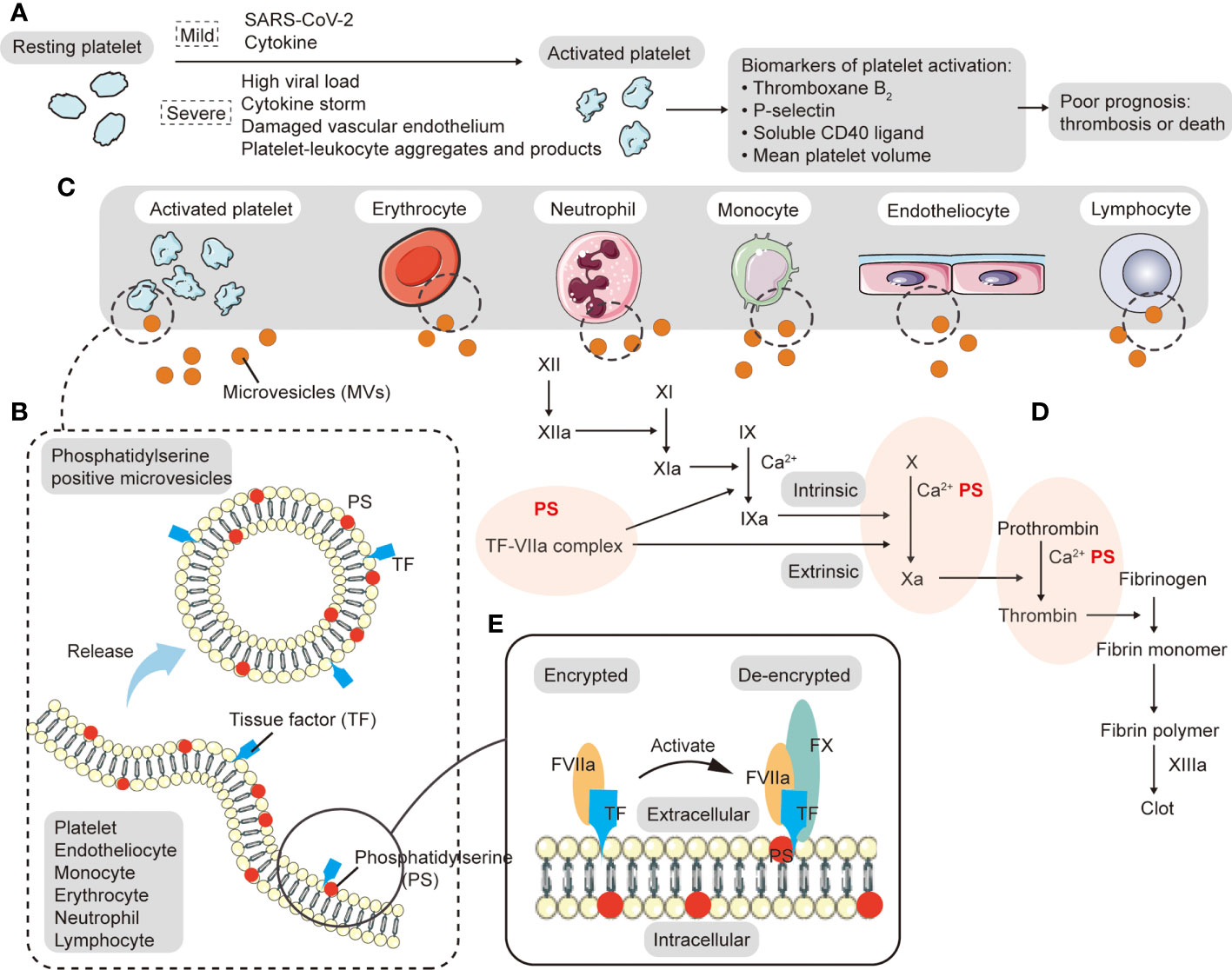
Figure 1 Origin and procoagulant activity of microvesicles (MVs). (A) During acute local inflammation, invasive SARS-CoV-2 triggers the immune response in which neutrophils and monocytes are recruited and cytokines activate platelets. When COVID-19 worsens, platelets are easily activated by high viral load, cytokine storm, damaged vascular endothelium, and platelet-leukocyte aggregates, forming widespread (micro-) blood clots in the body, which can lead to poor circulation, hypoxemia and multiple organ failure. Platelet activation markers (such as thromboxane B2, p-selectin, soluble CD40 ligand, and mean platelet volume) are associated with increased mortality and the risk of thrombosis. (B) MVs are plasma-membrane-derived particles that can be generated by direct budding or fission when cytoskeletal proteins are disrupted. Both tissue factor (TF) and phosphatidylserine (PS) on the surface of MVs can promote the generation of the procoagulant state. (C) Various types of cells release MVs in the circulation, including platelets, endothelial cells, red blood cells, neutrophils, lymphocytes, and monocytes, among which platelets are the most important. (D) In addition to providing a phospholipid surface for the synthesis of the intrinsic and extrinsic tenase complex (FIXa-FVIIa-Ca2+-PS) and prothrombinase complex (FXa-FVa-Ca2+-PS), PS can also facilitate the decryption of TF and promote TF-FVIIa complex formation to participate in extrinsic coagulation. The TF-FVIIa complex catalyzes the activation of FX and FIX. (E) PS can facilitate the decryption of TF.
In COVID-19, the classic cause of respiratory dysfunction is local or diffuse pulmonary inflammation. As the disease progresses chest radiographic findings change from patchy shadows in the lateral lung to bilateral pulmonary GGO (53). However, there are also studies reporting that patients with COVID-19 hypoxemia have good lung compliance but abnormal pulmonary blood perfusion (54, 55). In addition, autopsy results showed microthrombi around normal alveoli (9). These results emphasize the importance of thrombogenesis induced by activated platelets. In severe cases, pulmonary (micro)thrombosis occurs, and pulmonary capillary pressure increases, leading to pulmonary hypertension. The pressure difference between the two sides of the blood-air barrier pushes more plasma components into the alveolar cavity. Since the cardiopulmonary system is connected by blood vessels, pulmonary hypertension caused by vascular thrombosis can be transmitted to the heart. At the same time, cytokine storm and endothelial dysfunction also play a synergistic role in the formation of extrapulmonary complications. Of concern is that cardiovascular disease may further aggravate hypoxemia. In the acute phase, the compensatory manifestations of increased heart rate and enhanced myocardial contractility can aggravate myocardial oxygen consumption and accelerate the occurrence of myocardial dysfunction. In the decompensated phase, normal myocardial contraction and relaxation are impaired. With pulmonary circulation congestion and insufficient systemic circulation blood volume, patients may develop systemic ischemia and hypoxia, accompanied by decreased return blood volume.
Under physiological conditions, vascular endothelial cells have smooth surfaces, which inhibit platelet adhesion and fully cover the underlying collagen fibers. The endothelium releases prostacyclin (PGI2) and nitric oxide (NO) to inhibit platelet activation and prevents platelet amplification by NTPDase-1 (46). Elevated levels of endothelial stress products are often detected in the circulating blood of COVID-19 patients. Goshua et al. found that 80% of non-ICU patients had higher concentrations of vWF antigen than normal. ICU patients were associated with further elevated markers of vascular endothelial and platelet activation, including vWF antigen (565% vs. 278%) and soluble P-selectin (15.9 ng/mL vs. 11.2 ng/mL) (48). Another study indicated that among ICU patients with COVID-19, levels of endothelium biomarkers (such as soluble E-selectin, soluble P-selectin, angiogenin (Ang) 2, soluble intercellular adhesion molecule-1, and vWF) were higher in non-survivors (56). Fogarty et al. evaluated patients recovering from COVID-19 and found that vWF antigen, vWF propeptide, factor VIII and soluble thrombomodulin were significantly elevated (57). Persistent endothelial injury may be observed even in patients who are not hospitalized. These results suggest that activation of vascular endothelial cells may persist. In another study, Rauch et al. co-cultured human pulmonary microvascular endothelial cells (HPMVEC) in vitro with plasma from patients in acute and convalescent phase and observed that plasma from COVID-19 patients significantly reduced the activity of HPMVEC (58). Among them, the cytotoxicity of ICU patient plasma was the strongest, indicating that infected plasma can trigger microvascular endothelial cell damage.
In respiratory diseases, the alveolar side of the blood-air barrier can initiate the immune response through direct interaction with pathogens, while the vascular endothelial side is often affected by inflammation (59). At first, local damage occurs at the weak part of the blood-air barrier, followed by the aggregation of immune cells and cytokines. Cascading inflammatory reactions form a cytokine storm, which increases vascular endothelial permeability, accelerates the influx of inflammatory factors, and gradually increases the scope of vascular endothelial injury. A series of autopsy results of COVID-19 deaths revealed that viral components, such as SARS-CoV-2 inclusion bodies, were found in vascular endothelial cells. Studies have shown that immunocompromised patients are at risk for virus persistence, co-infection, and reinfection (60, 61), and the virus has the possibility of hiding in the body resulting in long-term symptoms (62). SARS-CoV-2 invades vascular endothelial cells and binds to ACE2 receptors, inducing a procoagulant state. ACE/ACE2 imbalance inhibits the conversion of angiotensin (Ang) II to Ang1-7. Ang II, which is generated by Ang I through ACE, then accumulates, causes vasoconstriction, endothelial activation, and thrombosis, affecting hemodynamic homeostasis (63–66). An experimental model of ACE2 knockout mice promotes vascular inflammation and platelet aggregation, while increased ACE2 can provide protection for the vascular endothelium (65).
After vascular endothelial cells are damaged, the factors (PGI2, NO, NTPDase-1), which inhibit platelet activation, aggregation, and expansion, decrease. Exposed basal membrane collagen enhances platelet adhesion by binding with platelet membrane GP Ib/IX/V complex via bridging molecule vWF. Numerous studies have shown that high levels of vWF are common in all stages of COVID-19 (48, 56, 57). Damaged endothelium can also promote the coagulation cascade downstream of platelets by expressing tissue factor (TF), entrapping other blood cells to form firm clumps. Moreover, the release of TF pathway inhibitors is reduced, promoting local coagulation. In a baboon model of sepsis, TF-induced coagulation cascades played a key role, and inhibition of TF effectively reduced multiple organ failure and mortality (67). Many studies have also demonstrated that the up-regulation of TF expression is related to the formation of immune thrombosis in COVID-19 (68, 69). A cross-sectional study found that thrombin-antithrombin III complex concentrations and FVIII activity were elevated in 90% of non-ICU patients, and significantly increased in ICU patients (48). From the perspective of hemodynamics, local vasoconstriction and slow blood flow are conducive to platelet adhesion when pulmonary vascular endothelial cells are damaged.
Pulmonary inflammation is the most common pathological change in COVID-19. In a follow-up study testing COVID-19 patients for 34 inflammatory-related substances, patients with mild symptoms showed higher levels of chemokine RANTES (CCL5), while patients with later-stage disease had significant increases in cytokines such as interleukin (IL) 6 and interferon-γ (IFN-γ) (70). Diffuse alveolar damage can be seen in many patients with severe and critical COVID-19. Inflammatory infiltration of the pulmonary interstitium and deposition of intravascular platelet-fibrin thrombi are important features. In COVID-19, the co-occurrence of inflammation and platelet thrombus on both sides of the blood-air barrier also suggests the close relationship between platelets and the immune system. A retrospective study by Chen et al. found that in severe COVID-19, inflammatory markers (lactate dehydrogenase, C-reactive protein, ferritin, procalcitonin, tumor necrosis factor α (TNF-α), IL-2R, IL-6, and IL-10) were significantly elevated and T-cell subsets (CD4+ T cells and CD8+ T cells) were reduced (71). Another autopsy study showed interstitial mononuclear inflammatory infiltration bilaterally in the liver and especially the lungs. The results indicated that the concentration of pro-inflammatory CCR6+ Th17 increased, enhancing cytotoxicity in CD8+ T cells (72). In the whole-body tissue atlas of COVID-19 established by Dorward et al., the lungs presented with diffuse alveolar damage and bronchopneumonia. Patchy vasculitis of the intima of small and medium pulmonary arteries was also seen, mainly infiltrated by MRP8+ monocytes and mixed with T cells and macrophages (73). A similar phenomenon was seen in SARS. Histopathology of severe SARS indicated a large number of macrophages in the alveoli and lung interstitium (74). Serological measurements have shown that cytokines (Th1-type interferon, inflammatory cytokines IL-1, IL-6, and IL-12) and chemokines (neutrophil chemokine IL-8, monocyte chemokine protein-1, and Th1 chemokine IFN-γ-inducible protein-10) are significantly increased (75). Accumulation of pathogenic monocytes/macrophages and elevated cytokine/chemokine was also detected in a mouse model of SARS-CoV (76). GGO caused by pulmonary inflammatory exudation is a common imaging manifestation in the acute phase of COVID-19 that can also appear in convalescent patients. In a cohort study of thoracic high-resolution computed tomography, GGO remained the most common symptom in previously hospitalized COVID-19 patients at six months of recovery (5). At another follow-up at 12 months, 76% of the severe group still had GGO (4).
Inflammation interacts with platelets, inducing a vicious cycle (77, 78). Inflammation/immunity is thought to be a potential promoter of platelet activation, causing the transition of normal platelets to a procoagulant phenotype and eventually forming thrombosis. Using in vitro experiments, Hottz et al. showed that monocyte subsets with high CD16 and low HLA-DR expression interacted with platelets to promote TF expression and secrete more TNF-α and IL-1β, amplifying inflammation in severe COVID-19 (79). Studies have indicated that toll-like receptor 4 signaling stimulates neutrophils to bind to activated platelets and then release neutrophil extracellular traps (77). Cell-free DNA activates the coagulation cascade; and histone components convert platelets, red blood cells, and endothelial cells to a procoagulant state (80). These need to be noted in COVID-19 patients with bacterial infection or sepsis. Both in vivo and in vitro experiments have proved that TNF-α/TNF-r1 signal and IL-1 can promote the expression of vascular endothelial vWF, and facilitate platelets adhesion to endothelial cells via GP Ib/lX/V. Then it transitions to a hypercoagulable state (81). In addition, inflammatory cytokines can also enhance the coagulation cascade downstream of the platelet-thrombus. For example, IL-1 and TNF-α can stimulate the synthesis of coagulation factors and activate monocytes and endothelial cells (82). Inflammation or cytokine storm can also indirectly promote the production of procoagulant platelets by damaging local or enlarged vascular endothelial cells. In addition, platelets enhance inflammatory responses, such as regulating immune cell function, promoting platelet-neutrophil aggregation, storing immunomodulatory molecules (transforming growth factor-β, IL-1, platelet-derived growth factor, and CC-chemokine ligand 5), and expressing Toll-like receptors to bind microorganisms (83, 84). The role of platelet-mediated vascular inflammation has been demonstrated in atherosclerosis and myocardial infarction (85, 86).
Dyspnea and hypoxemia can be used to assess disease progression and prognosis in COVID-19. Shortness of breath and dyspnea are common symptoms at admission. The majority of hospitalized patients present with mild hypoxemia, but some patients do not develop respiratory distress until partial arterial oxygen pressure (PaO2) drops to 60 mmHg (or lower). This indicates that even if the clinical symptoms are mild at admission, the patient’s condition may actually be more severe. In a study in Seattle, when patients were admitted, 53% had dyspnea, 86% had an abnormal initial chest X-ray, and 41% developed hypoxia (1). In another retrospective study, mortality was higher in patients with initial symptoms of respiratory distress, and hypoxemia was an independent predictor of in-hospital mortality (87). The long-term effects of SARS-CoV-2 on the lungs require continuous monitoring of the patients’ respiratory symptoms, lung imaging, and lung function tests. Mo et al. analyzed the lung function of discharged COVID-19 survivors. Of those studied, 30.4% of patients with mild pneumonia and 84.2% of patients with severe pneumonia showed impaired diffusion volume (2). A similar pattern was observed in patients 30 days after discharge. In patients discharged after severe disease, lung function was seriously impaired, with a higher proportion of DLCO and imaging abnormalities (3). A comparison of lung function at 6 and 12 months of discharge showed no significant improvement in dyspnea and diffuse lung injury, with 54% of the severe group having diffuse lung injury one year after discharge, compared to 23% of patients with milder illness (4, 5). Pulmonary fibrosis is another important cause of poor pulmonary function in convalescence. Pulmonary fibrosis is closely related to protein deposition in the extracellular stroma and fibroblast migration into muscle cells. Elevated levels of growth factor receptor B1 and respiratory virus infection are involved in this process (5, 88). Hypoxia can lead to inflammation-related gene transcription and aggravate endothelial injury through hypoxia inducible factor (HIF) and NF-Bĸ pathway (89). HIF-1α promotes the production of neutrophil extracellular traps, and cell-free DNA can activate the contact clotting pathway by providing negatively charged surfaces. Histone components can bind to platelets, red blood cells, and endothelial cells and then activate a procoagulant phenotype (80). In a hypoxic rat model, bleeding time was significantly shortened in the hypoxic exposure group. Coagulation tests showed reduced prothrombin time and activated partial thromboplastin time, as well as higher platelet reactivity and stronger interaction with fibrinogen (90). In other pulmonary syndromes such as chronic obstructive pulmonary disease and sleep apnea, hypoxia has been implicated in platelet hyperresponsiveness and aggregation (91, 92). The blood clots cause poor blood flow in the pulmonary microcirculation, so the vicious cycle of hypoxia and thrombosis can exacerbate pulmonary dysfunction.
MVs are plasma-membrane-derived particles that can be generated by direct budding or fission when cytoskeletal proteins are disrupted (Figure 1B), and they are mainly derived from activated, injured and dead cells. In a high intracellular Ca2+ environment, phospholipid translocation enzymes (floppase and flippase) are inactivated. PS is exposed to the outer surface of the cell membrane by scramblase, resulting in a loss of plasma membrane asymmetry and release of MVs (93–95). Various types of cells release MVs in the circulation, including platelets, endothelial cells, red blood cells, neutrophils, lymphocytes, and monocytes (Figure 1C) (96, 97). During acute local inflammation, invasive SARS-CoV-2 triggers the immune response in which neutrophils and monocytes are recruited and cytokines activate platelets. With viral clearance and cytokine production, neutrophils, monocytes, lymphocytes, and platelets undergo activation, damage, and death, releasing MVs, while lymphocytes primarily produce killer toxins and promote antibody production. Rausch et al. found that the number of PS+ peripheral blood mononuclear cells (PBMCs) in patients within weeks of the initial diagnosis of COVID-19 was significantly higher than that in healthy controls or recovered patients (98). When COVID-19 worsens, platelets are easily activated by high viral load, cytokine storm, damaged vascular endothelium, and platelet-leukocyte aggregates, forming widespread (micro-) blood clots in the body, which can lead to poor circulation, hypoxemia and multiple organ failure (Figure 1A). These platelets have the characteristics of large average volume, high activity and short life (as shown by an increased proportion of immature platelets). In response to stimulation by massive amounts of cytokines, platelets release large numbers of platelet extracellular vesicles (pEVs). Such pEVs are the main circulating MVs in prethrombotic and inflammatory diseases (99). Zaid et al. showed that pEVs concentrations were higher in non-severe or severe COVID-19 patients than in healthy controls (100). By grouping surface antigens, Balbi et al. found that in SARS-CoV-2 infection, most MVs originated from either the endothelium or platelets (101). Flaumenhaft et al. demonstrated that megakaryocytes also produce MVs directly, mainly CD41+, CD42b+ and PS-expressed MVs (102). MVs can also be found in endotheliocytes and erythrocytes that have been damaged in the thrombo-inflammatory environment and then cleared by various immune cells. Similarly elevated levels of MVs have been seen in other lung diseases and viral infections. For example, compared with hydrostatic edema, ARDS had a higher concentration of alveolar epithelial MVs which showed stronger procoagulant effects (103). High levels of pEVs are also seen in other viral infections, such as influenza A and dengue fever. They promote pEVs release through an FcgRIIa-related mechanism and a CLEC-2-dependent manner, respectively (104, 105).
Both TF and PS on the surface of MVs can promote the generation of the procoagulant state (Figure 1B), which plays an important role in the activation of the coagulation cascade and the formation of arterial/venous thrombosis. The TF-FVIIa complex catalyzes the activation of FX and FIX. A study analyzed MVs TF activity in COVID-19 patients, and found that SARS-CoV-2 may induce TF+ MVs release in moderate and severe disease. Results also indicated that MVs TF activity was correlated with coagulation markers (D-dimer, international normalized ratio, prothrombin, fibrinogen. and antithrombin levels), fibrinolytic levels (plasminase-antifibrinolytic complex), and endothelial activation markers (vWF) (106). In addition to providing a phospholipid surface for the synthesis of the intrinsic pathway factor Xase complex (FIXa-FVIIa-Ca2+-PS) and prothrombinase complex (FXa-FVa-Ca2+-PS) (Figure 1D), PS can also facilitate the decryption of TF and promote TF-FVIIa complex formation to participate in extrinsic coagulation (Figure 1E). Studies have shown that high PS content of about 30% enables the TF-FVIIa complex to exert the highest procoagulant activity (107). MVs have been associated with the initial formation of platelet thrombi in many animal models. For example, monocyte-derived TF+ MVs were involved in the accumulation of platelet-rich thrombi after laser-induced endothelial injury in mice (108). Another in vitro study showed that short duration (2-4 h) exposure to monocyte-derived MVs can lead to PS exposure on the surface of human umbilical vein endothelial cells and induce procoagulant activity and apoptosis of endothelial cells (109). Increased MVs may also predict the occurrence of thrombotic events. One study has shown that compared with healthy controls, non-severe COVID-19 group, and sepsis ICU group, platelet membrane potential depolarization, cytoplasmic Ca2+, and PS externalization were significantly higher in COVID-19 ICU patients, and were correlated with organ failure and increased D-Dimer (110). In addition, the frequency of PS+ and MVs-bound PBMCs in recovered patients was also statistically significantly higher than that in healthy donors, indicating that PS+ MVs+ PBMCs may be a predictor of long-term adverse prognosis in COVID-19 (98). In a prospective study conducted by Amabile et al., circulating MVs were an independent predictor of myocardial infarction and stroke in patients with end-stage renal failure (111). Pulmonary thrombotic events appear to accelerate the development of severe conditions. There is a close relationship between MVs, PS, platelets, and coagulation factors, and therefore special attention is needed.
In COVID-19, the most commonly used means of inhibiting hypercoagulability and preventing thrombotic events is anticoagulant therapy, such as unfractionated heparin, low molecular weight heparin, and direct oral anticoagulant drugs. As a part of comprehensive antithrombotic therapy, the ultimate goal of antiplatelet therapy is to control the downstream coagulation cascade by inhibiting platelet adhesion, aggregation, and release so as to inhibit thrombosis. In a multivariate analysis, Santoro et al. found that in-hospital treatment with antiplatelet drugs (e.g., aspirin, clopidogrel, ticlopidine, prasugrel, or ticagrelor) reduces mortality, even in subgroups of patients in ICU (112). Results from a retrospective study showed that patients treated with aspirin had lower cumulative morbidity during hospitalization than those without antiplatelet drugs, whether routinely used in critically ill patients or recommended for all hospitalized patients (113). Another randomized controlled trial indicated a decrease in the incidence of thrombosis (4.6% vs. 5.3%) and a 1.2% improvement in survival in the aspirin group (150 mg daily) compared with the usual care group (114). Some antiplatelet agents also have antiviral, anti-inflammatory, and anti-cytokine storm mitigation effects. For example, aspirin irreversibly inhibits cycloxygenase and inhibits inflammatory effects related to the nuclear transcription of Kappa B (115). P2Y12 inhibitors exert anti-inflammatory effects by attenuating cytokine release, platelet-leukocyte interaction, and the formation of neutrophil extracellular traps (116). Under the combined action of anti-platelet and anti-inflammatory treatments, platelets, other blood cells, vascular endothelial, and alveolar epithelial cells are less damaged. The decrease in phosphatidylserine exposure and MVs release inhibits the decryption of tissue factor and the formation of intrinsic and extrinsic tenase and prothrombinase complexes, thus indirectly inhibiting the coagulation cascade triggered by phosphatidylserine and blocking the formation of thrombosis.
As for the controversies about the efficacy of antiplatelet drugs, different conclusions may be drawn based on the bias in clinical trials, such as patients with different disease severity, the phase and duration of drug use, combinations of other drugs, diverse primary outcomes, and so on (Table 1). Analysis of a retrospective study showed no association of antiplatelet therapy (aspirin and/or P2Y12 receptor antagonists) with survival in hospitalized patients with COVID-19 (119). In a randomized controlled study, in-hospital antiplatelet therapy was less likely to improve organ support-free days within 21 days in critically ill patients, but it improved the probability of survival to discharge (97%) and survival over 90 days (99.7%) (120). Berger et al. focused on non-critically ill hospitalized patients with COVID-19, and found no improvement in the organ support–free days within 21 days and no significant differences in the incidence of thrombotic events or in-hospital deaths between the additional P2Y12 inhibitor group and the control group with therapeutic anticoagulation alone (121). Considering that the lifespan of platelets is only 7-9 days, the late intervention of antiplatelet therapy may miss the opportunity to modulate platelet activation and fail to achieve the expected efficacy. Analysis of an observational cohort study showed that patients receiving aspirin (81 mg daily), clopidogrel (75 mg daily), dipyridamole (200 mg BID), ticagrelor (90 mg BID), or prasugrel (10 mg daily) before admission could reduce the incidence of pulmonary embolism and death (117). In another study, patients who used aspirin (median dose: 81 mg; median treatment duration: 6 days) early in treatment (24 hours after admission or seven days before admission) had lower rates of mechanical ventilation (35.7% vs. 48.4%) and ICU admission (38.8% vs. 51.0%) (118). However, Corrochan et al. rejected the efficacy of early antiplatelet agents and found that pre-admission treatment with anticoagulants and antiplatelet agents did not significantly improve mortality and thrombotic events (122). Results in studies on the early use of antiplatelet agents vary widely. In addition to differences in study design, a variety of measures of adverse outcomes, and discussion of single results versus combined results can have an impact. Another reason for emphasizing early treatment is that by inhibiting (micro)thrombosis, hypoxia due to insufficient pulmonary microcirculation can be avoided. At the same time, the pulmonary vascular pressure can be maintained in the normal range to avoid pulmonary edema caused by the large pressure difference between the two sides of the blood-air barrier. Therefore, it can reduce the incidence of lung consolidation and sequela. Currently, in convalescent patients, no guidelines recommend routine use of antiplatelet agents in patients without thrombotic risk factors.
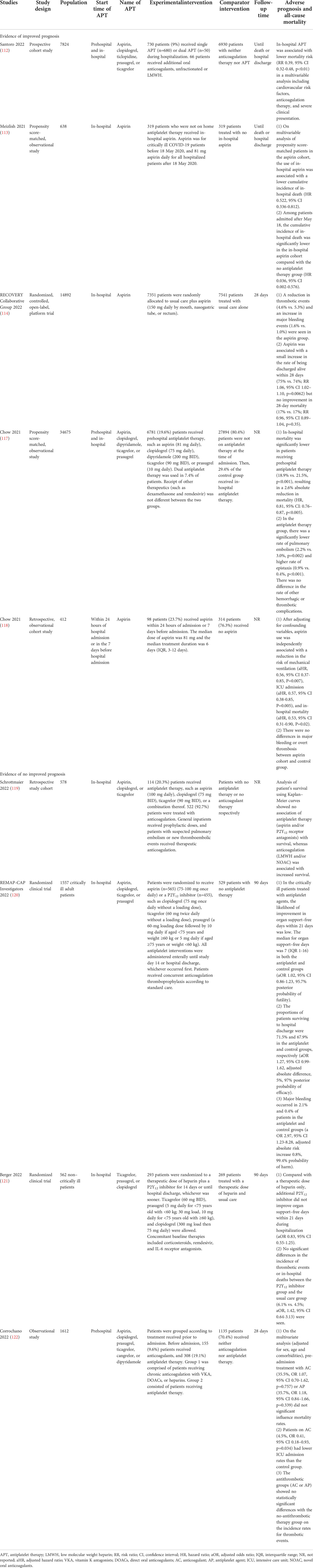
Table 1 Summary of studies on antiplatelet therapy in COVID-19.
Since the role of anticoagulants in improving hypercoagulability and preventing thrombosis in COVID-19 has been widely recognized, antiplatelet therapy is more often used as a supplementary treatment. Many guidelines recommend therapeutic doses of low molecular weight heparin for hospitalized, non-critically ill patients, and some narrow the patient population based on oxygen needs or bleeding risk. Prophylactic-intensity anticoagulant therapy for critically ill patients has been widely recommended (123–127). The formation of thrombi or extensive microthrombi consumes a large number of coagulation factors and platelets, leading to a hypofibrinolytic state during which antiplatelet or anticoagulation therapy would be less effective. Thus studies that administer therapeutic anticoagulation later in disease progression may not see a significant extension of survival time or improvement in survival rate. Coagulation abnormalities may exist for a long time, from the acute stage to the sequelae stage. We believe that it is of great importance to carry out comprehensive antithrombotic therapy in the early stage of COVID-19 to prevent thrombosis or remove (micro) thrombosis before there is a significant depletion of coagulation factors. If untreated early, hypercoagulation or thrombosis may cause a rapid increase in D-dimer, along with higher levels of fibrinogen, vWF, and P-selectin (14). When entering the period of severe and critical illness, cytokine storms, severe endothelial damage, intrapulmonary and extrapulmonary thrombosis, and consumption of platelets and clotting factors together lead to a significant risk of bleeding. Some studies of long COVID have detected higher D-dimer levels than normal (128). In addition, multiple studies have shown that patients may experience impaired fibrinolytic function during long COVID, with elevated plasminogen activator inhibitor-1 levels and decreased thrombi solubility (129–132). Buonsenso et al. found that anticoagulant therapy improved the quality of life in patients with poor pulmonary microcirculation on SPECT/CT during long COVID, suggesting extensive cell death, multiple tissue injury, and organ failure (133). The above data indicates that the improvement of hypercoagulability is very promising for the improvement of sequelae. However, the current guidelines do not recommend routine prophylactic doses of anticoagulant after discharge, with the exception of the National Institute for Health and Care Excellence (NICE) guidelines which recommend prophylaxis last for at least 7 days after discharge (125). Exposed PS may be another potential target for inhibiting hypercoagulable states. Studies have shown that PS can act as a signal to promote a hypercoagulable state in COVID-19 (98). In addition to being sensitive probes and effective PS blockers, lactadherin and annexin V act as anticoagulants by blocking activation of the clotting cascade in vitro and in vivo. Lactadherin inhibits intrinsic pathway factor Xase complex and prothrombinase complex by competing with coagulation factors V and VIII for PS binding sites. It also affects the formation of TF-FVIIa complex and inhibits the activation of FII. In contrast, annexin V is unable to inhibit factor Xase complex and has weak inhibition of the TF-FVIIa complex (134, 135). Dasgupta et al. demonstrated lactadherin-mediated clearance of PS+ pEVs in the circulation and observed an increase in the number of pEVs and thrombin production of more than 2 times in lactadherin-deficient mice (136). Therefore, the study of improving the procoagulant state by inhibiting PS can be viewed as a new direction for the treatment of COVID-19.
Low platelet counts and high platelet activity have been seen in both mild and severe COVID-19 patients and correlated with disease severity. For survivors, studies have also observed abnormal platelet counts and platelet activity from the acute phase to the recovery phase. This indicates that platelet-related risk factors for exacerbation may persist, which may not only aggravate disease progression and enhance the risk of thrombosis/embolism events and death, but also increase the incidence of sequelae in discharged patients. Therefore, antiplatelet therapy is still very meaningful, even while the efficacy for patients at different stages may vary. It is ideal to control platelet activation at an early stage, which plays a role in relieving the disease, inhibiting the formation of a hypercoagulable state, reducing thrombotic events and mortality, and improving sequelae.
MX prepared figures and wrote the manuscript. XW, HJ, LL, CW, and YW provided data and comments. JS came up with the project, designed the study, and contributed to successive drafts. VN reviewed this manuscript. All authors read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
We thank all participants for their contribution to our study and the reviewers for the suggestions provided.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Buckner FS, McCulloch DJ, Atluri V, Blain M, McGuffin SA, Nalla AK, et al. Clinical features and outcomes of 105 hospitalized patients with COVID-19 in Seattle, Washington. Clin Infect Dis (2020) 71(16):2167–73. doi: 10.1093/cid/ciaa632
2. Mo X, Jian W, Su Z, Chen M, Peng H, Peng P, et al. Abnormal pulmonary function in COVID-19 patients At time of hospital discharge. Eur Respir J (2020) 55(6):2001217. doi: 10.1183/13993003.01217-2020
3. Huang Y, Tan C, Wu J, Chen M, Wang Z, Luo L, et al. Impact of coronavirus disease 2019 on pulmonary function in early convalescence phase. Respir Res (2020) 21(1):163. doi: 10.1186/s12931-020-01429-6
4. Huang L, Yao Q, Gu X, Wang Q, Ren L, Wang Y, et al. 1-year outcomes in hospital survivors with COVID-19: A longitudinal cohort study. Lancet (2021) 398(10302):747–58. doi: 10.1016/S0140-6736(21)01755-4
5. Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet (2021) 397(10270):220–32. doi: 10.1016/S0140-6736(20)32656-
6. Caricchio R, Gallucci M, Dass C, Zhang X, Gallucci S, Fleece D, et al. Preliminary predictive criteria for COVID-19 cytokine storm. Ann Rheum Dis (2021) 80(1):88–95. doi: 10.1136/annrheumdis-2020-218323
7. Mangalmurti N, Hunter CA. Cytokine storms: Understanding COVID-19. Immunity (2020) 53(1):19–25. doi: 10.1016/j.immuni.2020.06.017
8. Iba T, Connors JM, Levy JH. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflammation Res (2020) 69(12):1181–9. doi: 10.1007/s00011-020-01401-6
9. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
10. Mei H, Luo L, Hu Y. Thrombocytopenia and thrombosis in hospitalized patients with COVID-19. J Hematol Oncol (2020) 13(1):161. doi: 10.1186/s13045-020-01003-z
11. Barrett TJ, Bilaloglu S, Cornwell M, Burgess HM, Virginio VW, Drenkova K, et al. Platelets contribute to disease severity in COVID-19. J Thromb Haemost (2021) 19(12):3139–53. doi: 10.1111/jth.15534
12. Cohen A, Harari E, Cipok M, Laish-Farkash A, Bryk G, Yahud E, et al. Immature platelets in patients hospitalized with covid-19. J Thromb Thromb (2021) 51(3):608–16. doi: 10.1007/s11239-020-02290-6
13. Welder D, Jeon-Slaughter H, Ashraf B, Choi SH, Chen W, Ibrahim I, et al. Immature platelets as a biomarker for disease severity and mortality in COVID-19 patients. Br J Haematol (2021) 194(3):530–6. doi: 10.1111/bjh.17656
14. Grobler C, Maphumulo SC, Grobbelaar LM, Bredenkamp JC, Laubscher GJ, Lourens PJ, et al. Covid-19: The rollercoaster of Fibrin(Ogen), d-dimer, Von willebrand factor, p-selectin and their interactions with endothelial cells, platelets and erythrocytes. Int J Mol Sci (2020) 21(14):5168. doi: 10.3390/ijms21145168
15. Yang X, Yang Q, Wang Y, Wu Y, Xu J, Yu Y, et al. Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost (2020) 18(6):1469–72. doi: 10.1111/jth.1484
16. Liao D, Zhou F, Luo L, Xu M, Wang H, Xia J, et al. Haematological characteristics and risk factors in the classification and prognosis evaluation of COVID-19: A retrospective cohort study. Lancet Haematol (2020) 7(9):e671–8. doi: 10.1016/S2352-3026(20)30217-9
17. Chen W, Li Z, Yang B, Wang P, Zhou Q, Zhang Z, et al. Delayed-phase thrombocytopenia in patients with coronavirus disease 2019 (COVID-19). Br J Haematol (2020) 190(2):179–84. doi: 10.1111/bjh.16885
18. Townsend L, Fogarty H, Dyer A, Martin-Loeches I, Bannan C, Nadarajan P, et al. Prolonged elevation of d-dimer levels in convalescent COVID-19 patients is independent of the acute phase response. J Thromb Haemost (2021) 19(4):1064–70. doi: 10.1111/jth.15267
19. Venkatesan P. Do vaccines protect from long COVID? Lancet Respir Med (2022) 10(3):e30. doi: 10.1016/S2213-2600(22)00020-0
20. Ledford H. Do vaccines protect against long COVID? what the data say. Nature (2021) 599(7886):546–8. doi: 10.1038/d41586-021-03495-2
21. Pavord S, Scully M, Hunt BJ, Lester W, Bagot C, Craven B, et al. Clinical features of vaccine-induced immune thrombocytopenia and thrombosis. N Engl J Med (2021) 385(18):1680–9. doi: 10.1056/NEJMoa2109908
22. Sangli S, Virani A, Cheronis N, Vannatter B, Minich C, Noronha S, et al. Thrombosis with thrombocytopenia after the messenger RNA-1273 vaccine. Ann Intern Med (2021) 174(10):1480–2. doi: 10.7326/L21-0244
23. Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med (2021) 384(22):2092–101. doi: 10.1056/NEJMoa2104840
24. Hippisley-Cox J, Patone M, Mei XW, Saatci D, Dixon S, Khunti K, et al. Risk of thrombocytopenia and thromboembolism after covid-19 vaccination and SARS-CoV-2 positive testing: Self-controlled case series study. BMJ (2021) 374:n1931. doi: 10.1136/bmj.n1931
25. Scully M, Singh D, Lown R, Poles A, Solomon T, Levi M, et al. Pathologic antibodies to platelet factor 4 after ChAdOx1 nCoV-19 vaccination. N Engl J Med (2021) 384(23):2202–11. doi: 10.1056/NEJMoa2105385
26. Sadoff J, Davis K, Douoguih M. Thrombotic thrombocytopenia after Ad26.COV2.S vaccination - response from the manufacturer. N Engl J Med (2021) 384(20):1965–6. doi: 10.1056/NEJMc2106075
27. Ho-Tin-Noé B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost (2011) Suppl 1(Suppl 1):56–65. doi: 10.1111/j.1538-7836.2011.04317.x
28. Braun LJ, Stegmeyer RI, Schäfer K, Volkery S, Currie SM, Kempe B, et al. Platelets docking to vWF prevent leaks during leukocyte extravasation by stimulating tie-2. Blood (2020) 136(5):627–39. doi: 10.1182/blood.2019003442
29. Weyrich AS, Zimmerman GA. Platelets in lung biology. Annu Rev Physiol (2013) 75:569–91. doi: 10.1146/annurev-physiol-030212-183752
30. TM SCHEININ, AP KOIVUNIEMI. Megakaryocytes in the pulmonary circulation. Blood (1963) 22:82–7. doi: 10.1182/blood.V22.1.82.82
31. Ouzegdouh Y, Capron C, Bauer T, Puymirat E, Diehl JL, Martin JF, et al. The physical and cellular conditions of the human pulmonary circulation enable thrombopoiesis. Exp Hematol (2018) 63:22–27.e3. doi: 10.1016/j.exphem.2018.04.001
32. Trowbridge EA, Martin JF, Slater DN. Evidence for a theory of physical fragmentation of megakaryocytes, implying that all platelets are produced in the pulmonary circulation. Thromb Res (1982) 28(4):461–75. doi: 10.1016/0049-3848(82)90163-3
33. Battina HL, Alentado VJ, Srour EF, Moliterno AR, Kacena MA. Interaction of the inflammatory response and megakaryocytes in COVID-19 infection. Exp Hematol (2021) 104:32–9. doi: 10.1016/j.exphem.2021.09.005
34. Zhu A, Real F, Capron C, Rosenberg AR, Silvin A, Dunsmore G, et al. Infection of lung megakaryocytes and platelets by SARS-CoV-2 anticipate fatal COVID-19. Cell Mol Life Sci (2022) 79(7):365. doi: 10.1007/s00018-022-04318-x
35. Pearse DB, Brower RG, Adkinson NF Jr, Sylvester JT. Spontaneous injury in isolated sheep lungs: Role of perfusate leukocytes and platelets. J Appl Physiol (1985) 1989) 66(3):1287–96. doi: 10.1152/jappl.1989.66.3.1287
36. Heffner JE, Cook JA, Halushka PV. Human platelets modulate edema formation in isolated rabbit lungs. J Clin Invest (1989) 84(3):757–64. doi: 10.1172/JCI114233
37. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med (2000) 342(18):1334–49. doi: 10.1056/NEJM200005043421806
38. Hui P, Cook DJ, Lim W, Fraser GA, Arnold DM. The frequency and clinical significance of thrombocytopenia complicating critical illness: A systematic review. Chest (2011) 139(2):271–8. doi: 10.1378/chest.10-2243
39. Middleton EA, Weyrich AS, Zimmerman GA. Platelets in pulmonary immune responses and inflammatory lung diseases. Physiol Rev (2016) 96(4):1211–59. doi: 10.1152/physrev.00038.2015
40. Lax S, Rayes J, Wichaiyo S, Haining EJ, Lowe K, Grygielska B, et al. Platelet CLEC-2 protects against lung injury via effects of its ligand podoplanin on inflammatory alveolar macrophages in the mouse. Am J Physiol Lung Cell Mol Physiol (2017) 313(6):L1016–29. doi: 10.1152/ajplung.00023.2017
41. Valdebenito S, Bessis S, Annane D, Lorin de la Grandmaison G, Cramer-Bordé E, Prideaux B, et al. COVID-19 lung pathogenesis in SARS-CoV-2 autopsy cases. Front Immunol (2021) 12:735922. doi: 10.3389/fimmu.2021.735922
42. Yang L, Xie X, Tu Z, Fu J, Xu D, Zhou Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct Target Ther (2021) 6(1):255. doi: 10.1038/s41392-021-00679-0
43. Archer SL, Sharp WW, Weir EK. Differentiating COVID-19 pneumonia from acute respiratory distress syndrome and high altitude pulmonary edema: Therapeutic implications. Circulation (2020) 142(2):101–4. doi: 10.1161/CIRCULATIONAHA.120.047915
44. Gattinoni L, Chiumello D, Caironi P, Busana M, Romitti F, Brazzi L, et al. COVID-19 pneumonia: Different respiratory treatments for different phenotypes? Intensive Care Med (2020) 46(6):1099–102. doi: 10.1007/s00134-020-06033-2
45. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev (2013) 93(1):327–58. doi: 10.1152/physrev.00016.2011
46. Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med (2007) 357(24):2482–94. doi: 10.1056/NEJMra07101
47. Mackman N. Triggers, targets and treatments for thrombosis. Nature (2008) 451(7181):914–8. doi: 10.1038/nature06797
48. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-Associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol (2020) 7(8):e575–82. doi: 10.1016/S2352-3026(20)30216-
49. Barrett TJ, Lee AH, Xia Y, Lin LH, Black M, Cotzia P, et al. Platelet and vascular biomarkers associate with thrombosis and death in coronavirus disease. Circ Res (2020) 127(7):945–7. doi: 10.1161/CIRCRESAHA.120.317803
50. McCafferty C, Van Den Helm S, Letunica N, Attard C, Karlaftis V, Cai T, et al. Increased platelet activation in SARS-CoV-2 infected non-hospitalised children and adults, and their household contacts. Br J Haematol (2021) 195(1):90–4. doi: 10.1111/bjh.17629
51. Garcia C, Au Duong J, Poëtte M, Ribes A, Payre B, Mémier V, et al. Platelet activation and partial desensitization are associated with viral xenophagy in patients with severe COVID-19. Blood Adv (2022) 6(13):3884–98. doi: 10.1182/bloodadvances.2022007143
52. Alarabi AB, Mohsen A, Mizuguchi K, Alshbool FZ, Khasawneh FT. Co-Expression analysis to identify key modules and hub genes associated with COVID-19 in platelets. BMC Med Genomics (2022) 15(1):83. doi: 10.1186/s12920-022-01222-y
53. Oudkerk M, Büller HR, Kuijpers D, van Es N, Oudkerk SF, McLoud T, et al. Diagnosis, prevention, and treatment of thromboembolic complications in COVID-19: Report of the national institute for public health of the Netherlands. Radiology (2020) 297(1):E216–22. doi: 10.1148/radiol.2020201629
54. Bhatia P, Mohammed S. Severe hypoxemia in early COVID-19 pneumonia. Am J Respir Crit Care Med (2020) 202(4):621–2. doi: 10.1164/rccm.202004-1313LE
55. Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D. COVID-19 does not lead to a “Typical” acute respiratory distress syndrome. Am J Respir Crit Care Med (2020) 201(10):1299–300. doi: 10.1164/rccm.202003-0817LE
56. Vassiliou AG, Keskinidou C, Jahaj E, Gallos P, Dimopoulou I, Kotanidou A, et al. ICU Admission levels of endothelial biomarkers as predictors of mortality in critically ill COVID-19 patients. Cells (2021) 10(1):186. doi: 10.3390/cells10010186
57. Fogarty H, Townsend L, Morrin H, Ahmad A, Comerford C, Karampini E, et al. Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J Thromb Haemost (2021) 19(10):2546–53. doi: 10.1111/jth.15490
58. Rauch A, Dupont A, Goutay J, Caplan M, Staessens S, Moussa M, et al. Endotheliopathy is induced by plasma from critically ill patients and associated with organ failure in severe COVID-19. Circulation (2020) 142(19):1881–4. doi: 10.1161/CIRCULATIONAHA.120.050907
59. Radeva MY, Waschke J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol (Oxf) (2018) 222(1):e12860. doi: 10.1111/apha.12860
60. Tarhini H, Recoing A, Bridier-Nahmias A, Rahi M, Lambert C, Martres P, et al. Long-term severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infectiousness among three immunocompromised patients: From prolonged viral shedding to SARS-CoV-2 superinfection. J Infect Dis (2021) 223(9):1522–7. doi: 10.1093/infdis/jiab075
61. Wajnberg A, Amanat F, Firpo A, Altman DR, Bailey MJ, Mansour M, et al. Robust neutralizing antibodies to SARS-CoV-2 infection persist for months. Science (2020) 370(6521):1227–30. doi: 10.1126/science.abd772
62. de Melo GD, Lazarini F, Levallois S, Hautefort C, Michel V, Larrous F, et al. COVID-19-Related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci Transl Med (2021) 13(596):eabf8396. doi: 10.1126/scitranslmed.abf839
63. Maiuolo J, Mollace R, Gliozzi M, Musolino V, Carresi C, Paone S, et al. The contribution of endothelial dysfunction in systemic injury subsequent to SARS-CoV-2 infection. Int J Mol Sci (2020) 21(23):9309. doi: 10.3390/ijms21239309
64. Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol (2020) 20(7):389–91. doi: 10.1038/s41577-020-0343-0
65. Thomas MC, Pickering RJ, Tsorotes D, Koitka A, Sheehy K, Bernardi S, et al. Genetic ACE2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ Res (2010) 107(7):888–97. doi: 10.1161/CIRCRESAHA.110.219279
66. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med (2005) 11(8):875–9. doi: 10.1038/nm1267
67. Creasey AA, Chang AC, Feigen L, Wün TC, Taylor FB Jr, Hinshaw LB. Tissue factor pathway inhibitor reduces mortality from escherichia coli septic shock. J Clin Invest (1993) 91(6):2850–60. doi: 10.1172/JCI116529
68. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood (2020) 136(11):1330–41. doi: 10.1182/blood.2020007252
69. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest (2020) 130(11):6151–7. doi: 10.1172/JCI14137
70. Zhao Y, Qin L, Zhang P, Li K, Liang L, Sun J, et al. Longitudinal COVID-19 profiling associates IL-1RA and IL-10 with disease severity and RANTES with mild disease. JCI Insight (2020) 5(13):e139834. doi: 10.1172/jci.insight.139834
71. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest (2020) 130(5):2620–9. doi: 10.1172/JCI137244
72. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med (2020) 8(4):420–2. doi: 10.1016/S2213-2600(20)30076-X
73. Dorward DA, Russell CD, Um IH, Elshani M, Armstrong SD, Penrice-Randal R, et al. Tissue-specific immunopathology in fatal COVID-19. Am J Respir Crit Care Med (2021) 203(2):192–201. doi: 10.1164/rccm.202008-3265OC
74. Nicholls JM, Poon LL, Lee KC, Ng WF, Lai ST, Leung CY, et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet (2003) 361(9371):1773–8. doi: 10.1016/s0140-6736(03)13413-7
75. Wong CK, Lam CW, Wu AK, Ip WK, Lee NL, Chan IH, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol (2004) 136(1):95–103. doi: 10.1111/j.1365-2249.2004.02415.x
76. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-Infected mice. Cell Host Microbe (2016) 19(2):181–93. doi: 10.1016/j.chom.2016.01.007
77. Delvaeye M, Conway EM. Coagulation and innate immune responses: Can we view them separately? Blood (2009) 114(12):2367–74. doi: 10.1182/blood-2009-05-199208
78. Qu Z, Chaikof EL. Interface between hemostasis and adaptive immunity. Curr Opin Immunol (2010) 22(5):634–42. doi: 10.1016/j.coi.2010.08.017
79. Hottz ED, Martins-Gonçalves R, Palhinha L, Azevedo-Quintanilha IG, de Campos MM, Sacramento CQ, et al. Platelet-monocyte interaction amplifies thromboinflammation through tissue factor signaling in COVID-19. Blood Adv (2022) 6(17):5085–99. doi: 10.1182/bloodadvances.2021006680
80. Noubouossie DF, Reeves BN, Strahl BD, Key NS. Neutrophils: Back in the thrombosis spotlight. Blood (2019) 133(20):2186–97. doi: 10.1182/blood-2018-10-862243
81. Nishimura S, Manabe I, Nagasaki M, Kakuta S, Iwakura Y, Takayama N, et al. In vivo imaging visualizes discoid platelet aggregations without endothelium disruption and implicates contribution of inflammatory cytokine and integrin signaling. Blood (2012) 119(8):e45–56. doi: 10.1182/blood-2011-09-381400
82. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol (2007) 7(10):803–15. doi: 10.1038/nri2171
83. Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol (2011) 11(4):264–74. doi: 10.1038/nri2956
84. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res (2018) 122(2):337–51. doi: 10.1161/CIRCRESAHA.117.310795
85. Lindemann S, Krämer B, Seizer P, Gawaz M. Platelets, inflammation and atherosclerosis. J Thromb Haemost (2007) 5 (Suppl 1):203–11. doi: 10.1111/j.1538-7836.2007.02517.x
86. Morrell CN, Pariser DN, Hilt ZT, Vega Ocasio D. The platelet Napoleon complex-small cells, but big immune regulatory functions. Annu Rev Immunol (2019) 37:125–44. doi: 10.1146/annurev-immunol-042718-041607
87. Xie J, Covassin N, Fan Z, Singh P, Gao W, Li G, et al. Association between hypoxemia and mortality in patients with COVID-19. Mayo Clin Proc (2020) 95(6):1138–47. doi: 10.1016/j.mayocp.2020.04.006
88. Cueto-Robledo G, Porres-Aguilar M, Puebla-Aldama D, Barragán-Martínez MDP, Jurado-Hernández MY, García-César M, et al. Severe pulmonary hypertension: An important sequel after severe post-acute COVID-19 pneumonia. Curr Probl Cardiol (2022) 47(3):101004. doi: 10.1016/j.cpcardiol.2021.101004
89. McElvaney OJ, McEvoy NL, McElvaney OF, Carroll TP, Murphy MP, Dunlea DM, et al. Characterization of the inflammatory response to severe COVID-19 illness. Am J Respir Crit Care Med (2020) 202(6):812–21. doi: 10.1164/rccm.202005-1583OC
90. Tyagi T, Ahmad S, Gupta N, Sahu A, Ahmad Y, Nair V, et al. Altered expression of platelet proteins and calpain activity mediate hypoxia-induced prothrombotic phenotype. Blood (2014) 123(8):1250–60. doi: 10.1182/blood-2013-05-501924
91. Sanner BM, Konermann M, Tepel M, Groetz J, Mummenhoff C, Zidek W. Platelet function in patients with obstructive sleep apnoea syndrome. Eur Respir J (2000) 16(4):648–52. doi: 10.1034/j.1399-3003.2000.16d14.x
92. Rångemark C, Hedner JA, Carlson JT, Gleerup G, Winther K. Platelet function and fibrinolytic activity in hypertensive and normotensive sleep apnea patients. Sleep (1995) 18(3):188–94. doi: 10.1093/sleep/18.3.188
93. Bevers EM, Williamson PL. Getting to the outer leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol Rev (2016) 96(2):605–45. doi: 10.1152/physrev.00020.2015
94. Yu SL, Gan XG, Huang JM, Cao Y, Wang YQ, Pan SH, et al. Oxalate impairs aminophospholipid translocase activity in renal epithelial cells Via oxidative stress: Implications for calcium oxalate urolithiasis. J Urol (2011) 186(3):1114–20. doi: 10.1016/j.juro.2011.04.106
95. Hugel B, Martínez MC, Kunzelmann C, Freyssinet JM. Membrane microparticles: Two sides of the coin. Physiol (Bethesda) (2005) 20:22–7. doi: 10.1152/physiol.00029.2004
96. Morel O, Toti F, Hugel B, Bakouboula B, Camoin-Jau L, Dignat-George F, et al. Procoagulant microparticles: Disrupting the vascular homeostasis equation? Arterioscler Thromb Vasc Biol (2006) 26(12):2594–604. doi: 10.1161/01.ATV.0000246775.14471.26
97. Owens AP 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res (2011) 108(10):1284–97. doi: 10.1161/CIRCRESAHA.110.233056
98. Rausch L, Lutz K, Schifferer M, Winheim E, Gruber R, Oesterhaus EF, et al. Binding of phosphatidylserine-positive microparticles by pbmcs classifies disease severity in COVID-19 patients. J Extracell Vesicles (2021) 10(14):e12173. doi: 10.1002/jev2.12173
99. Burnouf T, Goubran HA, Chou ML, Devos D, Radosevic M. Platelet microparticles: Detection and assessment of their paradoxical functional roles in disease and regenerative medicine. Blood Rev (2014) 28(4):155–66. doi: 10.1016/j.blre.2014.04.002
100. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets can associate with SARS-CoV-2 RNA and are hyperactivated in COVID-19. Circ Res (2020) 127(11):1404–18. doi: 10.1161/CIRCRESAHA.120.31770
101. Balbi C, Burrello J, Bolis S, Lazzarini E, Biemmi V, Pianezzi E, et al. Circulating extracellular vesicles are endowed with enhanced procoagulant activity in SARS-CoV-2 infection. EBioMedicine (2021) 67:103369. doi: 10.1016/j.ebiom.2021.103369
102. Flaumenhaft R, Dilks JR, Richardson J, Alden E, Patel-Hett SR, Battinelli E, et al. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood (2009) 113(5):1112–21. doi: 10.1182/blood-2008-06-163832
103. Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol (2009) 297(6):L1035–41. doi: 10.1152/ajplung.00214.2009
104. Boilard E, Paré G, Rousseau M, Cloutier N, Dubuc I, Lévesque T, et al. Influenza virus H1N1 activates platelets through FcΓRIIA signaling and thrombin generation. Blood (2014) 123(18):2854–63. doi: 10.1182/blood-2013-07-515536
105. Sung PS, Huang TF, Hsieh SL. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality Via CLEC5A/TLR2. Nat Commun (2019) 10(1):2402. doi: 10.1038/s41467-019-10360-4
106. Rosell A, Havervall S, von Meijenfeldt F, Hisada Y, Aguilera K, Grover SP, et al. Patients with COVID-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality-brief report. Arterioscler Thromb Vasc Biol (2021) 41(2):878–82. doi: 10.1161/ATVBAHA.120.315547
107. Neuenschwander PF, Bianco-Fisher E, Rezaie AR, Morrissey JH. Phosphatidylethanolamine augments factor viia-tissue factor activity: Enhancement of sensitivity to phosphatidylserine. Biochemistry (1995) 34(43):13988–93. doi: 10.1021/bi00043a004
108. Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time In vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med (2002) 8(10):1175–81. doi: 10.1038/nm782
109. Aharon A, Tamari T, Brenner B. Monocyte-derived microparticles and exosomes induce procoagulant and apoptotic effects on endothelial cells. Thromb Haemost (2008) 100(5):878–85. doi: 10.1160/th07-11-0691
110. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Häberle H, et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood (2021) 137(8):1061–71. doi: 10.1182/blood.2020008762
111. Amabile N, Guérin AP, Tedgui A, Boulanger CM, London GM. Predictive value of circulating endothelial microparticles for cardiovascular mortality in end-stage renal failure: A pilot study. Nephrol Dial Transplant (2012) 27(5):1873–80. doi: 10.1093/ndt/gfr573
112. Santoro F, Nuñez-Gil IJ, Vitale E, Viana-Llamas MC, Reche-Martinez B, Romero-Pareja R, et al. Antiplatelet therapy and outcome in COVID-19: the health outcome predictive evaluation registry. Heart (2022) 108(2):130–6. doi: 10.1136/heartjnl-2021-319552
113. Meizlish ML, Goshua G, Liu Y, Fine R, Amin K, Chang E, et al. Intermediate-dose anticoagulation, aspirin, and in-hospital mortality in COVID-19: A propensity score-matched analysis. Am J Hematol (2021) 96(4):471–9. doi: 10.1002/ajh.26102
114. Collaborative Group RECOVERY. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet (2022) 399(10320):143–51. doi: 10.1016/S0140-6736(21)01825-0
115. Kopp E, Ghosh S. Inhibition of NF-kappa b by sodium salicylate and aspirin. Science (1994) 265(5174):956–9. doi: 10.1126/science.8052854
116. Mansour A, Bachelot-Loza C, Nesseler N, Gaussem P, Gouin-Thibault I. P2Y12 inhibition beyond thrombosis: Effects on inflammation. Int J Mol Sci (2020) 21(4):1391. doi: 10.3390/ijms21041391
117. Chow JH, Yin Y, Yamane DP, Davison D, Keneally RJ, Hawkins K, et al. Association of prehospital antiplatelet therapy with survival in patients hospitalized with COVID-19: A propensity score-matched analysis. J Thromb Haemost (2021) 19(11):2814–24. doi: 10.1111/jth.15517
118. Chow JH, Khanna AK, Kethireddy S, Yamane D, Levine A, Jackson AM, et al. Aspirin use is associated with decreased mechanical ventilation, intensive care unit admission, and in-hospital mortality in hospitalized patients with coronavirus disease 2019. Anesth Analg (2021) 132(4):930–41. doi: 10.1213/ANE.0000000000005292
119. Schrottmaier WC, Pirabe A, Pereyra D, Heber S, Hackl H, Schmuckenschlager A, et al. Platelets and antiplatelet medication in COVID-19-Related thrombotic complications. Front Cardiovasc Med (2022) 8:802566. doi: 10.3389/fcvm.2021.802566
120. REMAP-CAP Writing Committee for the REMAP-CAP Investigators, Bradbury CA, Lawler PR, Stanworth SJ, McVerry BJ, McQuilten Z, et al. Effect of antiplatelet therapy on survival and organ support-free days in critically ill patients with COVID-19: A randomized clinical trial. JAMA (2022) 327(13):1247–59. doi: 10.1001/jama.2022.2910
121. Berger JS, Kornblith LZ, Gong MN, Reynolds HR, Cushman M, Cheng Y, et al. Effect of P2Y12 inhibitors on survival free of organ support among non-critically ill hospitalized patients with COVID-19: A randomized clinical trial. JAMA (2022) 327(3):227–36. doi: 10.1001/jama.2021.23605
122. Corrochano M, Acosta-Isaac R, Mojal S, Miqueleiz S, Rodriguez D, Quijada-Manuitt MÁ, et al. Impact of pre-admission antithrombotic therapy on disease severity and mortality in patients hospitalized for COVID-19. J Thromb Thromb (2022) 53(1):96–102. doi: 10.1007/s11239-021-02507-2
123. Moores LK, Tritschler T, Brosnahan S, Carrier M, Collen JF, Doerschug K, et al. Thromboprophylaxis in patients with COVID-19: A brief update to the CHEST guideline and expert panel report. Chest (2022) 162(1):213–25. doi: 10.1016/j.chest.2022.02.006
124. World Health Organization. Living guidance for clinical management of COVID-19 (2021). Available at: https://www.who.int/publications/i/item/WHO-2019-nCoV-clinical-2021-2.
125. The National Institute for Health and Care Excellence. COVID-19 rapid guideline: managing COVID-19 (2022). Available at: https://www.nice.org.uk/guidance/ng191/chapter/Recommendations.
126. American Society of Hematology. ASH guidelines on use of anticoagulation in patients with COVID-19 (2022). Available at: https://www.hematology.org/education/clinicians/guidelines-and-quality-care/clinical-practice-guidelines/venous-thromboembolism-guidelines/ash-guidelines-on-use-of-anticoagulation-in-patients-with-covid-19.
127. National Institutes of Health. Coronavirus disease 2019 (COVID-19) treatment guideines (2022). Available at: https://www.covid19treatmentguidelines.nih.gov.
128. Pasini E, Corsetti G, Romano C, Scarabelli TM, Chen-Scarabelli C, Saravolatz L, et al. Serum metabolic profile in patients with long-covid (PASC) syndrome: Clinical implications. Front Med (Lausanne) (2021) 8:714426. doi: 10.3389/fmed.2021.714426
129. Kruse JM, Magomedov A, Kurreck A, Münch FH, Koerner R, Kamhieh-Milz J, et al. Thromboembolic complications in critically ill COVID-19 patients are associated with impaired fibrinolysis. Crit Care (2020) 24(1):676. doi: 10.1186/s13054-020-03401-8
130. Meizoso JP, Moore HB, Moore EE. Fibrinolysis shutdown in COVID-19: Clinical manifestations, molecular mechanisms, and therapeutic implications. J Am Coll Surg (2021) 232(6):995–1003. doi: 10.1016/j.jamcollsurg.2021.02.019
131. Bachler M, Bösch J, Stürzel DP, Hell T, Giebl A, Ströhle M, et al. Impaired fibrinolysis in critically ill COVID-19 patients. Br J Anaesth (2021) 126(3):590–8. doi: 10.1016/j.bja.2020.12.010
132. Nougier C, Benoit R, Simon M, Desmurs-Clavel H, Marcotte G, Argaud L, et al. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J Thromb Haemost (2020) 18(9):2215–9. doi: 10.1111/jth.15016
133. Couzin-Frankel J. Clues to long COVID. Science (2022) 376(6599):1261–5. doi: 10.1126/science.add4297
134. Shi J, Gilbert GE. Lactadherin inhibits enzyme complexes of blood coagulation by competing for phospholipid-binding sites. Blood (2003) 101(7):2628–36. doi: 10.1182/blood-2002-07-1951
135. Shi J, Pipe SW, Rasmussen JT, Heegaard CW, Gilbert GE. Lactadherin blocks thrombosis and hemostasis In vivo: Correlation with platelet phosphatidylserine exposure. J Thromb Haemost (2008) 6(7):1167–74. doi: 10.1111/j.1538-7836.2008.03010.x
Keywords: COVID-19, platelet, microvesicles, phosphatidylserine, antiplatelet, thrombosis
Citation: Xiang M, Wu X, Jing H, Liu L, Wang C, Wang Y, Novakovic VA and Shi J (2022) The impact of platelets on pulmonary microcirculation throughout COVID-19 and its persistent activating factors. Front. Immunol. 13:955654. doi: 10.3389/fimmu.2022.955654
Received: 29 May 2022; Accepted: 15 August 2022;
Published: 28 September 2022.
Edited by:
Morgane Bomsel, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Emma Lefrancais, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceCopyright © 2022 Xiang, Wu, Jing, Liu, Wang, Wang, Novakovic and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jialan Shi, SmlhbGFuX3NoaUBkZmNpLmhhcnZhcmQuZWR1; c2hpNzM2NjFAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.