 Min Zhang1,2
Min Zhang1,2 Jinming Zhou
Jinming Zhou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 September 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.954129
This article is part of the Research Topic A year in review: Discussions in Autoimmune and Autoinflammatory Disorders View all 12 articles
The cGAS-STING signaling plays an integral role in the host immune response, and the abnormal activation of cGAS-STING is highly related to various autoimmune diseases. Therefore, targeting the cGAS-STING-TBK1 axis has become a promising strategy in therapy of autoimmune diseases. Herein, we summarized the key pathways mediated by the cGAS-STING-TBK1 axis and various cGAS-STING-TBK1 related autoimmune diseases, as well as the recent development of cGAS, STING, or TBK1 selective inhibitors and their potential application in therapy of cGAS-STING-TBK1 related autoimmune diseases. Overall, the review highlights that inhibiting cGAS-STING-TBK1 signaling is an attractive strategy for autoimmune disease therapy.
Autoimmune diseases including various chronic inflammatory illnesses have affected the health of around 3%-10% of people in the world (1). The aberrant responses of the immune system to self are thought as the major factor leading to autoimmune diseases. Although the innate immune system which detects and responds to the pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) serves as the organism’s first line of defense against foreign invasion, the dysregulation and over-activation of the innate immune system will lead to various inflammatory illnesses (2, 3). Moreover, the autoinflammation induced by the abnormal innate immune signaling can achieve the establishment of adaptive immune responses, thus leading to the progress of autoimmunity. The endosomal or cytosolic nucleic-acid sensing involved in innate immunity is one of the initial triggers of autoimmunity. The nuclear acid recognition receptors, including retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated gene 5 (MDA5), Toll-like receptors (TLR3, 7, 8, and 9), and the cyclic GMP-AMP synthase–stimulator of interferon genes–tank-binding kinase 1 (cGAS-STING-TBK1) axis, have been directly related to the pathogenesis of various autoimmune diseases (4–9).
The cGAS-STING signaling pathway combines DNA sensing with the induction of a strong innate immune defense program, playing a crucial role in the host immune response (10). Through the recognition of the exogenous DNA from virus and bacterial or own damaged DNA, cGAS catalyzes the synthesis of cyclic GMP-AMP (cGAMP) from adenosine triphosphate (ATP) and guanosine triphosphate (GTP). cGAMP further interacts with STING and activates downstream pathways to induce the expression of type I interferons (IFNs), interferon-stimulator genes (ISGs), and other pro-inflammatory cytokines, thus extensively activating the host immune system and further inhibiting and eliminating tumors or viruses (7, 11–14). The dysregulation of this broad and powerful recognition system (cGAS-STING) can also disrupt the dynamic homeostasis of cells and organs by inducing aberrant innate immune responses and a variety of inflammatory triggers (15, 16). The persistent or chronic inflammatory signaling that links to the activation of cGAS-STING signaling is prone to developing the autoimmune disease including Aicardi-Goutières syndrome (AGS), systemic lupus erythematosus (SLE), STING-associated vasculopathy of infancy (SAVI), and amyotrophic lateral sclerosis (ALS), etc. (17–21) Interestingly, the cGAS-STING pathway inhibitors such as H-151 effectively improved the symptoms of the autoimmune diseases ALS and psoriasis by decreasing the inflammatory signaling in animal models, thereby becoming a promising therapeutic agent for autoimmune diseases (17, 22). Moreover, Ablasser’s lab reported that H-151 inhibited the inflammation in SARS-COV-2 driven disease COVID-19 (23). Herein, we focus on the key pathways mediated by cGAS-STING and various cGAS-STING-TBK1 signaling related autoimmune diseases, as well as the recent development of cGAS, STING, or TBK1 selective inhibitors.
Human cGAS is a 60 kD protein from the nucleotidyl transferase (NTase) family, consisting of a non-conserved N-terminal structural domain (1-160) and a highly conserved C-terminal NTase structural domain (161-522) (Figure 1A) (24). The N-terminal domain is identified to play a role in stabilizing or suppressing cGAS protein (25).While the NTase domain contains three dsDNA binding sites and is essential for dsDNA recognition and the synthesis of the second messenger 2’3’-cGAMP. The cGAS can bind non-sequence-dependently to dsDNA through the phosphate backbone, leading to significant conformational changes in the NTase structural domain of cGAS and a structural switch in the catalytic pocket, which initiates the catalytic synthesis of GTP and ATP to 2’3’-cGAMP (Figures 1B, C) (10, 15, 26). The synthesis of 2’3’-cGAMP is a critical step in the triggering of the STING-mediated immune system (14, 27). The binding of long-stranded dsDNA to cGAS which forms a ladder-like network is thought necessary to the activation of the cGAS-STING signaling pathway, thus becoming a pattern that effectively prevents the activation of STING by the short-stranded DNA (28, 29). cGAS binds to the negatively charged acidic patch formed by histones H2A and H2B through its DNA binding site. High-affinity nucleosome binding prevents dsDNA recruitment and keeps cGAS in an inactive conformation (30–34).
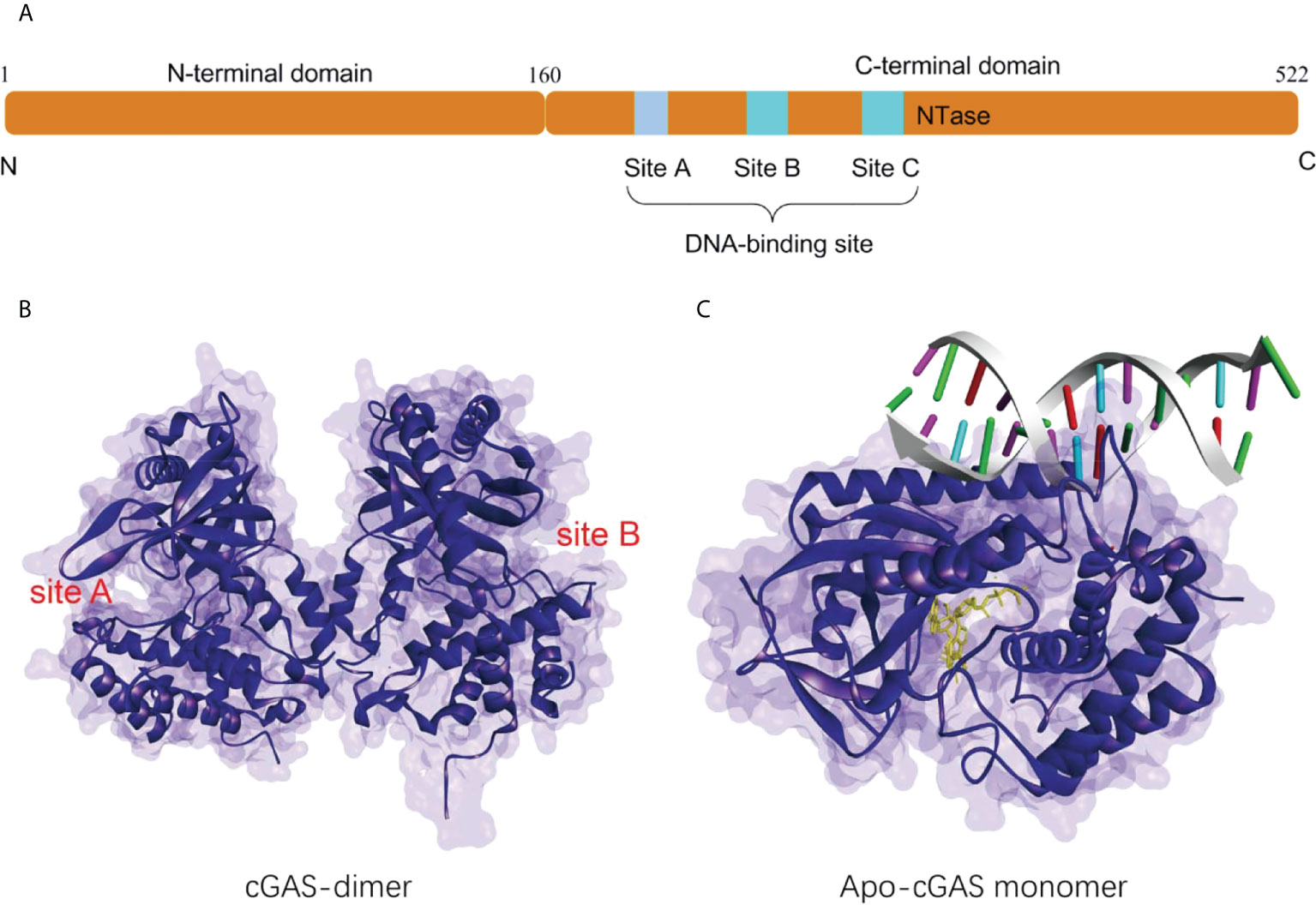
Figure 1 (A) Schematic organization of the structural domain of human cGAS; (B) Structure of the human cGAS dimer (PDB-ID: 4LEV); (C) Structure of porcine cGAS (blue ribbon, apo-cGAS monomer) in complex with DNA, ATP and GTP (yellow ribbon, PDB ID: 4KB6).
The human TBK1 kinase consists of 729 amino acids, including an N-terminal kinase domain (KD), a ubiquitin-like domain (ULD), an alpha-helical scaffold dimerization domain (SDD), and a C-terminal adaptor-binding domain (CTD) (Figure 2A) (35, 36). Extensive interactions between KD, ULD, and SDD form the dense TBK1 dimer. The KD of TBK1 consists of N-terminal and C-terminal leaflets with an active ATP binding site at the interface. Ser172 residue on the activation loop is the phosphorylation site for TBK1 kinase (37). When TBK1 is phosphorylated, the αC-helix of the kinase structural domain rotates to the inward active position, facilitating the formation of a critical salt-bridge interaction between Glu55 of the αC-helix and Lys38 at the active site. However, when TBK1 is in the inactive conformation, the activation loop is disrupted and the αC-helix is positioned in an inactive position outside the ATP-binding structural domain (35). In the structure of the TBK1 dimer, the activation of TBK1 is mainly controlled by trans-autophosphorylation, in which two KDs limit the cis-autophosphorylation activity of TBK1 (38). A highly conserved PLRT/SD motif in the C-terminal tail (CTT) of STING mediates the recruitment of TBK1 by binding directly to the dimeric interface of TBK1. Further analysis of the crystal structure of STING and TBK1 showed that the dimeric TBK1 binds to two monomers of the CTT of STING, with each STING monomer simultaneously binding to two TBK1 monomers to form a 2:2 complex (Figures 2B, C) (39). The 2’,3’-cGAMP binding initiates the STING activation by forming a stable oligomer, and the conserved PLPLRT/SD protein motif in STING-CTT can dimerize the TBK1 interface to induce the phosphorylation and activation of STING and TBK1 through hydrophobic binding. Further recruitment and phosphorylation of interferon regulatory factor 3 (IRF3) and TBK1 leads to the involvement of downstream signaling components and the inducible regulation of IFN-I transcription, which is the hallmark signal for the initiation of the cGAS-STING-TBK1 signaling pathway (35, 39–42).
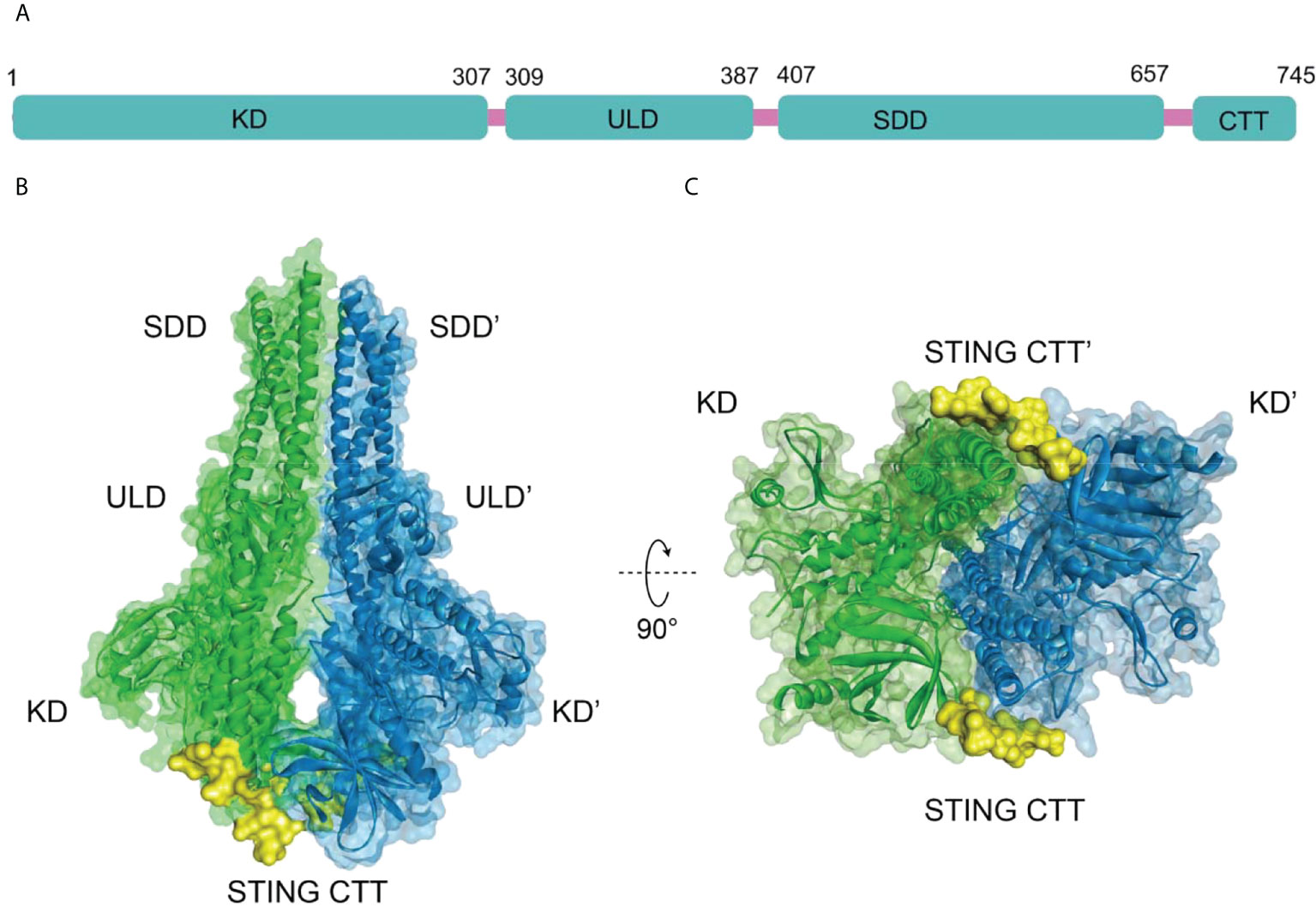
Figure 2 (A) Schematic organization of the structural domain of human TBK1, (B) Structure of human TBK1 in complex with chicken STING CTT (PDB-ID: 6NT9); (C) Bottom view of TBK1 structure.
Human STING (MITA) is a transmembrane protein located on the endoplasmic reticulum (ER) and consists of an N-terminal transmembrane structural domain (NTD) containing four transmembrane helices TM1 (residues 21-41), TM2 (residues 47-67), TM3 (residues 87-106), TM4 (residues 116-136) and a globular C-terminal structural domain (CTD, residues 157-379) (Figure 3A). STING is highly expressed in immunomodulatory-related cells and tissues such as bone marrow, spleen, and peripheral blood leukocytes (43–46). The STING-CTD is comprised of a ligand-binding domain (LBD, residues 157-335), an IRF3-binding domain (residues 362-366), and a TBK1-binding motif (TBM, residues 369-377) (47–49). Through biophysical technology, especially X-ray crystallography, the STING-CTD is identified as a butterfly-like dimer with a ligand binding site located at the groove of the interface (Figure 3B).
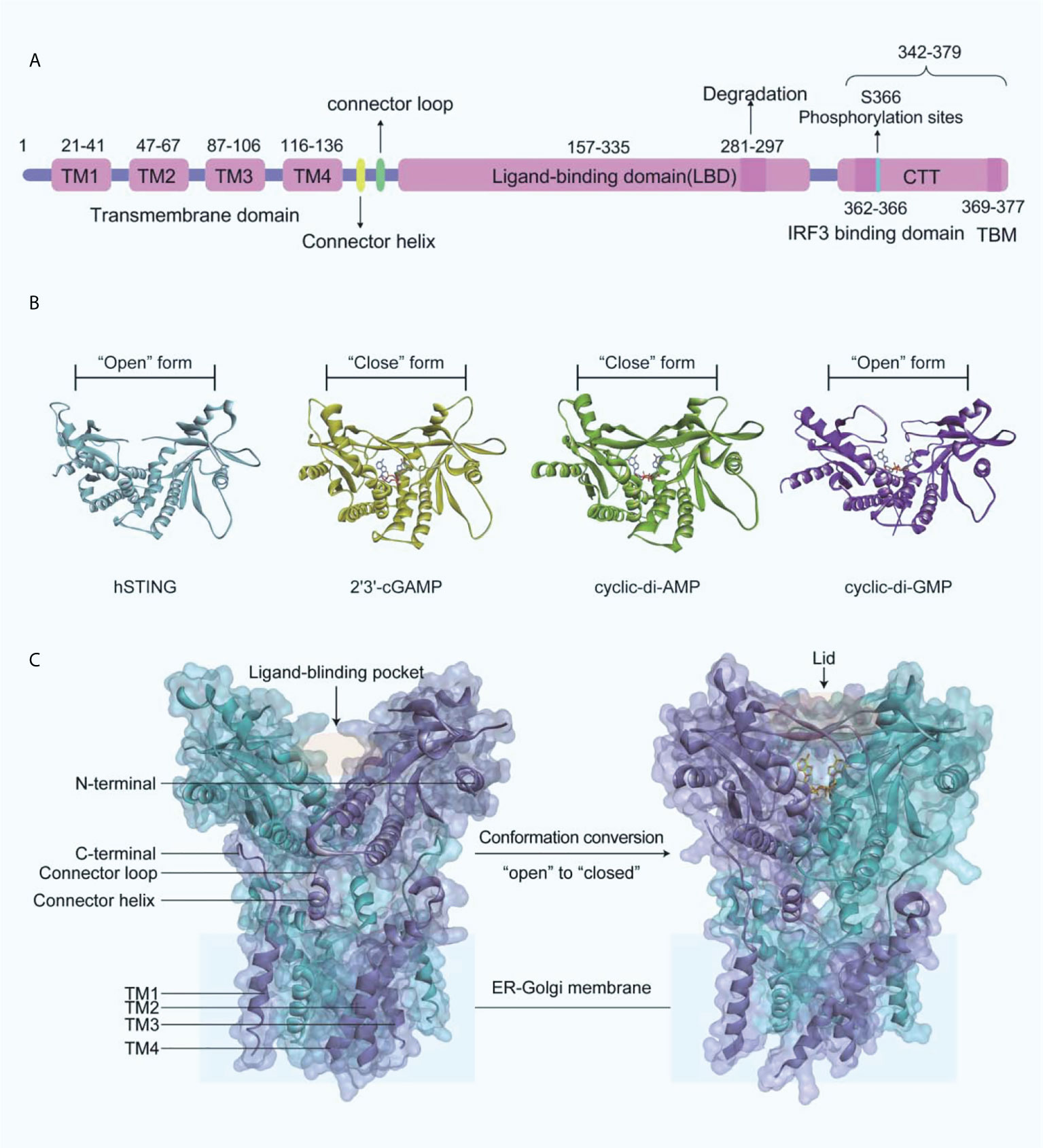
Figure 3 (A) Schematic organization of the structural domain of human STING; (B) Crystal structures of human STING CTD (blue ribbon, apo-STING, PDB-ID: 4EMU; yellow ribbon, STING bound 2’3’-GAMP, PDB-ID: 4LOH; green ribbon, STING bound CDA, PDB-ID: 4F5D; purple ribbon, STING bound CDG, PDB-ID: 4F5Y); (C) On the left is the crystal structure of full-length human STING in open conformation (PDB-ID: 6NT5), and on the right is the crystal structure of chicken STING and 2’3’-cGAMP in closed conformation (PDB-ID: 6NT7).
The endogenously produced 2’3’-cGAMP is detected by STING, which in turn binds to STING CTD in a dimeric form (45). Subsequently, STING performs extensive conformational changes, with an inward 180°C flip of the V-type STING LBD dimer, an “open” to “closed” transition of conformation to form a “lid” covering the 2’3’-cGAMP binding site (Figure 3C) (45, 50). The formation of STING polymers via a C148-mediated disulfide bond is essential to the activation of STING, while the “opening” or “closing” of the LBD regulates its activity by affecting the affinity with the ligand to the protein (50). It is important to note that cyclic-di-GMP (CDG) binding does not produce the conformational changes induced by 2’3’-cGAMP or cyclic-di-AMP (CDA), while CDG-bound STING may also lead to the activation of the cGAS-STING-IRF3 pathway (Figure 3B). What is more, recent research indicated that the potent STING agonist diABZI did not promote the closure of the lid region of STING either (51).
The subsequent translocation of STING from the ER to the Golgi apparatus is mediated primarily by coat protein complex II (COPII) vesicles (52), which is dependent on GTPase SAR1A and COPII complex components, including SEC24C and ARF-GTPase ARF1. After transporting to the Golgi apparatus, STING is palmitoylated at two cysteine residues Cys88 and Cys91, which is necessary for the recruitment of TBK1 and IFNs transcription (53, 54). However, TBK1 recruiting to STING alone does not induce the activation of IRF3 at the CTT of STING (residues 342-379) (Figure 3A). The residues Leu333 and Arg334 at STING-CTD play critical roles in c-GAMP-induced autophagy and phosphorylation of TBK1 and IRF3 (52). TBK1 phosphorylates IRF3, which subsequently induces the dimerization and translocates into the nucleus, thereby driving the transcriptional expression of IFNs (55). After the initiation of downstream signaling, STING is degraded in endolysosomes, and the residues 281-297 are required for the transport-mediated STING degradation (56).
The activation of the cGAS-STING axis will induce the modification of IRF3 and its translocation to the nucleus, thereby driving the transcriptional expression of IFNs (57–59). Simultaneously, the binding of IFNs to its receptor activates Janus kinases (JAKs), including JAK1 and tyrosine kinase 2 (TYK2), which in turn phosphorylate the receptor (20). This process allows DNA-binding protein signal transducer and activator of transcription 1 (STAT1) and 2 (STAT2) to bind to the receptor, thereby phosphorylating and dimerizing them. The dimer then translocates to the nucleus where it upregulates the transcription of IFN-responsive genes, including the transcription of IFNs dependent on interferon regulatory factor 9 (IRF9) (Figure 4). The synthesis and release of IFN and its binding to the IFN receptor further upregulated the interferon genes in a positive feedback loop (20).
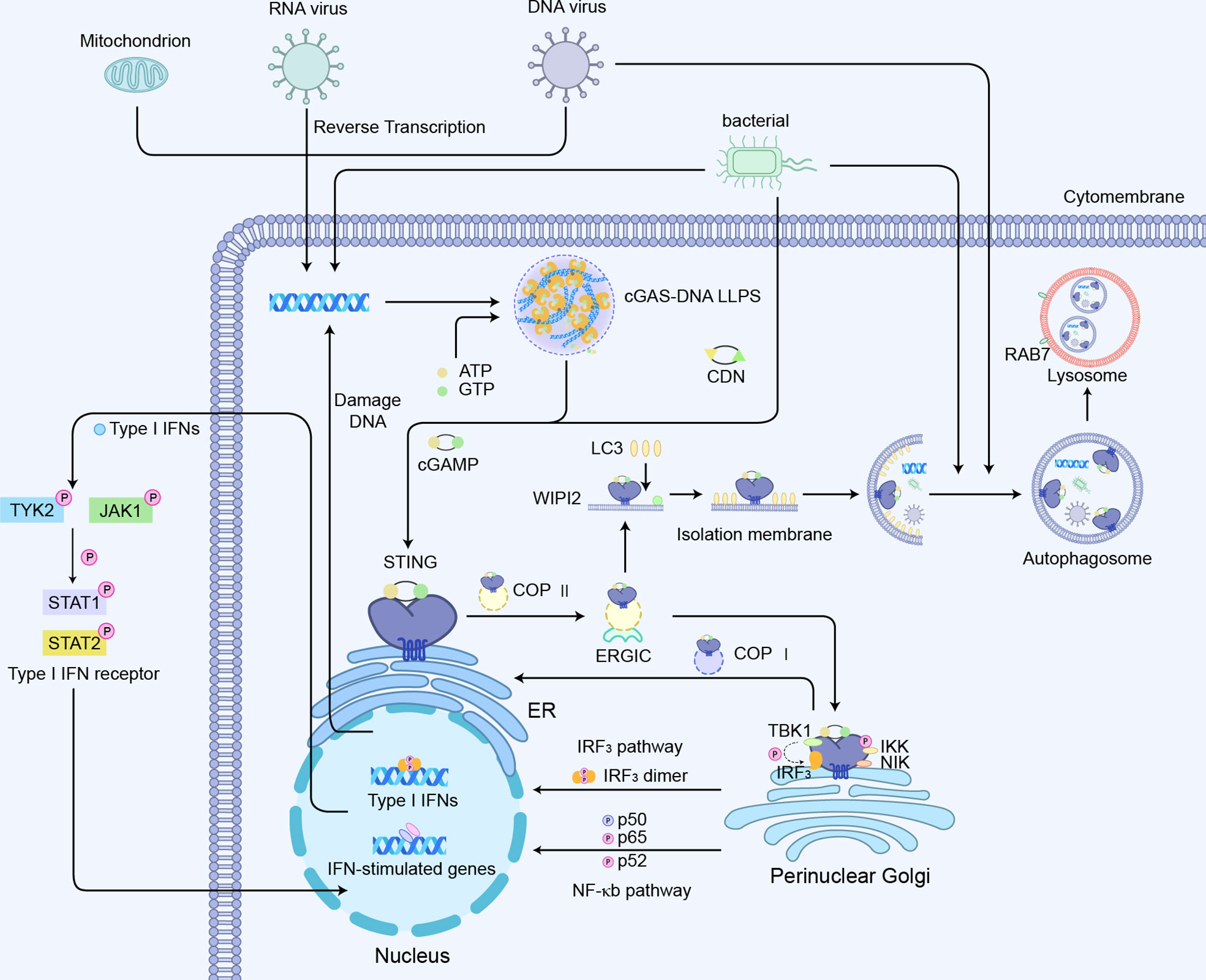
Figure 4 cGAS-STING-mediated signaling pathways:1) cGAS-STING-IRF3 pathway; 2) cGAS-STING mediated NF-κb pathway activation; 3) cGAS -STING induced autophagy process.
To avoid a severe inflammatory response to the induced transcription of excess IFN, there is also an associated negative feedback mechanism. When the cGAS-STING-mediated immune response is continuously activated, STING is induced to be degraded in the endosome. Following the activation and translocation of STING, it is phosphorylated by serine/threonine protein kinase 1/autophagy-associated protein 1 (ULK1/ATG1) to inhibit the sustained induction into IFNs and inflammatory disease (60). ULK1-2 function can be regulated by AMP-activated protein kinases (AMPK) or mammalian targets of rapamycin (mTOR) which is activated under cellular stress conditions. Cytoplasmic dsDNA and/or CDN are found to activate ULK1-2, which initiates a negative feedback loop controlling STING overexpression through restricting STING translocation from the Golgi apparatus by autophagy-associated protein 9a (ATG9a) and decreasing the association of STING and TBK1 (61). NOD-like receptor C3 (NLRC3) binds to STING and prevents its translocation from the ER to the Golgi apparatus, thereby reducing the IFNs response (62). Also, the movement of STING outside the ER facilitates its recruitment to LC3 autophagic vesicles through a WD repeat structural domain phosphoinositides interacting protein 2 (WIPI2)-dependent mechanism (52). LC3 coordinates the negative regulation of STING by transporting STING complexes, DNA, and pathogens to autophagy vesicles for lysosomal dependent degradation, a process that requires the RAS-associated protein Rab-7a (RAB7) GTPase (52, 63). Moreover, recent findings have revealed a mechanism that moves STING from the Golgi to the ER to downregulate the cellular activation of STING. Specifically, the adaptor protein SURF-4 interacts with STING on the Golgi apparatus to promote STING encapsulation into coat protein complex I (COPI) vesicles for retrograde transporting STING from the Golgi apparatus to the ER, thereby inhibiting sustained STING activation (64, 65). Upon the entry of IRF3 into the nucleus and the activation of ISGs, the STING-TBK1-IRF3 complex is dissociated and drives E3 ubiquitin ligase RNF5/TRIM30α mediated K48-linked polyubiquitinated STING, which promotes the degradation of STING via the proteasome pathway (66, 67).
Another major signaling module involved in the regulation of STING is nuclear factor kappa B (NF-κB)-mediated transcriptional activation, which promotes the expression of several pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 (Figure 4) (43). The CTT motif of STING is necessary for triggering the IRF3-dependent transcription of IFNs, whereas the STING-dependent NF-κB pathway is not entirely relied on the CTT of STING (68). STING-mediated NF-κB activation indicates much less sensitivity to the knockout of TBK1 (69). TBK1 alone is dispensable for STING-induced NF-κB responses in human immune cells, while acts redundantly with IκB kinase ϵ (IKKϵ) to drive NF-κB upon STING activation (68, 70). Consistently, the ancestral STING homologs in insects and early postlarvae completely lack CTT signaling, but could still achieve a host defense by promoting NF-κB responses. Using a tamoxifen-induced TBK1 deficiency model in adult mice, it was observed that TBK1 deficiency had little effect on cytokines of NF-κB following the administration of the mouse STING agonist DMXAA (69). Interestingly, the nuclear DNA damage would induce the non-canonical activation of STING by ATM and IFI16, leading to the activation of NF-κB signaling (71). Also, the genotoxic DNA damage induced by camptothecin drove IL-6 production through non-canonical STING signaling in the STING-expressing cancer cells (72). The stimulation of cGAS-STING also promotes a non-canonical NF-κB response by triggering p52 nuclear translocation (Figure 4) (73, 74). This signaling restricts IFN-I and the classic NF-κB pathway as regulators of the negative feedback mechanism of STING (75). Therefore, the explicit mechanisms of STING interacting with NF-κB pathway components are still required to be verified.
Previous work suggested that autophagy induction via STING trafficking is a primal function of the cGAS-STING pathway (52). The cGAS–STING pathway can induce canonical autophagy through liquid-phase separation of the cGAS–DNA complex, the interaction of cGAS and Beclin-1, and STING-triggered ER stress–mTOR signaling. Moreover, both cGAS and STING can trigger non-canonical autophagy via LC3-interacting regions and binding with LC3. What is more, autophagy induced by the cGAS–STING pathway plays crucial roles in balancing innate immune responses, maintaining intracellular environmental homeostasis, and restricting tumor growth (76). Conventional autophagy dependent on the ULK complex and TBK1 is involved in STING-mediated LC3 autophagy vesicle formation, and the activation of STING can also trigger non-canonical autophagy responses mediated by the PI3P effector WIPI2 and the ATG5-12-16L1 complex (Figure 4) (52, 60, 61, 77, 78). STING activation is indispensable for autophagic induction LC3 interacting regions (LIRs), and mutants of STING abolish its interaction with LC3 and its activation of autophagy (78). Of note, autophagy components also feedback on the regulation of STING activity through assisting STING intracellular trafficking capabilities as well as its lysosomal degradation (79). The direct interaction between Beclin-1 autophagy protein and cGAS not only inhibits STING signaling and decreases IFNs expression but also promotes the autophagy-mediated degradation of cytosolic DNA to avoid excess cGAS-STING activation (80). Similarly, key genes involved in the mechanism of autophagy, such as ULK-1 and Atg9, have appeared to suppress the STING/TBK1/IRF3 pathway, effectively inhibiting sustained immune response and excessive inflammation (60, 61). Moreover, autophagy receptor CCDC50 modulates STING-directed IFNs signaling activity by delivering the K63-polyubiquitinated STING to autolysosomes for the degradation (81).
AGS is an early-onset systemic inflammatory disorder that manifests clinically as neurological dysfunction and frostbite-like skin lesions. The nuclei acid exonuclease TREX1 was the first gene found to be related to AGS (Figure 5A) (82). TREX1 prevents excessive accumulation of endogenous auto-DNA and prevents aberrant activation of DNA-mediated cGAS-STING signaling, while structurally inactivated TREX1 leads to the IFN-dependent autoimmune disease AGS (19). In addition, it has also been reported that mice with mutations in three RNaseH2 enzyme complexes (RNaseH2 A, RNaseH2 B, and RNaseH2 C) exhibit increased IFN signaling and inflammation, and ultimately cause AGS-like symptoms (83).The failure of mutated RNaseH2 to degrade RNA/DNA hybrids led to the excessive activation of cGAS-STING signaling, which induced AGS. The lethality of some mice with dysfunctional mutated RNaseH2 was rescued by the knockout of STING (84). Similarly, sterile α motif and histidine-aspartate domain-containing protein 1 (SAMHD1) promotes the degradation of nascent DNA in human cell lines by stimulating the exonuclease activity of meiotic recombination 11 homolog A (MRE11A), and the deletion of SAMHD1 lead to the accumulation of genomic DNA in the cytoplasm and triggers AGS (85).
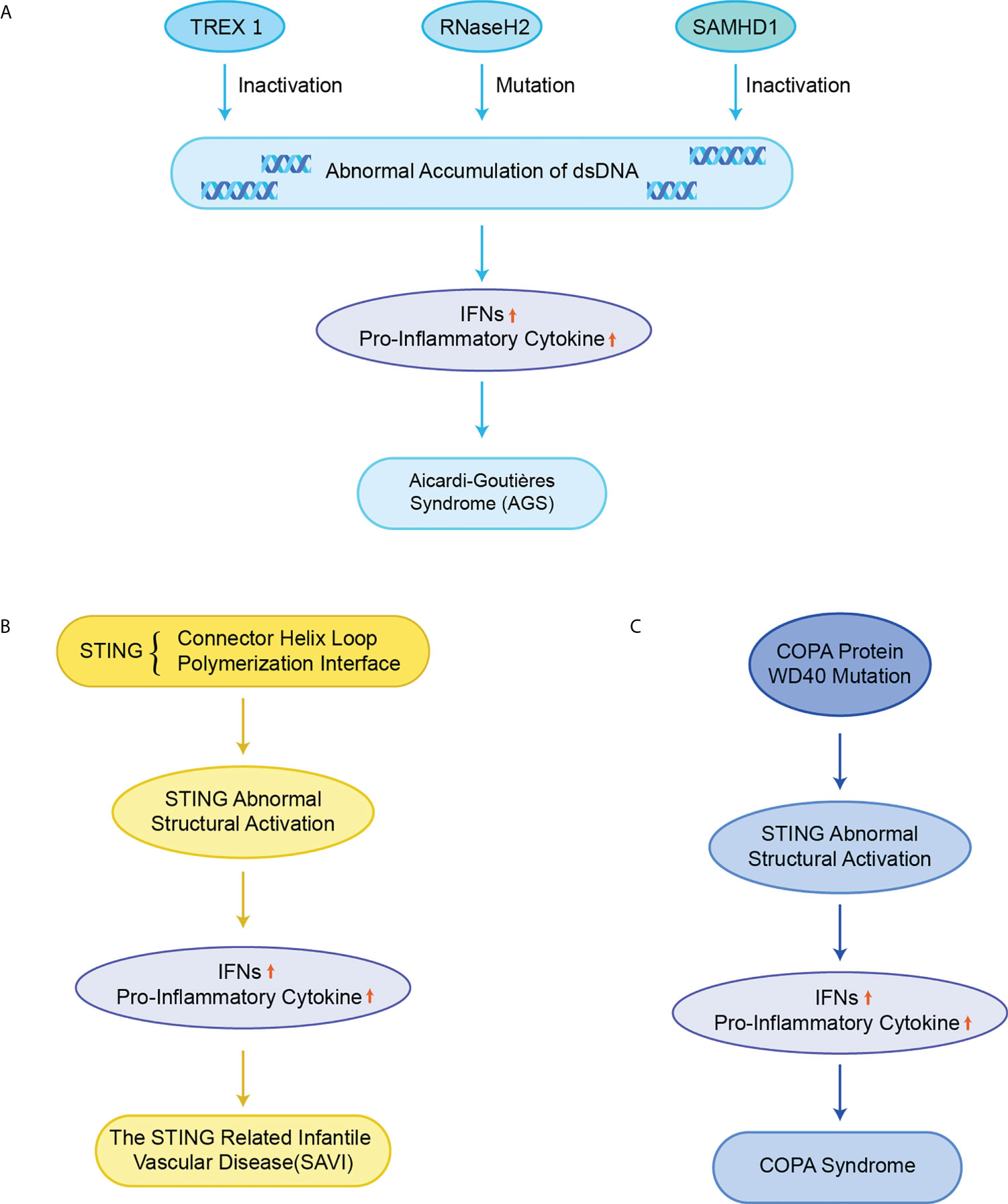
Figure 5 STING in monogenic autoinflammatory syndromes (A) Inactivation of TREX1, RNaseH2 and SAMHD1 leads to abnormal accumulation of dsDNA in normal cells, which over-activate the cGAS-STING signaling pathway, upregulates the expression of IFNs and pro-inflammatory cytokines, and ultimately triggers AGS; (B) Mutations in STING in the connector helix loop (N154S, V155M, and V147L) and the polymerization interface (G207E, R281Q, R284G, and R284S) lead to structural activation of STING, upregulating the expression of IFNs and pro-inflammatory cytokines, and ultimately causing SAVI; (C) Missense mutations in the structural domain of COPA WD40 impair endoplasmic reticulum binding and target protein sequencing, leading to structural activation of STING, upregulating the expression of IFNs and pro-inflammatory cytokines, and ultimately causing COPA syndrome.
Mutations in exon 5 of STING lead to functional activation of STING, resulting in the excessive STING-induced IFN signaling, causing a disorder termed SAVI including recurrent fever, ulcerative skin lesions, vasculitis, and interstitial lung disease (Figure 5B) (18, 20). Mutant residues are located in two separate regions on STING, the connector helix loop (N154S, V155M, G158A, G166E, H72N, and V147M/L) and the polymerization interface (C206Y/G, G207E, F279L, R281Q/W, and R284G/S) (86–89). Mutations in the regions can spontaneously rotate around the connected helix loop by inducing the LBD allosteric activation, or by promoting the STING polymerization, thereby triggering the ligand-independent activation of STING (50, 58). An obvious feature of the mouse SAVI model is the severe lymphopenia and immunodeficiency due to the abnormal lymphocyte development and aberrant intrinsic T cells (90). However, it has likewise been reported that the inhibition of IFN signaling did not affect disease pathogenesis in the N153S STING mouse model of SAVI. Instead, the T-cell depletion protected N153S mice from lung disease progressions, which may explain why JAK inhibitors targeting the IFN-α receptor (IFNAR) are not always successful in the treatment of SAVI patients (91).
The Ca2+ sensor stromal interaction molecule 1 (STIM1) has been reported to be a promising target for the treatment of SAVI, where STIM1 directly interacts with STING and inhibits the transport of STING from the ER to the Golgi apparatus (92). A peptide ISD017 has been reported to block the activity of STING in vivo and improve the disease progression of a mouse model of lupus in a STIM1-dependent manner (93). The activity of three disease-associated STING variants, V147L, N154S, and V155M, also can be inhibited by STIM1 in part by blocking their translocation to the ERGIC. In the SAVI model, the activation of STING leads to cellular T-cell defects by modulating T cell proliferation and differentiation (90, 94), and plays a key role in the initiation and progression of SAVI (95).
Pathogenic COPA variants can lead to immune dysregulation in Mendelian syndrome. COPA is a subunit of COPI that mediates STING from the Golgi apparatus to ER transport, and the dysfunction of the target thereby leads to the structural activation of STING (Figure 5C) (96). In a mouse model of COPA syndrome (CopaE241K/+), IFN-driven inflammation of the mice could be rescued through crossing with STING-deficient mice (STING1gt/gt). In addition, the embryonic lethality in Purex COPAE241K/E241K mice could be rescued by the knockout of STING (64). JAK inhibitors can improve clinical performance and IFN levels, but the effect is very limited (97). In mouse models with functionally acquired STING mutations, the development of lung lesions is dependent on T cells instead of IFN-I (98). This result may explain the poor therapeutic effect of JAK inhibitors in human COPA syndrome. In addition, the small molecule STING inhibitor H-151 has also been reported to improve the inflammation in COPA syndrome (99). The inhibition of STING has emerged as an efficient way for the treatment of COPA syndrome.
While the association of STING with neurodegenerative diseases has been poorly investigated in previous studies, the activation of the immune system is a prominent feature of several neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), frontotemporal dementia (FTD), multiple sclerosis (MS), and ALS. Several recent studies have revealed the relationship between STING and autoimmune neurodegenerative diseases (100). Although IFNs are also produced by neurons and astrocytes, STING is mainly expressed in microglia to elicit the IFN responses in the brain (101, 102). In chronic neurodegenerative disease states, aberrantly activated STING signaling induces the expression of IFNs and increases the phenotype of microglia and astrocytes, thereby accelerating the development of neuroinflammation (103, 104).
The cytoplasmic accumulation of TDP-43 is a hallmark of ALS (Figure 6A) (105). TDP-43 induces mtDNA release via voltage-dependent anion channel 1 (VDAC1), which subsequently induces IFNs and inflammatory cytokine expression in a cGAS-STING dependent manner (17). STING gene deletion or the use of small molecular inhibitors of STING significantly improved ALS symptoms and prolonged the life span of mice. Besides, the expansion of the hexanucleotide repeat sequence (GGGGCC) in the mice lacking chromosome 9 open reading frame 72 (C9orf72) gene is the most common cause of familial ALS (106). Dendritic cells isolated from mice lacking the C9orf72 protein showed marked early activation of IFN-I responses, and mice showed age-dependent lymphoid hypertrophy and autoinflammation. The C9orf72-deficient mice were more likely to develop experimental autoimmune encephalitis. Also, bone marrow cells lacking C9orf72 showed signs of hyper-activation upon being exposed to STING agonists and reduced autolysosomal degradation of STING. The C9orf72- deficient ALS patients had higher levels of IFN-I signaling than patients with sporadic ALS and could improve symptoms with STING inhibitors treatment.
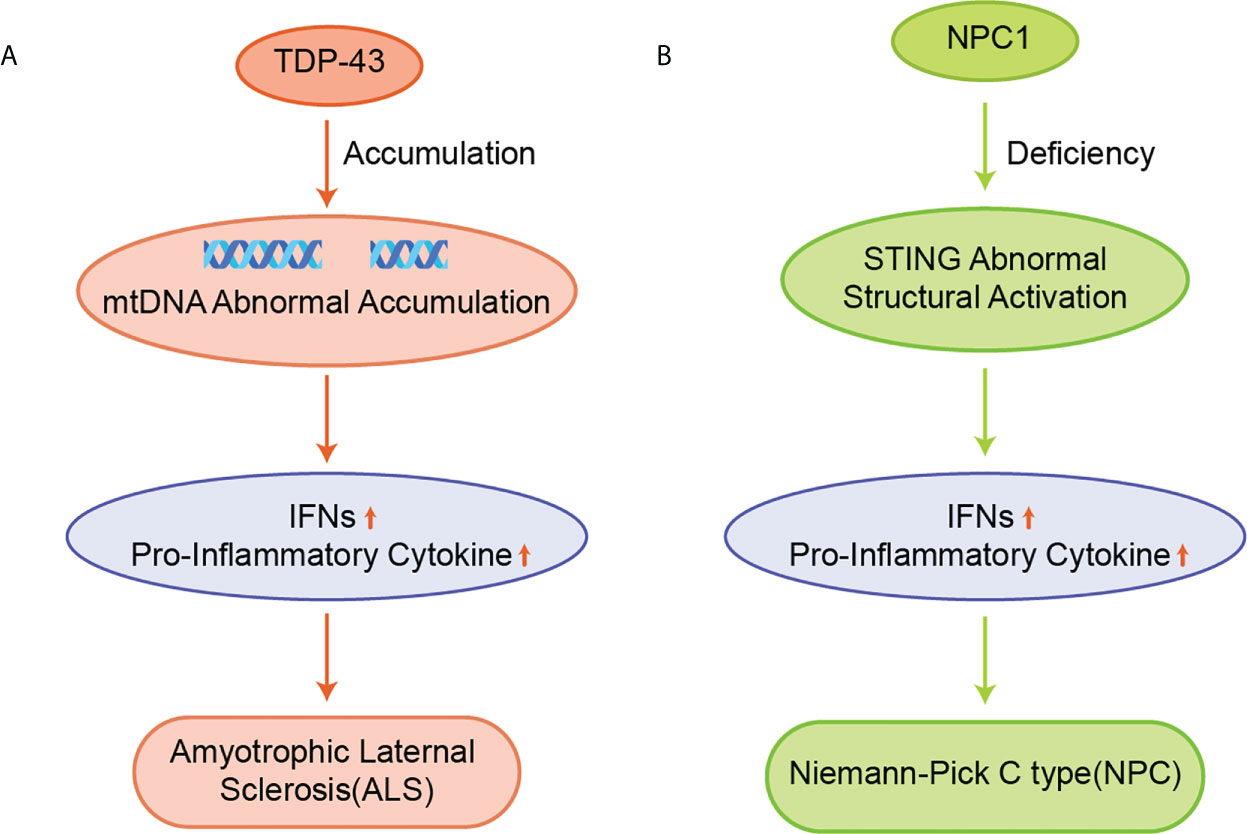
Figure 6 (A) Accumulation of TDP-43 leads to aberrant accumulation of mtDNA, which induces the expression of IFNs and pro-inflammatory cytokines in a cGAS/STING dependent manner and promotes the development of ALS; (B) NPC1 leads to the 2’3’-cGAMP-independent activation of STING, upregulating the expression of IFNs and pro-inflammatory cytokines, and ultimately causing NPC.
NPC is a chronic neuroautoimmune disease caused by a deficiency of Niemann-Pick C1 (NPC1), which leads to the impaired metabolism of neurospherin phospholipids (Figure 6B) (107). NPC1 is an auxiliary protein for transporting STING to the lysosome for its degradation. NPC1 deficiency leads to the accumulation of cholesterol and other lipids in the lysosome, resulting in the decrease of ER cholesterol levels and the activation of SREBP2-SCAP translocation from the ER to the Golgi apparatus (108). The STING protein is recruited by the SREBP2-SCAP complex, which triggers 2’3’-cGAMP-independent STING activation by hijacking STING to transport from the ER to the Golgi apparatus in NPC1 KO cells, thereby leading to a progressive loss of Purkinje in NPC1-/- mice, thus resulting in impaired motor function and reduced survival (63, 109).
MS is considered a progressive autoimmune disease which is caused by inflammation and neurological damage from immune system attacks on myelin (110). Early studies reported that the antiviral drug ganciclovir induced the suppression of MS experimental autoimmune encephalomyelitis (EAE) model in a STING-dependent manner (109). STING acts as a regulator of microglial cell reactivity and neuroinflammation, which improves the pathology of EAE in mice by reducing immune cell infiltration and inhibiting the proliferation of microglia or the immune cells of the central nervous system. Besides, The oral administration of Bowman-Birk inhibitor (BBI), a serine protease inhibitor derived from soy was reported to inhibit EAE (111). The inhibition is dependent on STING and IFN-β secreted by macrophages, and the absence of IFNAR in bone marrow cells restricts the inhibition of EAE by BBI.
While it is true that neurodegenerative diseases occur after aberrant activation of the cGAS-STING pathway, the role of this pathway in different neurological diseases is still needed to be further investigated. The inhibition of the STING signaling may be a potential way in therapy of STING-associated neurodegenerative diseases. However, we must also note that activation of cGAS-STING signaling is not a single facilitator in neurodegenerative disease. IFNs, the downstream expression products are also negative regulators of some inflammation in the peripheral or central nervous system (109, 112). Therefore, how to effectively regulate the cGAS-STING signaling using a cGAS or STING modulator is key to the treatment of neurological autoimmune diseases.
SLE is also an autoimmune disease that has been reported to be related to STING. The elevated serum 2’3’-cGAMP levels in SLE, leading to the redundant STING activation, have been reported in approximately 15% of all SLE patients (21). The exact cause of SLE is not clear yet, but elevated dsDNA levels were identified in cells from SLE patients, and apoptosis-derived membrane vesicles (AdMVs) in the serum of SLE patients had high inducing ISGs (Figure 7A) (113). Defective clearance of apoptotic cells produces dsDNA-containing AdMVs, which in turn induces ISGs via the cGAS-STING pathway. Next, ISGs activate an immune response that leads to tissue damage in various organs, resulting in further production of AdMVs and a positive feedback loop of ISGs.
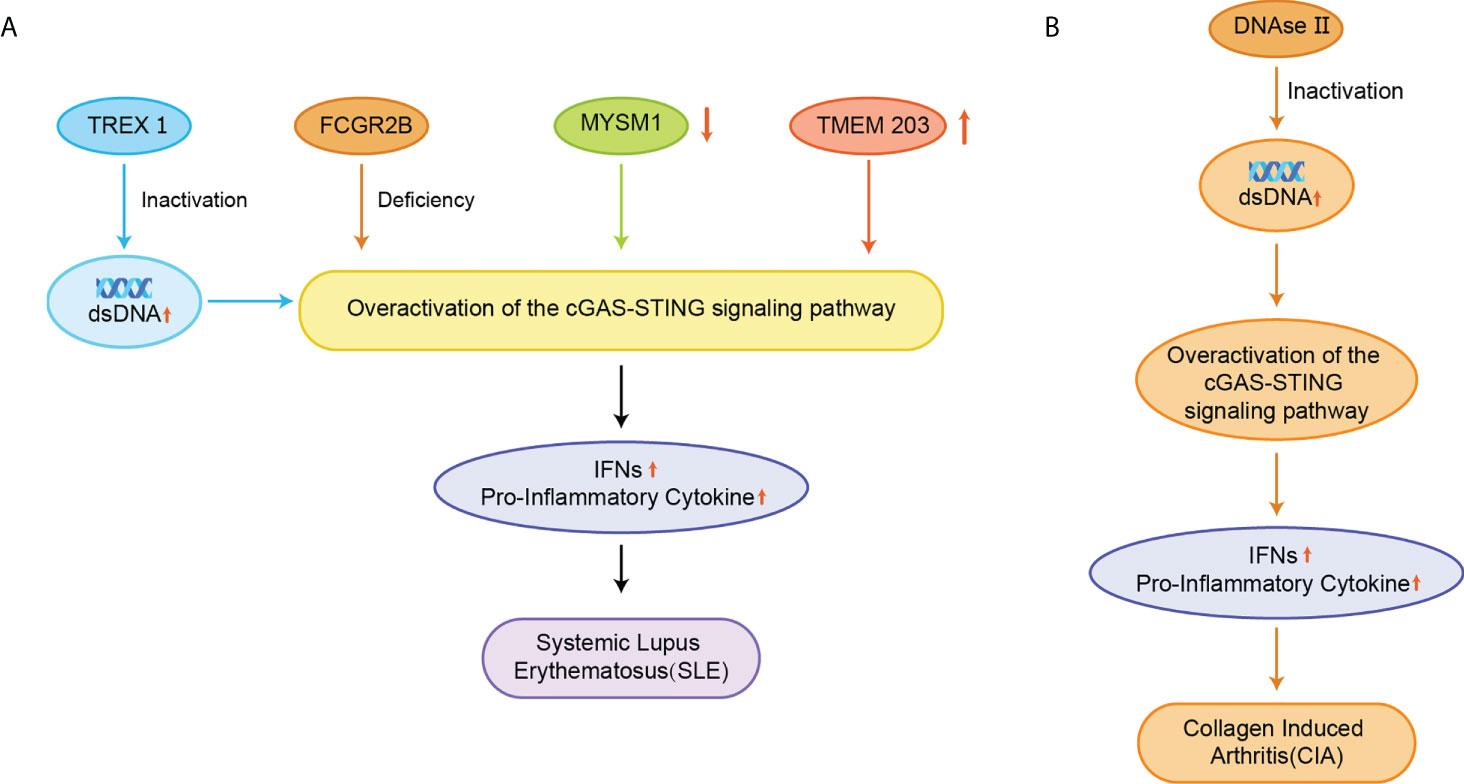
Figure 7 (A) Inactivation of TREX1 resulted in the accumulation of AdMVs in serum, deletion of the FCGR2B gene, decreased MYSM1 production in PBMC, and elevated TMEM203 signaling in T cells were associated with overexpression of the cGAS-STING signaling pathway, upregulation of IFNs and pro-inflammatory cytokine expression, and induction of SLE; (B) Inactivation of DNase II causes abnormal accumulation of dsDNA, which over-activates the cGAS-STING signaling pathway and triggers CIA.
Besides, a comprehensive genetic analysis has identified FCGR2B as a susceptibility gene in patients with SLE, and the mutation of the FCGR2B gene leads to the induction of SLE (Figure 7A) (114). The disruption of STING signaling relieves the lupus development in FCGR2B-deficient mice, and the transplantation of STING-activated bone marrow-derived dendritic cells into the mice with both FCGR2B and STING defects restores the lupus phenotype (115). MYSM1 interacts with STING and cleaves the ubiquitination of the STING at Lys63 to inhibit cGAS-STING signaling (Figure 7A) (116). In PBMCs from patients with SLE, the expression of MYSM1 was reduced, while the level of IFN-I and pro-inflammatory cytokines were increased.
During the development of SLE, the mTOR signaling is activated, and blocking the mTOR pathway using rapamycin has emerged as a new strategy for treating SLE in animal models and patients (117–119). A phase 1/2 clinical trial of rapamycin showed the improvement in disease in SLE patients over a 12-month treatment period (120). Inhibition of mTOR by rapamycin prevented IFN-I production by SLE monocytes and promoted autophagy-mediated degradation of STING (121). Transmembrane protein 203 (TMEM203) is an intracellular regulator of STING-mediated signaling that interacts with STING to activate the cGAS-STING signaling pathway (122). The signaling of TMEM203 is elevated in T cells isolated from SLE patients and correlates with disease severity, and inhibiting TMEM203 may also be a potential therapeutic option for the treatment of SLE (Figure 7A) (123).
Although both SLE elicitation and development appear to be associated with STING and IFN-I upregulation, there are contradictory results in different mouse models of lupus. It suggested that STING could also be a negative regulatory factor in SLE. The deficiency of STING failure to constrain aberrantly activated TLR signaling cascades responsible for the disease (124). In addition, the knockdown of IFNAR in MRL/LPR mice exacerbates lymphocyte proliferation, autoantibody production, and organ damage (125, 126). Therefore, the role of STING in the pathogenesis of SLE still needs to be further investigated.
The pathogenesis of RA is associated with dsDNA accumulation (127). Deoxyribonuclease II (DNase II) can degrade DNA by hydrolyzing its phosphodiester bonds to prevent its abnormal accumulation. The lack of DNase II prevents this process and promotes STING activation and IFN-dependent systemic auto-inflammation, such as collagen-induced arthritis (CIA) (Figure 7B) (128, 129). STING gene-deficient mice had significantly higher levels of anti-collagen antibodies and showed better survival rates than wild-type (WT) mice (128). STING promotes the expression of IFN-inducible genes and the expansion of dendritic cells in CIA. In the CIA model, STING plays a negative regulatory role in B cells when BCR is involved. The inhibition of STING promoted anti-collagen antibody production and B-cell survival, and STING-deficient mice did not spontaneously develop similar autoimmune symptoms (7).
SS is a chronic autoimmune disease affecting multiple organ systems and is characterized by elevated IFN-I levels, which have likewise been reported to be associated with STING (130). Subcutaneous administration of DMXAA to female C57BL/6 mice induced features similar to those of SS patients, such as hypoglandular function and autoantibody production. Activation of STING induced an increase in the expression of IFN-β, IL-6, TNF-α, and IFN-γ in salivary glands and the recruitment of type 1 innate lymphoid cells (ILC1) to the lungs, thereby causing persistent inflammation in the lung (131).
Hall et al. performed a saturation transferred differential 1H NMR screening for the Pfizer fragments library using the cGAS crystallized structure, and a low-affinity fragment tetrazolo[1,5-a]pyrimidine (Kd = 171 μM) was identified with weak inhibition of cGAS (IC50 = 78 μM) (132–134). Further optimization for this compound led to compound 1 (Kd = 0.2 μM, IC50 = 4.9 μM) (Figure 8). However, compound 1 lacked inhibitory activity in cellular assays for high levels of intracellular ATP and GTP.
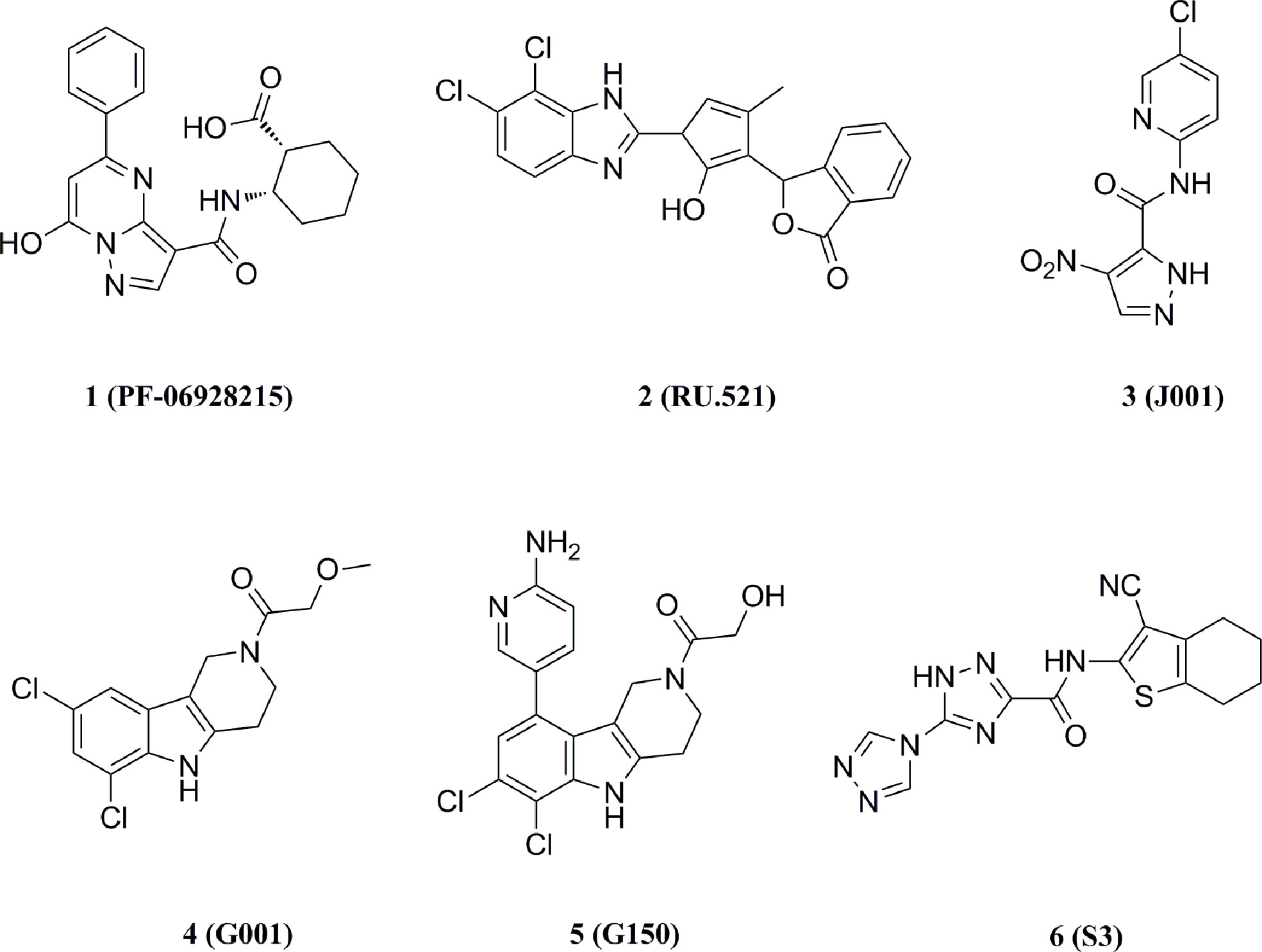
Figure 8 Structures of cGAS inhibitors targeting catalytic site.
Moreover, Vincent et al. performed a high-throughput screen of 123306 compounds and identified four compounds that exhibited good activities (135). The compounds occupied the active center of mouse cGAS and formed key stacking interactions with Agr376 and Try436 at the catalytic site. Based on the binding mode, they subsequently obtained the high-affinity cGAS ligand 2 (Kd = 36 nM) with the best inhibitory activity in cellular assays (IC50 = 0.70 μM) (Figure 8). Meanwhile, the tested results in other signaling pathways showed that compound 2 was a selective inhibitor of cGAS and reduced the mRNA level of IFN-β in bone marrow derived macrophages (BMDM) of AGS model TREX1-/- mice, thus indicating the potential for the treatment of autoimmune diseases.
Lama et al. performed an ATP-coupled high-throughput assay for the identification of small molecule inhibitors of h-cGAS (136). Two cross-species active compounds, 3 and 4 (G chemotype backbone), were obtained after multiple rounds of screening (Figure 8). Their analogue compound 5 exhibited the good inhibitory activity against both THP-1 cells (IC50 = 1.96 μM) and primary human macrophages (IC50 = 0.62 μM) (Figure 8). Compound 5 showed selective inhibition of cGAS in a series of inhibition tests of other innate immune pathways. The structural biology data identified its analogs binding to the cGAS active site. However, the G backbone compounds do not fully occupy the ATP and GTP binding pockets of cGAS and fail to give a clear structure-activity relationship (SAR), and further optimization studies on this backbone are still required.
The crystal structures of cGAS have been solved, which provides quite useful information for structural-based drug design. Based on the high-resolution crystal structure (1.8 Å) of cGAS and compound 1 complex (Figure 9), four effective fragments were identified by virtual screening and thermal shift analysis by Zhao et al. (137) Subsequently, the inhibitory activity of 59 compounds was evaluated using PPiase-coupled assays. One of these compounds did not show any activity in the thermal shift assay and was found to have better inhibitory activity. A similarity search based on this compound was performed and compound 6 (IC50 = 4.9 μM) was identified by PPiase coupling assay (Figure 8).
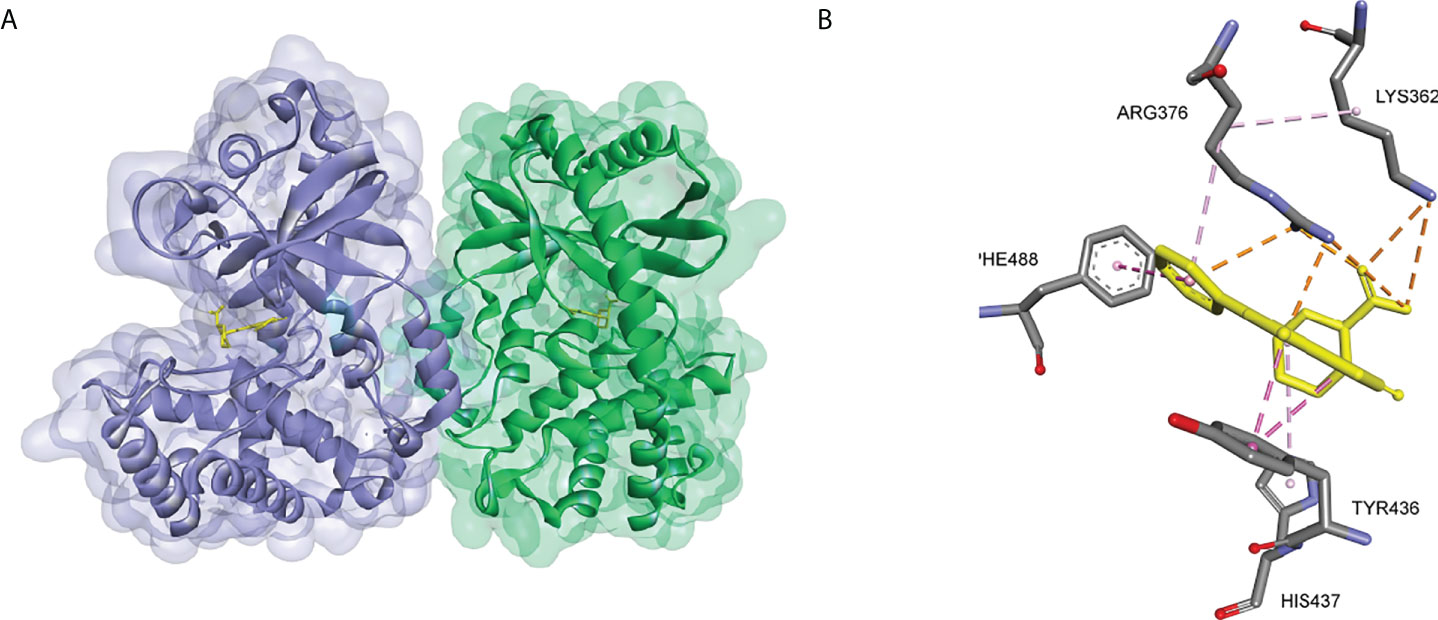
Figure 9 (A) Crystal structure of the cGAS inhibitor PF-06928215 bound to a cGAS dimer (PDB-ID: 4LRC); (B) Residues of the cGAS active center interacting with PF-06928215 and the interactions (PDB-ID: 4LRC).
Anti-malarial drugs such as compound 7 and compound 8 have been reported to disrupt the binding of cGAS to dsDNA and selectively block cGAS-double-stranded β interactions to inhibit the IFN expression (Figure 10) (138, 139). In turn, a second-generation compound 9 was obtained and the potential for this type of compounds in therapy of autoimmune diseases such as AGS or SLE was validated in the AGS model TREX1-/- mice (Figure 10) (140).
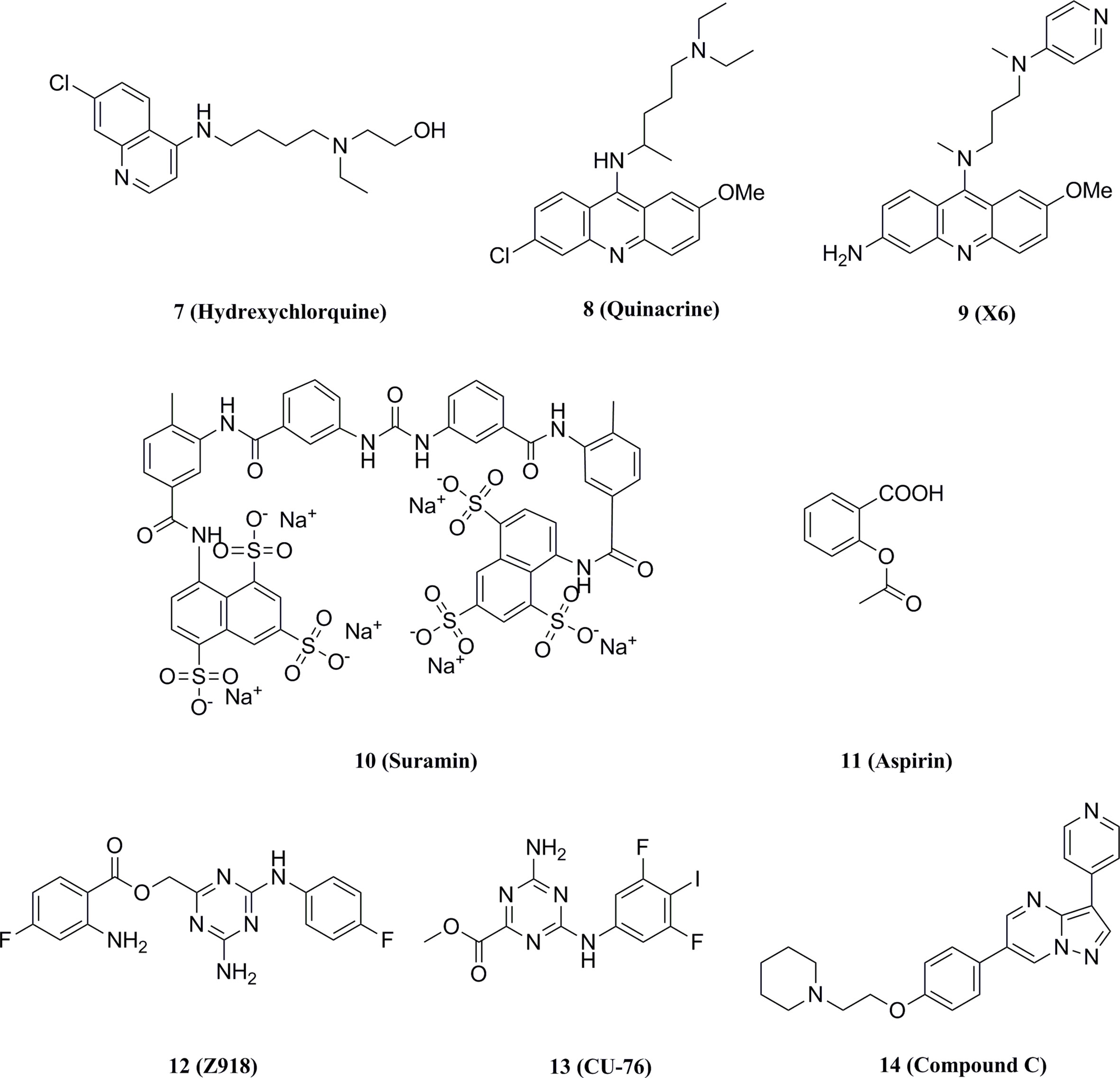
Figure 10 Structures of other types of cGAS inhibitors.
Wang et al. screened a library of 268 compounds to obtain the cGAS inhibitor Suramin, which interfered with the formation of the cGAS-dsDNA complex by competing with the dsDNA binding site of cGAS (Figure 10) (141). It was also proposed that the potential mechanism of action of compound 10 was that its anionic sulfate acted as a phosphate mimetic, binding to the positively charged region on cGAS (138). Besides, Dai et al. found that acetylation of any of the three cGAS residues K384, K394, or K414 affected the binding of cGAS to DNA (142). Further studies revealed that compound 11 which might acetylate the residues significantly reduced ISGs in peripheral blood mononuclear cells of AGS patients and attenuated the auto-DNA-induced autoimmune symptoms in TREX1-/- mice (Figure 10).
Padilla-Salinas et al. performed a virtual screen towards a potentially druggable pocket around Lys347 and Lys394 in h-cGAS to develop protein-protein interface inhibitors of the cGAS dimer itself (143). Only one hit (12) was active to inhibit h-cGAS in vitro with the IC50 of 100 μM (Figure 10). Subsequent optimization resulted in a highly potent inhibitor 13 (IC50 = 0.24 μM) which selectively inhibited the activity of cGAS (Figure 10). Molecular docking suggested that this series of compounds might bind to a binding pocket other than the dsDNA binding site or the catalytic site, while the crystal structure of compound 13 and the cGAS complex could not be solved to verify the precise binding mode. In addition, Aduro Biotech has disclosed several classes of cGAS inhibitors which have shown a good inhibitory activity at both protein and cell levels. However, the underlying mechanism of cGAS inhibition by these compounds is still required for further elucidation (144–146).
In addition, an oligonucleotide A151 was reported to inhibit cGAS activity (147). A151 contains four TTAGGG motif repeats that can act as an inhibitor of cGAS by interacting with the dsDNA binding domain. In cellular experiments, A151 effectively abolished the activation of cGAS by cytoplasmic DNA, thereby inhibiting the production of IFN-I by human monocytes and preventing endogenous DNA accumulation in TREX1-deficient monocytes. The inhibitory activity of A151 is dependent on the nucleotide sequence and phosphate backbone structure, but its specific binding site to cGAS remains to be further explored (148). Through a screen of 2’OMe ASOs and further sequence mutant, Valentin et al. recently characterized key features within the 20-mer ASOs regulating cGAS and TLR9 inhibition and identified a highly potent cGAS inhibitor, which exhibited more potently than A151 (149).
As a potent inhibitor of AMPK, Lai et al. found that compound 14 also inhibited dsDNA-dependent induction of IFN-I (150–152). Further experiments showed that IFN-β was down-regulated by compound 14 through the inhibition of cGAS rather than the inhibition of STING or TBK1. The IFN-β expression was also inhibited by compound 14 in knockdown AMPK cell lines. Subsequent experiments showed that compound 14 improved the autoimmune phenotype of TREX-/- cells. However, they did not resolve the crystal structure of the complex formed by compound 14, cGAS, and dsDNA. They suggested that compound 14 did not bind directly to the cGAS active site, but rather inhibited the upstream genes of the cGAS-mediated pathway. Besides, Huffman et al. develop a stereoselective butyrolactone coupling with the rapid construction of C-C bonds (153). By this method, four inhibitors for chemical screening of cGAS-STING pathway-targeted cell phenotypes were identified based on a 250,000 compound library.
Besides, cGAS binds DNA in a sequence-independent manner through multivalent interactions mediated by its catalytic core and its positively charged disordered N-terminal domain and induces liquid-liquid phase separation (LLPS) of cGAS-DNA bimolecular condensates (154, 155). Recent works indicated that the natural product epigallocatechin gallate (EGCG) directly impacted DNA-induced cGAS-LLPS in vitro, which might represent a novel opportunity to control some self-autoimmune diseases driven by cGAS (156, 157).
15 (IC50 = 6.0 nM) was the earliest TBK1 inhibitor reported in 2009 (Figure 11) (158). This compound was originally developed as an inhibitor of 3 phosphoinositide-dependent protein kinase 1 (PDK1, IC50 = 111 nM), but has also shown strong inhibition of several other kinases (136). Biological assays had shown that 15 inhibited the inflammatory response induced by gram-positive bacteria and the infection of cells with multiple drug-resistant strains of herpes simplex virus type 1. In addition, 15 inhibited the proliferation of oral squamous cell carcinoma (OSCC) by inducing apoptosis and M-phase blockade (159). However, off-targeting effects of 15 on other kinases limited its further development. Further optimization of 15 resulted in 16 (TBK1 IC50 = 19.0 nM, IKKϵ IC50 = 160.0 nM), which showed good selectivity for IKKα, IKKβ, etc (Figure 11) (160). The co-X-ray crystal structure of TBK1 with 16 shows that it binds to TBK1 in a similar pattern but forms fewer interactions with the kinase compared to 15, resulting in reduced potency and off-target effects (37).
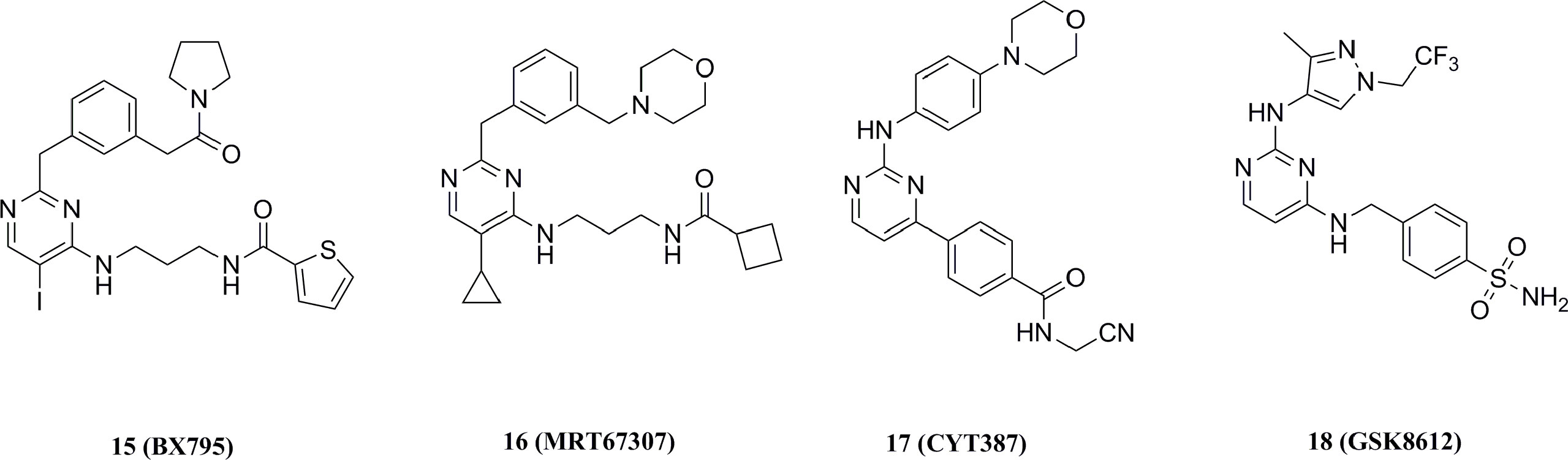
Figure 11 Aminopyrimidine structural TBK1 small molecular inhibitors.
Likewise, the JAK1/2 kinase inhibitor 17 for the treatment of myelofibrosis exhibited inhibitory activity for TBK1 (IC50 = 58 nM, Figure 11) (161, 162). 18 is also a highly selective TBK1 inhibitor (pIC50 = 6.8), and this compound effectively inhibits TBK1-mediated IRF3 phosphorylation and IFNα/β production in addition to its high water solubility and cell permeability (Figure 11) (163). Since all of these early TBK1 inhibitors carry a central aminopyrimidine backbone, SAR studies based on this backbone will further deepen the understanding of the pharmacophores for such type of TBK1 inhibitors.
19 (TBK1 IC50 = 0.8 μM, IKKε IC50 = 5.8 μM) is a drug approved for the treatment of mouth sores and asthma. Biological studies have shown that 19 increases energy expenditure by increasing thermogenesis, improving insulin sensitivity, and reducing body weight and steatosis in mice (Figure 12) (164, 165). In addition, 19 has been found to alleviate acetaminophen-induced liver fibrosis and acute liver injury in mice by inhibiting TBK1/IKKε (166). However, the low solubility and moderate potency of 19 limited its further development. Further structural modifications were performed to the C3-carboxylic acid and C7-isopropyl substituents of 19. Among the analogs, only the tetrazole-substituted compound 21 containing C3-carboxylic acid showed strong inhibition of TBK1 (IC50 = 0.4 μM) and IKKε (IC50 = 0.2 μM), but the cellular activity of this compound was low (Figure 12). Among the other analogs, C7-cyclohexyl analog 22 produced the highest levels of IL-6 secretion in 3T3-L1 cells, but none of these compounds had a synergistic effect.
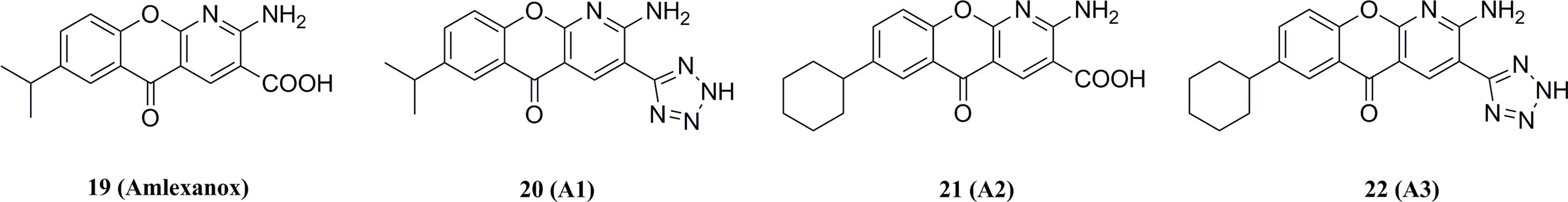
Figure 12 Amlexanox and its derivatives.
PROTAC (Proteolysis Targeting Chimeras) is an emerging and popular technology in the field of drug discovery in recent years (167). Based on this technique, the Crews group selected a TBK1 bound inhibitor 2,4-diaminopyrimidine-like structure and VHL (Von Hippel Lindau) ligand as a linkage model for PROTAC construction (168). After the optimization, the highly efficient TBK1 inhibitor 23 (TBK1 DC50 = 12 nM, Dmax=96%) was obtained, with good selectivity for the related kinase IKKϵ. (Figure 13) The ability of PROTACs to display high potency and selectivity towards TBK1 was revealed by changing the linker length and modulating the binding affinity. The potential for PROTACs was further confirmed in several cancer cells, where TBK1 was almost completely degraded and had no effect on the proliferation of tested cancer cells.
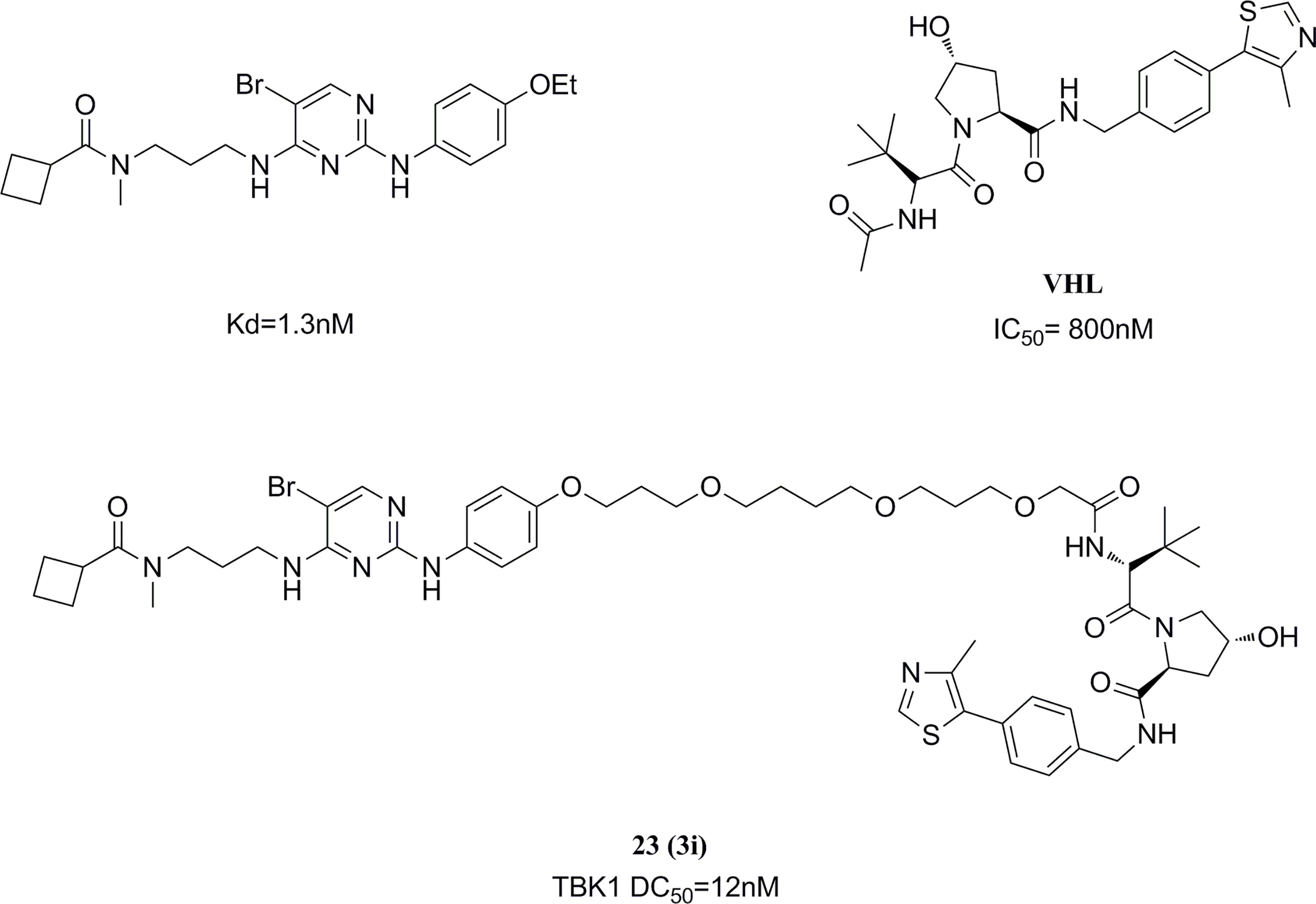
Figure 13 TBK1 targeting PROTAC molecule.
Wang et al. reported a series of imidazopyridines as TBK1 inhibitors, of which the representative compound 24 (IC50 = 9 nM) showed enhanced efficacy and good kinase selectivity (Figure 14) (169, 170). The structurally similar imidazopyridine derivative 25 (IC50 = 5 nM) synergized with the MEK inhibitor AZD6244 to induce apoptosis in drug-resistant NRAS-mutant melanoma cells (Figure 14) (171). In contrast, 26 (IC50 = 13 nM), also with an imidazopyridine backbone, was found to be a potent, low toxicity inhibitor of TBK1 with promising therapeutic effects in mice against autoimmune diseases such as systemic lupus erythematosus (Figure 14) (172). The compound also inhibited the growth of cancer cell lines in non-small cell lung cancer by inhibiting TBK1, thereby leading to a reduction in downstream AKT signaling. The benzimidazole compound 27 (IC50 = 2 nM) reported by Bayer is a highly selective TBK1 inhibitor, but its poor pharmacokinetic properties led it to exhibit poor anti-tumor activity in melanoma mice (Figure 14).
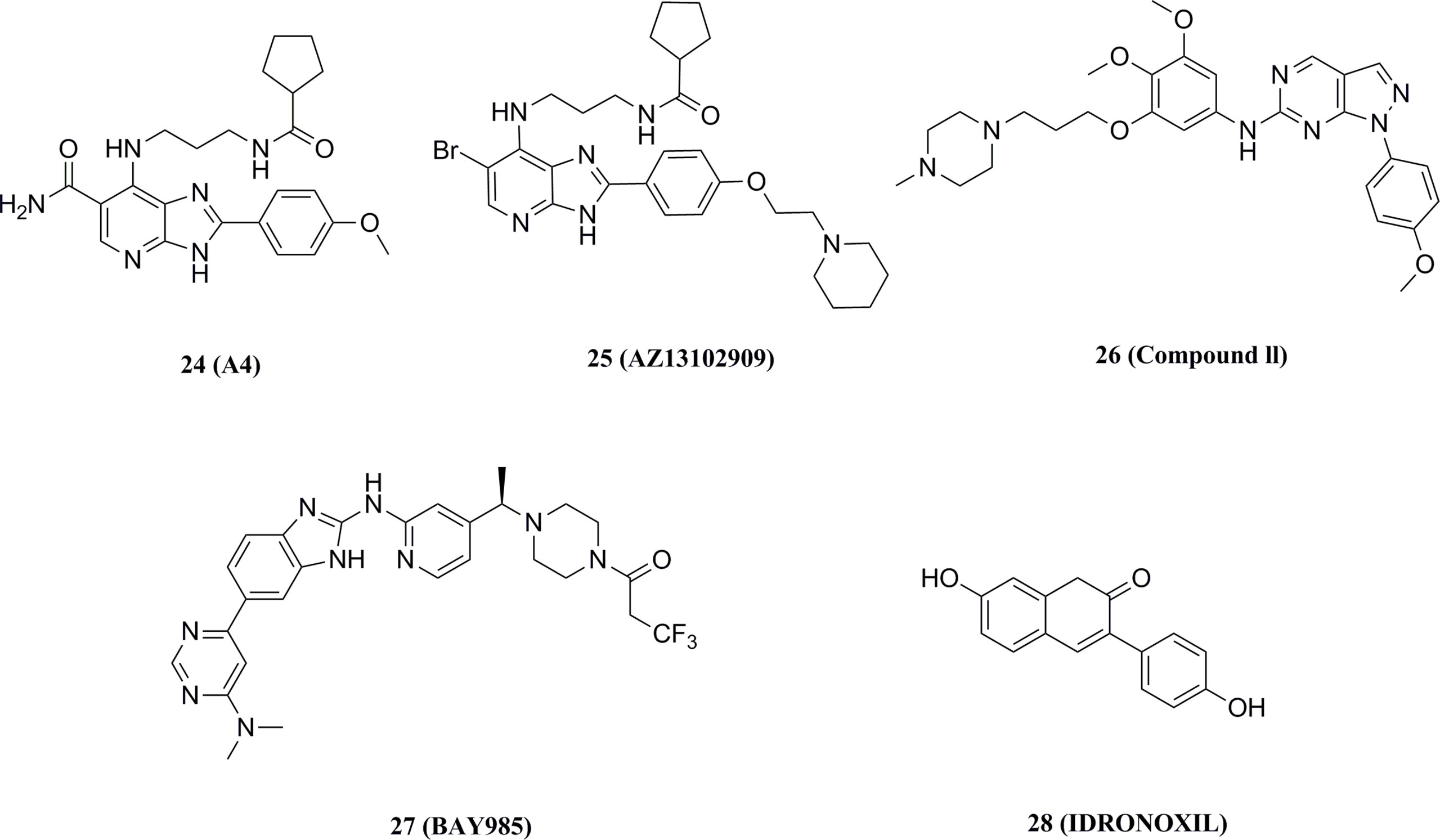
Figure 14 Other TBK1 small molecule inhibitors.
Recently, idronoxil 28 is found to be effective in inhibiting the STING signaling pathway. 28 was reported to disrupt the complex formed by TBK1 and STING, blocking the phosphorylation of Ser172 and leading to dual inhibition of the IRF3 and NF-κB transcriptional programs (Figure 14) (173). 28 has shown promising results in models for the treatment of COVID-19, providing a potential drug with direct access to the clinic for the treatment of inflammatory diseases.
In 2018, Siu et al. used the symmetry of the CDN binding domain to design small molecular inhibitors that were able to bind to the STING protein. Using mass spectrometry-based ligand screening techniques, they found a low-affinity hit (compound 29, R71H-G230A-R293Q HAQ STING IC50 = 7.3 μM) (Figure 15). Co-crystal of determination showed that STING adopted an inactive open conformation, with two molecules occupying the CDN ligand pocket (Figure 16). Based on the identification of several major hydrophobic interactions and polar contact between compound 29 and STING protein, compound 30 (HAQ STING IC50 = 0.08 μM) was identified by further SAR studies, which bound to STING similarly and could inhibit 2’3’-cGAMP-induced the secretion of IFN-β with an IC50 of 11 μM (Figure 15).
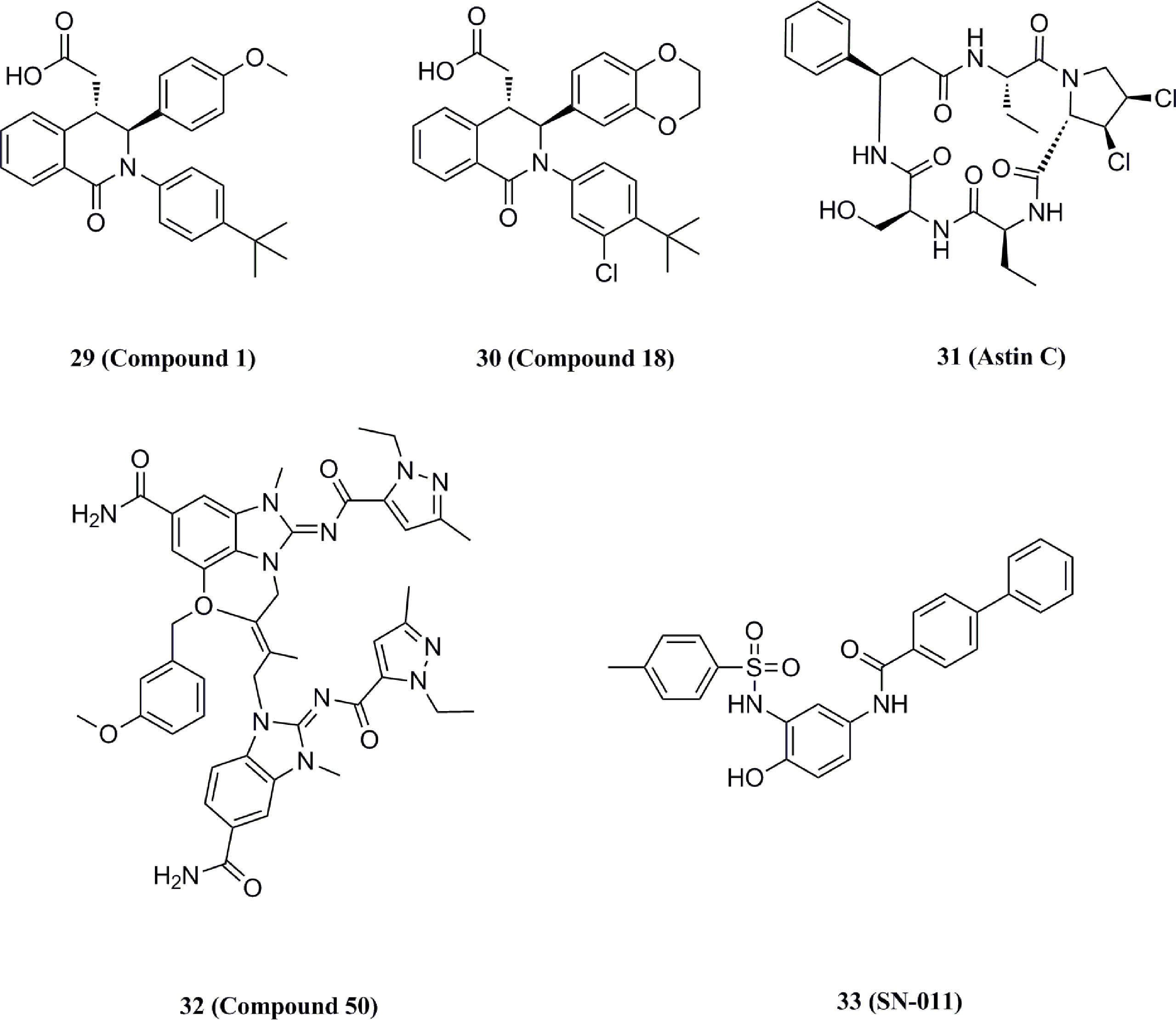
Figure 15 Structures of STING competitive binding antagonists.
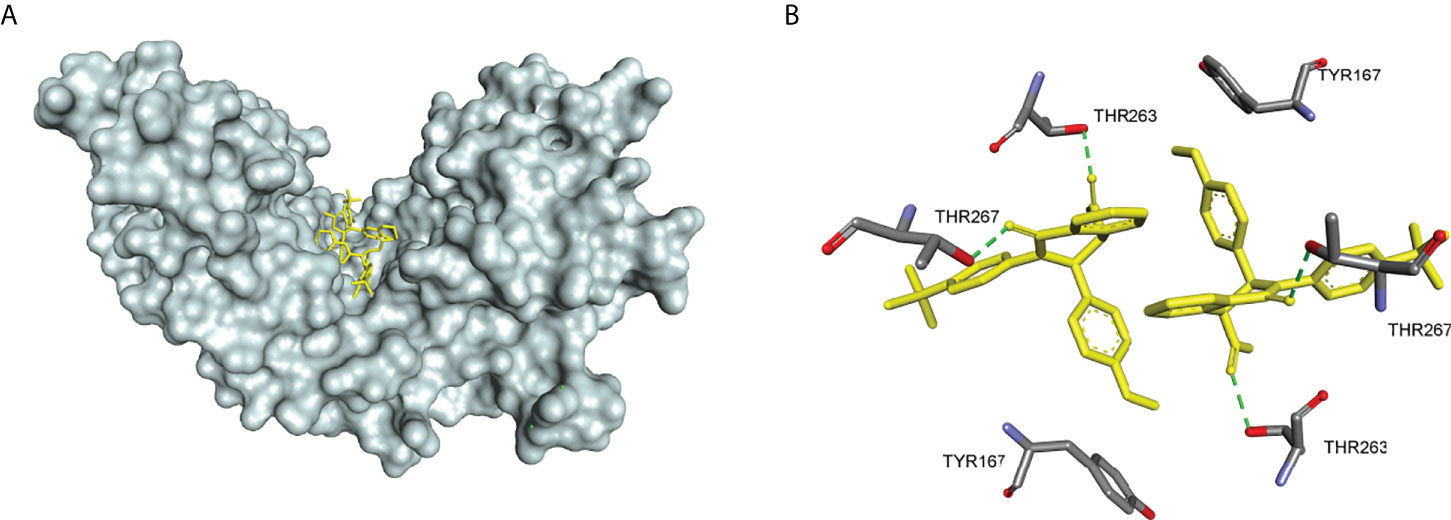
Figure 16 (A) X-ray structure of high-affinity ligand bound to STING protein (PDBID: 6MX3); (B) the interactions between compound 1 and Thr-263 and Thr-267, green dashed line indicating selected hydrogen bonding interactions.
Li et al. identified the natural product 31 from a composite-type cyclopeptides screen based on a reporter gene assay. Further experiments using biotin-labeled compound 31 and h-STING demonstrated the competitive binding of compound 31 to the CDN site, and the addition of high concentrations of CDN (10-fold) abolished the binding of biotin-labeled compound 31 with STING (Figure 15) (174). Subsequent mechanistic studies showed that compound 31 locked the recruitment of IRF3 to STING signaling vesicles without affecting the DNA sensing and TBK1 recruitment, thereby preventing the downstream signaling in the cGAS-STING pathway. Notably, GlaxoSmithKline disclosed a series of N-methylamide-based benzimidazole-like STING antagonists in a patent (Figure 15) (175). The analogs are derived from the previously reported agonist diABZI, which occupies the CDN site at the STING dimer interface. Notably, compounds in this group, such as compound 32, have shown promising inhibitory properties in both binding assays and cellular experiments (FRET pIC50 > 9.9, THP-1 pIC50 = 8.9%, hPBMC pIC50 = 7.1%) (51).
In 2021, Hong et al. obtain the STING small molecular inhibitor compound 33 (IC50 = 0.076 μM) by the virtual screening towards the STING CDN site and following SAR studies (Figure 15) (176). Compound 33 has a higher affinity with binding to the upper CDN binding pocket compared to endogenous 2’3’-cGAMP and locks the STING dimer in an open inactive conformation. This process prevents STING from the oligomerization, translocation, and activation of cytoplasmic DNA, thereby significantly reducing STING-driven IFN-I and pro-inflammatory cytokine expression. Subsequent cellular and animal experiments showed that the compound not only inhibited the over-activation of STING mutants from SAVI patients but also significantly alleviated auto-inflammatory symptoms and prevented the death in TREX1-/- mice. Meanwhile, this compound exhibited comparable inhibitory activity to the previously reported STING covalent inhibitor compound 36 without cytotoxicity, which provides strong support for the development of STING inhibitors for the treatment of STING-related autoimmune diseases.
In 2018, Haag et al. discovered the covalent STING inhibitor compound 36 through structural optimization based on the structures of compound 34 and compound 35, the mouse STING covalent inhibitors which were obtained through high-throughput screening (Figure 17) (177). Compound 36 and the analogs blocked the activated palmitoylation of STING by covalently binding to Cys91, thus preventing STING from assembling into a multimeric complex in the Golgi apparatus, thus inhibiting its downstream signaling (Figure 18). Importantly, compound 36 shows great potential for the treatment of autoimmune diseases. Compound 36 significantly reduced the systemic cytokine response in CMA-treated mice. Treatment with compound 36 in ALS-model mice effectively ameliorated the inflammatory signal caused by the accumulation of TDP-43, restoring neuronal number and motor function (17, 178). Subsequently, compound 36 was found to reduce the symptoms of the chronic inflammatory disease psoriasis by decreasing protein levels of the pro-inflammatory cytokines IL-17A, IL-23, and IL-6 in serum and skin lesions (22).
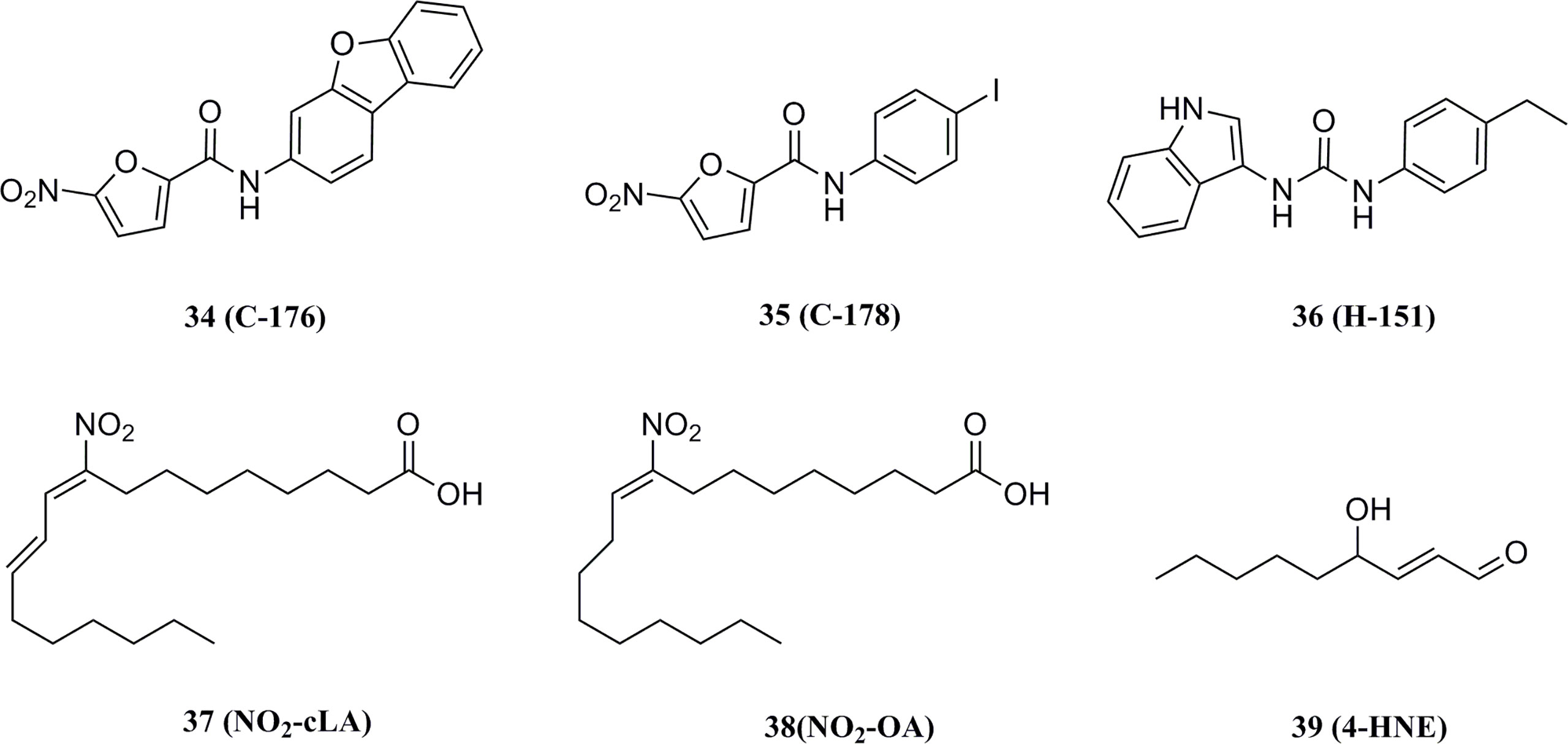
Figure 17 Structures of STING covalent binding antagonists.
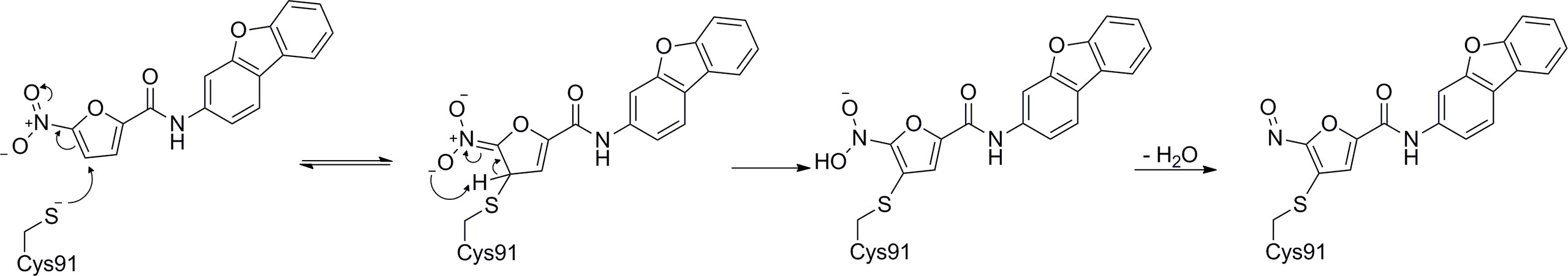
Figure 18 Possibly reaction mechanism of covalent small molecular inhibitors and STING proteins (using C-176 compound as an example).
Besides, Hansen et al. found that host infection with HSV results in the formation of nitro fatty acids in vivo and that endogenous nitro fatty acids (NO2-Fas) can covalently modify STING by Michael addition reaction to adjacent cysteines at positions 88 and 91 (Cys88/91) or N-terminal histidine (His16), thereby inhibiting the palmitoylation of STING and subsequent production of IFNs in host cells (Figure 17) (179). Similarly, the lipid peroxidation during viral infection leads to an increase in one of the major products, 4-hydroxynonenal (38), which promotes the carbonylation of STING, thereby inhibiting the transport of STING from the ER to the Golgi apparatus and suppressing STING activation (Figure 17) (180).
Based on the PROTAC technique, Liu et al. selected the previously reported STING inhibitor C-170 linked to pomalidomide (CRBN ligand) as the PROTACs targeting STING (Figure 19) (181). Among them, compound 40 (DC50 = 3.2 μM) induced the degradation of STING via the CRBN-dependent ubiquitin-proteasome pathway, and dose-dependently downregulated the levels of IFN-β, IL-6, and CXCL10 triggered by 2’3’-cGAMP in THP-1 cells. A partial biological evaluation of this compound as an anti-inflammatory agent was also performed. PROTAC has the advantages of reduced drug exposure, low toxicity, and overcoming drug resistance compared to conventional drugs, and this compound has been reported to provide an alternative strategy for the development of new STING inhibitors.
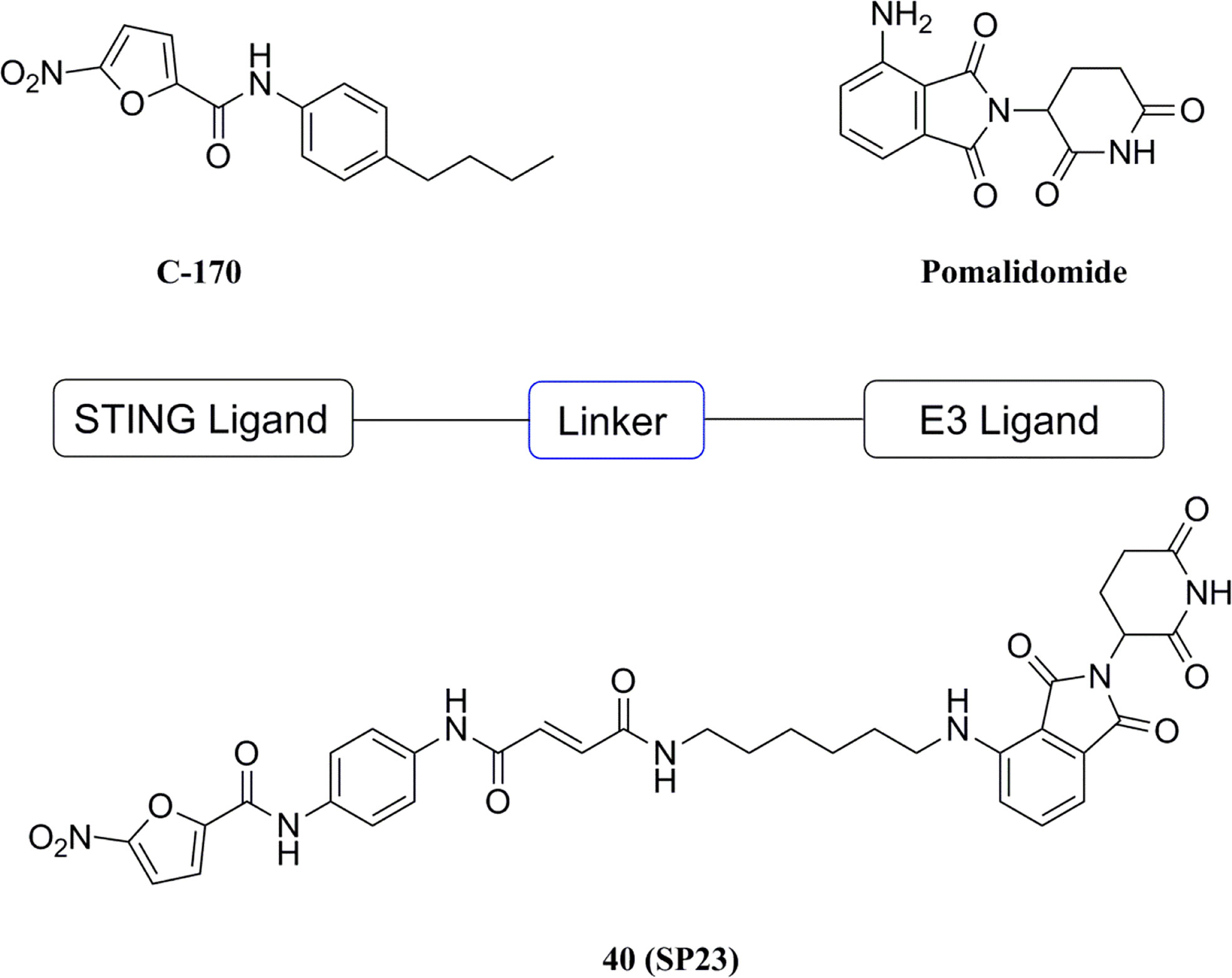
Figure 19 PROTAC molecule target STING.
The GAS-STING signaling plays a critical role in the innate immune response, and the abnormal activation of GAS-STING is linked to various autoimmune diseases (182). The genetic mutants which induce the continuous activation of STING or the cytoplasmic dsDNA accumulation contribute to several STING-relevant autoimmune diseases. While for some autoimmune diseases like SLE which are regarded as the systemic disease for multifactorial complex pathogenesis, the GAS-STING signaling activation is only one of the multiple factors. Moreover, quite a few neurodegenerative diseases including ALS and NPC belong to STING-relevant autoimmune diseases. Importantly, the knock-outing of the STING gene would ameliorate the pathological features of the STING-relevant autoimmune disease, which indicate that the cGAS-STING-TBK1 axis is a promising therapeutic target for various autoimmune diseases.
The inhibitors of cGAS-STING-TBK1 signaling were reported to decrease the protein levels of the inflammatory cytokines and the inflammatory signaling at the cellular and animal levels. The covalent STING inhibitor compound 36 significantly decreased systemic cytokine responses in CMA (the STING agonist) treated mice, thereby attenuating symptoms of autoinflammatory disease in vivo. Moreover, another CDN binding STING inhibitor compound 33 treatment shows the comparable suppression of IFN-β and ISGs expression in the TREX1−/−mice. Therefore, the STING antagonists have become the potential therapeutic agents for STING-relevant autoimmune diseases. Besides the direct inhibition of the STING signaling, targeting the upstream and downstream nodes of the STING activation pathway is also an alternative way to develop the drug against autoimmune diseases. For instance, inhibitors targeting cGAS significantly downregulated IFN expression in TREX1-/- PMBCs in AGS model mice, showing excellent potential for drug development in the treatment of autoimmune diseases caused by DNA accumulation such as AGS and SLE. However, drugs targeting cGAS did not show promising results in the SAVI model, and treatment of cGAS-independent autoimmune diseases may still have to focus on small molecular inhibitors targeting STING. Nerveless, cGAS inhibitors still can be a good complement to therapeutic regimens for the treatment of DNA-dependent autoimmune diseases. Besides, it was reported that TBK1 inhibitor BX795 could downregulate IFN-I activation in PBMCs of SS, SLE, and MS patients (183, 184). Compared to blocking common natural immune targets, such as TBK1 or IFNs, inhibition of cGAS-STING has less risk of immunosuppression and opportunistic infections without keeping the other PRR systems intact. Moreover, STING inhibitors may also be more potent than existing therapeutic agents (e.g., JAK inhibitors and IFN receptor antibodies) because the latter do not limit the maladaptive effects of other cytokines such as TNF-α and IL-6 (185).
Three types of STING inhibitors have been reported including covalent inhibitors forming specific covalent linkage to Cys91, Cys88/91, or His16, with compound 36 as the representative compound (177). The second of type inhibitors just like 28 can disrupt STING/TBK1 interactions (173). The third type of STING antagonists compete with 2’3’-cGAMP at the STING CDN binding site such as 33 (176). Currently, both the first type and the third type show much more potent activity (the IC50 is at the nanomolar level) than the second type inhibitor (the IC50 is at the micromolar level). It was reported that 33 had lower cytotoxicity and higher specificity than compound 36, which indicated that the competed inhibitor at CDN binding site would lead to better specificity and less toxicity. Interestingly, compound 32 which is derived from the STING agonist diABZI exhibits the potent inhibitory of cGAS-STING signaling. The molecular dynamics studies should be performed to investigate how the structural modification in such a scaffold affects the conformational changes of STING CTD and lead to the agonistic or antagonistic activity of the diABZI analogs, which would shed light on the modulation mechanism of STING activation. Moreover, the crystal structures of the complexes of STING and the antagonists have been solved (PDB code: 6MX3), which would facilitate rational drug design based on the complex structure. In fact, the discovery of compound 33 was achieved through the optimization of the active hit which was obtained via the molecular docking towards the CDN site using the virtual chemical database (ZINC).
MZ, YZ, XZ collect and analysis the reference data. MZ designed the experiments and wrote the original manuscript. JZ reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version. All authors contributed to the article and approved the submitted version.
This work was in part supported by the National key research and development program (2018YFE0107600, ZJM), Nature Science Foundation of China (22077115, 81672559). National Mega-Project for Significant new drug discovery (2018ZX09711003-002-002, ZJM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Cooper GS, Bynum MLK, Somers EC. Recent insights in the epidemiology of autoimmune diseases: Improved prevalence estimates and understanding of clustering of diseases. J Autoimm (2009) 33:197–207. doi: 10.1016/j.jaut.2009.09.008
2. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
3. Bot A, Patterson J. In this issue: Sensing immune danger through unfolded protein response plus pathogen recognition receptors; and immune modulation for cancer and HIV-1 disease. Int Rev Immunol (2011) 30:1–3. doi: 10.3109/08830185.2011.542103
4. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol (2009) 10:266–72. doi: 10.1038/ni.1702
5. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol (2010) 11:997–1004. doi: 10.1038/ni.1932
6. Zheng C, Goodrum F. Evasion of cytosolic DNA-stimulated innate immune responses by herpes simplex virus 1. J Virol (2018) 92:e00099–17. doi: 10.1128/JVI.00099-17
7. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461:788–92. doi: 10.1038/nature08476
8. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50. doi: 10.1016/j.immuni.2011.05.006
9. Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol (2014) 35:88–93. doi: 10.1016/j.it.2013.10.010
10. Ablasser A, Chen ZJ. cGAS in action: Expanding roles in immunity and inflammation. Science (2019) 363:eaat8657. doi: 10.1126/science.aat8657
11. Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep (2013) 3:1355–61. doi: 10.1016/j.celrep.2013.05.009
12. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell (2013) 153:1094–107. doi: 10.1016/j.cell.2013.04.046
13. Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, et al. Chen, Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell (2013) 51:226–35. doi: 10.1016/j.molcel.2013.05.022
14. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi: 10.1126/science.1232458
15. Bai J, Liu F. The cGAS-cGAMP-STING pathway: A molecular link between immunity and metabolism. Diabetes (2019) 68:1099–108. doi: 10.2337/dbi18-0052
16. Motwani M, Pesiridis S, Fitzgerald KA. DNA Sensing by the cGAS–STING pathway in health and disease. Nat Rev Genet (2019) 20:657–74. doi: 10.1038/s41576-019-0151-1
17. Yu C-H, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell (2020) 183:636–649.e18. doi: 10.1016/j.cell.2020.09.020
18. Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci (2011) 1238:91–8. doi: 10.1111/j.1749-6632.2011.06220.x
19. Crow YJ. Chapter 166 - aicardi–goutières syndrome. In: Dulac O, Lassonde M, Sarnat HB, editors. Handbook of clinical neurology, vol. 113pp 1629–35. Handbook of Clinical Neurology: Elsevier (2013).
20. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, et al. Activated STING in a vascular and pulmonary syndrome. New Engl J Med (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
21. An J, Durcan L, Karr RM, Briggs TA, Rice GI, Teal TH, et al. Expression of cyclic GMP-AMP synthase in patients with systemic lupus erythematosus. Arthritis Rheumatol (2017) 69:800–7. doi: 10.1002/art.40002
22. Pan Y, You Y, Sun L, Sui Q, Liu L, Yuan H, et al. The STING antagonist h-151 ameliorates psoriasis via suppression of STING/NF-κB-mediated inflammation. Br J Pharmacol 178(24):4907–22. doi: 10.1111/bph.15673
23. Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature (2022) 603:145–51. doi: 10.1038/s41586-022-04421-w
24. Barnett KC, Coronas-Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, et al. Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self- and viral DNA. Cell (2019) 176:1432–1446.e11. doi: 10.1016/j.cell.2019.01.049
25. Gentili M, Lahaye X, Nadalin F, Nader GPF, Puig Lombardi E, Herve S, et al. The n-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep (2019) 26:2377–93.e13. doi: 10.1016/j.celrep.2019.01.105
26. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2′,5′);pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell (2013) 153:1094–107. doi: 10.1016/j.cell.2013.04.046
27. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2013) 339:826–30. doi: 10.1126/science.1229963
28. Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, et al. Structure of the human cGAS–DNA complex reveals enhanced control of immune surveillance. Cell (2018) 174:300–311.e11. doi: 10.1016/j.cell.2018.06.026
29. Andreeva L, Hiller B, Kostrewa D, Lässig C, de Oliveira Mann CC, Jan Drexler D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein–DNA ladders. Nature (2017) 549:394–8. doi: 10.1038/nature23890
30. Zhao B, Xu P, Rowlett CM, Jing T, Shinde O, Lei Y, et al. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature (2020) 587:673–7. doi: 10.1038/s41586-020-2749-z
31. Pathare GR, Decout A, Glück S, Cavadini S, Makasheva K, Hovius R, et al. Structural mechanism of cGAS inhibition by the nucleosome. Nature (2020) 587:668–72. doi: 10.1038/s41586-020-2750-6
32. Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, et al. Structural basis for the inhibition of cGAS by nucleosomes. Science (2020) 370:455–8. doi: 10.1126/science.abd0237
33. Boyer JA, Spangler CJ, Strauss JD, Cesmat AP, Liu P, McGinty RK, et al. Structural basis of nucleosome-dependent cGAS inhibition. Science (2020) 370:450–4. doi: 10.1126/science.abd0609
34. Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, et al. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature (2020) 587:678–82. doi: 10.1038/s41586-020-2748-0
35. Larabi A, Devos JM, Ng SL, Ng SL, Round A, Maniatis T, et al. Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep (2013) 3:734–46. doi: 10.1016/j.celrep.2013.01.034
36. Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain (2017) 10:5. doi: 10.1186/s13041-017-0287-x
37. Tu D, Zhu Z, Zhou AY, Yun C-H, Lee K-E, Toms AV, et al. Eck, structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep (2013) 3:747–58. doi: 10.1016/j.celrep.2013.01.033
38. Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, et al. Molecular basis of tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci (2012) 109:9378. doi: 10.1073/pnas.1121552109
39. Zhao B, Du F, Xu P, Shu C, Sankaran B, Bell SL, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature (2019) 569:718–22. doi: 10.1038/s41586-019-1228-x
40. Li J, Li J, Miyahira A, Sun J, Liu Y, Cheng G, et al. Crystal structure of the ubiquitin-like domain of human TBK1. Protein Cell (2012) 3:383–91. doi: 10.1007/s13238-012-2929-1
41. Zhao C, Zhao W. TANK-binding kinase 1 as a novel therapeutic target for viral diseases. Expert Opin Ther Targets (2019) 23:437–46. doi: 10.1080/14728222.2019.1601702
42. Zhang T, Qian Y, Wang S, Huang G, Zhang L, Xue Z. Influence of the heat dissipation mode of long-flute cutting tools on temperature distribution during HFCVD diamond films. Crystals 9 (2019) 9(8):394. doi: 10.3390/cryst9080394
43. Paludan SR, Bowie AG. Bowie, Immune sensing of DNA. Immunity (2013) 38:870–80. doi: 10.1016/j.immuni.2013.05.004
44. Watson RO, Manzanillo PS, Cox JS. Extracellular m. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell (2012) 150:803–15. doi: 10.1016/j.cell.2012.06.040
45. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signaling. Nature (2008) 455:674–8. doi: 10.1038/nature07317
46. Konno H, Ishikawa H, Ma Z, Barber GN. PS2-54 sting regulates intracellular DNA–mediated, type I interferon-dependent innate immunity. Cytokine (2010) 52:61–2. doi: 10.1016/j.cyto.2010.07.259
47. Huang Y-H, Liu X-Y, Du X-X, Jiang Z-F, Su X-D. The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat Struct Mol Biol (2012) 19:728–30. doi: 10.1038/nsmb.2333
48. Shu C, Yi G, Watts T, Kao CC, Li P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol (2012) 19:722–4. doi: 10.1038/nsmb.2331
49. Ouyang S, Song X, Wang Y, Ru H, Shaw N, Jiang Y, et al. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity (2012) 36:1073–86. doi: 10.1016/j.immuni.2012.03.019
50. Ergun SL, Fernandez D, Weiss TM, Li L. STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell (2019) 178:290–301.e10. doi: 10.1016/j.cell.2019.05.036
51. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang S-Y, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature (2018) 564:439–43. doi: 10.1038/s41586-018-0705-y
52. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
53. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING requires palmitoylation at the golgi. Nat Commun (2016) 7:11932. doi: 10.1038/ncomms11932
54. Zhong B, Yang Y, Li S, Wang Y-Y, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity (2008) 29:538–50. doi: 10.1016/j.immuni.2008.09.003
55. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
56. Wang Q, Liu X, Cui Y, Tang Y, Chen W, Li S, et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity (2014) 41:919–33. doi: 10.1016/j.immuni.2014.11.011
57. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature (2013) 498:380–4. doi: 10.1038/nature12306
58. Shang G, Zhang C, Chen ZJ, Bai X-C, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature (2019) 567:389–93. doi: 10.1038/s41586-019-0998-5
59. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature (2011) 478:515–8. doi: 10.1038/nature10429
60. Konno H, Konno K, Glen N. Barber, cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell (2013) 155:688–98. doi: 10.1016/j.cell.2013.09.049
61. Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA (2009) 106:20842–6. doi: 10.1073/pnas.0911267106
62. Mangan MS, Latz. E. NLRC3 puts the brakes on STING. Immunity (2014) 40:305–6. doi: 10.1016/j.immuni.2014.02.007
63. Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12–5-16L1. Mol Cell (2014) 55:238–52. doi: 10.1016/j.molcel.2014.05.021
64. Deng Z, Chong Z, Law CS, Mukai K, Ho FO, Martinu T, et al. A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J Exp Med (2020) 217:e20201045. doi: 10.1084/jem.20201045
65. Mukai K, Ogawa E, Uematsu R, Kuchitsu Y, Uemura T, Waguri S, et al. Homeostatic regulation of STING by golgi-to-ER membrane traffic. bioRxiv (2020) 12:61. doi: 10.1101/2020.05.20.107664
66. Zhong B, Zhang L, Lei C, Li Y, Mao A-P, Yang Y, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity (2009) 30:397–407. doi: 10.1016/j.immuni.2009.01.008
67. Wang Y, Lian Q, Yang B, Yan S, Zhou H, He L, et al. TRIM30α is a negative-feedback regulator of the intracellular DNA and DNA virus-triggered response by targeting STING. PloS Pathog (2015) 11:1–18. doi: 10.1371/journal.ppat.1005012
68. de Oliveira Mann CC, Orzalli MH, King DS, Kagan JC, Lee ASY, Kranzusch PJ. Modular architecture of the STING c-terminal tail allows interferon and NF-κB signaling adaptation. Cell Rep (2019) 27:1165–1175.e5. doi: 10.1016/j.celrep.2019.03.098
69. Balka KR, Louis C, Saunders TL, Smith AM, Calleja DJ, D’Silva DB, et al. TBK1 and IKKϵ act redundantly to mediate STING-induced NF-κB responses in myeloid cells. Cell Rep (2020) 31:107492. doi: 10.1016/j.celrep.2020.03.056
70. Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg M-C, et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med (2017) 214:1769–85. doi: 10.1084/jem.20161674
71. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jonsson KL, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-kappaB signaling after nuclear DNA damage. Mol Cell (2018) 71:745–760 e5. doi: 10.1016/j.molcel.2018.07.034
72. Al-Asmari SS, Rajapakse A, Ullah TR, Pepin G, Croft LV, Gantier MP. Pharmacological targeting of STING-dependent IL-6 production in cancer cells. Front Cell Dev Biol (2021) 9:709618. doi: 10.3389/fcell.2021.709618
73. Hou Y, Liang H, Rao E, Zheng W, Huang X, Deng L, et al. Non-canonical NF-κB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity (2018) 49:490–503.e4. doi: 10.1016/j.immuni.2018.07.008
74. Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature (2018) 553:467–72. doi: 10.1038/nature25432
75. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
76. Zheng W, Xia N, Zhang J, Chen N, Meurens F, Liu Z, et al. How the innate immune DNA sensing cGAS-STING pathway is involved in autophagy. Int J Mol Sci (2021) 22(24):13232. doi: 10.3390/ijms222413232
77. Prabakaran T, Bodda C, Krapp C, Zhang B-C, Christensen MH, Sun C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J (2018) 37:e97858. doi: 10.15252/embj.201797858
78. Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Different (2019) 26:1735–49. doi: 10.1038/s41418-018-0251-z
79. Hu Q, Knight PH, Ren Y, Ren H, Zheng J, Wu X, et al. The emerging role of stimulator of interferons genes signaling in sepsis: Inflammation, autophagy, and cell death. Acta Physiol (2019) 225:e13194. doi: 10.1111/apha.13194
80. Liang Q, Seo GJ, Choi YJ, Kwak M-J, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA sensor and beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe (2014) 15:228–38. doi: 10.1016/j.chom.2014.01.009
81. Hou P, Lin Y, Li Z, Lu R, Wang Y, Tian T, et al. Autophagy receptor CCDC50 tunes the STING-mediated interferon response in viral infections and autoimmune diseases. Cell Mol Immunol (2021) 18:2358–71. doi: 10.1038/s41423-021-00758-w
82. Crow YJ, Manel N. Aicardi–goutières syndrome and the type I interferonopathies. Nat Rev Immunol (2015) 15:429–40. doi: 10.1038/nri3850
83. Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, et al. Mutations in genes encoding ribonuclease H2 subunits cause aicardi-goutières syndrome and mimic congenital viral brain infection. Nat Genet (2006) 38:910–6. doi: 10.1038/ng1842
84. Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, et al. RNase H2 catalytic core aicardi-goutières syndrome–related mutant invokes cGAS–STING innate immune-sensing pathway in mice. J Exp Med (2016) 213:329–36. doi: 10.1084/jem.20151464
85. Coquel F, Silva M-J, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature (2018) 557:57–61. doi: 10.1038/s41586-018-0050-1
86. Saldanha RG, Balka KR, Davidson S, Wainstein BK, Wong M, Macintosh R, et al. A mutation outside the dimerization domain causing atypical STING-associated vasculopathy with onset in infancy. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01535
87. Sadighi Akha AA, Tschumper RC, Mills JR, Isham CR, Witty EE, Viswanatha DS, et al. A rare case of selective igκ chain deficiency: Biologic and clinical implications. J Allergy Clin Immunol (2020) 146:1208–1210.e6. doi: 10.1016/j.jaci.2020.02.023
88. Melki I, Rose Y, Uggenti C, Van Eyck L, Frémond M-L, Kitabayashi N, et al. Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J Allergy Clin Immunol (2017) 140:543–552.e5. doi: 10.1016/j.jaci.2016.10.031
89. David C, Frémond M-L. Lung inflammation in STING-associated vasculopathy with onset in infancy (SAVI). Cells (2022) 11:318. doi: 10.3390/cells11030318
90. Bennion BG, Croft CA, Ai TL, Qian W, Menos AM, Miner CA, et al. STING gain-of-Function disrupts lymph node organogenesis and innate lymphoid cell development in mice. Cell Rep (2020) 31:107771. doi: 10.1016/j.celrep.2020.107771
91. Siedel H, Roers A, Rösen-Wolff A, Luksch H. Type I interferon-independent T cell impairment in a Tmem173 N153S/WT mouse model of STING associated vasculopathy with onset in infancy (SAVI). Clin Immunol (2020) 216:108466. doi: 10.1016/j.clim.2020.108466
92. Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol (2019) 20:152–62. doi: 10.1038/s41590-018-0287-8
93. Prabakaran T, Troldborg A, Kumpunya S, Alee I, Marinković E, Windross SJ, et al. A STING antagonist modulating the interaction with STIM1 blocks ER-to-Golgi trafficking and inhibits lupus pathology. EBioMedicine (2021) 66:103314. doi: 10.1016/j.ebiom.2021.103314
94. Warner JD, Irizarry-Caro RA, Bennion BG, Ai TL, Smith AM, Miner CA, et al. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med (2017) 214:3279–92. doi: 10.1084/jem.20171351
95. Frémond M-L, Hadchouel A, Berteloot L, Melki I, Bresson V, Barnabei L, et al. Overview of STING-associated vasculopathy with onset in infancy (SAVI) among 21 patients. J Allergy Clin Immunol: In Pract (2021) 9:803–818.e11. doi: 10.1016/j.jaip.2020.11.007
96. Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, et al. COPA mutations impair ER-golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet (2015) 47:654–60. doi: 10.1038/ng.3279
97. Frémond M-L, Legendre M, Fayon M, Clement A, Filhol-Blin E, Richard N, et al. Use of ruxolitinib in COPA syndrome manifesting as life-threatening alveolar haemorrhage. Thorax (2020) 75:92. doi: 10.1136/thoraxjnl-2019-213892
98. Luksch H, Stinson WA, Platt DJ, Qian W, Kalugotla G, Miner CA, et al. STING-associated lung disease in mice relies on T cells but not type I interferon. J Allergy Clin Immunol (2019) 144:254–266.e8. doi: 10.1016/j.jaci.2019.01.044
99. Steiner A, Hrovat-Schaale K, Prigione I, Yu C-H, Laohamonthonkul P, Harapas CR, et al. Deficiency in coatomer complex I causes aberrant activation of STING signalling. Nat Commun (2022) 13:2321. doi: 10.1038/s41467-022-29946-6
100. Deczkowska A, Baruch K, Schwartz M. Type I/II interferon balance in the regulation of brain physiology and pathology. Trends Immunol (2016) 37:181–92. doi: 10.1016/j.it.2016.01.006
101. Yamada T, Horisberger MA, Kawaguchi N, Moroo I, Toyoda T. Immunohistochemistry using antibodies to α-interferon and its induced protein, MxA, in alzheimer’s and parkinson’s disease brain tissues. Neurosci Lett (1994) 181:61–4. doi: 10.1016/0304-3940(94)90560-6
102. Owens T, Khorooshi R, Wlodarczyk A, Asgari N. Interferons in the central nervous system: A few instruments play many tunes. Glia (2014) 62:339–55. doi: 10.1002/glia.22608
103. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging. aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science (2014) 346:89–93. doi: 10.1126/science.1252945
104. Nazmi A, Field RH, Griffin EW, Haugh O, Hennessy E, Cox D, et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia (2019) 67:1254–76. doi: 10.1002/glia.23592
105. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (2006) 314:130–3. doi: 10.1126/science.1134108
106. McCauley ME, O’Rourke JG, Yáñez A, Markman JL, Ho R, Wang X, et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature (2020) 585:96–101. doi: 10.1038/s41586-020-2625-x
107. Wheeler S, Sillence DJ. Niemann–pick type c disease: cellular pathology and pharmacotherapy. J Neurochem (2020) 153:674–92. doi: 10.1111/jnc.14895
108. Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on cholesterol homeostasis: The central role of scap. Annu Rev Biochem (2018) 87:783–807. doi: 10.1146/annurev-biochem-062917-011852
109. Mathur V, Burai R, Vest RT, Bonanno LN, Lehallier B, Zardeneta ME, et al. Activation of the STING-dependent type I interferon response reduces microglial reactivity and neuroinflammation. Neuron (2017) 96:1290–302.e6. doi: 10.1016/j.neuron.2017.11.032
110. Caprariello AV, Rogers JA, Morgan ML, Hoghooghi V, Plemel JR, Koebel A, et al. Biochemically altered myelin triggers autoimmune demyelination. Proc Natl Acad Sci USA (2018) 115:5528–33. doi: 10.1073/pnas.1721115115
111. Casella G, Rasouli J, Mason K, Boehm A, Kumar G, Hwang D, et al. A serine protease inhibitor suppresses autoimmune neuroinflammation by activating the STING/IFN-β axis in macrophages. Cell Mol Immunol (2020) 17:1278–80. doi: 10.1038/s41423-020-0405-z
112. Axtell RC, Steinman L. Type 1 interferons cool the inflamed brain. Immunity (2008) 28:600–2. doi: 10.1016/j.immuni.2008.04.006
113. Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS–STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheumat Dis (2018) 77:1507. doi: 10.1136/annrheumdis-2018-212988
114. Zhu X-W, Wang Y, Wei Y-H, Zhao P-P, Wang X-B, Rong J-J, et al. Comprehensive assessment of the association between FCGRs polymorphisms and the risk of systemic lupus erythematosus: Evidence from a meta-analysis. Sci Rep (2016) 6:31617. doi: 10.1038/srep31617
115. Thim-uam A, Prabakaran T, Tansakul M, Makjaroen J, Wongkongkathep P, Chantaravisoot N, et al. STING mediates lupus via the activation of conventional dendritic cell maturation and plasmacytoid dendritic cell differentiation. iScience (2020) 23:101530. doi: 10.1016/j.isci.2020.101530
116. Tian M, Liu W, Zhang Q, Huang Y, Li W, Wang W, et al. MYSM1 represses innate immunity and autoimmunity through suppressing the cGAS-STING pathway. Cell Rep (2020) 33:108297. doi: 10.1016/j.celrep.2020.108297
117. Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol (2016) 12:169–82. doi: 10.1038/nrrheum.2015.172
118. Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin reduces disease activity and normalizes T cell activation–induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheumat (2006) 54:2983–8. doi: 10.1002/art.22085
119. Warner LM, Adams LM, Sehgal SN. Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arthritis Rheumat (1994) 37:289–97. doi: 10.1002/art.1780370219
120. Lai Z-W, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet (2018) 391:1186–96. doi: 10.1016/S0140-6736(18)30485-9
121. Murayama G, Chiba A, Kuga T, Makiyama A, Yamaji K, Tamura N, et al. Inhibition of mTOR suppresses IFNα production and the STING pathway in monocytes from systemic lupus erythematosus patients. Rheumatology (2020) 59:2992–3002. doi: 10.1093/rheumatology/keaa060
122. Shambharkar PB, Bittinger M, Latario B, Xiong Z, Bandyopadhyay S, Davis V, et al. TMEM203 is a novel regulator of intracellular calcium homeostasis and is required for spermatogenesis. PloS One (2015) 10:e0127480–e0127480. doi: 10.1371/journal.pone.0127480
123. Li Y, James SJ, Wyllie DH, Wynne C, Czibula A, Bukhari A, et al. TMEM203 is a binding partner and regulator of STING-mediated inflammatory signaling in macrophages. Proc Natl Acad Sci (2019) 116:16479–88. doi: 10.1073/pnas.1901090116
124. Sharma S, Campbell AM, Chan J, Schattgen SA, Orlowski GM, Nayar R, et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc Natl Acad Sci (2015) 112:E710–7. doi: 10.1073/pnas.1420217112
125. Hron JD, Peng SL. Type I IFN protects against murine lupus. J Immunol (2004) 173:2134. doi: 10.4049/jimmunol.173.3.2134
126. Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol (2010) 184:1840. doi: 10.4049/jimmunol.0902592
127. Wang J, Li R, Lin H, Qiu Q, Lao M, Zeng S, et al. Accumulation of cytosolic dsDNA contributes to fibroblast-like synoviocytes-mediated rheumatoid arthritis synovial inflammation. Int Immunopharmacol (2019) 76:105791. doi: 10.1016/j.intimp.2019.105791
128. Tansakul M, Thim-uam A, Saethang T, Makjaroen J, Wongprom B, Pisitkun T, et al. Deficiency of STING promotes collagen-specific antibody production and b cell survival in collagen-induced arthritis. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01101
129. Baum R, Sharma S, Organ JM, Jakobs C, Hornung V, Burr DB, et al. STING contributes to abnormal bone formation induced by deficiency of DNase II in mice. Arthritis Rheumatol (2017) 69:460–71. doi: 10.1002/art.39863
130. Bodewes ILA, Al-Ali S, van Helden-Meeuwsen CG, Maria NI, Tarn J, Lendrem DW, et al. Systemic interferon type I and type II signatures in primary sjögren’s syndrome reveal differences in biological disease activity. Rheumatology (2018) 57:921–30. doi: 10.1093/rheumatology/kex490
131. Papinska J, Bagavant H, Gmyrek GB, Deshmukh US. Pulmonary involvement in a mouse model of sjögren’s syndrome induced by STING activation. Int J Mol Sci (2020) 21:4512. doi: 10.3390/ijms21124512
132. Hall JD, Wang H, Byrnes LJ, Shanker S, Wang K, Efremov IV, et al. Binding screen for cystic fibrosis transmembrane conductance regulator correctors finds new chemical matter and yields insights into cystic fibrosis therapeutic strategy. Protein Sci (2016) 25:360–73. doi: 10.1002/pro.2821
133. Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep (2014) 6:421–30. doi: 10.1016/j.celrep.2014.01.003
134. Hall J, Brault A, Vincent F, Weng S, Wang H, Dumlao D, et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PloS One (2017) 12:e0184843/1-e0184843/16. doi: 10.1371/journal.pone.0184843
135. Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun (2017) 8:750. doi: 10.1038/s41467-017-00833-9
136. Lama L, Adura C, Xie W, Tomita D, Kamei T, Kuryavyi V, et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat Commun (2019) 10:2261. doi: 10.1038/s41467-019-08620-4
137. Zhao W, Xiong M, Yuan X, Li M, Sun H, Xu Y. In silico screening-based discovery of novel inhibitors of human cyclic GMP–AMP synthase: A cross-validation study of molecular docking and experimental testing. J Chem Inf Model (2020) 60:3265–76. doi: 10.1021/acs.jcim.0c00171
138. An J, Woodward JJ, Sasaki T, Minie M, Elkon KB. Cutting edge: Antimalarial drugs inhibit IFN-β production through blockade of cyclic GMP-AMP synthase–DNA interaction. J Immunol (2015) 194:4089. doi: 10.4049/jimmunol.1402793
139. An J, Minie M, Sasaki T, Woodward JJ, Elkon KB. Antimalarial drugs as immune modulators: New mechanisms for old drugs. Annu Rev Med (2017) 68:317–30. doi: 10.1146/annurev-med-043015-123453
140. An J, Woodward JJ, Lai W, Minie M, Sun X, Tanaka L, et al. Inhibition of cyclic GMP-AMP synthase using a novel antimalarial drug derivative in Trex1-deficient mice. Arthritis Rheumatol (2018) 70:1807–19. doi: 10.1002/art.40559
141. Wang M, Sooreshjani MA, Mikek C, Opoku-Temeng C, Sintim HO. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-β levels. Future Medicin Chem (2018) 10:1301–17. doi: 10.4155/fmc-2017-0322
142. Dai J, Huang Y-J, He X, Zhao M, Wang X, Liu Z-S, et al. Acetylation blocks cGAS activity and inhibits self-DNA-Induced autoimmunity. Cell (2019) 176:1447–1460.e14. doi: 10.1016/j.cell.2019.01.016
143. Padilla-Salinas R, Sun L, Anderson R, Yang X, Zhang S, Chen ZJ, et al. Discovery of small-molecule cyclic GMP-AMP synthase inhibitors. J Organ Chem (2020) 85:1579–600. doi: 10.1021/acs.joc.9b02666
144. Ndubaku CO, Katibah GE, Roberts TC, Sung L, Ciblat S, Raeppel F, et al. Preparation of pyrazolopyrimidinone compounds as inhibitors of the cGAS/STING pathway and of the cellular cytokine secretion and their uses in the treatment of autoimmune, inflammatory and neurodegenerative disorders. USA: Aduro Biotech, Inc. (2019). p. 366.
145. Katibah GE, Kim JY, Ndubaku CO, Roberts TC, Tjandra M. Triazine compounds as cGAS inhibitor and their preparation. USA: Aduro Biotech, Inc. (2019). p. 165.
146. Ndubaku CO, Roberts TC, Johnson T, Ciblat S, Ramtohul YK, Latimer BK. Preparation of imidazopyridazinone compounds for treating cGAS/STING pathway-mediated diseases. USA: Aduro BioTech, Inc. (2020). p. 68.
147. Kaminski JJ, Schattgen SA, Tzeng T-C, Bode C, Klinman DM, Fitzgerald KA. Synthetic oligodeoxynucleotides containing suppressive TTAGGG motifs inhibit AIM2 inflammasome activation. J Immunol (2013) 191:3876. doi: 10.4049/jimmunol.1300530
148. Steinhagen F, Zillinger T, Peukert K, Fox M, Thudium M, Barchet W, et al. Suppressive oligodeoxynucleotides containing TTAGGG motifs inhibit cGAS activation in human monocytes. Eur J Immunol (2018) 48:605–11. doi: 10.1002/eji.201747338
149. Valentin R, Wong C, Alharbi AS, Pradeloux S, Morros MP, Lennox KA, et al. Sequence-dependent inhibition of cGAS and TLR9 DNA sensing by 2′-o-methyl gapmer oligonucleotides. Nucleic Acids Res (2021) 49:6082–99. doi: 10.1093/nar/gkab451
150. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest (2001) 108:1167–74. doi: 10.1172/JCI13505
151. Jin J, Mullen TD, Hou Q, Bielawski J, Bielawska A, Zhang X, et al. AMPK inhibitor compound c stimulates ceramide production and promotes bax redistribution and apoptosis in MCF7 breast carcinoma cells. J Lipid Res (2009) 50:2389–97. doi: 10.1194/jlr.M900119-JLR200
152. Lai J, Luo X, Tian S, Zhang X, Huang S, Wang H, et al. Compound c reducing interferon expression by inhibiting cGAMP accumulation. Front Pharmacol 11 (2020) 11:88. doi: 10.3389/fphar.2020.00088
153. Huffman BJ, Chen S, Schwarz JL, Plata RE, Chin EN, Lairson LL, et al. Electronic complementarity permits hindered butenolide heterodimerization and discovery of novel cGAS/STING pathway antagonists. Nat Chem (2020) 12:310–7. doi: 10.1038/s41557-019-0413-8
154. Du M, Chen ZJ. DNA-Induced liquid phase condensation of cGAS activates innate immune signaling. Science (2018) 361:704–9. doi: 10.1126/science.aat1022
155. Mehta S, Zhang J. Liquid–liquid phase separation drives cellular function and dysfunction in cancer. Nat Rev Cancer (2022) 22:239–52. doi: 10.1038/s41568-022-00444-7
156. Zhao M, Xia T, Xing J-Q, Yin L-H, Li X-W, Pan J, et al. The stress granule protein G3BP1 promotes pre-condensation of cGAS to allow rapid responses to DNA. EMBO Rep (2022) 23:e53166. doi: 10.15252/embr.202153166
157. Liu ZS, Cai H, Xue W, Wang M, Xia T, Li WJ, et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol (2019) 20:18–28. doi: 10.1038/s41590-018-0262-4
158. Clark K, Plater L, Peggie M, Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IκB kinase ϵ: A distinct upstream kinase mediates ser-172 phosphorylation and activation*. J Biol Chem (2009) 284:14136–46. doi: 10.1074/jbc.M109.000414
159. Bai L-Y, Chiu C-F, Kapuriya NP, Shieh T-M, Tsai Y-C, Wu C-Y, et al. BX795, a TBK1 inhibitor, exhibits antitumor activity in human oral squamous cell carcinoma through apoptosis induction and mitotic phase arrest. Eur J Pharmacol (2015) 769:287–96. doi: 10.1016/j.ejphar.2015.11.032
160. Clark K, Peggie M, Plater L, Sorcek RJ, Young ERR, Madwed JB, et al. Novel cross-talk within the IKK family controls innate immunity. Biochem J (2011) 434:93–104. doi: 10.1042/BJ20101701
161. Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia (2009) 23:1441–5. doi: 10.1038/leu.2009.50
162. Ng AHS. Nationalism and the intangible effects of violence in malik sajad’s munnu: A boy from Kashmir. South Asian Rev (2018) 39:159–74. doi: 10.1080/02759527.2018.1515803
163. Thomson DW, Poeckel D, Zinn N, Rau C, Strohmer K, Wagner AJ, et al. Discovery of GSK8612, a highly selective and potent TBK1 inhibitor. ACS Medicin Chem Lett (2019) 10:780–5. doi: 10.1021/acsmedchemlett.9b00027
164. Reilly SM, Chiang S-H, Decker SJ, Chang L, Uhm M, Larsen MJ, et al. An inhibitor of the protein kinases TBK1 and IKK-ε improves obesity-related metabolic dysfunctions in mice. Nat Med (2013) 19:313–21. doi: 10.1038/nm.3082
165. Beyett TS, Gan X, Reilly SM, Chang L, Gomez AV, Saltiel AR, et al. Carboxylic acid derivatives of amlexanox display enhanced potency toward TBK1 and IKK<em<ϵ</em< and reveal mechanisms for selective inhibition. Mol Pharmacol (2018) 94:1210. doi: 10.1124/mol.118.112185
166. Zhou Z, Qi J, Zhao J, Lim CW, Kim J-W, Kim B. Dual TBK1/IKKε inhibitor amlexanox attenuates the severity of hepatotoxin-induced liver fibrosis and biliary fibrosis in mice. J Cell Mol Med (2020) 24:1383–98. doi: 10.1111/jcmm.14817
167. Maniaci C, Ciulli A. Bifunctional chemical probes inducing protein–protein interactions. Curr Opin Chem Biol (2019) 52:145–56. doi: 10.1016/j.cbpa.2019.07.003
168. Crew AP, Raina K, Dong H, Qian Y, Wang J, Vigil D, et al. Identification and characterization of Von hippel-Lindau-Recruiting proteolysis targeting chimeras (PROTACs) of TANK-binding kinase 1. J Medicin Chem (2018) 61:583–98. doi: 10.1021/acs.jmedchem.7b00635
169. Johannes JW, Chuaqui C, Cowen S, Devereaux E, Gingipalli L, Molina A, et al. Discovery of 6-aryl-azabenzimidaoles that inhibit the TBK1/IKK-ϵ kinases. Bioorgan Medicin Chem Lett (2014) 24:1138–43. doi: 10.1016/j.bmcl.2013.12.123
170. Wang T, Block MA, Cowen S, Davies AM, Devereaux E, Gingipalli L, et al. Discovery of azabenzimidazole derivatives as potent, selective inhibitors of TBK1/IKK kinases. Bioorgan Medicin Chem Lett (2012) 22:2063–9. doi: 10.1016/j.bmcl.2012.01.018
171. Vu HL, Aplin AE. Targeting TBK1 inhibits migration and resistance to MEK inhibitors in mutant NRAS melanoma. Mol Cancer Res (2014) 12:1509–19. doi: 10.1158/1541-7786.MCR-14-0204
172. Hasan M, Dobbs N, Khan S, White MA, Wakeland EK, Li Q-Z, et al. Cutting edge: Inhibiting TBK1 by compound II ameliorates autoimmune disease in mice. J Immunol (2015) 195:4573. doi: 10.4049/jimmunol.1500162
173. Gantier M, Ullah T, Johansen M, Balka K, Ambrose R, Gearing L, et al. Pharmacological inhibition of TBK1/IKKϵ blunts COVID-19 immunopathology. (2022). doi: 10.21203/rs.3.rs-1336801/v1
174. Li S, Hong Z, Wang Z, Li F, Mei J, Huang L, et al. The cyclopeptide astin c specifically inhibits the innate immune CDN sensor STING. Cell Rep (2018) 25:3405–3421.e7. doi: 10.1016/j.celrep.2018.11.097
175. Fosbenner DT, Graybill TL, Kang J, King BW, Lan Y, Leister LK, et al. Preparation of heterocyclic amides as modulators of stimulator of interferon genes (STING), GlaxoSmithKline intellectual property development limited, UK. (2019). p. 372.
176. Hong Z, Mei J, Li C, Bai G, Maimaiti M, Hu H, et al. STING inhibitors target the cyclic dinucleotide binding pocket. Proc Natl Acad Sci (2021) 118:e2105465118. doi: 10.1073/pnas.2105465118
177. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING with covalent small-molecule inhibitors. Nature (2018) 559:269–73. doi: 10.1038/s41586-018-0287-8
178. Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, et al. The cGAS–STING pathway drives type I IFN immunopathology in COVID-19. Nature (2022) 603:145–51. doi: 10.1038/s41586-022-04421-w
179. Hansen AL, Buchan GJ, Rühl M, Mukai K, Salvatore SR, Ogawa E, et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc Natl Acad Sci (2018) 115:E7768. doi: 10.1073/pnas.1806239115
180. Jia M, Qin D, Zhao C, Chai L, Yu Z, Wang W, et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunol (2020) 21:727–35. doi: 10.1038/s41590-020-0699-0
181. Liu J, Yuan L, Ruan Y, Deng B, Yang Z, Ren Y, et al. Novel CRBN-recruiting proteolysis-targeting chimeras as degraders of stimulator of interferon genes with In vivo anti-inflammatory efficacy. J Medicin Chem (2022) 65(9):6593–611. doi: 10.1021/acs.jmedchem.1c01948
182. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
183. Bodewes ILA, Huijser E, van Helden-Meeuwsen CG, Tas L, Huizinga R, Dalm VASH, et al. TBK1: A key regulator and potential treatment target for interferon positive sjögren’s syndrome, systemic lupus erythematosus and systemic sclerosis. J Autoimmun (2018) 91:97–102. doi: 10.1016/j.jaut.2018.02.001
184. Steiner A, Schaale KH, Prigione I, De Nardo D, Dagley LF, Yu C-H, et al. Activation of STING due to COPI-deficiency. bioRxiv (2020). doi: 10.1101/2020.07.09.194399
Keywords: CGAS, STING, autoimmune disease, inhibitor, TBK1
Citation: Zhang M, Zou Y, Zhou X and Zhou J (2022) Inhibitory targeting cGAS-STING-TBK1 axis: Emerging strategies for autoimmune diseases therapy. Front. Immunol. 13:954129. doi: 10.3389/fimmu.2022.954129
Received: 27 May 2022; Accepted: 22 August 2022;
Published: 12 September 2022.
Edited by:
Steven O'Reilly, STipe Therapeutics, DenmarkReviewed by:
Hadida Yasmin, Cooch Behar Panchanan Barma University, IndiaCopyright © 2022 Zhang, Zou, Zhou and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinming Zhou, emhvdWppbm1pbmdAempudS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.