 Isaac T. W. Harley
Isaac T. W. Harley Kristen Allison
Kristen Allison R. Hal Scofield
R. Hal Scofield
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 24 August 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.953439
This article is part of the Research TopicGenetic Basis of Tolerance Induction Defects Underlying the Development of Autoimmune PathologiesView all 11 articles
Most B cells produced in the bone marrow have some level of autoreactivity. Despite efforts of central tolerance to eliminate these cells, many escape to periphery, where in healthy individuals, they are rendered functionally non-responsive to restimulation through their antigen receptor via a process termed anergy. Broad repertoire autoreactivity may reflect the chances of generating autoreactivity by stochastic use of germline immunoglobulin gene segments or active mechanisms may select autoreactive cells during egress to the naïve peripheral B cell pool. Likewise, it is unclear why in some individuals autoreactive B cell clones become activated and drive pathophysiologic changes in autoimmune diseases. Both of these remain central questions in the study of the immune system(s). In most individuals, autoimmune diseases arise from complex interplay of genetic risk factors and environmental influences. Advances in genome sequencing and increased statistical power from large autoimmune disease cohorts has led to identification of more than 200 autoimmune disease risk loci. It has been observed that autoantibodies are detectable in the serum years to decades prior to the diagnosis of autoimmune disease. Thus, current models hold that genetic defects in the pathways that control autoreactive B cell tolerance set genetic liability thresholds across multiple autoimmune diseases. Despite the fact these seminal concepts were developed in animal (especially murine) models of autoimmune disease, some perceive a disconnect between human risk alleles and those identified in murine models of autoimmune disease. Here, we synthesize the current state of the art in our understanding of human risk alleles in two prototypical autoimmune diseases – systemic lupus erythematosus (SLE) and type 1 diabetes (T1D) along with spontaneous murine disease models. We compare these risk networks to those reported in murine models of these diseases, focusing on pathways relevant to anergy and central tolerance. We highlight some differences between murine and human environmental and genetic factors that may impact autoimmune disease development and expression and may, in turn, explain some of this discrepancy. Finally, we show that there is substantial overlap between the molecular networks that define these disease states across species. Our synthesis and analysis of the current state of the field are consistent with the idea that the same molecular networks are perturbed in murine and human autoimmune disease. Based on these analyses, we anticipate that murine autoimmune disease models will continue to yield novel insights into how best to diagnose, prognose, prevent and treat human autoimmune diseases.
Upwards of 75% of bone marrow produced B cells express B cell antigen receptors (BCRs) that bind self-antigen (1–8). Several mechanisms conspire to remove these autoreactive BCRs from the diverse repertoire needed to provide effective protective humoral immunity without autoimmunity. These mechanisms act both centrally by receptor editing and clonal deletion and peripherally by anergy (7). Central tolerance mechanisms typically remove clones from the wild type repertoire with the most avid interaction with autoantigens. However, peripheral tolerance or anergy is the operative mechanism that silences most autoreactive B cells (3–6). Anergy arises as a consequence of chronic antigen receptor stimulation in the absence of second signals (4, 7, 8). It is defined by non-responsiveness to re-stimulation through the BCR. Importantly, in several B-cell dependent human autoimmune diseases, most individuals with clinically apparent autoimmune disease develop serologically detectable autoantibodies prior to clinical diagnosis (9–13). While we would define B cell dependence as the ability of a B cell depleting therapy to prevent or treat human disease, the inclusion of type 1 diabetes and multiple sclerosis as a B-cell dependent diseases is not universally accepted. However, paired with the clinical efficacy of B-cell targeted therapies either in prevention or treatment of diverse autoimmune pathologies (11, 13–29) these observations implicate dysregulation of central tolerance mechanisms, peripheral tolerance mechanisms or both in the etiopathogenesis of these diseases. Evidence supporting regulatory defects in both central (30–33) and peripheral (31, 32) tolerance mechanisms have been described in numerous human autoimmune pathologies. Central B cell tolerance defects have been described in human SLE (34–36), T1D (37), RA (38, 39) and Sjogren’s Syndrome (40). Peripheral B cell tolerance defects have been described in T1D (41), Autoimmune Thyroid Disease (AITD) (42), SLE (43–46), RA (47–49) and anti-neutrophil cytoplasmic antibody (ANCA)–associated vascluitis (AAV) (50). Current immunologic paradigms hold that immune systems have been selected to balance response to pathogens with damage to self (51–53). If this dominant theoretical framework of immunology is correct, the observation that such high levels of autoreactivity are the norm in some ways challenges our teleology of (auto-)immunity. Indeed, this apparent paradox is perhaps not surprising, as our aim is to reduce a complex system that has evolved to specifically, efficiently and flexibly respond to a universe of molecules with a range of approximately quintillion possibilities (54) to a simple and understandable set of rules.
There are obvious (and non-obvious) differences and drawbacks inherent in extrapolating principles to human pathologies from animal model systems (55). Nevertheless, our understanding of the mechanisms that regulate both central (33) and peripheral B cell tolerance (3, 56, 57)as well as the development of autoreactive B-cell dependent autoimmune pathologies (58–61) has been informed by frameworks developed in murine animal models. Indeed, our current models of the etiopathogenesis of human autoimmune pathology largely consist of a consilience of inductions from both observation and experimentation on living humans, model systems comprised of human tissues/cells and study of murine model systems. However, several have challenged the use of animal models to understand autoimmune pathologies (55). One reason cited for this challenge is that advanced tools for studying human immune responses (62–66) (i.e. CyToF, single cell RNA-sequencing, spectral flow cytometry) now allow more precise definition of human immune responses. Another reason cited for this challenge are high-profile failures in translating findings from animal model of autoimmune disease to humans (67, 68) (some oft cited failures in translation include: oral tolerance with insulin in type 1 diabetes prevention (69), subcutaneous administration of partial agonists to induce antigen-specific T cell tolerance in multiple sclerosis (70–72), the use of interferon gamma (73) and inhibition of TNF-alpha (74, 75) in multiple sclerosis). Importantly, the most often cited high-profile failures in translation have arisen from observations in the EAE (Experimental Autoimmune/Allergic Encephalitis) murine model of multiple sclerosis. Notwithstanding the difference between mice and human beings, challenges in translation are perhaps not surprising, given that clinically defined human phenotypes may well represent congeries of etiopathogenic and pathogenetic mechanisms (76–78). That is, in these diseases each individual actually takes a single path to disease development out of many possible routes. Likewise, each murine model system of autoimmune pathology may well represent a single pathogenetic route to disease development.
Here we synthesize the recent advances in our understanding of the complex genetic basis of two paradigmatic human B-cell dependent autoimmune diseases: Systemic Lupus Erythematosus (SLE) and Type 1 Diabetes Mellitus (T1D). SLE is the prototypical protean multi-system autoimmune disease, whereas type 1 diabetes is the prototypical organ-specific autoimmune disease invariably leading to pancreatic beta-cell destruction. Importantly, both of these disease states have long been modeled with mouse strains that spontaneously develop disease features that closely resemble several of the key phenotypes and pathophysiologies of the human diseases being modeled. Because of the long history of investigation of the cellular and molecular mechanisms of these models, we expect that models of these two diseases are likely to have a more complete list of the genetic contributors and understanding of the relevant cellular and molecular mechanisms leading to murine autoimmune disease.
To address this overlap, we also synthesize what is known regarding the function of putative causal genes across murine models of both systemic autoimmune pathologies (SLE and T1D) and autoreactive B cell tolerance. We discuss several plausible potential explanations for the non-monotonic relationship between currently known human and murine autoimmune risk alleles. Through this analysis, we show that the molecular networks comprised of putative human and murine risk alleles for B-cell dependent autoimmunity and autoimmune pathology substantially overlap. Finally, we propose a framework for steps toward more successful translation of findings from murine model systems to clinical application in humans.
In humans both SLE and T1D have heritable component with sibling recurrence risk ratios (lambda S) indicating a substantive genetic contribution (Lambda S SLE = 20, Lambda S T1D = 15) (79). Both are incompletely penetrant, with the monozygotic twin concordance rate estimated to be at most 40-50% but likely substantially lower for both diseases (79). Thus, for both of these autoimmune pathologies, non-heritable factors also impact disease development. These non-heritable risk factors are often assumed to represent exposure to one or more environmental triggers. Other stochastic events, such as somatic mutation or particular antigen receptor rearrangement towards a pathologic autoantigen could also plausibly contribute. In SLE the non-heritable component has been estimated to account for ~56% of disease risk (80) and in T1D, this has been estimated at ~34% (81).
In terms of epidemiology, SLE is both more prevalent and more severe in several populations of predominately non-European ancestry than in populations with European ancestry (82). A recent cause of death analysis puts these differences in stark contrast (83). Whereas SLE is the 10th leading cause of death in all female persons aged 15-24 in the US, it is the 5th leading cause of death in African American and Hispanic female persons. Similarly, a recent population-based registry reported approximately 30% mortality within 10 years of diagnosis in Black SLE patients, whereas white SLE patients from the same population exhibited approximately 10% mortality. These differences are likely due to a complex mixture of factors. Potential contributions to these disparities likely include systematic population level differences in access to healthcare and possibly also genetic variants that are exclusive to a particular ancestral group (84, 85). However, population level genetic differences explain only 16% of genetic variability in human populations (86). Therefore, systemic population level differences in access to care may have a greater impact on outcome differences in SLE. A recent report estimates that SLE occurs in US male persons at a rate of 8 to 53 per 100 000 and US female persons at a rate of 84 to 270 per 100 000, depending on the population (87). Importantly, SLE exhibits sexual dimorphism, occurring more commonly in female persons at rate of 9:1 (87). A caveat to the studies referenced above is that they rely on medical record abstraction and administrative data analysis methods that by their nature preclude obtaining sex, gender, race and ethnicity self-identification.
In terms of epidemiology, T1D is reported to be more prevalent in persons who self-identify as non-Hispanic white, followed by non-Hispanic black, Hispanic and other racial/ethnic identities (0.35 to 2.55 per 1 000) with approximately equal prevalence in boys and girls in the US (1.93 per 1 000) (88). T1D incidence increases with age, peaking between 10-14 years of age. Notably, cases with onset < six months of age are not entirely uncommon (89). However, for reasons that remain incompletely clear, the overall incidence of T1D is increasing according to several studies performed in the US (90–92). As a result, based on anticipated demographic shifts, the prevalence is projected to increase from 2.13 per 1 000 in 2010 to 5.20 per 1 000 by 2050 (88). Increasing incidence in recent decades is not unique to type 1 diabetes amongst other autoimmune diseases (93).
When taken together with the observations that different geographies have different rates of autoimmune diseases (94) and autoimmunity (at least the rate of antinuclear antibody seropositivity) has also increased over the same time course (95), these data have been interpreted to strongly imply a changing autoimmunity/autoimmune disease risk environmental exposure has change in recent decades, as the kinetics seem too fast for a genetic explanation.
Several environmental factors have been associated with SLE, including smoking, silica exposure, exogenous sex hormones and infection, especially prior Epstein-Barr virus infection (96, 97). Similarly, in T1D, microbiome, micronutrient, diet, early life metabolism and immune stimuli (infection and vaccination) have been implicated with risk for incident disease (98).
In sum, both SLE and T1D in humans are complex diseases where both genetic and environmental factors contribute both to disease development and disease manifestations.
Both SLE and autoimmune type 1 diabetes pose practical challenges in disease definition, diagnosis and classification that should be considered when evaluating the utility and applicability of any disease model. One cannot evaluate whether a model recapitulates human disease pathogenesis if the definition of disease is unclear.
The particular nomenclature of autoimmune type 1 diabetes may strike the reader as oddly redundant, but it makes the point that type 1 diabetes is a clinical diagnosis. This diagnosis is made in part through typical seropositive autoimmunity to several pancreatic islet expressed proteins (insulin, ZnT8, IA-2, GAD65) (9) in the setting of insulin deficiency. This clinical scenario has been alternately referred to as type 1a diabetes or as immune-mediated type 1 diabetes (99–101). However, a small proportion of individuals clinically diagnosed with type 1 diabetes in large cohort studies have been found to have an alternative etiology for their disease that is non-autoimmune. These individuals commonly have either childhood onset monogenic type 2 diabetes (102) or fulminant onset diabetes with non-autoimmune beta-cell destruction. This latter category of disease has been alternatively referred to as type 1b diabetes, idiopathic type 1 diabetes or nonautoimmune diabetes plus IS (Insulin Sensitivity) (99–101). In some type 1 diabetes cohorts this proportion may be as high as 10% (103). Prior decades of careful phenotyping and molecular characterization has led to description of several subphenotypes of what would have previously considered either type 1 diabetes (young onset, insulin sensitive and autoimmune) or type 2 diabetes (later onset, insulin resistant non-autoimmune). These include latent autoimmune diabetes of adults (LADA), type 1.5 diabetes, ketosis-prone type 2 diabetes and maturity-onset diabetes of the young. See (104) for an excellent review of the nosological challenges of clinical diabetes classification. Our distinction in nomenclature seeks to differentiate monogenic causes of clinical type 1 diabetes with pathologic autoimmunity from monogenic causes of diabetes that clinically resemble autoimmune type 1 diabetes, but arise from non-autoimmune causes. This distinction is clinically important, as management is substantially different (insulin replacement vs. sulfonylureas and other therapies) (105). Indeed, cohorts clinically diagnosed and treated as type 1 diabetics with potential alternative etiologic explanations have been described (106). There is a growing body of literature that using polygenic risk scores (106) and/or sequencing panels of non-autoimmune monogenic risk alleles can help distinguish these two phenotypes. This approach may even be cost effective in select situations (107). Further highlighting the potential for case misclassification in type 1 diabetes cohorts, several recent studies applied type 1 diabetes polygenic risk scores (PRS) to define individuals with clinical type 1 diabetes with low genetic risk (108–110). As expected, these analyses identified rare T1D risk variants in or near genes with well-known effects on immune responses. In addition, these studies identified several rare risk variants in genes with metabolic function or impacts on obesity and no known function in immune responses. Taken together, they suggest that many of the type 1 diabetes cohorts used for GWAS studies likely include a mixture of individuals with autoimmune type 1 diabetes (T1aD) and individuals with non-autoimmune type 1 diabetes (T1bD).
By the same token, SLE is a clinical diagnosis. In order to develop homogeneous patient populations for clinical studies, several iterations of classification criteria have been developed (111–115). The most recent revision was published in 2019 (115). However, most studies of SLE in the past two decades defined SLE cases according to the 1997 revised classification criteria (113). It has been observed that the 1997 criteria lead to 330 possible combinations of clinical manifestations that could satisfy SLE classification (76). Thus, despite being unified by anti-nucleic acid/anti-nucleoprotein autoimmunity (116), human SLE remains a clinically heterogenous disease state. Since particular patients differ in which features of SLE they manifest, attention must be paid to which features of human SLE a particular murine model recapitulates.
It is becoming increasingly clear that in most humans who develop autoimmune disease, disease most commonly arises from a complex interplay between many polygenic risk factors and one or more environmental triggers (79). Decreased cost of genotyping and the increasing size of autoimmune disease genetic cohorts has led to a seemingly ever-increasing list of disease risk loci. Indeed, for several common autoimmune diseases, the number of risk genetic loci across the genome now exceeds 200 (117). Each of these loci makes at most a modest contribution to relative risk of disease (odd ratio < 1.2) (117) and most are favored to act by regulating target causal genes (118–120). Together these risk alleles are thought to set a liability threshold that allows the development of autoimmune pathology in certain circumstances. These rules for human autoimmune pathologies appear to generally apply in the case of SLE and T1D with some subtle differences (caveats)?. One notable difference is that of association genetic association with the Major Histocompatibility Complex (MHC)/Human Leukocyte Antigen(HLA) Locus. In T1D, specific HLA alleles are associated with disease. Together, three amino acid variants account for nearly 30% of the phenotypic variance in T1D in European ancestry populations (121). This is similar to the case in RA, where specific HLA alleles have been shown to facilitate binding and presentation of the classic RA autoantigen, citrullinated peptides (122). In SLE, on the other hand, the major contribution to genetic association with the MHC/HLA locus has been mapped to Complement component 4 (C4A & C4B) gene copy number (123). Both C4A and C4B are genes that lie within the SLE association interval within the MHC/HLA locus. It has been shown that, in contrast to RA and T1D, the contribution of amino acid sequence variants to the SLE association at the MHC/HLA locus is minimal. HLA is not uninvolved in SLE etiopathogenesis, as there are additional contributions to SLE risk at this complex genetic locus that are attributable to regulation of MHC class II expression (123). However, the bulk of the risk from HLA in SLE arises from regulation of the complement system and not specific MHC alleles (123).
In terms of genetic structure, SLE is most commonly polygenic (117), but numerous monogenic forms of SLE have been described, 51 of which we are aware (124–196). Monogenic SLE presents more commonly with childhood onset and a severe disease phenotype (117, 124–126). It appears that in addition a minority of childhood onset cases, currently estimated at approximately 15% exhibit a probable mix of monogenic and polygenic genetic etiologies (197, 198). Ongoing studies suggest that rare or private mutations also partially contribute to risk in multipatient SLE pedigrees. However, the extent to which such mutations contribute to SLE risk is still being defined (199). To synthesize what is known about polygenic causes of SLE, we applied a previously described approach to published SLE risk variants in the NHGRI-EBI GWAS catalog (117). First, we grouped SLE risk variants listed in the GWAS catalog (200) into loci/regions, then integrated published results the from Open Targets Genetics (201) Locus to Gene (L2G) (202) algorithm. L2G is a machine learning pipeline that predicts a causal gene by integrating several sources of evidence. These sources include distance from causal credible set variants to gene, molecular QTL co-localisation, chromatin interaction data and where applicable variant pathogenicity prediction from the variant effect predictor algorithm. This evidence is then weighted by gold-standard functionally demonstrated causal variants from different GWAS studies. For loci where L2G was able to be confidently annotate a likely causal gene, that gene was included in the molecular network. This list is not comprehensive. Our approach to region definition obscures several known regions with multiple independent genetic effects. Despite this, we find 182 polygenic human SLE risk loci. By applying the L2G automated machine learning pipeline and manual annotation our final list includes 109 loci with assignable putative causal genes within these loci (Supplementary Table 2A).
In contradistinction to SLE, only very few (8 of which we are aware – Supplementary Table 1B) monogenic causes of autoimmune type 1 diabetes have been described (203–213). Monogenic autoimmune T1D arises in genetic syndromes of polyendocrinopathy. These autoimmune diseases are characterized by autoimmunity that adversely impacts multiple endocrine organs, not merely the pancreas. Only eight monogenic routes to autoimmune diabetes have been described provides a contrast to SLE. This may be in part due to the diffuse, systemic nature of SLE versus the more narrow target organ range of T1D. While SLE exhibits considerable clinical and phenotypic heterogeneity (214) that is unified around anti-nucleic acid/anti-nucleoprotein autoimmunity (116), type 1 diabetes leads to autoimmune pancreatic beta cell destruction. So, it may merely be that in this case there are more opportunities to develop an immune dysregulation syndrome resembling one or more features of SLE, as the manifestations of SLE are both numerous and diverse.
In individuals with T1D, the disease more commonly arises from the aggregate effects of polygenic risk alleles, just as with SLE. Indeed, in the comprehensive review of monogenic autoimmune type 1 diabetes to date reflects the experience of approximately 500 individuals worldwide (203). Thus, monogenic genetic effects or rare genetic effects of large effect size do not likely explain a significant proportion of type 1 diabetes patients and this also appears to be the case in several autoimmune diseases (215). To explore this risk gene network we applied the same approach to define a high confidence causal polygenic risk gene network in human type 1 diabetes. This analysis of type 1 diabetes risk loci from the GWAS catalog yields a list of 131 polygenic human T1D risk loci. The L2G algorithm was able to confidently identify 63 putative causal genes within these loci (Supplementary Table 2B). Again, our approach likely obscures the presence of multiple independent signals in a particular region. A recent GWAS meta-analysis of T1D reported that 33% of the independent association signals occurred in loci with multiple independent association signals within the same locus. These independent signals within the same locus might exert their biological effects on disease risk through the same gene. Alternately, these multiple independent signals might exert their biological effects on disease risk through multiple independent genes.
IL2RA stands out as an algorithmically defined putative causal genes that is also present in the list of monogenic autoimmune type 1 diabetes genes (Supplementary Table 1B) as has been observed by others (216). Like SLE (Figure 1), the monogenic and polygenic type 1 diabetes risk networks overlap at this hub node (Figure 2). This suggests that these hub nodes may be particularly attractive as targets that span disease states based on their central location in both monogenic and polygenic disease molecular networks. In sum, the overlap between polygenic and monogenic disease genetic networks in both human autoimmune Type 1 Diabetes and SLE indicates that the monogenic forms of these diseases perturb the same diseases networks as polygenic disease.
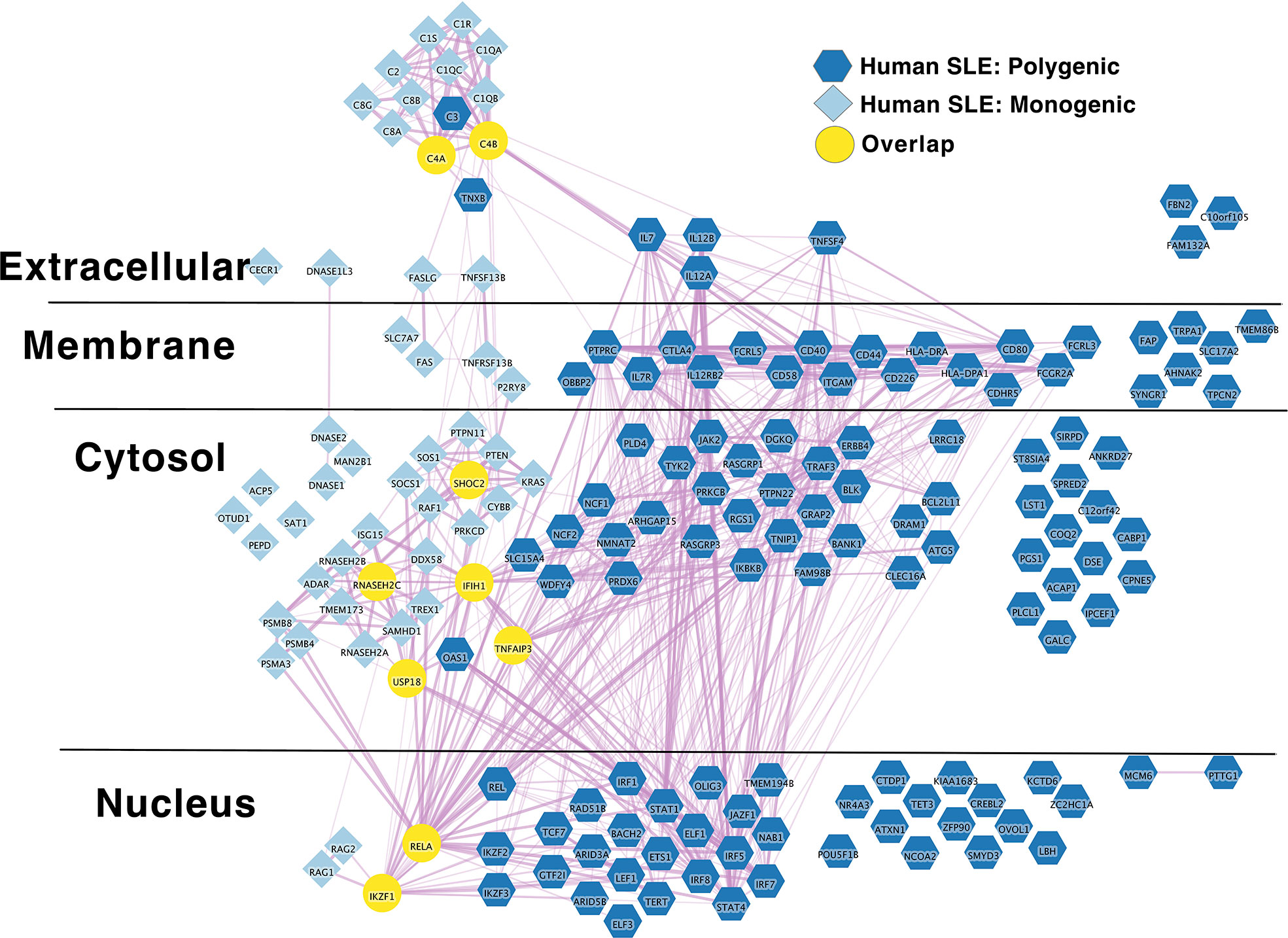
Figure 1 Monogenic and Polygenic human SLE risk gene networks overlap at hub genes. Light blue diamond – Monogenic human SLE genes; dark blue hexagon – Polygenic human SLE genes; Yellow circles – overlapping genes. Downloadable/Interactive network diagram can be found at: https://doi.org/10.18119/N9231T.
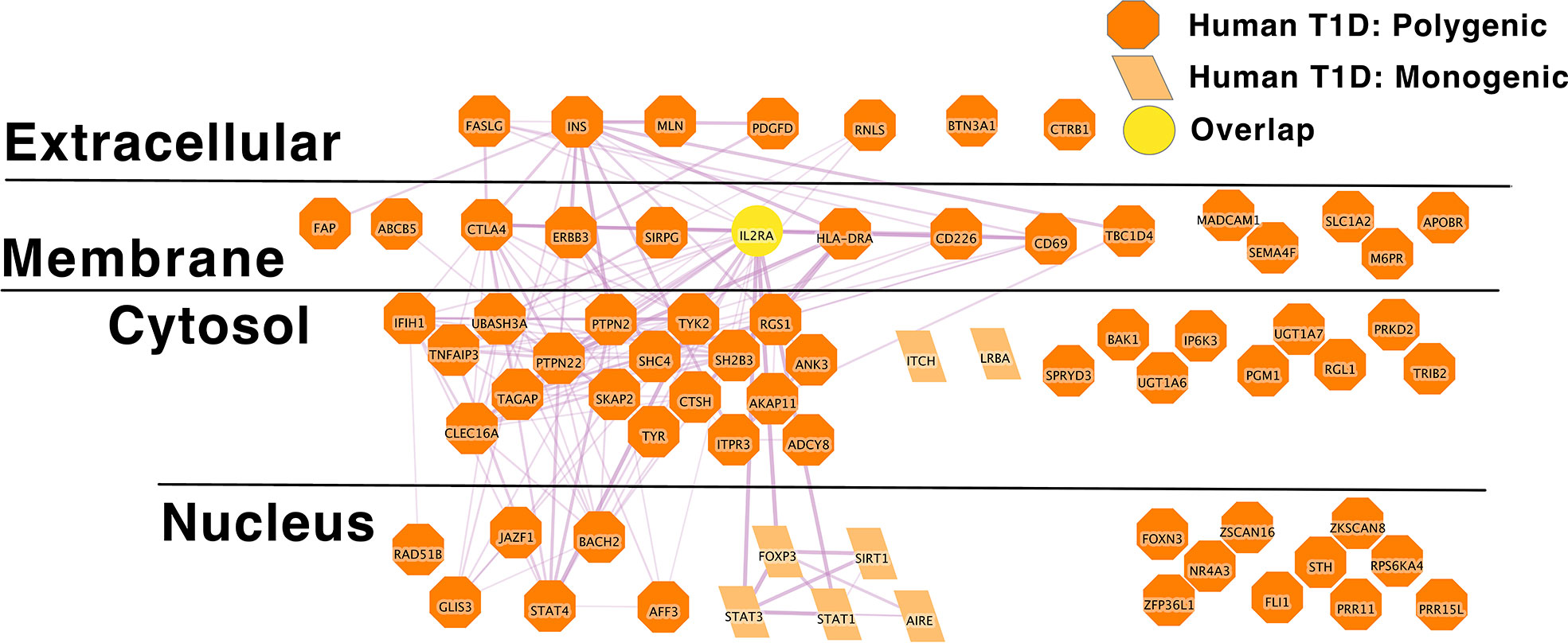
Figure 2 IL2RA is the link between Monogenic and Polygenic human type 1 diabetes risk gene networks; light orange parallelogram– human monogenic autoimmune type 1 diabetes gene; dark orange octagon– human polygenic autoimmune type 1 diabetes gene; Yellow circles – overlapping genes.Downloadable/Interactive network diagram can be found at: https://doi.org/10.18119/N94W34.
A few general points concerning polygenic genetic structure should be considered. One objection that has been raised to polygenic structure in complex human disease is that sporadic cases are common. Sporadic refers to cases without a known family history of disease. However, statistical genetic models predict that sporadic cases of complex genetic disease will commonly occur even in disease with a polygenic genetic structure (217). Second, the bulk of polygenic risk alleles reported to date in common autoimmune disease only have small effects. In human SLE, as an example, only a handful of common genetic risk factors (four that we know of) impact disease relative risk from 2-10-fold (117). Applying knowledge of population prevalence, the genetic factor with the largest effect would change the absolute risk of SLE from approximately 0.1% to 0.4% (117). This kind of polygenic genetic architecture is present in many human phenotypes. This observation prompted the proposal of the Omnigenic model of complex traits (218). In this model, larger effect size variants (>1.1-fold increase in relative risk) operate within core disease pathways. However, thousands of loci with infinitesimally small effect size spanning the entire genome change absolute genetic liability (218). In this model, the entire genome is ultimately involved in disease risk, with each variation outside of the core disease pathway adding a very tiny amount of residual risk. In simple terms, it seems perhaps tautological to state that the whole genome is involved in any given trait, even if only slightly changing the trait. It is worth noting that predictions of this model appear to hold in other complex human genetic traits, such as height (219).
As an aside, the omnigenic model provides a potential explanation for why autoimmune disease genes have not been eliminated via natural selection. If most of the hundreds of core risk alleles are inherited independently (low correlation or linkage disequilibrium) and they each have a small effect, then selective pressure would not be expected to be strong in individuals with polygenic autoimmune disease. By way of analogy, being related to someone who wins the lottery does not make winning the lottery more likely for you, unless you buy more lottery tickets. On the other hand, many monogenic disease genes represent either de novo mutations or recent founder effects. Therefore, monogenic mutations have not had a very long to be subject to natural selection. These observations when combined with theoretical frameworks describing the balance between host collateral damage from immune responses and microbe clearance (51–53) may also explain the retention of these alleles in the wider gene pool. That is, there are several ways in which immune responses can be balanced to avoid damage to host. Genetic variation that modulates an immune response that is too weak or too strong for one context, may, in another context or in another generation better strike that balance.
If the omnigenic model is correct and thousands of risk loci are involved in determination of common polygenic traits, then sample sizes of > 1 000 000 affected individuals may be needed to develop risk scores that capture enough variants to explain the majority of variation in genetic risk (220). For most autoimmune diseases, these samples exceed the total number of affected individuals living on entire continents. If true, it would make systematically dissecting genetic network interaction with environmental disease triggers so complicated as to be potentially intractable. Our aim is to deconstruct disease processes, in order to improve our ability to diagnose, prognose, prevent and treat autoimmune diseases. Therefore, we must reduce the complexity of the systems we aim to deconstruct. In this way, we can build conceptual models of autoimmune disease development and maintenance that we can actually comprehend.
One approach is murine models. Such models may strike an appropriate balance between over-simplification and a sufficient degree of biological complexity such that core disease relevant cellular and molecular networks are conserved. Thus, findings can be expected to translate to humans. When proper controls and careful attention to potential confounders is observed, mouse models of disease have been very powerful in advancing our understanding of autoimmune pathologies (59).
Having an intermediate model of sufficient biological complexity is likely necessary for many types of causal evidence that allow inference regarding mechanism in cellular and molecular disease networks. In many cases this kind of inference cannot be achieved for either ethical or technical reasons in humans and are inadequately modeled in vitro. Many therapies that are promising in vitro do not stand up to testing in the more complex biological system that a whole organism in vivo represents. One recent example of relevance to autoimmune disease is that of hydroxychloroquine (a mainstay of SLE and Rheumatoid Arthritis therapy (221)) in the treatment of COVID-19. Indeed, hydroxychloroquine robustly inhibited SARS-CoV-2 (and other coronaviruses) in vitro (222), but was shown to be ineffective in prevention of SARS-CoV-2 infection and treatment of COVID-19 in randomized controlled trials in humans (223–226). While it is a moot point now that the high-quality human data exist, an intermediate in vivo model system may have been able to predict and understand this therapeutic failure and thereby reprioritized COVID-19 patients for more suitable trials.
Several criticisms of mouse models of human autoimmune pathologies specifically and human disease writ large (with the use of SOD1-deficient mice in Amyotrophic Lateral Sclerosis representing a high-profile model with several issues of phenotypic non-correspondence) have been raised [notably (55, 67, 227, 228)]. See section 5 for our attempt at a comprehensive list of some key variables to consider in modeling human autoimmune disease in mice.
One major criticism that has been raised for why mouse models of human autoimmune disease are ‘lousy’ is failures in translation from experimental autoimmune/allergic encephalomyelitis into successful therapy for multiple sclerosis. However, we would submit that careful attention to both the details of the murine and human pathology and careful reexamination of models in light of the clinical, phenotypic, cellular and molecular features of the human diseases we seek to model would have predicted successful therapeutic targets even in this ‘lousiest’ of autoimmune disease models.
Failed trials of TNF-alpha inhibitors as well as oral and IV tolerance autoantigen-specific tolerance protocols that succeeded in mice, but failed in MS patients are often cited. Incidentally, TNF-alpha inhibition did not merely fail, but was subsequently discovered to be a risk factor for incident demyelination, just as it is a cause of drug-induced lupus. It is worth noting that despite many high-profile therapeutic failures, reassessment of successes, failures and refinement of models have led to several successful novel therapeutic approaches for MS treatment in the interim (68). Subsequently, phenomenally successful trials of B cell-depleting monoclonal antibodies directed against CD20 were performed in MS. In fact, B cells are so important in this autoimmune disease, that B cell depletion using anti-CD20 monoclonal antibodies is now the mainstay of therapy. This is not necessarily a conclusion that would have been reached by solely relying on data from the EAE model (229–234), even though careful experimentation ultimately revealed an important contributory role for B cells once early studies demonstrated the efficacy of anti-CD20 therapies in human MS (235). Subsequent work by many groups has demonstrated that antigen presenting B cells play a central role in the pathogenesis of human MS (236). Building on the principle of the oral tolerance studies in MS, re-enforcing tolerance in formerly anergic B cells remains an active area of investigation (237). More recent data has further advanced our understanding of the role of B cells in MS, as prior Epstein-Barr virus infection (but not other common latent viral infections) was shown to be an independent risk factor for MS development (238), leading commenters to infer that “These findings provide compelling data that implicate EBV as the trigger for the development of MS” (239). These data led to pan-proteome analysis of the auto-specificities of the pathognomonic oligoclonal bands found in the CSF of MS patients. Crossreactivity was shown between a human CNS autoantigen, GlialCAM and the EB viral latency transcription factor EBNA-1 (240). Indeed, as a final attempt to prove etiopathogenesis of EBV in MS – using a modified version of Koch’s postulates, the authors of the latter paper immunized EAE mice and concluded that “EBNA1 immunization aggravates EAE”. In doing so, they have nominated yet another potential therapeutic approach for MS that relies, in part, on the EAE model, the prevention of EB virus infection. In retrospect, the story of the EAE model seems to us more like the typical pattern of advances in science where models are challenged by data and refined so that the model predictions better fit the observed data. Indeed, it now appears that the use of proper controls, challenging murine models with ideas from human data and vice versa has an aggregate effect of reducing the influence of potential confounders. In so doing this approach would be expected to lead to a more accurate model autoimmune etiopathogeneis than either approach would have been able to do on its own (60). (many important potential variables are detailed in section 5.)
Thus, despite oft being cited as a model of autoimmune disease with high profile failures in translation, careful attention to the human processes being modeled by the EAE model continues to yield insight into MS pathology. In a similar manner, we expect that careful attention to potential confounders of lupus and T1D models, the use of multiple models and iterative comparison to intermediate human disease phenotypes would be expected to yield important insight into these human autoimmune pathologies.
To better understand the relationship between human autoimmune pathology and murine models of autoimmune disease, we compared their respective gene networks. We have focused on making our comparison in long-standing murine disease models of two human autoimmune diseases that are fairly-well characterized in terms of correspondence across spontaneous disease models. For models of both diseases, excellent reviews of the convergent and divergent immunopathogenic bases for disease development between mice and humans have been written and we refer the interested reader to read them: [murine lupus (60, 61, 241, 242): murine type 1 diabetes (243, 244)].
The prevailing model of autoimmune disease risk is that the genetic networks regulating lymphocyte tolerance are core to autoimmune disease and span multiple autoimmunities (56, 57, 245, 246). That is, human genetic risk alleles shared across multiple autoimmune diseases perturb the normal function of lymphocyte self-tolerance networks. To begin both to evaluate this model more systematically and to more fully understand the differences between the murine and human autoimmune disease genetic risk networks, we reviewed the literature and collected lists of putative causal genes in murine models of SLE and type 1 diabetes, as well as genes whose disruption lead to B cell central or peripheral tolerance defects (247–450). Together, each of these sets of genes comprise a molecular network and many of the genes in each network overlap with those in the other networks (Figure 3). Taken together, these data point towards an important role of B cell central and peripheral tolerance regulatory networks in murine models of type 1 diabetes and SLE.
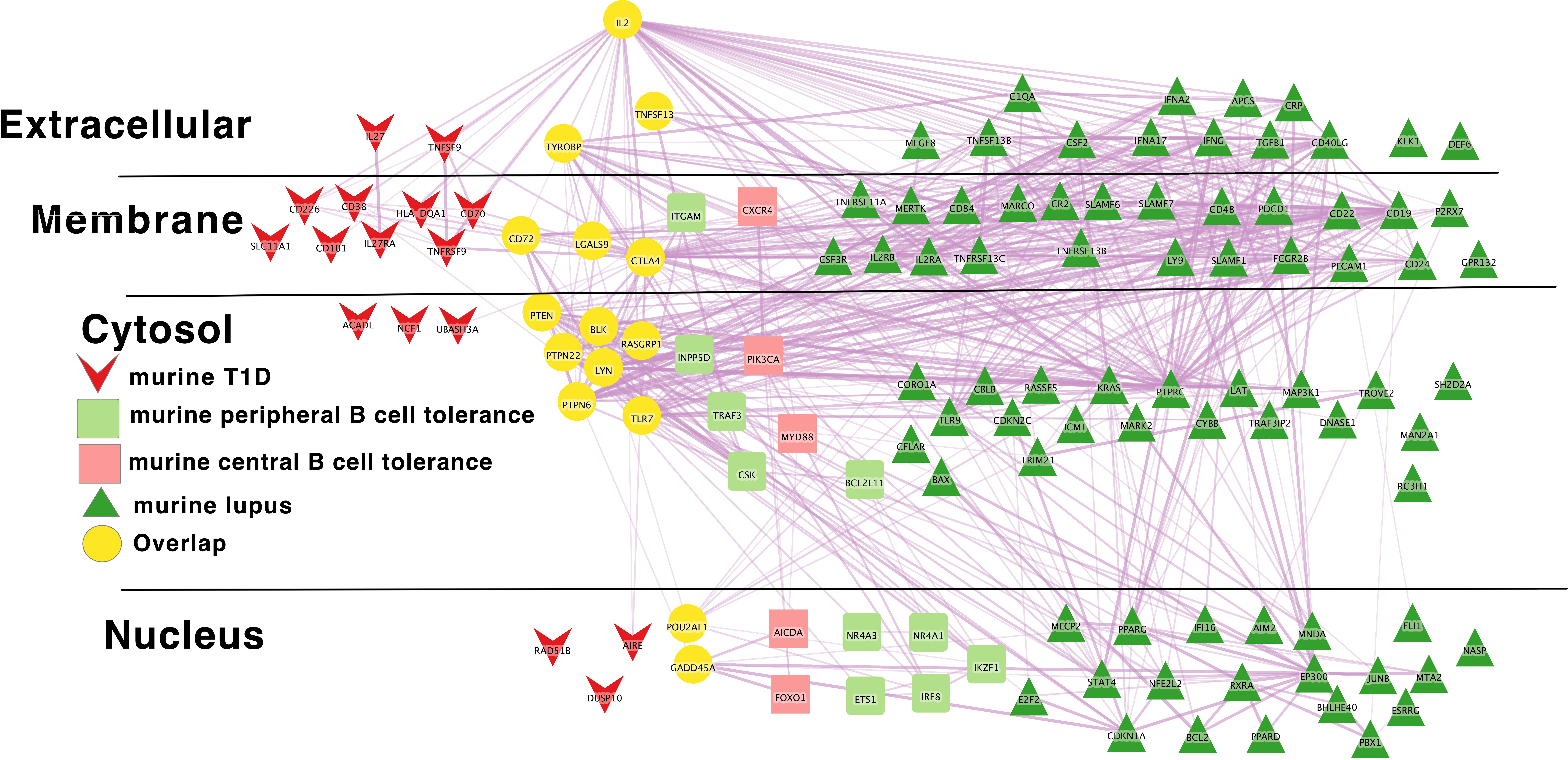
Figure 3 Murine autoimmune diabetes and lupus networks are densely connected to peripheral autoreactive B cell tolerance networks; dark green triangle – murine lupus gene; light green rounded rectangle – murine peripheral B cell tolerance gene; Yellow circles – overlapping genes. Downloadable/Interactive network diagram can be found at: https://doi.org/10.18119/N9161J.
To understand how autoimmune disease gene networks overlap, we merged the murine and human risk gene networks for SLE and T1D in several ways. Our goal was to evaluate whether the published studies support the prevailing model – that the genes regulating tolerance induction and escape of autoreactive B cells are central to the risk gene network of these seropositive autoimmune diseases. First, we combined risk genes from monogenic human SLE (Supplemental Table 1A), polygenic human SLE (Supplemental Table 2A) and murine Lupus genes (Supplemental Table 3) into a single network (Figure 4). Second, we combined gene from monogenic human T1D (Supplemental Table 1B), polygenic human T1D (Supplemental Table 2B) and murine autoimmune diabetes (Supplemental Table 3) genes into a single network (Figure 5). Finally, we combined both of the disease-specific networks (from Figures 4, 5) along with both B cell central (Supplemental Table 3) and peripheral tolerance (Supplemental Table 3) gene networks into a single network (Figure 6). Strikingly each of these gene sets formed a distinct protein-protein interaction network with greater overlap than expected by chance (Table 1). Further, the human monogenic and polygenic and murine genetic networks overlap 16-fold to 63-fold more than would be expected by chance (Table 2). Likewise, these networks overlap with one another or the overall B cell tolerance and murine disease networks between 15-fold and 86-fold more often than expected by chance (Table 3).
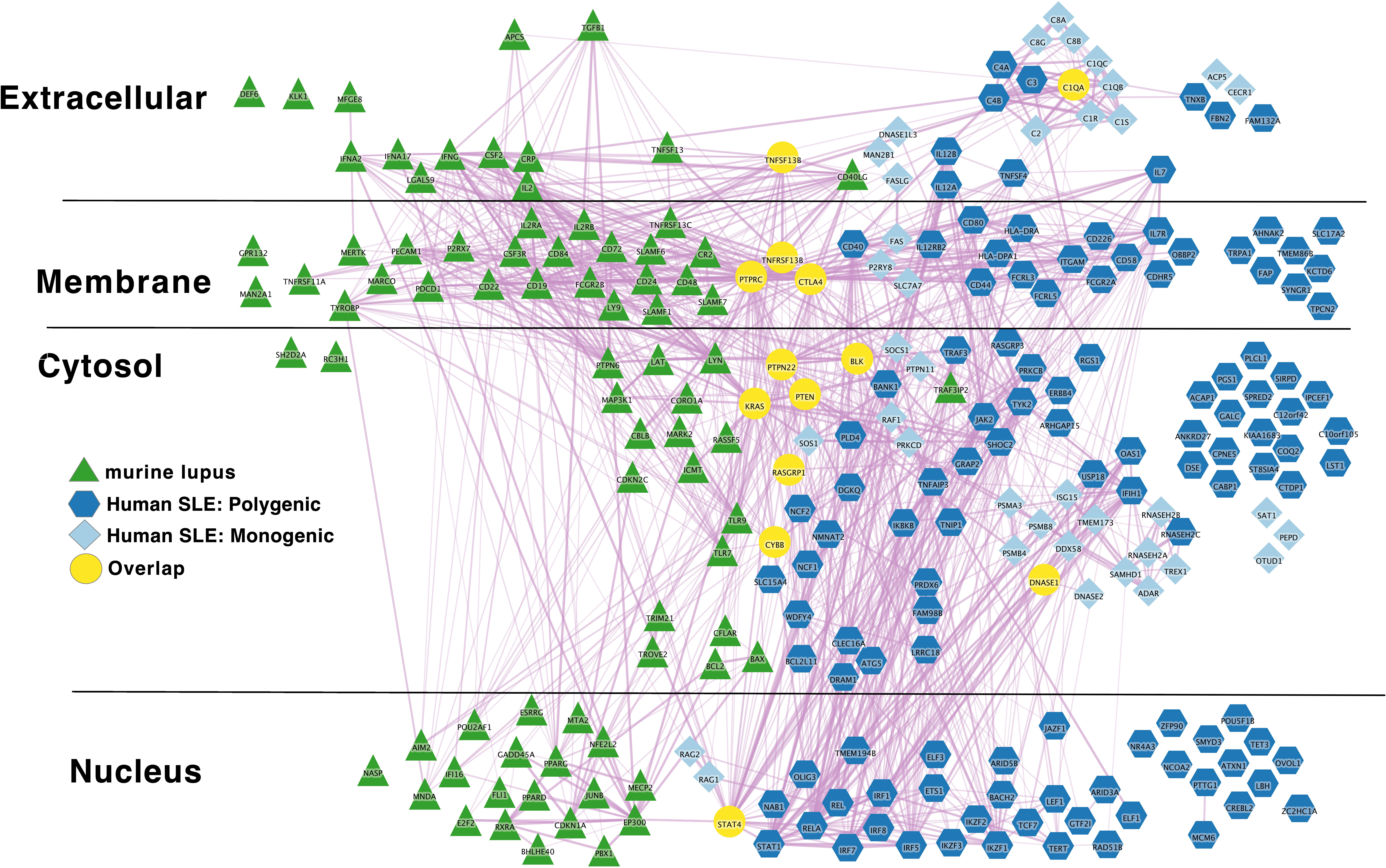
Figure 4 Murine Lupus risk genes connect to Polygenic Human SLE risk genes at the periphery of the core network in a manner similar to the monogenic risk SLE network; Light blue diamond – Monogenic human SLE gene; dark blue hexagon – Polygenic human SLE gene; dark green triangle – murine lupus gene; yellow circles – Overlapping genes. Downloadable/Interactive network diagram can be found at: https://doi.org/10.18119/N9WC8P.
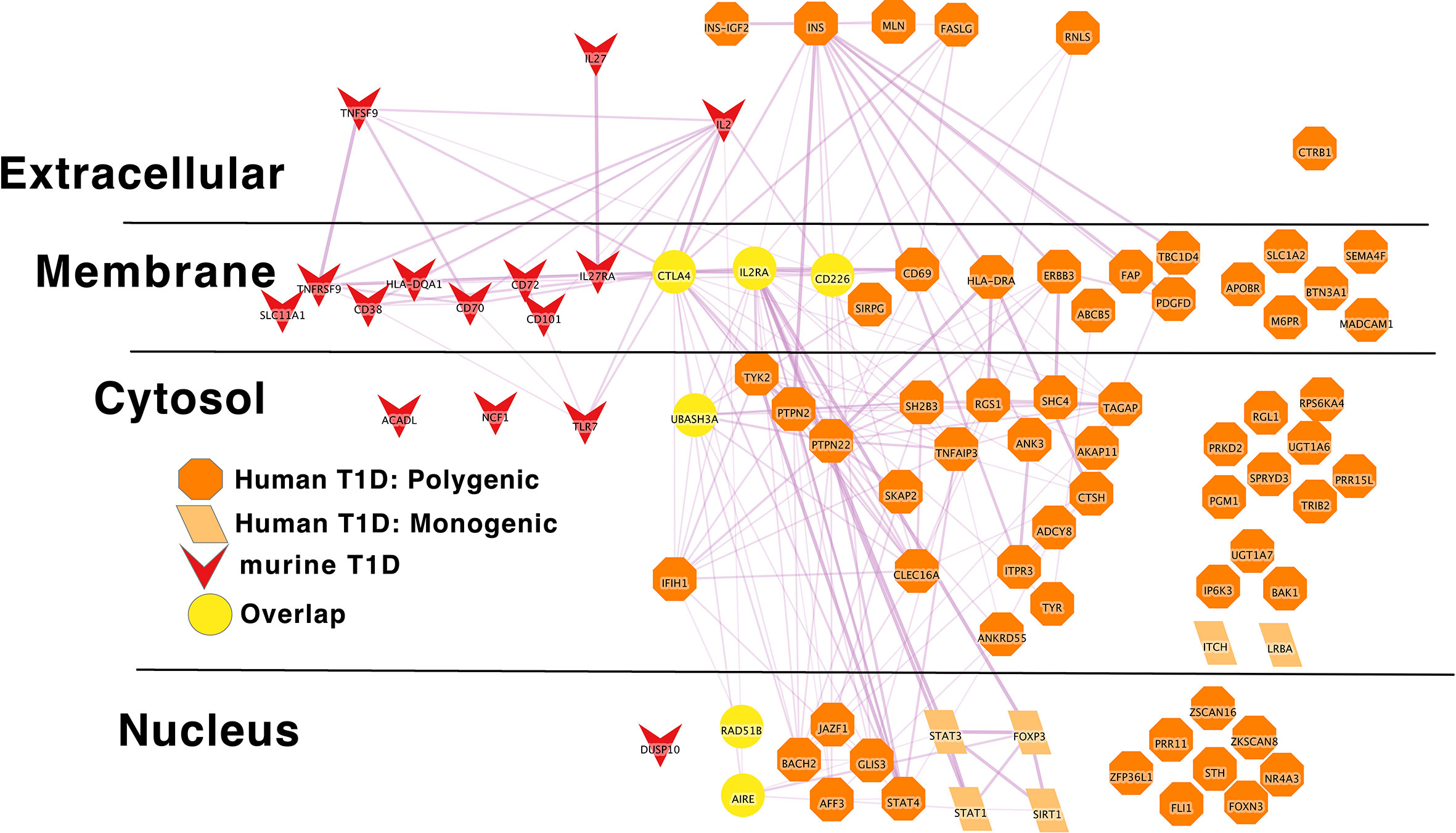
Figure 5 Murine autoimmune diabetes risk genes connect to Polygenic Human T1D risk genes at the periphery of the core network in a manner similar to the monogenic risk T1D network dark red inverted triangle – murine autoimmune type 1 diabetes gene; light orange parallelogram– human monogenic autoimmune type 1 diabetes gene; dark orange octagon– human polygenic autoimmune type 1 diabetes gene. Downloadable/Interactive network diagram can be found at: https://doi.org/10.18119/N9RP6S.
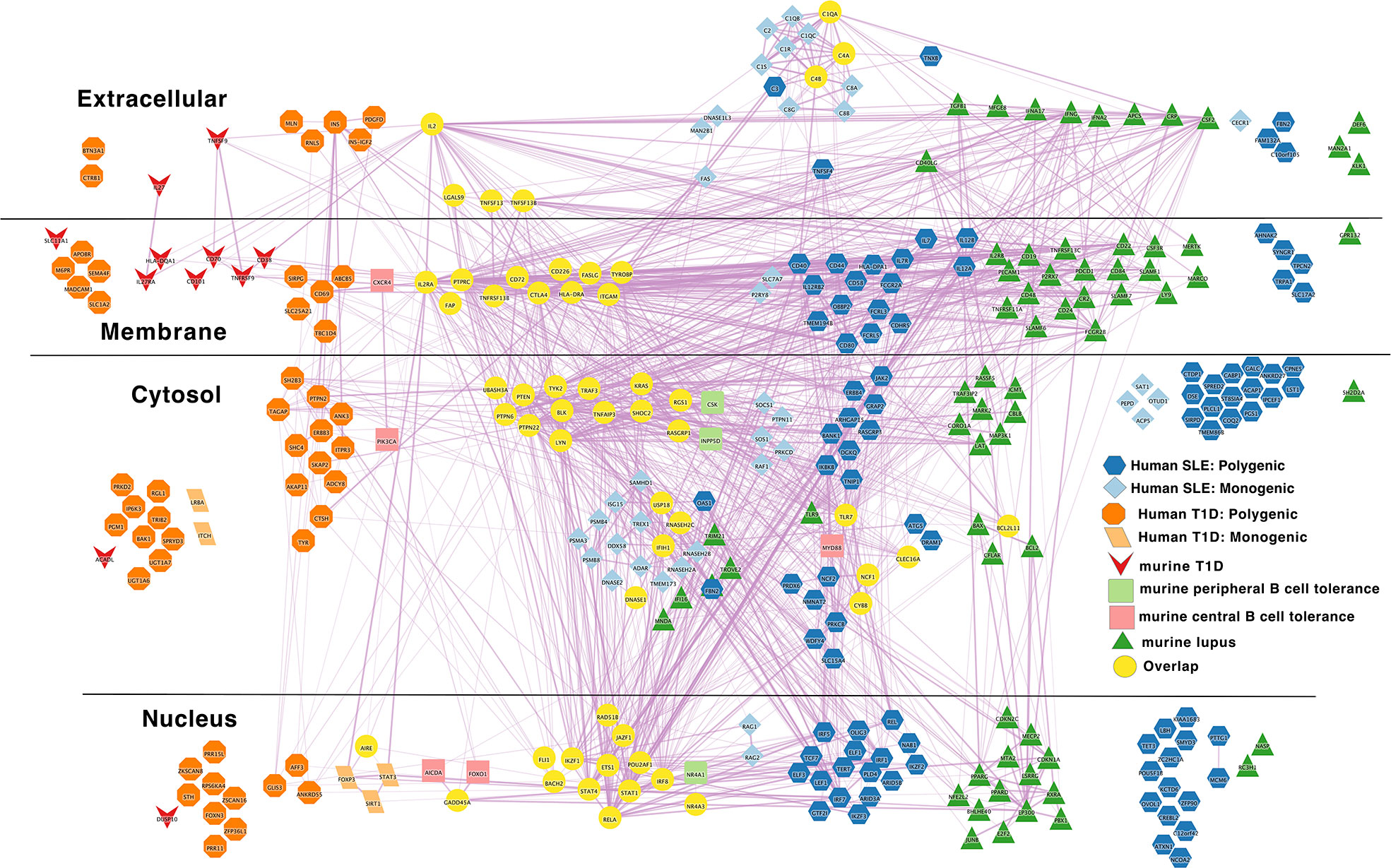
Figure 6 Murine autoimmune disease model genes center around the autoreactive B cell peripheral tolerance network in the middle of combined human autoimmune disease polygenic risk networks. Light blue diamond – Monogenic human SLE gene; dark blue hexagon – Polygenic human SLE gene; dark green triangle – murine lupus gene; light green rounded rectangle – murine peripheral B cell tolerance gene; light red rectangle – murine central B cell tolerance gene; dark red inverted triangle – murine autoimmune type 1 diabetes gene; light orange parallelogram– human monogenic autoimmune type 1 diabetes gene; dark orange octagon– human polygenic autoimmune type 1 diabetes gene; yellow circles – Overlapping genes. Downloadable/Interactive network diagram can be found at: https://doi.org/10.18119/N9MW3G.
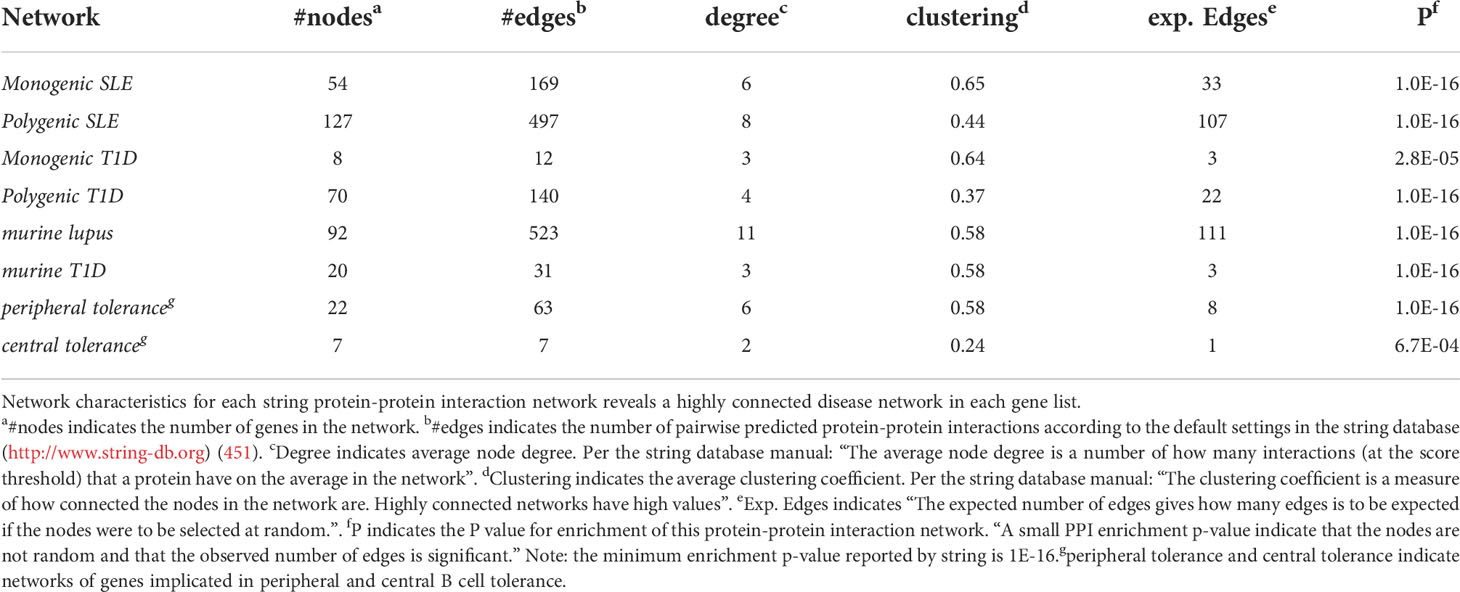
Table 1 Network characteristics.
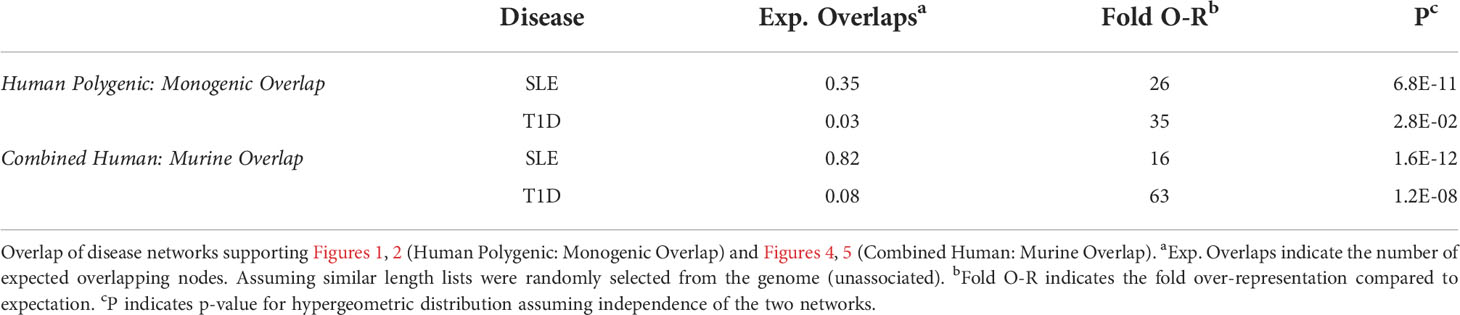
Table 2 Disease Network Overlap.
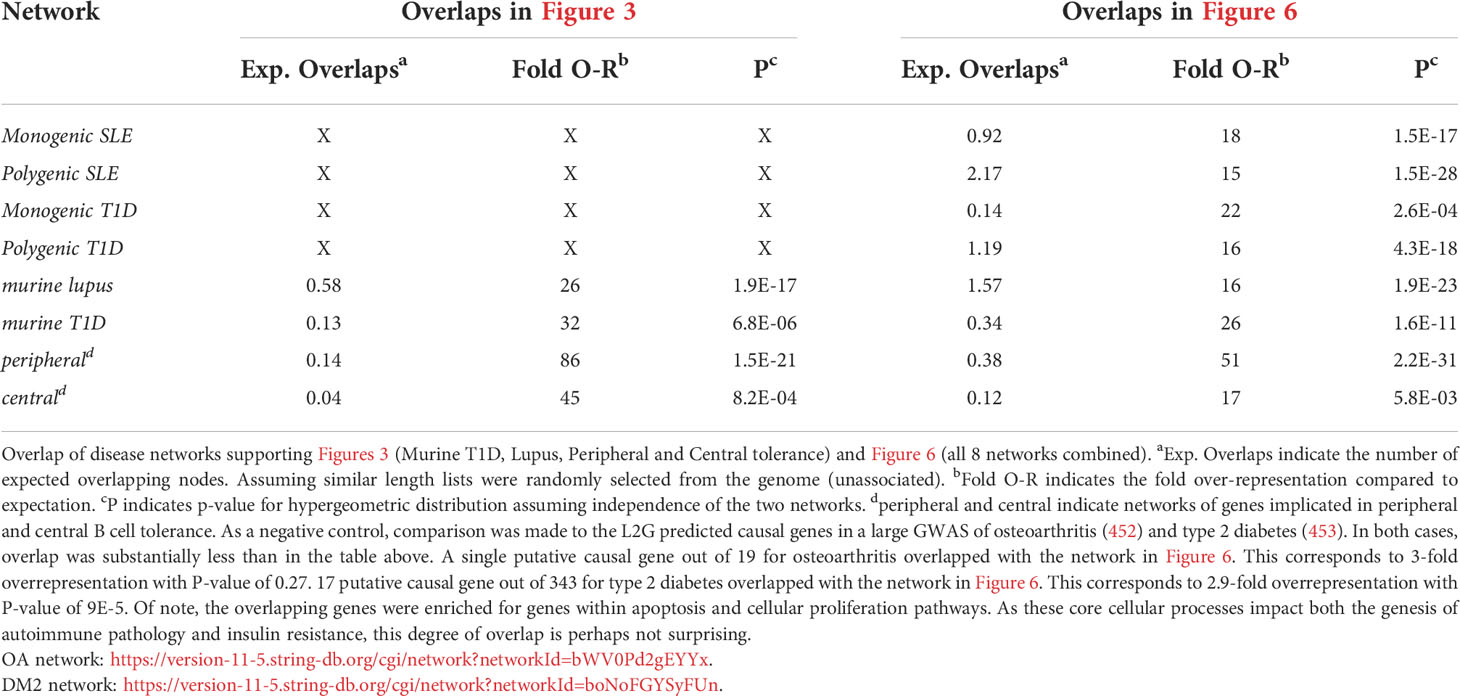
Table 3 Overlaps of disease networks supporting Figures 3 (Murine T1D, Lupus, Peripheral and Central tolerance) and Figure 6 (all 8 networks combined).
Overall, this analysis reveals a densely interconnected core autoimmunity gene network centered around genes that regulate B cell peripheral tolerance. This observation provides some degree of support for the prevailing model in the field, that the genes regulating tolerance induction and escape of autoreactive B cells are central to the risk gene network of these seropositive autoimmune diseases. Intermixed within this core are the murine type 1 diabetes and lupus gene networks. While this approach has utility in providing a high-level overview of autoimmune disease risk regulatory networks, it does have some drawbacks. In each particular network, there are several putative causal genes that are not well connected to the central network. Certainly, it is possible that these genes have yet to be discovered function in the genesis of autoimmunity. However, there are other potential explanations for lack of connection to this central network. In some cases, these may represent misattribution of causality. For example, while the L2G algorithm nominated PTTG1 as a putative causal gene for SLE, we have previously shown that altered function of the microRNA, MIR146A, likely better explains the observed association with SLE at this locus (454). Alternately, these genes may impact lupus function in a way that has not yet been represented in the molecular networks of the STRING database. For example, recent work has established DNASE1L3 as casual for SLE. First, non-synonymous coding changes in DNASE1L3 explain the bulk of the genetic association with SLE near the PXK locus (455). Second, germline mutations in this gene have been described as a monogenic route to lupus (198, 456–458). Third, titers of autoantibodies against this enzyme correlate with disease flare in patients with lupus nephritis (459). Fourth, functional studies implicate the function of this secreted, extracellular DNAse in digesting the nucleic acids present in autoantigenic debris from dying cells (460–462). Thus, while the role of DNASE1L3 in SLE risk is becoming abundantly clear, the STRING database (451) has not yet codified this new understanding. At the same time, there may be other information missing from the gene network as we have defined it. At this same locus, DNASE1L3-PXK, an additional contribution to genetic association with SLE is seen (455). This additional association is due to variation near PXK, a phox-homology kinase implicated in B-cell receptor endocytosis (463). There is evidence for a potential role of PXK in modulating B-cell receptor signaling and generating autoreactivity. However, the automated algorithmic approach that we used did not place PXK within the polygenic SLE risk network. While this approach provides a useful overview of the interrelationships between gene networks, by its nature, it also provides an incomplete picture of disease risk due to incomplete information.
On a more granular level, these analyses revealed overlapping networks between monogenic and polygenic SLE. This overlap was between complement, cytosolic nucleic acid sensors, Ikaros and NF-kB pathways (Figure 1). In terms of monogenic and polygenic autoimmune type 1 diabetes, not surprisingly, there is limited overlap (Figure 2). However, there is still more than expected by chance. This includes a preponderance of key transcriptional regulators (STAT1, STAT3, FOXP3, AIRE) that are central regulators of T lymphocyte development in monogenic T1D. Close inspection of these networks shows that they do not overlap at AIRE. This lack of overlap highlights one of the drawbacks of the automated, algorithmic approach to putative causal gene definition. A rare variation in AIRE, rs74203920, was recently reported in a large GWAS of human autoimmune type 1 diabetes (464). This non-synonymous variation results in an amino acid change that is predicted to be deleterious. It has a minor allele frequency of ~2% in individuals with European continental ancestry in the 1000 Genomes project. Further, using Bayesian statistical approaches, the authors report a posterior probability of association > 99% (464). There are examples of non-synonymous coding changes in GWAS genes whose biological effects on disease risk may be through modulation of gene expression (465). However, it seems most parsimonious to conclude that AIRE is, in fact, the likely causal gene at this T1D risk locus. That our approach using L2G did not identify this particular variant and it therefore did not overlap with the monogenic T1D risk network highlights one of the drawbacks of this approach in terms of misattrubtion. It further suggests that our overlaps are more likely to represent a lower bound on the overlap between the true disease risk networks than an upper bound.
Turning to the network that combnines murine lupus, murine T1D and murine B cell tolerance gene networks, we also find substantial overlap. This overlap occurs within several pathways: IL2 (IL2), BCR signaling (BLK, Lyn etc.), tolerance response to nucleic acid (CD72, TLR7), tolerance to self-nucleic acid and control of viral infection. These overlaps serve as unifying pathways in these models of autoimmune pathology (Figure 3). Overlap of murine and human lupus occurs at B-cell signaling hubs involving BAFF, APRIL and B cell antigen receptor signaling. Of note, despite its central importance in SLE etiopathogenesis (117), TLR7 is absent from the human disease networks, though its signaling intermediates remain. Likewise, LYN is absent from the human disease networks despite its identification as a likely causal gene for SLE in GWAS follow-up studies. (Figure 4) Thus, our analysis likely underestimates the true extent of overlap between these various gene networks. Similar to Lupus, type 1 diabetes in mouse and humans is unified by T-cell tolerance regulators (CTLA4, IL2RA, CD226, AIRE, etc.) (Figure 5). Finally, peripheral B cell tolerance is the most over-represented compared to no association when looking at the unified network of all these states of pathologic autoimmunity (Figure 6). The substantial overlap between these different networks is consistent with a prominent role of particular environmental drivers in specifying the target organ focus of autoimmunity.
One question that arises is whether these associations represent an increase over what would be expected by chance. Indeed, overlap between the gene networks in type 2 diabetes (453) and osteoarthritis (452) are much less with these non-autoimmune traits than any of the autoimmune pathology networks (Table 3). Another question is how to address cell type specificity of these networks. One might assume that these gene networks only operate in concert within specific cell types. PTPN22 may serve as a counterexample to this – a recent review highlighted evidence for six independent mechanisms of the PTPN22R620W variant each operating in different cellular lineages (466). It may be that some autoimmune disease risk alleles do act in a cell type and cellular context-specific way. However, for many complex human traits, the genetic structure predicted by the omnigenic model appears to be the case. That is, hundreds to thousands of genetic variants of (mostly) very small effect size act in aggregate to set a genetic liability threshold. The central nodes in these disease gene networks have the largest effect size and therefore likely a lower statistical power requirement to demonstrate association. Thus, like many pharmacotherapies (467), it may well be that these core disease genes have multiple mechanisms through which they modulate disease risk. Hence, they are centrally located and have outsized effect sizes. Certainly, BLK, Lyn and the BAFF family genes in these networks could be argued to have effects selective to the B cell lineage. However, both BAFF (468) and Lyn (469) have well described actions outside of B cells. Likewise, BLK exhibits high expression in human plasmacytoid dendritic cells (470, 471) and the most strongly associated eQTL variants are within human fibroblasts. Both of these cell lineages are independent from B cells and have direct relevance to SLE etiopathogenesis. A role for these three genes acting to increase SLE risk within B cells is certainly more parsimonious. Alternately, it has been argued that several of the polygenic risk variants for human type 1 diabetes exhibit opposite action in effector and regulatory T cells (472). That is, several risk variants increase the likelihood of activation in effector T cells and simultaneously increase the likelihood of inhibition in regulatory T cells. Thus, even with specific cellular mechanisms, the risk alleles of the strongest effect size may be the most likely to have multiple mechanisms whereby they alter disease risk. Cogent arguments can be made for the cellular specificity of gene networks acting within a disease state. However, much work remains to be done to convincingly demonstrate cell-type specificity of genetic effects, over against disease risk networks that span and exert their effects within multiple cellular lineages.
What are the explanations for challenges in translatability of autoimmune disease mouse models?
We have discussed spontaneous, induced and humanized murine autoimmune disease models above in general terms. Here we focus on key potential differences that in our estimation are likely to affect several spontaneous models of lupus, such as those derived from the NZB/NZW F1 (BW) mice and the NOD mouse model of type 1 diabetes.
The use of recombinant inbred mice more closely resembles consanguinity that is seen more commonly the parents of individuals with childhood onset autosomal recessive disease. In this way, these murine models may offer more opportunities to develop monogenic mutations and sub-strain differences can profoundly alter physiology (473). One example sticks out in particular. The most commonly used lab mouse strain, C57BL6/J, developed a loss of function mutation in Nnt, the gene encoding for the nicotinamide nucleotide transhydrogenase (473). This mutant Nnt diverges from another commonly used lab mouse strain C57BL6/NJ. Unfortunately, Nnt mutation inadvertently serves as a model of familial glucocorticoid deficiency, which has been described in mice and humans who have mutant NNT (474). This could conceivably confound interpretation of results obtained using models that have not controlled for this mutation in lupus in particular, where glucocorticoids are a mainstay of therapy. As another example, a body of literature describing functions previously attributed to caspase-1 are in fact due caspase-11 deficiency due to inadvertent gene-targeting leading to generation of caspase-1/caspase-11 double knockout mice (475).
Sixty-five million years of evolutionary history seems like a long time. Certainly, it is long enough to develop changes in how genes respond to the environment. As a stark example, Gout is a disease of higher primates. It is one of the most common forms of inflammatory arthritis and is estimated to affect 1 in 200 people worldwide. Gout occurs when uric acid levels are too high and uric acid crystals precipitate out of the serum, driving acute and chronic inflammation. Gout is thought to have arisen ~ twenty-two million years ago when one of a series of loss of function mutations in uricase (which converts uric acid to the much more water-soluble allantoin) and URAT1 and important renal uric acid transporter. As this system non-redundantly regulates blood pressure, it stands to reason that changes across similarly complex immune networks could have also developed differences in some critical regulatory genes. Indeed, many immune phenotypes that diverge between mice and humans have been described (476). Two select examples of gene to phenotype non-correspondence include MyD88 and STAT5B. MyD88 deficiency leads to early life susceptibility to only pyogenic infections in humans whereas it leads to long lasting susceptibility to a broad array of infections in mice (477). STAT5B deficiency leads to different phenotypes in terms of Treg generation, IL2R signaling and in vivo T cell effector function in mice as compared with humans (478).
While humans are housed in varied circumstances, housing of mice is somewhat uniform. Environmental enrichment (EE) makes mouse housing more “fun” and leads to reductions in a variety of depressive/anxious behaviors and indicators of stress response in mice (479). At the same time, there is evidence that EE substantively impacts the antitumor response of NK cells and immunotherapy treated anti-cancer T cells (480). Thus, differences in the monotony and variety of environment may be a factor that alters immune system responses and could impact autoimmune disease pathways.
When given the option, mice, like humans tend to inhabit places with comfortable ambient temperature or change their environment to maintain their own core temperature in the thermoneutral zone. Humans do this by wearing clothes, whereas mice tend to fill their burrows with bedding and insulation. Observation of mice in the wild indicates that during their light cycle, mice tend to maintain a thermoneutral zone of 30-32 degrees Celsius. For historical reasons and for the comfort of clothed humans, most mouse facilities house mice at room temperature 19-25 degrees Celsius. Thus, mice are subjected to chronic “cold stress” which carries with it attendant increased sympathetic nervous system/beta-adrenergic tone and changes in whole organism metabolism and physiology (481). Removal of this cold stress through thermoneutral housing has been demonstrated to impact several immune phenotypes, including notably, induction of oral tolerance (482–485). Further there is growing evidence that the parasympathetic nervous system impacts autoimmune disease. For example, vagal nerve (parasympathetic) (486) stimulation has led to improvement of systemic inflammatory parameters in short-term trials (487, 488).
Mice are typically handled in the vivarium during daylight hours, a period during which they commonly sleep in the wild. Several autoimmune diseases are associated with sleep disturbance (489) due to incompletely clear mechanisms. Indeed, less than 7 hours of sleep is associated with the onset of human SLE in longitudinal cohort studies (490). Further, several reports indicate that systematically sleep deprived NZB/NZWF (1) mice develop increased lupus activity (491, 492). Thus, differences in circadian cycles may be an additional factor to consider when modeling human autoimmune pathologies in mice.
Our immune system gene networks have subject to selective pressure for the sixty-five million years since divergence from mice. At the same time, the mutualistic relationship with our microbiota has been under pressure from our immune system and vice versa. This may be another important meta-genomic divergence that leads to non-correspondence of murine models of human disease (59). Following our reductionist tendencies, the character and make up of mouse microbiota is being intensively defined and simplified as specific-pathogen-free facilities are increasingly used (493, 494). Normalizing the microbiome to one that more closely resembles wild mice leads to several substantial changes in immune response (495–498). Thus, colonization with comparatively non-immunogenic microbiota may be yet another factor that needs to be accounted for when modeling human autoimmune disease in mice.
Most therapies given to people with autoimmune disorders are usually administered to counter a matured, often chronic disease. While prevention trials are underway in several human autoimmune diseases (221), many therapies employed in mouse models are preventive in nature. That is, intervention occurs prior to the onset of disease.
Many lab rodent diets contain substantial proportions of alfalfa meal (499, 500). Alfalfa sprout consumption was long ago associated with incident lupus-like disease in higher primates and attributed to the presence of canavanine, a non-canonical arginine-related amino acid (501). Subsequent studies have also found epidemiological evidence of association with lupus (502), to the point that a commonly used Lupus patient education website recommends avoidance of alfalfa sprouts (503). Curiously, anti-cyclic citrullinated peptide antibodies (against peptides with the non-canonical arginine related amino acid citrulline) are commonly seen in individuals with rheumatoid arthritis as well as those with clinical features of both SLE and RA (504). Recent work has also implicated peptide processing that leads to hybrid-insulin peptide formation, generating a neoepitope as etiologic in type 1 Diabetes (505). Protein dietary and metabolic changes could theoretically alter the generation of neoepitopes in alfalfa fed mice and more broadly appear to have an important role in the genesis of several autoimmune pathologies.
Established disease in humans almost always means confounders – behavior, medications, adherence, understanding, communication, health literacy, numerical literacy, risk perception and risk calculus [COVID-19 pandemic as a global example (506)], to name a few. There is a situation when established disease in humans tends to go along with fewer confounders – early life. However, ethical and practical issues usually prevent trials in children for diseases that also develop in adults. Maybe it isn’t that mice are simple, but that humans are just too complicated?
Most research animal facilities, have strict policies against taking mice out of the viviarium for a walk in the sun. This likely lowers the risk for the skin manifestations of lupus, which are importantly mediated by UV. While the artificial environment of the vivarium can be addressed artificially with transient UV exposure (507), vitamin D is also an independent protective factor for lupus flares and the development of several autoimmune disease (508–511).
The absence of extensive hair follicles, dermal and epidermal layers that are twice as thick and the absence of a specialized muscle layer (Panniculus carnosus) all distinguish human from murine skin (512–514). If histological differences do not pose a sufficient challenge in modeling human skin pathologies in mice, it has been observed that only ~30% of the top skin-expressed genes overlap between mouse and human skin (515). Taken together, these differences pose several problems in modeling SLE, as autoimmune response in the skin is the first disease manifestation in many affected humans.
In addition to implication in MS (discussed above), EBV infection in humans is associated with SLE. There are mechanistic links implicating molecular mimicry by EBNA-1 (516) and substantial enrichment of EBNA-2, the latency transcription factor, at GWAS loci for SLE and other autoimmune diseases (516). There are also examples of allele specific binding of EB viral transcription factors to causal risk alleles. How might this confound translatability of murine model data? The closest gammaherpes virus to EBV that infects mice is murine gamma-herpesvirus 68. While murine gamma-herpesvirus 68 does infect mice, it lacks several features of EBV (517). If one of those divergent features omits a critical step in the EBV-dependent development of autoimmune disease, then this divergence would impact our ability to model autoimmune disease development in a way that parallels what is suspected to occur in humans.
In this section we point out some differences to consider when interpreting murine model data in light of human autoimmune pathology. There are several features of humans that make modeling an inherently error-prone process. These complicating features are in addition to the potential intractability of understanding gene X environment interactions, if the omnigenic model proves true. Despite these drawbacks, murine models of autoimmune diseases have advanced our understanding of the gene networks that regulate autoimmune pathologies. At the same time, efforts at translation require both careful attention to potential confounders and continual reexamination of our models in light of the clinical, phenotypic, cellular and molecular features of the human diseases we seek to model.
Simply put, the need for improved understanding and more diverse and less toxic therapeutic options for SLE and Type 1 diabetes is dire. The discrepant severity of SLE outcomes between populations simply cannot be accepted in a just society. To the extent that our lack of understanding contributes to this discrepancy, it needs to be corrected. In a similar manner, Type 1 diabetes disproportionately afflicts some of the most vulnerable members of our society with a burden of chronic disease and a concomitant burden of co-morbidity and mortality. Despite life-saving advances in therapy in the prior decades, the incidence of this disease is rising. So, we must better understand its genesis in order to more effectively intervene.
We need to understand disease mechanisms and define causal genetic immunophenotypes in humans. For this understanding to be certain regarding causal relationships, parallel understanding of mechanism in model systems is required for effective trial design. Mice have proven to be excellent sacrificial companions on our collective journey of disease deconstruction for both SLE and T1D. They have facilitated perturbations of genes and environmental triggers, allowing assessment of the impacts on murine intermediate immune cellular and molecular phenotypes and correlates of pathology. It continues to be prudent to advance therapies that can prove efficacy in these model systems along the path toward clinical application. However, careful attention to the details of both the model system and the disease processes being modeled is necessary to fully evaluate both therapeutic candidate successes and failures. Nearly 90% of trialed pharmaco-therapeutic candidates do not advance to the FDA approval (518). These rates are better for biologics than for small molecules at each stage of drug development, possibly due to the more specifically targeted nature of biologic therapies versus small molecules (519, 520). This failure is despite the best efforts of many who are employed by pharmaceutical companies. Our ability to fully understand these incredibly complex biological systems remains incomplete. Thus, it is perhaps not surprising that there have been several high-profile failures to develop autoimmune disease therapy.
How best to evaluate therapeutic leads for autoimmune diseases? Our proposed approach follows. Cellular/molecular phenotypes and pathological correlates of disease would need to be ameliorated by candidate therapeutic leads in murine systems to a reasonable degree of certainty in terms of causality. At the same time parallel approaches could be validated in human in vitro (cell lines), ex vivo (primary cells) or in vivo (hu-mice) reductionist model systems and shown to return the cellular/molecular phenotypes and pathologic correlates move to a healthier status with any therapeutic lead. Therapies that pass this bar could be trialed in first in human trials after primate evaluation or if repurposing (if already FDA approved), moved directly to phase 3 trials. Human trials based on the cellular, molecular and pathologic frameworks derived from model systems would need to include assessment of correlates of the postulated mechanism. Additionally, evaluation of any competing mechanisms would assist post-hoc evaluation of whether a given trial represented a true trial of therapy. Indeed, two recent (the first two since the 1950s) FDA-approved therapies for SLE, belimumab (anti-BAFF) and anifrolumab (anti-IFNAR1), both took approaches similar to the approach that we lay out. Following identification of antigen-presentation by B cells (521–528) as key in the genesis of murine autoimmune type 1 diabetes there is now a focus on B cell tolerance pathways in human T1D (41, 529–533). Further characterization of the role of B cell tolerance (534) and efforts to manipulate pathogenic autoantigen-reactive B cells in type 1 diabetes promise (530) to bring therapeutic successes in this disease, where T cells have long been the subject of focus. Our analysis highlights a potential role for autoreactive B cell tolerance in the development of multiple autoimmune pathologies. In doing so, it adds to a growing body of work that supports viewing seropositive autoimmunity as an endophenotype of multiple autoimmune diseases (535–541). As our efforts to more broadly understand autoimmune disease polygenic genetic risk network impacts on B cell function advance, we anticipate that murine disease models will continue to be critically important to furthering understanding of autoimmune diseases and advancing the goal of improved outcomes for patients.
IH conceived of the article, carried out the analyses, drafted and revised the manuscript. KA contributed to data analysis and interpretation and critically revised the manuscript. RS contributed to data analysis, interpretation and critically revised the manuscript All authors contributed to the article and approved the submitted version.
IH receives funding from the Rheumatology Research Foundation in the form of a Scientist Development Award. He is also partly funded by the Pfizer Global Grants Foundation Rheumatology program #51849703, but those funds did not support his work on this project. RS receives funding from NIH R01DE029303, VA I01 CX001877 and VA I01 BX001451.
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used to support the claim that BLK eQTLs occur in fibroblasts were obtained from the GTEx Portal http://www.gtexportal.org/ on 2022-05-10. https://www.gtexportal.org/home/gene/BLK. The BioGPS resource (470), human primary cell atlas (471) was used to identify non B-cell expression of BLK. http://ds.biogps.org/?dataset=BDS_00013&gene=640. [Accessed 2022-05-10].
IH is partly funded by the Pfizer Global Grants Foundation Rheumatology program #51849703, but those funds did not support his work on this project.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The contents of this manuscript do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.953439/full#supplementary-material
Supplementary Table 1A | Monogenic Routes to Human Lupus – Gene(s) refers to the gene or genes that when mutated has been reported to lead to lupus or a lupus-like phenotype. Gene name refers to the HGNC (HUGO Gene Nomenclature Committee [https://www.genenames.org/]) official full name for that gene. Locus refers to chromosome and cytoband for that gene. Protein refers to the common protein name for a particular gene. Inheritance indicates the mode of inheritance (if reported) according to the abbreviations at the end of the table. Pathway refers to the reported pathway disrupted by the mutation. Phenotype refers to the phenotype observed, whether part of another defined genetic syndrome, such as Noonan syndrome or whether part of bona fide SLE or another lupus-like phenotype. Reference refers to numbered reference in the bibliography of this publication. PMID or link refers to the pubmed.gov identifier (PMID) for the publication or publications establishing the gene as a monogenic route to lupus or a review of several publications.
Supplementary Table 1B | Monogenic Routes to Human autoimmune Type 1 Diabetes – Gene(s) refers to the gene or genes that when mutated has been reported to lead to lupus or a lupus-like phenotype. Gene name refers to the HGNC (HUGO Gene Nomenclature Committee [https://www.genenames.org/]) official full name for that gene. Locus refers to chromosome and cytoband for that gene. Protein refers to the common protein name for a particular gene. Inheritance indicates the mode of inheritance (if reported) according to the abbreviations at the end of the table. Pathway refers to the reported pathway disrupted by the mutation. Phenotype refers to the phenotype observed according to online mendelian mutation in man (OMIM) [https://omim.org/]. Reference refers to numbered reference in the bibliography of this publication. PMID or link refers to the pubmed.gov identifier (PMID) for the publication or publications establishing the gene as a monogenic route to type 1 diabetes or a review of several publications.
Supplementary Table 2A | Human SLE Polygenic risk loci from GWAS catalog and putative causal gene(s) as identified by OpenTargetsGenetics L2G pipeline – Variant & Risk allele: the genetic variant with smallest reported P-value for association with SLE by GWAS and the corresponding risk allele as reported by the EBI/NHGRI GWAS catalog [https://www.ebi.ac.uk/gwas/]. P-value: the p-value reported for that variant. P-value annotation: commentary on the P-value reported for that variant as reported in the EBI GWAS catalog (i.e. association in a specific population, conditional logistic regression based on covariates, etc.) RAF – risk allele frequency, if reported in the EBI GWAS catalog. OR – reported odds ratio for the risk allele as reported in the EBI GWAS catalog. Beta – effect size or natural logarithm of the odds ratio. CI – 95% confidence interval of the estimated odds ratio (or beta where reported). Mapped gene – contiguous or adjacent gene mapped to the location of the lead genetic variant. Reported Trait – trait for the GWAS that reported the lead marker from the EBI GWAS catalog. Only “Systemic lupus erythematosus” is included in this table over against, i.e. “lupus nephritis”. Trait(s) – trait or subphenotype. Only “systemic lupus erythematosus” is included in this table over against, i.e. “neonatal lupus”. Background trait – indicator of background trait (i.e. in the case of lupus nephritis in a cohort of SLE patients, SLE would represent the background trait), if present. Study accession – GWAS catalog study identifier. PubMed ID – pubmed ID of the study reporting association. First Author (First author of the study in question). Location – chromosome:position of the lead variant on human genome build 38 (hg38). P – P value converted from format in EBI GWAS catalog to scientific notation. Chromosome – chromosome of lead variant. Position (hg38) position in base pairs of the variant of the lead variant on human genome build 38 (hg38). Region – numerical value of this GWAS region as associated with SLE. Putative Causal gene as predicted by open targets genetics L2G algorithm. Opentargets – link to opentargets genetics prediction and evidence supporting this prediction for that region. “NR” or “‘-” indicates value not reported in the EBI GWAS catalog.
Supplementary Table 2B | Human T1D Polygenic risk variants from GWAS catalog and putative causal gene(s) as identified by OpenTargetsGenetics L2G pipeline – columns are identical to Supplementary Table 2A, except that they apply to type 1 diabetes and not Systemic lupus erythematosus.
Supplementary Table 3 | Genes involved in lupus, type 1 diabetes, peripheral and central B cell tolerance from mouse models – Murine locus – genetic locus, gene name or common protein name of the gene. Gene location – murine chromosome and cytoband of the gene in question. GL Murine – gene location on chromosome in centimorgans. Human orthologue – where identifiable, the human orthlogue to the murine gene in question. Gene location (human) chromosome and cytoband of that human gene. Gene name – HGNC gene name of the gene where applicable. (HGNC = HUGO Gene Nomenclature Committee [https://www.genenames.org/]). Protein – abbreviation or common name for the protein encoded by the gene. Pathway – pathway implicated in the function of this gene in the corresponding class. Model – murine model where gene was implicated. Phenotype – phenotype resulting from gene alteration. Class – murine disease/model state implicated: either lupus, T1D, peripheral tolerance, central tolerance or some combination. PMID – PubMed ID of the publications supporting the link of the gene with a particular class.
Supplementary Table 4 | hyperlinks to Networks. This table consists of references to networks in this paper along with links to permanent versions of the networks and analyses that were used to develop the conclusions of this paper. Disease network – disease network as referred to in this paper. Color – color encoding of the disease network in question in –. Shape – shape encoding of the disease network in question in –. URL (NDEX) – universal resource locator for network @ http://www.ndexbio.org URL(string-db.org) – universal resource locator for network @ https://www.string-db.org/URL (Enrichr) – universal resource locator for network @ Enrichr pathway and geneset enrichment analysis: https://maayanlab.cloud/Enrichr/.
1. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC, et al. Predominant autoantibody production by early human b cell precursors. Science (2003) 301(5638):1374–7. doi: 10.1126/science.1086907
2. Grandien A, Fucs R, Nobrega A, Andersson J, Coutinho A. Negative selection of multireactive b cell clones in normal adult mice. Eur J Immunol (1994) 24(6):1345–52. doi: 10.1002/eji.1830240616
3. Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic b cells? Nat Rev Immunol (2007) 7(8):633–43. doi: 10.1038/nri2133
4. Cambier JC, Getahun A. B cell activation versus anergy; the antigen receptor as a molecular switch. Immunol Lett (2010) 128(1):6–7. doi: 10.1016/j.imlet.2009.09.006
5. Gauld SB, Benschop RJ, Merrell KT, Cambier JC. Maintenance of b cell anergy requires constant antigen receptor occupancy and signaling. Nat Immunol (2005) 6(11):1160–7. doi: 10.1038/ni1256
6. Gauld SB, Merrell KT, Cambier JC. Silencing of autoreactive b cells by anergy: a fresh perspective. Curr Opin Immunol (2006) 18(3):292–7. doi: 10.1016/j.coi.2006.03.015
7. Nemazee D. Receptor editing in lymphocyte development and central tolerance. Nat Rev Immunol (2006) 6(10):728–40. doi: 10.1038/nri1939
8. Tan C, Hiwa R, Mueller JL, Vykunta V, Hibiya K, Noviski M, et al. NR4A nuclear receptors restrain b cell responses to antigen when second signals are absent or limiting. Nat Immunol (2020) 21:1267–79. doi: 10.1038/s41590-020-0765-7
9. Dayan CM, Besser REJ, Oram RA, Hagopian W, Vatish M, Bendor-Samuel O, et al. Preventing type 1 diabetes in childhood. Science (2021) 373(6554):506–10. doi: 10.1126/science.abi4742
10. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med (2003) 349(16):1526–33. doi: 10.1056/NEJMoa021933
11. Sabatino JJ Jr., Probstel AK, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci (2019) 20(12):728–45. doi: 10.1038/s41583-019-0233-2
12. Deane KD, Holers VM. Rheumatoid arthritis pathogenesis, prediction, and prevention: An emerging paradigm shift. Arthritis Rheumatol (2021) 73(2):181–93. doi: 10.1002/art.41417
13. Sospedra M. B cells in multiple sclerosis. Curr Opin Neurol (2018) 31(3):256–62. doi: 10.1097/WCO.000000000000563
14. Stohl W, Schwarting A, Okada M, Scheinberg M, Doria A, Hammer AE, et al. Efficacy and safety of subcutaneous belimumab in systemic lupus erythematosus: A fifty-Two-Week randomized, double-blind, placebo-controlled study. Arthritis Rheumatol (2017) 69(5):1016–27. doi: 10.1002/art.40049
15. Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzova D, et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits b lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum (2011) 63(12):3918–30. doi: 10.1002/art.30613
16. Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet (2011) 377(9767):721–31. doi: 10.1016/S0140-6736(10)61354-2
17. Klubo-Gwiezdzinska J, Lange M, Cochran E, Semple RK, Gewert C, Brown RJ, et al. Combined immunosuppressive therapy induces remission in patients with severe type b insulin resistance: A prospective cohort study. Diabetes Care (2018) 41(11):2353–60. doi: 10.2337/dc18-0884
18. Linsley PS, Greenbaum CJ, Rosasco M, Presnell S, Herold KC, Dufort MJ. Elevated T cell levels in peripheral blood predict poor clinical response following rituximab treatment in new-onset type 1 diabetes. Genes Immun (2019) 20(4):293–307. doi: 10.1038/s41435-018-0032-1
19. Pescovitz MD, Greenbaum CJ, Bundy B, Becker DJ, Gitelman SE, Goland R, et al. B-lymphocyte depletion with rituximab and beta-cell function: two-year results. Diabetes Care (2014) 37(2):453–9. doi: 10.2337/dc13-0626
20. Yu L, Herold K, Krause-Steinrauf H, McGee PL, Bundy B, Pugliese A, et al. Rituximab selectively suppresses specific islet antibodies. Diabetes (2011) 60(10):2560–5. doi: 10.2337/db11-0674
21. Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, et al. Rituximab, b-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med (2009) 361(22):2143–52. doi: 10.1056/NEJMoa0904452
22. Herold KC, Pescovitz MD, McGee P, Krause-Steinrauf H, Spain LM, Bourcier K, et al. Increased T cell proliferative responses to islet antigens identify clinical responders to anti-CD20 monoclonal antibody (rituximab) therapy in type 1 diabetes. J Immunol (2011) 187(4):1998–2005. doi: 10.4049/jimmunol.1100539
23. Gerlag DM, Safy M, Maijer KI, Tang MW, Tas SW, Starmans-Kool MJF, et al. Effects of B-cell directed therapy on the preclinical stage of rheumatoid arthritis: the PRAIRI study. Ann Rheum Dis (2019) 78(2):179–85. doi: 10.1136/annrheumdis-2017-212763
24. De Vita S, Zaja F, Sacco S, De Candia A, Fanin R, Ferraccioli G., et al. Efficacy of selective b cell blockade in the treatment of rheumatoid arthritis: evidence for a pathogenetic role of b cells. Arthritis Rheum (2002) 46(8):2029–33. doi: 10.1002/art.10467
25. Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of b-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med (2004) 350 (25):2572–81. doi: 10.1056/NEJMoa032534
26. Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum (2006) 54(9):2793–806. doi: 10.1002/art.22025
27. Keystone E, Emery P, Peterfy CG, Tak PP, Cohen S, Genovese MC, et al. Rituximab inhibits structural joint damage in patients with rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitor therapies. Ann Rheum Dis (2009) 68(2):216–21. doi: 10.1136/ard.2007.085787
28. Vital EM, Dass S, Buch MH, Rawstron AC, Emery P. An extra dose of rituximab improves clinical response in rheumatoid arthritis patients with initial incomplete b cell depletion: a randomised controlled trial. Ann Rheum Dis (2015) 74(6):1195–201. doi: 10.1136/annrheumdis-2013-204544
29. Bredemeier M, Campos GG, de Oliveira FK. Updated systematic review and meta-analysis of randomized controlled trials comparing low- versus high-dose rituximab for rheumatoid arthritis. Clin Rheumatol (2015) 34(10):1801–5. doi: 10.1007/s10067-015-2977-z
30. Meffre E, O'Connor KC. Impaired b-cell tolerance checkpoints promote the development of autoimmune diseases and pathogenic autoantibodies. Immunol Rev (2019) 292(1):90–101. doi: 10.1111/imr.12821
31. Cashman KS, Jenks SA, Woodruff MC, Tomar D, Tipton CM, Scharer CD, et al. Understanding and measuring human b-cell tolerance and its breakdown in autoimmune disease. Immunol Rev (2019) 292(1):76–89. doi: 10.1111/imr.12820
32. Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol (2008) 20(6):632–8. doi: 10.1016/j.coi.2008.09.001
33. Pelanda R, Greaves SA, Alves da Costa T, Cedrone LM, Campbell ML, Torres RM. B-cell intrinsic and extrinsic signals that regulate central tolerance of mouse and human b cells. Immunol Rev (2022) 307(1):12–26. doi: 10.1111/imr.13062
34. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective b cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med (2005) 201(5):703–11. doi: 10.1084/jem.20042251
35. Suzuki N, Harada T, Mihara S, Sakane T. Characterization of a germline vk gene encoding cationic anti-DNA antibody and role of receptor editing for development of the autoantibody in patients with systemic lupus erythematosus. J Clin Invest (1996) 98 (8):1843–50. doi: 10.1172/jci118985
36. Dörner T, Foster SJ, Farner NL, Lipsky PE. Immunoglobulin kappa chain receptor editing in systemic lupus erythematosus. J Clin Invest (1998) 102 (4):688–94. doi: 10.1172/jci3113
37. Panigrahi AK, Goodman NG, Eisenberg RA, Rickels MR, Naji A, Luning Prak ET. RS rearrangement frequency as a marker of receptor editing in lupus and type 1 diabetes. J Exp Med (2008) 205 (13):2985–94. doi: 10.1084/jem.20082053
38. Samuels J, Ng YS, Coupillaud C, Paget D, Meffre E. Impaired early b cell tolerance in patients with rheumatoid arthritis. J Exp Med (2005) 201 (10):1659–67. doi: 10.1084/jem.20042321
39. Menard L, Samuels J, Ng YS, Meffre E. Inflammation-independent defective early b cell tolerance checkpoints in rheumatoid arthritis. Arthritis Rheum (2011) 63 (5):1237–45. doi: 10.1002/art.30164
40. Glauzy S, Sng J, Bannock JM, Gottenberg JE, Korganow AS, Cacoub P, et al. Defective early b cell tolerance checkpoints in sjögren's syndrome patients. Arthritis Rheumatol (2017) 69 (11):2203–8. doi: 10.1002/art.40215
41. Smith MJ, Packard TA, O'Neill SK, Henry Dunand CJ, Huang M, Fitzgerald-Miller L, et al. Loss of anergic b cells in prediabetic and new-onset type 1 diabetic patients. Diabetes (2015) 64 (5):1703–12. doi: 10.2337/db13-1798
42. Smith MJ, Rihanek M, Coleman BM, Gottlieb PA, Sarapura VD, Cambier JC, et al. Activation of thyroid antigen-reactive b cells in recent onset autoimmune thyroid disease patients. J Autoimmun (2018) 89:82–9. doi: 10.1016/j.jaut.2017.12.001
43. Malkiel S, Jeganathan V, Wolfson S, Manjarrez Orduño N, Marasco E, Aranow C, et al. Checkpoints for autoreactive b cells in the peripheral blood of lupus patients assessed by flow cytometry. Arthritis Rheumatol (2016) 68 (9):2210–20. doi: 10.1002/art.39710
44. Cappione A 3rd, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, et al. Germinal center exclusion of autoreactive b cells is defective in human systemic lupus erythematosus. J Clin Invest (2005) 115 (11):3205–16. doi: 10.1172/jci24179
45. Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol (2015) 16 (7):755–65. doi: 10.1038/ni.3175
46. Watanabe A, Su KY, Kuraoka M, Yang G, Reynolds AE, Schmidt AG, et al. Self-tolerance curtails the b cell repertoire to microbial epitopes. JCI Insight (2019) 4 (10):e122551. doi: 10.1172/jci.insight.122551
47. Liubchenko GA, Appleberry HC, Striebich CC, Franklin KE, Derber LA, Holers VM, et al. Rheumatoid arthritis is associated with signaling alterations in naturally occurring autoreactive b-lymphocytes. J Autoimmun (2013) 40:111–21. doi: 10.1016/j.jaut.2012.09.001
48. Thorarinsdottir K, Camponeschi A, Jonsson C, Granhagen Önnheim K, Nilsson J, Forslind K, et al. CD21(-/low) b cells associate with joint damage in rheumatoid arthritis patients. Scand J Immunol (2019) 90 (2):e12792. doi: 10.1111/sji.12792
49. Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement receptor 2/CD21- human naive b cells contain mostly autoreactive unresponsive clones. Blood (2010) 115 (24):5026–36. doi: 10.1182/blood-2009-09-243071
50. Berti A, Hillion S, Hummel AM, Son YM, Chriti N, Peikert T, et al. Circulating autoreactive proteinase 3+ b cells and tolerance checkpoints in ANCA-associated vasculitis. JCI Insight (2021) 6:e150999. doi: 10.1172/jci.insight.150999
51. Casadevall A, Pirofski LA. The damage-response framework of microbial pathogenesis. Nat Rev Microbiol (2003) 1 (1):17–24. doi: 10.1038/nrmicro732
52. Pirofski LA, Casadevall A. The damage-response framework of microbial pathogenesis and infectious diseases. Adv Exp Med Biol (2008) 635:135–46. doi: 10.1007/978-0-387-09550-9_11
53. Pirofski LA, Casadevall A. The damage-response framework as a tool for the physician-scientist to understand the pathogenesis of infectious diseases. J Infect Dis (2018) 218(suppl_1):S7–S11. doi: 10.1093/infdis/jiy083
54. Elhanati Y, Sethna Z, Marcou Q, Callan CG Jr, Mora T, Walczak AM. Inferring processes underlying b-cell repertoire diversity. Philos Trans R Soc Lond B Biol Sci (2015) 370(1676):20140243. doi: 10.1098/rstb.2014.0243
55. Davis MM. A prescription for human immunology. Immunity (2008) 29(6):835–8. doi: 10.1016/j.immuni.2008.12.003
56. Franks SE, Cambier JC. Putting on the brakes: Regulatory kinases and phosphatases maintaining B cell anergy. Front Immunol (2018) 9:665. doi: 10.3389/fimmu.2018.00665
57. Smith MJ, Cambier JC, Gottlieb PA. Endotypes in T1D: B lymphocytes and early onset. Curr Opin Endocrinol Diabetes Obes (2020) 27(4):225–30. doi: 10.1097/MED.0000000000000547
58. Hinman RM, Smith MJ, Cambier JC. B cells and type 1 diabetes in mice and men. Immunol Lett (2014) 160(2):128–32. doi: 10.1016/j.imlet.2014.01.010
59. Wekerle H, Flugel A, Fugger L, Schett G, Serreze D. Autoimmunity's next top models. Nat Med (2012) 18(1):66–70. doi: 10.1038/nm.2635
60. Morel L. Mouse models of human autoimmune diseases: essential tools that require the proper controls. PloS Biol (2004) 2(8):E241. doi: 10.1371/journal.pbio.0020241
61. Morel L. Genetics of SLE: evidence from mouse models. Nat Rev Rheumatol (2010) 6(6):348–57. doi: 10.1038/nrrheum.2010.63
62. Cao Y, Qiu Y, Tu G, Yang C. Single-cell RNA sequencing in immunology. Curr Genomics (2020) 21(8):564–75. doi: 10.2174/1389202921999201020203249
63. Chen H, Ye F, Guo G. Revolutionizing immunology with single-cell RNA sequencing. Cell Mol Immunol (2019) 16(3):242–9. doi: 10.1038/s41423-019-0214-4
64. Kimball AK, Oko LM, Bullock BL, Nemenoff RA, van Dyk LF, Clambey ET. A beginner's guide to analyzing and visualizing mass cytometry data. J Immunol (2018) 200(1):3–22. doi: 10.4049/jimmunol.1701494
65. den Braanker H, Bongenaar M, Lubberts E. How to prepare spectral flow cytometry datasets for high dimensional data analysis: A practical workflow. Front Immunol (2021) 12:768113. doi: 10.3389/fimmu.2021.768113
66. Bonilla DL, Reinin G, Chua E. Full spectrum flow cytometry as a powerful technology for cancer immunotherapy research. Front Mol Biosci (2020) 7:612801. doi: 10.3389/fmolb.2020.612801
67. von Herrath MG, Nepom GT. Lost in translation: barriers to implementing clinical immunotherapeutics for autoimmunity. J Exp Med (2005) 202(9):1159–62. doi: 10.1084/jem.20051224
68. Wiendl H, Hohlfeld R. Multiple sclerosis therapeutics: unexpected outcomes clouding undisputed successes. Neurology (2009) 72(11):1008–15. doi: 10.1212/01.wnl.0000344417.42972.54
69. Diabetes Prevention Trial–Type 1 Diabetes Study, G. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med (2002) 346(22):1685–91. doi: 10.1056/NEJMoa012350
70. Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med (2000) 6(10):1167–75. doi: 10.1038/80516
71. Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. the altered peptide ligand in relapsing MS study group. Nat Med (2000) 6:1176–82. doi: 10.1038/80525
72. Genain CP, Zamvil SS. Specific immunotherapy: one size does not fit all. Nat Med (2000) 6:1098–100. doi: 10.1038/80424
73. Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet (1987) 1(87):893–5. doi: 10.1016/s0140-6736(87)92863-7
74. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. the lenercept multiple sclerosis study group and the university of British Columbia MS/MRI analysis group. Neurology (1999) 53(3):457–65. doi: 10.1212/WNL.53.3.457
75. van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, von Blomberg BM, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology (1996) 47(6):1531–4. doi: 10.1212/wnl.47.6.1531
76. Rekvig OP. Systemic lupus erythematosus: Definitions, contexts, conflicts, enigmas. Front Immunol (2018) 9:387. doi: 10.3389/fimmu.2018.00387
77. Rekvig OP. The dsDNA, anti-dsDNA antibody, and lupus nephritis: What we agree on, what must be done, and what the best strategy forward could be. Front Immunol (2019) 10:1104. doi: 10.3389/fimmu.2019.01104
78. Rekvig OP. Autoimmunity and SLE: Factual and semantic evidence-based critical analyses of definitions, etiology, and pathogenesis. Front Immunol (2020) 11:569234. doi: 10.3389/fimmu.2020.569234
79. Gutierrez-Arcelus M, Rich SS, Raychaudhuri S. Autoimmune diseases - connecting risk alleles with molecular traits of the immune system. Nat Rev Genet (2016) 17(3):160–74. doi: 10.1038/nrg.2015.33
80. Kuo CF, Grainge MJ, Valdes AM, See LC, Luo SF, Yu KH, et al. Familial aggregation of systemic lupus erythematosus and coaggregation of autoimmune diseases in affected families. JAMA Internal Med (2015) 175(9):1518–26. doi: 10.1001/jamainternmed.2015.3528
81. Kuo CF, Chou IJ, Grainge MJ, Luo SF, See LC, Yu KH, et al. Familial aggregation and heritability of type 1 diabetes mellitus and coaggregation of chronic diseases in affected families. Clin Epidemiol (2018) 10:1447–55. doi: 10.2147/CLEP.S172207
82. Barber MRW, Drenkard C, Falasinnu T, Hoi A, Mak A, Kow NY, et al. Global epidemiology of systemic lupus erythematosus. Nat Rev Rheumatol (2021) 17(9):515–32. doi: 10.1038/s41584-021-00668-1
83. Yen EY, Singh RR. Brief report: Lupus-an unrecognized leading cause of death in young females: A population-based study using nationwide death certificates, 2000-2015. Arthritis Rheumatol (2018) 70(8):1251–5. doi: 10.1002/art.40512
84. Alarcon-Riquelme ME, Ziegler-Estrada A, Sanchez-Rodriguez E, et al. Genome-wide association study in an Amerindian ancestry population reveals novel systemic lupus erythematosus risk loci and the role of European admixture. Arthritis Rheumatol (2016) 68(4):932–43. doi: 10.1002/art.39504
85. Sanchez E, Comeau ME, Freedman BI, Kelly JA, Kaufman KM, Langefeld CD, et al. Identification of novel genetic susceptibility loci in African American lupus patients in a candidate gene association study. Arthritis Rheum (2011) 63(11):3493–501. doi: 10.1002/art.30563
86. Tishkoff SA, Verrelli BC. Patterns of human genetic diversity: implications for human evolutionary history and disease. Annu Rev Genomics Hum Genet (2003) 4:293–340. doi: 10.1146/annurev.genom.4.070802.110226
87. Izmirly PM, Parton H, Wang L, McCune WJ, Lim SS, Drenkard C, et al. Prevalence of systemic lupus erythematosus in the united states: Estimates from a meta-analysis of the centers for disease control and prevention national lupus registries. Arthritis Rheumatol (2021) 73(6):991–6. doi: 10.1002/art.41632
88. Imperatore G, Mayer-Davis EJ, Orchard TJ, Zhong VW. Prevalence and incidence of type 1 diabetes among children and adults in the United States and comparison with non-U.S. Countries. In: rd, Cowie CC, Casagrande SS, Menke A, Cissell MA, Eberhardt MS, et al. editors. Diabetes in America 3rd Edition.Bethesda (MD):National Institute of Diabetes and Digestive and Kidney Diseases (US) (2018)
89. Johnson MB, Patel KA, De Franco E, Hagopian W, Killian M, McDonald TJ, et al. Type 1 diabetes can present before the age of 6 months and is characterised by autoimmunity and rapid loss of beta cells. Diabetologia (2020) 63(12):2605–15. doi: 10.1007/s00125-020-05276-4
90. Lipman TH, Levitt Katz LE, Ratcliffe SJ, Murphy KM, Aguilar A, Rezvani I, et al. Increasing incidence of type 1 diabetes in youth: twenty years of the Philadelphia pediatric diabetes registry. Diabetes Care (2013) 36(6):1597–603. doi: 10.2337/dc12-0767
91. Vehik K, Hamman RF, Lezotte D, Norris JM, Klingensmith G, Bloch C, et al. Increasing incidence of type 1 diabetes in 0- to 17-year-old Colorado youth. Diabetes Care (2007) 30(3):503–9. doi: 10.2337/dc06-1837
92. Lawrence JM, Imperatore G, Dabelea D, Mayer-Davis EJ, Linder B, Saydah S, et al. Trends in incidence of type 1 diabetes among non-Hispanic white youth in the U.S., 2002-2009. Diabetes (2014) 63(11):3938–45. doi: 10.2337/db13-1891
93. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med (2002) 347(12):911–20. doi: 10.1056/NEJMra020100
94. Selmi C. The worldwide gradient of autoimmune conditions. Autoimmun Rev (2010) 9(5):A247–250. doi: 10.1016/j.autrev.2010.02.004
95. Dinse GE, Parks CG, Weinberg CR, Co CA, Wilkerson J, Zeldin DC, et al. Increasing prevalence of antinuclear antibodies in the united states. Arthritis Rheumatol (2020) 72(6):1026–35. doi: 10.1002/art.41214
96. Leffers HCB, Lange T, Collins C, Ulff-Moller CJ, Jacobsen S. The study of interactions between genome and exposome in the development of systemic lupus erythematosus. Autoimmun Rev (2019) 18(4):382–92. doi: 10.1016/j.autrev.2018.11.005
97. Parks CG, de Souza Espindola Santos A, Barbhaiya M, Costenbader KH. Understanding the role of environmental factors in the development of systemic lupus erythematosus. Best Pract Res Clin Rheumatol (2017) 31(3):306–20. doi: 10.1016/j.berh.2017.09.005
98. Quinn LM, Wong FS, Narendran P. Environmental determinants of type 1 diabetes: From association to proving causality. Front Immunol (2021) 12:737964. doi: 10.3389/fimmu.2021.737964
99. Imagawa A, Hanafusa T, Miyagawa J, Matsuzawa Y. A novel subtype of type 1 diabetes mellitus characterized by a rapid onset and an absence of diabetes-related antibodies. Osaka IDDM study group. N Engl J Med (2000) 342:301–7. doi: 10.1056/NEJM200002033420501
100. American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care (2013) 36 Suppl 1:S67–74. doi: 10.2337/dc13-S067
101. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care (1997) 20:1183–97. doi: 10.2337/diacare.20.7.1183
102. Zhang H, Colclough K, Gloyn AL, Pollin TI. Monogenic diabetes: a gateway to precision medicine in diabetes. J Clin Invest (2021) 131(3):e142244. doi: 10.1172/JCI142244
103. Dabelea D, Pihoker C, Talton JW, D'Agostino RB Jr, Fujimoto W, Klingensmith GJ, et al. Etiological approach to characterization of diabetes type: the SEARCH for diabetes in youth study. Diabetes Care (2011) 34(7):1628–33. doi: 10.2337/dc10-2324
104. Redondo MJ, Hagopian WA, Oram R, Steck AK, Vehik K, Weedon M, et al. The clinical consequences of heterogeneity within and between different diabetes types. Diabetologia (2020) 63(10):2040–8. doi: 10.1007/s00125-020-05211-7
105. Hoffman LS, Fox TJ, Anastasopoulou C, Jialal I. Maturity Onset Diabetes in the Young. Statpearls. Treasure Island (FL): StatPearls Publishing (2022).
106. Yu MG, Keenan HA, Shah HS, Frodsham SG, Pober D, He Z, et al. Residual beta cell function and monogenic variants in long-duration type 1 diabetes patients. J Clin Invest (2019) 129(8):3252–63. doi: 10.1172/JCI127397
107. Naylor R. Economics of genetic testing for diabetes. Curr Diabetes Rep (2019) 19(5):23. doi: 10.1007/s11892-019-1140-7
108. Qu J, Qu HQ, Bradfield JP, Glessner JT, Chang X, Tian L, et al. Association of DLL1 with type 1 diabetes in patients characterized by low polygenic risk score. Metabolism (2021) 114:154418. doi: 10.1016/j.metabol.2020.154418
109. Qu HQ, Qu J, Bradfield J, Marchand L, Glessner J, Chang X, et al. Genetic architecture of type 1 diabetes with low genetic risk score informed by 41 unreported loci. Commun Biol (2021) 4(1):908. doi: 10.1038/s42003-021-02368-8
110. Qu J, Qu HQ, Bradfield JP, Glessner JT, Chang X, Tian L, et al. Insights into non-autoimmune type 1 diabetes with 13 novel loci in low polygenic risk score patients. Sci Rep (2021) 11(1):16013. doi: 10.1038/s41598-021-94994-9
111. Cohen AS, Canoso JJ. Criteria for the classification of systemic lupus erythematosus–status 1972. Arthritis Rheum (1972) 15(5):540–3. doi: 10.1002/art.1780150512
112. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum (1982) 25(11):1271–7. doi: 10.1002/art.1780251101
113. Hochberg MC. Updating the American college of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum (1997) 40(9):1725. doi: 10.1002/art.1780400928
114. Petri M, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum (2012) 64(8):2677–86. doi: 10.1002/art.34473
115. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. European League against Rheumatism/American college of rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol (2019) 71:1400–12. doi: 10.1002/art.40930
116. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. European League against Rheumatism/American college of rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis (2019) 78(9):1151–9. doi: 10.1136/annrheumdis-2018-214819
117. Harley ITW, Sawalha AH. Systemic lupus erythematosus as a genetic disease. Clin Immunol (2022) 236:108953. doi: 10.1016/j.clim.2022.108953
118. Cano-Gamez E, Trynka G. From GWAS to function: Using functional genomics to identify the mechanisms underlying complex diseases. Front Genet (2020) 11:424. doi: 10.3389/fgene.2020.00424
119. Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature (2010) 466(7307):714–9. doi: 10.1038/nature09266
120. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science (2012) 337(6099):1190–5. doi: 10.1126/science.1222794
121. Hu X, Deutsch AJ, Lenz TL, Onengut-Gumuscu S, Han B, et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat Genet (2015) 47(8):898–905. doi: 10.1038/ng.3353
122. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet (2012) 44(3):291–6. doi: 10.1038/ng.1076
123. Kamitaki N, Sekar A, Handsaker RE, de Rivera H, Tooley K, Morris DL, et al. Complement genes contribute sex-biased vulnerability in diverse disorders. Nature (2020) 582(7813):577–81. doi: 10.1038/s41586-020-2277-x
124. Demirkaya E, Sahin S, Romano M, Zhou Q, Aksentijevich I. New horizons in the genetic etiology of systemic lupus erythematosus and lupus-like disease: Monogenic lupus and beyond. J Clin Med (2020) 9(3):712. doi: 10.3390/jcm9030712
125. Alperin JM, Ortiz-Fernández L, Sawalha AH. Monogenic lupus: A developing paradigm of disease. Front Immunol (2018) 9:2496. doi: 10.3389/fimmu.2018.02496
126. Omarjee O, Picard C, Frachette C, Moreews M, Rieux-Laucat F, Soulas-Sprauel P, et al. Monogenic lupus: Dissecting heterogeneity. Autoimmun Rev (2019) 18:102361. doi: 10.1016/j.autrev.2019.102361
127. Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet (2011) 43(2):127–31. doi: 10.1038/ng.748
128. Skrabl-Baumgartner A, Plecko B, Schmidt WM, König N, Hershfield M, Gruber-Sedlmayr U, et al. Autoimmune phenotype with type I interferon signature in two brothers with ADA2 deficiency carrying a novel CECR1 mutation. Pediatr Rheumatol Online J (2017) 15:67. doi: 10.1186/s12969-017-0193-x
129. Schepp J, Bulashevska A, Mannhardt-Laakmann W, Cao H, Yang F, Seidl M, et al. Deficiency of adenosine deaminase 2 causes antibody deficiency. J Clin Immunol (2016) 36:179–86. doi: 10.1007/s10875-016-0245-x
130. Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, et al. Mutations in ADAR1 cause aicardi-goutières syndrome associated with a type I interferon signature. Nat Genet (2012) 44:1243–8. doi: 10.1038/ng.2414
131. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in trex1, rnaseh2a, rnaseh2b, rnaseh2c, samhd1, adar, and ifih1. Am J Med Genet A (2015) 167A:296–312. doi: 10.1002/ajmg.a.36887
132. Lood C, Gullstrand B, Truedsson L, Olin AI, Alm GV, Rönnblom L, et al. C1q inhibits immune complex-induced interferon-alpha production in plasmacytoid dendritic cells: a novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum (2009) 60:3081–90. doi: 10.1002/art.24852
133. Demirkaya E, Zhou Q, Smith CK, Ombrello MJ, Deuitch N, Tsai WL, et al. Brief report: Deficiency of complement 1r subcomponent in early-onset systemic lupus erythematosus: The role of disease-modifying alleles in a monogenic disease. Arthritis Rheumatol (2017) 69:1832–9. doi: 10.1002/art.40158
134. Bienaimé F, Quartier P, Dragon-Durey MA, Frémeaux-Bacchi V, Bader-Meunier B, Patey N, et al. Lupus nephritis associated with complete C1s deficiency efficiently treated with rituximab: a case report. Arthritis Care Res (Hoboken) (2010) 62:1346–50. doi: 10.1002/acr.20163
135. Miller EC, Atkinson JP. Overcoming C2 deficiency. Clin Immunol (2012) 144:269–71. doi: 10.1016/j.clim.2012.07.005
136. Blanchong CA, Chung EK, Rupert KL, Yang Y, Yang Z, Zhou B, et al. Genetic, structural and functional diversities of human complement components C4A and C4B and their mouse homologues, slp and C4. Int Immunopharmacol (2001) 1:365–92. doi: 10.1016/s1567-5769(01)00019-4
137. Pickering RJ, Rynes RI, LoCascio N, Monahan JB, Sodetz JM. Identification of the alpha-gamma subunit of the eighth component of complement (C8) in a patient with systemic lupus erythematosus and absent C8 activity: patients and family studies. Clin Immunol Immunopathol (1982) 23:323–34. doi: 10.1016/0090-1229(82)90118-0
138. Jasin HE. Absence of the eighth component of complement in association with systemic lupus erythematosus-like disease. J Clin Invest (1977) 60:709–15. doi: 10.1172/jci108823
139. Lemaigre C, Suarez F, Martellosio JP, Barbarin C, Brunet K, Chomel JC, et al. Late onset of chronic granulomatous disease revealed by paecilomyces lilacinus cutaneous infection. J Clin Immunol (2022) 42:60–3. doi: 10.1007/s10875-021-01140-1
140. Jang MA, Kim EK, Now H, Nguyen NT, Kim WJ, Yoo JY, et al. Mutations in DDX58, which encodes RIG-I, cause atypical singleton-merten syndrome. Am J Hum Genet (2015) 96:266–74. doi: 10.1016/j.ajhg.2014.11.019
141. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet (2001) 28:313–4. doi: 10.1038/91070
142. Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat Commun (2017) 8:2176. doi: 10.1038/s41467-017-01932-3
143. Kawane K, Fukuyama H, Kondoh G, Takeda J, Ohsawa Y, Uchiyama Y, et al. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science (2001) 292:1546–9. doi: 10.1126/science.292.5521.1546
144. Agrebi N, Ben-Mustapha I, Matoussi N, Dhouib N, Ben-Ali M, Mekki N, et al. Rare splicing defects of FAS underly severe recessive autoimmune lymphoproliferative syndrome. Clin Immunol (2017) 183:17–23. doi: 10.1016/j.clim.2017.06.009
145. Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD, et al. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest (1996) 98:1107–13. doi: 10.1172/jci118892
146. Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet (2014) 46:503–9. doi: 10.1038/ng.2933
147. Van Nieuwenhove E, Garcia-Perez JE, Helsen C, Rodriguez PD, van Schouwenburg PA, Dooley J, et al. A kindred with mutant IKAROS and autoimmunity. J Allergy Clin Immunol (2018) 142:699–702.e612. doi: 10.1016/j.jaci.2018.04.008
148. Hoshino A, Okada S, Yoshida K, Nishida N, Okuno Y, Ueno H, et al. Abnormal hematopoiesis and autoimmunity in human subjects with germline IKZF1 mutations. J Allergy Clin Immunol (2017) 140:223–31. doi: 10.1016/j.jaci.2016.09.029
149. Hermann M, Bogunovic D. ISG15: In sickness and in health. Trends Immunol (2017) 38:79–93. doi: 10.1016/j.it.2016.11.001
150. Leventopoulos G, Denayer E, Makrythanasis P, Papapolychroniou C, Fryssira H. Noonan syndrome and systemic lupus erythematosus in a patient with a novel KRAS mutation. Clin Exp Rheumatol (2010) 28:556–7.
151. Quaio CR, Carvalho JF, da Silva CA, Bueno C, Brasil AS, Pereira AC, et al. Autoimmune disease and multiple autoantibodies in 42 patients with RASopathies. Am J Med Genet A (2012) 158a:1077–82. doi: 10.1002/ajmg.a.35290
152. Bader-Meunier B, Cavé H, Jeremiah N, Magerus A, Lanzarotti N, Rieux-Laucat F, et al. Are RASopathies new monogenic predisposing conditions to the development of systemic lupus erythematosus? case report and systematic review of the literature. Semin Arthritis Rheum (2013) 43:217–9. doi: 10.1016/j.semarthrit.2013.04.009
153. Urushihara M, Kagami S, Yasutomo K, Ito M, Kondo S, Kitamura A, et al. Sisters with alpha-mannosidosis and systemic lupus erythematosus. Eur J Pediatr (2004) 163:192–5. doi: 10.1007/s00431-004-1404-2
154. Lu D, Song J, Sun Y, Qi F, Liu L, Jin Y, et al. Mutations of deubiquitinase OTUD1 are associated with autoimmune disorders. J Autoimmun (2018) 94:156–65. doi: 10.1016/j.jaut.2018.07.019
155. He Y, Gallman AE, Xie C, Shen Q, Ma J, Wolfreys FD, et al. P2RY8 variants in lupus patients uncover a role for the receptor in immunological tolerance. J Exp Med (2022) 219(1):e20211004. doi: 10.1084/jem.20211004
156. Klar A, Navon-Elkan P, Rubinow A, Branski D, Hurvitz H, Christensen E, et al. Prolidase deficiency: it looks like systemic lupus erythematosus but it is not. Eur J Pediatr (2010) 169:727–32. doi: 10.1007/s00431-009-1102-1
157. Kurien BT, D'Sousa A, Bruner BF, Gross T, James JA, Targoff IN, et al. Prolidase deficiency breaks tolerance to lupus-associated antigens. Int J Rheum Dis (2013) 16:674–80. doi: 10.1111/1756-185x.12254
158. Shrinath M, Walter JH, Haeney M, Couriel JM, Lewis MA, Herrick AL, et al. Prolidase deficiency and systemic lupus erythematosus. Arch Dis Child (1997) 76:441–4. doi: 10.1136/adc.76.5.441
159. Di Rocco M, Fantasia AR, Taro M, Loy A, Forlino A, Martini A, et al. Systemic lupus erythematosus-like disease in a 6-year-old boy with prolidase deficiency. J Inherit Metab Dis 30(5):814. doi: 10.1007/s10545-007-0496-z (2007)
160. Falik-Zaccai TC, Khayat M, Luder A, Frenkel P, Magen D, Brik R, et al. A broad spectrum of developmental delay in a large cohort of prolidase deficiency patients demonstrates marked interfamilial and intrafamilial phenotypic variability. Am J Med Genet B Neuropsychiatr Genet (2010) 153b:46–56. doi: 10.1002/ajmg.b.30945
161. Butbul Aviel Y, Mandel H, Avitan Hersh E, Bergman R, Adiv OE, Luder A, et al. Prolidase deficiency associated with systemic lupus erythematosus (SLE): single site experience and literature review. Pediatr Rheumatol Online J (2012) 10:18. doi: 10.1186/1546-0096-10-18
162. Belot A, Kasher PR, Trotter EW, Foray AP, Debaud AL, Rice GI, et al. Protein kinase cδ deficiency causes mendelian systemic lupus erythematosus with b cell-defective apoptosis and hyperproliferation. Arthritis Rheum (2013) 65:2161–71. doi: 10.1002/art.38008
163. Al-Mayouf SM, AlSaleem A, AlMutairi N, AlSonbul A, Alzaid T, Alazami AM, et al. Monogenic interferonopathies: Phenotypic and genotypic findings of CANDLE syndrome and its overlap with C1q deficient SLE. Int J Rheum Dis (2018) 21:208–13. doi: 10.1111/1756-185x.13228
164. Al-Mayouf SM, AlTassan RS, AlOwain MA. Systemic lupus erythematosus in a girl with PTEN variant and transaldolase deficiency: a novel phenotype. Clin Rheumatol (2020) 39:3511–5. doi: 10.1007/s10067-020-05205-1
165. Tirosh I, Spielman S, Barel O, Ram R, Stauber T, Paret G, et al. Whole exome sequencing in childhood-onset lupus frequently detects single gene etiologies. Pediatr Rheumatol Online J (2019) 17:52. doi: 10.1186/s12969-019-0349-y
166. Lee T, Le EN, Glass DA 2nd, Bowen CD, Dominguez AR. Systemic lupus erythematosus in a patient with PTEN hamartoma tumour syndrome. Br J Dermatol (2014) 170:990–2. doi: 10.1111/bjd.12767
167. Heindl M, Händel N, Ngeow J, Kionke J, Wittekind C, Kamprad M. Autoimmunity, intestinal lymphoid hyperplasia, and defects in mucosal b-cell homeostasis in patients with PTEN hamartoma tumor syndrome. Gastroenterology (2012) 142:1093–1096.e1096. doi: 10.1053/j.gastro.2012.01.011
168. Sagar V, Bond JR, Chowdhary VR. A 50-Year-Old woman with cowden syndrome and joint pains. Arthritis Care Res (Hoboken) (2015) 67:1604–8. doi: 10.1002/acr.22616
169. Eissing M, Ripken L, Schreibelt G, Westdorp H, Ligtenberg M, Netea-Maier R, et al. PTEN hamartoma tumor syndrome and immune dysregulation. Transl Oncol (2019) 12:361–7. doi: 10.1016/j.tranon.2018.11.003
170. Chen K, Wu W, Mathew D, Zhang Y, Browne SK, Rosen LB, et al. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG1/2 mutations. J Allergy Clin Immunol (2014) 133:880–882.e810. doi: 10.1016/j.jaci.2013.11.038
171. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest (2015) 125:4135–48. doi: 10.1172/jci80477
172. Gennery A. Recent advances in understanding RAG deficiencies. F1000Res (2019) 8:148. doi: 10.12688/f1000research.17056.1
173. Walter JE, Lo MS, Kis-Toth K, Tirosh I, Frugoni F, Lee YN, et al. Impaired receptor editing and heterozygous RAG2 mutation in a patient with systemic lupus erythematosus and erosive arthritis. J Allergy Clin Immunol (2015) 135:272–3. doi: 10.1016/j.jaci.2014.07.063
174. Laura Barnabei HL, Castela M, Jeremiah N, Stolzenberg M-C, Chentout L V, Jacques S, et al. (2020).
175. Mackenzie KJ, Carroll P, Lettice L, Tarnauskaitė Ž, Reddy K, Dix F, et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J (2016) 35:831–44. doi: 10.15252/embj.201593339
176. Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A (2011) 155a:235–7. doi: 10.1002/ajmg.a.33778
177. Abdel-Salam GM, El-Kamah GY, Rice GI, El-Darouti M, Gornall H, Szynkiewicz M, et al. Chilblains as a diagnostic sign of aicardi-goutières syndrome. Neuropediatrics (2010) 41:18–23. doi: 10.1055/s-0030-1255059
178. Xu L ZJ, Sun Q, Geng L, Deng Y, Kamen D, et al. Does loss-of-function variants in Sat1 cause x-linked pediatric lupus. Arthritis Rheumatol 71(suppl 10):2810.
179. Estève E, Krug P, Hummel A, Arnoux JB, Boyer O, Brassier A, et al. Renal involvement in lysinuric protein intolerance: contribution of pathology to assessment of heterogeneity of renal lesions. Hum Pathol (2017) 62:160–9. doi: 10.1016/j.humpath.2016.12.021
180. Parsons H, Snyder F, Bowen T, Klassen J, Pinto A. Immune complex disease consistent with systemic lupus erythematosus in a patient with lysinuric protein intolerance. J Inherit Metab Dis (1996) 19:627–34. doi: 10.1007/bf01799838
181. Gattorno M, Di Rocco M, Buoncompagni A, Picco P, Meroni PL, Martini A, et al. Neonatal lupus and a seronegative mother. Lancet (2004) 363:1038. doi: 10.1016/s0140-6736(04)15839-x
182. Kamoda T, Nagai Y, Shigeta M, Kobayashi C, Sekijima T, Shibasaki M, et al. Lysinuric protein intolerance and systemic lupus erythematosus. Eur J Pediatr (1998) 157:130–1. doi: 10.1007/s004310050784
183. Aoki M, Fukao T, Fujita Y, Watanabe M, Teramoto T, Kato Y, et al. Lysinuric protein intolerance in siblings: complication of systemic lupus erythematosus in the elder sister. Eur J Pediatr (2001) 160:522–3. doi: 10.1007/pl00008455
184. Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun (2020) 11:5341. doi: 10.1038/s41467-020-18925-4
185. Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature (2020) 583:90–5. doi: 10.1038/s41586-020-2265-1
186. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
187. Duan R, Liu Q, Li J, Bian X, Yuan Q, Li Y, et al. A De novo frameshift mutation in TNFAIP3 impairs A20 deubiquitination function to cause neuropsychiatric systemic lupus erythematosus. J Clin Immunol (2019) 39:795–804. doi: 10.1007/s10875-019-00695-4
188. Su G, Lai J, Zhu J, Zhang D, Hou J, Xu Y, et al. Analysis of five cases of monogenic lupus related to primary immunodeficiency diseases. Inflammation Res (2021) 70:1211–6. doi: 10.1007/s00011-021-01479-6
189. Zhang D, Su G, Zhou Z, Lai J. Clinical characteristics and genetic analysis of A20 haploinsufficiency. Pediatr Rheumatol Online J (2021) 19:75. doi: 10.1186/s12969-021-00558-6
190. Shaheen ZR, Williams SJA, Binstadt BA. Case report: A novel TNFAIP3 mutation causing haploinsufficiency of A20 with a lupus-like phenotype. Front Immunol (2021) 12:629457. doi: 10.3389/fimmu.2021.629457
191. Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis (2018) 77:728–35. doi: 10.1136/annrheumdis-2017-212403
192. Yang J, Chandrasekharappa SC, Vilboux T, Smith AC, Peterson EJ. Immune complex-mediated autoimmunity in a patient with smith-magenis syndrome (del 17p11.2). J Clin Rheumatol (2014) 20:291–3. doi: 10.1097/rhu.0000000000000118
193. Theodorou E, Nezos A, Antypa E, Ioakeimidis D, Koutsilieris M, Tektonidou M, et al. B-cell activating factor and related genetic variants in lupus related atherosclerosis. J Autoimmun (2018) 92:87–92. doi: 10.1016/j.jaut.2018.05.002
194. González-Serna D, Ortiz-Fernández L, Vargas S, García A, Raya E, Fernández-Gutierrez B, et al. Association of a rare variant of the TNFSF13B gene with susceptibility to rheumatoid arthritis and systemic lupus erythematosus. Sci Rep (2018) 8:8195. doi: 10.1038/s41598-018-26573-4
195. Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol (2015) 35:235–43. doi: 10.1007/s10875-015-0147-3
196. Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med (2016) 213:1163–74. doi: 10.1084/jem.20151529
197. Belot A, Rice GI, Omarjee SO, Rouchon Q, Smith EM, Moreews M, et al. Contribution of rare and predicted pathogenic gene variants to childhood-onset lupus: a large, genetic panel analysis of British and French cohorts. Lancet Rheumatol (2020) 2:e99–e109. doi: 10.1016/S2665-9913(19)30142-0
198. Almlöf JC, Nystedt S, Leonard D, Eloranta ML, Grosso G, Sjöwall C, et al. Whole-genome sequencing identifies complex contributions to genetic risk by variants in genes causing monogenic systemic lupus erythematosus. Hum Genet (2019) 138(2):141–50. doi: 10.1007/s00439-018-01966-7
199. Delgado-Vega AM, Martinez-Bueno M, Oparina NY, Lopez Herraez D, Kristjansdottir H, Steinsson K, et al. Whole exome sequencing of patients from multicase families with systemic lupus erythematosus identifies multiple rare variants. Sci Rep (2018) 8:8775. doi: 10.1038/s41598-018-26274-y
200. Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res (2019) 47(D1):D1005–12. doi: 10.1093/nar/gky1120
201. Ghoussaini M, Mountjoy E, Carmona M, Peat G, Schmidt EM, Hercules A, et al. Open targets genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res (2021) 49(D1):D1311–20. doi: 10.1093/nar/gkaa840
202. Mountjoy E, Schmidt EM, Carmona M, Schwartzentruber J, Peat G, Miranda A, et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat Genet (2021) 53(11):1527–33. doi: 10.1038/s41588-021-00945-5
203. Johnson MB, Hattersley AT, Flanagan SE. Monogenic autoimmune diseases of the endocrine system. Lancet Diabetes Endocrinol (2016) 4(10):862–72. doi: 10.1016/S2213-8587(16)30095-X
204. Warshauer JT, Belk JA, Chan AY, Wang J, Gupta AR, Shi Q, et al. A human mutation in STAT3 promotes type 1 diabetes through a defect in CD8+ T cell tolerance. J Exp Med (2021) 218(8):e20210759. doi: 10.1084/jem.20210759
205. Warshauer JT, Bluestone JA, Anderson MS. New frontiers in the treatment of type 1 diabetes. Cell Metab (2020) 31(1):46–61. doi: 10.1016/j.cmet.2019.11.017
206. Sanyoura M, Lundgrin EL, Subramanian HP, Yu M, Sodadasi P, Greeley SAW, et al. Novel compound heterozygous LRBA deletions in a 6-month-old with neonatal diabetes. Diabetes Res Clin Pract (2021) 175:108798. doi: 10.1016/j.diabres.2021.108798
207. Zhang Y, Liu H, Ai T, Xia W, Chen T, Zhang L, et al. A delayed diagnosis of atypical immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: A case report. Med (Baltimore) (2021) 100(12):e25174. doi: 10.1097/MD.0000000000025174
208. Strakova V, Elblova L, Johnson MB, Dusatkova P, Obermannova B, Petruzelkova L, et al. Screening of monogenic autoimmune diabetes among children with type 1 diabetes and multiple autoimmune diseases: is it worth doing? J Pediatr Endocrinol Metab (2019) 32(10):1147–53. doi: 10.1515/jpem-2019-0261
209. Hwang JL, Park SY, Ye H, Sanyoura M, Pastore AN, Carmody D, et al. FOXP3 mutations causing early-onset insulin-requiring diabetes but without other features of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Diabetes (2018) 19(3):388–92. doi: 10.1111/pedi.12612
210. Johnson MB, De Franco E, Lango Allen H, Al Senani A, Elbarbary N, Siklar Z, et al. Recessively inherited LRBA mutations cause autoimmunity presenting as neonatal diabetes. Diabetes (2017) 66(8):2316–22. doi: 10.2337/db17-0040
211. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet (2014) 46(8):812–4. doi: 10.1038/ng.3040
212. Biason-Lauber A, Boni-Schnetzler M, Hubbard BP, Bouzakri K, Brunner A, Cavelti-Weder C, et al. Identification of a SIRT1 mutation in a family with type 1 diabetes. Cell Metab (2013) 17(3):448–55. doi: 10.1016/j.cmet.2013.02.001
213. Chuprin A, Avin A, Goldfarb Y, Herzig Y, Levi B, Jacob A, et al. The deacetylase Sirt1 is an essential regulator of aire-mediated induction of central immunological tolerance. Nat Immunol (2015) 16:737–45. doi: 10.1038/ni.3194
214. Coit P, Ruffalo L, Sawalha AH. Clinical subgroup clustering analysis in a systemic lupus erythematosus cohort from Western Pennsylvania. Eur J Rheumatol (2022) 9:3–7. doi: 10.5152/eurjrheum.2020.21225
215. Hunt KA, Mistry V, Bockett NA, Ahmad T, Ban M, Barker JN, et al. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature (2013) 498(7453):232–5. doi: 10.1038/nature12170
216. Johnson MB, Cerosaletti K, Flanagan SE, Buckner JH. Genetic mechanisms highlight shared pathways for the pathogenesis of polygenic type 1 diabetes and monogenic autoimmune diabetes. Curr Diabetes Rep (2019) 19(5):20. doi: 10.1007/s11892-019-1141-6
217. Yang J, Visscher PM, Wray NR. Sporadic cases are the norm for complex disease. Eur J Hum Genet (2010) 18:1039–43. doi: 10.1038/ejhg.2009.177
218. Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: From polygenic to omnigenic. Cell (2017) 169:1177–86. doi: 10.1016/j.cell.2017.05.038
219. Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet (2010) 42:565–9. doi: 10.1038/ng.608
220. O'Connor LJ. The distribution of common-variant effect sizes. Nat Genet (2021) 53:1243–9. doi: 10.1038/s41588-021-00901-3
221. Olsen NJ, James JA, Arriens C, Ishimori ML, Wallace DJ, Kamen DL, et al. Study of anti-malarials in incomplete lupus erythematosus (SMILE): study protocol for a randomized controlled trial. Trials (2018) 19(1):694. doi: 10.1186/s13063-018-3076-7
222. Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis (2020) 71(15):732–9. doi: 10.1093/cid/ciaa237
223. Group, R. C, Horby P, Mafham M, Linsell L, Bell JL, Staplin N, et al. Effect of hydroxychloroquine in hospitalized patients with covid-19. N Engl J Med (2020) 383(21):2030–40. doi: 10.1056/NEJMoa2022926
224. Cavalcanti AB, Zampieri FG, Rosa RG, Azevedo LCP, Veiga VC, Avezum A, et al. Hydroxychloroquine with or without azithromycin in mild-to-Moderate covid-19. N Engl J Med (2020) 383(21):2041–52. doi: 10.1056/NEJMoa2019014
225. Mitja O, Corbacho-Monne M, Ubals M, Alemany A, Suner C, Tebe C, et al. A cluster-randomized trial of hydroxychloroquine for prevention of covid-19. N Engl J Med (2021) 384:417–27. doi: 10.1056/NEJMoa2021801
226. Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for covid-19. N Engl J Med (2020) 383:517–25. doi: 10.1056/NEJMoa2016638
227. Perlman RL. Mouse models of human disease: An evolutionary perspective. Evol Med Public Health (2016) 2016:170–6. doi: 10.1093/emph/eow014
229. Wolf SD, Dittel BN, Hardardottir F, Janeway CA Jr. Experimental autoimmune encephalomyelitis induction in genetically b cell-deficient mice. J Exp Med (1996) 184(6):2271–8. doi: 10.1084/jem.184.6.2271
230. Cross AH, Trotter JL, Lyons J. B cells and antibodies in CNS demyelinating disease. J Neuroimmunol (2001) 112:1–14. doi: 10.1016/s0165-5728(00)00409-4
231. Du C, Sriram S. Increased severity of experimental allergic encephalomyelitis in lyn-/- mice in the absence of elevated proinflammatory cytokine response in the central nervous system. J Immunol (2002) 168:3105–12. doi: 10.4049/jimmunol.168.6.3105
232. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol (2002) 3(10):944–50. doi: 10.1038/ni833
233. Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol (1988) 130(3):443–54.
234. Lyons JA, San M, Happ MP, Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol (1999) 29(11):3432–9. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2
235. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory b cells inhibit EAE initiation in mice while other b cells promote disease progression. J Clin Invest (2008) 118(10):3420–30. doi: 10.1172/JCI36030
236. Zamvil SS, Hauser SL. Antigen presentation by b cells in multiple sclerosis. N Engl J Med (2021) 384(4):378–81. doi: 10.1056/NEJMcibr2032177
237. Wemlinger SM, Parker Harp CR, Yu B, Hardy IR, Seefeldt M, Matsuda J, et al. Preclinical analysis of candidate anti-human CD79 therapeutic antibodies using a humanized CD79 mouse model. J Immunol (2022) 208(7):1566–84. doi: 10.4049/jimmunol.2101056
238. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science (2022) 375(6578):296–301. doi: 10.1126/science.abj8222
239. Robinson WH, Steinman L. Epstein-Barr Virus and multiple sclerosis. Science (2022) 375(6578):264–5. doi: 10.1126/science.abm7930
240. Lanz TV, Brewer RC, Ho PP, Moon JS, Jude KM, Fernandez D, et al. Clonally expanded b cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature (2022) 603(7900):321–7. doi: 10.1038/s41586-022-04432-7
241. Richard ML, Gilkeson G. Mouse models of lupus: what they tell us and what they don't. Lupus Sci Med (2018) 5(1):e000199. doi: 10.1136/lupus-2016-000199
242. Halkom A, Wu H, Lu Q. Contribution of mouse models in our understanding of lupus. Int Rev Immunol (2020) 39(4):174–87. doi: 10.1080/08830185.2020.1742712
243. Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol (2011) 33:67–87. doi: 10.1007/s00281-010-0204-1
244. Ridgway WM, Peterson LB, Todd JA, Rainbow DB, Healy B, Burren OS, et al. Gene-gene interactions in the NOD mouse model of type 1 diabetes. Adv Immunol (2008) 100:151–75. doi: 10.1016/S0065-2776(08)00806-7
245. Smith MJ, Hinman RM, Getahun A, Kim S, Packard TA, Cambier JC, et al. Silencing of high-affinity insulin-reactive b lymphocytes by anergy and impact of the NOD genetic background in mice. Diabetologia (2018) 61(12):2621–32. doi: 10.1007/s00125-018-4730-z
246. Cambier JC. Autoimmunity risk alleles: hotspots in b cell regulatory signaling pathways. J Clin Invest (2013) 123(5):1928–31. doi: 10.1172/JCI69289
247. Tan Q, Tai N, Li Y, Pearson J, Pennetti S, Zhou Z, et al. Activation-induced cytidine deaminase deficiency accelerates autoimmune diabetes in NOD mice. JCI Insight (2018) 3(2):255–64. doi: 10.1172/jci.insight.95882
248. Gillmore JD, Hutchinson WL, Herbert J, Bybee A, Mitchell DA, Hasserjian RP, et al. Autoimmunity and glomerulonephritis in mice with targeted deletion of the serum amyloid p component gene: SAP deficiency or strain combination? Immunology (2004) 112:255–64. doi: 10.1111/j.1365-2567.2004.01860.x
249. Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF rescues self-reactive b cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity (2004) 20:785–98. doi: 10.1016/j.immuni.2004.05.010
250. Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, et al. BAFF mediates survival of peripheral immature b lymphocytes. J Exp Med (2000) 192:1453–66. doi: 10.1084/jem.192.10.1453
251. Tischner D, Woess C, Ottina E, Villunger A. Bcl-2-regulated cell death signalling in the prevention of autoimmunity. Cell Death Dis (2010) 1:e48. doi: 10.1038/cddis.2010.27
252. Ko K, Wang J, Perper S, Jiang Y, Yanez D, Kaverina N, et al. Bcl-2 as a therapeutic target in human tubulointerstitial inflammation. Arthritis Rheumatol (2016) 68:2740–51. doi: 10.1002/art.39744
253. Zhan Y, Carrington EM, Ko HJ, Vikstrom IB, Oon S, Zhang JG, et al. Bcl-2 antagonists kill plasmacytoid dendritic cells from lupus-prone mice and dampen interferon-α production. Arthritis Rheumatol (2015) 67:797–808. doi: 10.1002/art.38966
254. Liphaus BL, Kiss MH, Carrasco S, Goldenstein-Schainberg C. Increased fas and bcl-2 expression on peripheral blood T and b lymphocytes from juvenile-onset systemic lupus erythematosus, but not from juvenile rheumatoid arthritis and juvenile dermatomyositis. Clin Dev Immunol (2006) 13:283–7. doi: 10.1080/17402520600877786
255. Sun H, Lu B, Li RQ, Flavell RA, Taneja R. Defective T cell activation and autoimmune disorder in Stra13-deficient mice. Nat Immunol (2001) 2:1040–7. doi: 10.1038/ni721
256. Oliver PM, Vass T, Kappler J, Marrack P. Loss of the proapoptotic protein, bim, breaks b cell anergy. J Exp Med (2006) 203:731–41. doi: 10.1084/jem.20051407
257. Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Köntgen F, et al. Proapoptotic bcl-2 relative bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science (1999) 286:1735–8. doi: 10.1126/science.286.5445.1735
258. Wright JA, Bazile C, Clark ES, Carlesso G, Boucher J, Kleiman E, et al. Impaired b cell apoptosis results in autoimmunity that is alleviated by ablation of btk. Front Immunol (2021) 12:705307. doi: 10.3389/fimmu.2021.705307
259. Samuelson EM, Laird RM, Papillion AM, Tatum AH, Princiotta MF, Hayes SM, et al. Reduced b lymphoid kinase (Blk) expression enhances proinflammatory cytokine production and induces nephrosis in C57BL/6-lpr/lpr mice. PloS One (2014) 9:e92054. doi: 10.1371/journal.pone.0092054
260. Samuelson EM, Laird RM, Maue AC, Rochford R, Hayes SM. Blk haploinsufficiency impairs the development, but enhances the functional responses, of MZ b cells. Immunol Cell Biol (2012) 90:620–9. doi: 10.1038/icb.2011.76
261. Lindner JM, Kayo H, Hedlund S, Fukuda Y, Fukao T, Nielsen PJ, et al. Cutting edge: The transcription factor Bob1 counteracts b cell activation and regulates miR-146a in b cells. J Immunol (2014) 192:4483–6. doi: 10.4049/jimmunol.1303022
262. Haywood ME, Rogers NJ, Rose SJ, Boyle J, McDermott A, Rankin JM, et al. Dissection of BXSB lupus phenotype using mice congenic for chromosome 1 demonstrates that separate intervals direct different aspects of disease. J Immunol (2004) 173:4277–85. doi: 10.4049/jimmunol.173.7.4277
263. Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology (1998) 199:265–85. doi: 10.1016/s0171-2985(98)80032-6
264. Botto M. C1q knock-out mice for the study of complement deficiency in autoimmune disease. Exp Clin Immunogenet (1998) 15:231–4. doi: 10.1159/000019076
265. Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor cbl-b. Nature (2000) 403:211–6. doi: 10.1038/35003228
266. Yi Y, McNerney M, Datta SK. Regulatory defects in cbl and mitogen-activated protein kinase (extracellular signal-related kinase) pathways cause persistent hyperexpression of CD40 ligand in human lupus T cells. J Immunol (2000) 165:6627–34. doi: 10.4049/jimmunol.165.11.6627
267. Mattner J, Mohammed JP, Fusakio ME, Giessler C, Hackstein CP, Opoka R, et al. Genetic and functional data identifying Cd101 as a type 1 diabetes (T1D) susceptibility gene in nonobese diabetic (NOD) mice. PloS Genet (2019) 15:e1008178. doi: 10.1371/journal.pgen.1008178
268. Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent b-cell responses and b-1 cell development in CD19-deficient mice. Nature (1995) 376:352–5. doi: 10.1038/376352a0
269. Zhou LJ, Smith HM, Waldschmidt TJ, Schwarting R, Daley J, Tedder TF, et al. Tissue-specific expression of the human CD19 gene in transgenic mice inhibits antigen-independent b-lymphocyte development. Mol Cell Biol (1994) 14:3884–94. doi: 10.1128/mcb.14.6.3884-3894.1994
270. O'Keefe TL, Williams GT, Davies SL, Neuberger MS. Hyperresponsive b cells in CD22-deficient mice. Science (1996) 274:798–801. doi: 10.1126/science.274.5288.798
271. Clark EA, Giltiay NV. CD22: A regulator of innate and adaptive b cell responses and autoimmunity. Front Immunol (2018) 9:2235. doi: 10.3389/fimmu.2018.02235
272. Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, et al. Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity (1998) 8:497–508. doi: 10.1016/s1074-7613(00)80554-3
273. Shapiro MR, Yeh WI, Longfield JR, Gallagher J, Infante CM, Wellford S, et al. CD226 deletion reduces type 1 diabetes in the NOD mouse by impairing thymocyte development and peripheral T cell activation. Front Immunol (2020) 11:2180. doi: 10.3389/fimmu.2020.02180
274. Wandstrat AE, Nguyen C, Limaye N, Chan AY, Subramanian S, Tian XH, et al. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity (2004) 21:769–80. doi: 10.1016/j.immuni.2004.10.009
275. de Salort J, Cuenca M, Terhorst C, Engel P, Romero X. Ly9 (CD229) cell-surface receptor is crucial for the development of spontaneous autoantibody production to nuclear antigens. Front Immunol (2013) 4:225. doi: 10.3389/fimmu.2013.00225
276. Chen J, Chen YG, Reifsnyder PC, Schott WH, Lee CH, Osborne M, et al. Targeted disruption of CD38 accelerates autoimmune diabetes in NOD/Lt mice by enhancing autoimmunity in an ADP-ribosyltransferase 2-dependent fashion. J Immunol (2006) 176:4590–9. doi: 10.4049/jimmunol.176.8.4590
277. Higuchi T, Aiba Y, Nomura T, Matsuda J, Mochida K, Suzuki M, et al. Cutting edge: Ectopic expression of CD40 ligand on b cells induces lupus-like autoimmune disease. J Immunol (2002) 168:9–12. doi: 10.4049/jimmunol.168.1.9
278. Manea ME, Mueller RB, Dejica D, Sheriff A, Schett G, Herrmann M, et al. Increased expression of CD154 and FAS in SLE patients' lymphocytes. Rheumatol Int (2009) 30:181–5. doi: 10.1007/s00296-009-0933-4
279. Blossom S, Chu EB, Weigle WO, Gilbert KM. CD40 ligand expressed on b cells in the BXSB mouse model of systemic lupus erythematosus. J Immunol (1997) 159:4580–6.
280. McArdel SL, Terhorst C, Sharpe AH. Roles of CD48 in regulating immunity and tolerance. Clin Immunol (2016) 164:10–20. doi: 10.1016/j.clim.2016.01.008
281. Keszei M, Latchman YE, Vanguri VK, Brown DR, Detre C, Morra M, et al. Auto-antibody production and glomerulonephritis in congenic Slamf1-/- and Slamf2-/- [B6.129] but not in Slamf1-/- and Slamf2-/- [BALB/c.129] mice. Int Immunol (2011) 23:149–58. doi: 10.1093/intimm/dxq465
282. Koh AE, Njoroge SW, Feliu M, Cook A, Selig MK, Latchman YE, et al. The SLAM family member CD48 (Slamf2) protects lupus-prone mice from autoimmune nephritis. J Autoimmun (2011) 37:48–57. doi: 10.1016/j.jaut.2011.03.004
283. Lee KM, Forman JP, McNerney ME, Stepp S, Kuppireddi S, Guzior D, et al. Requirement of homotypic NK-cell interactions through 2B4(CD244)/CD48 in the generation of NK effector functions. Blood (2006) 107:3181–8. doi: 10.1182/blood-2005-01-0185
284. Morel L, Blenman KR, Croker BP, Wakeland EK. The major murine systemic lupus erythematosus susceptibility locus, Sle1, is a cluster of functionally related genes. Proc Natl Acad Sci USA (2001) 98:1787–92. doi: 10.1073/pnas.98.4.1787
285. Ye C, Low BE, Wiles MV, Brusko TM, Serreze DV, Driver JP, et al. CD70 inversely regulates regulatory T cells and invariant NKT cells and modulates type 1 diabetes in NOD mice. J Immunol (2020) 205:1763–77. doi: 10.4049/jimmunol.2000148
286. Xu M, Hou R, Sato-Hayashizaki A, Man R, Zhu C, Wakabayashi C, et al. Cd72(c) is a modifier gene that regulates fas(lpr)-induced autoimmune disease. J Immunol (2013) 190:5436–45. doi: 10.4049/jimmunol.1203576
287. Rojas A, Xu F, Rojas M, Thomas JW. Structure and function of CD72 in the non-obese diabetic (NOD) mouse. Autoimmunity (2003) 36:233–9. doi: 10.1080/0891693031000141059
288. Wang A, Batteux F, Wakeland EK. The role of SLAM/CD2 polymorphisms in systemic autoimmunity. Curr Opin Immunol (2010) 22:706–14. doi: 10.1016/j.coi.2010.10.014
289. Wong EB, Soni C, Chan AY, Domeier PP, Shwetank, Abraham T, et al. B cell-intrinsic CD84 and Ly108 maintain germinal center b cell tolerance. J Immunol (2015) 194:4130–43. doi: 10.4049/jimmunol.1403023
290. Balomenos D, Martín-Caballero J, García MI, Prieto I, Flores JM, Serrano M, et al. The cell cycle inhibitor p21 controls T-cell proliferation and sex-linked lupus development. Nat Med (2000) 6:171–6. doi: 10.1038/72272
291. Xu Z, Vallurupalli A, Perry D, Baker H, Croker BP, et al. Cyclin-dependent kinase inhibitor Cdkn2c regulates b cell homeostasis and function in the NZM2410-derived murine lupus susceptibility locus Sle2c1. J Immunol (2011) 186:6673–82. doi: 10.4049/jimmunol.1002544
292. Potula HH, Xu Z, Zeumer L, Sang A, Croker BP, Morel L. Cyclin-dependent kinase inhibitor Cdkn2c deficiency promotes B1a cell expansion and autoimmunity in a mouse model of lupus. J Immunol (2012) 189:2931–40. doi: 10.4049/jimmunol.1200556
293. Qiao G, Li Z, Minto AW, Shia J, Yang L, Bao L, et al. Altered thymic selection by overexpressing cellular FLICE inhibitory protein in T cells causes lupus-like syndrome in a BALB/c but not C57BL/6 strain. Cell Death Differ (2010) 17:522–33. doi: 10.1038/cdd.2009.143
294. Shenoy S, Mohanakumar T, Chatila T, Tersak J, Duffy B, Wang R, et al. Defective apoptosis in lymphocytes and the role of IL-2 in autoimmune hematologic cytopenias. Clin Immunol (2001) 99:266–75. doi: 10.1006/clim.2001.5017
295. Haraldsson MK, Louis-Dit-Sully CA, Lawson BR, Sternik G, Santiago-Raber ML, Gascoigne NR, et al. The lupus-related Lmb3 locus contains a disease-suppressing coronin-1A gene mutation. Immunity (2008) 28:40–51. doi: 10.1016/j.immuni.2007.11.023
296. Robey FA, Jones KD, Steinberg AD. C-reactive protein mediates the solubilization of nuclear DNA by complement. vitro J Exp Med (1985) 161:1344–56. doi: 10.1084/jem.161.6.1344
297. Szalai AJ, Weaver CT, McCrory MA, van Ginkel FW, Reiman RM, Kearney JF, et al. Delayed lupus onset in (NZB x NZW)F1 mice expressing a human c-reactive protein transgene. Arthritis Rheum (2003) 48:1602–11. doi: 10.1002/art.11026
298. Enzler T, Gillessen S, Manis JP, Ferguson D, Fleming J, Alt FW, et al. Deficiencies of GM-CSF and interferon gamma link inflammation and cancer. J Exp Med (2003) 197:1213–9. doi: 10.1084/jem.20021258
299. Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science (1994) 264:713–6. doi: 10.1126/science.8171324
300. Gubbels Bupp MR, Woodfin AE. Evaluating the role of candidate gene, csf3r, for sex-linked susceptibility to lupus-like disease in mice. J Immunol (2019) 202:50.11–1.
301. Lu W, Skrzypczynska KM, Weiss A. Acute csk inhibition hinders b cell activation by constraining the PI3 kinase pathway. Proc Natl Acad Sci USA (2021) 118(43):e2108957118. doi: 10.1073/pnas.2108957118
302. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
303. Alves da Costa T, Peterson JN, Lang J, Shulman J, Liang X, Freed BM, et al. Central human b cell tolerance manifests with a distinctive cell phenotype and is enforced via CXCR4 signaling in hu-mice. Proc Natl Acad Sci USA (2021) 118(16):e2021570118. doi: 10.1073/pnas.2021570118
304. Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med (2012) 4:157ra141. doi: 10.1126/scitranslmed.3004801
305. Fanzo JC, Yang W, Jang SY, Gupta S, Chen Q, Siddiq A, et al. Loss of IRF-4-binding protein leads to the spontaneous development of systemic autoimmunity. J Clin Invest (2006) 116:703–14. doi: 10.1172/jci24096
306. Napirei M, Ricken A, Eulitz D, Knoop H, Mannherz HG. Expression pattern of the deoxyribonuclease 1 gene: lessons from the Dnase1 knockout mouse. Biochem J (2004) 380:929–37. doi: 10.1042/bj20040046
307. Foray AP, Candon S, Hildebrand S, Marquet C, Valette F, Pecquet C, et al. De novo germline mutation in the dual specificity phosphatase 10 gene accelerates autoimmune diabetes. Proc Natl Acad Sci USA (2021), 118(47):e2112032118. doi: 10.1073/pnas.2112032118
308. Murga M, Fernández-Capetillo O, Field SJ, Moreno B, Borlado LR, Fujiwara Y, et al. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity (2001) 15:959–70. doi: 10.1016/s1074-7613(01)00254-0
309. Forster N, Gallinat S, Jablonska J, Weiss S, Elsässer HP, Lutz W, et al. p300 protein acetyltransferase activity suppresses systemic lupus erythematosus-like autoimmune disease in mice. J Immunol (2007) 178:6941–8. doi: 10.4049/jimmunol.178.11.6941
310. Perry DJ, Yin Y, Telarico T, Baker HV, Dozmorov I, Perl A, et al. Murine lupus susceptibility locus Sle1c2 mediates CD4+ T cell activation and maps to estrogen-related receptor γ. J Immunol (2012) 189:793–803. doi: 10.4049/jimmunol.1200411
311. Mayeux J, Skaug B, Luo W, Russell LM, John S, Saelee P, et al. Genetic interaction between Lyn, Ets1, and btk in the control of antibody levels. J Immunol (2015) 195:1955–63. doi: 10.4049/jimmunol.1500165
312. Luo W, Mayeux J, Gutierrez T, Russell L, Getahun A, Müller J, et al. A balance between b cell receptor and inhibitory receptor signaling controls plasma cell differentiation by maintaining optimal Ets1 levels. J Immunol (2014) 193:909–20. doi: 10.4049/jimmunol.1400666
313. Willcocks LC, Carr EJ, Niederer HA, Rayner TF, Williams TN, Yang W, et al. A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc Natl Acad Sci USA (2010) 107:7881–5. doi: 10.1073/pnas.0915133107
314. Fukuyama H, Nimmerjahn F, Ravetch JV. The inhibitory fcgamma receptor modulates autoimmunity by limiting the accumulation of immunoglobulin g+ anti-DNA plasma cells. Nat Immunol (2005) 6:99–106. doi: 10.1038/ni1151
315. Zhang L, Eddy A, Teng YT, Fritzler M, Kluppel M, Melet F, et al. An immunological renal disease in transgenic mice that overexpress fli-1, a member of the ets family of transcription factor genes. Mol Cell Biol (1995) 15:6961–70. doi: 10.1128/mcb.15.12.6961
316. Amin RH, Schlissel MS. Foxo1 directly regulates the transcription of recombination-activating genes during b cell development. Nat Immunol (2008) 9:613–22. doi: 10.1038/ni.1612
317. Salvador JM, Hollander MC, Nguyen AT, Kopp JB, Barisoni L, Moore JK, et al. Mice lacking the p53-effector gene Gadd45a develop a lupus-like syndrome. Immunity (2002) 16:499–508. doi: 10.1016/s1074-7613(02)00302-3
318. Li Y, Zhao M, Yin H, Gao F, Wu X, Luo Y, et al. Overexpression of the growth arrest and DNA damage-induced 45alpha gene contributes to autoimmunity by promoting DNA demethylation in lupus T cells. Arthritis Rheum (2010) 62:1438–47. doi: 10.1002/art.27363
319. Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, et al. Genomic instability in Gadd45a-deficient mice. Nat Genet (1999) 23:176–84. doi: 10.1038/13802
320. Smith LK, Fawaz K, Treanor B. Galectin-9 regulates the threshold of b cell activation and autoimmunity. Elife (2021) 10:e64557. doi: 10.7554/eLife.64557
321. Le LQ, Kabarowski JH, Weng Z, Satterthwaite AB, Harvill ET, Jensen ER, et al. Mice lacking the orphan G protein-coupled receptor G2A develop a late-onset autoimmune syndrome. Immunity (2001) 14:561–71. doi: 10.1016/s1074-7613(01)00145-5
322. Tsui HW, Siminovitch KA, de Souza L, Tsui FW. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet (1993) 4:124–9. doi: 10.1038/ng0693-124
323. Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, et al. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell (1993) 73:1445–54. doi: 10.1016/0092-8674(93)90369-2
324. Doyle HA, Gee RJ, Mamula MJ. A failure to repair self-proteins leads to T cell hyperproliferation and autoantibody production. J Immunol (2003) 171:2840–7. doi: 10.4049/jimmunol.171.6.2840
325. Xin H, D'Souza S, Jørgensen TN, Vaughan AT, Lengyel P, Kotzin BL, et al. Increased expression of Ifi202, an IFN-activatable gene, in B6.Nba2 lupus susceptible mice inhibits p53-mediated apoptosis. J Immunol (2006) 176:5863–70. doi: 10.4049/jimmunol.176.10.5863
326. Panchanathan R, Xin H, Choubey D. Disruption of mutually negative regulatory feedback loop between interferon-inducible p202 protein and the E2F family of transcription factors in lupus-prone mice. J Immunol (2008) 180:5927–34. doi: 10.4049/jimmunol.180.9.5927
327. Asefa B, Klarmann KD, Copeland NG, Gilbert DJ, Jenkins NA, Keller JR. The interferon-inducible p200 family of proteins: a perspective on their roles in cell cycle regulation and differentiation. Blood Cells Mol Dis (2004) 32:155–67. doi: 10.1016/j.bcmd.2003.10.002
328. Mondini M, Vidali M, Airò P, De Andrea M, Riboldi P, Meroni PL, et al. Role of the interferon-inducible gene IFI16 in the etiopathogenesis of systemic autoimmune disorders. Ann N Y Acad Sci (2007) 1110:47–56. doi: 10.1196/annals.1423.006
329. Li J, Liu Y, Xie C, Zhu J, Kreska D, Morel L, et al. Deficiency of type I interferon contributes to Sle2-associated component lupus phenotypes. Arthritis Rheum (2005) 52:3063–72. doi: 10.1002/art.21307
330. Seery JP, Carroll JM, Cattell V, Watt FM. Antinuclear autoantibodies and lupus nephritis in transgenic mice expressing interferon gamma in the epidermis. J Exp Med (1997) 186:1451–9. doi: 10.1084/jem.186.9.1451
331. Ehrenstein MR, O'Keefe TL, Davies SL, Neuberger MS. Targeted gene disruption reveals a role for natural secretory IgM in the maturation of the primary immune response. Proc Natl Acad Sci USA (1998) 95:10089–93. doi: 10.1073/pnas.95.17.10089
332. Ehrenstein MR, Cook HT, Neuberger MS. Deficiency in serum immunoglobulin (Ig)M predisposes to development of IgG autoantibodies. J Exp Med (2000) 191:1253–8. doi: 10.1084/jem.191.7.1253
333. Schwickert TA, Tagoh H, Schindler K, Fischer M, Jaritz M, Busslinger M. Ikaros prevents autoimmunity by controlling anergy and toll-like receptor signaling in b cells. Nat Immunol (2019) 20:1517–29. doi: 10.1038/s41590-019-0490-2
334. Schorle H, Holtschke T, Hünig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature (1991) 352:621–4. doi: 10.1038/352621a0
335. Crispín JC, Tsokos GC. Transcriptional regulation of IL-2 in health and autoimmunity. Autoimmun Rev (2009) 8:190–5. doi: 10.1016/j.autrev.2008.07.042
336. Ciecko AE, Foda B, Barr JY, Ramanathan S, Atkinson MA, Serreze DV, et al. Interleukin-27 is essential for type 1 diabetes development and sjögren syndrome-like inflammation. Cell Rep (2019) 29:3073–3086.e3075. doi: 10.1016/j.celrep.2019.11.010
337. Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity (1995) 3:521–30. doi: 10.1016/1074-7613(95)90180-9
338. Suzuki H, Kündig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science (1995) 268:1472–6. doi: 10.1126/science.7770771
339. Akerlund J, Getahun A, Cambier JC. B cell expression of the SH2-containing inositol 5-phosphatase (SHIP-1) is required to establish anergy to high affinity, proteinacious autoantigens. J Autoimmun (2015) 62:45–54. doi: 10.1016/j.jaut.2015.06.007
340. Pathak S, Ma S, Shukla V, Lu R. A role for IRF8 in b cell anergy. J Immunol (2013) 191:6222–30. doi: 10.4049/jimmunol.1301169
341. Ding C, Ma Y, Chen X, Liu M, Cai Y, Hu X, et al. Integrin CD11b negatively regulates BCR signalling to maintain autoreactive b cell tolerance. Nat Commun (2013) 4:2813. doi: 10.1038/ncomms3813
342. Pflegerl P, Vesely P, Hantusch B, Schlederer M, Zenz R, Janig E, et al. Epidermal loss of JunB leads to a SLE phenotype due to hyper IL-6 signaling. Proc Natl Acad Sci USA (2009) 106:20423–8. doi: 10.1073/pnas.0910371106
343. Meixner A, Zenz R, Schonthaler HB, Kenner L, Scheuch H, Penninger JM, et al. Epidermal JunB represses G-CSF transcription and affects haematopoiesis and bone formation. Nat Cell Biol (2008) 10:1003–11. doi: 10.1038/ncb1761
344. Li QZ, Zhou J, Yang R, Yan M, Ye Q, Liu K, et al. The lupus-susceptibility gene kallikrein downmodulates antibody-mediated glomerulonephritis. Genes Immun (2009) 10:503–8. doi: 10.1038/gene.2009.7
345. Maneva-Radicheva L, Amatya C, Parker C, Ellefson J, Radichev I, Raghavan A, et al. Autoimmune diabetes is suppressed by treatment with recombinant human tissue kallikrein-1. PloS One (2014) 9:e107213. doi: 10.1371/journal.pone.0107213
346. Moustardas P, Yamada-Fowler N, Apostolou E, Tzioufas AG, Turkina MV, Spyrou G. Deregulation of the kallikrein protease family in the salivary glands of the sjögren's syndrome ERdj5 knockout mouse model. Front Immunol (2021) 12:693911. doi: 10.3389/fimmu.2021.693911
347. Katzav A, Kloog Y, Korczyn AD, Niv H, Karussis DM, Wang N, et al. Treatment of MRL/lpr mice, a genetic autoimmune model, with the ras inhibitor, farnesylthiosalicylate (FTS). Clin Exp Immunol (2001) 126:570–7. doi: 10.1046/j.1365-2249.2001.01674.x
348. Rapoport MJ, Sharabi A, Aharoni D, Bloch O, Zinger H, Dayan M, et al. Amelioration of SLE-like manifestations in (NZBxNZW)F1 mice following treatment with a peptide based on the complementarity determining region 1 of an autoantibody is associated with a down-regulation of apoptosis and of the pro-apoptotic factor JNK kinase. Clin Immunol (2005) 117:262–70. doi: 10.1016/j.clim.2005.09.003
349. Sommers CL, Park CS, Lee J, Feng C, Fuller CL, Grinberg A, et al. A LAT mutation that inhibits T cell development yet induces lymphoproliferation. Science (2002) 296:2040–3. doi: 10.1126/science.1069066
350. Aguado E, Richelme S, Nuñez-Cruz S, Miazek A, Mura AM, Richelme M, et al. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science (2002) 296:2036–40. doi: 10.1126/science.1069057
351. Morel L, Yu Y, Blenman KR, Caldwell RA, Wakeland EK. Production of congenic mouse strains carrying genomic intervals containing SLE-susceptibility genes derived from the SLE-prone NZM2410 strain. Mamm Genome (1996) 7:335–9. doi: 10.1007/s003359900098
352. Watson ML, Rao JK, Gilkeson GS, Ruiz P, Eicher EM, Pisetsky DS, et al. Genetic analysis of MRL-lpr mice: relationship of the fas apoptosis gene to disease manifestations and renal disease-modifying loci. J Exp Med (1992) 176:1645–56. doi: 10.1084/jem.176.6.1645
353. Wang N, Keszei M, Halibozek P, Yigit B, Engel P, Terhorst C. Slamf6 negatively regulates autoimmunity. Clin Immunol (2016) 173:19–26. doi: 10.1016/j.clim.2016.06.009
354. Keszei M, Detre C, Rietdijk ST, Muñoz P, Romero X, Berger SB, et al. A novel isoform of the Ly108 gene ameliorates murine lupus. J Exp Med (2011) 208:811–22. doi: 10.1084/jem.20101653
355. Kumar KR, Li L, Yan M, Bhaskarabhatla M, Mobley AB, Nguyen C, et al. Regulation of b cell tolerance by the lupus susceptibility gene Ly108. Science (2006) 312:1665–9. doi: 10.1126/science.1125893
356. Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, et al. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell (1995) 83:301–11. doi: 10.1016/0092-8674(95)90171-x
357. Nishizumi H, Taniuchi I, Yamanashi Y, Kitamura D, Ilic D, Mori S, et al. Impaired proliferation of peripheral b cells and indication of autoimmune disease in lyn-deficient mice. Immunity (1995) 3:549–60. doi: 10.1016/1074-7613(95)90126-4
358. Whyburn LR, Halcomb KE, Contreras CM, Lowell CA, Witte ON, Satterthwaite AB. Reduced dosage of bruton's tyrosine kinase uncouples b cell hyperresponsiveness from autoimmunity in lyn-/- mice. J Immunol (2003) 171:1850–8. doi: 10.4049/jimmunol.171.4.1850
359. Green RS, Stone EL, Tenno M, Lehtonen E, Farquhar MG, Marth JD. Mammalian n-glycan branching protects against innate immune self-recognition and inflammation in autoimmune disease pathogenesis. Immunity (2007) 27:308–20. doi: 10.1016/j.immuni.2007.06.008
360. Chui D, Oh-Eda M, Liao YF, Panneerselvam K, Lal A, Marek KW, et al. Alpha-mannosidase-II deficiency results in dyserythropoiesis and unveils an alternate pathway in oligosaccharide biosynthesis. Cell (1997) 90:157–67. doi: 10.1016/s0092-8674(00)80322-0
361. Sawalha AH, Richardson B. MEK/ERK pathway inhibitors as a treatment for inflammatory arthritis might result in the development of lupus: comment on the article by thiel et al. Arthritis Rheum (2008) 58:1203–4. doi: 10.1002/art.23382
362. Rogers NJ, Lees MJ, Gabriel L, Maniati E, Rose SJ, Potter PK, et al. A defect in Marco expression contributes to systemic lupus erythematosus development via failure to clear apoptotic cells. J Immunol (2009) 182:1982–90. doi: 10.4049/jimmunol.0801320
363. Hurov JB, Stappenbeck TS, Zmasek CM, White LS, Ranganath SH, Russell JH, et al. Immune system dysfunction and autoimmune disease in mice lacking emk (Par-1) protein kinase. Mol Cell Biol (2001) 21:3206–19. doi: 10.1128/mcb.21.9.3206-3219.2001
364. Zalosnik MI, Fabio MC, Bertoldi ML, Castañares CN, Degano AL. MeCP2 deficiency exacerbates the neuroinflammatory setting and autoreactive response during an autoimmune challenge. Sci Rep (2021) 11:10997. doi: 10.1038/s41598-021-90517-8
365. Webb R, Wren JD, Jeffries M, Kelly JA, Kaufman KM, Tang Y, et al. Variants within MECP2, a key transcription regulator, are associated with increased susceptibility to lupus and differential gene expression in patients with systemic lupus erythematosus. Arthritis Rheum (2009) 60:1076–84. doi: 10.1002/art.24360
366. Pan Y, Sawalha AH. Epigenetic regulation and the pathogenesis of systemic lupus erythematosus. Transl Res (2009) 153:4–10. doi: 10.1016/j.trsl.2008.10.007
367. Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature (2001) 411:207–11. doi: 10.1038/35075603
368. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (2004) 304:1147–50. doi: 10.1126/science.1094359
369. Baek WY, Woo JM, Kim HA, Jung JY, Suh CH. Polymorphisms of MFGE8 are associated with susceptibility and clinical manifestations through gene expression modulation in koreans with systemic lupus erythematosus. Sci Rep (2019) 9:18565. doi: 10.1038/s41598-019-55061-6
370. Gonzalez-Martin A, Adams BD, Lai M, Shepherd J, Salvador-Bernaldez M, Salvador JM, et al. The microRNA miR-148a functions as a critical regulator of b cell tolerance and autoimmunity. Nat Immunol (2016) 17:433–40. doi: 10.1038/ni.3385
371. Lu X, Kovalev GI, Chang H, Kallin E, Knudsen G, Xia L, et al. Inactivation of NuRD component Mta2 causes abnormal T cell activation and lupus-like autoimmune disease in mice. J Biol Chem (2008) 283:13825–33. doi: 10.1074/jbc.M801275200
372. Kuraoka M, Snowden PB, Nojima T, Verkoczy L, Haynes BF, Kitamura D, et al. BCR and endosomal TLR signals synergize to increase AID expression and establish central b cell tolerance. Cell Rep (2017) 18:1627–35. doi: 10.1016/j.celrep.2017.01.050
373. Ju J, Xu J, Zhu Y, Fu X, Morel L, Xu Z. A variant of the histone-binding protein sNASP contributes to mouse lupus. Front Immunol (2019) 10:637. doi: 10.3389/fimmu.2019.00637
374. Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, et al. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol (2010) 185:5247–58. doi: 10.4049/jimmunol.1001472
375. Li J, Stein TD, Johnson JA. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol Genomics (2004) 18:261–72. doi: 10.1152/physiolgenomics.00209.2003
376. Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, et al. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int (2001) 60:1343–53. doi: 10.1046/j.1523-1755.2001.00939.x
377. Elliott JI, McVey JH, Higgins CF. The P2X7 receptor is a candidate product of murine and human lupus susceptibility loci: a hypothesis and comparison of murine allelic products. Arthritis Res Ther (2005) 7:R468–475. doi: 10.1186/ar1699
378. Niu Y, Sengupta M, Titov AA, Choi SC, Morel L. The PBX1 lupus susceptibility gene regulates CD44 expression. Mol Immunol (2017) 85:148–54. doi: 10.1016/j.molimm.2017.02.016
379. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11:141–51. doi: 10.1016/s1074-7613(00)80089-8
380. Curran CS, Gupta S, Sanz I, Sharon E. PD-1 immunobiology in systemic lupus erythematosus. J Autoimmun (2019) 97:1–9. doi: 10.1016/j.jaut.2018.10.025
381. Wilkinson R, Lyons AB, Roberts D, Wong MX, Bartley PA, Jackson DE. Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) acts as a regulator of b-cell development, b-cell antigen receptor (BCR)-mediated activation, and autoimmune disease. Blood (2002) 100:184–93. doi: 10.1182/blood-2002-01-0027
382. Greaves SA, Peterson JN, Strauch P, Torres RM, Pelanda R. Active PI3K abrogates central tolerance in high-avidity autoreactive b cells. J Exp Med (2019) 216:1135–53. doi: 10.1084/jem.20181652
383. Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med (2009) 15:1266–72. doi: 10.1038/nm.2048
384. Liu YH, Tsai YS, Lin SC, Liao NS, Jan MS, Liang CT, et al. Quantitative PPARγ expression affects the balance between tolerance and immunity. Sci Rep (2016) 6:26646. doi: 10.1038/srep26646
385. Roszer T, Menéndez-Gutiérrez MP, Lefterova MI, Alameda D, Núñez V, Lazar MA, et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol (2011) 186:621–31. doi: 10.4049/jimmunol.1002230
386. Setoguchi K, Misaki Y, Terauchi Y, Yamauchi T, Kawahata K, Kadowaki T, et al. Peroxisome proliferator-activated receptor-gamma haploinsufficiency enhances b cell proliferative responses and exacerbates experimentally induced arthritis. J Clin Invest (2001) 108:1667–75. doi: 10.1172/jci13202
387. Browne CD, Del Nagro CJ, Cato MH, Dengler HS, Rickert RC. Suppression of phosphatidylinositol 3,4,5-trisphosphate production is a key determinant of b cell anergy. Immunity (2009) 31:749–60. doi: 10.1016/j.immuni.2009.08.026
388. Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired fas response and autoimmunity in pten+/- mice. Science (1999) 285:2122–5. doi: 10.1126/science.285.5436.2122
389. Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet (1998) 19:348–55. doi: 10.1038/1235
390. Menard L, Saadoun D, Isnardi I, Ng YS, Meyers G, Massad C, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive b cells in humans. J Clin Invest (2011) 121:3635–44. doi: 10.1172/jci45790
391. Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet (2011) 43:902–7. doi: 10.1038/ng.904
392. Dai X, James RG, Habib T, Singh S, Jackson S, Khim S, et al. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest (2013) 123:2024–36. doi: 10.1172/jci66963
393. Zikherman J, Hermiston M, Steiner D, Hasegawa K, Chan A, Weiss A. PTPN22 deficiency cooperates with the CD45 E613R allele to break tolerance on a non-autoimmune background. J Immunol (2009) 182:4093–106. doi: 10.4049/jimmunol.0803317
394. Getahun A, Beavers NA, Larson SR, Shlomchik MJ, Cambier JC. Continuous inhibitory signaling by both SHP-1 and SHIP-1 pathways is required to maintain unresponsiveness of anergic b cells. J Exp Med (2016) 213:751–69. doi: 10.1084/jem.20150537
395. Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes b-1a cell development and causes systemic autoimmunity. Immunity (2007) 27:35–48. doi: 10.1016/j.immuni.2007.04.016
396. Green MC, Shultz LD. Motheaten, an immunodeficient mutant of the mouse. i. genetics and pathology. J Hered (1975) 66:250–8. doi: 10.1093/oxfordjournals.jhered.a108625
397. Majeti R, Xu Z, Parslow TG, Olson JL, Daikh DI, Killeen N, et al. An inactivating point mutation in the inhibitory wedge of CD45 causes lymphoproliferation and autoimmunity. Cell (2000) 103:1059–70. doi: 10.1016/s0092-8674(00)00209-9
398. Lamont KR, Hasham MG, Donghia NM, Branca J, Chavaree M, Chase B, et al. Attenuating homologous recombination stimulates an AID-induced antileukemic effect. J Exp Med (2013) 210:1021–33. doi: 10.1084/jem.20121258
399. Ratiu JJ, Racine JJ, Hasham MG, Wang Q, Branca JA, Chapman HD, et al. Genetic and small molecule disruption of the AID/RAD51 axis similarly protects nonobese diabetic mice from type 1 diabetes through expansion of regulatory b lymphocytes. J Immunol (2017) 198:4255–67. doi: 10.4049/jimmunol.1700024
400. Guo B, Rothstein TL. RasGRP1 is an essential signaling molecule for development of B1a cells with autoantigen receptors. J Immunol (2016) 196:2583–90. doi: 10.4049/jimmunol.1502132
401. Priatel JJ, Chen X, Zenewicz LA, Shen H, Harder KW, Horwitz MS, et al. Chronic immunodeficiency in mice lacking RasGRP1 results in CD4 T cell immune activation and exhaustion. J Immunol (2007) 179:2143–52. doi: 10.4049/jimmunol.179.4.2143
402. Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat Immunol (2003) 4:741–8. doi: 10.1038/ni950
403. Kinashi T, Katagiri K. Regulation of lymphocyte adhesion and migration by the small GTPase Rap1 and its effector molecule, RAPL. Immunol Lett (2004) 93:1–5. doi: 10.1016/j.imlet.2004.02.008
404. Katagiri K, Ueda Y, Tomiyama T, Yasuda K, Toda Y, Ikehara S, et al. Deficiency of Rap1-binding protein RAPL causes lymphoproliferative disorders through mislocalization of p27kip1. Immunity (2011) 34:24–38. doi: 10.1016/j.immuni.2010.12.010
405. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi: 10.1038/nature03555
406. Schulte-Pelkum J, Fritzler M, Mahler M. Latest update on the Ro/SS-a autoantibody system. Autoimmun Rev (2009) 8:632–7. doi: 10.1016/j.autrev.2009.02.010
407. Kurien BT, Dsouza A, Igoe A, Lee YJ, Maier-Moore JS, Gordon T, et al. Immunization with 60 kD ro peptide produces different stages of preclinical autoimmunity in a sjögren's syndrome model among multiple strains of inbred mice. Clin Exp Immunol (2013) 173:67–75. doi: 10.1111/cei.12094
408. Kurien BT, Porter A, Dorri Y, Iqbal S, D'Souza A, Singh A, et al. Degree of modification of Ro60 by the lipid peroxidation by-product 4-hydroxy-2-nonenal may differentially induce sjögren syndrome or systemic lupus erythematosus in BALB/c mice. Free Radic Biol Med (2011) 50:1222–33. doi: 10.1016/j.freeradbiomed.2010.10.687
409. Scofield RH, Kaufman KM, Baber U, James JA, Harley JB, Kurien BT. Immunization of mice with human 60-kd ro peptides results in epitope spreading if the peptides are highly homologous between human and mouse. Arthritis Rheum (1999) 42:1017–24. doi: 10.1002/1529-0131(199905)42:5<1017::Aid-anr22>3.0.Co;2-7
410. Maier-Moore JS, Kurien BT, D'Souza A, Bockus L, Asfa S, Dorri Y, et al. Passive transfer of antibodies to the linear epitope 60 kD ro 273-289 induces features of sjögren's syndrome in naive mice. Clin Exp Immunol (2015) 180(1):19–27. doi: 10.1111/cei.12480
411. Núñez V, Alameda D, Rico D, Mota R, Gonzalo P, Cedenilla M, et al. Retinoid X receptor alpha controls innate inflammatory responses through the up-regulation of chemokine expression. Proc Natl Acad Sci USA (2010) 107(23):10626–31. doi: 10.1073/pnas.0913545107
412. Pepys MB. Serum amyloid p component (not serum amyloid protein). Nat Med (1999) 5(8):852–3. doi: 10.1038/11272
413. Bickerstaff MC, Botto M, Hutchinson WL, Herbert J, Tennent GA, Bybee A, et al. Serum amyloid p component controls chromatin degradation and prevents antinuclear autoimmunity. Nat Med (1999) 5(6):694–7. doi: 10.1038/9544
414. Paul E, Carroll MC. SAP-less chromatin triggers systemic lupus erythematosus. Nat Med (1999) 5:607–8. doi: 10.1038/9450
415. Drappa J, Kamen LA, Chan E, Georgiev M, Ashany D, Marti F, et al. Impaired T cell death and lupus-like autoimmunity in T cell-specific adapter protein-deficient mice. J Exp Med (2003) 198(5):809–21. doi: 10.1084/jem.20021358
416. Xu Z, Xu J, Ju J, Morel L. A Skint6 allele potentially contributes to mouse lupus. Genes Immun (2017) 18(3):111–7. doi: 10.1038/gene.2017.8
417. Boackle SA, Holers VM, Chen X, Szakonyi G, Karp DR, Wakeland EK, et al. Cr2, a candidate gene in the murine Sle1c lupus susceptibility locus, encodes a dysfunctional protein. Immunity (2001) 15(5):775–85. doi: 10.1016/s1074-7613(01)00228-x
418. Cuda CM, Zeumer L, Sobel ES, Croker BP, Morel L. Murine lupus susceptibility locus Sle1a requires the expression of two sub-loci to induce inflammatory T cells. Genes Immun (2010) 11(7):542–53. doi: 10.1038/gene.2010.23
419. Cuda CM, Li S, Liang S, Yin Y, Potula HH, Xu Z, et al. Pre-b cell leukemia homeobox 1 is associated with lupus susceptibility in mice and humans. J Immunol (2012) 188(2):604–14. doi: 10.4049/jimmunol.1002362
420. Chen Y, Perry D, Boackle SA, Sobel ES, Molina H, Croker BP, et al. Several genes contribute to the production of autoreactive b and T cells in the murine lupus susceptibility locus Sle1c. J Immunol (2005) 175(2):1080–9. doi: 10.4049/jimmunol.175.2.1080
421. Zeumer L, Sang A, Niu H, Morel L. Murine lupus susceptibility locus Sle2 activates DNA-reactive b cells through two sub-loci with distinct phenotypes. Genes Immun (2011) 12(3):199–207. doi: 10.1038/gene.2010.69
422. Mohan C, Morel L, Yang P, Wakeland EK. Genetic dissection of systemic lupus erythematosus pathogenesis: Sle2 on murine chromosome 4 leads to b cell hyperactivity. J Immunol (1997) 159(1):454–65.
423. Xu Z, Duan B, Croker BP, Wakeland EK, Morel L. Genetic dissection of the murine lupus susceptibility locus Sle2: contributions to increased peritoneal b-1a cells and lupus nephritis map to different loci. J Immunol (2005) 175(2):936–43. doi: 10.4049/jimmunol.175.2.936
424. Mohan C, Yu Y, Morel L, Yang P, Wakeland EK. Genetic dissection of sle pathogenesis: Sle3 on murine chromosome 7 impacts T cell activation, differentiation, and cell death. J Immunol (1999) 162(11):6492–502.
425. Xu Z, Duan B, Croker BP, Morel L. STAT4 deficiency reduces autoantibody production and glomerulonephritis in a mouse model of lupus. Clin Immunol (2006) 120(2):189–98. doi: 10.1016/j.clim.2006.03.009
426. Dang H, Geiser AG, Letterio JJ, Nakabayashi T, Kong L, Fernandes G, et al. SLE-like autoantibodies and sjögren's syndrome-like lymphoproliferation in TGF-beta knockout mice. J Immunol (1995) 155(6):3205–12.
427. Geiser AG, Letterio JJ, Kulkarni AB, Karlsson S, Roberts AB, Sporn MB, et al. Transforming growth factor beta 1 (TGF-beta 1) controls expression of major histocompatibility genes in the postnatal mouse: aberrant histocompatibility antigen expression in the pathogenesis of the TGF-beta 1 null mouse phenotype. Proc Natl Acad Sci USA (1993) 90(21):9944–8. doi: 10.1073/pnas.90.21.9944
428. Debreceni IL, Chimenti MS, Serreze DV, Geurts AM, Chen YG, Lieberman SM, et al. Toll-like receptor 7 is required for lacrimal gland autoimmunity and type 1 diabetes development in Male nonobese diabetic mice. Int J Mol Sci 21(24):9478, doi: 10.3390/ijms21249478 (2020).
429. Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA (2006) 103(26):9970–5. doi: 10.1073/pnas.0603912103
430. Fairhurst AM, Hwang SH, Wang A, Tian XH, Boudreaux C, Zhou XJ, et al. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol (2008) 38(7):1971–8. doi: 10.1002/eji.200838138
431. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S, et al. Autoreactive b cell responses to RNA-related antigens due to TLR7 gene duplication. Science (2006) 312(5780):1669–72. doi: 10.1126/science.1124978
432. Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ, et al. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med (2005) 202(2):321–31. doi: 10.1084/jem.20050338
433. Alankus B, Ecker V, Vahl N, Braun M, Weichert W, Macher-Göppinger S, et al. Pathological RANK signaling in b cells drives autoimmunity and chronic lymphocytic leukemia. J Exp Med (2021) 218(2)::e20200517. doi: 10.1084/jem.20200517
434. Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, Grewal IS, et al. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity (2003) 18(2):279–88. doi: 10.1016/s1074-7613(03)00025-6
435. Boneparth A, Woods M, Huang W, Akerman M, Lesser M, Davidson A, et al. The effect of BAFF inhibition on autoreactive b-cell selection in murine systemic lupus erythematosus. Mol Med (2016) 22:173–82. doi: 10.2119/molmed.2016.00022
436. Huang W, Moisini I, Bethunaickan R, Sahu R, Akerman M, Eilat D, et al. BAFF/APRIL inhibition decreases selection of naive but not antigen-induced autoreactive b cells in murine systemic lupus erythematosus. J Immunol (2011) 187(12):6571–80. doi: 10.4049/jimmunol.1101784
437. Forsberg MH, Foda B, Serreze DV, Chen YG. Combined congenic mapping and nuclease-based gene targeting for studying allele-specific effects of Tnfrsf9 within the Idd9.3 autoimmune diabetes locus. Sci Rep (2019) 9(1):4316. doi: 10.1038/s41598-019-40898-8
438. Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med (1999) 190(11):1697–710. doi: 10.1084/jem.190.11.1697
439. Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates b cell growth. J Exp Med (1999) 189(11):1747–56. doi: 10.1084/jem.189.11.1747
440. Gorelik L, Gilbride K, Dobles M, Kalled SL, Zandman D, Scott ML, et al. Normal b cell homeostasis requires b cell activation factor production by radiation-resistant cells. J Exp Med (2003) 198(6):937–45. doi: 10.1084/jem.20030789
441. Foda BM, Ciecko AE, Serreze DV, Ridgway WM, Geurts AM, Chen YG, et al. The CD137 ligand is important for type 1 diabetes development but dispensable for the homeostasis of disease-suppressive CD137(+) FOXP3(+) regulatory CD4 T cells. J Immunol (2020) 204(11):2887–99. doi: 10.4049/jimmunol.1900485
442. Chen Z, Krinsky A, Woolaver RA, Wang X, Chen SMY, Popolizio V, et al. TRAF3 acts as a checkpoint of b cell receptor signaling to control antibody class switch recombination and anergy. J Immunol (2020) 205(3):830–41. doi: 10.4049/jimmunol.2000322
443. Li X. Act1 modulates autoimmunity through its dual functions in CD40L/BAFF and IL-17 signaling. Cytokine (2008) 41(2):105–13. doi: 10.1016/j.cyto.2007.09.015
444. Qian Y, Giltiay N, Xiao J, Wang Y, Tian J, Han S, et al. Deficiency of Act1, a critical modulator of b cell function, leads to development of sjögren's syndrome. Eur J Immunol (2008) 38(8):2219–28. doi: 10.1002/eji.200738113
445. Johnson AC, Davison LM, Giltiay NV, Vareechon C, Li X, Jørgensen TN, et al. Lack of T cells in Act1-deficient mice results in elevated IgM-specific autoantibodies but reduced lupus-like disease. Eur J Immunol (2012) 42(7):1695–705. doi: 10.1002/eji.201142238
446. Zhang CJ, Wang C, Jiang M, Gu C, Xiao J, Chen X, et al. Act1 is a negative regulator in T and b cells via direct inhibition of STAT3. Nat Commun (2018) 9(1):2745. doi: 10.1038/s41467-018-04974-3
447. Pisitkun P, Ha HL, Wang H, Claudio E, Tivy CC, Zhou H, et al. Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity (2012) 37(6):1104–15. doi: 10.1016/j.immuni.2012.08.014
448. Espinosa A, Dardalhon V, Brauner S, Ambrosi A, Higgs R, Quintana FJ, et al. Loss of the lupus autoantigen Ro52/Trim21 induces tissue inflammation and systemic autoimmunity by disregulating the IL-23-Th17 pathway. J Exp Med (2009) 206(8):1661–71. doi: 10.1084/jem.20090585
449. Nakano-Yokomizo T, Tahara-Hanaoka S, Nakahashi-Oda C, Nabekura T, Tchao NK, Kadosaki M, et al. The immunoreceptor adapter protein DAP12 suppresses b lymphocyte-driven adaptive immune responses. J Exp Med (2011) 208(8):1661–71. doi: 10.1084/jem.20101623
450. Chen YG, Ciecko AE, Khaja S, Grzybowski M, Geurts AM, Lieberman SM, et al. UBASH3A deficiency accelerates type 1 diabetes development and enhances salivary gland inflammation in NOD mice. Sci Rep (2020) 10:12019. doi: 10.1038/s41598-020-68956-6
451. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res (2019) 47(D1):D607–13. doi: 10.1093/nar/gky1131
452. Tachmazidou I, Hatzikotoulas K, Southam L, Esparza-Gordillo J, Haberland V, Zheng J, et al. Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK biobank data. Nat Genet (2019) 51(2):230–6. doi: 10.1038/s41588-018-0327-1
453. Vujkovic M, Keaton JM, Lynch JA, Miller DR, Zhou J, Tcheandjieu C, et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet (2020) 52(7):680–91. doi: 10.1038/s41588-020-0637-y
454. Hou G, Harley ITW, Lu X, Zhou T, Xu N, Yao C, et al. SLE non-coding genetic risk variant determines the epigenetic dysfunction of an immune cell specific enhancer that controls disease-critical microRNA expression. Nat Commun (2021) 12:135. doi: 10.1038/s41467-020-20460-1
455. Coke LN, Wen H, Comeau M, Ghanem MH, Shih A, Metz CN, et al. Arg206Cys substitution in DNASE1L3 causes a defect in DNASE1L3 protein secretion that confers risk of systemic lupus erythematosus. Ann Rheum Dis (2021) 80(6):782–7. doi: 10.1136/annrheumdis-2020-218810
456. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet (2011) 43(12):1186–8. doi: 10.1038/ng.975
457. Carbonella A, Mancano G, Gremese E, Alkuraya FS, Patel N, Gurrieri F, et al. An autosomal recessive DNASE1L3-related autoimmune disease with unusual clinical presentation mimicking systemic lupus erythematosus. Lupus (2017) 26:768–72. doi: 10.1177/0961203316676382
458. Batu ED, Koşukcu C, Taşkıran E, Sahin S, Akman S, Sözeri B, et al. Whole exome sequencing in early-onset systemic lupus erythematosus. J Rheumatol (2018) 45(12):1671–9. doi: 10.3899/jrheum.171358
459. Hartl J, Serpas L, Wang Y, Rashidfarrokhi A, Perez OA, Sally B, et al. Autoantibody-mediated impairment of DNASE1L3 activity in sporadic systemic lupus erythematosus. J Exp Med (2021) 218(5):e20201138. doi: 10.1084/jem.20201138
460. Soni C, Perez OA, Voss WN, Pucella JN, Serpas L, Mehl J, et al. Plasmacytoid dendritic cells and type I interferon promote extrafollicular b cell responses to extracellular self-DNA. Immunity (2020) 52(6):1022–1038.e1027. doi: 10.1016/j.immuni.2020.04.015
461. Wan Z, Pascual V. Breaching self-tolerance by targeting the gatekeeper. J Exp Med (2021) 218(5):e20210322. doi: 10.1084/jem.20210322
462. Sisirak V, Sally B, D'Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell (2016) 166(1):88–101. doi: 10.1016/j.cell.2016.05.034
463. Vaughn SE, Foley C, Lu X, Patel ZH, Zoller EE, Magnusen AF, et al. Lupus risk variants in the PXK locus alter b-cell receptor internalization. Front Genet (2014) 5:450. doi: 10.3389/fgene.2014.00450
464. Chiou J, Geusz RJ, Okino ML, Han JY, Miller M, Melton R, et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature (2021) 594(7863):398–402. doi: 10.1038/s41586-021-03552-w
465. Maiti AK, Kim-Howard X, Motghare P, Pradhan V, Chua KH, Sun C, et al. Combined protein- and nucleic acid-level effects of rs1143679 (R77H), a lupus-predisposing variant within ITGAM. Hum Mol Genet (2014) 23(15):4161–76. doi: 10.1093/hmg/ddu106
466. Rawlings DJ, Dai X, Buckner JH. The role of PTPN22 risk variant in the development of autoimmunity: finding common ground between mouse and human. J Immunol (2015) 194(7):2977–84. doi: 10.4049/jimmunol.1403034
467. Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y. Hydroxychloroquine: from malaria to autoimmunity. Clin Rev Allergy Immunol (2012) 42(2):145–53. doi: 10.1007/s12016-010-8243-x
468. Chan CC, Harley ITW, Pfluger PT, Trompette A, Stankiewicz TE, Allen JL, et al. A BAFF/APRIL axis regulates obesogenic diet-driven weight gain. Nat Commun (2021) 12(1):2911. doi: 10.1038/s41467-021-23084-1
469. Charles N, Hardwick D, Daugas E, Illei GG, Rivera J. Basophils and the T helper 2 environment can promote the development of lupus nephritis. Nat Med (2010) 16(6):701–7. doi: 10.1038/nm.2159
470. Wu C, Jin X, Tsueng G, Afrasiabi C, Su AI. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res (2016) 44(D1):D313–316. doi: 10.1093/nar/gkv1104
471. Mabbott NA, Baillie JK, Brown H, Freeman TC, Hume DA. An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genomics (2013) 14:632. doi: 10.1186/1471-2164-14-632
472. Chelsea Gootjes JJZ, Bart O. Roep and tatjana nikolic. functional impact of risk gene variants on the autoimmune responses in type 1 diabetes. Front Immunol (2022) 13:886736. doi: 10.3389/fimmu.2022.886736
473. Harley IT, Giles DA, Pfluger PT, Burgess SL, Walters S, Hembree J, et al. Differential colonization with segmented filamentous bacteria and lactobacillus murinus do not drive divergent development of diet-induced obesity in C57BL/6 mice. Mol Metab (2013) 2(3):171–83. doi: 10.1016/j.molmet.2013.04.004
474. Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet (2012) 44:740–2. doi: 10.1038/ng.2299
475. Sun Q, Scott MJ. Caspase-1 as a multifunctional inflammatory mediator: noncytokine maturation roles. J Leukoc Biol (2016) 100:961–7. doi: 10.1189/jlb.3MR0516-224R
476. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
477. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science (2008) 321:691–6. doi: 10.1126/science.1158298
478. Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, et al. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol (2006) 177:2770–4. doi: 10.4049/jimmunol.177.5.2770
479. Xiao R, Ali S, Caligiuri MA, Cao L. Enhancing effects of environmental enrichment on the functions of natural killer cells in mice. Front Immunol (2021) 12:695859. doi: 10.3389/fimmu.2021.695859
480. Liu C, Yang Y, Chen C, Li L, Li J, Wang X, et al. Environmental eustress modulates beta-ARs/CCL2 axis to induce anti-tumor immunity and sensitize immunotherapy against liver cancer in mice. Nat Commun (2021) 12:5725. doi: 10.1038/s41467-021-25967-9
481. Karp CL. Unstressing intemperate models: how cold stress undermines mouse modeling. J Exp Med (2012) 209:1069–74. doi: 10.1084/jem.20120988
482. Noah TK, Lee JB, Brown CA, Yamani A, Tomar S, Ganesan V, et al. Thermoneutrality alters gastrointestinal antigen passage patterning and predisposes to oral antigen sensitization in mice. Front Immunol (2021) 12:636198. doi: 10.3389/fimmu.2021.636198
483. Giles DA, Moreno-Fernandez ME, Stankiewicz TE, Graspeuntner S, Cappelletti M, Wu D, et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nat Med (2017) 23:829–38. doi: 10.1038/nm.4346
484. Stemmer K, Kotzbeck P, Zani F, Bauer M, Neff C, Muller TD, et al. Thermoneutral housing is a critical factor for immune function and diet-induced obesity in C57BL/6 nude mice. Int J Obes (Lond) (2015) 39:791–7. doi: 10.1038/ijo.2014.187
485. Giles DA, Ramkhelawon B, Donelan EM, Stankiewicz TE, Hutchison SB, Mukherjee R, et al. Modulation of ambient temperature promotes inflammation and initiates atherosclerosis in wild type C57BL/6 mice. Mol Metab (2016) 5:1121–30. doi: 10.1016/j.molmet.2016.09.008
486. Breit S, Kupferberg A, Rogler G, Hasler G. Vagus nerve as modulator of the brain-gut axis in psychiatric and inflammatory disorders. Front Psychiatry (2018) 9:44. doi: 10.3389/fpsyt.2018.00044
487. Koopman FA, Chavan SS, Miljko S, Grazio S, Sokolovic S, Schuurman PR, et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc Natl Acad Sci USA (2016) 113:8284–9. doi: 10.1073/pnas.1605635113
488. Drewes AM, Brock C, Rasmussen SE, Moller HJ, Brock B, Deleuran BW, et al. Short-term transcutaneous non-invasive vagus nerve stimulation may reduce disease activity and pro-inflammatory cytokines in rheumatoid arthritis: results of a pilot study. Scand J Rheumatol (2021) 50:20–7. doi: 10.1080/03009742.2020.1764617
489. Sangle SR, Tench CM, D'Cruz DP. Autoimmune rheumatic disease and sleep: a review. Curr Opin Pulm Med (2015) 21:553–6. doi: 10.1097/MCP.0000000000000215
490. Young KA, Munroe ME, Harley JB, Guthridge JM, Kamen DL, Gilkensen GS, et al. Less than 7 hours of sleep per night is associated with transitioning to systemic lupus erythematosus. Lupus (2018) 27:1524–31. doi: 10.1177/0961203318778368
491. Palma BD, Tufik S. Increased disease activity is associated with altered sleep architecture in an experimental model of systemic lupus erythematosus. Sleep (2010) 33:1244–8. doi: 10.1093/sleep/33.9.1244
492. Palma BD, Hipolide DC, Tufik S. Effects on prolactin secretion and binding to dopaminergic receptors in sleep-deprived lupus-prone mice. Braz J Med Biol Res (2009) 42:299–304. doi: 10.1590/s0100-879x2009000300012
493. Zhang C, Franklin CL, Ericsson AC. Consideration of gut microbiome in murine models of diseases. Microorganisms (2021) 9:1062. doi: 10.3390/microorganisms9051062
494. Zhang C, Burch M, Wylie K, Herter B, Franklin CL, Ericsson AC. Characterization of the eukaryotic virome of mice from different sources. Microorganisms (2021) 9(10):2064. doi: 10.3390/microorganisms9102064
495. Coughlan L. Caught in a trap: How pre-clinical studies in laboratory mice exaggerate vaccine responses. Cell Rep Med (2021) 2:100484. doi: 10.1016/j.xcrm.2021.100484
496. Fay EJ, Balla KM, Roach SN, Shepherd FK, Putri DS, Wiggen TD, et al. Natural rodent model of viral transmission reveals biological features of virus population dynamics. J Exp Med (2022) 219(2):e20211220. doi: 10.1084/jem.20211220
497. Huggins MA, Sjaastad FV, Pierson M, Kucaba TA, Swanson W, Staley C, et al. Microbial exposure enhances immunity to pathogens recognized by TLR2 but increases susceptibility to cytokine storm through TLR4 sensitization. Cell Rep (2019) 28:1729–1743.e1725. doi: 10.1016/j.celrep.2019.07.028
498. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature (2016) 532:512–6. doi: 10.1038/nature17655
499. LabDiet. Standard diets (2022). [Website]. St. Louis, MO 63144(2022) [cited 2022 2/23/2022]. Available at: https://www.labdiet.com/Products/StandardDiets/index.html.
500. LabDiet. Laboratory rodent diet (2021). Available at: https://www.labdiet.com/cs/groups/lolweb/@labdiet/documents/web_content/mdrf/mdi4/~edisp/ducm04_028021.pdf.
501. Malinow MR, Bardana EJ Jr., Pirofsky B, Craig S, McLaughlin P. Systemic lupus erythematosus-like syndrome in monkeys fed alfalfa sprouts: role of a nonprotein amino acid. Science (1982) 216:415–7. doi: 10.1126/science.7071589
502. Akaogi J, Barker T, Kuroda Y, Nacionales DC, Yamasaki Y, Stevens BR, et al. Role of non-protein amino acid l-canavanine in autoimmunity. Autoimmun Rev (2006) 5:429–35. doi: 10.1016/j.autrev.2005.12.004
503. Johns Hopkins Lupus Center. 5 things to avoid if you have lupus. Available at: https://www.hopkinslupus.org/lupus-info/lifestyle-additional-information/avoid/.
504. Antonini L, Le Mauff B, Marcelli C, Aouba A, de Boysson H. Rhupus: a systematic literature review. Autoimmun Rev (2020) 19:102612. doi: 10.1016/j.autrev.2020.102612
505. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351:711–4. doi: 10.1126/science.aad2791
506. Madanay FL, McDevitt RC, Ubel PA. Hydroxychloroquine for COVID-19: Variation in regional political preferences predicted new prescriptions after President trump's endorsement. J Health Polit Policy Law (2022) 47(4):429–51. doi: 10.1215/03616878-9716698
507. Wolf SJ, Estadt SN, Theros J, Moore T, Ellis J, Liu J, et al. Ultraviolet light induces increased T cell activation in lupus-prone mice via type I IFN-dependent inhibition of T regulatory cells. J Autoimmun (2019) 103:102291. doi: 10.1016/j.jaut.2019.06.002
508. Zou J, Thornton C, Chambers ES, Rosser EC, Ciurtin C. Exploring the evidence for an immunomodulatory role of vitamin d in juvenile and adult rheumatic disease. Front Immunol (2020) 11:616483. doi: 10.3389/fimmu.2020.616483
509. Hahn J, Cook NR, Alexander EK, Friedman S, Walter J, Bubes V, et al. Vitamin d and marine omega 3 fatty acid supplementation and incident autoimmune disease: VITAL randomized controlled trial. Bmj (2022) 376:e066452. doi: 10.1136/bmj-2021-066452
510. Ritterhouse LL, Crowe SR, Niewold TB, Kamen DL, Macwana SR, Roberts VC, et al. Vitamin d deficiency is associated with an increased autoimmune response in healthy individuals and in patients with systemic lupus erythematosus. Ann Rheum Dis (2011) 70:1569–74. doi: 10.1136/ard.2010.148494
511. Ritterhouse LL, Lu R, Shah HB, Robertson JM, Fife DA, Maecker HT, et al. Vitamin d deficiency in a multiethnic healthy control cohort and altered immune response in vitamin d deficient European-American healthy controls. PloS One (2014) 9:e94500. doi: 10.1371/journal.pone.0094500
512. Zomer HD, Trentin AG. Skin wound healing in humans and mice: Challenges in translational research. J Dermatol Sci (2018) 90:3–12. doi: 10.1016/j.jdermsci.2017.12.009
513. Gangwar RS, Gudjonsson JE, Ward NL. Mouse models of psoriasis: A comprehensive review. J Invest Dermatol (2022) 142:884–97. doi: 10.1016/j.jid.2021.06.019
514. Gudjonsson JE, Johnston A, Dyson M, Valdimarsson H, Elder JT. Mouse models of psoriasis. J Invest Dermatol (2007) 127:1292–308. doi: 10.1038/sj.jid.5700807
515. Gerber PA, Buhren BA, Schrumpf H, Homey B, Zlotnik A, Hevezi P. The top skin-associated genes: a comparative analysis of human and mouse skin transcriptomes. Biol Chem (2014) 395:577–91. doi: 10.1515/hsz-2013-0279
516. Harley JB, Harley IT, Guthridge JM, James JA. The curiously suspicious: a role for Epstein-Barr virus in lupus. Lupus (2006) 15:768–77. doi: 10.1177/0961203306070009
517. Ungerleider NA, Jain V, Wang Y, Maness NJ, Blair RV, Alvarez X, et al. Comparative analysis of gammaherpesvirus circular RNA repertoires: Conserved and unique viral circular RNAs. J Virol (2019) 93(6):e01952–18. doi: 10.1128/JVI.01952-18
518. Lowe D. The latest on drug failure and approval rates (2019). Available at: https://www.science.org/content/blog-post/latest-drug-failure-and-approval-rates.
519. Dowden H, Munro J. Trends in clinical success rates and therapeutic focus. Nat Rev Drug Discov (2019) 18:495–6. doi: 10.1038/d41573-019-00074-z
520. Smietana K, Siatkowski M, Moller M. Trends in clinical success rates. Nat Rev Drug Discov (2016) 15:379–80. doi: 10.1038/nrd.2016.85
521. Kendall PL, Case JB, Sullivan AM, Holderness JS, Wells KS, Liu E, et al. Tolerant anti-insulin b cells are effective APCs. J Immunol (2013) 190:2519–26. doi: 10.4049/jimmunol.1202104
522. Felton JL, Maseda D, Bonami RH, Hulbert C, Thomas JW. Anti-insulin b cells are poised for antigen presentation in type 1 diabetes. J Immunol (2018) 201:861–73. doi: 10.4049/jimmunol.1701717
523. Packard TA, Smith MJ, Conrad FJ, Johnson SA, Getahun A, Lindsay RS, et al. B cell receptor affinity for insulin dictates autoantigen acquisition and b cell functionality in autoimmune diabetes. J Clin Med (2016) 5(11):98. doi: 10.3390/jcm5110098
524. Serreze DV, Silveira PA. The role of b lymphocytes as key antigen-presenting cells in the development of T cell-mediated autoimmune type 1 diabetes. Curr Dir Autoimmun (2003) 6:212–27. doi: 10.1159/000066863
525. Mariño E, Tan B, Binge L, Mackay CR, Grey ST. B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes (2012) 61:2893–905. doi: 10.2337/db12-0006
526. Wang Q, Racine JJ, Ratiu JJ, Wang S, Ettinger R, Wasserfall C, et al. Transient BAFF blockade inhibits type 1 diabetes development in nonobese diabetic mice by enriching immunoregulatory b lymphocytes sensitive to deletion by anti-CD20 cotherapy. J Immunol (2017) 199:3757–70. doi: 10.4049/jimmunol.1700822
527. Leeth CM, Racine J, Chapman HD, Arpa B, Carrillo J, Carrascal J, et al. B-lymphocytes expressing an ig specificity recognizing the pancreatic ss-cell autoantigen peripherin are potent contributors to type 1 diabetes development in NOD mice. Diabetes (2016) 65:1977–87. doi: 10.2337/db15-1606
528. Hu CY, Rodriguez-Pinto D, Du W, Ahuja A, Henegariu O, Wong FS, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest (2007) 117:3857–67. doi: 10.1172/JCI32405
529. Habib T, Long SA, Samuels PL, Brahmandam A, Tatum M, Funk A, et al. Dynamic immune phenotypes of b and T helper cells mark distinct stages of T1D progression. Diabetes (2019) 68:1240–50. doi: 10.2337/db18-1081
530. Bluestone JA, Buckner JH, Herold KC. Immunotherapy: Building a bridge to a cure for type 1 diabetes. Science (2021) 373:510–6. doi: 10.1126/science.abh1654
531. Smith MJ, Rihanek M, Wasserfall C, Mathews CE, Atkinson MA, Gottlieb PA, et al. Loss of b-cell anergy in type 1 diabetes is associated with high-risk HLA and non-HLA disease susceptibility alleles. Diabetes (2018) 67:697–703. doi: 10.2337/db17-0937
532. Sudhir PR, Lin TD, Zhang Q. HLA allele-specific quantitative profiling of type 1 diabetic b lymphocyte immunopeptidome. J Proteome Res (2022) 21:250–64. doi: 10.1021/acs.jproteome.1c00842
533. Smith MJ, Ford BR, Rihanek M, Coleman BM, Getahun A, Sarapura VD, et al. Elevated PTEN expression maintains anergy in human b cells and reveals unexpectedly high repertoire autoreactivity. JCI Insight (2019) 4(3):e123384. doi: 10.1172/jci.insight.123384
534. Piekos SN, Gaddam S, Bhardwaj P, Radhakrishnan P, Guha RV, Oro AE. Biomedical data commons (BMDC) prioritizes b-lymphocyte non-coding genetic variants in type 1 diabetes. PloS Comput Biol (2021) 17:e1009382. doi: 10.1371/journal.pcbi.1009382
535. Rojas M, Ramírez-Santana C, Acosta-Ampudia Y, Monsalve DM, Rodriguez-Jimenez M, Zapata E, et al. New insights into the taxonomy of autoimmune diseases based on polyautoimmunity. J Autoimmun (2022) 126:102780. doi: 10.1016/j.jaut.2021.102780
536. Molano-González N, Rojas M, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Rodríguez Y, et al. Cluster analysis of autoimmune rheumatic diseases based on autoantibodies. new insights for polyautoimmunity. J Autoimmun (2019) 98:24–32. doi: 10.1016/j.jaut.2018.11.002
537. Barturen G, Babaei S, Català-Moll F, Martínez-Bueno M, Makowska Z, Martorell-Marugán J, et al. Integrative analysis reveals a molecular stratification of systemic autoimmune diseases. Arthritis Rheumatol (2021) 73:1073–85. doi: 10.1002/art.41610
538. Soret P, Le Dantec C, Desvaux E, Foulquier N, Chassagnol B, Hubert S, et al. A new molecular classification to drive precision treatment strategies in primary sjögren's syndrome. Nat Commun (2021) 12:3523. doi: 10.1038/s41467-021-23472-7
539. Barturen G, Beretta L, Cervera R, Van Vollenhoven R, Alarcón-Riquelme ME. Moving towards a molecular taxonomy of autoimmune rheumatic diseases. Nat Rev Rheumatol (2018) 14:75–93. doi: 10.1038/nrrheum.2017.220
540. Acosta-Herrera M, Kerick M, González-Serna D, Wijmenga C, Franke A, Gregersen PK, et al. Genome-wide meta-analysis reveals shared new loci in systemic seropositive rheumatic diseases. Ann Rheum Dis (2019) 78:311–9. doi: 10.1136/annrheumdis-2018-214127
Keywords: systemic lupus erythematosus (SLE), autoimmune type 1 diabetes mellitus (T1D), polygenic, monogenic, genome-wide association study (GWAS), autoimmune disease mouse model, central and peripheral tolerance (anergy), B cell receptor (BCR) signaling pathway
Citation: Harley ITW, Allison K and Scofield RH (2022) Polygenic autoimmune disease risk alleles impacting B cell tolerance act in concert across shared molecular networks in mouse and in humans. Front. Immunol. 13:953439. doi: 10.3389/fimmu.2022.953439
Received: 26 May 2022; Accepted: 19 July 2022;
Published: 24 August 2022.
Edited by:
David Serreze, Jackson Laboratory, United StatesReviewed by:
Kay L. Medina, Mayo Clinic, United StatesCopyright © 2022 Harley, Allison and Scofield. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isaac T. W. Harley, aXNhYWMuaGFybGV5QGN1YW5zY2h1dHouZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.