 Konstantin Kotschenreuther
Konstantin Kotschenreuther Shuaifeng Yan1†
Shuaifeng Yan1† David M. Kofler
David M. Kofler- 1Laboratory of Molecular Immunology, Division of Rheumatology and Clinical Immunology, Department I of Internal Medicine, Faculty of Medicine, University Hospital Cologne, University of Cologne, Cologne, Germany
- 2Center for Integrated Oncology Aachen Bonn Cologne Duesseldorf, Cologne, Germany
Regulatory T (Treg) cells are garnering increased attention in research related to autoimmune diseases, including rheumatoid arthritis (RA). They play an essential role in the maintenance of immune homeostasis by restricting effector T cell activity. Reduced functions and frequencies of Treg cells contribute to the pathogenesis of RA, a common autoimmune disease which leads to systemic inflammation and erosive joint destruction. Treg cells from patients with RA are characterized by impaired functions and by an altered phenotype. They show increased plasticity towards Th17 cells and a reduced suppressive capacity. Besides the suppressive function of Treg cells, their effectiveness is determined by their ability to migrate into inflamed tissues. In the past years, new mechanisms involved in Treg cell migration have been identified. One example of such a mechanism is the phosphorylation of vasodilator-stimulated phosphoprotein (VASP). Efficient migration of Treg cells requires the presence of VASP. IL-6, a cytokine which is abundantly present in the peripheral blood and in the synovial tissue of RA patients, induces posttranslational modifications of VASP. Recently, it has been shown in mice with collagen-induced arthritis (CIA) that this IL-6 mediated posttranslational modification leads to reduced Treg cell trafficking. Another protein which facilitates Treg cell migration is G-protein-signaling modulator 2 (GPSM2). It modulates G-protein coupled receptor functioning, thereby altering the cellular activity initiated by cell surface receptors in response to extracellular signals. The almost complete lack of GPSM2 in Treg cells from RA patients contributes to their reduced ability to migrate towards inflammatory sites. In this review article, we highlight the newly identified mechanisms of Treg cell migration and review the current knowledge about impaired Treg cell homeostasis in RA.
Introduction
Rheumatoid Arthritis (RA) is a chronic systemic inflammatory autoimmune disease characterized by a symmetric polyarthritis with subsequent joint destruction and deformity. While typically associated with cartilage destruction and bone erosion of afflicted synovial joints, RA can also present with a myriad of extra-articular manifestations (1). Symptom constellations seen in RA patients can lead to severe disability, with a significant reduction in quality of life as well as employability (2, 3). Considering the overall lifetime risk of developing RA in US-women is 3.6% and 1.7% in US-men (4) as well as an estimated worldwide prevalence of 0.46% (5), the aggregate reduction in quality of life and productivity, as well as increased utilization of healthcare resources have significant societal as well as economic impacts.
With continued research, our understanding of RA and its underlying mechanisms has grown significantly. While the pathogenesis of RA is complex and involves the interplay of many factors, an increasing amount of attention has been given to the role of regulatory T cells (Tregs), in disease development and progression. As a specialized subset of helper T cells, Tregs play an important role in maintaining homeostasis and self-tolerance, thereby preventing the development of autoimmune diseases such as RA. With this review we aim to synthesize the growing body of knowledge available on Treg cell migration and homeostasis as it pertains to RA. An understanding of this topic is not only relevant to the pathogenesis but also potential treatment modalities of RA.
Rheumatoid arthritis
The multifactorial pathogenesis of RA has been the subject of numerous publications as the exact mechanisms continue to be uncovered. It has been proposed that various environmental factors play a role in the susceptibility to RA, with smoking being identified as the most important environmental risk factor associated with the development of RA others of which include low vitamin D intake and levels as well as occupational dust exposure (6–9). Interestingly, mice with chronic exposure to cigarette smoke showed reduced levels of Tregs in peripheral blood via flow cytometry (10). By means of genome-wide association studies (GWAS) with subsequent meta-analyses, RA susceptibility has also been linked to major histocompatibility complex (MHC) genetic variants with different serological phenotypes of RA being associated with distinct human leukocyte antigen (HLA) gene variations. For example, it has been found that the HLA-DRB1 genotype, associated with increased CXCR4 expression on cluster of differentiation 4 positive (CD4+) T cells, a protein which is involved in cellular migration, leads to sustained autoimmunity and local inflammation thereby conferring an increased risk for RA development (11, 12). MicroRNA (miRNA), which is small non-coding RNA involved in the regulation of post-transcriptional gene expression has been proposed as an epigenetic process involved in RA pathogenesis by means of modulating T- and B-cell subtype development and differentiation. While certainly not the only miRNA domain with relevance to RA, it has been shown that microRNAs and their interplay with transcriptional factors can modulate the peripheral blood Th17/Treg balance (13). This is of particular note as RA is heavily associated with a disturbed Th17/Treg balance skewed in favor of the pro-inflammatory Th17-cells.
It is presumed that the presence of the aforementioned risk factors in combination with a triggering event such as infection or injury is what ultimately causes the development of autoreactive T- and B- cells and subsequent development of RA. While the exact pathomechanisms underlying RA and its associated manifestations are highly complex and multi-factorial, a significant amount of attention has been given to CD4+ T-helper cells (Th-cells), which represent the most abundant lymphocyte population in the synovial infiltrate. Naïve CD4+ T cells can develop into distinct subsets with specific phenotypes and functions (Figure 1). Observations have shown abnormalities in intracellular signaling as well as aging of T helper cells in patients with RA, thereby contributing to the chronic autoimmune response associated with RA (18).
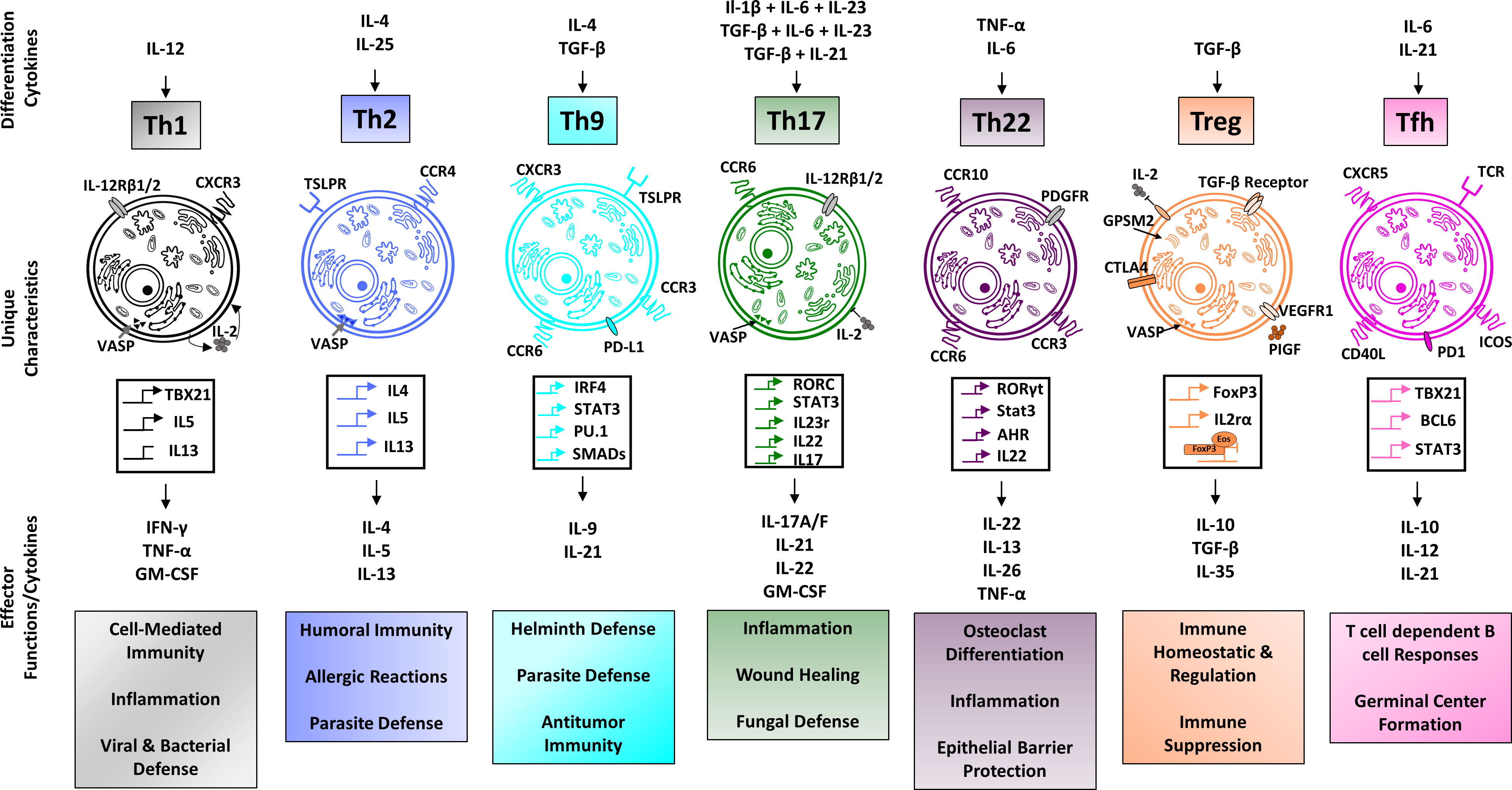
Figure 1 Overview of CD4+ T cell subsets. Regulatory T cells, along with the Th2 phenotype are classically associated with anti-inflammatory functioning, whereas Th1 and Th17 cells are typically associated with pro-inflammatory functions. Th9 cells are implicated in host defense against helminth infections, Th22 cells in host defense against bacterial pathogens and Tfh cells play an important role for the development of lasting immune memory. The individual characteristics and functions of CD4+ T cell subsets are made possible through distinct gene profiles resulting in a subtype-specific expression of receptors and cytokine production. Different environmental stimuli, such as TGF-β in the case of regulatory T cells induce the differentiation of naive CD4+ T cells into their respective subtypes (14–17).
RA presents clinically with the bilateral insidious onset of polyarticular symmetric arthritis. Patients experience pain and soft effusion-based swelling in affected joints, primarily the metacarpophalangeal, metatarsophalangeal and proximal interphalangeal joints as well as the wrist. RA, however, is not limited to small joints and can also affect ankle, knee, elbow and shoulder joints. While joint involvement is the classical manifestation associated with RA, inadequate treatment can lead to the development of numerous extra-articular manifestations including, but certainly not limited to cardiovascular disease, interstitial lung disease and liver dysfunction (1). In addition, when considering the mean loss of life expectancy in patients diagnosed with RA is 4.97 years (19) the gravity of the disease becomes clear.
Regulatory T cells
CD4+ regulatory T cells are a distinct T cell subpopulation involved in the maintenance and regulation of self-tolerance and homeostasis. First discovered in 1995, Tregs specifically express the transcription factor forkhead Box P3 (FoxP3) in the nucleus as well as CD25 and CTLA-4 on their cell surface (20). Although FoxP3 expression is significant for the differentiation of CD4+ T cells to the regulatory T cell phenotype, numerous epigenetic changes have also proven to be crucial for effective Treg functioning. However, FoxP3 holds a pivotal role to regulatory T cell function, as evidenced by the X-Linked syndrome of immune dysregulation, polyendocrinopathy, and enteropathy (IPEX) caused by a mutation in the FoxP3 gene leading to large amounts of polyclonal T-cell activation and tissue infiltration (21, 22). While numerous studies have been able to implicate dysfunctional Tregs in autoimmune disease and inflammatory conditions, increasing evidence suggests the role of Tregs goes far beyond simply acting as a checkpoint on overarching inflammatory processes. As our understanding of regulatory T cells and their role in disease progresses, increased attention is being given to this cell-type as a potential therapeutic target benefitting disease control. As a key regulator of immune function, both down- and up-regulation of immune responses by means of regulatory T cell modulation could be feasible therapeutic angles for treating numerous diseases.
Although FoxP3 has been shown to be key to Treg differentiation and functioning, further genes play a pivotal role in Treg development and functioning. Treg genome regulators such as SATB1 (Special AT-rich sequence binding protein 1) bind to genomic sites to unwind chromatin and activate super-enhancers. The importance of this genomic organizer to regulatory T cells has been illustrated by experiments showing SATB1-deficient mice not being able to generate Tregs in the thymus, as well as the failure of CD25+ FoxP3- CD4+ T cells deficient in SATB1 to differentiate into FoxP3+ Tregs under appropriate conditions for differentiation (23). These activated super-enhancers subsequently demethylate and thereby activate the hallmark Treg genes among which are FOXP3, IL2RA (encoding CD25), CTLA4, IKZF2 (encoding Helios) and IKZF4 (encoding Eos), collectively known as Treg -specific demethylated regions (Treg-DRs). While super-enhancer dependent demethylation is able to upregulate gene expression, FoxP3 expression has been associated with the downregulation of IL2, IFN-γ and Zap70 genes, which interestingly do not possess any of these Treg-DRs, thereby suggesting a FoxP3-independent mechanism of Treg-DR regulation (24). Both epigenetic changes and FoxP3 expression are regulated in an independent manner by means of TCR activation; however, both are required for a definite differentiation to the Treg lineage.
Although mainly produced in the thymus, Tregs also develop in the periphery, such as in the intestinal mucosa. Interestingly, in comparison to other T cell subtypes, Tregs leave the thymus in an antigen-primed state, characterized by the expression of CD25, CTLA4 and CD5 (25). In addition to their activated state, upon leaving the thymus, Tregs have also been shown to have a high T cell receptor (TCR) affinity for self-antigens (26). By means of their primed state and high TCR affinity for self-antigens, Tregs are activated to exert suppressive effects at up to ~100-fold lower peptide/MHC concentrations when compared to other T cell subsets that recognize the same antigen (27). These two characteristics ensure a rapid Treg response towards self-antigens, subsequently preventing other immune cells from being activated and causing an aberrant immunologic response. Once in the periphery, FoxP3+ Tregs find themselves in a continuous state of high proliferation under physiologic conditions, most likely due to continued recognition of self-antigens as well as antigens derived from ubiquitously present microbes (28, 29). Peripherally located naïve CD4+ T cells can differentiate to FoxP3+ Tregs under both inflammatory and non-inflammatory conditions following specific antigen exposure (30, 31). While thymic Tregs (tTregs) are classically associated with the recognition of self-antigens, peripherally induced Tregs (pTregs) are involved in the recognition of foreign antigens such as bacterial, allergy and food antigens (32). This differentiation is further reflected in the differential expression of Nrp-1, being high in tTregs and low in pTregs as well as in the TCR repertoire, which shows limited overlap among the two subsets (33). Interestingly, it has been shown tTregs, in the absence of pTregs, are incapable of suppressing chronic inflammation and autoimmunity (34).
Although classically associated with the suppression of overarching immune-responses, Tregs have garnered attention for numerous other functions. While Treg cells play an important role in various autoimmune diseases and contribute to dysregulated immune response in malignant diseases (35–40), they have also been implicated in controlling fetal-maternal tolerance (41–43), immunometabolic disease including obesity and atherosclerosis (44–46), degenerative diseases with inflammatory components as well as tissue regeneration (47). Currently, FoxP3, CD25, and CD45RA are used to identify and divide human circulating FoxP3+ CD4+ T cells into distinct subsets. Resting or naïve Tregs are characterized by CD45RA, as well as low levels of CD25 and FoxP3 expression. Effector Tregs are characterized by no expression of CD45RA but high levels of both FoxP3 and CD25 expression. Lastly, CD45RA negative cells with low CD25 and FoxP3 expression are not considered to be Tregs (28). As we continue to uncover new functions and mechanisms of Tregs, it is becoming clear just how vital this cell type is to maintain physiologic homeostasis in numerous organ systems. With their wide range of functions, Tregs continue to be an interesting target for further research both in the understanding, and treatment of RA as well as numerous other conditions.
Multiple mechanisms have been hypothesized by which Tregs exert their effector functions, all of which are dependent on FoxP3 expression. It is of note that Tregs have been shown to possess a large degree of lineage stability, maintaining their FoxP3 expression and subsequent inhibitory effects under the influence of many different immune stimuli (48). Of note is the ability of Tregs to express transcription factors and chemokine receptors typically associated with other T cell lineages, with the subsequent production of IFN-γ (Th-1 like T-regs), IL-17 (Th-17 like Tregs) and IL-13 (Th-2 like T-regs) (49–52). Interestingly, male scurfy strain mice that are hemizygous for the X-linked FoxP3sf mutation, essentially depriving them of Treg functioning, develop a CD4+ T cell mediated lymphoproliferative disease characterized by wasting and multi-organ infiltration by lymphocytes (53). This has been highlighted by the work of Fontenot et al. who were able to show that retroviral-driven Foxp3 expression in CD4+ CD25- T-cells resulted in protection from disease onset and progression classically associated with the scurfy strain (54). The suppressive effects are postulated to be exerted via cell-contact dependent as well as humoral factors including cytokines (IL-2, IL-10, TGF-β), cell surface molecules (CTLA-4, CD25, TIGIT) and intracellular molecules (granzymes, cAMP). Just as numerous as the mechanisms by which they potentially exert their functions are the cells that Tregs take effect on, including CD4+ and CD8+ T cells, NK cells, B-cells, monocytes and dendritic cells (DCs) (55). Much interest has been given to Tregs and their relation to interleukin 2 (IL-2), which has also been referred to as T cell Growth Factor (TCGF). IL-2 together with its receptor (IL-2R) is involved in the maintenance of self-tolerance as well as immunity and has become the target of biologic immune-modulators used in the treatment of autoimmune and rheumatic disease (56). As FoxP3+ Tregs barely produce any IL-2 due to downregulation of the IL-2 gene by FoxP3 expression, they are dependent on exogenous IL-2 for survival (57). While studies have come to differing conclusions on the matter, a role of Tregs as IL-2 “reservoir” has been postulated. Due to Tregs having the highest expression level of the IL-2 receptor α-chain among T cells, which is responsible for maintaining the receptor in a high-affinity state Tregs can effectively starve other immune cells and APCs of IL-2, thereby inhibiting their activation and preventing an active immune response (58–60). Interestingly, effector T cells produce IL-2 when in the activated state to sustain the immunologic response, which in turn activates regulatory T-cells in order to prevent an overarching immune reaction. In addition to IL-2, other “pro-inflammatory” cytokines such as IL-6 and TNF-α have been shown to induce Treg expansion, indicating the natural role of Tregs in controlling immune reactions at sites of inflammation (61). CTLA-4 expression is a further means of Treg functioning, causing the downregulation of both CD80 and CD86 expression in APCs, thereby preventing co-stimulatory signaling of CD28 to activate effector T cells (62). The importance of CTLA-4 mediated effector functioning has been shown by the conditional deletion of CTLA-4 in adult mice, causing spontaneous lymphoproliferation, hypergammaglobulinemia, as well as various autoimmune inflammatory manifestations with accompanying organ-specific antibodies (63).
Although only a selection of Treg functionalities and mechanisms of action have been touched upon in this section, it is evident that Tregs can provide an attractive target in the prevention or management of numerous diseases. Due to their ability to significantly alter immune functioning through down-regulation of effector cells, Tregs provide us with a powerful means to potentially modulate immune function. For example, the induction of Tregs in humans with IL-2 administration to treat disease caused by overreaching immune reactions has shown promising results (64, 65).
Tregs in rheumatoid arthritis
As we continue to expand on our knowledge in regard to regulatory T cells, they are becoming of ever more interest to the understanding of rheumatoid arthritis. With their immunosuppressive role, regulatory T-cells not only play a key role in the pathogenesis of RA but can also provide valuable insights into possible treatment modalities. Although controversial results have been published, the general consensus is that regulatory T-cell levels are reduced in patients with Rheumatoid Arthritis, which is in line with the general anti-inflammatory effect attributed to Tregs (66, 67). While difficult to pinpoint, the difference in experimental results has been attributed primarily to the absence of universal characteristics that define Tregs. While the most common method of identification for Tregs in flow cytometry is CD3+ CD4+ CD25high/CD127low, attempts are being made to identify better mechanisms to reliably and reproducibly identify Tregs. Helios has been proposed as one of these mechanisms, as its expression in CD4+ FoxP3+ T cells is negatively correlated to RA disease activity, significantly more so than other cell-surface markers such as CTLA-4. Further work has shown that diverging results may not only result from a lack of homogenous Treg identification, but also on the different expression of specific Treg subpopulations that require individual antibody-staining for flow cytometry. Moreover, some Treg subpopulations show specific features which are characteristics of other CD4+ T-cell subsets. Under certain inflammatory settings, Treg cells can develop into Th1-like, Th2-like, Th17-like, or Tfh-like Treg cells (68–72) (Figure 2). Th1-like Treg cells acquire a Th1-like phenotype and express the pro-inflammatory cytokine IFN-γ while they lose their suppressive capacity (49, 75). Similar, Th2-like and Th17-like Treg cells express the transcription factors GATA-3 or RORγt and secrete IL-13 or IL-17 (71, 76). Th2- and Th17-like Treg cells have been reported to maintain their suppressive functions (71, 76). Interestingly, Th17-like Treg cells have been observed in humans under physiological conditions (71). In RA, the frequency of Th1-like Treg cells is increased but deficient in function (75).
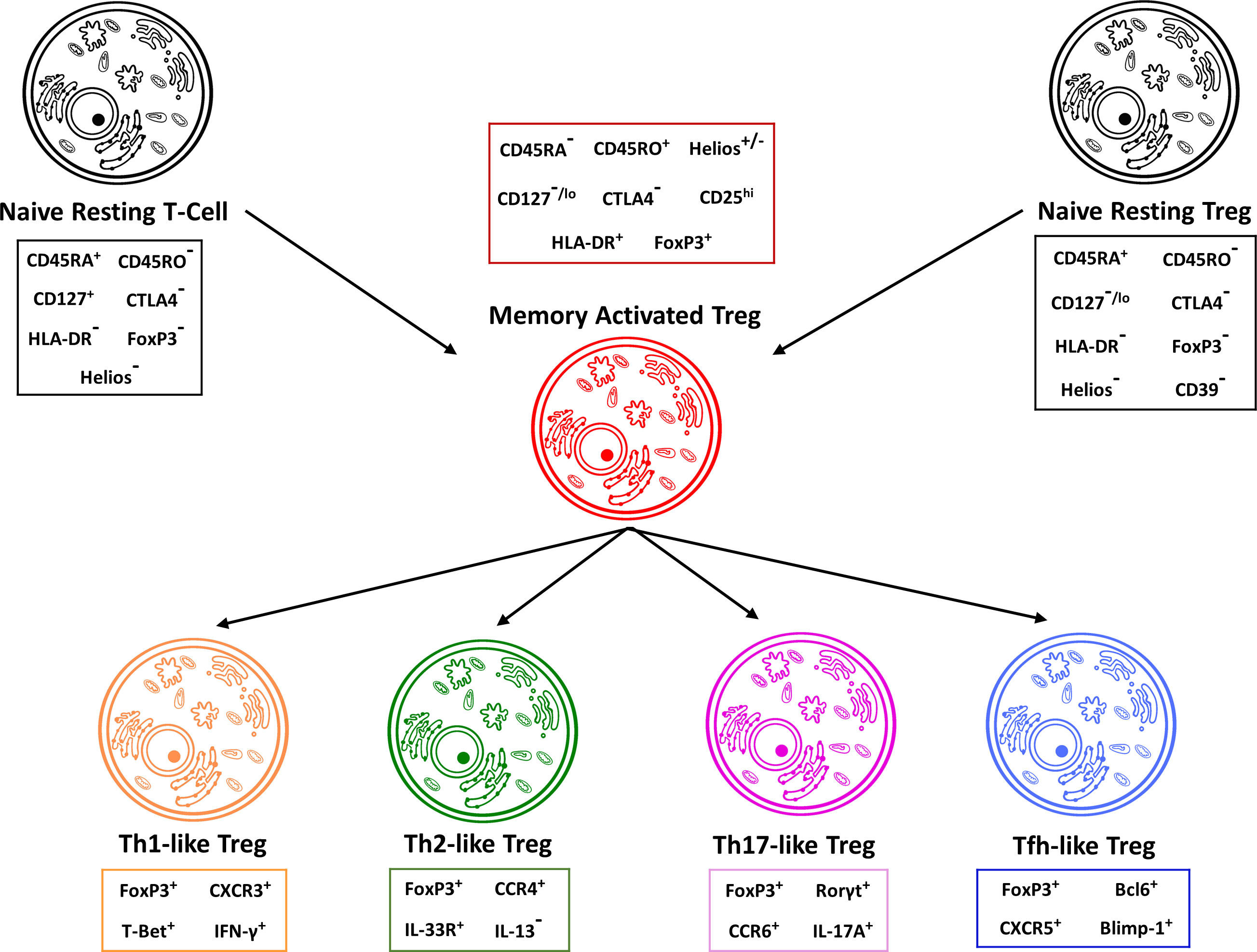
Figure 2 Treg cells can be divided in distinct subgroups. Treg cells can further be subdivided into memory activated, Th1-like, Th2-like, Th17-like and Tfh-like regulatory T cells. Both naïve CD4+ T cells as well as naïve resting regulatory T cells can differentiate into memory activated Tregs, which then further delineate into their specific regulatory T cell phenotypes. These phenotypes can be differentiated through different expression profiles for genes, transcription factors and cytokine production. In addition, the individual regulatory T cell phenotypes have unique immune capabilities and functions. Th17-like Tregs for example are capable of producing IL-17A and have been shown to be over-represented in patients suffering from RA (73, 74).
The aforementioned Helios as a member of the Ikaros transcription factor family, is a nuclear factor associated with the early development of regulatory T-cells. Studies have shown a significant importance of Helios in controlling the stability and function of Tregs (77). Interestingly, most of the FoxP3+ CD4+ T-cells in the synovium of inflammatory arthritis patients are Helios+. In addition, these Helios+ Tregs prove to be poor producers of effector cytokines including IL-10, IFN-γ and TNF-α. Helios expression seemed to not only affect cytokine production but was also associated with a higher Ki67 index indicating higher levels of cellular proliferation when compared to Helios- Tregs. The overall expression of FoxP3 gene expression as well as TSDR (regulatory T cell-specific demethylated region) demethylation is decreased in RA individuals when compared to healthy individuals, in contrast to Helios, which shows an overall higher expression in RA patients as compared to healthy individuals. These results suggest that the nuclear transcription factor Helios may act to suppress the inhibitory functions normally carried out by Tregs in patients with RA (78). While these results certainly demonstrate the importance of Helios in regards to RA, it is not the only Ikaros zinc finger transcription factor (IkZF) with implications for RA.
Eos, another of the IkZF factors, has garnered attention due to its high expression in Treg populations and function as a vital component of the FoxP3-mediated gene expression complex. Specifically, Eos forms a protein complex with C-terminal Binding Protein and FoxP3 to promote gene silencing, thereby acting as a co-repressor of vital importance to the maintenance of the Treg phenotype as well as its suppressive capabilities. This was further established by means of Eos expression knockdown in mice which resulted in a worsening of inflammation in colitis (79). In addition, the Eos/FoxP3 repression complex has been shown to target and subsequently repress the IL-2 gene locus. Interestingly, Helios has also been attributed with the ability to repress the IL-2 gene locus, signifying redundance in the IkZF Factor family functioning. Just as with Helios, Eos has also been shown to be downregulated as a consequence of increased DNA methylation in Tregs of RA patients. This suggests both Helios and Eos play a possible role in the aberrancy of Treg functioning, contributing to the development and progression of RA.
Aiolos, another member of the Ikaros family of transcription factors, is induced in CD4+ T cells by Ahr in the presence of TGF-β1 and has been shown to be upregulated in RA (37, 80). Not only has Aiolos been shown to be upregulated in RA patients, it is also correlated to a higher prevalence of a lymphoid pathotype, as well as higher ACPA and RF prevalence indicating more severe disease. Interestingly, Ikaros and Aiolos synovial expression was directly correlated with synovial cell infiltration as well as systemic inflammation (81). Aiolos levels increase during the β- and positive selection of thymocytes along with Helios and Ikaros. In contrast to Helios and Ikaros, Aiolos levels remained elevated after β- and positive selection throughout the continued thymocyte development (82). Interestingly, it has been reported that treatment with TNF-α inhibitors induces Aiolos in CD4+ T cells (83). With Aiolos being able to transactivate the Bcl-2 promoter, it has the potential to prolong cellular viability by preventing apoptosis. In contrast, IL-2 starvation induces Ras-Aiolos association, thereby inhibiting Aiolos functioning with the subsequent inhibition of BCL-2 expression leading to cellular apoptosis (84). In contrast to Helios and Ikaros which show transient expression in T cells, Aiolos levels remain elevated even as cells continue to mature, which is congruent with its function in preventing apoptosis (80). Interestingly, the expression of Aiolos in Tregs that lack Helios expression has been associated with a “pro-inflammatory Treg subtype” that is capable of producing and secreting IFN-γ, IL-2, and IL-17. Tregs not expressing Aiolos on the other hand, are better suited for suppression of effector T-cells. Whereas Helios and Eos are co-expressed in Treg cells, Aiolos is expressed in Helios- Treg cells (85).
It has been shown that there is a significant reduction in the proportion of Tregs in the peripheral blood of patients with active RA when compared to RA patients in remission, which is an interesting contrast to a higher proportion of Tregs in the synovial fluid of RA patients when compared to healthy controls. This might be in part due to the anoxic environment created through synovial inflammatory processes in active RA (86). Interestingly, it has been demonstrated that hypoxia and the subsequent induction of hypoxia-inducible factor 1 alpha (HIF-1α) expression promotes the induction of FoxP3 transcription, which in turn promotes the generation of Tregs. In addition, CD4+ CD25+ T cells from inflamed joints express high levels of CTLA-4, GITR, CD69 and MHC class 2 molecules, indicating an activated state. This at first may seem in stark contrast to the observed overarching inflammatory reaction seen in the joints of RA patients. However, it has been shown that RA patients have defective synovial Treg functioning with an inability to suppress not only the production of pro-inflammatory cytokines such as TNF-α and IFN-γ by other CD4+ T-cells as well as monocytes, but also a reduced suppression of T effector cell proliferation (87). While only part of a much larger picture, this data can suggest that the dysfunction more so than the absence of Tregs plays a role in the pathophysiologic processes dictating RA. We have directly been able to study the effects of regulatory T-cells in RA by proxy of collagen-induced arthritis (CIA) in mice. CD25-depleted mice induced with collagen to promote the development of RA showed significantly more severe disease when compared to control mice. This increased disease severity was underpinned by increased antibody titers against collagen as well as an increased proliferation of collagen-specific T cells. Furthermore, the adoptive transfer of CD4+ CD25+ T cells into CD25-depleted mice showed an attenuation of disease severity, highlighting the importance that Tregs have in controlling aberrant inflammatory responses in joints (88). In addition, the severity of RA symptoms as well as high levels of RF and anti-CCP antibodies has been associated with a lower number of activated Tregs in humans (89).
The production of these RF and anti-CCP antibodies occurs through B-cells, which require CXCR5+ T follicular helper cells (Tfh) for activation. Interestingly, Tfh cells require activation of the transcription factor Bcl-6, which is upregulated in CD4+ T-cells of RA patients, as well as IL-6 and IL-21 for their differentiation (90). The subsequent unregulated activation of antibody producing B cells and associated humoral responses are linked to the development of RA, as well as B cell lymphomas in RA patients (91, 92). In addition, Tfh cells, which are usually located in B cell follicles to regulate B cell survival, are overly abundant in the synovium of RA patients as compared to healthy individuals (93). Expansion of the Tfh population in RA is driven by enhanced IL-6/pSTAT3 signaling and leads to a shift in the ratio between Tfh cells and Tfh-like Treg cells, which share many phenotypic characteristics with Tfh cells, but lack CD40L and IL-21 expression (94). A subset of Tfh cells is derived from natural Treg cells and is characterized by the expression of FoxP3 (95). These cells are referred to as T follicular regulatory cells (Tfr) (95). Blimp-1 dependent (96) ability to suppress the formation of germinal centers as well as limit humoral responses makes Tfr cells an important counter-force to the Tfh-associated RA-promoting actions. Both activated Tfh and Tfr cells migrate to germinal centers, where Tfh cells secrete IL-21 and promote a B cell response through CD40L/CD40 interactions, whereas Tfr cells secrete IL-10 and suppress B cell responses via CTLA4 and GITR (97). A deficiency in CXCR5+ Treg cells, for example, has been associated with an increase in germinal center B-cells and the associated pro-inflammatory characteristics (95, 98).
Th17/Treg balance
Although regulatory T cells exert their effect on a multitude of different cell types, the pro-inflammatory CD4+ Th17 phenotype, which is characterized by expression of the retinoic acid-related orphan receptor (ROR-γt) and the production of the IL-17 cytokine family is of particular interest when trying to understand the implications of regulatory T cells in RA. The pro-inflammatory Th17 cells, along with pro-inflammatory Th1 cells have been shown to be resistant to CD6 down-regulation in RA, which is associated with an aggravation of autoimmune inflammation as well as increased cell proliferation and survival (99, 100). While long presumed to be a pro-inflammatory Th1-mediated disease, increasing attention is being given to Th17 cells as they pertain to the development and progression of RA. When considering that the inflammatory activity in arthritis has been attributed to Th17 cells in the synovial fluid (101), as well as the number of Th17 cells in the serum of RA patients being positively correlated with the disease activity score in 28 joints (DAS28), anti-CCP antibody and C-reactive protein levels (102), the importance of interaction between Tregs and Th17 cells in RA becomes very apparent. Through the production and secretion of numerous cytokines such as IL-17A, IL-17F and IL-22, Th17 cells have been shown to stimulate synovial fibroblasts as well as macrophages to the large-scale production of pro-inflammatory mediators such as IL-1, IL-6, TNF-α and PGE2, thereby worsening synovial inflammation (103). In addition, Th17 cells stimulate synovial stromal and innate lymphoid cells to secrete GM-CSF thereby initiating and elevating joint inflammation (104, 105).
While Th17 and Treg cells significantly differ in their functionality, they do share similarities such as the ability of TGF-β to induce their development and differentiation, which is interesting when considering their opposing functionality. While this at first may seem counterintuitive, it can be proposed that this is an innate attempt to prevent overarching inflammatory reactions that could be caused by aberrant-isolated Th17 differentiation (106, 107). The balance between Treg cells and Th17 cells is modified by various factors, including vitamin A, glycolysis, salt concentrations, and cytokines (Figure 3). While TGF-β is required for initial differentiation, many other factors play a role in determining the ultimate fate of Th17 and Treg cells. Furthermore, the Insulin-Like Growth Factor 1 Receptor (IGF1R) has been shown to be an important regulator of Th17 vs. Treg cell differentiation. IGF1R signaling possess the ability to enhance activation of the AKT-mTOR pathway in order to potentiate Th17 development in addition to suppressing Th17 apoptosis, while simultaneously hindering Treg development. IGF ligands possess the ability to skew Th17 programming towards the expression of pro-inflammatory genes with concurrent inhibition of anti-inflammatory genes; one proposed mechanism by which this is accomplished is an increase in HIF-1α dependent gene expression (108). Interestingly, continued research has been able to show that lifestyle choices, such as a high-salt diet or cannabinoid consumption might be able to skew this ratio in favor of Th17 cells (109, 110). Numerous studies have been able to show that the Th17/Treg ratio is skewed in favor of Th17 cells in RA when compared to healthy controls. Szodoray et al. tracked the Th17/Treg ratio in patients as they progressed from undifferentiated connective tissue disease (UCTD) to a definitive systemic autoimmune disease (SIAD). From healthy controls to UCTD to definitive SIAD the Th17/Treg ratio continually increased, showing a significant increase of the Th17/Treg ratio in the progression from a healthy state to autoimmune disease (111). These findings were in line with results demonstrating a positive correlation between the percentage of Th17 cells and several clinical markers of RA activity such as DAS28, ESR, CRP, anti-CCP RF and ANA. In contrast, a negative correlation between Treg percentages and DAS28, ESR, CRP, anti-CCP, and ANA was observed (112). These observations are congruent with the functions attributed to Th17 and Treg cells respectively, and further demonstrate the importance the Th17/Treg ratio plays in RA patients. However, clinical trials with the monoclonal antibody secukinumab have revealed a limited efficiency of IL-17A inhibition in patients with RA (113). The limitations of secukinumab in RA may be explained by the fact that the expression of IL-17A and its receptors in the synovium of RA patients is very heterogeneous (114). Moreover, secukinumab does not neutralize IL-17F, which is less active than IL-17A when used alone but is as efficient as IL-17A in the presence of TNF-α (115). A combined inhibition of IL-17A and IL-17F might therefore be more effective in RA as compared to IL-17A inhibition alone.
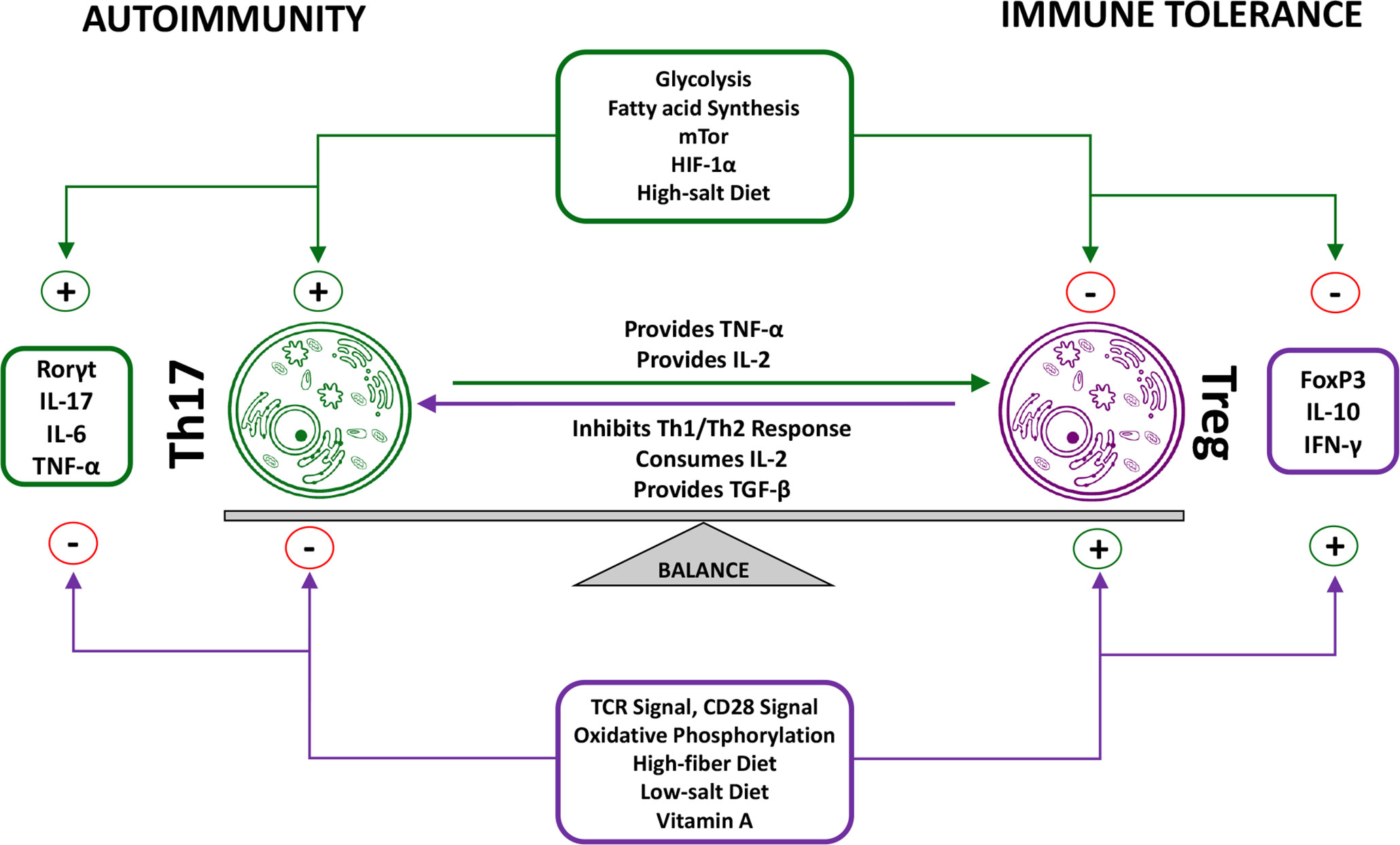
Figure 3 Reciprocal functions of Treg cells and Th17 cells. The balance between Treg cells and Th17 cells is of great importance for proper immune functioning. A dysregulation of this balance in favor of the pro-inflammatory Th17 phenotype has been observed in RA patients. Although various factors influence this balance through isolated effects on either Treg or Th17 cells, numerous factors have been identified that simultaneously take effect on both Tregs and Th17 cells. Furthermore, Tregs and Th17 cells also directly take effect on one another. Th17 functioning physiologically causes an increase in the Treg population, which in turn prevents overarching immune reactions through inhibition of Th17-specific as well as other immune pro-inflammatory functions (15, 16, 74).
While the Th17/Treg ratio is of interest when considering how regulatory T-cells could inhibit Th17 functioning, thereby reducing inflammatory signaling, there are further points of interest that need to be taken into consideration when it comes to RA. Komatsu et al. were able to demonstrate that FoxP3+ T-cells possess the ability to lose their Foxp3 expression (after which they are called exFoxp3 cells) and undergo transdifferentiation into Th17 cells under the influence of IL-6 derived from synovial fibroblasts. These exFoxp3 Th17 cells play a key role in the pathogenesis of autoimmune arthritis and are present in the synovium of patients with active RA. Interestingly, when compared to Th17 cells derived from naïve T cells, exFoxp3 Th17 cells specifically and highly express the transcription factor SOX4, which in turn positively regulates ROR-γt, thereby enhancing lymphoid cell survival (116, 117). In addition to enhanced survival, the high expression of molecules involved in cellular proliferation as well as a high frequency of Ki-67+ cells among the exFoxp3 Th17 population indicate a high proliferative index under arthritic conditions (118). Not only are exFoxP3 Th17 cells longer lived and more active in regards to proliferation, their osteoclastogenic activity is more pronounced when compared to non-exFoxP3 Th17 cells. It has been proposed that the promotion of osteoclastogenesis by exFoxP3 cells is based on an increased expression of RANKL as compared to regular Th17 cells, with exFoxP3 Th17 cells being able to directly induce osteoclastogenesis, independent of fibroblast presence. The ability of exFoxP3 Th17 cells to accumulate and proliferate in inflamed tissue where they stimulate osteoclastogenesis is significant for the progression of RA and an important aspect to understanding the dynamic between Th17 and Treg cells and how they contribute to RA.
The Th17/Treg ratio has become an area of interest when it comes to developing possible therapeutics to treat not only RA, but a wide array of autoimmune diseases that involve a Th17/Treg dysregulation. Certain medications already exist that take influence the Th17/Treg balance such as the IL-6 Receptor Inhibitor Tocilizumab (119), however, there is much therapeutic potential left to be unlocked by attempting to directly target Th17- and Treg-related cytokines, cytokine receptors, intracellular signaling pathways, as well as the modulation of Treg and Th17 specific transcription factors. With the exploration of this therapeutic potential, the far-reaching roles of both Treg and Th17 cells beyond RA pathogenesis have to be kept in mind. For example, Th17 cells and their cytokines IL-17 and IL-22 play a crucial role in the maintenance of immune homeostasis at mucosal surfaces, with IL-17 neutralization being accompanied by high rates of serious adverse events and fungal infections (120). Although the latter effect was observed in patients with Crohn’s Disease, it can be hypothesized that these effects would also be seen in other conditions as this effect is not based on the disease in and of itself, but rather therapy mediated dysregulation of Th17 functioning.
Mechanisms of T cell migration
Much regard is given to T cells and their effector functions in the synovium that ultimately result in the development of active RA. In order to exert their functions however, T cells must migrate to their respective sites of action. While originating primarily in the thymus in younger years, naïve T cell maintenance throughout life is primarily sustained through peripheral T cell division (121). Interestingly, with active thymic output in childhood 10-30% of all CD4+ T cells in blood, lymphoid tissue, and mucosal sites are Tregs as compared to only about 5% in adults (122). Even decades after thymic output ceases, naïve T cells comprise a significant proportion (20-50%) of total T cells in multiple lymph nodes. Interestingly, post neonatal thymectomy adults do not have increased incidences of autoimmunity or allergy when compared to age matched controls and are able to maintain Treg frequencies, indicating that while it does hold an important role in T cell development, the thymus is only part of the bigger T cell picture (123, 124). Regardless of age, Tregs in blood as well as lymphoid tissues are CD45RA+CCR7+, indicating a naïve phenotype as compared to mucosal Tregs, which are CD45RA- and more closely resemble conventional memory T cells (125).
The infiltration of the synovium by CD4+ T cells, but not CD8+ T cells or B-cells is necessary for the development of clinically active RA (126). This is of note as a profound understanding of the mechanisms leading to synovial infiltration can be of value in researching potential therapeutic modalities aiming to prevent the onset of RA, rather than combating symptoms of inflammation in manifest disease. Increasing evidence suggests that while not the priming site for naïve T cells, CD4+ T cell commitment occurs at the inflamed joints under the influence of cytokines produced by an uncharacteristically large number of activated macrophages and dendritic cells (127). Dendritic cells from RA patients secrete higher levels of inflammatory chemokines such as CCL17, which is involved in the recruitment of CCR4+ cells to the inflamed joint (128). Furthermore, monocyte derived dendritic cells from RA patients have been shown to possess an increased capability of Th17 differentiation induction as compared to healthy controls while simultaneously lacking the ability to efficiently induce FoxP3+ Treg cells (129). Interestingly, the inability to form succinate from succinyl-CoA due to SUCLG2 repression characteristic of RA in the tricarboxylic acid (TCA-) Cycle with subsequent accumulation of Acetyl-CoA has been shown to skew naïve CD4+ T cells to the short-lived effector subtype (SLECs). These SLECs are characterized not only by their hypermobility and ability to rapidly enter synovial tissue with induction of aggressive synovitis, but also by their propensity to proliferate (130).
For T cells to exert their effector functions at sites of inflammation, T cell trafficking consisting of rolling, adhesion and transmigration must occur. For this to take place, cells must express adhesion molecules and chemo-attractans such as selectins (L-selectin for leukocyte populations) as well as the P-selectin glycoprotein ligand-1 (PSGL-1) (131). L-selectin and PSGL-1 interaction allows for leukocyte-leukocyte interactions permitting the tethering and adhesion of leukocyte clusters to the endothelium via endothelial specific selectins (P- and E-selectins) of blood vessels prior to extravasation. In addition, lymphocyte function-associated antigen 1 (LFA-1) is of importance for Treg migration, being expressed in a high affinity state in inflammation seeking Tregs when compared to recirculating Tregs (132). Of note is not only the increased expression of E-selectins at inflamed synovial sites, but also the increased serum levels of P- and L-selectins in RA patients (Figure 4) (134, 135). This suggests an increased ability of leukocytes to migrate to the synovium in patients with RA as compared to healthy individuals. After initial leukocyte-endothelial interaction via selectins, integrins are responsible for the firm adhesion to, and arrest of leukocytes at the endothelium. Leukocyte integrins are able to interact with endothelial surface ICAM-1 or VCAM-1, which is a prerequisite for subsequent cellular extravasation. Different immune cell subtypes express individual repertoires of integrins, which are further upregulated in the presence of proinflammatory cytokines such as IL-1 or TNF-α (136). This individualized expression permits the specific localization of T cell subtypes in the inflamed joint. While essential to the extravasation and subsequent navigation to inflamed synovial sites, interactions between leukocyte integrins and their ligands also induce cellular proliferation, cytokine production, and angiogenesis, thereby significantly contributing to disease development (137). The use of specific antagonists to integrins as well as their ligands, has been able to prevent inflammation and angiogenesis in the CIA mouse model; furthermore high levels of soluble and endothelium-bound ICAM-1 have been identified in RA patients, as well as being linked to disease activity in CIA model mice (138–140). Finally, transendothelial chemotactic concentration gradients of molecules such as PECAM-1, ICAM-2, JAMA and ESAM to name a few, allow trans- and paracellular leukocyte migration beyond the endothelium into inflamed tissues (141). This process is regulated by the differential and specific expression of cell trafficking molecules unique to individual cellular subsets, for example, LFA-1 expression, which plays a key role in the emigration of T cells from the vasculature, is upregulated in effector T cells when compared to naïve T cells.
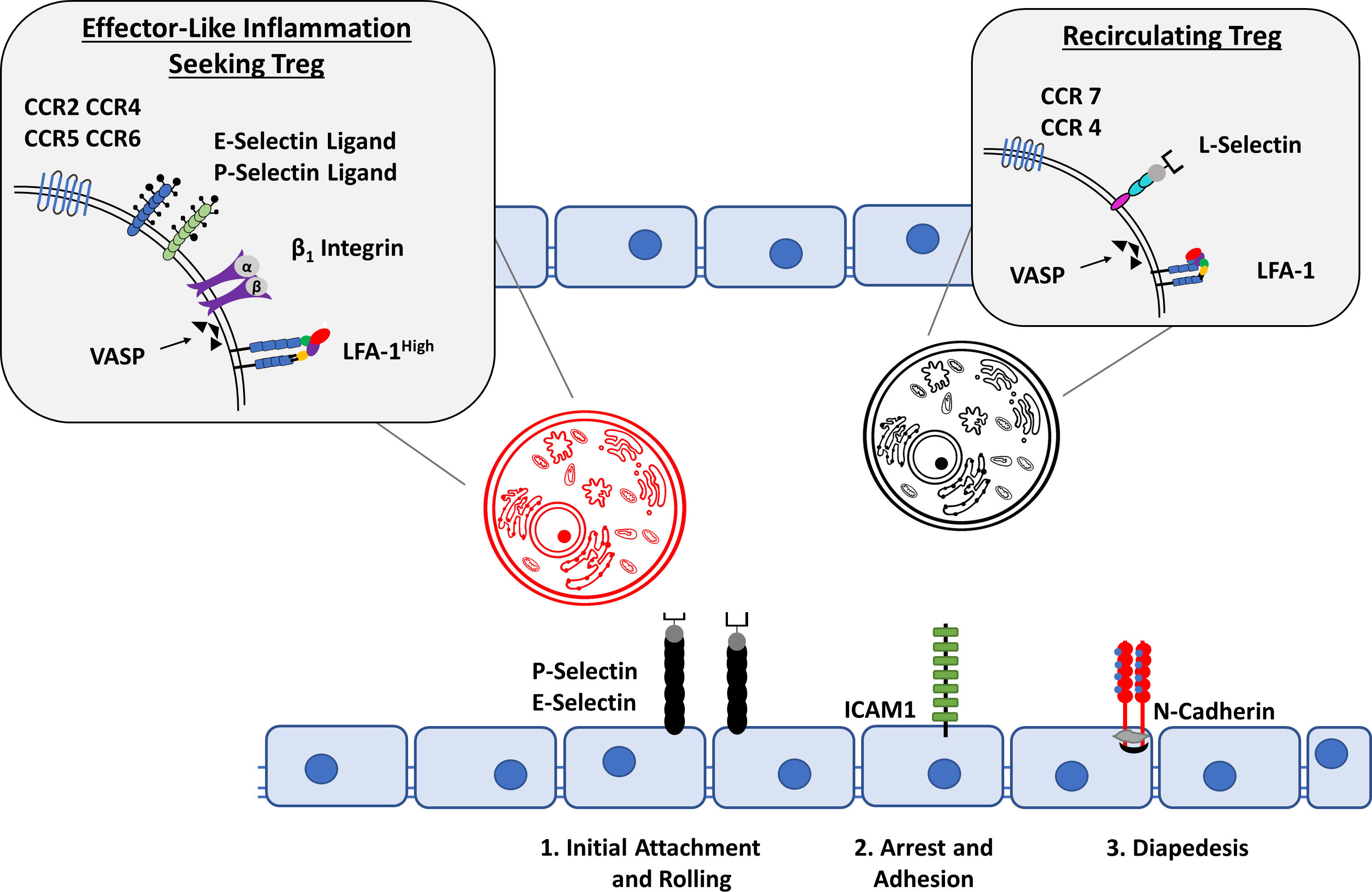
Figure 4 Treg cell migration into inflammatory sites. A dysregulation of Treg migration has been implicated in the pathogenesis of RA. Depending on the expression of cell-surface molecules, Tregs can be divided into recirculating Tregs and inflammation seeking Tregs. In contrast to the expression of CCR7, CCR4, L-Selectin and LFA-1 in a low-affinity state allows recirculating Tregs to remain in the bloodstream, activated inflammation seeking Tregs are able to migrate to their sites of action through the expression of E- and P- selectin Ligands, CCR2, CCR4, CCR5, CCR6, β1 Integrin as well as LFA-1 in a high affinity state. The Interaction with P- and E- selectin, ICAM1 and N-Cadherin among other molecules on the activated endothelium allows for the classical steps of cellular migration: Initial attachment and rolling, followed by arrest and adhesion which eventually leads to cellular diapedesis (14, 133).
While the migration of T cells in general is of continued interest in research pertaining to RA considering the potential therapeutic value, increased attention is also being given specifically to Treg migration mechanisms, which are diverse and dependent on the cells developmental stage, role, and target tissue. Tregs express numerous receptors for inflammatory chemokines as well as adhesion molecules which are not only vital for access to sites of inflammation, but also under heavy regulation. As migration is an energy-intensive process, Tregs are reliant on glucokinase-mediated glycolysis, which is supported by the inhibition of motility by means of glucose starvation, and the upregulation of the insulin receptor in supporting their increased energy consumption (142). Glycolytic feedback control through PI3K-Akt pathways has also been demonstrated with the activation of Akt causing a downregulation of L-selectin, CCR7 and sphingosine-1-phosphate receptor 1, implying that the activation of Akt could cause the failure of leukocyte homing to secondary lymphatic organs, instead promoting migration to peripheral tissue such as the synovium (143). Glycolysis interference through extracellular sodium-lactate and lactic acid has also been shown to trap CD4+ and CD8+ T cells at sites of infection through repression of their mobility (144). In contrast, inhibition of fatty acid oxidation had no impact on Treg migration (133). The PDK-1 signaling-dependent loss of phosphatase and tensin homolog (PTEN) can also take effect on Treg migration through decreasing the expression of CD62L as well as CCR7 (145). The Tumor Necrosis Factor Receptor 2 (TNFR2) has been shown to promote a remarkable degree of cellular migration, activation and proliferation through downstream initiation of the reciprocal PI3K/Akt pathway as well as canonical and non-canonical NF-kB activation (146). The serine threonine kinase mTOR has been identified as a critical component for regulatory T cell migration as well as stability. A specific deletion of mTOR in mouse Tregs led to the spontaneous development of severe and systemic inflammation, impaired Treg migration, as well as a loss of FoxP3 expression. Compared with controls, mTOR-deficient Tregs expressed increased levels of CD62L, CCR7 and S1P1, as well as decreased levels of CD69, which inhibits the migration from lymphoid tissue to the blood. Furthermore, mTor-deficient Tregs expressed lower levels of CD44 and CCR4 as well as slightly lower levels of CCR6 and CXCR3, possibly impairing access to non-lymphoid tissues. This suggests that a deficiency or downregulation of mTOR can cause a defect in entry to nonlymphoid tissues and promotes the recirculation to and within lymphoid organs (147).
Altered chemokine and cytokine milieu in the synovium
Given their central role in the specific recruitment and activation of T cells as well as other immune cells, chemokines have gained attention as potential treatment targets in RA. As a subgroup of the cytokine family, chemokines are chemotactic and immune modulatory molecules secreted in order to control not only the positioning of, but also the development, homeostasis and activity of numerous immune-competent cells. Interestingly, varying chemokine production profiles have been identified throughout different RA disease stages. CCL4, CXCL4, CXCL7 and CXCL13 expression are characteristic of early RA, whereas CCL3 and CCL9 were indicative of later disease stages (148, 149). Chemokines present in the synovial fluid of RA patients are characterized by high levels of citrullination, especially CXCL5 and CCL2, which causes an altered activity profile with subsequent increased recruitment of monocytes. In addition, the citrullination of CXCL5 has been shown to have a high correlation with RA disease activity (150).
Chemokines have been shown to play a central role in Th1 cell migration to the synovium, which is of note when considering the importance of Th1 cells to the inflammatory processes in RA (151). In addition to Th1 cells, chemokines such as CCL20, which are strongly expressed in the inflamed joint, allow Th17 migration to the synovium with subsequent activation. Tregs enjoy a widespread distribution throughout lymphoid and non-lymphoid tissues which is vital to their immunosuppressive function. This propensity to be distributed throughout the body can be altered by means of CCR4 chemokine receptor disruption with the subsequent development of pathogenic inflammatory responses (152). Interestingly, the relational expression of certain chemokines, such as CCR6 with other chemokines has been observed to correlate with the specific production of cytokines. CCR4+/CCR6+ Th cells express high IL-17A levels whereas CXCR3+/CCR6+ cells show low IL-17A expression, but elevated IFN-γ levels. CCR6+/CCR10+ Th cells express high levels of IL-22, thus indicating a connection to the Th22 phenotype, which is characterized by its IL-22 expression. This, however, does not seem to be a uni-directional relationship with IL-17 as well as IFN-γ having been shown to upregulate the expression of numerous chemokines. Interestingly, it has been shown that the presence of a dinucleotide polymorphism in the CCR6 gene is linked to RA susceptibility (153). However, further cytokines such as IL-1β and TNF-α have also been shown to induce chemokine expression, especially CXCL8 and CCL13, both of which act as chemoattractants for various immune cells including T cells and neutrophils (154).
CCL2, also known as monocyte chemotactic protein 1 (MCP1) is produced by synovial chondrocytes and fibroblasts and possesses the ability to recruit CCR2+ T cells, NK cells, basophils and macrophages to the synovium. While shown to be upregulated in the synovium of RA patients, the mechanisms underlying CCL2 functioning in RA are complex. This is supported by varying outcomes of CCL2 targeting studies where monoclonal antibodies specific to CCL2 were able to reduce ankle swelling in CIA as well as MRL-lpr mice. In contrast, CCL2 monoclonal antibody treatment aggravated RA during the progression phase in a murine CIA mouse model (155–157). CCL3 and CCL5 are two of the chemokines whose expression is induced by the activation of T cells via IL-1β and TNF-α (158). While the receptor for CCL3 and CCL5, CCR5, is not commonly expressed in PBMCs of RA patients, it has been found to be highly expressed in the RA synovium, either due to an upregulation of expression, increase in CCR5+ cells in the synovium, or an interplay of both (159). The importance of CCR5 becomes apparent when considering a CCR5 antagonist initiated days before the clinical arthritis manifestation was able to attenuate leukocyte migration to joints thereby reducing disease severity (160). Furthermore, increased levels of CCL3, CCL4 and CXCL10 can be identified in the synovial fluid and plasma of RA patients, whereas CCL5 only seems to be elevated in the plasma but reduced in contrast in the synovium of RA patients (67, 161–164). CXCR3, which is involved in the migration of Th1 cells through interaction with its ligands CXCL9, CXCL10 and CXCL11, has also been shown to correlate with IFN-γ production in its expression, and can reduce susceptibility to inflammatory autoimmune disease when knocked out in mice (165, 166). Furthermore, M1 macrophages have been shown to secrete CXCL5, CXCL8, CXCL9, CXCL10 and CXCL13 which further promotes leukocyte recruitment thereby aggravating joint destruction in RA (167, 168). CCR7 and its ligand CCL21 have also been implicated in the migration of not only T cells but also APCs to the synovium, thereby contributing to RA pathogenesis. CCL21 is of further interest in the regard that it is able to drive osteoclastogenesis in RA through M1 macrophage polarization of Th17 cells as well as the induction of neovascularization (169). CCL20, on the other hand, is secreted by chondrocytes, synoviocytes and Th17 cells in the joints, working in synergy with RANKL to promote the resorption and destruction of bone, as well as contributing to the chemotaxis towards joints of T cells, monocytes, and CD1α+ dendritic cells (170–173). CXCL12 was also shown to induce osteoclastogenesis by means of TNF-α-dependent RANKL upregulation in CD4+ T cells and synovial fibroblasts. CXCL12 and its receptor CXCR4, are correlated with the presence of CD4+ T cells in the synovium, indicating an additional role in T cell migration and joint destruction.
RA is characterized by dysregulated cytokine production and the serum concentration of several cytokines is altered in RA (Table 1). As one of the main pro-inflammatory cytokines, TNF-α plays a central role in RA. While the production of TNF-α is classically attributed to macrophages, numerous cells are capable of producing TNF-α. The TNF-α receptor is also widely expressed, including on immune cells. RA is characterized by increased levels of TNF-α in the serum of patients, which carries a central role in the development of the observed inflammation. TNF-α is of great importance due to its ability to stimulate the proliferation and differentiation of T cells, B-cells and NK-cells as well as inducing the production of other pro-inflammatory cytokines such as IL-1, IL-6, IL-8 and matrix metaloproteinases (MMPs). Furthermore, it upregulates the production of GM-CSF, prostaglandins, and collagenases as well as ICAM-1 by synovial fibroblasts (199–202) The importance of TNF-α for regulatory T cells in RA is highlighted by studies showing an increased Treg suppressive activity as well as an increase in Treg/Teff balance under treatment with the TNF-α antagonist Infliximab as well as a reduction in Treg numbers in TTG mice overexpressing hTNF-α. TNFR2+ Tregs isolated from human peripheral blood have been found to exhibit a more potent suppressive effect than TNFR2- Tregs, further suggesting an interplay between TNF-α and Tregs with relevance to RA pathogenesis. The expression of TNFR2 on Tregs increased with the progression of disease, suggesting a possible shedding function by Tregs to neutralize unbound TNF-α (203, 204).
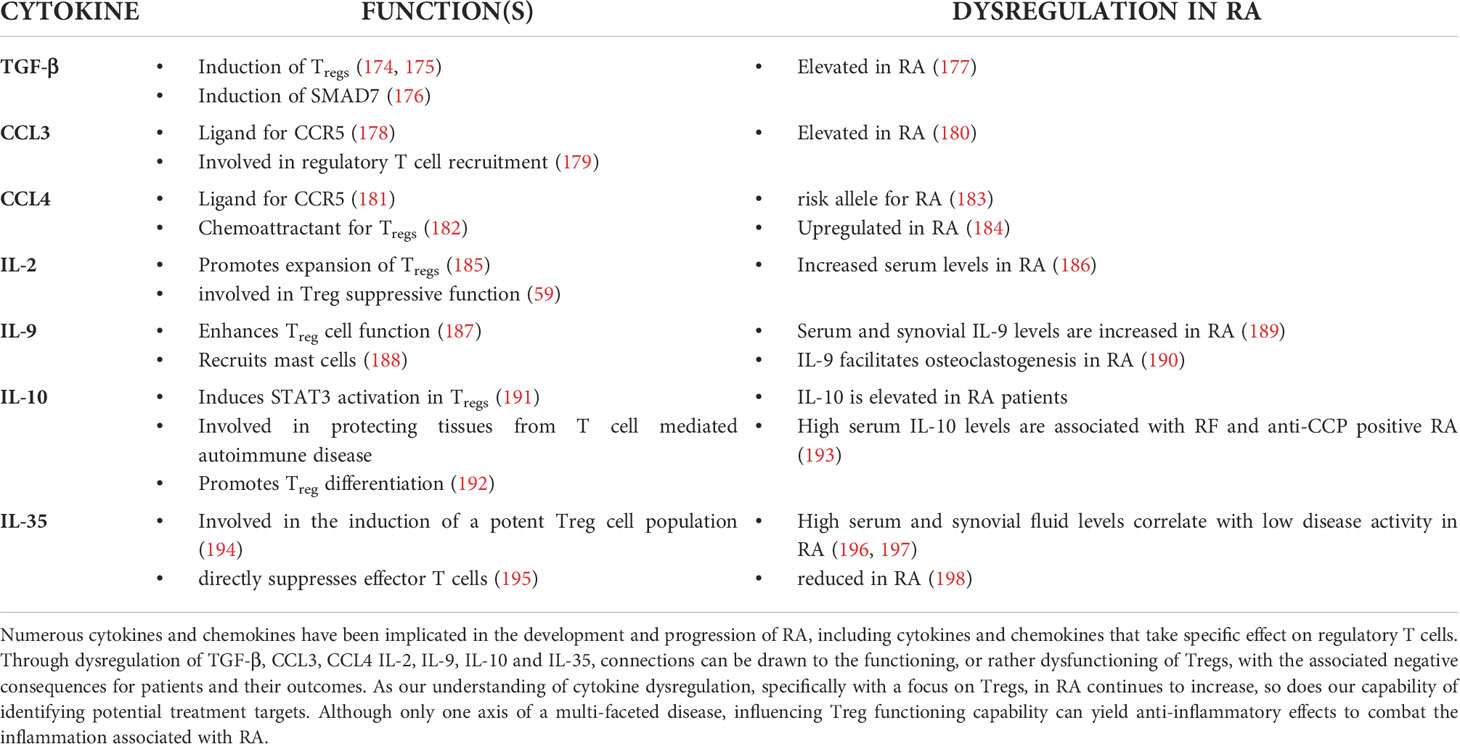
Table 1 Abnormal cytokine expression in RA.
Transforming growth factor-Beta (TGF-β) which is expressed by regulatory T cells, is critical in maintaining self-tolerance and immune homeostasis through its involvement in the regulation of cellular proliferation, differentiation, migration and survival (205). TGF-β is able to induce the expression of FoxP3, thereby promoting the differentiation of CD4+ T cells to the Treg phenotype (206). In addition, Tregs are the primary producers of TGF-β1, as well as being the only cell capable of activating this cytokine via expression of cell surface docking receptor glycoprotein A repetitions predominant (GARP) and α-V integrins (207). TGF-β is produced in an inactivated pre-pro-TGF-β precursor form which requires additional stimuli for the liberation and activation of TGF-β prior to exertion of its functions either through cell-surface binding to a heterodimeric receptor complex consisting of type 1 and type 2 trans-membrane serine/threonine kinase subunits, or acting in a soluble form (208). Intracellular signal transduction is subsequently accomplished by SMAD proteins, however SMAD-independent mechanisms of signal transduction have also been identified (209). Subsequently, TGF-β possesses the ability to suppress T cell proliferation via inhibition of IL-2 production. In addition, TGF-β modulates the T cell proliferation by alteration of cell cycle regulator expression, specifically causing an upregulation of the cyclin-dependent kinase inhibitors p15, p21 and p27, as well as downregulation of cell cycle-promoting factors such as CDK2, cyclin E and D2, as well as c-myc (210–215). TGF-β is further able to inhibit effector cytokine production by pro-inflammatory Th1 cells through various mechanisms, without affecting the anti-inflammatory effector cytokine production characteristic of the Th2 T cell phenotype (216). TGF-β also affects the differentiation of naïve T cells to different T-helper subsets, further potentiating its immune-modulatory effect (217). In addition to its effects on CD4+ T cells, TGF-β also controls CD8+ T cell effector functioning and proliferation by means of inhibiting the production of effector molecules such as IFN-γ and granzyme (218–221).
Interleukin-4 (IL-4) is an interleukin classically associated with anti-inflammatory effects which promotes the differentiation of naïve T cells to the Th2 phenotype, a phenotype which in turn further produces IL-4. With studies having come to different conclusions on IL-4 in RA, some suggesting no-to-minute expression in synovial fluid and other studies suggesting a relevant expression, it remains to be seen what role IL-4 plays in joint inflammation (222, 223). The need for additional studies is highlighted by the differential effects of IL-4 administration in RA models, with IL-4 administration being able to attenuate proteoglycan induced arthritis by means of inhibiting pro-inflammatory cytokine production, although having no disease modulating effect in collagen induced arthritis mice. While the aforementioned results paint a varied picture, IL-4 gene polymorphisms have been found to increase the risk of developing RA in European as well as Chinese individuals making them suitable for the use as a genetic marker to assess the susceptibility to, as well as the subsequent severity of RA (224–226). The role of IL-4 in the homeostasis of Tregs is clearer, with IL-4 playing an important role for the suppressive function of Tregs. IL-4 knockout mice showed a lower expression of granzyme B, which is important for Treg-mediated immune regulation, as well as promoting Treg survival, which has been shown through increased cell death percentages in Tregs treated with anti-IL-4 antibodies during activation (227).
High levels of IL-7 have been found in the synovial fluid of RA patients when compared to osteoarthritis patients. IL-7, after binding the IL-7R, causes phosphorylation events that cause downstream effects by means of altering Janus kinases (JAK1, JAK3) and STATs (STAT-5a, STAT-5b). JAKs are a group of intracellular non-receptor tyrosine kinases involved in cytokine-mediated signaling via the JAK-STAT pathway. While inducing the expression of numerous cytokines, including Il-1a, IL-1b, IL-6, IL-8, TNF-α and macrophage inflammatory protein in monocytes, IL-7 has also been shown to increase the responsiveness of CD4+ T cells, thereby lowering the ability of regulatory T cells to suppress them (228).
Although classically associated with Th9 cells, IL-9 is also produced in high quantities by activated Tregs. The produced IL-9 is essential for mast cell recruitment to tolerant tissue and subsequent mast cell functioning, making IL-9 the functional link between Tregs and mast cells (229, 230). In addition, IL-9 is able to prolong neutrophil survival, increase MMP-9 production, and promote the differentiation of Th17 cells, thus cementing its role in the pathogenesis of RA (231). These findings are consistent with the work of Ciccia et al. showing that an increased expression of IL-9 and IL-9R is found in synovial tissue of RA patients as well as correlating with the severity of tissue inflammation (232). Interestingly IL-9 has also been credited with playing a role in the resolution of chronic inflammation in an RA mouse model (233). It has been hypothesized that IL-9 may play different roles in the setting of inflammation depending on its localization and origin. With the expression of CCR3 and CCR6 responsible for trafficking to inflammatory sites, Th9 cells are considered inflammation promoting in autoimmune settings, which lies in stark contrast to Tregs, which are classically associated with the attenuation of an inflammatory state (234).
Human cytokine synthesis inhibitory factor (CSIF), more commonly known as IL-10 functions to inhibit the formation of pro-inflammatory cytokines such as TNF-α, IL-1α, IL-1β, IL-6, IL-8, IL-12 and GM-CSF, as well as reducing the expression of HLA-DR and B7 molecules subsequently inhibiting macrophage antigen presentation in the synovial fluid and peripheral blood of RA patients, which in turn attenuates inflammation (235). Furthermore, IL-10 is able to suppress the production of IL-2, thereby causing a broad inhibition of the T cell immune axis which has been reflected in studies showing an attenuation of disease in CIA mice after IL-10 injections (236). In accordance with this, SNPs associated with lower IL-10 mRNA expression are overrepresented in patients with RA (237). IL-10 also holds importance for the differentiation of Tregs, with studies showing that IL-10-producing B-cells possess the ability to induce Treg differentiation from naïve CD4+ T cells (238). In addition to being induced by IL-10, regulatory T cells also possess the ability to produce and secrete IL-10.
The IL-17 cytokine family, most prominently represented by IL-17A, which is produced by Th17 cells, promotes the production of pro-inflammatory cytokines such as GM-CSF, IL-6 and IL-8 from fibroblasts as well as epithelial and endothelial cells. Furthermore, IL-17 is able to promote neutrophil recruitment leading to the aggravation of inflammatory responses (239). First discovered in synovial fluid in 1999, various in vivo studies of RA models as well as human in vitro studies were able to support IL-17s critical role in promoting inflammation in RA, with newer studies linking IL-17 gene polymorphisms to early RA onset (240). Furthermore, IL-17 was able to increase IL-6, IL-8, CCL2, CXCL1, VEGF and MMP-1 production by RA synoviocytes. It has been found that upon activation, regulatory T cells are able to induce differentiation of CD4+CD25- naïve T cells as well as other Treg cells to the Th17 phenotype in the presence of IL-6, thereby promoting the production of IL-17 and subsequently promoting inflammation. This effect was observed independently of TGF-β presence, suggesting that the environment can determine Treg fate and plays a significant role in determining a regulatory T cells role in either attenuating inflammation, or promoting it via increased IL-17 production (241). Although numerous studies have been able to establish the importance of Th17 cells and their associated IL-17 in RA thereby making it a potential therapeutic target of interest, IL-17 inhibition has not delivered the hoped-for results. While IL-17A blockade has been shown to be an effective RA treatment when compared to a placebo with ACR20 responses as indicators of efficacy, the effects were only modest (242). Furthermore, when combining TNF with IL-17 inhibition, no significant change in treatment outcome was observed as compared to anti-TNF (adalimumab) treatment alone (243). These results need to be considered when deciding on how much importance should be attributed to IL-17 as a potential therapeutic target in RA.
Interleukin-35, considered to be part of the IL-12 family, is an inhibitory cytokine produced by regulatory T cells (244). Serum IL-35 levels as well as mRNA expression have been shown to be lower in RA patients as compared to healthy controls, with serum levels being negatively correlated to the erythrocyte sedimentation rate and DAS28 of RA patients. In contrast, a positive correlation was found between serum IL-35 levels and Treg frequencies, which seems logical considering Tregs themselves are capable of producing IL-35. Osteoclast formation and bone loss induced by TNF-α, as well as the production of the pro-inflammatory cytokines IL-17A and IFN-γ have been shown to be inhibited by IL-35 (245). This suggests that IL-35 enjoys a protective role in regards to RA development, and that its dysregulation is involved in the pathogenesis and clinical manifestation of RA (246).
IL-6 induced post-translational modification of VASP
Interleukin-6, which has both pro- and anti-inflammatory properties, is produced by numerous cells including T- and B-cells. Associated with the IL-6 specific receptor (IL-6Ra) as well as gp130, a signal transducer, downstream signaling of IL-6 interaction in target cells occurs via JAKs. While classical transmembrane receptor associated signaling is considered to carry anti-inflammatory effects, IL-6 trans signaling associated with the soluble form of the IL-6R is given pro-inflammatory characteristics (247). This in turn has given rise to therapeutics that target JAKs in order to attenuate pro-inflammatory effects conveyed by an overabundance of this cytokine. While certainly effective in the treatment of RA, we were able to show that in comparison to conventional biological DMARDs and MTX, which showed the ability to restore Treg numbers in the peripheral blood of RA patients, JAK inhibitors had no effect on Treg levels. This suggests that the Treg number is only one of many factors contributing to RA as a disease (119). High levels of IL-6 have been identified in both blood and synovial fluid of RA patients contributing to inflammation through inducing neutrophils to secrete reactive oxygen intermediates and proteolytic enzymes, as well as inducing osteoclast differentiation via a RANKL-independent mechanism. As with IL-4, multiple IL-6 gene polymorphisms have been linked to increased susceptibility as well as clinically more aggressive RA (248). Interestingly, IL-6 has been shown to repress Treg differentiation while simultaneously inducing the development of Th17 cells, thereby skewing the Treg/Th17 ratio towards a more pro-inflammatory state (249). Furthermore, the IL-6R is upregulated on Th17 cells in contrast to other CD4+ T cells from RA patients, especially so in untreated RA patients, suggesting a role of IL-6 in the retention of transcriptional as well as functional identity of Th17 cells.
Through its ability to reduce the phosphorylation levels of vasodilator-stimulated phosphoprotein (VASP), an important regulator of T cell migration, IL-6 is able to interfere with Treg migration to sites of inflammation, thereby preventing the exertion of Treg suppressive effects at target sites. mIL-6R mediated IL-6 signaling regulates the expression level of VASP that is phosphorylated at Ser157. Recently, Yan et al. were able to show in a mouse model a negative correlation between relative p-VASP expression and IL-6R expression in a CIA transgenic human IL-6 mouse model. It has been shown in HMEC-1 cells, that VASP levels were reduced following in-vitro cultivation in the presence of IL-6, thereby establishing a link between T cell migration and IL-6 influence (250). Interestingly, this interference was unique to regulatory T cells, with CD4+ effector T cell migration not being affected. This effect was only observed in RA patients, not in healthy controls (251). VASP, as part of the Ena/VASP family, is a cytoskeletal effector protein involved in the coordination of monomeric actin recruitment to the barbed end of the actin filament, thereby preventing actin filament capping as well as playing a role in linear actin polymerization and filament bundling, giving it a vital role to cellular migration (252, 253). Several phosphorylation sites have been identified in the three proteins that comprise the Ena/VASP family, which are regulated by kinases such as PKA, PKG, and PKD1 (254–256). The N-terminal EVH1 domain of VASP regulates cellular localization, whereas the C-terminal EVH2 is involved in tetramerization by binding F-actin thereby facilitating actin polymerization, which in turn is dependent on differential phosphorylation states (257, 258). Phosphorylation levels of VASP were significantly reduced in CIA mice as compared to healthy controls, and subsequently restored through IL-6 receptor blockade (251). These observations were also made in PBMCs of RA patients, presenting with an upregulation of IL-6 as well as a decreased phosphorylation of VASP in untreated RA patients as compared to RA patients treated with IL-6 receptor blockers (251). IL-6 receptor blocker treatment, in addition, was able to increase the frequency of Tregs in the peripheral blood of RA patients (251).
Proteomic analysis was able to demonstrate the effects of altered VASP phosphorylation levels with enrichment of proteins involved in integrin signaling, L1 signal transduction, MAP2K and MAPK activation, mitochondrial protein import, protein localization as well as integrin cell surface interactions. Of specific note is the differential protein expression with regard to integrin signaling, whereby patients with low p-VASP expression showed an upregulation in these proteins when compared to IL-6 receptor blocker-treated RA patients with high p-VASP expression. This suggests not only the importance of integrin signaling in the pathogenesis of RA, but also possible therapeutic approaches in regard to altering cellular migration via means of IL-6 receptor blockade (251). VASP phosphorylation also has effects on the genomic level, with low p-VASP expression being implicated in the alteration of pathways involved in integrin mediated signaling, integrin binding, leukocyte migration, cell-substrate adhesion, cell matrix adhesion, positive regulation of epithelial cell migration, regulation of substrate adhesion-dependent cell spreading, as well as the regulation of tissue remodeling. These results go to show that the phosphorylation levels of VASP, which are modulated by different IL-6/IL-6R levels, carry importance for both the proteomic as well as genomic homeostasis in RA patients.
While required for T cell diapedesis and trafficking, normal T cell development and the trafficking of naïve T cell populations to the lymph node and spleen occurs independent of VASP. Deletion of VASP in T cells results in an impairment of the alpha 4 integrin (CD49d) with subsequent impairment of activated T cell diapedesis, suggesting the effects of VASP on transendothelial migration being CD49d-dependent. Although diapedesis is inhibited through impaired VASP functioning, T cell adhesion to, as well as crawling on endothelium is not affected (259). First strides are being made in investigating the therapeutic potential of VASP inhibition in the prevention of cancer metastasis development. VASP might also prove to be an interesting target in the treatment of RA by means of promoting regulatory t cell migration to sites of inflammation, as well as potentially inhibiting the migration of pro-inflammatory acting effector T cells (260).
Lack of GPSM2
G-protein-signaling modulator 2 (GPSM2) also known as LGN, is involved in the modulation of G-protein-coupled receptor functioning, thereby altering the cellular response initiated by cell-surface receptors in response to extracellular signals. The N-terminal half contains 10 copies of leu-gly-asn amino acid repeats, hence the alternative LGN naming, as well as four GoLoco motifs on the C-terminal end which are involved in guanine nucleotide exchange (260). GPSM2 is classically associated with the regulation of cell division and the cell cycle as well as the development of normal hearing (261). GPSM2 mutations have been shown to cause non-syndromic hearing loss and deafness as well as contributing to mechanisms underlying common brain malformations, as well as finding implications in the cellular migration of malignant cells, the latter of which indicates a role of GPSM2 in aberrant cellular migration (262, 263).
Recently, the understanding of GPSM2 functioning was expanded by observations implicating GPSM2 in the pathogenesis of RA via means of altered Treg migration. Meyer et al. were able to show that phosphorylation of serine/threonine kinases in CD4+ T cells is significantly altered as compared to healthy controls. The phosphorylation level of GPSM2 is reduced in CD4+ T cells from RA patients and is significantly downregulated in experimental autoimmune arthritis following immunization of mice with collagen type II (264). Interestingly, treatment with anti-IL-6 receptor antibodies restores the phosphorylation level of GPSM2 in CD4+ T cells from RA patients (264). The changed phosphorylation level after treatment with anti-IL-6 receptor antibodies could be related to reduced IL-6 signaling or could be caused by a general reduction of inflammation (264). Phosphorylation of GPSM2 was shown to be significantly downregulated in untreated RA patients as compared to healthy controls and RA patients treated with IL-6 receptor inhibitors, making it a potential therapeutic target in the treatment of RA. This notion is further supported by the finding that GPSM2 expression is not only downregulated, but completely lost in CD4+ T cells four weeks following the immunization of mice with collagen type 2 in the induction of experimental autoimmune arthritis. While it was shown that loss of GPSM2 expression correlated with increased paw size in CIA mice, there was no correlation to radiographic signs of bone erosion (264).
The vital role of GPSM2 in cellular migration, and specifically Treg migration was established through the ability to significantly inhibit Treg migration through GPSM2-specific antibodies in healthy individuals. Interestingly, blockade of GPSM2 in patients with active untreated RA had no effect on Treg migration. This lack of effect in untreated RA patients can possibly be attributed to the lack of GPSM2 expression in the setting of untreated RA as established in the CIA mouse models. In contrast, Tregs from RA patients treated with IL-6 receptor inhibitors showed increased Treg migration when compared with untreated RA patients and healthy controls, as well as the ability to alter Treg migration via antibody-mediated GPSM2 inhibition. While implicated in regulatory T cell migration, specific blockade of GPSM2 showed no significant influence on overall CD4+ T cell migration. This data suggests an interplay between IL-6 receptor signaling, GPSM2 expression and subsequent inhibition of cellular migration, which can be attenuated by means of long-term IL-6 receptor inhibition. These findings have been able to implicate GPSM2 as a promoter of cellular migration specifically in Tregs, without taking effect on other CD4+ T cell subsets. While Treg migration is significantly reduced through a blockade of GPSM2, it is not completely eliminated, suggesting a certain amount of redundancy within the regulation of Treg migration that can compensate for a loss of GPSM2. GPSM2 thereby is another axis of the IL-6 receptor effects implicated in regulatory T cell homeostasis and migration, along with, among others, modulation of the NF-kB signaling pathway (264).
Discussion
Regulatory T cells have been able to attract increased attention in research related to autoimmune diseases, to which rheumatoid arthritis is no exception. Tregs, through their immune inhibitory properties, prove to be of great importance when it comes to understanding the pathogenesis and discovering possible treatment targets for RA. As our population continues to age and the prevalence of RA continues to increase in the western world, the significance of advancing our understanding of and treatment strategies for RA becomes more apparent. With this systematic literature review, we provide an overview of the latest understanding of the homeostasis and migration of Treg cells in RA.
As a CD4+ T cell subset with anti-inflammatory properties and immune inhibitory effects, Treg cells provide an important counterpart to overarching inflammatory processes. Dysregulation or inability to carry out the innate functions of Tregs, therefore, can lay the foundation for the development of diseases in the autoimmune spectrum such as RA. Regulatory T cells have been shown to play a vital role as an anti-inflammatory axis in the physiologic as well as diseased state, the modulation of which could prove effective in both prevention and treatment of RA. By means of affecting both environmental cytokine profiles and the functioning of numerous immune-competent cell types, Tregs can be considered a central player in the aberrant processes leading to the development and progression of not only RA, but various other diseases.
Numerous chemokines and cytokines, both affecting regulatory T cells, have been implicated in the development and progression of RA. These include both pro-inflammatory cytokines such as IL-6, IL-17, IL-1 and TNF-α, as well as anti-inflammatory cytokines such as IL-4 and TGF-β, to name a few. Chemokines such as CXCL5 and CCL2, among others, with relevance to regulatory T cells, have also been implicated in RA through their increased citrullination in the synovium, with CXCL5 citrullination levels enjoying a close correlation to disease activity. While taking direct effect on pro- and anti-inflammatory effector functions, altered chemokine and cytokine levels also affect regulatory T cell migration to sites of inflammation. RA is characterized by an altered ability of regulatory T cells to migrate to sites of inflammation, which in part can be explained by a lack of GPSM2 and VASP activity due to post-translational modifications and altered phosphorylation states.
Many of these already serve as therapeutic targets through monoclonal antibodies targeting TNF-α (Infliximab, Adalimumab, Golimumab), the IL-6 receptor (Tocilizumab) and the IL-1 receptor (Anakinra). As our understanding of RA and regulatory T cells continues to grow, so too does the list of potential targets for the therapeutics of tomorrow. Furthermore, this growing understanding might one day be able to help us prevent RA onset before a need for therapeutics arises.
Author contributions
KK, YS, and DK performed literature research and prepared the manuscript. KK prepared the figures. KK and DK wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kumar B, Das MP, Misra AK. A cross-sectional study of association of serostatus and extra-articular manifestations in patients with rheumatoid arthritis in a teaching hospital. J Family Med Prim Care (2020) 9(6):2789–93. doi: 10.4103/jfmpc.jfmpc_99_20
2. Myasoedova E, Davis JM, Achenbach SJ, Matteson EL, Crowson CS. OP0009 rising prevalence of functional disability in patients with rheumatoid arthritis over 20 years. Ann Rheumatic Dis (2018) 77(Suppl 2):54–4. doi: 10.1136/annrheumdis-2018-eular.3788
3. Gunnarsson C, Chen J, Rizzo JA, Ladapo JA, Naim A, Lofland JH. The employee absenteeism costs of rheumatoid arthritis: Evidence from US national survey data. J Occup Environ Med (2015) 57(6):635–42. doi: 10.1097/JOM.0000000000000461
4. Crowson CS, Matteson EL, Myasoedova E, Michet CJ, Ernste FC, Warrington KJ, et al. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis rheumatism (2011) 63(3):633–9. doi: 10.1002/art.30155
5. Almutairi K, Nossent J, Preen D, Keen H, Inderjeeth C. The global prevalence of rheumatoid arthritis: a meta-analysis based on a systematic review. Rheumatol Int (2021) 41(5):863–77. doi: 10.1007/s00296-020-04731-0
6. Khuder SA, Peshimam AZ, Agraharam S. Environmental risk factors for rheumatoid arthritis. Rev Environ Health (2002) 17(4):307–15. doi: 10.1515/REVEH.2002.17.4.307
7. Song GG, Bae SC, Lee YH. Association between vitamin d intake and the risk of rheumatoid arthritis: a meta-analysis. Clin Rheumatol (2012) 31(12):1733–9. doi: 10.1007/s10067-012-2080-7
8. Sugiyama D, Nishimura K, Tamaki K, Tsuji G, Nakazawa T, Morinobu A, et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis (2010) 69(1):70–81. doi: 10.1136/ard.2008.096487
9. Di Giuseppe D, Discacciati A, Orsini N, Wolk A. Cigarette smoking and risk of rheumatoid arthritis: a dose-response meta-analysis. Arthritis Res Ther (2014) 16(2):R61. doi: 10.1186/ar4498
10. Wang H, Peng W, Weng Y, Ying H, Li H, Xia D, et al. Imbalance of Th17/Treg cells in mice with chronic cigarette smoke exposure. Int Immunopharmacol (2012) 14(4):504–12. doi: 10.1016/j.intimp.2012.09.011
11. Contento RL, Molon B, Boularan C, Pozzan T, Manes S, Marullo S, et al. CXCR4-CCR5: a couple modulating T cell functions. Proc Natl Acad Sci USA. (2008) 105(29):10101–6. doi: 10.1073/pnas.0804286105
12. Nagafuchi Y, Shoda H, Sumitomo S, Nakachi S, Kato R, Tsuchida Y, et al. Immunophenotyping of rheumatoid arthritis reveals a linkage between HLA-DRB1 genotype, CXCR4 expression on memory CD4+ T cells and disease activity. Sci Rep (2016) 6(1):29338. doi: 10.1038/srep29338
13. Dong L, Wang X, Tan J, Li H, Qian W, Chen J, et al. Decreased expression of microRNA-21 correlates with the imbalance of Th17 and treg cells in patients with rheumatoid arthritis. J Cell Mol Med (2014) 18(11):2213–24. doi: 10.1111/jcmm.12353
14. Huehn J, Hamann A. Homing to suppress: address codes for treg migration. Trends Immunol (2005) 26(12):632–6. doi: 10.1016/j.it.2005.10.001
15. Zhu L, Hua F, Ding W, Ding K, Zhang Y, Xu C. The correlation between the Th17/Treg cell balance and bone health. Immun Ageing (2020) 17:30. doi: 10.1186/s12979-020-00202-z
16. Braza F, Durand M, Degauque N, Brouard S. Regulatory T cells in kidney transplantation: New directions? Am J Transplant (2015) 15(9):2288–300. doi: 10.1111/ajt.13395
17. Chatzileontiadou DSM, Sloane H, Nguyen AT, Gras S, Grant EJ. The many faces of CD4(+) T cells: Immunological and structural characteristics. Int J Mol Sci (2020) 22(1):1–27. doi: 10.3390/ijms22010073
18. Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci USA (2009) 106(11):4360–5. doi: 10.1073/pnas.0811332106
19. Chiu Y-M, Lu Y-P, Lan J-L, Chen D-Y, Wang J-D. Lifetime risks, life expectancy, and health care expenditures for rheumatoid arthritis: A nationwide cohort followed up from 2003 to 2016. Arthritis Rheumatol (2021) 73(5):750–8. doi: 10.1002/art.41597
20. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64.
21. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-Linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27(1):18–20. doi: 10.1038/83707
22. Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest (2006) 116(6):1713–22. doi: 10.1172/JCI25112
23. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol (2017) 18(2):173–83. doi: 10.1038/ni.3646
24. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T Cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for treg cell development. Immunity (2012) 37(5):785–99. doi: 10.1016/j.immuni.2012.09.010
25. Itoh M, Takahashi T, Sakaguchi N, Kuniyasu Y, Shimizu J, Otsuka F, et al. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol (1999) 162(9):5317–26.
26. Wyss L, Stadinski BD, King CG, Schallenberg S, McCarthy NI, Lee JY, et al. Affinity for self antigen selects treg cells with distinct functional properties. Nat Immunol (2016) 17(9):1093–101. doi: 10.1038/ni.3522
27. Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol (1998) 10(12):1969–80. doi: 10.1093/intimm/10.12.1969
28. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
29. Fisson S, Darrasse-Jeze G, Litvinova E, Septier F, Klatzmann D, Liblau R, et al. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J Exp Med (2003) 198(5):737–46. doi: 10.1084/jem.20030686
30. Seddon B, Mason D. Peripheral autoantigen induces regulatory T cells that prevent autoimmunity. J Exp Med (1999) 189(5):877–82. doi: 10.1084/jem.189.5.877
31. Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature (2020) 581(7809):475–9. doi: 10.1038/s41586-020-2193-0
32. McPherson SW, Heuss ND, Pierson MJ, Gregerson DS. Retinal antigen-specific regulatory T cells protect against spontaneous and induced autoimmunity and require local dendritic cells. J Neuroinflamm (2014) 11:205. doi: 10.1186/s12974-014-0205-4
33. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature (2011) 478(7368):250–4. doi: 10.1038/nature10434
34. Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity (2011) 35(1):109–22. doi: 10.1016/j.immuni.2011.03.029
35. Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity (2011) 34(2):237–46. doi: 10.1016/j.immuni.2011.01.016
36. Gilbert RS, Kobayashi R, Sekine S, Fujihashi K. Functional transforming growth factor-beta receptor type II expression by CD4+ T cells in peyer's patches is essential for oral tolerance induction. PloS One (2011) 6(11):e27501. doi: 10.1371/journal.pone.0027501
37. Cassani B, Villablanca EJ, Quintana FJ, Love PE, Lacy-Hulbert A, Blaner WS, et al. Gut-tropic T cells that express integrin alpha4beta7 and CCR9 are required for induction of oral immune tolerance in mice. Gastroenterology (2011) 141(6):2109–18. doi: 10.1053/j.gastro.2011.09.015
38. Kofler DM, Mayr C, Wendtner CM. Current status of immunotherapy in b cell malignancies. Curr Drug Targets (2006) 7(10):1371–4. doi: 10.2174/138945006778559120
39. Bamidis AD, Koehler P, di Cristanziano V, Rasche K, Demirel B, Bacher P, et al. First manifestation of adult-onset still's disease after COVID-19. Lancet Rheumatol (2021) 3(5):e319–21. doi: 10.1016/S2665-9913(21)00072-2
40. Wang J, Ke XY. The four types of tregs in malignant lymphomas. J Hematol Oncol (2011) 4:50. doi: 10.1186/1756-8722-4-50
41. Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol (2004) 5(3):266–71. doi: 10.1038/ni1037
42. Jasper MJ, Tremellen KP, Robertson SA. Primary unexplained infertility is associated with reduced expression of the T-regulatory cell transcription factor Foxp3 in endometrial tissue. Mol Hum Reprod (2006) 12(5):301–8. doi: 10.1093/molehr/gal032
43. Winger EE, Reed JL. Low circulating CD4(+) CD25(+) Foxp3(+) T regulatory cell levels predict miscarriage risk in newly pregnant women with a history of failure. Am J Reprod Immunol (2011) 66(4):320–8. doi: 10.1111/j.1600-0897.2011.00992.x
44. Mor A, Planer D, Luboshits G, Afek A, Metzger S, Chajek-Shaul T, et al. Role of naturally occurring CD4+ CD25+ regulatory T cells in experimental atherosclerosis. Arterioscler Thromb Vasc Biol (2007) 27(4):893–900. doi: 10.1161/01.ATV.0000259365.31469.89
45. Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med (2006) 12(2):178–80. doi: 10.1038/nm1343
46. Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes (2011) 60(11):2954–62. doi: 10.2337/db11-0358
47. Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature (2019) 565(7738):246–50. doi: 10.1038/s41586-018-0824-5
48. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science (2010) 329(5999):1667–71. doi: 10.1126/science.1191996
49. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol (2009) 10(6):595–602. doi: 10.1038/ni.1731
50. Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, et al. The transcription factors blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol (2011) 12(4):304–11. doi: 10.1038/ni.2006
51. Dong S, Maiella S, Xhaard A, Pang Y, Wenandy L, Larghero J, et al. Multiparameter single-cell profiling of human CD4+FOXP3+ regulatory T-cell populations in homeostatic conditions and during graft-versus-host disease. Blood (2013) 122(10):1802–12. doi: 10.1182/blood-2013-02-482539
52. Ochoa-Reparaz J, Rynda A, Ascon MA, Yang X, Kochetkova I, Riccardi C, et al. IL-13 production by regulatory T cells protects against experimental autoimmune encephalomyelitis independently of autoantigen. J Immunol (2008) 181(2):954–68. doi: 10.4049/jimmunol.181.2.954
53. Sharma R, Sung SS, Fu SM, Ju ST. Regulation of multi-organ inflammation in the regulatory T cell-deficient scurfy mice. J BioMed Sci (2009) 16:20. doi: 10.1186/1423-0127-16-20
54. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
55. Schildknecht A, Brauer S, Brenner C, Lahl K, Schild H, Sparwasser T, et al. FoxP3+ regulatory T cells essentially contribute to peripheral CD8+ T-cell tolerance induced by steady-state dendritic cells. Proc Natl Acad Sci USA (2010) 107(1):199–203. doi: 10.1073/pnas.0910620107
56. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Dis (2019) 78(2):209–17. doi: 10.1136/annrheumdis-2018-214229
57. Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med (2005) 201(5):723–35. doi: 10.1084/jem.20041982
58. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8(12):1353–62. doi: 10.1038/ni1536
59. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in treg cell function. Nat Immunol (2016) 17(11):1322–33. doi: 10.1038/ni.3540
60. Pekalski ML, Ferreira RC, Coulson RM, Cutler AJ, Guo H, Smyth DJ, et al. Postthymic expansion in human CD4 naive T cells defined by expression of functional high-affinity IL-2 receptors. J Immunol (2013) 190(6):2554–66. doi: 10.4049/jimmunol.1202914
61. Skartsis N, Peng Y, Ferreira LMR, Nguyen V, Ronin E, Muller YD, et al. IL-6 and TNFalpha drive extensive proliferation of human tregs without compromising their lineage stability or function. Front Immunol (2021) 12:783282. doi: 10.3389/fimmu.2021.783282
62. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity (2014) 41(6):1013–25. doi: 10.1016/j.immuni.2014.12.006
63. Klocke K, Sakaguchi S, Holmdahl R, Wing K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci USA (2016) 113(17):E2383–92. doi: 10.1073/pnas.1603892113
64. Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med (2010) 207(9):1871–8. doi: 10.1084/jem.20100209
65. Bonnet B, Vigneron J, Levacher B, Vazquez T, Pitoiset F, Brimaud F, et al. Low-dose IL-2 induces regulatory T cell-mediated control of experimental food allergy. J Immunol (2016) 197(1):188–98. doi: 10.4049/jimmunol.1501271
66. Vitales-Noyola M, Layseca-Espinosa E, Baranda L, Abud-Mendoza C, Nino-Moreno P, Monsivais-Urenda A, et al. Analysis of sodium chloride intake and Treg/Th17 lymphocytes in healthy individuals and patients with rheumatoid arthritis or systemic lupus erythematosus. J Immunol Res (2018) 2018:9627806. doi: 10.1155/2018/9627806
67. Tang TT, Song Y, Ding YJ, Liao YH, Yu X, Du R, et al. Atorvastatin upregulates regulatory T cells and reduces clinical disease activity in patients with rheumatoid arthritis. J Lipid Res (2011) 52(5):1023–32. doi: 10.1194/jlr.M010876
68. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and bcl-6 suppress germinal center reactions. Nat Med (2011) 17(8):983–8. doi: 10.1038/nm.2426
69. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature (2009) 458(7236):351–6. doi: 10.1038/nature07674
70. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17(8):975–82. doi: 10.1038/nm.2425
71. Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood (2009) 113(18):4240–9. doi: 10.1182/blood-2008-10-183251
72. Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U.S.A. (2009) 106(12):4793–8. doi: 10.1073/pnas.0900408106
73. Cretney E, Kallies A, Nutt SL. Differentiation and function of Foxp3(+) effector regulatory T cells. Trends Immunol (2013) 34(2):74–80. doi: 10.1016/j.it.2012.11.002
74. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
75. Zhang R, Miao J, Zhang K, Zhang B, Luo X, Sun H, et al. Th1-like treg cells are increased but deficient in function in rheumatoid arthritis. Front Immunol (2022) 13:863753. doi: 10.3389/fimmu.2022.863753
76. Van Gool F, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, et al. A mutation in the transcription factor Foxp3 drives T helper 2 effector function in regulatory T cells. Immunity (2019) 50(2):362–377.e6. doi: 10.1016/j.immuni.2018.12.016
77. Kim HJ, Barnitz RA, Kreslavsky T, Brown FD, Moffett H, Lemieux ME, et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science (2015) 350(6258):334–9. doi: 10.1126/science.aad0616
78. Zafari P, Yari K, Mostafaei S, Iranshahi N, Assar S, Fekri A, et al. Analysis of Helios gene expression and Foxp3 TSDR methylation in the newly diagnosed rheumatoid arthritis patients. Immunol Invest (2018) 47(6):632–42. doi: 10.1080/08820139.2018.1480029
79. Pan F, Yu H, Dang EV, Barbi J, Pan X, Grosso JF, et al. Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science (2009) 325(5944):1142–6. doi: 10.1126/science.1176077
80. Cai X, Liu X, Du S, Xu X, Liu A, Ge X, et al. Overexpression of aiolos in peripheral blood mononuclear cell subsets from patients with systemic lupus erythematosus and rheumatoid arthritis. Biochem Genet (2016) 54(1):73–82. doi: 10.1007/s10528-015-9702-0
81. Rivellese F, Congia M, Mauro D, Bombardieri M, Lewis M, Pitzalis C. THU0070 the transcription factors ikaros and aiolos are expressed in the synovial membrane of early rheumatoid arthritis patients in association with synovial lymphoid aggregates. Ann Rheumatic Dis (2018) 77(Suppl 2):259–9. doi: 10.1136/annrheumdis-2018-eular.6442
82. Mitchell JL, Seng A, Yankee TM. Expression patterns of ikaros family members during positive selection and lineage commitment of human thymocytes. Immunology (2016) 149(4):400–12. doi: 10.1111/imm.12657
83. Evans HG, Roostalu U, Walter GJ, Gullick NJ, Frederiksen KS, Roberts CA, et al. TNF-α blockade induces IL-10 expression in human CD4+ T cells. Nat Commun (2014) 5:3199. doi: 10.1038/ncomms4199
84. Romero F, Martinez AC, Camonis J, Rebollo A. Aiolos transcription factor controls cell death in T cells by regulating bcl-2 expression and its cellular localization. EMBO J (1999) 18(12):3419–30. doi: 10.1093/emboj/18.12.3419
85. Raffin C, Pignon P, Celse C, Debien E, Valmori D, Ayyoub M. Human memory Helios- FOXP3+ regulatory T cells (Tregs) encompass induced tregs that express aiolos and respond to IL-1beta by downregulating their suppressor functions. J Immunol (2013) 191(9):4619–27. doi: 10.4049/jimmunol.1301378
86. Biniecka M, Kennedy A, Fearon U, Ng CT, Veale DJ, O'Sullivan JN. Oxidative damage in synovial tissue is associated with in vivo hypoxic status in the arthritic joint. Ann Rheumatic Dis (2010) 69(6):1172–8. doi: 10.1136/ard.2009.111211
87. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood (2006) 108(1):253–61. doi: 10.1182/blood-2005-11-4567
88. Morgan ME, Sutmuller RP, Witteveen HJ, van Duivenvoorde LM, Zanelli E, Melief CJ, et al. CD25+ cell depletion hastens the onset of severe disease in collagen-induced arthritis. Arthritis Rheum (2003) 48(5):1452–60. doi: 10.1002/art.11063
89. Avdeeva A, Rubtsov Y, Dyikanov D, Popkova T, Nasonov E. Regulatory T cells in patients with early untreated rheumatoid arthritis: Phenotypic changes in the course of methotrexate treatment. Biochimie (2020) 174:9–17. doi: 10.1016/j.biochi.2020.03.014
90. Baecklund E, Backlin C, Iliadou A, Granath F, Ekbom A, Amini RM, et al. Characteristics of diffuse large b cell lymphomas in rheumatoid arthritis. Arthritis Rheum (2006) 54(12):3774–81. doi: 10.1002/art.22277
91. Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C, et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol 2012 (2012) p:827480. doi: 10.1155/2012/827480
92. Engelmann R, Brandt J, Eggert M, Karberg K, Krause A, Neeck G, et al. IgG1 and IgG4 are the predominant subclasses among auto-antibodies against two citrullinated antigens in RA. Rheumatol (Oxford) (2008) 47(10):1489–92. doi: 10.1093/rheumatology/ken336
93. Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol (2019) 20(7):928–42. doi: 10.1038/s41590-019-0378-1
94. Niu Q, Huang ZC, Wu XJ, Jin YX, An YF, Li YM, et al. Enhanced IL-6/phosphorylated STAT3 signaling is related to the imbalance of circulating T follicular helper/T follicular regulatory cells in patients with rheumatoid arthritis. Arthritis Res Ther (2018) 20(1):200. doi: 10.1186/s13075-018-1690-0
95. Wollenberg I, Agua-Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol (2011) 187(9):4553–60. doi: 10.4049/jimmunol.1101328
96. Yang G, Yang X, Zhang J, Li G, Zheng D, Peng A, et al. Transcriptional repressor Blimp1 regulates follicular regulatory T-cell homeostasis and function. Immunology (2018) 153(1):105–17. doi: 10.1111/imm.12815
97. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls b cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity (2014) 41(6):1026–39. doi: 10.1016/j.immuni.2014.12.005
98. Ramiscal RR, Vinuesa CG. T-Cell subsets in the germinal center. Immunol Rev (2013) 252(1):146–55. doi: 10.1111/imr.12031
99. Bruck C, Golumba-Nagy V, Yan S, Esser RL, Thiele J, Stahl D, et al. Th1 and Th17 cells are resistant towards T cell activation-induced downregulation of CD6. Clin Immunol (2022) 238:109025. doi: 10.1016/j.clim.2022.109025
100. Meyer A, Kofler DM. Failure of a T cell regulator: CD6 contributes to the aggravation of autoimmune inflammation. Cell Mol Immunol (2019) 16(9):733–4. doi: 10.1038/s41423-018-0089-9
101. Zizzo G, De Santis M, Bosello SL, Fedele AL, Peluso G, Gremese E, et al. Synovial fluid-derived T helper 17 cells correlate with inflammatory activity in arthritis, irrespectively of diagnosis. Clin Immunol (2011) 138(1):107–16. doi: 10.1016/j.clim.2010.10.002
102. Zhong W, Zhao L, Liu T, Jiang Z. IL-22-producing CD4+T cells in the treatment response of rheumatoid arthritis to combination therapy with methotrexate and leflunomide. Sci Rep (2017) 7:41143. doi: 10.1038/srep41143
103. Neumann E, Lefevre S, Zimmermann B, Gay S, Muller-Ladner U. Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol Med (2010) 16(10):458–68. doi: 10.1016/j.molmed.2010.07.004
104. Hirota K, Hashimoto M, Ito Y, Matsuura M, Ito H, Tanaka M, et al. Autoimmune Th17 cells induced synovial stromal and innate lymphoid cell secretion of the cytokine GM-CSF to initiate and augment autoimmune arthritis. Immunity (2018) 48(6):1220–1232 e5. doi: 10.1016/j.immuni.2018.04.009
105. Ganesan R, Rasool M. Interleukin 17 regulates SHP-2 and IL-17RA/STAT-3 dependent Cyr61, IL-23 and GM-CSF expression and RANKL mediated osteoclastogenesis by fibroblast-like synoviocytes in rheumatoid arthritis. Mol Immunol (2017) 91:134–44. doi: 10.1016/j.molimm.2017.09.003
106. Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med (2001) 194(5):629–44. doi: 10.1084/jem.194.5.629
107. Das J, Ren G, Zhang L, Roberts AI, Zhao X, Bothwell AL, et al. Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med (2009) 206(11):2407–16. doi: 10.1084/jem.20082286
108. DiToro D, Harbour SN, Bando JK, Benavides G, Witte S, Laufer VA, et al. Insulin-like growth factors are key regulators of T helper 17 regulatory T cell balance in autoimmunity. Immunity (2020) 52(4):650–667 e10. doi: 10.1016/j.immuni.2020.03.013
109. Kotschenreuther K, Waque I, Yan S, Meyer A, Haak T, von Tresckow J, et al. Cannabinoids drive Th17 cell differentiation in patients with rheumatic autoimmune diseases. Cell Mol Immunol (2021) 18(3):764–6. doi: 10.1038/s41423-020-0437-4
110. Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature (2013) 496(7446):513–7. doi: 10.1038/nature11984
111. Szodoray P, Nakken B, Barath S, Csipo I, Nagy G, El-Hage F, et al. Altered Th17 cells and Th17/regulatory T-cell ratios indicate the subsequent conversion from undifferentiated connective tissue disease to definitive systemic autoimmune disorders. Hum Immunol (2013) 74(12):1510–8. doi: 10.1016/j.humimm.2013.08.003
112. Taha HA, Hozayen WG, Okasha AM, Ahmed AE, Shata MAA, Abdel-Naem EA, et al. Investigating the balance between Th17/Treg cells in rheumatoid arthritis and its association with disease activity. J Child Sci (2019) 09(01):e75–83. doi: 10.1055/s-0039-1693158
113. Tahir H, Deodhar A, Genovese M, Takeuchi T, Aelion J, Van den Bosch F, et al. Secukinumab in active rheumatoid arthritis after anti-TNFα therapy: A randomized, double-blind placebo-controlled phase 3 study. Rheumatol Ther (2017) 4(2):475–88. doi: 10.1007/s40744-017-0086-y
114. van Baarsen LG, Lebre MC, van der Coelen D, Aarrass S, Tang MW, Ramwadhdoebe TH, et al. Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res Ther (2014) 16(4):426. doi: 10.1186/s13075-014-0426-z
115. Hot A, Miossec P. Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Ann Rheum Dis (2011) 70(5):727–32. doi: 10.1136/ard.2010.143768
116. Ramezani-Rad P, Geng H, Hurtz C, Chan LN, Chen Z, Jumaa H, et al. SOX4 enables oncogenic survival signals in acute lymphoblastic leukemia. Blood (2013) 121(1):148–55. doi: 10.1182/blood-2012-05-428938
117. Malhotra N, Narayan K, Cho OH, Sylvia KE, Yin C, Melichar H, et al. A network of high-mobility group box transcription factors programs innate interleukin-17 production. Immunity (2013) 38(4):681–93. doi: 10.1016/j.immuni.2013.01.010
118. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-Hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20(1):62–8. doi: 10.1038/nm.3432
119. Meyer A, Wittekind PS, Kotschenreuther K, Schiller J, von Tresckow J, Haak TH, et al. Regulatory T cell frequencies in patients with rheumatoid arthritis are increased by conventional and biological DMARDs but not by JAK inhibitors. Ann Rheum Dis (2021) 80(12):e196. doi: 10.1136/annrheumdis-2019-216576
120. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61(12):1693–700. doi: 10.1136/gutjnl-2011-301668
121. den Braber I, Mugwagwa T, Vrisekoop N, Westera L, Mogling R, de Boer AB, et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity (2012) 36(2):288–97. doi: 10.1016/j.immuni.2012.02.006
122. Thome JJ, Grinshpun B, Kumar BV, Kubota M, Ohmura Y, Lerner H, et al. Longterm maintenance of human naive T cells through in situ homeostasis in lymphoid tissue sites. Sci Immunol (2016) 1(6):1–23. doi: 10.1126/sciimmunol.aah6506
123. Silva SL, Albuquerque A, Amaral AJ, Li QZ, Mota C, Cheynier R, et al. Autoimmunity and allergy control in adults submitted to complete thymectomy early in infancy. PloS One (2017) 12(7):e0180385. doi: 10.1371/journal.pone.0180385
124. Silva SL, Albuquerque AS, Serra-Caetano A, Foxall RB, Pires AR, Matoso P, et al. Human naive regulatory T-cells feature high steady-state turnover and are maintained by IL-7. Oncotarget (2016) 7(11):12163–75. doi: 10.18632/oncotarget.7512
125. Thome JJ, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, et al. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat Med (2016) 22(1):72–7. doi: 10.1038/nm.4008
126. Thomas R, McIlraith M, Davis LS, Lipsky PE. Rheumatoid synovium is enriched in CD45RBdim mature memory T cells that are potent helpers for b cell differentiation. Arthritis Rheum (1992) 35(12):1455–65. doi: 10.1002/art.1780351209
127. Yabe R, Chung SH, Murayama MA, Kubo S, Shimizu K, Akahori Y, et al. TARM1 contributes to development of arthritis by activating dendritic cells through recognition of collagens. Nat Commun (2021) 12(1):94. doi: 10.1038/s41467-020-20307-9
128. Radstake TRDJ, van der Voort R, ten Brummelhuis M, de Waal Malefijt M, Looman M, Figdor CG, et al. Increased expression of CCL18, CCL19, and CCL17 by dendritic cells from patients with rheumatoid arthritis, and regulation by fc gamma receptors. Ann Rheumatic Dis (2005) 64(3):359–67. doi: 10.1136/ard.2003.017566
129. Estrada-Capetillo L, Hernandez-Castro B, Monsivais-Urenda A, Alvarez-Quiroga C, Layseca-Espinosa E, Abud-Mendoza C, et al. Induction of Th17 lymphocytes and treg cells by monocyte-derived dendritic cells in patients with rheumatoid arthritis and systemic lupus erythematosus. Clin Dev Immunol 2013 (2013) p:584303. doi: 10.1155/2013/584303
130. Wu B, Qiu J, Zhao TV, Wang Y, Maeda T, Goronzy IN, et al Succinyl-CoA ligase deficiency in pro-inflammatory and tissue-invasive T cells. Cell Metab (2020) 32(6):967–980 e5. doi: 10.1016/j.cmet.2020.10.025
131. Sumariwalla PF, Malfait AM, Feldmann M. P-selectin glycoprotein ligand 1 therapy ameliorates established collagen-induced arthritis in DBA/1 mice partly through the suppression of tumour necrosis factor. Clin Exp Immunol (2004) 136(1):67–75. doi: 10.1111/j.1365-2249.2004.02421.x
132. Wohler J, Bullard D, Schoeb T, Barnum S. LFA-1 is critical for regulatory T cell homeostasis and function. Mol Immunol (2009) 46(11-12):2424–8. doi: 10.1016/j.molimm.2009.04.004
133. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, et al. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity (2017) 47(5):875–889 e10. doi: 10.1016/j.immuni.2017.10.017
134. Ates A, Kinikli G, Turgay M, Duman M. Serum-soluble selectin levels in patients with rheumatoid arthritis and systemic sclerosis. Scand J Immunol (2004) 59(3):315–20. doi: 10.1111/j.0300-9475.2004.01389.x
135. Tak PP, Taylor PC, Breedveld FC, Smeets TJ, Daha MR, Kluin PM, et al. Decrease in cellularity and expression of adhesion molecules by anti-tumor necrosis factor alpha monoclonal antibody treatment in patients with rheumatoid arthritis. Arthritis Rheum (1996) 39(7):1077–81. doi: 10.1002/art.1780390702
136. Milam SB, Magnuson VL, Steffensen B, Chen D, Klebe RJ. IL-1 beta and prostaglandins regulate integrin mRNA expression. J Cell Physiol (1991) 149(2):173–83. doi: 10.1002/jcp.1041490202
137. Boisvert M, Gendron S, Chetoui N, Aoudjit F. Alpha2 beta1 integrin signaling augments T cell receptor-dependent production of interferon-gamma in human T cells. Mol Immunol (2007) 44(15):3732–40. doi: 10.1016/j.molimm.2007.04.003
138. de Fougerolles AR, Sprague AG, Nickerson-Nutter CL, Chi-Rosso G, Rennert PD, Gardner H, et al. Regulation of inflammation by collagen-binding integrins alpha1beta1 and alpha2beta1 in models of hypersensitivity and arthritis. J Clin Invest (2000) 105(6):721–9. doi: 10.1172/JCI7911
139. Klimiuk PA, Sierakowski S, Latosiewicz R, Cylwik JP, Cylwik B, Skowronski J, et al. Soluble adhesion molecules (ICAM-1, VCAM-1, and e-selectin) and vascular endothelial growth factor (VEGF) in patients with distinct variants of rheumatoid synovitis. Ann Rheum Dis (2002) 61(9):804–9. doi: 10.1136/ard.61.9.804
140. Bullard DC, Hurley LA, Lorenzo I, Sly LM, Beaudet AL, Staite ND. Reduced susceptibility to collagen-induced arthritis in mice deficient in intercellular adhesion molecule-1. J Immunol (1996) 157(7):3153–8.
141. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
142. Fischer HJ, Sie C, Schumann E, Witte AK, Dressel R, van den Brandt J, et al. The insulin receptor plays a critical role in T cell function and adaptive immunity. J Immunol (2017) 198(5):1910–20. doi: 10.4049/jimmunol.1601011
143. Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature (2006) 442(7100):299–302. doi: 10.1038/nature04882
144. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'Acquisto F, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PloS Biol (2015) 13(7):e1002202. doi: 10.1371/journal.pbio.1002202
145. Finlay DK, Sinclair LV, Feijoo C, Waugh CM, Hagenbeek TJ, Spits H, et al. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. J Exp Med (2009) 206(11):2441–54. doi: 10.1084/jem.20090219
146. Fu W, Hu W, Yi YS, Hettinghouse A, Sun G, Bi Y, et al. TNFR2/14-3-3ϵ signaling complex instructs macrophage plasticity in inflammation and autoimmunity. J Clin Invest (2021) 131(16):1–17. doi: 10.1172/JCI144016
147. Vallion R, Divoux J, Glauzy S, Ronin E, Lombardi Y, Lubrano di Ricco M, et al. Regulatory T cell stability and migration are dependent on mTOR. J Immunol (2020) 205(7):1799–809. doi: 10.4049/jimmunol.1901480
148. Yeo L, Adlard N, Biehl M, Juarez M, Smallie T, Snow M, et al. Expression of chemokines CXCL4 and CXCL7 by synovial macrophages defines an early stage of rheumatoid arthritis. Ann Rheum Dis (2016) 75(4):763–71. doi: 10.1136/annrheumdis-2014-206921
149. Greisen SR, Schelde KK, Rasmussen TK, Kragstrup TW, Stengaard-Pedersen K, Hetland ML, et al. CXCL13 predicts disease activity in early rheumatoid arthritis and could be an indicator of the therapeutic 'window of opportunity'. Arthritis Res Ther (2014) 16(5):434. doi: 10.1186/s13075-014-0434-z
150. Yoshida K, Korchynskyi O, Tak PP, Isozaki T, Ruth JH, Campbell PL, et al. Citrullination of epithelial neutrophil-activating peptide 78/CXCL5 results in conversion from a non-monocyte-recruiting chemokine to a monocyte-recruiting chemokine. Arthritis Rheumatol (2014) 66(10):2716–27. doi: 10.1002/art.38750
151. Shadidi KR, Aarvak T, Henriksen JE, Natvig JB, Thompson KM. The chemokines CCL5, CCL2 and CXCL12 play significant roles in the migration of Th1 cells into rheumatoid synovial tissue. Scand J Immunol (2003) 57(2):192–8. doi: 10.1046/j.1365-3083.2003.01214.x
152. Yu K, Chen Z, Khatri I, Gorczynski RM. CCR4 dependent migration of Foxp3+ treg cells to skin grafts and draining lymph nodes is implicated in enhanced graft survival in CD200tg recipients. Immunol Lett (2011) 141(1):116–22. doi: 10.1016/j.imlet.2011.09.002
153. Kochi Y, Okada Y, Suzuki A, Ikari K, Terao C, Takahashi A, et al. A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat Genet (2010) 42(6):515–9. doi: 10.1038/ng.583
154. Iwamoto T, Okamoto H, Kobayashi S, Ikari K, Toyama Y, Tomatsu T, et al. A role of monocyte chemoattractant protein-4 (MCP-4)/CCL13 from chondrocytes in rheumatoid arthritis. FEBS J (2007) 274(18):4904–12. doi: 10.1111/j.1742-4658.2007.06013.x
155. Ogata H, Takeya M, Yoshimura T, Takagi K, Takahashi K. The role of monocyte chemoattractant protein-1 (mcp-1) in the pathogenesis of collagen-induced arthritis in rats. J Pathol (1997) 182(1):106–14. doi: 10.1002/(SICI)1096-9896(199705)182:1<106::AID-PATH816>3.0.CO;2-A
156. Gong JH, Ratkay LG, Waterfield JD, Clark-Lewis I. An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J Exp Med (1997) 186(1):131–7. doi: 10.1084/jem.186.1.131
157. Bruhl H, Cihak J, Schneider MA, Plachy J, Rupp T, Wenzel I, et al. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol (2004) 172(2):890–8. doi: 10.4049/jimmunol.172.2.890
158. Rathanaswami P, Hachicha M, Sadick M, Schall TJ, McColl SR. Expression of the cytokine RANTES in human rheumatoid synovial fibroblasts. differential regulation of RANTES and interleukin-8 genes by inflammatory cytokines. J Biol Chem (1993) 268(8):5834–9. doi: 10.1016/S0021-9258(18)53395-0
159. Haringman JJ, Smeets TJM, Reinders-Blankert P, Tak PP. Chemokine and chemokine receptor expression in paired peripheral blood mononuclear cells and synovial tissue of patients with rheumatoid arthritis, osteoarthritis, and reactive arthritis. Ann Rheumatic Dis (2006) 65(3):294–300. doi: 10.1136/ard.2005.037176
160. Yang Y-F, Mukai T, Gao P, Yamaguchi N, Ono S, Iwaki H, et al. A non-peptide CCR5 antagonist inhibits collagen-induced arthritis by modulating T cell migration without affecting anti-collagen T cell responses. Eur J Immunol (2002) 32(8):2124–32. doi: 10.1002/1521-4141(200208)32:8<2124::AID-IMMU2124>3.0.CO;2-S
161. Katrib A, Tak PP, Bertouch JV, Cuello C, McNeil HP, Smeets TJ, et al. Expression of chemokines and matrix metalloproteinases in early rheumatoid arthritis. Rheumatol (Oxford) (2001) 40(9):988–94. doi: 10.1093/rheumatology/40.9.988
162. Eriksson C, Rantapaa-Dahlqvist S, Sundqvist KG. Changes in chemokines and their receptors in blood during treatment with the TNF inhibitor infliximab in patients with rheumatoid arthritis. Scand J Rheumatol (2013) 42(4):260–5. doi: 10.3109/03009742.2012.754937
163. Bao J, Liu W, Bao YX. Recombinant human interleukin receptor antagonist influences serum chemokines in patients with rheumatoid arthritis. Cent Eur J Immunol (2014) 39(2):170–3. doi: 10.5114/ceji.2014.43717
164. Sucur A, Jajic Z, Artukovic M, Matijasevic MI, Anic B, Flegar D, et al. Chemokine signals are crucial for enhanced homing and differentiation of circulating osteoclast progenitor cells. Arthritis Res Ther (2017) 19(1):142. doi: 10.1186/s13075-017-1337-6
165. Garcia-Lopez MA, Sanchez-Madrid F, Rodriguez-Frade JM, Mellado M, Acevedo A, Garcia MI, et al. CXCR3 chemokine receptor distribution in normal and inflamed tissues: expression on activated lymphocytes, endothelial cells, and dendritic cells. Lab Invest (2001) 81(3):409–18. doi: 10.1038/labinvest.3780248
166. Mohan K, Issekutz TB. Blockade of chemokine receptor CXCR3 inhibits T cell recruitment to inflamed joints and decreases the severity of adjuvant arthritis. J Immunol (2007) 179(12):8463–9. doi: 10.4049/jimmunol.179.12.8463
167. Lee D, Kim DW, Yoon S, Nam AR, Lee KH, Nam KH, et al. CXCL5 secreted from macrophages during cold exposure mediates white adipose tissue browning. J Lipid Res (2021) 62:100117. doi: 10.1016/j.jlr.2021.100117
168. Chen W, Wang Y, Zhou Y, Xu Y, Bo X, Wu J. M1 macrophages increase endothelial permeability and enhance p38 phosphorylation via PPAR-γ/CXCL13-CXCR5 in sepsis. Int Arch Allergy Immunol (2022) p:1–10. doi: 10.1159/000524272
169. Van Raemdonck K, Umar S, Palasiewicz K, Volkov S, Volin MV, Arami S, et al. CCL21/CCR7 signaling in macrophages promotes joint inflammation and Th17-mediated osteoclast formation in rheumatoid arthritis. Cell Mol Life Sci (2020) 77(7):1387–99. doi: 10.1007/s00018-019-03235-w
170. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med (2007) 204(12):2803–12. doi: 10.1084/jem.20071397
171. Lisignoli G, Piacentini A, Cristino S, Grassi F, Cavallo C, Cattini L, et al. CCL20 chemokine induces both osteoblast proliferation and osteoclast differentiation: Increased levels of CCL20 are expressed in subchondral bone tissue of rheumatoid arthritis patients. J Cell Physiol (2007) 210(3):798–806. doi: 10.1002/jcp.20905
172. Matsui T, Akahoshi T, Namai R, Hashimoto A, Kurihara Y, Rana M, et al. Selective recruitment of CCR6-expressing cells by increased production of MIP-3 alpha in rheumatoid arthritis. Clin Exp Immunol (2001) 125(1):155–61. doi: 10.1046/j.1365-2249.2001.01542.x
173. Haudenschild DR, Nguyen B, Chen J, D'Lima DD, Lotz MK. Rho kinase-dependent CCL20 induced by dynamic compression of human chondrocytes. Arthritis Rheum (2008) 58(9):2735–42. doi: 10.1002/art.23797
174. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi: 10.1084/jem.20030152
175. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med (2005) 201(7):1061–7. doi: 10.1084/jem.20042276
176. Zhou G, Sun X, Qin Q, Lv J, Cai Y, Wang M, et al. Loss of Smad7 promotes inflammation in rheumatoid arthritis. Front Immunol (2018) 9:2537. doi: 10.3389/fimmu.2018.02537
177. Pohlers D, Beyer A, Koczan D, Wilhelm T, Thiesen HJ, Kinne RW. Constitutive upregulation of the transforming growth factor-beta pathway in rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther (2007) 9(3):R59. doi: 10.1186/ar2217
178. Mueller A, Strange PG. CCL3, acting via the chemokine receptor CCR5, leads to independent activation of janus kinase 2 (JAK2) and gi proteins. FEBS Lett (2004) 570(1-3):126–32. doi: 10.1016/j.febslet.2004.04.100
179. Wang R, Feng W, Wang H, Wang L, Yang X, Yang F, et al. Blocking migration of regulatory T cells to leukemic hematopoietic microenvironment delays disease progression in mouse leukemia model. Cancer Lett (2020) 469:151–61. doi: 10.1016/j.canlet.2019.10.032
180. Zhang G, Liu HB, Zhou L, Cui XQ, Fan XH. CCL3 participates in the development of rheumatoid arthritis by activating AKT. Eur Rev Med Pharmacol Sci (2018) 22(20):6625–32. doi: 10.26355/eurrev_201810_16137
181. Ren M, Guo Q, Guo L, Lenz M, Qian F, Koenen RR, et al. Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme. EMBO J (2010) 29(23):3952–66. doi: 10.1038/emboj.2010.256
182. Bystry RS, Aluvihare V, Welch KA, Kallikourdis M, Betz AG. B cells and professional APCs recruit regulatory T cells via CCL4. Nat Immunol (2001) 2(12):1126–32. doi: 10.1038/ni735
183. Kuo SJ, Huang CC, Tsai CH, Hsu HC, Su CM, Tang CH. Chemokine c-c motif ligand 4 gene polymorphisms associated with susceptibility to rheumatoid arthritis. BioMed Res Int 2018 (2018) p:9181647. doi: 10.1155/2018/9181647
184. Zhang L, Yu M, Deng J, Lv X, Liu J, Xiao Y, et al. Chemokine signaling pathway involved in CCL2 expression in patients with rheumatoid arthritis. Yonsei Med J (2015) 56(4):1134–42. doi: 10.3349/ymj.2015.56.4.1134
185. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood (2006) 108(5):1571–9. doi: 10.1182/blood-2006-02-004747
186. Li B, Guo Q, Wang Y, Su R, Gao C, Zhao J, et al. Increased serum interleukin-2 levels are associated with abnormal peripheral blood natural killer cell levels in patients with active rheumatoid arthritis. Mediators Inflammation (2020) 2020:6108342. doi: 10.1155/2020/6108342
187. Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci USA (2009) 106(31):12885–90. doi: 10.1073/pnas.0812530106
188. Eller K, Wolf D, Huber JM, Metz M, Mayer G, McKenzie AN, et al. IL-9 production by regulatory T cells recruits mast cells that are essential for regulatory T cell-induced immune suppression. J Immunol (2011) 186(1):83–91. doi: 10.4049/jimmunol.1001183
189. Dantas AT, Marques CD, da Rocha Junior LF, Cavalcanti MB, Goncalves SM, Cardoso PR, et al. Increased serum interleukin-9 levels in rheumatoid arthritis and systemic lupus erythematosus: Pathogenic role or just an epiphenomenon? Dis Markers (2015) 2015:519638. doi: 10.1155/2015/519638
190. Kar S, Gupta R, Malhotra R, Sharma V, Farooque K, Kumar V, et al. Interleukin-9 facilitates osteoclastogenesis in rheumatoid arthritis. Int J Mol Sci (2021) 22(19):1–17. doi: 10.3390/ijms221910397
191. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity (2011) 34(4):566–78. doi: 10.1016/j.immuni.2011.03.018
192. Hsu P, Santner-Nanan B, Hu M, Skarratt K, Lee CH, Stormon M, et al. IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J Immunol (2015) 195(8):3665–74. doi: 10.4049/jimmunol.1402898
193. Cush JJ, Splawski JB, Thomas R, McFarlin JE, Schulze-Koops H, Davis LS, et al. Elevated interleukin-10 levels in patients with rheumatoid arthritis. Arthritis Rheum (1995) 38(1):96–104. doi: 10.1002/art.1780380115
194. Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol (2010) 11(12):1093–101. doi: 10.1038/ni.1952
195. Olson BM, Sullivan JA, Burlingham WJ. Interleukin 35: a key mediator of suppression and the propagation of infectious tolerance. Front Immunol (2013) 4:315. doi: 10.3389/fimmu.2013.00315
196. Li Y, Yao L, Liu S, Wu J, Xia L, Shen H, et al. Elevated serum IL-35 levels in rheumatoid arthritis are associated with disease activity. J Investig Med (2019) 67(3):707–10. doi: 10.1136/jim-2018-000814
197. Senolt L, Sumova B, Jandova R, Hulejova H, Mann H, Pavelka K, et al. Interleukin 35 synovial fluid levels are associated with disease activity of rheumatoid arthritis. PloS One (2015) 10(7):e0132674. doi: 10.1371/journal.pone.0132674
198. Ning X, Jian Z, Wang W. Low serum levels of interleukin 35 in patients with rheumatoid arthritis. Tohoku J Exp Med (2015) 237(2):77–82. doi: 10.1620/tjem.237.77
199. Edrees AF, Misra SN, Abdou NI. Anti-tumor necrosis factor (TNF) therapy in rheumatoid arthritis: correlation of TNF-alpha serum level with clinical response and benefit from changing dose or frequency of infliximab infusions. Clin Exp Rheumatol (2005) 23(4):469–74.
200. Haworth C, Brennan FM, Chantry D, Turner M, Maini RN, Feldmann M. Expression of granulocyte-macrophage colony-stimulating factor in rheumatoid arthritis: regulation by tumor necrosis factor-alpha. Eur J Immunol (1991) 21(10):2575–9. doi: 10.1002/eji.1830211039
201. Sakurada S, Kato T, Okamoto T. Induction of cytokines and ICAM-1 by proinflammatory cytokines in primary rheumatoid synovial fibroblasts and inhibition by n-acetyl-L-cysteine and aspirin. Int Immunol (1996) 8(10):1483–93. doi: 10.1093/intimm/8.10.1483
202. Callaghan MM, Lovis RM, Rammohan C, Lu Y, Pope RM. Autocrine regulation of collagenase gene expression by TNF-alpha in U937 cells. J Leukoc Biol (1996) 59(1):125–32. doi: 10.1002/jlb.59.1.125
203. Biton J, Semerano L, Delavallee L, Lemeiter D, Laborie M, Grouard-Vogel G, et al. Interplay between TNF and regulatory T cells in a TNF-driven murine model of arthritis. J Immunol (2011) 186(7):3899–910. doi: 10.4049/jimmunol.1003372
204. Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, Oppenheim JJ. Co-Expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur J Immunol (2010) 40(4):1099–106. doi: 10.1002/eji.200940022
205. Ishigame H, Zenewicz LA, Sanjabi S, Licona-Limón P, Nakayama M, Leonard WJ, et al. Excessive Th1 responses due to the absence of TGF-β signaling cause autoimmune diabetes and dysregulated treg cell homeostasis. Proc Natl Acad Sci USA (2013) 110(17):6961–6. doi: 10.1073/pnas.1304498110
206. Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol (2004) 172(9):5149–53. doi: 10.4049/jimmunol.172.9.5149
207. Edwards JP, Hand TW, Morais da Fonseca D, Glass DD, Belkaid Y, Shevach EM. The GARP/Latent TGF-β1 complex on treg cells modulates the induction of peripherally derived treg cells during oral tolerance. Eur J Immunol (2016) 46(6):1480–9. doi: 10.1002/eji.201546204
208. Ostroukhova M, Seguin-Devaux C, Oriss TB, Dixon-McCarthy B, Yang L, Ameredes BT, et al. Tolerance induced by inhaled antigen involves CD4(+) T cells expressing membrane-bound TGF-beta and FOXP3. J Clin Invest (2004) 114(1):28–38. doi: 10.1172/JCI200420509
209. Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates smad-independent TGF-beta responses. EMBO J (2002) 21(14):3749–59. doi: 10.1093/emboj/cdf366
210. Coffey RJ Jr., Bascom CC, Sipes NJ, Graves-Deal R, Weissman BE, Moses HL. Selective inhibition of growth-related gene expression in murine keratinocytes by transforming growth factor beta. Mol Cell Biol (1988) 8(8):3088–93. doi: 10.1128/mcb.8.8.3088-3093.1988
211. Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci U.S.A. (1995) 92(12):5545–9. doi: 10.1073/pnas.92.12.5545
212. Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature (1994) 371(6494):257–61. doi: 10.1038/371257a0
213. Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, et al. p27Kip1, a cyclin-cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev (1994) 8(1):9–22. doi: 10.1101/gad.8.1.9
214. Ruegemer JJ, Ho SN, Augustine JA, Schlager JW, Bell MP, McKean DJ, et al. Regulatory effects of transforming growth factor-beta on IL-2- and IL-4-dependent T cell-cycle progression. J Immunol (1990) 144(5):1767–76.
215. Wolfraim LA, Walz TM, James Z, Fernandez T, Letterio JJ. p21Cip1 and p27Kip1 act in synergy to alter the sensitivity of naive T cells to TGF-beta-mediated G1 arrest through modulation of IL-2 responsiveness. J Immunol (2004) 173(5):3093–102. doi: 10.4049/jimmunol.173.5.3093
216. Ludviksson BR, Seegers D, Resnick AS, Strober W. The effect of TGF-beta1 on immune responses of naive versus memory CD4+ Th1/Th2 T cells. Eur J Immunol (2000) 30(7):2101–11. doi: 10.1002/1521-4141(200007)30:7<2101::AID-IMMU2101>3.0.CO;2-P
217. Sad S, Mosmann TR. Single IL-2-secreting precursor CD4 T cell can develop into either Th1 or Th2 cytokine secretion phenotype. J Immunol (1994) 153(8):3514–22.
218. Bonig H, Banning U, Hannen M, Kim YM, Verheyen J, Mauz-Korholz C, et al. Transforming growth factor-beta1 suppresses interleukin-15-mediated interferon-gamma production in human T lymphocytes. Scand J Immunol (1999) 50(6):612–8. doi: 10.1046/j.1365-3083.1999.00635.x
219. Ranges GE, Figari IS, Espevik T, Palladino MA Jr. Inhibition of cytotoxic T cell development by transforming growth factor beta and reversal by recombinant tumor necrosis factor alpha. J Exp Med (1987) 166(4):991–8. doi: 10.1084/jem.166.4.991
220. Smyth MJ, Strobl SL, Young HA, Ortaldo JR, Ochoa AC. Regulation of lymphokine-activated killer activity and pore-forming protein gene expression in human peripheral blood CD8+ T lymphocytes. inhibition by transforming growth factor-beta. J Immunol (1991) 146(10):3289–97.
221. Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol (2005) 174(9):5215–23. doi: 10.4049/jimmunol.174.9.5215
222. Rivas D, Mozo L, Zamorano J, Gayo A, Torre-Alonso JC, Rodríguez A, et al. Upregulated expression of IL-4 receptors and increased levels of IL-4 in rheumatoid arthritis patients. J Autoimmun (1995) 8(4):587–600. doi: 10.1016/0896-8411(95)90010-1
223. Miossec P, Naviliat M, Dupuy d'Angeac A, Sany J, Banchereau J. Low levels of interleukin-4 and high levels of transforming growth factor beta in rheumatoid synovitis. Arthritis Rheum (1990) 33(8):1180–7. doi: 10.1002/art.1780330819
224. Finnegan A, Mikecz K, Tao P, Glant TT. Proteoglycan (aggrecan)-induced arthritis in BALB/c mice is a Th1-type disease regulated by Th2 cytokines. J Immunol (1999) 163(10):5383–90.
225. Joosten LA, Lubberts E, Durez P, Helsen MM, Jacobs MJ, Goldman M, et al. Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis. protective effect of interleukin-4 and interleukin-10 treatment on cartilage destruction. Arthritis Rheum (1997) 40(2):249–60. doi: 10.1002/art.1780400209
226. Park HK, Kim SK, Kweon HY, Lee KG, Arasu MV, Kim YO. Promoter polymorphism (-590, T/C) of interleukin 4 (IL4) gene is associated with rheumatoid arthritis: An updated meta-analysis. Saudi J Biol Sci (2017) 24(2):444–9. doi: 10.1016/j.sjbs.2016.01.013
227. Yang WC, Hwang YS, Chen YY, Liu CL, Shen CN, Hong WH, et al. Interleukin-4 supports the suppressive immune responses elicited by regulatory T cells. Front Immunol (2017) 8:1508. doi: 10.3389/fimmu.2017.01508
228. Churchman SM, El-Jawhari JJ, Burska AN, Parmar R, Goeb V, Conaghan PG, et al. Modulation of peripheral T-cell function by interleukin-7 in rheumatoid arthritis. Arthritis Res Ther (2014) 16(6):511. doi: 10.1186/s13075-014-0511-3
229. Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature (2006) 442(7106):997–1002. doi: 10.1038/nature05010
230. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta 'reprograms' the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol (2008) 9(12):1341–6. doi: 10.1038/ni.1659
231. Chowdhury K, Kumar U, Das S, Chaudhuri J, Kumar P, Kanjilal M, et al. Synovial IL-9 facilitates neutrophil survival, function and differentiation of Th17 cells in rheumatoid arthritis. Arthritis Res Ther (2018) 20(1):18. doi: 10.1186/s13075-017-1505-8
232. Ciccia F, Guggino G, Rizzo A, Manzo A, Vitolo B, La Manna MP, et al. Potential involvement of IL-9 and Th9 cells in the pathogenesis of rheumatoid arthritis. Rheumatol (Oxford) (2015) 54(12):2264–72. doi: 10.1093/rheumatology/kev252
233. Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat Med (2017) 23(8):938–44. doi: 10.1038/nm.4373
234. Kara EE, Comerford I, Bastow CR, Fenix KA, Litchfield W, Handel TM, et al. Distinct chemokine receptor axes regulate Th9 cell trafficking to allergic and autoimmune inflammatory sites. J Immunol (2013) 191(3):1110–7. doi: 10.4049/jimmunol.1203089
235. Mottonen M, Isomaki P, Saario R, Toivanen P, Punnonen J, Lassila O. Interleukin-10 inhibits the capacity of synovial macrophages to function as antigen-presenting cells. Br J Rheumatol (1998) 37(11):1207–14. doi: 10.1093/rheumatology/37.11.1207
236. Apparailly F, Verwaerde C, Jacquet C, Auriault C, Sany J, Jorgensen C. Adenovirus-mediated transfer of viral IL-10 gene inhibits murine collagen-induced arthritis. J Immunol (1998) 160(11):5213–20.
237. Hajeer AH, Lazarus M, Turner D, Mageed RA, Vencovsky J, Sinnott P, et al. IL-10 gene promoter polymorphisms in rheumatoid arthritis: SHORT REPORT. Scandinavian J Rheumatol (1998) 27(2):142–5. doi: 10.1080/030097498441029
238. Mielle J, Audo R, Hahne M, Macia L, Combe B, Morel J, et al. IL-10 producing b cells ability to induce regulatory T cells is maintained in rheumatoid arthritis. Front Immunol (2018) 9:961. doi: 10.3389/fimmu.2018.00961
239. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T Cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med (1996) 183(6):2593–603. doi: 10.1084/jem.183.6.2593
240. Amin A, Sheikh N, Mukhtar M, Saleem T, Akhtar T, Fatima N, et al. Association of interleukin-17 gene polymorphisms with the onset of rheumatoid arthritis. Immunobiology (2021) 226(1):152045. doi: 10.1016/j.imbio.2020.152045
241. Gu AD, Wang Y, Lin L, Zhang SS, Wan YY. Requirements of transcription factor smad-dependent and -independent TGF-β signaling to control discrete T-cell functions. Proc Natl Acad Sci USA (2012) 109(3):905–10. doi: 10.1073/pnas.1108352109
242. Kunwar S, Dahal K, Sharma S. Anti-IL-17 therapy in treatment of rheumatoid arthritis: a systematic literature review and meta-analysis of randomized controlled trials. Rheumatol Int (2016) 36(8):1065–75. doi: 10.1007/s00296-016-3480-9
243. Genovese MC, Weinblatt ME, Aelion JA, Mansikka HT, Peloso PM, Chen K, et al. ABT-122, a bispecific dual variable domain immunoglobulin targeting tumor necrosis factor and interleukin-17A, in patients with rheumatoid arthritis with an inadequate response to methotrexate: A randomized, double-blind study. Arthritis Rheumatol (2018) 70(11):1710–20. doi: 10.1002/art.40580
244. Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity (2002) 16(6):779–90. doi: 10.1016/S1074-7613(02)00324-2
245. Li Y, Yuan L, Jiang S, Liu S, Xia L, Shen H, et al. Interleukin-35 stimulates tumor necrosis factor-alpha activated osteoblasts differentiation through wnt/beta-catenin signaling pathway in rheumatoid arthritis. Int Immunopharmacol (2019) 75:105810. doi: 10.1016/j.intimp.2019.105810
246. Zhang X, Zhang X, Zhuang L, Xu C, Li T, Zhang G, et al. Decreased regulatory T-cell frequency and interleukin-35 levels in patients with rheumatoid arthritis. Exp Ther Med (2018) 16(6):5366–72. doi: 10.3892/etm.2018.6885
247. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci (2012) 8(9):1237–47. doi: 10.7150/ijbs.4989
248. Chen J, Zhang A, Yang Y, Si Y, Hao D. Assessment of interleukin 6 gene polymorphisms with rheumatoid arthritis. Gene (2021) 765:145070. doi: 10.1016/j.gene.2020.145070
249. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi: 10.1038/nature04753
250. Henes J, Schmit MA, Morote-Garcia JC, Mirakaj V, Kohler D, Glover L, et al. Inflammation-associated repression of vasodilator-stimulated phosphoprotein (VASP) reduces alveolar-capillary barrier function during acute lung injury. FASEB J (2009) 23(12):4244–55. doi: 10.1096/fj.09-138693
251. Yan S, Golumba-Nagy V, Kotschenreuther K, Thiele J, Refaian N, Shuya D, et al. Membrane-bound IL-6R is upregulated on Th17 cells and inhibits treg cell migration by regulating post-translational modification of VASP in autoimmune arthritis. Cell Mol Life Sci (2021) 79(1):3. doi: 10.1007/s00018-021-04076-2
252. Hansen SD, Mullins RD. VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J Cell Biol (2010) 191(3):571–84. doi: 10.1083/jcb.201003014
253. Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, et al. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell (2002) 109(4):509–21. doi: 10.1016/S0092-8674(02)00731-6
254. Gau D, Veon W, Shroff SG, Roy P. The VASP-profilin1 (Pfn1) interaction is critical for efficient cell migration and is regulated by cell-substrate adhesion in a PKA-dependent manner. J Biol Chem (2019) 294(17):6972–85. doi: 10.1074/jbc.RA118.005255
255. Samuel S, Zhang K, Tang YD, Gerdes AM, Carrillo-Sepulveda MA. Triiodothyronine potentiates vasorelaxation via PKG/VASP signaling in vascular smooth muscle cells. Cell Physiol Biochem (2017) 41(5):1894–904. doi: 10.1159/000471938
256. Döppler HR, Bastea LI, Lewis-Tuffin LJ, Anastasiadis PZ, Storz P. Protein kinase D1-mediated phosphorylations regulate vasodilator-stimulated phosphoprotein (VASP) localization and cell migration. J Biol Chem (2013) 288(34):24382–93. doi: 10.1074/jbc.M113.474676
257. Lambrechts A, Kwiatkowski AV, Lanier LM, Bear JE, Vandekerckhove J, Ampe C, et al. cAMP-dependent protein kinase phosphorylation of EVL, a Mena/VASP relative, regulates its interaction with actin and SH3 domains. J Biol Chem (2000) 275(46):36143–51. doi: 10.1074/jbc.M006274200
258. Benz PM, Blume C, Seifert S, Wilhelm S, Waschke J, Schuh K, et al. Differential VASP phosphorylation controls remodeling of the actin cytoskeleton. J Cell Sci (2009) 122(Pt 21):3954–65. doi: 10.1242/jcs.044537
259. Estin ML, Thompson SB, Traxinger B, Fisher MH, Friedman RS, Jacobelli J. Ena/VASP proteins regulate activated T-cell trafficking by promoting diapedesis during transendothelial migration. Proc Natl Acad Sci USA (2017) 114(14):E2901–10. doi: 10.1073/pnas.1701886114
260. Barone M, Muller M, Chiha S, Ren J, Albat D, Soicke A, et al. Designed nanomolar small-molecule inhibitors of Ena/VASP EVH1 interaction impair invasion and extravasation of breast cancer cells. Proc Natl Acad Sci USA (2020) 117(47):29684–90. doi: 10.1073/pnas.2007213117
261. Doherty D, Chudley AE, Coghlan G, Ishak GE, Innes AM, Lemire EG, et al. GPSM2 mutations cause the brain malformations and hearing loss in chudley-McCullough syndrome. Am J Hum Genet (2012) 90(6):1088–93. doi: 10.1016/j.ajhg.2012.04.008
262. He XQ, Zhang YF, Yu JJ, Gan YY, Han NN, Zhang MX, et al. High expression of G-protein signaling modulator 2 in hepatocellular carcinoma facilitates tumor growth and metastasis by activating the PI3K/AKT signaling pathway. Tumour Biol (2017) 39(3):1010428317695971. doi: 10.1177/1010428317695971
263. Dang SC, Qian XB, Jin W, Cui L, Chen JX, Gu M. G-Protein-signaling modulator 2 expression and role in a CD133(+) pancreatic cancer stem cell subset. Onco Targets Ther (2019) 12:785–94. doi: 10.2147/OTT.S187670
264. Meyer A, Yan S, Golumba-Nagy V, Esser RL, Barbarino V, Blakemore SJ, et al. Kinase activity profiling reveals contribution of G-protein signaling modulator 2 deficiency to impaired regulatory T cell migration in rheumatoid arthritis. J Autoimmun (2021) 124:102726. doi: 10.1016/j.jaut.2021.102726
Keywords: Treg - regulatory T cells, rheumatoid arthritis, T cell migration, T cell homeostasis, collagen-induced arthritis (CIA)
Citation: Kotschenreuther K, Yan S and Kofler DM (2022) Migration and homeostasis of regulatory T cells in rheumatoid arthritis. Front. Immunol. 13:947636. doi: 10.3389/fimmu.2022.947636
Received: 19 May 2022; Accepted: 20 July 2022;
Published: 09 August 2022.
Edited by:
Tsutomu Takeuchi, Keio University School of Medicine, JapanReviewed by:
Iain Comerford, University of Adelaide, AustraliaShingo Nakayamada, University of Occupational and Environmental Health Japan, Japan
Copyright © 2022 Kotschenreuther, Yan and Kofler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David M. Kofler, ZGF2aWQua29mbGVyQHVrLWtvZWxuLmRl
†These authors have contributed equally to this work