
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 August 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.953849
This article is part of the Research Topic The Mechanism and Novel Strategies of Overcoming Resistance of Hematological Malignancies to CAR T-cell Killing View all 6 articles
 Antonio Valeri1,2
Antonio Valeri1,2 Almudena García-Ortiz1,2
Almudena García-Ortiz1,2 Eva Castellano1,2
Eva Castellano1,2 Laura Córdoba1,2Elena Maroto-Martín1,2
Laura Córdoba1,2Elena Maroto-Martín1,2 Jessica Encinas1,2
Jessica Encinas1,2 Alejandra Leivas1,2
Alejandra Leivas1,2 Paula Río3
Paula Río3 Joaquín Martínez-López1,2*
Joaquín Martínez-López1,2*Despite the impressive results of autologous CAR-T cell therapy in refractory B lymphoproliferative diseases, CAR-NK immunotherapy emerges as a safer, faster, and cost-effective approach with no signs of severe toxicities as described for CAR-T cells. Permanently scrutinized for its efficacy, recent promising data in CAR-NK clinical trials point out the achievement of deep, high-quality responses, thus confirming its potential clinical use. Although CAR-NK cell therapy is not significantly affected by the loss or downregulation of its CAR tumor target, as in the case of CAR-T cell, a plethora of common additional tumor intrinsic or extrinsic mechanisms that could also disable NK cell function have been described. Therefore, considering lessons learned from CAR-T cell therapy, the emergence of CAR-NK cell therapy resistance can also be envisioned. In this review we highlight the processes that could be involved in its development, focusing on cytokine addiction and potential fratricide during manufacturing, poor tumor trafficking, exhaustion within the tumor microenvironment (TME), and NK cell short in vivo persistence on account of the limited expansion, replicative senescence, and rejection by patient’s immune system after lymphodepletion recovery. Finally, we outline new actively explored alternatives to overcome these resistance mechanisms, with a special emphasis on CRISPR/Cas9 mediated genetic engineering approaches, a promising platform to optimize CAR-NK cell function to eradicate refractory cancers.
Over the last decade, autologous CAR-T therapy has revolutionized the treatment of hematological tumors as reflected in six different CAR-T treatments that have received marketing authorization so far to treat multiple myeloma (MM) and CD19+ B cell malignancies, and are now routinely used in the clinic (1–7). Despite their undoubtedly clinical success in the relapsed and refractory setting, CAR-T real-world clinical experience reveals challenges such as cumbersome manufacturing and high-grade toxicities (8) as well as sub-optimal long-term disease control for many patients (1, 3, 9), associated with different mechanisms of resistance that have been extensively reviewed in Shah et al. (10) and in this article collection. Moreover, outcomes for patients who finally progress after CAR-T cell therapy are dismal (11). These limitations highlight the need to investigate alternative immune effector cells as potential vehicles for CAR engineering.
CAR-NK cells emerge as strong candidates due to the unique biological properties and multiple mechanisms of action of conventional Natural Killer (NK) cells. NK cells are innate effector lymphocytes but can also exhibit features of memory-like or adaptive response (12–14). The main function of NK cells is to identify and rapidly discriminate and kill virally infected, stressed, or senescent cells and control several types of tumor cells and metastases (15–17). Human NK cells have been traditionally subclassified into immature immunomodulatory NK cells (CD56brightCD16-/dim) and the mature NK cell (CD56dimCD16bright) subset, which mediates the cytolytic function (18, 19). In contrast to T cells, adoptive NK or CAR-NK therapy does not cause serious adverse events, such as on-target off-tumor toxicities, cytokine release syndrome (CRS), or immune effector cell‐associated neurotoxicity syndrome (ICANS), which may increase hospitalization length and raise therapy cost (20–22). The short NK lifespan in vivo and the different spectrum of cytokines and growth factors released during NK cell killing (e.g, TNF-α, IFN-γ, GM-CSF, and IL-3), are probably responsible for these advantages (23, 24). Allogeneic NK products surpass the expensive and lengthy procedure of autologous CAR-T manufacturing (25). Besides, they are also exempt from ex vivo expansion failures reported in heavily pre-treated patients (10-30%) (1) and tumor contamination events occurring during autologous CAR-T cell productions (26). Allogenic NK and CAR-NK cells constitute an “off‐the‐shelf” product for immunotherapy that can be applied to different patients and generated from multiple sources (20–22, 27). This potential arises due to their minimal risk to cause graft‐versus‐host disease (GvHD). NK cells are functionally similar to CD8+ T cells but do not require prior sensitization and lack TCR expression thereby their responses are not human leukocyte antigen (HLA)-restricted. Instead, NK cell function depends on the balance between activating and inhibitory signaling generated by several germline‐encoded receptors (see review in Sivore et al. and Xie et al.) (Figure 1) (28, 29). Thus, NK cells retain CAR-independent killing capacity through these innate receptors even in a tumor escape scenario characterized by CAR antigen loss or down-regulation. NK cells could eliminate tumor cells through CD16-mediated antibody-dependent cell-mediated cytotoxicity (ADCC), direct target killing by cytolytic granules, (e.g. perforin and granzymes), or via engagement of death receptors (e.g. FASL or TRAIL) (30). Additionally, NK cells efficiently produce cytokines and chemokines that modulate other immune mediators of cytotoxicity (31). Therefore, re-directing NK cells to express a CAR potentially synergizes to kill heterogeneous tumors and reduce the risk of relapse due to CAR-dependent mechanisms.
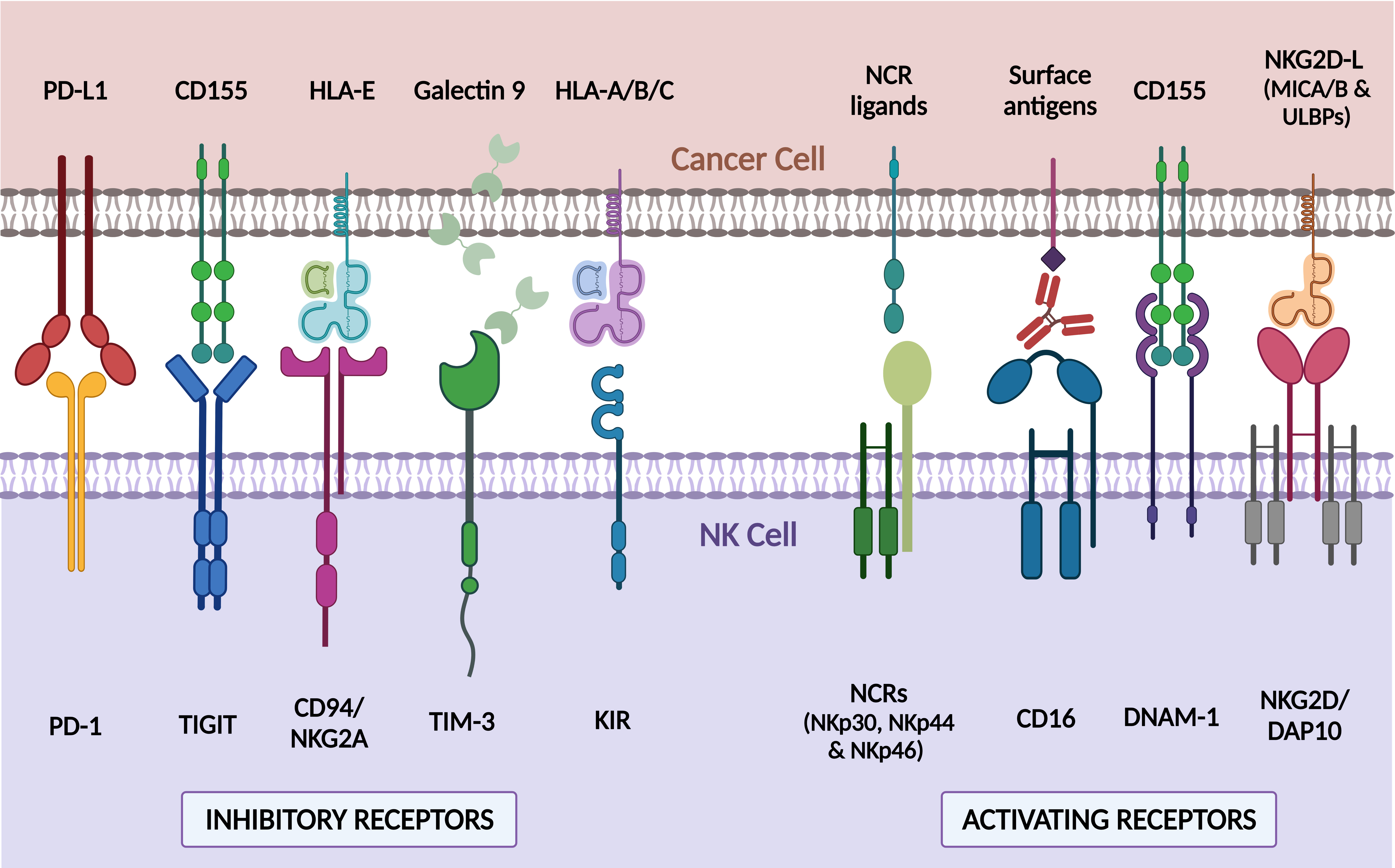
Figure 1 Major endogenous NK cell receptors and their associated ligands in tumor cells. NK cell function is modulated through different surface receptors which bind to ligands expressed on cancer cells. Receptors such as NCR (NKp30, NKp44, and NKp46), CD16, DNAM-1, or NKG2D/DAP10 trigger an activating NK signaling that results in a potent cytotoxic response against ligand-expressing cells. On the contrary, other receptors like PD-1, TIGIT, CD94/NKG2A, TIM-3, and KIRs, turn off NK response when bind to their cognate ligands. The combination of positive and negative signals regulates NK cell response to target cells. PD-1, Programmed Death 1; TIGIT, T cell immunoglobulin and ITIM domain; TIM-3, T cell immunoglobulin and mucin-domain containing-3; KIR, Killer-cell immunoglobulin-like receptor; NCRs, natural cytotoxicity receptors; DNAM-1, DNAX accessory molecule; NKG2D/DAP10, natural killer group 2D/DNAX-activation protein 10; PD-L1, Programmed Death ligand-1; HLA-E, HLA class I histocompatibility antigen, alpha chain E; HLA-A/B/C, HLA class I histocompatibility antigen, alpha chain A/B/C; NKG2D-L, NKG2D ligands; MICA/B, MHC class I polypeptide-related sequence A/B; ULBPs, UL16 binding proteins. Created with BioRender.com.
A large number of CAR-NK preclinical studies have been revealed to be effective in cancer therapy, particularly in the treatment of hematological malignancies (see Gong et al. and Daher et al., for exhaustive review) (32, 33). Up to now, 31 clinical trials are registered with 11 different CAR targets (CD19, CD20, CD22, NKG2D-L, CD33, and BCMA on the top) to address the clinical efficacy of CAR-NK cells in hematologic tumors (Table 1) (22, 34). The most promising data reported arise from first-in-human phase I/II CAR-NK clinical trials based on primary umbilical cord blood (UCB) CAR-NK and induced pluripotent stem cell (iPSC)-derived CAR-NK cell products (35). Rapid and impressive responses (ORR: 73% CR: 64%) were achieved with a bicistronic CD19-28-ζ CAR/IL-15 UCB NK cells in chronic lymphocytic leukemia (CLL) and lymphoma refractory and relapsed setting (NCT03056339). However, the durability of the response in this study could not be completely assessed in some patients because other therapies were administered 30 days after the infusion of CAR-NK cells (22, 36). In the same way, interim analysis in the single-dose cohort treated with FT596, a multi-engineered effector generated using a construct containing a CD19-targeting CAR, a high-affinity uncleavable CD16 (hnCD16) Fc receptor, and an IL-15/IL-15R fusion, revealed an estimated ORR of 69% and CR of 56% in combination with an anti-CD20 antibody (37). Recently, new data from a few patients have been released from NKX-101 (NKG2D CAR-NK in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS)) (3/5 patients achieved CR) and NKX-019 (CD19 CAR-NK in CD19+ B acute lymphocytic leukemia (ALL) and lymphoma) (5/6 patients in CR) (preliminary dose-finding data report, Nkarta, April 2022). While we look forward to seeing efficacy confirmation in the interim analysis of related ongoing products, such as TK-007 (CD19-28-ζ CAR/IL-15 UCB NK cells) or FT576 (iPSC-derived BCMA CAR-NK in MM), these early results suggest similar high-quality responses using CAR-NK cells as compared to CAR-T cells. Importantly, in contrast to CAR-T cell therapy, no evidence of severe CRS, ICANS, hemophagocytic lymphohistiocytosis (HLH), or life-threatening GvHD was observed in any of the aforementioned trials using CAR-NK cells.
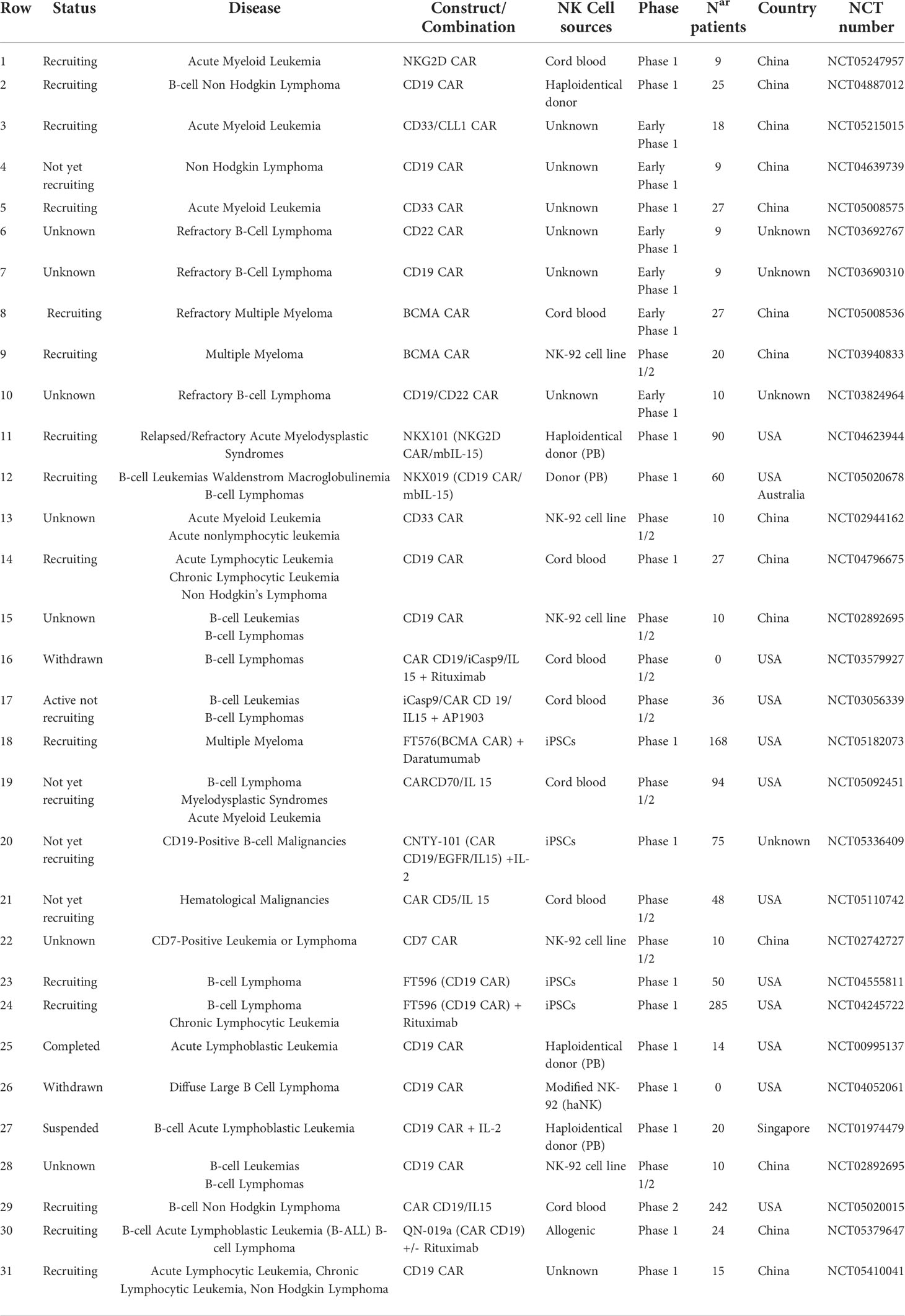
Table 1 Current CAR-NK therapy clinical trials in hematological tumors.
Despite the multiple advantages, CAR-NK therapy still has to confront additional shortcomings that provoke resistance and impact on its efficacy, as seen in CAR-T cells (38). Herein, we will discuss these mechanisms, especially focusing on the CAR-NK dysfunctionalities that lead to immune surveillance evasion by hematologic tumors. We will also review current strategies, mainly based on genome editing, to overcome CAR-NK functional exhaustion and limited migration, and harness CAR-NK effectors replication and persistence in vivo.
One of the main concerns regarding adoptive cell therapy is the need for a great number of enhanced functional effector cells with potential proliferative capacity for optimal clinical responses. Thus, optimizing the source, the cell cytokine-priming, and the expansion protocols can determine CAR-NK cell cytotoxicity and maintenance in vivo.
The most common sources used as platforms to develop CAR-NK therapy are NK cells from peripheral blood (PB) or UCB, NK cell lines (such as NK92), and stem cell-derived NK cells, generated from iPSCs, human embryonic stem cells (hESCs) or CD34+ hematopoietic stem cells (39). PB-NK cells are mature cells with high cytotoxic activity and extended expansion potential but show challenges regarding genetic modification. In contrast, UCB-NK cells contain mainly immature NK cells that after an expansion process acquire a cytotoxic status with equivalent functionality to PB-NK cells (40). Due to donor variability, both PB and UCB-NK cells are heterogeneous products. NK92 cell line provides unlimited homogeneous effectors with easy manufacturing expansion and genetic manipulation but mandatory irradiation before infusion for safety concerns impedes their persistence. In fact, CD33-CAR NK-92 cells do not appear to be effective against AML in the first CAR-NK92 clinical trial (NCT02944162) (34). Alternatively, iPSC-derived NK cells are a homogeneous, unlimited, and easy-to-edit option but their manufacturing is lengthy and requires specialized expertise (41).
Given their clinical suitability and that GvHD was not expected, first approaches for NK adoptive cell therapy were performed with autologous PB-NK cells. In spite of the proven safety, no clinical efficacy was observed due to self-tolerance mediated by HLA-matching (42, 43). Besides, NK cell repertoire in hematological malignancies patients is reduced and functionally altered by the tumor and previous aggressive treatments (44). Pioneer studies from Ruggeri et al. reported that host HLA-I-donor KIR mismatch can promote NK cell graft-versus-leukemia effect (GvL) in transplant setting (“missing-self”), therefore allogenic NK cells have been preferentially selected because exhibit additional advantages to being ready-to-use product (27). Since then, many studies have been focused on haploidentical NK therapy also in non-transplant context, demonstrating safety and modest clinical responses (45). Nevertheless, autologous NK cell immunotherapy is re-emerging in MM clinical consolidation setting with promising data of efficacy released (46). Taken together, these findings highlight that NK cell sources can impact adoptive NK cell therapy clinical outcomes and that the addition of a suitable CAR could unleash NK cell functionality even in inhibitory KIR/HLA-I compatible settings (“induced-self”).
Regarding NK cell priming and expansion strategies, most of them are based on the use of soluble cytokines and artificial antigen-presenting cells (aAPC) with membrane-bound molecules such as cytokines and/or costimulatory ligands (in-depth reviewed by Gurney et al. and Liu et al. (47, 48)). Common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21, and others like IL-12 or IL-18, alone or in combinations are the most commonly studied (49–51). The use of irradiated feeder cells like K562 genetically modified to express membrane-bound IL-15 or IL-21 (mbIL-15/mbIL-21) and 4-1BBL greatly increases fold-expansion rates while maintaining the cytotoxic potential of NK and CAR-NK cells (21, 22, 52–55). Other presenting cells such as Epstein-Barr Virus transformed Lymphoblastoid Cell Lines (EBV-LCLs) have also been studied. Yang et al. engineered a human B-lymphoblastoid cell line with mbIL-21 that provided higher NK cell expansion compared to conventional K562.mbIL-21 and a more favorable phenotype regarding functionality and proliferative capacity (56). aAPCs have demonstrated to efficiently expand NK cells and to be clinically safe (57), but cell-free approaches such as liposomal particles with mbIL-21 and 4-1BBL (58), membrane patches from K562.mbIL21.41BBL (59) or hyaluronic acid-based biomaterials (60) are also under investigation.
Despite some authors suggest optimal landscapes to boost NK cell proliferation (61), a harmonization between NK/CAR-NK expansion protocols is yet to be established. Cytokines such as IL-2, IL-15, and IL-21 play key roles in NK cell functionality and development (49, 62, 63), thus their exposure should be exhaustively addressed as may critically contribute to product efficacy. NK cells can become “addicted” to supraphysiological cytokine exposure, suffering a dramatic drop after interleukin withdrawal when infused into patients, limiting their persistence and efficacy in vivo (64). Molecular mechanisms leading to NK cell decline in the absence of interleukin stimulus are caspase 3 activation, decrease in BCL-2/BIM ratio, and induction of a proapoptotic splice variant of BIM (65). Consequently, in vivo administration of these cytokines was proposed to circumvent ex vivo signaling dependence.
Implications of systemic IL-2 supply to potentiate NK cell expansion were evaluated by Miller’s lab. The infusion of haploidentical NK cells with concomitant IL-2 support after a lymphodepleting chemotherapy obtained disappointing results as NK cell growth was inhibited by host regulatory T cells (Tregs) given that their IL-2Rα provides them with a higher affinity for IL-2 (66). Hence, they depleted Tregs with IL-2-diphtheria toxin fusion protein, prompting NK cell expansion immediately after lymphodepletion, achieving heighten CR rates (NCT00274846 and NCT01106950) (67). Systemic IL-2 administration has related toxicities such as capillary leak syndrome (68, 69), arising the need for using other cytokines with high NK selectivity, such as IL-15.
IL-15 shares similarities with IL-2 but has a high affinity for its IL-15Rα, thus stimulating NK cells but not Tregs (70). The short half-life of IL-15 (71) has promoted the development of alternative related molecules to overcome this drawback. Clinical Trials using either rhIL-15 (72) or IL-15 engineered molecules (N-803, formerly known as ALT-803 (NCT01885897, NCT02384954) (73–75) and NKTR-255 (76, 77)) demonstrated great NK and CD8+ T cell expansion and minimal effect over CD4+ T cells or Tregs but performed insufficient potency themselves (78–80). Moreover, Cooley et al. described CRS in around half of the patients receiving subcutaneous (but not intravenous) IL-15, who also had high IL-6 levels, suggesting that IL-15 stores could trigger proinflammatory cytokines release by myeloid cells (81). Other undesired effects such as neutropenia in nonhuman primates (82) or leukemia in mice (83, 84) have been associated with rhIL-15 systemic administration.
IL-21 is another common gamma-chain cytokine that has demonstrated biasing NK cells for a mature functional phenotype with augmented granule release (85–87), heightened IFN-γ secretion (88), and manageable toxicity in phase I-II clinical trials (89). Still, its effects seem to depend on exposure conditions (90) and high IL-21 has been related to apoptosis in vitro (85). Hence, its systemic supply should be accurately controlled.
As systemic administration of cytokines themselves also entails undesired effects, the newest engineering approaches are focused on in situ delivery and harnessing cytokine signalization to prolong NK/CAR-NK cell persistence while maintaining their optimized functionality. For instance, Liu et al. developed an IL-15 autocrine secreting CD19 CAR UCB-NK cell that showed enhanced cytotoxicity in vitro (91) and CAR-NK cell persistence in a phase I/II clinical trial without systemic IL-15 level increase in the patients (NCT03579927) (22). To further improve their candidate, they ablated CISH, a gene that encodes the cytokine checkpoint CIS (Cytokine inducible Src homology 2 containing protein), which restrains IL-2 and IL-15 signaling (92), obtaining optimal proliferation rates by increasing CAR-NK cell metabolic fitness via glycolysis potentiation (93). Zhu et al. also demonstrated benefits in persistence and antitumor effect of CISH-depleted iPSC NK cells in an AML mouse model (94). Cytokine signalization components have been modified to boost CAR-NK activity by incorporating the inducible MyD88/CD40 (iMC) system as an independent co-stimulator of an IL-15 secreting CAR-NK to enhance cell persistence and tumor control (95, 96). Other IL-15 armored CAR-NK cells developed by Zicheng Du et al. (97) and Christodoulou et al. (98) showed the same trend for CAR-NK cell persistence but in the latter, much lesser potency was achieved and dramatic systemic toxicity was observed. Therefore, other cytokine supply methods are being evaluated.
For instance, engineering NK cells to express mbIL-15 may provide benefits averting the aforementioned undesired effects of systemic IL-15. Additionally, mbIL-15 has demonstrated higher functionality in mice compared to the soluble form even at physiological levels (99). Imamura et al. developed mbIL-15 human NK cells with autonomous potential growth, activated antiapoptotic pathways, and enhanced antitumor effect toward hematological and solid cancers both in vitro and in vivo (100). Going a step beyond, CD19 CAR-NK cells have been engineered to express IL-15/IL-15 receptor α (IL-15/IL-15Rα) fusion protein, endowing them with enhanced persistence regarding their IL-15 secreting counterpart and potentially sustaining tumor control (101). Similarly, IL-15/IL-15Rα has been included in iPSC-derived CAR-NK cells against MM (FT576) (102) or B cell malignancies (FT596) (103), being the latter under assessment in a phase I clinical trial (NCT04245722) (37).
Differentiation into a memory-like setting is a unique strategy to enhance in vivo expansion, persistence, and antitumor responses. Romee, Fehniger, and colleagues demonstrated that brief priming with an IL-12, IL-15, and IL-18 cocktail reprogrammed allogeneic NK cells to a cytokine-induced memory-like (CIML) phenotype that endowed them with heightened expansion and persistence in vivo, higher interferon-γ (IFN-γ) production, and enhanced cytotoxicity against AML and other malignancies (13, 104–106). These acquired advantages have been reproduced in different preclinical studies and phase I/II clinical trials demonstrating a suitable safety profile and promising efficacy, achieving 56% OR rate and 44% CR rate in AML and MDS patients (NCT01898793). CIML-NK clinical trials are also ongoing in a haploidentical hematopoietic cell transplantation (HCT) context (NCT02782546) (107) or donor-derived adoptive therapy in post-HCT AML relapse setting (NCT04024761, NCT03068819) (108, 109). Moreover, CIML-NK combinatorial approaches with chemotherapy (e.g. Ara-C), IL-15 superagonist (NCT02782546) (107), NK-cell engagers (NCT04074746) (110), or a CD38-antibody recruiting molecule (NCT04634435) (111) are also being developed. Regarding CAR-NK context, CIML-NK modified to express CD19 CAR exhibited synergism on CAR activation and demonstrated the aforementioned CIML-NK advantages in an NK-resistant lymphoma model (112). Very recently, Romee’s lab showed that arming CIML NK cells with TCR-like CAR against intracellular neoepitope nucleophosphmin-1 (NPM1) improves anti-AML responses and could be considered as a treatment for NPM1c-mutated HLA-A2+ AML patients (113).
NK cell ex vivo expansion can entail an undesired phenomenon known as fratricide, by which cells recognize receptors or ligands on the surface of their siblings and trigger a cytotoxic activity against them. Several mechanisms can lead to fratricide during NK or CAR-NK cell expansion.
Among them, the well-known Fas/FasL axis is one of the most relevant mechanisms. FasL-mediated cytotoxicity plays a key role in NK cell functionality since it triggers caspase-dependent apoptosis when binds to its receptor Fas in target cells. Fas can also be physiologically expressed by NK cells as a homeostatic mechanism to restrain NK cell activity, termed activation-induced cell death (AICD), but it has been reported that its expression can be abnormally increased during NK cell expansion, especially when cultured in the presence of IL-2 (114), continuous IL-15 treatment (115) or specific feeder cells such as K562-mIL21 (56), leading to fratricide. Moreover, apart from a “self-killing” effect, the enhanced expression of Fas during CAR-NK cell expansion concurrently with a FasL overexpression that has been described in tumor cells such as malignant plasma cells (116) or in the tumor microenvironment (TME) (117), may contribute to tumor escape to adoptive cell therapy.
Another receptor potentially causing fratricide among NK cells is NKG2D. NKG2D is a natural receptor mainly expressed by NK, CD8+ T, and γδ T cells that recognizes several stress-induced ligands (NKG2D-Ls; MHC class I chain-related molecule A/B (MICA/B) and UL16-binding protein (ULBP) 1-6), frequently expressed by cancerous or virally infected cells. There are increasing data describing NKG2D-L expression by activated NK cells but its origin and implications regarding NK cell functionality remain controversial (118). Some studies described that NKG2D-Ls can be transferred from cancerous cells to NK cells after NKG2D/NKG2D-L ligation in a process known as trogocytosis (119, 120), although other authors associated this expression to interleukins such as IL-2 (121) or IL-12, IL-15 and IL-18 (122), depicting a non-fratricidal role but a recently activated mature phenotype.
In the context of CAR-NK cells, fratricide can also appear due to CAR-ligand/antigen recognition in CAR-NK cell surface as previously described in CAR-T cells (123–126). For instance, CD38 CAR-NK cells undergo fratricide since NK cells naturally express CD38 and its expression can be upregulated during ex vivo expansion in the presence of IL-2 or engineered feeder cells. Therefore, NK cells can destroy their siblings either after exposure to anti-CD38 antibody-based therapy (via ADCC) or by their recognition by an anti-CD38 CAR (127, 128). In a similar way, other antigens such as CD70 or CD33, which seem promising candidates to target hematological malignancies with CAR-NK cells, can be upregulated in NK cells during ex vivo expansion, depending on the employed stimulation protocol, entailing CD70 CAR and CD33 CAR-NK-mediated fratricide, respectively (129, 130).
Taken all together, it is strongly necessary to consider the most appropriate expansion method, the use of inhibitors or monoclonal antibodies during CAR-NK manufacturing, currently described for CAR-T cells (123), or even selecting NK donors with specific SNPs that avoid antibody or CAR recognition, as has been reported for CD33 (130). These strategies are emerging, together with CRISPR/Cas9-based gene editing that will be presented later in this review, to avoid fratricidal events that result in lower yields and diminished efficacy of CAR-NK cells in vivo.
Donor NK cell recognition and rejection by the host immune system may potentially reduce allogenic CAR-NK cell persistence in the clinical setting. The primarily effectors responsible for these mechanisms are alloreactive T cells, which recognize non-self HLA molecules on allogeneic NK cells. Higher levels of exhausted T cells after lymphodepletion have been associated with a longer persistence of transferred haploidentical NK cells in leukemia patients (131). Lymphodepleting chemotherapy induces a transient reduction of the host immune system that improves adoptive cell engraftment. Alongside decreasing T and NK cells, lymphodepleting drugs also diminish cell populations that act as sinks for cytokines and/or have immunosuppressive properties, such as Tregs and myeloid-derived suppressor cells (MDSC), generating a more favorable microenvironment for adoptive cell expansion (132, 133). Miller et al. reported that high-dose of cyclophosphamide and fludarabine are required to achieve NK cell engraftment and expansion (134). Endogenous IL-15 increases after lymphodepleting treatment and has been associated with initial NK cell in vivo proliferation (134, 135). However, IL-15 together with IL-7 are essential for T cell homeostatic proliferation, which occurred after severe T cell depletion (133). Therefore, IL-15 may also contribute to CD8+ T cell allorejection (81). Exogenous cytokine support has been proposed to lengthen NK therapy persistence. Nevertheless, the IL-15 superagonist complex N-803 reduces clinical responses in AML patients treated with haploidentical ML-NK cells because of CD8+ T lymphocytes stimulation (NCT03050216 and NCT01898793) (136). Alternatively, autocrine secretion from bicistronic CAR constructs containing cytokines may provide a better approach. In that sense, IL-15-expressing CD19-CAR UCB NK cells have been detected for long-term post-infusion in non-Hodgkin’s lymphoma or CLL patients despite HLA-mismatching (22). Still, the optimal support to create the appropriate cytokine milieu that improves NK cell persistence minimizing T-cell-mediated allorejection has yet to be established.
Multiple NK cell infusions do not solve this issue because the persistence of NK cells from a second infusion is even shorter, suggesting a quicker allogenic response (137). Recently, two studies have reported that an immune-compatible clinical setting generated in the early post-HCT period may improve the persistence of allogeneic CIML-NK cells obtained from the same donor, due to the match of infused NK cells with graft-derived lymphocytes and absence of host alloreactive T cells (107, 109).
Currently, additional strategies are being developed to prevent host system rejection. Several approaches studied in other cell types are based on the expression of molecules that block the attack over the infused cell such as immune checkpoints (138), or on providing cells with receptors that favor the elimination of alloreactive T cells (139). In human PB-NK cells, Hoerster et al. have disrupted HLA class I expression by targeting the β-2-microglobulin gene (β2M) to circumvent CD8+ T cell alloreactivity. Simultaneously, a single-chain HLA-E molecule, which binds the inhibitory receptor NKG2A, has been overexpressed to avoid NK cell fratricide and host NK cell rejection by “missing-self” recognition (140). These modifications have been incorporated in CNTY-101 (Figure 2), a multi-engineered iPSC-derived CD19 CAR-NK product, in which class II major histocompatibility complex transactivator (CIITA) gene has also been disrupted to confer resistance to CD4+ T cell allogeneic response (141). Preclinical studies with both NK products reported that these genetic modifications do not compromise their antitumor potential.
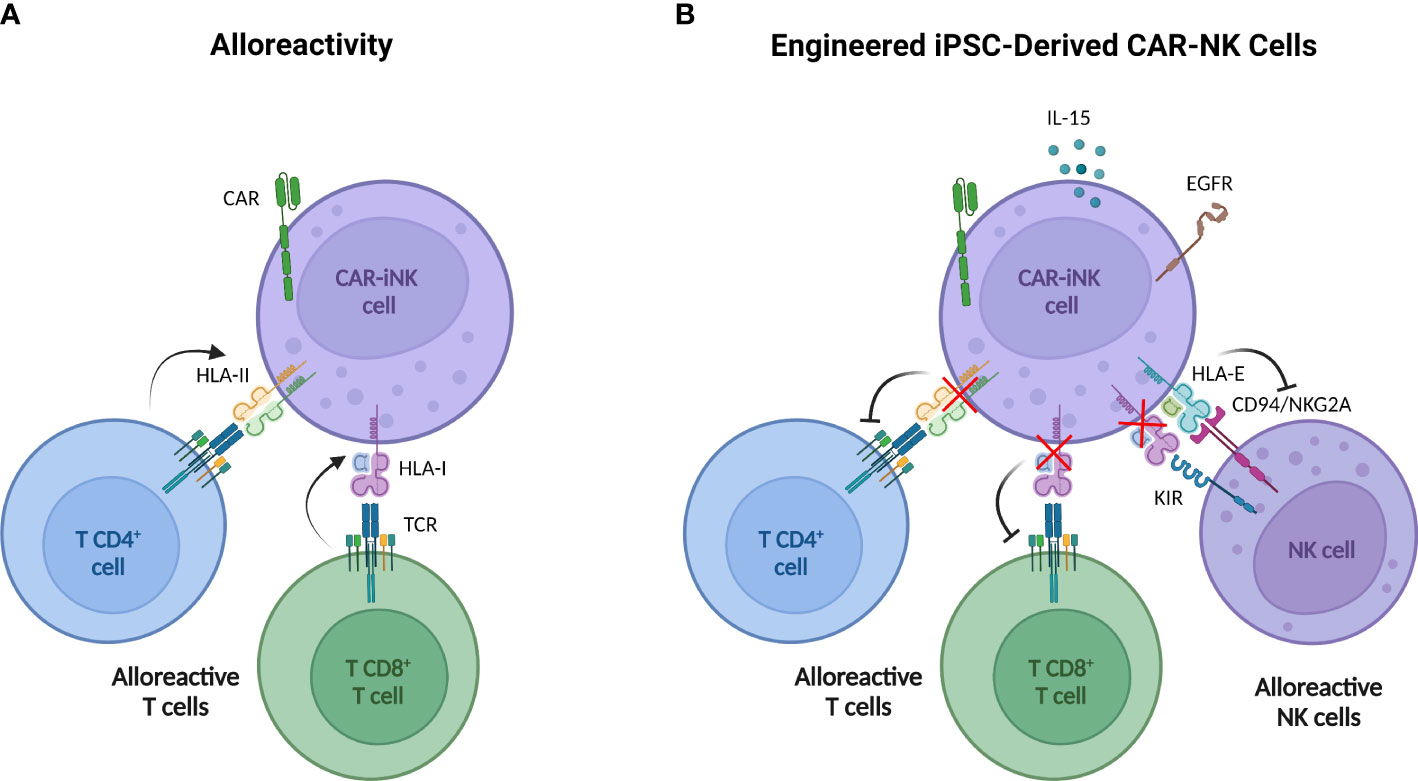
Figure 2 Multi-engineered iPSC-derived CAR NK cells designed to overcome T cell alloreactivity. (A) iPSC-derived CAR NK cells may be rejected by host alloreactive T cells due to the recognition of non-self HLA I by CD8+ T cells and HLA II by CD4+ T cells. (B) Engineered iPSC-derived CAR NK cells incorporate six modifications through three gene-editing steps. 1) The β-2-microglobulin (β2M) disruption to avoid HLA-I expression with the simultaneous insertion of a transgene encoding HLA-E protein (tethered with β2M and a peptide) impedes NK cell killing activity by “missing-self” recognition. 2) The CIITA knock-out to deplete the HLA-II expression concurrently with knock-in of the epidermal growth factor receptor (EGFR) safety switch and the interleukin 15 (IL-15). Safety switch strategy allows the elimination of iPSC-derived CAR NK by the administration of anti-EGFR antibodies and IL-15 secretion improves cell persistence. 3) CD19 CAR knock-in. Created with BioRender.com.
NK cell short in vivo lifespan in the absence of cytokine support reduces off-tumor toxicities and malignancy risk but narrows the therapeutic window, abrogating long-lasting immunotherapy responses (142). In vivo persistence and proliferation of NK cells following adoptive transfer have been previously shown to correlate with clinical responses (143, 144). Therefore, low persistence in vivo could cause early relapses due to the disappearance of CAR-NK therapy. In addition, a short lifespan limits NK cell proliferation and expansion ex vivo during manufacturing, making it harder to achieve sufficient cell numbers for immunotherapy doses (55) and diminishing the time for NK cell optimization by genetic engineering. Consequently, an extension in the effector longevity may boost CAR-NK cell efficacy.
Unlike T effectors that can persist from months to a decade (145), human NK lifespan is not clearly defined, varies between subsets, and can be markedly manipulated in vitro. We and other groups reported that human primary NK or CAR-NK cells co-cultured with K562 aAPCs lines and cytokines typically promote log-phase NK cell expansion for up to 4 to 6 weeks without evidence of senescence (55, 142, 146). Primary NK cells activated by these feeder cells can eventually become unresponsive to stimulation and undergo senescence in a limit record of 15 weeks of continuous proliferation (147).
In vivo, mature NK cells require continuous cytokine support, without which they are detectable in the circulation for only 1-2 weeks (148). Expanded and activated human NK or CAR-NK only survived between 4-5 weeks in xenografted immunodeficient mice without any stimulation (142, 146) and up to 68 days when CAR-NK is engineered to express IL-15 (91).
In clinical use, Liu et al. found CD19-CAR UCB-NK cells by flow cytometry limited to the first three weeks even with lymphodepletion and IL-15-autocrine support. Nevertheless, DNA copies of the infused CAR were detected up to 12 months measured by real time-PCR in patients suffering CD19+ lymphoid tumors (22). Similarly, autologous NKAEs were detected by multiparametric flow cytometry around four weeks after infusion in MM clinical trial performed in a consolidation setting (46). In line with augmented lifespan bolstered by cytokines, CIML NK cells increased persistence up to 2-3 months analyzed by mass cytometry (107, 109), albeit CIML NK cells were administered in an “immune-permissive” microenvironment and combined with an IL-15 superagonist (107). Human CAR-NK effectors do not typically clonally expand in vivo like antigen-specific-T cells or virus-specific adaptive NK cells (149). They frequently peak in circulation between the first and second week post-transfer from where they progressively decline (22, 43, 107, 134). Together, these studies suggest that, although permissive to lifespan modification by exogenous cytokines or HLA matching, NK and CAR-NK are short-lived cells impacted by senescence that inexorably arises ex vivo as a consequence of expansion methods, and subsequently in vivo, where these effectors proliferate in a narrow window and do not persist long in patients.
Cellular senescence is a universal process considered a hallmark of aging and can be triggered in non-tumoral cells in response to different intrinsic and extrinsic stressors, as well as developmental signals. In particular, replicative senescence is related to loss of proliferative capacity and functional deficit characterized by the shortening of telomeres, the detection of genomic DNA double-strand breaks, the activation of repair machinery, and the arrest of the cell cycle to stop replication and prevent genomic instability (150). Eventually, senescent cells can surpass cell cycle checkpoints and enter in a crisis phase with augmented chromosomal and genomic instability, inducing apoptosis (151). T cell immunosenescence is a well-studied phenomenon observed during aging and prolonged in vitro cultures and differs from immune exhaustion by repeated stimulation. Terminal differentiated T effectors are characterized by CD28- CD27- KLRG1+ CD57+ CD45RA+ phenotype with shortened telomeres, active metabolic reprogramming, higher production of pro-inflammatory molecules (senescence-associated secretory phenotype), and less replicative ability (see review in Kasakovski et al.) (152). Although a phenotype of highly mature NK cell based on CD57 expression has been proposed, the NK cell immunosenescence field is still in its infancy with no phenotype and function clearly established (153, 154). Terminal NK cells are dysfunctional, identified by decreased NK effector functions, such as impaired ADCC, as well as reduced cytokine secretion like IFN-γ and expression of perforin and granzyme (155). Among the factors involved in NK cell longevity control, telomere length is critical because its shortening after multiple rounds of cell divisions (Hayflick limit) exposes the unstable chromosomal ends, initiates fusion-bridge-breakage cycles, and leads to genomic instability and replicative senescence. Human NK cells display telomere shortening and a reduction in telomerase activity with age (156). Cellular differentiation impacts telomere shortening, leading to the more mature CD56dim NK cells having shorter relative telomere length than the immature CD56bright subset (157).
Regarding adoptive NK therapy, telomere length depends on the NK source or the activation/expansion method selected. For example, telomere length in iPSC-derived NK cells is much longer compared to those expanded from PB (158). Yang et al. reported up-regulation of positive telomere length regulator genes such as ZNF257, LRRC34, NAF1, and human telomerase reverse transcriptase (hTERT) in NK cells expanded and activated with 721.221 feeder line with IL-2 and IL-15 (56). hTERT expression and activity are strictly regulated in somatic cells and can be reprogrammed by common gamma-chain cytokines, c-Myc (159) or fine-tuned by miRNAs (160, 161). Indeed, all IL-2, IL-15, and IL-21 have been shown to up-regulate telomerase activity in NK cells, thereby preventing telomere loss and allowing cells to extend replication. IL-2 increases telomerase activity in NK cells upregulating hTERT mRNA levels (162). MbIL-21 increased NK cell longevity by maintaining telomere length in K562 co-cultures (55, 163). In addition, IL-15 induces hTERT expression and cellular growth in NK culture ex vivo at lower doses than IL-2 (164). IL-21 is known to signal primarily through the STAT3 component of the JAK/STAT pathway, whereas IL-15 signals mainly through STAT5 (165).
Ectopic expression of hTERT by genetic engineering may be an effective strategy to improve CAR-NK cell persistence and thereby their therapeutic potential, paralleling seminal studies in CAR-T cells (166) where the maintenance of telomere length and replicative ability is associated with engraftment efficacy and antitumor efficiency (167). In a pioneer study from Campana´s lab, hTERT transfected NK and CD19 CAR-NK cells (expanded with K562-mb15-41BBL) restored replicative ability and could be cultured for almost one year with continued cytotoxicity against leukemic cell lines and exhibited normal karyotype (analyzed at day 186). However, transfected NK cells were not able to grow autonomously in nonobese diabetic severe‐combined‐immunodeficient γc−/− (NSG) mice and still eventually developed delayed senescence in vitro (147). More recently, Streltsova and coworkers corroborated that stable hTERT ectopic expression, even when gamma-retrovirus is used, increases the proliferation and lifespan of expanded and activated (K562-mbIL21+IL2) NK cells rather than complete immortalization (168). The safety of this strategy is a critical question because so far, the inability of ectopically expressed hTERT to cause oncogenic transformation of NK cells has not been firmly established. Further research is needed to attempt more refined approaches to overexpress hTERT in terms of expression control, for instance, inducible promoters or transient expression, as reported in CAR-T cells, which lead to improved proliferation and persistence in murine xenograft tumor models of human B-cell lymphomas (169). Another possible strategy is the implementation of safety switches in hTERT constructs to assure safety. Direct reprogramming of other components of telomere machinery by genetic engineering or by their stabilizers and/or manipulating telomere elongation factors could accelerate the translation of these strategies into clinical reality.
As a result of overstimulation, tumor progression induces a reversible exhausted status in NK cells characterized by impaired effector functions and altered phenotype, similar to previously described in T cells. The expression of tumor-associated immune checkpoints reduces NK cell killing activity via direct cell-cell interactions. NK cell ex vivo expansion also fosters the expression of some immune checkpoint receptors, potentially decreasing CAR-NK therapy efficacy (Figure 3). On top of that, patients’ NK cells and infused CAR-NK cells encounter a hostile microenvironment in the tumor niche, generated by immunosuppressive cells and soluble factors, which leads to NK cell suppression. Consequently, many efforts are underway to identify and neutralize the negative TME factors that may limit CAR therapy effectiveness.
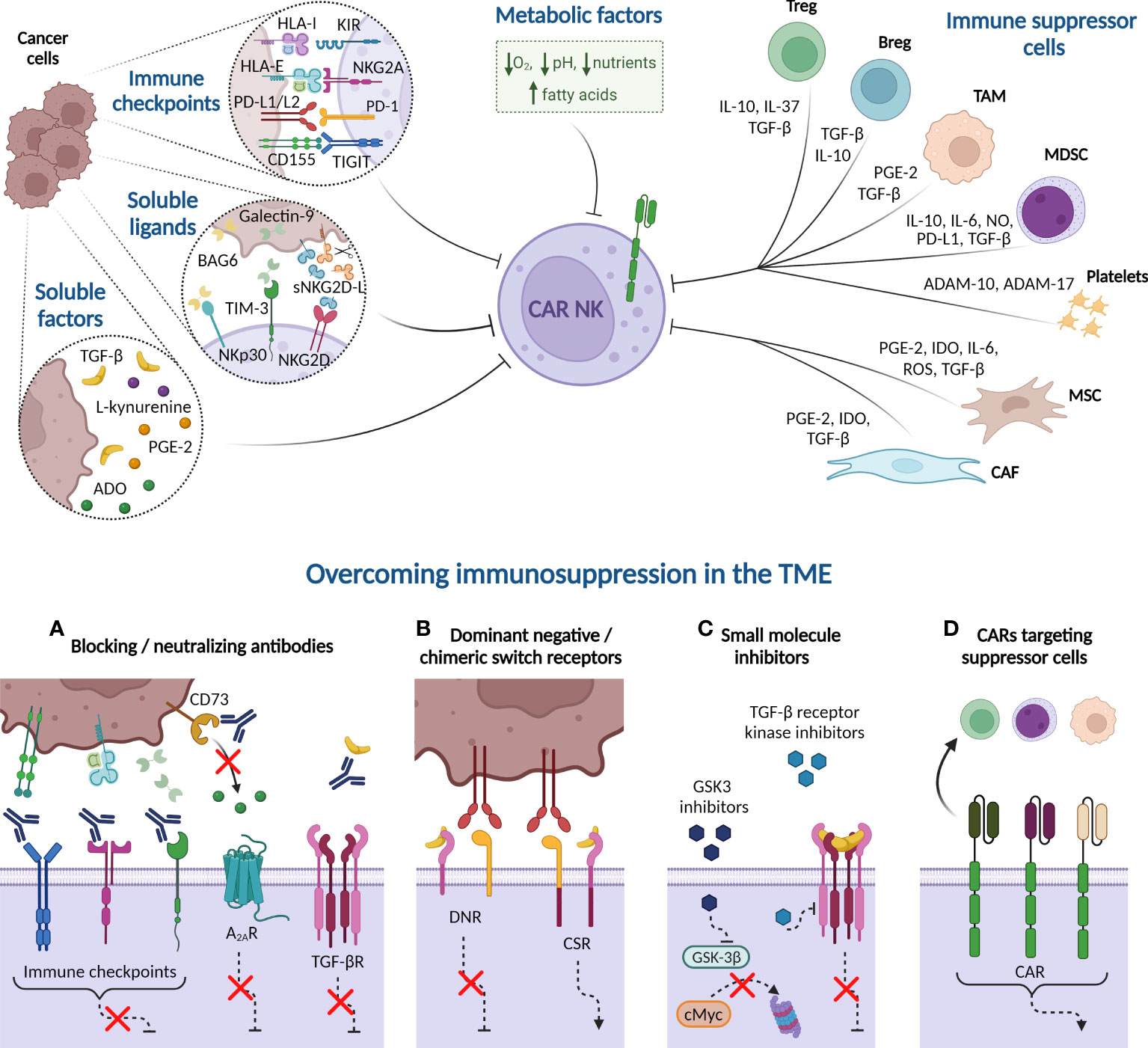
Figure 3 Immune suppressive tumor microenvironment (TME) factors potentially involved in CAR-NK cell dysfunction. Cancer cells express immune checkpoint ligands in their plasma membrane that mediate an inhibitory interaction with NK cells. Besides, tumor cells may suppress NK cell function by releasing soluble ligands to the milieu, such as BAG-6, galectin-9, and soluble NKG2D-L (sNKG2D-L), as well as other soluble factors, including cytokines, such as transforming growth factor-β (TGF-β), enzymes and metabolites. Many of these soluble factors are also produced by immune cells present in the TME, such as Tregs, Bregs, tumor-associated macrophages (TAM), and myeloid-derived suppressor cells (MDSC). Platelets, in turn, secrete the metalloproteinases ADAM-10 and ADAM-17 that prompt NKG2D-L shedding. Other non-immune cells, such as derived-mesenchymal stromal cells (MSC) and cancer-associated fibroblasts (CAF), also produce indoleamine 2, 3 dioxygenase (IDO) or reactive oxygen species (ROS) that reduce NK cell activity. Additionally, hypoxia, high concentrations of fatty acids, nutrient deprivation, and acidity, among other metabolic factors, contribute to generate a complex immunosuppressive TME that hampers the NK cell effectiveness against hematologic malignancies. Several strategies can overcome the immunosuppression mechanism from TME. (A) Blocking antibodies targeting immune checkpoints prevent the inhibition of NK cell cytotoxicity. Other receptors, such as adenosine A2A receptor (A2AR) also disable NK cell function when binds to extracellular adenosine (ADO). Blockade of CD73 ectoenzyme, which synthetizes ADO, reduces the levels of this metabolite in the TME, therefore increasing NK cell killing activity. Furthermore, anti-TGF-β neutralizing antibodies impede the NK cell suppressive effect unleashed by the interaction of this cytokine with its receptor (TGF-βR). (B) Dominant-negative receptor (DNR) expression hinders the inhibitory signaling triggered by PD-1 and TGF-βR in the presence of PD-L1/L2 or TGF-β, respectively. Chimeric switch receptors (CSR) constitutes another approach based on replacing these negative signals by activating ones, through intracellular domains exchange, reverting the outcomes in NK cell activity. (C) Small molecule inhibitors directed against GSK-3β impact on NK cell metabolism and improve their cytotoxic potency. Other inhibitors are engineered to inhibit the kinase activity of TGF-βR. (D) CAR constructs are designed against molecules expressed in immune suppressor cells to eliminate them from TME. HLA-I, HLA class I histocompatibility antigen; KIR, Killer-cell immunoglobulin-like receptor; HLA-E, HLA class I histocompatibility antigen, alpha chain E; PD-L1, Programmed Death ligand-1; PD-1, Programmed Death 1; TIGIT, T cell immunoglobulin and ITIM domain; BCL2-associated Athanogene 6 (BAG-6); sNKG2D-L, soluble natural killer group 2D ligands; TIM-3, T cell immunoglobulin and mucin-domain containing-3; PGE-2, prostaglandin E2; NO, nitric oxide; A Disintegrin And Metalloproteinase (ADAM). Created with BioRender.com.
Classical inhibitory receptors of NK cells, such as KIRs and NKG2A act as immune checkpoints (see major immune checkpoints in Figure 1). In many malignancies, tumor cells downregulate the expression of classical HLA-I molecules, preventing Ag-dependent recognition by T cells but allowing NK cell “missing-self” activation (170, 171). By contrast, in some hematological tumors such as MM, classical HLA-I expression is elevated in advanced stages (172), inducing KIR-mediated NK cell inhibition. In preclinical studies, the use of anti-KIR antibodies recovers NK cell killing activity (173, 174), but the administration of pan-KIR2D antibodies, such as IPH2101 or lirilumab (IPH2102), has not shown single-agent activity in clinical trials of MM (175) or AML (176). Unexpectedly, IPH2101 infusion in smoldering MM patients induces NK cell anergy due to the removal of KIR2D surface molecules through trogocytosis (175). In addition, a phase II trial reports that lirilumab administration as maintenance therapy for elderly AML patients fails to improve leukemia-free survival (LFS) (NCT01687387) (176). For that reason, combined therapies with drugs, such as IMiDs, or other immune checkpoints blocking antibodies are being examined (177). IPH2101/lenalidomide dual therapy has shown a synergistic effect against MM in preclinical studies and preliminary evidence of efficacy in phase I clinical trial (NCT01217203) (174, 178). Similarly, the same synergic strategy is being evaluated in studies of anti-KIR antibodies combined with anti-PD-1 and anti-CTLA-4 blocking antibodies against solid tumors (NCT01750580, NCT01714739, NCT03203876). Many studies have focused on the NKG2A receptor that triggers inhibitory signaling upon binding HLA-E, a non-classical HLA-I molecule overexpressed in several tumors (179). Approximately half of peripheral blood NK cells from healthy donors express NKG2A (180, 181) and their levels increase after ex vivo stimulation with cytokines (182). NKG2A trapping in the endoplasmic reticulum/Golgi by smartly designed protein expression blockers (PEBLs) (183) or the treatment with anti-NKG2A blocking antibodies, broadens the oncolytic activity of NK cells against hematological tumors such as AML, CLL or lymphomas (184). Monalizumab (IPH2201), a humanized monoclonal anti-NKG2A blocking antibody, has also shown promising preclinical results against hematologic and solid tumors (185, 186), which have prompted to evaluate its efficacy in clinical trials either as monotherapy or combined with other treatments (187). Other strategies to inhibit the NKG2A/HLA-E axis are based on NKG2A downregulation in NK cells by dasatinib (188), or the reduction of HLA-E levels in tumor cells, by drugs such as bortezomib, dinaciclib or selinexor, tested in vitro on MM, AML, and lymphoma, respectively (189–191). Mechanistically, dasatinib treatment inhibits p38 mitogen-activated protein kinase (MAPK), diminishing the import of NKG2A transcription factor GATA-3 to the cell nuclei (188). Meanwhile, HLA-E downregulation by bortezomib is induced through endoplasmatic reticulum-stress unleashed by proteasome inhibition (189). Constant de novo protein synthesis is essential for maintaining HLA-E surface expression levels. Consequently, selinexor induced degradation of nuclear export protein exportin-1 (XPO1), which regulates the transport of ribosomal subunits from nucleus to cytoplasm, decreases the number of HLA-E molecules in plasma membrane, mainly because of the reduction of HLA-E binding substrates (191). Although the action mechanism of dinaciclib has not yet been elucidated, the administration of this cyclin-dependent kinase (CDK) inhibitor prior to NK cell infusion further boosts their killing activity in an AML mouse model (190). Due to their antitumor function, bortezomib, dinaciclib, and selinexor are being used in different hematological pathologies, but besides, their administration as a pretreatment before adoptive cell therapy could enhance CAR-NK cell efficacy.
Combining CAR-T cell therapy with Programmed Death (PD)-1/PD-L1 axis blockade has improved clinical responses in hematological tumors (192). Although NK cell inhibition by PD-1/PD-L1 checkpoint has been reported (193, 194), ex vivo expanded NK cells exhibit very low PD-1 expression levels, hence the combination with pembrolizumab does not improve in vitro cytotoxicity (195–197). However, some studies suggest that this immune checkpoint may become important post-NK therapy infusion because expanded NK cells increase PD-1 expression in the presence of tumor cells, and IFN-γ produced by NK cells augments PD-L1 expression in a lung cancer mouse model (198). In addition, trogocytosis has been described recently as a new mechanism whereby NK cells obtain PD-1 from tumor cells (199). A phase II clinical trial (NCT04847466) combining PD-L1 CAR-NK cells with pembrolizumab and N-803 is currently being studied in gastric and head and neck cancer. Similar to NKG2A, NK cell ex vivo expansion upregulates the expression of other exhaustion receptors, such as T cell Ig and mucin-containing domain-3 (TIM-3) (200) and T cell immunoreceptor with Ig and ITIM domains (TIGIT) (197). There is controversy regarding the role of TIM-3 in NK cell activity. Although most studies define TIM-3 as an NK cell checkpoint inhibitor, some papers show that TIM-3 interaction with its ligands (including Galectin-9) unleashes IFN-γ production or associate TIM-3 expression with a functionally licensed NK cell phenotype with higher cytotoxic activity (201, 202). A functional threshold that controls the activatory or inhibitory NK cell function of this receptor has been proposed to explain this discrepancy. Meanwhile, TIGIT recognizes poliovirus receptor (PVR or CD155), Nectin-2 (CD112), or Nectin-3 overexpressed in hematologic cancers (203, 204). The blockade of TIM-3 or TIGIT in preclinical studies improves NK cell cytotoxic potency against solid and hematologic malignancies (205–209) and currently neutralizing antibodies are being tested in several clinical trials (e.g. NCT04623216, NCT03489343, NCT04150965, NCT04354246, and NCT05289492). Nevertheless, the results obtained from these studies should be analyzed considering that TIM-3 and TIGIT are also expressed in T cells, making difficult to determine the precise involvement of the NK population in the patient response. Nectin-2 is also recognized by PVRIG (CD112R) a novel T cell inhibitory receptor that does not bind CD155 (210). Studies in AML suggest that PVRIG impacts NK cell cytotoxic activity against CD112highCD155low tumors, whereas, in those with overexpression of both ligands, TIGIT inhibitory signaling is predominant (211). Unlike in T cells, expression levels and inhibition relevance of other receptors such as LAG-3 or CTLA-4 remain unclear in NK cells (212, 213).
Additional molecules are still being included within the immune checkpoint category. For instance, signal regulatory protein α (SIRPα) is a myeloid-lineage receptor recently described in T and NK cells, whose expression increases after IL-2 stimulation (214, 215). SIRPα deficiency or blockade enhances NK cell cytotoxicity against CD47-expressing tumor cells (214), which include hematological malignancies, such as diffuse large B-cell lymphoma (216). Recently, it has been described that many tumor proteins are hypersialylated, a modification that confers cancer cell resistance to the cytotoxic activity of different immune populations through their recognition by inhibitory sialic acid-binding immunoglobulin-like lectin (Siglec) receptors (217). Deletion of Siglec-7 or blockade of Siglec-9 restores NK cell cytotoxic activity against MM (218) or CML cells, respectively (219).
Taken together, not all the immune checkpoints are induced at the same levels in ex vivo expanded NK cells neither are they similarly relevant in the modulation of NK cell antitumor activity. The balance of activating and inhibitory signals regulates NK cell function; therefore, more efforts are needed to evaluate the impact of each immune checkpoint expression in the presence of CAR stimulation to direct the strategies to improve CAR-NK therapy.
Soluble factors from TME contribute to heightening NK cell inhibition. Tumor cells release soluble ligands that can bind activating and inhibitory receptors expressed in NK cells to promote their dysfunction. Soluble NKG2D-L (sNKG2D-L) generated by proteolytic shedding decrease the expression of NKG2D, reducing NK cell antitumor potency (220, 221). Interestingly, sNKG2D-L do not impair the effectiveness of NKG2D-CAR NK92MI nor NKG2D-CAR T cells against MM and leukemia cells, respectively (222, 223). Specific antibodies directed against the MICA α3domain inhibit the shedding of this NKG2D-L, allowing NK cell-mediated cytotoxicity and thus avoiding tumor evasion (224). CAR constructs designed to recognize the same MICA/B domain have shown efficacy against leukemia in iPSC-derived NK cells and are currently being studied in a dual-CAR in combination with BCMA specificity for MM (225, 226). Likewise, the presence of BCL2-associated Athanogene 6 (BAG-6), one of the NKp30 ligands, in the tumor cell membrane or exosomes stimulates NK cell antitumor activity whereas its soluble form hampers NK cell function (227, 228). Other soluble ligands such as galectin-9 have been found at high levels in the blood plasma of AML patients (208). Galectin-9 effect has not been studied in CAR-NK cells yet, but its blockade reestablishes CAR-T cell antitumor responses (229).
Additionally, well-known soluble factors present in the TME of most cancers, such as certain interleukins, enzymes, and metabolites impact NK cell effectiveness. Most of them are released not only by tumor cells but also by immune suppressor cells that coexist in the tumor niche. That is the case of IL-37, produced by Tregs, which exerts an inhibitory action over canonical NK cells while adaptive NK cells are highly resistant (230). High concentrations of other suppressive cytokines, such as IL-6, IL-10, and TGF-β have been widely reported in hematologic tumors (231–234). Concerning NK cells, TGF-β favors tumor development by decreasing activating receptor expression, cytokine production, and metabolism (235–237). Alongside the anti-TGF-β neutralizing antibodies or engineered NK cells to knock-down TGF-β receptor expression, other strategies such as small molecule receptor kinase inhibitors can be implemented for CAR-NK cell therapy in hematological cancers (238, 239). For instance, ex vivo expanded NK cells restored their in vitro anti-AML activity by the addition of TGF-β receptor kinase inhibitor LY2157299 (235). TGF-β is affected by prostaglandin E2 (PGE-2), which is secreted by stromal cells derived from lymph nodes (LN) or bone marrow (BM) as well as by some tumor cells, such as leukemic blasts, sustaining MM and leukemic cell proliferation (240–242). The binding of PGE-2 to its receptor in monocytes induces an “MDSC-like” phenotype that enhances TGF-β production. In this manner, PGE-2 can interfere with NK cell function in two different ways, directly, joining to its receptors in NK cells, and indirectly, enhancing TGF-β concentrations in the TME (243, 244). Furthermore, TGF-β raises the expression of CD39 and CD73 in cancerous and other cell types (245, 246). These ectoenzymes are involved in adenosine (ADO) production from ATP/AMP, promoting tumor proliferation. ADO inhibits NK cell activity and expansion and reduces its metabolism (247–249). In fact, combination therapy of NKG2D-CAR-NK cells with anti-CD73 blocking antibody increase NK-92 cell killing potency against CD73+ cancer cells, reducing ADO levels in the TME (250). By the same token, the activity of indoleamine 2,3-dioxygenase (IDO) downregulates NKp46 and NKG2D and restricts NK cell cytotoxicity (251). IDO is an enzyme that degrades tryptophan to L-kynurenine and its inhibition reestablishes NK cell activity (252). IDO expression has been associated with poor prognosis in AML, diffuse large B-cell lymphoma as well as solid tumors (253–256). Apart from the effect of the tryptophan-derived suppressive catabolite L-kynurenine (251), IDO contributes to decreasing NK cell proliferation by depriving tryptophan in the tumor milieu. Similarly, high concentrations of arginase, released by immune suppressor cells, decrease arginine levels, essential for T and NK functions, which can be restored by arginase inhibitors (257, 258).
In addition to the aforementioned strategies to prevent NK cell inhibitory interactions with cancer cells by immune checkpoints or with immunosuppressive soluble factors, dominant-negative receptors have also been designed (259–261). The expression of a dominant-negative TGF-βRII (DNRII) in UCB-NK cells allows the maintenance of the NKG2D and DNAM-1 expression levels and glioblastoma cell-specific lysis in the presence of TGF-β (260). Other approaches are based on converting inhibitory into activating signals by chimeric switch receptors (CSR), also known as chimeric costimulatory converting receptor (CCCR). Tested so far in the context of solid tumors, CSR directed to PD-1, B7-H3 or TGF-β increases the anti-tumorigenic capacity of NK92 cells (262–264). Moreover, TGF-βRII-NKG2D receptor expression promotes NK92 chemotaxis to the TGF-β-expressing tumor cells and inhibits the differentiation of CD4+ T cells to Tregs (264).
Immune suppressor cells present in the TME promote tumor proliferation while reducing NK cell function through direct cell-cell contact or by releasing soluble factors. The detrimental effect of Tregs on NK cells through these mechanisms and the previously mentioned competition for IL-2 are widely known (67, 265, 266). Other immune cells that contribute to NK cell suppression such as Bregs, MDSCs, and tumor-associated macrophages (TAM), mainly M2 phenotype, are highly represented in the TME (267–271). An additional impediment to NK cell function is mediated by platelets which stunt NK cell recognition (272, 273). In addition, platelets favor NKG2D-L shedding due to A Disintegrin And Metalloproteinase (ADAM)-10 and ADAM-17 release. Platelets’ blockade enables NK cell access to the tumor and recovers activating receptor expression on the NK cell membrane (274–279). Furthermore, non-immune cells present in the TME, such as derived-mesenchymal stromal cells (MSC) and cancer-associated fibroblasts (CAF), strengthen NK dysfunction because they impair degranulation, reduce perforin and cytokine secretion and hinder antitumor efficacy (280, 281).
Such evidence have led to developing CAR therapies to eliminate these suppressor cells. In hematological cancers, CAR-NK cell products have been designed to target Tregs (directed against CD25 or CD38) (282, 283) or MSCs (against CD38) (284) to restore tumor-infiltrating NK cell activity. Interestingly, other CAR-NK cell strategies take advantage of the ligand shared expression between cancer cells and MDSCs, such as NKG2D-CAR-NK cells and PD-L1-CAR-NK cells that can kill both cell types (285, 286). Similarly, dual FAP/SLAMF7-CAR-T therapies are novel approaches for targeting CAFs, surmounting the suppressor function of these cells over CAR effectors, and eliminating MM cells at the same time (287). Meanwhile, TAM-specific CAR-T cells, targeting folate receptor β (FRβ), reduce the number of TAMs in the TME while decreasing tumor cells’ proliferation rate. Therefore, this adoptive treatment has been suggested to be administered before tumor-directed CAR-T cells (288). These two last approaches in CAR-T cells could also be implemented to create a more benign milieu for CAR-NK cell therapy.
Hypoxia and metabolic factors, such as nutrient deprivation, and acidity also generate an unfavorable microenvironment that compromises NK cell antitumor activity. Hypoxia is a well-described protumor factor in solid cancers and a feature of LN and BM microenvironment (289). Low O2 concentrations hamper the upregulation of activating receptors in NK cells (290, 291) while increasing MICA shedding (292, 293) and granzyme B degradation by autophagy in malignant cells (294). Additionally, hypoxia induces CD73 expression through hypoxia-inducible factor-1α (HIF-1α) (295), contributing to achieving high ADO levels in the TME. Upon stimulation, NK cells suffer a metabolic reprogramming regulated by mammalian target of rapamycin complex 1 (mTORC1), elevating oxidative phosphorylation (OXPHOS) and glycolysis and increasing the expression of glucose and amino acid transporters (296–299). Tumor cells require a greater amount of nutrients to proliferate, thus they compete for these resources with surrounding immune cells. Depriving NK cells of glucose or certain amino acids hinders the NK cell metabolism by inhibiting mTORC1 and cMyc pathways (296, 300). cMyc is key for NK cell metabolic responses because is involved in regulating the expression of glucose transporters and glycolytic enzymes (300). The levels of cMyc are initially controlled by mTOR and sustained through amino acid availability, to counteract its rapid rates of proteolysis by glycogen synthase kinase (GSK)-3β (301). Furthermore, GSK-3β inhibition increase ex vivo expanded NK cell antitumor activity in an AML mouse model (302), adding another pathway to aim CAR-NK cell improvement. Because cancer cells obtain energy mainly through high rates of glycolysis converting glucose into lactic acid, the latter is accumulated in the TME contributing to its acidification. Lactic acid diminishes NK cell cytokine production and induces apoptosis, in addition to increasing the number of MDSC (303, 304). In a lymphoma mouse model, oral delivery of bicarbonate restores NK cell IFN-γ production and tumor infiltration, although other tumor-dependent mechanisms preclude NK cell cytotoxicity (305). Additionally, tumor cells increase lipid metabolism, and fatty acid exposure hampers cytokine production and cytotoxic activity of NK cells (306). High levels of intracellular lipids trigger PPAR-γ/PPAR-δ signaling that aids NK cells in adapting to the altered milieu but inhibits the sterol regulatory element binding protein (SREBP). SREBP has been reported to be essential to regulate glucose metabolism (307, 308).
Considering the above, soluble factors, suppressor cells, as well as metabolic factors can induce NK cell dysfunction and should be taken into account when designing a new CAR-NK therapy. Overcoming the TME could guarantee longer and deeper CAR-NK responses in cancer patients. Studies with CAR-T cells and non-engineered NK cells have provided relevant information to take the first step, but specific assays with CAR-NK cells in hematologic tumors must be performed to maximize the results.
One of the challenges of CAR immunotherapy lies in limited trafficking and homing ability to reach the BM and LNs which is ultimately related to the exerted efficacy of adoptively transferred NK cells. Clinical studies suggest that improved trafficking of adoptively infused NK cells into BM niches is associated with better control of the disease in AML patients (143, 144), which could apply to other malignancies residing in the BM. A variety of strategies to maintain and/or enforce the expression of chemokine or adhesion receptors in CAR-NK cells are now being explored in preclinical models to improve their localization into the BM and LN.
CXCR4, CXCR3, CCR3, CCR5, and CX3CR1 are the main chemokine receptors expressed by NK populations that contribute to NK distribution in response to chemokines present in the TME (309). CXCL12 chemokine, the CXCR4 ligand, is highly expressed in MM by endothelial and BM stromal cells (310, 311), as well as in leukemia and lymphoma (312). Similarly, the BM niche of ALL (313), AML (314), lymphoma (315, 316), and MM patients is frequently characterized by upregulation of CXCL9 and/or CXCL10 (317). Of note, CXCL9 and CXCL10 (CXCR3 ligands) are considered immunosuppressive chemokines involve in MM resistance mechanisms (318). Additionally, macrophage migration inhibitory factor (MIF) can also bind to CXCR4 and CXCR7 and it was also found in high levels in MM BM (319). CCL19 and CCL21, chemokine ligands of CCR7, participate in the entry of CLL cells (320) into LNs, where they are found (321, 322).
Taking into account the high levels of chemokines found in the TME of hematologic malignancies, modifying the expression of chemokine receptors in adoptively transferred NK cells seems to be a suitable strategy. CXCR4 expression levels are higher in UCB-derived NK cells than those in PB-NK cells, suggesting a better BM homing ability (323). CXCR4 is generally downregulated during ex vivo activation of NK cells (317, 324–327), resulting in decreased homing to BM. Thus, some strategies are studying how to improve NK trafficking to this tumor niche. Human CD19-CAR-NK cells modified to overexpress CXCR4 through bicistronic lentiviral transduction augmented more than two-fold the migration to CD19+ tumor cells compared to huCAR19 NK cells (328). Ectopic expression of gain-of-function (GOF) mutation in CXCR4 (CXCR4R334X) via mRNA transfection on ex vivo expanded NK cells resulted in improved chemotaxis toward CXCL12 (also known as SDF-1α) and superior in vivo migration to BM (324). Similarly, the overexpression of the CXCR4R334X receptor via electroporation on ex vivo expanded BCMA-CAR-NK cells was effective in enhancing the in vivo migration toward the BM and also eliminating myeloma cells in mice with prolonged survival as compared with only anti-BCMA-CAR-NK cells (327). Conversely, CXCR3 can be responsible for NK mobilization outside the BM (317, 329). This negative role of CXCR3 can be reversed by genetic deletion of Cxcr3 gene or by using an anti-CXCR3 blocking mAb to increase BM NK cell infiltration (317).
CCR7 is known to promote NK cell migration to LNs in response to chemokine ligands CCL19 and CCL21 (330). Its downregulation following ex vivo expansion has also been reported (325) but can be restored upon treatment with IL-18 (331). Besides, the transfer of CCR7 receptor from antigen-presenting cell (APC) to NK cell through trogocytosis can occur when co-cultured (332). In this line, LN homing of adoptively transferred NK cells can be enhanced by the ex vivo acquisition of CCR7 via trogocytosis from K562 feeder cells in athymic nude mice (333). Similarly, CCR7 mRNA-electroporation has been used to promote migration toward CCL19 and CCL21 chemokines (330, 334). CCR7 transfected anti-CD19 CAR-NK cells enhance their capacity to kill CD19+ tumors up to 5-fold increase and their migratory capability in response to CCL19 and CCL21 chemokines up to 6-fold increase (334). Schomer et al. have recently corroborated in CCR7 engineered CD19 CAR t-haNK cells an improved migration toward LN chemokine CCL19 compared to only CD19-CAR t-haNK cells in an in vivo xenografted NSG lymphoma model (335).
A novel strategy knocking-down CCR5 in ex vivo expanded NK cells reduces sequestration by the liver following i.v. infusion of adoptive immunotherapy which, in turn, favors their presence in the circulation (325). Given that CCR5 expression augments upon ex vivo expansion of adoptively transferred NK cells, this strategy bolsters the capacity to redirect NK trafficking in vivo. Levy et al. have opened the way to explore whether combining the overexpression of CXCR4 and CCR5 deletion would boost NK cell immunotherapy migration to the tumors residing in the BM (325), or even into the LN.
Apart from strategies modulating the expression of chemokine receptors, BM homing can be enhanced by promoting the interaction of NK cells with adhesion molecules like E-selectin. For instance, the treatment of NK-92MI cells with human fucosyltransferase 6 (FUT6) and GDP-fucose creates cell-surface E-selectin ligand sialyl Lewis X (sLeX) to improve migration to BM and increase the killing of B-lymphoma cells (336). Besides, while augmenting NK cell expansion in culture, nicotinamide (NAM) leads to CD62L (L-selectin) upregulation (337) resulting in improved in vivo migration of adoptively transferred NK cells to multiple organs including the BM (338, 339).
Most migration studies are being carried out for solid cancers as NK penetration into the tumor site is more critical than in liquid malignancies. Nevertheless, many of the chemokine ligand-receptor axes that are studied in solid tumors can be implemented in hematologic malignancies. An NK-cell-recruiting protein-conjugated antibody (NRP-body) that includes a cleavable CXCL16 molecule, the CXCR6 ligand, has been used to increase the trafficking and infiltration of expanded NK cells into pancreatic tumors (340). Additionally, CXCR1 and CXCR2 chemokine receptors display a high affinity for IL-8 (CXCL8), secreted by tumor cells in different cancer types. Although NK cells lose expression of these receptors upon activation and expansion (341, 342), their upregulation promotes enhanced attraction to tumor sites. Ng et al. show that CAR-NK cells expressing an NKG2D-CAR and CXCR1 chemokine receptor augmented their migration ability toward IL-8-secreting ovarian tumors and enhanced in vivo tumor control (341). Similarly, CXCR2-transduced NK cells have an increased ability to migrate toward renal cell carcinoma tumors in a ligand-specific manner, resulting in increased killing of target cells (342). As long as CXCL16 is found in the BM at high levels (343) and CXCL8, the CXCR1, and CXCR2 ligand is significantly elevated in MM (344, 345), CLL (346), AML, and MDS patients (347), the targeting of CXCL16-CXCR6 and IL8-CXCR2/CXCR1 pathways should be studied in depth for hematologic malignancies.
Genome editing promotes desired modifications in the specific locus of the genome by the use of engineered nucleases that activate endogenous DNA repair mechanisms. Although different engineered nucleases have been developed, the CRISPR/Cas9 system has completely revolutionized the field (348, 349).
CRISPR/Cas9 type II system, the most widely applied, is composed by an RNA-guided endonuclease (Cas9 nuclease) and a guide RNA (gRNA) that will direct the Cas9 nuclease to the desired locus. Cas9 and gRNA complex can specifically bind to the target sequence when protospacer adjacent motif (PAM) sequence is present (350) and generate a double-strand break (DSB) in the aimed region. Then, this DSB can be repaired by the endogenous DNA repair mechanisms of the cells, and consequently lead to the interruption of the gene, when Non-Homologous End Joining (NHEJ) or microhomology-mediated end joining (MMEJ) mechanisms occur, or the correction of a specific sequence when a donor DNA molecule with homology regions is present by homology-directed repair (HDR) (351). Importantly, NHEJ is the preferred mechanism to repair DSBs in mammalian cells, complicating the application of gene editing mediated by HDR in human cells (352).
Moreover, gene modification of NK cells using gene therapy delivery vectors such as plasmids and lentiviral vectors is particularly arduous, due to innate immune mechanisms that confer them resistance to genetic modifications (353, 354). This indicates that knock-in approaches, in which the delivery of a donor template requires the use of different types of vectors or plasmids will be challenging in these cells. In contrast, knock-out strategies using pre-transcribed gRNA and Cas9 protein in ribonucleoprotein complexes can potentially avoid problems associated with NK resistance to viral transduction and double-strand DNA (355–357).
The application of gene editing constitutes nowadays the great hope to improve NK efficiency and persistence against different types of tumors. To this aim different studies have been focused on identifying negative regulators that could be targeted to modulate immune function and enhance NK and CAR-NK potency, either by increasing cytotoxicity, improving metabolism and in vivo persistence, or by overcoming mechanisms of functional exhaustion triggered by inhibitory immune checkpoints and TME (Figure 4).
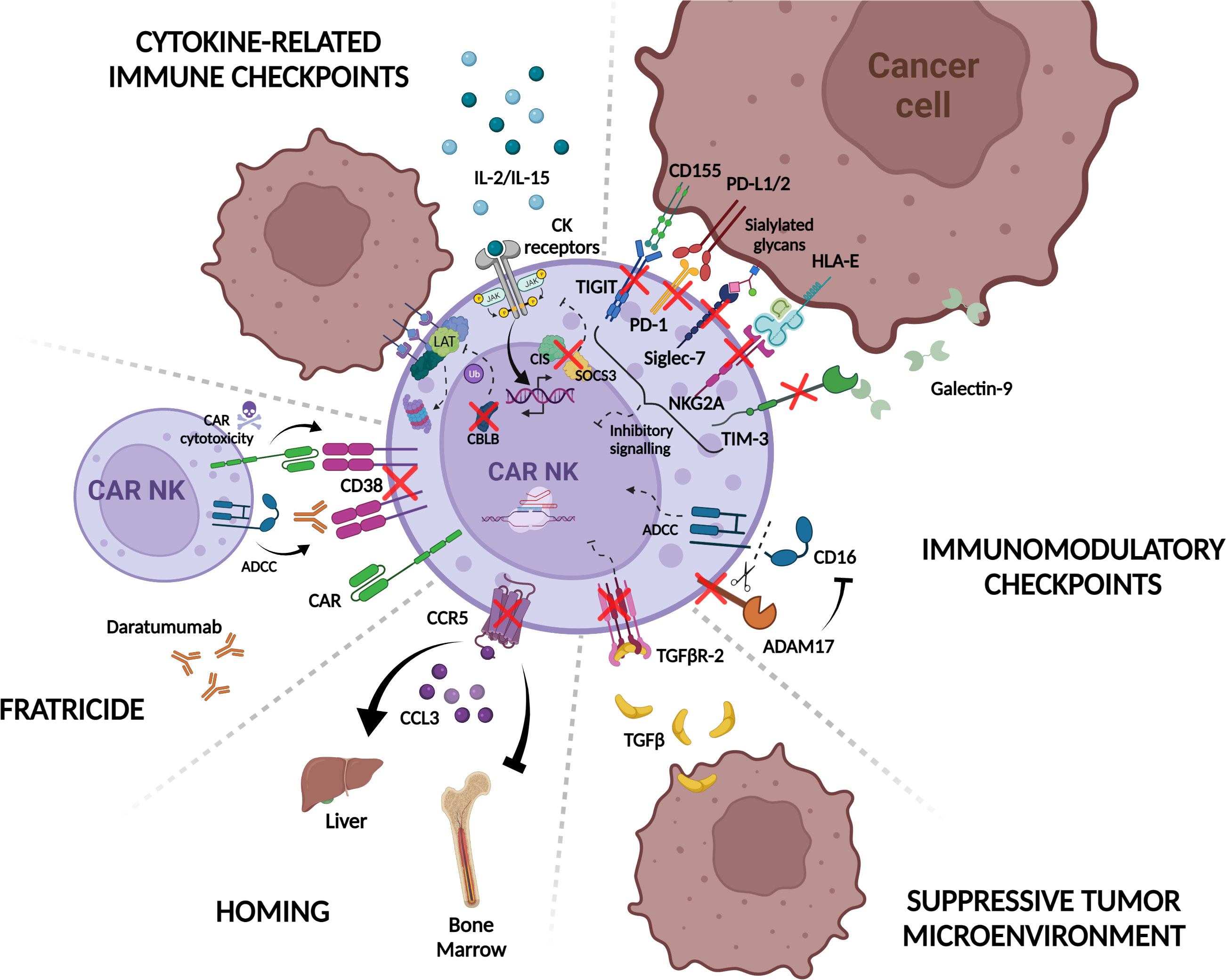
Figure 4 CRISPR/Cas9 knock-out strategies to improve CAR-NK immunotherapy. CAR-NK function and cytotoxicity are modulated by intrinsic mechanisms in NK cells. For example, engagement of tumor ligands with NK immunomodulatory checkpoint receptors such as TIGIT, PD-1, NKG2A, TIM-3, and Siglec-7, inhibit CAR-NK cell response to target cells. ADAM17 also restrains NK cell ADCC response by shedding CD16 receptor from the NK cell surface. Additionally, in response to cytokine signaling, the expression of internal checkpoints including CLBL, SOCS3, and CIS regulate NK activation and immune synapsis formation with tumor cells. The suppressive tumor microenvironment contributes as well to CAR-NK inhibition through the release of suppressive factors like TGF-β. In this context, CAR-NK potency is attenuated and less effective against tumor cells. CAR-NK cell homing is also regulated by chemokine receptors such as CCR5 that mediates homing to the liver in response to CCL3 reducing CAR-NK efficacy against bone marrow-resident tumors. Another problem that CAR-NK manufacturing can encounter is fratricide either by expression of the CAR-targeted molecule on the surface of the effector cell or the use of monoclonal antibodies that bind to NK cells and induce “self-killing” through ADCC. Ablation of different NK cell proteins implicated in these pathways by the use of the CRISPR/Cas9 system (red crosses) would overcome the aforementioned limitations and result in more potent, persistent, and tumor-directed CAR-NK effectors for their use in adoptive immunotherapy. PD-1, Programmed Death 1; TIGIT, T cell immunoglobulin and ITIM domain; TIM-3, T cell immunoglobulin and mucin-domain containing-3; NKG2A, natural killer group 2A; PD-L1/2, Programmed Death ligand-1/2; HLA-E, HLA class I histocompatibility antigen, alpha chain E; ADAM-17, A disintegrin and metalloprotease 17; TGF- β, Transforming growth factor beta; TGFβR-2, Transforming growth factor beta receptor type 2; CCL3 Chemokine (C-C motif) ligand 3; CCR5, C-C chemokine receptor type 5; CAR, chimeric antigen receptor; ADCC, Antibody-dependent cellular cytotoxicity; CK, cytokines; CIS, cytokine-inducible SH2-containing protein; SOCS3, suppressors of cytokine signaling; LAT, linker for activation of T cell; CBLB, Casitas B-lineage lymphoma protooncogene B Created with BioRender.com.
One of the main strategies to improve CAR-NK persistence is focused on targeting inhibitory immune checkpoints such as PD-1/PD-L1 axis and in fact, PD-1 knockout (KO) in NK cells increased their antitumor activity in a xenograft model of ovarian cancer (358). The same authors tested the efficacy of PD-1 KO NK cells against CML and AML cell lines in vitro (358). Despite controversy about the low expression of PD-1 in ex vivo expanded NK cells, the enhanced response with PD-1 blocking antibodies in MM (359), suggests that disruption of this receptor could be a promising strategy to potentiate CAR-NK efficacy against hematological malignancies.
TIM-3 is another checkpoint receptor expressed by NK cells as a marker of dysfunction when TIM-3 positive NK cells encounter tumors expressing TIM3 ligands such as glioblastoma (360, 361) or AML blasts (208). Consequently, TIM-3 KO NK cells mediated improved cytotoxicity in vitro (362). A similar CRISPR/Cas9-based strategy has been applied for Siglec-7 receptor, which triggers NK inhibition when binds to certain sialylated glycans expressed on tumor cells, resulting in enhanced NK antitumor efficacy against Siglec-7 ligand-expressing MM cell lines (218).
Several groups have shown that TIGIT can inhibit immune response mediated by T and NK cells, leading to tumor escape. Thus, blocking TIGIT with mAbs is translated to an increase in NK persistence and antitumor capacity (363). More recently, TIGIT KO by gene editing has also been performed (355, 356). Although functional studies in TIGIT KO NK cells have not been described in this study one can envision that similar results could be obtained.
Targeting inhibitory signals in NK cells, by the use of KIR (NCT01714739 and NCT01750580) or NKG2A inhibitors (NCT02331875) is another interesting strategy to increase NK efficiency against hematological and solid tumors. Moving to CRISPR/Cas9 mediated editing approaches, targeting HLA-E/NKG2A axis by knocking out NKG2A-encoding killer cell lectin like receptor C1 (KLRC1) in CIML NK or NK cells has been described to induce a heightened response measured by IFN-γ production or increase cytotoxicity against AML or primary MM cells, respectively (364, 365). However, in the context of CAR-NK cells, it is still not clear whether ablation of this inhibitory receptor would provide an advantageous feature to the effector cells, considering the results indicating that NKG2A-KO CAR-NK92 cells and iPSC-derived CAR-NK cells do not show enhanced in vitro cytotoxicity against both solid and liquid tumor cell lines (263, 366). These results suggest that NKG2A not only constitutes an inhibitory receptor but also impacts during NK “education” process (366).
Alternatively, strategies focused on blocking regulators of NK activators such as ADAM-17 that cleaves CD16a, the NK cell-mediated ADCC activating receptor, have been tested to enhance antitumor response against a Burkitt’s lymphoma cell line in combination with monoclonal antibodies (358, 367).
To avoid NK cell exhaustion, targeting cytokine-related immune checkpoints is another interesting approach. Several proteins including Suppressors of cytokine signaling (SOCS1–7 and CIS) downregulate cytokine signaling via JAK/STAT pathway in NK cells. These receptors are induced by cytokines such as IL-2 and IL-15 (368, 369) which are commonly used in NK in vitro expansion methods as it has been mentioned above, and consequently, their interruption could increase activity and persistence of NK cells. Studies from different groups have shown that the ablation of CISH in NK cells increased their cytotoxic properties (357) and even improved their metabolic fitness (94). In the context of CAR-NK cells, Rezvani’s group also showed an enhanced expansion capacity and cytotoxicity against leukemic cell lines when CD19 CAR-NK cells co-expressing hIL15 were used and demonstrated that this effect is in part related to an increase in the metabolic activity of CAR-NK cells (93). In a similar way, targeting SOCS3, another suppressor of cytokine signaling, resulted in NK cells with higher proliferation capacity and antitumor capacity (370). Another cytokine-induced intracellular checkpoint that has been targeted in NK cells is CLBL (Casitas B-lineage lymphoma pro-oncogene-b), an E3 ubiquitin ligase that mediates degradation of LAT protein, impairing the immunological synapse between NK cells and target cells (371). CLBL KO in placental-derived NK cells increased their cytotoxic potential against liquid tumors in vivo (372).
One of the greatest challenges in CAR therapy, especially in solid tumors, is to skip the immunosuppressive microenvironment generated around the tumor. Although many different cell types and molecules can contribute to this effect, as we previously discussed, TGF-β seems to be a key regulator of TME (373) and most of the efforts in this area have been directed to target this signaling pathway. Several groups have already successfully edited NK cells to disrupt the TGFβ-R2 (TGF-β receptor type 2) in NK and CAR-NK cells which conferred them resistance to TGF-β inhibition in vitro (374) and therefore enhanced tumor control against difficult to treat tumors such as prostate adenocarcinoma (375) or glioblastoma (376). Importantly, this study also showed that knocking out CD9 and CD103, surface ligands in NK cells interacting with αv Integrins, can inhibit TGF-β1 release by glioblastoma stem cells and increase NK cytotoxic effect (376).
Another drawback to consider in CAR-T and NK therapy is that in some cases tumor antigens targeted by CAR molecules or monoclonal antibodies are not restricted to malignant cells, but they are also expressed in the therapeutic NK cells, resulting in fratricide and consequently, in a decreased response to treatment due to death of effector cells. Elimination of the CAR-targeted receptor in CAR-NK cells is crucial to develop an effective immunotherapy product, particularly in the context of hematologic tumors. This is the case of CD70-CAR-NK cells that constitute a promising therapy for both solid and hematologic malignancies but during in vitro feeder cell-dependent expansion and activation, CD70 is upregulated in NK cells which results in fratricide. By eliminating CD70 in NK cells using CRISPR/Cas9, fratricide-resistant cells were obtained without affecting their cytotoxic potency (129). Following a similar strategy, Gurney et al. have demonstrated that CD38 knock-out by CRISPR/Cas9 in CD38-CAR-NK cells results in a decreased cell death due to fratricide and a more potent cytotoxic response of CAR-NK cells against AML primary cells (127). In MM, where CD38 is highly expressed in malignant plasma cells, daratumumab treatment together with a concomitant CD38 expression in NK cells leads to a marked decrease in NK cell numbers due to fratricide. Consequently, CD38 KO NK cells blocked daratumumab-induced fratricide, showing an improved metabolic profile and consequently enhanced cytotoxic activity against CD38 expressing MM cell lines and primary cells (377, 378). In this setting, it is worth mentioning that a clinical trial in MM is already ongoing to test FT576, iPSC-derived BCMA CAR NK cells in which CD38 has been ablated to avoid mAb-mediated fratricide, in combination with other drugs (NCT05182073). These results pave the way to optimize other CAR-NK cells that are being developed for BM malignancies and target antigens that are also expressed in NK cells such as the NKG2D-L MICA or CD7 (97, 146, 379) as previously shown in CAR-T cells (124, 125).
As it has been mentioned above chemokine signaling plays an important role in CAR-NK biodistribution and determines antitumor efficacy depending on tumor location. In this context, CRISPR gene editing can be applied to modulate NK cell trafficking. Levy et al. showed that CCR5 disruption using CRISPR/Cas9 modified NK cell migration in vivo, which reduced trafficking to the liver and increased BM homing (325). This approach seems promising to re-direct CAR-NK cells and increase their potency against BM-resident hematological malignancies. Similar knock-out and even knock-in strategies could be applied to target or express other chemokine receptors on the surface of CAR-NKs and redirect them to the tumor site. This strategy has been followed in NK-92 cells where overexpression of chemokine receptor CXC chemokine receptor 2 (CXCR2) and IL-2 by HDR mediated gene editing increased NK92 migration to tumor sites and improved tumor growth inhibition in vivo in a human colon cancer model (380).
One of the advantages of using CRISPR/Cas9 system in comparison with previously designed nucleases is the potential of this editing tool to target several loci at the same time. This greatly increases the possibility to optimize NK and CAR-NK cell cytotoxicity and persistence by multiplex targeting. Some examples include the combination of CISH and TGFBR2 targeting in NK cells (381) which increases cytotoxic activity against different hematological tumors. However, this type of strategies must be considered in detail since targeting several genes can also decrease NK survival and increase the risk of off-targets, including the risk of translocations (355, 356) as has been previously observed in CAR-T cells (382).
Thanks to the CRISPR/Cas9 system versatility, knock-in strategies focused on the delivery of a specific CAR molecule in a desired region of the genome can be implemented to increase the safety of the strategy. Although the efficiency of this strategy is still suboptimal in comparison with knock out approaches due to the intrinsic lower efficiency of HDR versus NHEJ (352) and the NK reluctance to the different donor template delivery systems previously mentioned, recent studies demonstrated the feasibility to deliver CAR molecules to NK cells using CRISPR/Cas9 tools, resulting in an increased killing capacity of the effector cells when an EGFR-CAR was used against prostatic adenocarcinoma (375) and a CD33-CAR in an AML model (383). This will also allow the combination of different modifications to improve CAR-NK functionality to target challenging tumor cell types.
The recent description of new gene-editing tools that do not generate double-strand breaks in the genome, such as Base editing (384, 385) and Prime editing (386) are a promising strategy for the multiplex gene-editing approaches in CAR-NK cells. In both cases wild type Cas9 is substituted by a catalytically impaired Cas9 protein (dead Cas9 or Cas9 nickase) that will not generate DSBs in the genome, minimizing the risk of potential off-targets or translocations and improving the safety of the therapeutic product.
Over the last years, preclinical studies and preliminary clinical evidence indicate that “off-the-shelf” allogeneic CAR-NK therapy is a novel platform with a better safety profile than autologous CAR-T due to the low incidence of adverse events. Fast and high-quality responses achieved in a limited number of clinical trials with available results point to the efficacy of CAR-NK cell therapy in treating CD19+ relapsed/refractory tumors. Results of several ongoing clinical trials are awaited to clarify the broad applicability and long-term responses of CAR-NK in monotherapy or combinatorial approaches. Preclinical studies also anticipate the existence of CAR-independent challenges that hinder CAR-NK long-term function, leading to tumor resistance. Many approaches described in this review are under investigation to deeply understand these mechanisms and their involved targets, which differ between NK cell sources, triggering exhaustion and limited expansion and persistence in vivo. Although “optional” in allogeneic NK cells compared to allogeneic CAR-T therapy, CRISPR/Cas9 gene-editing constitutes a key strategy to obtain multi-engineered antitumor effectors to surpass these obstacles and outperform undesired effects of other combinatorial approaches, such as mAbs or inhibitors. Multiplex gene-edited CAR-NK products are exponentially growing in the field as an optimized “all in one” solution and have already become a clinical reality, offering an alternative to patients with limited treatment opportunities.
All authors contributed to the article and approved the submitted version. AV and JM-L conceptualized and designed the manuscript. All authors were involved in the literature collection and writing. AV, AG-O, and EC designed and created the figures, and AV and EM-M generated the clinical trial table. All authors contributed to the article and approved the submitted version.
This work was supported by the Research Institute Hospital 12 de Octubre (i+12) and by Instituto de Salud Carlos III (ISCIII)/Red Española de Terapias Avanzadas RICORS/TERAV -RD21/0017/0027 and RD21/0017/0030- Supported by European Union-NextGenerationEU. Plan de Recuperación Transformación y Resiliencia and grants PI18/01519 from Instituto de Salud Carlos III (ISCIII) and from the CRIS foundation to JM-L. AV and EM-M is supported by Research Institute Hospital 12 de Octubre (i+12). AG-O is supported by HIGEA 2019/0123 AIE project to JM-L. EC is supported by a Fellowship from Spanish Ministry of Science, Innovation and Universities (FPU18/02963). LC is supported by ICI20/00022 project to JM-L. JE is supported by a Fellowship from Instituto de Salud Carlos III (ISCIII) (IFI18/00034). AL is supported by Advanced Therapies National Network RICORS (Rd21/0017/0030).
JM-L has received grant support from BMS; and has performed consultancy work for BMS, Janssen, Novartis, GSK, Incyte, Roche, and Astellas. PR has licensed medicinal products and receives research funding and equity from Rocket Pharmaceuticals, Inc., Patents&Royalties, Research Funding.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse Large b-cell lymphoma. N Engl J Med (2019) 380(1):45–56. doi: 10.1056/NEJMoa1804980
2. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
3. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory Large b-cell lymphoma (Zuma-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol (2019) 20(1):31–42. doi: 10.1016/s1470-2045(18)30864-7
4. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. Kte-X19 car T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med (2020) 382(14):1331–42. doi: 10.1056/NEJMoa1914347
5. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory Large b-cell lymphomas (Transcend nhl 001): A multicentre seamless design study. Lancet (2020) 396(10254):839–52. doi: 10.1016/s0140-6736(20)31366-0
6. Munshi NC, Anderson LD Jr., Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med (2021) 384(8):705–16. doi: 10.1056/NEJMoa2024850
7. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a b-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (Cartitude-1): A phase 1b/2 open-label study. Lancet (2021) 398(10297):314–24. doi: 10.1016/s0140-6736(21)00933-8
8. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol (2022) 22(2):85–96. doi: 10.1038/s41577-021-00547-6
9. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-bcma car T-cell therapy Bb2121 in relapsed or refractory multiple myeloma. N Engl J Med (2019) 380(18):1726–37. doi: 10.1056/NEJMoa1817226
10. Shah NN, Fry TJ. Mechanisms of resistance to car T cell therapy. Nat Rev Clin Oncol (2019) 16(6):372–85. doi: 10.1038/s41571-019-0184-6
11. Chow VA, Gopal AK, Maloney DG, Turtle CJ, Smith SD, Ujjani CS, et al. Outcomes of patients with Large b-cell lymphomas and progressive disease following Cd19-specific car T-cell therapy. Am J Hematol (2019) 94(8):E209–E13. doi: 10.1002/ajh.25505
12. Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated Nkg2c+ natural killer cells with potent function. Blood (2012) 119(11):2665–74. doi: 10.1182/blood-2011-10-386995
13. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med (2016) 8(357):357ra123. doi: 10.1126/scitranslmed.aaf2341
14. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature (2009) 457(7229):557–61. doi: 10.1038/nature07665
15. Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, et al. Senescent cells evade immune clearance via hla-E-Mediated nk and Cd8(+) T cell inhibition. Nat Commun (2019) 10(1):2387. doi: 10.1038/s41467-019-10335-5
16. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by nk cells. Cancer Cell (2017) 32(2):135–54. doi: 10.1016/j.ccell.2017.06.009
17. Shimasaki N, Jain A, Campana D. Nk cells for cancer immunotherapy. Nat Rev Drug Discovery (2020) 19(3):200–18. doi: 10.1038/s41573-019-0052-1
18. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity (2017) 47(5):820–33. doi: 10.1016/j.immuni.2017.10.008
19. Melsen JE, Lugthart G, Lankester AC, Schilham MW. Human circulating and tissue-resident Cd56(Bright) natural killer cell populations. Front Immunol (2016) 7:262. doi: 10.3389/fimmu.2016.00262
20. Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. Nk cells mediate reduction of gvhd by inhibiting activated, alloreactive T cells while retaining gvt effects. Blood (2010) 115(21):4293–301. doi: 10.1182/blood-2009-05-222190
21. Shah N, Li L, McCarty J, Kaur I, Yvon E, Shaim H, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol (2017) 177(3):457–66. doi: 10.1111/bjh.14570
22. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of car-transduced natural killer cells in Cd19-positive lymphoid tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
23. Zhang Y, Wallace DL, de Lara CM, Ghattas H, Asquith B, Worth A, et al. In vivo kinetics of human natural killer cells: The effects of ageing and acute and chronic viral infection. Immunology (2007) 121(2):258–65. doi: 10.1111/j.1365-2567.2007.02573.x
24. Klingemann H. Are natural killer cells superior car drivers? Oncoimmunology (2014) 3:e28147. doi: 10.4161/onci.28147
25. Tyagarajan S, Spencer T, Smith J. Optimizing car-T cell manufacturing processes during pivotal clinical trials. Mol Ther Methods Clin Dev (2020) 16:136–44. doi: 10.1016/j.omtm.2019.11.018
26. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic b cell. Nat Med (2018) 24(10):1499–503. doi: 10.1038/s41591-018-0201-9
27. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295(5562):2097–100. doi: 10.1126/science.1068440
28. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human nk cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol (2019) 16(5):430–41. doi: 10.1038/s41423-019-0206-4
29. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. Car-nk cells: A promising cellular immunotherapy for cancer. EBioMedicine (2020) 59:102975. doi: 10.1016/j.ebiom.2020.102975
30. Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandstrom N, et al. Nk cells switch from granzyme b to death receptor-mediated cytotoxicity during serial killing. J Exp Med (2019) 216(9):2113–27. doi: 10.1084/jem.20181454
31. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol (2006) 6(11):836–48. doi: 10.1038/nri1961
32. Daher M, Melo Garcia L, Li Y, Rezvani K. Car-nk cells: The next wave of cellular therapy for cancer. Clin Transl Immunol (2021) 10(4):e1274. doi: 10.1002/cti2.1274
33. Gong Y, Klein Wolterink RGJ, Wang J, Bos GMJ, Germeraad WTV. Chimeric antigen receptor natural killer (Car-nk) cell design and engineering for cancer therapy. J Hematol Oncol (2021) 14(1):73. doi: 10.1186/s13045-021-01083-5
34. Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-Man clinical trial of car nk-92 cells: Safety test of Cd33-car nk-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res (2018) 8(6):1083–9.
35. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human ipsc-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell (2018) 23(2):181–92.e5. doi: 10.1016/j.stem.2018.06.002
36. Karadimitris A. Cord blood car-nk cells: Favorable initial efficacy and toxicity but durability of clinical responses not yet clear. Cancer Cell (2020) 37(4):426–7. doi: 10.1016/j.ccell.2020.03.018
37. Bachanova V, Ghobadi A, Patel K, Park JH, Flinn IW, Shah P, et al. Safety and efficacy of Ft596, a first-in-Class, multi-antigen targeted, off-the-Shelf, ipsc-derived Cd19 car nk cell therapy in Relapsed/Refractory b-cell lymphoma. Blood (2021) 138:823. doi: 10.1182/blood-2021-151185
38. Young RM, Engel NW, Uslu U, Wellhausen N, June CH. Next-generation car T-cell therapies. Cancer Discov (2022) 12(7):1625–33. doi: 10.1158/2159-8290.cd-21-1683.
39. Zhang L, Meng Y, Feng X, Han Z. Car-nk cells for cancer immunotherapy: From bench to bedside. Biomark Res (2022) 10(1):12. doi: 10.1186/s40364-022-00364-6
40. Alnabhan R, Madrigal A, Saudemont A. Differential activation of cord blood and peripheral blood natural killer cells by cytokines. Cytotherapy (2015) 17(1):73–85. doi: 10.1016/j.jcyt.2014.08.003
41. Tarannum M, Romee R, Shapiro RM. Innovative strategies to improve the clinical application of nk cell-based immunotherapy. Front Immunol (2022) 13:859177. doi: 10.3389/fimmu.2022.859177
42. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res an Off J Am Assoc Cancer Res (2011) 17(19):6287–97. doi: 10.1158/1078-0432.ccr-11-1347
43. Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother (Hagerstown Md 1997) (2015) 38(1):24–36. doi: 10.1097/cji.0000000000000059
44. Veluchamy JP, Kok N, van der Vliet HJ, Verheul HMW, de Gruijl TD, Spanholtz J. The rise of allogeneic natural killer cells as a platform for cancer immunotherapy: Recent innovations and future developments. Front Immunol (2017) 8:631. doi: 10.3389/fimmu.2017.00631
45. Lupo KB, Matosevic S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers (2019) 11(6). doi: 10.3390/cancers11060769
46. Nahi H, Chrobok M, Meinke S, Gran C, Marquardt N, Afram G, et al. Autologous nk cells as consolidation therapy following stem cell transplantation in multiple myeloma. Cell Rep Med (2022) 3(2):100508. doi: 10.1016/j.xcrm.2022.100508
47. Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. Nk cell-based cancer immunotherapy: From basic biology to clinical development. J Hematol Oncol (2021) 14(1):7. doi: 10.1186/s13045-020-01014-w
48. Gurney M, Kundu S, Pandey S, O'Dwyer M. Feeder cells at the interface of natural killer cell activation, expansion and gene editing. Front Immunol (2022) 13:802906. doi: 10.3389/fimmu.2022.802906
49. Heinze A, Grebe B, Bremm M, Huenecke S, Munir TA, Graafen L, et al. The synergistic use of il-15 and il-21 for the generation of nk cells from Cd3/Cd19-depleted grafts improves their ex vivo expansion and cytotoxic potential against neuroblastoma: Perspective for optimized immunotherapy post haploidentical stem cell transplantation. Front Immunol (2019) 10:2816. doi: 10.3389/fimmu.2019.02816
50. Oberschmidt O, Morgan M, Huppert V, Kessler J, Gardlowski T, Matthies N, et al. Development of automated separation, expansion, and quality control protocols for clinical-scale manufacturing of primary human nk cells and alpharetroviral chimeric antigen receptor engineering. Hum Gene Ther Methods (2019) 30(3):102–20. doi: 10.1089/hgtb.2019.039
51. Choi YH, Lim EJ, Kim SW, Moon YW, Park KS, An HJ. Il-27 enhances il-15/Il-18-Mediated activation of human natural killer cells. J Immunother Cancer (2019) 7(1):168. doi: 10.1186/s40425-019-0652-7
52. Ciurea SO, Kongtim P, Soebbing D, Trikha P, Behbehani G, Rondon G, et al. Decrease post-transplant relapse using donor-derived expanded nk-cells. Leukemia (2022) 36(1):155–64. doi: 10.1038/s41375-021-01349-4
53. Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, et al. Phase 1 clinical trial using Mbil21 ex vivo-expanded donor-derived nk cells after haploidentical transplantation. Blood (2017) 130(16):1857–68. doi: 10.1182/blood-2017-05-785659
54. Zhao XY, Jiang Q, Jiang H, Hu LJ, Zhao T, Yu XX, et al. Expanded clinical-grade membrane-bound il-21/4-1bbl nk cell products exhibit activity against acute myeloid leukemia in vivo. Eur J Immunol (2020) 50(9):1374–85. doi: 10.1002/eji.201948375
55. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound il-21 promotes sustained ex vivo proliferation of human natural killer cells. PloS One (2012) 7(1):e30264. doi: 10.1371/journal.pone.0030264
56. Yang Y, Badeti S, Tseng HC, Ma MT, Liu T, Jiang JG, et al. Superior expansion and cytotoxicity of human primary nk and car-nk cells from various sources via enriched metabolic pathways. Mol Ther Methods Clin Dev (2020) 18:428–45. doi: 10.1016/j.omtm.2020.06.014
57. Leivas A, Perez-Martinez A, Blanchard MJ, Martín-Clavero E, Fernández L, Lahuerta JJ, et al. Novel treatment strategy with autologous activated and expanded natural killer cells plus anti-myeloma drugs for multiple myeloma. Oncoimmunology (2016) 5(12):e1250051. doi: 10.1080/2162402x.2016.1250051
58. Vasu S, Jaglowski SM. Running the tank to empty: How far can the car go? Blood (2019) 133(15):1617–8. doi: 10.1182/blood-2019-02-900761
59. Oyer JL, Pandey V, Igarashi RY, Somanchi SS, Zakari A, Solh M, et al. Natural killer cells stimulated with Pm21 particles expand and biodistribute in vivo: Clinical implications for cancer treatment. Cytotherapy (2016) 18(5):653–63. doi: 10.1016/j.jcyt.2016.02.006
60. Ahn YH, Ren L, Kim SM, Seo SH, Jung CR, Kim DS, et al. A three-dimensional hyaluronic acid-based niche enhances the therapeutic efficacy of human natural killer cell-based cancer immunotherapy. Biomaterials (2020) 247:119960. doi: 10.1016/j.biomaterials.2020.119960
61. Vidard L, Dureuil C, Baudhuin J, Vescovi L, Durand L, Sierra V, et al. Cd137 (4-1bb) engagement fine-tunes synergistic il-15- and il-21-Driven nk cell proliferation. J Immunol (2019) 203(3):676–85. doi: 10.4049/jimmunol.1801137
62. Wu Y, Tian Z, Wei H. Developmental and functional control of natural killer cells by cytokines. Front Immunol (2017) 8:930. doi: 10.3389/fimmu.2017.00930
63. Granzin M, Wagner J, Kohl U, Cerwenka A, Huppert V, Ullrich E. Shaping of natural killer cell antitumor activity by ex vivo cultivation. Front Immunol (2017) 8:458. doi: 10.3389/fimmu.2017.00458
64. Pfefferle A, Jacobs B, Haroun-Izquierdo A, Kveberg L, Sohlberg E, Malmberg KJ. Deciphering natural killer cell homeostasis. Front Immunol (2020) 11:812. doi: 10.3389/fimmu.2020.00812
65. Jacobs B, Pfefferle A, Clement D, Berg-Larsen A, Saetersmoen ML, Lorenz S, et al. Induction of the bim short splice variant sensitizes proliferating nk cells to il-15 withdrawal. J Immunol (2019) 202(3):736–46. doi: 10.4049/jimmunol.1801146
66. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase ii study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy (2011) 13(1):98–107. doi: 10.3109/14653249.2010.515582
67. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using il-2 diphtheria toxin fusion protein. Blood (2014) 123(25):3855–63. doi: 10.1182/blood-2013-10-532531
68. Assier E, Jullien V, Lefort J, Moreau JL, Di Santo JP, Vargaftig BB, et al. Nk cells and polymorphonuclear neutrophils are both critical for il-2-Induced pulmonary vascular leak syndrome. J Immunol (2004) 172(12):7661–8. doi: 10.4049/jimmunol.172.12.7661
69. MacDonald A, Wu TC, Hung CF. Interleukin 2-based fusion proteins for the treatment of cancer. J Immunol Res (2021) 2021:7855808. doi: 10.1155/2021/7855808
70. Fehniger TA. Mystery solved: Il-15. J Immunol (2019) 202(11):3125–6. doi: 10.4049/jimmunol.1900419
71. Stoklasek TA, Schluns KS, Lefrançois L. Combined il-15/Il-15ralpha immunotherapy maximizes il-15 activity in vivo. J Immunol (2006) 177(9):6072–80. doi: 10.4049/jimmunol.177.9.6072
72. Conlon KC, Potter EL, Pittaluga S, Lee CR, Miljkovic MD, Fleisher TA, et al. Il15 by continuous intravenous infusion to adult patients with solid tumors in a phase I trial induced dramatic nk-cell subset expansion. Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25(16):4945–54. doi: 10.1158/1078-0432.CCR-18-3468
73. Margolin K, Morishima C, Velcheti V, Miller JS, Lee SM, Silk AW, et al. Phase I trial of alt-803, a novel recombinant Il15 complex, in patients with advanced solid tumors. Clin Cancer Res an Off J Am Assoc Cancer Res (2018) 24(22):5552–61. doi: 10.1158/1078-0432.CCR-18-0945
74. Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, et al. First-in-Human phase 1 clinical study of the il-15 superagonist complex alt-803 to treat relapse after transplantation. Blood (2018) 131(23):2515–27. doi: 10.1182/blood-2017-12-823757
75. Foltz JA, Hess BT, Bachanova V, Bartlett NL, Berrien-Elliott MM, McClain E, et al. Phase I trial of n-803, an Il15 receptor agonist, with rituximab in patients with indolent non-Hodgkin lymphoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2021) 27(12):3339–50. doi: 10.1158/1078-0432.CCR-20-4575
76. Shah N, Perales MA, Turtle CJ, Cairo MS, Cowan AJ, Saeed H, et al. Phase I study protocol: Nktr-255 as monotherapy or combined with daratumumab or rituximab in hematologic malignancies. Future Oncol (2021) 17(27):3549–60. doi: 10.2217/fon-2021-0576
77. Miyazaki T, Maiti M, Hennessy M, Chang T, Kuo P, Addepalli M, et al. Nktr-255, a novel polymer-conjugated rhil-15 with potent antitumor efficacy. J Immunother Cancer (2021) 9(5). doi: 10.1136/jitc-2020-002024
78. Myers JA, Miller JS. Exploring the nk cell platform for cancer immunotherapy. Nat Rev Clin Oncol (2021) 18(2):85–100. doi: 10.1038/s41571-020-0426-7
79. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and Cd8 T cells, and cytokine production during first-in-Human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol (2015) 33(1):74–82. doi: 10.1200/JCO.2014.57.3329
80. Miller JS, Morishima C, McNeel DG, Patel MR, Kohrt HEK, Thompson JA, et al. A first-in-Human phase I study of subcutaneous outpatient recombinant human Il15 (Rhil15) in adults with advanced solid tumors. Clin Cancer Res an Off J Am Assoc Cancer Res (2018) 24(7):1525–35. doi: 10.1158/1078-0432.CCR-17-2451
81. Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, et al. First-in-Human trial of rhil-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv (2019) 3(13):1970–80. doi: 10.1182/bloodadvances.2018028332
82. Berger C, Berger M, Hackman RC, Gough M, Elliott C, Jensen MC, et al. Safety and immunologic effects of il-15 administration in nonhuman primates. Blood (2009) 114(12):2417–26. doi: 10.1182/blood-2008-12-189266
83. Fehniger TA, Suzuki K, Ponnappan A, VanDeusen JB, Cooper MA, Florea SM, et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype Cd8+ T cells. J Exp Med (2001) 193(2):219–31. doi: 10.1084/jem.193.2.219
84. Mishra A, Liu S, Sams GH, Curphey DP, Santhanam R, Rush LJ, et al. Aberrant overexpression of il-15 initiates Large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell (2012) 22(5):645–55. doi: 10.1016/j.ccr.2012.09.009
85. Li Q, Ye LJ, Ren HL, Huyan T, Li J, Shi JL, et al. Multiple effects of il-21 on human nk cells in ex vivo expansion. Immunobiology (2015) 220(7):876–88. doi: 10.1016/j.imbio.2015.01.009
86. Brady J, Hayakawa Y, Smyth MJ, Nutt SL. Il-21 induces the functional maturation of murine nk cells. J Immunol (2004) 172(4):2048–58. doi: 10.4049/jimmunol.172.4.2048
87. Frederiksen KS, Lundsgaard D, Freeman JA, Hughes SD, Holm TL, Skrumsager BK, et al. Il-21 induces in vivo immune activation of nk cells and Cd8(+) T cells in patients with metastatic melanoma and renal cell carcinoma. Cancer Immunol Immunother CII (2008) 57(10):1439–49. doi: 10.1007/s00262-008-0479-4
88. Coquet JM, Skak K, Davis ID, Smyth MJ, Godfrey DI. Il-21 modulates activation of nkt cells in patients with stage iv malignant melanoma. Clin Transl Immunol (2013) 2(10):e6. doi: 10.1038/cti.2013.7
89. Croce M, Rigo V, Ferrini S. Il-21: A pleiotropic cytokine with potential applications in oncology. J Immunol Res (2015) 2015:696578. doi: 10.1155/2015/696578
90. Lim DP, Jang YY, Kim S, Koh SS, Lee JJ, Kim JS, et al. Effect of exposure to interleukin-21 at various time points on human natural killer cell culture. Cytotherapy (2014) 16(10):1419–30. doi: 10.1016/j.jcyt.2014.04.008
91. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood nk cells engineered to express il-15 and a Cd19-targeted car show long-term persistence and potent antitumor activity. Leukemia (2018) 32(2):520–31. doi: 10.1038/leu.2017.226
92. Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. Cis is a potent checkpoint in nk cell-mediated tumor immunity. Nat Immunol (2016) 17(7):816–24. doi: 10.1038/ni.3470
93. Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood car-nk cells. Blood (2021) 137(5):624–36. doi: 10.1182/blood.2020007748
94. Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic reprograming via deletion of cish in human ipsc-derived nk cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell (2020) 27(2):224–37.e6. doi: 10.1016/j.stem.2020.05.008
95. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of Myd88 and Cd40 in car T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discovery (2017) 7(11):1306–19. doi: 10.1158/2159-8290.CD-17-0263
96. Wang X, Jasinski DL, Medina JL, Spencer DM, Foster AE, Bayle JH. Inducible Myd88/Cd40 synergizes with il-15 to enhance antitumor efficacy of car-nk cells. Blood Adv (2020) 4(9):1950–64. doi: 10.1182/bloodadvances.2020001510
97. Du Z, Ng YY, Zha S, Wang S. Piggybac system to Co-express Nkg2d car and il-15 to augment the in vivo persistence and anti-aml activity of human peripheral blood nk cells. Mol Ther Methods Clin Dev (2021) 23:582–96. doi: 10.1016/j.omtm.2021.10.014
98. Christodoulou I, Ho WJ, Marple A, Ravich JW, Tam A, Rahnama R, et al. Engineering car-nk cells to secrete il-15 sustains their anti-aml functionality but is associated with systemic toxicities. J Immunother Cancer (2021) 9(12). doi: 10.1136/jitc-2021-003894
99. Kobayashi H, Dubois S, Sato N, Sabzevari H, Sakai Y, Waldmann TA, et al. Role of trans-cellular il-15 presentation in the activation of nk cell-mediated killing, which leads to enhanced tumor immunosurveillance. Blood (2005) 105(2):721–7. doi: 10.1182/blood-2003-12-4187
100. Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SM, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood (2014) 124(7):1081–8. doi: 10.1182/blood-2014-02-556837
101. Soldierer M, Bister A, Haist C, Thivakaran A, Cengiz SC, Sendker S, et al. Genetic engineering and enrichment of human nk cells for car-enhanced immunotherapy of hematological malignancies. Front Immunol (2022) 13:847008. doi: 10.3389/fimmu.2022.847008
102. Bjordahl R, Gaidarova S, Goodridge JP, Mahmood S, Bonello G, Robinson M, et al. Ft576: A novel multiplexed engineered off-the-Shelf natural killer cell immunotherapy for the dual-targeting of Cd38 and bcma for the treatment of multiple myeloma. Blood (2019) 134(Supplement_1):3214. doi: 10.1182/blood-2019-131373
103. Goodridge JP, Mahmood S, Zhu H, Gaidarova S, Blum R, Bjordahl R, et al. Ft596: Translation of first-of-Kind multi-antigen targeted off-the-Shelf car-nk cell with engineered persistence for the treatment of b cell malignancies. Blood (2019) 134(Supplement_1):301. doi: 10.1182/blood-2019-129319
104. Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U.S.A. (2009) 106(6):1915–9. doi: 10.1073/pnas.0813192106
105. Uppendahl LD, Felices M, Bendzick L, Ryan C, Kodal B, Hinderlie P, et al. Cytokine-induced memory-like natural killer cells have enhanced function, proliferation, and in vivo expansion against ovarian cancer cells. Gynecol Oncol (2019) 153(1):149–57. doi: 10.1016/j.ygyno.2019.01.006
106. Marin ND, Krasnick BA, Becker-Hapak M, Conant L, Goedegebuure SP, Berrien-Elliott MM, et al. Memory-like differentiation enhances nk cell responses to melanoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2021) 27(17):4859–69. doi: 10.1158/1078-0432.CCR-21-0851
107. Berrien-Elliott MM, Foltz JA, Russler-Germain DA, Neal CC, Tran J, Gang M, et al. Hematopoietic cell transplantation donor-derived memory-like nk cells functionally persist after transfer into patients with leukemia. Sci Transl Med (2022) 14(633):eabm1375. doi: 10.1126/scitranslmed.abm1375
108. Shapiro RM, Birch GC, Hu G, Vergara Cadavid J, Nikiforow S, Baginska J, et al. Expansion, persistence, and efficacy of donor memory-like nk cells infused for post-transplant relapse. J Clin Invest (2022) 132(11). doi: 10.1172/JCI154334
109. Bednarski JJ, Zimmerman C, Berrien-Elliott MM, Foltz JA, Becker-Hapak M, Neal CC, et al. Donor memory-like nk cells persist and induce remissions in pediatric patients with relapsed aml after transplant. Blood (2022) 139(11):1670–83. doi: 10.1182/blood.2021013972
110. Kerbauy LN, Marin ND, Kaplan M, Banerjee PP, Berrien-Elliott MM, Becker-Hapak M, et al. Combining Afm13, a bispecific Cd30/Cd16 antibody, with cytokine-activated blood and cord blood-derived nk cells facilitates car-like responses against Cd30(+) malignancies. Clin Cancer Res an Off J Am Assoc Cancer Res (2021) 27(13):3744–56. doi: 10.1158/1078-0432.CCR-21-0164
111. Bunin A, McGrath K, Rossi AM, Welsch M, Vidal C, Trinh D, et al. A novel class of bifunctional immunotherapeutic that exploits a universal antibody binding terminus (Uabt) to recruit endogenous antibodies to cell expressing Cd38 demonstrate in vivo efficacy in three distinct animal models. Blood (2019) 134(Supplement_1):1820. doi: 10.1182/blood-2019-130838
112. Gang M, Marin ND, Wong P, Neal CC, Marsala L, Foster M, et al. Car-modified memory-like nk cells exhibit potent responses to nk-resistant lymphomas. Blood (2020) 136(20):2308–18. doi: 10.1182/blood.2020006619
113. Dong H, Ham JD, Hu G, Xie G, Vergara J, Liang Y, et al. Memory-like nk cells armed with a neoepitope-specific car exhibit potent activity against Npm1 mutated acute myeloid leukemia. Proc Natl Acad Sci U.S.A. (2022) 119(25):e2122379119. doi: 10.1073/pnas.2122379119
114. Medvedev AE, Johnsen AC, Haux J, Steinkjer B, Egeberg K, Lynch DH, et al. Regulation of fas and fas-ligand expression in nk cells by cytokines and the involvement of fas-ligand in Nk/Lak cell-mediated cytotoxicity. Cytokine (1997) 9(6):394–404. doi: 10.1006/cyto.1996.0181
115. Felices M, Lenvik AJ, McElmurry R, Chu S, Hinderlie P, Bendzick L, et al. Continuous treatment with il-15 exhausts human nk cells via a metabolic defect. JCI Insight (2018) 3(3). doi: 10.1172/jci.insight.96219
116. Silvestris F, Tucci M, Cafforio P, Dammacco F. Fas-l up-regulation by highly malignant myeloma plasma cells: Role in the pathogenesis of anemia and disease progression. Blood (2001) 97(5):1155–64. doi: 10.1182/blood.v97.5.1155
117. Rossin A, Miloro G, Hueber AO. Trail and fasl functions in cancer and autoimmune diseases: Towards an increasing complexity. Cancers (2019) 11(5). doi: 10.3390/cancers11050639
118. Trembath AP, Markiewicz MA. More than decoration: Roles for natural killer group 2 member d ligand expression by immune cells. Front Immunol (2018) 9:231. doi: 10.3389/fimmu.2018.00231
119. Lopez-Cobo S, Romera-Cardenas G, Garcia-Cuesta EM, Reyburn HT, Vales-Gomez M. Transfer of the human Nkg2d ligands Ul16 binding proteins (Ulbp) 1-3 is related to lytic granule release and leads to ligand retransfer and killing of ulbp-recipient natural killer cells. Immunology (2015) 146(1):70–80. doi: 10.1111/imm.12482
120. Nakamura K, Nakayama M, Kawano M, Amagai R, Ishii T, Harigae H, et al. Fratricide of natural killer cells dressed with tumor-derived Nkg2d ligand. Proc Natl Acad Sci U.S.A. (2013) 110(23):9421–6. doi: 10.1073/pnas.1300140110
121. Brennan K, McSharry BP, Keating S, Petrasca A, O'Reilly VP, Keane J, et al. Human natural killer cell expression of Ulbp2 is associated with a mature functional phenotype. Hum Immunol (2016) 77(10):876–85. doi: 10.1016/j.humimm.2016.06.018
122. Sharma N, Trinidad CV, Trembath AP, Markiewicz MA. Nkg2d signaling between human nk cells enhances tace-mediated tnf-alpha release. J Immunol (2017) 199(8):2865–72. doi: 10.4049/jimmunol.1700647
123. Breman E, Demoulin B, Agaugue S, Mauen S, Michaux A, Springuel L, et al. Overcoming target driven fratricide for T cell therapy. Front Immunol (2018) 9:2940. doi: 10.3389/fimmu.2018.02940
124. Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An "Off-the-Shelf" fratricide-resistant car-T for the treatment of T cell hematologic malignancies. Leukemia (2018) 32(9):1970–83. doi: 10.1038/s41375-018-0065-5
125. Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. Cd7-edited T cells expressing a Cd7-specific car for the therapy of T-cell malignancies. Blood (2017) 130(3):285–96. doi: 10.1182/blood-2017-01-761320
126. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. Car T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature (2019) 568(7750):112–6. doi: 10.1038/s41586-019-1054-1
127. Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, et al. Cd38 knockout natural killer cells expressing an affinity optimized Cd38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica (2022) 107(2):437–45. doi: 10.3324/haematol.2020.271908
128. Wang Y, Zhang Y, Hughes T, Zhang J, Caligiuri MA, Benson DM, et al. Fratricide of nk cells in daratumumab therapy for multiple myeloma overcome by ex vivo-expanded autologous nk cells. Clin Cancer Res an Off J Am Assoc Cancer Res (2018) 24(16):4006–17. doi: 10.1158/1078-0432.CCR-17-3117
129. Choi E, Chang J-W, Krueger J, Lahr WS, Pomeroy E, Walsh M, et al. Engineering Cd70-directed car-nk cells for the treatment of hematological and solid malignancies. Blood (2021) 138(Supplement 1):1691. doi: 10.1182/blood-2021-148649
130. Hejazi M, Zhang C, Bennstein SB, Balz V, Reusing SB, Quadflieg M, et al. Cd33 delineates two functionally distinct nk cell populations divergent in cytokine production and antibody-mediated cellular cytotoxicity. Front Immunol (2021) 12:798087. doi: 10.3389/fimmu.2021.798087
131. Williams RL, Cooley S, Bachanova V, Blazar BR, Weisdorf DJ, Miller JS, et al. Recipient T cell exhaustion and successful adoptive transfer of haploidentical natural killer cells. Biol Blood Marrow Transplant (2018) 24(3):618–22. doi: 10.1016/j.bbmt.2017.11.022
132. Wang Z, Liu Y, Zhang Y, Shang Y, Gao Q. Mdsc-decreasing chemotherapy increases the efficacy of cytokine-induced killer cell immunotherapy in metastatic renal cell carcinoma and pancreatic cancer. Oncotarget (2016) 7(4):4760–9. doi: 10.18632/oncotarget.6734
133. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific Cd8+ T cells. J Exp Med (2005) 202(7):907–12. doi: 10.1084/jem.20050732
134. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical nk cells in patients with cancer. Blood (2005) 105(8):3051–7. doi: 10.1182/blood-2004-07-2974
135. Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, et al. Lymphoma remissions caused by anti-Cd19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol (2017) 35(16):1803–13. doi: 10.1200/jco.2016.71.3024
136. Berrien-Elliott MM, Becker-Hapak M, Cashen AF, Jacobs M, Wong P, Foster M, et al. Systemic il-15 promotes allogeneic cell rejection in patients treated with natural killer cell adoptive therapy. Blood (2022) 139(8):1177–83. doi: 10.1182/blood.2021011532
137. Curti A, Ruggeri L, D'Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical kir ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood (2011) 118(12):3273–9. doi: 10.1182/blood-2011-01-329508
138. Rong Z, Wang M, Hu Z, Stradner M, Zhu S, Kong H, et al. An effective approach to prevent immune rejection of human esc-derived allografts. Cell Stem Cell (2014) 14(1):121–30. doi: 10.1016/j.stem.2013.11.014
139. Quach DH, Becerra-Dominguez L, Rouce RH, Rooney CM. A strategy to protect off-the-Shelf cell therapy products using virus-specific T-cells engineered to eliminate alloreactive T-cells. J Transl Med (2019) 17(1):240. doi: 10.1186/s12967-019-1988-y
140. Hoerster K, Uhrberg M, Wiek C, Horn PA, Hanenberg H, Heinrichs S. Hla class I knockout converts allogeneic primary nk cells into suitable effectors for "Off-the-Shelf" immunotherapy. Front Immunol (2020) 11:586168. doi: 10.3389/fimmu.2020.586168
141. Borges L, Wallet MA, Bullaughey C-L, Naso MF, Gurung B, Keating S, et al. Development of multi-engineered ipsc-derived car-nk cells for the treatment of b-cell malignancies. Blood (2021) 138(Supplement 1):1729. doi: 10.1182/blood-2021-148438
142. Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res (2009) 69(9):4010–7. doi: 10.1158/0008-5472.Can-08-3712
143. Grzywacz B, Moench L, McKenna D Jr., Tessier KM, Bachanova V, Cooley S, et al. Natural killer cell homing and persistence in the bone marrow after adoptive immunotherapy correlates with better leukemia control. J Immunother (2019) 42(2):65–72. doi: 10.1097/cji.0000000000000250
144. Björklund AT, Carlsten M, Sohlberg E, Liu LL, Clancy T, Karimi M, et al. Complete remission with reduction of high-risk clones following haploidentical nk-cell therapy against mds and aml. Clin Cancer Res an Off J Am Assoc Cancer Res (2018) 24(8):1834–44. doi: 10.1158/1078-0432.Ccr-17-3196
145. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of Cd4(+) car T cells. Nature (2022) 602. doi: 10.1038/s41586-021-04390-6
146. Leivas A, Valeri A, Córdoba L, García-Ortiz A, Ortiz A, Sánchez-Vega L, et al. Nkg2d-Car-Transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J (2021) 11(8):146. doi: 10.1038/s41408-021-00537-w
147. Fujisaki H, Kakuda H, Imai C, Mullighan CG, Campana D. Replicative potential of human natural killer cells. Br J Haematol (2009) 145(5):606–13. doi: 10.1111/j.1365-2141.2009.07667.x
148. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi: 10.1038/ni1582
149. Adams NM, Grassmann S, Sun JC. Clonal expansion of innate and adaptive lymphocytes. Nat Rev Immunol (2020) 20(11):694–707. doi: 10.1038/s41577-020-0307-4
150. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol (2013) 75:685–705. doi: 10.1146/annurev-physiol-030212-183653
151. Wright WE, Pereira-Smith OM, Shay JW. Reversible cellular senescence: Implications for immortalization of normal human diploid fibroblasts. Mol Cell Biol (1989) 9(7):3088–92. doi: 10.1128/mcb.9.7.3088-3092.1989
152. Kasakovski D, Xu L, Li Y. T Cell senescence and car-T cell exhaustion in hematological malignancies. J Hematol Oncol (2018) 11(1):91. doi: 10.1186/s13045-018-0629-x
153. Lopez-Vergès S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, et al. Cd57 defines a functionally distinct population of mature nk cells in the human Cd56dimcd16+ nk-cell subset. Blood (2010) 116(19):3865–74. doi: 10.1182/blood-2010-04-282301
154. Streltsova MA, Erokhina SA, Kanevskiy LM, Lee DA, Telford WG, Sapozhnikov AM, et al. Analysis of nk cell clones obtained using interleukin-2 and gene-modified K562 cells revealed the ability of "Senescent" nk cells to lose Cd57 expression and start expressing Nkg2a. PloS One (2018) 13(12):e0208469. doi: 10.1371/journal.pone.0208469
155. Judge SJ, Murphy WJ, Canter RJ. Characterizing the dysfunctional nk cell: Assessing the clinical relevance of exhaustion, anergy, and senescence. Front Cell Infect Microbiol (2020) 10:49. doi: 10.3389/fcimb.2020.00049
156. Fali T, Papagno L, Bayard C, Mouloud Y, Boddaert J, Sauce D, et al. New insights into lymphocyte differentiation and aging from telomere length and telomerase activity measurements. J Immunol (2019) 202(7):1962–9. doi: 10.4049/jimmunol.1801475
157. Ouyang Q, Baerlocher G, Vulto I, Lansdorp PM. Telomere length in human natural killer cell subsets. Ann New York Acad Sci (2007) 1106:240–52. doi: 10.1196/annals.1392.001
158. Scaria G, Argall T, Bendzick L, Kaufman DS. Increased telomere length in natural killer cells generated from human induced pluripotent stem cells. Blood (2014) 124(21):5816–. doi: 10.1182/blood.V124.21.5816.5816
159. Wu KJ, Grandori C, Amacker M, Simon-Vermot N, Polack A, Lingner J, et al. Direct activation of tert transcription by c-myc. Nat Genet (1999) 21(2):220–4. doi: 10.1038/6010
160. Yang Y, Yang JJ, Tao H, Jin WS. Microrna-21 controls htert via pten in human colorectal cancer cell proliferation. J Physiol Biochem (2015) 71(1):59–68. doi: 10.1007/s13105-015-0380-5
161. Mitomo S, Maesawa C, Ogasawara S, Iwaya T, Shibazaki M, Yashima-Abo A, et al. Downregulation of mir-138 is associated with overexpression of human telomerase reverse transcriptase protein in human anaplastic thyroid carcinoma cell lines. Cancer Sci (2008) 99(2):280–6. doi: 10.1111/j.1349-7006.2007.00666.x
162. Kawauchi K, Ihjima K, Yamada O. Il-2 increases human telomerase reverse transcriptase activity transcriptionally and posttranslationally through phosphatidylinositol 3'-Kinase/Akt, heat shock protein 90, and mammalian target of rapamycin in transformed nk cells. J Immunol (2005) 174(9):5261–9. doi: 10.4049/jimmunol.174.9.5261
163. Kweon S, Phan MT, Chun S, Yu H, Kim J, Kim S, et al. Expansion of human nk cells using K562 cells expressing Ox40 ligand and short exposure to il-21. Front Immunol (2019) 10:879. doi: 10.3389/fimmu.2019.00879
164. Watkinson F, Nayar SK, Rani A, Sakellariou CA, Elhage O, Papaevangelou E, et al. Il-15 upregulates telomerase expression and potently increases proliferative capacity of nk, nkt-like, and Cd8 T cells. Front Immunol (2020) 11:594620. doi: 10.3389/fimmu.2020.594620
165. Yamada O, Ozaki K, Akiyama M, Kawauchi K. Jak-stat and jak-Pi3k-Mtorc1 pathways regulate telomerase transcriptionally and posttranslationally in atl cells. Mol Cancer Ther (2012) 11(5):1112–21. doi: 10.1158/1535-7163.Mct-11-0850
166. Rufer N, Migliaccio M, Antonchuk J, Humphries RK, Roosnek E, Lansdorp PM. Transfer of the human telomerase reverse transcriptase (Tert) gene into T lymphocytes results in extension of replicative potential. Blood (2001) 98(3):597–603. doi: 10.1182/blood.v98.3.597
167. Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol (2005) 175(10):7046–52. doi: 10.4049/jimmunol.175.10.7046
168. Streltsova MA, Ustiuzhanina MO, Barsov EV, Kust SA, Velichinskii RA, Kovalenko EI. Telomerase reverse transcriptase increases proliferation and lifespan of human nk cells without immortalization. Biomedicines (2021) 9(6). doi: 10.3390/biomedicines9060662
169. Bai Y, Kan S, Zhou S, Wang Y, Xu J, Cooke JP, et al. Enhancement of the in vivo persistence and antitumor efficacy of Cd19 chimeric antigen receptor T cells through the delivery of modified tert mrna. Cell Discovery (2015) 1:15040. doi: 10.1038/celldisc.2015.40
170. Restifo NP, Esquivel F, Kawakami Y, Yewdell JW, Mulé JJ, Rosenberg SA, et al. Identification of human cancers deficient in antigen processing. J Exp Med (1993) 177(2):265–72. doi: 10.1084/jem.177.2.265
171. Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of mhc class I antigen presentation. Front Immunol (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
172. Carbone E, Neri P, Mesuraca M, Fulciniti MT, Otsuki T, Pende D, et al. Nkg2d, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood (2005) 105(1):251–8. doi: 10.1182/blood-2004-04-1422
173. Romagné F, André P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7f9, a novel human anti-kir receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood (2009) 114(13):2667–77. doi: 10.1182/blood-2009-02-206532
174. Benson DM Jr., Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. Iph2101, a novel anti-inhibitory kir antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood (2011) 118(24):6387–91. doi: 10.1182/blood-2011-06-360255
175. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of Kir2d with the monoclonal antibody Iph2101 induces contraction and hyporesponsiveness of nk cells in patients with myeloma. Clin Cancer Res an Off J Am Assoc Cancer Res (2016) 22(21):5211–22. doi: 10.1158/1078-0432.CCR-16-1108
176. Vey N, Dumas P-Y, Recher C, Gastaud L, Lioure B, Bulabois C-E, et al. Randomized phase 2 trial of lirilumab (Anti-kir monoclonal antibody, mab) as maintenance treatment in elderly patients (Pts) with acute myeloid leukemia (Aml): Results of the effikir trial. Blood (2017) 130(Supplement 1):889. doi: 10.1182/blood.V130.Suppl_1.889.889
177. Khan M, Arooj S, Wang H. Nk cell-based immune checkpoint inhibition. Front Immunol (2020) 11:167. doi: 10.3389/fimmu.2020.00167
178. Benson DM Jr., Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-kir antibody Iph2101 and lenalidomide in patients with Relapsed/Refractory multiple myeloma. Clin Cancer Res an Off J Am Assoc Cancer Res (2015) 21(18):4055–61. doi: 10.1158/1078-0432.ccr-15-0304
179. Wieten L, Mahaweni NM, Voorter CE, Bos GM, Tilanus MG. Clinical and immunological significance of hla-e in stem cell transplantation and cancer. Tissue Antigens (2014) 84(6):523–35. doi: 10.1111/tan.12478
180. Mahapatra S, Mace EM, Minard CG, Forbes LR, Vargas-Hernandez A, Duryea TK, et al. High-resolution phenotyping identifies nk cell subsets that distinguish healthy children from adults. PloS One (2017) 12(8):e0181134. doi: 10.1371/journal.pone.0181134
181. Manser AR, Uhrberg M. Age-related changes in natural killer cell repertoires: Impact on nk cell function and immune surveillance. Cancer Immunol Immunother CII (2016) 65(4):417–26. doi: 10.1007/s00262-015-1750-0
182. Tognarelli S, Wirsching S, von Metzler I, Rais B, Jacobs B, Serve H, et al. Enhancing the activation and releasing the brakes: A double hit strategy to improve nk cell cytotoxicity against multiple myeloma. Front Immunol (2018) 9:2743. doi: 10.3389/fimmu.2018.02743
183. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor Nkg2a overcomes tumor resistance to nk cells. J Clin Invest (2019) 129(5):2094–106. doi: 10.1172/JCI123955
184. Ruggeri L, Urbani E, André P, Mancusi A, Tosti A, Topini F, et al. Effects of anti-Nkg2a antibody administration on leukemia and normal hematopoietic cells. Haematologica (2016) 101(5):626–33. doi: 10.3324/haematol.2015.135301
185. McWilliams EM, Mele JM, Cheney C, Timmerman EA, Fiazuddin F, Strattan EJ, et al. Therapeutic Cd94/Nkg2a blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology (2016) 5(10):e1226720. doi: 10.1080/2162402X.2016.1226720
186. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-Nkg2a mab is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and nk cells. Cell (2018) 175(7):1731–43.e13. doi: 10.1016/j.cell.2018.10.014
187. Borst L, van der Burg SH, van Hall T. The Nkg2a–Hla-E axis as a novel checkpoint in the tumor microenvironment. Clin Cancer Res (2020) 26(21):5549–56. doi: 10.1158/1078-0432.ccr-19-2095
188. Chang MC, Cheng HI, Hsu K, Hsu YN, Kao CW, Chang YF, et al. Nkg2a down-regulation by dasatinib enhances natural killer cytotoxicity and accelerates effective treatment responses in patients with chronic myeloid leukemia. Front Immunol (2018) 9:3152. doi: 10.3389/fimmu.2018.03152
189. Carlsten M, Namazi A, Reger R, Levy E, Berg M, St Hilaire C, et al. Bortezomib sensitizes multiple myeloma to nk cells via er-Stress-Induced suppression of hla-e and upregulation of Dr5. Oncoimmunology (2019) 8(2):e1534664. doi: 10.1080/2162402X.2018.1534664
190. Yun HD, Schirm DK, Felices M, Miller JS, Eckfeldt CE. Dinaciclib enhances natural killer cell cytotoxicity against acute myelogenous leukemia. Blood Adv (2019) 3(16):2448–52. doi: 10.1182/bloodadvances.2019000064
191. Fisher JG, Walker CJ, Doyle AD, Johnson PW, Forconi F, Cragg MS, et al. Selinexor enhances nk cell activation against malignant b cells via downregulation of hla-e. Front Oncol (2021) 11:785635. doi: 10.3389/fonc.2021.785635
192. Stock S, Kluever AK, Endres S, Kobold S. Enhanced chimeric antigen receptor T cell therapy through Co-application of synergistic combination partners. Biomedicines (2022) 10(2). doi: 10.3390/biomedicines10020307
193. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of nk cells to immunotherapy mediated by pd-1/Pd-L1 blockade. J Clin Invest (2018) 128(10):4654–68. doi: 10.1172/jci99317
194. Benson DM Jr., Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The pd-1/Pd-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for ct-011, a novel monoclonal anti-Pd-1 antibody. Blood (2010) 116(13):2286–94. doi: 10.1182/blood-2010-02-271874
195. Calvo T, Reina-Ortiz C, Giraldos D, Gascon M, Woods D, Asenjo J, et al. Expanded and activated allogeneic nk cells are cytotoxic against b-chronic lymphocytic leukemia (B-cll) cells with sporadic cases of resistance. Sci Rep (2020) 10(1):19398. doi: 10.1038/s41598-020-76051-z
196. Jung D, Baek YS, Lee IJ, Kim KY, Jang H, Hwang S, et al. Ex vivo expanded allogeneic natural killer cells have potent cytolytic activity against cancer cells through different receptor-ligand interactions. J Exp Clin Cancer Res (2021) 40(1):333. doi: 10.1186/s13046-021-02089-0
197. Judge SJ, Dunai C, Aguilar EG, Vick SC, Sturgill IR, Khuat LT, et al. Minimal pd-1 expression in mouse and human nk cells under diverse conditions. J Clin Invest (2020) 130(6):3051–68. doi: 10.1172/jci133353
198. Poznanski SM, Ritchie TM, Fan IY, El-Sayes A, Portillo AL, Ben-Avi R, et al. Expanded human nk cells from lung cancer patients sensitize patients' Pdl1-negative tumors to Pd1-blockade therapy. J Immunother Cancer (2021) 9(1). doi: 10.1136/jitc-2020-001933
199. Hasim MS, Marotel M, Hodgins JJ, Vulpis E, Makinson OJ, Asif S, et al. When killers become thieves: Trogocytosed pd-1 inhibits nk cells in cancer. Sci Adv (2022) 8(15):eabj3286. doi: 10.1126/sciadv.abj3286
200. Jiang W, Li F, Jiang Y, Li S, Liu X, Xu Y, et al. Tim-3 blockade elicits potent anti-multiple myeloma immunity of natural killer cells. Front Oncol (2022) 12:739976. doi: 10.3389/fonc.2022.739976
201. Rakova J, Truxova I, Holicek P, Salek C, Hensler M, Kasikova L, et al. Tim-3 Levels Correlate with Enhanced Nk Cell Cytotoxicity and Improved Clinical Outcome in Aml Patients. Oncoimmunology (2021) 10(1):1889822. doi: 10.1080/2162402x.2021.1889822.
202. Gleason MK, Lenvik TR, McCullar V, Felices M, O'Brien MS, Cooley SA, et al. Tim-3 Is an Inducible Human Natural Killer Cell Receptor That Enhances Interferon Gamma Production in Response to Galectin-9. Blood (2012) 119(13):3064–72. doi: 10.1182/blood-2011-06-360321.
203. Kaito Y, Hirano M, Futami M, Nojima M, Tamura H, Tojo A, et al. Cd155 and Cd112 as possible therapeutic targets of Flt3 inhibitors for acute myeloid leukemia. Oncol Lett (2022) 23(2):51. doi: 10.3892/ol.2021.13169
204. Pende D, Spaggiari GM, Marcenaro S, Martini S, Rivera P, Capobianco A, et al. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: Evidence for the involvement of the poliovirus receptor (Cd155) and nectin-2 (Cd112). Blood (2005) 105(5):2066–73. doi: 10.1182/blood-2004-09-3548
205. Brauneck F, Seubert E, Wellbrock J, Schulze Zur Wiesch J, Duan Y, Magnus T, et al. Combined blockade of tigit and Cd39 or A2ar enhances nk-92 cell-mediated cytotoxicity in aml. Int J Mol Sci (2021) 22(23). doi: 10.3390/ijms222312919
206. Daly J GM, Gurney M, O’Dwyer M. Knockout of Cd96 or tigit using Crispr/Cas9 enhances nk cell-induced cytotoxicity and cytokine production in the presence of Cd155-expressing myeloma cells. (2021). European Hematology Association Library. 06/09/21; 325699; EP941.
207. Maas RJ, Hoogstad-van Evert JS, van der Meer JM, Mekers V, Rezaeifard S, Korman AJ, et al. Tigit blockade enhances functionality of peritoneal nk cells with altered expression of dnam-1/Tigit/Cd96 checkpoint molecules in ovarian cancer. Oncoimmunology (2020) 9(1):1843247. doi: 10.1080/2162402x.2020.1843247
208. Gonçalves Silva I, Yasinska IM, Sakhnevych SS, Fiedler W, Wellbrock J, Bardelli M, et al. The Tim-3-Galectin-9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine (2017) 22:44–57. doi: 10.1016/j.ebiom.2017.07.018
209. Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim-3 expression in peripheral nk cells predicts a poorer prognosis and Tim-3 blockade improves nk cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol (2015) 29(2):635–41. doi: 10.1016/j.intimp.2015.09.017
210. Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, et al. Identification of Cd112r as a novel checkpoint for human T cells. J Exp Med (2016) 213(2):167–76. doi: 10.1084/jem.20150785
211. Li J, Whelan S, Kotturi MF, Meyran D, D'Souza C, Hansen K, et al. Pvrig is a novel natural killer cell immune checkpoint receptor in acute myeloid leukemia. Haematologica (2021) 106(12):3115–24. doi: 10.3324/haematol.2020.258574
212. Narayanan S AP, Bijin VA, Kaliaperumal N, Lim SG, Wang CI, Fairhurst AM, et al. Lag3 is a central regulator of nk cell cytokine production. bioRxiv (2020). doi: 10.1101/2020.01.31.928200
213. Lang S, Vujanovic NL, Wollenberg B, Whiteside TL. Absence of B7.1-Cd28/Ctla-4-Mediated Co-stimulation in human nk cells. Eur J Immunol (1998) 28(3):780–6. doi: 10.1002/(SICI)1521-4141(199803)28:03<780::AID-IMMU780>3.0.CO;2-8
214. Deuse T, Hu X, Agbor-Enoh S, Jang MK, Alawi M, Saygi C, et al. The sirpα-Cd47 immune checkpoint in nk cells. J Exp Med (2021) 218(3). doi: 10.1084/jem.20200839
215. Myers LM, Tal MC, Torrez Dulgeroff LB, Carmody AB, Messer RJ, Gulati G, et al. A functional subset of Cd8(+) T cells during chronic exhaustion is defined by sirpα expression. Nat Commun (2019) 10(1):794. doi: 10.1038/s41467-019-08637-9
216. Kazama R, Miyoshi H, Takeuchi M, Miyawaki K, Nakashima K, Yoshida N, et al. Combination of Cd47 and signal-regulatory protein-α constituting the "Don't eat me signal" is a prognostic factor in diffuse Large b-cell lymphoma. Cancer Sci (2020) 111(7):2608–19. doi: 10.1111/cas.14437
217. Wisnovsky S, Möckl L, Malaker SA, Pedram K, Hess GT, Riley NM, et al. Genome-wide crispr screens reveal a specific ligand for the glycan-binding immune checkpoint receptor siglec-7. Proc Natl Acad Sci U.S.A. (2021) 118(5). doi: 10.1073/pnas.2015024118
218. Daly J, Sarkar S, Natoni A, Stark JC, Riley NM, Bertozzi CR, et al. Targeting hypersialylation in multiple myeloma represents a novel approach to enhance nk cell-mediated tumor responses. Blood Adv (2022) 6(11):3352–66. doi: 10.1182/bloodadvances.2021006805
219. Choi H, Ho M, Adeniji OS, Giron L, Bordoloi D, Kulkarni AJ, et al. Development of siglec-9 blocking antibody to enhance anti-tumor immunity. Front Oncol (2021) 11:778989. doi: 10.3389/fonc.2021.778989
220. Hilpert J, Grosse-Hovest L, Grünebach F, Buechele C, Nuebling T, Raum T, et al. Comprehensive analysis of Nkg2d ligand expression and release in leukemia: Implications for Nkg2d-mediated nk cell responses. J Immunol (2012) 189(3):1360–71. doi: 10.4049/jimmunol.1200796
221. Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, et al. Mhc class I chain-related protein a antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci U.S.A. (2008) 105(4):1285–90. doi: 10.1073/pnas.0711293105
222. Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric Nkg2d receptor. Cancer Res (2006) 66(11):5927–33. doi: 10.1158/0008-5472.CAN-06-0130
223. Maroto-Martín E, Encinas J, García-Ortiz A, Ugalde L, Alonso R, Leivas A, et al. Exploring NKG2D and BCMA-CAR NK-92 for adoptive cellular therapy to multiple myeloma. Clin Lymphoma Myeloma Leuk. (2019) 19(10):e24–5.
224. Ferrari de Andrade L, Tay RE, Pan D, Luoma AM, Ito Y, Badrinath S, et al. Antibody-mediated inhibition of mica and micb shedding promotes nk cell-driven tumor immunity. Science (2018) 359(6383):1537–42. doi: 10.1126/science.aao0505
225. Reiser J, Mathavan K, Mahmood S, Pan Y, Hancock B, Blum R, et al. Dual chimeric antigen receptor approach combining novel tumor targeting strategies circumvents antigen escape in multiple myeloma. Blood (2021) 138(Supplement 1):1718. doi: 10.1182/blood-2021-154025
226. Bjordahl R, Goulding J, Pribadi M, Blum R, Chang C, Pan Y, et al. Development of a novel Mica/B-specific car as a pan-tumor targeting strategy for off-the-Shelf, cell-based cancer immunotherapy. Blood (2020) 136(Supplement 1):5–6. doi: 10.1182/blood-2020-141095
227. Schuller A, Doshi A, Cantin S, Secinaro M, Wang Y, Prickett L, et al. Inhibition of arginase in combination with anti-PDL1 leads to increased infiltration and activation of CD8+ T cells, NK cells, and CD103+ dendritic cells in mouse syngeneic tumor models. Cancer Res (2020) 80(Suppl 16):4523. doi: 10.1158/1538-7445.AM2020-4523
228. Reiners KS, Topolar D, Henke A, Simhadri VR, Kessler J, Sauer M, et al. Soluble ligands for nk cell receptors promote evasion of chronic lymphocytic leukemia cells from nk cell anti-tumor activity. Blood (2013) 121(18):3658–65. doi: 10.1182/blood-2013-01-476606
229. Yazdanifar M, Zhou R, Grover P, Williams C, Bose M, Moore LJ, et al. Overcoming immunological resistance enhances the efficacy of a novel anti-Tmuc1-Car T cell treatment against pancreatic ductal adenocarcinoma. Cells (2019) 8(9). doi: 10.3390/cells8091070
230. Sarhan D, Hippen KL, Lemire A, Hying S, Luo X, Lenvik T, et al. Adaptive nk cells resist regulatory T-cell suppression driven by Il37. Cancer Immunol Res (2018) 6(7):766–75. doi: 10.1158/2326-6066.cir-17-0498
231. Bataille R, Jourdan M, Zhang XG, Klein B. Serum levels of interleukin 6, a potent myeloma cell growth factor, as a reflect of disease severity in plasma cell dyscrasias. J Clin Invest (1989) 84(6):2008–11. doi: 10.1172/jci114392
232. Gupta M, Han JJ, Stenson M, Maurer M, Wellik L, Hu G, et al. Elevated serum il-10 levels in diffuse Large b-cell lymphoma: A mechanism of aberrant Jak2 activation. Blood (2012) 119(12):2844–53. doi: 10.1182/blood-2011-10-388538
233. Wang H, Wang L, Chi PD, Wang WD, Chen XQ, Geng QR, et al. High level of interleukin-10 in serum predicts poor prognosis in multiple myeloma. Br J Cancer (2016) 114(4):463–8. doi: 10.1038/bjc.2016.11
234. Mozas P, Rivas-Delgado A, Rivero A, Dlouhy I, Nadeu F, Balagué O, et al. High serum levels of il-2r, il-6, and tnf-α are associated with higher tumor burden and poorer outcome of follicular lymphoma patients in the rituximab era. Leukemia Res (2020) 94:106371. doi: 10.1016/j.leukres.2020.106371
235. Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting tgf-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in aml and colon cancer models. PloS One (2018) 13(1):e0191358. doi: 10.1371/journal.pone.0191358
236. Rouce RH, Shaim H, Sekine T, Weber G, Ballard B, Ku S, et al. The tgf-Beta/Smad pathway is an important mechanism for nk cell immune evasion in childhood b-acute lymphoblastic leukemia. Leukemia (2016) 30(4):800–11. doi: 10.1038/leu.2015.327
237. Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. Tgf-beta inhibits the activation and functions of nk cells by repressing the mtor pathway. Sci Signal (2016) 9(415):ra19. doi: 10.1126/scisignal.aad1884
238. Kim BG, Malek E, Choi SH, Ignatz-Hoover JJ, Driscoll JJ. Novel therapies emerging in oncology to target the tgf-β pathway. J Hematol Oncol (2021) 14(1):55. doi: 10.1186/s13045-021-01053-x
239. Daher M, Basar R, Shaim H, Gokdemir E, Uprety N, Kontoyiannis A, et al. The tgf-β/Smad signaling pathway as a mediator of nk cell dysfunction and immune evasion in myelodysplastic syndrome. Blood (2017) 130(Supplement 1):53. doi: 10.1182/blood.V130.Suppl_1.53.53
240. Lu ZY, Bataille R, Poubelle P, Rapp MJ, Harousseau JL, Klein B. An interleukin 1 receptor antagonist blocks the il-1-Induced il-6 paracrine production through a prostaglandin E2-related mechanism in multiple myeloma. Stem Cells (1995) 13 Suppl 2:28–34.
241. Truffinet V, Donnard M, Vincent C, Faucher JL, Bordessoule D, Turlure P, et al. Cyclooxygenase-1, but not -2, in blast cells of patients with acute leukemia. Int J Cancer (2007) 121(4):924–7. doi: 10.1002/ijc.22786
242. Naderi EH, Skah S, Ugland H, Myklebost O, Sandnes DL, Torgersen ML, et al. Bone marrow stroma-derived Pge2 protects bcp-all cells from DNA damage-induced P53 accumulation and cell death. Mol Cancer (2015) 14(1):14. doi: 10.1186/s12943-014-0278-9
243. Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E(2) (Pge (2)) suppresses natural killer cell function primarily through the Pge(2) receptor Ep4. Cancer Immunol Immunother CII (2011) 60(11):1577–86. doi: 10.1007/s00262-011-1064-9
244. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-E2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res an Off J Am Assoc Cancer Res (2014) 20(15):4096–106. doi: 10.1158/1078-0432.CCR-14-0635
245. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. Cd39/Cd73 upregulation on myeloid-derived suppressor cells via tgf-β-Mtor-Hif-1 signaling in patients with non-small cell lung cancer. Oncoimmunology (2017) 6(6):e1320011. doi: 10.1080/2162402x.2017.1320011
246. Chen S, Fan J, Zhang M, Qin L, Dominguez D, Long A, et al. Cd73 expression on effector T cells sustained by tgf-β facilitates tumor resistance to anti-4-1bb/Cd137 therapy. Nat Commun (2019) 10(1):150. doi: 10.1038/s41467-018-08123-8
247. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A2a receptors potently suppresses the metastasis of Cd73+ tumors. Proc Natl Acad Sci U.S.A. (2013) 110(36):14711–6. doi: 10.1073/pnas.1308209110
248. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-Inhibition of Cd73 and A2ar adenosine signaling improves anti-tumor immune responses. Cancer Cell (2016) 30(3):391–403. doi: 10.1016/j.ccell.2016.06.025
249. Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, et al. A2ar adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res (2018) 78(4):1003–16. doi: 10.1158/0008-5472.CAN-17-2826
250. Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic targeting enhances immunotherapy of Cd73(+) solid tumors with piggybac-engineered chimeric antigen receptor natural killer cells. J Immunother Cancer (2018) 6(1):136. doi: 10.1186/s40425-018-0441-8
251. Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite l-kynurenine inhibits the surface expression of Nkp46- and Nkg2d-activating receptors and regulates nk-cell function. Blood (2006) 108(13):4118–25. doi: 10.1182/blood-2006-03-006700
252. Wang D, Saga Y, Mizukami H, Sato N, Nonaka H, Fujiwara H, et al. Indoleamine-2,3-Dioxygenase, an immunosuppressive enzyme that inhibits natural killer cell function, as a useful target for ovarian cancer therapy. Int J Oncol (2012) 40(4):929–34. doi: 10.3892/ijo.2011.1295
253. Wells G, Kennedy PT, Dahal LN. Investigating the role of indoleamine 2,3-dioxygenase in acute myeloid leukemia: A systematic review. Front Immunol (2021) 12:651687. doi: 10.3389/fimmu.2021.651687
254. Ninomiya S, Hara T, Tsurumi H, Hoshi M, Kanemura N, Goto N, et al. Indoleamine 2,3-dioxygenase in tumor tissue indicates prognosis in patients with diffuse Large b-cell lymphoma treated with r-chop. Ann Hematol (2011) 90(4):409–16. doi: 10.1007/s00277-010-1093-z
255. Mangaonkar A, Mondal AK, Fulzule S, Pundkar C, Park EJ, Jillella A, et al. A novel immunohistochemical score to predict early mortality in acute myeloid leukemia patients based on indoleamine 2,3 dioxygenase expression. Sci Rep (2017) 7(1):12892. doi: 10.1038/s41598-017-12940-0
256. Wang S, Wu J, Shen H, Wang J. The prognostic value of ido expression in solid tumors: A systematic review and meta-analysis. BMC Cancer (2020) 20(1):471. doi: 10.1186/s12885-020-06956-5
257. Alwin Schuller AD, Cantin S, Secinaro M, Wang Y, Prickett L, Tentarelli S, et al. Inhibition of arginase in combination with anti-Pdl1 leads to increased infiltration and activation of Cd8+ T cells, nk cells, and Cd103+ dendritic cells in mouse syngeneic tumor models. Cancer Res (2020). doi: 10.1158/1538-7445.AM2020-4523
258. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by cb-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer (2017) 5(1):101. doi: 10.1186/s40425-017-0308-4
259. Mensali N, Dillard P, Fayzullin A, Köksal H, Gaudernack G, Kvalheim G, et al. "Built-in" pd-1 blocker to rescue nk-92 activity from pd-L1-Mediated tumor escape mechanisms. FASEB J (2021) 35(9):e21750. doi: 10.1096/fj.202100025R
260. Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative tgf-beta receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy (2017) 19(3):408–18. doi: 10.1016/j.jcyt.2016.12.005
261. Yang B, Liu H, Shi W, Wang Z, Sun S, Zhang G, et al. Blocking transforming growth factor-beta signaling pathway augments antitumor effect of adoptive nk-92 cell therapy. Int Immunopharmacol (2013) 17(2):198–204. doi: 10.1016/j.intimp.2013.06.003
262. Lu C, Guo C, Chen H, Zhang H, Zhi L, Lv T, et al. A novel chimeric Pd1-Nkg2d-41bb receptor enhances antitumor activity of Nk92 cells against human lung cancer H1299 cells by triggering pyroptosis. Mol Immunol (2020) 122:200–6. doi: 10.1016/j.molimm.2020.04.016
263. Grote S, Urena-Bailen G, Chan KC, Baden C, Mezger M, Handgretinger R, et al. In vitro evaluation of Cd276-car nk-92 functionality, migration and invasion potential in the presence of immune inhibitory factors of the tumor microenvironment. Cells (2021) 10(5). doi: 10.3390/cells10051020
264. Wang Z, Guo L, Song Y, Zhang Y, Lin D, Hu B, et al. Augmented anti-tumor activity of nk-92 cells expressing chimeric receptors of tgf-βr ii and Nkg2d. Cancer Immunol Immunother CII (2017) 66(4):537–48. doi: 10.1007/s00262-017-1959-1
265. Chang WC, Li CH, Chu LH, Huang PS, Sheu BC, Huang SC. Regulatory T cells suppress natural killer cell immunity in patients with human cervical carcinoma. Int J Gynecol Cancer (2016) 26(1):156–62. doi: 10.1097/IGC.0000000000000578
266. Geng X, Li M, Cui B, Lu C, Liu X, Zhang P, et al. Cd4+Cd25+Foxp3+ regulatory T cells suppress Nkg2d-mediated nk cell cytotoxicity in peripheral blood. Med (Baltimore) (2019) 98(22):e15722. doi: 10.1097/MD.0000000000015722
267. Zhang L, Tai YT, Ho M, Xing L, Chauhan D, Gang A, et al. Regulatory b cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood Cancer J (2017) 7(3):e547. doi: 10.1038/bcj.2017.24
268. Krneta T, Gillgrass A, Poznanski S, Chew M, Lee AJ, Kolb M, et al. M2-polarized and tumor-associated macrophages alter nk cell phenotype and function in a contact-dependent manner. J Leukoc Biol (2017) 101(1):285–95. doi: 10.1189/jlb.3A1215-552R
269. Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing fc receptor-mediated natural killer cell function. Clin Cancer Res (2018) 24(8):1891–904. doi: 10.1158/1078-0432.CCR-17-0691
270. Cencini E, Fabbri A, Sicuranza A, Gozzetti A, Bocchia M. The role of tumor-associated macrophages in hematologic malignancies. Cancers (2021) 13(14). doi: 10.3390/cancers13143597
271. Giallongo C, Tibullo D, Parrinello NL, La Cava P, Di Rosa M, Bramanti V, et al. Granulocyte-like myeloid derived suppressor cells (G-mdsc) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (Msc). Oncotarget (2016) 7(52):85764–75. doi: 10.18632/oncotarget.7969
272. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and Fibrin(Ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood (2005) 105(1):178–85. doi: 10.1182/blood-2004-06-2272
273. Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res (1999) 59(6):1295–300.
274. Cluxton CD, Spillane C, O'Toole SA, Sheils O, Gardiner CM, O'Leary JJ. Suppression of natural killer cell Nkg2d and Cd226 anti-tumour cascades by platelet cloaked cancer cells: Implications for the metastatic cascade. PloS One (2019) 14(3):e0211538. doi: 10.1371/journal.pone.0211538
275. Placke T, Orgel M, Schaller M, Jung G, Rammensee HG, Kopp HG, et al. Platelet-derived mhc class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res (2012) 72(2):440–8. doi: 10.1158/0008-5472.CAN-11-1872
276. Placke T, Salih HR, Kopp HG. Gitr ligand provided by thrombopoietic cells inhibits nk cell antitumor activity. J Immunol (2012) 189(1):154–60. doi: 10.4049/jimmunol.1103194
277. Clar KL, Hinterleitner C, Schneider P, Salih HR, Maurer S. Inhibition of nk reactivity against solid tumors by platelet-derived rankl. Cancers (2019) 11(3). doi: 10.3390/cancers11030277
278. Kopp HG, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates Nkg2d thereby inhibiting natural killer cell antitumor reactivity. Cancer Res (2009) 69(19):7775–83. doi: 10.1158/0008-5472.CAN-09-2123
279. Maurer S, Kropp KN, Klein G, Steinle A, Haen SP, Walz JS, et al. Platelet-mediated shedding of Nkg2d ligands impairs nk cell immune-surveillance of tumor cells. Oncoimmunology (2018) 7(2):e1364827. doi: 10.1080/2162402X.2017.1364827
280. Sarhan D, Wang J, Sunil Arvindam U, Hallstrom C, Verneris MR, Grzywacz B, et al. Mesenchymal stromal cells shape the mds microenvironment by inducing suppressive monocytes that dampen nk cell function. JCI Insight (2020) 5(5). doi: 10.1172/jci.insight.130155
281. Frassanito MA, Rao L, Moschetta M, Ria R, Di Marzo L, De Luisi A, et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: In vitro and in vivo studies. Leukemia (2014) 28(4):904–16. doi: 10.1038/leu.2013.254
282. Dehbashi M, Hojati Z, Motovali-Bashi M, Ganjalikhany MR, Cho WC, Shimosaka A, et al. A novel car expressing nk cell targeting Cd25 with the prospect of overcoming immune escape mechanism in cancers. Front Oncol (2021) 11:649710. doi: 10.3389/fonc.2021.649710
283. Stikvoort A, van der Schans J, Sarkar S, Poels R, Ruiter R, Naik J, et al. Cd38-specific chimeric antigen receptor expressing natural killer khyg-1 cells: A proof of concept for an "Off the shelf" therapy for multiple myeloma. Hemasphere (2021) 5(7):e596. doi: 10.1097/HS9.0000000000000596
284. Holthof LC, Stikvoort A, van der Horst HJ, Gelderloos AT, Poels R, Li F, et al. Bone marrow mesenchymal stromal cell-mediated resistance in multiple myeloma against nk cells can be overcome by introduction of Cd38-car or trail-variant. Hemasphere (2021) 5(5):e561. doi: 10.1097/HS9.0000000000000561
285. Fabian KP, Padget MR, Donahue RN, Solocinski K, Robbins Y, Allen CT, et al. Pd-L1 targeting high-affinity nk (T-hank) cells induce direct antitumor effects and target suppressive mdsc populations. J Immunother Cancer (2020) 8(1). doi: 10.1136/jitc-2019-000450
286. Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. Nk cells expressing a chimeric activating receptor eliminate mdscs and rescue impaired car-T cell activity against solid tumors. Cancer Immunol Res (2019) 7(3):363–75. doi: 10.1158/2326-6066.CIR-18-0572
287. Sakemura R, Hefazi M, Siegler EL, Cox MJ, Larson DP, Hansen MJ, et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to cart-cell therapy in multiple myeloma. Blood (2022) 139(26):3708–21. doi: 10.1182/blood.2021012811
288. Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O'Connor RS, et al. Car-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun (2021) 12(1):877. doi: 10.1038/s41467-021-20893-2
289. Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature (2014) 508(7495):269–73. doi: 10.1038/nature13034
290. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in nk-Cell-Mediated target cell killing without affecting adcc. Eur J Immunol (2013) 43(10):2756–64. doi: 10.1002/eji.201343448
291. Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, van Gelder M, Bos GM, et al. Hypoxia induced impairment of nk cell cytotoxicity against multiple myeloma can be overcome by il-2 activation of the nk cells. PloS One (2013) 8(5):e64835. doi: 10.1371/journal.pone.0064835
292. Yamada N, Yamanegi K, Ohyama H, Hata M, Nakasho K, Futani H, et al. Hypoxia downregulates the expression of cell surface mica without increasing soluble mica in osteosarcoma cells in a hif-1α-Dependent manner. Int J Oncol (2012) 41(6):2005–12. doi: 10.3892/ijo.2012.1630
293. Barsoum IB, Hamilton TK, Li X, Cotechini T, Miles EA, Siemens DR, et al. Hypoxia induces escape from innate immunity in cancer cells via increased expression of Adam10: Role of nitric oxide. Cancer Res (2011) 71(24):7433–41. doi: 10.1158/0008-5472.Can-11-2104
294. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme b degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U.S.A. (2013) 110(43):17450–5. doi: 10.1073/pnas.1304790110
295. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5'-Nucleotidase (Cd73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest (2002) 110(7):993–1002. doi: 10.1172/jci15337
296. Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, et al. Mtorc1-dependent metabolic reprogramming is a prerequisite for nk cell effector function. J Immunol (2014) 193(9):4477–84. doi: 10.4049/jimmunol.1401558
297. Keating SE, Zaiatz-Bittencourt V, Loftus RM, Keane C, Brennan K, Finlay DK, et al. Metabolic reprogramming supports ifn-Γ production by Cd56bright nk cells. J Immunol (2016) 196(6):2552–60. doi: 10.4049/jimmunol.1501783
298. Jensen H, Potempa M, Gotthardt D, Lanier LL. Cutting edge: Il-2-Induced expression of the amino acid transporters Slc1a5 and Cd98 is a prerequisite for Nkg2d-mediated activation of human nk cells. J Immunol (2017) 199(6):1967–72. doi: 10.4049/jimmunol.1700497
299. Terrén I, Orrantia A, Mosteiro A, Vitallé J, Zenarruzabeitia O, Borrego F. Metabolic changes of interleukin-12/15/18-Stimulated human nk cells. Sci Rep (2021) 11(1):6472. doi: 10.1038/s41598-021-85960-6
300. Loftus RM, Assmann N, Kedia-Mehta N, O'Brien KL, Garcia A, Gillespie C, et al. Amino acid-dependent cmyc expression is essential for nk cell metabolic and functional responses in mice. Nat Commun (2018) 9(1):2341. doi: 10.1038/s41467-018-04719-2
301. Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem (2003) 278(51):51606–12. doi: 10.1074/jbc.M310722200
302. Parameswaran R, Ramakrishnan P, Moreton SA, Xia Z, Hou Y, Lee DA, et al. Repression of Gsk3 restores nk cell cytotoxicity in aml patients. Nat Commun (2016) 7:11154. doi: 10.1038/ncomms11154
303. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. Ldha-associated lactic acid production blunts tumor immunosurveillance by T and nk cells. Cell Metab (2016) 24(5):657–71. doi: 10.1016/j.cmet.2016.08.011
304. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and nk cells. J Immunol (2013) 191(3):1486–95. doi: 10.4049/jimmunol.1202702
305. Pötzl J, Roser D, Bankel L, Hömberg N, Geishauser A, Brenner CD, et al. Reversal of tumor acidosis by systemic buffering reactivates nk cells to express ifn-Γ and induces nk cell-dependent lymphoma control without other immunotherapies. Int J Cancer (2017) 140(9):2125–33. doi: 10.1002/ijc.30646
306. Kobayashi T, Lam PY, Jiang H, Bednarska K, Gloury R, Murigneux V, et al. Increased lipid metabolism impairs nk cell function and mediates adaptation to the lymphoma environment. Blood (2020) 136(26):3004–17. doi: 10.1182/blood.2020005602
307. Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol (2018) 19(12):1330–40. doi: 10.1038/s41590-018-0251-7
308. Assmann N, O'Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, et al. Srebp-controlled glucose metabolism is essential for nk cell functional responses. Nat Immunol (2017) 18(11):1197–206. doi: 10.1038/ni.3838
309. Bernardini G, Antonangeli F, Bonanni V, Santoni A. Dysregulation of Chemokine/Chemokine receptor axes and nk cell tissue localization during diseases. Front Immunol (2016) 7:402. doi: 10.3389/fimmu.2016.00402
310. Pellegrino A, Ria R, Pietro GD, Cirulli T, Surico G, Pennisi A, et al. Bone marrow endothelial cells in multiple myeloma secrete cxc-chemokines that mediate interactions with plasma cells. Br J Haematol (2005) 129(2):248–56. doi: 10.1111/j.1365-2141.2005.05443.x
311. Zannettino ACW, Farrugia AN, Kortesidis A, Manavis J, To LB, Martin SK, et al. Elevated serum levels of stromal-derived factor-1α are associated with increased osteoclast activity and osteolytic bone disease in multiple myeloma patients. Cancer Res (2005) 65(5):1700–9. doi: 10.1158/0008-5472.CAN-04-1687
312. Barbieri F, Bajetto A, Thellung S, Würth R, Florio T. Drug design strategies focusing on the Cxcr4/Cxcr7/Cxcl12 pathway in leukemia and lymphoma. Expert Opin Drug Discovery (2016) 11(11):1093–109. doi: 10.1080/17460441.2016.1233176
313. Magalhães-Gama F, Kerr MWA, de Araújo ND, Ibiapina HNS, Neves JCF, Hanna FSA, et al. Imbalance of chemokines and cytokines in the bone marrow microenvironment of children with b-cell acute lymphoblastic leukemia. J Oncol (2021) 2021:5530650. doi: 10.1155/2021/5530650
314. Kittang AO, Hatfield K, Sand K, Reikvam H, Bruserud Ø. The chemokine network in acute myelogenous leukemia: Molecular mechanisms involved in leukemogenesis and therapeutic implications. Curr Topics Microbiol Immunol (2010) 341:149–72. doi: 10.1007/82_2010_25
315. Ruiduo C, Ying D, Qiwei W. Cxcl9 promotes the progression of diffuse Large b-cell lymphoma through up-regulating β-catenin. Biomed Pharmacother = Biomed Pharmacotherapie (2018) 107:689–95. doi: 10.1016/j.biopha.2018.07.171
316. Venetz D, Ponzoni M, Schiraldi M, Ferreri AJ, Bertoni F, Doglioni C, et al. Perivascular expression of Cxcl9 and Cxcl12 in primary central nervous system lymphoma: T-cell infiltration and positioning of malignant b cells. Int J Cancer (2010) 127(10):2300–12. doi: 10.1002/ijc.25236
317. Bonanni V, Antonangeli F, Santoni A, Bernardini G. Targeting of Cxcr3 improves anti-myeloma efficacy of adoptively transferred activated natural killer cells. J Immunother Cancer (2019) 7(1):290. doi: 10.1186/s40425-019-0751-5
318. Chiu J, Ernst DM, Keating A. Acquired natural killer cell dysfunction in the tumor microenvironment of classic Hodgkin lymphoma. Front Immunol (2018) 9:267. doi: 10.3389/fimmu.2018.00267
319. Zheng Y, Wang Q, Li T, Qian J, Lu Y, Li Y, et al. Role of myeloma-derived mif in myeloma cell adhesion to bone marrow and chemotherapy response. J Natl Cancer Institute (2016) 108(11). doi: 10.1093/jnci/djw131
320. Till KJ, Lin K, Zuzel M, Cawley JC. The chemokine receptor Ccr7 and α4 integrin are important for migration of chronic lymphocytic leukemia cells into lymph nodes. Blood (2002) 99(8):2977–84. doi: 10.1182/blood.V99.8.2977
321. Redondo-Muñoz J, José Terol M, García-Marco JA, García-Pardo A. Matrix metalloproteinase-9 is up-regulated by Ccl21/Ccr7 interaction via extracellular signal-regulated kinase-1/2 signaling and is involved in Ccl21-driven b-cell chronic lymphocytic leukemia cell invasion and migration. Blood (2008) 111(1):383–6. doi: 10.1182/blood-2007-08-107300
322. Bekiaris V, Gaspal F, Kim M-Y, Withers DR, McConnell FM, Anderson G, et al. Cd30 is required for Ccl21 expression and Cd4 T cell recruitment in the absence of lymphotoxin signals. J Immunol (2009) 182(8):4771–5. doi: 10.4049/jimmunol.0803481
323. Luevano M, Daryouzeh M, Alnabhan R, Querol S, Khakoo S, Madrigal A, et al. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum Immunol (2012) 73(3):248–57. doi: 10.1016/j.humimm.2011.12.015
324. Levy E, Reger R, Segerberg F, Lambert M, Leijonhufvud C, Baumer Y, et al. Enhanced bone marrow homing of natural killer cells following mrna transfection with gain-of-Function variant Cxcr4(R334x). Front Immunol (2019) 10:1262. doi: 10.3389/fimmu.2019.01262
325. Levy ER, Clara JA, Reger RN, Allan DSJ, Childs RW. Rna-seq analysis reveals Ccr5 as a key target for crispr gene editing to regulate in vivo nk cell trafficking. Cancers (Basel) (2021) 13(4). doi: 10.3390/cancers13040872
326. Beider K, Nagler A, Wald O, Franitza S, Dagan-Berger M, Wald H, et al. Involvement of Cxcr4 and il-2 in the homing and retention of human nk and nk T cells to the bone marrow and spleen of Nod/Scid mice. Blood (2003) 102(6):1951–8. doi: 10.1182/blood-2002-10-3293
327. Ng YY, Du Z, Zhang X, Chng WJ, Wang S. Cxcr4 and anti-bcma car Co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene Ther (2022) 29(5):475–83. doi: 10.1038/s41417-021-00365-x
328. Jamali A, Hadjati J, Madjd Z, Mirzaei HR, Thalheimer FB, Agarwal S, et al. Highly efficient generation of transgenically augmented car nk cells overexpressing Cxcr4. Front Immunol (2020) 11:2028. doi: 10.3389/fimmu.2020.02028
329. Ponzetta A, Benigni G, Antonangeli F, Sciumè G, Sanseviero E, Zingoni A, et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res (2015) 75(22):4766–77. doi: 10.1158/0008-5472.Can-15-1320
330. Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M, et al. Efficient mrna-based genetic engineering of human nk cells with high-affinity Cd16 and Ccr7 augments rituximab-induced adcc against lymphoma and targets nk cell migration toward the lymph node-associated chemokine Ccl19. Front Immunol (2016) 7:105. doi: 10.3389/fimmu.2016.00105
331. Mailliard RB, Alber SM, Shen H, Watkins SC, Kirkwood JM, Herberman RB, et al. Il-18-Induced Cd83+Ccr7+ nk helper cells. J Exp Med (2005) 202(7):941–53. doi: 10.1084/jem.20050128
332. Marcenaro E, Cantoni C, Pesce S, Prato C, Pende D, Agaugué S, et al. Uptake of Ccr7 and acquisition of migratory properties by human kir+ nk cells interacting with monocyte-derived dc or ebv cell lines: Regulation by Kir/Hla-class I interaction. Blood (2009) 114(19):4108–16. doi: 10.1182/blood-2009-05-222265
333. Somanchi SS, Somanchi A, Cooper LJ, Lee DA. Engineering lymph node homing of ex vivo-expanded human natural killer cells via trogocytosis of the chemokine receptor Ccr7. Blood (2012) 119(22):5164–72. doi: 10.1182/blood-2011-11-389924
334. Ingegnere T, Mariotti FR, Pelosi A, Quintarelli C, De Angelis B, Tumino N, et al. Human car nk cells: A new non-viral method allowing high efficient transfection and strong tumor cell killing. Front Immunol (2019) 10:957. doi: 10.3389/fimmu.2019.00957
335. Schomer NT, Jiang ZK, Lloyd MI, Klingemann H, Boissel L. Ccr7 expression in Cd19 chimeric antigen receptor-engineered natural killer cells improves migration toward Ccl19-expressing lymphoma cells and increases tumor control in mice with human lymphoma. Cytotherapy (2022). doi: 10.1016/j.jcyt.2022.02.006
336. Hong S, Yu C, Wang P, Shi Y, Cao W, Cheng B, et al. Glycoengineering of nk cells with glycan ligands of Cd22 and selectins for b-cell lymphoma therapy. Angewandte Chemie (International ed English) (2021) 60(7):3603–10. doi: 10.1002/anie.202005934
337. Bachanova V, McKenna DH, Luo X, Defor TE, Cooley S, Warlick E, et al. First-in-Human phase I study of nicotinamide-expanded related donor natural killer cells for the treatment of Relapsed/Refractory non-Hodgkin lymphoma and multiple myeloma. Biol Blood Marrow Transplant (2019) 25(Supplement 3):S175–S6. doi: 10.1016/j.bbmt.2018.12.317
338. Frei GM, Persi N, Lador C, Peled A, Cohen YC, Nagler A, et al. Nicotinamide, a form of vitamin B3, promotes expansion of natural killer cells that display increased in vivo survival and cytotoxic activity. Blood (2011) 118(21):4035. doi: 10.1182/blood.V118.21.4035.4035
339. Frei GM, Berg M, Peled T, Reger RN, Kotecha R, Onishi T, et al. Improved Homing to Bone Marrow, Spleen and Lung of Adoptively Infused Nk Cells Expanded Ex Vivo with the Small Molecule Nicotinamide Using Feeder-Free Conditions. Blood (2013) 122(21):897. doi: 10.1182/blood.V122.21.897.897.
340. Lee J, Kang TH, Yoo W, Choi H, Jo S, Kong K, et al. An antibody designed to improve adoptive nk-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol Res (2019) 7(2):219–29. doi: 10.1158/2326-6066.Cir-18-0317
341. Ng YY, Tay JCK, Wang S. Cxcr1 expression to improve anti-cancer efficacy of intravenously injected car-nk cells in mice with peritoneal xenografts. Mol Ther - Oncolytics (2020) 16:75–85. doi: 10.1016/j.omto.2019.12.006
342. Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human nk cells to express Cxcr2 improves migration to renal cell carcinoma. J Immunother Cancer (2017) 5(1):73. doi: 10.1186/s40425-017-0275-9
343. Nakayama T, Hieshima K, Izawa D, Tatsumi Y, Kanamaru A, Yoshie O. Cutting edge: Profile of chemokine receptor expression on human plasma cells accounts for their efficient recruitment to target tissues. J Immunol (2003) 170(3):1136–40. doi: 10.4049/jimmunol.170.3.1136
344. Goodyear OC, Essex S, Seetharam A, Basu S, Moss P, Pratt G. Neoplastic plasma cells generate an inflammatory environment within bone marrow and markedly alter the distribution of T cells between lymphoid compartments. Oncotarget (2017) 8(18):30383–94. doi: 10.18632/oncotarget.16628
345. Kohsari M, Khadem-Ansari MH, Rasmi Y, Sayyadi H. Serum levels of interleukin-8 and soluble interleukin-6 receptor in patients with stage-I multiple myeloma: A case-control study. Asian Pacific J Cancer Prev APJCP (2020) 21(1):127–32. doi: 10.31557/apjcp.2020.21.1.127
346. Wierda WG, Johnson MM, Do KA, Manshouri T, Dey A, O'Brien S, et al. Plasma interleukin 8 level predicts for survival in chronic lymphocytic leukaemia. Br J Haematol (2003) 120(3):452–6. doi: 10.1046/j.1365-2141.2003.04118.x
347. Schinke C, Giricz O, Li W, Shastri A, Gordon S, Barreyro L, et al. Il8-Cxcr2 pathway inhibition as a therapeutic strategy against mds and aml stem cells. Blood (2015) 125(20):3144–52. doi: 10.1182/blood-2015-01-621631
348. Dever DP, Porteus MH. The changing landscape of gene editing in hematopoietic stem cells: A step towards Cas9 clinical translation. Curr Opin Hematol (2017) 24(6):481–8. doi: 10.1097/MOH.0000000000000385
349. Fix SM, Jazaeri AA, Hwu P. Applications of crispr genome editing to advance the next generation of adoptive cell therapies for cancer. Cancer Discovery (2021) 11(3):560–74. doi: 10.1158/2159-8290.CD-20-1083
350. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-Rna-Guided DNA endonuclease in adaptive bacterial immunity. Science (2012) 337(6096):816–21. doi: 10.1126/science.1225829
351. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell (2012) 47(4):497–510. doi: 10.1016/j.molcel.2012.07.029
352. Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of hr in mid s phase. Mol Cell (2012) 47(2):320–9. doi: 10.1016/j.molcel.2012.05.052
353. Robbins GM, Wang M, Pomeroy EJ, Moriarity BS. Nonviral genome engineering of natural killer cells. Stem Cell Res Ther (2021) 12(1):350. doi: 10.1186/s13287-021-02406-6
354. Schmidt P, Raftery MJ, Pecher G. Engineering nk cells for car therapy-recent advances in gene transfer methodology. Front Immunol (2020) 11:611163. doi: 10.3389/fimmu.2020.611163
355. Huang RS, Lai MC, Shih HA, Lin S. A robust platform for expansion and genome editing of primary human natural killer cells. J Exp Med (2021) 218(3). doi: 10.1084/jem.20201529
356. Huang RS, Shih HA, Lai MC, Chang YJ, Lin S. Enhanced nk-92 cytotoxicity by crispr genome engineering using Cas9 ribonucleoproteins. Front Immunol (2020) 11:1008. doi: 10.3389/fimmu.2020.01008
357. Rautela J, Surgenor E, Huntington ND. Drug target validation in primary human natural killer cells using crispr rnp. J Leukoc Biol (2020) 108(4):1397–408. doi: 10.1002/JLB.2MA0620-074R
358. Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, et al. A genetically engineered primary human natural killer cell platform for cancer immunotherapy. Mol Ther (2020) 28(1):52–63. doi: 10.1016/j.ymthe.2019.10.009
359. Guo Y, Feng X, Jiang Y, Shi X, Xing X, Liu X, et al. Pd1 blockade enhances cytotoxicity of in vitro expanded natural killer cells towards myeloma cells. Oncotarget (2016) 7(30):48360–74. doi: 10.18632/oncotarget.10235
360. Ndhlovu LC, Lopez-Verges S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood (2012) 119(16):3734–43. doi: 10.1182/blood-2011-11-392951
361. da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of nk-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res (2014) 2(5):410–22. doi: 10.1158/2326-6066.CIR-13-0171
362. Morimoto T, Nakazawa T, Matsuda R, Nishimura F, Nakamura M, Yamada S, et al. Crispr-Cas9-Mediated Tim3 knockout in human natural killer cells enhances growth inhibitory effects on human glioma cells. Int J Mol Sci (2021) 22(7). doi: 10.3390/ijms22073489
363. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor tigit prevents nk cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol (2018) 19(7):723–32. doi: 10.1038/s41590-018-0132-0
364. Berrien-Elliott MM, Cashen AF, Cubitt CC, Neal CC, Wong P, Wagner JA, et al. Multidimensional analyses of donor memory-like nk cells reveal new associations with response after adoptive immunotherapy for leukemia. Cancer Discovery (2020) 10(12):1854–71. doi: 10.1158/2159-8290.CD-20-0312
365. Bexte T, Alzubi J, Reindl LM, Wendel P, Schubert R, Salzmann-Manrique E, et al. Crispr-Cas9 based gene editing of the immune checkpoint Nkg2a enhances nk cell mediated cytotoxicity against multiple myeloma. Oncoimmunology (2022) 11(1):2081415. doi: 10.1080/2162402x.2022.2081415
366. Kanaya M, Philippon C, Cieslar-Pobuda A, Cichocki F, Saetersmoen M, Mahmood S, et al. Car19 ipsc-derived nk cells utilize the innate functional potential mediated through Nkg2a-driven education and override the hla-e check point to effectively target b cell lymphoma. Blood (2020) 136(Supplement 1):34–5. doi: 10.1182/blood-2020-138527
367. Yamamoto K, Blum R, Kaufman DS. Adam17-deficient pluripotent stem cell-derived natural killer cells possess improved antibody-dependent cellular cytotoxicity and antitumor activity. Blood (2020) 136(Supplement 1):2. doi: 10.1182/blood-2020-137766
368. Inagaki-Ohara K, Hanada T, Yoshimura A. Negative regulation of cytokine signaling and inflammatory diseases. Curr Opin Pharmacol (2003) 3(4):435–42. doi: 10.1016/s1471-4892(03)00070-5
369. Yoshimura A, Nishinakamura H, Matsumura Y, Hanada T. Negative regulation of cytokine signaling and immune responses by socs proteins. Arthritis Res Ther (2005) 7(3):100–10. doi: 10.1186/ar1741
370. Naeimi Kararoudi M, Elmas E, Lamb M, Chakravarti N, Trikha P, Lee DA. Disruption of Socs3 promotes the anti-cancer efficacy of primary nk cells. Blood (2018) 132(Supplement 1):5687. doi: 10.1182/blood-2018-99-116621
371. Matalon O, Barda-Saad M. Cbl ubiquitin ligases mediate the inhibition of natural killer cell activity. Commun Integr Biol (2016) 9(6):e1216739. doi: 10.1080/19420889.2016.1216739
372. Guo X, Mahlakoiv T, Ye Q, Somanchi S, He S, Rana H, et al. Cblb ablation with Crispr/Cas9 enhances cytotoxicity of human placental stem cell-derived nk cells for cancer immunotherapy. J Immunother Cancer (2021) 9(3). doi: 10.1136/jitc-2020-001975
373. Derynck R, Turley SJ, Akhurst RJ. Tgfbeta biology in cancer progression and immunotherapy. Nat Rev Clin Oncol (2021) 18(1):9–34. doi: 10.1038/s41571-020-0403-1
374. Naeimi Kararoudi M, Dolatshad H, Trikha P, Hussain SA, Elmas E, Foltz JA, et al. Generation of knock-out primary and expanded human nk cells using Cas9 ribonucleoproteins. J Vis Exp (2018) 136). doi: 10.3791/58237
375. Zhang L, Zuris JA, Viswanathan R, Edelstein JN, Turk R, Thommandru B, et al. Ascas12a ultra nuclease facilitates the rapid generation of therapeutic cell medicines. Nat Commun (2021) 12(1):3908. doi: 10.1038/s41467-021-24017-8
376. Shaim H, Shanley M, Basar R, Daher M, Gumin J, Zamler DB, et al. Targeting the alphav Integrin/Tgf-beta axis improves natural killer cell function against glioblastoma stem cells. J Clin Invest (2021) 131(14). doi: 10.1172/JCI142116
377. Clara JA, Levy ER, Reger R, Barisic S, Chen L, Cherkasova E, et al. High-affinity Cd16 integration into a Crispr/Cas9-edited Cd38 locus augments Cd38-directed antitumor activity of primary human natural killer cells. J Immunother Cancer (2022) 10(2). doi: 10.1136/jitc-2021-003804
378. Naeimi Kararoudi M, Nagai Y, Elmas E, Pereira MdeSF, Ali SA, Imus PH, et al. Cd38 deletion of human primary nk cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood (2020) 136(21):2416–27. doi: 10.1182/blood.2020006200
379. You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, et al. A novel Cd7 chimeric antigen receptor-modified nk-92mi cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res (2019) 9(1):64–78.
380. Gao L, Yang L, Zhang S, Ge Z, Su M, Shi Y, et al. Engineering nk-92 cell by upregulating Cxcr2 and il-2 via crispr-Cas9 improves its antitumor effects as cellular immunotherapy for human colon cancer. J Interferon Cytokine Res (2021) 41(12):450–60. doi: 10.1089/jir.2021.0078
381. Borges CM, Wasko K, Nasser JM, Donahue K, Pfautz A, Antony LP, et al. Preclinical development of edit-201, a multigene edited healthy donor nk cell with enhanced anti-tumor function and superior serial killing activity in an immunosuppressive environment. Blood (2020) 136(Supplement 1):33. doi: 10.1182/blood-2020-139988
382. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. Crispr-engineered T cells in patients with refractory cancer. Science (2020) 367(6481). doi: 10.1126/science.aba7365
383. Naeimi Kararoudi M, Likhite S, Elmas E, Schwartz M, Sorathia K, Yamamoto K, et al. Cd33 targeting primary car-nk cells generated by crispr mediated gene insertion show enhanced anti-aml activity. Blood (2020) 136(Supplement 1):3. doi: 10.1182/blood-2020-142494
384. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of a*T to G*C in genomic DNA without DNA cleavage. Nature (2017) 551(7681):464–71. doi: 10.1038/nature24644
385. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature (2016) 533(7603):420–4. doi: 10.1038/nature17946
Keywords: chimeric antigen receptor (CAR), CAR NK cells, hematologic tumor, genome editing, CRISPR/Cas9, tumor microenvironment, tumor resistance, CAR persistence
Citation: Valeri A, García-Ortiz A, Castellano E, Córdoba L, Maroto-Martín E, Encinas J, Leivas A, Río P and Martínez-López J (2022) Overcoming tumor resistance mechanisms in CAR-NK cell therapy. Front. Immunol. 13:953849. doi: 10.3389/fimmu.2022.953849
Received: 26 May 2022; Accepted: 11 July 2022;
Published: 03 August 2022.
Edited by:
Min Xiao, Huazhong University of Science and Technology, ChinaReviewed by:
Reona Sakemura, Mayo Clinic, United StatesCopyright © 2022 Valeri, García-Ortiz, Castellano, Córdoba, Maroto-Martín, Encinas, Leivas, Río and Martínez-López. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joaquín Martínez-López, am1hcnRpMDFAdWNtLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.