 Umeharu Ohto
Umeharu Ohto
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 15 September 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.953530
This article is part of the Research TopicInnate Immunity and Intercellular CommunicationView all 12 articles
Innate immunity is a primary defense system against microbial infections. Innate immune pattern recognition receptors (PRRs) play pivotal roles in detection of invading pathogens. When pathogens, such as bacteria and viruses, invade our bodies, their components are recognized by PRRs as pathogen-associated molecular patterns (PAMPs), activating the innate immune system. Cellular components such as DNA and RNA, acting as damage-associated molecular patterns (DAMPs), also activate innate immunity through PRRs under certain conditions. Activation of PRRs triggers inflammatory responses, interferon-mediated antiviral responses, and the activation of acquired immunity. Research on innate immune receptors is progressing rapidly. A variety of these receptors has been identified, and their regulatory mechanisms have been elucidated. Nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) constitute a major family of intracellular PRRs and are involved in not only combating pathogen invasion but also maintaining normal homeostasis. Some NLRs are known to form multi-protein complexes called inflammasomes, a process that ultimately leads to the production of inflammatory cytokines and induces pyroptosis through the proteolytic cascade. The aberrant activation of NLRs has been found to be associated with autoimmune diseases. Therefore, NLRs are considered targets for drug discovery, such as for antiviral drugs, immunostimulants, antiallergic drugs, and autoimmune disease drugs. This review summarizes our recent understanding of the activation and regulation mechanisms of NLRs, with a particular focus on their structural biology. These include NOD2, neuronal apoptosis inhibitory protein (NAIP)/NLRC4, NLR family pyrin domain containing 1 (NLRP1), NLRP3, NLRP6, and NLRP9. NLRs are involved in a variety of diseases, and their detailed activation mechanisms based on structural biology can aid in developing therapeutic agents in the future.
Bacteria and viruses invading our bodies are recognized as foreign substrates and, therefore, activate the immune system. Immune responses are classified into innate and acquired immunity. In the early stages of microbial invasion, innate immunity is triggered first (1, 2). Pathogen-associated molecular patterns (PAMPs) are recognized by an innate immune receptor called pattern recognition receptor (PRR) (3). Signals are transmitted downstream, ultimately triggering inflammatory responses, interferon-mediated antiviral responses, and the activation of acquired immunity (4). The discovery of toll-like receptors (TLRs) in the late 1990s led to an explosion of research on innate immune receptors, resulting in the identification of a variety of innate immune receptors (5). Each of these receptors was found to be involved in the recognition of unique PAMPs (6). Previously, innate immunity was considered a nonspecific immune response; however, it has now been recognized as a specific immune response due to PRRs (7). In addition to recognizing PAMPs, innate immune receptors may be activated by self-derived molecular patterns (damage-associated molecular patterns, DAMPs) released from necrotic cells, which are known to cause autoimmune diseases (8). Therefore, innate immune receptors are potential drug targets, such as for antiviral drugs, immunostimulants, antiallergic drugs, and drugs for autoimmune diseases (9).
In addition to the aforementioned TLRs, representative innate immune receptors have been identified as nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) (10), retinoic acid-inducible gene I (RIG I)-like receptors (11), absent in melanoma 2 (AIM2)-like receptors (12), and cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) (13). TLRs are located on the plasma membrane surface and endosomal membranes, whereas other receptors are located in the cytoplasm. Moreover, TLRs recognize PAMPs and DAMPs that have entered the cell. Recently, a rapid progress in the study of intracellular sensors that exist in the cytoplasm and activate innate immunity by recognizing foreign substances, such as pathogen-derived DNA and RNA, has been observed (14, 15). Structural biology studies using X-ray crystallography and cryo-electron microscopy (EM) have made remarkable progress in recent years. Moreover, these studies have played a major role in elucidating the mechanisms by which innate immune receptors recognize PAMPs and DAMPs and activate innate immunity. This review focuses on NLRs, a family of innate immune receptors that exist primarily in the cytoplasm, and introduces the mechanisms of activity regulation and signal transduction revealed by structural biology studies conducted over the past decade (Table 1).

Table 1 Summary of structural studies of NLRs.
To date, 22 NLRs with various functional roles have been identified in humans (31). NLR typically consists of three functional domains, namely N-terminal signaling, central [NAIP, CIITA, HETE, TP1 (NACHT)], and leucine-rich repeat (LRR) domains (32–35) (Figure 1A). The N-terminal signaling domain is responsible for signal transduction through interactions with downstream adaptor proteins. The central NACHT domain has ATPase activity and is assumed to be self-oligomerized through this domain on activation. The NACHT domain is further classified into nucleotide-binding domain (NBD), helical domain 1 (HD1), winged-helix domain (WHD), and HD2 subdomains. The LRR domain on the C-terminal side is believed to be involved in ligand recognition and functional regulation. NLRs are classified into subfamilies according to the type of N-terminal signaling domain: those with pyrin domain (PYD) are called the NLR pyrin domain containing (NLRP) family and those with caspase recruitment domain (CARD) are called the NLR CARD containing (NLRC) family. An NLRP uses its PYD to form a scaffold that interacts with the adaptor, apoptosis-associated, speck-like protein containing a CARD (ASC) through PYD-PYD interactions to recruit procaspase-1. Procaspase-1 is activated to caspase-1, which further cleaves pro-interleukin (IL)-1β and pro-IL-18, resulting in the generation of mature IL-1β and IL-18, respectively, and triggering an inflammatory response. Caspase-1 also induces pyroptosis by cleaving gasdermin D. In addition to the caspase-1-mediated canonical inflammasomes, noncanonical inflammasomes involving caspase-4/5 in human and caspase-11 in mice have been identified and are known to respond to cytosolic LPS (37). An NLRC, however, is thought to activate caspase-1 through direct CARD-CARD interactions in addition to the ASC-mediated activation of caspase-1. Diverse PAMPs and DAMPs have been found to activate NLRs. Some NLRs are known to activate innate immunity by forming high-molecular-weight multi-protein complexes called inflammasomes to signal downstream.
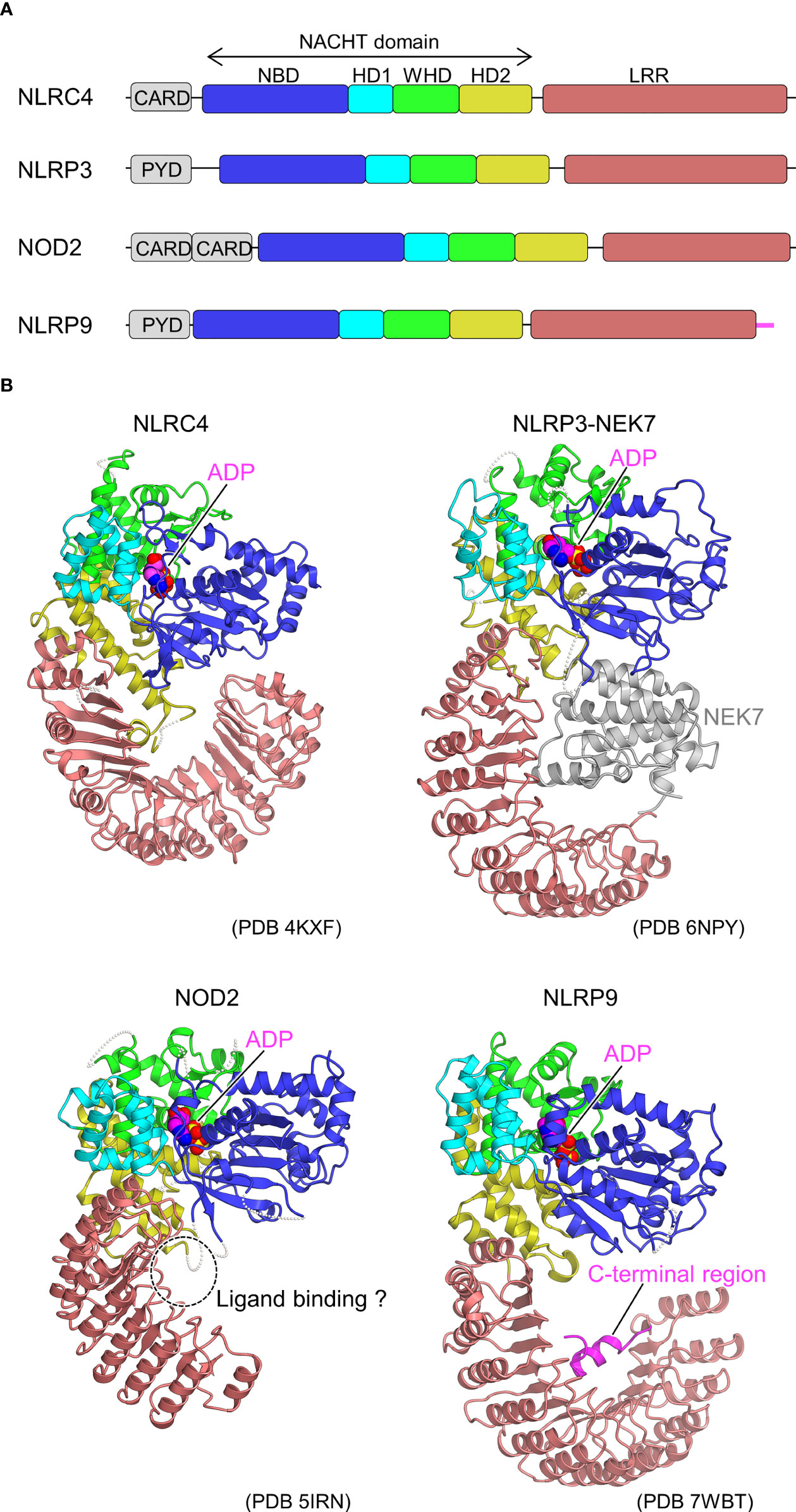
Figure 1 Domain organization and structures of inactive NLRs. (A) Domain organization of NLRs. Each of the domains and sub domains are indicated by the different colors and are correspondingly indicated in Figure 1B. (B), Structures of inactive NLRs. Structures of inactive NLRC4 (PDB 4KXF) (16), NLRP3-NEK7 complex (PDB 6NPY) (36), NOD2 (PDB 5IRN) (29), and NLRP9 (PDB 7WBT) (30) are shown with the domains colored as per (A). Bound ADP molecules are shown in space filling representations. In the NLRP3-NEK7 complex, bound NEK7 is shown in gray. The potential ligand-binding site in NOD2 and the C-terminal region of NLRP9 are indicated. Structural figures were generated using CueMol throughout this review (http://www.cuemol.org).
In the neuronal apoptosis inhibitory protein (NAIP)/NLRC4 pathway, flagellin, a component of bacterial flagella, and the bacterial rod protein PrgJ bind to NAIPs, whereupon NLRC4 binds as an adapter to form active inflammasome (38–43).
In 2013, the first crystal structure of NLR was reported for the inactive form of NLRC4, lacking the CARD domain (16) (Figure 1B). The inactivated conformation of NLRC4 was a monomeric, autoinhibited conformation, in which the region of the NACHT domain involved in self-association was covered by the LRR domain. Thus, the LRR domain of NLRC4 functions to sequester NLRC4 in a monomeric state. It was also found that ADP bound to the NACHT domain stabilizes the closed and autoinhibited conformation of NLRC4 by mediating the interactions between the subdomains of the NACHT domain. This was consistent with previous biochemical experiments showing that the deletion of the LRR domain leads to self-activation without NAIP or FliC (39).
Subsequently, cryo-EM analysis revealed that the inflammasome structure of NAIP2-NLRC4 is induced by the bacterial rod protein PrgJ, as reported almost simultaneously by two groups (17, 18) (Figure 2). A low-resolution structure of the NAIP5-NLRC4 helical oligomer induced by flagellin was also reported using cryo-EM tomography (19). Afterwards, cryo-EM structures of the flagellin-NAIP5-NLRC4 were reported, revealing the detailed ligand recognition mechanism of NAIP5 as well as how it leads to the oligomerization of NLRC4 (20, 21). The NAIP2-NLRC4 or NAIP5-NLRC4 oligomer induced by ligand forms a ring-like structure consisting of 10–12 molecules, including one NAIP molecule (Figure 2B). This oligomer is formed by unidirectional chain oligomerization of NLRC4 molecules, starting with the ligand-bound NAIP molecule. In the upper part of the ring, CARD assembly may provide a scaffold for CARD-CARD interactions with downstream caspase-1 (Figure 2B). Upon activation, closed NLRC4 is converted to an open conformation by binding to open NLRC4 via the NACHT domain (Figure 2A). During this process, rigid body motion at the linkage between HD1 and WHD of the NACHT domain is observed. This mechanism amplifies the signal by catalytically converting the closed structure to an open structure in a self-propagating manner. Moreover, this mechanism contrasts with the activation mechanism of the apoptotic protease-activating factor-1 (Apaf-1) apoptosome (octameric ring structure), which is related to the NLR. In the case of Apaf-1, each subunit must be activated by its own ligand, that is, a stoichiometric number of ligands is required for all subunits activation (44).
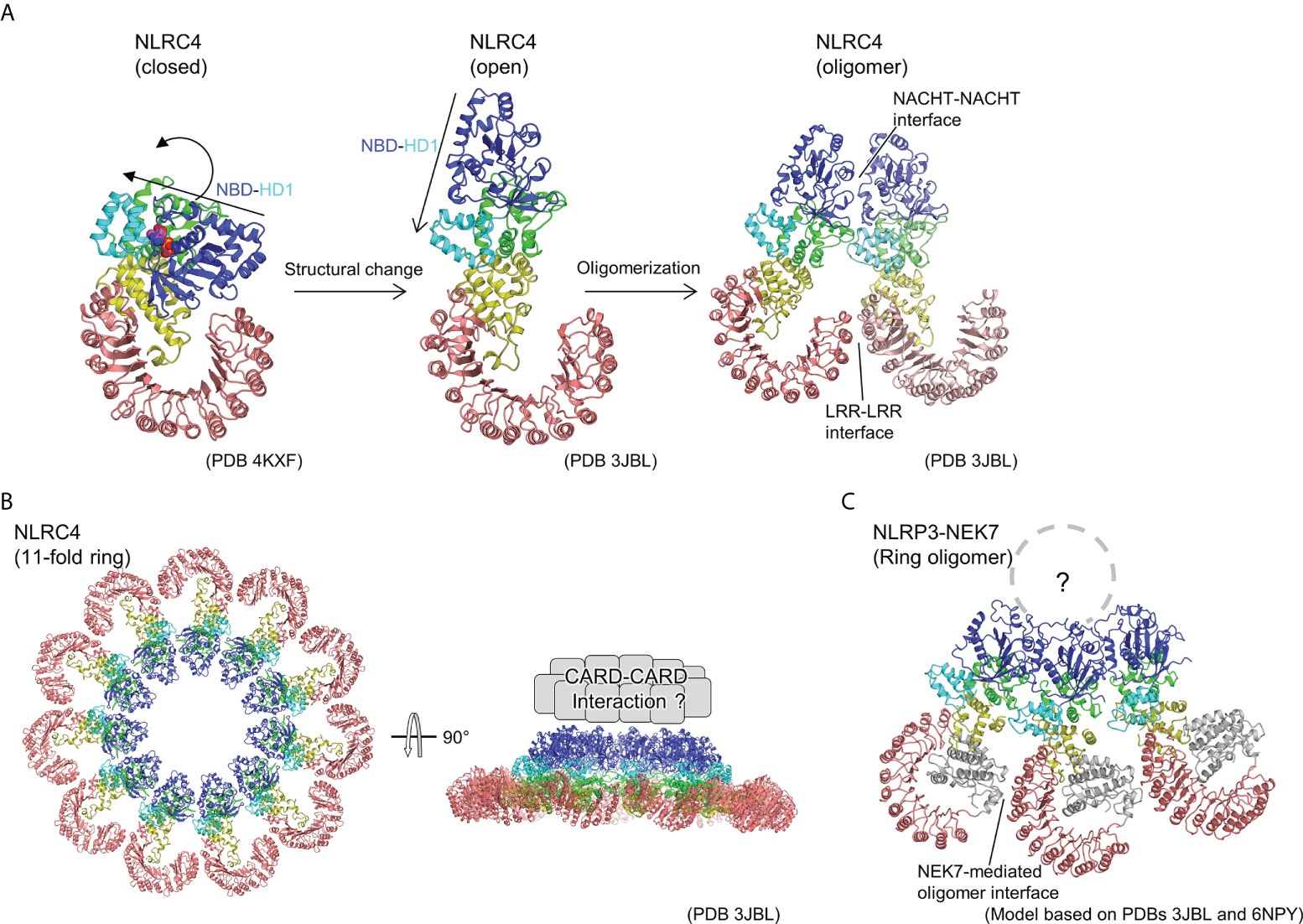
Figure 2 Ring-shaped active oligomer of NLRC4. (A) Structural changes underlying NLRC4 oligomerization. The NLRC4 monomer undergoes a structural change from a closed (PDB 4KXF) (16) to an open form (PDB 3JBL) (17), causing the NBD-HD1 part to undergo a large rotational movement relative to the other parts. This opens the NACHT domain and the corresponding activated NLRC4 molecules to form a laterally aligned dimer and subsequently form ring-shaped oligomer. (B) Structure of 11-fold ring-shaped NLRC4 oligomer (PDB 3JBL) (17). Top (left) and side (right) views are shown. The CARD domains are predicted to be concentrated at the top of the ring as shown schematically in the side view (right). (C) Hypothetical structure of NLR3-NEK7 oligomer. The structure of the inactive form of the NLRP3-NEK7 complex (6NPY) (36) was split into the NBD-HD1 and the WHD-HD2-LRR parts, and each was fitted into the corresponding part of the 11-fold ring oligomer of NLRC4 (PDB 3JBL) (17).
NLRP3 is one of the most well-studied inflammasome-forming NLRs. Moreover, its activators are diverse. For instance, nigericin, uric acid, amyloid-beta fibrils, extracellular ATP, and reactive oxygen species are a few activators of NLRP3 (20, 35). Some of these are thought to trigger NLRP3 activation by lowering intracellular K+ concentrations (45). NLRP3 activation requires two steps of stimulation: “priming” and “activation” (45–47). Priming stimuli include the TLR ligands Pam3CSK4, Poly(I:C), lipopolysaccharide (LPS), and R848, which activate TLRs to upregulate NLRP3 and caspase-1 expression and provide the soil for NLRP3 activation. In addition, priming causes post-translational modifications in NLRP3, such as phosphorylation (48), which are thought to be important for NLRP3 activation. The activating factors include nigericin, extracellular ATP, as well as silica, cholesterol, and uric acid crystals that destabilize lysosomes. As mentioned previously, a wide variety of NLRP3 activators exists, and the direct triggers of NLRP3 activation remain unclear. In addition, it has been reported that NLRP3 activation involves interactions with a variety of proteins. These include SGT1, HSP90 (49), thioredoxin-interacting protein (TXNIP) (50), mitochondrial antiviral-signaling protein (MAVS) (51), never in mitosis A-related kinase 7 (NEK7) (52–54), MAP/microtubule affinity-regulating kinase 4 (MARK4) (55), macrophage migration inhibitory factor (MIF) (56), DEAD box RNA helicase (DDX) 3X (57), and receptor of activated protein C kinase 1 (RACK1) (58). However, the mechanisms by which these factors regulate NLRP3 activation remain unclear.
As a starting point for the structural biology studies of NLRP3, the cryo-EM structure of inactivated human NLRP3 (PYD domain deleted) bound to NEK7 was first revealed (36) (Figure 1B). The overall structure was similar to the previously reported structures of NLRC4 (16) and NOD2 (29) in the inactivated form. The kinase C-lobe of NEK7 binds to the concave side of the LRR of NLRP3. Only the C-lobe of NEK7 was visible in the cryo-EM map, but the N-lobe did not clash with NLRP3 even when full-length NEK7 was superimposed. NLRP3 binds to NEK7 at multiple interaction sites (LRR, HD2, and NBD). This binding is suggested to involve electrostatic interactions between the positively charged surface of NEK7 and the negatively charged surface of NLRP3. NEK7 is known to form a complex with NEK9 to participate in mitosis (59); however, the NEK7 surface used for this complexation overlaps in part with the surface used for binding to NLRP3. Therefore, it was expected that once NEK7 binds to NLRP3, it cannot bind to NEK9 and vice versa.
As mentioned previously, upon activation, NLRC4 multimerizes and activates by opening the NACHT domain via a large rigid body rotation between HD1 and WHD (Figure 2A) (17, 18). Imitating the oligomeric structure of NLRC4, an oligomeric model of the NLRP3-NEK7 complex was constructed (Figure 2C), where NEK7 was found to be located at the boundary with the neighboring molecule in the oligomer. To investigate the importance of this modeled oligomeric interface, the authors of this paper performed experiments using mutants of NLRP3 and NEK7 and demonstrated that both mutants reduce NLRP3 activation, indicating that this NEK7–NLRP3 interface may be used when NLRP3 is activated (36). In the case of NLRC4, in addition to the contacts at the NACHT site, interactions at the LRR-LRR sites are observed during the formation of ring-shaped oligomers (Figure 2A, B) (17, 18). However, the LRR-LRR interaction in the NLRP3 oligomer is not possible between adjacent monomers because the LRR of NLRP3 is smaller than that of NLRC4. Considering the result of the mutational experiment showing the importance of hypothetical NEK7-NLRP3 interface describe earlier, NEK7-mediated bridging of adjacent LRRs of NLRP3 may reinforce the oligomerization of NLRP3. In other words, in NLRP3, as in the case of NLRC4, the interaction between NACHTs in the inner ring layer and that between LRRs via NEK7 in the outer ring layer are thought to contribute to oligomer formation.
Although experimental structural information on the activated oligomer of NLRP3 is not yet available, three groups have reported cryo-EM structures of the full-length NLRP3 oligomer in its inactivated form recently (Figure 3) (23–25). Paradoxically, this inactivated oligomer formation has been shown to be important in the regulation of NLRP3 activation (25). Mouse NLRP3 forms 12-, 14-, and 16-mer (Figure 3A) (23, 25), whereas human NLRP3 forms 10-mer (Figure 3B) (24) hollow, cage-like oligomeric structures with NACHT on the top surface and LRR-LRR interactions on the sides. The density of the PYD domains could not be clearly confirmed, but they were considered to be disordered and located inside or at the top of the cage.
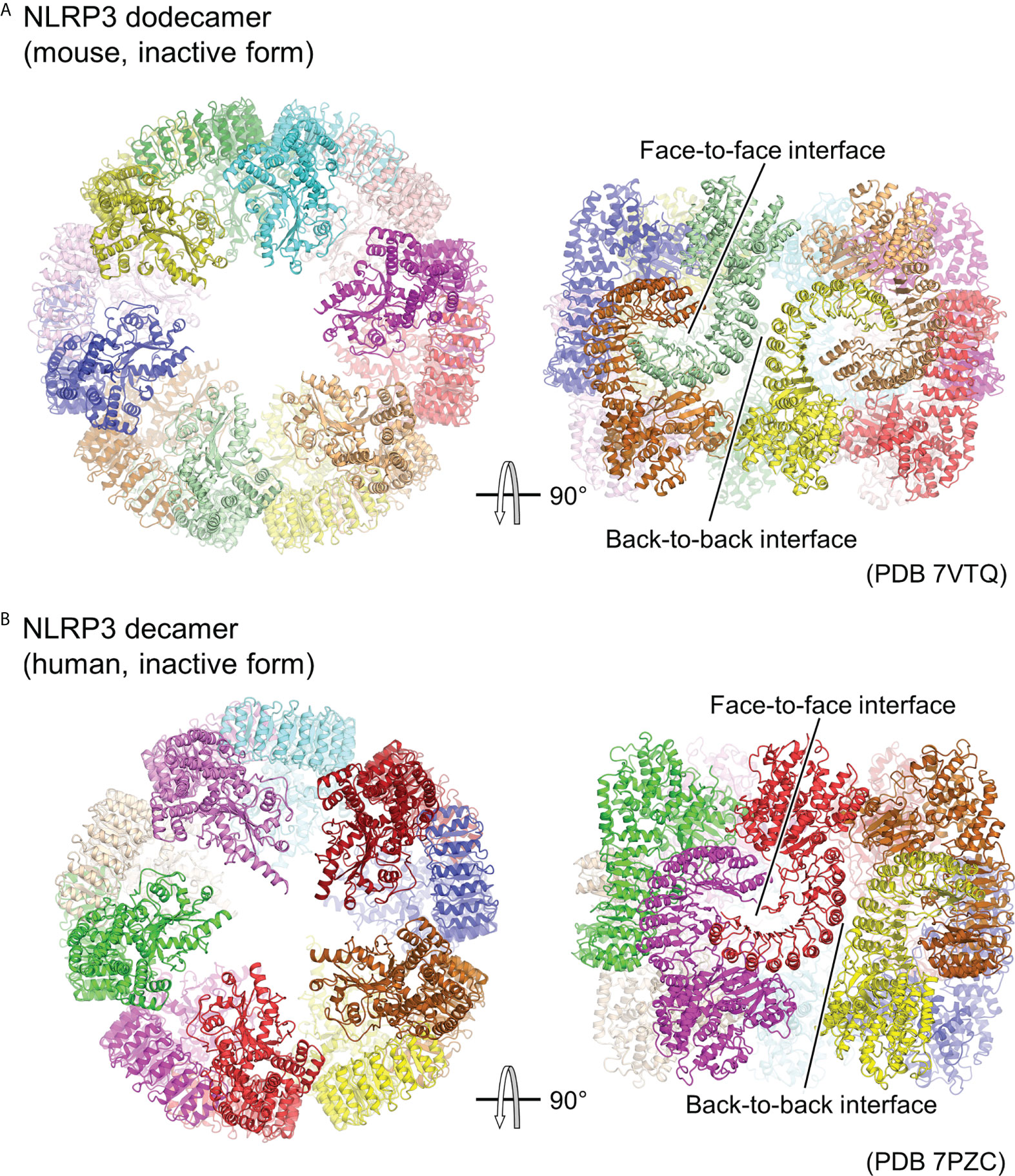
Figure 3 Cage-shaped inactive oligomer of NLRP3. Top and side views of the structure of (A) full-length mouse NLRP3 dodecamer (PDB 7VTQ) (23) and (B) Human NLRP3 decamer (PDB 7PZC) (24). Each protomer is shown in a different color. The LRR-mediated oligomer interfaces are indicated.
LRR-LRR interactions on the side of the cage are the main contributors to the multimer formation. The interactions at this site are “face-to-face” or “head-to-head,” in which neighboring LRRs interact closely with each other (Figure 3). These interactions are mainly due to electrostatic complementarity and hydrophobicity, respectively. In contrast, the NACHTs on the upper and lower surfaces of the cage are proximal to each other, but there is little direct interaction between them. As a result, in all the reported oligomer structures, the density of the LRR portions on the sides of the cage is clear, whereas that of the NACHT portions on the top and bottom surfaces of the cage is relatively obscure.
The structure of the NLRP3 protomer in the oligomer matches well with the previously reported structure of NLRP3 in the inactivated NLRP3-NEK7 structure (36). The LRR-LRR and NLRP3-NEK7 interaction interfaces overlap, suggesting that this cage-like NLRP3 oligomer cannot accommodate NEK7. Moreover, this suggests that the cage-like NLRP3 oligomer is reorganized when NEK7 binds to and activates NLRP3. Furthermore, it has been shown that adding NEK7 to the NLRP3 oligomer partially dissociates the oligomer (36). NEK7 is a centrosomal kinase that mainly localizes to the microtubule-organizing center (60, 61), where NLRP3 does not encounter NEK7 in resting cells, suggesting that spatial isolation is one of the mechanisms preventing NLRP3 from being unintentionally activated (25).
Although the density of PYD was not clearly identified in the cage-like oligomeric structure, it is likely that PYD contributes to the formation of this cage-like oligomer, as it does not form when PYD is deleted (23, 25). Moreover, the PYD-deleted form of human NLRP3 forms a hexamer, while intact human NLRP3 forms a cage-like decamer (29, 59). The cage-like NLRP3 oligomers did not induce downstream ASC filament formation (25), suggesting that the PYDs in the oligomers were confined or spatially constrained within the cage, thereby inhibiting filament formation (23–25). This has been proposed as one of the mechanisms limiting NLRP3 activation.
The cage-like NLRP3 oligomer has been shown to have an affinity for membranes (23, 25). Oligomers of NLRP3 have been detected in membrane-extracted fractions from HEK293T cells overexpressing NLRP3 or from immortalized bone marrow-derived macrophages that express NLRP3 endogenously by LPS stimulation (25). Lipid strip assay results have shown that NLRP3 has an affinity for acidic lipids such as phosphatidylinositides, phosphatidic acid, phosphatidyl serine, and cardiolipin (23, 25). This corresponds well with the localization of NLRP3 to acidic lipids in the trans-Golgi network (TGN) (62). Furthermore, thorough functional assay results indicated that the cage-like NLRP3 oligomer is essential for TGN dispersion and NLRP3 activation (25).
In summary, the following mechanism has been proposed (23–25): NLRP3 is localized as a cage-like oligomer on the TGN and mitochondrial membranes in the resting state, where its activation is inhibited by the confinement or structural restriction of PYD. NLRP3 is then activated by activation signals such as due to nigericin, which induces a conformational change to form an activated oligomer.
The cryo-EM structures of the artificial hexamer of human NLRP3 (PYD-deficient), full-length mouse NLRP3 dodecamer (23), and full-length human NLRP3 decamer (24) as well as the crystal structure of the NACHT domain of human NLRP3 (26) have been determined in the presence of the NLRP3 inhibitor MCC950 or its analogs (63–66). This revealed the inhibitor binding mode and the mechanism of inhibition of NLRP3 activation (Figure 4). The inhibitor binds to the bottom of the cavity in the NACHT domain. This cavity is composed of all the domains and subdomains of NLRP3. Although the inhibitor binds spatially close to the ADP binding site, the binding sites are separated by an interaction between NBD, HD1, and WHD, allowing the inhibitor to access NLRP3 from the NBD-HD2-LRR side, whereas ADP accesses NLRP3 from the opposite side. The closed conformation of NACHT domains is generally characterized by tight packing between NACHT subdomains via ADP binding (16, 29, 30, 36). Like ADP, the inhibitor binds to NLRP3 and mediates its interaction with its subdomain as well as with LRR. This suggests that inhibitors stabilize the closed conformation of the NACHT domain of NLRP3, thereby preventing the NACHT domain from changing to an open conformation and being activated (23, 24, 26).
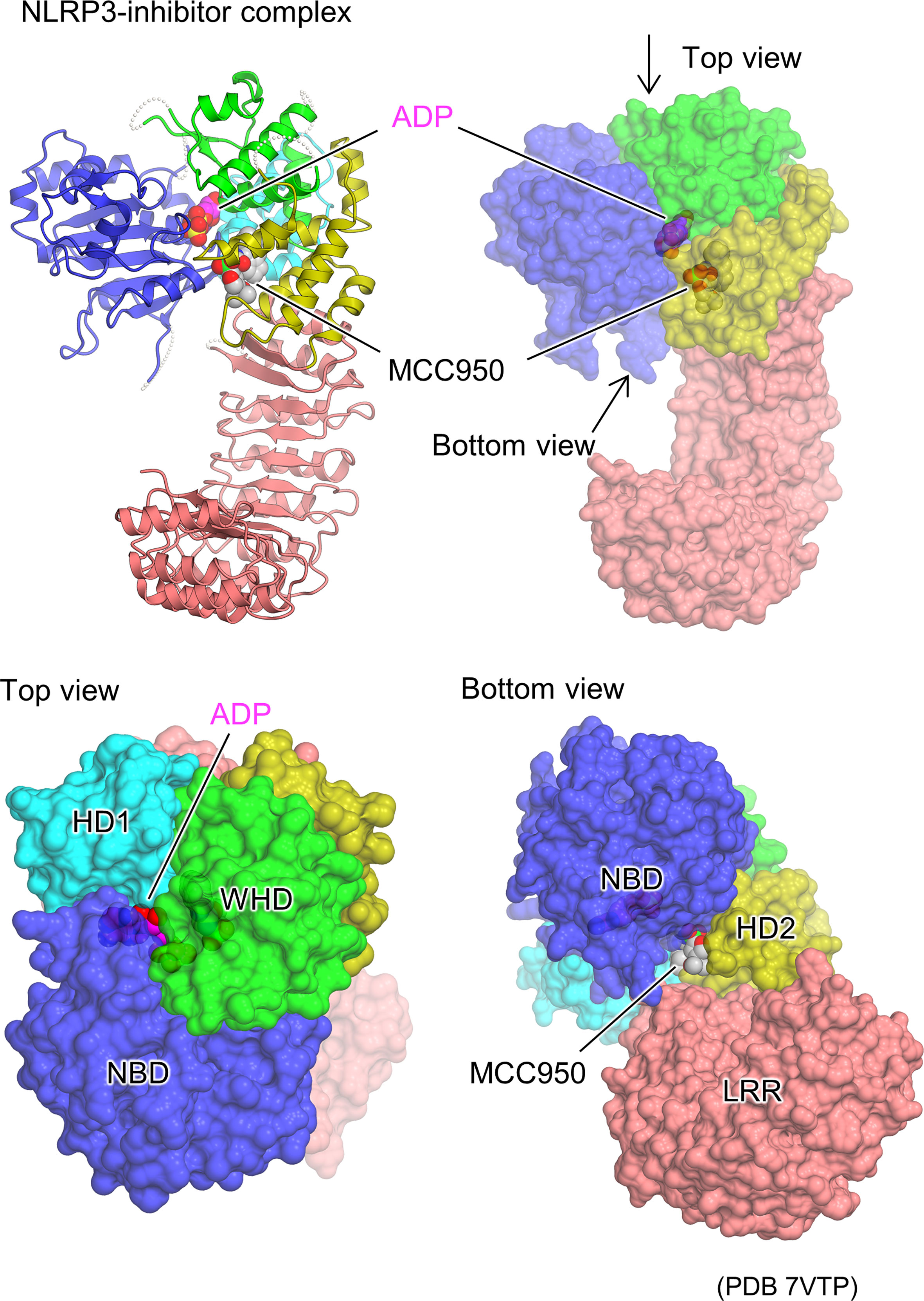
Figure 4 Structural basis of NLRP3 inhibitor binding. Protomer structure of the human NLRP3 (PYD deleted) hexamer (PDB 7VTP) (23) with bound molecules of the ADP and NLRP3 inhibitor, MCC950, is illustrated as space filling representations. Ribbon representation (top left) and surface representations from three different views (top right, bottom left, and bottom right) are demonstrated.
Human-NLRP1 is an NLR with an atypical domain configuration with PYD, NACHT, LRR, a function to find domain (FIIND), and CARD domains from the N-terminal to the C-terminal side (Figure 5A) (67, 68). FIIND is further divided into ZU5 (found in ZO-1 and UNC5) and UPA (found in UNC5, PIDD, and ankyrins) subdomains. Autoproteolysis between these two subdomains is important for NLRP1 activation (69, 70). Gain-of-function mutations in NLRP1 are known to cause inflammatory diseases, particularly in the skin (67, 71). NLRs generally signal through their N-terminal PYD or CARD domains, but previous studies have shown that the C-terminal CARD domain is responsible for signaling in NLRP1 (69). The trigger for the activation of NLRP1 has been unknown for many years. However, recently, it was shown that the activation is triggered by the cleavage of human NLRP1 via the enteroviral 3C protease at the linker between PYD and NACHT (Q130-G131) (Figure 5A) (72, 73). The resulting N-terminal glycine activates the N-glycine-mediated degradation pathway, which degrades the autoinhibitory NACHT-LRR domain and releases a C-terminal fragment (UPA-CARD) to activate NLRP1 (74–76). The CARD domain of the free C-terminal fragment forms filaments, through which ASC or procaspase-1 is recruited to form the inflammasome (77, 78). Similarly, mouse NLRP1B is cleaved near its N-terminal side by bacterial lethal toxin proteases, resulting in the initiation of N-terminal degradation and release of the C-terminal activating fragment (79–81). In addition, ubiquitination of NLRP1B by bacterial pathogen Shigella flexneri IpaH7.8 E3 ubiquitin ligase has shown to activate NLRP1B (75). Dipeptidyl peptidase (DPP)8 and DPP9 are cytoplasmic dipeptidyl peptidases that bind directly to NLRP1 and inhibit its activation. Inhibition of NLRP1 by DPP8/DPP9 is counteracted by DPP8/DPP9 inhibitors; DPP8/DPP9 inhibitors activate NLRP1 (74, 82–85). Furthermore, human NLRP1 has been shown to be activated by recognition of virus-derived double-stranded RNA (dsRNA) (86).
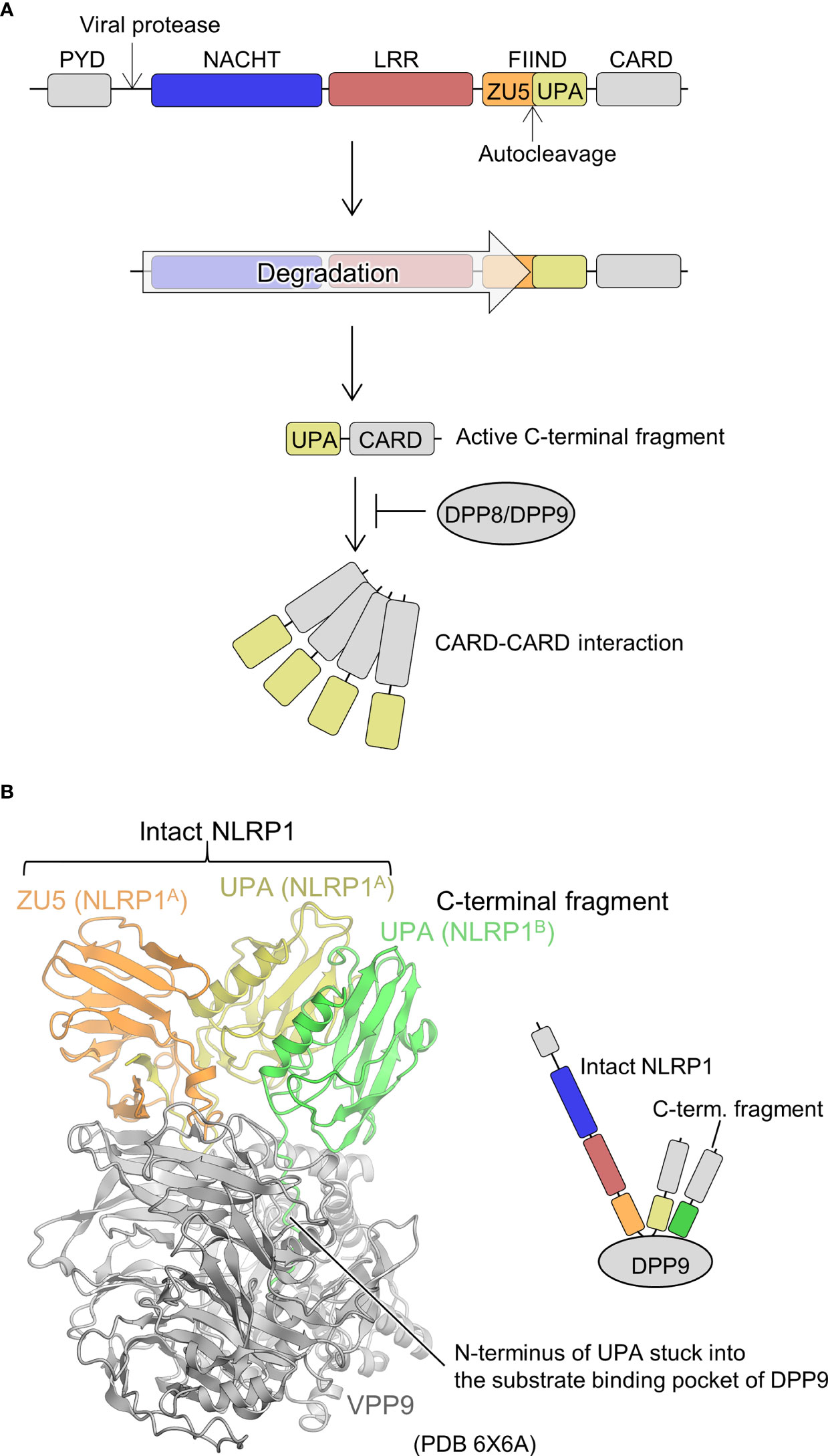
Figure 5 Mechanism underlying NLRP1 activation and DPP9-mediated suppression of NLRP1 activation. (A) NLRP1 activation mechanism. The domains are indicated in various colors and are correspondingly represented in Figure 5B. (B) Structure of the DPP9-NLRP1 complex (PDB 6X6A) (28). In the structure, the ZU5-UPA region from intact NLRP1 (denoted as NLRP1A) and UPA portion of the C-terminal fragment of NLRP1 (denoted as NLRP1B) bound to a DPP9 molecule are indicated. The schematic of the complex is represented in the right panel.
Regarding the structural biology of NLRP1, the structure of the region containing the central NACHT-LRR domain has not yet been elucidated. However, cryo-EM analysis has recently revealed a mechanism by which the C-terminal fragment released by the N-terminal degradation is repressed by DPP9 (Figure 5B) (27, 28). A ternary complex consisting of one molecule of full-length NLRP1 (NLRP1A) and one molecule of the C-terminal fragment of NLRP1 (NLRP1B) with one molecule of DPP9 was elucidated. The complex contained full-length NLRP1A, but only DPP9, the FIIND domain of NLRP1A (ZU5 and UPA), and the UPA portion of NLRP1B were resolved by cryo-EM analysis; other portions were not observed in the cryo-EM map. A peptide of approximately 10 residues on the N-terminal side of NLRP1B, generated by the auto-cleavage of the FIIND domain, was inserted into the substrate recognition pocket of DPP9. Thus, inhibitors of DPP9 that bind to this pocket competitively drive out NLRP1B, allowing the C-terminal fragment to escape capture by DPP9 and become active. In the complex structure, interactions between ZU5 of NLRP1A and DPP9, UPA of NLRP1B and DPP9, as well as UPAs of NLRP1A and NLRP1B were identified. It has been shown that mutations in the first two parts cause constitutive activation of NLRP1, while mutations in the latter inhibits NLRP1 activation. This suggests that not only the C-terminal fragment of NLRP1B, which binds to the substrate recognition pocket of DPP9, but also the ZU5 domain of full-length NLRP1A is important for the inhibition of activation of the C-terminal fragment of NLRP1B by DPP9. In other words, when a small amount of the C-terminal fragment is unintentionally generated, the presence of intact NLRP1 provides a checkpoint to prevent unintended activation of NLRP1 by the DPP9 inhibitory mechanism (27, 28). However, increased production of the C-terminal fragment of NLRP1, such as during viral infection, is thought to decrease intact NLRP1, rendering this DPP9 checkpoint dysfunctional, resulting in the release of the C-terminal fragment, which in turn leads to NLRP1 activation.
However, the mechanism of NLRP1 activation remains unclear. In other NLRs, oligomerization via the NACHT-LRR portion causes spatial proximity between the signaling domains, which is thought to trigger activation (17, 18). The NACHT-LRR portion of NLRP1 acts as a domain that inhibits the release of the C-terminal fragment in the functional degradation mechanism (75, 76) described above. Moreover, the NACHT-LRR portion of NLRP1 is involved in dsRNA recognition during NLRP1 activation by a recently reported virus-derived dsRNA (86). Further studies are required to elucidate the precise role of the NACHT-LRR portion of NLRP1.
NOD2 is a member of the NLRC family, and its mutations are closely associated with inflammatory diseases such as Crohn’s disease, Blau syndrome, and early-onset sarcoidosis (87, 88), requiring further functional explanation based on its structural biology. It has two CARD domains on its N-terminal side as signaling domains (Figure 1A). NOD2 is believed to be activated by muramyl dipeptide (MDP) from the bacterial cell wall (89, 90). In addition, diverse stimuli, including Salmonella typhimurium effector protein SipA and SopE have been identified to activate NOD2 (91, 92). Upon activation, NOD2 oligomerizes to bring its CARD domains into proximity, recruiting downstream RIPK2 through CARD-CARD interaction, and ultimately activating nuclear factor-κB and inducing an inflammatory response (89, 90).
To date, the crystal structure of the ADP-bound, inactivated form of NOD2 lacking the CARD domain has been determined (Figure 1B) (29). Similar to the inactivated forms of NLRC4 (16) and NLRP3 (36) (Figure 1B), the NACHT domain maintains a closed structure by binding ADP. Mutations that disrupt the interactions between NACHT subdomains increase NOD2 activation, indicating that the interactions between these subdomains are important in maintaining the inactivated conformation (29). The MDP-binding site inferred from previous mutation experiments (93) was located on the concave side of the LRR (29). Mutations in the residues at this site have been shown to decrease the MDP responsiveness of NOD2. It is thought that the binding of MDP to this site induces a conformational change that results in oligomer formation; however, the details have not been yet clarified. Disease-associated mutations are distributed throughout NOD2. Among these, gain-of-function mutations are particularly prevalent at residues located at the interface between the NACHT subdomains. Few studies reported that NOD2 functions by binding to the membrane (94), and some disease-associated mutations are located on positively charged surface residues of HD2, suggesting that NOD2 may bind to the membrane at this site (29).
NLRP9, a member of the NLRP family, together with DExH box RNA helicase (DHX) 9, recognizes rotavirus RNA in intestinal epithelial cells to form inflammasomes and is involved in resistance to rotavirus infection (95). Recently, the crystal and cryo-EM structures of an ADP-bound inactivated form of NLRP9 lacking the PYD domain have been reported (Figure 1B) (30), and both structures are nearly identical. ADP-bound NLRP9, like other inactive forms of NLRs (16, 29, 36), has a closed NACHT domain. Approximately 10 residues on the C-terminal side of NLRP9 have been found to fold back from the tip of LRR to the concave side of LRR, forming an extensive interaction with the concave side of LRR. As discussed, the concave surface of the LRR of each NLR has a distinctive function (23–25, 29, 36, 96). Moreover, it has been speculated that this region of NLRP9 may also play an important role in interactions with other proteins and oligomer formation. However, most mechanisms remain unclear, including the mechanism of inflammasome activation by NLRP9 and the recognition of virus-derived RNA in cooperation with DHX9 (95).
NLRP6 is a member of the NLRP family and, as with NLRP9, plays an important role in immune responses in intestinal epithelial cells (97, 98). Similar to NLRP9, it cooperates with DHX15, an RNA helicase, to bind to the RNA introduced by enteric viruses and induce interferon production through MAVS (98). It is also known to sense microbiota-associated metabolites and form ASC-dependent inflammasomes (97). For NLRP6, the structures of the PYD domain and its filament structure are known (99). However, the structure of the remaining NACHT-LRR portion remains unclear. Recently, it has become clear that liquid-liquid phase separation (LLPS), which has attracted much attention recently because of its involvement in various biological phenomena, plays an important role in the activation of NLRP6 (100). In vitro and intracellular experiments indicate that dsRNA induces LLPS formation of NLRP6 and that this LLPS formation is important for the activation of the NLRP6 inflammasome. The adaptor molecule ASC solidifies the LLPS of NLRP6 and activates the inflammasome. The poly-lysine sequence in the NACHT domain of NLRP6 has been shown to be important for LLPS formation. LLPS-mediated NLRP6 activation is a novel NLR inflammasome activation mechanism, and whether similar mechanisms exist for other NLRs must be further investigated in the future.
The past decade has provided a better understanding of the activation mechanisms of NLRs based on structural biology studies. The mechanism of ring-shaped oligomers as a starting point for downstream adaptor signaling, as evidenced structurally in NLRC4 and postulated in NLRP3, is now clear. However, there is little structural evidence regarding the activation mechanism of NLRs. For instance, how ATPase activities of NLR are involved in the activation, how the conformational change leading to the oligomerization is triggered, and further studies are essential to clarify the activation mechanisms of NLRs. In contrast, the mechanism by which the NACHT-LRR portion of NLRP1 is degraded and that by which the released C-terminal fragment serves as a scaffold for downstream adaptor molecules have been elucidated. Moreover, the mechanism by which NLRP6 condensed by LLPS serves as a scaffold for downstream adaptor molecules has also been revealed. Although these activation mechanisms promote recruitment of downstream adaptor molecules by increasing the local concentration of signaling domains, diverse NLR activation mechanisms are still being uncovered. NLRs are involved in a variety of diseases, and their detailed activation mechanisms based on structural biology should be further studied to aid in developing therapeutic agents.
The author confirms being the sole contributor of this work and has approved it for publication.
This work was supported by a Grant-in-Aid from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (Grant Nos. 22H02556 and 19H03164).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Beutler B. Innate immunity: an overview. Mol Immunol (2004) 40:845–59. doi: 10.1016/j.molimm.2003.10.005
2. Medzhitov R, Janeway C Jr. Innate immunity. N Engl J Med (2000) 343:338–44. doi: 10.1056/NEJM200008033430506
3. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
4. Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev (2009) 227:221–33. doi: 10.1111/j.1600-065X.2008.00731.x
5. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol (2003) 21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126
6. Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
7. Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev (2009) 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x
8. Waldner H. The role of innate immune responses in autoimmune disease development. Autoimmun. Rev (2009) 8:400–4. doi: 10.1016/j.autrev.2008.12.019
9. Ulevitch RJ. Therapeutics targeting the innate immune system. Nat Rev Immunol (2004) 4:512–20. doi: 10.1038/nri1396
10. Franchi L, Warner N, Viani K, Nunez G. Function of nod-like receptors in microbial recognition and host defense. Immunol Rev (2009) 227:106–28. doi: 10.1111/j.1600-065X.2008.00734.x
11. Loo YM, Gale M Jr. Immune signaling by RIG-i-like receptors. Immunity (2011) 34:680–92. doi: 10.1016/j.immuni.2011.05.003
12. Kumari P, Russo AJ, Shivcharan S, Rathinam VA. AIM2 in health and disease: Inflammasome and beyond. Immunol Rev (2020) 297:83–95. doi: 10.1111/imr.12903
13. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
14. Tan XJ, Sun LJ, Chen JQ, Chen ZJJ. Detection of microbial infections through innate immune sensing of nucleic acids. Annu Rev Microbiol (2018) 72:447–78. doi: 10.1146/annurev-micro-102215-095605
15. Miyake K, Shibata T, Ohto U, Shimizu T, Saitoh SI, Fukui R, et al. Mechanisms controlling nucleic acid-sensing toll-like receptors. Int Immunol (2018) 30:43–51. doi: 10.1093/intimm/dxy016
16. Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science (2013) 341:172–5. doi: 10.1126/science.1236381
17. Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science (2015) 350:404–9. doi: 10.1126/science.aac5789
18. Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science (2015) 350:399–404. doi: 10.1126/science.aac5489
19. Diebolder CA, Halff EF, Koster AJ, Huizinga EG, Koning RI. Cryoelectron tomography of the NAIP5/NLRC4 inflammasome: Implications for NLR activation. Structure (2015) 23:2349–57. doi: 10.1016/j.str.2015.10.001
20. Yang X, Yang F, Wang W, Lin G, Hu Z, Han Z, et al. Structural basis for specific flagellin recognition by the NLR protein NAIP5. Cell Res (2018) 28:35–47. doi: 10.1038/cr.2017.148
21. Tenthorey JL, Haloupek N, Lopez-Blanco JR, Grob P, Adamson E, Hartenian E, et al. The structural basis of flagellin detection by NAIP5: A strategy to limit pathogen immune evasion. Science (2017) 358:888–93. doi: 10.1126/science.aao1140
22. Shen C, Sharif H, Xia S, Wu H. Structural and mechanistic elucidation of inflammasome signaling by cryo-EM. Curr Opin Struct Biol (2019) 58:18–25. doi: 10.1016/j.sbi.2019.03.033
23. Ohto U, Kamitsukasa Y, Ishida H, Zhang Z, Murakami K, Hirama C, et al. Structural basis for the oligomerization-mediated regulation of NLRP3 inflammasome activation. Proc Natl Acad Sci USA (2022) 119:e2121353119. doi: 10.1073/pnas.2121353119
24. Hochheiser IV, Pilsl M, Hagelueken G, Moecking J, Marleaux M, Brinkschulte R, et al. Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature (2022) 604:184–9. doi: 10.1038/s41586-022-04467-w
25. Andreeva L, David L, Rawson S, Shen C, Pasricha T, Pelegrin P, et al. NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell (2021) 184:6299–6312 e6222. doi: 10.1016/j.cell.2021.11.011
26. Dekker C, Mattes H, Wright M, Boettcher A, Hinniger A, Hughes N, et al. Crystal structure of NLRP3 NACHT domain with an inhibitor defines mechanism of inflammasome inhibition. J Mol Biol (2021) 433:167309. doi: 10.1016/j.jmb.2021.167309
27. Huang M, Zhang X, Toh GA, Gong Q, Wang J, Han Z, et al. Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature (2021) 592:773–7. doi: 10.1038/s41586-021-03320-w
28. Hollingsworth LR, Sharif H, Griswold AR, Fontana P, Mintseris J, Dagbay KB, et al. DPP9 sequesters the c terminus of NLRP1 to repress inflammasome activation. Nature (2021) 592:778–83. doi: 10.1038/s41586-021-03350-4
29. Maekawa S, Ohto U, Shibata T, Miyake K, Shimizu T. Crystal structure of NOD2 and its implications in human disease. Nat Commun (2016) 7:11813. doi: 10.1038/ncomms11813
30. Kamitsukasa Y, Nakano K, Murakami K, Hirata K, Yamamoto M, Shimizu T, et al. The structure of NLRP9 reveals a unique c-terminal region with putative regulatory function. FEBS Lett (2022) 596:876–85. doi: 10.1002/1873-3468.14302
31. Kienes I, Weidl T, Mirza N, Chamaillard M, Kufer TA. Role of NLRs in the regulation of type I interferon signaling, host defense and tolerance to inflammation. Int J Mol Sci (2021) 22:1301. doi: 10.3390/ijms22031301
32. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
33. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi: 10.1038/nm.3893
34. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
35. Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol (2010) 10:210–5. doi: 10.1038/nri2725
36. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature (2019) 570:338–43. doi: 10.1038/s41586-019-1295-z
37. Matikainen S, Nyman TA, Cypryk W. Function and regulation of noncanonical caspase-4/5/11 inflammasome. J Immunol (2020) 204:3063–9. doi: 10.4049/jimmunol.2000373
38. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi: 10.1038/nature10510
39. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477:592–5. doi: 10.1038/nature10394
40. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA (2010) 107:3076–80. doi: 10.1073/pnas.0913087107
41. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta. via Ipaf. Nat Immunol (2006) 7:569–75. doi: 10.1038/ni1344
42. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol (2006) 7:576–82. doi: 10.1038/ni1346
43. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and ipaf. Nature (2004) 430:213–8. doi: 10.1038/nature02664
44. Yuan SJ, Topf M, Reubold TF, Eschenburg S, Akey CW. Changes in apaf-1 conformation that drive apoptosome assembly. Biochemistry (2013) 52:2319–27. doi: 10.1021/bi301721g
45. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
46. Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci (2014) 1319:82–95. doi: 10.1111/nyas.12458
47. Jin C, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol (2010) 30:628–31. doi: 10.1007/s10875-010-9440-3
48. Song N, Li T. Regulation of NLRP3 inflammasome by phosphorylation. Front Immunol (2018) 9:2305. doi: 10.3389/fimmu.2018.02305
49. Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol (2007) 8:497–503. doi: 10.1038/ni1459
50. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11:136–40. doi: 10.1038/ni.1831
51. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell (2013) 153:348–61. doi: 10.1016/j.cell.2013.02.054
52. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol (2016) 17:250–8. doi: 10.1038/ni.3333
53. Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, et al. A genome-wide CRISPR (Clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem (2016) 291:103–9. doi: 10.1074/jbc.C115.700492
54. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530:354–7. doi: 10.1038/nature16959
55. Li X, Thome S, Ma X, Amrute-Nayak M, Finigan A, Kitt L, et al. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat Commun (2017) 8:15986. doi: 10.1038/ncomms15986
56. Lang T, Lee JPW, Elgass K, Pinar AA, Tate MD, Aitken EH, et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat Commun (2018) 9:2223. doi: 10.1038/s41467-018-04581-2
57. Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature (2019) 573:590. doi: 10.1038/s41586-019-1551-2
58. Duan Y, Zhang L, Angosto-Bazarra D, Pelegrin P, Nunez G, He Y. RACK1 mediates NLRP3 inflammasome activation by promoting NLRP3 active conformation and inflammasome assembly. Cell Rep (2020) 33:108405. doi: 10.1016/j.celrep.2020.108405
59. Haq T, Richards MW, Burgess SG, Gallego P, Yeoh S, O'Regan L, et al. Mechanistic basis of Nek7 activation through Nek9 binding and induced dimerization. Nat Commun (2015) 6:8771. doi: 10.1038/ncomms9771
60. Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science (2020) 369:eaas8995. doi: 10.1126/science.aas8995
61. Kim S, Lee K, Rhee K. NEK7 is a centrosomal kinase critical for microtubule nucleation. Biochem Bioph Res Co (2007) 360:56–62. doi: 10.1016/j.bbrc.2007.05.206
62. Chen J, Chen ZJ. PtdIns4P on dispersed trans-golgi network mediates NLRP3 inflammasome activation. Nature (2018) 564:71–6. doi: 10.1038/s41586-018-0761-3
63. Tapia-Abellan A, Angosto-Bazarra D, Martinez-Banaclocha H, de Torre-Minguela C, Ceron-Carrasco JP, Perez-Sanchez H, et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol (2019) 15:560–4. doi: 10.1038/s41589-019-0278-6
64. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
65. Primiano MJ, Lefker BA, Bowman MR, Bree AG, Hubeau C, Bonin PD, et al. Efficacy and pharmacology of the NLRP3 inflammasome inhibitor CP-456,773 (CRID3) in murine models of dermal and pulmonary inflammation. J Immunol (2016) 197:2421–33. doi: 10.4049/jimmunol.1600035
66. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21:248–55. doi: 10.1038/nm.3806
67. Taabazuing CY, Griswold AR, Bachovchin DA. The NLRP1 and CARD8 inflammasomes. Immunol Rev (2020) 297:13–25. doi: 10.1111/imr.12884
68. Mitchell PS, Sandstrom A, Vance RE. The NLRP1 inflammasome: new mechanistic insights and unresolved mysteries. Curr Opin Immunol (2019) 60:37–45. doi: 10.1016/j.coi.2019.04.015
69. Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, et al. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J Biol Chem (2012) 287:31456–6. doi: 10.1074/jbc.A112.378323
70. D'Osualdo A, Weichenberger CX, Wagner RN, Godzik A, Wooley J, Reed JC, et al. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PloS One (2011) 6:e27396. doi: 10.1371/journal.pone.0027396
71. Fenini G, Karakaya T, Hennig P, Di Filippo M, Beer HD. The NLRP1 inflammasome in human skin and beyond. Int J Mol Sci (2020) 21(13):4788. doi: 10.3390/ijms21134788
72. Tsu BV, Beierschmitt C, Ryan AP, Agarwal R, Mitchell PS, Daugherty MD, et al. Diverse viral proteases activate the NLRP1 inflammasome. Elife (2021) 10:e60609. doi: 10.7554/eLife.60609
73. Robinson KS, Teo DET, Tan KS, Toh GA, Ong HH, Lim CK, et al. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science (2020) 370(6521):eaay2002. doi: 10.1126/science.aay2002
74. Xu H, Shi J, Gao H, Liu Y, Yang Z, Shao F, et al. The n-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO J (2019) 38:e101996. doi: 10.15252/embj.2019101996
75. Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE, et al. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science (2019) 364(6435):eaau1330. doi: 10.1126/science.aau1330
76. Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, et al. N-terminal degradation activates the NLRP1B inflammasome. Science (2019) 364:82–5. doi: 10.1126/science.aau1208
77. Robert Hollingsworth L, David L, Li Y, Griswold AR, Ruan J, Sharif H, et al. Mechanism of filament formation in UPA-promoted CARD8 and NLRP1 inflammasomes. Nat Commun (2021) 12:189. doi: 10.1038/s41467-020-20320-y
78. Gong Q, Robinson K, Xu C, Huynh PT, Chong KHC, Tan EYJ, et al. Structural basis for distinct inflammasome complex assembly by human NLRP1 and CARD8. Nat Commun (2021) 12:188. doi: 10.1038/s41467-020-20319-5
79. Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PloS Pathog (2012) 8:e1002638. doi: 10.1371/journal.ppat.1002638
80. Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I, et al. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PloS One (2012) 7:e49741. doi: 10.1371/journal.pone.0049741
81. Chavarria-Smith J, Vance RE. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PloS Pathog (2013) 9(6):e1003452. doi: 10.1371/journal.ppat.1003452
82. Zhong FL, Robinson K, Teo DET, Tan KY, Lim C, Harapas CR, et al. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J Biol Chem (2018) 293:18864–78. doi: 10.1074/jbc.RA118.004350
83. Okondo MC, Rao SD, Taabazuing CY, Chui AJ, Poplawski SE, Johnson DC, et al. Inhibition of Dpp8/9 activates the Nlrp1b inflammasome. Cell Chem Biol (2018) 25:262–267 e265. doi: 10.1016/j.chembiol.2017.12.013
84. Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med (2018) 24:1151–6. doi: 10.1038/s41591-018-0082-y
85. Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat Chem Biol (2017) 13:46–53. doi: 10.1038/nchembio.2229
86. Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. Human NLRP1 is a sensor for double-stranded RNA. Science (2021) 371:482. doi: 10.1126/science.abd0811
87. Abraham C, Cho JH. Functional consequences of NOD2 (CARD15) mutations. Inflammation Bowel Dis (2006) 12:641–50. doi: 10.1097/01.MIB.0000225332.83861.5f
88. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to crohn’s disease. Nature (2001) 411:603–6. doi: 10.1038/35079114
89. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. implications for crohn’s disease. J Biol Chem (2003) 278:5509–12. doi: 10.1074/jbc.C200673200
90. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278:8869–72. doi: 10.1074/jbc.C200651200
91. Keestra AM, Winter MG, Klein-Douwel D, Xavier MN, Winter SE, Kim A, et al. A salmonella virulence factor activates the NOD1/NOD2 signaling pathway. mBio (2011) 2(6):e00266-11. doi: 10.1128/mBio.00266-11
92. Keestra AM, Winter MG, Auburger JJ, Frassle SP, Xavier MN, Winter SE, et al. Manipulation of small rho GTPases is a pathogen-induced process detected by NOD1. Nature (2013) 496:233. doi: 10.1038/nature12025
93. Tanabe T, Chamaillard M, Ogura Y, Zhu L, Qiu S, Masumoto J, et al. Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J (2004) 23:1587–97. doi: 10.1038/sj.emboj.7600175
94. Barnich N, Aguirre JE, Reinecker HC, Xavier R, Podolsky DK. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-kappa b activation in muramyl dipeptide recognition. J Cell Biol (2005) 170:21–6. doi: 10.1083/jcb.200502153
95. Zhu S, Ding SY, Wang PH, Wei Z, Pan W, Palm NW, et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature (2017) 546:667. doi: 10.1038/nature22967
96. Reubold TF, Hahne G, Wohlgemuth S, Eschenburg S. Crystal structure of the leucine-rich repeat domain of the NOD-like receptor NLRP1: implications for binding of muramyl dipeptide. FEBS Lett (2014) 588:3327–32. doi: 10.1016/j.febslet.2014.07.017
97. Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell (2015) 163:1428–43. doi: 10.1016/j.cell.2015.10.048
98. Wang P, Zhu S, Yang L, Cui S, Pan W, Jackson R, et al. Nlrp6 regulates intestinal antiviral innate immunity. Science (2015) 350:826–30. doi: 10.1126/science.aab3145
99. Shen C, Lu A, Xie WJ, Ruan J, Negro R, Egelman EH, et al. Molecular mechanism for NLRP6 inflammasome assembly and activation. Proc Natl Acad Sci U.S.A. (2019) 116:2052–7. doi: 10.1073/pnas.1817221116
Keywords: innate immunity, pathogen-associated molecular patterns, damage-associated molecular patterns, pattern recognition receptors, NOD-like receptors (NLRs), inflammasome
Citation: Ohto U (2022) Activation and regulation mechanisms of NOD-like receptors based on structural biology. Front. Immunol. 13:953530. doi: 10.3389/fimmu.2022.953530
Received: 26 May 2022; Accepted: 29 August 2022;
Published: 15 September 2022.
Edited by:
Hang Yin, Tsinghua University, ChinaReviewed by:
Jijie Chai, Tsinghua University, ChinaCopyright © 2022 Ohto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Umeharu Ohto, dW1lamlAbW9sLmYudS10b2t5by5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.