 Jun Wang
Jun Wang Miriam Erlacher
Miriam Erlacher Juncal Fernandez-Orth
Juncal Fernandez-Orth
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 11 August 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.951937
This article is part of the Research TopicThe Interplay Between the Immune System and Bone Marrow Function in Inborn Errors of Immunity and Marrow FailureView all 5 articles
Hematopoiesis is a remarkable system that plays an important role in not only immune cell function, but also in nutrient transport, hemostasis and wound healing among other functions. Under inflammatory conditions, steady-state hematopoiesis switches to emergency myelopoiesis to give rise to the effector cell types necessary to fight the acute insult. Sustained or aberrant exposure to inflammatory signals has detrimental effects on the hematopoietic system, leading to increased proliferation, DNA damage, different forms of cell death (i.e., apoptosis, pyroptosis and necroptosis) and bone marrow microenvironment modifications. Together, all these changes can cause premature loss of hematopoiesis function. Especially in individuals with inherited bone marrow failure syndromes or immune-mediated aplastic anemia, chronic inflammatory signals may thus aggravate cytopenias and accelerate disease progression. However, the understanding of the inflammation roles in bone marrow failure remains limited. In this review, we summarize the different mechanisms found in mouse models regarding to inflammatory bone marrow failure and discuss implications for future research and clinical practice.
Steady-state hematopoiesis is identified as a hierarchically orchestrated and highly regulated process from embryonic development to adulthood, to produce and replenish the whole blood system from the bone marrow (BM). The hematopoietic homeostasis relies on the division and self-renewing of the hematopoietic stem cells (HSCs) (1), with enormous self-renewal capacities, that differentiate through the lineage-committed progenitors to the different mature blood cells (2). However, severe systematic infections or injuries (e.g. sepsis, chronic inflammatory disease or spinal cord injury) evoking inflammatory signals can give rise to emergency hematopoiesis (3, 4). In this process, pathogens can directly be sensed by pattern recognition receptors, such as Toll-like receptors (TLRs) expressed on hematopoietic stem and progenitor cells (HSPCs) (5). Signaling is then amplified by inflammatory cytokines (interferons (IFN) and tumor necrosis factor (TNF)) secreted at peripheral, immune active sites and/or locally in the BM (6, 7). As a consequence of these stimuli, downstream signaling cascades are initiated and induce the mobilization of HSPCs to replenish consumed short-lived mature hematopoietic immune effector cells and enhance the host defenses (8, 9). Forced repeated cycling of HSCs, however, can result in their exhaustion and eventually in hematopoietic failure, especially when occurring in the context of genetic conditions predisposing to BM failure (BMF). Indeed, elevated levels of IFN and TNF have been found overexpressed in a plethora of syndromes like aplastic anemia (AA) or Fanconi anemia (FA), however, their expression are not sufficient to decipher the pathological role of them between inflammation and disease progression (10–13). Thus, different animal models are helping to identify the link between the specific molecules causing inflammation and BMF, paving the way for novel therapeutic and preventive approaches. In this perspective, we discuss recent findings about how inflammatory signals (including IFN, TLRs, TGF-β, TNF and ILs) as well as inflammatory DNA damage and their interactions can provoke BMF.
Interferons (IFNs) are inflammatory cytokines that were named based on their role in interfering with viral infections (14). They are divided into three different families (I, II and III) with several classes among them. Recently, a new IFN ligand-receptor was identified that may be considered as type IV IFN (15, 16). Type I IFNs are not only released by immune cells upon the interaction with pathogens (i.e. viral, bacterial, fungal, and parasitic infections) but can also be induced endogenously in other cell types by sensing self-ligands, TLR agonists, host factors and cytokines such as TNF (17–25). IFNs are used to clinically treat patients suffering viral diseases and autoimmune diseases as well as different malignancies like chronic myeloid leukemia (CML).
Once IFNs are produced and secreted, they bind to their respective receptors and activate different signal transduction pathways that induce specific transcriptional responses leading to the formation of an antiviral phase in affected cells. For further details, we refer to recent reviews (26–29). Apart from the roles already mentioned, IFNs (principally α and γ) are known to be involved during both developmental and adult hematopoiesis (30, 31). In this sense, IFN-α facilitates embryonic HSC maturation in the aorta-gonad-mesonephros (AGM) during developmental hematopoiesis (32), whereas IFN-γ positively regulates HSC development (33). Beyond their role during developmental hematopoiesis, IFNs are known to play a role as antiproliferative and apoptotic mediators in many different cell types (34, 35). In HSCs, they both induce cell division and impair the self-renewal capacity (8, 9, 36). It has been recently suggested that beside the protective effects of IFNs in the hematopoietic system, they can also compromise the HSPC self-renewal capacity and long-term survival (8, 9, 36, 37), resulting in suppression of blood cell production during adult hematopoiesis (15, 38). Moreover, some studies showed that preservation of murine long-term stem cells (LT-HSCs; defined as Lin-,c-Kit+,Sca1+, CD150+,CD48-), capable to reconstitute the whole hematopoietic system, is critically compromised by IFNγ (39). This is explained by the fact that IFNγ perturbs thrombopoietin (TPO), inducing phosphorylation of the signal transducer and activator of transcription 5 (STAT5) whose expression is associated with genes implicated in HSC proliferation (37, 39). All these examples show the relevance of the IFN family during infection and immune-mediated processes and the impact of this signaling pathway in HSC differentiation. Due to the important role of IFNs on HSC functions, it is crucial to determine the effects of both chronic and acute IFN exposure on HSCs.
Persistent production of type I and II IFN happening during infection processes has been shown to lead to HSCs exhaustion and eventually, hematopoietic failure (9, 40). This process has been deeply studied through several infection models in mice, which are helping to understand the mechanisms underlying the link between inflammation and BMF to further determine the effect of IFNs on stem cell balance. One of these models is the polyinosinic-polycytidylic acid (poly(I:C)) model, in which a synthetic viral nucleic acid imitates viral infections inducing IFN-α production together with pro-inflammatory cytokines (AP-1 and NF-κB). In 1981, Gidali J. et al. (41) first tested the effect of type 1 IFN both in vitro and in vivo on murine HSPCs by analyzing the number of colony forming units (CFU) in S-phase. In both cases, IFN remarkably reduced the number of CFUs without altering the colony subtypes. Although the effect of poly(I:C) is known to mimic the effects of IFN response, it needs to take into consideration that the effects of poly(I:C) cannot be only delimited to IFN itself, as some other signaling pathways are known to be altered. Long-term exposure to type I IFN signaling leads to a functional reduction and finally loss of HSCs in which the IFN-α-dependent transcriptional regulator Irf2 plays an important role in limiting the excess of IFN signaling (42). Also in IFN-γ adenylate-uridylate–rich element (ARE)–deleted (del) mice, IFN-γ alone resulted in BMF by disrupting the generation of common myeloid progenitors and lineage differentiation (43). The poly(I:C) model is not the only model that leads to HSCs exhaustion via IFN signaling. Several infections shed light on the influence of IFN on hematopoiesis. Ehrlichia muris acute infection changes transiently the activation of LT-HSCs and progenitors from dormancy to activity by IFN-γ signaling in order to induce an innate immune response (44–47). A long-term Mycobacterium tuberculosis antigen stimulation leads to a continuous IFN-γ signaling with harmful effects on proliferation and differentiation of HSCs (48). Similarly, upon recurrent infection with Mycobacterium avium, mice become pancytopenic due to a reduction in the number of myeloid-biased HSCs induced by IFN-γ signaling (36, 40). This myeloid differentiation effect is driven by the overexpression of the Batf2 transcription factor (36). HSC functional impairment is also observed in the chronic Lymphocytic choriomeningitis virus (LCMV) mouse model. Type I and II IFN signaling mediate the depletion of the supportive BM mesenchymal CXCL12-abundant reticular cells network (49, 50). On the other hand, in both IFN-/- and IFNGR1-/- mice, HCSs show a better reconstitution capability during homeostasis and infectious stress conditions, revealing that IFN-α signaling drives hematopoietic collapse by directly sensitization of HSPCs to undergo cell death and enhanced HSC quiescence (51).
All these models show the relevance of the IFN family during infection processes and the impact of this signaling pathway on HSC differentiation. In addition to the role of IFN in infection models, type I and II IFNs have also a strong impact on BMF syndromes by driving hematopoietic collapse (52).
Hematopoietic exhaustion caused by chronic IFN-γ signaling is severely accelerated in inherited BMF syndromes (IBMFs). FA is the most common inherited BMF syndrome caused by mutations in one of the 23 genes encoding DNA repair proteins of the Fanconi pathway (53). Several FA mouse models have been established by targeting the disruption of FA genes, such as Fanca-/-, Fancc-/-, Fancg-/- (53–57). In these models, it was demonstrated that FA hematopoietic progenitors are highly sensitive to IFN-γ-induced apoptosis via the Fas apoptotic pathway (58, 59). In a model of immune-mediated AA, in which splenocytes were injected intraperitoneally, IFN-γ-dependent HSC loss and hematopoietic failure were driven by macrophages working as sensors of IFN-γ (60).
Hence, the different animal models developed in the recent years regarding immune-related mechanisms (61–63) and hematopoiesis have helped to understand the pathogenesis of the different congenital and acquired BM failure syndromes (64–67) and might yield further insights into the development of novel therapeutic strategies that will target or even prevent hematopoietic failure in these syndromes.
In contrast to chronic IFN effects, during acute infections, the production of IFN-γ leads to a transient activation and proliferation of otherwise quiescent HSCs (42, 68, 69), due to the downregulation of different quiescent-enforcing mechanisms (38) among other processes. Indeed, only one single poly I:C injection leading to a short exposure of type 1 IFN, transiently enhances not only HSC proliferation but also early progenitors, followed by quiescence-enforcing mechanisms in vivo, helping HSCs to reestablish quiescence, protecting and maintaining so the HSC pool from the IFNs-dependent effects (9, 38).
Another pivotal pro-inflammatory cytokine is the tumor necrosis factor (TNF). It belongs to the TNF superfamily, which consists of 19 different members (70), and is produced by antigen-stimulated macrophages and monocytes. By exerting its function via 2 different receptors (TNFR1 and TNFR2), it is responsible for many different signalling processes in the cell, like cellular proliferation, survival, differentiation or apoptosis (71, 72). Among others, it plays important roles in regulating cell functions as immune responses, hematopoiesis and tumorigenesis (73, 74). Although the functions of TNF-α during inflammation are well characterized, the roles during hematopoiesis and HSC homeostasis are poorly described and remain controversial (75–78). From acting as potent inhibitor to promote proliferation, most probably TNF is required for HSC emergence during the development of the embryo after the activation of different signaling pathways, like the activation of the NF-κB pathway via TLR4-MyD88 signaling (79). Studies performed in zebrafish revealed that TNF promotes HSC survival and myeloid differentiation by activating a specific p65/NF-κB-dependent gene program that primarily prevents necroptosis (76). Dysregulations on TNF production has been related to directly inhibit growth and induce apoptosis of HSCs, as well as indirectly change the bone marrow microenvironment critical for HSC homeostasis. TNF enhanced expression has been also observed in the pathogenesis of several BMF syndromes, like FA (10, 80–83). Studies using Fanca−/−, Fancc−/− and Fancg−/− mice revealed that TNF production is abnormally high in macrophages, contributing to enhance TNF-induced apoptosis, which relies on the apoptosis signal-regulating kinase 1 (ASK1) (84, 85). Furthermore, HSCs and progenitor cells from Fancc−/− mice showed that TNF overproduction leads to bone marrow hypoplasia. After long-term exposure, clonal evolution and eventually myeloid leukemia arises secondary to BMF (83). Similarly, by using murine models for immune-mediated AA and genetically modified mice deficient in TNF or TNF receptors it could be observed, that after infusing TNF-/- donor lymph node (LN) cells into CByB6F1 recipients or injection of FVB LN cells into TNFR-/- recipients BM failure was induced. This reveals the importance of this cytokine and these cells in the pathogenesis of the disease (81). Some other studies have revealed that hematopoietic deficiency of the receptor-interacting serine/threonine-protein kinase 1 (RIPK1) results in RIPK3-activation, which leads to necroptosis, and loss of HSPCs and subsequently, to BM failure (86).
In the first line of defense of the innate immune system are a family of pattern recognition receptors (PRRs) that detect pathogen associated signatures derived from all kind of microorganisms. In mammals, when PRRs recognize pathogens, they activate downstream a cascade of different signaling pathways by producing IFN1 as well as other mediators in order to display an effective immune response to an acute infection or injury (87). Among the different classes of PRRs, toll-like receptors (TLRs) are known to play a key role in immunity, mediating a rapid inflammatory reaction and appropriate T-cell activation in response to infection and tissue damage. Comprising 10 different members (TLR1-TLR10) in human and 12 in mice (TLR1-TLR10, TLR11-TLR13), TLRs are not only found in most effector immune and stromal cells but also in hematopoietic and progenitor cells as well as endothelial cells (87–91). Besides their well-known job in effector immune cells, TLRs influence HSCs in terms of proliferation and differentiation in response to ‘danger’ signals (e.g. various infections, as well as purified or synthetic TLR ligands induce the release of proinflammatory cytokines like TNF) (92), helping the hematopoietic system to recognize stress events and inducing emergency hematopoiesis (87, 93, 94). Hence, the immuno-surveillance effects of TLRs expressed on HSCs induce the activation of quiescent HSCs pushing them to proliferate and differentiate into myeloid cells. However, a persistent or dysregulated TLR signaling induced by daily injections of LPS for 4-6 weeks affect stem cell balance, leading to ineffective hematopoiesis, loss of HSCs and consequently, to BMF (95). Different mouse models have been developed to further define which bone marrow populations are affected and their relative contribution to the disease pathogenesis (6, 95–98). Some studies revealed TLRs signaling mediated inflammatory pathogenesis in the context of inherited BMF (80). Fanconi gene products protect hematopoietic cells from damage and modulate TLR responses in macrophages (98). TLR8 and the canonical downstream signaling intermediates interleukin 1 receptor-associated kinase (IRAK) and IkappaB kinase-alpha/beta induce TNF production of THP-1 cells and macrophages, which contributes to the hematopoietic defects seen in Fancc-/- mice (80). Another study on immune-mediated AA suggests that TLR2 and TLR4 individually do not play an essential role in the induction of hematopoietic failure, but depends on IFN-γ and TNF (99). By exposing mice to a TLR-2 agonist, PAM3CSK4, the augmentation of TLR2 signaling leads to an increase on the phenotype of HSPCs but accompanied with a reduction of bone marrow HSC function (96). Treatment with granulocyte colony-stimulation factors (G-CSF) leads to the induction of TLR expression and signaling resulting in expansion and increase of HSCs but with HSC repopulation defects in mice lacking TLR2, TLR4 or the TLR signaling adaptor MyD88 (98).
In the early 80s, a polypeptide named Sarcoma Growth Factor (SGF) was discovered in transformed rat kidney fibroblasts cultures. This polypeptide was composed by both Transforming Growth Factor-α (TGF-α) and Transforming Growth Factor-β (TGF-β) (100). The TGF-β superfamily comprises a big number of proteins being involved in not only fibroblast growth and collagen production, but also inhibiting cell proliferation among other functions (101, 102). TGF-β has been seen to contribute to hematopoietic suppression in Fanconi Anemia as well as other myelodysplastic diseases (MDS) (103–105). Mice models disrupting TGF-β signaling in FA HSPCs by using a neutralizing antibody called 1D11 (106), significantly improved the proliferation and survival of these cells reducing toxic non-homologous end-joining (NHEJ) machinery and increasing homologous recombinant (HR) activity (107). Indeed, pharmacological inhibition of the TGF-β pathway have shown efficacy in preclinical human and murine models (103, 108, 109).
Interleukins (ILs) are a group of cytokines with complex immunomodulatory roles produced by leukocytes, lymphocytes and in some particular cases by other type of cells (110). They are involved in many different functions, like proliferation, immune cell differentiation and activation as well playing an important role in the pathophysiology of several disorders (111). ILs comprise more than 50 different members and related proteins, which can be divided into four main groups depending on their structural features. IL-1 is one of the main inflammatory mediators but it is also known to be involved in the regulation of HSCs and HSPCs, including radioprotection, cell growth and/or differentiation as well as altering cell adhesion and migration (112). Indeed, it has been observed that administration of IL-1 prior lethal doses of radiation protects mice from fatal hematopoietic syndrome (113), which is associated to the role of IL-1 on cell cycle activation through the expansion of HSPCs and myeloid precursor cells (114–116). Moreover, chronic IL-1 exposure leads to impairment of HSCs function (117). However, the mechanisms leading to these effects are still largely unknown. Another IL playing a role in hematopoiesis is IL-6. The IL-6 family comprises ten different cytokines: IL-6, LIF, CNTF, CLCF1, OSM, CT-1, IL-11, IL-27, IL-35 and IL-39 (118–120), involved in chronic inflammation, autoimmunity and cancer among other functions (118). Studies have shown that T cells lacking IL-6 led to pancytopenia and BMF as well as deletions on the IL-6 gene were inducing a variable degree of immune-mediated BMF, however, these studies do not indicate a significant or a direct role of IL-6 in murine BMF (99).
In response to an increase in the number of inflammatory signals, HSCs are forced to exit their homeostatic quiescent state and proliferate to generate more cells. In this scenario, the chances to acquire and accumulate cellular mutations increase, as more cellular and DNA base pair divisions are happening. Indeed, long-term exposure to IFNα, TLR, or TNF mimicking chronic inflammatory stimuli resulted in an increased mitochondrial reactive oxygen species (ROS)-induced DNA damage in HSCs, which is another mechanism that may cause BMF (52). Moreover, chronic poly(I:C) injections inducing an IFN-I response leads to an increased mitochondrial ROS-induced DNA damage in WT HSCs, resulting in BMF in Fanca-/- mice with a nonfunctional FA DNA repair pathway (52). In addition, the process of DNA damage in HSCs induced by prolonged LPS stimulation or Salmonella typhimurium infection is mediated via TLR4-TRIF-ROS-p38 pathway, but not the classic MyD88 signaling (7). TNF-induced accumulation of ROS and oxidative DNA damage leads to premature senescence in HSCs and progenitor cells of WT mice. Furthermore, TNF-treated Fancc–/– mice showed chromosomal aberrations together with impaired oxidative DNA-damage repair pathway (121). Therefore, it needs to be taken into consideration that all these inflammatory signals can also lead to DNA damage, promoting the depletion of HSCs. In some syndromes defective DNA repair pathways can hence contribute to the pathogenesis of BMF.
Programmed cell death (PCD) is an important process that keeps the homeostasis of hematopoiesis among other systems. Being apoptosis and necroptosis the two main forms of PCD, the mode of action of the two processes differs, as apoptosis is not immune-related, while necroptosis leads to inflammation through the secretion of DAMPS (122). As already mentioned, HSPCs respond to DAMPs producing different cytokines to overcome hematopoiesis damage and keep the homeostasis. By the development of several mouse models, it has been observed that an increased in necroptotic death cell in the bone marrow leads to loss of HSPCs while proliferation of SLAM-HSCs happens, inducing stem cell exhaustion and finally BMF (123). Apart from the inflammatory roles of TNF, this cytokine is able to initiate both apoptosis (caspase-8-dependent apoptosis) and RIPK1 kinase dependent necroptosis (124). Several publications have revealed the role of RIPK1 in immune homeostasis and emergency hematopoiesis. Indeed, after an infection induced by an Ehrlichia pathogen, RIPK1 is activated, diminishing caspase 8 expression and leading to BMF and hematopoietic suppression after IFNα/β induction (51). Additional work has also shown that IFNα/β is required for the increase of IL-18 expression during infection processes leading to loss of short-term HSCs. Absence of IL-18 was shown to prevent BM aplasia and increase HSCs/HSPCs (125).
Furthermore, it has been shown that RIPK3 plays an important role in generating necroptotic DAMPs as well as promoting the production of inflammatory cytokines (126). Beside apoptosis and necroptosis, Caspase-1 dependent death, also named as pyroptosis, has been shown to trigger HSPCs cytopenias upon NLRP1a inflammasome activation (127). Thus, all these different mechanisms suggest that both necroptosis and pyroptosis might be good therapeutic targets to prevent BMF.
Altogether, these studies indicate that different inflammatory signaling pathways play an important role in the regulation of hematopoiesis (Figure 1, Table 1). Long-term exposure to inflammation leads to impairment of HSCs function and self-renew, increasing the chances to develop BMF. Inflammation accelerates hematopoietic failure significantly in mouse models of inherited BMF syndromes such as FA and of immune-mediated AA. It is thus conceivable that inflammatory signals affect the time point of cytopenia onset also in patients with such diseases. It needs to be also mentioned, that all inherited BM failure syndromes as well as immune-mediated AA predispose to leukemia. There is increasing evidence that both infectious diseases and inflammation contribute to the development of hematological neoplasia (128). It is thus conceivable that inflammation does not only promote hematopoietic failure but also consecutive malignant transformation (e.g. by DNA damage induced by ROS). However, a more detailed understanding of the roles of key inflammatory signaling and their interactions in hematopoiesis could open attractive novel ways to develop therapies aimed at modulating the inflammatory immune response to prevent BMF. Antagonizing proinflammatory IFNs, TLR, TNF, and/or ROS may have therapeutic benefits in patients with BMF. The elimination of the key molecules by either neutralizing antibodies or deleting/silencing the genes to abrogated the negative effects of inflammatory factors on HSCs proliferation may lead to restore the ability of the progenitor cells to reconstitute impaired bone marrow, preventing so, fatal consequences derived from BMF. Likewise, inhibition of ROS production may potentially rescue suppressed hematopoietic cell function. For example, Fisetin, a dietary flavonoid, has displayed anti-oxidant activities, which can alleviate CLP-induced multiple organs injury by reducing the expression of TNF and dose-dependently inhibiting the phosphorylation of p38 MAPK, MK2 (129). Rapamycin is an effective therapy in mouse models of immune-mediated BMF by reducing IFN-γ and TNF, stimulating the expansion of functional regulatory T cells, eliminating effector CD8+ T cells and preserving hematopoietic stem and progenitor cells (130). Alternatively, downstream effects of inflammation might be targeted. Inhibition of necroptosis and/or pyroptotic cell death might be particularly attractive to prevent hematopoietic failure in inherited BMF syndromes and immune-mediated AA.
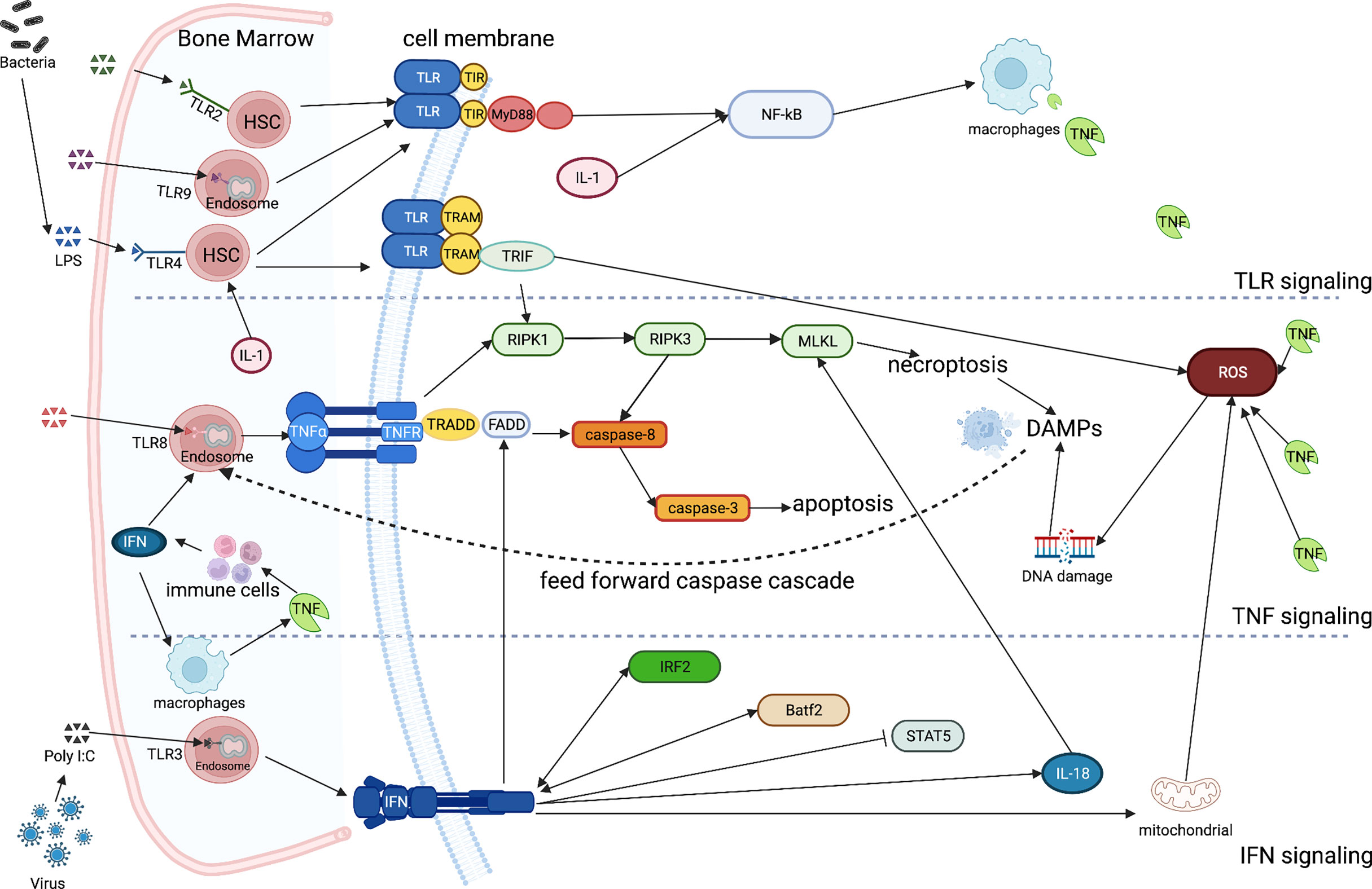
Figure 1 Simplified inflammatory signaling of TLR, TNF, IFN and ILs in bone marrow failure While most of the TLRs are located on the surface of HSCs, some others keep intracellularly (TLR3/8/9). They all directly sense the inflammatory stimuli and induce macrophages to release the excess of TNF through the toll-interleukin-1 receptor (TIR) and the myeloid differentiation primary response 88 (MyD88)-NF-κB pathway. IL-1 directly regulates HSC fate targeting also the NF- κB pathway. TLR4 can specifically activate the TIR-domain-containing the adapter-inducing interferon-β (TRIF) via the TRIF-related adaptor molecule (TRAM) cross talking with the TNF signaling, paving the way for necroptosis trough the RIPK1/3- MLKL pathway. The products resulting from necroptosis constitute the DAMPs and feed-forward the caspase cascade. A large amount of TNF may also cause the accumulation of ROS, stimulating immune cells to release IFNs. After the activation of TNFR, TNFR1-associated death domain protein (TRADD) is bound, recruiting the adaptor Fas-associated death domain (FADD) triggering the caspase-8/3 receptor complex, inducing at the end apoptosis. The IFN signal, which is regulated by Irf2 and Batf2, leads also to apoptosis via the Fas pathway (FADD), being also perturb the phosphorylation state of STAT5. Moreover, IFN may also lead to an increased mitochondrial ROS, inducing DNA damage. Finally, IFNs are required for an increased in the compromising IL-18 expression, which mediates the MLKL-dependent cell death, compromising hematopoiesis during infection. (Created with BioRender.com).
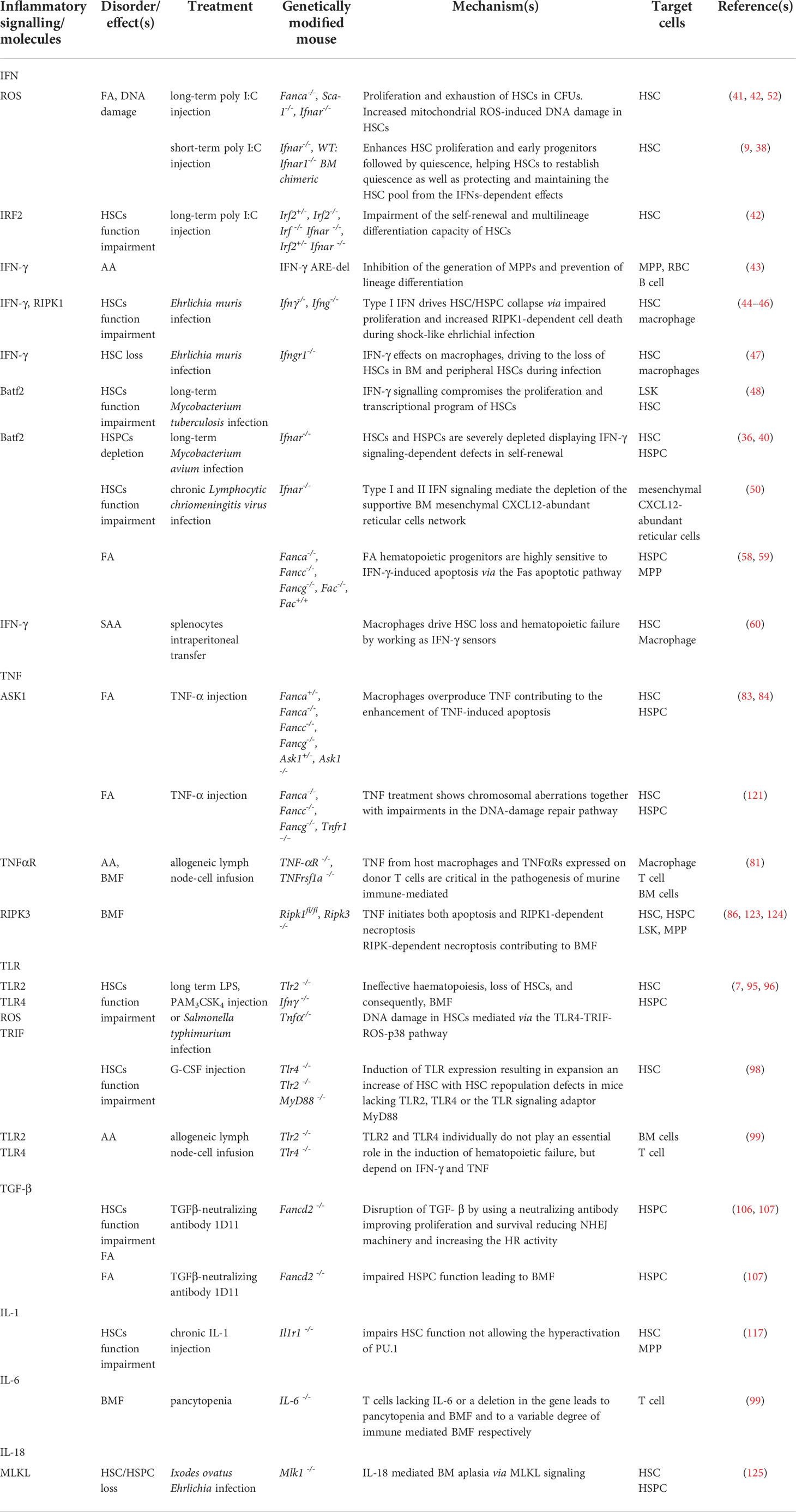
Table 1 Inflammatory signalling in the mouse models with implication in the bone marrow failure.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
JW is supported by “Local Excellent Medical Graduated Student” of Shennong Medical Facility. ME received support from the European Research Council (ERC Starting Grant no. 638145 “ApoptoMDS” to ME), the German Federal Ministry of Education and Research (BMBF), Berlin (“MyPred - Network for young individuals with syndromes predisposing to myeloid malignancies” no. 01GM1911A), the EJP-RD program (RiboEurope consortium) and by the Collaborative Research Center (CRC) 1479 “Oncogene-driven immune escape”. JF-O is supported by the Deutsche Forschungsgemeinchaft (no. GZ:FE 2257/1-1).
The authors thank all the members of the group for the critical reading of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wei Q, Frenette PS. Niches for hematopoietic stem cells and their progeny. Immunity (2018) 48(4):632–48. doi: 10.1016/j.immuni.2018.03.024
2. Tseng YJ, Chapple R, Hu T, Rajapakshe K, Hoegenauer K, Coarfa C, et al. Hematopoietic hierarchy under steady-state and stress conditions. Blood (2019) 134(Supplement_1):1181–1. doi: 10.1182/blood-2019-125110
3. Skirecki T, Drechsler S, Jeznach A, Hoser G, Jafarmadar M, Kawiak J, et al. An early myelosuppression in the acute mouse sepsis is partly outcome-dependent. Front Immunol (2021) 12:708670. doi: 10.3389/fimmu.2021.708670
4. Carpenter RS, Marbourg JM, Brennan FH, Mifflin KA, Hall JCE, Jiang RR, et al. Spinal cord injury causes chronic bone marrow failure. Nat Commun (2020) 11:3702. doi: 10.1038/s41467-020-17564-z
5. Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol (2014) 14(5):302–14. doi: 10.1038/nri3660
6. Megías J, Yáñez A, Moriano S, O’Connor JE, Gozalbo D, Gil ML. Direct toll-like receptor-mediated stimulation of hematopoietic stem and progenitor cells occurs in vivo and promotes differentiation toward macrophages. Stem Cells (2012) 30(7):1486–95. doi: 10.1002/stem.1110
7. Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, et al. Pathogen-induced tlr4-trif innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell (2017) 21(2):225–40.e5. doi: 10.1016/j.stem.2017.06.013
8. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature (2010) 465(7299):793–7. doi: 10.1038/nature09135
9. Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNα activates dormant haematopoietic stem cells in vivo. Nature (2009) 458(7240):904–8. doi: 10.1038/nature07815
10. Dufour C, Corcione A, Svahn J, Haupt R, Poggi V, Béka’ssy AN, et al. TNF-alpha and IFN-gamma are overexpressed in the bone marrow of fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood (2003) 102(6):2053–9. doi: 10.1182/blood-2003-01-0114
11. Erlacher M, Strahm B. Missing cells: pathophysiology, diagnosis, and management of (pan)cytopenia in childhood. Front Pediatr (2015) 3:64. doi: 10.3389/fped.2015.00064
12. Dubey S, Shukla P, Nityanand S. Expression of interferon-gamma and tumor necrosis factor-alpha in bone marrow T cells and their levels in bone marrow plasma in patients with aplastic anemia. Ann Hematol (2005) 84(9):572–7. doi: 10.1007/s00277-005-1022-8
13. Sloand E, Kim S, Maciejewski JP, Tisdale J, Follmann D, Young NS. Intracellular interferon-gamma in circulating and marrow T cells detected by flow cytometry and the response to immunosuppressive therapy in patients with aplastic anemia. Blood (2002) 100(4):1185–91. doi: 10.1182/blood-2002-01-0035
14. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
15. Lee AJ, Ashkar AA. The dual nature of type i and type ii interferons. Front Immunol (2018) 9:2061. doi: 10.3389/fimmu.2018.02061
16. Chen SN, Gan Z, Hou J, Yang YC, Huang L, Huang B, et al. Identification and establishment of type IV interferon and the characterization of interferon-υ including its class II cytokine receptors IFN-υR1 and IL-10R2. Nat Commun (2022) 13(1):999. doi: 10.1038/s41467-022-28645-6
17. Stifter SA, Feng CG. Interfering with immunity: detrimental role of type i ifns during infection. J Immunol (2015) 194(6):2455–65. doi: 10.4049/jimmunol.1402794
18. Espinosa V, Dutta O, McElrath C, Du P, Chang YJ, Cicciarelli B, et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci Immunol (2017) 2(16):eaan5357. doi: 10.1126/sciimmunol.aan5357
19. Boxx GM, Cheng G. The roles of type I interferon in bacterial infection. Cell Host Microbe (2016) 19(6):760–9. doi: 10.1016/j.chom.2016.05.016
20. Duncan CJA, Randall RE, Hambleton S. Genetic lesions of type i interferon signalling in human antiviral immunity. Trends Genet (2021) 37(1):46–58. doi: 10.1016/j.tig.2020.08.017
21. Wells AI, Coyne CB. Type III interferons in antiviral defenses at barrier surfaces. Trends Immunol (2018) 39(10):848–58. doi: 10.1016/j.it.2018.08.008
22. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
23. Venkatesh D, Ernandez T, Rosetti F, Batal I, Cullere X, Luscinskas FW, et al. TNF receptor 2 induces IRF1 transcription factor-dependent interferon-β autocrine signaling to promote monocyte recruitment. Immunity (2013) 38(5):1025–37. doi: 10.1016/j.immuni.2013.01.012
24. Stanifer ML, Guo C, Doldan P, Boulant S. Importance of type i and iii interferons at respiratory and intestinal barrier surfaces. Front Immunol (2020) 11:608645. doi: 10.3389/fimmu.2020.608645
25. He X, Xia L, Tumas KC, Wu J, Su XZ. Type i interferons and malaria: a double-edge sword against a complex parasitic disease. Front Cell Infect Microbiol (2020) 10:594621. doi: 10.3389/fcimb.2020.594621
26. Pervolaraki K, Talemi SR, Albrecht D, Bormann F, Bamford C, Mendoza JL, et al. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PloS Pathog (2018) 14(11):e1007420. doi: 10.1371/journal.ppat.1007420
27. Bolen CR, Ding S, Robek MD, Kleinstein SH. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology (2014) 59(4):1262–72. doi: 10.1002/hep.26657
28. Stanifer ML, Pervolaraki K, Boulant S. Differential regulation of type i and type iii interferon signaling. Int J Mol Sci (2019) 20(6):E1445. doi: 10.3390/ijms20061445
29. Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the jak-STAT pathway and the mitogen-activated protein kinases. J Virol (2007) 81(14):7749–58. doi: 10.1128/JVI.02438-06
30. Demerdash Y, Kain B, Essers MAG, King KY. Yin and yang: The dual effects of interferons on hematopoiesis. Exp Hematol (2021) 96:1–12. doi: 10.1016/j.exphem.2021.02.002
31. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med (2014) 371(26):2488–98. doi: 10.1056/NEJMoa1408617
32. Kim PG, Canver MC, Rhee C, Ross SJ, Harriss JV, Tu HC, et al. Interferon-α signaling promotes embryonic HSC maturation. Blood (2016) 128(2):204–16. doi: 10.1182/blood-2016-01-689281
33. Sawamiphak S, Kontarakis Z, Stainier DYR. Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Dev Cell (2014) 31(5):640–53. doi: 10.1016/j.devcel.2014.11.007
34. Schmeisser H, Bekisz J, Zoon KC. New function of type I IFN: induction of autophagy. J Interferon Cytokine Res (2014) 34(2):71–8. doi: 10.1089/jir.2013.0128
35. Bekisz J, Sato Y, Johnson C, Husain SR, Puri RK, Zoon KC. Immunomodulatory effects of interferons in malignancies. J Interferon Cytokine Res (2013) 33(4):154–61. doi: 10.1089/jir.2012.0167
36. Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, et al. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep (2016) 17(10):2584–95. doi: 10.1016/j.celrep.2016.11.031
37. de Bruin AM, Demirel Ö, Hooibrink B, Brandts CH, Nolte MA. Interferon-γ impairs proliferation of hematopoietic stem cells in mice. Blood (2013) 121(18):3578–85. doi: 10.1182/blood-2012-05-432906
38. Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med (2014) 211(2):245–62. doi: 10.1084/jem.20131043
39. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-γ on hematopoiesis. Blood (2014) 124(16):2479–86. doi: 10.1182/blood-2014-04-568451
40. Florez MA, Matatall KA, Jeong Y, Ortinau L, Shafer PW, Lynch AM, et al. Interferon gamma mediates hematopoietic stem cell activation and niche relocalization through bst2. Cell Rep (2020) 33(12):108530. doi: 10.1016/j.celrep.2020.108530
41. Gidáli J, Fehér I, Tálas M. Proliferation inhibition of murine pluripotent haemopoietic stem cells by interferon or poly i:C. Cell Proliferation (1981) 14(1):1–7. doi: 10.1111/j.1365-2184.1981.tb00505.x
42. Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med (2009) 15(6):696–700. doi: 10.1038/nm.1973
43. ching LF, Karwan M, Saleh B, Hodge DL, Chan T, Boelte KC, et al. IFN-γ causes aplastic anemia by altering hematopoietic stem/progenitor cell composition and disrupting lineage differentiation. Blood (2014) 124(25):3699–708. doi: 10.1182/blood-2014-01-549527
44. MacNamara KC, Jones M, Martin O, Winslow GM. Transient activation of hematopoietic stem and progenitor cells by IFNγ during acute bacterial infection. PloS One (2011) 6(12):e28669. doi: 10.1371/journal.pone.0028669
45. MacNamara KC, Oduro K, Martin O, Jones DD, McLaughlin M, Choi K, et al. Infection-induced myelopoiesis during intracellular bacterial infection is critically dependent upon ifn-γ signaling. J Immunol (2011) 186(2):1032–43. doi: 10.4049/jimmunol.1001893
46. Zhang Y, Jones M, McCabe A, Winslow GM, Avram D, MacNamara KC. MyD88-signaling in cd4 t cells promotes ifnγ production and hematopoietic progenitor cell expansion in response to intracellular bacterial infection. J Immunol (2013) 190(9):4725–35. doi: 10.4049/jimmunol.1203024
47. McCabe A, Zhang Y, Thai V, Jones M, Jordan MB, MacNamara KC. Macrophage-lineage cells negatively regulate the hematopoietic stem cell pool in response to IFNγ at steady state and during infection. Stem Cells (2015) 33(7):2294–305. doi: 10.1002/stem.2040
48. Li F, Liu X, Niu H, Lv W, Han X, Zhang Y, et al. Persistent stimulation with mycobacterium tuberculosis antigen impairs the proliferation and transcriptional program of hematopoietic cells in bone marrow. Mol Immunol (2019) 112:115–22. doi: 10.1016/j.molimm.2019.05.001
49. Zhou X, Ramachandran S, Mann M, Popkin DL. Role of lymphocytic choriomeningitis virus (lcmv) in understanding viral immunology: past, present and future. Viruses (2012) 4(11):2650–69. doi: 10.3390/v4112650
50. Isringhausen S, Mun Y, Kovtonyuk L, Kräutler NJ, Suessbier U, Gomariz A, et al. Chronic viral infections persistently alter marrow stroma and impair hematopoietic stem cell fitness. J Exp Med (2021) 218(12):e20192070. doi: 10.1084/jem.20192070
51. Smith JNP, Zhang Y, Li JJ, McCabe A, Jo HJ, Maloney J, et al. Type I IFNs drive hematopoietic stem and progenitor cell collapse via impaired proliferation and increased RIPK1-dependent cell death during shock-like ehrlichial infection. PloS Pathog (2018) 14(8):e1007234. doi: 10.1371/journal.ppat.1007234
52. Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature (2015) 520(7548):549–52. doi: 10.1038/nature14131
53. Li X, Yang Y, Yuan J, Hong P, Freie B, Orazi A, et al. Continuous in vivo infusion of interferon-gamma (IFN-γ) preferentially reduces myeloid progenitor numbers and enhances engraftment of syngeneic wild-type cells in fancc-/- mice. Blood (2004) 104(4):1204–9. doi: 10.1182/blood-2004-03-1094
54. Pulliam-Leath AC, Ciccone SL, Nalepa G, Li X, Si Y, Miravalle L, et al. Genetic disruption of both fancc and fancg in mice recapitulates the hematopoietic manifestations of fanconi anemia. Blood (2010) 116(16):2915–20. doi: 10.1182/blood-2009-08-240747
55. Si Y, Ciccone S, Yang FC, Yuan J, Zeng D, Chen S, et al. Continuous in vivo infusion of interferon-gamma (IFN-γ) enhances engraftment of syngeneic wild-type cells in fanca–/– and fancg–/– mice. Blood (2006) 108(13):4283–7. doi: 10.1182/blood-2006-03-007997
56. Parmar K, D’Andrea A, Niedernhofer LJ. Mouse models of fanconi anemia. Mutat Res (2009) 668(1–2):133–40. doi: 10.1016/j.mrfmmm.2009.03.015
57. Bakker ST, de Winter JP, te Riele H. Learning from a paradox: recent insights into fanconi anaemia through studying mouse models. Dis Model Mech (2013) 6(1):40–7. doi: 10.1242/dmm.009795
58. Haneline LS, Broxmeyer HE, Cooper S, Hangoc G, Carreau M, Buchwald M, et al. Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from fac-/- mice. Blood (1998) 91(11):4092–8. doi: 10.1182/blood.V91.11.4092
59. Whitney MA, Royle G, Low MJ, Kelly MA, Axthelm MK, Reifsteck C, et al. Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the fanconi anemia c gene. Blood (1996) 88(1):49–58. doi: 10.1182/blood.V88.1.49.49
60. McCabe A, Smith JNP, Costello A, Maloney J, Katikaneni D, MacNamara KC. Hematopoietic stem cell loss and hematopoietic failure in severe aplastic anemia is driven by macrophages and aberrant podoplanin expression. Haematologica (2018) 103(9):1451–61. doi: 10.3324/haematol.2018.189449
61. Elahi S. Hematopoietic responses to SARS-CoV-2 infection. Cell Mol Life Sci (2022) 79(3):187. doi: 10.1007/s00018-022-04220-6
62. Patil AM, Choi JY, Park SO, Uyangaa E, Kim B, Kim K, et al. Type I IFN signaling limits hemorrhage-like disease after infection with Japanese encephalitis virus through modulating a prerequisite infection of CD11b+Ly-6C+ monocytes. J Neuroinflam (2021) 18:136. doi: 10.1186/s12974-021-02180-5
63. Giudice V, Risitano AM, Selleri C. Infectious agents and bone marrow failure: a causal or a casual connection? Front Med (Lausanne) (2021) 8:757730. doi: 10.3389/fmed.2021.757730
64. Landelouci K, Sinha S, Pépin G. Type-I interferon signaling in fanconi anemia. Front Cell Infect Microbiol (2022) 12:820273. doi: 10.3389/fcimb.2022.820273
65. Zhu C, Lian Y, Wang C, Wu P, Li X, Gao Y, et al. Single-cell transcriptomics dissects hematopoietic cell destruction and T-cell engagement in aplastic anemia. Blood (2021) 138(1):23–33. doi: 10.1182/blood.2020008966
66. Votavova H, Belickova M. Hypoplastic myelodysplastic syndrome and acquired aplastic anemia: Immune-mediated bone marrow failure syndromes (Review). Int J Oncol (2021) 60(1):7. doi: 10.3892/ijo.2021.5297
67. Smith JNP, Kanwar VS, MacNamara KC. Hematopoietic stem cell regulation by type i and ii interferons in the pathogenesis of acquired aplastic anemia. Front Immunol (2016) 7:330. doi: 10.3389/fimmu.2016.00330
68. Bakker ST, Passegué E. Resilient and resourceful: genome maintenance strategies in hematopoietic stem cells. Exp Hematol (2013) 41(11):915–23. doi: 10.1016/j.exphem.2013.09.007
69. Pietras EM, Warr MR, Passegué E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol (2011) 195(5):709–20. doi: 10.1083/jcb.201102131
70. Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood (2012) 119(3):651–65. doi: 10.1182/blood-2011-04-325225
71. Silke J, Hartland EL. Masters, marionettes and modulators: intersection of pathogen virulence factors and mammalian death receptor signaling. Curr Opin Immunol (2013) 25(4):436–40. doi: 10.1016/j.coi.2013.05.011
72. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol (2015) 15(6):362–74. doi: 10.1038/nri3834
73. Idriss HT, Naismith JH. TNF alpha and the TNF receptor superfamily: structure-function relationship(s). Microsc Res Tech (2000) 50(3):184–95. doi: 10.1002/1097-0029(20000801)50:3<184::AID-JEMT2>3.0.CO;2-H
74. Horiuchi T, Mitoma H, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatol (Oxford) (2010) 49(7):1215–28. doi: 10.1093/rheumatology/keq031
75. Pronk CJH, Veiby OP, Bryder D, Jacobsen SEW. Tumor necrosis factor restricts hematopoietic stem cell activity in mice: involvement of two distinct receptors. J Exp Med (2011) 208(8):1563–70. doi: 10.1084/jem.20110752
76. Yamashita M, Passegué E. TNFα coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell (2019) 25(3):357–72.e7. doi: 10.1016/j.stem.2019.05.019
77. He H, Xu P, Zhang X, Liao M, Dong Q, Cong T, et al. Aging-induced IL27Ra signaling impairs hematopoietic stem cells. Blood (2020) 136(2):183–98. doi: 10.1182/blood.2019003910
78. Rezzoug F, Huang Y, Tanner MK, Wysoczynski M, Schanie CL, Chilton PM, et al. TNF-alpha is critical to facilitate hemopoietic stem cell engraftment and function. J Immunol (2008) 180(1):49–57. doi: 10.4049/jimmunol.180.1.49
79. Gogoleva VS, Atretkhany KSN, Dygay AP, Yurakova TR, Drutskaya MS, Nedospasov SA. Current perspectives on the role of tnf in hematopoiesis using mice with humanization of tnf/lt system. Front Immunol (2021) 12:661900. doi: 10.3389/fimmu.2021.661900
80. Vanderwerf SM, Svahn J, Olson S, Rathbun RK, Harrington C, Yates J, et al. TLR8-dependent TNF-(alpha) overexpression in fanconi anemia group c cells. Blood (2009) 114(26):5290–8. doi: 10.1182/blood-2009-05-222414
81. Sun W, Wu Z, Lin Z, Hollinger M, Chen J, Feng X, et al. Macrophage TNF-α licenses donor T cells in murine bone marrow failure and can be implicated in human aplastic anemia. Blood (2018) 132(26):2730–43. doi: 10.1182/blood-2018-05-844928
82. Chen Y, Zou Z, Wu Z, Zhao Z, Luo X, Xie C, et al. TNF-α-induced programmed cell death in the pathogenesis of acquired aplastic anemia. Expert Rev Hematol (2015) 8(4):515–26. doi: 10.1586/17474086.2015.1049593
83. Li J, Sejas DP, Zhang X, Qiu Y, Nattamai KJ, Rani R, et al. TNF-α induces leukemic clonal evolution ex vivo in fanconi anemia group c murine stem cells. J Clin Invest (2007) 117(11):3283–95. doi: 10.1172/JCI31772
84. Bijangi-Vishehsaraei K, Saadatzadeh MR, Werne A, McKenzie KAW, Kapur R, Ichijo H, et al. Enhanced TNF-α–induced apoptosis in fanconi anemia type c–deficient cells is dependent on apoptosis signal-regulating kinase 1. Blood (2005) 106(13):4124–30. doi: 10.1182/blood-2005-05-2096
85. Kennedy RD, D’Andrea AD. The fanconi anemia/brca pathway: new faces in the crowd. Genes Dev (2005) 19(24):2925–40. doi: 10.1101/gad.1370505
86. Roderick JE, Hermance N, Zelic M, Simmons MJ, Polykratis A, Pasparakis M, et al. Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc Natl Acad Sci (2014) 111(40):14436–41. doi: 10.1073/pnas.1409389111
87. Monlish DA, Bhatt ST, Schuettpelz LG. The role of toll-like receptors in hematopoietic malignancies. Front Immunol (2016) 7:390. doi: 10.3389/fimmu.2016.00390
88. Crack PJ, Bray PJ. Toll-like receptors in the brain and their potential roles in neuropathology. Immunol Cell Biol (2007) 85(6):476–80. doi: 10.1038/sj.icb.7100103
89. Fitzner N, Clauberg S, Essmann F, Liebmann J, Kolb-Bachofen V. Human skin endothelial cells can express all 10 TLR genes and respond to respective ligands. Clin Vaccine Immunol (2008) 15(1):138–46. doi: 10.1128/CVI.00257-07
90. Schmid MA, Takizawa H, Baumjohann DR, Saito Y, Manz MG. Bone marrow dendritic cell progenitors sense pathogens via toll-like receptors and subsequently migrate to inflamed lymph nodes. Blood (2011) 118(18):4829–40. doi: 10.1182/blood-2011-03-344960
91. Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood (2012) 119(13):2991–3002. doi: 10.1182/blood-2011-12-380113
92. Chen M, Deng H, Zhao Y, Miao X, Gu H, Bi Y, et al. Toll-like receptor 2 modulates pulmonary inflammation and tnf-α release mediated by mycoplasma pneumoniae. Front Cell Infect Microbiol (2022) 12:824027. doi: 10.3389/fcimb.2022.824027
93. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity (2006) 24(6):801–12. doi: 10.1016/j.immuni.2006.04.008
94. Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature (2003) 424(6950):743–8. doi: 10.1038/nature01889
95. Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol (2011) 186(9):5367–75. doi: 10.4049/jimmunol.1003438
96. Herman AC, Monlish DA, Romine MP, Bhatt ST, Zippel S, Schuettpelz LG. Systemic TLR2 agonist exposure regulates hematopoietic stem cells via cell-autonomous and cell-non-autonomous mechanisms. Blood Cancer J (2016) 6(6):e437–7. doi: 10.1038/bcj.2016.45
97. Ichii M, Shimazu T, Welner RS, Garrett KP, Zhang Q, Esplin BL, et al. Functional diversity of stem and progenitor cells with b-lymphopoietic potential. Immunol Rev (2010) 237(1):10–21. doi: 10.1111/j.1600-065X.2010.00933.x
98. Schuettpelz LG, Borgerding JN, Christopher MJ, Gopalan PK, Romine MP, Herman AC, et al. G-CSF regulates hematopoietic stem cell activity, in part, through activation of toll-like receptor signaling. Leukemia (2014) 28(9):1851–60. doi: 10.1038/leu.2014.68
99. Solorzano S, Kim J, Chen J, Feng X, Young NS. Minimal role of interleukin 6 and toll-like receptor 2 and 4 in murine models of immune-mediated bone marrow failure. PloS One (2021) 16(3):e0248343. doi: 10.1371/journal.pone.0248343
100. de Larco JE, Todaro GJ. Growth factors from murine sarcoma virus-transformed cells. Proc Natl Acad Sci (1978) 75(8):4001–5. doi: 10.1073/pnas.75.8.4001
101. Tucker RF, Shipley GD, Moses HL, Holley RW. Growth inhibitor from bsc-1 cells closely related to platelet type β transforming growth factor. Science (1984) 226(4675):705–7. doi: 10.1126/science.6093254
102. Roberts AB, Anzano MA, Wakefield LM, Roche NS, Stern DF, Sporn MB. Type beta transforming growth factor: a bifunctional regulator of cellular growth. Proc Natl Acad Sci (1985) 82(1):119–23. doi: 10.1073/pnas.82.1.119
103. Suragani RNVS, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med (2014) 20(4):408–14. doi: 10.1038/nm.3512
104. Zhou L, McMahon C, Bhagat T, Alencar C, Yu Y, Fazzari M, et al. Reduced SMAD7 leads to overactivation of TGF-beta signaling in MDS that can be reversed by a specific inhibitor of TGF-beta receptor I kinase. Cancer Res (2011) 71(3):955–63. doi: 10.1158/0008-5472.CAN-10-2933
105. Zingariello M, Martelli F, Ciaffoni F, Masiello F, Ghinassi B, D’Amore E, et al. Characterization of the TGF-β1 signaling abnormalities in the Gata1low mouse model of myelofibrosis. Blood (2013) 121(17):3345–63. doi: 10.1182/blood-2012-06-439661
106. Brenet F, Kermani P, Spektor R, Rafii S, Scandura JM. TGFβ restores hematopoietic homeostasis after myelosuppressive chemotherapy. J Exp Med (2013) 210(3):623–39. doi: 10.1084/jem.20121610
107. Zhang H, Kozono DE, O’Connor KW, Vidal-Cardenas S, Rousseau A, Hamilton A, et al. TGF-β inhibition rescues hematopoietic stem cell defects and bone marrow failure in fanconi anemia. Cell Stem Cell (2016) 18(5):668–81. doi: 10.1016/j.stem.2016.03.002
108. Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discovery (2012) 11(10):790–811. doi: 10.1038/nrd3810
109. Grafe I, Yang T, Alexander S, Homan EP, Lietman C, Jiang MM, et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat Med (2014) 20(6):670–5. doi: 10.1038/nm.3544
111. Brocker C, Thompson D, Matsumoto A, Nebert DW, Vasiliou V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum Genomics (2010) 5(1):30–55. doi: 10.1186/1479-7364-5-1-30
112. Orelio C, Haak E, Peeters M, Dzierzak E. Interleukin-1–mediated hematopoietic cell regulation in the aorta-gonad-mesonephros region of the mouse embryo. Blood (2008) 112(13):4895–904. doi: 10.1182/blood-2007-12-123836
113. Oppenheim J, Neta R, Tiberghien P, Gress R, Kenny J, Longo D. Interleukin-1 enhances survival of lethally irradiated mice treated with allogeneic bone marrow cells. Blood (1989) 74(6):2257–63. doi: 10.1182/blood.V74.6.2257.2257
114. Brugger W, Mocklin W, Heimfeld S, Berenson R, Mertelsmann R, Kanz L. Ex vivo expansion of enriched peripheral blood CD34+ progenitor cells by stem cell factor, interleukin-1 beta (IL-1 beta), IL-6, IL-3, interferon-gamma, and erythropoietin. Blood (1993) 81(10):2579–84. doi: 10.1182/blood.V81.10.2579.2579
115. Fibbe W, van Damme J, Billiau A, Goselink H, Voogt P, van Eeden G, et al. Interleukin 1 induces human marrow stromal cells in long-term culture to produce granulocyte colony-stimulating factor and macrophage colony- stimulating factor. Blood (1988) 71(2):430–5. doi: 10.1182/blood.V71.2.430.430
116. Heimfeld S, Hudak S, Weissman I, Rennick D. The in vitro response of phenotypically defined mouse stem cells and myeloerythroid progenitors to single or multiple growth factors. Proc Natl Acad Sci (1991) 88(21):9902–6. doi: 10.1073/pnas.88.21.9902
117. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol (2016) 18(6):607–18. doi: 10.1038/ncb3346
118. Jones SA, Jenkins BJ. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol (2018) 18(12):773–89. doi: 10.1038/s41577-018-0066-7
119. Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, et al. The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol (2012) 13(3):290–9. doi: 10.1038/ni.2227
120. Wang X, Wei Y, Xiao H, Liu X, Zhang Y, Han G, et al. A novel IL-23p19/Ebi3 (IL-39) cytokine mediates inflammation in lupus-like mice. Eur J Immunol (2016) 46(6):1343–50. doi: 10.1002/eji.201546095
121. Zhang X, Sejas DP, Qiu Y, Williams DA, Pang Q. Inflammatory ROS promote and cooperate with the fanconi anemia mutation for hematopoietic senescence. J Cell Sci (2007) 120(Pt 9):1572–83. doi: 10.1242/jcs.003152
122. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity (2013) 38(2):209–23. doi: 10.1016/j.immuni.2013.02.003
123. Wagner PN, Shi Q, Salisbury-Ruf CT, Zou J, Savona MR, Fedoriw Y, et al. Increased Ripk1-mediated bone marrow necroptosis leads to myelodysplasia and bone marrow failure in mice. Blood (2019) 133(2):107–20. doi: 10.1182/blood-2018-05-847335
124. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol (2010) 11(10):700–14. doi: 10.1038/nrm2970
125. Howard JE, Smith JNP, Fredman G, MacNamara KC. IL-18R-mediated HSC quiescence and MLKL-dependent cell death limit hematopoiesis during infection-induced shock. Stem Cell Rep (2021) 16(12):2887–99. doi: 10.1016/j.stemcr.2021.10.011
126. Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. RIPK1 regulates RIPK3-MLKL-Driven systemic inflammation and emergency hematopoiesis. Cell (2014) 157(5):1175–88. doi: 10.1016/j.cell.2014.04.019
127. Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity (2012) 37(6):1009–23. doi: 10.1016/j.immuni.2012.08.027
128. Hauer J, Fischer U, Borkhardt A. Toward prevention of childhood ALL by early-life immune training. Blood (2021) 138(16):1412–28. doi: 10.1182/blood.2020009895
129. Zhang Hf, Zhang H, Wu Xp, Guo Yl, Cheng Wd, Qian F. Fisetin alleviates sepsis-induced multiple organ dysfunction in mice via inhibiting p38 MAPK/MK2 signaling. Acta Pharmacol Sin (2020) 41(10):1348–56. doi: 10.1038/s41401-020-0462-y
Keywords: hematopoiesis, inflammation, bone marrow failure, mouse models, disease
Citation: Wang J, Erlacher M and Fernandez-Orth J (2022) The role of inflammation in hematopoiesis and bone marrow failure: What can we learn from mouse models? Front. Immunol. 13:951937. doi: 10.3389/fimmu.2022.951937
Received: 24 May 2022; Accepted: 25 July 2022;
Published: 11 August 2022.
Edited by:
Markus G. Seidel, Medical University of Graz, AustriaReviewed by:
Seth Corey, Cleveland Clinic, United StatesCopyright © 2022 Wang, Erlacher and Fernandez-Orth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juncal Fernandez-Orth, anVuY2FsLmZlcm5hbmRlei5vcnRoQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.