 Davide Scozzi
Davide Scozzi Fuyi Liao1
Fuyi Liao1 Daniel Kreisel
Daniel Kreisel- 1Department of Surgery, Washington University in St. Louis, St. Louis, MO, United States
- 2Department of Surgery, University of Maryland, Baltimore, MD, United States
- 3Department of Pathology and Immunology, Washington University in St. Louis, St. Louis, MO, United States
Acute lung injury (ALI) is a heterogeneous inflammatory condition associated with high morbidity and mortality. Neutrophils play a key role in the development of different forms of ALI, and the release of neutrophil extracellular traps (NETs) is emerging as a common pathogenic mechanism. NETs are essential in controlling pathogens, and their defective release or increased degradation leads to a higher risk of infection. However, NETs also contain several pro-inflammatory and cytotoxic molecules than can exacerbate thromboinflammation and lung tissue injury. To reduce NET-mediated lung damage and inflammation, DNase is frequently used in preclinical models of ALI due to its capability of digesting NET DNA scaffold. Moreover, recent advances in neutrophil biology led to the development of selective NET inhibitors, which also appear to reduce ALI in experimental models. Here we provide an overview of the role of NETs in different forms of ALI discussing existing gaps in our knowledge and novel therapeutic approaches to modulate their impact on lung injury.
Introduction
ALI is an inflammatory condition characterized by the acute onset of lung tissue damage and pulmonary dysfunction originating from infectious or sterile insults (1). Typical features of ALI are the alveolar accumulation of protein-rich fluid and activated immune cells due to pulmonary endothelial barrier disruption and increased vascular permeability (2). The pathological alterations of ALI are responsible for a clinical syndrome characterized by extensive non-cardiogenic pulmonary edema and decreasing oxygenation, also known as acute respiratory distress syndrome (ARDS) (3). ARDS is associated with high morbidity and mortality (~40%), and its increasing incidence, particularly related to the recent COVID-19 pandemic, represents a significant global burden (4).
Airway neutrophilia has been historically considered a hallmark of ARDS (5). However, the underlying mechanisms that control neutrophil contribution to ALI are not fully understood. Although neutrophils play well-established functions in regulating pulmonary injury through the generation of reactive oxygen species (ROS) or conducting phagocytosis and degranulation, recent reports have highlighted a critical role for NETosis in ALI pathogenesis. NETs consist of a mix of nuclear chromatin (6), mitochondrial DNA (7–9) and neutrophil granule proteins (10) that primarily absolve a defensive role against lung infections. On the other hand, accumulating evidence indicate that NETosis is also increased in lung sterile inflammatory conditions and that exuberant NET release promotes microvascular dysfunction, thromboinflammation, and direct cellular injury (11). Moreover, high levels of NETs in the peripheral blood or bronchoalveolar lavage (BAL) of critically ill subjects are frequently associated with the worst ARDS outcomes (12, 13).
In this review, we will discuss general mechanisms of NETosis and NET-mediated tissue damage (Figure 1) with a specific focus on the contribution of NETs to different forms of infective and sterile ALI (Figure 2).
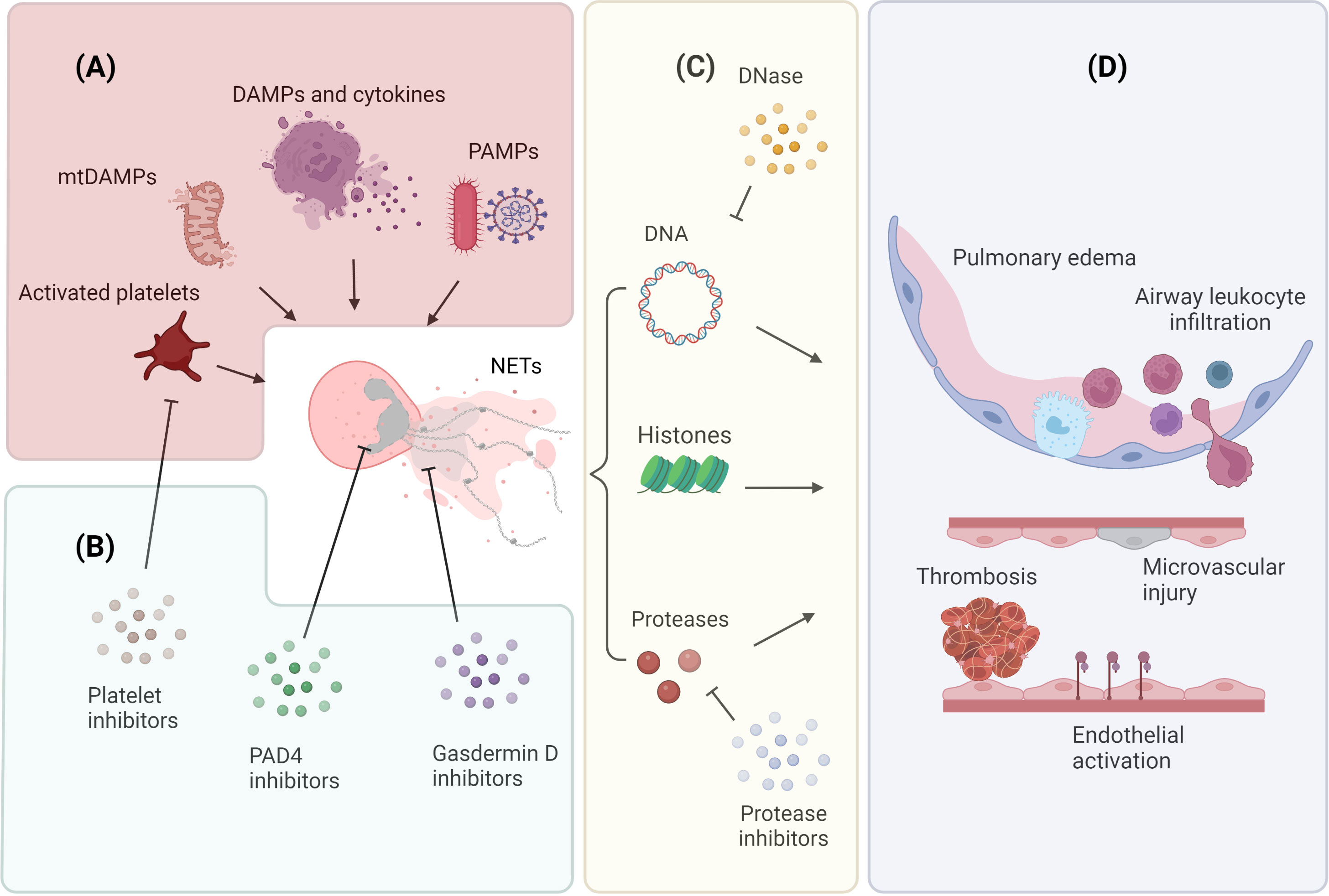
Figure 1 Mechanisms of NET release and NET-mediated lung injury (A) Neutrophils release NETs in response to endogenous and exogenous stimuli. Endogenous factors include DAMPs, pro-inflammatory cytokines, mtDAMPs, and molecules released by activated platelets; exogenous factors include PAMPs associated with microbial infections. (B) Inhibition of NET generation and release. The contribution of platelets to NETosis can be attenuated by using platelet activation inhibitors; neutrophil chromatin decondensation can be targeted by using PAD4 inhibitors; neutrophil membrane permeabilization can be prevented by using gasderimin D inhibitors. (C) NETs comprise a DNA scaffold decorated with granule proteases and histone proteins. NET DNA scaffold can be digested by DNase; NET proteolytic activity can be abrogated by specific protease inhibitors. (D) NETs release contributes to the pathogenesis of ALI. NETs facilitate the formation of thrombi, promote endothelial cell activation, and induce microvascular injury. These microvascular alterations result in increased vascular permeability, intra-alveolar accumulation of protein-rich fluid, and infiltration of inflammatory cells. Image created by DS using BioRender (https://biorender.com/).
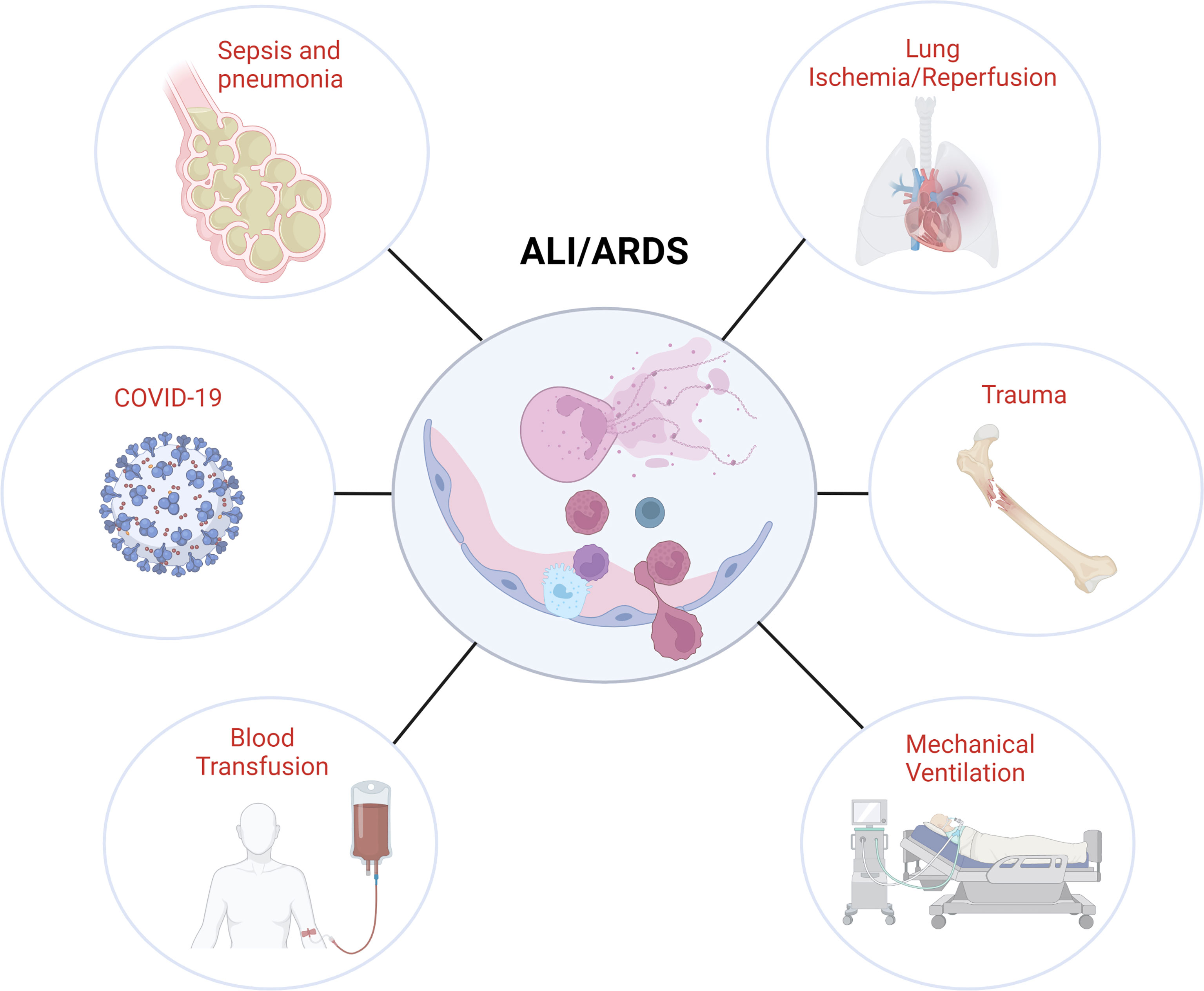
Figure 2 Association of NETs with different ALI/ARDS etiologies. Image created by DS using BioRender (https://biorender.com/).
Non-vital and vital NETosis
NETosis was first described in 2004 when Brinkman and coworkers observed that activated neutrophils could kill bacteria by releasing nuclear chromatin decorated with proteins usually confined to their granules (14). Following studies characterized the NADPH oxidase 2 (NOX2) Ros-dependent pathway involved in the release of NETs and defined NETosis as a new mechanism of neutrophil programmed cell death distinct from apoptosis and necrosis (15, 16). NETosis was originally described as a slow multistep process that leads directly to senescence, where chromatin decondensation and nuclear envelope rupture allow for mixing nuclear material with cytoplasm content. Pore-forming proteins like gasdermin D then permeabilize the plasma membrane allowing for the release of NETs into the extracellular space (17). However, this view of NETosis as a terminal process has been recently challenged by the demonstration of an alternative, non-lytic and NADPH-independent mechanism of NET release, which appears to be particularly rapid in response to bacterial infection and leaves behind a neutrophil that can still exclude vital dyes (18). This form of NETosis is initiated by a sudden rise in intracellular calcium that leads to the expulsion of nuclear chromatin and granule proteins, resulting in an anucleated cytoplast still capable of migration and phagocytosis (11, 18, 19). Finally, a third type of fast, vital and ROS-dependent NETosis was recently described in response to granulocyte-macrophage colony-stimulating factor or lipopolysaccharide (LPS) leading to the release of mitochondrial DNA (mtDNA) mixed with granule proteins (7).
Almost two decades after the first observations, it is now commonly accepted that NET release can occur in response to multiple stimuli via several and frequently interconnected activation pathways (17, 20–22) that are not necessarily associated with neutrophil death (7, 18, 19). Regardless of the stimuli and the activation mechanisms, NET composition has intrinsic pro-inflammatory characteristics due to the presence of granule-derived proteases, histones and cell-free DNA (23).
NET composition
Serine proteases
Under homeostatic conditions, neutrophil azurophilic granules contain different serine proteases, including neutrophil elastase (NE), cathepsin G (CatG), proteinase 3 (PR3), and neutrophil serine protease 4 (NSP4). Due to their antimicrobial and immunomodulatory function, these pre-stored catalytically active mediators play a major contribution in the physiological response to infection (24). However, their uncontrolled extracellular release may have unintended consequences by causing damage to the surrounding healthy tissue (25). Proteomic analysis and studies on the functional activity of human NETs indicate that NE is the most abundant non-histone protein and the predominant responsible for the NET “proteolytic signature” (10, 26). NE is a well-established mediator of alveolo-capillary permeability where it is thought to trigger microvascular injury through primarily catalyzing endothelial cell cadherin proteolysis (27–29). In a mouse model of LPS induced endotoxemia, treatment with the NE selective inhibitor Sivelestat attenuated pulmonary endothelial injury reducing endothelial glycocalyx damage and preserving thrombomodulin and syndecan-1 expression (30). Similar findings were observed in work by Okeke and colleagues, where Sivelestat treatment reduced NET-mediated injury on human umbilical vein endothelial cells (HUVECs) (31). Interestingly, Sivelestat was able to reverse the NET-mediated upregulation of Intercellular Adhesion Molecule-1 (ICAM-1) on HUVECs, suggesting that NETs may propagate the inflammatory response of endothelial cells through NE (31). Furthermore, reduced NET deposition after Sivelestat treatment also suggested that NE may be directly involved in the release of NETs (31). This observation would be consistent with previous findings describing the role of NE nuclear translocation in facilitating histone degradation and chromatin decondensation (32). However, in mice genetically deficient for NE, the stimulation with the well-known NETosis inducer, phorbol 12-myristate 13-acetate (PMA), does not abrogate the release of NETs (33). In addition, the effect of NE inhibition appears to vary among neutrophils from different healthy donors, suggesting that NE may not always be necessary for the execution of NETosis (34).
Other proteases such as PR3 and Cat G are known to enhance inflammatory reactions by modulating the balance between pro and anti-inflammatory proteins (35). However, despite their documented proteolytic activity in human NETs, the precise contribution to mediating ALI is currently less clear.
In summary, besides the explicit role of NE in NET-mediated lung microvascular damage, the overall contribution of neutrophil serine proteases to NET-dependent inflammation in ALI is still undefined.
Histones
Histones are generally located within the cell nucleus, where they play an essential role in organizing and regulating DNA (36). During NETosis, the enzyme peptidyl arginine deiminase 4 (PAD4) alters histone intermolecular interactions by converting arginine residues into citrulline (37). Citrullination decreases histone protein stability and facilitates chromatin decondensation and chromosomal DNA expulsion (38, 39). PAD4-dependent histones citrullination has been frequently reported as a critical component of NETosis in response to numerous physiological stimuli (40), and treatment with PAD4 inhibitors such as Cl-amidine (41) and GSK484 (42) have been shown positive effects in attenuating ALI. Interestingly, recent works indicate that there are conditions where NETosis can occur even in the absence of PAD4 activity, for example, in response to Candida albicans, Klebsiella pneumoniae or cholesterol crystals (43–45). These evidence suggest that during NETosis, chromatin decondensation may be not exclusively dependent on histone citrullination and that alternative mechanisms, such as direct histone cleavage by NE nuclear translocation, may also be involved (32). How different NET stimuli activate alternative mechanisms of chromatin decondensation and what is the level of redundancy and interaction between such pathways remains unclear, indicating the need for a better characterization of the upstream signaling involved in NET formation and release.
Extracellular histones account for ~70% of all NET-associated proteins (10), and their presence highly contributes to NET-mediated thromboinflammation (36, 46). For example, histone engagement of TLR2 and TLR4 can induce platelet activation resulting in the release of thrombin (47). In addition, histones can activate αIIbβ3 integrin on the platelet surface, inducing subsequent fibrinogen-mediated platelet aggregation (48). Histones also appear to stimulate TLR9-dependent responses leading to mitochondrial ROS production and NLR Family Pyrin Domain Containing 3 (NLRP3) inflammasome activation (49). In a mouse model of ALI secondary to severe trauma, high circulating histones were associated with signs of edema, increased alveolar wall thickening, and occasional hemorrhage (50). However, inhibition with TLR4 and TLR2-neutralizing antibodies did not show protective effects on cultured endothelial cells suggesting that TLR activation may not be the major pathway for histone toxicity. In this regard, extracellular histones have been shown to cause endothelial cell damage through direct interactions with the plasma membrane phospholipids, which lead to increased transmembrane conductance (50). Although DNase treatment is generally considered effective in degrading NET DNA scaffold, questions remain on the fate of NET proteins, particularly histones, after DNase digestion. For example, Saffarzadeh and coworkers show that DNase-mediated NET degradation was insufficient to abolish NET cytotoxicity, suggesting that histone proteins, after DNA digestion, can still mediate most of the NET-related tissue damage (51). Similar findings are also present in work by Kolaczkowska and colleagues where, in a model of bloodstream infection with methicillin-resistant Staphylococcus aureus (MRSA), they demonstrate that significant amount of active histones and NE remained attached to the vasculature wall after DNase treatment (52).
In summary, extracellular histones play a potent and heterogeneous contribution in NET-mediated thromboinflammation and tissue injury. Reducing the release, neutralizing, or blocking histone signal transduction may represent in the future a novel pharmacological approach for ALI treatment.
DNA
DNA forms the NET backbone, and for a long time, its singular function has been thought limited to maintaining NET structural integrity. However, recent findings suggest that DNA may also play a direct role in promoting thromboinflammation. Besides providing a scaffold for the recruitment of platelets, leukocytes, and coagulation factors (53), cell-free DNA derived from NETs has been shown to mediate thrombin generation through the Factor XII- or Factor XI-dependent coagulation pathways in patients with sepsis (54). Accordingly, the digestion or precipitation of DNA networks markedly diminishes NET pro-coagulant effects (55). In addition to nuclear DNA, NETs can also be enriched with DNA derived from mitochondria (mtDNA) (7–9). mtDNA are circular molecules of double-stranded DNA that encode for some of the genes that form the electron transport chain as well as drive mitochondrial-specific protein synthesis (56). However, given their ancient bacterial origin (57), NET-associated mtDNA release can act as a damaged associated molecular pattern (DAMP) by stimulating the CpG DNA sensor TLR9 (58, 59) as well as triggering NLRP3 inflammasome pathway activation on immune cells (60). In addition, the NET-mediated release of oxidized mtDNA has been shown to stimulate type I interferon (IFN) signaling through a signaling pathway dependent on the DNA sensor stimulator of interferon genes (STING), an intracellular pattern recognition receptor (9). Moreover, recent findings indicate that mtDNA can also directly activate neutrophils to release NETs through TLR9 engagement (61–63).
In summary, besides acting as a scaffold, NET DNA may directly stimulate innate immune responses that promote pulmonary inflammation.
NETs and ALI etiologies
Sepsis and pneumonia
Sepsis is a common and deadly inflammatory condition defined as a dysregulated host response to infection associated with multi-organ dysfunction (64). With the development of ARDS, lung involvement is frequently observed in critically ill subjects affected with sepsis (65). Moreover, infectious pneumonia is one of the leading causes of sepsis and the most frequent cause of ALI (66–68). Although pathogens themselves can directly damage the lung, a significant contribution to the injury comes from the exuberant activation of the host immune cells in response to pathogen-associated molecular patterns (PAMPs) and DAMPs, collectively defined as “alarmins” (69–71).
NETs have been firstly described as a potent defensive mechanism able to block and inactivate pathogens preventing their growth and dissemination (14, 72). The cationic NETs components provide an ideal scaffold for electrostatic interactions with the negatively charged surface of microorganisms exposing them to a high local concentration of cytotoxic molecules. Moreover, even in the absence of direct killing, experiments in primary human neutrophils showed that NETs can indirectly increase the complement ability to destroy pathogens such as Pseudomonas aeruginosa and Staphylococcus aureus (73). Considering the major role played by NETs in controlling infections, it is not surprising that several microbes have evolved specific adaptive strategies to counteract this defensive mechanism (74). For example, Streptococcus pneumoniae, the most common cause of community-acquired pneumonia (CAP), expresses endonucleases that degrade the NET DNA scaffold allowing for bacterial escape (75). In alternative, Bordetella pertussis, the causative agent of whooping cough, expresses adenylate cyclase toxin (ACT) which inhibits the release of NETs by increasing cAMP levels and reducing intracellular ROS production (76). In humans, the critical role of NETs in protecting from lung infections is evident in subjects affected by Chronic Granulomatous Disease (CDG), a systemic disorder caused by a genetic deficiency in NADPH oxidase and consequent impairment in NET formation. Initially described as “a fatal granulomatous disease of childhood’’, CGD used to be associated with recurrent respiratory infections and high mortality rate before the introduction of effective antimicrobial treatments (77). The protective role of NETs, particularly in the early phase of infection, in controlling pathogen growth and dissemination is also evident from experimental observations (78–84). In a mouse model of polymicrobial sepsis by cecal ligation puncture (CLP), Czaikoski and coworkers report that DNase treatment increased systemic bacterial burden and decreased survival (78). Consistent results were also reported by Meng and coworkers that, using DNase treatment in a similar mouse model of sepsis, described a profound and sustainable reduction of NET-mediated bactericidal activity associated with worst inflammation, more severe organ injury and higher early mortality (79). Finally, better outcomes in sepsis were reported by Lee and coworkers in mice deficient for phospholipase D2 (PLD2) that exhibit an intrinsic up-regulation of PAD4 activity associated with augmenting NET formation and bacterial killing (80).
Although the contribution of NETs in pathogen clearance is well established, aberrant NET release, particularly into the lung, has also been associated with the exacerbation of inflammation, immunothrombosis, and tissue injury (85–90). For example, NETs formed during LPS-induced ALI have been shown to directly cause organ damage and exacerbate an inflammatory response characterized by leukocyte accumulation, diffuse alveolar damage and cytokine release which could be improved by DNase treatment (85). Further details into the mechanism of NeT-mediated damage during bacterial sepsis were provided by McDonald and coworkers that recently described the implication of NET–platelet–thrombin axis in the promotion of intravascular coagulation and showed that PAD4 deficient mice or mice that received DNase have reduced microvascular occlusion and organ dysfunction (86). Blocking NET release by pharmacological inhibition of Gasdermin D (91) or PAD4 (41, 92) similarly reduced signs of sepsis-related multi-organ dysfunction and ALI. Moreover, data on critically ill subjects affected with infectious pneumonia and sepsis indicate that higher circulating NETs are associated with the worst clinical outcomes and directly correlate with organ dysfunction (12, 69, 93–96). Finally, a meta-analysis of randomized controlled trials on the use of Sivelestat for the treatment of ARDS in subjects with sepsis showed improved lung function even in the absence of a significant increased overall survival (97).
A possible explanation for the complex interplay between antimicrobial and pro-inflammatory NET function has been recently proposed by Lafrançais and coworkers (95). In this study, the authors confirmed the presence of higher levels of NETs in ARDS subjects with infectious etiology that correlated with the worst clinical outcomes. Moreover, in a mouse model of bacterial pneumonia, they found that NET release exacerbated signs of ALI, which could be attenuated in PAD4 deficient mice or by DNase administration. Interestingly, increased bacterial growth, particularly observed in mice with a genetic defect in NETosis, partially counteracted these beneficial effects, suggesting that the early release of NETs is important in initially control the infection and that ARDS in sepsis may represent the maladaptive consequence of this primarily defense mechanism (95). This view is consistent with previous findings showing that early DNase administration in a CLP model of sepsis increased signs of ALI but that the same treatment delivered in later phases was beneficial in reducing the level of systemic inflammation and increased survival (98).
In conclusion, the experimental evidence summarized in Table 1, and the clinical observations from sepsis subjects with ARDS, indicate that NETs play a dual role in ALI associated with sepsis and pneumonia. On the one hand, the early release of NETs at the site of infection is essential for controlling pathogen growth, and defects in NETosis or early NET degradation are associated with recurrent pneumonia and disseminated infections. However, NET exuberant release, particularly in a later phase dominated by post-infectious inflammatory events, appears largely detrimental and concurrently exacerbates tissue damage and ALI. Possible therapeutic strategies directed to modulate NET-mediated lung damage during infection should consider this dynamic relation between the early contribution to pathogen control and the late post-infectious exacerbation of systemic inflammation.
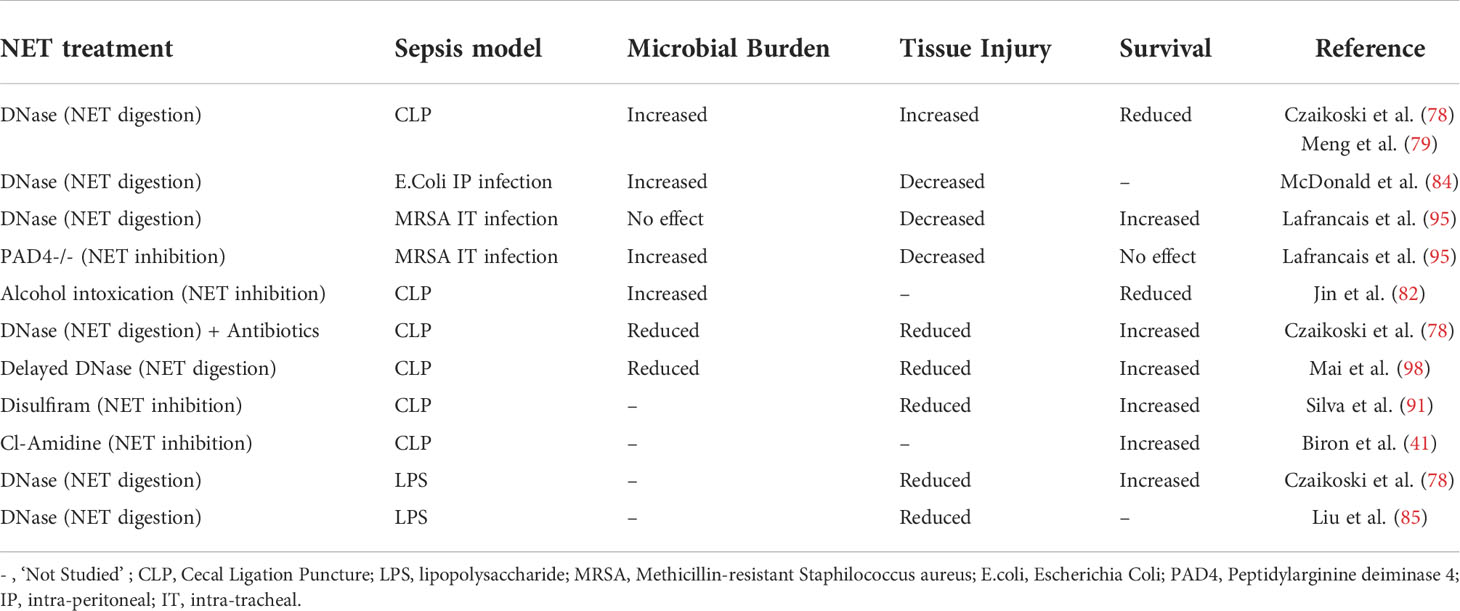
Table 1 The effects of NET treatment in experimental models of sepsis.
COVID-19
COVID-19 is a global pandemic caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection, which in its most severe clinical presentation, is associated with ALI/ARDS and multiple organ failure (99–101). COVID-19 infection with a high neutrophil-to-lymphocyte ratio is generally associated with the worst clinical outcomes (102, 103). Neutrophil activation signature is a prominent feature of circulating leukocyte transcriptomes of severe cases (104, 105). Additionally, single-cell RNA sequencing (scRNA-seq) analysis of COVID-19 whole blood samples has revealed distinct neutrophil clusters associated with NET release and disease severity (106). In order to characterize COVID-19 immunopathogenesis, several studies have focused on neutrophil effector functions, and in particular, NETosis (107–111). SARS-CoV-2 infection has been shown to directly induce NETosis in healthy neutrophils (111). NETosis has also been reported as a predictor of COVID-19 severity (110, 112). Post-mortem examinations of lungs from COVID-19 patients reveal diffuse neutrophil infiltration with abundant NET deposition frequently associated with platelet accumulation and microvascular thrombosis (113–116). This inflammatory pattern of thrombosis is consistent with findings from Zuo and colleagues, who showed a positive correlation between markedly elevated cell-free DNA and D-dimer levels, a degradation product of fibrin (110). The pathogenic role of NET release has also been described in different thrombosis-mediated inflammatory states associated with ARDS (90, 117). One current view of the underlying causes of ALI-mediated pulmonary thrombosis is that NET web-like structures immobilize and activate platelets and leukocytes. Through mechanisms that have yet to be fully elucidated, this induces a feed-forward loop that triggers additional intravascular NETosis and higher local concentrations of histones and proteases that increase endothelial permeability and promote microvascular obstruction (117). Given the prominent role of immunothrombosis in the pathogenesis of COVID-19 ALI/ARDS, pharmacological NET inhibition or degradation could represent an intriguing approach to attenuate disease severity and improve survival. A recent study by Fisher and colleagues supports this notion by showing remarkable improvement in clinical outcomes in five severely ill COVID-19 subjects treated with off-label aerosolized recombinant human (rh) DNase (118). Similar findings were also observed in different single-center case series and cohort studies where nebulized rhDNase administration showed favorable clinical outcomes in the absence of drug-associated toxicities (119, 120). Although the results from these small observational studies are encouraging, larger and randomized clinical trials will be required to assess the long terms effects of DNase treatment on COVID-19 ARDS.
Transfusion-related acute lung injury (TRALI)
TRALI is a form of ALI that occurs within six hours of transfusion that cannot be explained by another ALI risk factor (121). Neutrophil activation and sequestration within the pulmonary capillaries in response to blood components is considered a central feature in the pathogenesis of TRALI (122). Recent insights into the mechanisms of neutrophil-mediated tissue damage indicate that NETs form in the lung and contribute to the pathogenesis of this disease (123–125). Using an established mouse model of TRALI (126), Caudrillier and coworkers show that platelet sequestration increases with NET proximity and that aspirin treatment decreases NET formation and NET-associated platelets (123). Moreover, human platelets activated with the PAR-1 agonist, thrombin receptor-activating peptide (TRAP), induced robust NET release via Thromboxane A2 (123). Further investigation into how platelets drive NETosis revealed dependence on the canonical Raf/MEK/ERK signaling pathway that is also critical to PMA-induced NADPH-dependent NETosis (127). Despite the reported association of NETs with TRALI, a different study by Thomas and colleagues found that platelet depletion did not completely abrogate NETosis and that FcγR engagement by anti-neutrophil antibodies was sufficient to promote NET release (124). Moreover, this study showed that DNase administration effectively prevented NET-mediated ALI only when delivered through inhalation, probably because of the predominant accumulation of NETs within alveolar spaces (124). NET inhibition rather than digestion also appears to be a promising approach to preventing TRALI as indicated by a recent study where the administration of Disulfiram, an FDA-approved gasdermin D inhibitor for the treatment of alcohol abuse, prevented signs of lung injury (125).
In summary, NETs appear to have a pathogenic role in TRALI, and preclinical observations indicate that NET inhibition or digestion may improve TRALI outcomes. However, conflicting reports as to the specific contribution of platelet activation to NET-mediated tissue injury in TRALI further highlight the need for studies to dissect the molecular mechanisms of NETosis in vivo.
Lung ischemia-reperfusion injury (LIRI)
LIRI is a primarily sterile inflammatory disease occurring in lungs that sustain ischemic damage followed by reperfusion. LIRI is typically associated with pulmonary embolisms and various cardiothoracic surgical procedures, including lung transplantation (LTx) (128). The physiopathological mechanisms of LIRI are incredibly complex (129), and the specific contribution of neutrophils is still debated (130, 131). The prevailing view is that LIRI is a bimodal process, with an early phase primarily dependent on pulmonary macrophages and activated platelets followed by later neutrophil-mediated damage (132–134).
A recent study by Sayah and colleagues demonstrated a pathogenic role for NETs in primary graft dysfunction (PGD), a common form of LTx-related LIRI occurring within 72 hours. after surgery. Using a mouse model of syngeneic orthotopic left lung transplant (OLT), this group detected the presence of intra-graft NETs in association with platelet activation. Pretreatment with aspirin or intrabronchial administration of DNase were both able to attenuate signs of PGD in the OLT model (135). A later study by our group extended these findings by visualizing intragraft NETosis by intravital 2-photon microscopy in an allogeneic mouse model of OLT. Interestingly, the systemic administration of DNase improved lung function but at the cost of increasing alloimmune responses and graft rejection due to the release of inflammatory NET fragments. However, directly targeting NET generation by the genetic deletion or pharmacological inhibition of PAD4 prevented alloimmune responses and allograft rejection (136). The unintended consequences associated with NET digestion have been increasingly described in different animal models, raising the question of whether it is more effective to prevent NETosis in lieu of DNase treatment (137). For example, NETs have been described to merge in high-density structures named aggNETs, which are thought to prevent bystander tissue injury as well as contribute to wound healing and inflammation resolution through driving the proteolytic degradation of cytokines and histones (138–140). Most recently, our group has provided further insight into the mechanisms of NET-mediated graft injury by showing that necroptosis, occurring in response to warm-ischemia, drives early neutrophil accumulation into the sub-pleural capillary network resulting in NET-mediated microvascular damage (59). Necroptosis is a Receptor Interacting Serine/Threonine Kinase 3 activation-mediated form of cell death that leads to the extracellular release of various alarmins, including mitochondrial-derived DAMPs (mtDAMPs) (141). Additionally, our group and others have shown that LIRI leads to the release of mtDAMPs, which guide neutrophil intragraft trafficking, stimulate ROS production and trigger NETosis (61, 142).
Altogether, the observations from the LIRI models and human LTx recipients with PGD support the notion that DAMPs released from the ischemic tissues induce innate immune responses resulting in NETosis and tissue injury. Interfering with the mechanisms that activate NETosis rather than enhancing the degradation of NETs appears a more desirable option for attenuating LTx-related ALI without increasing the risk of allograft rejection.
Ventilator-induced lung injury (VILI)
VILI is a form of ALI triggered by mechanical ventilation (143). Neutrophil recruitment in response to VILI is driven by L-selectin expression and is promoted by stretch-induced inflammatory events (144) that lead to the release of neutrophil chemokines such as CXCL1 (KC) and CXCL2/3 (MIP-2) (145). Short periods of mechanical ventilation have been shown to be associated with the release of pro-inflammatory cytokines, including TNF-α, IL-1β and IL-8 (146, 147), which are well-known activators of NETosis (148). Accordingly, recent studies have focused on the possible contribution of NETs to VILI pathogenesis. Using a “double-hit” model of intra-tracheal LPS challenge followed by high tidal mechanical ventilation, Yildiz and colleagues showed that VILI is associated with significant NET release. DNase, also administrated intratracheally, reduced BAL markers of NETs and improved lung compliance but did not affect other measures of lung injury including oxygenation, inflammatory cell infiltration and lung permeability (149). The singular improvement in lung compliance led the authors to hypothesize that DNase treatment decreased airway secretion viscosity rather than imparting VILI protection (149). In contrast to these findings, Rossaint and coworkers found that targeting NETs with DNase effectively protected mice from ALI in a single hit model of VILI (150). Moreover, the group further demonstrated that VILI-induced NETosis is a platelet-dependent process requiring simultaneous stimulation of integrins and G-protein–coupled receptors on neutrophils (150). Using the same single hit model of VILI, Li and coworkers confirmed the beneficial effect of DNase treatment in preventing ALI and showed that TLR4 signaling is implicated in the release of NETs in VILI (151).
In summary, the current evidence shows that NETs are released during VILI and that targeting NETs may be beneficial in preventing injury in this setting. The differing effects observed with DNase treatment may be related to the use of alternative experimental models suggesting further pre-clinical evaluation is likely necessary to better characterize the effects of NETosis on VILI.
Trauma
Trauma is defined as an injury caused by an external physical force, as it may result in the consequence of blunt force impact, penetrating injuries, or extensive thermal and chemical burns. Although advancements in surgery and resuscitation have substantially increased the chance of surviving a major trauma, some patients still develop systemic inflammatory response syndrome (SIRS) and infections (152). ARDS is a well-known complication of major trauma, and it represents one of its leading causes of death (153). Trauma is commonly associated with the loss of natural barrier function, which places large demands on neutrophils to scavenge tissue debris and control the diffusion of pathogens. However, due to the large microvascular bed and the long transit time, the lung represents a preferred site for the accumulation of activated neutrophils after trauma (154). NET release has been demonstrated in several experimental models of trauma (63, 155–157), and elevated levels of circulating NET markers have been associated with the worst clinical outcomes in subjects developing trauma-related ARDS (50). Interestingly, ARDS resulting from trauma seems to be overall associated with better clinical outcomes when compared to other forms of ARDS, suggesting the presence of peculiar immunological features (158–161). In this regard, trauma is a known driver of emergency hematopoiesis leading to a high number of circulating immature neutrophils (162) with impaired effector functions (163, 164). In addition, trauma induces the expansion of specific neutrophil subsets that are either unresponsive or display immunosuppressive activity, increasing the risk of secondary infections (165–167). Recent work by Hazeldine and colleagues has suggested that reduced neutrophil activity in the trauma setting is related to their ability to form NETs (168). This group demonstrated that a few minutes after the injury, neutrophils exhibit an activated phenotype characterized by enhanced glucose metabolism and NETosis. However, hours later, neutrophils became unresponsive, and their ability to form NETs dramatically decreased. To explain this apparent change in neutrophil function, the authors hypothesize that mtDAMPs, such as N-formylated peptides, desensitize neutrophils to further stimulation, effectively reducing their ability to release NETs (168). Recent findings from Itagaki and colleagues support the existence of this regulatory pathway. They showed that blocking the N-formylated peptide receptor-1 (FPR1) preserves neutrophil function after trauma (169). Other evidence, however, point to the pro-inflammatory role of mtDAMPs in activating neutrophils (58, 170, 171), increasing NETosis (61, 62), and predisposing to the development of SIRS (172). Further complicating this picture, studies of trauma subjects show reduced endogenous DNase activity and increased circulating cell-free DNA levels associated with post-injury complications (164, 173, 174). Despite the general state of immune unresponsiveness and the increased risk of infection, these studies suggest that restoring DNase activity is likely beneficial after trauma.
In summary, ALI associated with trauma appears to display peculiar immunological features, possibly related to the sudden and massive release of DAMPs and the alteration of regulatory mechanisms that promote DAMP clearance. How different DAMPs control neutrophil metabolism, differentiation and NETosis as well as shape host responses to trauma remains to be determined.
Discussion
Increasing evidence suggests that NETs play an essential role in infectious and sterile ALI and that circulating NET markers can predict worst clinical outcomes in ARDS subjects (5). Most of the current literature has focused on the use of DNase in preventing NET-mediated ALI (175). However, although DNase has shown promising results in reducing signs of lung injury and inflammation associated with NET release, several questions about its global use to treat ARDS conditions remain. For example, does DNase-mediated NET degradation increase the risk of infection? In addition, do the rapid release of histones and proteases following DNAse treatment have unintended effects on outcomes? Preventing formation rather than digesting NETs may offer a more suitable alternative strategy to treat some forms of ALI. In this regard, selective inhibitors have been developed to prevent the generation and release of NETs at different stages of NETosis (17, 37). Nevertheless, it remains to be determined if NET formation inhibitors will be effective for most forms of ALI and, like DNase, could potentially dysregulate host defense. It also remains to be determined if NET inhibitors will be effective for all forms of NETosis, given the apparent redundancy in the pathways that generate NETs. Platelet activation and coagulation factors appear to be highly interconnected with NET release leading to a state of thromboinflammation (176). Although this mechanism appears to be extremely relevant in the ARDS associated with COVID-19, its contribution still needs to be precisely characterized for other ALI etiologies. In addition, the molecular details that control the crosstalk between innate immunity and coagulation remain poorly understood. DAMPs, particularly those derived from mitochondria, appear to be strongly involved in the activation of NETosis (61, 62). Interfering with their signals may represent a promising strategy to reduce exuberant NET release and prevent neutrophil desensitization to microbial PAMPs. However, to safely leverage these observations, pathways that control the delicate balance between neutrophil activation and desensitization will need further investigation.
In conclusion, future studies that better define the mechanisms involved in the formation, release, and degradation of NETs could form the basis of therapeutic strategies that target ALI without disabling pathogen surveillance.
Author contributions
DS, FL, AK, DK, and AG contributed to conception and design of the study. DS wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was funded by the NIH (NIH R01HL094601, NIH P01AI11650, NIH R01HL157407).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kulkarni HS, Lee JS, Bastarache JA, Kuebler WM, Downey GP, Albaiceta GM, et al. Update on the features and measurements of experimental acute lung injury in animals: An official American thoracic society workshop report. Am J Respir Cell Mol Biol (2022) 66:e1–e14. doi: 10.1165/rcmb.2021-0531ST
2. Millar FR, Summers C, Griffiths MJ, Toshner MR, Proudfoot AG. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax (2016) 71:462–73. doi: 10.1136/thoraxjnl-2015-207461
3. Fanelli V, Vlachou A, Ghannadian S, Simonetti U, Slutsky AS, Zhang H. Acute respiratory distress syndrome: New definition, current and future therapeutic options. J Thorac Dis (2013) 5:326–34. doi: 10.3978/j.issn.2072-1439.2013.04.05
4. Hendrickson KW, Peltan ID, Brown SM. The epidemiology of acute respiratory distress syndrome before and after coronavirus disease 2019. Crit Care Clin (2021) 37:703–16. doi: 10.1016/j.ccc.2021.05.001
5. Yang S-C, Tsai Y-F, Pan Y-L, Hwang T-L. Understanding the role of neutrophils in acute respiratory distress syndrome. BioMed J (2021) 44:439–46. doi: 10.1016/j.bj.2020.09.001
6. Neubert E, Meyer D, Rocca F, Günay G, Kwaczala-Tessmann A, Grandke J, et al. Chromatin swelling drives neutrophil extracellular trap release. Nat Commun (2018) 9:3767. doi: 10.1038/s41467-018-06263-5
7. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
8. McIlroy DJ, Jarnicki AG, Au GG, Lott N, Smith DW, Hansbro PM, et al. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J Crit Care (2014) 29:1133.e1–5. doi: 10.1016/j.jcrc.2014.07.013
9. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22:146–53. doi: 10.1038/nm.4027
10. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against candida albicans. PloS Pathog (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
11. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol (2018) 18:134–47. doi: 10.1038/nri.2017.105
12. Mikacenic C, Moore R, Dmyterko V, West TE, Altemeier WA, Liles WC, et al. Neutrophil extracellular traps (NETs) are increased in the alveolar spaces of patients with ventilator-associated pneumonia. Crit Care (2018) 22:358. doi: 10.1186/s13054-018-2290-8
13. Pan B, Alam HB, Chong W, Mobley J, Liu B, Deng Q, et al. CitH3: A reliable blood biomarker for diagnosis and treatment of endotoxic shock. Sci Rep (2017) 7:8972. doi: 10.1038/s41598-017-09337-4
14. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
15. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176:231–41. doi: 10.1083/jcb.200606027
16. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ (2011) 18:581–8. doi: 10.1038/cdd.2011.1
17. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin d plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol (2018) 3:eaar6689. doi: 10.1126/sciimmunol.aar6689
18. Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to staphylococcus aureus. J Immunol (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
19. Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking. vivo Nat Med (2012) 18:1386–93. doi: 10.1038/nm.2847
20. Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife (2017) 6:e24437. doi: 10.7554/eLife.24437
21. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res (2011) 21:290–304. doi: 10.1038/cr.2010.150
22. Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol (2016) 46:223–9. doi: 10.1002/eji.201545605
23. Kaplan MJ, Radic M. Neutrophil extracellular traps (NETs): Double-edged swords of innate immunity. J Immunol (2012) 189:2689–95. doi: 10.4049/jimmunol.1201719
24. Stapels DAC, Geisbrecht BV, Rooijakkers SHM. Neutrophil serine proteases in antibacterial defense. Curr Opin Microbiol (2015) 0:42–8. doi: 10.1016/j.mib.2014.11.002
25. Pham CTN. Neutrophil serine proteases fine-tune the inflammatory response. Int J Biochem Cell Biol (2008) 40:1317–33. doi: 10.1016/j.biocel.2007.11.008
26. O’Donoghue AJ, Jin Y, Knudsen GM, Perera NC, Jenne DE, Murphy JE, et al. Global substrate profiling of proteases in human neutrophil extracellular traps reveals consensus motif predominantly contributed by elastase. PloS One (2013) 8:e75141. doi: 10.1371/journal.pone.0075141
27. Carden D, Xiao F, Moak C, Willis BH, Robinson-Jackson S, Alexander S. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am J Physiology-Heart Circulatory Physiol (1998) 275:H385–92. doi: 10.1152/ajpheart.1998.275.2.H385
28. Hermant B, Bibert S, Concord E, Dublet B, Weidenhaupt M, Vernet T, et al. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J Biol Chem (2003) 278:14002–12. doi: 10.1074/jbc.M300351200
29. Suttorp N, Nolte A, Wilke A, Drenckhahn D. Human neutrophil elastase increases permeability of cultured pulmonary endothelial cell monolayers. Int J Microcirc Clin Exp (1993) 13:187–203.
30. Suzuki K, Okada H, Takemura G, Takada C, Kuroda A, Yano H, et al. Neutrophil elastase damages the pulmonary endothelial glycocalyx in lipopolysaccharide-induced experimental endotoxemia. Am J Pathol (2019) 189:1526–35. doi: 10.1016/j.ajpath.2019.05.002
31. Okeke EB, Louttit C, Fry C, Najafabadi AH, Han K, Nemzek J, et al. Inhibition of neutrophil elastase prevents neutrophil extracellular trap formation and rescues mice from endotoxic shock. Biomaterials (2020) 238:119836. doi: 10.1016/j.biomaterials.2020.119836
32. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol (2010) 191:677–91. doi: 10.1083/jcb.201006052
33. Martinod K, Witsch T, Farley K, Gallant M, Remold-O’Donnell E, Wagner DD. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemostasis (2016) 14:551–8. doi: 10.1111/jth.13239
34. Tokuhiro T, Ishikawa A, Sato H, Takita S, Yoshikawa A, Anzai R, et al. Oxidized phospholipids and neutrophil elastase coordinately play critical roles in NET formation. Front Cell Dev Biol (2021) 9:718586. doi: 10.3389/fcell.2021.718586
35. Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, et al. Extracellular neutrophil proteases are efficient regulators of IL-1, IL-33, and IL-36 cytokine activity but poor effectors of microbial killing. Cell Rep (2018) 22:2937–50. doi: 10.1016/j.celrep.2018.02.062
36. Hoeksema M, van Eijk M, Haagsman HP, Hartshorn KL. Histones as mediators of host defense, inflammation and thrombosis. Future Microbiol (2016) 11:441–53. doi: 10.2217/fmb.15.151
37. Rohrbach A, Slade D, Thompson P, Mowen K. Activation of PAD4 in NET formation. Front Immunol (2012) 3:360. doi: 10.3389/fimmu.2012.00360
38. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med (2010) 207:1853–62. doi: 10.1084/jem.20100239
39. Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol (2008) 180:1895–902. doi: 10.4049/jimmunol.180.3.1895
40. Tatsiy O, McDonald PP. Physiological stimuli induce PAD4-dependent, ROS-independent NETosis, with early and late events controlled by discrete signaling pathways. Front Immunol (2018) 9:2036. doi: 10.3389/fimmu.2018.02036
41. Biron BM, Chung C-S, O’Brien XM, Chen Y, Reichner JS, Ayala A. Cl-amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun (2017) 9:22–32. doi: 10.1159/000448808
42. Zhan Y, Ling Y, Deng Q, Qiu Y, Shen J, Lai H, et al. HMGB1-mediated neutrophil extracellular trap formation exacerbates intestinal Ischemia/Reperfusion-induced acute lung injury. J Immunol (2022) 208:968–78. doi: 10.4049/jimmunol.2100593
43. Guiducci E, Lemberg C, Küng N, Schraner E, Theocharides APA, LeibundGut-Landmann S. Candida albicans-induced NETosis is independent of peptidylarginine deiminase 4. Front Immunol (2018) 9:1573. doi: 10.3389/fimmu.2018.01573
44. Claushuis TAM, van der Donk LEH, Luitse AL, van Veen HA, van der Wel NN, van Vught LA, et al. Role of peptidylarginine deiminase 4 in neutrophil extracellular trap formation and host defense during klebsiella pneumoniae–induced pneumonia-derived sepsis. J Immunol (2018) 201:1241–52. doi: 10.4049/jimmunol.1800314
45. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Neutrophil extracellular traps license macrophages and Th17 cells for cytokine production in atherosclerosis. Science (2015) 349:316–20. doi: 10.1126/science.aaa8064
46. Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis (2017) 8:e2812–2. doi: 10.1038/cddis.2017.52
47. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
48. Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood (2011) 118:3708–14. doi: 10.1182/blood-2011-01-332676
49. Huang H, Chen H-W, Evankovich J, Yan W, Rosborough BR, Nace GW, et al. Histones activate the NLRP3 inflammasome in kupffer cells during sterile inflammatory liver injury. J Immunol (2013) 191:2665–79. doi: 10.4049/jimmunol.1202733
50. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med (2013) 187:160–9. doi: 10.1164/rccm.201206-1037OC
51. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PloS One (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
52. Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee W-Y, Sanz M-J, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun (2015) 6:6673. doi: 10.1038/ncomms7673
53. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U.S.A. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
54. Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SHC, Weitz JI, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol (2014) 34:1977–84. doi: 10.1161/ATVBAHA.114.304114
55. Yang J, Wu Z, Long Q, Huang J, Hong T, Liu W, et al. Insights into immunothrombosis: The interplay among neutrophil extracellular trap, Von willebrand factor, and ADAMTS13. Front Immunol (2020) 11:610696. doi: 10.3389/fimmu.2020.610696
56. Taanman J-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim Biophys Acta (BBA) - Bioenergetics (1999) 1410:103–23. doi: 10.1016/S0005-2728(98)00161-3
57. Gray MW. Mitochondrial evolution. Cold Spring Harb Perspect Biol (2012) 4:a011403. doi: 10.1101/cshperspect.a011403
58. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi: 10.1038/nature08780
59. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via P38 map kinase. Shock (2010) 34:55–9. doi: 10.1097/SHK.0b013e3181cd8c08
60. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
61. Mallavia B, Liu F, Lefrançais E, Cleary SJ, Kwaan N, Tian JJ, et al. Mitochondrial DNA stimulates TLR9-dependent neutrophil extracellular trap formation in primary graft dysfunction. Am J Respir Cell Mol Biol (2020) 62:364–72. doi: 10.1165/rcmb.2019-0140OC
62. Itagaki K, Kaczmarek E, Lee YT, Tang IT, Isal B, Adibnia Y, et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PloS One (2015) 10:e0120549. doi: 10.1371/journal.pone.0120549
63. Liu L, Mao Y, Xu B, Zhang X, Fang C, Ma Y, et al. Induction of neutrophil extracellular traps during tissue injury: Involvement of STING and toll-like receptor 9 pathways. Cell Prolif (2019) 52:e12579. doi: 10.1111/cpr.12579
64. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315:801–10. doi: 10.1001/jama.2016.0287
65. Hu Q, Hao C, Tang S. From sepsis to acute respiratory distress syndrome (ARDS): Emerging preventive strategies based on molecular and genetic researches. Biosci Rep (2020) 40:BSR20200830. doi: 10.1042/BSR20200830
66. Rhee C, Jones TM, Hamad Y, Pande A, Varon J, O’Brien C, et al. Prevalence, underlying causes, and preventability of sepsis-associated mortality in US acute care hospitals. JAMA Network Open (2019) 2:e187571. doi: 10.1001/jamanetworkopen.2018.7571
67. Lee K-Y. Pneumonia, acute respiratory distress syndrome, and early immune-modulator therapy. Int J Mol Sci (2017) 18:388. doi: 10.3390/ijms18020388
68. De Freitas Caires N, Gaudet A, Portier L, Tsicopoulos A, Mathieu D, Lassalle P. Endocan, sepsis, pneumonia, and acute respiratory distress syndrome. Crit Care (2018) 22:280. doi: 10.1186/s13054-018-2222-7
69. Denning N-L, Aziz M, Gurien SD, Wang P. DAMPs and NETs in sepsis. Front Immunol (2019) 10:2536. doi: 10.3389/fimmu.2019.02536
70. Kang J-W, Kim S-J, Cho H-I, Lee S-M. DAMPs activating innate immune responses in sepsis. Ageing Res Rev (2015) 24:54–65. doi: 10.1016/j.arr.2015.03.003
71. Raymond SL, Holden DC, Mira JC, Stortz JA, Loftus TJ, Mohr AM, et al. Microbial recognition and danger signals in sepsis and trauma. Biochim Biophys Acta (BBA) - Mol Basis Dis (2017) 1863:2564–73. doi: 10.1016/j.bbadis.2017.01.013
72. O’Brien XM, Biron BM, Reichner JS. Consequences of extracellular trap formation in sepsis. Curr Opin Hematol (2017) 24:66–71. doi: 10.1097/MOH.0000000000000303
73. Azzouz L, Cherry A, Riedl M, Khan M, Pluthero FG, Kahr WHA, et al. Relative antibacterial functions of complement and NETs: NETs trap and complement effectively kills bacteria. Mol Immunol (2018) 97:71–81. doi: 10.1016/j.molimm.2018.02.019
74. Storisteanu DML, Pocock JM, Cowburn AS, Juss JK, Nadesalingam A, Nizet V, et al. Evasion of neutrophil extracellular traps by respiratory pathogens. Am J Respir Cell Mol Biol (2017) 56:423–31. doi: 10.1165/rcmb.2016-0193PS
75. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An endonuclease allows streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol (2006) 16:401–7. doi: 10.1016/j.cub.2006.01.056
76. Eby JC, Gray MC, Hewlett EL. Cyclic AMP-mediated suppression of neutrophil extracellular trap formation and apoptosis by the bordetella pertussis adenylate cyclase toxin. Infect Immun (2014) 82:5256–69. doi: 10.1128/IAI.02487-14
77. Arnold DE, Heimall JR. A review of chronic granulomatous disease. Adv Ther (2017) 34:2543–57. doi: 10.1007/s12325-017-0636-2
78. Czaikoski PG, Mota JMSC, Nascimento DC, Sônego F, Castanheira FV, Melo PH, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PloS One (2016) 11:e0148142. doi: 10.1371/journal.pone.0148142
79. Meng W, Paunel-Görgülü A, Flohé S, Hoffmann A, Witte I, MacKenzie C, et al. Depletion of neutrophil extracellular traps In vivo results in hypersusceptibility to polymicrobial sepsis in mice. Crit Care (2012) 16:R137. doi: 10.1186/cc11442
80. Lee SK, Kim SD, Kook M, Lee HY, Ghim J, Choi Y, et al. Phospholipase D2 drives mortality in sepsis by inhibiting neutrophil extracellular trap formation and down-regulating CXCR2. J Exp Med (2015) 212:1381–90. doi: 10.1084/jem.20141813
81. Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, et al. Impaired neutrophil extracellular trap (NET) formation: A novel innate immune deficiency of human neonates. Blood (2009) 113:6419–27. doi: 10.1182/blood-2008-07-171629
82. Jin L, Batra S, Jeyaseelan S. Diminished neutrophil extracellular trap (NET) formation is a novel innate immune deficiency induced by acute ethanol exposure in polymicrobial sepsis, which can be rescued by CXCL1. PloS Pathog (2017) 13:e1006637. doi: 10.1371/journal.ppat.1006637
83. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13:463–9. doi: 10.1038/nm1565
84. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe (2012) 12:324–33. doi: 10.1016/j.chom.2012.06.011
85. Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H, et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci Rep (2016) 6:37252. doi: 10.1038/srep37252
86. McDonald B, Davis RP, Kim S-J, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood (2017) 129:1357–67. doi: 10.1182/blood-2016-09-741298
87. Camicia G, Pozner R, de Larrañaga G. Neutrophil extracellular traps in sepsis. Shock (2014) 42:286–94. doi: 10.1097/SHK.0000000000000221
88. Colón DF, Wanderley CW, Franchin M, Silva CM, Hiroki CH, Castanheira FVS, et al. Neutrophil extracellular traps (NETs) exacerbate severity of infant sepsis. Crit Care (2019) 23:113. doi: 10.1186/s13054-019-2407-8
89. Yang S, Qi H, Kan K, Chen J, Xie H, Guo X, et al. Neutrophil extracellular traps promote hypercoagulability in patients with sepsis. Shock (2017) 47:132–9. doi: 10.1097/SHK.0000000000000741
90. Zhang H, Zhou Y, Qu M, Yu Y, Chen Z, Zhu S, et al. Tissue factor-enriched neutrophil extracellular traps promote immunothrombosis and disease progression in sepsis-induced lung injury. Front Cell Infect Microbiol (2021) 11:677902. doi: 10.3389/fcimb.2021.677902
91. Silva CMS, Wanderley CWS, Veras FP, Sonego F, Nascimento DC, Gonçalves AV, et al. Gasdermin d inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood (2021) 138:2702–13. doi: 10.1182/blood.2021011525
92. Li Y, Liu Z, Liu B, Zhao T, Chong W, Wang Y, et al. Citrullinated histone H3 – a novel target for treatment of sepsis. Surgery (2014) 156:229–34. doi: 10.1016/j.surg.2014.04.009
93. Margraf S, Lögters T, Reipen J, Altrichter J, Scholz M, Windolf J. Neutrophil-derived circulating free DNA (Cf-DNA/NETs): A potential prognostic marker for posttraumatic development of inflammatory second hit and sepsis. Shock (2008) 30:352–8. doi: 10.1097/SHK.0b013e31816a6bb1
94. Maruchi Y, Tsuda M, Mori H, Takenaka N, Gocho T, Huq MA, et al. Plasma myeloperoxidase-conjugated DNA level predicts outcomes and organ dysfunction in patients with septic shock. Crit Care (2018) 22:176. doi: 10.1186/s13054-018-2109-7
95. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight (2018) 3:98178. doi: 10.1172/jci.insight.98178
96. Ebrahimi F, Giaglis S, Hahn S, Blum CA, Baumgartner C, Kutz A, et al. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: Secondary analysis of a randomised controlled trial. Eur Respir J (2018) 51:1701389. doi: 10.1183/13993003.01389-2017
97. Pu S, Wang D, Liu D, Zhao Y, Qi D, He J, et al. Effect of sivelestat sodium in patients with acute lung injury or acute respiratory distress syndrome: A meta-analysis of randomized controlled trials. BMC Pulm Med (2017) 17:148. doi: 10.1186/s12890-017-0498-z
98. Mai SHC, Khan M, Dwivedi DJ, Ross CA, Zhou J, Gould TJ, et al. Delayed but not early treatment with DNase reduces organ damage and improves outcome in a murine model of sepsis. Shock (2015) 44:166–72. doi: 10.1097/SHK.0000000000000396
99. Acosta MAT, Singer BD. Pathogenesis of COVID-19-Induced ARDS: Implications for an ageing population. Eur Respir J (2020) 56(3):2002049. doi: 10.1183/13993003.02049-2020
100. Yuki K, Fujiogi M, Koutsogiannaki S. COVID-19 pathophysiology: A review. Clin Immunol (2020) 215:108427. doi: 10.1016/j.clim.2020.108427
101. Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect Dis (2020) 20:1135–40. doi: 10.1016/S1473-3099(20)30434-5
102. Li X, Liu C, Mao Z, Xiao M, Wang L, Qi S, et al. Predictive values of neutrophil-to-Lymphocyte ratio on disease severity and mortality in COVID-19 patients: A systematic review and meta-analysis. Crit Care (2020) 24:647. doi: 10.1186/s13054-020-03374-8
103. Liu J, Liu Y, Xiang P, Pu L, Xiong H, Li C, et al. Neutrophil-To-Lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J Trans Med (2020) 18:206. doi: 10.1186/s12967-020-02374-0
104. Meizlish ML, Pine AB, Bishai JD, Goshua G, Nadelmann ER, Simonov M, et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv (2021) 5:1164–77. doi: 10.1182/bloodadvances.2020003568
105. Aschenbrenner AC, Mouktaroudi M, Krämer B, Oestreich M, Antonakos N, Nuesch-Germano M, et al. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med (2021) 13:7. doi: 10.1186/s13073-020-00823-5
106. Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell (2020) 182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001
107. Reusch N, De Domenico E, Bonaguro L, Schulte-Schrepping J, Baßler K, Schultze JL, et al. Neutrophils in COVID-19. Front Immunol (2021) 12:652470. doi: 10.3389/fimmu.2021.652470
108. Cavalcante-Silva LHA, Carvalho DCM, Lima É de A, Galvão JGFM, da Silva JS de F, de Sales-Neto JM, et al. Neutrophils and COVID-19: The road so far. Int Immunopharmacol (2021) 90:107233. doi: 10.1016/j.intimp.2020.107233
109. Arcanjo A, Logullo J, Menezes CCB, de Souza Carvalho Giangiarulo TC, dos Reis MC, de Castro GMM, et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep (2020) 10:19630. doi: 10.1038/s41598-020-76781-0
110. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight (2020) 5:138999. doi: 10.1172/jci.insight.138999
111. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2–triggered neutrophil extracellular traps mediate COVID-19 pathologySARS-CoV-2 directly triggers ACE-dependent NETs. J Exp Med (2020) 217:e20201129. doi: 10.1084/jem.20201129
112. Janiuk K, Jabłońska E, Garley M. Significance of NETs formation in COVID-19. Cells (2021) 10:151. doi: 10.3390/cells10010151
113. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: Neutrophil extracellular trapsneutrophil extracellular traps in COVID-19. J Exp Med (2020) 217:e20200652. doi: 10.1084/jem.20200652
114. Parra-Medina R, Herrera S, Mejia J. Systematic review of microthrombi in COVID-19 autopsies. Actq Haematol (2021), 144(5):476–83. doi: 10.1159/000515104
115. Fang X-Z, Wang Y-X, Xu J-Q, He Y-J, Peng Z-K, Shang Y. Immunothrombosis in acute respiratory dysfunction of COVID-19. Front Immunol (2021) 12:651545. doi: 10.3389/fimmu.2021.651545
116. Middleton EA, He X-Y, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood (2020) 136:1169–79. doi: 10.1182/blood.2020007008
117. Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in acute respiratory distress syndrome: Cross talks between inflammation and coagulation. RES (2017) 93:212–25. doi: 10.1159/000453002
118. Fisher J, Mohanty T, Karlsson CAQ, Khademi SMH, Malmström E, Frigyesi A, et al. Proteome profiling of recombinant DNase therapy in reducing NETs and aiding recovery in COVID-19 patients. Mol Cell Proteomics (2021) 20:100113. doi: 10.1016/j.mcpro.2021.100113
119. Weber AG, Chau AS, Egeblad M, Barnes BJ, Janowitz T. Nebulized in-line endotracheal dornase Alfa and albuterol administered to mechanically ventilated COVID-19 patients: A case series. Mol Med (2020) 26:91. doi: 10.1186/s10020-020-00215-w
120. Toma A, Darwish C, Taylor M, Harlacher J, Darwish R. The use of dornase Alfa in the management of COVID-19-Associated adult respiratory distress syndrome. Crit Care Res Pract (2021) 2021:e8881115. doi: 10.1155/2021/8881115
121. Toy P, Lowell C. TRALI - definition, mechanisms, incidence and clinical relevance. Best Pract Res Clin Anaesthesiol (2007) 21:183–93. doi: 10.1016/j.bpa.2007.01.003
122. Rebetz J, Semple JW, Kapur R. The pathogenic involvement of neutrophils in acute respiratory distress syndrome and transfusion-related acute lung injury. TMH (2018) 45:290–8. doi: 10.1159/000492950
123. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest (2012) 122:2661–71. doi: 10.1172/JCI61303
124. Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood (2012) 119:6335–43. doi: 10.1182/blood-2012-01-405183
125. Adrover JM, Carrau L, Daßler-Plenker J, Bram Y, Chandar V, Houghton S, et al. Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and SARS-CoV-2 infection. JCI Insight (2022) 7. doi: 10.1172/jci.insight.157342
126. Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest (2009) 119:3450–61. doi: 10.1172/JCI38432
127. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol (2011) 7:75–7. doi: 10.1038/nchembio.496
128. Weyker PD, Webb CAJ, Kiamanesh D, Flynn BC. Lung ischemia reperfusion injury: A bench-To-Bedside review. Semin Cardiothorac Vasc Anesth (2013) 17:28–43. doi: 10.1177/1089253212458329
129. den Hengst WA, Gielis JF, Lin JY, Van Schil PE, De Windt LJ, Moens AL. Lung ischemia-reperfusion injury: A molecular and clinical view on a complex pathophysiological process. Am J Physiol Heart Circ Physiol (2010) 299(5):H1283–99. doi: 10.1152/ajpheart.00251.2010
130. Steimle CN, Guynn TP, Morganroth ML, Bolling SF, Carr K, Deeb GM. Neutrophils are not necessary for ischemia-reperfusion lung injury. Ann Thorac Surg (1992) 53:64–72. doi: 10.1016/0003-4975(92)90758-v
131. Deeb GM, Grum CM, Lynch MJ, Guynn TP, Gallagher KP, Ljungman AG, et al. Neutrophils are not necessary for induction of ischemia-reperfusion lung injury. J Appl Physiol (1985) (1990) 68:374–81. doi: 10.1152/jappl.1990.68.1.374
132. de Perrot M, Liu M, Waddell TK, Keshavjee S. Ischemia–reperfusion–induced lung injury. Am J Respir Crit Care Med (2003) 167:490–511. doi: 10.1164/rccm.200207-670SO
133. Eppinger MJ, Jones ML, Deeb GM, Bolling SF, Ward PA. Pattern of injury and the role of neutrophils in reperfusion injury of rat lung. J Surg Res (1995) 58:713–8. doi: 10.1006/jsre.1995.1112
134. Fiser SM, Tribble CG, Long SM, Kaza AK, Cope JT, Laubach VE, et al. Lung transplant reperfusion injury involves pulmonary macrophages and circulating leukocytes in a biphasic response. J Thorac Cardiovasc Surg (2001) 121:1069–75. doi: 10.1067/mtc.2001.113603
135. Sayah DM, Mallavia B, Liu F, Ortiz-Muñoz G, Caudrillier A, DerHovanessian A, et al. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med (2015) 191:455–63. doi: 10.1164/rccm.201406-1086OC
136. Scozzi D, Wang X, Liao F, Liu Z, Zhu J, Pugh K, et al. Neutrophil extracellular trap fragments stimulate innate immune responses that prevent lung transplant tolerance. Am J Transplant (2019) 19:1011–23. doi: 10.1111/ajt.15163
137. Zhu S, Yu Y, Ren Y, Xu L, Wang H, Ling X, et al. The emerging roles of neutrophil extracellular traps in wound healing. Cell Death Dis (2021) 12:984. doi: 10.1038/s41419-021-04294-3
138. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med (2014) 20:511–7. doi: 10.1038/nm.3547
139. Hahn J, Schauer C, Czegley C, Kling L, Petru L, Schmid B, et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J (2019) 33:1401–14. doi: 10.1096/fj.201800752R
140. Knopf J, Leppkes M, Schett G, Herrmann M, Muñoz LE. Aggregated NETs sequester and detoxify extracellular histones. Front Immunol (2019) 10:2176. doi: 10.3389/fimmu.2019.02176
141. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity (2013) 38:209–23. doi: 10.1016/j.immuni.2013.02.003
142. Scozzi D, Ibrahim M, Liao F, Lin X, Hsiao H-M, Hachem R, et al. Mitochondrial damage associated molecular patterns released by lung transplants are associated with primary graft dysfunction. Am J Transplant (2019) 19:1464–77. doi: 10.1111/ajt.15232
143. Carrasco Loza R, Villamizar Rodríguez G, Medel Fernández N. Ventilator-induced lung injury (VILI) in acute respiratory distress syndrome (ARDS): Volutrauma and molecular effects. Open Respir Med J (2015) 9:112–9. doi: 10.2174/1874306401509010112
144. Choudhury S, Wilson MR, Goddard ME, O’Dea KP, Takata M. Mechanisms of early pulmonary neutrophil sequestration in ventilator-induced lung injury in mice. Am J Physiology-Lung Cell Mol Physiol (2004) 287:L902–10. doi: 10.1152/ajplung.00187.2004
145. Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, et al. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest (2002) 110:1703–16. doi: 10.1172/JCI15849
146. Bohrer B, Silveira RC, Neto EC, Procianoy RS. Mechanical ventilation of newborns infant changes in plasma pro- and anti-inflammatory cytokines. J Pediatr (2010) 156:16–9. doi: 10.1016/j.jpeds.2009.07.027
147. Bose CL, Laughon MM, Allred EN, O’Shea TM, Van Marter LJ, Ehrenkranz RA, et al. Systemic inflammation associated with mechanical ventilation among extremely preterm infants. Cytokine (2013) 61:315–22. doi: 10.1016/j.cyto.2012.10.014
148. Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines induced neutrophil extracellular traps formation: Implication for the inflammatory disease condition. PloS One (2012) 7:e48111. doi: 10.1371/journal.pone.0048111
149. Yildiz C, Palaniyar N, Otulakowski G, Khan MA, Post M, Kuebler WM, et al. Mechanical ventilation induces neutrophil extracellular trap formation. Anesthesiology (2015) 122:864–75. doi: 10.1097/ALN.0000000000000605
150. Rossaint J, Herter JM, Van Aken H, Napirei M, Döring Y, Weber C, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap–mediated sterile inflammation. Blood (2014) 123:2573–84. doi: 10.1182/blood-2013-07-516484
151. Li H, Pan P, Su X, Liu S, Zhang L, Wu D, et al. Neutrophil extracellular traps are pathogenic in ventilator-induced lung injury and partially dependent on TLR4. BioMed Res Int (2017) 2017:8272504. doi: 10.1155/2017/8272504
152. Lord JM, Midwinter MJ, Chen Y-F, Belli A, Brohi K, Kovacs EJ, et al. The systemic immune response to trauma: An overview of pathophysiology and treatment. Lancet (2014) 384:1455–65. doi: 10.1016/S0140-6736(14)60687-5
153. Watkins TR, Nathens AB, Cooke CR, Psaty BM, Maier RV, Cuschieri J, et al. Acute respiratory distress syndrome after trauma: Development and validation of a predictive model. Crit Care Med (2012) 40:2295–303. doi: 10.1097/CCM.0b013e3182544f6a
154. Pillay J, Hietbrink F, Koenderman L, Leenen LPH. The systemic inflammatory response induced by trauma is reflected by multiple phenotypes of blood neutrophils. Injury (2007) 38:1365–72. doi: 10.1016/j.injury.2007.09.016
155. Li X, Luck ME, Herrnreiter CJ, Cannon AR, Choudhry MA. IL-23 promotes neutrophil extracellular trap formation and bacterial clearance in a mouse model of alcohol and burn injury. ImmunoHorizons (2022) 6:64–75. doi: 10.4049/immunohorizons.2100109
156. Surolia R, Li FJ, Wang Z, Kashyap M, Srivastava RK, Traylor AM, et al. NETosis in the pathogenesis of acute lung injury following cutaneous chemical burns. JCI Insight (2021) 6(10):e147564. doi: 10.1172/jci.insight.147564
157. Laggner M, Lingitz M-T, Copic D, Direder M, Klas K, Bormann D, et al. Severity of thermal burn injury is associated with systemic neutrophil activation. Sci Rep (2022) 12:1654. doi: 10.1038/s41598-022-05768-w
158. Calfee CS, Eisner MD, Ware LB, Thompson BT, Parsons PE, Wheeler AP, et al. Trauma-associated lung injury differs clinically and biologically from acute lung injury due to other clinical disorders. Crit Care Med (2007) 35:2243–50. doi: 10.1097/01.CCM.0000280434.33451.87
159. Treggiari MM, Hudson LD, Martin DP, Weiss NS, Caldwell E, Rubenfeld G. Effect of acute lung injury and acute respiratory distress syndrome on outcome in critically ill trauma patients. Crit Care Med (2004) 32:327–31. doi: 10.1097/01.CCM.0000108870.09693.42
160. Bakowitz M, Bruns B, McCunn M. Acute lung injury and the acute respiratory distress syndrome in the injured patient. Scandinavian J Trauma Resuscitation Emergency Med (2012) 20:54. doi: 10.1186/1757-7241-20-54
161. Salim A, Martin M, Constantinou C, Sangthong B, Brown C, Kasotakis G, et al. Acute respiratory distress syndrome in the trauma intensive care unit: Morbid but not mortal. Arch Surg (2006) 141:655–8. doi: 10.1001/archsurg.141.7.655
162. Fuchs A, Monlish DA, Ghosh S, Chang S-W, Bochicchio GV, Schuettpelz LG, et al. Trauma induces emergency hematopoiesis through IL-1/MyD88 dependent production of G-CSF. J Immunol (2019) 202:3020–32. doi: 10.4049/jimmunol.1801456
163. Hampson P, Dinsdale RJ, Wearn CM, Bamford AL, Bishop JRB, Hazeldine J, et al. Neutrophil dysfunction, immature granulocytes, and cell-free DNA are early biomarkers of sepsis in burn-injured patients: A prospective observational cohort study. Ann Surg (2017) 265:1241–9. doi: 10.1097/SLA.0000000000001807
164. Hazeldine J, Dinsdale RJ, Naumann DN, Acharjee A, Bishop JRB, Lord JM, et al. Traumatic injury is associated with reduced deoxyribonuclease activity and dysregulation of the actin scavenging system. Burns Trauma (2021) 9:tkab001. doi: 10.1093/burnst/tkab001
165. Hesselink L, Spijkerman R, van Wessem KJP, Koenderman L, Leenen LPH, Huber-Lang M, et al. Neutrophil heterogeneity and its role in infectious complications after severe trauma. World J Emergency Surg (2019) 14:24. doi: 10.1186/s13017-019-0244-3
166. Janicova A, Becker N, Xu B, Simic M, Noack L, Wagner N, et al. Severe traumatic injury induces phenotypic and functional changes of neutrophils and monocytes. J Clin Med (2021) 10:4139. doi: 10.3390/jcm10184139
167. Mortaz E, Zadian SS, Shahir M, Folkerts G, Garssen J, Mumby S, et al. Does neutrophil phenotype predict the survival of trauma patients? Front Immunol (2019) 10:2122. doi: 10.3389/fimmu.2019.02122
168. Hazeldine J, Dinsdale RJ, Harrison P, Lord JM. Traumatic injury and exposure to mitochondrial-derived damage associated molecular patterns suppresses neutrophil extracellular trap formation. Front Immunol (2019) 10:685. doi: 10.3389/fimmu.2019.00685
169. Itagaki K, Kaczmarek E, Kwon WY, Chen L, Vlková B, Zhang Q, et al. FPR1 blockade prevents receptor regulation by mitochondrial DAMPs and preserves neutrophil function after trauma. Crit Care Med (2020) 48:e123–32. doi: 10.1097/CCM.0000000000004094
170. Hazeldine J, Hampson P, Opoku FA, Foster M, Lord JM. N-formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury (2015) 46:975–84. doi: 10.1016/j.injury.2015.03.028
171. Raoof M, Zhang Q, Itagaki K, Hauser CJ. Mitochondrial peptides are potent immune activators that activate human neutrophils Via FPR-1. J Trauma Acute Care Surg (2010) 68:1328–34. doi: 10.1097/TA.0b013e3181dcd28d
172. Itagaki K, Riça I, Konecna B, Kim HI, Park J, Kaczmarek E, et al. Role of mitochondria-derived danger signals released after injury in systemic inflammation and sepsis. Antioxid Redox Signal (2021) 35:1273–90. doi: 10.1089/ars.2021.0052
173. Dinsdale RJ, Hazeldine J, Al Tarrah K, Hampson P, Devi A, Ermogenous C, et al. Dysregulation of the actin scavenging system and inhibition of DNase activity following severe thermal injury. Br J Surg (2020) 107:391–401. doi: 10.1002/bjs.11310
174. Meng W, Paunel-Görgülü A, Flohé S, Witte I, Schädel-Höpfner M, Windolf J, et al. Deoxyribonuclease is a potential counter regulator of aberrant neutrophil extracellular traps formation after major trauma. Mediators Inflamm (2012) 2012:e149560. doi: 10.1155/2012/149560
175. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: Potential anti-NETs therapeutics. Clinic Rev Allerg Immunol (2021) 61:194–211. doi: 10.1007/s12016-020-08804-7
Keywords: NETs (neutrophil extracellular traps), ALI (acute lung injury), ARDS (acute respiratory distress syndrome), sterile inflammatory response, infections and sepsis, COVID-19, DAMPs (damage-associated molecular patterns), Thromboinflammation
Citation: Scozzi D, Liao F, Krupnick AS, Kreisel D and Gelman AE (2022) The role of neutrophil extracellular traps in acute lung injury. Front. Immunol. 13:953195. doi: 10.3389/fimmu.2022.953195
Received: 25 May 2022; Accepted: 28 June 2022;
Published: 29 July 2022.
Edited by:
Kim Maree O’Sullivan, Monash University, AustraliaReviewed by:
Tamara Muñoz-Caro, Universidad Santo Tomás, ChileAdriana R Silva, Oswaldo Cruz Foundation (Fiocruz), Brazil
Copyright © 2022 Scozzi, Liao, Krupnick, Kreisel and Gelman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew E. Gelman, YWdlbG1hbkB3dXN0bC5lZHU=