
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 22 July 2022
Sec. Antigen Presenting Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.948297
This article is part of the Research TopicInsights in Antigen Presenting Cell Biology: 2022View all 7 articles
 Silvia D’Amico1
Silvia D’Amico1 Patrizia Tempora1
Patrizia Tempora1 Ombretta Melaiu1,2
Ombretta Melaiu1,2 Valeria Lucarini1
Valeria Lucarini1 Loredana Cifaldi2,3
Loredana Cifaldi2,3 Franco Locatelli1,4
Franco Locatelli1,4 Doriana Fruci1*
Doriana Fruci1*Despite the significant clinical advances with the use of immune checkpoint inhibitors (ICIs) in a wide range of cancer patients, response rates to the therapy are variable and do not always result in long-term tumor regression. The development of ICI-resistant disease is one of the pressing issue in clinical oncology, and the identification of new targets and combination therapies is a crucial point to improve response rates and duration. Antigen processing and presentation (APP) pathway is a key element for an efficient response to ICI therapy. Indeed, malignancies that do not express tumor antigens are typically poor infiltrated by T cells and unresponsive to ICIs. Therefore, improving tumor immunogenicity potentially increases the success rate of ICI therapy. In this review, we provide an overview of the key elements of the APP machinery that can be exploited to enhance tumor immunogenicity and increase the efficacy of ICI-based immunotherapy.
The impressive clinical results achieved with immunotherapy in terms of remission and improved survival have given new optimism for treating patients with cancer (1, 2). However, the number of patients in whom the benefict of treatment are lasting and resolving is limited.
Identifying the mechanisms adopted by the tumor to escape immunologic surveillance becomes of paramount importance to improve the efficacy of immunotherapy. The cancer immune cycle describes the sequence of events by which an antitumor immune response results in the effective killing of cancer cells (3). In the first step, tumor antigens present in the tumor microenvironment are captured by antigen-presenting cells (APC), such as dendritic cells (DCs), which migrate to draining lymph nodes to present antigens to naïve T lymphocytes via MHC class I and MHC class II molecules. This results in the activation of immature T cells into effector T cells which subsequently reach the tumor site through the bloodstream to specifically recognize and eliminate tumor cells (3). Because each step of the cycle contributes to tumor cell killing, inhibition of one or more of these processes may cause attenuation of antitumor responses or immune escape (4). In this regard, failure of ICI therapy could also result from a defect in any of the above-mentioned steps, i.e., insufficient generation of antitumor T cells, inadequate function of tumor-specific T cells (5, 6), or impaired T-cell memory formation (7, 8). Recently, it has been shown that impaired migration of DCs from tumor tissues to the regional lymph nodes caused defective antigen presentation and priming of T cells, leading to uncontrolled tumor development and resistance to ICIs in several cancer types (9–12). Mechanisms related to immune escape in the cancer immune cycle as well as therapies to overcome immune escape and resistance to ICI therapies have been widely addressed (8, 13, 14).
ICI-based immunotherapy acts by blocking the inhibitory action of immune checkpoints and restoring the function of tumor-infiltrating immune cells (15). CD8+ T cells are one of the main immune cell populations targeted by ICI-based therapy. These cells continuously scrutinize the integrity of the proteome by scanning peptides presented on the cell surface bound to HLA class I molecules (Figure 1). The integrity of this process, called antigen processing and presentation (APP) pathway, is critical to ensure generation of effective CD8+ T cells to fight cancer (16). The APP pathway includes several closely related proteins, whose level of expression has been shown to influence quantitatively and qualitatively the repertoire of tumor antigens presented to the immune system. Changes in the expression of individual components of the APP machinery have been associated with the lack or reduced presentation of tumor antigens, as well as variation in the level of tumor-infiltrating CD8+ T cells and response to ICI-based therapy (17–19). Recently, Maggs and colleagues (20) provided a detailed overview of the structural and nonstructural mechanisms underlying HLA class I APP defects in malignant cells. In this review, we deepen the contribution of APP machinery to the cancer immunopeptidome, with a focus on how they can be exploited to enhance tumor immunogenicity.
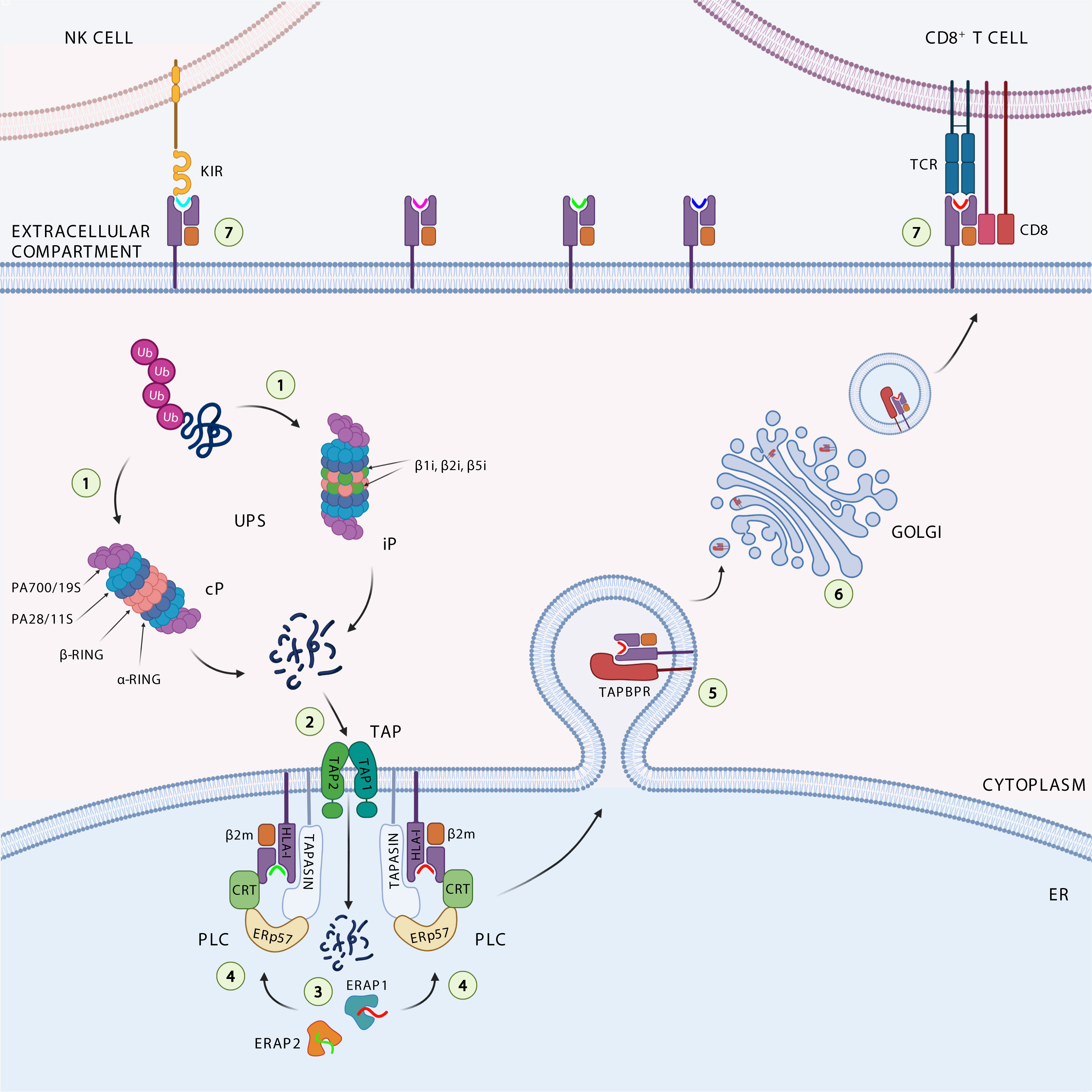
Figure 1 Antigen processing and presentation pathway. The ubiquitinated proteins are degraded (1) by the catalytic subunits of the constitutive proteasome (cP) or immunoproteasome (iP) into peptides. The peptides generated are translocated (2) through the transporter associated with antigen processing (TAP1 and TAP2) complex into the endoplasmic reticulum (ER), where they can be further trimmed (3) by the ER aminopeptidases ERAP1 and ERAP2. Loading onto the HLA class I heavy chain-associated β2-microglobulin (β2m) (4) is a multistep process facilitated by the peptide loading complex (PLC), composed of TAP, ERp57, and the chaperones tapasin and calreticulin (CRT). Binding of high-affinity peptide induces dissociation of the HLA class I complex from PLC, its stabilization by interaction with TAP-binding protein related (TAPBPR) (5), and subsequent trafficking (6) to the cell surface to be presented to cytotoxic CD8+ T cells and NK cells (7).
Tumor antigens are small pieces of protein able to provoke an immune attack against cancer. They can be derived from mutated or unmutated cellular proteins and are referred to as tumor-specific antigens or tumor-associated antigens, respectively (21). Several lines of evidence indicate that potential sources of tumor antigens could also arise from the translation of presumably non-coding transcripts, such as introns, untranslated regions, intergenic sequences, or out-of-frame exons (22–25). In general, peptides presented by HLA class I molecules are not necessarily derived from the most highly expressed proteins, such as housekeeping proteins (histones, ribosomal proteins, metabolic enzymes, and cytoskeletal proteins) (22, 23, 26). Indeed, unstable proteins, i.e., those that are rapidly degraded by the proteosome, are also represented in the immunopeptidome. Such proteins, although hardly detectable by mass spectrometry analysis, are in fact able to generate immunogenic peptides (22, 23, 26). Regardless of their origin, peptides compete with each other for presentation by HLA class I molecules (22, 26, 27). It has been estimated that the immunopeptidome, composed of thousands of short peptide antigens of 8-12 amino acids, contains at most 1% of the peptide sequences found in individual cells.
The intracellular antigen processing pathway almost exclusively deals with the ubiquitinin proteasome system (UPS) activity, by which endogenous proteins are first ubiquitinated and subsequently degraded by the proteasome, a large cytosolic proteolytic complex which digests unneeded or damaged proteins (Figure 1) (28). Proteasome complex consists of 14 structural subunits and 2 copies of 3 catalytic subunits β1, β2, and β5 (29). Following stimulation with interferon-γ (IFNγ), due to cellular stress and inflammation, the constitutive proteasome is converted in the immunoproteasome, which differs in the catalytic subunits β1i (LMP2 or Psmb9), β2i (MECL1 or Psmb10) and β5i (LMP7 or Psmb8) (30). The immunoproteasome shows increased peptide cleavage activity after basic or hydrofobic residues, allowing the production of peptides with higher affinity for HLA class I molecules (31–33). A fraction of these peptides are transported into the lumen of the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP), a heterodimeric complex belonging to the ABC transporter superfamily. TAP is composed of two subunits (TAP1/TAP2), both consisting of a transmembrane region, a substrate binding site, and an ATP-binding domain, and preferentially interacts with peptides (one at a time) ranging from 8 to 16 amino acids in length (34). These properties give TAP the ability to perform an initial selection of peptides available for HLA class I binding. It has been estimated that about 25% of antigenic peptides reaching the cell surface bound to HLA class I molecules are shortened at the N-terminal end by ER aminopetidases ERAP1 and ERAP2 (ERAAP in mouse), which are known to generate and destroy peptides (35–37). Peptides with the correct length and sequence to bind HLA class I alleles are loaded onto β2m-associated HLA class I heavy chain dimers with the help of the peptide-loading complex (PLC) that consists of TAP, ER resident protein 57 (ERp57), and chaperone molecules calreticulin and tapasin. The peptide-HLA class I-β2m complex formed is further stabilized by the binding with the tapasin-binding protein related (TAPBPR), the newest member of the APP machinery (Figure 1) (38). Tapasin and TAPBPR work together sequentially, with tapasin involved in loading the peptide into the HLA class I-β2m dimer within the PLC in the ER, and TAPBPR assigned to control the stability of the peptide-HLA class I-β2m complex in the Golgi during its transport across the plasma membrane (39). The resulting trimeric complex travels to the plasma membrane to be presented to cytotoxic CD8+ T cells and NK cells.
Defective expression of APP components has detected in most of the tumors. Many evidence indicates that impaired functionality of the APP machinery in tumor cells, in addition to causing a change in the repertoire of antigens presented on the cell surface, results in a change in the level of tumor-infiltrating CD8+ T cells and the response to ICI-based therapy.
Several studies have shown that mice lacking the single catalytic subunits of the immunoproteasome have relatively modest changes in the immunopeptidome (40–42). However, mice completely lacking immunoproteasome had defects in the presentation of several MHC class I epitopes with an immunopeptidome 50% difference from wild type mice (43). These differences were sufficient to stimulate robust rejection of wild type splenocytes transplanted in triple knock-out mice (43), thus suggesting that the immunoproteasome plays a primary role in antigen presentation. Based on its ability to modulate the expression of pro-tumorigenic cytokines and chemokines or increase the presentation of tumor antigens, the immunoproteasome shows both pro- and anti-tumor properties, respectively (44). The pro-tumoral role of the immunoproteasome is evident in the case of colitis-associated carcinogenesis, where increased expression of β5i/LMP7 and β1i/LMP2 has been observed in cronically inflamed colons (45). As a result, β5i/LMP7-deficient mice show reduced tumor formation and production of pro-tumor chemokines (e.g. CXCL1, CXCL2, and CXCL3) (45). In contrast to the inflammatory environment of the gut, increased expression of β5i/LMP7 and β1i/LMP2 subunits in melanoma cells resulted in an altered immunopeptidome which correlated with increased reactivity of tumor-infiltrating CD8+ T cells, improved survival and response to ICIs (46). Indeed, increased immunoproteasome expression has been attributed to IFNγ secretion by tumor-infiltrating CD8+ T cells in several tumor types, including melanoma, colorectal cancer, gastric cancer, and breast cancer (46–50). Similarly, in non-small cell lung cancer (NSCLC), high immunoproteasome expression was correlated with good prognosis (51). In this scenario, induction of immunoproteasome expression could be an interesting approach to improve the effect of ICI-based therapy in specific tumor settings.
Several viruses and tumors exploit the down-regulation or inhibition of TAP as a strategy to evade CD8+ T-cell control (52). In this context, it has been observed that TAP-deficient cells display a severe reduction of HLA class I surface expression, but still sufficient to have a partial antigen presentation and functional T-cell recognition through alternative processing pathways (53). In TAP-deficient tumor cells, although the presentation of conventional tumor-specific antigens may decrease, an alternative class of non-mutated antigens emerges within the immunopeptidome. These novel antigens, called T cell epitopes associated with impaired peptide processing (TEIPPs), derive from normal housekeeping proteins and are not loaded on HLA class I molecules in healthy cells. Accordingly, in mouse and human models, CD8+ T cells of healthy individuals specific for some TEIPP antigens reside in the naïve state, indicating that they are not triggered during an immune response (53, 54). Conversely, as demonstrated by a number of studies, TEIPPs are efficient in elicit a functional antitumor CD8+ T cell response (53, 55, 56). Marijt and colleagues identified a human HLA-A2-binding TEIPP derived from the LDL Receptor Related Protein Associated Protein 1 (LRPAP1) signal sequence (LRPAP121-30) common to different type of tumors, including melanoma, lymphoma, colon and renal cell carcinoma (53). The authors demonstrated that LRPAP121-30-specific CD8+ T cells selectively recognized TAP-defective cancers, but not the TAP-expressing counterparts on healthy tissue (53). More recently, the same research group designed a synthetic long peptide vaccine by optimizing a peptide sequence based on the previously identified LRPAP1-derived TEIPP (57). The resulting vaccine was able to induce CD8+ T-cell immunity and cross-presentation by monocyte-derived DC, resulting in tumor control (57). Interestingly, Garrido and colleagues used a clinical applicable method to increase the antigenicity of tumor cells by downregulating TAP expression in situ. The authors demonstrated that administration of TAP siRNA conjugated to a broad-spectrum tumor-targeting nucleolin aptamer resulted in: i) inhibition of tumor growth in multiple transplanted, orthotopic and autochthonous mouse tumor models of different origin and genetic background without measurable toxicity, ii) enhancement of the antitumor effect of PD-1 antibody, and iii) induction of TAP-independent peptide presentation in human tumor cells (55). Thus, treatment with the siRNA-conjugated nucleolin-TAP aptamer represents a widely applicable approach to increase the antigenicity of tumor lesions and improve the efficacy of ICI-based therapies. These studies offer different strategies to exploit TAP inhibition as an approach to make tumor cells responsive to ICI-based therapy. If in the first case, a TAP-deficient tumor proves to be more sensitive to a therapy based on TEIPP targeting, in the second one the alteration of antigen processing is used as a novel therapeutic approach to target tumor cells.
Silencing of TAPBPR in HeLa cells causes an increase in the overall number of peptides presented on the cell surface by HLA class I molecules compared with control cells (39). This suggests that inhibition of TAPBPR, by reducing affinity requirements for peptide binding to HLA class I molecules, could expand the repertoire of antigens presented on the cell surface, thereby eliciting an immune response. Recently, Ilca and colleagues demonstrated that the plasma membrane-targeted or recombinant soluble form of TAPBPR retain their peptide-editing capacities (58). Addition of recombinant TAPBPR to the extracellular environment promotes the exchange of peptides bound to HLA class I molecules directly at the plasma membrane. The authors suggest that a similar approach could be useful to enrich tumor cells with highly immunogenic peptides, inducing immune recognition of tumors and thus potentially improving immunotherapy (58). It is now clear that, as with other components of the APP, the binding affinity of TAPBPR varies among HLA class I allotypes with a strong preference for HLA-A (59). This implies that patients with different HLA class I typing might be susceptible to TAPBPR chaperone-mediated peptide editing to different degrees. This could play a role in disease susceptibility, but also influence patients’ response to possible TAPBPR-mediated therapy (60).
The first evidence that loss of ERAP1 function causes a profound alteration of the immunopeptidome able of eliciting potent immune responses comes from pioneering studies in mouse models of Shastri and other groups (61–64). The authors demonstrated that wild-type mice respond vigorously to the injected ERAAP-/- splenocytes by activating the response of CD8+ T cells that specifically recognize peptides normally destroyed by ERAAP (61). Given the key role of ER aminopeptidases in the generation of the cancer immunopeptidome, several studies have explored the possibility of targeting ER aminopeptidases for generating protective anticancer responses. The first evidence comes from Cifaldi and colleagues, who demostrate that ERAAP silencing leads to rejection of murine RMA T-cell lymphoma in syngeneic mice (65). This tumor rejection was due to both T cells and NK cells, and was dependent on the repertoire of peptides bound to MHC class I molecules. Indeed, replacement of endogenous peptides with high-affinity peptides was sufficient to restore a protective effect through recognition of the stable peptide-MHC class I complex by NK cell inhibitory receptors (65). Similarly, inhibition of human ERAP1 was also able to regulate NK cell activity by controlling the interaction of peptide-HLA class I complexes with NK cell inhibitory receptors (66, 67). James and colleagues assessed the role of ERAAP expression in a syngeneic model of colorectal cancer and demonstrated that the immunodominant peptide GSW11 is trimmed and destroyed by ERAAP (Table 1). As expected, inhibition of ERAAP by either gene silencing or drug treatment, caused an increase of GSW11 presentation and reduced tumor growth in syngeneic mice (68).
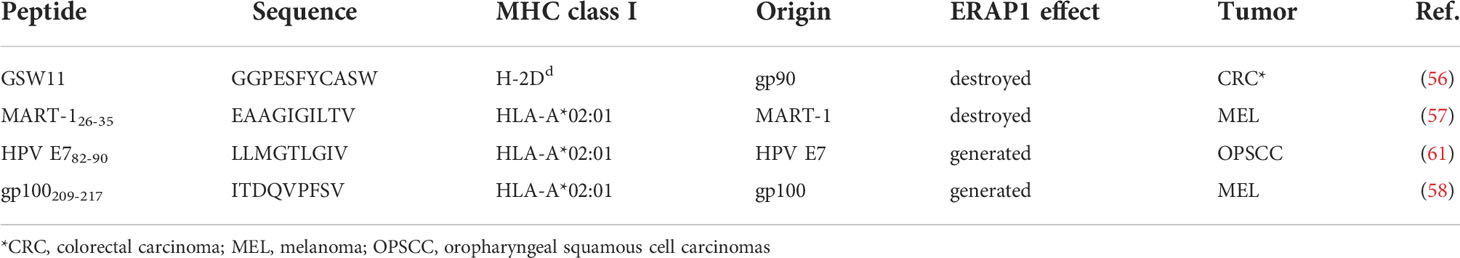
Table 1 Tumor antigens affected by ERAAP/ERAP1.
The involvement of ERAP1 in the generation and destruction of tumor antigens has been endorsed by many other studies. Keller and colleagues demonstrated that constitutive expression of ERAP1 and proteasome activator 28 (PA28) were sufficient to inhibit generation of the MART-126-35 epitope (69) (Table 1). The authors show that both genetic and pharmacological inhibition of ERAP1 strongly increase MART-126-35 presentation in human melanoma cells and IFN-γ release by MART-126-35-specific CD8+ T cells (69). Textoris-Taube and colleagues demonstrate that ERAP1, but not ERAP2, is involved in the generation of the glycoprotein 100 (gp100)209-217 immunogenic epitope derived from melanoma differentiation antigen (gp100)PMEL17, and promotes the activation of gp100209-217-specific CD8+ T cells by melanoma cells (Table 1) (70). Administration of the gp100 peptide combined with ipilimumab, the CTLA-4 antagonist, showed no improvement in disease progression in a proportion of patients with metastatic melanoma compared with those treated with ipilimumab alone, most likely due to insufficient epitope presentation (71, 72). It is possible that the allelic status of ERAP1 known to affect its enzymatic activity and ability to generate and/or destroy antigenic peptides, may contribute to the variability of immune responses between individuals. Indeed, Reeves and colleagues demonstrated that the functional activity of different ERAP1 allotypes is positively correlated with the amount of tumour-infiltrating CD8+ T cells in HPV+ oropharyngeal squamous cell carcinomas (OPSCC) through the generation of HPV E6/E7 epitopes (73). This is the first evidence that different ERAP1 allotypes affect the HPV-16 epitope presentation and anti-HPV T-cell responses (Table 1) (73).
These studies collectively suggest that inhibition of ERAP1 activity, by resulting in a novel immunopeptidome, could represent a viable therapeutic strategy to enhance protective anti-tumor immune responses. Consistently, Koumantou and colleagues demonstrated that pharmacological inhibition of ERAP1 in a melanoma cell line induced profound changes in both the quality and quantity of one half of the peptides presented, specifically increasing the presentation of peptides with high binding affinity for HLA class I molecules (74). Moreover, inhibition of ERAAP by nucleolin-targeted siRNA was able to elicit an efficient antitumor response by sensitizing transplantable 4T1 breast carcer cell model to anti-PD-1 immunotherapy (55).
Proteomic studies revealed that ERAP2, when expressed, also contributes to the immunopeptidome (75). In the tumor contest, Temponeras and colleagues showed that pharmacological inhibition of ERAP2 alters the immunopeptidome in the MOLT-4 human leukemia cell line, with more than 20% of peptides detected as novel, or significantly up-regulated (76). Most of these peptides were 9mers with sequence motifs consistent with optimal binding motifs for at least one of the HLA class I alleles carried by MOLT-4 (76). Such peptides might be able to induce novel cytotoxic CD8+ T-cell responses against tumor cells and synergize with ICI-based therapy.
The immunopeptidome is the representation on the cell surface of what is actively translated and degraded within the cell, i.e., the element through which immune cells are able to detect and eliminate cancer cells (77–79). Indeed, loss of expression of HLA class I and APP components is one of the best-known mechanisms exploited by tumors to evade immune surveillance. The resulting reduced tumor antigen presentation has been associated with resistance to ICI therapy (20). However, there is a small percentage of patients in which ICI can still work despite of APP alterations, suggesting that, especially for tumors with high mutation burden, antigen presentation is not completely abolished and can represent an Achilles’ heel of the tumour (80).
Nevertheless, alterations in components of the APP machinery in tumors can potentially change the repertoire of peptides presented by HLA class I complexes. Accordingly, preclinical studies suggest that cells with an altered immunopeptidome due to ERAP1 or TAP deficiency still elicit strong T cell responses (55, 61). These observations indicate that a similar alteration of the immunopeptidome, by promoting the presentation of novel tumor antigens, could increase tumor immunogenicity making anticancer therapeutic protocols more effective (Figure 2). In this regard, there is much evidence in favor of the hypothesis that functional alteration of any component of the APP that can result in a novel immunopeptidome, could increase reactivity of tumor-infiltrating CD8+ T cells and sensitize tumors to ICI therapy (Figure 2). Interestingly, two recent in vivo CRISPR-Cas9 genome editing studies have demonstrated that deletion of APP-related genes is able to increase the efficacy of ICI-based immunotherapy in melanoma and renal carcinoma (81, 82). The influence of APP components on cancer cell sensitivity to immune pressure differs between the two cancer models, with ERAP1, calreticulin and TAPBPR more relevant in melanoma, and β2m in renal carcinoma (81, 82). The ability of APP component depletions to sensitize two tumor models to ICI immunotherapy (81, 82), strengthens the idea that their pharmacological inhibition may have therapeutic value in these tumors. In melanoma, it has been shown that about 12% of tumor-infiltrating T cells recognise non-mutated tumour antigens, i.e. peptides encoded by canonical exons (83).
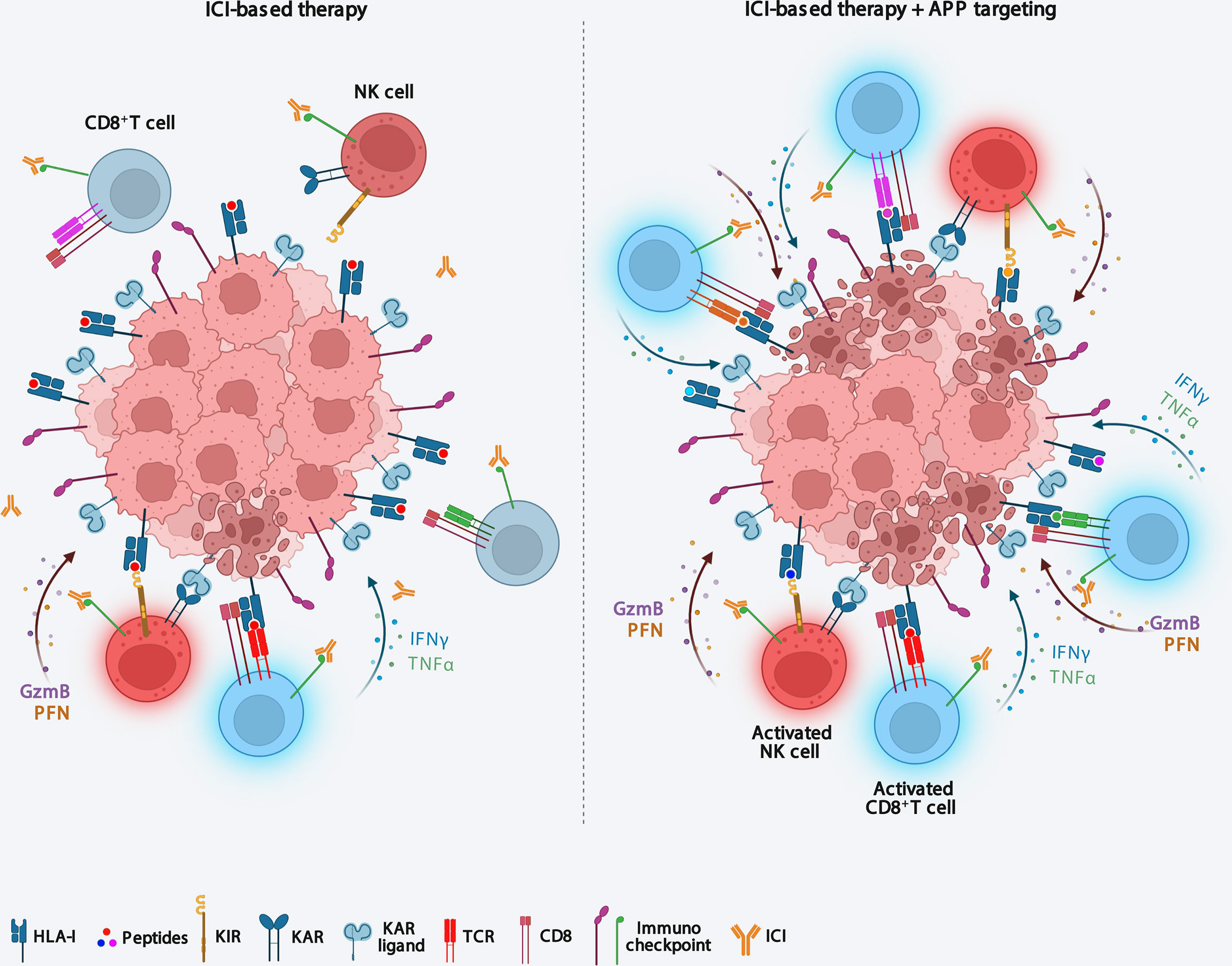
Figure 2 Targeting antigen processing and presentation machinery immunosensitizes ICI-resistent tumors. Downmodulation of APP components causes both an increase in tumor immunogenicity and the recall of functional effector CD8+ T cells and NK cells into the tumor microenvironment, thus making tumors sensitive to ICI-based immunotherapy. APP, antigen processing and presentation; ICI, Immune checkpoint inhibitor; KIR, killer inhibitory receptor; KAR, killer activating receptor.
Perturbation of APP in tumors may represent a strategy to elicit the presentation of both non-mutated tumor antigens with strong antitumor potential, as demonstrated by TEIPPs, and immunogenic epitopes usually destroyed by ERAP1 (53, 55, 57, 68, 69). This subset of peptides, considered as ‘altered self’, represents a very interesting category, as it is potentially shared by several tumors genetically or pharmacologically inhibited for APP components (55, 84).
Overall, it is clear that APP perturbation, by increasing tumor immunogenicity and widening the range of tumor antigens presented could provide an excellent opportunity to stimulate the immune response and increase the efficacy of to ICI therapy. Thus, future investigations and additional pre-clinical studies in this area are needed to reveal new and exciting anticancer therapeutic opportunities.
This work was supported by grants awarded by Associazione Italiana Ricerca sul Cancro (AIRC) IG18495 and IG24345, H2020-MSCA-ITN-2020 Capstone-954992, Ministero della Salute Ricerca. Ricerca Finalizzata PE-2011-02351866, and Ricerca Corrente (DF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Melaiu O, Lucarini V, Giovannoni R, Fruci D, Gemignani F. News on immune checkpoint inhibitors as immunotherapy strategies in adult and pediatric solid tumors. Semin Cancer Biol (2022) 79:18–43. doi: 10.1016/j.semcancer.2020.07.001
2. Wolchok JD. Checkpoint blockade: The end of the beginning. Nat Rev Immunol (2021) 21(10):621. doi: 10.1038/s41577-021-00617-9
3. Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
4. Tang S, Ning Q, Yang L, Mo Z, Tang S. Mechanisms of immune escape in the cancer immune cycle. Int Immunopharmacol (2020) 86:106700. doi: 10.1016/j.intimp.2020.106700
5. Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: Molecular mechanisms and functional significance. Adv Immunol (2000) 74:181–273. doi: 10.1016/s0065-2776(08)60911-6
6. Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med (2005) 201(8):1257–68. doi: 10.1084/jem.20042028
7. O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to Pd1/Pdl1 checkpoint inhibition. Cancer Treat Rev (2017) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
8. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
9. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for Cd103(+)/Cd141(+) dendritic cells bearing Ccr7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell (2016) 30(2):324–36. doi: 10.1016/j.ccell.2016.06.003
10. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of Cd103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic pd-L1 and braf inhibition. Immunity (2016) 44(4):924–38. doi: 10.1016/j.immuni.2016.03.012
11. Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing type-2 dendritic cells to drive protective antitumor Cd4(+) T cell immunity. Cell (2019) 177(3):556–71.e16. doi: 10.1016/j.cell.2019.02.005
12. Wangmo D, Premsrirut PK, Yuan C, Morris WS, Zhao X, Subramanian S. Ackr4 in tumor cells regulates dendritic cell migration to tumor-draining lymph nodes and T-cell priming. Cancers (Basel) (2021) 13(19):5655 doi: 10.3390/cancers13195021
13. Jia Y, Liu L, Shan B. Future of immune checkpoint inhibitors: Focus on tumor immune microenvironment. Ann Transl Med (2020) 8(17):1095. doi: 10.21037/atm-20-3735
14. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer (2018) 118(1):9–16. doi: 10.1038/bjc.2017.434
15. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
16. Tundo GR, Sbardella D, Oddone F, Kudriaeva AA, Lacal PM, Belogurov AA Jr., et al. At The cutting edge against cancer: A perspective on immunoproteasome and immune checkpoints modulation as a potential therapeutic intervention. Cancers (Basel) (2021) 13(19):4852 doi: 10.3390/cancers13194852
17. Zilionyte K, Bagdzeviciute U, Mlynska A, Urbstaite E, Paberale E, Dobrovolskiene N, et al. Functional antigen processing and presentation mechanism as a prerequisite factor of response to treatment with dendritic cell vaccines and anti-Pd-1 in preclinical murine Llc1 and Gl261 tumor models. Cancer Immunol Immunother (2022). doi: 10.1007/s00262-022-03190-9
18. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired hla class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discovery (2017) 7(12):1420–35. doi: 10.1158/2159-8290.CD-17-0593
19. Seliger B, Ferrone S. Hla class I antigen processing machinery defects in cancer cells-frequency, functional significance, and clinical relevance with special emphasis on their role in T cell-based immunotherapy of malignant disease. Methods Mol Biol (2020) 2055:325–50. doi: 10.1007/978-1-4939-9773-2_15
20. Maggs L, Sadagopan A, Moghaddam AS, Ferrone S. Hla class I antigen processing machinery defects in antitumor immunity and immunotherapy. Trends Cancer (2021) 7(12):1089–101. doi: 10.1016/j.trecan.2021.07.006
21. Leko V, Rosenberg SA. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell (2020) 38(4):454–72. doi: 10.1016/j.ccell.2020.07.013
22. Chen J, Brunner AD, Cogan JZ, Nunez JK, Fields AP, Adamson B, et al. Pervasive functional translation of noncanonical human open reading frames. Science (2020) 367(6482):1140–6. doi: 10.1126/science.aay0262
23. Ouspenskaia T, Law T, Clauser KR, Klaeger S, Sarkizova S, Aguet F, et al. Unannotated proteins expand the mhc-I-Restricted immunopeptidome in cancer. Nat Biotechnol (2022) 40(2):209–17. doi: 10.1038/s41587-021-01021-3
24. Erhard F, Dolken L, Schilling B, Schlosser A. Identification of the cryptic hla-I immunopeptidome. Cancer Immunol Res (2020) 8(8):1018–26. doi: 10.1158/2326-6066.CIR-19-0886
25. Larouche JD, Trofimov A, Hesnard L, Ehx G, Zhao Q, Vincent K, et al. Widespread and tissue-specific expression of endogenous retroelements in human somatic tissues. Genome Med (2020) 12(1):40. doi: 10.1186/s13073-020-00740-7
26. Laumont CM, Daouda T, Laverdure JP, Bonneil E, Caron-Lizotte O, Hardy MP, et al. Global proteogenomic analysis of human mhc class I-associated peptides derived from non-canonical reading frames. Nat Commun (2016) 7:10238. doi: 10.1038/ncomms10238
27. Chong C, Muller M, Pak H, Harnett D, Huber F, Grun D, et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat Commun (2020) 11(1):1293. doi: 10.1038/s41467-020-14968-9
28. Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on mhc class I molecules. Cell (1994) 78(5):761–71. doi: 10.1016/s0092-8674(94)90462-6
29. Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20s and 26s proteasomes. Annu Rev Biochem (1996) 65:801–47. doi: 10.1146/annurev.bi.65.070196.004101
30. Tanaka K. Role of proteasomes modified by interferon-gamma in antigen processing. J Leukoc Biol (1994) 56(5):571–5. doi: 10.1002/jlb.56.5.571
31. Gaczynska M, Rock KL, Goldberg AL. Gamma-interferon and expression of mhc genes regulate peptide hydrolysis by proteasomes. Nature (1993) 365(6443):264–7. doi: 10.1038/365264a0
32. Driscoll J, Brown MG, Finley D, Monaco JJ. Mhc-linked lmp gene products specifically alter peptidase activities of the proteasome. Nature (1993) 365(6443):262–4. doi: 10.1038/365262a0
33. Aki M, Shimbara N, Takashina M, Akiyama K, Kagawa S, Tamura T, et al. Interferon-gamma induces different subunit organizations and functional diversity of proteasomes. J Biochem (1994) 115(2):257–69. doi: 10.1093/oxfordjournals.jbchem.a124327
34. Praest P, Luteijn RD, Brak-Boer IGJ, Lanfermeijer J, Hoelen H, Ijgosse L, et al. The influence of Tap1 and Tap2 gene polymorphisms on tap function and its inhibition by viral immune evasion proteins. Mol Immunol (2018) 101:55–64. doi: 10.1016/j.molimm.2018.05.025
35. Compagnone M, Cifaldi L, Fruci D. Regulation of Erap1 and Erap2 genes and their disfunction in human cancer. Hum Immunol (2019) 80(5):318–24. doi: 10.1016/j.humimm.2019.02.014
36. Lopez de Castro JA, Alvarez-Navarro C, Brito A, Guasp P, Martin-Esteban A, Sanz-Bravo A. Molecular and pathogenic effects of endoplasmic reticulum aminopeptidases Erap1 and Erap2 in mhc-I-Associated inflammatory disorders: Towards a unifying view. Mol Immunol (2016) 77:193–204. doi: 10.1016/j.molimm.2016.08.005
37. Nagarajan NA, de Verteuil DA, Sriranganadane D, Yahyaoui W, Thibault P, Perreault C, et al. Eraap shapes the peptidome associated with classical and nonclassical mhc class I molecules. J Immunol (2016) 197(4):1035–43. doi: 10.4049/jimmunol.1500654
38. Boyle LH, Hermann C, Boname JM, Porter KM, Patel PA, Burr ML, et al. Tapasin-related protein tapbpr is an additional component of the mhc class I presentation pathway. Proc Natl Acad Sci USA (2013) 110(9):3465–70. doi: 10.1073/pnas.1222342110
39. Hermann C, van Hateren A, Trautwein N, Neerincx A, Duriez PJ, Stevanovic S, et al. Tapbpr alters mhc class I peptide presentation by functioning as a peptide exchange catalyst. Elife (2015) 4, e09617 doi: 10.7554/eLife.09617
40. Fehling HJ, Swat W, Laplace C, Kuhn R, Rajewsky K, Muller U, et al. Mhc class I expression in mice lacking the proteasome subunit lmp-7. Science (1994) 265(5176):1234–7. doi: 10.1126/science.8066463
41. Nussbaum AK, Rodriguez-Carreno MP, Benning N, Botten J, Whitton JL. Immunoproteasome-deficient mice mount largely normal Cd8+ T cell responses to lymphocytic choriomeningitis virus infection and DNA vaccination. J Immunol (2005) 175(2):1153–60. doi: 10.4049/jimmunol.175.2.1153
42. Yewdell J, Lapham C, Bacik I, Spies T, Bennink J. Mhc-encoded proteasome subunits Lmp2 and Lmp7 are not required for efficient antigen presentation. J Immunol (1994) 152(3):1163–70.
43. Kincaid EZ, Che JW, York I, Escobar H, Reyes-Vargas E, Delgado JC, et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat Immunol (2011) 13(2):129–35. doi: 10.1038/ni.2203
44. Tubio-Santamaria N, Ebstein F, Heidel FH, Kruger E. Immunoproteasome function in normal and malignant hematopoiesis. Cells (2021) 10(7):1577. doi: 10.3390/cells10071577
45. Vachharajani N, Joeris T, Luu M, Hartmann S, Pautz S, Jenike E, et al. Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit Lmp7. Oncotarget (2017) 8(31):50447–59. doi: 10.18632/oncotarget.14579
46. Kalaora S, Lee JS, Barnea E, Levy R, Greenberg P, Alon M, et al. Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat Commun (2020) 11(1):896. doi: 10.1038/s41467-020-14639-9
47. Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW, et al. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J Clin Oncol (2012) 30(21):2678–83. doi: 10.1200/JCO.2011.37.8539
48. Mlecnik B, Bifulco C, Bindea G, Marliot F, Lugli A, Lee JJ, et al. Multicenter international society for immunotherapy of cancer study of the consensus immunoscore for the prediction of survival and response to chemotherapy in stage iii colon cancer. J Clin Oncol (2020) 38(31):3638–51. doi: 10.1200/JCO.19.03205
49. Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res (2013) 73(12):3499–510. doi: 10.1158/0008-5472.CAN-13-0371
50. Lee J, Kim DM, Lee A. Prognostic role and clinical association of tumor-infiltrating lymphocyte, programmed death ligand-1 expression with neutrophil-lymphocyte ratio in locally advanced triple-negative breast cancer. Cancer Res Treat (2019) 51(2):649–63. doi: 10.4143/crt.2018.270
51. Tripathi SC, Peters HL, Taguchi A, Katayama H, Wang H, Momin A, et al. Immunoproteasome deficiency Is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc Natl Acad Sci USA (2016) 113(11):E1555–64. doi: 10.1073/pnas.1521812113
52. Iorgulescu JB, Braun D, Oliveira G, Keskin DB, Wu CJ. Acquired mechanisms of immune escape in cancer following immunotherapy. Genome Med (2018) 10(1):87. doi: 10.1186/s13073-018-0598-2
53. Marijt KA, Blijleven L, Verdegaal EME, Kester MG, Kowalewski DJ, Rammensee HG, et al. Identification of non-mutated neoantigens presented by tap-deficient tumors. J Exp Med (2018) 215(9):2325–37. doi: 10.1084/jem.20180577
54. Doorduijn EM, Sluijter M, Querido BJ, Oliveira CC, Achour A, Ossendorp F, et al. Tap-independent self-peptides enhance T cell recognition of immune-escaped tumors. J Clin Invest (2016) 126(2):784–94. doi: 10.1172/JCI83671
55. Garrido G, Schrand B, Rabasa A, Levay A, D’Eramo F, Berezhnoy A, et al. Tumor-targeted silencing of the peptide transporter tap induces potent antitumor immunity. Nat Commun (2019) 10(1):3773. doi: 10.1038/s41467-019-11728-2
56. Durgeau A, Virk Y, Gros G, Voilin E, Corgnac S, Djenidi F, et al. Human preprocalcitonin self-antigen generates tap-dependent and -independent epitopes triggering optimised T-cell responses toward immune-escaped tumours. Nat Commun (2018) 9(1):5097. doi: 10.1038/s41467-018-07603-1
57. Marijt KA, Griffioen L, Blijleven L, van der Burg SH, van Hall T. Cross-presentation of a tap-independent signal peptide induces Cd8 T immunity to escaped cancers but necessitates anchor replacement. Cancer Immunol Immunother (2022) 71(2):289–300. doi: 10.1007/s00262-021-02984-7
58. Ilca FT, Neerincx A, Wills MR, de la Roche M, Boyle LH. Utilizing tapbpr to promote exogenous peptide loading onto cell surface mhc I molecules. Proc Natl Acad Sci USA (2018) 115(40):E9353–E61. doi: 10.1073/pnas.1809465115
59. Ilca FT, Drexhage LZ, Brewin G, Peacock S, Boyle LH. Distinct polymorphisms in hla class I molecules govern their susceptibility to peptide editing by tapbpr. Cell Rep (2019) 29(6):1621–32 e3. doi: 10.1016/j.celrep.2019.09.074
60. Hafstrand I, Aflalo A, Boyle LH. Why tapbpr? implications of an additional player in mhc class I peptide presentation. Curr Opin Immunol (2021) 70:90–4. doi: 10.1016/j.coi.2021.04.011
61. Hammer GE, Gonzalez F, James E, Nolla H, Shastri N. In the absence of aminopeptidase eraap, mhc class I molecules present many unstable and highly immunogenic peptides. Nat Immunol (2007) 8(1):101–8. doi: 10.1038/ni1409
62. Yan J, Parekh VV, Mendez-Fernandez Y, Olivares-Villagomez D, Dragovic S, Hill T, et al. In vivo role of er-associated peptidase activity in tailoring peptides for presentation by mhc class ia and class ib molecules. J Exp Med (2006) 203(3):647–59. doi: 10.1084/jem.20052271
63. York IA, Brehm MA, Zendzian S, Towne CF, Rock KL. Endoplasmic reticulum aminopeptidase 1 (Erap1) trims mhc class I-presented peptides in vivo and plays an important role in immunodominance. Proc Natl Acad Sci USA (2006) 103(24):9202–7. doi: 10.1073/pnas.0603095103
64. Firat E, Saveanu L, Aichele P, Staeheli P, Huai J, Gaedicke S, et al. The role of endoplasmic reticulum-associated aminopeptidase 1 in immunity to infection and in cross-presentation. J Immunol (2007) 178(4):2241–8. doi: 10.4049/jimmunol.178.4.2241
65. Cifaldi L, Lo Monaco E, Forloni M, Giorda E, Lorenzi S, Petrini S, et al. Natural killer cells efficiently reject lymphoma silenced for the endoplasmic reticulum aminopeptidase associated with antigen processing. Cancer Res (2011) 71(5):1597–606. doi: 10.1158/0008-5472.CAN-10-3326
66. Cifaldi L, Romania P, Falco M, Lorenzi S, Meazza R, Petrini S, et al. Erap1 regulates natural killer cell function by controlling the engagement of inhibitory receptors. Cancer Res (2015) 75(5):824–34. doi: 10.1158/0008-5472.CAN-14-1643
67. D’Amico S, D’Alicandro V, Compagnone M, Tempora P, Guida G, Romania P, et al. Erap1 controls the interaction of the inhibitory receptor Kir3dl1 with hla-B51:01 by affecting natural killer cell function. Front Immunol (2021) 12:778103. doi: 10.3389/fimmu.2021.778103
68. James E, Bailey I, Sugiyarto G, Elliott T. Induction of protective antitumor immunity through attenuation of eraap function. J Immunol (2013) 190(11):5839–46. doi: 10.4049/jimmunol.1300220
69. Keller M, Ebstein F, Burger E, Textoris-Taube K, Gorny X, Urban S, et al. The proteasome immunosubunits, Pa28 and er-aminopeptidase 1 protect melanoma cells from efficient mart-126-35 -specific T-cell recognition. Eur J Immunol (2015) 45(12):3257–68. doi: 10.1002/eji.201445243
70. Textoris-Taube K, Cammann C, Henklein P, Topfstedt E, Ebstein F, Henze S, et al. Er-aminopeptidase 1 determines the processing and presentation of an immunotherapy-relevant melanoma epitope. Eur J Immunol (2020) 50(2):270–83. doi: 10.1002/eji.201948116
71. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
72. McDermott D, Haanen J, Chen TT, Lorigan P, O’Day S, investigators MDX. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase iii trial (Mdx010-20). Ann Oncol (2013) 24(10):2694–8. doi: 10.1093/annonc/mdt291
73. Reeves E, Wood O, Ottensmeier CH, King EV, Thomas GJ, Elliott T, et al. Hpv epitope processing differences correlate with Erap1 allotype and extent of Cd8(+) T-cell tumor infiltration in opscc. Cancer Immunol Res (2019) 7(7):1202–13. doi: 10.1158/2326-6066.CIR-18-0498
74. Koumantou D, Barnea E, Martin-Esteban A, Maben Z, Papakyriakou A, Mpakali A, et al. Editing the immunopeptidome of melanoma cells using a potent inhibitor of endoplasmic reticulum aminopeptidase 1 (Erap1). Cancer Immunol Immunother (2019) 68(8):1245–61. doi: 10.1007/s00262-019-02358-0
75. Lopez de Castro JA. How Erap1 and Erap2 shape the peptidomes of disease-associated mhc-I proteins. Front Immunol (2018) 9:2463. doi: 10.3389/fimmu.2018.02463
76. Temponeras I, Stamatakis G, Samiotaki M, Georgiadis D, Pratsinis H, Panayotou G, et al. Erap2 inhibition induces cell-surface presentation by molt-4 leukemia cancer cells of many novel and potentially antigenic peptides. Int J Mol Sci (2022) 23(3):1913. doi: 10.3390/ijms23031913
77. Pearson H, Daouda T, Granados DP, Durette C, Bonneil E, Courcelles M, et al. Mhc class I-associated peptides derive from selective regions of the human genome. J Clin Invest (2016) 126(12):4690–701. doi: 10.1172/JCI88590
78. Caron E, Vincent K, Fortier MH, Laverdure JP, Bramoulle A, Hardy MP, et al. The mhc I immunopeptidome conveys to the cell surface an integrative view of cellular regulation. Mol Syst Biol (2011) 7:533. doi: 10.1038/msb.2011.68
79. Yewdell JW, Holly J. Drips get molecular. Curr Opin Immunol (2020) 64:130–6. doi: 10.1016/j.coi.2020.05.009
80. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat Rev Cancer (2021) 21(5):298–312. doi: 10.1038/s41568-021-00339-z
81. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo crispr screening identifies Ptpn2 as a cancer immunotherapy target. Nature (2017) 547(7664):413–8. doi: 10.1038/nature23270
82. Dubrot J, Lane-Reticker SK, Kessler EA, Ayer A, Mishra G, Wolfe CH, et al. In vivo screens using a selective crispr antigen removal lentiviral vector system reveal immune dependencies in renal cell carcinoma. Immunity (2021) 54(3):571–85.e6. doi: 10.1016/j.immuni.2021.01.001
83. Murata K, Nakatsugawa M, Rahman MA, Nguyen LT, Millar DG, Mulder DT, et al. Landscape mapping of shared antigenic epitopes and their cognate tcrs of tumor-infiltrating T lymphocytes in melanoma. Elife (2020) 9, e53244. doi: 10.7554/eLife.53244
Keywords: immunopeptidome, tumor antigens, antigen processing, HLA class I, immune checkpoint inhibitors, TEIPPs, ERAP1/2, cancer immunotherapy
Citation: D’Amico S, Tempora P, Melaiu O, Lucarini V, Cifaldi L, Locatelli F and Fruci D (2022) Targeting the antigen processing and presentation pathway to overcome resistance to immune checkpoint therapy. Front. Immunol. 13:948297. doi: 10.3389/fimmu.2022.948297
Received: 19 May 2022; Accepted: 29 June 2022;
Published: 22 July 2022.
Edited by:
Peter M. Van Endert, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2022 D’Amico, Tempora, Melaiu, Lucarini, Cifaldi, Locatelli and Fruci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Doriana Fruci, ZG9yaWFuYS5mcnVjaUBvcGJnLm5ldA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.