 Wenping Liu
Wenping Liu Jibo Wang
Jibo Wang- Department of Rheumatology & Clinical Immunology, The Affiliated Hospital of Qingdao University, Qingdao, China
Systemic lupus erythematosus (SLE) is a typical autoimmune disease with a complex pathogenesis and genetic predisposition. With continued understanding of this disease, it was found that SLE is related to the interferon gene signature. Most studies have emphasized the important role of IFN-α in SLE, but our previous study suggested a nonnegligible role of IFN-γ in SLE. Some scholars previously found that IFN-γ is abnormally elevated as early as before the classification of SLE and before the emergence of autoantibodies and IFN-α. Due to the large overlap between IFN-α and IFN-γ, SLE is mostly characterized by expression of the IFN-α gene after onset. Therefore, the role of IFN-γ in SLE may be underestimated. This article mainly reviews the role of IFN-γ in SLE and focuses on the nonnegligible role of IFN-γ in SLE to gain a more comprehensive understanding of the disease.
1 Introduction
Systemic lupus erythematosus (SLE) is a typical autoimmune disease that can affect various tissues and organs throughout the body. SLE is characterized by excessive activation of the immune system, resulting in increases in autoantibodies and immune complexes and organ dysfunction (1). At present, the pathogenesis of SLE is unclear. Sun exposure or viral infection can induce the disease in individuals with genetic susceptibilities, and women are the most vulnerable group (2). With continued development of sequencing technology, SLE was found to have a distinct interferon (IFN) gene signature (3), which is found in approximately 75% of adult patients and 90% of pediatric patients (4). Interferon is a cytokine produced in response to viral infection and has various effects, such as regulating immunity, antiviral and antitumor activities (5). According to the primary protein sequence, cognate receptor, gene locus, and cell type responsible for its production, IFNs are mainly divided into three types. Type I IFNs include IFN-α subtypes and IFN-β, -ϵ, -κ, and -ω. Type II IFNs include IFN-γ. Type III IFNs include IFN-λ (6). Many studies have shown the dominance of IFN-α in SLE (7), but some studies have also indicated that the IFN-γ gene signature may occur early in SLE (8) and may have an important role in lupus nephritis (LN) (9). Some studies found that the levels of IFN-γ and its related genes were closely related to the activation of type I IFNs in SLE patients (10, 11). More importantly, treatment against IFN-α seems to have a limited effect on SLE (12, 13), while treatment against IFN-γ could be more attractive (14). Our previous studies have emphasized the important role of IFN-γ in the initial and active stages of SLE (15). Therefore, this review will focus on IFN-γ and SLE to contribute to the understanding and treatment of SLE.
2 IFN-γ and its signal transduction
IFN-γ is a pleiotropic type II IFN that is mainly produced by effector Th1 CD4+ T cells, cytotoxic CD8+ T cells and NK cells and to a lesser extent by other cell types, such as dendritic cells (DCs), macrophages and B cells (16). IFN-γ binds to the IFN-γ receptor (IFNGR), which is expressed on most cells and activates janus kinase 1 (JAK1) and JAK2 through the canonical pathway (Figure 1), leading to the phosphorylation of STAT1 homodimers and binding to the IFN-γ activation site (GAS) followed by subsequent gene transcribe (17). In addition, IFN-γ can also play a role in signal transduction through noncanonical pathways (18, 19). There is significant overlap (crosstalk) between type I and type II inducible genes, and signaling pathways can be shared between the two. Each interferon type induces the production of the other, ultimately resulting in stimulation from both sides and a mixed signature (17). Therefore, it is difficult to tell the difference between the two.
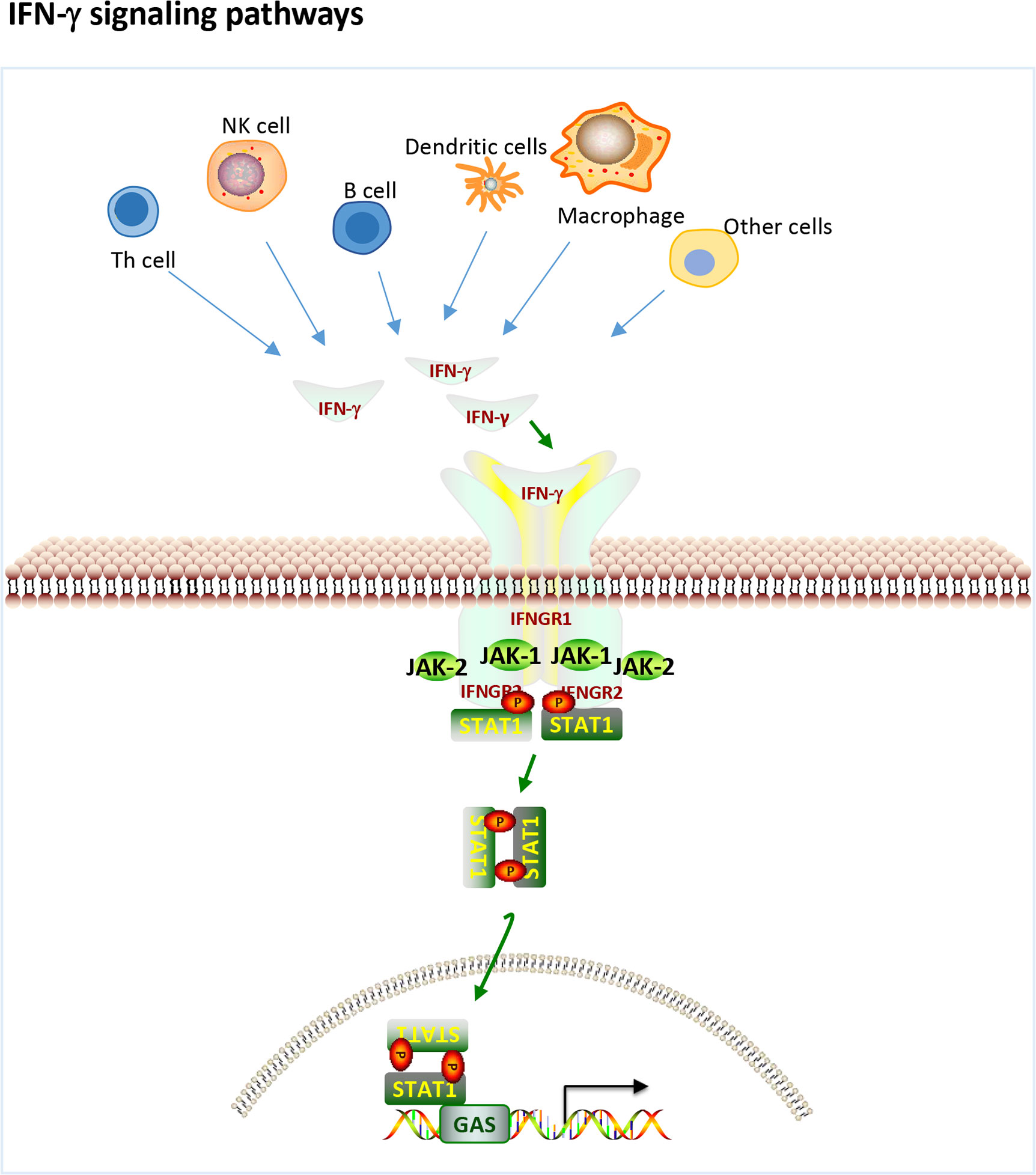
Figure 1 IFN-γ production and canonical signaling pathways. Th1 CD4+ T cells, cytotoxic CD8+ T cells and NK cells and to a lesser extent other cell types, such as dendritic cells (DCs), macrophages and B cells can produce IFN-γ. IFN-γ binds to the IFN-γ receptor (IFNGR) to activate JAK1 and JAK2 leading to the phosphorylation of STAT1 homodimers and binding to the IFN-gamma activation site (GAS) followed by subsequent gene transcribe.
3 IFN-γ signature in SLE
Studies have shown that the level of IFN-γ in the serum of patients with SLE is higher than that in healthy individuals (20–23), and there is abnormal accumulation of IFN-γ in the body long before the diagnosis of SLE and before the appearance of autoantibodies and IFN-α (8). Both the mRNA and protein levels of IFN-γ were significantly higher in SLE patients than in healthy donors (9, 24), and the mRNA levels of type II IFN-inducible genes (IRF1, GBP1, CXCL9, CXCL10, and SERPING1) were elevated in SLE patients. In addition, the relative expression levels of the important transcription factors TBX21 and EOMES (25), which promote IFN-γ gene expression, were also elevated in SLE patients. We previously found an IFN-γ signature when analyzing the genetic signature of active SLE onset (15). Liu et al. evaluated the relationship between the IFN-γ signaling pathway and disease activity-related indicators and found that IFN-γ titers had good correlations with disease activity (25, 26). Manman et al. found that IFNG expression and the IFN-II score were positively correlated with SLEDAI scores and anti-dsDNA antibody levels but negatively correlated with serum complement third-component levels (25). Moreover, some studies also showed that the levels of IFN-γ in the serum of patients with LN were higher than those of patients with SLE without LN (27), and IFN-γ was detected in the renal tissue of patients with LN. Single-cell transcriptome analysis of kidney-infiltrating immune cells revealed that all patients produced IFN-γ (9). Furthermore, transgenic mice overexpressing IFN-γ developed autoantibodies against dsDNA and proliferative glomerulonephritis (28). Overall, the IFN-γ signaling pathway is activated in SLE patients, and IFNG levels and IFNII scores can be used as indicators of SLE disease activity to guide clinical treatment.
4 Genetics and epigenetics of IFN-γ in SLE
SLE is a disease with genetic characteristics. DNA sequence differences and epigenetic differences such as DNA methylation and acetylation can alter gene expression and play an important role in SLE (29). Single nucleotide polymorphisms (SNPs) are the most common genetic polymorphisms. Multiple IFN-γ related SNPs have been identified as risk loci in SLE. The greatest risk of developing SLE was detected in individuals with a Met14/Val14 genotype of IFNGR1 or a Gln64/Gln64 genotype of IFNGR2 (30). IFN-γ gene polymorphisms associated with susceptibility to SLE (31). Marut’s study found an association between the IFN-γ gene polymorphism (+874A) and the manifestations of SLE arthritis (32). A SNP of STAT4 (rs7574865) (33) was found to be associated with SLE in the IFN-STAT signaling pathway. In addition, many SNPs of interferon regulated factors (IRFs) have also been found to be associated with the risk of developing SLE, including IRF3 rs2304206 (34), IRF5 rs200464 (35), IRF7 rs1131665 (36), IRF8 rs11644034 and rs2280381 (37) polymorphism. More important, epigenetic regulation is an important mechanism of transcriptional activation in SLE pathogenesis. Epigenetics refers to the genetic regulation of changes in gene expression caused by changes in DNA methylation, histone acetylation, and chromatin accessibility without changing the nucleotide sequence of DNA (38). The enhanced response of Th1 cells in SLE is accompanied by transcriptional activation of the intracellular IFGN locus (39, 40), which is mainly due to various epigenetic changes such as H3K4 tri-methylation (41), H4-Ac catalyzed by histone acetyltransferase (42), and chromatin conformational remodeling (43). On the other hand, IFN-γ can also induce extensive remodeling of the epigenome (44). For example, IFN-γ induces IRF1-STAT1 and histone acetylation to mark promoters and enhancers of TNF and IL6 loci, resulting in increased inflammatory responses in response to subsequent over-induction by TLR ligand stimulation (45). Additionally, acetylation (H3K27Ac) is associated with gene expression, while trimethylation (H3K27me3) is associated with gene silencing (46). It was found that IFN-γ stably silences a small group of genes of anti-inflammatory including MERTK, PPARG and RANK by maintaining H3K27me3 at gene promoters (47). Thus, IFN-γ treatment made these genes refractory to the induction of glucocorticoids and IL-4. All these epigenetic changes involving IFN-γ promote and solidify the inflammation development of SLE.
5 Mode of action of IFN-γ in SLE
IFN-γ is a major proinflammatory cytokine that regulates the functions of several important immune system cells (Figure 2), including B cells and T cells (48, 49), and contributes significantly to the development of SLE.
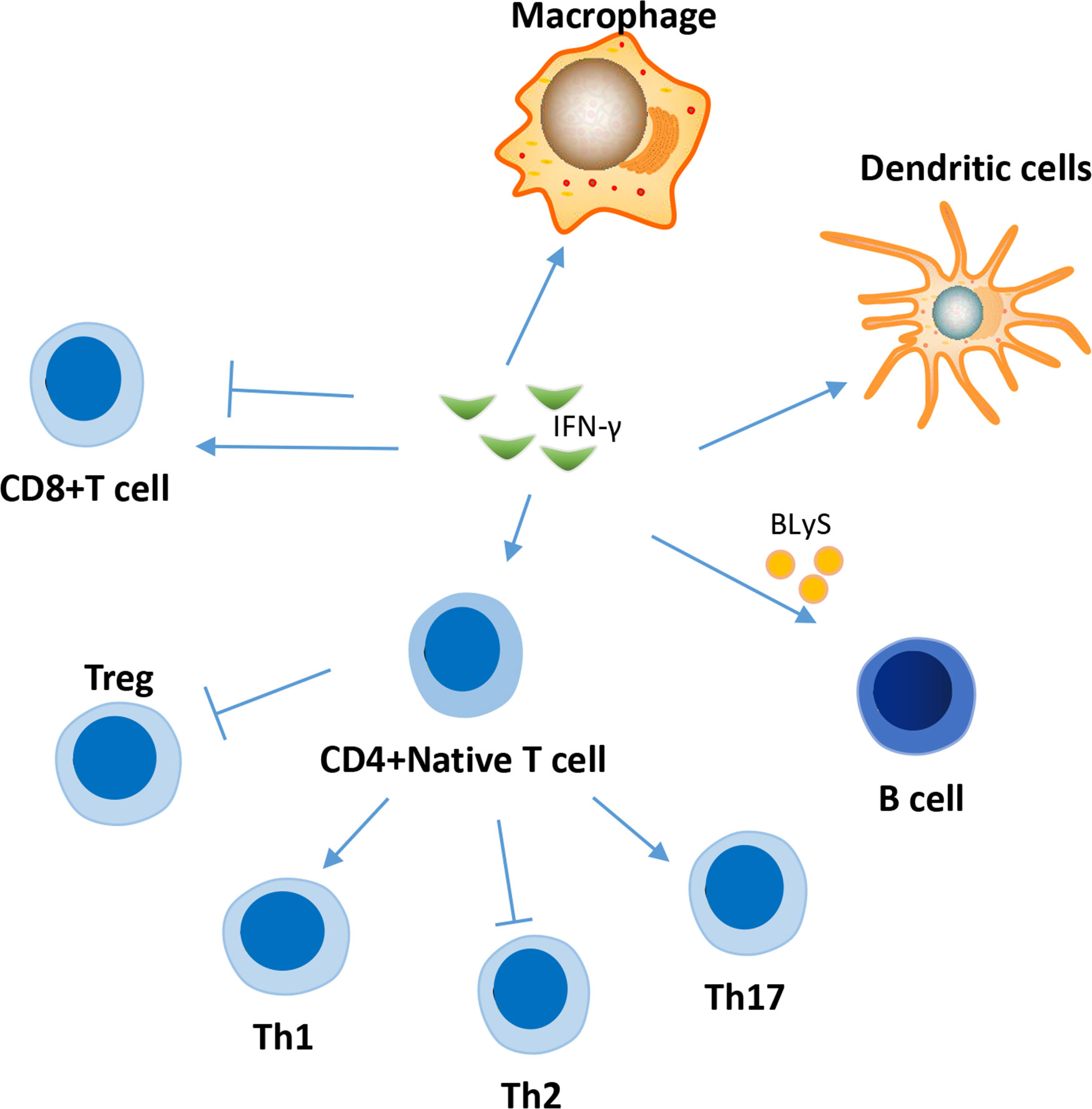
Figure 2 Effects of IFN-γ on several immune cells in the pathogenesis of SLE. IFN-γ affect the function of a variety of immune cells in the pathogenesis of SLE, involving T cell, B cell, macrophage, dendritic cell and et al. The effect of IFN-γ on CD8 cells in SLE is two-sided. IFN-γ can promote the differentiation of naive CD4+ T cells into inflammatory Th1 and Th17 cells, while inhibiting their differentiation into Treg cells and Th2 cells.
5.1 IFN-γ affects T cell function in SLE
5.1.1 CD4+T cell
Imbalance of Th1 and Th2 cells is common in the pathogenesis of SLE (50). Earlier studies suggested that the production of large amounts of antibodies in SLE was associated with Th2 responses in peripheral blood (51), but growing evidence suggests the importance of the T helper 1 (Th1) response in SLE (52). The immune response in proliferative LN has been shown to be biased toward the Th1 axis (53, 54). Th1 cells can secrete IFN-γ to promote SLE-related pathology, while IFN-γ in turn enhances the pathogenic role of Th1 cells. IFN-γ plays a vital role in the differentiation and maturation of Th1 cells (55). Although IL-12 is a typical cytokine that is necessary for the activation of Th1 cells, the role of IFN-γ is still very important (56). IFN-γ and STATI can activate the downstream transcriptional target T-bet, and the transcription factor T-bet is the main regulator of the Th1 phenotype and can stabilize the Th1 phenotype (57). More importantly, Th1 polarization in the absence of IFN-γ induction is incomplete (58, 59). In addition, IFN-γ signaling is actively involved in inhibiting CD4+ T-cell differentiation into Th2 (60), which is one of the reasons for the imbalance of Th1/Th2. Although in vitro studies have shown that IFN-γ has an inhibitory effect on Th17 cells (61, 62), increased Th17 cells and IL-17 cytokines have also been found in SLE patients (63). The ratio of Th17 and Th1 cells in SLE patients were both higher than that in healthy controls. And it was found that Th17 cells play an important role in SLE histopathological damage (64, 65). Shah’ study found that the frequency of IL-17+ cells was directly correlated with the frequency of IFN-γ+ cells (66). Anyhow, elevated IFN-γ in SLE resulting in a skewed phenotype of CD4+ T cell populations toward Th1 and Th17, which play an important role in the pathogenesis of SLE.
5.1.2 Treg cell
Treg cells act as immunosuppressors, and defects in function or numbers are thought to contribute to SLE pathogenesis due to their role in maintaining peripheral immune tolerance (67). Recent studies have shown that in addition to the ability of IFN-γ to directly inhibit Treg cell function (68–70), the inhibition of effector T-cell activation by Treg cells is suppressed in an IFN-γ-enriched environment, and this inhibition requires the expression of IFNGR on Treg cells (71). Of course, there have been some studies showing that although the function and number of regulatory T cells in SLE patients are defective, this effect is due to the resistance of effector T cells to inhibit SLE, rather than defects in Treg cell functions (72). In conclusion, the mechanism by which IFN-γ inhibits Treg cell functions remains to be elucidated.
5.1.3 CD8+ T cells
CD8+ T cells are cytotoxic cells that kill infected or damaged cells by releasing cytotoxins such as granzymes and perforin. There appears to be some inconsistency in the role of CD8+ T cells in SLE (73). On the one hand, CD8+ T cells in the peripheral blood of SLE patients often have reduced granzyme B and perforin production and exhibit impaired cytolytic function (74), which impairs the removal of autoreactive B cells and increases autoantibodies, accelerating the onset of lupus. Furthermore, the decrease in cytolytic capacity was associated with poor control of Epstein–Barr virus infection and susceptibility to infection (75, 76), which are more common in SLE. On the other hand, in contrast to the decreased cytolytic functions of circulating CD8+ T cells, CD8+ T cells extracted from sites of inflammation mostly showed enhanced effector functions (77, 78), leading to tissue damage. The effects of IFN-γ on CD8+ T cells are also multifaceted. IFN-γ signaling directly regulates several aspects of CD8+ T-cell biology. Most importantly, IFN-γ is required for cytolytic capacity of CD8+ T cells (79). In fact, early experiments using recombinant proteins showed that full cytolytic capacity was not achieved until CD8+ T cells were exposed to IFN. IFN-γ signaling in CD8+ T cells upregulates the expression of IL-2 receptor, the transcription factor T-bet, and granzymes. IL-2 responsiveness is critical for the generation of cytolytic CD8+ T cells, while granzymes are responsible for mediating the cytolysis of CD8+ T-cell targets (80). IFN-γ also regulates CD8+ T-cell proliferation after antigen exposure (81). Conversely, IFN-γ-restricted CD8+ T-cell effector responses were found in some studies (82). In short, the effect of IFN-γ on CD8+ T cells remains unclear.
5.2 IFN-γ affects B cell function in SLE
5.2.1 B cell production
B cells play an important role in the pathogenesis of SLE (83). These cells are mediators of inflammation, enhancing inflammation and leading to direct tissue and cell damage by producing pathogenic antibodies. IFN-γ signaling can promote B-cell division during the early proliferative response following primary antigen exposure (84). IFN-γ can stimulate T cells (85), and antigen presenting cells (APCs) to produce B lymphocyte-stimulating factor (BLyS) (86), which is essential for B-cell differentiation, proliferation and survival, regulates B-cell generation and maturation (87, 88), and has been identified as a therapeutic target for SLE.
5.2.2 Germinal centers formation in B cells
Furthermore, IFN-γ can induce the formation of germinal centers (GC) and B cells (89). IFN-γ integrates with BCR-, TLR- and/or CD40-dependent signaling to promote expression of the B-cell-intrinsic key transcription factor of B-cell lymphoma 6 protein (BCL-6) in mouse and human primary B cells (90). BCL-6 is critical in GC reactions (91). Lack of B-cell IFN-γR signaling significantly reduced all autoantibody isotypes by eliminating spontaneous GC formation (90). Furthermore, Chodisetti et al. found that type II but not type I IFN signaling was essential for TLR7-mediated promotion of autoreactive B cells and systemic autoimmunity (92). IFNγ and its downstream signaling molecules STAT1 and T-bet have nonredundant roles in B cell-mediated promotion of TLR7-driven development of AFC, GC and SLE, and type I IFN signaling contributes modestly to these processes (90, 92, 93).
5.2.3 IgG class switching
In addition to IFN-γ-mediated activation of STAT1 in B cells to induce autoantibody production (89), IFN-γ plays an important role in antibody class switching. IFN-γ is able to promote B-cell IgG class switching to more pathogenic (mouse IgG2a and IgG3) autoantibodies (94–97) and promote the activation of IgGFc receptors and complement (98), contributing to disease severity. In addition, IFN-γ is involved in the development of lupus-associated hypergamma globulinemia (99, 100). IFNG also activates CD11b+ cells (101), enabling these cells to bind to antibody-coated target cells, thereby promoting inflammation and exacerbating the development of SLE.
5.3 IFN-γ affects dendritic cells
Dendritic cells (DCs) are the most typical APCs, which can activate naive T cells and trigger T cell responses that lead to tissue damage in SLE (102). Among the numerous DCs, CD11b+ DC subset appears to be specialized in MHC class II-mediated antigen presentation in vivo (103). Like the mouse CD11b+ DC subset, human BDCA1 DCs may act as a subset of DCs that exclusively present antigens through MHC class II molecules (104). Upregulation of CD11b+ DCs has been found to have a central role in the pathological development of LN and a major role in driving end-organ disease (105). IFN-γ plays a critical role in the maturation and differentiation of DCs, and affects the entire process of antigen processing and presentation. IFN-γ is considered to be an important stimulator of MHC class II gene expression (106). The ability to upregulate MHC class II is unique to IFN-γ, which induces the expression of class II transactivator (CIITA) (107), a master regulator of MHC transcription, and promotes the assembly of the MHC II enhancer. IFN-γ can upregulate the expression of CD40, CD80, CD83 and CD86 molecules on DCs to induce DCs maturation (108). Besides, IFN-γ can up-regulate the expression of immunoproteasome components of LMP1 and LMP7 (109) and the expression of transporter proteins (TAPs) associated with antigen processing (110), which plays an important role in antigen presentation process involving MHC II.
5.4 IFN-γ affects macrophages
Aberrant activation and unbalanced polarization of macrophages have been shown to be involved in the pathogenesis of SLE (111). IFN-γ can enhance the quantity, quality and pool of peptides bound by class I and class II MHC (106, 109, 112–114), furthermore, IFN-γ can activate the transcription of class I and class II MHC molecules, induce the expression of MHC class I and class II antigens in macrophages, endothelial cells or epithelial cells, promote the local presentation of antigens, activate macrophages (115) and induce macrophage polarization to the M1 phenotype (116, 117), which can enhance the secretion of cytokines (IL-1 and TNF) (118) and increase the release of reactive oxygen species intermediates and Nitricoxide (NO) (119). These M1 macrophages exhibit proinflammatory functions and play an important role in organ damage in SLE (120). And IFN-γ regulates the synthesis of chemokines such as CXCL10 (121). These effects have a marked effect on the activation of inflammatory cell populations.
6 Targeting IFN-γ in SLE
Satisfactory outcomes were observed after the application of anti-IFN-γ in a mouse lupus model (Table 1). Ozmen et al. found that treating NZB/W mice with soluble murine IFN-γ receptors inhibited the onset of glomerulonephritis (123). Werwitzke et al. treated lupus-prone NZB/NZW F1 mice with recombinant soluble Fc gamma receptor II (CD32), and found that it inhibited chronic murine lupus pathologyin vivo (128). Lawson et al. performed intramuscular injection of a cDNA plasmid encoding IFN-γR/Fc into MRL-Fas(lpr) lupus mice, and found that lupus development and progression could be delayed, even if the treatment was initiated at a late stage (126). Besides, in (NZB)/(NZW)F1 mice, a favorable effect was observed in the treatment of lupus nephritis using an IFN-γ monoclonal antibody in vivo (122). Furthermore, lack of IFN-RII protects MRL/lpr mice from developing severe autoimmune-related lymphadenopathy, autoantibodies, and kidney disease (127). Deletion of the IFN-γ receptor prevents autoantibody production and glomerulonephritis in lupus-prone (NZB x NZW) F1 mice (124). Additionally, Schrott et al. found that chronic soluble IFN-γ receptor treatment attenuated behavioral abnormalities in autoimmune mice (125). Human clinical trials targeting IFN-γ have yielded some results (Table 1). AMG 811 is a fully human (IgG1) anti-IFN-γ antibody. In patients with mild to moderate SLE, a single dose of AMG 811 was well tolerated and could normalize IFN-regulated gene expression, resulting in a dose-dependent decrease in serum CXCL-10 levels (14, 129). AMG 811 treatment led to changes in IFN-γ-associated biomarkers and was well tolerated, but no significant clinical benefit was observed in patients with discoid lupus erythematosus (DLE) (130). Encouragingly positive phase Ib trials have shown the efficacy of blocking the IFN-γ pathway to treat extrarenal lupus (131). Collectively, these findings suggest that IFN-γ is a central cytokine in LN, and further studies of LN should examine IFN-γ inhibition given the acceptable safety profile of its direct blockade.
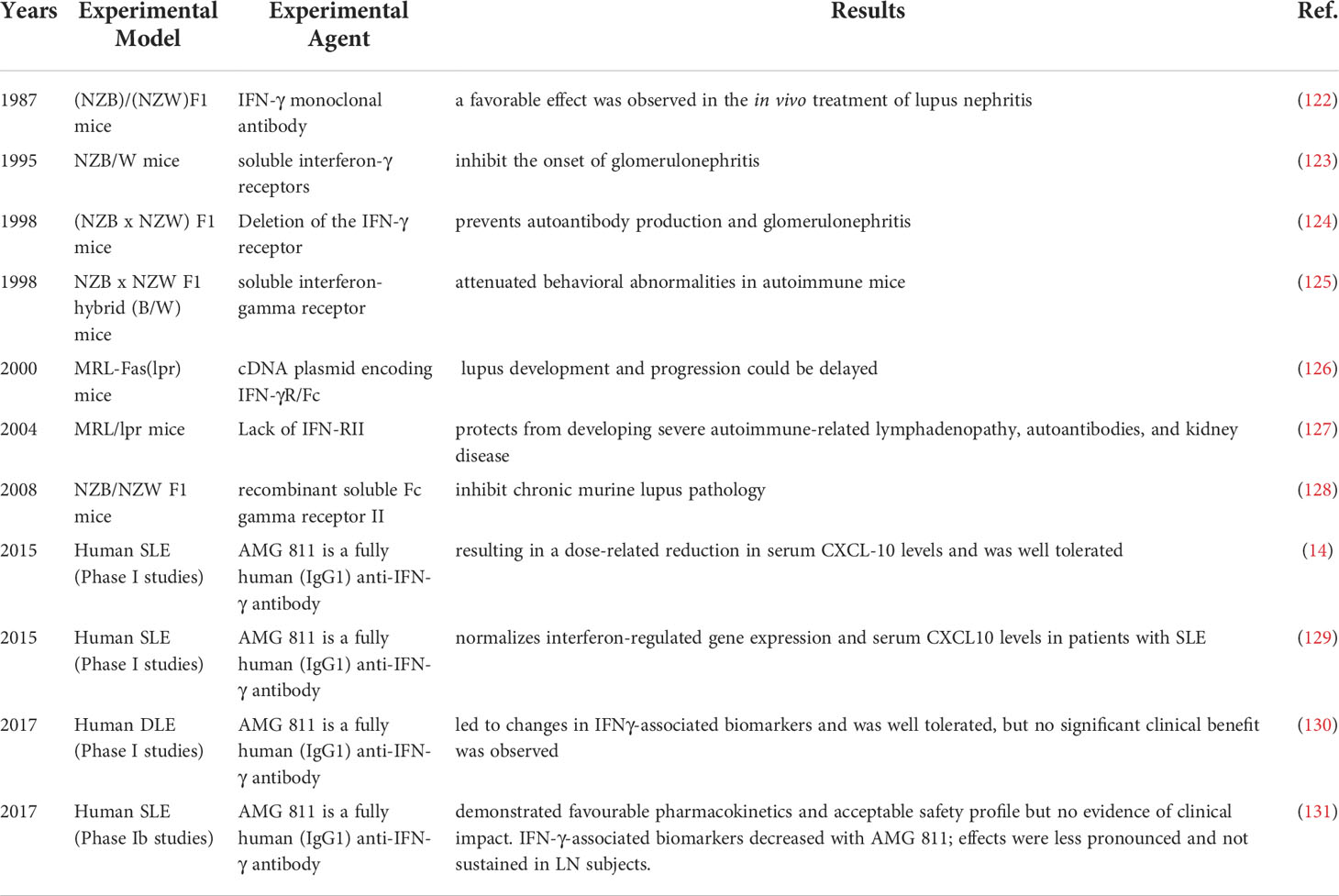
Table 1 Therapy effect of targeting IFN-γ on SLE.
7 JAK inhibitors that block IFN-γ in SLE
Janus kinases (JAKs) are intracellular non-receptor tyrosine kinases that play key roles in the signaling pathways of many cytokines. This also provides a basis for the application of JAK inhibitors in the treatment of SLE. Both type I and type II interferon conduct signal transduction through the JAK-STAT signaling pathway (132), and there are many overlaps downstream of both. Therefore, blocking the JAK pathway has both therapeutic effects on both type I IFN and type II IFN mediated disease processes. Studies have found that JAKs inhibitors can inhibit the IFN signaling in human DCs, reduce CD80/CD86 expression and T cell stimulation ability (133), and reduce the production of various inflammatory cytokines including IFN-γ (134) in SLE mice. It can also restore the balance of naive CD4+ T cells and effector/memory cell populations in SLE mice (135). Besides, evidence from a lupus model suggests that tofacitinib (a JAK inhibitor) reduces levels of anti-dsDNA and proteinuria, and relieve symptom of nephritis and rash (136, 137). Moreover, in clinical studies, JAKs inhibitors were also found to significantly improve the signs and symptoms of active SLE, with a high remission rate of 67% for arthritis or rash in SLE patients (138, 139).
8 Conclusion
There are genetic features of IFN-γ in SLE, especially in the initial and active stages of the disease, suggesting that IFN-γ plays an important role in the pathogenesis of SLE. IFN-γ is an important contributor to immune regulation in the body, which may be one of the roles that cannot be ignored in the pathogenesis of SLE. Moreover, current animal studies support the feasibility of targeted IFN-γ therapy in SLE; however, no obvious effect of targeted IFN therapy has been found in human clinical trials, although some of the inflammatory indicators showed significant changes compared with those in the placebo group. Whereas, these trials are designed to focus more on drug safety than efficacy. It is worth noting that due to the important role of IFN-γ in the response to infection with some viruses, such as herpesvirus and Salmonella, during targeted IFN-γ therapy also needs to be examined in more extensive population experiments. Nevertheless, the pathogenic role of IFN-γ in SLE is of interest and treatment target IFN-γ is more promising.
Author contributions
WL: Organize literature and original draft writing; SZ: Search literature and writing editing; JW: Conception and writing review. All authors contributed to the article and approved the submitted version.
Acknowledgments
We gratefully thank all authors.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pons-Estel GJ, Alarcón GS, Scofield L, Reinlib L, Cooper GS. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum (2010) 39:257. doi: 10.1016/j.semarthrit.2008.10.007
2. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers (2016) 2:16039. doi: 10.1038/nrdp.2016.39
3. Nakano M, Iwasaki Y, Fujio K. Transcriptomic studies of systemic lupus erythematosus. Inflamm Regener (2021) 41:11. doi: 10.1186/s41232-021-00161-y
4. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med (2003) 197:711–23. doi: 10.1084/jem.20021553
5. Zhang S-Y, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev (2008) 226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x
7. Rönnblom L, Alm GV. An etiopathogenic role for the type I IFN system in SLE. Trends Immunol (2001) 22:427–31. doi: 10.1016/s1471-4906(01)01955-x
8. Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM, et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis (2016) 75:2014–21. doi: 10.1136/annrheumdis-2015-208140
9. Fava A, Buyon J, Mohan C, Zhang T, Belmont HM, Izmirly P, et al. Integrated urine proteomics and renal single-cell genomics identify an IFN-γ response gradient in lupus nephritis. JCI Insight (2020) 5:138345. doi: 10.1172/jci.insight.138345
10. Greene JA, DeVecchio JL, Gould MP, Auletta JJ, Heinzel FP. In vivo and in vitro regulation of type I IFN synthesis by synergistic effects of CD40 and type II IFN. J Immunol (2006) 176:5995–6003. doi: 10.4049/jimmunol.176.10.5995
11. Weihua X, Ling W, Kalvakolanu DV. Regulation of interferon-alpha/beta-stimulated gene expression through the gamma-activated transcriptional element. Antiviral Res (1999) 40:145–53. doi: 10.1016/s0166-3542(98)00056-4
12. Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, et al. A phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis (2016) 75:196–202. doi: 10.1136/annrheumdis-2014-206090
13. Amezcua-Guerra LM, Ferrusquía-Toriz D, Castillo-Martínez D, Márquez-Velasco R, Chávez-Rueda AK, Bojalil R. Limited effectiveness for the therapeutic blockade of interferon α in systemic lupus erythematosus: a possible role for type III interferons. Rheumatol (Oxford) (2015) 54:203–5. doi: 10.1093/rheumatology/keu020
14. Welcher AA, Boedigheimer M, Kivitz AJ, Amoura Z, Buyon J, Rudinskaya A, et al. Blockade of interferon-γ normalizes interferon-regulated gene expression and serum CXCL10 levels in patients with systemic lupus erythematosus. Arthritis Rheumatol (2015) 67:2713–22. doi: 10.1002/art.39248
15. Liu W, Li M, Wang Z, Wang J. IFN-γ mediates the development of systemic lupus erythematosus. BioMed Res Int (2020) 2020:7176515. doi: 10.1155/2020/7176515
16. Billiau A, Matthys P. Interferon-gamma: a historical perspective. Cytokine Growth Factor Rev (2009) 20:97–113. doi: 10.1016/j.cytogfr.2009.02.004
17. Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264:1415–21. doi: 10.1126/science.8197455
18. Ramana CV, Gil MP, Han Y, Ransohoff RM, Schreiber RD, Stark GR. Stat1-independent regulation of gene expression in response to IFN-gamma. Proc Natl Acad Sci U.S.A. (2001) 98:6674–9. doi: 10.1073/pnas.111164198
19. Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol (2002) 23:96–101. doi: 10.1016/s1471-4906(01)02118-4
20. Viallard JF, Pellegrin JL, Ranchin V, Schaeverbeke T, Dehais J, Longy-Boursier M, et al. Th1 (IL-2, interferon-gamma (IFN-gamma)) and Th2 (IL-10, IL-4) cytokine production by peripheral blood mononuclear cells (PBMC) from patients with systemic lupus erythematosus (SLE). Clin Exp Immunol (1999) 115:189–95. doi: 10.1046/j.1365-2249.1999.00766.x
21. Paradowska-Gorycka A, Wajda A, Stypinska B, Walczuk E, Rzeszotarska E, Walczyk M, et al. Variety of endosomal TLRs and interferons (IFN-α, IFN-β, IFN-γ) expression profiles in patients with SLE, SSc and MCTD. Clin Exp Immunol (2021) 204:49–63. doi: 10.1111/cei.13566
22. al-Janadi M, al-Balla S, al-Dalaan A, Raziuddin S. Cytokine profile in systemic lupus erythematosus, rheumatoid arthritis, and other rheumatic diseases. J Clin Immunol (1993) 13:58–67. doi: 10.1007/BF00920636
23. Yang BC, Wang YS, Lin LC, Liu MF. Induction of apoptosis and cytokine gene expression in T-cell lines by sera of patients with systemic lupus erythematosus. Scand J Immunol (1997) 45:96–102. doi: 10.1046/j.1365-3083.1997.d01-371.x
24. Csiszár A, Nagy G, Gergely P, Pozsonyi T, Pócsik E. Increased interferon-gamma (IFN-gamma), IL-10 and decreased IL-4 mRNA expression in peripheral blood mononuclear cells (PBMC) from patients with systemic lupus erythematosus (SLE). Clin Exp Immunol (2000) 122:464–70. doi: 10.1046/j.1365-2249.2000.01369.x
25. Liu M, Liu J, Hao S, Wu P, Zhang X, Xiao Y, et al. Higher activation of the interferon-gamma signaling pathway in systemic lupus erythematosus patients with a high type I IFN score: relation to disease activity. Clin Rheumatol (2018) 37:2675–84. doi: 10.1007/s10067-018-4138-7
26. Liu TF, Jones BM. Impaired production of IL-12 in system lupus erythematosus. II: IL-12 production in vitro is correlated negatively with serum IL-10, positively with serum IFN-gamma and negatively with disease activity in SLE. Cytokine (1998) 10:148–53. doi: 10.1006/cyto.1997.0269
27. Miyake K, Nakashima H, Akahoshi M, Inoue Y, Nagano S, Tanaka Y, et al. Genetically determined interferon-gamma production influences the histological phenotype of lupus nephritis. Rheumatol (Oxford) (2002) 41:518–24. doi: 10.1093/rheumatology/41.5.518
28. Seery JP, Carroll JM, Cattell V, Watt FM. Antinuclear autoantibodies and lupus nephritis in transgenic mice expressing interferon gamma in the epidermis. J Exp Med (1997) 186:1451–9. doi: 10.1084/jem.186.9.1451
29. Costa-Reis P, Sullivan KE. Genetics and epigenetics of systemic lupus erythematosus. Curr Rheumatol Rep (2013) 15:369. doi: 10.1007/s11926-013-0369-4
30. Nakashima H, Inoue H, Akahoshi M, Tanaka Y, Yamaoka K, Ogami E, et al. The combination of polymorphisms within interferon-gamma receptor 1 and receptor 2 associated with the risk of systemic lupus erythematosus. FEBS Lett (1999) 453:187–90. doi: 10.1016/s0014-5793(99)00701-2
31. Kim K, Cho S-K, Sestak A, Namjou B, Kang C, Bae S-C. Interferon-gamma gene polymorphisms associated with susceptibility to systemic lupus erythematosus. Ann Rheum Dis (2010) 69:1247–50. doi: 10.1136/ard.2009.117572
32. Tangwattanachuleeporn M, Sodsai P, Avihingsanon Y, Wongpiyabovorn J, Wongchinsri J, Hirankarn N. Association of interferon-gamma gene polymorphism (+874A) with arthritis manifestation in SLE. Clin Rheumatol (2007) 26:1921–4. doi: 10.1007/s10067-007-0699-6
33. Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med (2007) 357:977–86. doi: 10.1056/NEJMoa073003
34. Santana-de Anda K, Gómez-Martín D, Monsivais-Urenda AE, Salgado-Bustamante M, González-Amaro R, Alcocer-Varela J. Interferon regulatory factor 3 as key element of the interferon signature in plasmacytoid dendritic cells from systemic lupus erythematosus patients: novel genetic associations in the Mexican mestizo population. Clin Exp Immunol (2014) 178:428–37. doi: 10.1111/cei.12429
35. Wang J-M, Huang A-F, Yuan Z-C, Su L-C, Xu W-D. Association of IRF5 rs2004640 polymorphism and systemic lupus erythematosus: A meta-analysis. Int J Rheum Dis (2019) 22:1598–606. doi: 10.1111/1756-185X.13654
36. Kawasaki A, Furukawa H, Kondo Y, Ito S, Hayashi T, Kusaoi M, et al. Association of PHRF1-IRF7 region polymorphism with clinical manifestations of systemic lupus erythematosus in a Japanese population. Lupus (2012) 21:890–5. doi: 10.1177/0961203312439333
37. Lessard CJ, Adrianto I, Ice JA, Wiley GB, Kelly JA, Glenn SB, et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am J Hum Genet (2012) 90:648–60. doi: 10.1016/j.ajhg.2012.02.023
38. Zouali M. Epigenetics in lupus. Ann NY Acad Sci (2011) 1217:154–65. doi: 10.1111/j.1749-6632.2010.05831.x
39. Aune TM, Collins PL, Chang S. Epigenetics and T helper 1 differentiation. Immunology (2009) 126:299–305. doi: 10.1111/j.1365-2567.2008.03026.x
40. Aune TM, Collins PL, Collier SP, Henderson MA, Chang S. Epigenetic activation and silencing of the gene that encodes IFN-γ. Front Immunol (2013) 4:112. doi: 10.3389/fimmu.2013.00112
41. Zhang Z, Shi L, Dawany N, Kelsen J, Petri MA, Sullivan KE. H3K4 tri-methylation breadth at transcription start sites impacts the transcriptome of systemic lupus erythematosus. Clin Epigenet (2016) 8:14. doi: 10.1186/s13148-016-0179-4
42. Zhou W, Chang S, Aune TM. Long-range histone acetylation of the ifng gene is an essential feature of T cell differentiation. Proc Natl Acad Sci U.S.A. (2004) 101:2440–5. doi: 10.1073/pnas.0306002101
43. Eivazova ER, Aune TM. Dynamic alterations in the conformation of the ifng gene region during T helper cell differentiation. Proc Natl Acad Sci U.S.A. (2004) 101:251–6. doi: 10.1073/pnas.0303919101
44. Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol (2019) 20:1574–83. doi: 10.1038/s41590-019-0466-2
45. Qiao Y, Giannopoulou EG, Chan CH, Park S-H, Gong S, Chen J, et al. Synergistic activation of inflammatory cytokine genes by interferon-γ-induced chromatin remodeling and toll-like receptor signaling. Immunity (2013) 39:454–69. doi: 10.1016/j.immuni.2013.08.009
46. Chen T, Dent SYR. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet (2014) 15:93–106. doi: 10.1038/nrg3607
47. Qiao Y, Kang K, Giannopoulou E, Fang C, Ivashkiv LB. IFN-γ induces histone 3 lysine 27 trimethylation in a small subset of promoters to stably silence gene expression in human macrophages. Cell Rep (2016) 16:3121–9. doi: 10.1016/j.celrep.2016.08.051
48. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol (2004) 75:163–89. doi: 10.1189/jlb.0603252
49. Tau G, Rothman P. Biologic functions of the IFN-γ receptors. Allergy (1999) 54:1233–51. doi: 10.1034/j.1398-9995.1999.00099.x
50. Chan RW-Y, Lai FM-M, Li EK-M, Tam L-S, Chow K-M, Li PK-T, et al. Imbalance of Th1/Th2 transcription factors in patients with lupus nephritis. Rheumatol (Oxford) (2006) 45:951–7. doi: 10.1093/rheumatology/kel029
51. Klinman DM, Steinberg AD. Inquiry into murine and human lupus. Immunol Rev (1995) 144:157–93. doi: 10.1111/j.1600-065x.1995.tb00069.x
52. Akahoshi M, Nakashima H, Tanaka Y, Kohsaka T, Nagano S, Ohgami E, et al. Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus. Arthritis Rheum (1999) 42:1644–8. doi: 10.1002/1529-0131(199908)42:8<1644::AID-ANR12>3.0.CO;2-L
53. Masutani K, Akahoshi M, Tsuruya K, Tokumoto M, Ninomiya T, Kohsaka T, et al. Predominance of Th1 immune response in diffuse proliferative lupus nephritis. Arthritis Rheum (2001) 44:2097–106. doi: 10.1002/1529-0131(200109)44:9<2097::AID-ART360>3.0.CO;2-6
54. Tucci M, Lombardi L, Richards HB, Dammacco F, Silvestris F. Overexpression of interleukin-12 and T helper 1 predominance in lupus nephritis. Clin Exp Immunol (2008) 154:247–54. doi: 10.1111/j.1365-2249.2008.03758.x
55. Cope A, Le Friec G, Cardone J, Kemper C. The Th1 life cycle: molecular control of IFN-γ to IL-10 switching. Trends Immunol (2011) 32:278–86. doi: 10.1016/j.it.2011.03.010
56. Bradley LM, Dalton DK, Croft M. A direct role for IFN-gamma in regulation of Th1 cell development. J Immunol (1996) 157:1350–8. doi: 10.1006/cimm.1996.0224
57. Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-Bet is a STAT1-induced regulator of IL-12R expression in naïve CD4+ T cells. Nat Immunol (2002) 3:549–57. doi: 10.1038/ni794
58. Hattori K, Nishikawa M, Watcharanurak K, Ikoma A, Kabashima K, Toyota H, et al. Sustained exogenous expression of therapeutic levels of IFN-gamma ameliorates atopic dermatitis in NC/Nga mice via Th1 polarization. J Immunol (2010) 184:2729–35. doi: 10.4049/jimmunol.0900215
59. Morandi B, Bougras G, Muller WA, Ferlazzo G, Münz C. NK cells of human secondary lymphoid tissues enhance T cell polarization via IFN-gamma secretion. Eur J Immunol (2006) 36:2394–400. doi: 10.1002/eji.200636290
60. Oriss TB, McCarthy SA, Morel BF, Campana MA, Morel PA. Crossregulation between T helper cell (Th)1 and Th2: inhibition of Th2 proliferation by IFN-gamma involves interference with IL-1. J Immunol (1997) 158:3666–72.
61. Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol (2006) 18:349–56. doi: 10.1016/j.coi.2006.03.017
62. Lexberg MH, Taubner A, Albrecht I, Lepenies I, Richter A, Kamradt T, et al. IFN-γ and IL-12 synergize to convert in vivo generated Th17 into Th1/Th17 cells. Eur J Immunol (2010) 40:3017–27. doi: 10.1002/eji.201040539
63. Yang J, Chu Y, Yang X, Gao D, Zhu L, Yang X, et al. Th17 and natural treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum (2009) 60:1472–83. doi: 10.1002/art.24499
64. Koga T, Otomo K, Mizui M, Yoshida N, Umeda M, Ichinose K, et al. Calcium/Calmodulin-dependent kinase IV facilitates the recruitment of interleukin-17-Producing cells to target organs through the CCR6/CCL20 axis in Th17 cell-driven inflammatory diseases. Arthritis Rheumatol (2016) 68:1981–8. doi: 10.1002/art.39665
65. Schmidt T, Paust H-J, Krebs CF, Turner J-E, Kaffke A, Bennstein SB, et al. Function of the Th17/interleukin-17A immune response in murine lupus nephritis. Arthritis Rheumatol (2015) 67:475–87. doi: 10.1002/art.38955
66. Shah K, Lee W-W, Lee S-H, Kim SH, Kang SW, Craft J, et al. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res Ther (2010) 12:R53. doi: 10.1186/ar2964
67. Bonelli M, Smolen JS, Scheinecker C. Treg and lupus. Ann Rheum Dis (2010) 69 Suppl 1:i65–66. doi: 10.1136/ard.2009.117135
68. Olalekan SA, Cao Y, Hamel KM, Finnegan A. B cells expressing IFN-γ suppress treg-cell differentiation and promote autoimmune experimental arthritis. Eur J Immunol (2015) 45:988–98. doi: 10.1002/eji.201445036
69. Chang J-H, Kim Y-J, Han S-H, Kang C-Y. IFN-gamma-STAT1 signal regulates the differentiation of inducible treg: potential role for ROS-mediated apoptosis. Eur J Immunol (2009) 39:1241–51. doi: 10.1002/eji.200838913
70. Kelchtermans H, De Klerck B, Mitera T, Van Balen M, Bullens D, Billiau A, et al. Defective CD4+CD25+ regulatory T cell functioning in collagen-induced arthritis: an important factor in pathogenesis, counter-regulated by endogenous IFN-gamma. Arthritis Res Ther (2005) 7:R402–415. doi: 10.1186/ar1500
71. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, et al. Interferon-γ drives treg fragility to promote anti-tumor immunity. Cell (2017) 169:1130–1141.e11. doi: 10.1016/j.cell.2017.05.005
72. Chen P-M, Tsokos GC. T Cell abnormalities in the pathogenesis of systemic lupus erythematosus: an update. Curr Rheumatol Rep (2021) 23:12. doi: 10.1007/s11926-020-00978-5
73. Chen P-M, Tsokos GC. The role of CD8+ T-cell systemic lupus erythematosus pathogenesis: an update. Curr Opin Rheumatol (2021) 33:586–91. doi: 10.1097/BOR.0000000000000815
74. Comte D, Karampetsou MP, Yoshida N, Kis-Toth K, Kyttaris VC, Tsokos GC. SLAMF7 engagement restores defective effector CD8+ T cells activity in response to foreign antigens in systemic lupus erythematosus. Arthritis Rheumatol (2017) 69:1035–44. doi: 10.1002/art.40038
75. Kang I, Quan T, Nolasco H, Park S-H, Hong MS, Crouch J, et al. Defective control of latent Epstein-Barr virus infection in systemic lupus erythematosus. J Immunol (2004) 172:1287–94. doi: 10.4049/jimmunol.172.2.1287
76. Larsen M, Sauce D, Deback C, Arnaud L, Mathian A, Miyara M, et al. Exhausted cytotoxic control of Epstein-Barr virus in human lupus. PloS Pathog (2011) 7:e1002328. doi: 10.1371/journal.ppat.1002328
77. Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol (2019) 20:902–14. doi: 10.1038/s41590-019-0398-x
78. Couzi L, Merville P, Deminière C, Moreau J-F, Combe C, Pellegrin J-L, et al. Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis. Arthritis Rheum (2007) 56:2362–70. doi: 10.1002/art.22654
79. Siegel JP. Effects of interferon-gamma on the activation of human T lymphocytes. Cell Immunol (1988) 111:461–72. doi: 10.1016/0008-8749(88)90109-8
80. Maraskovsky E, Chen WF, Shortman K. IL-2 and IFN-gamma are two necessary lymphokines in the development of cytolytic T cells. J Immunol (1989) 143:1210–4.
81. Haring JS, Corbin GA, Harty JT. Dynamic regulation of IFN-gamma signaling in antigen-specific CD8+ T cells responding to infection. J Immunol (2005) 174:6791–802. doi: 10.4049/jimmunol.174.11.6791
82. Driver JP, Racine JJ, Ye C, Lamont DJ, Newby BN, Leeth CM, et al. Interferon-γ limits diabetogenic CD8+ T-cell effector responses in type 1 diabetes. Diabetes (2017) 66:710–21. doi: 10.2337/db16-0846
83. Szelinski F, Lino AC, Dörner T. B cells in systemic lupus erythematosus. Curr Opin Rheumatol (2022) 34:125–32. doi: 10.1097/BOR.0000000000000865
84. Jurado A, Carballido J, Griffel H, Hochkeppel HK, Wetzel GD. The immunomodulatory effects of interferon-gamma on mature b-lymphocyte responses. Experientia (1989) 45:521–6. doi: 10.1007/BF01990501
85. Harigai M, Kawamoto M, Hara M, Kubota T, Kamatani N, Miyasaka N. Excessive production of IFN-gamma in patients with systemic lupus erythematosus and its contribution to induction of b lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B. J Immunol (2008) 181:2211–9. doi: 10.4049/jimmunol.181.3.2211
86. Scapini P, Carletto A, Nardelli B, Calzetti F, Roschke V, Merigo F, et al. Proinflammatory mediators elicit secretion of the intracellular b-lymphocyte stimulator pool (BLyS) that is stored in activated neutrophils: implications for inflammatory diseases. Blood (2005) 105:830–7. doi: 10.1182/blood-2004-02-0564
87. Yan M, Marsters SA, Grewal IS, Wang H, Ashkenazi A, Dixit VM. Identification of a receptor for BLyS demonstrates a crucial role in humoral immunity. Nat Immunol (2000) 1:37–41. doi: 10.1038/76889
88. Do RK, Hatada E, Lee H, Tourigny MR, Hilbert D, Chen-Kiang S. Attenuation of apoptosis underlies b lymphocyte stimulator enhancement of humoral immune response. J Exp Med (2000) 192:953–64. doi: 10.1084/jem.192.7.953
89. Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, et al. IFN-γ receptor and STAT1 signaling in b cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med (2016) 213:715–32. doi: 10.1084/jem.20151722
90. Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, et al. B cell IFN-γ receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J Exp Med (2016) 213:733–50. doi: 10.1084/jem.20151724
91. Cattoretti G, Chang CC, Cechova K, Zhang J, Ye BH, Falini B, et al. BCL-6 protein is expressed in germinal-center b cells. Blood (1995) 86:45–53. doi: 10.1182/blood.V86.1.45.bloodjournal86145
92. Chodisetti SB, Fike AJ, Domeier PP, Singh H, Choi NM, Corradetti C, et al. Type II but not type I IFN signaling is indispensable for TLR7-promoted development of autoreactive b cells and systemic autoimmunity. J Immunol (2020) 204:796–809. doi: 10.4049/jimmunol.1901175
93. Harris DP, Goodrich S, Gerth AJ, Peng SL, Lund FE. Regulation of IFN-gamma production by b effector 1 cells: essential roles for T-bet and the IFN-gamma receptor. J Immunol (2005) 174:6781–90. doi: 10.4049/jimmunol.174.11.6781
94. Xu W, Zhang JJ. Stat1-dependent synergistic activation of T-bet for IgG2a production during early stage of b cell activation. J Immunol (2005) 175:7419–24. doi: 10.4049/jimmunol.175.11.7419
95. Snapper CM, Paul WE. Interferon-gamma and b cell stimulatory factor-1 reciprocally regulate ig isotype production. Science (1987) 236:944–7. doi: 10.1126/science.3107127
96. Snapper CM, Peschel C, Paul WE. IFN-gamma stimulates IgG2a secretion by murine b cells stimulated with bacterial lipopolysaccharide. J Immunol (1988) 140:2121–7. doi: 10.0000/PMID3127461
97. Peng SL, Szabo SJ. Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci USA (2002) 99:5545–50. doi: 10.1073/pnas.082114899
98. Cassatella MA, Bazzoni F, Calzetti F, Guasparri I, Rossi F, Trinchieri G. Interferon-gamma transcriptionally modulates the expression of the genes for the high affinity IgG-fc receptor and the 47-kDa cytosolic component of NADPH oxidase in human polymorphonuclear leukocytes. J Biol Chem (1991) 266:22079–82. doi: 10.1016/S0021-9258(18)54534-8
99. Balomenos D, Rumold R, Theofilopoulos AN. Interferon-gamma is required for lupus-like disease and lymphoaccumulation in MRL-lpr mice. J Clin Invest (1998) 101:364–71. doi: 10.1172/JCI750
100. Haas C, Ryffel B, Le Hir M. IFN-gamma is essential for the development of autoimmune glomerulonephritis in MRL/Ipr mice. J Immunol (1997) 158:5484–91.
101. Shaabani N, Honke N, Dolff S, Görg B, Khairnar V, Merches K, et al. IFN-γ licenses CD11b(+) cells to induce progression of systemic lupus erythematosus. J Autoimmun (2015) 62:11–21. doi: 10.1016/j.jaut.2015.05.007
102. Chan VS-F, Nie Y-J, Shen N, Yan S, Mok M-Y, Lau C-S. Distinct roles of myeloid and plasmacytoid dendritic cells in systemic lupus erythematosus. Autoimmun Rev (2012) 11:890–7. doi: 10.1016/j.autrev.2012.03.004
103. Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, et al. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat Immunol (2014) 15:161–7. doi: 10.1038/ni.2795
104. Robbins SH, Walzer T, Dembélé D, Thibault C, Defays A, Bessou G, et al. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome Biol (2008) 9:R17. doi: 10.1186/gb-2008-9-1-r17
105. Celhar T, Hopkins R, Thornhill SI, De Magalhaes R, Hwang S-H, Lee H-Y, et al. RNA Sensing by conventional dendritic cells is central to the development of lupus nephritis. Proc Natl Acad Sci USA (2015) 112:E6195–6204. doi: 10.1073/pnas.1507052112
106. Haas C, Ryffel B, Aguet M, Le Hir M. MHC antigens in interferon gamma (IFN gamma) receptor deficient mice: IFN gamma-dependent up-regulation of MHC class II in renal tubules. Kidney Int (1995) 48:1721–7. doi: 10.1038/ki.1995.470
107. Dong Y, Rohn WM, Benveniste EN. IFN-gamma regulation of the type IV class II transactivator promoter in astrocytes. J Immunol (1999) 162:4731–9.
108. Xue M, Zhu L, Meng Y, Wang L, Sun H, Wang F, et al. Detailed modulation of phenotypes and functions of bone marrow dendritic cells (BMDCs) by interferon-gamma (IFN-γ). Int Immunopharmacol (2013) 17:366–72. doi: 10.1016/j.intimp.2013.07.002
109. Gaczynska M, Rock KL, Spies T, Goldberg AL. Peptidase activities of proteasomes are differentially regulated by the major histocompatibility complex-encoded genes for LMP2 and LMP7 Proc Natl Acad Sci USA (1994) 91:9213-7. doi: 10.1073/pnas.91.20.9213
110. Monaco JJ. A molecular model of MHC class-i-restricted antigen processing. Immunol Today (1992) 13:173–9. doi: 10.1016/0167-5699(92)90122-N
111. Ma C, Xia Y, Yang Q, Zhao Y. The contribution of macrophages to systemic lupus erythematosus. Clin Immunol (2019) 207:1–9. doi: 10.1016/j.clim.2019.06.009
112. Pamer E, Cresswell P. Mechanisms of MHC class I–restricted antigen processing. Annu Rev Immunol (1998) 16:323–58. doi: 10.1146/annurev.immunol.16.1.323
113. Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu Rev Immunol (1994) 12:259–93. doi: 10.1146/annurev.iy.12.040194.001355
114. Wolf PR, Ploegh HL. How MHC class II molecules acquire peptide cargo: biosynthesis and trafficking through the endocytic pathway. Annu Rev Cell Dev Biol (1995) 11:267–306. doi: 10.1146/annurev.cb.11.110195.001411
115. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med (1983) 158:670–89. doi: 10.1084/jem.158.3.670
116. Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine (2018) 30:303–16. doi: 10.1016/j.ebiom.2018.02.009
117. Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-gamma: the major mediator of resistance against toxoplasma gondii. Science (1988) 240:516–8. doi: 10.1126/science.3128869
118. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi: 10.1172/JCI59643
119. Liew FY, Millott S, Parkinson C, Palmer RM, Moncada S. Macrophage killing of leishmania parasite in vivo is mediated by nitric oxide from l-arginine. J Immunol (1990) 144:4794–7.
120. Ahamada MM, Jia Y, Wu X. Macrophage polarization and plasticity in systemic lupus erythematosus. Front Immunol (2021) 12:734008. doi: 10.3389/fimmu.2021.734008
121. Aota K, Kani K, Yamanoi T, Nakashiro K-I, Ishimaru N, Azuma M. Distinct regulation of CXCL10 production by cytokines in human salivary gland ductal and acinar cells. Inflammation (2018) 41:1172–81. doi: 10.1007/s10753-018-0764-0
122. Jacob CO, van der Meide PH, McDevitt HO. In vivo treatment of (NZB X NZW)F1 lupus-like nephritis with monoclonal antibody to gamma interferon. J Exp Med (1987) 166:798–803. doi: 10.1084/jem.166.3.798
123. Ozmen L, Roman D, Fountoulakis M, Schmid G, Ryffel B, Garotta G. Experimental therapy of systemic lupus erythematosus: the treatment of NZB/W mice with mouse soluble interferon-gamma receptor inhibits the onset of glomerulonephritis. Eur J Immunol (1995) 25:6–12. doi: 10.1002/eji.1830250103
124. Haas C, Ryffel B, Le Hir M. IFN-gamma receptor deletion prevents autoantibody production and glomerulonephritis in lupus-prone (NZB x NZW)F1 mice. J Immunol (1998) 160:3713–8. doi: 10.1172/JCI10167
125. Schrott LM, Crnic LS. Attenuation of behavioral abnormalities in autoimmune mice by chronic soluble interferon-gamma receptor treatment. Brain Behav Immun (1998) 12:90–106. doi: 10.1006/brbi.1998.0522
126. Lawson BR, Prud’homme GJ, Chang Y, Gardner HA, Kuan J, Kono DH, et al. Treatment of murine lupus with cDNA encoding IFN-gammaR/Fc. J Clin Invest (2000) 106:207–15. doi: 10.1172/JCI10167
127. Hron JD, Peng SL. Type I IFN protects against murine lupus. J Immunol (2004) 173:2134–42. doi: 10.4049/jimmunol.173.3.2134
128. Werwitzke S, Trick D, Sondermann P, Kamino K, Schlegelberger B, Kniesch K, et al. Treatment of lupus-prone NZB/NZW F1 mice with recombinant soluble fc gamma receptor II (CD32). Ann Rheum Dis (2008) 67:154–61. doi: 10.1136/ard.2006.068981
129. Chen P, Vu T, Narayanan A, Sohn W, Wang J, Boedigheimer M, et al. Pharmacokinetic and pharmacodynamic relationship of AMG 811, an anti-IFN-γ IgG1 monoclonal antibody, in patients with systemic lupus erythematosus. Pharm Res (2015) 32:640–53. doi: 10.1007/s11095-014-1492-2
130. Werth VP, Fiorentino D, Sullivan BA, Boedigheimer MJ, Chiu K, Wang C, et al. Brief report: Pharmacodynamics, safety, and clinical efficacy of AMG 811, a human anti-interferon-γ antibody, in patients with discoid lupus erythematosus. Arthritis Rheumatol (2017) 69:1028–34. doi: 10.1002/art.40052
131. Boedigheimer MJ, Martin DA, Amoura Z, Sánchez-Guerrero J, Romero-Diaz J, Kivitz A, et al. Safety, pharmacokinetics and pharmacodynamics of AMG 811, an anti-interferon-γ monoclonal antibody, in SLE subjects without or with lupus nephritis. Lupus Sci Med (2017) 4:e000226. doi: 10.1136/lupus-2017-000226
132. Platanias LC. Mechanisms of type-i- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5:375–86. doi: 10.1038/nri1604
133. Kubo S, Yamaoka K, Kondo M, Yamagata K, Zhao J, Iwata S, et al. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann Rheum Dis (2014) 73:2192–8. doi: 10.1136/annrheumdis-2013-203756
134. Lu LD, Stump KL, Wallace NH, Dobrzanski P, Serdikoff C, Gingrich DE, et al. Depletion of autoreactive plasma cells and treatment of lupus nephritis in mice using CEP-33779, a novel, orally active, selective inhibitor of JAK2. J Immunol (2011) 187:3840–53. doi: 10.4049/jimmunol.1101228
135. Wang S, Yang N, Zhang L, Huang B, Tan H, Liang Y, et al. Jak/STAT signaling is involved in the inflammatory infiltration of the kidneys in MRL/lpr mice. Lupus (2010) 19:1171–80. doi: 10.1177/0961203310367660
136. Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, et al. Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol (2017) 69:148–60. doi: 10.1002/art.39818
137. Lee J, Park Y, Jang SG, Hong S-M, Song Y-S, Kim M-J, et al. Baricitinib attenuates autoimmune phenotype and podocyte injury in a murine model of systemic lupus erythematosus. Front Immunol (2021) 12:704526. doi: 10.3389/fimmu.2021.704526
138. Dörner T, Tanaka Y, Dow ER, Koch AE, Silk M, Ross Terres JA, et al. Mechanism of action of baricitinib and identification of biomarkers and key immune pathways in patients with active systemic lupus erythematosus. Ann Rheum Dis (2022) annrheumdis-2022-222335. doi: 10.1136/annrheumdis-2022-222335
139. Dörner T, van Vollenhoven RF, Doria A, Jia B, Ross Terres JA, Silk ME, et al. Baricitinib decreases anti-dsDNA in patients with systemic lupus erythematosus: results from a phase II double-blind, randomized, placebo-controlled trial. Arthritis Res Ther (2022) 24:112. doi: 10.1186/s13075-022-02794-x
Keywords: IFN-γ, systemic lupus erythematosus, autoimmune, biologic therapy, immune cells
Citation: Liu W, Zhang S and Wang J (2022) IFN-γ, should not be ignored in SLE. Front. Immunol. 13:954706. doi: 10.3389/fimmu.2022.954706
Received: 27 May 2022; Accepted: 25 July 2022;
Published: 10 August 2022.
Edited by:
William Stohl, University of Southern California, United StatesReviewed by:
Susan John, King’s College London, United KingdomYujun Sheng, First Affiliated Hospital of Anhui Medical University, China
Copyright © 2022 Liu, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jibo Wang, d2FuZ2ppYm8yMDA1QDEyNi5jb20=