
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 30 August 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.955035
This article is part of the Research TopicJAK Inhibition in Autoimmune and Inflammatory DiseasesView all 11 articles
 Maddison Lensing1,2
Maddison Lensing1,2 Ali Jabbari1,2,3*
Ali Jabbari1,2,3*Alopecia Areata (AA) is a common autoimmune disease characterized by non-scarring hair loss ranging from patches on the scalp to complete hair loss involving the entire body. Disease onset is hypothesized to follow the collapse of immune privilege of the hair follicle, which results in an increase in self-peptide/MHC expression along the follicular epithelium. Hair loss is associated with infiltration of the hair follicle with putatively self-reactive T cells. This process is thought to skew the hair follicle microenvironment away from a typically homeostatic immune state towards one of active inflammation. This imbalance is mediated in part by the dominating presence of specific cytokines. While interferon-γ (IFNγ) has been identified as the key player in AA pathogenesis, many other cytokines have also been shown to play pivotal roles. Mechanistic studies in animal models have highlighted the contribution of common gamma chain (γc) cytokines such as IL-2, IL-7, and IL-15 in augmenting disease. IFNγ and γc cytokines signal through pathways involving receptor activation of Janus kinases (JAKs) and signal transducers and activators of transcription (STATs). Based on these findings, JAK/STAT pathways have been targeted for the purposes of therapeutic intervention in the clinical setting. Case reports and series have described use of small molecule JAK inhibitors leading to hair regrowth among AA patients. Furthermore, emerging clinical trial results show great promise and position JAK inhibitors as a treatment strategy for patients with severe or recalcitrant disease. Demonstrated efficacy from large-scale clinical trials of the JAK inhibitor baricitinib led to the first-in-disease FDA-approved treatment for AA in June of 2022. This review aims to highlight the JAK/STAT signaling pathways of various cytokines involved in AA and how targeting those pathways may impact disease outcomes in both laboratory and clinical settings.
Alopecia Areata (AA) is a prevalent autoimmune disease that carries a lifetime risk of approximately 2% (1). AA is characterized by nonscarring hair loss that typically appears as small patches on the scalp. Severe cases of AA can result in complete hair loss of the scalp (alopecia totalis, AT) or complete loss of hair on the entire body (alopecia universalis, AU). While this disease can affect people of all ages, the mean age range of the first instance of hair loss is 25 to 37 years of age (1). The physical loss of hair in AA patients is often accompanied with increased psychological burden, which may manifest as depression, anxiety, and/or an impaired quality of life when compared to healthy individuals (2). The cause of AA is obscure but is believed to arise from a combination of environmental influences and genetic factors, such as variants within loci or certain genes implicated in autoimmunity and T cell signaling (3). There is no cure for AA, and off-label treatments, such as topical or locally-injected agents or systemic immunosuppressants, are commonly used, although historically these agents have been shown to have variable or poor efficacy. In the past decade, a new strategy has emerged for treating AA which acts to dampen ongoing inflammation by inhibition of Janus kinase (JAK) signaling. As of June 2022, the FDA has approved one JAK inhibitor for the treatment of AA, marking the first and currently only on-label treatment for adults with AA. JAKs are signaling mediators that function to convert the engagement of cytokine receptors with cognate cytokine to downstream effects. Many cytokine pathways that contribute to the pathogenesis of AA, such as interferon-γ (IFNγ) and those in the common gamma chain (γc) family of cytokines, act by way of JAK signaling. Counteracting the effects of these cytokines with JAK inhibitors (JAKis) has shown efficacy in treating AA.
AA is a disease that affects the hair follicles, which are known to follow a cyclic physiologic pattern. Broadly, there are three phases: anagen, a growth phase; catagen, a transition phase; and telogen, a rest phase. The anagen hair follicle is known to reside in a state of immune privilege (IP), which is characterized by low levels of MHC protein expression in its inferior portion, a relative lack of danger or stress ligands, the presence of anti-inflammatory cytokines/hormones (TGFβ, IL-10, α-MSH) and a paucity of immune cell infiltrates (4, 5). This IP state is thought to help protect the follicle from unwanted immune-mediated attack during this time. Maintaining this evasive state may be particularly important during the anagen phase, where a large number of tissue-specific peptides and antigens are generated. However, the microenvironment of the hair follicle exhibits dramatic changes in the AA disease state. In hair follicles in AA lesions, there is an increase in expression of MHC Class I and MHC Class II, upregulation of danger ligands (such as NKG2D activating ligands MICA and ULBP molecules), increased levels of proinflammatory cytokines, and a robust immune cell infiltrate (4, 6) (Figure 1). Immune cell infiltrates are comprised predominantly of CD4+ and CD8+ T cells. CD8+ T cells in close association with the hair follicle were found to express NKG2D, an activating receptor commonly associated with the natural killer (NK) cell lineage (6). This population of CD8+ NKG2D+ T cells was found to be sufficient for induction of disease in the cell-transfer murine model of AA (7). Activated CD8+ T cells are potent producers of pro-inflammatory cytokines, such as IFNγ, which skew the hair follicle microenvironment towards a proinflammatory state. Additionally, CD8+ T cell cytotoxic activity is known to be enhanced in the presence of cytokines such as those of the γc family. CD4+ T cells are also capable of producing cytokines that may contribute to the inflammatory state of the hair follicle during AA. Activated CD4+ T cells are known to produce the γc cytokine IL-2, which may enhance CD8+ T cell function. Additionally, a subset of CD4+ T cells known as T helper type 1 (Th1) cells produce IFNγ in an activated state, which may contribute to the inflammatory setting of the AA hair follicle. Substantial evidence demonstrates that a variety of potent cytokines and their respective signaling pathways are contributing to the pathogenesis of AA by driving functional and phenotypic changes in populations of T cells and in the hair follicle.

Figure 1 The collapse of immune privilege in the anagen hair follicle during alopecia areata.
A large number of cytokine receptors lack intrinsic kinase activity, necessitating intermediaries, including tyrosine kinases, to transmit their downstream signals. By engaging their receptors, many types of growth factors and cytokines elicit their effects through use of the JAK molecules (Figure 2). There are four members of the JAK family of tyrosine kinases: JAK1, JAK2, JAK3, and TYK2. They are unique from other families of tyrosine kinases by virtue of their domain structure involving seven similar JAK Homology (JH) domains. All members contain a kinase domain (JH1) at their C-terminus region, and the phosphorylation of a conserved tyrosine residue within this region stimulates their catalytic activity (8). At their N-terminal region, JAKs contain a Band-4.1, ezrin, radixin, moesin (FERM) domain that mediates their interactions with cytokine receptors (9). Inactive JAKs may be found uncomplexed in the cytosol or pre-associated via their membrane-proximal cytoplasmic region of cytokine receptor subunits at their Box1 and Box2 motifs. The sequence of these two motifs determines with which cytokine receptor the JAK molecule will interact (10). Cytokine ligation results in conformational changes in the receptor that lead to dimerization of receptor subunits and in the juxtapositioning of the pre-associated JAKs or the recruitment of free JAKs to their docking sites. This proximity allows for the auto- or trans-phosphorylation of conserved tyrosine (Tyr) residues of the kinase domain and subsequent activation of the JAKs. The activated JAKs then go on to phosphorylate Tyr residues found on the distal intracellular region of the cytokine receptor, creating a docking site for the Src homology-2(SH2) domains of recruited signal transducers and activators of transcription (STATs).
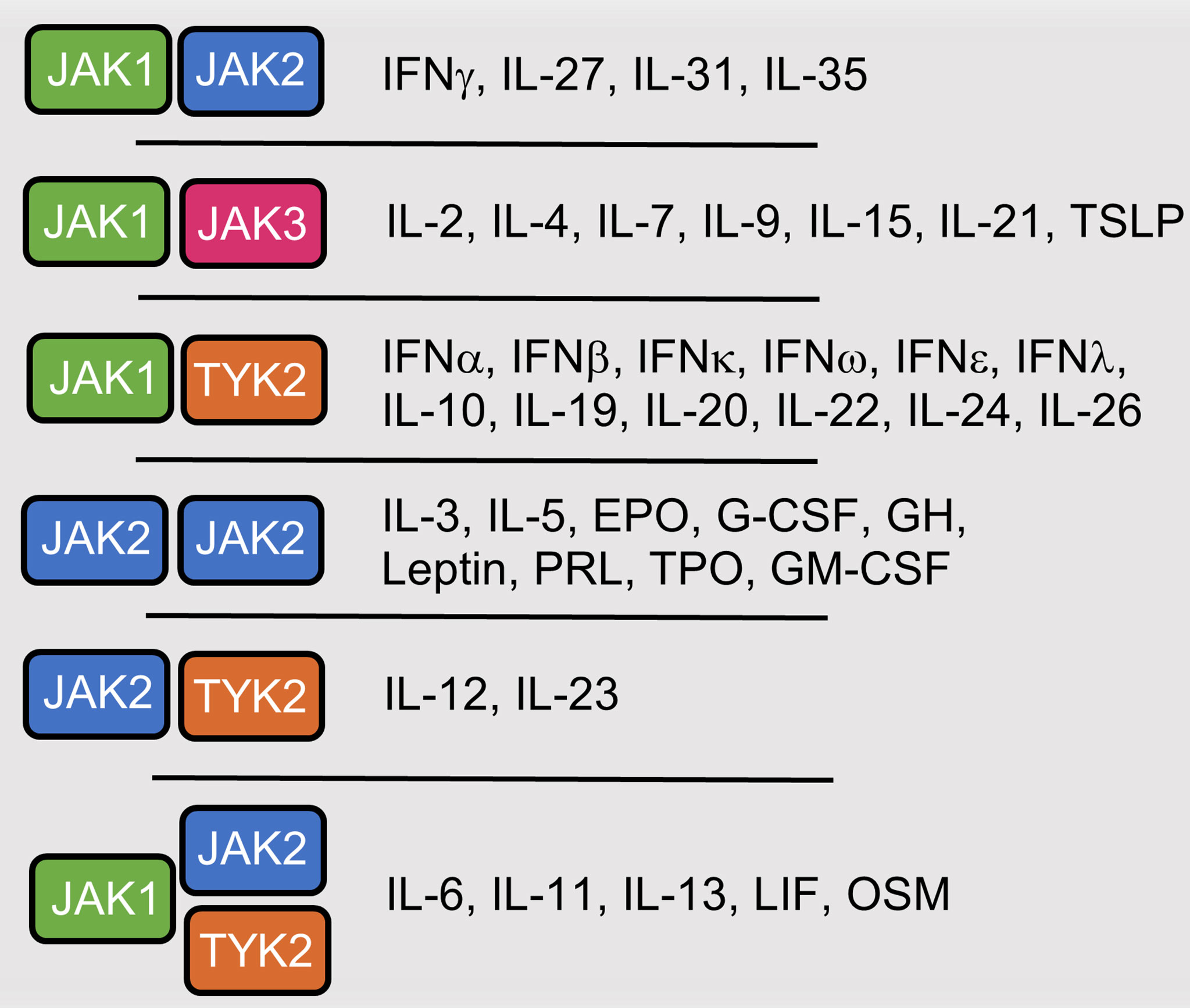
Figure 2 Cytokines and hormones that utilize JAK-mediated signaling. Interferon (IFN), Interleukin (IL), Thymic stromal lymphopoietin (TSLP), Erythropoietin (EPO), Granulocyte colony-stimulating factor (G-CSF), Growth Hormone (GH), Prolactin (PRO), Thrombopoietin (TPO), Granulocyte-macrophage colony-stimulating factor (GM-CSF), Leukemia inhibitory factor (LIF), Oncostatin M (OSM).
There are seven different members of the STAT family, STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. They share six similar domains related to their function. All STATs contain a centrally located DNA-binding domain, which allows for recognition of, and binding to, DNA sequences of target genes. This is followed by the SH2 domain, which can recognize tyrosine-phosphorylated (p-Tyr) sites on cytokine receptor subunits to facilitate STAT docking; each cytokine receptor subunit contains different STAT binding motifs, and the sequence surrounding the p-Tyr dictates which STAT will bind with highest affinity (10). At the C-termini of STATs are the transcription activation domain (TAD), which contains serine phosphorylation sites that direct transcriptional activity and contain binding sites for nuclear cofactors (11). Upon their recruitment to docking sites on cytokine receptors, STATs are phosphorylated by JAKs at a tyrosine residue located between the SH2 domain and TAD. This phosphorylation results in the SH2 domain of each STAT releasing from the receptor subunit and instead binding to the p-Tyr site on the other STAT monomer, resulting in the formation of a STAT dimer in active conformation with exposed DNA-binding sites. Dimers then translocate into the nucleus and bind to target genes within the DNA that possess STAT-binding sites, which ultimately results in inflammatory gene transcription, and the production of pro-inflammatory cytokines (10).
Pro-inflammatory cytokines have been shown to play key roles in the pathogenesis of AA. Mechanistic studies highlighting the importance of these cytokines in disease onset and development have been performed using murine models of alopecia. The C3H/HeJ (referred to C3H herein) strain of mice spontaneously develops hair loss that recapitulates many features of human AA. In particular, C3H AA is a non-scarring form of alopecia, exhibits waxing and waning cycles, and is marked by lymphocytic infiltrates around the hair follicle, including CD8+ and CD4+ T cells (12). Additionally, previously unaffected mice can be induced to develop AA in a reproducible manner by using two different methods. One method consists of grafting a piece of skin from an alopecic mouse, e.g. a mouse that spontaneously developed AA, onto the dorsal region of a naïve recipient mouse (13). Another method involves collecting the skin-draining lymph nodes from a previously-affected mouse, activating and expanding the T cell populations in vitro, and then injecting the resulting cell population intra-dermally into naïve recipient mice (14). The development of these induction methods, resulting in tractable murine AA models that rapidly develop disease in a reproducible manner, have made it feasible to study and unravel the pathogenesis of this disease and has furthered our knowledge obtained from human AA patients, which had been largely limited to observational or in vitro studies. In particular, the roles of IFNγ and γc family members, including IL-2, IL-7, and IL-15, have been specifically investigated. The findings from these studies will be discussed in more detail below.
IFNγ is a 17 kDa protein that is biologically active as a homodimer; it is produced at high levels by activated CD4+ T helper type 1 (Th1) and CD8+ T cells, γδ T cells, and NK cells, and to a lesser extent by NKT cells, B cells, and antigen-presenting cells (15). The secretion of IFNγ is often associated with host defense towards foreign pathogens and is known to aid in tumor surveillance and the anti-tumor response. IFNγ is the sole member of the Type II IFN class of cytokines and signals through a tetrameric receptor comprised of two IFNGR1 chains and two IFNGR2 chains. Nearly all types of mammalian cells express the IFNγ receptor and are thus responsive to the effects of IFNγ to some extent. The level of IFNγ sensitivity is largely controlled by the level of expression of the IFNGR2 chain (16). Typically, IFNGR1 is readily expressed at relatively moderate levels, while the IFNGR2 chain can be upregulated from its low-level baseline expression during cases of activation or based on cell differentiation status (15). IFNGR1 chains have binding sites for JAK1, and IFNGR2 chains have binding sites for JAK2. Cytokine recognition by the IFN-γ receptor leads to autophosphorylation of JAK2 proteins; these active JAK2 molecules then go onto trans-phosphorylate JAK1 proteins. Activated JAK1 molecules each phosphorylate a tyrosine residue on their respective IFNGR1 chain, leading to the dual recruitment and activation of STAT1 monomers (16). Active STAT1 homodimers, also known as gamma-activated factors (GAFs), translocate to the nucleus and bind to the gamma-activated sequence (GAS) to drive target gene transcription (17).
In the setting of AA, sera from patients contain significantly higher levels of IFNγ when compared to healthy patients (18–20). IFNγ is hypothesized to play an important role in the collapse of the immune privileged state of the hair follicle. The ability of IFNγ to cause IP collapse has been demonstrated in experiments where anagen-stage hair follicles collected from a healthy scalp were cultured in the presence of IFNγ, resulting in increased expression of MHC I molecules (21). In a murine skin-graft induction model of AA, IFNγ has been shown to play a critical role in disease onset, as IFNγ-deficient mice fail to develop disease after grafting (22). Mice treated with an IFNγ neutralizing antibody starting at the time of grafting were prevented from developing disease, had a reduction in skin-infiltrating CD8+ NKG2D+ T cells, and failed to upregulate MHC I and MHC II molecules in the skin (7). Interestingly, groups have shown that intradermal injection of IFNγ alone or in combination with poly[I:C] into anagen-induced skin of naïve mice can drive the emergence of AA (23, 24). Overall, IFNγ has been identified as one of the main cytokines contributing to disease and does so, at least in part, by driving changes in the follicular epithelium to disrupt hair follicle immune privilege.
IL-2 is a 15 kDa protein that is mainly produced by activated CD4+ T cells and, to a lesser extent, by activated CD8+ T cells and DCs, NK cells, and NKT cells (25). IL-2 can signal through a high-affinity trimeric receptor (Kd ~10-11 M) or an intermediate-affinity dimeric receptor (Kd ~10-9 M) (26). The trimeric receptor consists of a unique IL-2Rα (CD25) subunit, the shared IL-2/15Rβ (CD122) subunit, and the γc (CD132) subunit, while the dimeric receptor consists of only the β and γc subunits. In order to form the trimeric receptor, IL-2 first weakly binds to IL-2Rα (Kd ~10-8 M); this initial interaction causes a conformational change in IL-2 that allows for association with the β subunit, recruitment of the γc subunit, and leads to the formation of a stable IL-2/receptor complex (27). Expression of the high-affinity, trimeric IL-2R is observed in CD4+ FoxP3+ regulatory T cells (Tregs) and activated conventional CD4+ or CD8+ T cells, suggesting that these populations of cells are exquisitely responsive to the effects of IL-2. The intermediate-affinity, dimeric IL-2R can be found on CD8+ memory T cells and NK cells, indicating they may also respond to IL-2. The dimeric receptor can also engage IL-15, and this promiscuity results in competition of the two cytokines for this receptor.
IL-2Rα is not known to engage any intracellular signaling molecules, whereas the IL-2Rβ subunit engages JAK1, and the γc subunit engages JAK3. Activation of JAK1/3 leads to subsequent activation and formation of STAT5/5 or STAT3/5 dimers that translocate to the nucleus to drive gene transcription programs that promote cellular expansion, survival, and maintenance. Additionally, STAT5 plays an important role in influencing T cell lineage fate. STAT5 is known to drive the expression of the Th1/Tc1 fate determining transcription factor T-bet which is crucial for acquisition of effector phenotype and functions, such as inducing CXCR3 expression and IFNγ production (28). There is also a STAT5 binding site in the CNS2 region of the FOXP3 gene, which implicates STAT5 as a stabilizer of Foxp3 expression, which is important for the potent immunosuppressive phenotype and function of Tregs (29). Thus, IL-2 is a pleotropic cytokine that can enhance both effector and regulatory T cell populations that may work against each other in the context of an autoimmune disease such as AA.
In AA patients, increased mRNA expression of IL-2 is detected in the deep dermis of the skin, which contains the bulb of the hair follicle, when compared to levels of the upper dermis (30). Furthermore, active lesions of AA patients contain higher amounts of IL-2 mRNA than inactive areas, suggesting that IL-2 may be contributing to the localized areas of disease. A GWAS of AA patients and healthy controls revealed disease associations with SNPs in both the IL-2 gene and the IL-2Rα (CD25) gene (31). Various groups have profiled the serum of AA patients and observed higher levels of IL-2 in circulation in comparison to healthy patients (19, 20, 32). The crucial role of IL-2 has also been demonstrated with the murine skin-graft induction model. Graft-mediated induction experiments with IL-2+/- mice, which possess only one functional IL-2 allele and produce 50% of typical IL-2 amounts, revealed that these mice were partially resistant to disease induction and had reduced leukocyte infiltration into skin lesions (33). Other reports demonstrated that mice treated with an anti-IL-2 neutralizing antibody at the time of grafting failed to develop disease, had a marked reduction of pathogenic CD8+ NKG2D+ T cells infiltrating the skin, and prevented the upregulation of MHC Class I and Class II in skin (7). Overall, IL-2 plays an essential function in T cell proliferation and homeostasis. The important roles of IL-2 in AA outlined above provides growing support for the role of γc cytokine family members in this disease and its relevant animal models.
IL-7 is a 17 kDa protein that is produced by various non-hematopoietic cells, such as stromal cells in the lymphoid organs or epithelial cells in the thymus or intestine (34). IL-7 signals through a dimeric receptor consisting of a unique IL-7Rα (CD127) subunit and the γc subunit. Expression of the IL-7 receptor can be found on naïve and memory αβ T Cells and is crucial for their development, survival, proliferation, and overall homeostasis. The IL-7Rα subunit preferentially binds JAK1, while the γc subunit binds JAK3. Activation of JAK1/3 leads to the recruitment and activation of STAT5, prompting the formation of a STAT5 homodimer that activates target genes (35). Genes transcribed after IL-7 signaling include the anti-apoptotic gene, BCL2, and genes that promote cell-cycle progression and differentiation, such as MYC and PIM1 (36).
In AA patients, T cells infiltrating the HF were shown to have increased expression of IL-7Rα in comparison to T cells in control scalp samples (37). Furthermore, PBMCs isolated from AA patients stimulated in the presence of IL-7 resulted in significantly higher IFNγ production by CD8+ T cells in comparison to control patients (37). GWAS data has revealed one SNP in the IL-7Rα region associated with AA patients (31, 37). Together this suggests that T cells in both the circulation and skin of AA patients are more responsive to IL-7, and this may enhance their effector phenotype that promotes ongoing disease in these patients.
Murine studies of alopecia have demonstrated similar findings regarding the role of IL-7 and have also begun to unravel mechanisms of action. Gene expression analysis has revealed higher levels of IL-7 in AA skin when compared to skin of unaffected (UA) mice (37). The increase in IL-7 is likely due to increased production by keratinocytes, as they are known to upregulate IL-7 expression in the presence of IFNγ, which is likely released by CD8+ T lymphocytes infiltrating alopecic skin (37, 38). This suggests that a feedback loop exists between IL-7 production and IFNγ production of resident and infiltrating immune cells within the skin that may enhance the local pro-inflammatory environment. Mice treated with exogenous IL-7 complexed to an anti-IL-7 antibody, which expands the lifespan of IL-7 in vivo, experienced significantly quicker disease onset than mice receiving isotype control in a skin-graft induction setting. Additionally, mice treated with an anti-IL-7Rα blocking antibody at the time of skin-graft induction were completely resistant to development of disease. Clinical relevance of IL-7 blockade was demonstrated by the treatment of mice with anti-IL-7Rα after the first instance of hair loss. This treatment led to hair-regrowth in 60% of mice and reduced the inflammatory profiles of skin (37). While IL-7 is one of the more recent cytokines shown to contribute to the pathogenesis of AA, it again highlights the pertinent involvement of γc family members in enhancing this disease.
IL-15 is a 13kDa protein that is structurally similar to IL-2 and is produced by a variety of cells types, ranging from innate cells such as monocytes or dendritic cells to epithelial cells of the kidney, lung, heart, muscle and skin (39). IL-15 typically signals through a heterotrimeric receptor that consists of a unique IL-15Rα (CD215) subunit, the shared IL-2/15Rβ subunit, and the γc subunit. The IL-15Rα is expressed by lymphoid cells (including CD8+ T lymphocytes, NK Cells and γδ T lymphocytes), myeloid cells (including monocytes, macrophages, and dendritic cells), and nonhematopoietic cells (including colonic epithelial cells, microglial cells, or keratinocytes) (40). As discussed earlier (in the section on IL-2), expression of the β/γc subunits are often restricted to T lymphocytes or NK cells. IL-15 binds with highest affinity to the IL-15Rα (Kd ~ 10-11 M), which can then associate with the remaining β/γc subunits that are expressed on the same cell, known as cis-presentation (40). Interestingly, trans-presentation can also occur, in which a cell expressing the IL-15Rα subunit binds IL-15 and then presents it to another nearby cell expressing only the β and γc subunits to initiate IL-15-mediated signaling of the recipient cell. Of note, trans-presentation does not occur with IL-2, likely due to the lower binding of affinity of IL-2 with IL-2Rα (27). The IL-15/IL-15Rα complex has been shown to undergo endosomal recycling in certain cells, such as monocytes, which is a distinguishing feature from IL-2 (41). The high-affinity complex for IL-15 signaling involves engagement of all three (α/β/γc) subunits. However, IL-15 can also signal through an intermediate-affinity complex consisting of only the latter IL-2/15Rβ and γc subunits (Kd ~ 10-9 M); as mentioned earlier, this results in competition of this receptor complex with IL-2 due to the redundant subunits (40). While the IL-15Rα subunit is not known to engage any JAKs, the remaining two subunits make use of JAK/STAT signaling networks to influence cellular actions. The shared IL-2/15Rβ subunit binds JAK1 leading to STAT3 or STAT5 activation, while the γc subunit binds JAK3 and leads to STAT5 activation. The STAT3/5 heterodimers or STAT5 homodimers lead to transcription of the anti-apoptotic gene BCL2 and the proto-oncogenes MYC, FOS, and JUN, among others (42). IL-15 signaling is known to support CD8+ memory T cell survival, drive the expansion and maintenance of T cells and NK cells, and also enhance CD8+ T cell production of granzymes, IFNγ, and tumor necrosis factor α (42).
AA patients demonstrated increased levels of serum IL-15 compared to healthy controls, and patients with more severe forms of AA (AT or AU) exhibited significantly more IL-15 in comparison to patients with a few patches of hair loss (43, 44). Additionally, SALT scores, an AA clinical scoring system based on percentage of hair loss on the scalp, positively correlated with levels of circulating IL-15 (45). IL-15 levels in the serum have also been observed to correlate with disease duration (46). In comparison to healthy controls, both AA patients and AA mice have increased expression of IL-15 and IL-15Rα in the outer root sheath of the hair follicle (7). It is hypothesized that expression of the IL-15/IL-15Rα complex by skin cells engages the β/γ subunits on infiltrating NKG2D+ CD8+ T cells and enhances their IFNγ production. This local increase in IFNγ can then act back on the skin epithelium to enhance expression of IL-15 and expression of NKG2D ligands, leading to further recruitment/activation of effector CD8+ T cells. This IL-15/IFNγ feedback loop between CD8+ T cells and the epithelium surrounding the hair follicle is thought to be a driving force to the diseased state (7). This concept was further supported using the murine skin graft induction model, in which mice treated with IL-2/15Rβ blocking antibody were protected from disease, had significantly reduced numbers of skin-infiltrating NKG2D+ CD8+ T cells, failed to upregulate MHC molecules in the skin and presented a skin gene signature similar to an unaffected mouse (7). Overall, these findings suggest that IL-15 may contribute to ongoing disease in patients, likely by enhancing effector functions of pathogenic CD8+ T cells.
JAKs phosphorylate their target substrates by transferring the terminal phosphate group from a high-energy adenosine triphosphate (ATP) molecule to the hydroxyl (-OH) residue of a tyrosine. This occurs when an ATP molecule binds to an open site within the ~300 base pair kinase domain of the active JAK. This ATP binding pocket is surrounding by the N-terminal lobe and the C-terminal lobe of the kinase domain. The N lobe helps to orient and anchor the ATP, while the C lobe binds the ATP and initiates the phospho-transfer (47). Interest in JAK inhibition as a therapeutic strategy was invigorated in 2005 when patients with myeloproliferative disorders (MPD) were found to have a JAK2 point mutation leading to its constitutively active state (48). This spurred the development of an inhibitor with activity against JAK2, and in 2011 the FDA approved the use of ruxolitinib for the treatment of patients with MPD. Shortly after, in 2012, tofacitinib was approved for patients with rheumatoid arthritis that were resistant to other treatments (49). JAKis are small membrane-permeable molecules that work by outcompeting ATP for binding in the pocket of the kinase domain, which ultimately prevents the JAK from phosphorylating its target substrate. However, due to similarities in the structure of the ATP-binding sites across the JAKs, the first generation of JAKis inhibit more than one JAK, typically in a hierarchical order based on binding site affinity. Second generation JAKis have been developed to more selectively inhibit individual JAK family members, with many currently being tested in clinical trials for a variety of autoimmune diseases (50). The preclinical rationale and the first demonstration of efficacy for JAKis to treat AA were published in 2014 (7, 51); these publications heralded the explosion in interest and clinical trials examining the efficacy and safety of JAKis for patients with AA as well as other dermatological conditions. Select clinical trials are described in Table 2.
The first generation of JAKis affect more than one JAK family member and have shown promising results in the context of AA; the efficacy of first generation JAKis in AA may be due to the inhibition of multiple cytokines/JAK signaling pathways that are contributing in parallel, and are partially redundant, in disease pathogenesis. The first generation of JAKis are comprised of ruxolitinib, tofacitinib, baricitinib, and oclacitinib (52). Of this group, the first three have been explored as treatments for AA and will be discussed further.
Ruxolitinib is commonly regarded as a JAK1/JAK2 inhibitor (Table 1). The activity profile of ruxolitinib makes it an interesting therapeutic candidate for AA given how it can dampen both γc family cytokine signaling (JAK1/JAK3) and also IFNγ signaling(JAK1/JAK2). Thus, ruxolitinib is likely to act upon immune cells, which are responsive to γc family cytokine signaling, and act at the level of the hair follicle, which is responsive to IFNγ signaling. Ruxolitinib was among the first of the JAKis that demonstrated efficacy for AA. In 2014, three patients with moderate/severe AA treated with oral ruxolitinib exhibited nearly complete hair regrowth after 3-5 months of treatment. Ruxolitinib treatment in these patients resulted in fewer CD8+ and CD4+ T cells infiltrating the hair follicle and its periphery (7). The positive outcome from this initial study led to numerous investigations testing the efficacy of ruxolitinib further.
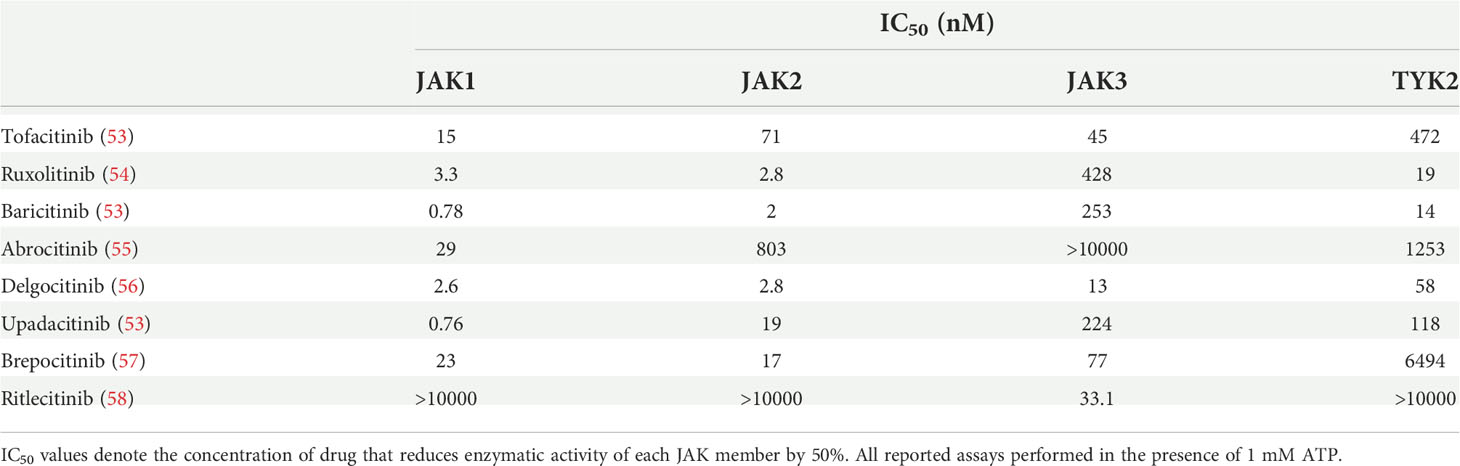
Table 1 JAK inhibitors with documented efficacy for treating AA.
From 2016 onward, case reports have appeared highlighting the use of ruxolitinib to treat AA, sometimes under special circumstances such as AA patients presenting with additional autoimmune diseases as well as in pediatric AA patients. An early case report demonstrating ruxolitinib efficacy for AA involved a patient presenting with both AA and vitiligo, a cutaneous autoimmune disease that causes depigmentation of the skin and, like AA, is mediated by CD8+ T cells and IFNγ. The patient received treatment for 20 weeks, and experienced significant regrowth of hair (85% of scalp coverage from a baseline 63% coverage) after only 12 weeks of treatment, and this regrowth was maintained for at least 12 weeks after treatments ceased (59). A more recent case report demonstrated one of the first uses for ruxolitinib to treat AA in a preadolescent patient. A 9-year-old patient with AT was treated twice daily with 20 mg of oral ruxolitinib, and 4 months of treatment yielded nearly complete regrowth of hair on scalp and eyebrows. The dosage was tapered to 10 mg every other day, a level that maintained efficacy with no adverse effects (60).
Early cases reports identified that hair regrowth after cessation of ruxolitinib is not sustained long-term, as patients tended to experience hair loss relapse after ending treatment. Case reports of ruxolitinib for treating AA have involved testing different treatments regimens, such as changing the duration and altering dosage, to prolong positive outcomes. A report involving two patients, one presenting with AT and one presenting with AU, were given daily ruxolitinib treatments for 13-14 months, and then tapered to less frequent dosing. The patients exhibited near complete regrowth by 6-8 months of treatment, and the tapered dosing led to maintenance of hair regrowth (61). An additional study demonstrated efficacy of lower dosing ruxolitinib to reverse hair loss in severe AA patients. Eight patients were treated twice daily with 10-25 mg of ruxolitinib for a range of 5-31 months. Interestingly, six of the enrolled patients had undergone prior tofacitinib therapy with varied results. Five of the enrolled patients attained near complete hair regrowth, with four of them having received the lower dosage of 10 mg ruxolitinib twice daily (62).
A few clinical trials testing the use of both systemic oral ruxolitinib and localized topical ruxolitinib to treat AA have been completed. A phase 2, open-label trial with oral ruxolitinib involved a cohort of twelve patients with moderate/severe AA that received daily ruxolitinib for a duration of 3-6 months. 9 of 12 patients had at least 50% regrowth of hair and continuous reduction in SALT scores over time while receiving treatment. Three months after treatment cessation, all nine responsive patients noted hair shedding, with three of those patients experiencing marked hair loss. Gene expression profiling revealed that ruxolitinib treatment normalized the aberrant IFNγ and CD8+ T cell-based gene signatures in the scalp, demonstrating that AA lesion biopsies taken after 12 weeks of treatment clustered closer to healthy skin biopsies than baseline AA biopsies (Table 2) (64).

Table 2 Completed or currently active clinical trials involving the use of JAK inhibitors to treat AA.
A phase 2, double-blind, placebo-controlled trial examining topical ruxolitinib cream was conducted in a cohort of 16 AU patients that received twice daily ruxolitinib applications for 12 weeks. 5 of 16 patients exhibited partial regrowth in areas treated with topical ruxolitinib. However, hair loss was noted after cessation of treatments at 6-week and 12-week follow-up time points (74). Soon after, another phase 2, double-blind, vehicle-controlled trial appraised topical ruxolitinib cream in a cohort of 78 patients with moderate AA. Patients received daily applications to affected areas for a duration of 24 weeks. Both the ruxolitinib cream and vehicle treated groups resulted in 5 of 39 patients showing reduced SALT scores over the 24-week period. This study concluded that topical ruxolitinib does not significantly affect AA patients (Table 2) (66).
To accompany the numerous case reports and trials, the protective effects of ruxolitinib have been demonstrated using an in vitro hair follicle culture model. Human dermal papilla cells (hDPCs) pre-treated with ruxolitinib had significantly lower MHC Class II expression following IFNγ exposure when compared to resting hDPCs exposed to IFNγ. Furthermore, hDPCs treated with ruxolitinib showed a reversal in the expression of IFNγ-inducible genes (such as caspase-1, IL-15, IL-1β) following stimulation with IFNγ. These findings suggest that ruxolitinib plays a role in helping maintain an immune privileged state of the hair follicle, likely by dampening the downstream effects of IFNγ signaling, in addition to its effects on immune effectors (75).
Of note, a deuterated form of ruxolitinib, CTP-543, has recently been developed for the treatment of AA. Efficacy for CTP-543 has been supported by data from a phase 2, double-blind, placebo-controlled trial involving 149 adult patients with severe AA that received 4 mg, 8 mg, or 12 mg of CTP-543 twice daily or a placebo for a duration of 24 weeks. The primary endpoint was the frequency of patients achieving a SALT50 score, which is a 50% or greater reduction in SALT scores in comparison to baseline. The trial found that patients in the higher dosage groups (8 mg and 12 mg) had significantly more patients reach the endpoint (47% and 58%) in comparison to placebo (9%). The frequency of patients experiencing adverse effects were similar among all treatment groups, and only one patient (12 mg dosage) experienced a serious adverse effect (76). The positive outcomes from this trial led to the initiation of two larger-scale studies, THRIVE-AA1 and THRIVE-AA2, examining further efficacy and safety (Table 2). THRIVE-AA1 contained 706 patients receiving 12 mg CTP-543, 8 mg CTP-543 or placebo twice daily for 24 weeks, with a primary endpoint of achieving a SALT score ≤ 20. 42% of patients in the 12 mg group and 30% in the 8 mg group met this endpoint, compared to 1% of the placebo group. Additionally, 53% of patients in the 12 mg group and 42% in the 8 mg group were satisfied or very satisfied with their results after 24 weeks of treatment compared to 5% in the placebo group. Serious adverse effects were reported by 1 patient receiving 12 mg, 4 patients receiving 8 mg, and 4 patients in the placebo group (73). THRIVE-AA2 contained 517 patients randomly assigned to 12 mg CTP-543, 8 mg CTP-543, or placebo. Similar to THRIVE-AA1, the primary endpoint was to achieve a SALT score of ≤ 20. 38.3% of patients in the 12 mg group and 33% of patients in the 8 mg group met this endpoint, compared to 0.8% in the placebo group. 52% and 47% of patients in the 12 mg and 8 mg groups were satisfied or very satisfied with their results compared to 2% of patients in the placebo group (74). Together, the two THRIVE trials enrolled over 1200 patients. The positive results from these massive trials indicate that CTP-543 may be the next inhibitor granted approval for treating adult patients with AA.
Tofacitinib is typically classified as a pan-JAK inhibitor with preference towards the JAK1/JAK3 pair (Table 1). The inhibitory properties of tofacitinib show it to be effective in dampening signals from the γc family cytokines (JAK1/JAK3) and IFNγ (JAK1/JAK2), indicating that it can act on immune cells and on the hair follicle. To date, tofacitinib has likely been prescribed for patients with AA more than any other JAKi.
Tofacitinib efficacy for AA was initially suggested in 2014, in which a patient presenting with both plaque psoriasis and AU exhibited complete regrowth of scalp hair after 3 months of treatment and full regrowth of all body hair after 8 months of treatment (51). Since then, many case studies have appeared supporting the efficacy of tofacitinib to treat AA. An early report involved a patient with persistent AA who received daily tofacitinib for 4 months and demonstrated near complete regrowth of hair. Transcriptional analysis of skin biopsies taken 4 weeks into treatment revealed a decrease in IFN and cytotoxic T cell gene signatures when compared to baseline biopsies. Additionally, serum levels of Cxcl10, an IFN-inducible chemokine, were noticeably lower after 4 weeks of tofacitinib treatment. However, upon ceasing treatment, the patient presented with near complete loss of hair at the 16-week timepoint (77). A more recent entry in the literature reported the efficacy of treating a patient that presented with AU, atopic dermatitis, and ulcerative colitis simultaneously. Daily tofacitinib treatments led to marked scalp hair regrowth by 4 months of treatment, along with significant improvement in itch and erythema, and colonic biopsies suggested a remissive state. The treatment dose for this patient was tapered, and, at the 10-month follow-up, all three disease states remained well controlled. This report highlights a potential advantage of pan-JAKi therapy for patients exhibiting multiple diseases of inflammatory nature (78).
There have also been ample reports promoting the efficacy of tofacitinib to treat AA in the pediatric population, a group with increased risk of developing more severe and chronic forms when compared with adults (79). Increasing the pool of safe and effective therapeutics for pediatric patients would be especially beneficial given the psychological burdens this disease carries in children (80). An initial report involved 4 patients (5-7 years of age) with AT or AU who were treated with oral tofacitinib. Drug dosing was based on concurrent clinical trials testing tofacitinib in juvenile idiopathic arthritis patients. Two AA patients demonstrated complete regrowth of hair by 3-6 months of daily treatments, another had 62% of regrowth occur, and the remaining patient exhibited no substantial regrowth (81). Shortly after, another case series was communicated detailing 11 patients (7-11 years of age) that exhibited a range of AA, from involving eyebrows only to AU. The patients received daily oral tofacitinib for at least 6 months, starting at a relatively low dose (2.5-7.5 mg daily), which was increased depending on patient tolerance and responsiveness. Nine of the patients responded well, with a mean SALT score change of 67.8%, and two remaining patients experienced no noticeable hair regrowth. The treatment appeared well tolerated, with most adverse events being characterized as mild and transient (82). These studies support that systemic tofacitinib therapy may be an effective option for treating pediatric AA.
A few clinical trials investigating the efficacy of daily oral tofacitinib to treat AA have been completed. An open-label trial was conducted with 66 patients, with the majority (71.2%) having AU at the time of enrollment. After 3 months of treatment, 32% of the patients had strongly responded by exhibiting significant improvements in SALT scores (>50% change), and the treatment was tolerated with limited adverse effects. Immunofluorescent staining of a biopsy from a strongly responding patient had a marked reduction in STAT3 expression within the follicular epithelium after 2 months of treatment, suggesting that blocking this pathway is associated with a positive outcome. 20 patients were evaluated at 3 months after cessation of treatment, and these patients had all experienced hair loss, suggesting that transient treatment with tofacitinib does not result in a durable response (Table 2) (63). Another open-label trial examining tofacitinib for AA involved 12 patients. Eleven patients completed the study, having received daily treatments over 6-18 months. 8 of 11 patients reached the primary efficacy endpoint of a SALT score reduction of > 50%. Upon cessation, six of the eight responding patients experienced hair loss as early as 4 weeks post treatment, while one patient presented with maintained hair regrowth even up to 6 months post treatment (Table 2) (67).
The use of tofacitinib as a topical therapeutic agent has also been investigated. An initial study examined the efficacy of topical 2% tofacitinib to treat three pediatric patients exhibiting patchy AA or AT. They received twice daily applications, and 2 of 3 patients (one with patchy AA and the other with AT) experienced 80% and 95% regrowth of scalp hair. However, a third patient with AT failed to exhibit any regrowth (83). Shortly after, a clinical trial was completed which tested the efficacy of daily application of 2% tofacitinib ointment in ten adult patients with moderate/severe AA (≥ 2 patches of scalp hair loss). 3 of 10 patients had hair regrowth occur, with an average SALT score change of 34.6% and minimal adverse effects (Table 2) (68). Larger, placebo-controlled studies are needed to confirm the benefits for topical delivery of tofacitinib, and, although the data above appear to show an efficacy signal, improved topical delivery methods will likely be required. Overall, however, there is substantial support for the use of systemic tofacitinib as a therapeutic option for patients with AA.
Baricitinib is considered a dual JAK1/JAK2 inhibitor given the similarly strong binding interactions with those two proteins (Table 1). Efficacy of baricitinib for AA was initially suggested in 2015, when a CANDLE patient was being treated with this drug; the patient exhibited comorbid AA and demonstrated complete hair regrowth after 9 months of treatment (84). This clinical data was further supported by studies done in the murine model of AA in the same report. Systemic administration of baricitinib led to reduced frequency of mice developing disease, reduction of CD8+ T cell infiltrates and a reduction of MHC Class I and Class II expression in the skin, suggesting baricitinib may help prevent the collapse of an immune privileged state of the hair follicle. Additionally, topical baricitinib applied onto lesions of AA mice led to nearly complete regrowth of hair (84). A later published case-report further indicated efficacy for baricitinib in the treatment of AA. This report involved an adult patient with progressive AT who received daily oral baricitinib. She was initially unresponsive to a starting dose of 2 mg; however, upon increasing the dosage to 4 mg, she experienced remarkable hair regrowth. 8 months of treatment resulted in 97% of regrowth on the scalp as well as regrowth on eyebrows and eyelashes. After 13 months of treatment, regrowth had been maintained, and no adverse effects were noted (85).
In the past few years, larger clinical trials testing baricitinib to treat AA have been completed. A phase 2, double blind, placebo-controlled trial randomized 110 patients with severe AA into four test groups: placebo, 1 mg, 2 mg, or 4 mg of baricitinib daily. After 12 weeks of treatments, interim SALT scores were assessed, and the 2 mg and 4 mg groups presented with the highest frequency of patients achieving SALT30 scores (29.6% and 33.3% respectively) (86). These two higher treatment groups were selected for continued usage, with patients initially assigned to the 1 mg dosing being transitioned to a 4 mg dosage for the remainder of the trial. A later interim analysis at 36 weeks of treatment aimed to identify the frequency of patients with SALT scores ≤ 20. The 2 mg and 4 mg groups had significantly higher frequencies of patients, 33% and 51.9% respectively, meet the criteria in comparison to the placebo group(3.6%) (Table 2). Overall, this study demonstrated that longer durations of treatment with higher dosages of baricitinib were effective at stimulating scalp hair regrowth and was well tolerated, with most adverse effects being classified as mild (86).
The positive results from the previous trial spurred the formation of two large-scale phase 3 trials looking at 2 mg or 4 mg of oral baricitinib to treat severe AA. The two trials, termed BRAVE-AA1, and BRAVE-AA2 contained 654 and 546 patients, respectively. Each trial randomly assigned patients to daily treatments consisting of placebo, 2 mg, or 4 mg of baricitinib, with a primary outcome of achieving a SALT score of ≤ 20. After 36 weeks of treatment, the 4 mg group had 35.9-38.8% of patients achieve the primary outcome, while the 2mg group had 19.4-22.8% and the placebo had only 3.3-6.2% of patients reach the primary outcome (Table 2) (72). Favorable results from the BRAVE-AA1 and BRAVE-AA2 trials led to the first-in-disease systemic FDA-approved treatment for AA. On June 13th 2022, the FDA approved a once-daily regimen of baricitinib (Olumiant) available as 1 mg, 2 mg, or 4 mg tablets for adults with severe AA (87). Approved FDA labeling of Olumiant includes boxed warnings for potential serious adverse effects. This moment marks an important time in history for AA patients and their clinicians alike who now have a more accessible treatment option available. This is especially crucial given how insurance approval has historically been an issue for AA patients seeking off-label usage of JAKis (88).
Second generation JAKis were designed to specifically inhibit a single JAK family isoform, which may allow for more selective intervention of inflammatory diseases based on the contributing cytokines and their respective JAK signaling molecules. In addition, their more limited range of inhibitory effects may reduce the potential for adverse events. Despite their relatively recent development, studies have already begun to emerge describing efficacy for the resolution of hair loss in AA patients. The results from select studies will be discussed further below.
Abrocitinib (PF-04965842) is a JAK1-specific inhibitor that has shown efficacy in treating patients with atopic dermatitis (AD), which is a disease marked by a higher risk of developing AA (Table 1) (89). A 2022 case report demonstrated efficacy of abrocitinib for AA, when a patient presenting with both AD and AU began a daily oral regimen of 200 mg. Within 12 weeks, patchy hair regrowth was noted, and full regrowth of scalp hair was observed after one year of treatment. The patient’s AD lesions were noticeably improved as well, suggesting that this selective JAKi may be a promising option for patients with dual AD and AA diagnoses (90). Another recent report highlighted two AD patients presenting with severe AA that had noticeable hair-regrowth occur upon extended duration of abrocitinib therapy. Both patients were enrolled into a long-term clinical trial termed JADE EXTEND after showing responsiveness to abrocitinib, noted by improvement in AD, in an initial shorter treatment window. The patients had noticeable hair regrowth occur at 12-14 weeks into the trial that maintained throughout the course of treatment (91).
Upadacitinib (ABT-494) is a JAK1-specific inhibitor which was initially shown to be effective in treating AD (Table 1) (92). A case report describing two patients with AD and comorbid AA revealed potential efficacy of upadacitinib for treating AA. One patient had AU (SALT score 100), while the other patient had a resistant patch of AA (SALT score 43). Near complete hair regrowth was observed for both patients after 4 months of treatment with 30 mg daily (93). A more recent case report again indicated efficacy in treating a patient presenting with dual AD and AA (SALT score 89.2). The patient had failed multiple therapies for both diseases, but upon switching to 30mg of daily upadacitinib, the patient demonstrated remarkable clinical improvements in both AD and AA, with no reported adverse effects. Larger, placebo-controlled studies will be needed to determine true efficacy in patients with AA (94).
Brepocitinib (PF-06700841) is an inhibitor of JAK1 and TYK2 that was initially shown to be effective in treating patients with plaque psoriasis (Table 1) (95). Efficacy for brepocitinib for treating AA was demonstrated in the recently reported ALLEGRO trial, where it was tested in parallel with ritlecitinib, which is discussed further below. 47 patients with severe AA were treated daily with 60 mg of oral brepocitinib for 4 weeks, and then tapered to a 30 mg daily dose for 20 additional weeks. After 24 weeks of treatment, 64% of patients achieved the primary outcome of a SALT30 score, which is a 30% or greater reduction in SALT scoring in comparison to baseline. Two patients discontinued use after experiencing serious adverse effects (69). Lesional biopsies taken from patients receiving brepocitinib (or ritlecitinib) at the 12- and 24-week timepoints revealed significant decreases in CD3+ and CD8+ T cell counts in comparison to baseline levels. Transcriptomic analysis from biopsies also showed a significant decrease in inflammatory gene signatures (immune genes, Th1, Th2 and IL-12/IL-23) in the patients receiving either brepocitinib or ritlecitinib when compared to placebo (Table 2) (96). The positive outcome from this initial trial warrant further investigations into the efficacy of brepocitinib for treating AA, although it appears that further investigation and development by the manufacturer have focused on ritlecitinib over brepocitinib for the treatment of AA.
Ritlecitinib (PF-06651600) is an inhibitor specific for JAK3 and the tyrosine kinase expressed in hepatocellular carcinoma (TEC) family of kinases (Table 1) (97). It acts through irreversible, covalent interactions with the cysteine 909 residue found on the active site of these kinases. The remaining members of the JAK family all possess a serine residue at this site, which accounts for the selectivity of ritlecitinib for JAK3 (58). This feature is likely clinically relevant given the use of JAK1, JAK2, and TYK2 broadly by wide-ranging receptors responsible for blood and tissue homeostasis and protective responses against pathogens. Additionally, targeting JAK3 spares signaling of immunoregulatory cytokines such as IL-10R (TYK2/JAK1), IL-27R (JAK1/JAK2), and IL-35R (JAK1/JAK2) (98), whose activity may contribute to the prevention of autoimmunity to the hair follicle, potentially making a selective JAK3 inhibitor a more potent strategy than pan-JAK inhibition. As previously mentioned, JAK3 associates exclusively with receptors for γc cytokines; because downstream signaling from γc receptors are most often transmitted in cells of the immune system, ritlecitinib represents a JAK inhibitor that may act specifically through the modulation of the immune compartment without directly influencing the hair follicle itself.
Ritlecitinib is the only irreversible JAKi being assessed for clinical use for AA patients. Efficacy of ritlecitinib for AA was demonstrated during a recently reported clinical trial (phase 2, double-blind, placebo-controlled) termed ALLEGRO, where a primary outcome was designated as a 30% improvement in SALT score. 48 patients with severe AA (≥ 50% hair loss on scalp) were treated with a loading regimen of 200 mg daily of ritlecitinib for 4 weeks, and then treated with 50 mg daily for 20 weeks. By the 24th week of treatment, 50% of patients receiving ritlecitinib had reached the primary outcome, while only 3% of patients receiving placebo reached that endpoint. The number of patients experiencing adverse effects were similar between the groups; overall ritlecitinib was generally well tolerated by patients and may be an effective agent for restoring hair growth in patients with severe AA (69). Phase 3 and long-term extension trials are underway for ritlecitinib in patients with AA (Table 2).
While JAKis have demonstrated great promise for reversing the immune-mediated hair loss in AA, they also present a risk for potentially severe side effects. This notion is not necessarily surprising given the redundancy of JAK proteins for cytokine, growth factor, and hormone receptor signaling. Commonly documented minor adverse events experienced by AA patients taking JAKis include acne, headache, nausea, urinary tract infections, respiratory tract infections, anemia, thrombocytopenia, neutropenia, and elevated creatinine levels (99). Patients using JAKis also commonly exhibit elevated LDL levels, which is a known risk factor for cardiovascular disease (99, 100). Serious adverse effects most often reported in AA trial patients receiving JAKis include varicella zoster emergence, pneumonia, tuberculosis, sepsis, and the development of non-melanoma skin cancer (99).
Beyond AA, more extensive safety analyses have been reported for patients using JAKis to treat other diseases such as rheumatoid arthritis, atopic dermatitis, psoriatic arthritis, ulcerative colitis, and myeloproliferative disorders. Results from these studies should be carefully considered until more large-scale safety trials are completed specific for the AA population. A recent meta-analysis comprised of over 126,000 JAKi patient cases for the above-mentioned diseases found that the most common adverse effects experienced were infections (herpes, influenza, fungal, and mycobacterial), thrombosis and pulmonary embolism, and neoplasms (101).
The FDA has issued boxed warnings to all JAK inhibitors that are being used for inflammatory diseases. These cautions constitute the highest level of safety warning assigned by the FDA, and are intended to alert consumers of major potential risks associated with the drug (102). In 2020, a large-scale phase-4 safety study compared the side effects of RA patients receiving 5-10 mg twice daily oral tofacitinib to patients receiving TNFα inhibitor injections for a mean duration of about 40 months. This robust study contained more than 1450 patients in each treatment group, and primary endpoints were to assess rate of cancers (excluding nonmelanoma skin cancer) and major adverse cardiovascular events (MACE) including death from cardiovascular causes, nonfatal stroke, and nonfatal myocardial infarction. A goal of this study was to examine whether tofacitinib met noninferiority criteria for the two primary endpoints; meeting this criterion indicates that there is no difference between the treatments based on the boundaries of the confidence intervals associated with the hazard ratio (103).
The incidence of cancers reported during follow-up appointments (median of 4 years), was 4.2% for tofacitinib users and 2.9% for TNFα users, with a hazard ratio of 1.48 (tofacitinib:TNFα). The most common cancer experienced among tofacitinib users was lung cancer, while breast cancer was most common among patients receiving the TNFα inhibitor. The frequency of tofacitinib users experiencing MACE was 3.4% compared to 2.5% of patients receiving TNFα inhibitor, with a hazard ratio of 1.33 (tofacitinib:TNFα). The most common MACE experienced was nonfatal myocardial infarction among tofacitinib users and nonfatal stroke among patients receiving the TNFα inhibitor. When compared to TNFα inhibitors, tofacitinib failed to meet noninferiority criteria for both incidence of cancers and MACE (103).The study determined that there is indeed a difference in adverse events experienced between users of tofacitinib and TNFα inhibitors. Of note, the efficacy was similar among all patients regardless of treatment, suggesting that in the context of RA, tofacitinib performs similarly to a well-documented biologic, but also presents a significantly greater safety risk (103).
Overall, JAKis have potential to cause off-target harm in addition to their therapeutic benefit. Thus, usage of JAKis should only be considered once the risks are clearly communicated to and comprehended by patients. Nevertheless, as more large-scale clinical trials are completed, more FDA approvals of JAKis to treat AA are likely in the not-too-distant future.
The past decade has been an exciting time for the identification and testing of new therapeutics for patients with AA. The therapeutic pool is changing from use of broad-acting immunosuppressants to more inferred and targeted approaches. These advances are possible due to the identification of novel disease mechanisms in basic science laboratories and pre-clinical AA animal models. Recent therapeutic approaches are targeting the actions of pro-inflammatory cytokines, which have been identified as crucial players and determinants of the onset, progression, and severity of AA. JAKis have taken the spotlight for now. However, despite the recent FDA approval of the first JAKi to treat AA, side effects and incomplete efficacy for refractory patients begs the question, “What else is there?” As more mechanisms of this enigmatic disease are unraveled, the arsenal of therapeutic options for AA will surely continue to grow.
Both authors were responsible for writing and revising the manuscript.
This work was supported by the Department of Veterans Affairs (VA Merit Award I01 BX004907 to AJ), the National Institutes of Health (R01 AR077194 and K08 AR069111 to AJ), and the University of Iowa Department of Dermatology.
AJ has served as a consultant for and has received institutional payments for study-related costs from Pfizer.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Villasante Fricke AC, Miteva M. Epidemiology and burden of alopecia areata: a systematic review. Clin Cosmet Investig Dermatol (2015) 8:397–403. doi: 10.2147/CCID.S53985
2. Jankovic S, Peric J, Maksimovic N, Cirkovic A, Marinkovic J, Jankovic J, et al. Quality of life in patients with alopecia areata: a hospital-based cross-sectional study. J Eur Acad Dermatol Venereol (2016) 30(5):840–6. doi: 10.1111/jdv.13520
3. Connell SJ, Jabbari A. The current state of knowledge of the immune ecosystem in alopecia areata. Autoimmun Rev (2022) 21(5):103061. doi: 10.1016/j.autrev.2022.103061
4. Paus R, Ito N, Takigawa M, Ito T. The hair follicle and immune privilege. J Investig Dermatol Symp Proc (2003) 8(2):188–94. doi: 10.1046/j.1087-0024.2003.00807.x
5. Bertolini M, McElwee K, Gilhar A, Bulfone-Paus S, Paus R. Hair follicle immune privilege and its collapse in alopecia areata. Exp Dermatol (2020) 29(8):703–25. doi: 10.1111/exd.14155
6. Ito T, Ito N, Saatoff M, Hashizume H, Fukamizu H, Nickoloff BJ, et al. Maintenance of hair follicle immune privilege is linked to prevention of NK cell attack. J Invest Dermatol (2008) 128(5):1196–206. doi: 10.1038/sj.jid.5701183
7. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med (2014) 20(9):1043–9. doi: 10.1038/nm.3645
8. Szente BE. CHAPTER 4 - cytokine signaling. In: Thomson AW, Lotze MT, editors. The cytokine handbook, 4th ed., vol. p . London: Academic Press (2003). p. 85–97.
9. Yamaoka K, Saharinen P, Pesu M, Holt VE 3rd, Silvennoinen O, O'Shea JJ. The janus kinases (Jaks). Genome Biol (2004) 5(12):253. doi: 10.1186/gb-2004-5-12-253
10. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci (2018) 27(12):1984–2009. doi: 10.1002/pro.3519
11. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther (2021) 6(1):402. doi: 10.1038/s41392-021-00791-1
12. Sundberg JP, Cordy WR, King LE Jr. Alopecia areata in aging C3H/HeJ mice. J Invest Dermatol (1994) 102(6):847–56. doi: 10.1111/1523-1747.ep12382416
13. McElwee KJ, Boggess D, King LE Jr., Sundberg JP. Experimental induction of alopecia areata-like hair loss in C3H/HeJ mice using full-thickness skin grafts. J Invest Dermatol (1998) 111(5):797–803. doi: 10.1046/j.1523-1747.1998.00380.x
14. Wang EHC, Khosravi-Maharlooei M, Jalili RB, Yu R, Ghahary A, Shapiro J, et al. Transfer of alopecia areata to C3H/HeJ mice using cultured lymph node-derived cells. J Invest Dermatol (2015) 135(10):2530–2. doi: 10.1038/jid.2015.176
15. Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol (2018) 9:847. doi: 10.3389/fimmu.2018.00847
16. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol (2004) 75(2):163–89. doi: 10.1189/jlb.0603252
17. Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity (2009) 31(4):539–50. doi: 10.1016/j.immuni.2009.09.002
18. Tomaszewska K, Kozlowska M, Kaszuba A, Lesiak A, Narbutt J, Zalewska-Janowska A. Increased serum levels of IFN-gamma, IL-1beta, and IL-6 in patients with alopecia areata and nonsegmental vitiligo. Oxid Med Cell Longev (2020) 2020:5693572. doi: 10.1155/2020/5693572
19. Teraki Y, Imanishi K, Shiohara T. Cytokines in alopecia areata: contrasting cytokine profiles in localized form and extensive form (alopecia universalis). Acta Derm Venereol (1996) 76(6):421–3. doi: 10.2340/0001555576421423
20. Barahmani N, Lopez A, Babu D, Hernandez M, Donley SE, Duvic M. Serum T helper 1 cytokine levels are greater in patients with alopecia areata regardless of severity or atopy. Clin Exp Dermatol (2010) 35(4):409–16. doi: 10.1111/j.1365-2230.2009.03523.x
21. Ito T, Ito N, Bettermann A, Tokura Y, Takigawa M, Paus R. Collapse and restoration of MHC class-i-dependent immune privilege: exploiting the human hair follicle as a model. Am J Pathol (2004) 164(2):623–34. doi: 10.1016/S0002-9440(10)63151-3
22. Freyschmidt-Paul P, McElwee KJ, Hoffmann R, Sundberg JP, Vitacolonna M, Kissling S, et al. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol (2006) 155(3):515–21. doi: 10.1111/j.1365-2133.2006.07377.x
23. Gilhar A, Kam Y, Assy B, Kalish RS. Alopecia areata induced in C3H/HeJ mice by interferon-gamma: evidence for loss of immune privilege. J Invest Dermatol (2005) 124(1):288–9. doi: 10.1111/j.0022-202X.2004.23580.x
24. Shin JM, Choi DK, Sohn KC, Koh JW, Lee YH, Seo YJ, et al. Induction of alopecia areata in C3H/HeJ mice using polyinosinic-polycytidylic acid (poly[I:C]) and interferon-gamma. Sci Rep (2018) 8(1):12518. doi: 10.1038/s41598-018-30997-3
25. Malek TR. The biology of interleukin-2. Annu Rev Immunol (2008) 26(1):453–79. doi: 10.1146/annurev.immunol.26.021607.090357
26. Lin JX, Leonard WJ. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene (2000) 19(21):2566–76. doi: 10.1038/sj.onc.1203523
27. Malek TR, Castro I. Interleukin-2 receptor signaling: At the interface between tolerance and immunity. Immunity (2010) 33(2):153–65. doi: 10.1016/j.immuni.2010.08.004
28. Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol (2011) 12(6):551–9. doi: 10.1038/ni.2030
29. Mahmud SA, Manlove LS, Farrar MA. Interleukin-2 and STAT5 in regulatory T cell development and function. JAKSTAT (2013) 2(1):e23154. doi: 10.4161/jkst.23154
30. Zhang X, Zhao Y, Ye Y, Li S, Qi S, Yang Y, et al. Lesional infiltration of mast cells, langerhans cells, T cells and local cytokine profiles in alopecia areata. Arch Dermatol Res (2015) 307(4):319–31. doi: 10.1007/s00403-015-1539-1
31. Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature (2010) 466(7302):113–7. doi: 10.1038/nature09114
32. Kasumagic-Halilovic E, Cavaljuga S, Ovcina-Kurtovic N, Zecevic L. Serum levels of interleukin-2 in patients with alopecia areata: Relationship with clinical type and duration of the disease. Skin Appendage Disord (2018) 4(4):286–90. doi: 10.1159/000486462
33. Freyschmidt-Paul P, McElwee KJ, Hoffmann R, Sundberg JP, Kissling S, Hummel S, et al. Reduced expression of interleukin-2 decreases the frequency of alopecia areata onset in C3H/HeJ mice. J Invest Dermatol (2005) 125(5):945–51. doi: 10.1111/j.0022-202X.2005.23888.x
34. Gao J, Zhao L, Wan YY, Zhu B. Mechanism of action of IL-7 and its potential applications and limitations in cancer immunotherapy. Int J Mol Sci (2015) 16(5):10267–80. doi: 10.3390/ijms160510267
35. Gonzalez-Garcia S, Garcia-Peydro M, Alcain J, Toribio ML. Notch1 and IL-7 receptor signalling in early T-cell development and leukaemia. Curr Top Microbiol Immunol (2012) 360:47–73. doi: 10.3390/ph14050443
36. Lodewijckx I, Cools J. Deregulation of the interleukin-7 signaling pathway in lymphoid malignancies. Pharm (Basel) (2021) 14(5):443. doi: 10.3390/ph14050443
37. Dai Z, Wang EHC, Petukhova L, Chang Y, Lee EY, Christiano AM. Blockade of IL-7 signaling suppresses inflammatory responses and reverses alopecia areata in C3H/HeJ mice. Sci Adv (2021) 7(14):eabd1866. doi: 10.1126/sciadv.abd1866
38. Ariizumi K, Meng Y, Bergstresser PR, Takashima A. IFN-gamma-dependent IL-7 gene regulation in keratinocytes. J Immunol (1995) 154(11):6031–9. http://www.jimmunol.org/content/154/11/6031
39. Budagian V, Bulanova E, Paus R, Bulfone-Paus S. IL-15/IL-15 receptor biology: a guided tour through an expanding universe. Cytokine Growth Factor Rev (2006) 17(4):259–80. doi: 10.1016/j.cytogfr.2006.05.001
40. Hamid Q, Ito I, Muro S. INTERLEUKINS | IL-15. In: : Laurent GJ, Shapiro SD, editors. Encyclopedia of respiratory medicine, vol. p . Oxford: Academic Press (2006). p. 385–90.
41. Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Ralpha recycles and presents IL-15 In trans to neighboring cells. Immunity (2002) 17(5):537–47. doi: 10.1016/s1074-7613(02)00429-6
42. Mishra A, Sullivan L, Caligiuri MA. Molecular pathways: interleukin-15 signaling in health and in cancer. Clin Cancer Res (2014) 20(8):2044–50. doi: 10.1158/1078-0432.CCR-12-3603
43. Ebrahim AA, Salem RM, El Fallah AA, Younis ET. Serum interleukin-15 is a marker of alopecia areata severity. Int J Trichol (2019) 11(1):26–30. doi: 10.4103/ijt.ijt_80_18
44. Askin O, Yucesoy SN, Coskun E, Engin B, Serdaroglu S. Evaluation of the level of serum interleukins (IL-2, IL-4, IL-15 andIL-17) and its relationship with disease severity in patients with alopecia areata. Bras Dermatol (2021) 96(5):551–7. doi: 10.1016/j.abd.2021.03.006
45. El Aziz Ragab MA, Hassan EM, El Niely D, Mohamed MM. Serum level of interleukin-15 in active alopecia areata patients and its relation to age, sex, and disease severity. Postepy Dermatol Alergol (2020) 37(6):904–8. doi: 10.5114/ada.2020.102103
46. Tabara K, Kozlowska M, Jedrowiak A, Bienias W, Kaszuba A. Serum concentrations of selected proinflammatory cytokines in children with alopecia areata. Postepy Dermatol Alergol (2019) 36(1):63–9. doi: 10.5114/ada.2019.82826
47. Schenk PW, Snaar-Jagalska BE. Signal perception and transduction: the role of protein kinases. Biochim Biophys Acta (BBA) - Mol Cell Res (1999) 1449(1):1–24. doi: 10.1016/S0167-4889(98)00178-5
48. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature (2005) 434(7037):1144–8. doi: 10.1038/nature03546
49. Furumoto Y, Gadina M. The arrival of JAK inhibitors: advancing the treatment of immune and hematologic disorders. BioDrugs (2013) 27(5):431–8. doi: 10.1007/s40259-013-0040-7
50. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O'Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discovery (2017) 17(1):78–. doi: 10.1038/nrd.2017.267
51. Craiglow BG, King BA. Killing two birds with one stone: oral tofacitinib reverses alopecia universalis in a patient with plaque psoriasis. J Invest Dermatol (2014) 134(12):2988–90. doi: 10.1038/jid.2014.260
52. Damsky W, King BA. JAK inhibitors in dermatology: The promise of a new drug class. J Am Acad Dermatol (2017) 76(4):736–44. doi: 10.1016/j.jaad.2016.12.005
53. Dowty ME, Lin TH, Jesson MI, Hegen M, Martin DA, Katkade V, et al. Janus kinase inhibitors for the treatment of rheumatoid arthritis demonstrate similar profiles of in vitro cytokine receptor inhibition. Pharmacol Res Perspect (2019) 7(6):e00537. doi: 10.1002/prp2.537
54. Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood (2010) 115(15):3109–17. doi: 10.1182/blood-2009-04-214957
55. Vazquez ML, Kaila N, Strohbach JW, Trzupek JD, Brown MF, Flanagan ME, et al. Identification of n-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfo namide (PF-04965842): A selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem (2018) 61(3):1130–52. doi: 10.1021/acs.jmedchem.7b01598
56. Tanimoto A, Ogawa Y, Oki C, Kimoto Y, Nozawa K, Amano W, et al. Pharmacological properties of JTE-052: a novel potent JAK inhibitor that suppresses various inflammatory responses in vitro and in vivo. Inflammation Res (2015) 64(1):41–51. doi: 10.1007/s00011-014-0782-9
57. Fensome A, Ambler CM, Arnold E, Banker ME, Brown MF, Chrencik J, et al. Dual inhibition of TYK2 and JAK1 for the treatment of autoimmune diseases: Discovery of (( s)-2,2-Difluorocyclopropyl)((1 R,5 s)-3-(2-((1-methyl-1 h-pyrazol-4-yl)amino)pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octan-8-yl)methanone (PF-06700841). J Med Chem (2018) 61(19):8597–612. doi: 10.1021/acs.jmedchem.8b00917
58. Telliez JB, Dowty ME, Wang L, Jussif J, Lin T, Li L, et al. Discovery of a JAK3-selective inhibitor: Functional differentiation of JAK3-selective inhibition over pan-JAK or JAK1-selective inhibition. ACS Chem Biol (2016) 11(12):3442–51. doi: 10.1021/acschembio.6b00677
59. Harris JE, Rashighi M, Nguyen N, Jabbari A, Ulerio G, Clynes R, et al. Rapid skin repigmentation on oral ruxolitinib in a patient with coexistent vitiligo and alopecia areata (AA). J Am Acad Dermatol (2016) 74(2):370–1. doi: 10.1016/j.jaad.2015.09.073
60. Peterson DM, Vesely MD. Successful treatment of alopecia totalis with ruxolitinib in a preadolescent patient. JAAD Case Rep (2020) 6(4):257–9. doi: 10.1016/j.jdcr.2020.02.007
61. Vandiver A, Girardi N, Alhariri J, Garza LA. Two cases of alopecia areata treated with ruxolitinib: a discussion of ideal dosing and laboratory monitoring. Int J Dermatol (2017) 56(8):833–5. doi: 10.1111/ijd.13598
62. Liu LY, King BA. Ruxolitinib for the treatment of severe alopecia areata. J Am Acad Dermatol (2019) 80(2):566–8. doi: 10.1016/j.jaad.2018.08.040
63. Kennedy Crispin M, Ko JM, Craiglow BG, Li S, Shankar G, Urban JR, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight (2016) 1(15):e89776. doi: 10.1172/jci.insight.89776
64. Mackay-Wiggan J, Jabbari A, Nguyen N, Cerise JE, Clark C, Ulerio G, et al. Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight (2016) 1(15):e89790. doi: 10.1172/jci.insight.89790
65. Mikhaylov D, Glickman JW, Del Duca E, Nia J, Hashim P, Singer GK, et al. A phase 2a randomized vehicle-controlled multi-center study of the safety and efficacy of delgocitinib in subjects with moderate-to-severe alopecia areata. Arch Dermatol Res (2022). doi: 10.1007/s00403-022-02336-0
66. Olsen EA, Kornacki D, Sun K, Hordinsky MK. Ruxolitinib cream for the treatment of patients with alopecia areata: A 2-part, double-blind, randomized, vehicle-controlled phase 2 study. J Am Acad Dermatol (2020) 82(2):412–9. doi: 10.1016/j.jaad.2019.10.016
67. Jabbari A, Sansaricq F, Cerise J, Chen JC, Bitterman A, Ulerio G, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch-type alopecia areata, totalis, and universalis. J Invest Dermatol (2018) 138(7):1539–45. doi: 10.1016/j.jid.2018.01.032
68. Liu LY, Craiglow BG, King BA. Tofacitinib 2% ointment, a topical janus kinase inhibitor, for the treatment of alopecia areata: A pilot study of 10 patients. J Am Acad Dermatol (2018) 78(2):403–4 e1. doi: 10.1016/j.jaad.2017.10.043
69. King B, Guttman-Yassky E, Peeva E, Banerjee A, Sinclair R, Pavel AB, et al. A phase 2a randomized, placebo-controlled study to evaluate the efficacy and safety of the oral janus kinase inhibitors ritlecitinib and brepocitinib in alopecia areata: 24-week results. J Am Acad Dermatol (2021) 85(2):379–87. doi: 10.1016/j.jaad.2021.03.050
70. Aclaris therapeutics announces phase 2 clinical trial of ATI-501 oral in patients with alopecia areata met primary endpoint [press release] Vol. 2019. Wayne, PA, US: Aclaris Therapeutics, Inc(2019).
71. Pfizer announces positive top-line results from phase 2b/3 trial of ritlecitinib in alopecia areata [press release] Vol. 2021. New York, NY, US: Pfizer, Inc. (2021).
72. King B, Ohyama M, Kwon O, Zlotogorski A, Ko J, Mesinkovska NA, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med (2022)386(18):1687–99. doi: 10.1056/NEJMoa2110343
73. Concert pharmaceuticals reports positive topline resuslts for first CTP-543 phase 3 clinical trial in alopecia areata [press release]. Lexington, MA, US: Concert Pharmaceuticals, Inc. (2022).
74. Concert pharmaceuticals reports positive topline results for second CTP-543 phase 3 clinical trial in alopecia areata [press release]. Lexington, MA, US: Concert Pharmaceuticals, Inc (2022).
75. Bokhari L, Sinclair R. Treatment of alopecia universalis with topical janus kinase inhibitors - a double blind, placebo, and active controlled pilot study. Int J Dermatol (2018) 57(12):1464–70. doi: 10.1111/ijd.14192
76. Kim JE, Lee YJ, Park HR, Lee DG, Jeong KH, Kang H. The effect of JAK inhibitor on the survival, anagen re-entry, and hair follicle immune privilege restoration in human dermal papilla cells. Int J Mol Sci (2020) 21(14):5137. doi: 10.3390/ijms21145137
77. King B, Mesinkovska N, Mirmirani P, Bruce S, Kempers S, Guttman-Yassky E, et al. Phase 2 randomized, dose-ranging trial of CTP-543, a selective janus kinase inhibitor, in moderate-to-severe alopecia areata. J Am Acad Dermatol (2022) 87(2):306–13. doi: 10.1016/j.jaad.2022.03.045
78. Jabbari A, Nguyen N, Cerise JE, Ulerio G, de Jong A, Clynes R, et al. Treatment of an alopecia areata patient with tofacitinib results in regrowth of hair and changes in serum and skin biomarkers. Exp Dermatol (2016) 25(8):642–3. doi: 10.1111/exd.13060
79. Honap S, Cookson H, Sharma E, Samaan MA, Irving PM. Tofacitinib for the treatment of ulcerative colitis, alopecia universalis, and atopic dermatitis: One drug, three diseases. Inflammation Bowel Dis (2021) 27(2):e13–e4. doi: 10.1093/ibd/izaa243
80. Ikeda T. A new classification of alopecia areata. Dermatologica (1965) 131(6):421–45. doi: 10.1159/000254503
81. Christensen T, Yang JS, Castelo-Soccio L. Bullying and quality of life in pediatric alopecia areata. Skin Appendage Disord (2017) 3(3):115–8. doi: 10.1159/000466704
82. Craiglow BG, King BA. Tofacitinib for the treatment of alopecia areata in preadolescent children. J Am Acad Dermatol (2019) 80(2):568–70. doi: 10.1016/j.jaad.2018.08.041
83. Jerjen R, Meah N, Trindade de Carvalho L, Wall D, Eisman S, Sinclair R. Treatment of alopecia areata in pre-adolescent children with oral tofacitinib: A retrospective study. Pediatr Dermatol (2021) 38(1):103–8. doi: 10.1111/pde.14422
84. Bayart CB, DeNiro KL, Brichta L, Craiglow BG, Sidbury R. Topical janus kinase inhibitors for the treatment of pediatric alopecia areata. J Am Acad Dermatol (2017) 77(1):167–70. doi: 10.1016/j.jaad.2017.03.024
85. Jabbari A, Dai Z, Xing L, Cerise JE, Ramot Y, Berkun Y, et al. Reversal of alopecia areata following treatment with the JAK1/2 inhibitor baricitinib. EBioMedicine (2015) 2(4):351–5. doi: 10.1016/j.ebiom.2015.02.015
86. Olamiju B, Friedmann A, King B. Treatment of severe alopecia areata with baricitinib. JAAD Case Rep (2019) 5(10):892–4. doi: 10.1016/j.jdcr.2019.07.005
87. King B, Ko J, Forman S, Ohyama M, Mesinkovska N, Yu G, et al. Efficacy and safety of the oral janus kinase inhibitor baricitinib in the treatment of adults with alopecia areata: Phase 2 results from a randomized controlled study. J Am Acad Dermatol (2021) 85(4):847–53. doi: 10.1016/j.jaad.2021.05.050
88. FDA Approves lilly and incyte's OLUMIANT® (baricitinib) as first and only systemic medicine for adults with severe alopecia areata [press release] Vol. 2022. Indianapolis, IN, US: Cision PR Newswire (2022).
89. Thompson HJ, Vavra T, Jabbari A. Factors associated with insurance coverage of tofacitinib for alopecia areata: A retrospective review from an academic institution. J Am Acad Dermatol (2020) 83(5):1509–10. doi: 10.1016/j.jaad.2020.06.028
90. de Lusignan S, Alexander H, Broderick C, Dennis J, McGovern A, Feeney C, et al. Atopic dermatitis and risk of autoimmune conditions: Population-based cohort study. J Allergy Clin Immunol (2022). doi: 10.1016/j.jaci.2022.03.030
91. Zhao J, Liu L. A case of atopic dermatitis with alopecia universalis in a patient treated with abrocitinib. JAAD Case Rep (2022) 22:99–100. doi: 10.1016/j.jdcr.2022.02.027
92. Bennett M, Moussa A, Sinclair R. Successful treatment of chronic severe alopecia areata with abrocitinib. Australas J Dermatol (2022)63(2):274–6. doi: 10.1111/ajd.13836
93. Guttman-Yassky E, Thaci D, Pangan AL, Hong HC, Papp KA, Reich K, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol (2020) 145(3):877–84. doi: 10.1016/j.jaci.2019.11.025
94. Gambardella A, Licata G, Calabrese G, De Rosa A, Alfano R, Argenziano G. Dual efficacy of upadacitinib in 2 patients with concomitant severe atopic dermatitis and alopecia areata. Dermatitis (2021) 32(1S):e85–e6. doi: 10.1097/DER.0000000000000780
95. Cantelli M, Martora F, Patruno C, Nappa P, Fabbrocini G, Napolitano M. Upadacitinib improved alopecia areata in a patient with atopic dermatitis: A case report. Dermatol Ther (2022) 35(4):e15346. doi: 10.1111/dth.15346
96. Banfield C, Scaramozza M, Zhang W, Kieras E, Page KM, Fensome A, et al. The safety, tolerability, pharmacokinetics, and pharmacodynamics of a TYK2/JAK1 inhibitor (PF-06700841) in healthy subjects and patients with plaque psoriasis. J Clin Pharmacol (2018) 58(4):434–47. doi: 10.1002/jcph.1046
97. Guttman-Yassky E, Pavel AB, Diaz A, Zhang N, Del Duca E, Estrada Y, et al. Ritlecitinib and brepocitinib demonstrate significant improvement in scalp alopecia areata biomarkers. J Allergy Clin Immunol (2022) 149(4):1318–28. doi: 10.1016/j.jaci.2021.10.036
98. Xu H, Jesson MI, Seneviratne UI, Lin TH, Sharif MN, Xue L, et al. PF-06651600, a dual JAK3/TEC family kinase inhibitor. ACS Chem Biol (2019) 14(6):1235–42. doi: 10.1021/acschembio.9b00188
99. Sung L. Chapter two - covalent drugs in development for immune-mediated diseases. In: Ward RA, Grimster NP, editors. Annual reports in medicinal chemistry, vol. 56. Academic Press;Cambridge, MA, US (2021). p. 33–74.
100. Gilhar A, Keren A, Paus R. JAK inhibitors and alopecia areata. Lancet (2019) 393(10169):318–9. doi: 10.1016/S0140-6736(18)32987-8
101. Reiner Z. Hypertriglyceridaemia and risk of coronary artery disease. Nat Rev Cardiol (2017) 14(7):401–11. doi: 10.1038/nrcardio.2017.31
102. Hoisnard L, Lebrun-Vignes B, Maury S, Mahevas M, El Karoui K, Roy L, et al. Adverse events associated with JAK inhibitors in 126,815 reports from the WHO pharmacovigilance database. Sci Rep (2022) 12(1):7140. doi: 10.1038/s41598-022-10777-w
Keywords: alopecia areata, JAK/STAT, JAK inhibition, cytokines, clinical trials
Citation: Lensing M and Jabbari A (2022) An overview of JAK/STAT pathways and JAK inhibition in alopecia areata. Front. Immunol. 13:955035. doi: 10.3389/fimmu.2022.955035
Received: 27 May 2022; Accepted: 02 August 2022;
Published: 30 August 2022.
Edited by:
Francesca Romana Spinelli, Sapienza University of Rome, ItalyReviewed by:
Joanna Czuwara, Medical University of Warsaw, PolandCopyright © 2022 Lensing and Jabbari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali Jabbari, YWxpLWphYmJhcmlAdWlvd2EuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.