 Huayu Guan
Huayu Guan Xiang Zhang
Xiang Zhang Ming Kuang
Ming Kuang Jun Yu
Jun Yu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 August 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.946628
This article is part of the Research Topic Hepatic Immune Response underlying Liver Cirrhosis and Portal Hypertension View all 19 articles
In healthy settings, the gut–liver axis allows host–microbiota communications and mediates immune homeostasis through bidirectional regulation. Meanwhile, in diseases, gut dysbiosis, combined with an impaired intestinal barrier, introduces pathogens and their toxic metabolites into the system, causing massive immune alternations in the liver and other extrahepatic organs. Accumulating evidence suggests that these immune changes are associated with the progression of many liver diseases, especially hepatic cirrhosis. Pathogen-associated molecular patterns that originated from gut microbes directly stimulate hepatocytes and liver immune cells through different pattern recognition receptors, a process further facilitated by damage-associated molecular patterns released from injured hepatocytes. Hepatic stellate cells, along with other immune cells, contribute to this proinflammatory and profibrogenic transformation. Moreover, cirrhosis-associated immune dysfunction, an imbalanced immune status characterized by systemic inflammation and immune deficiency, is linked to gut dysbiosis. Though the systemic inflammation hypothesis starts to link gut dysbiosis to decompensated cirrhosis from a clinical perspective, a clearer demonstration is still needed for the role of the gut–liver–immune axis in cirrhosis progression. This review discusses the different immune states of the gut–liver axis in both healthy and cirrhotic settings and, more importantly, summarizes the current evidence about how microbiota-derived immune remodeling contributes to the progression of hepatic cirrhosis via the gut–liver axis.
The gut–liver axis is the bidirectional communication between the intestine, its microbiota, and the liver. While receiving nutrient-rich blood from the gut through portal veins, the liver also directly contacts translocating bacteria and their various components and metabolites. Fortunately, in healthy settings, an intact and multilayered intestinal barrier restricts such direct host–microbiota contact and defends against excessive bacterial translocation. Another important interplay between the gut and the liver relies on bile acid metabolism. Synthesized in the liver and excreted into the gut along with other bioactive substances, primary bile acids are then converted into secondary bile acids by certain commensal microbes, especially Clostridium cluster XIV (1). About 95% of the bile acids are reabsorbed by the intestine, transported back to the liver, and secreted into the gut again, establishing a metabolic cycle called enterohepatic circulation. Within this cycle, bile acids modulate the composition of gut microbiota via selective pressure and, simultaneously, influence the metabolism and functionality of the liver. In addition to bile acids, many other host–microbiota–derived metabolites also take part in the bidirectional regulation utilizing similar routes, such as free fatty acids, choline, and ethanol derivatives (2). Through these complex interregulations, commensal bacteria and their metabolites help to maintain the metabolic and immune homeostasis of the liver. For instance, Akkermansia muciniphila, a Gram-negative anaerobic bacterium colonizing the mucus layer of the intestine, helps to alleviate intestinal inflammation and mitigate alcoholic and nonalcoholic liver damage (3, 4). Bile acids produced by certain bacteria can activate intestinal farnesoid X receptor (FXR) and thus promote fatty acid oxidation while reducing lipogenesis and lipid absorption in the liver, ameliorating hepatic inflammation and steatosis (5, 6). Another bacteria-derived metabolite, butyrate, helps to maintain gut barrier integrity and alleviate ethanol-induced liver injury (7). In short, the bidirectional communication between the host and the microbiota is essential not just to the health of the gut but also to that of the liver and probably the whole system.
Liver cirrhosis is a huge burden on public health worldwide. About one million deaths around the world are attributable to liver cirrhosis annually, making it the 11th most common cause of death and the third leading cause of death in people aged 45–64 years (8). According to WHO’s Global Burden of Diseases studies for 2019, liver cirrhosis was responsible for 560.4 age-standardized disability-adjusted life-years (DALYs) per 100,000 population globally, while liver cancer causes only 151.1 DALYs (9). The etiology of cirrhosis is rather complicated since various chronic liver diseases can lead to shrinkage of the liver parenchyma and overproduction of scar tissue. The most dominant causes of liver cirrhosis are hepatitis B and C, alcoholic liver disease (ALD), and nonalcoholic fatty liver disease (NAFLD) (8, 9). Despite the different pathological settings of these precirrhotic diseases, the trajectory of cirrhosis comes down to similar complications that mark the transition from compensated to decompensated cirrhosis. In a traditional perspective, variceal bleeding, ascites, and hepatic encephalopathy are the three major complications of cirrhosis, which result from portal hypertension, arterial vasodilation, and hyperammonemia, respectively. However, giving each complication or organ failure an independent pathophysiological mechanism does not seem to explain the complexity of decompensated cirrhosis well enough. Recent studies suggest that systemic inflammation and organ immunopathology are additional contributors to organ dysfunction, further verifying the notion that cirrhosis is a systemic disease (10, 11). For patients with acute decompensation of cirrhosis, the severity of systemic inflammation increases in parallel with the disease progression and the number of organ failures (10, 12, 13). Moreover, the significant correlations between systemic inflammation and portal hypertension, ascites, and hepatic encephalopathy indicate that systemic inflammation is the common pathophysiological mechanism for different complications of decompensated cirrhosis (13). Interestingly, gut microbes have long been considered the major source of systemic inflammation in cirrhosis (11, 13). Evidence indicates that gut dysbiosis is associated with the pathogenesis and progression of many precirrhotic diseases such as viral hepatitis, NAFLD, and ALD (14–19). Hepatic cirrhosis, as the advanced stage of these liver diseases, is linked to the altered composition and reduced diversity of gut microbiota despite etiology. Moreover, patients with cirrhotic conditions are prone to an impaired intestine barrier, pathological bacterial translocation, and systematic inflammation (20). Pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS) can stimulate immune cells and cytokine secretion in a Toll-like receptor (TLR)-NF-κB-dependent way, generating a proinflammatory and profibrogenic immune environment. Additionally, a bacterial infection is now regarded as the fourth major complication of decompensated cirrhosis because of its astonishingly high prevalence (21). The most common infection for cirrhosis patients, spontaneous bacterial peritonitis (SBP), is a perfect demonstration of how bacterial translocation constantly elicits inflammation and alters the host immunity (22). Therefore, it is tempting to speculate that gut microbiota contribute to the progression of hepatic cirrhosis through immune remodeling in a dysbiotic setting. Since bacteria are the most well-studied members of the gut microbiota and probably play a central role in microbiota–host interaction, this review will focus on gut bacteria but not viruses, fungi, archaea, or other microbes.
The human intestine contains 1014 microbes, over 99% of which are bacteria. With such a huge quantity of microorganisms living inside the intestinal lumen, our system needs a strong defense to protect us from the excessive input of viable bacteria and their toxic metabolites. The multilayered intestinal barrier may serve as the first line of defense. The inner surface of the intestine is covered with a mucus layer that physically separates the bacteria from the intestine wall and delivers tolerogenic signals (23, 24). Beneath the mucus layer lies the intestinal epithelium, which consists of enterocytes, goblet cells, tuft cells, Paneth cells, M cells, and different immune cells (20). Adjacent epithelial cells form junctional complexes between each other to limit paracellular trafficking of intestinal contents. These complexes consist of desmosomes, adherens junctions (AJs), and tight junctions (TJs) (25). Paneth cells secrete α-defensins, islet-derived protein III-gamma (RegIIIγ), and lysozyme to defend against pathogens (26). Intraepithelial lymphocytes, including αβT cells and γδT cells, are activated by various cellular or cytokine signals to battle bacterial infection (20). Mononuclear phagocytes such as dendritic cells (DCs), with their processes sticking into the intestinal lumen, take part in both antibacterial immunity and oral tolerance (27). In the lamina propria, plasma cells secrete sIgA into the mucus layer to reinforce the frontline defense, while Th17 cells help to strengthen the tight junctions and promote epithelial regeneration. Microbial signals sensed by DCs or group 3 innate lymphoid cells (ILC3) trigger the secretion of IL-17 and IL-22, which promote the release of antibacterial peptides, mucin, and sIgA by other cells (28, 29). The last and most critical defense of the intestinal barrier, the gut–vascular barrier, is composed of endothelial cells, pericytes, and enteric glial cells. This gut–vascular unit is also reinforced by junctional complexes, allowing antigens from food or commensal bacteria to pass for tolerance induction but not bacterial translocation (20, 23).
Interestingly, this host–microbiota regulation is reciprocal, with recent studies proving that the intestinal barrier can be modulated by the gut microbiota. For instance, adhesion of certain microbes like segmented filamentous bacteria to the intestinal epithelial cells triggers robust induction of Th17 cells (30). When bacteria penetrate the inner layer of mucus, a group of sentinel goblet cells can nonspecifically sense microbial molecules and secrete more Muc2 mucin to expel the pathogens by activating the NLRP6 inflammasome downstream of ROS synthesis (31). CX3CR1+ macrophages are localized around the intestinal lamina propria vasculature, forming an interdigitating network to defend against pathogens. The conversion of such macrophages from CCR2hiCX3CR1+ monocytes is mediated by the microbiota in a Nr4a1-dependent manner (32). Furthermore, innate lymphoid cells in the lamina propria can maintain long-term antibacterial activity after being trained with bacteria (33). Bacteria-derived metabolites also contribute to the homeostasis of the gut. Indole is a tryptophan derivative produced by commensal bacteria that regulate the IL-22 expression of ILC3 via aryl hydrocarbon receptor (AHR). IL-22, in turn, modulates the secretion of antibacterial RegIIIγ by Paneth cells, making indole a favorable signal to gut homeostasis (34). Other studies suggest that indole reinforces the gut barrier integrity by increasing TJ resistance (35). To conclude, the host defense does not develop all by itself but rather depends on the constant stimulations from commensal microbes. In turn, active surveillance by intestinal immunity keeps gut microbes in line. Such delicate mechanisms of bilateral regulation guarantee a balance between tolerance for autochthonous microbes and antibacterial activities against pathogens in the gut (Figure 1A).
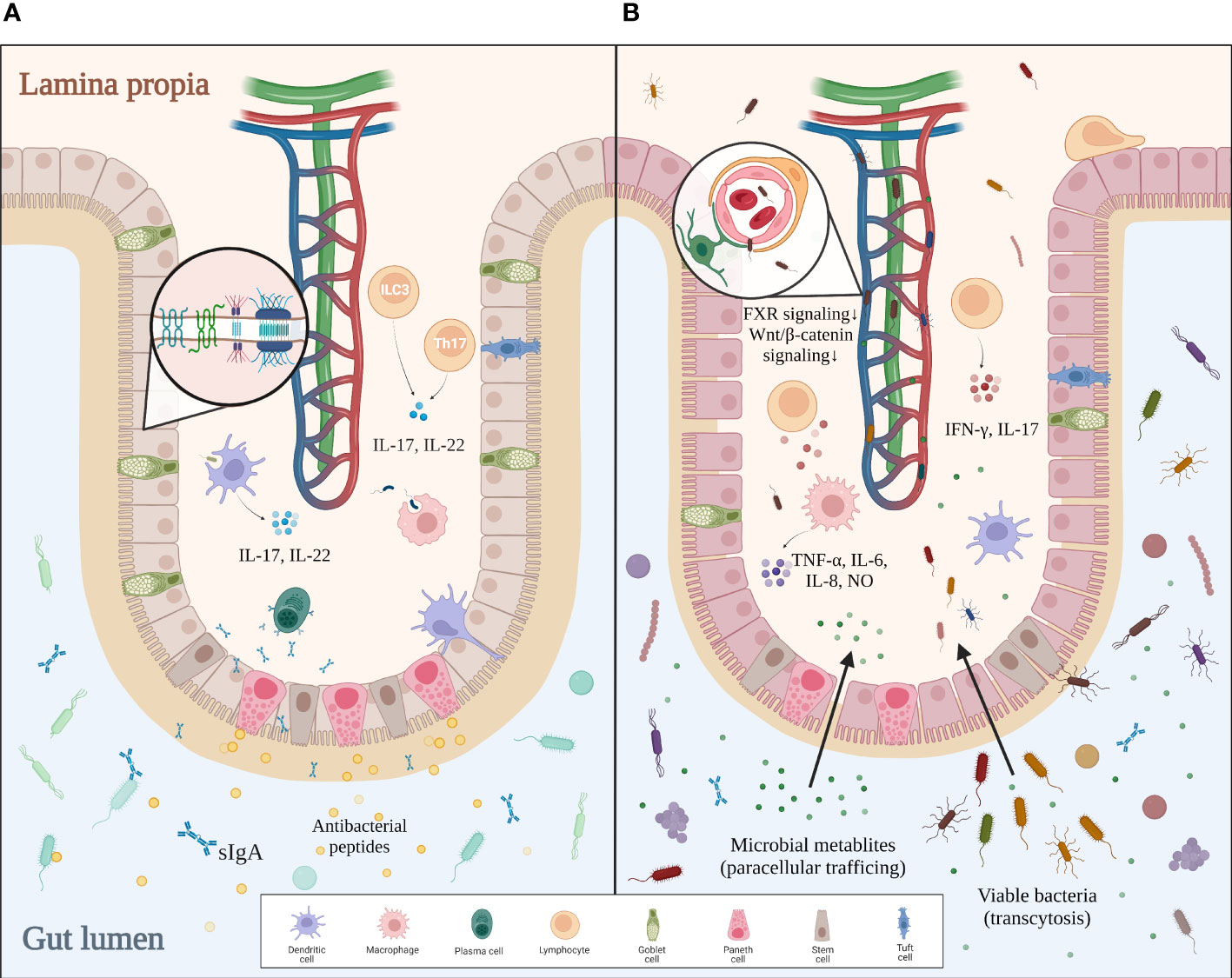
Figure 1 Comparison between intact and impaired gut barriers. (A) In healthy settings, the mucus layer serves as the first defense of the intestinal barrier against intraluminal bacteria. Epithelial cells tightly jointed together by junctional complexes limit the translocation of bacteria. Immune cells within the lamina propia not only actively remove invading pathogens by phagocytosis but also strengthen the gut barrier by secreting certain cytokines. Altogether, an intact gut barrier prevents pathological bacterial translocation. (B) In gut dysbiosis, bacterial overgrowth and disproportion can be found in the intestinal lumen. The mucus layer becomes thinner and looser, allowing pathogens to reach the epithelium. Disrupted junctional complexes and impaired gut–vascular barrier further promote pathological translocation. Additionally, dysregulated intestinal immunity aggravates inflammation and enterocyte injury, which eventually leads to a leaky gut.
The liver faces bacterial challenges constantly due to the unique anatomical and hemodynamic features of the portal system. If potent antibacterial immunity were induced each time the bacterial antigens reached the liver, there would be relentless inflammation and severe collateral damages to the system. Therefore, it is essential that liver parenchymal cells and other liver-resident cells form a fine-tuned immune network together and respond to these challenges in a well-balanced way.
Upon stimulation by gut-derived microbes, hepatocytes not only can secrete acute-phase proteins, complement proteins, and other bioactive substances to battle bacterial infection but also can play an important role in immune surveillance via expressing MHC I/II and costimulatory molecules. Liver sinusoidal endothelial cells (LSECs) allow the interaction between gut-derived molecules and the underlying hepatocytes and nonparenchymal cells via the special fenestrae (36). In addition to recruiting monocytes and lymphocytes in an ICAM-, VCAM-, or VAP-dependent manner, LSECs actively regulate the periportal distributions of myeloid and lymphoid cells via MYD88 signaling induced by gut commensal bacteria, resulting in more efficient prevention of systemic bacterial dissemination (37). Kupffer cells (KCs), a group of liver-resident macrophages that patrol the sinusoidal lumen, are important immune sentinels to detect, capture, and present bacterial antigens (38). Furthermore, KCs also regulate iron metabolism and prevent accumulative toxicity by removing damaged RBCs and hemoglobin from the bloodstream (39). Hepatic stellate cells (HSCs) crawl around the liver vasculature and store lipids and vitamin A in basal conditions. Whereas in the inflamed liver, HSCs transdifferentiate into fibrinogenic and immunomodulatory cells (40). Even though these different kinds of cells all express MHC and other antigen-presenting molecules, they are poor activators for T cells under most circumstances. Constant exposure to low-level LPS induces a refractory response in APCs, a phenomenon called endotoxin tolerance (41) and downregulation of costimulatory signals and upregulation of coinhibitory molecules like PD-L1 (38, 42). These alternations promote the development of regulatory T cells and cause incomplete activation, clonal anergy, and even premature death to the CD8+ T cells. In addition to direct contact, these antigen-presenting cells can also secrete inhibitory cytokines such as IL-10 and TGF-β, which dampen the activation and functions of CD4+ T cells and CD8+ T cells. Other studies suggest that Kupffer cells and HSCs prime the naïve CD4+ T cells toward Treg phenotype via secretion of prostaglandins (PGE2) and retinoic acid, respectively (38, 43). Furthermore, low-level LPS and proinflammatory cytokines like IFN-γ can induce the expression of indoleamine-2,3-dioxygenase (IDO) in Kupffer cells and DCs (42). This enzyme produces an immunosuppressive metabolite called kynurenine and contributes to the suppression of T-cell functions.
To conclude, gut-derived signals help to direct liver immunity to a tolerogenic phenotype that prevents immune overreaction in basal conditions. However, such immune tolerance does not exist without limitations. The liver still needs to efficiently mobilize immune cells and initiate an antibacterial response when a dangerous infection occurs. In fact, different receptors expressed by liver cells can distinguish antigens from commensal flora and those from pathogens. When excessive or dangerous signals are detected, APCs, including hepatocytes, LSECs, DCs, and Kupffer cells, can recruit neutrophils, natural killer cells, and lymphocytes to eliminate pathogens. In short, with the help of gut-derived signals, liver parenchymal and nonparenchymal cells construct a harmoniously coordinated network to maintain immune homeostasis (Figure 2A).
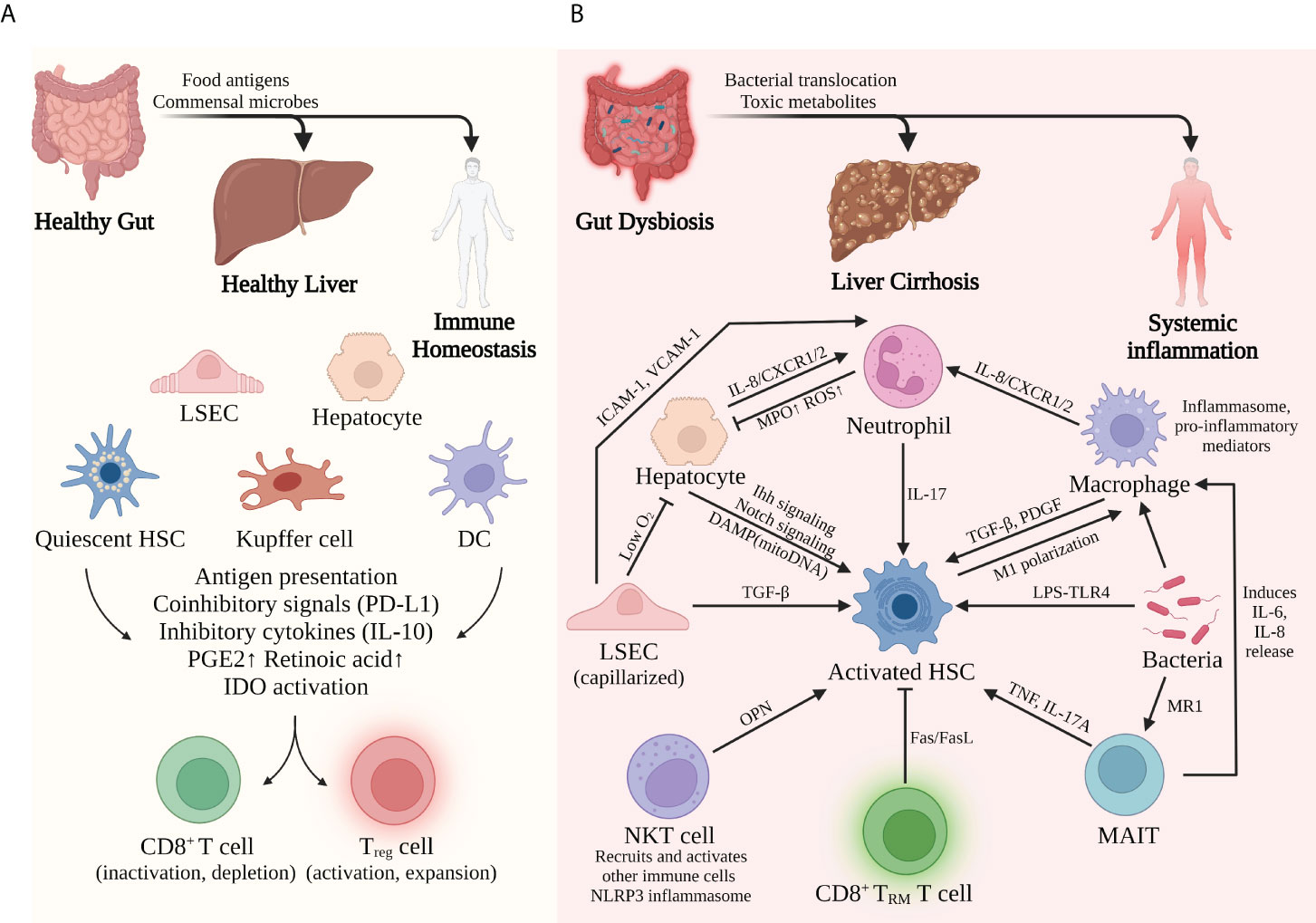
Figure 2 Liver immune environment in tolerogenic and immunogenic conditions. (A) In basal conditions, commensal bacteria and food antigens from a healthy gut help to maintain liver immune homeostasis. Constant exposure to LPS induces tolerance of APCs and therefore inactivation of CD8+ T cells but activation of Treg cells. (B) By contrast, bacterial dysbiosis and impaired gut barrier promote the pathological translocation of viable bacteria and their products, causing massive inflammation in the liver and the whole system. The complicated interplay among different immune cells facilitates the proinflammatory and profibrogenic transformation of the liver. Activation of hepatic stellate cells seems to be the common mechanism for this transformation.
A diverse and relatively stable gut microbiome is essential to maintain the immune homeostasis of the host. Once initiated by certain insulting etiologies, gut dysbiosis slowly develops at the early stage of liver disease and progresses as the disease progresses. Researchers have confirmed that gut dysbiosis is closely associated with the pathogenesis and progression of hepatic cirrhosis. To begin with, reduced overall gene richness was found in the stool samples of cirrhotic patients (44, 45). Furthermore, a decreased α-diversity of gut microbiota is another feature of cirrhotic patients (46–48). β-Diversity was also significantly altered in individuals with NAFLD-cirrhosis (48, 49). These findings all suggest a less diverse and less stable gut microbiome in cirrhotic patients. Meanwhile, the composition of the gut microbiota is also changed adversely. Firmicutes and Bacteroidetes are the most dominant phyla of the human gut microbiota, followed by Proteobacteria and Actinobacteria with much smaller abundances. In cirrhotic settings, Proteobacteria and Fusobacteria are enriched while Bacteroidetes are depleted (44, 49–51). At the family level, potentially beneficial autochthonous taxa like Lachnospiraceae, Ruminococcaceae, and Clostridiales XIV are reduced while potentially pathogenic taxa including Staphylococcaceae, Enterobacteriaceae, and Enterococcaceae are increased (52). At the genus level, cirrhotic patients display higher abundances of buccal microbes including Veillonella, Streptococcus, and Prevotella, indicating that oral commensals may invade the intestine in cirrhotic conditions (44–48). At the species level, potential pathogens like Ruminococcus gnavus, Veillonella parvula, and Streptococcus parasanguinis are enriched, while beneficial commensals like Eubacterium rectale and Faecalibacterium prausnitzii are depleted (46, 48). Another marked alternation of the gut microbiota in cirrhotic patients is the small intestinal bacterial overgrowth (SIBO), partly due to decreased bowel motility, delayed transit time, and use of acid inhibitors and antibiotics (53, 54). Instead of being exclusive to cirrhosis, SIBO appears to be prevalent in different precirrhotic diseases, indicating that it develops as the disease progresses (55, 56) and correlates with disease severity (57). Even though alcohol consumption, a high-fat diet, virus infection, autoimmunity, and other cirrhosis-related factors may all affect the gut microbiota in certain ways, cirrhotic patients share similar microbiota profiles despite etiology. This might suggest that gut dysbiosis not only has something to do with the unique pathophysiological changes of cirrhosis but also partly results from some common medical interventions for cirrhosis. One possible explanation is that impaired hepatic function changes the intestinal environment (e.g., reduced bile flow and complement synthesis), which is then aggravated by cirrhosis complications (e.g., impaired gut motility due to ascites) and medical interventions (e.g., proton-pump inhibitors for variceal hemorrhage prevention). Further investigation is needed to figure out how gut dysbiosis initiates in cirrhosis patients.
Bacterial translocation (BT) is defined as the translocation of bacteria and/or bacterial metabolites from the gut lumen to mesenteric lymph nodes or the portal bloodstream. Though BT exists in basal conditions and helps to build tolerance for commensal microbes, its quantity markedly increases in pathological settings, eliciting a proinflammatory response and even systemic infection. As discussed before, an intact intestinal barrier is essential to prevent pathological bacterial translocation and to maintain immune homeostasis. This line of defense no longer holds in cirrhotic settings, with evidence showing the structural and functional breakdown of the gut barrier in cirrhosis (58, 59). Firstly, the mucus layer becomes thinner and easier for bacteria to colonize, even in the relatively denser and supposedly sterile inner layer. Other structural distortions include enlarged interepithelial space, shortening and widening of microvilli, submucosal edema, and disorganization of interepithelial TJs (20, 53, 58). In fact, these structural changes are related to decreased expression of tight-junction proteins occludin and claudin-1 (60, 61). Also, evidence has proven that the impairment of epithelial TJs is related to dysregulated fermentation of the gut microbiota. Ethanol and its toxic derivative acetaldehyde can damage the TJs directly and increase gut permeability in ALD (61, 62). The reduction of butyrate and other protective metabolites also contributes to the damaged barrier (7, 63). It is thought that weakened TJs promote the paracellular trafficking of bacterial metabolites, while translocation of viable bacteria likely depends on transcytosis (27, 54, 64). Although research on the detailed mechanisms of bacterial transcytosis is still lacking, many lines of evidence suggest that intestinal immune dysregulation induced by dysbiosis contributes to the bacterial penetration of the gut barrier. Cirrhotic rat models exhibit an inflammatory pattern of immune dysregulation in intraepithelial lymphocyte (IEL) and lamina propria lymphocyte (LPL), with an increase in activated lymphocytes and IFN-γ and IL-17 production (58, 65). Furthermore, dysbiosis-induced inflammation impairs the gut barrier via TNF-α/TNFR1 signaling mediated by monocytes and macrophages (62). Another study on cirrhosis patients also reveals that activated macrophages secrete NO, IL-6, and IL-8 that undermine the gut barrier, most probably under bacterial stimulation (59). By contrast, reduced synthesis and release of defensin, RegIIIβ/γ, and sIgA suggest impaired antibacterial functions of Paneth cells, neutrophils, B cells, and other epithelial cells (66). These findings are in line with a report of B-cell dysfunction in cirrhosis patients (67). Intestinal DCs also show a less-activated phenotype with decreased TNF-α production, deficient phagocytosis, and impaired migration in cirrhotic rats with excessive bacterial translocation (68). As for the final defense of the intestinal barrier, the gut vascular barrier is also damaged by certain pathogens such as Salmonella typhimurium via dampened β-catenin-dependent signaling (69). In dysbiotic conditions, reduced FXR signaling due to dysregulation of bile acid metabolism also impairs the integrity of GVB (70). To conclude, gut dysbiosis leads to the accumulation of invasive pathogens and toxic metabolites, which directly impair the gut barrier and cause intestinal inflammation. Local inflammation not only damages enterocytes but also weakens the antibacterial ability of the gut barrier. Altogether, these changes facilitate pathological bacterial translocation (Figure 1B).
Translocated bacteria and gut-derived metabolites can directly interact with host cells. One of the most well-established mechanisms involves a group of receptors named pattern recognition receptors (PRRs), which are widely expressed on the surface of various hepatic and intestinal cells (38). PRRs include TLRs, nucleotide-binding oligomerization domain-like receptors (NLRs), C-type lectin receptors (CLRs), retinoic acid-inducible gene I-like receptors (RLRs), and absent in melanoma-2 (AIM2)-like receptors (ALRs) (71). Different PRRs can recognize different conserved molecular patterns of microbes (PAMPs) or damaged cells [damage-associated molecular patterns (DAMPs)]. For instance, a cell wall component of Gram negative bacteria, is a typical PAMP, while mitochondrial DNA released from injured cells belongs to DAMPs. Among these receptor–ligand interactions, TLR4-LPS is one of the most thoroughly studied and relevant pairs in cirrhosis. Upon recognition of LPS, TLR4 initiates downstream activation in both a MyD88-dependent manner and a TRIF-dependent manner. For MyD88-dependent signaling, TLR4-LPS interaction activates the MyD88-NF-κB pathway and leads to the production of proinflammatory cytokines such as TNF, IL-1, IL-6, and chemokines. For TRIF-dependent signaling (or MyD88-independent signaling), the TRIF-TBK1-IRF-3 axis is activated to secrete type I IFN (72). In short, interactions between PRRs and PAMPs/DAMPs promote the clearance of pathogens or damaged cells, thus causing inflammation-related damages in the gut and the liver in a cirrhotic setting.
In acute or chronic liver injury, especially liver cirrhosis, the delicate balance between tolerogenic and immunogenic responses is broken. Detrimental effects caused by an impaired gut barrier, bacterial translocation, alcohol consumption, and an unhealthy diet all contribute to the immune malfunction of the liver through the gut–liver axis. Chronic liver diseases such as ALD and NAFLD not only directly affect liver metabolism and immunity but also indirectly impaired liver function via a dysregulated intestinal barrier and gut dysbiosis. In response to these pathogenic conditions, hepatocytes and liver-resident immune cells may lose their normal functions and transform into proinflammatory, profibrogenic phenotypes that facilitate the progression of cirrhosis (Figure 2B).
Hepatic stellate cells (HSCs) are the primary precursors for myofibroblasts during liver fibrosis (40). Gut-derived LPS can stimulate TLR4 of quiescent HSCs and activate these cells in a MyD88-NF-κB-dependent manner, thus causing profibrogenic transformation and accelerating liver fibrosis (73). Moreover, profibrogenic cytokines such as TGF-β and IL-17 are potent activators for HSCs and collagen production, which are excessively secreted by other hepatic cells under inflamed conditions (40, 74). A mouse model of liver fibrosis suggests that MyD88 signaling in activated HSCs promotes macrophage M1 polarization in a CXCL10/CXCR3-dependent manner, thus promoting liver fibrosis and inflammation (75).
Hepatocytes, the major parenchymal cells of the liver, play a crucial part in liver immune surveillance in health. However, recent studies suggest that hepatocytes might also promote liver cirrhosis in the presence of PAMPs and DAMPs. Activation of the TLR4-NF-κB pathway in hepatocytes promotes Jagged1/Notch signaling in the NASH mouse model, thus inducing OPN-dependent HSC activation and progressive fibrosis (76, 77). DAMPs released from injured hepatocyte mitochondria, mainly mtDNA, can directly activate HSCs and promote liver fibrosis. Such mito-DAMPs are increased in both mouse models and human patients with NASH and advanced fibrosis (78). In addition, TAZ is a transcription factor markedly elevated in the hepatocytes of human and murine NASH livers, which can initiate HSC activation in an Indian hedgehog (Ihh)-dependent manner and promote inflammation and fibrosis in NASH (79).
Neutrophils are recruited to the liver via the adhesion molecules ICAM-1 and VCAM-1 expressed by LSECs. These innate immune cells counteract bacterial infection mainly by phagocytosis and releasing lysozyme, ROS, elastase, and myeloperoxidase (MPO). Moreover, special extracellular fibrous structures named neutrophil extracellular traps (NETs) are formed to trap and eliminate pathogens (38). Studies suggest that neutrophils are involved in several cirrhosis-related conditions and the progression of cirrhosis (80). For example, nonalcoholic steatohepatitis (NASH) patients display significant neutrophil infiltration and activation in the liver. These cells secrete excessive MPO that directly kills hepatocytes, activates HSCs, and subsequently promotes liver fibrosis. Likewise, in cirrhosis, neutrophil functions are also disturbed by the dysfunctional gut–liver axis, leading to inflammation-related hepatocyte injury and IL-17-dependent HSC activation (81). Furthermore, intrahepatic neutrophils in patients with acute-on-chronic liver failure (ACLF) have higher expression of CXCR1 and CXCR2, receptors that are crucial for neutrophil recruitment, inflammatory mediator production (e.g., IL-8, IL-6, IL-23, CCL-20, and ROS), and contact-dependent cell death (82).
Hepatic macrophages can be divided into several subsets, among which Kupffer cells and monocyte-derived macrophages are essential players in maintaining immune homeostasis. In cirrhotic settings, especially ACLF, excessive bacterial translocation from the disrupted intestinal barrier exhausts the scavenging ability of macrophages and causes type I IFN-mediated IL-10 expression, resulting in a high risk of bacterial infection for cirrhosis patients. These macrophages also express a high level of MER tyrosine kinase (MERTK), which dampens the response to PAMPs and therefore antibacterial activity (83, 84). In addition to impaired bacterial clearance, macrophages also contribute to cirrhosis progression via promoting inflammation and fibrogenesis. Upon recognition of DAMPs and PAMPs, activated macrophages secrete proinflammatory mediators such as TNF, IL-1β, IL-6, IL-8, and ROS and promote the activation and survival of HSCs and myofibroblasts via TGF-β1 and PDGF (84). Macrophage-derived inflammasome resulting from bacterial translocation and tissue damage also contributes to the inflammatory injury to the liver (85). Such a multiprotein complex can be activated by PAMPs, DAMPs, ROS, cholesterol crystals, and other PRR ligands (86). Activated inflammasome initiates caspase-1-dependent production of proinflammatory cytokines like IL-1β and IL-18, which subsequently enhance liver inflammation, fibrosis, and damage in ALD, NASH, and other settings of liver injury (87).
Normally, mucosal-associated invariant T cells (MAIT) are anti-infection effectors that can be activated by bacterial metabolites from the vitamin B2 biosynthesis pathway in an MR1-dependent way (88). On the other hand, chronic liver inflammation exerts deleterious effects on the disease progression. A recent study suggests that MAITs are enriched in the fibrotic septa of cirrhotic patients, making direct contact with fibrinogenic cells. In vitro experiments show that MAIT can enhance the proinflammatory properties of myofibroblasts and monocyte-derived macrophages, further promoting liver fibrosis progression (89). Such MAIT–myofibroblast interaction can be achieved via the secretion of TNF and IL-17A by MAIT.
LSECs, along with Kupffer cells, constitute the most powerful scavenging system in the liver. LSECs maintain immune homeostasis by inducing tolerance to harmless antigens from food or commensal bacteria and maintaining quiescence of HSC (90). However, in acute or chronic liver injury, LSECs undergo several aberrant alternations in terms of morphology and function that promote a proinflammatory, profibrogenic, and proapoptotic phenotype. One of the major alternations in the capillarization of LSECs is a transformation characterized by the loss of fenestrae and the development of the basal membrane. Capillarized LSECs can no longer provide enough oxygen for the underlying hepatocytes, causing cell apoptosis, necrosis, and eventually the release of DAMPs (91). DAMPs and LSEC-derived signals such as TGF-β activate the profibrogenic HSCs and promote the generation of collagen and the progression of liver fibrosis. Dysfunctional LSECs also gain sinusoidal vasoconstriction ability due to reduced eNOS activity and increased vasoconstrictors, causing detrimental changes in liver hemodynamics that favor the development of portal hypertension.
Upon stimulation of DAMPs such as HMGB1, liver DCs can be switched from a tolerogenic, IL-10-producing phenotype to an immunogenic and TNF-producing phenotype. In addition, these cells activate NK cells and prime T cells within portal tract-associated lymphoid tissues (PALTs), further facilitating the progression of inflammation and tissue injury in a fibrotic setting.
NKT cells, a group of innate lymphoid cells that recognize endogenous or exogenous glycolipid antigens in a CD1d-dependent manner, are important regulators of liver immunity. When activated by different antigens or cytokines, type I NKT cells can activate NK, DCs, B, and T cells and recruit neutrophils and monocytes to the liver, propagating the liver inflammation. A recent study suggests that NKT promotes inflammatory response with the engagement of NLRP3 inflammasome in a mouse model of liver fibrosis (92). Moreover, type I NKT cells contribute to liver fibrosis via activation of the Hedgehog pathway and HSCs via secretion of osteopontin (OPN) (73, 93, 94).
To conclude, the massive release of PAMPs and DAMPs due to the impaired gut barrier and subsequent inflammation completely transforms the liver’s immune landscape. Tolerogenic properties in healthy conditions are replaced by immunogenic and fibrinogenic properties in cirrhotic settings, which feature the expansion of proinflammatory cells and cytokines and the deposition of the extracellular matrix. It is apparent that activation of HSCs is the common and central mechanism of cirrhosis progression induced by different cells (Figure 2B), which is not surprising due to the collagen-producing function of activated HSCs. Moreover, during this transforming process, innate immunity seems to play the leading part, while adaptive immunity shows some protective effects. In a murine NASH model, CD8+ tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells in a Fas/FasL-dependent manner (95). However, the interplay between innate and adaptive immunity in the development of cirrhosis still needs in-depth investigation.
Cirrhosis-associated immune dysfunction (CAID) is the complex manifestation of an impaired immune system in the cirrhotic setting, mainly characterized by systemic inflammation and immune deficiency. CAID includes two phenotypes: the low-grade systemic inflammatory phenotype found in patients with compensated cirrhosis or decompensation without organ failure and the high-grade systemic inflammatory phenotype found in patients with ACLF (96). Evidence suggests that CAID is closely related to gut dysbiosis and impaired intestinal barrier, raising the idea that a dysfunctional gut–liver axis affects not only the immune environment of the gut and the liver but also the systemic immune functions.
Impaired gut barrier and bacterial dysbiosis excessively increase the bacteria and their components or metabolites within the blood flow. These PAMPs bind to PRRs of different organs and tissues, causing massive release of proinflammatory cytokines and activation of various immune cells and inflammasomes. Moreover, detrimental elements like endotoxins, alcohol, cholesterol, ROS, and the inflammation they induce will cause liver cell damage and thus the release of DAMPs into circulation, which further exacerbates the systemic inflammation. In the meantime, impaired liver functions render insufficient albumin, a protein that is supposed to neutralize excessive PAMPs during systemic inflammation. Likewise, deficient production of anti-inflammatory cytokines such as IL-10 by monocytes and Kupffer cells dampens the immune tolerance and promotes inflammation (96). Furthermore, stimulation by certain bacteria, immunogenic cell death, and oxidative stress in combination with excessive protein-folding demand during severe inflammation finally overwhelms the processing abilities of the endoplasmic reticulum, eliciting the unfolded protein response (UFR). UFR per se is a source of inflammation via secreting proinflammatory cytokines such as IL-6 and TNF. In turn, circulating cytokines like IL-1, IL-6, IL-8, and TNF can activate UFR in the liver, making a positive feedback loop that amplifies the systemic and hepatic inflammation (97, 98).
Prolonged inflammation causes damage not only to parenchymal cells but also to the circulating immune cells. Immune cells show upregulated markers of activation but hampered immune response (99). For instance, in cirrhosis without ACLF, CD14+CD16+ monocytes are enriched and express more HLA-DR and TNF, favoring a proinflammatory and profibrogenic phenotype. However, their functions, such as phagocytosis, chemotaxis, and T-cell activation, seem to be inhibited. When the disease progresses to ACLF, expression of HLA-DR and TNF by these cells markedly decreases, as anti-inflammatory cytokines such as IL-10 appear to increase (96, 100). Likewise, neutrophils experience impaired antibacterial activity in the progression of cirrhosis (101). Moreover, continuous activation renders lymphocytes susceptible to anergy, apoptosis, and exhaustion. IFN-γ produced by T cells decreases, while inhibitory signals such as PD-1 and TIM3 increase (102). Changes in liver structure and function also contribute to immune deficiency. Extracellular matrix deposition, sinusoidal capillarization, intrahepatic shunting, and loss of hepatocytes all hamper the immunosurveillance and pathogen clearance functions of the liver (96). A recent study suggests that IL-2, a proinflammatory cytokine secreted by various cells in response to bacterial invasion, can markedly impair follicular T helper cells and therefore hamper the humoral immunity in advanced cirrhosis (103). Immune efficiency worsens as cirrhosis progresses, making advanced patients, especially ACLF patients, susceptible to severe systemic infection (104, 105).
Liver cirrhosis is the advanced stage of various liver diseases, characterized by its diffuse, fibrinogenic, and nodule-forming changes pathologically. Cirrhosis patients are prone to gut dysbiosis, an impaired intestinal barrier, pathological bacterial translocation, and severe inflammation and fibrosis in the liver. It is not surprising to see how gut dysbiosis directly affects the progression of liver cirrhosis, given the close relationship they have in terms of anatomy, physiology, and metabolism. However, seeing how a dysregulated gut–liver axis can elicit such massive immune alternations in the fibrotic liver is still intriguing. The TLR4–LPS interaction seems to initiate most of the immune transformations in this process. A high level of PAMPs breaks the immune tolerance in the liver and causes prolonged inflammation and massive tissue damage. DAMPs released from injured cells further amplify the inflammation not only in the liver but also in the whole system. Eventually, immune paralysis occurs when the immune system gets overwhelmed by intense and continuous activation. During disease progression, hepatocytes and various nonparenchymal cells experience drastic changes in terms of phenotype and function. Among these changes, activation of HSCs appears to be the center of fibrosis progression, which is one of the pathological features of cirrhosis. However, HSC is not the entire story, as mounting evidence indicates that complex interplays exist among different immune cells.
Of note, due to the complexity of etiology, differences between various types of cirrhosis, such as virus-related cirrhosis, alcohol-related cirrhosis, NAFLD-related cirrhosis, and autoimmune-related cirrhosis, are not discussed in this review. It is of great importance to know that the etiology per se might, along with gut dysbiosis, contribute to the disease progression. For instance, alcohol alone can disrupt the gut barrier and cause inflammatory injury to the liver. Therefore, it might work in synergy with gut dysbiosis to reshape the immune environment of the liver. Relevant studies were extensively reviewed elsewhere (16, 63, 106, 107).
Given the great impact of gut dysbiosis on liver immunity, it is tempting to design therapy for cirrhosis patients targeting the gut–liver-immune axis. For microbiota modulation, fecal microbiota transplantation (FMT) and prebiotics/probiotics are proven to have beneficial effects on cirrhosis patients (53, 108, 109). Most interestingly, bacteriophages targeting pathogens such as Enterococcus faecalis can ameliorate alcoholic liver injury in an animal model, indicating a novel strategy to modulate gut microbiota (110). For gut barrier restoration, FXR agonists appear to be a promising choice (5, 70) while nonselective B-blockers (NSBBs) can reduce SIBO, and therefore BT, probably by (111) improving gut motility. Additionally, there is also new progress in modulating the liver-related immune response. Targeting TLR4 signaling and other cirrhosis-related proinflammatory pathways might be of great therapeutic potential (112, 113).
Though we are starting to comprehend the role of the gut–liver axis in the pathogenesis and immune remodeling of liver cirrhosis, multiple research perspectives remain largely elusive. Firstly, although researchers have identified some bacterial species that are correlated with liver cirrhosis, very few studies have discovered the mechanistic links between these specific species and cirrhosis progression. In addition to the common PAMP–PRR interaction, other components and metabolites of these species may have their own unique ways of communicating with the host. Secondly, intercellular communications in cirrhosis settings deserve more attention. The single-cell transcriptomic analysis begins to show its advantages in dissecting the complicated crosstalk among different immune cells in chronic liver diseases (95, 114). In the foreseeable future, multiomics studies, including transcriptomics, metagenomics, and metabolomics, will continue to provide fresh insights into the intrahepatic immune environment and host–microbiota interaction (115, 116). Thirdly, immune remodeling through the gut–liver axis goes far beyond the gut and the liver. Cirrhosis-related changes in other immune compartments such as the peritoneal cavity (22), lung, kidney, and brain (23) still warrant thorough investigations. Given the fact that gut-derived bacteria and metabolites are the major sources of systemic inflammation, a common condition associated with mortality for decompensated cirrhosis patients, it is very important to understand how the dysbiosis starts and how it affects the disease progression. Future studies need to focus on the cellular and molecular mechanisms of gut–liver–immune regulation and, hopefully, help patients benefit from these studies.
Manuscript writing was done by HG. Manuscript revision was done by JY, MK, XZ, and HG. All authors contributed to the article and approved the submitted version.
This project was supported by research funds from the RGC Theme-based Research Scheme (T12-703/19-R), the RGC Collaborative Research Fund (C7026-18G, C7065-18G), the Health and Medical Research Fund, Hong Kong (08191336), the National Natural Sciences Foundation of China (82103355) and CUHK direct grant.
All figures were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes (2013) 4(5):382–7. doi: 10.4161/gmic.25723
2. Giraud J, Saleh M. Host-microbiota interactions in liver inflammation and cancer. Cancers (Basel) (2021) 13(17):4342. doi: 10.3390/cancers13174342
3. Raftar SKA, Ashrafian F, Abdollahiyan S, Yadegar A, Moradi HR, Masoumi M, et al. The anti-inflammatory effects of akkermansia muciniphila and its derivates in HFD/CCL4-induced murine model of liver injury. Sci Rep (2022) 12(1):2453. doi: 10.1038/s41598-022-06414-1
4. Xia J, Lv L, Liu B, Wang S, Zhang S, Wu Z, et al. Akkermansia muciniphila ameliorates acetaminophen-induced liver injury by regulating gut microbial composition and metabolism. Microbiol Spectr (2022) 10(1):e0159621. doi: 10.1128/spectrum.01596-21
5. Simbrunner B, Trauner M, Reiberger T. Review article: therapeutic aspects of bile acid signalling in the gut-liver axis. Aliment Pharmacol Ther (2021) 54(10):1243–62. doi: 10.1111/apt.16602
6. Clifford BL, Sedgeman LR, Williams KJ, Morand P, Cheng A, Jarrett KE, et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab (2021) 33(8):1671–84.e4. doi: 10.1016/j.cmet.2021.06.012
7. Cresci GA, Glueck B, McMullen MR, Xin W, Allende D, Nagy LE. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J Gastroenterol Hepatol (2017) 32(9):1587–97. doi: 10.1111/jgh.13731
8. Gines P, Krag A, Abraldes JG, Sola E, Fabrellas N, Kamath PS. Liver cirrhosis. Lancet (2021) 398(10308):1359–76. doi: 10.1016/S0140-6736(21)01374-X
9. Jepsen P, Younossi ZM. The global burden of cirrhosis: A review of disability-adjusted life-years lost and unmet needs. J Hepatol (2021) 75 Suppl 1:S3–S13. doi: 10.1016/j.jhep.2020.11.042
10. Jalan R, D'Amico G, Trebicka J, Moreau R, Angeli P, Arroyo V. New clinical and pathophysiological perspectives defining the trajectory of cirrhosis. J Hepatol (2021) 75 Suppl 1:S14–26. doi: 10.1016/j.jhep.2021.01.018
11. Bernardi M, Moreau R, Angeli P, Schnabl B, Arroyo V. Mechanisms of decompensation and organ failure in cirrhosis: From peripheral arterial vasodilation to systemic inflammation hypothesis. J Hepatol (2015) 63(5):1272–84. doi: 10.1016/j.jhep.2015.07.004
12. Trebicka J, Fernandez J, Papp M, Caraceni P, Laleman W, Gambino C, et al. The PREDICT study uncovers three clinical courses of acutely decompensated cirrhosis that have distinct pathophysiology. J Hepatol (2020) 73(4):842–54. doi: 10.1016/j.jhep.2020.06.013
13. Arroyo V, Angeli P, Moreau R, Jalan R, Claria J, Trebicka J, et al. The systemic inflammation hypothesis: Towards a new paradigm of acute decompensation and multiorgan failure in cirrhosis. J Hepatol (2021) 74(3):670–85. doi: 10.1016/j.jhep.2020.11.048
14. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol (2016) 13(7):412–25. doi: 10.1038/nrgastro.2016.85
15. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R, et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol (2019) 71(6):1216–28. doi: 10.1016/j.jhep.2019.08.005
16. Bajaj JS. Alcohol, liver disease and the gut microbiota. Nat Rev Gastroenterol Hepatol (2019) 16(4):235–46. doi: 10.1038/s41575-018-0099-1
17. Maccioni L, Gao B, Leclercq S, Pirlot B, Horsmans Y, De Timary P, et al. Intestinal permeability, microbial translocation, changes in duodenal and fecal microbiota, and their associations with alcoholic liver disease progression in humans. Gut Microbes (2020) 12(1):1782157. doi: 10.1080/19490976.2020.1782157
18. Neag MA, Mitre AO, Catinean A, Buzoianu AD. Overview of the microbiota in the gut-liver axis in viral b and c hepatitis. World J Gastroenterol (2021) 27(43):7446–61. doi: 10.3748/wjg.v27.i43.7446
19. Milosevic I, Russo E, Vujovic A, Barac A, Stevanovic O, Gitto S, et al. Microbiota and viral hepatitis: State of the art of a complex matter. World J Gastroenterol (2021) 27(33):5488–501. doi: 10.3748/wjg.v27.i33.5488
20. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol (2020) 72(3):558–77. doi: 10.1016/j.jhep.2019.10.003
21. Bajaj JS, Kamath PS, Reddy KR. The evolving challenge of infections in cirrhosis. N Engl J Med (2021) 384(24):2317–30. doi: 10.1056/NEJMra2021808
22. Van der Merwe S, Chokshi S, Bernsmeier C, Albillos A. The multifactorial mechanisms of bacterial infection in decompensated cirrhosis. J Hepatol (2021) 75 Suppl 1:S82–S100. doi: 10.1016/j.jhep.2020.11.029
23. Brescia P, Rescigno M. The gut vascular barrier: a new player in the gut-liver-brain axis. Trends Mol Med (2021) 27(9):844–55. doi: 10.1016/j.molmed.2021.06.007
24. Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science (2013) 342(6157):447–53. doi: 10.1126/science.1237910
25. Camilleri M. Leaky gut: mechanisms, measurement and clinical implications in humans. Gut (2019) 68(8):1516–26. doi: 10.1136/gutjnl-2019-318427
26. Bel S, Pendse M, Wang Y, Li Y, Ruhn KA, Hassell B, et al. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science (2017) 357(6355):1047–52. doi: 10.1126/science.aal4677
27. Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol (2014) 60(1):197–209. doi: 10.1016/j.jhep.2013.07.044
28. Martínez-López M, Iborra S, Conde-Garrosa R, Mastrangelo A, Danne C, Mann ER, et al. Microbiota sensing by mincle-syk axis in dendritic cells regulates interleukin-17 and -22 production and promotes intestinal barrier integrity. Immunity (2019) 50(2):446–61.e9. doi: 10.1016/j.immuni.2018.12.020
29. Chun E, Lavoie S, Fonseca-Pereira D, Bae S, Michaud M, Hoveyda HR, et al. Metabolite-sensing receptor Ffar2 regulates colonic group 3 innate lymphoid cells and gut immunity. Immunity (2019) 51(5):871–84.e6. doi: 10.1016/j.immuni.2019.09.014
30. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell (2015) 163(2):367–80. doi: 10.1016/j.cell.2015.08.058
31. Birchenough GM, Nyström EE, Johansson ME, Hansson GC. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science (2016) 352(6293):1535–42. doi: 10.1126/science.aaf7419
32. Honda M, Surewaard BGJ, Watanabe M, Hedrick CC, Lee WY, Brown K, et al. Perivascular localization of macrophages in the intestinal mucosa is regulated by Nr4a1 and the microbiome. Nat Commun (2020) 11(1):1329. doi: 10.1038/s41467-020-15068-4
33. Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, Franchini F, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity (2019) 50(2):432–45.e7. doi: 10.1016/j.immuni.2018.12.018
34. Park JW, Kim SE, Lee NY, Kim JH, Jung JH, Jang MK, et al. Role of microbiota-derived metabolites in alcoholic and non-alcoholic fatty liver diseases. Int J Mol Sci (2021) 23(1):426. doi: 10.3390/ijms23010426
35. Bansal T, Alaniz RC, Wood TK, Jayaraman A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc Natl Acad Sci U S A. (2010) 107(1):228–33. doi: 10.1073/pnas.0906112107
36. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol (2013) 14(10):996–1006. doi: 10.1038/ni.2691
37. Gola A, Dorrington MG, Speranza E, Sala C, Shih RM, Radtke AJ, et al. Commensal-driven immune zonation of the liver promotes host defence. Nature (2021) 589(7840):131–6. doi: 10.1038/s41586-020-2977-2
38. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13(2):88–110. doi: 10.1038/nrgastro.2015.200
39. Gammella E, Buratti P, Cairo G, Recalcati S. Macrophages: central regulators of iron balance. Metallomics (2014) 6(8):1336–45. doi: 10.1039/c4mt00104d
40. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol (2021) 18(3):151–66. doi: 10.1038/s41575-020-00372-7
41. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol (2010) 10(11):753–66. doi: 10.1038/nri2858
42. Crispe IN. Immune tolerance in liver disease. Hepatology (2014) 60(6):2109–17. doi: 10.1002/hep.27254
43. Dunham RM, Thapa M, Velazquez VM, Elrod EJ, Denning TL, Pulendran B, et al. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J Immunol (2013) 190(5):2009–16. doi: 10.4049/jimmunol.1201937
44. Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature (2014) 513(7516):59–64. doi: 10.1038/nature13568
45. Solé C, Guilly S, Da Silva K, Llopis M, Le-Chatelier E, Huelin P, et al. Alterations in gut microbiome in cirrhosis as assessed by quantitative metagenomics: Relationship with acute-on-Chronic liver failure and prognosis. Gastroenterology (2021) 160(1):206–18.e13. doi: 10.1053/j.gastro.2020.08.054
46. Caussy C, Tripathi A, Humphrey G, Bassirian S, Singh S, Faulkner C, et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat Commun (2019) 10(1):1406. doi: 10.1038/s41467-019-09455-9
47. Ren Z, Li A, Jiang J, Zhou L, Yu Z, Lu H, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut (2019) 68(6):1014–23. doi: 10.1136/gutjnl-2017-315084
48. Oh TG, Kim SM, Caussy C, Fu T, Guo J, Bassirian S, et al. A universal gut-Microbiome-Derived signature predicts cirrhosis. Cell Metab (2020) 32(5):901. doi: 10.1016/j.cmet.2020.10.015
49. Behary J, Amorim N, Jiang XT, Raposo A, Gong L, McGovern E, et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun (2021) 12(1):187. doi: 10.1038/s41467-020-20422-7
50. Chen Y, Yang F, Lu H, Wang B, Lei D, Wang Y, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology (2011) 54(2):562–72. doi: 10.1002/hep.24423
51. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab (2017) 25(5):1054–62.e5. doi: 10.1016/j.cmet.2017.04.001
52. Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol (2014) 60(5):940–7. doi: 10.1016/j.jhep.2013.12.019
53. Tranah TH, Edwards LA, Schnabl B, Shawcross DL. Targeting the gut-liver-immune axis to treat cirrhosis. Gut (2021) 70(5):982–94. doi: 10.1136/gutjnl-2020-320786
54. Trebicka J, Macnaughtan J, Schnabl B, Shawcross DL, Bajaj JS. The microbiota in cirrhosis and its role in hepatic decompensation. J Hepatol (2021) 75 Suppl 1:S67–81. doi: 10.1016/j.jhep.2020.11.013
55. Shah A, Shanahan E, Macdonald GA, Fletcher L, Ghasemi P, Morrison M, et al. Systematic review and meta-analysis: Prevalence of small intestinal bacterial overgrowth in chronic liver disease. Semin Liver Dis (2017) 37(4):388–400. doi: 10.1055/s-0037-1608832
56. Ferolla SM, Armiliato GN, Couto CA, Ferrari TC. The role of intestinal bacteria overgrowth in obesity-related nonalcoholic fatty liver disease. Nutrients (2014) 6(12):5583–99. doi: 10.3390/nu6125583
57. Pande C, Kumar A, Sarin SK. Small-intestinal bacterial overgrowth in cirrhosis is related to the severity of liver disease. Aliment Pharmacol Ther (2009) 29(12):1273–81. doi: 10.1111/j.1365-2036.2009.03994.x
58. Muñoz L, Borrero MJ, Úbeda M, Conde E, Del Campo R, Rodríguez-Serrano M, et al. Intestinal immune dysregulation driven by dysbiosis promotes barrier disruption and bacterial translocation in rats with cirrhosis. Hepatology (2019) 70(3):925–38. doi: 10.1002/hep.30349
59. Du Plessis J, Vanheel H, Janssen CE, Roos L, Slavik T, Stivaktas PI, et al. Activated intestinal macrophages in patients with cirrhosis release NO and IL-6 that may disrupt intestinal barrier function. J Hepatol (2013) 58(6):1125–32. doi: 10.1016/j.jhep.2013.01.038
60. Assimakopoulos SF, Tsamandas AC, Tsiaoussis GI, Karatza E, Triantos C, Vagianos CE, et al. Altered intestinal tight junctions' expression in patients with liver cirrhosis: a pathogenetic mechanism of intestinal hyperpermeability. Eur J Clin Invest (2012) 42(4):439–46. doi: 10.1111/j.1365-2362.2011.02609.x
61. Wang Y, Tong J, Chang B, Wang B, Zhang D, Wang B. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol Med Rep (2014) 9(6):2352–6. doi: 10.3892/mmr.2014.2126
62. Chen P, Starkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology (2015) 61(3):883–94. doi: 10.1002/hep.27489
63. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol (2018) 15(7):397–411. doi: 10.1038/s41575-018-0011-z
64. Macutkiewicz C, Carlson G, Clark E, Dobrindt U, Roberts I, Warhurst G. Characterisation of escherichia coli strains involved in transcytosis across gut epithelial cells exposed to metabolic and inflammatory stress. Microbes Infect (2008) 10(4):424–31. doi: 10.1016/j.micinf.2008.01.001
65. Úbeda M, Lario M, Muñoz L, Borrero MJ, Rodríguez-Serrano M, Sánchez-Díaz AM, et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol (2016) 64(5):1049–57. doi: 10.1016/j.jhep.2015.12.010
66. Teltschik Z, Wiest R, Beisner J, Nuding S, Hofmann C, Schoelmerich J, et al. Intestinal bacterial translocation in rats with cirrhosis is related to compromised paneth cell antimicrobial host defense. Hepatology (2012) 55(4):1154–63. doi: 10.1002/hep.24789
67. Doi H, Iyer TK, Carpenter E, Li H, Chang KM, Vonderheide RH, et al. Dysfunctional b-cell activation in cirrhosis resulting from hepatitis c infection associated with disappearance of CD27-positive b-cell population. Hepatology (2012) 55(3):709–19. doi: 10.1002/hep.24689
68. Muñoz L, José Borrero M, Ubeda M, Lario M, Díaz D, Francés R, et al. Interaction between intestinal dendritic cells and bacteria translocated from the gut in rats with cirrhosis. Hepatology (2012) 56(5):1861–9. doi: 10.1002/hep.25854
69. Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science (2015) 350(6262):830–4. doi: 10.1126/science.aad0135
70. Sorribas M, Jakob MO, Yilmaz B, Li H, Stutz D, Noser Y, et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J Hepatol (2019) 71(6):1126–40. doi: 10.1016/j.jhep.2019.06.017
71. Fan Y, Li Y, Chu Y, Liu J, Cui L, Zhang D. Toll-like receptors recognize intestinal microbes in liver cirrhosis. Front Immunol (2021) 12:608498. doi: 10.3389/fimmu.2021.608498
72. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther (2021) 6(1):291. doi: 10.1038/s41392-021-00687-0
73. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med (2007) 13(11):1324–32. doi: 10.1038/nm1663
74. Paquissi FC. Immunity and fibrogenesis: The role of Th17/IL-17 axis in HBV and HCV-induced chronic hepatitis and progression to cirrhosis. Front Immunol (2017) 8:1195. doi: 10.3389/fimmu.2017.01195
75. Zhang J, Liu Y, Chen H, Yuan Q, Wang J, Niu M, et al. MyD88 in hepatic stellate cells enhances liver fibrosis via promoting macrophage M1 polarization. Cell Death Dis (2022) 13(4):411. doi: 10.1038/s41419-022-04802-z
76. Yu J, Zhu C, Wang X, Kim K, Bartolome A, Dongiovanni P, et al. Hepatocyte TLR4 triggers inter-hepatocyte Jagged1/Notch signaling to determine NASH-induced fibrosis. Sci Transl Med (2021) 13(599):eabe1692. doi: 10.1126/scitranslmed.abe1692
77. Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, et al. Hepatocyte notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci Transl Med (2018) 10(468):eaat0344. doi: 10.1126/scitranslmed.aat0344
78. An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun (2020) 11(1):2362. doi: 10.1038/s41467-020-16092-0
79. Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab (2016) 24(6):848–62. doi: 10.1016/j.cmet.2016.09.016
80. Liu K, Wang FS, Xu R. Neutrophils in liver diseases: pathogenesis and therapeutic targets. Cell Mol Immunol (2021) 18(1):38–44. doi: 10.1038/s41423-020-00560-0
81. Fabre T, Molina MF, Soucy G, Goulet JP, Willems B, Villeneuve JP, et al. Type 3 cytokines IL-17A and IL-22 drive TGF-beta-dependent liver fibrosis. Sci Immunol (2018) 3(28):eaar7754. doi: 10.1126/sciimmunol.aar7754
82. Khanam A, Trehanpati N, Riese P, Rastogi A, Guzman CA, Sarin SK. Blockade of neutrophil's chemokine receptors CXCR1/2 abrogate liver damage in acute-on-Chronic liver failure. Front Immunol (2017) 8:464. doi: 10.3389/fimmu.2017.00464
83. Bernsmeier C, Pop OT, Singanayagam A, Triantafyllou E, Patel VC, Weston CJ, et al. Patients with acute-on-chronic liver failure have increased numbers of regulatory immune cells expressing the receptor tyrosine kinase MERTK. Gastroenterology (2015) 148(3):603–15 e14. doi: 10.1053/j.gastro.2014.11.045
84. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17(5):306–21. doi: 10.1038/nri.2017.11
85. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol (2017) 66(5):1037–46. doi: 10.1016/j.jhep.2017.01.022
86. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol (2021) 18(1):45–56. doi: 10.1038/s41423-020-00558-8
87. Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol (2015) 12(7):387–400. doi: 10.1038/nrgastro.2015.94
88. Toubal A, Nel I, Lotersztajn S, Lehuen A. Mucosal-associated invariant T cells and disease. Nat Rev Immunol (2019) 19(10):643–57. doi: 10.1038/s41577-019-0191-y
89. Hegde P, Weiss E, Paradis V, Wan J, Mabire M, Sukriti S, et al. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat Commun (2018) 9(1):2146. doi: 10.1038/s41467-018-04450-y
90. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol (2018) 15(9):555–67. doi: 10.1038/s41575-018-0020-y
91. Gracia-Sancho J, Caparros E, Fernandez-Iglesias A, Frances R. Role of liver sinusoidal endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol (2021) 18(6):411–31. doi: 10.1038/s41575-020-00411-3
92. Nilsson J, Hörnberg M, Schmidt-Christensen A, Linde K, Nilsson M, Carlus M, et al. NKT cells promote both type 1 and type 2 inflammatory responses in a mouse model of liver fibrosis. Sci Rep (2020) 10(1):21778. doi: 10.1038/s41598-020-78688-2
93. Bandyopadhyay K, Marrero I, Kumar V. NKT cell subsets as key participants in liver physiology and pathology. Cell Mol Immunol (2016) 13(3):337–46. doi: 10.1038/cmi.2015.115
94. Kumar V. NKT-cell subsets: promoters and protectors in inflammatory liver disease. J Hepatol (2013) 59(3):618–20. doi: 10.1016/j.jhep.2013.02.032
95. Koda Y, Teratani T, Chu PS, Hagihara Y, Mikami Y, Harada Y, et al. CD8(+) tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun (2021) 12(1):4474. doi: 10.1038/s41467-021-24734-0
96. Albillos A, Martin-Mateos R, van der Merwe S, Wiest R, Jalan R, Álvarez-Mon M. Cirrhosis-associated immune dysfunction. Nat Rev Gastroenterol Hepatol (2022) 19(2):112–34. doi: 10.1038/s41575-021-00520-7
97. Tazi KA, Bièche I, Paradis V, Guichard C, Laurendeau I, Dargère D, et al. In vivo altered unfolded protein response and apoptosis in livers from lipopolysaccharide-challenged cirrhotic rats. J Hepatol (2007) 46(6):1075–88. doi: 10.1016/j.jhep.2007.01.034
98. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol (2016) 16(8):469–84. doi: 10.1038/nri.2016.62
99. Lebossé F, Gudd C, Tunc E, Singanayagam A, Nathwani R, Triantafyllou E, et al. CD8 + T cells from patients with cirrhosis display a phenotype that may contribute to cirrhosis-associated immune dysfunction. EBioMedicine (2019) 49:258–68. doi: 10.1016/j.ebiom.2019.10.011
100. Hackstein CP, Assmus LM, Welz M, Klein S, Schwandt T, Schultze J, et al. Gut microbial translocation corrupts myeloid cell function to control bacterial infection during liver cirrhosis. Gut (2017) 66(3):507–18. doi: 10.1136/gutjnl-2015-311224
101. Bernsmeier C, van der Merwe S, Perianin A. Innate immune cells in cirrhosis. J Hepatol (2020) 73(1):186–201. doi: 10.1016/j.jhep.2020.03.027
102. Markwick LJ, Riva A, Ryan JM, Cooksley H, Palma E, Tranah TH, et al. Blockade of PD1 and TIM3 restores innate and adaptive immunity in patients with acute alcoholic hepatitis. Gastroenterology (2015) 148(3):590–602.e10. doi: 10.1053/j.gastro.2014.11.041
103. Basho K, Zoldan K, Schultheiss M, Bettinger D, Globig AM, Bengsch B, et al. IL-2 contributes to cirrhosis-associated immune dysfunction by impairing follicular T helper cells in advanced cirrhosis. J Hepatol (2021) 74(3):649–60. doi: 10.1016/j.jhep.2020.10.012
104. Fernández J, Prado V, Trebicka J, Amoros A, Gustot T, Wiest R, et al. Multidrug-resistant bacterial infections in patients with decompensated cirrhosis and with acute-on-chronic liver failure in Europe. J Hepatol (2019) 70(3):398–411. doi: 10.1016/j.jhep.2018.10.027
105. Piano S, Singh V, Caraceni P, Maiwall R, Alessandria C, Fernandez J, et al. Epidemiology and effects of bacterial infections in patients with cirrhosis worldwide. Gastroenterology (2019) 156(5):1368–80.e10. doi: 10.1053/j.gastro.2018.12.005
106. Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology (2015) 148(1):30–6. doi: 10.1053/j.gastro.2014.10.042
107. Aron-Wisnewsky J, Vigliotti C, Witjes J, Le P, Holleboom AG, Verheij J, et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol (2020) 17(5):279–97. doi: 10.1038/s41575-020-0269-9
108. Bajaj JS, Khoruts A. Microbiota changes and intestinal microbiota transplantation in liver diseases and cirrhosis. J Hepatol (2020) 72(5):1003–27. doi: 10.1016/j.jhep.2020.01.017
109. Bajaj JS, Fagan A, Gavis EA, Kassam Z, Sikaroodi M, Gillevet PM. Long-term outcomes of fecal microbiota transplantation in patients with cirrhosis. Gastroenterology (2019) 156(6):1921–3.e3. doi: 10.1053/j.gastro.2019.01.033
110. Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature (2019) 575(7783):505–11. doi: 10.1038/s41586-019-1742-x
111. Reiberger T, Ferlitsch A, Payer BA, Mandorfer M, Heinisch BB, Hayden H, et al. Non-selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL-6 in patients with cirrhosis. J Hepatol (2013) 58(5):911–21. doi: 10.1016/j.jhep.2012.12.011
112. Engelmann C, Sheikh M, Sharma S, Kondo T, Loeffler-Wirth H, Zheng YB, et al. Toll-like receptor 4 is a therapeutic target for prevention and treatment of liver failure. J Hepatol (2020) 73(1):102–12. doi: 10.1016/j.jhep.2020.01.011
113. Zhang XW, Mi S, Li Z, Zhou JC, Xie J, Hua F, et al. Antagonism of interleukin-17A ameliorates experimental hepatic fibrosis by restoring the IL-10/STAT3-suppressed autophagy in hepatocytes. Oncotarget (2017) 8(6):9922–34. doi: 10.18632/oncotarget.14266
114. Deczkowska A, David E, Ramadori P, Pfister D, Safran M, Li B, et al. XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med (2021) 27(6):1043–54. doi: 10.1038/s41591-021-01344-3
115. Oakley F. Interrogating mechanisms of liver fibrosis with omics. Nat Rev Gastroenterol Hepatol (2022) 19(2):89–90. doi: 10.1038/s41575-021-00567-6
Keywords: gut-liver axis, microbiota, immune, gut microbiome, liver cirrhosis, immune homeostasis, immune remodeling
Citation: Guan H, Zhang X, Kuang M and Yu J (2022) The gut–liver axis in immune remodeling of hepatic cirrhosis. Front. Immunol. 13:946628. doi: 10.3389/fimmu.2022.946628
Received: 17 May 2022; Accepted: 11 July 2022;
Published: 08 August 2022.
Edited by:
Enis Kostallari, Mayo Clinic, United StatesReviewed by:
Mingxuan Zheng, Xuzhou Medical University, ChinaCopyright © 2022 Guan, Zhang, Kuang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Kuang, a3VhbmdtQG1haWwuc3lzdS5lZHUuY24=; Jun Yu, anVueXVAY3Voay5lZHUuaGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.