
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 18 August 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.952183
 Vanessa Chilunda1Jessica Weiselberg1Samuel Martinez-Meza1
Vanessa Chilunda1Jessica Weiselberg1Samuel Martinez-Meza1 Lwidiko E. Mhamilawa1,2,3Laura Cheney1,4
Lwidiko E. Mhamilawa1,2,3Laura Cheney1,4 Joan W. Berman1,5*
Joan W. Berman1,5*HIV-associated neurocognitive impairment (HIV-NCI) persists in 15-40% of people with HIV (PWH) despite effective antiretroviral therapy. HIV-NCI significantly impacts quality of life, and there is currently no effective treatment for it. The development of HIV-NCI is complex and is mediated, in part, by the entry of HIV-infected mature monocytes into the central nervous system (CNS). Once in the CNS, these cells release inflammatory mediators that lead to neuroinflammation, and subsequent neuronal damage. Infected monocytes may infect other CNS cells as well as differentiate into macrophages, thus contributing to viral reservoirs and chronic neuroinflammation. Substance use disorders in PWH, including the use of methamphetamine (meth), can exacerbate HIV neuropathogenesis. We characterized the effects of meth on the transcriptional profile of HIV-infected mature monocytes using RNA-sequencing. We found that meth mediated an upregulation of gene transcripts related to viral infection, cell adhesion, cytoskeletal arrangement, and extracellular matrix remodeling. We also identified downregulation of several gene transcripts involved in pathogen recognition, antigen presentation, and oxidative phosphorylation pathways. These transcriptomic changes suggest that meth increases the infiltration of mature monocytes that have a migratory phenotype into the CNS, contributing to dysregulated inflammatory responses and viral reservoir establishment and persistence, both of which contribute to neuronal damage. Overall, our results highlight potential molecules that may be targeted for therapy to limit the effects of meth on HIV neuropathogenesis.
HIV remains a major public health issue, with approximately 38 million people worldwide currently living with the virus (1). Antiretroviral therapy (ART) has dramatically improved the lifespan of people with HIV (PWH). However, a significant percentage of PWH develop comorbidities that significantly impact their quality of life. One such comorbidity is HIV-associated neurocognitive impairment (HIV-NCI), which occurs in about 15-40% of PWH despite suppressed viremia (2–8). HIV-NCI is a spectrum of disorders that can fluctuate over time and includes, but is not limited to, impaired executive function, working memory, and attention (2). People with HIV-NCI have a premature decline in overall physical and psychological health, with difficulties maintaining employment and social relationships and in performing instrumental activities of daily living (9–13). HIV-NCI is an independent risk factor for mortality, and currently, there are no effective treatments in the context of ART (14–16). The development of HIV-NCI is complex, resulting from both viral seeding and reseeding, and chronic inflammation and neuronal damage to the central nervous system (CNS).
Substance use disorder (SUD), another highly significant public health issue, also contributes to HIV-NCI in PWH. In a large cohort study, SUD was reported in approximately 48% of PWH, with 13% of those using methamphetamine (meth) (17). Meth is a stimulant that readily crosses the blood-brain barrier (BBB), and can cause neuronal damage directly, and indirectly by affecting microglia and astrocytes that provide neuronal support (18). People without HIV who use meth chronically have been shown to have increased neuronal injury and neurocognitive impairment compared to those who do not use meth (19–21). Studies that examined the impact of meth on neuronal health in PWH showed that those who use meth have decreased neuronal health compared to people without HIV who use meth and compared to PWH who do not use meth (22, 23). Other studies have shown that PWH who use meth have increased neurocognitive impairment compared to the other groups (24, 25). These data suggest that meth may exacerbate HIV neuropathogenesis in PWH.
HIV entry into the CNS, which is critical to development of HIV-NCI, occurs, in part, by the transmigration of HIV-infected monocytes across the BBB (26–30). These infected monocytes produce virus that can infect and activate resident CNS cells (31, 32). This results in production of host and viral factors that are neurotoxic and can also recruit additional monocytes from the peripheral blood into the CNS. Intermediate monocytes, a specific subset of monocytes that expresses surface CD14, the LPS co-receptor, and CD16, the FcγRIII receptor, are key mediators of this process (27). The CD14+CD16+ intermediate monocytes are referred to as mature monocytes throughout the text. These cells are preferentially infected with HIV and preferentially transmigrate across a BBB model compared to other monocyte subsets (28, 33, 34). These monocytes are increased in the peripheral blood of PWH and they are increased further in those who use substances, including meth, demonstrating that meth can affect these monocytes (35–37). The infected monocytes can differentiate into long-lived macrophages that help sustain viral persistence, neuroinflammation, and neuronal damage. In vivo studies showed that meth increased the population of CD14+CD16+ macrophages and the amount of virus in the brains of Simian Immunodeficiency Virus (SIV) infected macaques (38, 39). PWH who use meth were shown to have increased plasma and cerebrospinal fluid (CSF) viral loads despite adherence to ART (23, 40, 41). These studies suggest that meth may increase viral replication as well as the entry of HIV-infected mature monocytes into the CNS, thus contributing to viral seeding/reseeding and exacerbated neuroinflammation and subsequent neuronal damage.
The effects of meth on peripheral HIV-infected mature monocytes that can contribute to HIV neuropathogenesis in PWH have not been extensively characterized. In this study, we performed RNA sequencing of HIV-infected mature monocytes treated with or without meth. We demonstrate that meth treatment of HIV-infected mature monocytes increases expression of transcripts related to cytoskeletal rearrangement, cell movement, and extravasation. We also show that meth decreases expression of transcripts related to antigen presentation, pathogen recognition, and inflammatory responses. Additionally, we found changes in mitochondria related transcripts that could affect the bioenergetics of these cells. These changes could result in increased transmigration of mature monocytes into the CNS, cause dampening of the immune response, which can impair the ability of these cells to fight pathogens, and impact proper functioning of monocytes. These effects on mature monocytes can contribute to the pathogenesis of HIV-NCI in PWH who use meth.
Leukopaks from anonymous healthy donors were obtained from New York Blood Center. Institutional Review Board (IRB) approval for these studies was obtained from the Einstein Human Research Protection Program (HRPP) at Albert Einstein College of Medicine (IRB no. 1994-0003). Isolation and culture of primary mature monocytes was performed as previously described (28). Briefly, peripheral blood mononuclear cells (PBMC) were isolated from leukopaks using Ficoll-Paque PLUS (GE Healthcare, Uppsala, Sweden) density centrifugation. Monocytes were positively selected from the PBMC using Miltenyi Beads CD14 Selection Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to manufacturer’s instructions. Isolated CD14+ monocytes were resuspended at a concentration of 2x106 cells/ml in monocyte media containing RPMI 1640 1X (Gibco, Grand Island, New York) supplemented with 5% fetal bovine serum (Sigma-Aldrich, St. Louis, Missouri), 10% human serum (Sigma-Aldrich, St. Louis, Missouri), 1% HEPES (Sigma-Aldrich, St. Louis, Missouri), 1%Pen-Strep (Gibco), and 10ng/ml macrophage colony stimulating factor (M-CSF) (Peprotech, Rocky Hill, New Jersey). Cells were cultured non-adherently in Teflon coated flasks at 37°C with 5% CO2 for 2 days to increase the population of mature monocytes in culture (28, 33, 42). Using this culturing technique, we previously showed that the number of mature monocytes is increased from 5-10% to approximately 70-90% (33).
Mature monocytes were divided into 2 flasks, and cells in each flask were resuspended at 10x106 cells/ml in fresh monocyte media and infected for 8 h with 1 ug p24/ml of HIVADA isolate prepared as described (43). This technique facilitates virus uptake by the cells in vitro as established previously in our laboratory (43). Virus was removed from cells by centrifugation, cells were resuspended at 2x106 cells/ml in fresh media, and cultured non-adherently for 3 additional days to facilitate viral infection and replication. Three days post-infection, 50µM Methamphetamine hydrochloride, resuspended in sterile ddH20 (Sigma-Aldrich), was added to one of the flasks for 6 h. This time point is in the range within when meth reaches peak concentration in plasma of meth users (44). The meth concentration chosen is within the range of levels found in plasma of people who use meth (45). After treatment, cells from both flasks were centrifuged for use in flow cytometry and for RNA collection. Supernatants were also collected for HIV p24 quantification using a sensitive HIV p24 alphaLISA kit (Perkin Elmer, Waltham, Massachusetts) as per manufacturer’s protocol to confirm infection of the mature monocytes.
A portion of both meth treated and untreated HIV-infected cells were analyzed for CD14 and CD16 surface expression by flow cytometry. Cells were labeled on ice in the dark for 30 minutes using anti-human CD14 (0.05 ug per 2x105 cells) conjugated with allophycocyanin (APC) (BD Biosciences, San Jose, California; clone M5E2), and anti-human CD16 (0.05 ug per 2x105 cells) conjugated to phycoerythrin/Cy7 (PE-Cy7) (BD Biosciences; clone 3G8). Corresponding isotype matched antibodies and fluorescence minus one (FMO) were used as negative controls. Labeled cells were fixed with 2% paraformaldehyde in 1%BSA/PBS and acquired with the Attune NxT flow cytometer (ThermoFisher Scientific). A minimum of 10,000 events was acquired for each condition. Data analyses were performed using FlowJo software version 10.6.1 (Treestar, Ashland, Oregon).
RNA extraction was performed using Qiagen RNeasy Micro Kit (Qiagen, Hilden, Germany) as per manufacturer’s protocol. To ensure minimal organic contamination, 260/230 ratios were quantified with Nanodrop (ThermoFisher Scientific). RNA quality and concentration were confirmed by Genewiz (South Plainfield, New Jersey). RNA degradation was measured using TapeStation electrophoresis system (Agilent, Santa Clara, California) and RNA was quantified by Qubit fluorimeter (ThermoFisher Scientific). Good quality RNA was determined by an RNA Integrity Number (RIN) of greater than 6. All samples had RIN > 9 (data not shown).
Library preparation, RNA sequencing, and sequence alignment was performed at Genewiz. Briefly, an RNA-sequencing library was prepared using ribosomal RNA depletion followed by fragmentation and random priming, cDNA synthesis, end repair, 5’ phosphorylation, dA-tailing, adapter ligation, PCR enrichment and sequencing. Sequencing was performed using Illumina HiSeq (Illumina, San Diego, California) with a configuration 2 X 150bp. The sequenced reads were trimmed using Trimmomatic version 0.36. The trimmed reads were mapped onto the Homo sapiens GRCh38 reference genome available on ENSEMBL using the STAR aligner version 2.5.2b. The BAM files were generated, and gene hit counts were calculated by using featureCounts from the Subread package version 1.5.2.
Analysis was conducted using R version 4.0.2. We used ComBat_seq function for batch correction to account for the variability inherent in primary cells. Data were then normalized using the calcNormFactors function to account for library size differences among samples. EdgeR function was used to obtain differentially expressed genes (DEG) between meth treated HIV-infected mature monocytes and untreated HIV-infected cells. DEG were considered significant if Benjamini-Hochberg adjusted p-value was less than 0.05 and absolute log 2-fold change was greater than 0.25. To visualize the expression of transcripts between treated and untreated cells, we used ggplot, and heatmap.2 functions.
We performed downstream pathway and functional analyses of the significant DEG using Ingenuity Pathway Analysis (IPA) software version 01-20-04 (Qiagen). We used a p-value of < 0.05 and z-score of either >2 or <-2 to determine significantly predicted affected pathways and functions. GraphPad Prism Software version 9.3.1 (GraphPad Software, San Diego, California) was used to plot the pathway and functional analyses data.
Monocytes were isolated, cultured, infected, and treated as described. Some HIV-infected mature monocytes were also treated with meth for 24 h for protein analyses to capture proteins translated after the 6 h time point used for RNA analyses. Cells were lysed with RIPA buffer containing 1X Halt Protease Inhibitor and Phosphatase Inhibitor cocktail (Thermo Fisher Scientific). Lysate protein concentrations were quantified by Bradford Assay using Protein Assay Reagent Concentrate (Bio-Rad, Hercules, California). Equal amounts of protein from each condition for each donor were electrophoresed by SDS-PAGE under reducing conditions, with subsequent transfer to polyvinylidene difluoride (PVDF) membranes overnight at 4°C. Li-Cor Revert Total Protein Stain (Li-Cor, Lincoln, Nebraska) was used to quantify total protein optical density (O.D.), using the Li-Cor Odyssey Fc System for visualization, and Image Studio software (Li-Cor) version 5.2 for O.D. quantification. Membranes were blocked in SuperBlock Blocking Buffer (ThermoFisher) followed by overnight incubation at 4°C with mouse anti-MMP9 (1:500, ThermoFisher, #MA5-15886) or rabbit anti-Gelsolin (1:1000, Cell Signaling Technology, Danvers, Massachusetts, #12953) in 5% BSA-TBST. HRP-conjugated anti-mouse IgG (Cell Signaling Technology, #7076) was used as secondary antibody for MMP9, and HRP-conjugated anti-rabbit IgG (Cell Signaling Technology, #7074) was used for gelsolin, each at 1:1000 dilution in 5% milk-TBST. Blots were developed using Super Signal West Femto Chemiluminescent Substrate (ThermoFisher) for MMP9, or Western Lightning Plus ECL Oxidizing Reagent Plus (Perkin Elmer) for gelsolin, and visualized and analyzed the same as for total protein. The O.D. of target proteins were normalized to total protein O.D. Data were analyzed as the fold change of normalized O.D. of target proteins in HIV-infected meth treated cells over the normalized O.D. of target protein in untreated cells. Figures were prepared in GraphPad Prism Software version 9.3.1.
Cells infected and treated with meth as described were centrifuged at 1000rpm for 5 minutes at room temperature. The supernatants were collected and assayed using the MMP-9 DuoSet ELISA kit as per manufacturer’s instructions (R&D Systems, Minneapolis, Minnesota). The limit of detection for this kit is 31.25pg/ml. GraphPad Prism Software version 9.3.1 was used to prepare the figures and statistical analyses. The MMP-9 concentrations were normalized to 10 million cells to adjust for the different cell numbers in culture per experiment. Data were analyzed as the fold change of HIV-infected meth treated cells over untreated HIV-infected cells.
For MMP-9 ELISA and HIV p24 alphalisa experiments, D’Agostino-Pearson omnibus normality test was performed to test for gaussian distribution of the data. One sample t-test or paired t-test was used for the normally distributed data, with p < 0.05 considered statistically significant.
The mechanisms by which meth contributes to HIV-NCI in PWH are not incompletely characterized. We examined the effects of meth on the transcriptome of HIV-infected mature monocytes by RNA-sequencing to identify potential mechanisms by which meth contributes to neuropathogenesis in PWH. Primary monocytes from three independent healthy donors were positively isolated from leukopaks, cultured and infected with HIV, and treated with meth as described. RNA from HIV-infected and HIV-infected meth treated cells was then sequenced, mapped onto the genome, counted, and then we performed principal component analysis (PCA) to determine similarities in gene expression patterns among samples. Separate samples from the same donors clustered together (Figure 1A). To reduce the bias in downstream differential gene expression analyses that could be introduced by this similar clustering, we performed batch correction that accounts for the donor variation commonly seen in primary cells from different people (Figure 1B). We identified 121 upregulated transcripts and 137 downregulated transcripts in HIV-infected mature monocytes treated with meth relative to untreated infected cells (Figure 1C and Table S1). Some of the top upregulated transcripts are involved in cell movement signaling pathways including NCS1, a regulator of G-protein coupled receptor phosphorylation, and RASALI, a GTPase-activating molecule (Figure 1D). Meth also upregulated cytoskeletal binding molecules including PLEC (Figure 1D). Meth downregulated transcripts including ND6, a mitochondria molecule involved in oxidative phosphorylation, and TLR7, a toll like receptor (TLR) involved in antiviral immune response (Figure 1D). Meth also downregulated transcripts including HLA-DPA1 and HLA-DPB1 that are important in antigen presentation. This differential gene expression profile induced by meth could lead to changes in functions of HIV-infected mature monocytes contributing to increased cell migration, dysregulated immune response, and consequently neuropathogenesis in PWH who use meth.
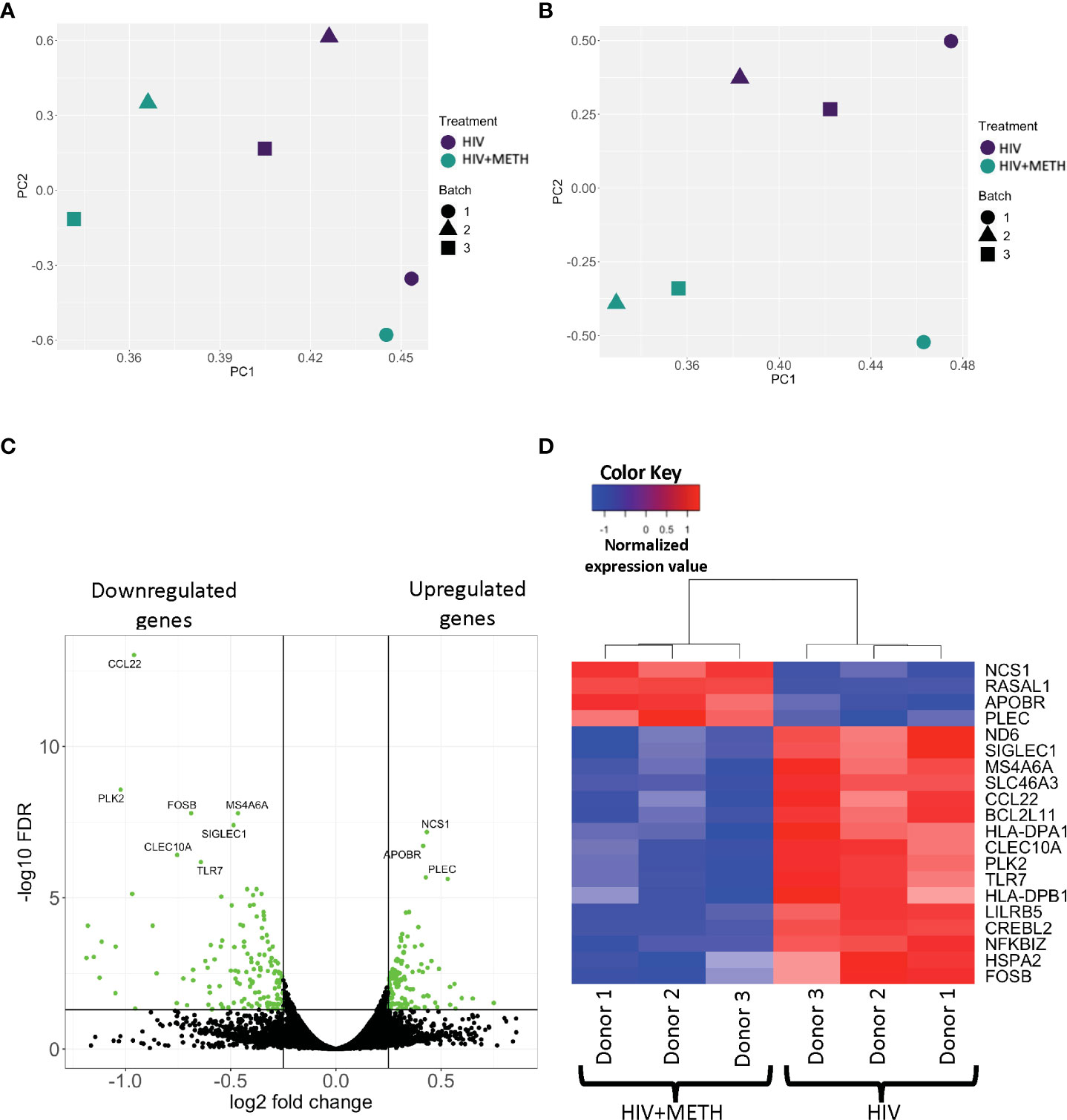
Figure 1 RNA sequencing analyses demonstrate differential gene expression in HIV-infected mature monocytes treated with meth compared to untreated HIV-infected cells. (A, B) Principal component analysis (PCA) plot of sample clustering based on gene expression patterns before (A) and after (B) batch correction. Each data point represents an individual sample, and the data shapes represent the individual donors. The y-axis and x-axis represent the first and second principal components, respectively. HIV-infected mature monocytes treated with meth are shown in blue and untreated HIV-infected mature monocytes are shown in green. (C) A volcano plot showing overall DEG between the HIV-infected mature monocytes treated with or without meth. Each dot represents a gene that was either statistically significantly different(green) or unchanged (black). The y-axis represents -log false discovery rate (FDR), and the x-axis represents log 2-fold change. (D) A heat map representing normalized gene expression levels of the top 20 DEG between HIV-infected mature monocytes treated with meth or untreated. Each column represents an individual sample, and each row represents a gene. The color scale represents lower (blue) to higher (red) gene expression levels.
To ensure that meth did not affect cell survival or mature monocyte surface markers, a portion of infected monocytes was analyzed by flow cytometry for CD14 and CD16. Meth treatment did not change the survival, nor the percentage of mature monocytes (Supplementary Figures 1A, B). We confirmed HIV infection by quantifying HIV p24 protein in cell supernatant with AlphaLISA. Meth increased the amount of HIV released from cells from each independent donor (Supplementary Figure 2). This is consistent with other studies that reported increased HIV infection in other cell types including macrophages (46, 47).
We performed IPA of DEG to identify predicted functions of mature monocytes that are impacted by meth. We found that meth downregulated pathogen recognition and binding molecules TLR5, TLR7, TLR8, and SIGLEC1, antigen presenting MHC transcripts HLA-DMA, HLA-DMB, and HLA-DPA1, and TNFSF9, NOD2, and CCL22, molecules involved in inflammatory responses (Table S2 and Figure 2A, B). Downregulation of these transcripts by meth could lead to dysregulated immune responses by impairing the ability of monocytes to respond to pathogens and other inflammatory related stimuli. Infection by virus was predicted to be activated in meth treated HIV-infected mature monocytes compared to untreated infected cells (Table S2 and Figure 2A). Some of the transcripts related to infection by virus that were decreased include TRIM22, NOD2, IFITM3, and SPHK1 (Figure 2C). These changes could lead to a decreased immune response to fight viruses, leading to increased viral infection in the meth treated HIV-infected mature monocytes that can contribute to viral reservoir persistence.
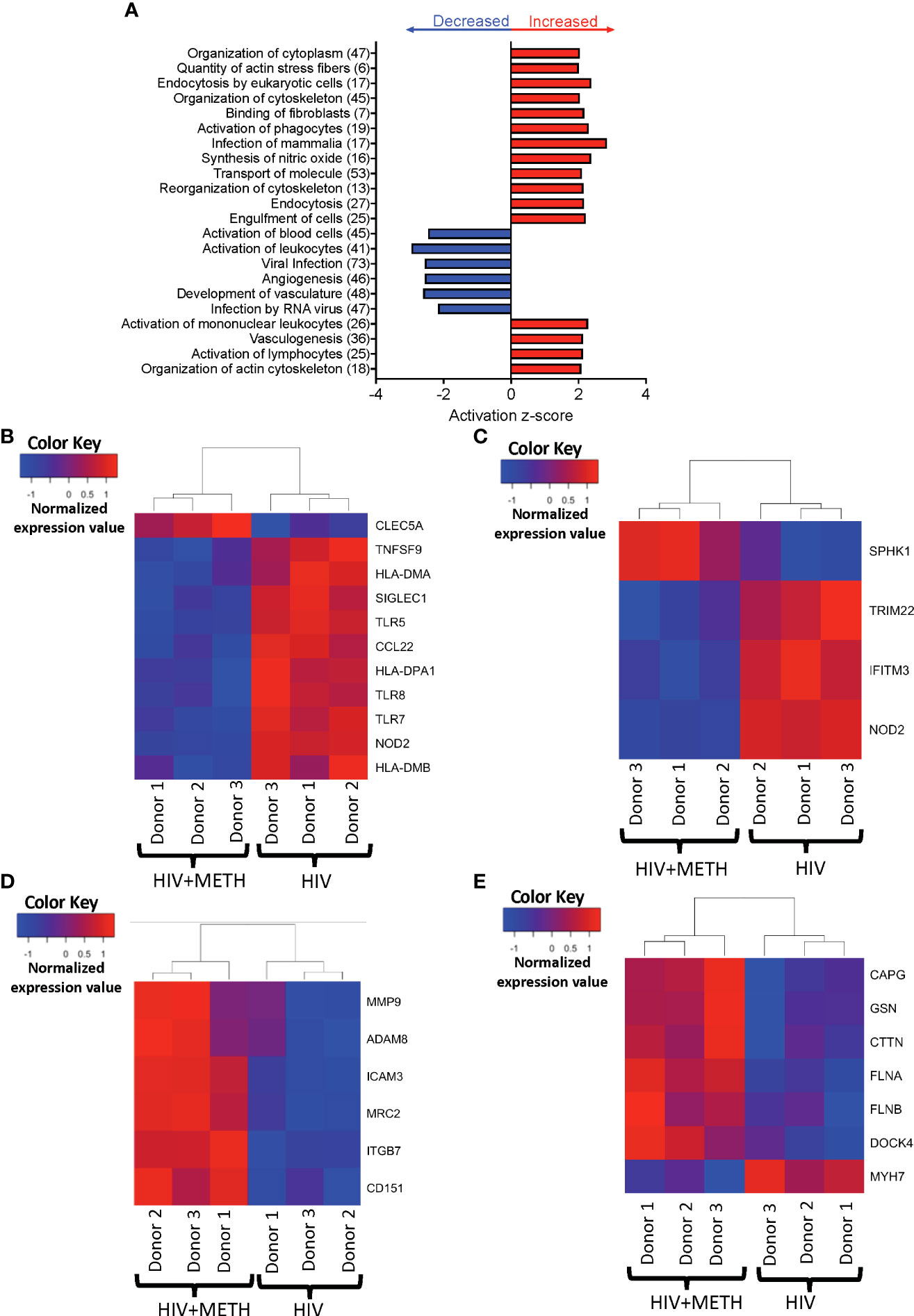
Figure 2 HIV-infected mature monocytes treated with meth compared to untreated HIV-infected cells have a predicted increase in viral infection and cytoskeletal rearrangement and decrease in cell activation. (A) Top functions predicted to be increased (red) or decreased (blue) in the meth treated compared to untreated HIV-infected cells. The y-axis represents the list of functions, and the numbers in the parenthesis represent the quantity of molecules that were differentially expressed after meth treatment. The x-axis represents activation z-score. (B–E) Heat maps representing normalized expression levels of some genes involved in (B) activation of leukocytes, (C) viral infection, (D) cell adhesion and matrix degradation, and (E) organization of cytoskeleton in HIV-infected mature monocytes treated with and without meth. Each column represents an individual sample, and each row represents a gene. The color scale represents lower (blue) to higher (red) gene expression levels. The dendrograms show unsupervised clustering of samples.
Our data showed meth increased transcripts involved in cell adhesion and matrix degradation, including MMP-9, MRC2, ADAM8, ITGB7, CD151 and ICAM3 (Table S1 and Figure 2D). We also found that meth increased expression of molecules involved in cytoskeletal arrangement including MYH7, MYH8, CTTN, GSN, FLNA, FLNB, DOCK4, and CAPG (Table S2 and Figure 2E). The proteins encoded by these transcripts are important in cytoskeletal rearrangement that is needed for cell motility and invasion. These changes may result in increased HIV-infected mature monocyte migration into the CNS. The entry of these cells into the CNS can lead to infection and activation of other CNS cells, and production of inflammatory mediators including host and viral proteins, that can contribute to neuronal damage and HIV-NCI.
We determined potential biological pathways that may be changed in HIV-infected mature monocytes treated with meth using IPA of the DEG. We found that pathways including neuroinflammation signaling, T-cell receptor signaling, and oxidative phosphorylation were predicted to be downregulated, whereas RHOGDI and MSP-RON signaling pathways were predicted to be upregulated (Figure 3A, Table S3). We further examined transcripts involved in neuroinflammation signaling pathways and signaling with T-cell receptor. We found that molecules such as Major Histocompatibility Complex (MHC) class II molecules including HLA-DMA, HLA-DMB, HLA-DPA1, HLA-DQB1, HLA-DRA, and HLA-DRB, and toll like receptor transcripts, TLR5, TLR7, and TLR8, were downregulated by meth (Figures 3B, C), which is consistent with predicted pathway changes. We also found meth downregulated CISD2 and VMP1 which are involved in the endosomal-lysosome pathway as part of antigen processing (Table S1). These suggest that meth may impair the ability of HIV-infected mature monocytes to recognize, process, and present antigens to T-cells. This could lead to viral immune evasion and persistence in the CNS. We also found that mitochondrial transcripts COX2, ND4, ND5, and ND6 were downregulated contributing to the predicted decrease in oxidative phosphorylation (Figure 3D). Additionally, we found downregulation of the ATP synthase transcript, ATP8, and upregulation of transcripts involved in glycolysis, such as ENO2 and PFKP (Table S1). These could lead to bioenergetic changes in the monocytes, and potentially affect monocyte functions such as fighting pathogens and immune surveillance. Molecules involved in RHOGDI signaling including CFL1, ITGB7, MYH7, MYH9, and RHOU, and MSP-RON pathways such as ST14 were upregulated by meth (Figure 3E and Table S2). The increased expression of these molecules could lead to downstream activation of signaling molecules that may contribute to changes in the biology of these monocytes.
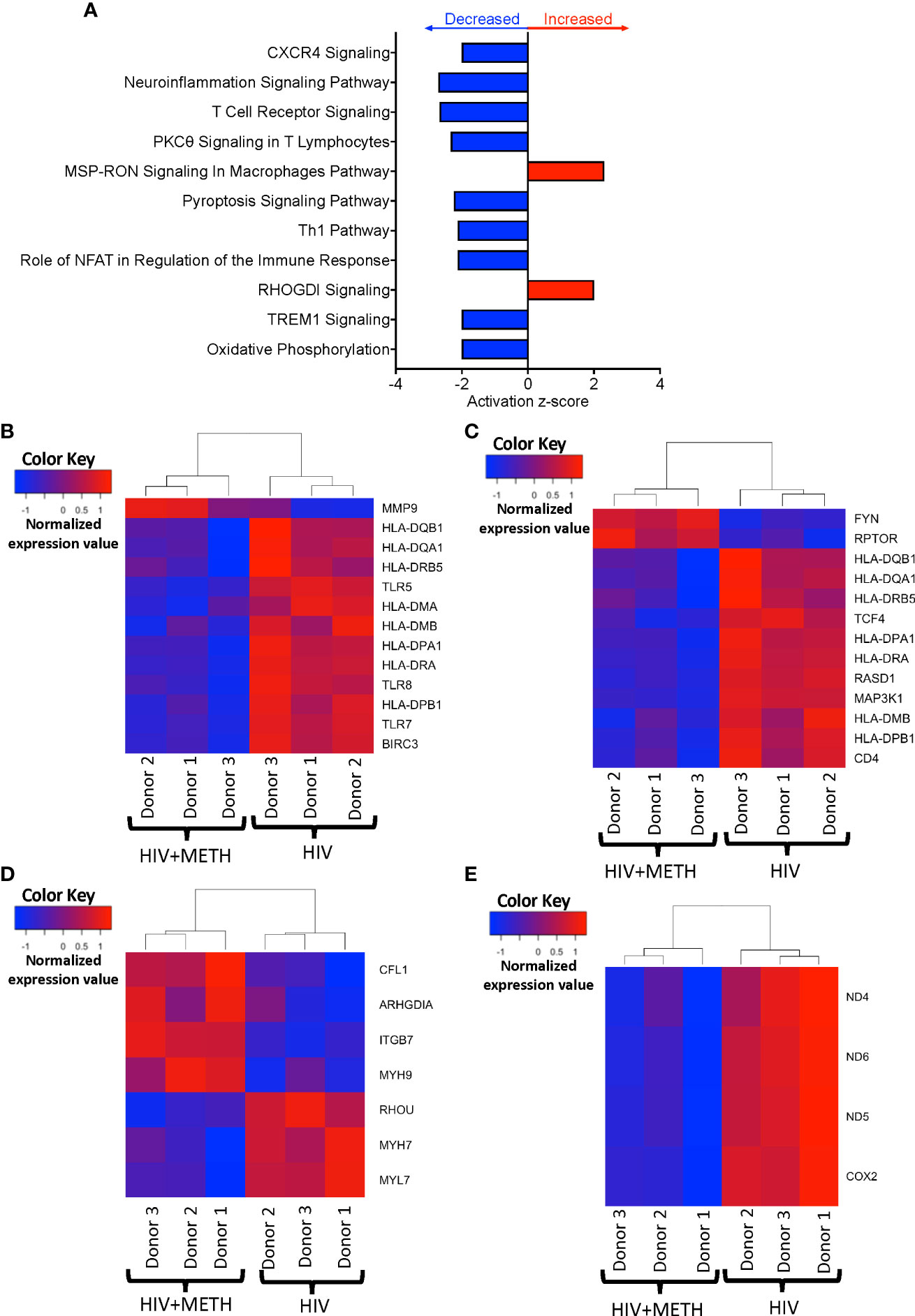
Figure 3 HIV-infected mature monocytes treated with meth have decreased expression of genes related to antigen presentation, pathogen recognition, and oxidative phosphorylation, and upregulation of genes related to cellular RHO-GD1 signaling pathways. (A) Top cellular pathways predicted to be increased (red) or decreased (blue) in meth treated compared to untreated HIV-infected cells. The y-axis represents the list of pathways, and the x-axis represents activation z-score. (B–E) Heat maps representing normalized expression levels of some genes involved in (B) neuroinflammation signaling pathways, (C) T-cell receptor signaling, (D) oxidative phosphorylation, and (E) RHO-GDI signaling in HIV-infected mature monocytes treated, or not, with meth. Each column represents an individual sample, and each row represents a gene. The color scale represents lower (blue) to higher (red) gene expression levels. The dendrograms show unsupervised clustering of samples.
IPA identified cytoskeletal rearrangement as a function predicted to be increased by meth in mature monocytes. One specific DEG that was increased by meth in this pathway is Matrix metalloproteinase-9 (MMP-9) (Table S1). We validated protein expression of MMP-9 because metalloproteinases have been shown to be important in neuropathogenesis (48). Animal studies showed that blocking MMP-9 reduced neuroinflammation in multiple sclerosis animal model (49). MMP-9 degrades extracellular matrices and may contribute to neuroinflammation by increasing monocyte migration into the CNS (50), and/or by activating chemokines such as CXCL8 that can contribute to leukocyte infiltration (49). To validate this RNA-seq finding, we determined protein expression of MMP-9 in HIV-infected mature monocytes treated with or without meth, by Western blotting and by ELISA. MMP-9 can exist as pro- and active forms. We found that intracellular levels of both pro- and active MMP-9 in HIV-infected mature monocytes treated with meth for 6 h appeared to be increased (Figures 4A, B). There was no difference in MMP-9 after 24 h of meth treatment (Figure 4B). While the increase in intracellular MMP-9 at 6 hours of meth was not reflected by an increase in extracellular MMP-9 by ELISA (Figure 4C), we found that extracellular MMP-9 was statistically increased after 24 h meth treatment relative to untreated infected cells (Figure 4C). It could be that after 24 h of meth treatment, HIV-infected mature monocytes are secreting more MMP-9 into the extracellular environment than what is being maintained intracellularly, such that increased protein level cannot be detected intracellularly at that time point. Increased MMP-9 could contribute to increased transmigration of mature monocytes across the BBB into the CNS by degrading the basement membrane, and by increasing recruitment of mature monocytes by increasing chemokine activation.
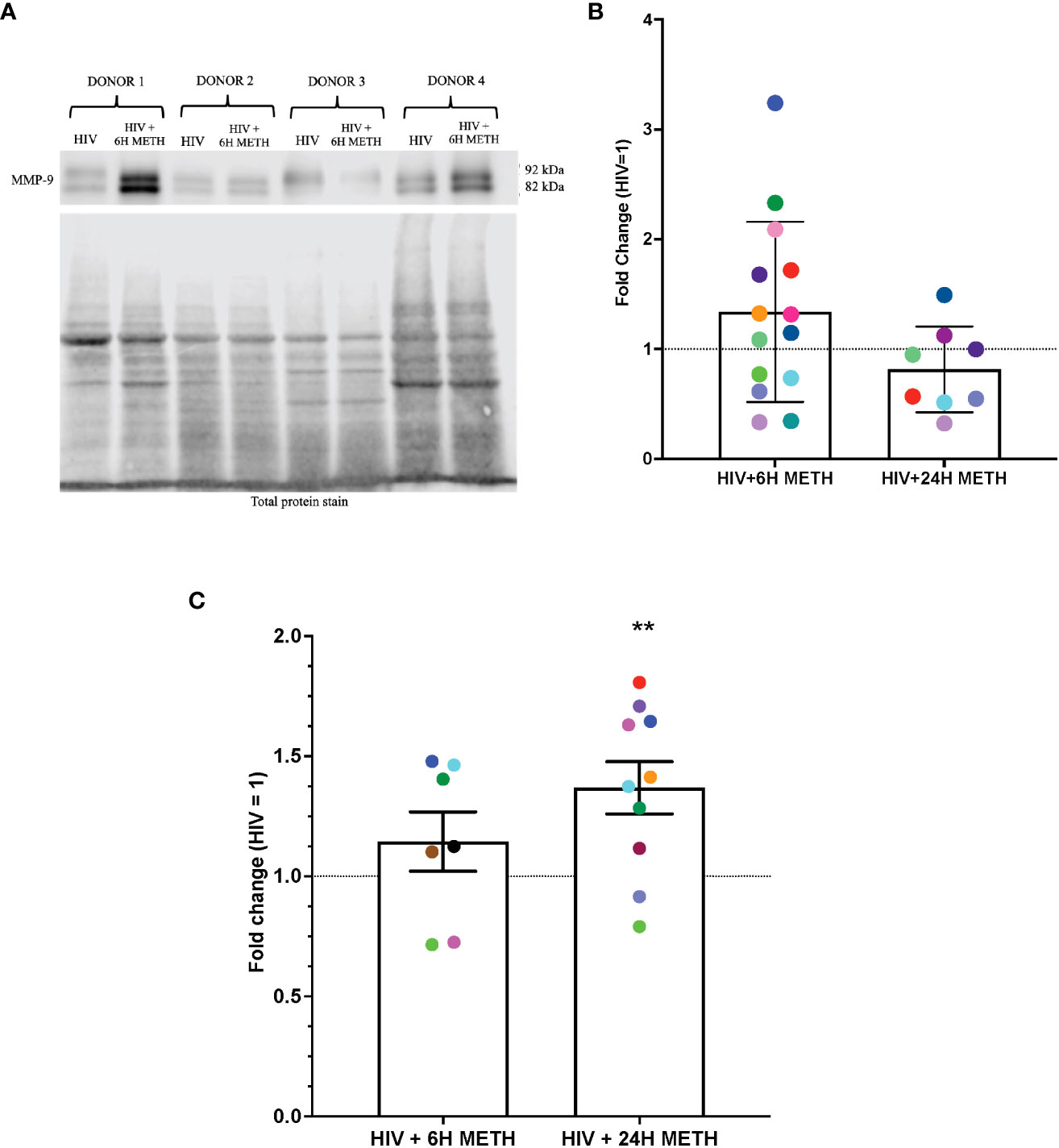
Figure 4 MMP-9 protein is increased in HIV-infected mature monocytes treated with meth. (A) A representative western blot with corresponding total protein stain of HIV-infected mature monocytes either untreated or treated for 6 h with meth. (B) Fold change of normalized MMP-9 protein from HIV-infected mature monocytes treated with meth over untreated HIV-infected mature monocytes. Each colored dot represents an individual donor. The columns and error bars depict mean and standard deviation (SD), respectively. n=7–14. (C) Fold change of normalized MMP-9 protein in supernatants of HIV-infected mature monocytes treated with meth over untreated HIV-infected cells. Each colored dot represents an individual donor. The columns and error bars depict mean and standard error of the mean (SEM), respectively; n=7-10; **, p<0.005 by the one sample t-test.
IPA analyses of DEG also showed that organization of actin cytoskeleton and cell movement of mature monocytes treated with meth was predicted to be increased. One transcript in this pathway that was increased by meth is GSN (Table S1), which encodes for Gelsolin, an actin binding protein involved in severing and capping actin filaments, an important step in cytoskeletal rearrangement needed for cell movement (51). We validated RNA-seq data by performing western blotting of gelsolin, in HIV-infected cells treated with or without meth. We validated gelsolin expression because it is one of the most abundant actin binding proteins that regulate cytoskeletal arrangement, a necessary process in monocyte migration (52). We found that 6 h of meth treatment increased intracellular gelsolin in 7 out of 9 donors (Figures 5A, B). While there is donor variability in gelsolin after 24 h meth treatment, there is a trend towards increased gelsolin (Figures 5A, B).
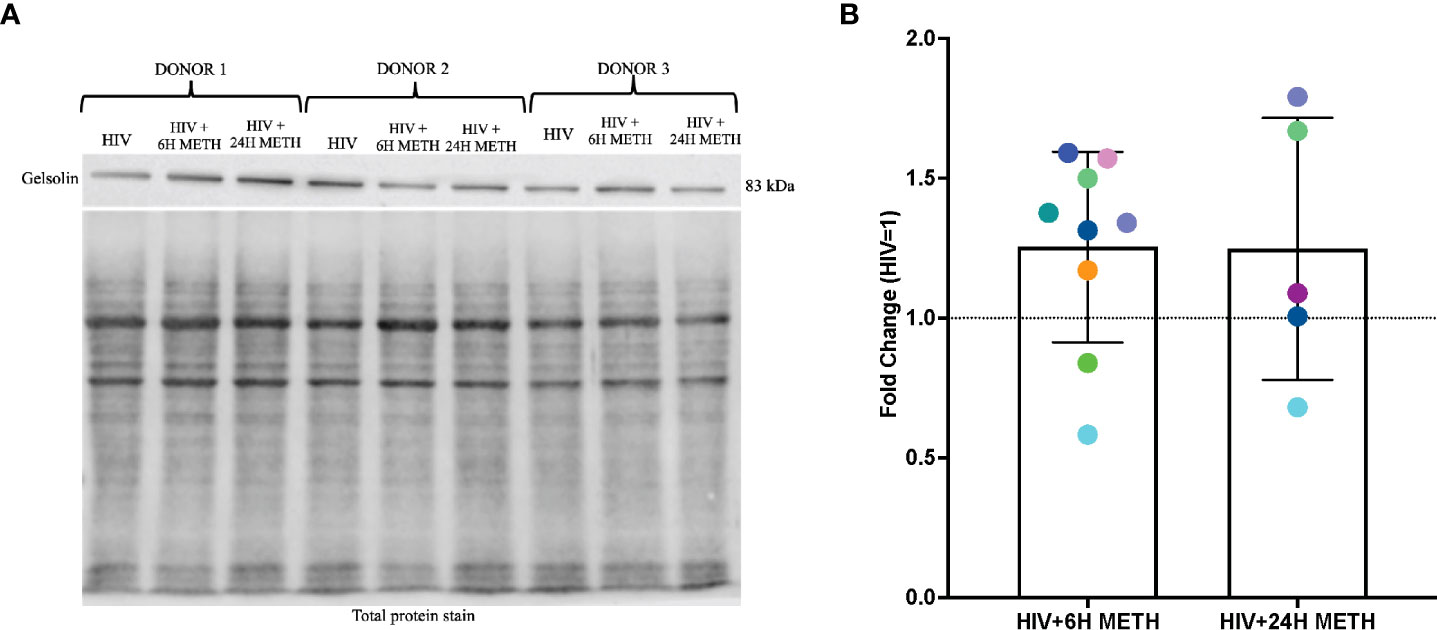
Figure 5 Gelsolin protein is increased in HIV-infected mature monocytes treated with meth. (A) A representative gelsolin western blot with corresponding total protein stain of HIV-infected mature monocytes either untreated or treated for 6 h or 24 with meth. (B) Fold change of gelsolin of HIV-infected mature monocytes treated with meth over untreated HIV-infected cells. Each colored dot represents an individual donor. The column and error bars depict mean and standard deviation (SD), respectively. n = 5-9.
In summary, our results show that meth may contribute to increased monocyte recruitment, through increased expression of molecules related to matrix degradation, cell adhesion and cytoskeletal rearrangement (Figure 6). We also identified downregulation of transcripts involved in antigen presentation and pathogen recognition, and predicted increase in viral infection (Figure 6). These changes can result in immune dysregulation and increased viral reservoirs once these cells enter the CNS. Meth also induced changes in signaling molecules, and downregulated molecules involved in oxidative phosphorylation (Figure 6). These changes could impact the functions of these cells, contributing to ongoing neuropathogenesis in PWH who use meth.
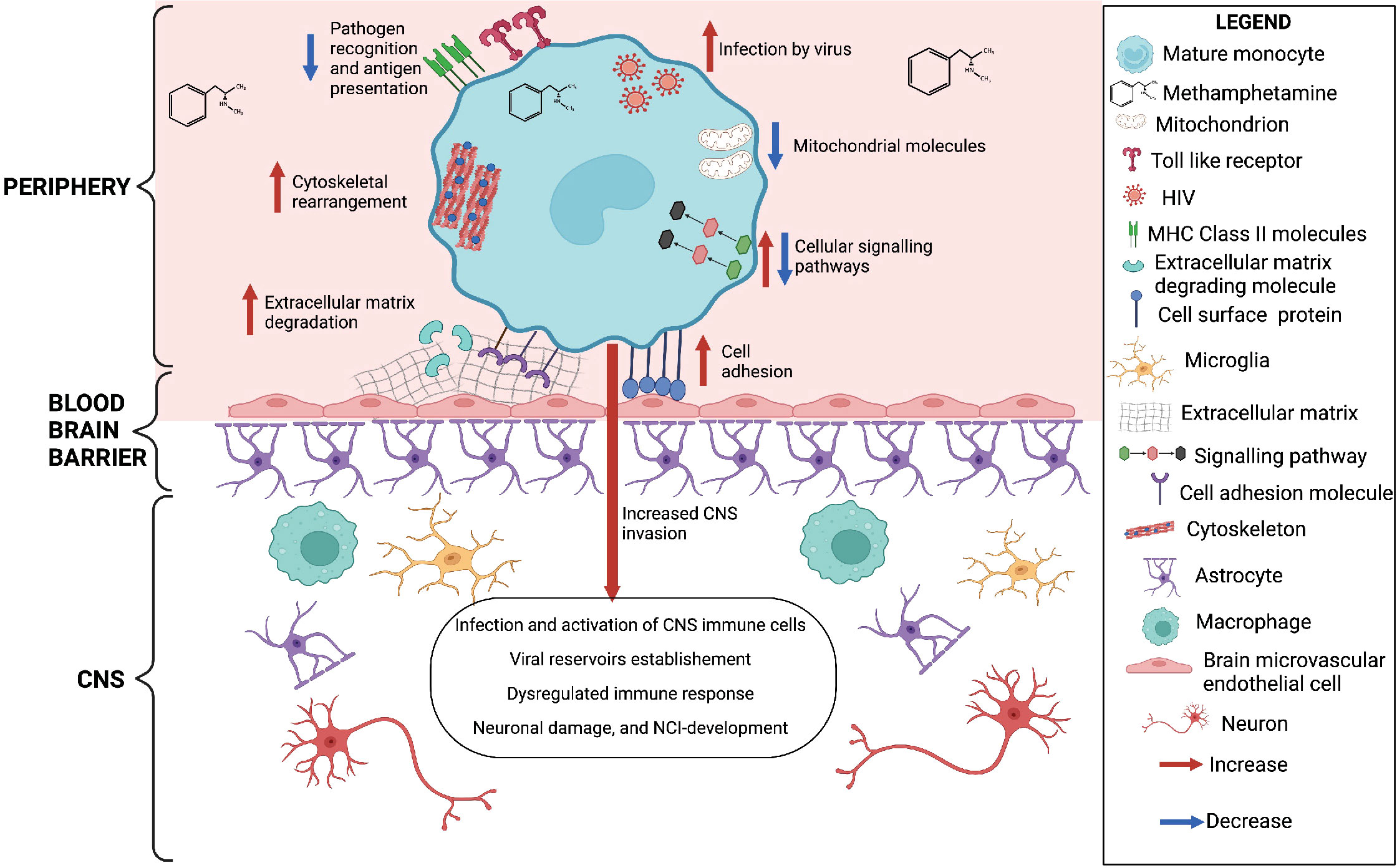
Figure 6 Meth-mediated effects on HIV-infected mature monocytes as identified by RNA sequencing and western blot analyses. Figure created with BioRender.com.
Despite the great success of ART, comorbidities including HIV-NCI persist in PWH who use meth (53). However, the mechanisms by which meth contributes to HIV-NCI have not been fully described. We characterized the impact of meth on HIV-infected mature monocytes using RNA sequencing analyses to identify molecules that are potentially involved in the development HIV-NCI in PWH who use meth. We identified differential gene expression in HIV-infected mature monocytes treated with meth compared to untreated HIV-infected cells. Some DEG include those related to increased cell adhesion, extravasation, cytoskeletal rearrangement, and viral infection of mature monocytes. We also found downregulation of DEG involved in antigen presentation, pathogen recognition, inflammatory response, and oxidative phosphorylation.
HIV-infected monocyte migration into the CNS contributes to viral entry and inflammation which contributes to HIV-NCI. Our study demonstrates that meth treatment of HIV-infected mature monocytes led to the upregulation of transcripts involved in matrix degradation and cell adhesion. MMP-9 is an extracellular protease that breaks down extracellular matrices and BBB basement membranes. This has been shown to lead to BBB disruption and immune cell recruitment into the CNS (50). We found increased MMP-9 RNA and protein in HIV-infected mature monocytes treated with meth compared to untreated HIV-infected cells alone. We also show that meth increases the amount of MMP-9 released into the extracellular environment by HIV-infected cells. Our data are in agreement with findings from other studies that showed that meth increased expression of MMP9 in mice, and of MMP-9 protein in brain microvascular endothelial cells (54, 55). In addition to our western blotting and ELISA data, the increased expression of MMP9 is also supported by our in vitro data showing that meth increases transmigration of HIV-infected mature monocytes across a human BBB model in response to CCL2, a chemokine elevated in the CNS of PWH (data not shown). We found increased levels of MRC2 and ADAM8 in meth treated HIV-infected cells. MRC2, a member of mannose receptor family, binds and internalizes collagen contributing to matrix degradation and turnover (56). ADAM8, a member of a disintegrin and metalloproteinase family, is involved in integrin activation, cell adhesion, migration, and break down of extracellular matrix components to promote cell movement (57). ADAM8 has also been shown to increase cell migration through upregulation of MMP-9 (58). The increased levels of MMP-9, MRC2 and ADAM8 after meth treatment may contribute to increased degradation of extracellular matrix and basement membrane components, and consequently migration of HIV-infected mature monocytes into the CNS. This may lead to accumulation of infected cells contributing to viral reservoirs and CNS damage.
Monocyte adhesion to brain microvascular endothelial cells is important during monocyte transmigration into the CNS. We show that meth increases expression of the cell adhesion-related transcripts ITGB7, ICAM3, CD151, and ICAM3. ITGB7 encodes a protein that heterodimerizes with either CD49d or CD103 to form α4β7 and αEβ7 integrins, respectively. These integrins bind with their ligands MADCAM-1 and E-cadherin, respectively, promoting cell adhesion (59). ITGB7 downregulation was reported to decrease adhesion and migration of multiple myeloma cells (60). In addition, ITGB7 is important in T-cell homing in the gut and is involved in the pathogenesis of inflammatory bowel disease (61). ICAM-3 is an adhesion molecule that can interact with LFA-1 extracellularly or ezrin/radixin/moesin intracellularly, contributing to tumor metastasis (62). CD151, a tetraspanin, interacts with integrins promoting cell matrix adhesion (63). In T-cells, CD151 interacts with integrins LFA-1 and VLA-4, and upon external stimuli, it can lead to actin rearrangement and cell migration (64). In mature monocytes, CD151 is involved in formation of multinucleated giant cells that can occur during inflammatory processes (65). CD151 also forms homotypic interactions that lead to increased cell movement and MMP-9 production (66). CD151 can interact with JAM-A, a molecule increased on the surface of HIV-infected mature monocytes, contributing to increased cell migration (33, 67). Changes in these molecules may contribute to cell adhesion, downstream signaling activation, and their consequent migration from the periphery into the CNS.
Actin cytoskeleton rearrangement is key for monocyte transmigration into the CNS across the BBB. Myosin II, a motor protein that binds F-actin, contributes to cell attachment, spreading and migration (68). We found that meth upregulates myosin II heavy chain molecule, MYH9, and there is a predicted increase in cytoskeletal rearrangement. We also show there is an increase in CFL1, which encodes cofilin. Cofilin is an actin binding protein that cleaves F-actin providing ends for polymerization and depolymerization and is important in chemotaxis and directional cell movement (69, 70). This could lead to increased actin rearrangement that contributes to cell movement. Our findings are in agreement with another study that showed increased expression of cofilin with activation of RhoA/ROCK signaling pathway leading to cytoskeletal rearrangement in rat brain microvascular endothelial cells treated with meth (55).
Meth treatment of HIV-infected mature monocytes also upregulated FLNA and GSN, which encode filamin A and gelsolin, respectively. Filamins are actin binding and cross-linking proteins that facilitate cell spreading and migration (71, 72). In neutrophils, a decrease in Filamin A resulted in decreased cell migration and reduced myosin-II activation (73). Gelsolin is an actin binding protein implicated in migration and invasion of cancer cells (74, 75). This protein severs and caps F-actin, which is needed for actin polymerization during cell movement (51). We found increased gelsolin RNA and demonstrated a trend towards increased gelsolin protein in HIV-infected mature monocytes treated with meth compared to untreated cells. One study showed that mouse macrophages that do not express gelsolin have decreased chemotaxis and migration, suggesting this molecule is important for myeloid cell migration (76). In mouse osteoclasts, gelsolin promotes podosome formation and cell motility (77). Meth-induced changes in the expression of actin binding proteins could contribute to changes in cytoskeletal arrangement mediating cell movement and entry into the CNS.
Several signaling pathways regulate monocyte functions such as movement and differentiation that may contribute to HIV neuropathogenesis. Our analyses showed changes in expression of transcripts related to signaling pathways and predicted an increase in RHOGDI signaling. We found increased expression of DOCK4, a guanine exchange factor, in meth treated HIV-infected cells. DOCK4 activates Rac1 and promotes actin reorganization and formation of lamellipodia at the leading edge of breast cancer cells (78). Our results also showed an increased expression of NCS1, a molecule that encodes a calcium binding molecule that regulates G protein coupled receptor phosphorylation and promotes cell movement (79). We found a predicted activation of the MSP-RON pathway in meth treated HIV-infected mature monocytes. Macrophage stimulating protein (MSP) is activated by matriptase, encoded by ST14, a transcript upregulated by meth. This activated form binds to Recepteur d’origine Nantais (RON), resulting in increased cell migration and matrix invasion (80). Matriptase mediated downstream protein kinase C (PKC) signaling that led to increased MMP-9 and metastasis of a breast cancer cell line (81). Dysregulation of these molecules can change signaling pathways in mature monocytes that facilitate their entry into the CNS.
Mature monocytes are involved in immune responses through their ability to process and present antigens, activate T-cells, and release inflammatory mediators in the CNS. Once in the CNS, monocytes can also differentiate into perivascular macrophages that contribute to antigen presentation and inflammatory responses (82). Our analyses showed that meth treatment of HIV-infected mature monocytes decreases expression of many antigen presenting MHC class II molecules (HLA-DMA, HLA-DMB, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRA). These are important for processing and presenting antigens derived from extracellular pathogens (83). Downregulation of MHC class II molecules can reduce the ability of the mature monocytes to present antigens to CD4+ T-cells, compromising immune response to pathogens. We also show decreased CISD2 and VMP transcripts, involved in endosomal-lysosomal pathways important for antigen presentation by MHC molecules (84–86). Our findings are consistent with a study that showed that meth decreases MHC Class II antigen processing and presentation by inhibiting the endosomal-lysosomal pathway in dendritic cells (87). Although a separate report showed that the ability of MHC Class II antigen processing and presentation is not impaired in total monocytes from PWH, our data indicate that in the presence of meth, this may be dysregulated in HIV-infected mature monocytes (88). This could inhibit clearance of pathogens in the CNS.
Toll like receptors (TLR) are pattern recognition receptors (PRR) present on immune cells including monocytes. TLR are important for inflammation and immune responses to pathogens. They are activated upon binding of pathogen-associated molecular patterns, resulting in downstream signaling that leads to interferon production. This can mediate recruitment of additional immune cells and activation of antiviral immune responses (89). Our data show that meth downregulates expression of TLR7, an endosomal receptor for single stranded RNA (89, 90). TLR7 was shown to trigger a Type I interferon response upon HIV RNA binding in dendritic cells (91, 92). Meth also decreases TLR8, another endosomal receptor for single stranded RNA (89, 90). TLR8 in monocytes recognize HIV, leading to inflammasome activation (91, 92). Meth-mediated downregulation of TLR7 and TLR8 in HIV-infected mature monocytes could be impaired, resulting in reduced ability to clear HIV after entering the CNS. In addition, activating TLR signaling pathways is a potential mechanism for reactivation of latent virus within viral reservoirs in eradication approaches (93–95). A TLR8 receptor agonist was shown to re-activate latent HIV in CD4 T-cells (95). Downregulation of TLR8 by meth in HIV-infected mature monocytes could reduce efficacy of this strategy in PWH who use meth.
Our functional analyses predicted an increase in viral infection of meth treated HIV-infected mature monocytes compared to untreated HIV-infected cells. We found increased levels of HIV release from cells after 6h of meth treatment, although we did not quantify intracellular HIV levels. The early changes in transcripts expression related to infection by viruses could contribute to increased HIV infection of meth treated mature monocytes at later timepoints. This is consistent with studies that reported increased HIV infection in meth treated macrophages, monocytes, microglia, and dendritic cells treated with meth for various amounts of time (46, 96–99). The prediction of increased viral infection may be due, in part, to downregulation of the antiviral related molecules IFITM3, and TRIM22. Interferon-induced transmembrane proteins (IFITM) can inhibit HIV entry and viral protein synthesis, and IFITM3 was reported to be involved in antiviral responses by trafficking vesicles containing viruses into the lysosome for degradation (100–102). Downregulation of IFITM3 by meth could result in increased viral infection of mature monocytes due to impaired ability to reduce viral entry and/or degradation of the virus. TRIM22, a member of tripartite motif proteins family, also inhibits HIV replication (103–106). Thus, downregulation of TRIM22 in mature monocytes can promote increased viral production.
Under homeostatic conditions, cells depend upon mitochondrial oxidative phosphorylation as the main source of energy. We found decreased expression of mitochondria Complex I gene transcripts, ND4, ND5, and ND6, and the ATP synthase transcript, ATP8, in meth treated HIV-infected mature monocytes, and a predicted decrease in the oxidative phosphorylation pathway. Downregulation of complex I and ATP synthase molecules could lead to decreased energy production through the oxidative phosphorylation pathway (107). Our results are consistent with a study that found that meth causes mitochondria dysfunction through loss of mitochondrial membrane potential, mitochondria permeability transition pore opening, or dysregulation of mitochondria complexes (108). Another study reported that short meth exposure decreases the oxygen consumption rate and ATP levels in astrocytes (109). Our data show upregulation of transcripts encoding enzymes involved in glycolysis, including ENO2 and PFKP in the meth treated cells. These results are similar to those from another study that showed increased protein levels of ENO2 in immature dendritic cells treated with meth (110). Meth-mediated changes in glycolysis related molecules could shift the metabolic profile of the cells to rely on energy produced by glycolysis. A switch to glycolysis results in release of metabolites that cause downstream signaling that can promote inflammation and cell migration (111, 112). This may contribute to influx of cells into the CNS leading to inflammation and consequent neuronal damage.
Mitochondria complexes I and III are major sources of reactive oxygen species (ROS) which are involved in the regulation of hypoxia, inflammation, and cell death (113). We observed a decrease of complex I transcripts expression after meth treatment. Our data also showed that meth caused a downregulation of the antioxidant transcript SELENOP, and upregulation of an oxidative stress marker, GGT1 (114, 115). These changes can lead to dysregulated ROS production, increased oxidative stress, and contribute to CNS inflammation. One study found decreased levels of Complex I proteins in PBMC from PWH and this correlated with inflammation (116). Additionally, increased ROS has been shown in HIV neuropathogenesis (117). The meth-induced changes indicate a potential ROS imbalance, that can contribute to cellular stress and homeostatic imbalance in the CNS.
Meth can induce the differential gene expression patterns in HIV-infected mature monocytes through activation of various downstream signaling pathways. Identifying these pathways will provide potential upstream targets to limit the impact of meth on specific monocyte functions. Meth is a lipophilic molecule that enters cells either by diffusing across the plasma membrane or by binding dopamine transporters, which are expressed on monocytes (118). Inside the cell, meth can bind to trace amine-associated receptor 1 (TAAR1), an intracellular G-protein coupled receptor (119–121). This can lead to downstream activation of RhoA, PKA or CREB signaling pathways (119–121). CREB is a transcriptional factor that regulates transcription of various genes including those of MHC class II molecules (122). CREB also regulates transcription of cofilin, CFL1, a molecule whose expression was upregulated in our HIV-infected monocytes treated with meth (123). Meth-mediated modulation of all these pathways may contribute to the differential gene expression patterns identified in our study, which may contribute to changes in cellular functions. Thus, targeting these pathways with inhibitors may limit the meth-mediated effects in HIV-infected monocytes.
To determine whether HIV alone affects expression of some of the DEG identified, we examined our previously published single cell RNA sequencing data to compare expression of the transcripts between HIV-infected and uninfected mature monocytes cultured in vitro (42). We showed that HIV alone did not increase expression of many of the cell movement related molecules including MMP9, ADAM8, GSN, and CFL1. However, we found that HIV downregulated expression of some MHC class II molecules including HLA-DMA, HLA-DMB, and HLA-DPA1 (42). This suggests that both HIV and meth downregulate these molecules that could impair the ability of the cells to present antigens, including viral proteins to protect them from being killed by cytotoxic T cells. This can contribute to persistence of viral reservoirs. Although we did not determine the effects of meth on expression of these molecules on uninfected mature monocytes, our data suggest that in the context of HIV, meth could worsen disease pathogenesis by further impairing some of their functions.
Our study did not determine the percentage of cells expressing the identified DEG. To address this, we used our published single cell RNA sequencing data, and found that there was heterogeneous expression of some molecules (42). For example, MMP9, GSN, ND4, and HLA-DMA were present in more than 70% of HIV-infected mature monocytes, whereas molecules such as ITGB7, FLNB, and TLR8 were present in fewer than 50% of the cells (42). Future studies can perform flow cytometry and microscopy studies to evaluate protein expression of these molecules in mature monocytes.
Our data identify potential mechanisms by which meth may exacerbate HIV-NCI in PWH. Some of the limitations of this study include the short period of time for which the cells were treated with meth. The gene expression changes found may not represent those that occur during chronic meth use. HIV-infected mature monocytes are a heterogeneous population comprised of HIV harboring and HIV-exposed cells. This study used bulk RNA sequencing. Therefore, we cannot distinguish the effects of meth on cells that are harboring HIV from the effects on cells exposed to HIV but not harboring the virus. It is also possible that some of the specific gene expression changes might not reflect protein or functional changes. Additional studies are needed to determine the effects of chronic meth exposure on HIV-infected mature monocytes. More assays need to be performed to confirm the RNA sequencing data and to determine the specific effect of meth on HIV harboring and HIV-exposed cells. We also did not perform comparisons among untreated uninfected cells, meth treated uninfected cells, untreated HIV-infected cells, and meth treated HIV-infected cells. Thus, we could not determine the effects of meth with and without HIV. Our future studies will address this. Another limitation of this study is we could not sort for specifically CD14+CD16+ monocytes due to the limited number of cells. However, we showed that the majority of cells were mature monocytes. Future studies will also confirm the identified DEG using flow cytometry and microscopy assays to specifically identify expression of the molecules on cells expressing both CD14 and CD16.
To our knowledge, this is the first study to characterize the impact of meth on HIV-infected mature monocytes, a key cell type in the neuropathogenesis of HIV-NCI. We found that meth may increase monocyte recruitment, viral seeding and reseeding, and immune dysregulation through changes in transcripts involved in matrix degradation, cell adhesion, cytoskeletal arrangement, signaling pathways, antigen presentation, pathogen recognition, viral infection, and oxidative phosphorylation. These data identify potential biological changes that occur in meth treated HIV-infected mature monocytes that can contribute to HIV-NCI in PWH who use meth. These changes could potentially be used for therapeutic targets to reduce infected monocyte entry into the CNS, viral seeding and reseeding, immune response dysregulation, and neuronal damage in PWH who use meth.
The data represented in this study are deposited in the GEO repository, accession number GSE210168.
The studies involving human participants were reviewed and approved by Einstein Human Research Protection Program (HRPP) at Albert Einstein College of Medicine (IRB no. 1994-0003). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
JB and VC designed this study. VC performed the computational data analysis and wrote the manuscript. VC, JW, SM-M, LM, LC, and JB designed and performed the validation experiments. VC, JW, SM-M, LM, LC, and JB contributed to discussions about manuscript content, writing, and editing of the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by NIH grants R01DA044584 (VC, LC, and JB), R01DA048609 (LC, SM-M, and JB), R01MH112391 (SM-M and JB) and 5T32AI007501 (JW).
We would also like to thank The Advanced Technologies and Biomarker Core under the Einstein-Rockefeller-CUNY Center for AIDS Research (P30AI124414).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.952183/full#supplementary-material
1. UNAIDS. Global HIV & AIDS statistics — fact sheet 2022 [cited 2022 march 27th]. Available at: https://www.unaids.org/en/resources/fact-sheet
2. Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-Associated neurocognitive disorder–pathogenesis and prospects for treatment. Nat Rev Neurol (2016) 12(4):234–48. doi: 10.1038/nrneurol.2016.27
3. Cysique LA, Brew BJ. Prevalence of non-confounded HIV-associated neurocognitive impairment in the context of plasma HIV RNA suppression. J Neurovirol (2011) 17(2):176–83. doi: 10.1007/s13365-011-0021-x
4. Spudich S, Robertson KR, Bosch RJ, Gandhi RT, Cyktor JC, Mar H, et al. Persistent HIV-infected cells in cerebrospinal fluid are associated with poorer neurocognitive performance. J Clin Invest (2019) 129(8):3339–46. doi: 10.1172/JCI127413
5. Heaton RK, Clifford DB, Franklin DR Jr., Woods SP, Ake C, Vaida F, et al. HIV-Associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER study. Neurology. (2010) 75(23):2087–96. doi: 10.1212/WNL.0b013e318200d727
6. Heaton RK, Franklin DR Jr., Deutsch R, Letendre S, Ellis RJ, Casaletto K, et al. Neurocognitive change in the era of HIV combination antiretroviral therapy: the longitudinal CHARTER study. Clin Infect Dis (2015) 60(3):473–80. doi: 10.1093/cid/ciu862
7. Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, et al. HIV-Associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol (2011) 17(1):3–16. doi: 10.1007/s13365-010-0006-1
8. Gott C, Gates T, Dermody N, Brew BJ, Cysique LA. Cognitive change trajectories in virally suppressed HIV-infected individuals indicate high prevalence of disease activity. PloS One (2017) 12(3):e0171887. doi: 10.1371/journal.pone.0171887
9. Alford K, Daley S, Banerjee S, Vera JH. Quality of life in people living with HIV-associated neurocognitive disorder: A scoping review study. PloS One (2021) 16(5):e0251944-e. doi: 10.1371/journal.pone.0251944
10. Heaton RK, Marcotte TD, Mindt MR, Sadek J, Moore DJ, Bentley H, et al. The impact of HIV-associated neuropsychological impairment on everyday functioning. J Int Neuropsychol Soc (2004) 10(3):317–31. doi: 10.1017/S1355617704102130
11. Jones JD, Kuhn T, Levine A, Sacktor N, Munro CA, Teplin LA, et al. Changes in cognition precede changes in HRQoL among HIV+ males: Longitudinal analysis of the multicenter AIDS cohort study. Neuropsychology. (2019) 33(3):370–8. doi: 10.1037/neu0000530
12. van Gorp WG, Baerwald JP, Ferrando SJ, McElhiney MC, Rabkin JG. The relationship between employment and neuropsychological impairment in HIV infection. J Int Neuropsychol Soc (1999) 5(6):534–9. doi: 10.1017/s1355617799566071
13. Tozzi V, Balestra P, Murri R, Galgani S, Bellagamba R, Narciso P, et al. Neurocognitive impairment influences quality of life in HIV-infected patients receiving HAART. Int J STD AIDS (2004) 15(4):254–9. doi: 10.1258/095646204773557794
14. Patel S, Parikh NU, Aalinkeel R, Reynolds JL, Dmello R, Schwartz SA, et al. United states national trends in mortality, length of stay (LOS) and associated costs of cognitive impairment in HIV population from 2005 to 2014. AIDS Behav (2018) 22(10):3198–208. doi: 10.1007/s10461-018-2128-z
15. Tozzi V, Balestra P, Serraino D, Bellagamba R, Corpolongo A, Piselli P, et al. Neurocognitive impairment and survival in a cohort of HIV-infected patients treated with HAART. AIDS Res Hum Retroviruses (2005) 21(8):706–13. doi: 10.1089/aid.2005.21.706
16. Vivithanaporn P, Heo G, Gamble J, Krentz HB, Hoke A, Gill MJ, et al. Neurologic disease burden in treated HIV/AIDS predicts survival: a population-based study. Neurology. (2010) 75(13):1150–8. doi: 10.1212/WNL.0b013e3181f4d5bb
17. Hartzler B, Dombrowski JC, Crane HM, Eron JJ, Geng EH, Christopher Mathews W, et al. Prevalence and predictors of substance use disorders among HIV care enrollees in the united states. AIDS behavior (2017) 21(4):1138–48. doi: 10.1007/s10461-016-1584-6
18. Yang X, Wang Y, Li Q, Zhong Y, Chen L, Du Y, et al. The main molecular mechanisms underlying methamphetamine- induced neurotoxicity and implications for pharmacological treatment. Front Mol Neurosci (2018) 11:186. doi: 10.3389/fnmol.2018.00186
19. Potvin S, Pelletier J, Grot S, Hebert C, Barr AM, Lecomte T. Cognitive deficits in individuals with methamphetamine use disorder: A meta-analysis. Addict Behav (2018) 80:154–60. doi: 10.1016/j.addbeh.2018.01.021
20. Wang T-Y, Fan T-T, Bao Y-P, Li X-D, Liang C-M, Wang R-J, et al. Pattern and related factors of cognitive impairment among chronic methamphetamine users. Am J Addictions (2017) 26(2):145–51. doi: 10.1111/ajad.12505
21. Ersche KD, Sahakian BJ. The neuropsychology of amphetamine and opiate dependence: implications for treatment. Neuropsychol Rev (2007) 17(3):317–36. doi: 10.1007/s11065-007-9033-y
22. Chang L, Ernst T, Speck O, Grob CS. Additive effects of HIV and chronic methamphetamine use on brain metabolite abnormalities. Am J Psychiatry (2005) 162(2):361–9. doi: 10.1176/appi.ajp.162.2.361
23. Chana G, Everall IP, Crews L, Langford D, Adame A, Grant I, et al. Cognitive deficits and degeneration of interneurons in HIV+ methamphetamine users. Neurology. (2006) 67(8):1486–9. doi: 10.1212/01.wnl.0000240066.02404.e6
24. Kesby JP, Heaton RK, Young JW, Umlauf A, Woods SP, Letendre SL, et al. Methamphetamine exposure combined with HIV-1 disease or gp120 expression: Comparison of learning and executive functions in humans and mice. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol (2015) 40(8):1899–909. doi: 10.1038/npp.2015.39
25. Rippeth JD, Heaton RK, Carey CL, Marcotte TD, Moore DJ, Gonzalez R, et al. Methamphetamine dependence increases risk of neuropsychological impairment in HIV infected persons. J Int Neuropsychol Soc (2004) 10(1):1–14. doi: 10.1017/S1355617704101021
26. Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci (2006) 26(4):1098–106. doi: 10.1523/JNEUROSCI.3863-05.2006
27. Ellery PJ, Tippett E, Chiu YL, Paukovics G, Cameron PU, Solomon A, et al. The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol (2007) 178(10):6581–9. doi: 10.4049/jimmunol.178.10.6581
28. Veenstra M, Leon-Rivera R, Li M, Gama L, Clements JE, Berman JW. Mechanisms of CNS viral seeding by HIV(+) CD14(+) CD16(+) monocytes: Establishment and reseeding of viral reservoirs contributing to HIV-associated neurocognitive disorders. MBio. (2017) 8(5):e01280-17. doi: 10.1128/mBio.01280-17
29. Fischer-Smith T, Croul S, Sverstiuk AE, Capini C, L'Heureux D, Regulier EG, et al. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol (2001) 7(6):528–41. doi: 10.1080/135502801753248114
30. Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis (2012) 206(2):275–82. doi: 10.1093/infdis/jis326
31. Churchill MJ, Gorry PR, Cowley D, Lal L, Sonza S, Purcell DF, et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J Neurovirol (2006) 12(2):146–52. doi: 10.1080/13550280600748946
32. Ko A, Kang G, Hattler JB, Galadima HI, Zhang J, Li Q, et al. Macrophages but not astrocytes harbor HIV DNA in the brains of HIV-1-Infected aviremic individuals on suppressive antiretroviral therapy. J Neuroimmune Pharmacol (2019) 14(1):110–9. doi: 10.1007/s11481-018-9809-2
33. Williams DW, Calderon TM, Lopez L, Carvallo-Torres L, Gaskill PJ, Eugenin EA, et al. Mechanisms of HIV entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-a, and ALCAM in diapedesis. PloS One (2013) 8(7):e69270. doi: 10.1371/journal.pone.0069270
34. Williams DW, Veenstra M, Gaskill PJ, Morgello S, Calderon TM, Berman JW. Monocytes mediate HIV neuropathogenesis: mechanisms that contribute to HIV associated neurocognitive disorders. Curr HIV Res (2014) 12(2):85–96. doi: 10.2174/1570162x12666140526114526
35. Williams DW, Anastos K, Morgello S, Berman JW. JAM-a and ALCAM are therapeutic targets to inhibit diapedesis across the BBB of CD14+CD16+ monocytes in HIV-infected individuals. J Leukoc Biol (2015) 97(2):401–12. doi: 10.1189/jlb.5A0714-347R
36. Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS. Unique monocyte subset in patients with AIDS dementia. Lancet (9053) 1997:349. doi: 10.1016/s0140-6736(96)10178-1
37. Calderon TM, Williams DW, Lopez L, Eugenin EA, Cheney L, Gaskill PJ, et al. Dopamine increases CD14(+)CD16(+) monocyte transmigration across the blood brain barrier: Implications for substance abuse and HIV neuropathogenesis. J Neuroimmune Pharmacol (2017) 12(2):353–70. doi: 10.1007/s11481-017-9726-9
38. Marcondes MCG, Flynn C, Watry DD, Zandonatti M, Fox HS. Methamphetamine increases brain viral load and activates natural killer cells in simian immunodeficiency virus-infected monkeys. Am J pathology. (2010) 177(1):355–61. doi: 10.2353/ajpath.2010.090953
39. Niu M, Morsey B, Lamberty BG, Emanuel K, Yu F, León-Rivera R, et al. Methamphetamine increases the proportion of SIV-infected Microglia/Macrophages, alters metabolic pathways, and elevates cell death pathways: A single-cell analysis. Viruses. (2020) 12(11):1297. doi: 10.3390/v12111297
40. Carey CL, Woods SP, Rippeth JD, Gonzalez R, Heaton RK, Grant I. Additive deleterious effects of methamphetamine dependence and immunosuppression on neuropsychological functioning in HIV infection. AIDS Behav (2006) 10(2):185–90. doi: 10.1007/s10461-005-9056-4
41. Plankey MW, Ostrow DG, Stall R, Cox C, Li X, Peck JA, et al. The relationship between methamphetamine and popper use and risk of HIV seroconversion in the multicenter AIDS cohort study. J Acquir Immune Defic Syndr (2007) 45(1):85–92. doi: 10.1097/QAI.0b013e3180417c99
42. León-Rivera R, Morsey B, Niu M, Fox HS, Berman JW. Interactions of monocytes, HIV, and ART identified by an innovative scRNAseq pipeline: Pathways to reservoirs and HIV-associated comorbidities. mBio. (2020) 11(4):e01037-20. doi: 10.1128/mBio.01037-20
43. León-Rivera R, Veenstra M, Donoso M, Tell E, Eugenin EA, Morgello S, et al. Central nervous system (CNS) viral seeding by mature monocytes and potential therapies to reduce CNS viral reservoirs in the cART era. mBio (2021) 12(2):e03633-20. doi: 10.1128/mBio.03633-20
44. Hart CL, Gunderson EW, Perez A, Kirkpatrick MG, Thurmond A, Comer SD, et al. Acute physiological and behavioral effects of intranasal methamphetamine in humans. Neuropsychopharmacology. (2008) 33(8):1847–55. doi: 10.1038/sj.npp.1301578
45. Melega WP, Cho AK, Harvey D, Laćan G. Methamphetamine blood concentrations in human abusers: Application to pharmacokinetic modeling. Synapse. (2007) 61(4):216–20. doi: 10.1002/syn.20365
46. Liang H, Wang X, Chen H, Song L, Ye L, Wang SH, et al. Methamphetamine enhances HIV infection of macrophages. Am J Pathol (2008) 172(6):1617–24. doi: 10.2353/ajpath.2008.070971
47. Liu Y, Meng F-Z, Wang X, Wang P, Liu J-B, Hu W-H, et al. Methamphetamine facilitates HIV infection of primary human monocytes through inhibiting cellular viral restriction factors. Cell Biosci (2021) 11(1):194. doi: 10.1186/s13578-021-00703-4
48. Mastroianni CM, Liuzzi GM. Matrix metalloproteinase dysregulation in HIV infection: implications for therapeutic strategies. Trends Mol Med (2007) 13(11):449–59. doi: 10.1016/j.molmed.2007.09.001
49. Hannocks MJ, Zhang X, Gerwien H, Chashchina A, Burmeister M, Korpos E, et al. The gelatinases, MMP-2 and MMP-9, as fine tuners of neuroinflammatory processes. Matrix Biol (2019) 75–76:102-13. doi: 10.1016/j.matbio.2017.11.007
50. Turner RJ, Sharp FR. Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front Cell Neurosci (2016) 10:56. doi: 10.3389/fncel.2016.00056
51. Sun HQ, Yamamoto M, Mejillano M, Yin HL. Gelsolin, a multifunctional actin regulatory protein*. J Biol Chem (1999) 274(47):33179–82. doi: 10.1074/jbc.274.47.33179
52. Feldt J, Schicht M, Garreis F, Welss J, Schneider UW, Paulsen F. Structure, regulation and related diseases of the actin-binding protein gelsolin. Expert Rev Mol Med (2019) 20:e7. doi: 10.1017/erm.2018.7
53. Lerner AM, Eisinger RW, Fauci AS. Comorbidities in persons with HIV: The lingering challenge. JAMA. (2020) 323(1):19–20. doi: 10.1001/jama.2019.19775
54. Liu Y, Brown S, Shaikh J, Fishback JA, Matsumoto RR. Relationship between methamphetamine exposure and matrix metalloproteinase 9 expression. Neuroreport. (2008) 19(14):1407–9. doi: 10.1097/WNR.0b013e32830dd606
55. Xue Y, He JT, Zhang KK, Chen LJ, Wang Q, Xie XL. Methamphetamine reduces expressions of tight junction proteins, rearranges f-actin cytoskeleton and increases the blood brain barrier permeability via the RhoA/ROCK-dependent pathway. Biochem Biophys Res Commun (2019) 509(2):395–401. doi: 10.1016/j.bbrc.2018.12.144
56. López-Guisa JM, Cai X, Collins SJ, Yamaguchi I, Okamura DM, Bugge TH, et al. Mannose receptor 2 attenuates renal fibrosis. J Am Soc Nephrol (2012) 23(2):236–51. doi: 10.1681/asn.2011030310
57. Romagnoli M, Mineva ND, Polmear M, Conrad C, Srinivasan S, Loussouarn D, et al. ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis. EMBO Mol Med (2014) 6(2):278–94. doi: 10.1002/emmm.201303373
58. Conrad C, Götte M, Schlomann U, Roessler M, Pagenstecher A, Anderson P, et al. ADAM8 expression in breast cancer derived brain metastases: Functional implications on MMP-9 expression and transendothelial migration in breast cancer cells. Int J Cancer. (2018) 142(4):779–91. doi: 10.1002/ijc.31090
59. Zhang H, Zheng Y, Pan Y, Lin C, Wang S, Yan Z, et al. A mutation that blocks integrin α4β7 activation prevents adaptive immune-mediated colitis without increasing susceptibility to innate colitis. BMC Biol (2020) 18(1):64. doi: 10.1186/s12915-020-00784-6
60. Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, et al. Integrin β7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood. (2011) 117(23):6202–13. doi: 10.1182/blood-2010-06-292243
61. Zhang HL, Zheng YJ, Pan YD, Xie C, Sun H, Zhang YH, et al. Regulatory T-cell depletion in the gut caused by integrin β7 deficiency exacerbates DSS colitis by evoking aberrant innate immunity. Mucosal Immunol (2016) 9(2):391–400. doi: 10.1038/mi.2015.68
62. Shen W, Zhang X, Du R, Fan Y, Luo D, Bao Y, et al. ICAM3 mediates tumor metastasis via a LFA-1-ICAM3-ERM dependent manner. Biochim Biophys Acta (BBA) - Mol Basis Dis (2018) 1864(8):2566–78. doi: 10.1016/j.bbadis.2018.05.002
63. Liu L, He B, Liu WM, Zhou D, Cox JV, Zhang XA. Tetraspanin CD151 promotes cell migration by regulating integrin trafficking. J Biol Chem (2007) 282(43):31631–42. doi: 10.1074/jbc.M701165200
64. Zelman-Toister E, Bakos E, Cohen S, Zigmond E, Shezen E, Grabovsky V, et al. CD151 regulates T-cell migration in health and inflammatory bowel disease. Inflammation Bowel Dis (2016) 22(2):257–67. doi: 10.1097/mib.0000000000000621
65. Champion TC, Partridge LJ, Ong S-M, Malleret B, Wong S-C, Monk PN. Monocyte subsets have distinct patterns of tetraspanin expression and different capacities to form multinucleate giant cells. Front Immunol (2018) 9:1247. doi: 10.3389/fimmu.2018.01247
66. Hong IK, Jin YJ, Byun HJ, Jeoung DI, Kim YM, Lee H. Homophilic interactions of tetraspanin CD151 up-regulate motility and matrix metalloproteinase-9 expression of human melanoma cells through adhesion-dependent c-jun activation signaling pathways. J Biol Chem (2006) 281(34):24279–92. doi: 10.1074/jbc.M601209200
67. Thölmann S, Seebach J, Otani T, Florin L, Schnittler H, Gerke V, et al. JAM-a interacts with α3β1 integrin and tetraspanins CD151 and CD9 to regulate collective cell migration of polarized epithelial cells. Cell Mol Life Sci (2022) 79(2):88. doi: 10.1007/s00018-022-04140-5
68. Betapudi V. Myosin II motor proteins with different functions determine the fate of lamellipodia extension during cell spreading. PloS One (2010) 5(1):e8560. doi: 10.1371/journal.pone.0008560
69. Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. (2004) 304(5671):743–6. doi: 10.1126/science.1094561
70. Sidani M, Wessels D, Mouneimne G, Ghosh M, Goswami S, Sarmiento C, et al. Cofilin determines the migration behavior and turning frequency of metastatic cancer cells. J Cell Biol (2007) 179(4):777–91. doi: 10.1083/jcb.200707009
71. Baldassarre M, Razinia Z, Burande CF, Lamsoul I, Lutz PG, Calderwood DA. Filamins regulate cell spreading and initiation of cell migration. PloS One (2009) 4(11):e7830. doi: 10.1371/journal.pone.0007830
72. Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol (2004) 6(11):1034–8. doi: 10.1038/ncb1104-1034
73. Roth H, Samereier M, Begandt D, Pick R, Salvermoser M, Brechtefeld D, et al. Filamin a promotes efficient migration and phagocytosis of neutrophil-like HL-60 cells. Eur J Cell Biol (2017) 96(6):553–66. doi: 10.1016/j.ejcb.2017.05.004
74. Deng R, Hao J, Han W, Ni Y, Huang X, Hu Q. Gelsolin regulates proliferation, apoptosis, migration and invasion in human oral carcinoma cells. Oncol Lett (2015) 9(5):2129–34. doi: 10.3892/ol.2015.3002
75. Marino N, Marshall J-C, Collins JW, Zhou M, Qian Y, Veenstra T, et al. Nm23-H1 binds to gelsolin and inactivates its actin-severing capacity to promote tumor cell motility and metastasis. Cancer Res (2013) 73(19):5949–62. doi: 10.1158/0008-5472.Can-13-0368
76. Gonçalves AF, Dias NG, Moransard M, Correia R, Pereira JA, Witke W, et al. Gelsolin is required for macrophage recruitment during remyelination of the peripheral nervous system. Glia. (2010) 58(6):706–15. doi: 10.1002/glia.20956
77. Chellaiah M, Kizer N, Silva M, Alvarez U, Kwiatkowski D, Hruska KA. Gelsolin deficiency blocks podosome assembly and produces increased bone mass and strength. J Cell Biol (2000) 148(4):665–78. doi: 10.1083/jcb.148.4.665
78. Hiramoto K, Negishi M, Katoh H. Dock4 is regulated by RhoG and promotes rac-dependent cell migration. Exp Cell Res (2006) 312(20):4205–16. doi: 10.1016/j.yexcr.2006.09.006
79. Grosshans HK, Fischer TT, Steinle JA, Brill AL, Ehrlich BE. Neuronal calcium sensor 1 is up-regulated in response to stress to promote cell survival and motility in cancer cells. Mol Oncol (2020) 14(6):1134–51. doi: 10.1002/1878-0261.12678
80. Huang L, Fang X, Shi D, Yao S, Wu W, Fang Q, et al. MSP-RON pathway: Potential regulator of inflammation and innate immunity. Front Immunol (2020) 11:569082. doi: 10.3389/fimmu.2020.569082
81. Kim J-M, Park J, Noh E-M, Song H-K, Kang SY, Jung SH, et al. Downregulation of matriptase suppresses the PAR−2/PLCγ2/PKC−mediated invasion and migration abilities of MCF−7 breast cancer cells. Oncol Rep (2021) 46(6):247. doi: 10.3892/or.2021.8198
82. Fani Maleki A, Rivest S. Innate immune cells: Monocytes, monocyte-derived macrophages and microglia as therapeutic targets for alzheimer's disease and multiple sclerosis. Front Cell Neurosci (2019) 13:355. doi: 10.3389/fncel.2019.00355
83. Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol (2015) 15(4):203–16. doi: 10.1038/nri3818
84. Chang NC, Nguyen M, Shore GC. BCL2-CISD2: An ER complex at the nexus of autophagy and calcium homeostasis? Autophagy (2012) 8(5):856–7. doi: 10.4161/auto.20054
85. Wang P, Kou D, Le W. Roles of VMP1 in autophagy and ER–membrane contact: Potential implications in neurodegenerative disorders. Front Mol Neurosci (2020) 13:42. doi: 10.3389/fnmol.2020.00042
86. Watts C. The endosome-lysosome pathway and information generation in the immune system. Biochim Biophys Acta (2012) 1824(1):14–21. doi: 10.1016/j.bbapap.2011.07.006
87. Tallóczy Z, Martinez J, Joset D, Ray Y, Gácser A, Toussi S, et al. Methamphetamine inhibits antigen processing, presentation, and phagocytosis. PloS pathogens. (2008) 4(2):e28–e. doi: 10.1371/journal.ppat.0040028
88. Woc-Colburn L, Smultea L, Ramachandra L, Canaday DH. Preserved MHC class II antigen processing in monocytes from HIV-infected individuals. PloS One (2010) 5(3):e9491. doi: 10.1371/journal.pone.0009491
89. Carty M, Guy C, Bowie AG. Detection of viral infections by innate immunity. Biochem Pharmacol (2021) 183:114316. doi: 10.1016/j.bcp.2020.114316
90. Yang J, Yan H. TLR5: beyond the recognition of flagellin. Cell Mol Immunol (2017) 14(12):1017–9. doi: 10.1038/cmi.2017.122
91. Chattergoon MA, Latanich R, Quinn J, Winter ME, Buckheit RW 3rd, Blankson JN, et al. HIV And HCV activate the inflammasome in monocytes and macrophages via endosomal toll-like receptors without induction of type 1 interferon. PloS Pathogens. (2014) 10(5):e1004082. doi: 10.1371/journal.ppat.1004082
92. O’Brien M, Manches O, Sabado RL, Jimenez Baranda S, Wang Y, Marie I, et al. Spatiotemporal trafficking of HIV in human plasmacytoid dendritic cells defines a persistently IFN-α–producing and partially matured phenotype. J Clin Invest (2011) 121(3):1088–101. doi: 10.1172/JCI44960
93. Alvarez-Carbonell D, Garcia-Mesa Y, Milne S, Das B, Dobrowolski C, Rojas R, et al. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology. (2017) 14(1):9. doi: 10.1186/s12977-017-0335-8
94. Novis CL, Archin NM, Buzon MJ, Verdin E, Round JL, Lichterfeld M, et al. Reactivation of latent HIV-1 in central memory CD4α T cells through TLR-1/2 stimulation. Retrovirology. (2013) 10:119–. doi: 10.1186/1742-4690-10-119
95. Meås HZ, Haug M, Beckwith MS, Louet C, Ryan L, Hu Z, et al. Sensing of HIV-1 by TLR8 activates human T cells and reverses latency. Nat Commun (2020) 11(1):147. doi: 10.1038/s41467-019-13837-4
96. Wires ES, Alvarez D, Dobrowolski C, Wang Y, Morales M, Karn J, et al. Methamphetamine activates nuclear factor kappa-light-chain-enhancer of activated b cells (NF-kappaB) and induces human immunodeficiency virus (HIV) transcription in human microglial cells. J Neurovirol (2012) 18(5):400–10. doi: 10.1007/s13365-012-0103-4
97. Toussi SS, Joseph A, Zheng JH, Dutta M, Santambrogio L, Goldstein H. Short communication: Methamphetamine treatment increases in vitro and in vivo HIV replication. AIDS Res Hum Retroviruses (2009) 25(11):1117–21. doi: 10.1089/aid.2008.0282
98. Reynolds JL, Law WC, Mahajan SD, Aalinkeel R, Nair B, Sykes DE, et al. Nanoparticle based galectin-1 gene silencing, implications in methamphetamine regulation of HIV-1 infection in monocyte derived macrophages. J Neuroimmune Pharmacol (2012) 7(3):673–85. doi: 10.1007/s11481-012-9379-7
99. Nair MP, Saiyed ZM, Nair N, Gandhi NH, Rodriguez JW, Boukli N, et al. Methamphetamine enhances HIV-1 infectivity in monocyte derived dendritic cells. J Neuroimmune Pharmacol (2009) 4(1):129–39. doi: 10.1007/s11481-008-9128-0
100. Lee W-YJ, Fu RM, Liang C, Sloan RD. IFITM proteins inhibit HIV-1 protein synthesis. Sci Rep (2018) 8(1):14551. doi: 10.1038/s41598-018-32785-5
101. Lu J, Pan Q, Rong L, Liu S-L, Liang C. The IFITM proteins inhibit HIV-1 infection. J Virol (2011) 85(5):2126–37. doi: 10.1128/JVI.01531-10
102. Spence JS, He R, Hoffmann H-H, Das T, Thinon E, Rice CM, et al. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat Chem Biol (2019) 15(3):259–68. doi: 10.1038/s41589-018-0213-2
103. Turrini F, Marelli S, Kajaste-Rudnitski A, Lusic M, Van Lint C, Das AT, et al. HIV-1 transcriptional silencing caused by TRIM22 inhibition of Sp1 binding to the viral promoter. Retrovirology. (2015) 12(1):104. doi: 10.1186/s12977-015-0230-0
104. Barr SD, Smiley JR, Bushman FD. The interferon response inhibits HIV particle production by induction of TRIM22. PloS Pathog (2008) 4(2):e1000007. doi: 10.1371/journal.ppat.1000007
105. Kajaste-Rudnitski A, Marelli SS, Pultrone C, Pertel T, Uchil PD, Mechti N, et al. TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, tat, and NF-kappaB-responsive long terminal repeat elements. J Virol (2011) 85(10):5183–96. doi: 10.1128/jvi.02302-10
106. Pagani I, Poli G, Vicenzi E. TRIM22. a multitasking antiviral factor. Cells. (2021) 10(8):1864. doi: 10.3390/cells10081864
107. Chaban Y, Boekema EJ, Dudkina NV. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim Biophys Acta (BBA) - Bioener (2014) 1837(4):418–26. doi: 10.1016/j.bbabio.2013.10.004
108. Potula R, Hawkins BJ, Cenna JM, Fan S, Dykstra H, Ramirez SH, et al. Methamphetamine causes mitrochondrial oxidative damage in human T lymphocytes leading to functional impairment. J Immunol (Baltimore Md 1950). (2010) 185(5):2867–76. doi: 10.4049/jimmunol.0903691
109. Borgmann K, Ghorpade A. Methamphetamine augments concurrent astrocyte mitochondrial stress, oxidative burden, and antioxidant capacity: Tipping the balance in HIV-associated neurodegeneration. Neurotoxicity Res (2018) 33(2):433–47. doi: 10.1007/s12640-017-9812-z
110. Reynolds JL, Mahajan SD, Sykes DE, Schwartz SA, Nair MPN. Proteomic analyses of methamphetamine (METH)-induced differential protein expression by immature dendritic cells (IDC). Biochim Biophys Acta (2007) 1774(4):433–42. doi: 10.1016/j.bbapap.2007.02.001
111. Soto-Heredero G, Gómez de las Heras MM, Gabandé-Rodríguez E, Oller J, Mittelbrunn M. Glycolysis – a key player in the inflammatory response. FEBS J (2020) 287(16):3350–69. doi: 10.1111/febs.15327
112. Marelli-Berg FM, Jangani M. Metabolic regulation of leukocyte motility and migration. J Leukoc Biol (2018) 104(2):285–93. doi: 10.1002/JLB.1MR1117-472R
113. Li X, Fang P, Mai J, Choi ET, Wang H, Yang X-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol (2013) 6(1):19. doi: 10.1186/1756-8722-6-19
114. Zhang Z-H, Song G-L. Roles of selenoproteins in brain function and the potential mechanism of selenium in alzheimer’s disease. Front Neurosci (2021) 15:646518. doi: 10.3389/fnins.2021.646518
115. Koenig G, Seneff S. Gamma-glutamyltransferase: A predictive biomarker of cellular antioxidant inadequacy and disease risk. Dis Markers (2015) 2015:818570. doi: 10.1155/2015/818570
116. Gangcuangco LMA, Mitchell BI, Siriwardhana C, Kohorn LB, Chew GM, Bowler S, et al. Mitochondrial oxidative phosphorylation in peripheral blood mononuclear cells is decreased in chronic HIV and correlates with immune dysregulation. PloS One (2020) 15(4):e0231761. doi: 10.1371/journal.pone.0231761
117. Buckley S, Byrnes S, Cochrane C, Roche M, Estes JD, Selemidis S, et al. The role of oxidative stress in HIV-associated neurocognitive disorders. Brain behavior Immun - Health (2021) 13:100235–. doi: 10.1016/j.bbih.2021.100235
118. Mackie P, Lebowitz J, Saadatpour L, Nickoloff E, Gaskill P, Khoshbouei H. The dopamine transporter: An unrecognized nexus for dysfunctional peripheral immunity and signaling in parkinson's disease. Brain behavior immunity (2018) 70:21–35. doi: 10.1016/j.bbi.2018.03.020
119. Underhill SM, Hullihen PD, Chen J, Fenollar-Ferrer C, Rizzo MA, Ingram SL, et al. Amphetamines signal through intracellular TAAR1 receptors coupled to Gα13 and GαS in discrete subcellular domains. Mol Psychiatry (2021) 26(4):1208–23. doi: 10.1038/s41380-019-0469-2
120. Cisneros IE, Ghorpade A. Methamphetamine and HIV-1-induced neurotoxicity: role of trace amine associated receptor 1 cAMP signaling in astrocytes. Neuropharmacology. (2014) 85:499–507. doi: 10.1016/j.neuropharm.2014.06.011
121. Panas MW, Xie Z, Panas HN, Hoener MC, Vallender EJ, Miller GM. Trace amine associated receptor 1 signaling in activated lymphocytes. J neuroimmune Pharmacol Off J Soc NeuroImmune Pharmacol (2012) 7(4):866–76. doi: 10.1007/s11481-011-9321-4
122. Moreno CS, Beresford GW, Louis-Plence P, Morris AC, Boss JM. CREB regulates MHC class II expression in a CIITA-dependent manner. Immunity. (1999) 10(2):143–51. doi: 10.1016/S1074-7613(00)80015-1
Keywords: methamphetamine, monocytes, HIV, neuroinflammation, migration, viral reservoirs
Citation: Chilunda V, Weiselberg J, Martinez-Meza S, Mhamilawa LE, Cheney L and Berman JW (2022) Methamphetamine induces transcriptional changes in cultured HIV-infected mature monocytes that may contribute to HIV neuropathogenesis. Front. Immunol. 13:952183. doi: 10.3389/fimmu.2022.952183
Received: 24 May 2022; Accepted: 29 July 2022;
Published: 18 August 2022.
Edited by:
Julie Olson, University of Minnesota Twin Cities, United StatesReviewed by:
Luis R. Martinez, University of Florida, United StatesCopyright © 2022 Chilunda, Weiselberg, Martinez-Meza, Mhamilawa, Cheney and Berman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joan W. Berman, Sm9hbi5iZXJtYW5AZWluc3RlaW5tZWQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.