 Alvaro Torres-Gomez1,2*
Alvaro Torres-Gomez1,2* Tara Fiyouzi1,2
Tara Fiyouzi1,2 Claudia Guerra-Espinosa1
Claudia Guerra-Espinosa1 Beatriz Cardeñes2
Beatriz Cardeñes2 Irene Clares2Víctor Toribio3
Irene Clares2Víctor Toribio3 Pedro A. Reche1,2
Pedro A. Reche1,2 Carlos Cabañas1,2,3
Carlos Cabañas1,2,3 Esther M. Lafuente1,2*
Esther M. Lafuente1,2*- 1Department of Immunology, Ophthalmology and Otorhinolaryngology, School of Medicine, Universidad Complutense de Madrid, Madrid, Spain
- 2Instituto de Investigación Sanitaria Hospital 12 de Octubre (i+12), Inflammatory Diseases and Immune Disorders (Lymphocyte Immunobiology Unit), Madrid, Spain
- 3Tissue and Organ Homeostasis Program (Cell-Cell Communication and Inflammation Unit), Centre for Molecular Biology "Severo Ochoa", Consejo Superior de Investigaciones Científicas (CSIC)-Universidad Autónoma de Madrid (UAM), Madrid, Spain
Activation of the integrin phagocytic receptors CR3 (αMβ2, CD11b/CD18) and CR4 (αXβ2, CD11c/CD18) requires Rap1 activation and RIAM function. RIAM controls integrin activation by recruiting Talin to β2 subunits, enabling the Talin-Vinculin interaction, which in term bridges integrins to the actin-cytoskeleton. RIAM also recruits VASP to phagocytic cups and facilitates VASP phosphorylation and function promoting particle internalization. Using a CRISPR-Cas9 knockout approach, we have analyzed the requirement for RIAM, VASP and Vinculin expression in neutrophilic-HL-60 cells. All knockout cells displayed abolished phagocytosis that was accompanied by a significant and specific reduction in ITGAM (αM), ITGAX (αX) and ITGB2 (β2) mRNA, as revealed by RT-qPCR. RIAM, VASP and Vinculin KOs presented reduced cellular F-actin content that correlated with αM expression, as treatment with the actin filament polymerizing and stabilizing drug jasplakinolide, partially restored αM expression. In general, the expression of αX was less responsive to jasplakinolide treatment than αM, indicating that regulatory mechanisms independent of F-actin content may be involved. The Serum Response Factor (SRF) was investigated as the potential transcription factor controlling αMβ2 expression, since its coactivator MRTF-A requires actin polymerization to induce transcription. Immunofluorescent MRTF-A localization in parental cells was primarily nuclear, while in knockouts it exhibited a diffuse cytoplasmic pattern. Localization of FHL-2 (SRF corepressor) was mainly sub-membranous in parental HL-60 cells, but in knockouts the localization was disperse in the cytoplasm and the nucleus, suggesting RIAM, VASP and Vinculin are required to maintain FHL-2 close to cytoplasmic membranes, reducing its nuclear localization and inhibiting its corepressor activity. Finally, reexpression of VASP in the VASP knockout resulted in a complete reversion of the phenotype, as knock-ins restored αM expression. Taken together, our results suggest that RIAM, VASP and Vinculin, are necessary for the correct expression of αMβ2 and αXβ2 during neutrophilic differentiation in the human promyelocytic HL-60 cell line, and strongly point to an involvement of these proteins in the acquisition of a phagocytic phenotype.
Introduction
The leukocytic integrins αMβ2 (CD11b/CD18) and αXβ2 (CD11c/CD18), also known as CR3 and CR4, respectively, are the main receptors of the complement fragment iC3b and their activation results in phagocytosis of iC3b-opsonized targets (1, 2). Both integrins can bind additional ligands such as fibrinogen (3), or intercellular adhesion molecule-1 (ICAM-1) on endothelial cells, participating in the phase of firm adhesion and fast migration during PMN extravasation to inflammation sites (4, 5).
Membrane expression of αMβ2 and αXβ2 greatly increases upon leukocyte activation. In unstimulated neutrophils, these integrins localize mainly in endomembranes (75% at gelatinase granules, 20% with secretory vesicles) and only a minor portion is expressed at the plasma membrane. Stimulation with chemoattracting agents (fMLP, LTB-4) and/or certain cytokines (IL-8, granulocyte-macrophage colony stimulating factor, platelet-activating factor, TNF-α) induces a rapid translocation of αMβ2 and αXβ2 to the plasma membrane, increasing its expression by 6 to 7-fold without apparently involving transcriptional regulation (6). αMβ2 and αXβ2 require a conformational change in order to be fully active and capable of binding their ligands with high affinity. This high affinity state can be induced by signaling pathways initiated from other receptors (pattern recognition receptors, chemokine receptors, anaphylatoxin receptor, among others) (7). These inside-out pathways converge in a central node, represented by the active form of the small GTPase Rap1 (Rap1-GTP) which interacts with its effector protein RIAM, promoting its translocation to the plasma membrane (8, 9). RIAM itself interacts with Talin releasing its auto-inhibition and triggering its recruitment to the plasma membrane, where Talin may bind to the proximal NPxY motif present in the cytoplasmic tail of the integrin β2 subunit (10, 11). This interaction results in αMβ2 activation, which acquires the “open headpiece” conformation that allows for high-affinity ligand interaction (12). Upon ligand binding, αMβ2 triggers outside-in signals that recruit protein complexes consisting of adaptor molecules, kinases, phosphatases, and actin polymerases, that regulate cell adhesion and phagocytosis, leading to crucial phenotypical and functional changes in myeloid leukocytes (13). Talin, RIAM, VASP and Vinculin have been identified as being key components of the Integrin Adhesion Complex (IAC), a subset of integrin proximal proteins of the integrin adhesome (14, 15). Signaling stemming from this complex determines cell adhesion and migration, but also other fundamental processes such as cell growth and differentiation (16).
We have previously reported that the adaptor molecule RIAM and its interacting partner VASP participate in this outside-in signaling pathway during complement-mediated phagocytosis (17). By interfering with RIAM expression in neutrophil-like HL-60 cells (RIAM KD) we have demonstrated that VASP recruitment at phagocytic cups is impaired and so is VASP phosphorylation at Ser157, which has been associated to its actin polymerization activity. In line with this observation, RIAM KD cells presented reduced F-actin content at phagocytic cups during complement-dependent phagocytosis. Knocking out VASP in HL-60 cells using CRISPR/Cas9 technology also resulted in a drastic decrease in phagocytic capacity with significant decrease in particle association. We concluded that RIAM worked as a relay for integrin complement receptors in outside-in signaling, coordinating integrin activation and cytoskeletal rearrangements via its interaction with VASP (17).
The importance of RIAM and Talin in integrin activation is highlighted by the effect of their deletion, where knockout mice display a phenotype reminiscent of Leukocyte Adhesion Deficiency (LAD) syndromes, with a pronounced neutrophilic (4-fold increase) and monocytic leukocytosis for RIAM-/- mice that is even more pronounced for Talin1-/- mice (>30-fold increase). LAD syndromes are characterized by recurrent infections due to ineffective neutrophil and macrophage function, and disrupted myeloid cell differentiation with extreme neutrophilia (~5 to 10-fold increases), caused by the egress of immature precursors (18–20). In the case of LAD-I patients, leukocyte expression of integrin β2 is greatly diminished, or absent, with a concomitantly reduced or null expression of its binding partner subunits αL, αM and αX (21).
Since RIAM-/- had shown LAD-like defects, and we had observed an abolishment of phagocytosis when deleting its binding partner VASP, in this work we decided to dissect the effects of RIAM, VASP or Vinculin knockouts on αMβ2 and αXβ2 function and expression. By using CRISPR/Cas9 technology, we have generated RIAM-/-, VASP-/- and Vinculin-/- HL-60 cells. All these cell lines presented a common phenotype, a reduced expression of ITGAM (αM), ITGAX (αX) and ITGB2 (β2) mRNA with their correspondingly low level of surface protein expression, which was not due to transport deficiencies. This defect in integrin subunit expression was accompanied by a reduction in cellular F-actin. Treatment with the actin-stabilizing drug jasplakinolide, which induces actin polymerization, alleviated the defect in integrin expression. Similarly, integrin subunit expression could be renormalized by reexpression of VASP in HL-60 VASP-/- cells (HL-60 VASP KIs). Nuclear translocation and activation of the transcriptional co-activator MRTF-A, a cofactor to the Serum Response transcription Factor (SRF), is dependent on actin polymerization and was therefore a prime candidate to explain the observed phenotype. Analysis of MRTF-A subcellular distribution revealed differences in nuclear localization in comparison to parental HL-60 cells that could account for the defect in αM and αX expression. The reverse pattern was observed for the SRF co-repressor FHL-2, an antagonist for MRTF-A. We conclude that the expression of αMβ2 and αXβ2 integrins is dependent on the activity of RIAM, VASP and Vinculin, all of which have been previously shown to form complexes with each other. Since αMβ2 and αXβ2 are both markers for neutrophilic differentiation, a correct regulation of their expression could have implications in leukemic change and development during neutrophil differentiation.
Material and methods
Cell cultures
Human promyelocytic HL-60 (ATCC: CCL-240) and derived cell lines were cultured in 10 ml RPMI 1640 medium with 10% (v/v) fetal-calf serum (FCS), 1% (v/v) glutamine and 1% (w/v) penicillin-streptomycin (Lonza) (11, 17, 22) using Nunc™ 100 mm dishes (Thermo Scientific). Cells were differentiated into neutrophil-like HL-60 using 1 μM retinoic acid (Sigma) during at least 2 days. HEK 293T cells (ATCC: CRL-3216) were cultured in the same media in Nunc™ 100 mm dishes (Thermo Scientific) for adherent cells.
Phagocytosis assays
Phagocytosis assays were carried out as previously described (11, 17). Briefly, fresh sheep red blood cells (RBCs) (Thermo Scientific) labelled with 2 μM DDAO-AM (Invitrogen) were incubated with sub-agglutinating concentrations of polyclonal rabbit IgM anti-sheep RBC cells (MyBioSource) and later treated with 10% C5-deficient human serum (Sigma) for complement opsonization. Differentiated HL-60 cells starved for 3 h in serum-free RPMI, were treated with 320 nM LPS (Sigma) for 30 minutes or 1 mM MnCl2 for 5 minutes. Cells were incubated with complement-opsonized RBCs (C3-RBC) or unopsonized RBCs as negative control, for 30 min at 37°C in a 1:10 ratio, and unbound RBCs were washed thrice with ice-cold PBS. To determine particle internalization, cell-bound RBCs were exposed to a 30 s hypotonic shock with distilled H2O, and isotonicity restored with an equal volume of twice-concentrated PBS. Cells were analyzed in a BD FACSCalibur II flow cytometer (BD Biosciences), using the FlowJo package (BD Biosciences) and expressed as Association Index (AI), indicating the number of cells with attached and engulfed particles, or Phagocytic Index (PI) indicating cells with internalized particles (11). These indexes are all normalized with respect to the AI for unstimulated control cells.
Western blotting
Cell lysates were obtained from 10 million cells using GST Buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 2 mM MgCl2, 10% v/v glycerol, 1% v/v NP-40) supplemented with 1 mM PMSF, 25 mM NaF, 1 mM Na3VO4, and a protease inhibitor cocktail (Sigma). 50 μg protein were separated by SDS-PAGE electrotransferred to a nitrocellulose membrane, blocked and incubated with the following anti-human primary antibodies: rabbit IgG anti-VASP, (Cell Signaling), mouse IgG anti-α-Tubulin, (Sigma), mouse IgG anti-Vinculin (H-10 clone, Santa Cruz), sheep IgG anti-RIAM (R&D Systems) and mouse IgG anti-Talin (8D4 clone, Sigma). After washes, membranes were incubated with a secondary IRDye® IgG anti-rabbit, anti-goat/sheep or anti-mouse fluorescent antibodies (Li-Cor). All antibodies were used as per the manufacturer’s instructions. The signal was then measured in a Li-Cor Odyssey imaging system and quantified using the ImageStudio software (Li-Cor).
Gene knockout and gene transfection
Protein knockout lines were obtained using a CRISPR-Cas9 system and a double nickase strategy. Pairs of sgRNAs were designed using the Optimized CRISPR Design tool (23), and the highest scoring pairs were selected. To ensure the truncated proteins were non-functional, sgRNAs were directed towards the first common exon for all isoforms of VASP (exon 2), RIAM (exon 3) and Vinculin (exon 3).The corresponding pairs of sgRNAs for VASP (5’-CACCGGTAGATCTGGACGCGGCTGA-3’ and 5’-CACCGGCCAATTCCTTTCGCGTCGT-3’), APBB1IP (RIAM), (5’-CACCGATTTGTTCCATAACCAAGAG-3’ and 5’-CACCGCACTGGTATCAGCCAATATG-3’) VCL, (5’-CACCGTCAATTAGATAATCTCGAGC-3’ and 5’-CACCGGGGTCAAGGGGCATCCTCTC-3’) and their complementary oligonucleotide chains were ordered (Sigma), and cloned into BbsI-digested PX458 plasmid (23). Cell transfection was carried out using the Neon Transfection System (ThermoFisher) following manufacture instructions. For each nucleofection, 250 000 cells and mixture of 3 μg of the two sgRNA plasmids were employed. Cells were then transfected in a 10 μl volume using a single 35 ms and 1350 V pulse and left to recover for 24 h in RPMI 1640 10% FCS media without antibiotics. Cells were then sorted according to transient EGFP fluorescence using a FACS Aria Fusion cell sorter (BD Biosciences). EGFP-positive cells were diluted and cloned into p96 wells. Protein expression was then assessed through western blot and negative clones were selected. Sanger sequencing was used to analyze DNA editing in the selected clones, and sequences were compared to their respective genomic sequences. RIAM KO clone H9D2 presented 2 nt deletion (650 Del and 689 Del), VASP KO clone F6, presented a 46 nt deletion (394_439 Del), VASP KO clone F10 presented a 44 nt deletion (397_441 Del), Vcl KO clone A3 presented a 38 nt deletion (400_438 Del) and Vcl KO clone C4 presented a 35 nt deletion (405_440 Del). Once the transient EGFP expression was lost, we used VASP KO clones to generate the VASP rescue polyclonal cell lines F6 KI and F10 KI. This was done through retroviral transduction using the plasmid pMSCV-EGFP-VASP and HEK 293T packaging cells following a previously described protocol (17). Characterization of these cells was done via western blot.
Integrin expression analysis and jasplakinolide treatment
Integrin expression was monitored by flow cytometry and Geometric Mean Fluorescence Intensity (GMFI) was determined and normalized against the GMFI obtained for isotype controls. Staining was performed in a p96 U-bottom plate (Thermo Scientific) Cells were fixed with 2% formaldehyde for 10 minutes and blocked for 30 minutes with a PBS buffer containing 1% BSA and 100 μg/ml human gamma globulin (Sigma). When required, the cells were permeabilized with 0.1% Triton X-100 in PBS buffer for 10 minutes before blocking. The following hybridoma-derived monoclonal mouse antibodies were used to detect protein expression: BEAR-1 (integrin αM) (24), Ts1/11 (integrin αL) (25), HC1/1 (integrin αX) (26), BU15 mAb anti-CD11c (Immunotools), Ts2/16 (integrin β1) (27), Lia2/3 (integrin β2) (28), PAINS-10 (tetraspanin CD9) (29), Vj1/12 (CD59) (30) and 5A6 (tetraspanin CD81) (31). Cells were washed and stained with a donkey anti-mouse Alexa Fluor® 488 antibody (Life Technologies), as per the manufacturer’s instructions. To determine total cellular F-actin, Phalloidin-Alexa Fluor® 647 (Life Technologies) was used. When indicated, cells were treated with 1 µM jasplakinolide (Santa Cruz) 24 hours previous to completing the retinoic acid-induced differentiation.
Integrin transcript analysis through RT-qPCR
Quantification of mRNA levels were performed using the RT-qPCR service offered by the Genomics unit of the Parque Científico de Madrid, Madrid, Spain. Briefly, 6x106 retinoic acid differentiated HL-60 cells per assay were used. Quantification was performed with both triplicate biological replicates and technical replicates. mRNA was extracted using a miRNAEasy kit (Thermo Scientific). cDNA and RT-minus samples were prepared as per the service’s standardized protocols. PCR primers and TaqMan probes were acquired from Applied Biosystems (Thermo Fisher) for the following genes: ITGAM (αM integrin subunit), ITGAX (αX integrin subunit), ITGAL (αL integrin subunit), ITGB2 (β2 integrin subunit), APBB1IP (RIAM), and GAPDH and 18S rRNA were used as reference genes. Since calculated efficiencies for amplification of the gene of interest and reference genes were similar and close to 100%, fold change in mRNA expression was calculated using the Livak-Schmittgen ΔΔCT method (32).
Fluorescence microscopy
Differentiated HL-60 cells were seeded on PLL (Poly-L-Lysine) coated glass slides, then treated with 1 µM jasplakinolide (Santa Cruz) for 2 hours or left unstimulated, and fixed with 4% paraformaldehyde for 10 minutes. Cells were permeabilized with PBS 0.3% Triton X-100 for 10 minutes and incubated with the indicated primary antibodies: mouse anti-MRTF-A (G8 clone, SantaCruz), rabbit anti-FHL-2 (Abcam) or biotin-labelled mouse Ts2/16 (prepared in house using a Biotin labelling kit purchased from Sigma, as described elsewhere (29). Primary antibodies were used as per the manufacturer’s instructions (1:20 anti-MRTF-A, and 1:100 anti-FHL-2), or 10 µg/ml of biotin-labelled purified antibody. Cells were stained with secondary antibodies (donkey anti-rabbit or donkey anti-mouse Alexa Fluor® 647 conjugated antibodies, Life Technologies) and 488-conjugated streptavidin (Life Technologies). All antibodies were used as per the manufacturer’s instructions, in the presence of excess human gamma globulin (100 μg/ml) and 1% BSA as blocking agents. Imaging was performed using an LSM710 confocal laser scanning microscope coupled to an AxioImager M2 microscope (Zeiss) and analyzed using the ImageJ software package.
Statistical analysis
Figures were prepared to show either representative results or mean ± standard deviation (SD) of at least 3 independent experiments (repetitions are stated in the figure legends). Significance between means was determined using a multiple comparisons ANOVA followed by a post-hoc Dunnett’s test to identify differences between groups. P-values less than 0.05 were considered statistically significant. To signal the degree of significance, asterisks were used as follows: a single asterisk denotes a significance of p<0.05; a double asterisk, p<0.01, a triple asterisk p<0.005 and a quadruple asterisk, p<0.0001. Finally, ns is used to denote no significance. Statistical calculations, data handling and graphing were performed on Microsoft Excel 2016 (Microsoft, Redmond, WA, USA) and GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA).
Results
RIAM, VASP and Vinculin knockouts abolished phagocytosis
We previously determined that correct VASP expression is necessary for particle engulfment during phagocytosis, since VASP knockout resulted in an abolished phagocytosis and VASP overexpression significantly reduced phagocytic efficiency (17). Here, we further analyze the requirement for expression of VASP and VASP-interacting proteins, namely RIAM and Vinculin, in the process of complement-mediated phagocytosis.
Knockout of RIAM and Vinculin was performed using a double nick strategy and a transitory transfection using a CRISPR-Cas9 system. Western blot confirmed that all-trans retinoic acid differentiated HL-60 knockouts did not express detectable levels of the knocked-out proteins and demonstrated that genetic deletion of one of the genes from IAC does not affect the expression of the rest of components assessed (Figures 1A–C).
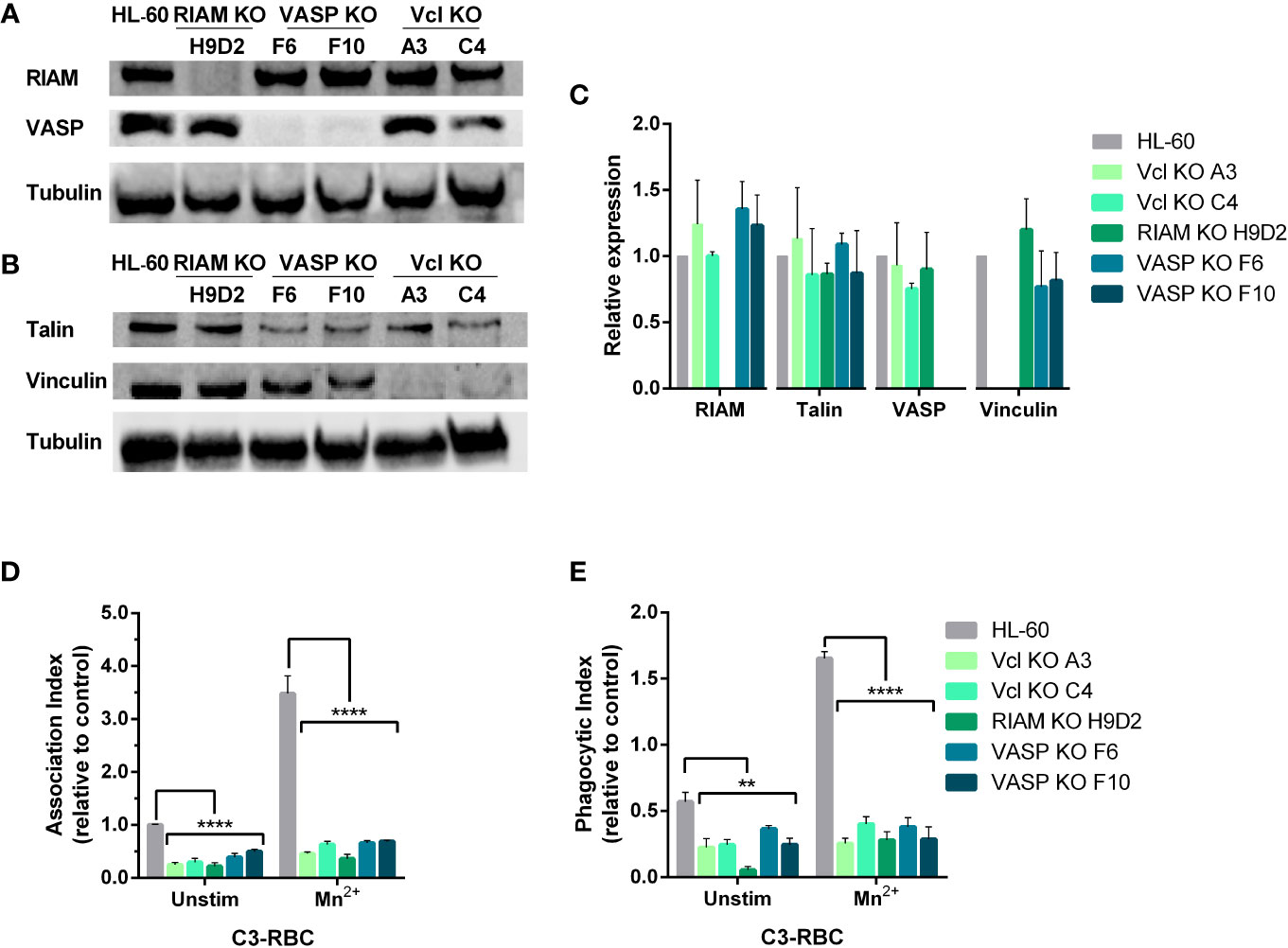
Figure 1 Knockout of either RIAM, VASP or Vinculin, abolishes phagocytosis. (A, B) Newly generated HL-60 Vinculin (Vcl), RIAM and VASP knockout monoclonal cell lines were tested for integrin related protein expression by western blot. (C) Quantification of protein expression in neutrophil-like HL-60 cells and derived knockouts analyzed by western blot. Results are represented as relative to HL-60 levels and are from 5 independent experiments. (D, E) Phagocytic cells were challenged with complement opsonized sheep red blood cells (C3-RBC) after being stimulated with 1 mM MnCl2 or left unstimulated, and Association (AI) and Phagocytic (PI) indexes were obtained. Data are normalized with respect to the AI of unstimulated C3-RBC-challenged HL-60 cells (control cells). Data are presented as mean ± SD, where the error bars denote standard deviation. Significance (ANOVA) has been calculated with respect to HL-60 controls, ** denotes p<0.01 and ****,p<0.0001.
Since our prior reports indicated that VASP knockout abolishes particle internalization (17), RIAM and Vinculin (Vcl) knockout clones were subjected to a phagocytosis assay (Figures 1D, E). We observed that similarly to the results obtained from VASP deficient cells, RIAM and Vinculin knockout clones presented a drastically diminished Association Index (AI) that was detected even at basal state, demonstrating a 70-75% for Vcl KOs, and an 80% reduction for RIAM KOs (Figure 1D). This diminished binding capacity in the absence of stimulation was suggestive of defects in the adhesive properties of these cells. When integrins were activated via outside-in signaling using MnCl2 (Mn2+), all knockouts showed defective particle association, as stimulation was barely able to increase AI values to that of unstimulated control HL-60 cells. This defect in activation seemed to be more pronounced in RIAM KOs, a result which is in line with previous reports using RIAM-specific shRNA in HL-60 cell lines (10, 11, 17).
With regards to particle internalization (Figure 1E), all knockouts showed severely affected PI values, where again RIAM KO stood out as the most affected, as phagocytosis was barely detectable in these conditions. Contrasting with parental HL-60 cells, most KOs were incapable of responding to Mn2+, and no significant change was observed between unstimulated and stimulated cells (1.7-fold increase in KOs cells versus the 3.5-fold increase in HL-60 parental cells). It is worth noting that the response detected in KO cells could result from limitations in determining PI in unstimulated conditions.
Overall, the effects observed in phagocytosis seem to indicate a profound alteration in integrin activation and in the cytoskeletal rearrangements necessary for particle internalization. RIAM and VASP are reported to have an impact on F-actin content (8, 11) and potentially modulate transcriptional activity. The absence of IAC components could also have an effect in integrin stability and integrin recycling. Moreover, we speculated that integrin expression could be also affected in knockout cells.
Expression of αMβ2 and αXβ2 integrins is reduced in RIAM, VASP and Vinculin knockout cells
To further characterize the phenotype observed in all knockouts, we analyzed the surface expression of αM, αX and αL subunits that form complement receptors CR3 and CR4 and integrin LFA1 respectively, together with β2 and β1 integrin subunits and other membrane integral proteins CD59, CD9 and CD81 as controls (Figure 2A and Supplemental Figure 1B). We observed that expression of the αM subunit was significantly reduced (74-50%) as well as the expression of αX which in some cases was virtually undetectable in KOs. The reduction in αX and αM expression was accompanied by a proportional reduction in β2 subunit expression in all KO clones (40-50%) (Figure 2A, Supplemental Figure 1A). No statistically significant differences were observed in the expression of αL subunit, β1 integrin or tetraspanins CD9, and CD81 (all integrin related molecules) or CD59, a molecule unrelated to integrins (Supplemental Figure 1B). We confirmed that this reduction was also maintained when total cellular integrin expression was analyzed in permeabilized cells (Supplemental Figure 1C), thus ruling out a defect in protein transport to the plasma membrane. Since the observed reductions in αM and αX were similar in all clones, this suggests a similar mechanism may be involved in controlling expression of both CR3/αMβ2 and CR4/αXβ2 complement receptors, that requires the expression of RIAM, VASP and Vinculin. Furthermore, this mechanism appears to be highly specific for these integrin subunits, as it only affects the alpha subunits αM and αX, whilst not affecting the closely related αL.
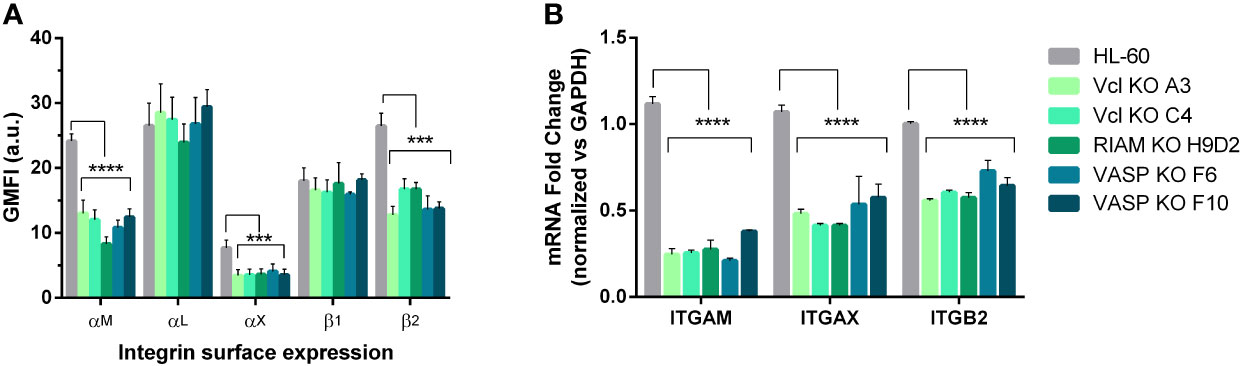
Figure 2 αM and αX expression is dependent on Vinculin, RIAM and VASP expression. (A) Vinculin (Vcl), RIAM and VASP knockout cell lines and HL-60 parental cells were differentiated into neutrophilic-like cells with 1 μM all-trans retinoic acid (RA) and stained with monoclonal antibodies specific for αM, αL, αX, β1, β2 integrin subunits. The geometric mean fluorescence intensity (GMFI) was obtained by flow cytometry and data represented as relative to HL-60 levels and are from 24 independent experiments done in duplicate. (B) Expression of ITGAM (αM), ITGAX (αX), and ITGB2 (β2) mRNA levels was determined by RT-qPCR in neutrophil-like RIAM, VASP and Vcl HL-60 knockouts. Results are represented as relative to GAPDH mRNA and are from 3 independent experiments done in triplicate. Data are presented as mean ± SD, where the error bars denote standard deviation. Significance (ANOVA) has been calculated with respect to HL-60 controls, *** denotes p<0.005, and ****, p<0.0001.
Next, we determined whether the decreased expression of αM and αX subunits was also detectable at mRNA level through RT-qPCR (Figure 2B). After neutrophilic differentiation, all HL-60 knockouts cell lines showed a statistically significant 60-80% reduction in ITGAM (αM gene) mRNA, and 40-50% for ITGAX (αX gene) mRNA expression with respect to parental HL-60 cells, confirming a downregulated transcription. We also observed a decrease in ITGB2 (β2 gene) mRNA levels that was comparable to the reduction observed in β2 expression at the plasma membrane. This downregulation was not observed for ITGAL (αL gene) or APBB1IP (RIAM) in the VASP knockout (Supplemental Figure 2A). This indicates that the amount of β2 subunit produced in RIAM, Vcl and VASP KOs was enough to yield normal levels of integrin αLβ2.
Defective upregulation of integrins αMβ2 and αXβ2 during differentiation correlates with reduced F-actin content
Next, we investigated whether the reduced expression of αM and αX in RIAM, VASP and Vinculin KO cells was caused by a failure to induce its expression during HL-60 differentiation. In HL-60 cells, treatment with retinoic acid induced a two-fold increase in αM and around a five-fold increase in αX expression (Figure 3), as well as an increase in RIAM and VASP expression (Supplemental Figures 2B, C). However, in the knockout clones, treatment with RA only induced a small increase in αM expression reaching a level comparable to undifferentiated wild-type cells (Figure 3A), and a modest 2.5-fold increase in αX expression compared to parental HL-60 cells (Figure 3B). This is suggestive of an impairment in the transcriptional activation of these integrins that takes place during neutrophilic differentiation, and is more acute in the case of αM.
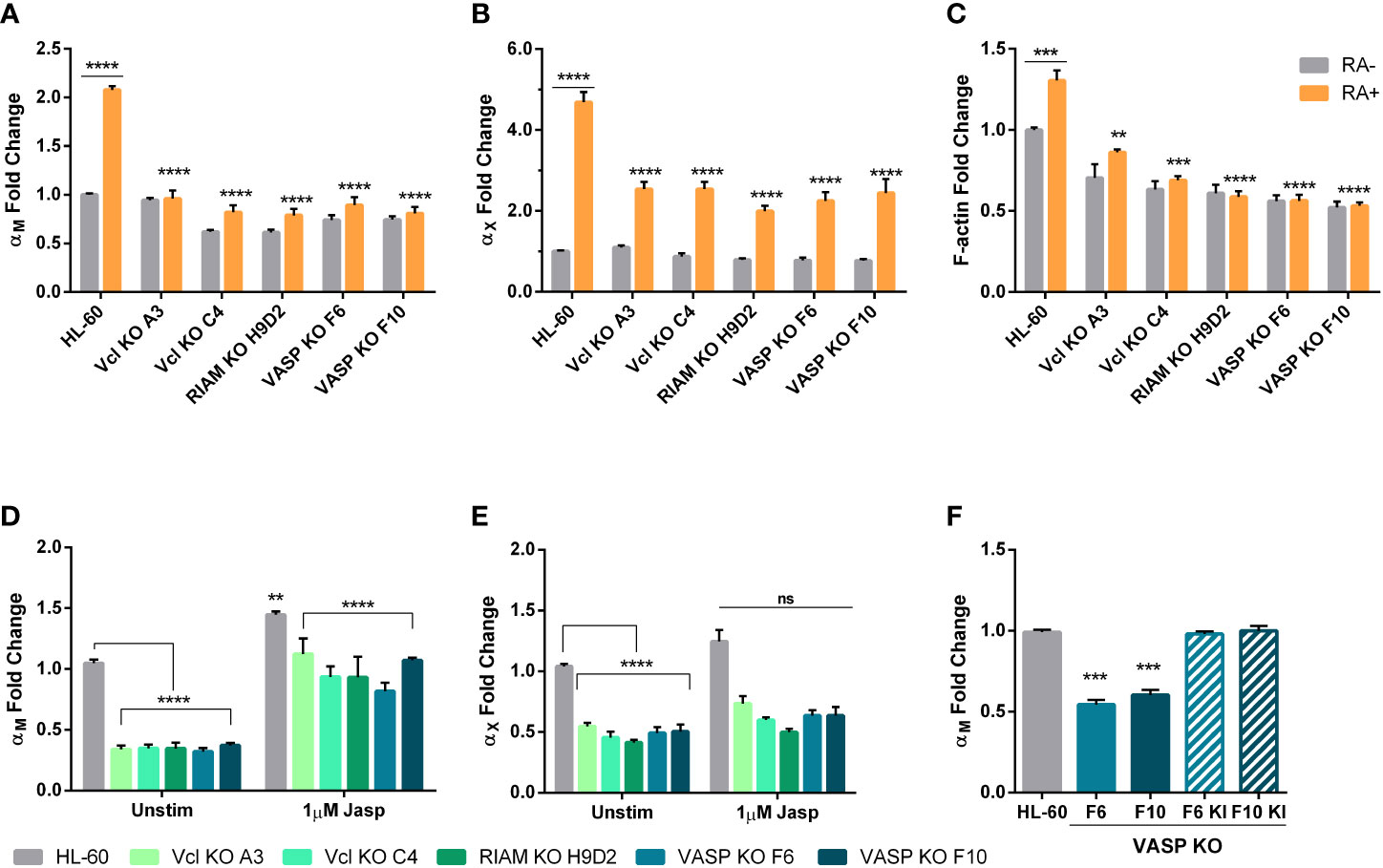
Figure 3 Upregulation of αM and αX expression during HL-60 neutrophilic differentiation depends on Vinculin, RIAM and VASP expression and is related cellular F- actin content. (A, B) Vinculin (Vcl), RIAM and VASP knockout cell lines and HL-60 parental cells were differentiated into neutrophilic-like cells with 1 μM all-trans retinoic acid treatment (RA+) or maintained undifferentiated (RA-), and expression of αM and αX integrins was analyzed by flow cytometry. (C) Cellular F-actin content was analyzed using fluorescently labeled phalloidin in HL-60 knockout cell lines and parental cells, in undifferentiated or differentiated cells. (D, E) Vinculin, RIAM and VASP knockout cell lines and HL-60 parental cells were treated with a 2 h 1 µM jasplakinolide stimulation, followed by a 24 h resting period during neutrophilic differentiation. Then, expression of αM and αX integrins was analyzed by flow cytometry. (F) Expression of αM integrin was analyzed in VASP F6 and F10 knockout clones and in VASP knock-in polyclonal cell lines F6 KI and F10 KI. Results are represented as GMFI relative to HL-60 wild type levels and are from at least 3 independent experiments done in triplicate. Data are presented as mean ± SD, where the error bars denote standard deviation. Significance (ANOVA) has been calculated with respect to HL-60 controls, ** denotes p<0.01, *** p<0.005, and **** p<0.0001, and ns denotes no significance.
Prior studies revealed that RIAM silencing resulted in a reduction in F-actin content (8). It is also well known that the G:F actin ratio determines the activation of transcriptional regulation programs. Hence, we analyzed the total cellular F-actin content in RIAM, VASP and Vinculin KO cells (Figure 3C). In all cases F-actin content was diminished, with RIAM and VASP knockout clones showing a strong reduction (40-50%). This is in agreement with the previously observed defects in phagocytic capacity and prior reports linking RIAM and VASP with the control of cytoskeletal rearrangements necessary for particle engulfment (11, 17). F-actin content was also reduced in Vinculin knockouts but more moderately (30-37%). Differentiation induced a modest yet significant increase (31%) of total F-actin content in wild type HL-60 cells, but not in RIAM VASP or Vinculin knockout cells, which were unresponsive to retinoic acid. Overall, this reduction in total F-actin content, correlated with the observed defect in αM expression.
Since F-actin content is capable of controlling gene expression, and it seemed to be the thread connecting all three knockouts, we tried to reverse the phenotype using the actin stabilizer jasplakinolide (Jasp), which induces actin polymerization. Indeed, this treatment induced a significant 3-fold increase in αM levels for all knockouts, causing them to reach levels comparable to those of unstimulated parental cells, while having a modest effect on control HL-60 cells (1.4-fold increase) (Figure 3D). However, the effect of jasplakinolide in reverting αM expression was partial, as knockouts could not reach the expression levels induced by this drug in parental cells. Nonetheless, this served as a proof-of-concept that F-actin levels are capable of controlling αM expression in neutrophils. For αX jasplakinolide treatment only induced a marginal 1.2-fold increase in expression, indicating a less relevant implication of cellular F-actin content in the control of αX expression (Figure 3E).
Finally, to ensure that the defect in αM expression was due to the gene deletion of the studied proteins, VASP was knocked-in in the two knockout clones, yielding the lines VASP F6 KI and F10 KI (Supplemental Figure 2D). When αM levels were assessed (Figure 3F), these cells were indistinguishable from parental HL-60 cells. This served as a demonstration that genetic reconstitution of VASP was able to revert the observed phenotype and that therefore the observed effect was not due to the experimental system used.
Distribution of the SRF co-regulators MRTF-A and FHL-2 is controlled by RIAM, VASP and Vinculin expression
The Serum Response Factor (SRF) transcription factor along with its co-activator MRTF-A are described as the main targets of actin dynamics (33–35). MRTF-A is sequestered in an inactive state in the cytoplasm by binding monomeric G-actin (36). Actin filament elongation reduces total cellular G-actin and leads to the dissociation of MRTF-A-G-actin complexes, and therefore allows MRTF-A nuclear import and subsequent activation of SRF-mediated transcription.
To test the hypothesis that the SRF pathway could be involved in regulating αM integrin expression, we studied MRTF-A subcellular localization in wild type and knockout lines using fluorescent microcopy (Figure 4A). For wild type cells, MRTF-A showed a punctate stain pattern, which co-localized with DAPI. This pattern implies a nuclear translocation, and is suggestive of an active transcription of SRF-controlled genes. In contrast, knockouts presented a diffuse staining pattern with MRTF-A predominantly distributed in the cytoplasm.
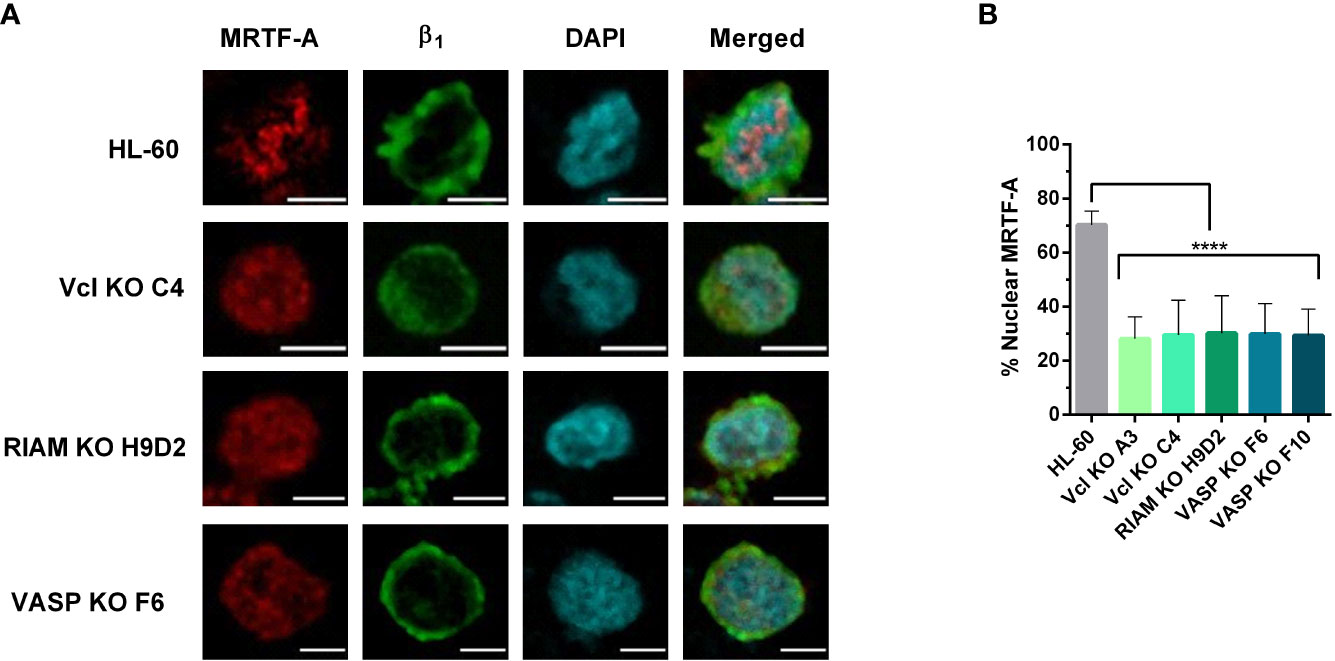
Figure 4 MRTF-A distribution is altered in Vinculin, RIAM and VASP knockouts. (A) HL-60 parental cells and the knockouts for Vinculin (Vcl), RIAM and VASP were differentiated into neutrophil-like cells, fixed, permeabilized and fluorescently labelled with an anti-MRTF-A mAb, DAPI for nuclear staining and anti-β1 integrin to delimit the plasma membrane. Images show representative results from 3 independent experiments analyzed by confocal fluorescence microscopy. Bars indicate 5 µm. (B) Quantification of MRFT-A nuclear distribution in these images is represented. Results are represented as relative to the wild-type nuclear ratio and are from 3 independent experiments with at least 50 cells. Data are presented as mean ± SD, where the error bars denote standard deviation. Significance (ANOVA) has been calculated with respect to HL-60 controls, **** denotes p<0.0001.
The extent of MRTF-A nuclear translocation was determined by fluorescent signal quantification. (Figure 4B). For each cell we defined the total fluorescence (or integrated density in ImageJ) in the MRTF-A channel for regions delimited by the cortical β1 integrin staining channel, as total cellular MRTF-A, and the fluorescence which co-localized with the nuclear DAPI stain, as the nuclear MRTF-A. While HL-60 cells showed a primarily nuclear localization of MRTF-A (~74%) the three HL-60 knockouts showed little nuclear translocation (<30%), indicating that knockout of these proteins results in a statistically significant and drastic reduction in MRTF-A translocation. These results are in agreement with our previous observations that knockouts present reduced levels of F-actin and suggest that defective SRF activity plays a role in the loss of αM expression observed for the knockouts.
Similarly, we studied the subcellular localization of the SRF corepressor FHL-2 (Figure 5). FHL-2 is known to compete with MRTF-A for SRF-binding and acts as a negative feedback loop, since MRTF-A is capable of inducing FHL-2 expression (37). Wild type cells showed FHL-2 enrichment in the cytoplasm in close proximity to β1 integrins, which is indicative of a membranous localization. However, for knockouts FHL-2 staining followed a cytoplasmic and nuclear distribution. The FHL-2 fluorescence was quantified (Figures 5B, C). Signals co-localizing with DAPI were assigned as nuclear FHL-2 and sub-membranous when localizing with integrin β1. Total cellular FHL-2 was also determined. Our data confirmed that for RIAM, VASP and Vinculin knockouts FHL-2 localization was mainly nuclear (~60%) with a minor proportion at the sub-membrane (~20%), while in HL-60 parental cells, FHL-2 was more abundant at the membrane (~50%) and only a 35% was nuclear. This result suggests that the expression of RIAM, VASP, and Vinculin may be required to retain FHL-2 close to the cytoplasmic membrane, preventing its corepressor activity and allowing proper integrin expression.

Figure 5 FHL-2 nuclear translocation is enhanced in Vinculin, RIAM and VASP knockouts. (A) HL-60 parental cells and the knockouts for Vcl, RIAM, VASP HL-60 cells were differentiated into neutrophilic-like cells with 1 μM all-trans retinoic acid for 48 h, fixed, permeabilized and fluorescently labelled using anti-FHL-2 and anti-β1 integrin antibodies and DAPI. Confocal microscopy images were analyzed using the ImageJ software package and are representative results from 3 independent experiments. Bars indicate 5 µm. (B, C) Subcellular distribution of FHL-2. The graphs represent the quantification of images. Results are represented as relative to the wild-type nuclear ratio and are from 3 independent experiments with at least 50 cells. Data are presented as mean ± SD, where the error bars denote standard deviation. Significance (ANOVA) has been calculated with respect to HL-60 controls, ***denotes p<0.005, and ****, p<0.0001.
Jasplakinolide treatment renormalizes SRF co-regulator subcellular localization
The data shown in Figure 3 indicates that jasplakinolide treatment in RIAM, VASP, and Vinculin KO cell lines was able to increase αM expression. Thereby, we assessed whether this treatment could also revert the subcellular localization of MRTF-A in knockout cells to resemble wild type cells (Figure 6). As expected, we observed that jasplakinolide treatment increases MRTF-A signal at the nucleus for HL-60 controls, since the MRTF-A closely co-localized with the nuclear DAPI stain. MRTF-A also displayed a clear nuclear localization in all knockout cells indicating that jasplakinolide treatment positively affected MRTF-A nuclear translocation, bypassing the lack of expression of the knocked-out proteins. Furthermore, for all jasplakinolide-treated cells, barely any cytoplasmic MRTF-A was observable.
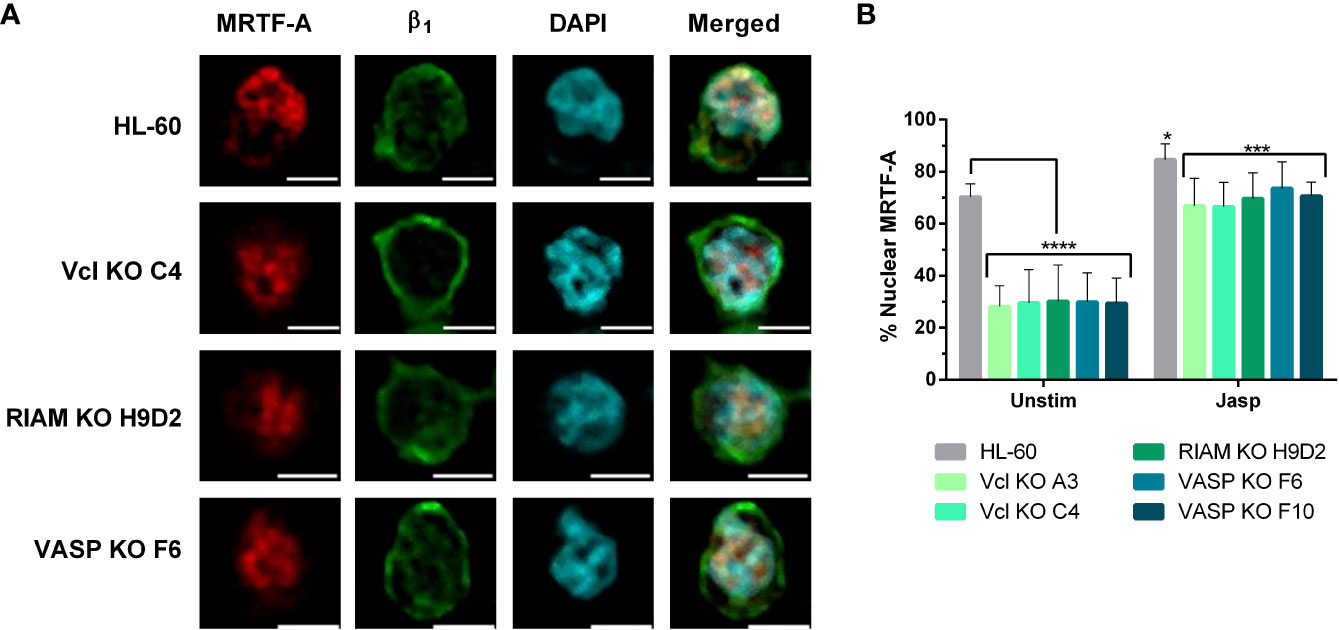
Figure 6 Jasplakinolide induces MRTF-A nuclear translocation in Vinculin, RIAM and VASP knockouts. (A) Differentiated HL-60 parental cells and HL-60 knockouts for Vcl, RIAM and VASP were adhered to slides, then subjected to a 1 µM 2 h jasplakinolide stimulation, fixed, permeabilized and fluorescently stained to determine MRTF-A localization. β1 integrin was used to delimit the plasma membrane and DAPI was used as a nuclear stain. Confocal microscopy images were analyzed using the ImageJ package and are representative results from 3 independent experiments. Bars indicate 5 µm. (B) Quantification images from the untreated cells shown on Figure 4 and jasplakinolide-treated cells on (A). Results are represented as relative to the wild-type nuclear ratio and are from 3 independent experiments with at least 50 cells. Data are presented as mean ± SD, where the error bars denote standard deviation. Significance (ANOVA) has been calculated with respect to HL-60 controls, * denotes p<0.05, ***,p<0.005, and ****,p<0.0001.
Next, we analyzed the extent of MRFT-A translocation by fluorescent signal quantification in jasplakinolide treated cells (Figure 6B) and compared to untreated cells, following the approach used in Figure 4B. In jasplakinolide-treated cells we observed a statistically significant increase in nuclear localization for MRTF-A in all cells, compared to untreated cells. Such increase was minor in HL-60 cells (1.2-fold), as most MRTF-A was nuclear prior to the jasplakinolide treatment, but quite significant in knockout cells (2.3 and 2.5-fold). This fold change correlated with the increase in αM expression for differentiated HL-60 cells observed in Figure 3D (2-fold), suggesting that the pathway F-actin-MRTF-A/SRF is critically involved in the expression of αM.
FHL-2 subcellular localization was also assessed in jasplakinolide treated cells (Figure 7). In parental cells, FHL-2 showed a sub-membranous localization as judged by its close proximity to β1 integrin. FHL-2 was also present in the nucleus for jasplakinolide treated HL-60 cells, although to a lesser extent, coinciding with reports showing that MRTF-A signaling induces FHL-2 expression acting as a negative feedback loop (37). Although some nuclear distribution was also retained, knockouts displayed an observable FHL-2 enrichment at the cytoplasm and at the sub-membranous zone induced by jasplakinolide treatment (Figure 7A).
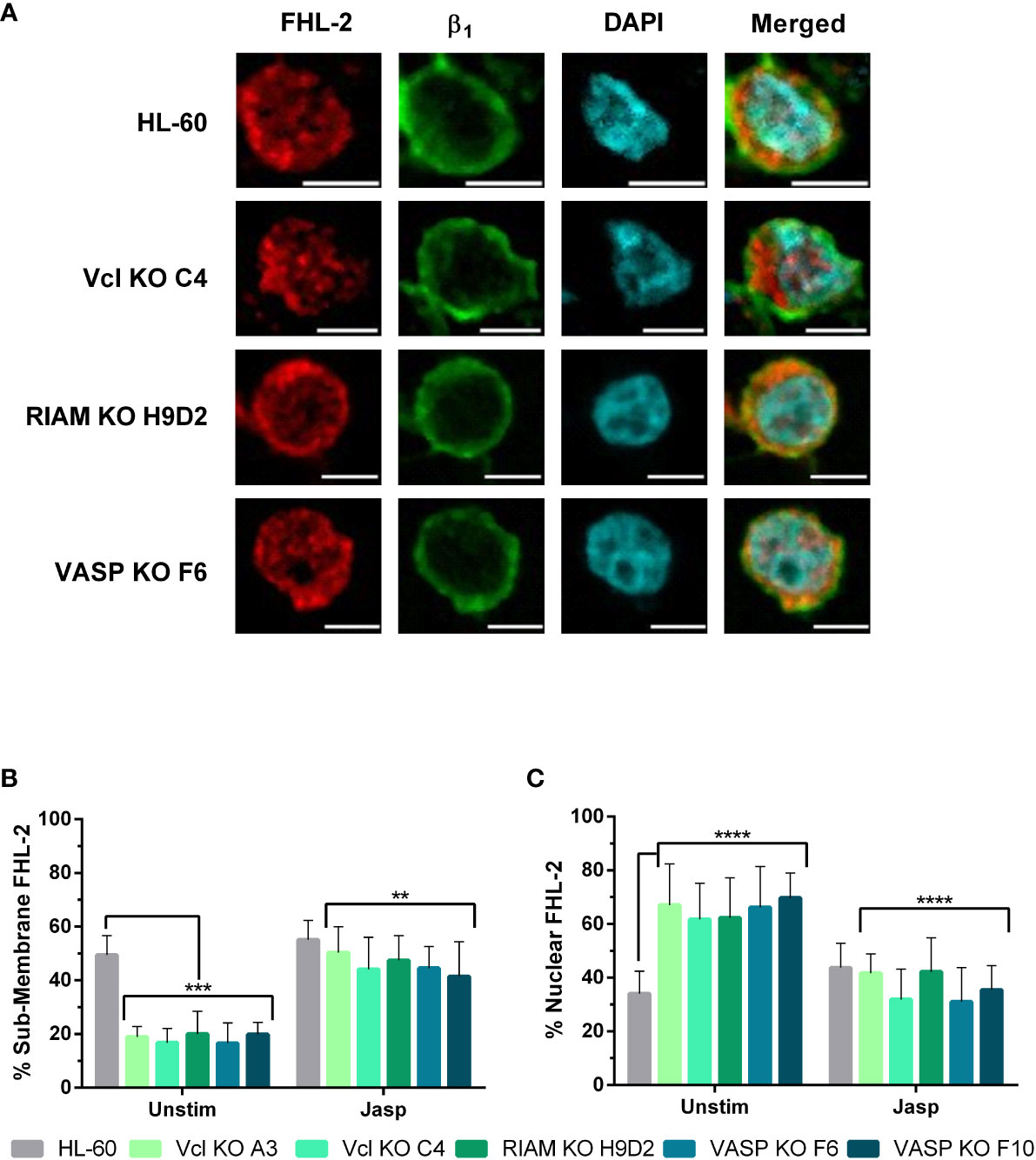
Figure 7 Jasplakinolide reduces nuclear localization of FHL-2 in Vinculin, RIAM and VASP knockouts. (A) Differentiated HL-60 parental cells and HL-60 knockouts for Vcl, RIAM and VASP were adhered to slides, then subjected to a 1 µ M 2 h jasplakinolide stimulation, fixed, permeabilized and fluorescently stained to determine FHL-2 localization. β1 integrin was used to delimit the plasma membrane and DAPI was used as a nuclear stain. Confocal microscopy images were analyzed using the ImageJ package and are representative results from 3 independent experiments. Bars indicate 5 μm. (B, C) Subcellular distribution of FHL-2. The graphs represent the quantification of images from untreated cells shown on Figure 5 and jasplakinolide-treated cells shown on (A). Results are represented as relative to the parental HL-60 membrane and nuclear ratio and are from 3 independent experiments with at least 50 cells. Data are presented as mean ± SD, where the error bars denote standard deviation. Significance (ANOVA) has been calculated with respect to HL-60 controls, ** denotes p<0.01, ***, p<0.005, and ****, p<0.0001.
We analyzed the extent of FHL-2 translocation by fluorescent signal quantification in jasplakinolide treated cells (Figures 7B, C), following the same criteria used in Figures 5B, C. Jasplakinolide treatment induced an increase in FHL-2 membrane distribution (55%) on HL-60 parental cells compared to untreated cells, leading to the equal distribution of this protein between the membrane and nuclear compartments. For all knockouts jasplakinolide also increased FHL-2 localization at the membrane (from 20% to ~50%) and decreased nuclear FHL-2, compared to untreated cells. This renormalization of FHL-2 subcellular distribution correlates with our previous results showing a similar correction of MRTF-A nuclear translocation (Figure 6) and integrin αM expression (Figure 3C) after jasplakinolide treatment.
Discussion
Myeloid cell function is critically dependent on the correct expression of cell adhesion molecules as well as downstream effectors controlling their activation. Disruption of integrin expression or defects in proteins involved in their activation cause immunodeficiencies, as is the case of Leukocyte Adhesion Deficiencies (LAD), which are due to defects in β2 integrin or Kindlin-3 expression or function (18–20). The results presented here in HL-60 cells outline that the upregulation of αM and αX observed during myeloid differentiation depends on the correct expression of RIAM, VASP and Vinculin, which are proteins involved in actin cytoskeletal dynamics and integrin signaling.
We demonstrate that deletion of either RIAM, VASP or Vinculin causes a profound defect on the expression of CR3 and CR4 receptors without affecting other IAC components, in line with previous reports. RIAM knockout in mice had no detectable influence on Talin-1, Kindlin-3, Rap1 or Cal-DAG expression in platelets, macrophages, or PMNs (38) and Vinculin deletion in MEFs does not affect the expression of Talin, Paxillin, FAK, Zyxin, or VASP (39).
Similarly, it has been reported that IAC component-deficient cells frequently display defects on the expression of specific integrin subunits in a cell dependent context (39–43). Talin-1 deletion affects β3 expression, but not β1 in osteoclasts (44), while Talin-1 and Talin-2 double knockout exhibited increased β3, α5 and a slight increase in αV, but retained normal levels of β1 in fibroblasts (45). In B-cells, Talin-1 deletion caused an altered B-cell differentiation and homing to peripheral lymph nodes, but they had normal levels of α4 and αL integrins. RIAM deletion in B-cells had no impact on α4 and αL expression, but cells showed defective homing and adhesion (46), whereas T-cell specific deletion of RIAM resulted in defective αE expression and increased α4 (47), and no changes in surface expression of integrins β1, β2, β7 (48). This is consistent with our results that again show integrin subunit specificity. Ena and VASP double knockout showed a specific decrease in α4, β1 and β7 subunits in T cells with a moderate and possibly compensatory increase in αL (49). hMENA silencing in lung and breast cancer cell lines caused a significant reduction in surface α3, α6 and β1, with diminished ITGB1 (β1 gene) mRNA levels (50). This closely matches our results which show that RIAM, VASP and Vinculin knockouts all caused a specific reduction of ITGAM (αM gene) and ITGAX (αX gene) mRNA levels. Moreover, hMENA expression was required to maintain normal cytoskeletal organization and G:F-actin ratios (50). This result closely resembles our own, where VASP, RIAM or Vinculin knockout cells all had deficient F-actin content.
Alterations in F-actin content and aberrant cytoskeletal morphologies have been described for RIAM (8, 11, 51) and Vinculin deficient cells (52, 53). Previous results from our group, showed that RIAM deficient cells display decreased F-actin content at the phagocytic cup and that this correlates with deficient VASP pSer157 phosphorylation and phagocytic cup recruitment, thereby explaining the deficiency in phagocytosis (17). Much like the results presented here, Ena-VASP-hMena triple knockout cells (mouse fibroblasts and melanoma cell lines) had aberrant lamellipodial morphology which was concordant with a loss of lamellipodial F-actin network organization and decreased F-actin content (54). Our results provide a link between the lack of expression of RIAM, VASP and Vinculin and a reduction in F-actin content, leading to a loss in αM expression which persists under all-trans retinoic acid treatment, which is known to induce their expression during neutrophilic differentiation in HL-60 cells. This is supported by the fact that the defect in αM was partially reversed by the actin stabilizing drug jasplakinolide inducing a 2.5-fold increase in expression, indicating that αM expression is heavily regulated by an F-actin dependent mechanism but does not exclude other additional F-actin-independent mechanisms downstream of RIAM, VASP and Vinculin.
In contrast with the results obtained for αM, jasplakinolide treatment had a minor effect on αX, inducing a non-significant 1.3-fold increase in expression for all cells, suggesting that αX expression is less dependent of F-actin, and that other pathways may be more critical. Interestingly, during neutrophilic differentiation granulocyte colony-stimulating factor (G-CSF) signaling to the MAPK/ERK pathway is required to activate AP-1 (55), which in turn regulates αX/CD11c expression (56). We have previously reported that RIAM has an impact on ERK1/2 phosphorylation dynamics (17, 57), which may result in activation of transcription factors like the aforementioned AP-1. Expression and activity of IAC proteins could have an impact on the nuclear translocation of transcription factors downstream of the MAPK/ERK pathway, independently of the SRF/MRTF-A pathway, thereby explaining our results.
We also demonstrate that knockout of RIAM, VASP or Vinculin results in a change in subcellular localization of the SRF coactivator MRTF-A, which is consistent with an increase in inactive cytoplasmic G-actin-bound MRTF-A. hMENA knockouts presented a reduction in SRF activity, an effect comparable to that observed using the SRF inhibitor CCG1423 (50). Similarly, it has been shown that VASP controls SRF activity and co-immunoprecipitates with mDIA1 in mouse fibroblasts (33). Mouse hematopoietic stem cells deficient in mDIA2 presented defective engraftment and migration, reduced F-actin content and inhibited transcription of SRF target genes, which included FHL-2, SRF, ITGA2, ITGAL, ITGAM and ITGB2 (58).
There are numerous reports on integrin-mediated functions being inhibited in SRF or MRTF-A deficient cells (35, 36) and that MRTF-A loss-of-function mutations lead to severe immunodeficiency (59). Interestingly a β2-Kindlin3-SRF-MRTF-A pathway is proposed to regulate dendritic cell function (34). Our results expand upon this discovery suggesting that disruption of integrin proximal components such as RIAM, VASP or Vinculin can cause severe defects in integrin expression through a dysregulation of the MRTF-A subcellular localization. We also demonstrate that modulation of the G:F-actin ratio using jasplakinolide renormalizes MRTF-A distribution, and that this effect goes in parallel with an increase in αM expression in all knockout cell lines.
We also demonstrated that the Four-and-a-Half LIM domain protein FHL-2, a SRF regulated gene itself, which has been described to compete with the coactivator MRTF-A for SRF binding (37), shifts localization in the absence of RIAM, VASP or Vinculin expression. Whereas FHL-2 is enriched at sub-membranous zones in HL-60 cells, knockouts show an increased nuclear FHL-2 signal, which can be reduced by jasplakinolide treatment. These results suggest that FHL-2 can be sequestered close to integrin adhesion complexes, whereas RIAM, VASP and Vinculin deletion significantly favors its nuclear translocation. In fact, FHL-2 has been described to directly bind several integrin subunits, including β2 through its N-terminal LIM domains (60, 61) and is capable of binding α-actin mainly through its C-terminal LIM domains (3 and 4) (62). FHL-2, much like Vinculin and RIAM (63, 64), is described to form part of a mechanosensitive pathway, regulating protein expression according to substrate rigidity and the strength of the adhesion (65). Although, a direct interaction between FHL-2 and VASP, Vinculin or RIAM has not been described yet, the evidence in the literature supports this idea. Both VASP and Vinculin directly interact with the focal adhesion proteins, Zyxin and Paxillin (66–73), and these interactions take place via their LIM domains. FHL-2 directly binds to Growth factor receptor-bound protein 7 (Grb7) in a tyrosine phosphorylation-dependent manner via its RA and PH domains (74). Since RIAM shares a similar molecular architecture, with an RA-PH module and Proline-rich regions (8–11, 57), and has therefore been compared to the Grb7 protein family (8, 9, 75), it is also possible that RIAM directly interacts with FHL-2. Alternatively, this interaction could also be indirect, through RIAM-mediated recruitment of VASP (17) or Vinculin (63, 76).
Finally, we show that the decrease in αMβ2 and αXβ2 integrin expression observed in RIAM, VASP or Vinculin cell knockouts occurs during all-trans retinoic acid induced neutrophilic differentiation of HL-60 cells. A failure to upregulate these integrin subunits might have important significance during the differentiation program of myeloid cells.
Myelopoiesis requires the hierarchical and sequential activation of transcriptional programs controlled mainly by the transcription factors PU.1 and CEBPα (77). PU.1 upregulates αM expression in promyelocytic cell lines in response to all-trans retinoic acid (78–80) and this effect has been shown to be in response to G-CSF-Stat3 signaling (81). Vav1 activation through tyrosine phosphorylation helps to drive PU.1 mediated upregulation of αM, although the authors found the formation of PU.1 complexes on the ITGAM promoter in Vav1 knockouts, suggesting that this may be a feedforward loop (82). Vav1 is upregulated after all-trans retinoic acid treatment (83, 84) and its tyrosine phosphorylation is induced downstream of integrin activation (10, 85–89). Therefore, the notion that the expression of RIAM, VASP and Vinculin might help to further drive this purported feedforward loop is given credence.
Based on this model, an initial transcription of integrin subunits could occur via myelopoiesis-specific transcription factor networks like PU.1, C/EBPα, C/EBPϵ and Gfi-1, which are also involved in the expression of G-CSF receptor. Concomitantly, VASP, RIAM and Vinculin could be upregulated, which concurs with our observations noting RIAM and VASP upregulation after all-trans retinoic acid treatment and the appearance of an upper band consistent with the phosphorylated form of VASP (Supplementary Figures 2B, C) (11). RIAM/VASP/Vinculin activity downstream of different receptors would increase F-actin content allowing MRTF-A/SRFα mediated upregulation of αM with concomitant FHL-2 sequestration at the membrane. Additionally, activation of other transcription factors induced by RIAM/VASP/Vinculin activity may contribute to regulate αM and αX expression (Figure 8). Deficiency in αMβ2 and αXβ2 expression has not been described in RIAM, VASP or Vinculin knockout mice (38, 45, 53, 90, 91). This discrepancy could be explained by the existence of compensatory mechanisms operating during myelopoiesis, involving homologous proteins and/or alternative signaling pathways, including those that promote F-actin polymerization. Our results therefore highlight the existence of a pathway that occurs during all-trans retinoic acid-induced differentiation of HL-60 cells that works to ensure the upregulation of αMβ2 and αXβ2 integrins and subsequently the acquisition of a phagocytic phenotype.
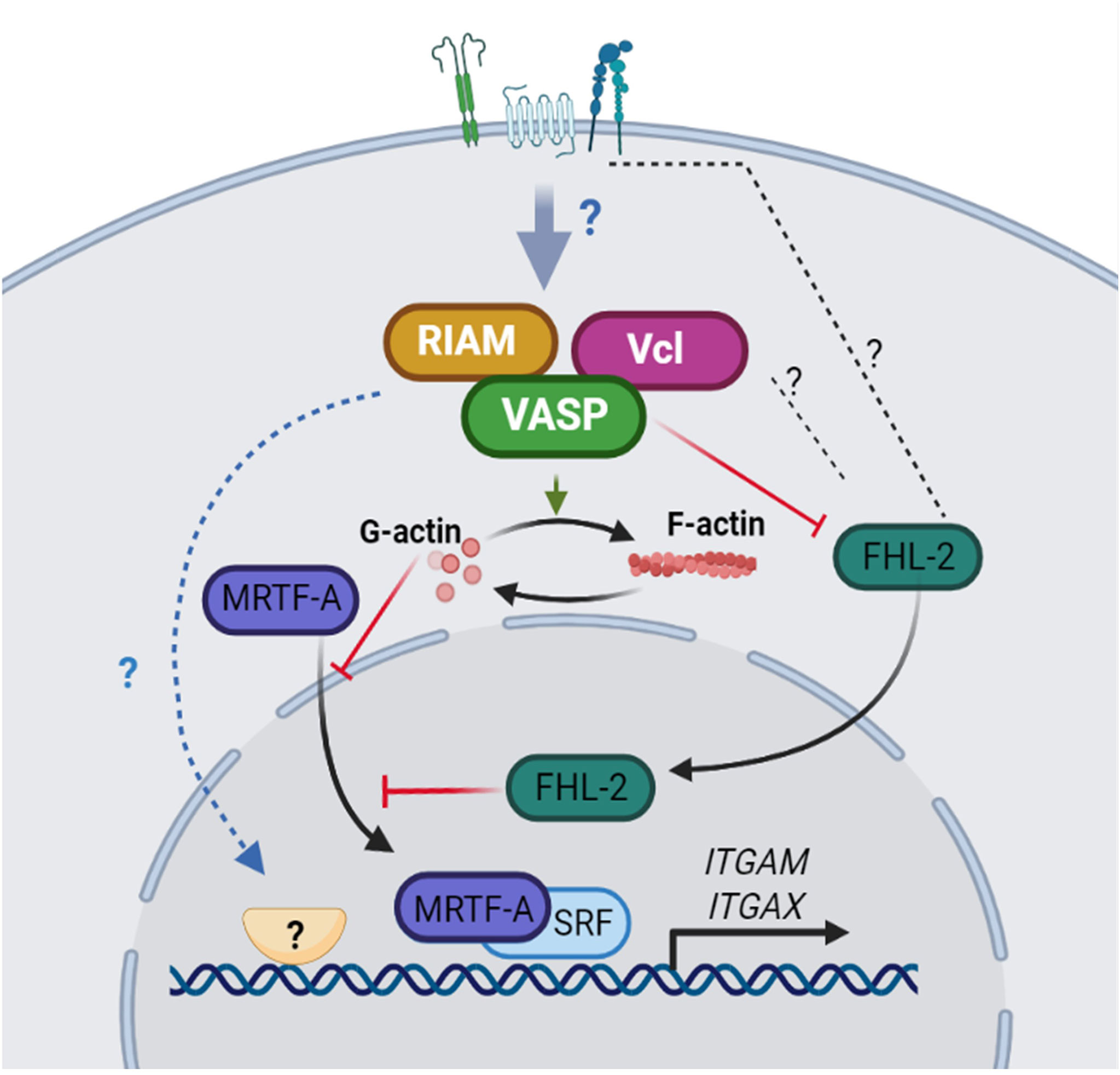
Figure 8 A model for integrin αM and αX upregulation dependent on RIAM, VASP and Vinculin expression. The expression of RIAM, VASP and Vinculin downstream of receptor signaling, contributes to increase the polymerization of actin. The reduced G-actin content allows MRTF-A translocation to the nucleus where, in combination with the SRF transcription factor promotes αM expression predominantly. Simultaneously, FHL-2 is sequestered in the plasma membrane, possibly by interacting with integrins or associated components, reducing FHL-2 co-repressor activity on SRF and allowing integrin expression. In addition, RIAM, VASP and Vinculin could regulate the activity of other potential transcription factors involved in the control αM and αX expression.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
AT-G, and EL, contributed to the conception and design of the study. Data acquisition and analysis was conducted by AT-G and EL, with experimental contributions by TF and BC (integrin expression analysis), CG-E and IC (western blot analysis) VT (CRISPR/Cas9 cloning). AT-G, and EL wrote the original draft. Scientific consultation was provided by PR. Final writing and editing were performed by AT-G, CC and EL. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by Ministerio Español de Economı́a y Competitividad (MINECO) grants: SAF2016-77096-R and PID2021-123199OB-I00 (E.L. and C.C.), the CAM (Comunidad Autónoma de Madrid) research agency through grant IND2020/BMD-17364 (P.A.R. and T.F.), and fellowship from MINECO to A. T-G. (FPU15/05349).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.951280/full#supplementary-material
References
1. Schlesinger LS, Horwitz MA. Phagocytosis of mycobacterium leprae by human monocyte-derived macrophages is mediated by complement receptors CR1 (CD35), CR3 (CD11b/CD18), and CR4 (CD11c/CD18) and IFN-gamma activation inhibits complement receptor function and phagocytosis of this bacterium. J Immunol (Baltimore Md 1950). (1991) 147(6):1983–94.
2. Ross GD, Vetvicka V. CR3 (CD11b, CD18): a phagocyte and NK cell membrane receptor with multiple ligand specificities and functions. Clin Exp Immunol (1993) 92(2):181–4. doi: 10.1111/j.1365-2249.1993.tb03377.x
3. Wright SD, Weitz JI, Huang AJ, Levin SM, Silverstein SC, Loike JD. Complement receptor type three (CD11b/CD18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proc Natl Acad Sci (1988) 85(20):7734–8. doi: 10.1073/pnas.85.20.7734
4. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med (2006) 203(12):2569–75. doi: 10.1084/jem.20060925
5. Vandendriessche S, Cambier S, Proost P, Marques PE. Complement receptors and their role in leukocyte recruitment and phagocytosis. Front Cell Dev Biol (2021) 9:624025. doi: 10.3389/fcell.2021.624025
6. Sengeløv H, Kjeldsen L, Diamond MS, Springer TA, Borregaard N. Subcellular localization and dynamics of mac-1 (alpha m beta 2) in human neutrophils. J Clin Invest (1993) 92(3):1467–76. doi: 10.1172/JCI116724
7. Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol (2009) 27:339–62. doi: 10.1146/annurev.immunol.021908.132554
8. Lafuente EM, van Puijenbroek AA, Krause M, Carman CV, Freeman GJ, Berezovskaya A, et al. RIAM, an Ena/VASP and profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Dev Cell (2004) 7(4):585–95. doi: 10.1016/j.devcel.2004.07.021
9. Patsoukis N, Bardhan K, Weaver JD, Sari D, Torres-Gomez A, Li L, et al. The adaptor molecule RIAM integrates signaling events critical for integrin-mediated control of immune function and cancer progression. Sci Signal (2017) 10(493). doi: 10.1126/scisignal.aam8298
10. Torres-Gomez A, Cabanas C, Lafuente EM. Phagocytic integrins: Activation and signaling. Front Immunol (2020) 11:738. doi: 10.3389/fimmu.2020.00738
11. Medrano-Fernandez I, Reyes R, Olazabal I, Rodriguez E, Sanchez-Madrid F, Boussiotis VA, et al. RIAM (Rap1-interacting adaptor molecule) regulates complement-dependent phagocytosis. Cell Mol Life Sci (2013) 70(13):2395–410. doi: 10.1007/s00018-013-1268-6
12. Lim J, Wiedemann A, Tzircotis G, Monkley SJ, Critchley DR, Caron E. An essential role for talin during alpha(M)beta(2)-mediated phagocytosis. Mol Biol Cell (2007) 18(3):976–85. doi: 10.1091/mbc.e06-09-0813
13. Bouti P, Webbers SDS, Fagerholm SC, Alon R, Moser M, Matlung HL, et al. β2 integrin signaling cascade in neutrophils: More than a single function. Front Immunol (2021) 11. doi: 10.3389/fimmu.2020.619925
14. Jaumouille V, Cartagena-Rivera AX, Waterman CM. Coupling of beta2 integrins to actin by a mechanosensitive molecular clutch drives complement receptor-mediated phagocytosis. Nat Cell Biol (2019) 21(11):1357–69. doi: 10.1038/s41556-019-0414-2
15. Lagarrigue F, Vikas Anekal P, Lee HS, Bachir AI, Ablack JN, Horwitz AF, et al. A RIAM/lamellipodin-talin-integrin complex forms the tip of sticky fingers that guide cell migration. Nat Commun (2015) 6:8492. doi: 10.1038/ncomms9492
16. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell (2002) 110(6):673–87. doi: 10.1016/S0092-8674(02)00971-6
17. Torres-Gomez A, Sanchez-Trincado JL, Toribio V, Torres-Ruiz R, Rodriguez-Perales S, Yanez-Mo M, et al. RIAM-VASP module relays integrin complement receptors in outside-in signaling driving particle engulfment. Cells (2020) 9(5):1166–1184. doi: 10.3390/cells9051166
18. Das J, Sharma A, Jindal A, Aggarwal V, Rawat A. Leukocyte adhesion defect: Where do we stand circa 2019? Genes Dis (2020) 7(1):107–14. doi: 10.1016/j.gendis.2019.07.012
19. Etzioni A. Leukocyte adhesion deficiency syndromes. In: Orange JS, Chinen J, editors. Encyclopedia of medical immunology: Immunodeficiency diseases, vol. p . New York, NY: Springer New York (2020). p. 425–8.
20. Fagerholm SC, Guenther C, Llort Asens M, Savinko T, Uotila LM. Beta2-integrins and interacting proteins in leukocyte trafficking, immune suppression, and immunodeficiency disease. Front Immunol (2019) 10:254. doi: 10.3389/fimmu.2019.00254
21. Almarza Novoa E, Kasbekar S, Thrasher AJ, Kohn DB, Sevilla J, Nguyen T, et al. Leukocyte adhesion deficiency-I: A comprehensive review of all published cases. J Allergy Clin Immunol Pract (2018) 6(4):1418–20.e10. doi: 10.1016/j.jaip.2017.12.008
22. Raab M, Wang H, Lu Y, Smith X, Wu Z, Strebhardt K, et al. T Cell receptor "inside-out" pathway via signaling module SKAP1-RapL regulates T cell motility and interactions in lymph nodes. Immunity (2010) 32(4):541–56. doi: 10.1016/j.immuni.2010.03.007
23. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA Targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol (2013) 31(9):827–32. doi: 10.1038/nbt.2647
24. Keizer GD, Borst J, Figdor CG, Spits H, Miedema F, Terhorst C, et al. Biochemical and functional characteristics of the human leukocyte membrane antigen family LFA-1, Mo-1 and p150,95. Eur J Immunol (1985) 15(11):1142–8. doi: 10.1002/eji.1830151114
25. Sanchez-Madrid F, Nagy JA, Robbins E, Simon P, Springer TA. A human leukocyte differentiation antigen family with distinct alpha-subunits and a common beta-subunit: the lymphocyte function-associated antigen (LFA-1), the C3bi complement receptor (OKM1/Mac-1), and the p150,95 molecule. J Exp Med (1983) 158(6):1785–803. doi: 10.1084/jem.158.6.1785
26. Cabanas C, Sanchez-Madrid F, Acevedo A, Bellon T, Fernandez JM, Larraga V, et al. Characterization of a CD11c-reactive monoclonal antibody (HC1/1) obtained by immunizing with phorbol ester differentiated U937 cells. Hybridoma. (1988) 7(2):167–76. doi: 10.1089/hyb.1988.7.167
27. Arroyo AG, Sanchez-Mateos P, Campanero MR, Martin-Padura I, Dejana E, Sanchez-Madrid F. Regulation of the VLA integrin-ligand interactions through the beta 1 subunit. J Cell Biol (1992) 117(3):659–70. doi: 10.1083/jcb.117.3.659
28. Campanero MR, del Pozo MA, Arroyo AG, Sanchez-Mateos P, Hernandez-Caselles T, Craig A, et al. ICAM-3 interacts with LFA-1 and regulates the LFA-1/ICAM-1 cell adhesion pathway. J Cell Biol (1993) 123(4):1007–16. doi: 10.1083/jcb.123.4.1007
29. Gutierrez-Lopez MD, Ovalle S, Yanez-Mo M, Sanchez-Sanchez N, Rubinstein E, Olmo N, et al. A functionally relevant conformational epitope on the CD9 tetraspanin depends on the association with activated beta1 integrin. J Biol Chem (2003) 278(1):208–18. doi: 10.1074/jbc.M207805200
30. Yanez-Mo M, Alfranca A, Cabanas C, Marazuela M, Tejedor R, Ursa MA, et al. Regulation of endothelial cell motility by complexes of tetraspan molecules CD81/TAPA-1 and CD151/PETA-3 with alpha3 beta1 integrin localized at endothelial lateral junctions. J Cell Biol (1998) 141(3):791–804. doi: 10.1083/jcb.141.3.791
31. Oren R, Takahashi S, Doss C, Levy R, Levy S. TAPA-1, the target of an antiproliferative antibody, defines a new family of transmembrane proteins. Mol Cell Biol (1990) 10(8):4007–15. doi: 10.1128/mcb.10.8.4007-4015.1990
32. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. (2001) 25(4):402–8. doi: 10.1006/meth.2001.1262
33. Grosse R, Copeland JW, Newsome TP, Way M, Treisman R. A role for VASP in RhoA-diaphanous signalling to actin dynamics and SRF activity. EMBO J (2003) 22(12):3050–61. doi: 10.1093/emboj/cdg287
34. Guenther C, Faisal I, Uotila LM, Asens ML, Harjunpaa H, Savinko T, et al. A beta2-Integrin/MRTF-A/SRF pathway regulates dendritic cell gene expression, adhesion, and traction force generation. Front Immunol (2019) 10:1138. doi: 10.3389/fimmu.2019.01138
35. Taylor A, Tang W, Bruscia EM, Zhang P-X, Lin A, Gaines P, et al. SRF is required for neutrophil migration in response to inflammation. Blood (2014) 123(19):3027–36. doi: 10.1182/blood-2013-06-507582
36. Xie L. MKL1/2 and ELK4 co-regulate distinct serum response factor (SRF) transcription programs in macrophages. BMC Genomics (2014) 15:301. doi: 10.1186/1471-2164-15-301
37. Philippar U, Schratt G, Dieterich C, Muller JM, Galgoczy P, Engel FB, et al. The SRF target gene Fhl2 antagonizes RhoA/MAL-dependent activation of SRF. Mol Cell (2004) 16(6):867–80. doi: 10.1016/j.molcel.2004.11.039
38. Klapproth S, Sperandio M, Pinheiro EM, Prunster M, Soehnlein O, Gertler FB, et al. Loss of the Rap1 effector RIAM results in leukocyte adhesion deficiency due to impaired beta2 integrin function in mice. Blood (2015) 126(25):2704–12. doi: 10.1182/blood-2015-05-647453
39. Thievessen I, Thompson PM, Berlemont S, Plevock KM, Plotnikov SV, Zemljic-Harpf A, et al. Vinculin-actin interaction couples actin retrograde flow to focal adhesions, but is dispensable for focal adhesion growth. J Cell Biol (2013) 202(1):163–77. doi: 10.1083/jcb.201303129
40. Law AL, Vehlow A, Kotini M, Dodgson L, Soong D, Theveneau E, et al. Lamellipodin and the Scar/WAVE complex cooperate to promote cell migration in vivo. J Cell Biol (2013) 203(4):673–89. doi: 10.1083/jcb.201304051
41. Bae YH, Mui KL, Hsu BY, Liu SL, Cretu A, Razinia Z, et al. A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci Signal (2014) 7(330):ra57. doi: 10.1126/scisignal.2004838
42. Manso AM, Okada H, Sakamoto FM, Moreno E, Monkley SJ, Li R, et al. Loss of mouse cardiomyocyte talin-1 and talin-2 leads to beta-1 integrin reduction, costameric instability, and dilated cardiomyopathy. Proc Natl Acad Sci USA. (2017) 114(30):E6250–E9. doi: 10.1073/pnas.1701416114
43. Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med (2008) 14(3):325–30. doi: 10.1038/nm1722
44. Zou W, Izawa T, Zhu T, Chappel J, Otero K, Monkley SJ, et al. Talin1 and Rap1 are critical for osteoclast function. Mol Cell Biol (2013) 33(4):830–44. doi: 10.1128/MCB.00790-12
45. Bromberger T, Klapproth S, Rohwedder I, Zhu L, Mittmann L, Reichel CA, et al. Direct Rap1/Talin1 interaction regulates platelet and neutrophil integrin activity in mice. Blood (2018) 132(26):2754–62. doi: 10.1182/blood-2018-04-846766
46. Su W, Wynne J, Pinheiro EM, Strazza M, Mor A, Montenont E, et al. Rap1 and its effector RIAM are required for lymphocyte trafficking. Blood (2015) 126(25):2695–703. doi: 10.1182/blood-2015-05-644104
47. Yazicioglu Y-F, Aksoylar H-I, Pal R, Patsoukis N, Boussiotis VA. The Rap1-RIAM pathway regulates the expression of integrins αEβ7(CD103) and α4β7, which guide T cell homing to intestinal compartments. Blood (2018) 132(Supplement 1):864. doi: 10.1182/blood-2018-99-119450
48. Sun H, Lagarrigue F, Wang H, Fan Z, Lopez-Ramirez MA, Chang JT, et al. Distinct integrin activation pathways for effector and regulatory T cell trafficking and function. J Exp Med (2021) 218(2):1–13. doi: 10.1084/jem.20201524
49. Estin ML, Thompson SB, Traxinger B, Fisher MH, Friedman RS, Jacobelli J. Ena/VASP proteins regulate activated T-cell trafficking by promoting diapedesis during transendothelial migration. Proc Natl Acad Sci U S A. (2017) 114(14):E2901–E10. doi: 10.1073/pnas.1701886114
50. Di Modugno F, Spada S, Palermo B, Visca P, Iapicca P, Di Carlo A, et al. hMENA isoforms impact NSCLC patient outcome through fibronectin/beta1 integrin axis. Oncogene. (2018) 37(42):5605–17. doi: 10.1038/s41388-018-0364-3
51. Patsoukis N, Lafuente EM, Meraner P, Kim J, Dombkowski D, Li L, et al. RIAM regulates the cytoskeletal distribution and activation of PLC-gamma1 in T cells. Sci Signal (2009) 2(99):ra79. doi: 10.1126/scisignal.2000409
52. Fukunaga T, Zou W, Warren JT, Teitelbaum SL. Vinculin regulates osteoclast function. J Biol Chem (2014) 289(19):13554–64. doi: 10.1074/jbc.M114.550731
53. Wilson ZS, Witt H, Hazlett L, Harman M, Neumann BM, Whitman A, et al. Context-dependent role of vinculin in neutrophil adhesion, motility and trafficking. Sci Rep (2020) 10(1):2142. doi: 10.1038/s41598-020-58882-y
54. Damiano-Guercio J, Kurzawa L, Mueller J, Dimchev G, Schaks M, Nemethova M, et al. Loss of Ena/VASP interferes with lamellipodium architecture, motility and integrin-dependent adhesion. Elife (2020) 9:1–31. doi: 10.7554/eLife.55351
55. de la Luz Sierra M, Sakakibara S, Gasperini P, Salvucci O, Jiang K, McCormick PJ, et al. The transcription factor Gfi1 regulates G-CSF signaling and neutrophil development through the ras activator RasGRP1. Blood (2010) 115(19):3970–9. doi: 10.1182/blood-2009-10-246967
56. Lopez-Rodriguez C, Kluin-Nelemans HC, Corbi AL. AP-1 regulates the basal and developmentally induced transcription of the CD11c leukocyte integrin gene. J Immunol (1996) 156(10):3780–7.
57. Hernández-Varas P, Coló GP, Bartolomé RA, Paterson A, Medraño-Fernández I, Arellano-Sánchez N, et al. Rap1-GTP-interacting adaptor molecule (RIAM) protein controls invasion and growth of melanoma cells. J Biol Chem (2011) 286(21):18492–504. doi: 10.1074/jbc.M110.189811
58. Mei Y, Han X, Liu Y, Yang J, Sumagin R, Ji P. Diaphanous-related formin mDia2 regulates beta2 integrins to control hematopoietic stem and progenitor cell engraftment. Nat Commun (2020) 11(1):3172. doi: 10.1038/s41467-020-16911-4
59. Record J, Malinova D, Zenner HL, Plagnol V, Nowak K, Syed F, et al. Immunodeficiency and severe susceptibility to bacterial infection associated with a loss-of-function homozygous mutation of MKL1. Blood (2015) 126(13):1527–35. doi: 10.1182/blood-2014-12-611012
60. Johannessen M, Moller S, Hansen T, Moens U, Van Ghelue M. The multifunctional roles of the four-and-a-half-LIM only protein FHL2. Cell Mol Life Sci (2006) 63(3):268–84. doi: 10.1007/s00018-005-5438-z
61. Wixler V, Geerts D, Laplantine E, Westhoff D, Smyth N, Aumailley M, et al. The LIM-only protein DRAL/FHL2 binds to the cytoplasmic domain of several alpha and beta integrin chains and is recruited to adhesion complexes. J Biol Chem (2000) 275(43):33669–78. doi: 10.1074/jbc.M002519200
62. Coghill ID, Brown S, Cottle DL, McGrath MJ, Robinson PA, Nandurkar HH, et al. FHL3 is an actin-binding protein that regulates alpha-actinin-mediated actin bundling: FHL3 localizes to actin stress fibers and enhances cell spreading and stress fiber disassembly. J Biol Chem (2003) 278(26):24139–52. doi: 10.1074/jbc.M213259200
63. Vigouroux C, Henriot V, Le Clainche C. Talin dissociates from RIAM and associates to vinculin sequentially in response to the actomyosin force. Nat Commun (2020) 11(1):3116. doi: 10.1038/s41467-020-16922-1
64. Omachi T, Ichikawa T, Kimura Y, Ueda K, Kioka N. Vinculin association with actin cytoskeleton is necessary for stiffness-dependent regulation of vinculin behavior. PloS One (2017) 12(4):e0175324. doi: 10.1371/journal.pone.0175324
65. Nakazawa N, Sathe AR, Shivashankar GV, Sheetz MP. Matrix mechanics controls FHL2 movement to the nucleus to activate p21 expression. Proc Natl Acad Sci U S A. (2016) 113(44):E6813–E22. doi: 10.1073/pnas.1608210113
66. Drees B, Friederich E, Fradelizi J, Louvard D, Beckerle MC, Golsteyn RM. Characterization of the interaction between zyxin and members of the ena/vasodilator-stimulated phosphoprotein family of proteins. J Biol Chem (2000) 275(29):22503–11. doi: 10.1074/jbc.M001698200
67. Hoffman LM, Jensen CC, Chaturvedi A, Yoshigi M, Beckerle MC. Stretch-induced actin remodeling requires targeting of zyxin to stress fibers and recruitment of actin regulators. Mol Biol Cell (2012) 23(10):1846–59. doi: 10.1091/mbc.e11-12-1057
68. Smith MA, Blankman E, Deakin NO, Hoffman LM, Jensen CC, Turner CE, et al. LIM domains target actin regulators paxillin and zyxin to sites of stress fiber strain. PloS One (2013) 8(8):e69378. doi: 10.1371/journal.pone.0069378
69. Uemura A, Nguyen TN, Steele AN, Yamada S. The LIM domain of zyxin is sufficient for force-induced accumulation of zyxin during cell migration. Biophys J (2011) 101(5):1069–75. doi: 10.1016/j.bpj.2011.08.001
70. Anderson CA, Kovar DR, Gardel ML, Winkelman JD. LIM domain proteins in cell mechanobiology. Cytoskeleton (Hoboken). (2021) 78(6):303–11. doi: 10.1002/cm.21677
71. Brown MC, Perrotta JA, Turner CE. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J Cell Biol (1996) 135(4):1109–23. doi: 10.1083/jcb.135.4.1109
72. Deakin NO, Turner CE. Paxillin comes of age. J Cell Sci (2008) 12115:2435–44. doi: 10.1242/jcs.018044
73. Lopez-Colome AM, Lee-Rivera I, Benavides-Hidalgo R, Lopez E. Paxillin: a crossroad in pathological cell migration. J Hematol Oncol (2017) 10(1):50. doi: 10.1186/s13045-017-0418-y
74. Siamakpour-Reihani S, Argiros HJ, Wilmeth LJ, Haas LL, Peterson TA, Johnson DL, et al. The cell migration protein Grb7 associates with transcriptional regulator FHL2 in a Grb7 phosphorylation-dependent manner. J Mol Recognit (2009) 22(1):9–17. doi: 10.1002/jmr.916
75. Holt LJ, Daly RJ. Adapter protein connections: the MRL and Grb7 protein families. Growth Factors. (2005) 23(3):193–201. doi: 10.1080/08977190500196267
76. Goult BT, Zacharchenko T, Bate N, Tsang R, Hey F, Gingras AR, et al. RIAM and vinculin binding to talin are mutually exclusive and regulate adhesion assembly and turnover. J Biol Chem (2013) 288(12):8238–49. doi: 10.1074/jbc.M112.438119
77. Ai Z, Udalova IA. Transcriptional regulation of neutrophil differentiation and function during inflammation. J Leukoc Biol (2020) 107(3):419–30. doi: 10.1002/JLB.1RU1219-504RR
78. Kastner P, Chan S. PU.1: a crucial and versatile player in hematopoiesis and leukemia. Int J Biochem Cell Biol (2008) 40(1):22–7. doi: 10.1016/j.biocel.2007.01.026
79. Pahl HL, Scheibe RJ, Zhang DE, Chen HM, Galson DL, Maki RA, et al. The proto-oncogene PU.1 regulates expression of the myeloid-specific CD11b promoter. J Biol Chem (1993) 268(7):5014–20. doi: 10.1016/S0021-9258(18)53496-7
80. Durual S, Rideau A, Ruault-Jungblut S, Cossali D, Beris P, Piguet V, et al. Lentiviral PU.1 overexpression restores differentiation in myeloid leukemic blasts. Leukemia (2007) 21(5):1050–9. doi: 10.1038/sj.leu.2404645
81. Panopoulos AD, Bartos D, Zhang L, Watowich SS. Control of myeloid-specific integrin alpha mbeta 2 (CD11b/CD18) expression by cytokines is regulated by Stat3-dependent activation of PU.1. J Biol Chem (2002) 277(21):19001–7. doi: 10.1074/jbc.M112271200
82. Brugnoli F, Lambertini E, Varin-Blank N, Piva R, Marchisio M, Grassilli S, et al. Vav1 and PU.1 are recruited to the CD11b promoter in APL-derived promyelocytes: role of Vav1 in modulating PU.1-containing complexes during ATRA-induced differentiation. Exp Cell Res (2010) 316(1):38–47.doi: 10.1016/j.yexcr.2009.09.004
83. Bertagnolo V, Brugnoli F, Mischiati C, Sereni A, Bavelloni A, Carini C, et al. Vav promotes differentiation of human tumoral myeloid precursors. Exp Cell Res (2005) 306(1):56–63. doi: 10.1016/j.yexcr.2004.12.001
84. Bertagnolo V, Grassilli S, Bavelloni A, Brugnoli F, Piazzi M, Candiano G, et al. Vav1 modulates protein expression during ATRA-induced maturation of APL-derived promyelocytes: a proteomic-based analysis. J Proteome Res (2008) 7(9):3729–36. doi: 10.1021/pr7008719
85. Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ. Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc Natl Acad Sci U S A. (2003) 100(23):13298–302. doi: 10.1073/pnas.2336149100
86. Xiao R, Xi XD, Chen Z, Chen SJ, Meng G. Structural framework of c-src activation by integrin beta3. Blood (2013) 121(4):700–6. doi: 10.1182/blood-2012-07-440644
87. Lowell CA, Fumagalli L, Berton G. Deficiency of src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J Cell Biol (1996) 133(4):895–910. doi: 10.1083/jcb.133.4.895
88. Giagulli C, Ottoboni L, Caveggion E, Rossi B, Lowell C, Constantin G, et al. The src family kinases hck and fgr are dispensable for inside-out, chemoattractant-induced signaling regulating beta 2 integrin affinity and valency in neutrophils, but are required for beta 2 integrin-mediated outside-in signaling involved in sustained adhesion. J Immunol (2006) 177(1):604–11. doi: 10.4049/jimmunol.177.1.604
89. Wu L, Bernard-Trifilo JA, Lim Y, Lim ST, Mitra SK, Uryu S, et al. Distinct FAK-src activation events promote alpha5beta1 and alpha4beta1 integrin-stimulated neuroblastoma cell motility. Oncogene. (2008) 27(10):1439–48. doi: 10.1038/sj.onc.1210770
90. Bromberger T, Klapproth S, Rohwedder I, Weber J, Pick R, Mittmann L, et al. Binding of Rap1 and riam to Talin1 fine-tune β2 integrin activity during leukocyte trafficking. Front Immunol (2021) 12:702345. doi: 10.3389/fimmu.2021.702345
Keywords: phagocytosis, cytoskeleton, vasp, integrin expression, CR3 (CD11b/CD18), Vinculin, CR4 (CD11c/CD18), riam
Citation: Torres-Gomez A, Fiyouzi T, Guerra-Espinosa C, Cardeñes B, Clares I, Toribio V, Reche PA, Cabañas C and Lafuente EM (2022) Expression of the phagocytic receptors αMβ2 and αXβ2 is controlled by RIAM, VASP and Vinculin in neutrophil-differentiated HL-60 cells. Front. Immunol. 13:951280. doi: 10.3389/fimmu.2022.951280
Received: 23 May 2022; Accepted: 23 August 2022;
Published: 27 September 2022.
Edited by:
Allan R Brasier, University of Wisconsin-Madison, United StatesReviewed by:
Frederic Lagarrigue, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceLai Wen, La Jolla Institute for Immunology (LJI), United States
Copyright © 2022 Torres-Gomez, Fiyouzi, Guerra-Espinosa, Cardeñes, Clares, Toribio, Reche, Cabañas and Lafuente. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Esther M. Lafuente, bWVsYWZ1ZW50ZUBtZWQudWNtLmVz; Alvaro Torres-Gomez, YXRvcnIwMUB1Y20uZXM=