
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 09 August 2022
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.945063
This article is part of the Research TopicNew Roles for CD4+ T cells in Type 2 Immune ResponsesView all 7 articles
 Kota Kokubo1†
Kota Kokubo1† Atsushi Onodera1,2†
Atsushi Onodera1,2† Masahiro Kiuchi1
Masahiro Kiuchi1 Kaori Tsuji1
Kaori Tsuji1 Kiyoshi Hirahara1*
Kiyoshi Hirahara1* Toshinori Nakayama1,3*
Toshinori Nakayama1,3*Type 2 helper T (Th2) cells, a subset of CD4+ T cells, play an important role in the host defense against pathogens and allergens by producing Th2 cytokines, such as interleukin-4 (IL-4), IL-5, and IL-13, to trigger inflammatory responses. Emerging evidence reveals that Th2 cells also contribute to the repair of injured tissues after inflammatory reactions. However, when the tissue repair process becomes chronic, excessive, or uncontrolled, pathological fibrosis is induced, leading to organ failure and death. Thus, proper control of Th2 cells is needed for complete tissue repair without the induction of fibrosis. Recently, the existence of pathogenic Th2 (Tpath2) cells has been revealed. Tpath2 cells produce large amounts of Th2 cytokines and induce type 2 inflammation when activated by antigen exposure or tissue injury. In recent studies, Tpath2 cells are suggested to play a central role in the induction of type 2 inflammation whereas the role of Tpath2 cells in tissue repair and fibrosis has been less reported in comparison to conventional Th2 cells. In this review, we discuss the roles of conventional Th2 cells and pathogenic Th2 cells in the sequence of tissue inflammation, repair, and fibrosis.
Type 2 helper T (Th2) cells are a subset of CD4+ T cells characterized by the production of Th2 cytokines, including interleukin-4 (IL-4), IL-5, and IL-13 (1). The Th1-Th2 paradigm was first described about 30 years ago (2), and for a long time before that, type 2 immunity was thought to be a simple counter-regulatory mechanism that antagonizes type 1 immunity (3). Now, however, Th2 cells are often observed in tissues in allergic patients and are known to play critical roles in the pathogenesis of allergic diseases (1, 4).
It was recently shown that Th2 cells include pathogenic Th2 (Tpath2) cells that highly express the receptor for IL-33 (a cytokine that is released during tissue injury) and produce large amounts of IL-5 (1). Thus, the “pathogenic Th population disease-inducing model” has been proposed, in which Tpath2 cells are important promoters of allergic inflammation in both mouse and human systems (1, 5). Th2 cells are also involved in the repair of tissues that are damaged by parasitic infections, house dust mite (HDM), and air pollution. IL-4 and IL-13, which are induced by Th2 cells, activate macrophages and epithelial cells and enhance the production of extracellular matrix (ECM), an element crucial for tissue repair (6–10). However, when the tissue repair process becomes chronic, excessive, or uncontrolled, it may induce the development of pathological fibrosis in various organ systems.
The mechanisms underlying the induction of fibrosis by Th2 cells have been reported to include deposition of ECM by fibroblasts in response to various cytokines and growth factors, including Th2 cytokines (11), but the detailed cellular and molecular mechanisms remain unclear. Currently, research to elucidate the mechanism underlying Th2 cell-mediated fibrosis is being actively conducted. Recently, we have reported that eosinophil reprogramming via IL-5 and amphiregulin production by Tpath2 cells (12) and a CD103low cell population among tissue-resident CD4+ T cells (13) were also found to promote tissue fibrosis.
We herein review the role of Th2 cells, including the conventional, pathogenic, and tissue-resident Th2 cells that we identified, in the sequence of tissue inflammation, repair, and fibrosis, and discuss how tissue fibrosis can be controlled.
Th2 cells produce so-called Th2 cytokines: IL-4, IL-5, and IL-13. These cytokines play an important role in humoral immunity and helminthic infection defense but are also central to the pathogenesis of many allergic inflammatory diseases.
Peripheral naïve CD4+ T cells, upon recognition of antigenic peptides by the T cell receptor (TCR), migrate into lymphoid tissues and functionally differentiate into different Th cell subsets. When CD4+ T cells receive IL-4 that is released by innate immune cells, such as group 2 innate immune system lymphocytes (ILC2s), mast cells, and basophils in lymphoid tissues, these CD4+ T cells differentiate into Th2 cells through IL-4-induced phosphorylation of STAT6 and the increased expression of GATA3 (14–19). GATA3 protein stability is also important for the functions of Th2 cells (20, 21). Through direct or indirect interaction with epigenetic modifiers, GATA3 is involved in chromatin remodeling at the Th2 cytokine gene loci: histone H3K9 acetylation (22, 23), histone H3K4 methylation (20), and DNA demethylation (24). In addition, epigenetic modifications of the Gata3 gene control the stable expression of GATA3 proteins (20, 25, 26). The TCR-mediated signaling pathway also contributes to Th2 cell differentiation (27). T cells that lack nuclear factor of activated T-cells 2 (NFAT2), encoded by the Nfatc1 gene, produce less IL-4, indicating that the TCR-dependent activation of NFAT2 plays a role in Th2 cell differentiation (28). Prolonged TCR signaling is proposed to enhance the pathogenicity of Th2 cells due to a lack of Klf2 gene reactivation (29, 30).
When airway epithelial tissues are damaged by helminth infection, HDM, or air pollution, the innate immune system is activated by the release of alarmin cytokines, such as thymic stromal lymphoid neoplastic factor (TSLP), IL-25, and IL-33 from the damaged epithelial cells. In addition to IL-4, these alarmin cytokines have been reported to be important for inducing Th2 responses under various conditions in vivo and in vitro (31). TSLP activates local dendritic cells to secrete CC chemokine ligand 22 (CCL22) and CCL17, which promotes Th2 cell differentiation. IL-25 induces the production of IL-4, IL-5, and IL-13 from antigen-presenting cells and promotes the differentiation of Th2 cells (31). In addition, the production of IL-5 from CD4+ T cells was enhanced in polyps of eosinophilic chronic rhinosinusitis (ECRS) patients by direct stimulation by increased IL-25, as Tpath2 cells also express the receptor for IL-25 (32). IL-33 activates NF-κB and MAP kinase via the IL-1 receptor-like 1 (ST2) and induces Th2 cytokine production in mast cells (33). Recent studies have shown that in addition to IL-4, the Wnt antagonist, platelet-derived Dickkopf-related protein 1 (DKK1)—in combination with TCR involvement—is an important contributor to Th2 cell differentiation in pathological type 2 cell-mediated inflammation (34). In addition to the well-known fact that platelets are an important source of platelet-derived growth factor (PDGF), there are mechanistic and physiological links among local tissue damage, platelet activation, and Th2 cell differentiation (Figure 1). Finally, the gene expression pattern as a Th2 subset that is acquired by the above-described mechanism is maintained by epigenetic modifications (25, 35).
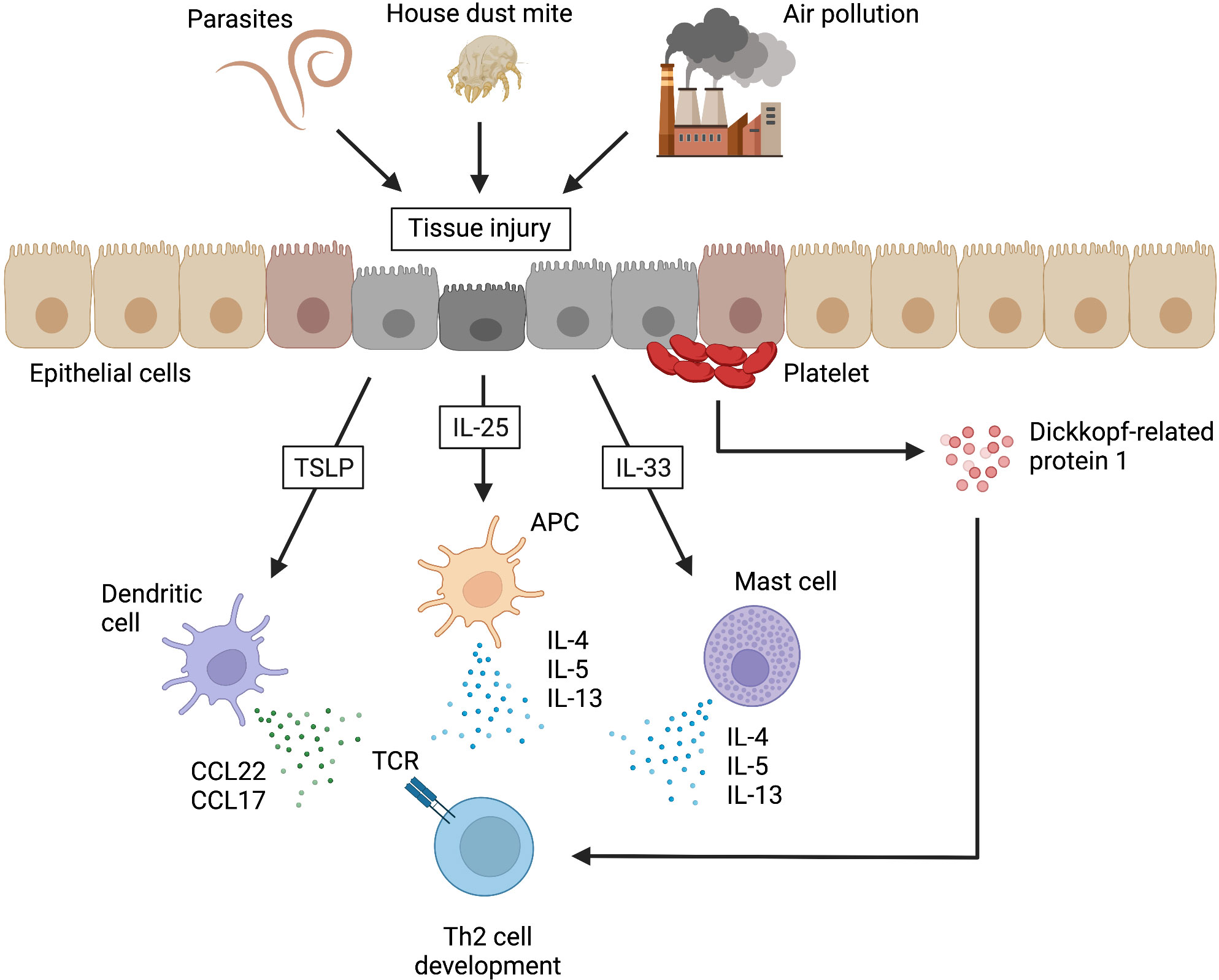
Figure 1 Alarmins that induce Th2 cell development in non-lymphoid tissues. When airway epithelial tissues are damaged by helminth infections, house dust mites, or air pollution, the damaged epithelial cells release alarmins (e.g., thymic stromal lymphoid neoplastic factor [TSLP], interleukin [IL]-25, and IL-33). TSLP activates local dendritic cells to secrete CC chemokine ligand 22 (CCL22) and CCL17, which promote the development of type 2 helper T (Th2) cells. IL-25 induces the production of IL-4, IL-5, and IL-13 from antigen-presenting cells (APCs), resulting in the promotion of Th2 cell development. IL-33 also promotes Th2 cell development by inducing the production of Th2 cytokines from mast cells. In addition, Dickkopf-related protein 1, which is a Wnt antagonist released from platelets at the site of injury, can similarly promote Th2 cell development. TCR, T cell receptor. This figure was created with BioRender.com.
Effector Th2 cells are primed in lymphoid tissues and eventually migrate into inflammatory tissues (36). CD69, a type II transmembrane glycoprotein with C-type lectin domains expressed on activated CD4+ T cells, is important for migration into inflammatory tissues (37). CD69 helps CD4+ T cells localize to inflammatory tissues by binding to a functional ligand for CD69, Myosin Light Chain 9 (Myl9), which is contained in the intravascular net-like structures that are formed in inflammatory tissues (38, 39). Th2 cells that migrate to inflammatory tissues are able to exert an effector function when exposed to factors in their environment. In particular, in the absence of signals from epithelial-derived cytokines (TSLP, IL25, and IL33), Th2 cells in the lung do not express IL-5 and IL-13, and are unable to exert full effector functions (40). Th2 cells that have an acquired effector function can produce Th2 cytokines, including IL-4, IL-13, and IL-5. IL-4 induces Th2 cell differentiation in an autocrine manner at inflammatory sites (41). IL-13 promotes allergen elimination from the epithelium by inducing the accumulation of mucin in the epithelium (42, 43). IL-5 recruits eosinophils to inflammatory sites and also promotes their survival (44). Eosinophil-derived neurotoxin (EDN) in granule proteins acts as an activator and chemoattractant of dendritic cells, and consequently, activated dendritic cells enhance Th2 responses. In addition, eosinophils can kill some parasites via the classical antibody-dependent cytotoxicity (ADCC) mechanism (44, 45). Furthermore, eosinophils are sources of Th2-inducing cytokines (mainly IL-4 and IL-13) in the very early stage of the immune response in lymph nodes and tissues and can induce Th2 differentiation by antigen presentation to CD4+ T cells (46–48) (Figure 2). ILC2s are also associated with the induction of type 2 inflammation by directly producing Th2 cytokines and activating Th2 cells via MHCII and IL-13 (49). ILC2s produce large amounts of Th2 cytokines upon stimulation by IL-25, IL-33 and TSLP, and also constitutively produce a certain amount of IL-5 (50, 51).
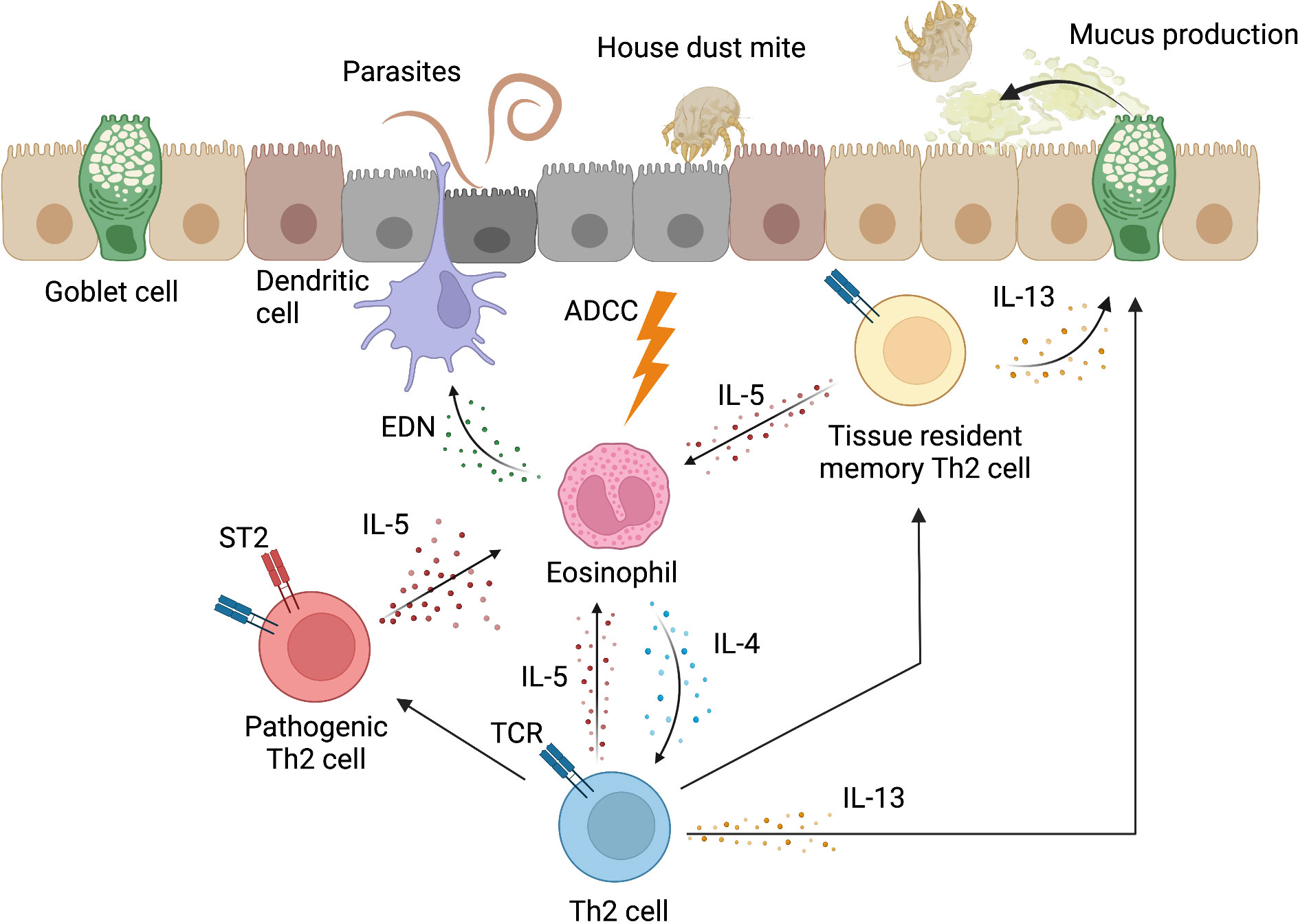
Figure 2 Inflammatory response induced by Th2 cells. In inflammatory tissues, Th2 cells induce an inflammatory response to eliminate antigens. Th2 cells recruit eosinophils to the site of inflammation through IL-5 production and promote their survival. Eosinophil-derived neurotoxin (EDN) acts as a chemoattractant and activator of dendritic cells. Eosinophils can also kill some parasites via the classical antibody-dependent cytotoxicity (ADCC) mechanism. In addition, eosinophils promote the development of Th2 cells through the production of IL-4. In addition to the induction of eosinophilic inflammation by IL-5, Th2 cells produce IL-13, which induces mucus production from epithelial cells, including goblet cells, and promotes the elimination of allergens from epithelial tissues. The Th2 cell population contains pathogenic Th2 (Tpath2) cells that highly express ST2, which is the receptor for IL-33. Tpath2 cells become high-producers of IL-5 when activated by IL-33. Tissue-resident memory Th2 cells are differentiated from a portion of Th2 cells, and play an important role in type 2 inflammatory responses in local tissues through the production of Th2 cytokines, such as IL-5 and IL-13. This figure was created with BioRender.com.
One of the major advances in the understanding of the induction of tissue inflammation by Th2 cells is the identification of Tpath2 cells (1, 52–57). Tpath2 cells express high levels of ST2, a component of the IL-33 receptor, and when activated by IL-33 released from damaged epithelial cells, they become a high producer of IL-5 (56). IL-33 is proposed to be required for optimal human pathogenic Th2 cell effector function (57). The IL-5 produced by Tpath2 cells exacerbates the pathology of eosinophilic inflammation in both mouse and humans (53). In fact, mepolizumab and benralizumab, which are humanized monoclonal antibodies targeting IL-5 and IL-5Rα, respectively, have both shown remarkable therapeutic effects in various types of eosinophilic diseases, including ECRS and asthma (58). In addition to IL-33, IL-7 maintains memory Tpath2 cells in an ectopic lymphoid tissue called “inducible bronchus-associated lymphoid tissue (iBALT)” in murine inflamed lung tissue (59). Recent technical advances, including the genome-wide analysis of single-cell transcriptomes, regulomes and proteomes, have highlighted the diversity and detailed characteristics of Tpath2 cells (60, 61). We herein summarize the current understanding of pathogenic Th2 cell markers other than ST2 and IL-5 (62).
A CC chemokine, CCL8, is highly expressed in the skin and functions as a ligand for the chemokine receptor, CCR8. CCR8+ memory-type Th2 cells have been identified as key pathogenic players in chronic skin inflammation in a mouse model of skin inflammation. In addition, a similar cell type that produces higher levels of IL-5 is found in healthy human donors, suggesting that CCR8 could be a marker for pathogenic Th2 cells (52). CRTH2 is a G-protein coupled receptor with which Tpath2 cells sense and react to prostaglandin D2, a pleiotropic mediator for activation, migration, and cytokine production in type 2 immune responses (62). Importantly, human pathogenic Th2 cells also express hematopoietic prostaglandin D synthase (HPGDS), which catalyzes the conversion of prostaglandin H2 to D2. Thus, Tpath2 cells are able to amplify their own activation and effector function via the HPGDS-PGD2-CRTH2 axis (53, 63). CD161 is a C-type lectin-like receptor and plays an inhibitory role on NK cell cytotoxicity. The ligand of CD161 is the C-type lectin domain family 2 member D (CLEC2D or LLT1), which is expressed in human epithelial cell lines isolated from the lung, suggesting that pathogenic Th2 cells might be activated via CD161 and contribute to inflammation in the lung (64). The receptor of IL-25, which consists of IL-17 receptor B (IL-17RB) and IL-17RA, also serves as a marker of the human pathogenic Th2 cells, since IL-25 stimulates Tpath2 proliferation and cytokine production (54).
Lipid metabolism-related molecules are also involved in the induction of pathogenic Th2 cells. For example, peroxisome proliferator-activated receptor gamma (PPAR-γ) is a fatty acid-activated nuclear receptor known for controlling adipogenesis, lipid and glucose metabolism, and inflammation. The PPARG gene is reproducibly reported as a marker associated with the human pathogenic Th2 phenotype (55, 65–67), while specific endogenous PPAR-γ ligands remain to be identified. In humans, the roles of PPAR-γ in pathogenic Th2 cells remain unclear. However, the specific functional role of PPAR-γ in pathogenic Th2 cells has been characterized in mice: the upregulation of IL-33 receptor ST2 and the activation of Tpath2 cells in allergic inflammation (68). Indeed, mice lacking PPAR-γ in T cells showed impaired immune responses in both allergic lung inflammation and parasite infection (69). Another example of the markers associated with lipid metabolism is free fatty acid receptor 3 (FFAR3), which encodes a G protein-coupled receptor for short chain fatty acids. Multiple studies have shown that human pathogenic Th2 cells express FFAR3, the expression levels of which were correlated with IL-5 levels and the number of tissue eosinophils (55, 65, 66).
As described above, the concept of Tpath2 cells has been recognized as fundamental for understanding the pathogenesis of inflammatory diseases, including allergic inflammation of the upper and lower airways, atopic dermatitis, and gastrointestinal inflammation in humans (39, 70).
While conventional Th2 cells have long been implicated in allergic airway inflammation, recent studies in human patients indicate that the disease is specifically driven by pathogenic Th2 cells (63, 66, 67). This idea is further supported by the successful clinical application of anti-IL-5 monoclonal antibodies in the treatment of asthma patients (56). In addition, other therapeutics targeting alarmins that activate pathogenic Th2 cells, such as anti-TSLP and anti-IL-33 treatments, are being developed for allergic asthma (71). Moreover, inhibitors of CRTH2 are currently being evaluated in clinical trials for allergic asthma (62). In ECRS patients, IL-5-producing pathogenic Th2 cells have also been identified in the upper airways (32).
Evidence that the pathogenic Th2 cell subset might play a role in atopic dermatitis (AD) came from a mouse model in which chronic skin inflammation was found to be driven by a specific subset of CCR8+ memory Th2 cells expressing high levels of IL-5 (52). Consistent with these findings, CCL18, a ligand of CCR8, is shown to be overexpressed in the skin of AD patients and to be well-correlated with disease activity (72). However, treatment with a monoclonal antibody to human interleukin-5 (mepolizumab) did not result in clinical success in patients with AD (73), whereas blocking of the Th2 cytokine signaling with a monoclonal antibody against the IL-4/IL-13 receptor alpha chain shows high efficacy in patients with severe AD (74).
Recently, a single cell analysis of T cells isolated from eosinophilic esophagitis (EoE) tissue in humans identified a pathogenic Th2 cell population that expresses HPGDS, CRTH2, and CD161. These pathogenic Th2 cells produce higher levels of IL-5 and IL-13, which are correlated with disease severity (65). Thus, EoE has been considered as one of the diseases associated with pathogenic Th2 cells. In another study, CRTH2hiCD161hiHPGDShiST2+ Tpath2 cells were proposed to be a pathogenic T-cell population in EoE patients (53). Furthermore, the effects of allergen-specific immunotherapy in EoE patients have revealed a correlation between the presence of CD45ROhiCRTH2hiCCD49dhiCCR4hiCXCR3loCCR6loCD27loST2+ Tpath2 cells and disease activity (55). G-protein-coupled receptor 15 (GPR15) is a known lymphocyte trafficking receptor and may serve as a pathogenic marker in inflammatory bowel disease: increased numbers of CD4+CD45RO+GPR15+ memory-type Th2 cells are found in the colon of patients with ulcerative colitis (75, 76).
Memory Th2 cells, which are differentiated from effector Th2 cells, are central to adaptive immunity against parasite infection and play an important role in the host defense through inflammatory responses (36). Memory Th2 cells have also been implicated in the pathogenesis of chronic inflammatory diseases (1). Evidence from recent studies indicates that memory Th2 cells, including pathogenic memory Th2 cells, are more heterogeneous in their function than previously recognized. Thus, a precise understanding of the nature of the involvement of memory Th cells in various chronic inflammatory diseases is quite important for the development of new therapeutic approaches for intractable immune-related disorders. More recently, it has become clear that there are two types of memory CD4+ T cells: circulating and non-circulating (77). The non-circulating cells are now called tissue-resident memory T (TRM) cells (78, 79). Unlike circulating memory T cells, which circulate throughout the body via blood vessels and lymphatic vessels, TRM cells are retained in non-lymphoid tissues, such as the lungs, intestines, and skin (78). In the lungs of mice in which inflammation is induced by HDM, Th2 TRM cells are transcriptionally and functionally distinct from circulating memory Th2 cells (80). Th2 TRM cells have increased expression of Il5 and Il13 (80). In addition, in the lungs of mice, CD4+ TRM cells that were induced by repeated exposure to Aspergillus fumigatus caused an inflammatory response (13). Thus, Th2 TRM cells play an important role in type 2 inflammatory responses in local tissues (81) (Figure 2).
In the skin, TRM provide frontline immune defense (82, 83). Recently, a transcriptome analysis of human CD4+ TRM from healthy skin revealed an interesting overlap of the TRM transcriptome with that of pathogenic Th2 cells (84). For example, human CD4+ TRM were found to express specific molecules, including markers described in the previous section—CCR8 and PPAR-γ—which are closely linked to pathogenic Th2 cells. The biological relevance of this remarkable similarity remains to be elucidated; however, the result indicates that both TRM and pathogenic Th2 cells share a similar differentiation program for adaptation to the tissue environment of the skin. Interestingly, in mice, PPAR-γ mediates metabolic adaptation of CD8+ TRM to the lipid-rich microenvironment of the skin (23). Since both TRM and pathogenic Th2 cells are linked to the chronic inflammation of barrier tissues, the similarity in the transcriptome could be a hint to future therapeutic or even curative strategies.
In addition to inducing type 2 inflammatory responses, type 2 cytokines that are produced by Th2 cells also directly or indirectly help the repair of injured tissues by targeting a wide variety of immune and non-immune cells, including fibroblasts, epithelial cells, macrophages, and endothelial cells (Figure 3). Tissue repair is a tightly coordinated and highly dynamic process that restores tissues injured by parasitic infections, HDM, air pollution, and other conditions to their original state. We herein summarize the outline of the current understanding of the tissue repair processes, which are thought to be mainly conventional Th2 cells.
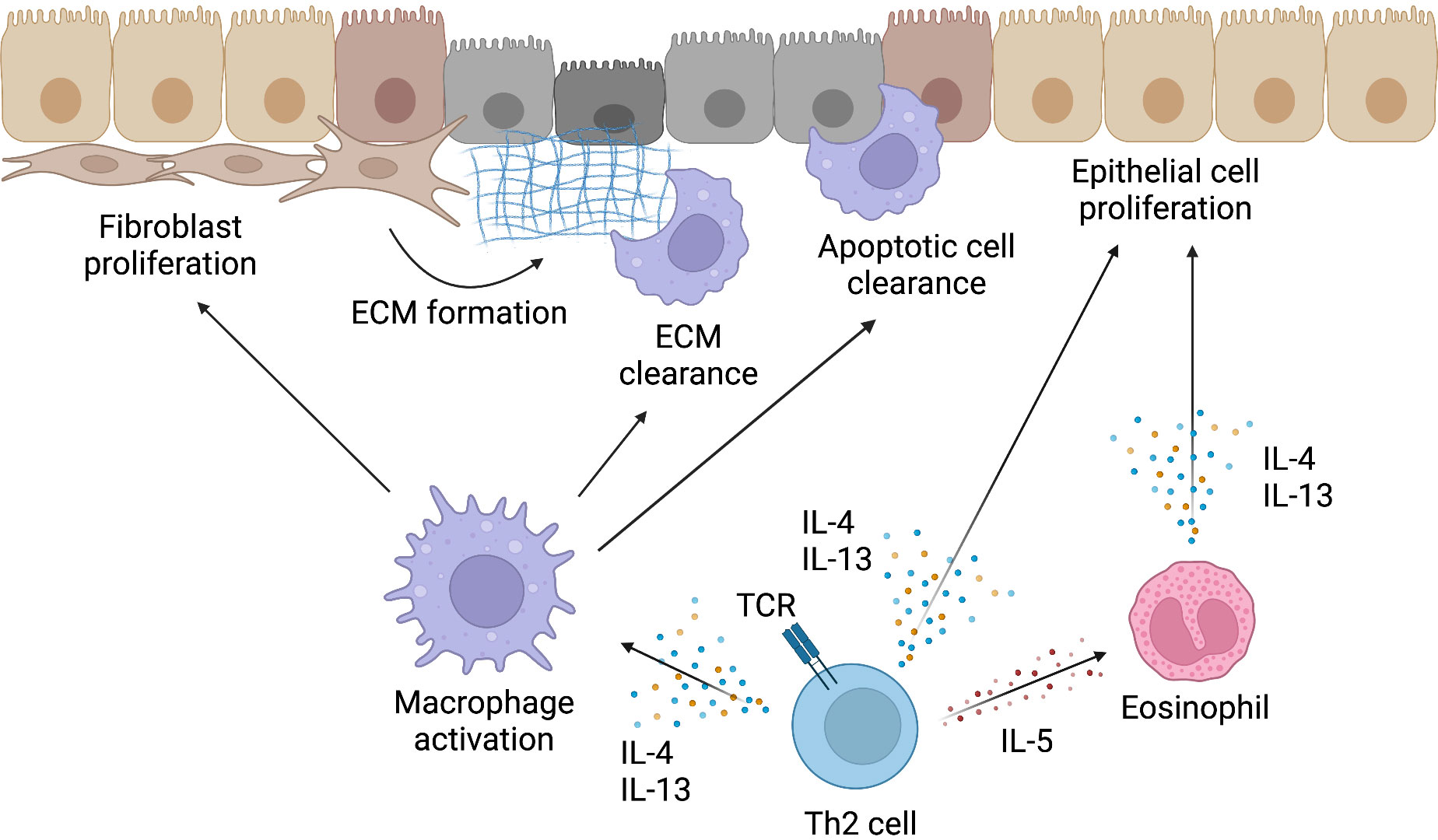
Figure 3 Mechanism of tissue repair by Th2 cells. Type 2 cytokines produced by Th2 cells not only induce type 2 inflammatory responses but also contribute to the repair of injured tissues. The activation of macrophages by IL-4 and IL-13 is important for the resolution of the type 2 inflammatory response and initiation of tissue repair. Activated macrophages induce fibroblast proliferation, remove extracellular matrix (ECM) formed by fibroblasts and other cells, and clear apoptotic cells in injured tissues. IL-4 and IL-13 produced by Th2 cells can also directly induce the proliferation of epithelial cells and contribute to the reconstruction of injured tissues. Furthermore, eosinophils, which are recruited to inflammatory sites by IL-5 produced by Th2 cells, can also produce IL-4 and IL-13. Thus, Th2 cells induce tissue repair both directly and indirectly. This figure was created with BioRender.com.
Basically, this process can be divided into three distinct but overlapping phases. These three stages are defined as “coagulation and inflammation”, “tissue formation”, and “tissue reconstruction” (85). In the “coagulation and inflammation” phase, the injury site is first isolated from outside the body by blood coagulation, and then the mobilization of inflammatory cells, such as Th2 cells, begins (85). The activation of macrophages by type 2 cytokines, such as IL-4 and IL-13, is important in the resolution of the subsequent stages of inflammation and in tissue formation and remodeling (6, 7).
In the second stage, “tissue formation”, pro-inflammatory signals, such as type 2 cytokines, weaken, and cell proliferation is initiated by growth factors, such as transforming growth factor-β (TGF-β) and basic fibroblast growth factor (FGF-2). At this stage, two processes are thought to be important: the removal of apoptotic cell debris by activated macrophages and the induction of fibroblast proliferation (86). In fact, wound healing is impaired when the number of macrophages is reduced (87). In particular, macrophages that receive type 2 cytokines (also referred as to an alternatively activated or M2 macrophages) are involved in tissue repair in the liver, central nervous system (CNS), heart, skeletal muscle, and lung (88). In mice, IL-4 produced by basophils and Th2 cells activates type 2 immune responses that convert the recruited inflammatory monocytes into wound-healing macrophages in the liver (89). These macrophages can replace the lost Kupffer cells in the liver and restore tissue homeostasis (89). Furthermore, the IL-4-activated macrophages are highly metabolically active and quickly consume critical amino acids in the injury site, which can slow the progression of fibrosis. This is due to the suppression of local CD4+ T cell proliferation and myofibroblast activation (90, 91). In the CNS of mice, depletion of intralesional IL-4-associated macrophages substantially delayed oligodendrocyte differentiation (92). In addition, multipotent adult progenitor cells protect against axonal dieback in spinal cord injury by expanding protective IL-4-associated macrophages in rats (93). In the heart, the critical contribution of IL-4- and IL-13-activated macrophages to cardiac repair was reported (94): Trib1-deficient mice that selectively lack IL-4-associated macrophages frequently experienced cardiac rupture after myocardial infarction. In the skeletal muscle in a mouse model of Duchenne muscular dystrophy, which is associated with IFN-γ-activated macrophages, IFN-γ-deficiency markedly improved the motor function in the late regenerative phase. It was associated with a skewing of macrophages toward an IL-4-associated anti-inflammatory phenotype (95). In humans, myogenic precursor cell activation by anti-inflammatory macrophages is proposed to be crucial to muscle cell regeneration following injury (96). Taken together, these results suggest a possible protective role of reparative macrophages in the skeletal muscle.
Eosinophils are also involved in tissue repair. For example, eosinophils induce hepatocyte proliferation by secreting IL-4 and IL-13 in an IL-4Rα-dependent manner during the tissue repair process in the liver (97). IL-4 and IL-13 also induce the proliferation of epithelial cells that express IL-4Rα, which promotes stem and progenitor cells in adult tissues to proliferate and differentiate (8, 9). Similar findings have also been reported in cases of skeletal muscle injury in a mouse model. In skeletal muscle injury, IL-4, which is derived from eosinophils, targets the regenerative function of muscle-resident adipocyte progenitors and fibroblasts that contribute to myogenesis (98). As described in Figure 2, IL-5, which is essential for the eosinophil function, is produced in large amounts by Tpath2 cells. Since Tpath2 cells are activated by IL-33 released from injured tissues in both mice and humans (39), the IL-33-Tpath2 axis is expected to play an important role in the repair of injured tissues by collaborating with macrophages and eosinophils. For example, IL-33 was shown to attenuate experimental autoimmune encephalomyelitis (EAE) in mice by suppressing IL-17 and IFN-γ production and by inducing reparative IL-4-associated macrophages, suggesting that type 2 cytokines produced by Tpath2 cells polarize anti-inflammatory macrophages (99). As targets of IL-33, ILC2s also contribute to tissue repair. In allergic (100–102) and infection models (40, 103), ILC2s, in addition to Th2 cells, have been reported to be a source of IL-13. In particular, ILC2s produce large amounts of IL-13 when stimulated by alarmin released during tissue injury (50). Lung ILC2s also produce amphiregulin in response to IL-33 and restore the respiratory function after influenza virus-induced lung injury (104).
In addition to the dynamic proliferation and differentiation of these cells, the formation of de novo ECM and the deposition of collagen supporting the migrated cells are also hallmarks of the “tissue formation” phase. In tissue repair, the ECM, which includes components such as collagen and fibronectin, is important because it mechanically stabilizes the damaged tissue, anchors growth factors, and serves as a scaffold for endothelial cells, immune cells, and fibroblasts and to migrate to the site of tissue damage or repair (10). The ECM can also bind growth factors, thus acting as a reservoir for growth factors, concentrating their activity close to the cell and protecting it from degradation (105). Through the action of tissue inhibitor of metalloproteinases (TIMPs) and matrix metalloproteinases (MMPs), myofibroblasts continuously regulate matrix deposition and turnover (106, 107). This process is organized by cell-to-cell and cell-to-ECM interactions.
The third stage, “tissue reconstruction,” is the final stage of tissue repair. In this stage, the tissue is reconstructed to its original form by the wound reconstruction process. Wound tissue reconstruction is a multi-step process. During this stage, which can last from several weeks to several months, myofibroblasts, macrophages, and endothelial cells present in the tissue undergo apoptosis, and newly formed blood vessels regress (85, 108). The newly formed collagen layer beneath the wound undergoes remodeling and reconstruction. During this phase, macrophages contribute to tissue repair by ingesting cellular debris and decomposing the excess ECM that has accumulated in and around the wound (108). Nonetheless, the depletion of skin macrophages during the tissue reconstruction phase does not significantly affect the outcome of tissue repair. This suggests that macrophages have redundant roles in tissue repair and that their function may overlap with the functions of other cell populations at this wound repair phase (109). However, how tissue repair is regulated by macrophages that are activated by type 2 cytokines and other cell types that characterize type 2 inflammation, such as mast cells, basophils, eosinophils, Th2 cells, and ILC2s, remains unclear.
Fibrosis is not a disease, rather it is the result of uncontrolled tissue repair responses that occur after many types of tissue injury, particularly caused by chronic inflammatory disease. Fibrosis is the widespread accumulation of fibrous connective tissue and is defined by the excessive accumulation of ECM components, such as collagen and fibronectin (110). In fact, the formation of fibrotic tissue is a normal and critical step in tissue repair in all organs in response to tissue injury caused by various triggers, such as infection, inflammation, autoimmune diseases, degenerative diseases, and tumors (111). When tissue is injured, the wound healing response is initiated through mechanisms that were described previously. If the injury is mild or non-repetitive, the deposition of ECM components increases only temporarily, allowing for efficient wound healing and facilitating the functional recovery of tissue structures (106, 107). However, if the damage is repeated or severe, wound healing mechanisms may become uncontrollable and ECM components may continue to accumulate, resulting in the destruction of tissue structures, organ dysfunction, and eventually organ failure (110, 112). In various diseases, such as hepatic cirrhosis, renal interstitial fibrosis, pulmonary fibrosis, myocardial infarction, graft-versus-host disease (GVHD), and systemic sclerosis, fibrotic remodeling impairs the organ function and causes high morbidity and mortality (111). Although the role of macrophages in fibrogenesis has been studied for many years, the mechanisms through which T cells regulate fibrogenesis are not fully understood.
Memory-type Tpath2 cells express high levels of the IL-33 receptor ST2 and play important roles in fibrogenesis in both HDM-induced murine lung inflammation and human airway inflammatory disease (5, 12) (Figure 4). In mouse, IL-33, which is mainly produced by epithelial cells undergoing tissue injury, activates memory-type Tpath2 cells and induces the production of amphiregulin, a member of the epidermal growth factor family (12). In HDM-induced murine lung inflammation, amphiregulin reprograms the eosinophil transcriptome via the epidermal growth factor receptor (EGFR) and promotes the production of osteopontin, which is a fibrosis-inducing protein (12, 113). Amphiregulin-producing memory-type Tpath2 cells and IL-5-producing memory-type Tpath2 cells were thought to be distinct subpopulations and to cooperate to induce eosinophilic inflammation and to establish airway fibrosis (12). In addition, an analysis of nasal polyps from patients with ECRS showed that the percentages of memory-type Tpath2 cells in nasal polyps were higher than those in peripheral blood (Figure 5). Furthermore, in nasal polyps of ECRS patients, amphiregulin-producing memory CD4+ T cells and osteopontin-producing eosinophils were found, with a dense fibrotic response (12, 114). Pharmacological blocking of EGFR signaling attenuates the expression of Spp1, encoding osteopontin, in both mouse and human eosinophils (12). Thus, the IL-33-amphiregulin-osteopontin axis induces a fibrotic response in chronic allergic inflammation in both mouse and human systems (12). In contrast, amphiregulin that is produced from activated lung ILC2s restores the respiratory function after influenza virus-induced lung injury (104). This suggests that amphiregulin has different functions depending on the cell type and biological background.
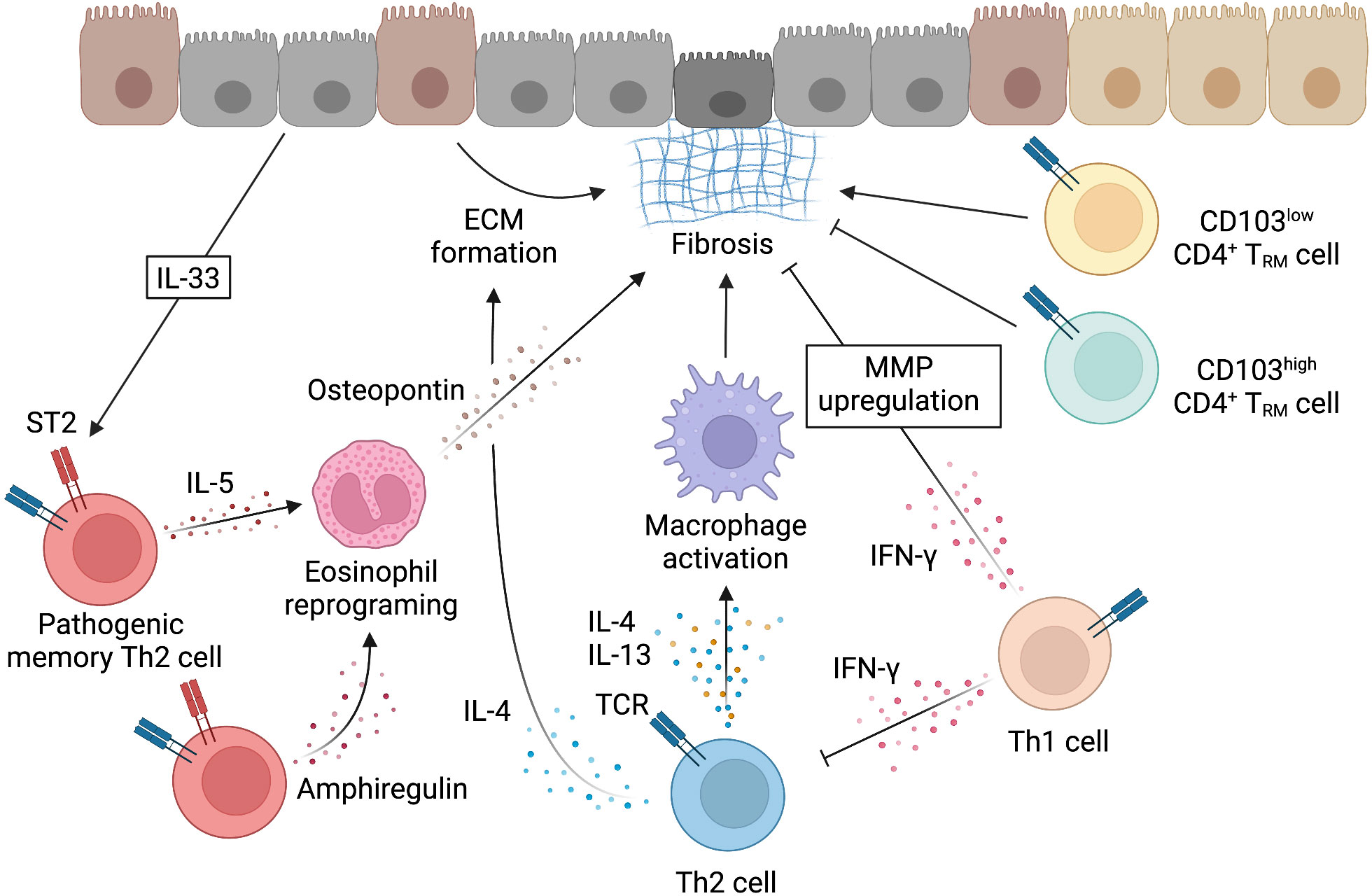
Figure 4 Mechanism underlying the induction of fibrosis by Th2 cells. Fibrosis can be induced when the tissue repair response by cells such as Th2 cells becomes uncontrollable. While macrophages that are activated by IL-4 and IL-13 work for tissue repair through the mechanisms described in Figure 3, fibrosis is induced by various mechanisms. IL-4 can also directly induce fibrosis by acting on epithelial cells. IL-33, which is produced by injured epithelial cells, activates memory-type pathogenic Th2 cells via ST2. Memory-type pathogenic Th2 cells include a subpopulation that produces IL-5 and a subpopulation that produces amphiregulin in response to IL-33 stimulation. Amphiregulin acts on eosinophils recruited by IL-5 and reprograms eosinophils to produce osteopontin and induce fibrosis. CD4+ T cells that are involved in fibrosis also include tissue-resident memory T (TRM) cells with the low or high expression of CD103. CD4+ TRM cells with the low expression of CD103 produce high levels of Th2 cytokines and promote fibrosis, whereas CD4+ TRM cells with the high expression of CD103 express high levels of Foxp3 and inhibit fibrosis. IFN-γ produced by Th1 cells has been reported to inhibit the induction of fibrosis by Th2 cells. IFN-γ may directly inhibit fibrosis by upregulating the expression of matrix metalloproteinases (MMP) that degrade ECM. This figure was created with BioRender.com.
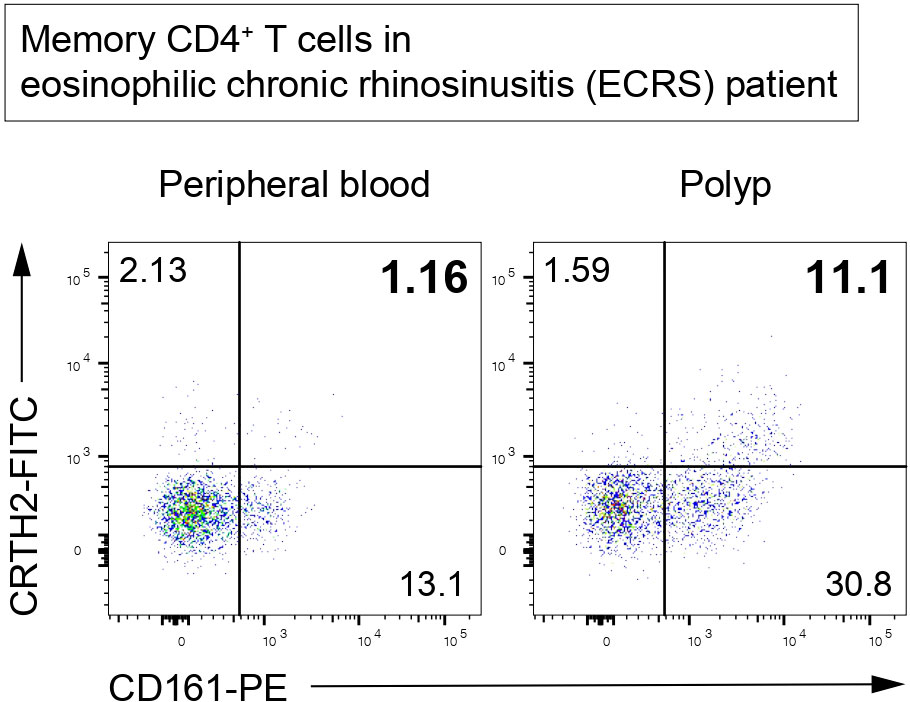
Figure 5 Memory-type Tpath2 cells in an ECRS patient. Representative plots of CD4+CD45RO+ (memory CD4+) T cells isolated from the peripheral blood and polyp of an ECRS patient. CRTH2-positive and CD161-positive cells are defined as Tpath2 cells (12).
Several cell types, including fibroblasts, epithelial cells, and macrophages, are known to induce fibrosis in response to Th2 cytokines (Figure 4). Fibroblasts play an important role in normal tissue repair and the induction of fibrosis by directly depositing ECM in response to various growth factors and cytokines, including Th2 cytokines (11). In particular, IL-13 signaling is important in the induction of fibrosis by fibroblasts (8). Pathological fibrosis is caused by direct signaling of IL-13 in PDGFRB+ fibroblasts, suggesting that the duration and magnitude of the IL-13 response may determine whether or not tissue repair evolves into fibrosis (8). In addition, several studies have suggested that senescent fibroblasts are resistant to apoptosis and thereby maintain inflammation and fibrosis by producing inflammatory cytokines, immunomodulators, proteases, and growth factors (115, 116). Therefore, senolytic and senotherapeutic drugs may have potential applications in the treatment of fibrosis and aging-related diseases (115, 116). In addition to being an important source of cytokines that trigger type 2 immune responses, epithelial cells—another cell type that induces fibrosis—can directly respond to IL-4 and IL-13 to induce important functions of type 2 immune responses, such as mucus secretion (9). IL-4Rα deficiency in epithelial cells was reported to completely diminish fibrosis during schistosomiasis and bile proliferation, which was associated with experimental liver fibrosis induced by the overexpression of IL-13 (8). Macrophages are also capable of inducing fibrosis in response to Th2 cytokines. Macrophages in tissues are important regulators of tissue fibrosis and play key roles in the initiation, maintenance, and resolution of tissue damage (87, 88, 117). Tissue macrophages are also important producers of chemokines that attract T cells and fibroblasts, which influence the formation of the fibrotic niche (118).
The macrophages that are activated by IL-4 and IL-13 have been reported to induce fibrosis by various mechanisms. First, IL-4 and IL-13 promote fibrosis as well as tissue repair by inducing fibroblast activation and the development of ECM-producing myofibroblasts through the promotion of TGF-β1 production from macrophages (87). Second, MMP12, which is also produced by macrophages, plays a role. MMP12 suppresses the expression of collagen-degrading proteins MMP2, MMP9, and MMP13, which cause an increased fibrotic response and decreased matrix degradation after infection (119). Furthermore, resistin-like alpha (RELMα) secreted by macrophages activates fibroblasts and upregulates lysyl hydroxylase 2, which controls mechanical cross-linking of collagen fibrils (120), resulting in the production of type 2 cytokines (120). In contrast, there was a report showing that macrophages may inhibit fibrosis and inflammation by competing for essential metabolites, such as l-arginine with T cells and other cells. In that report, mice with tissue macrophage-specific deletion of IL-4Rα showed increased inflammation after Schistosoma mansoni infection but little change in their liver fibrosis status (91, 121). In other words, the role of macrophages that are activated by IL-4 or IL-13 may vary depending on the organ or pathogenesis (122).
Eosinophils play an important role in fibrosis in the lungs and skin in chronic asthma and atopic dermatitis, respectively. In addition, vascular injury is induced by eosinophils after high-dose irradiation through various pro-fibrotic cytokines, including osteopontin, and eosinophil granule proteins (84, 123–125). In contrast, studies using eosinophil-deficient ΔdblGATA and TgPHIL mice demonstrated almost normal fibrosis, granulomatous inflammation, and type 2 cytokine production after infection (126). Thus, the importance of eosinophils in fibrosis remains controversial. It has also been reported that—in Th2 cell-driven allergic inflammation—a subset of eosinophils that are recruited to the tissue enhances inflammation, whereas another subset of eosinophils that is retained in the tissue prior to the induction of inflammation suppresses inflammation via the inhibition of bone marrow-derived dendritic cell (BMDC) maturation (127). Therefore, while the recruited eosinophil subset induces an inflammatory response and promotes the development of Th2 cell-dependent fibrosis, it is also possible that the tissue-retaining eosinophil population suppresses fibrosis via the suppression of inflammation (127). The existence of such functionally distinct populations of eosinophils may suggest the multi-functional roles of eosinophils in the induction and resolution of fibrosis.
Alarmins released from damaged tissues, such as IL-33, TSLP, and IL-25, have been reported to contribute not only to the induction of Th2 cell differentiation but also the initiation and progression of tissue fibrosis. For example, IL-33 induces the production of IL-13 by ILC2, macrophages (128), and eosinophils (129), as well as the activation of Tpath2 cells (130), as described above. It has been proposed that fibrosis is promoted through the induction of IL-13 production by eosinophils (129). In fact, IL-33 elevation has been reported in dermal tissue in dermatofibrosis (131, 132), lung and intestinal epithelium in patients with fibrotic colitis and pulmonary fibrosis (129, 133–135), and the liver of mice with liver fibrosis (133). TSLP is also detected in human and experimental colonic fibrosis (136), systemic sclerosis (137–139), pulmonary fibrosis (140, 141), and dermatofibrosis (142). IL-25, the third alarmin that promotes fibrosis, activates ILC2s to produce large amounts of the fibrosis-promoting cytokine IL-13 (143). In fact, the intranasal administration of recombinant IL-25 to mice causes airway inflammation and pulmonary fibrosis via the production of connective tissue growth factor (CTGF) and TGF-β1 (144). ILC2s that are activated by alarmins also produce large amounts of IL-5 (50). IL-5 recruits and activates eosinophils in bleomycin-induced pulmonary fibrosis (145). Attempts have been made to inhibit tissue fibrosis by inhibiting these alarmins. In mouse models of chronic liver fibrosis and acute pulmonary fibrosis, individual blocking of IL-33, TSLP, and IL-25 had no effect on IL-13-mediated pathology, but simultaneous blocking of these three alarmins significantly reduced fibrosis, eosinophilia, inflammation, and ILC2 mobilization (146). These results suggest that there may be redundancy in the fibrosis-inducing functions of IL-33, TSLP, and IL-25.
The elucidation of the mechanisms underlying tissue repair and fibrosis by Th2 cells and Th2 cytokines is an urgent issue, as COVID-19, the disease caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection, which is currently causing a global pandemic (147), is accompanied by fibrosis (148–151) and is particularly dangerous for patients with pulmonary fibrotic diseases (152). In patients with severe COVID-19, T helper lymphocytes were found to be biased toward a Th2-like phenotype (153). It has also been reported that miRNAs encoded by SARS-CoV-2 target IL-5 and IL-33 for transcriptional regulation, suggesting that Tpath2 cells may be associated with the pathology of COVID-19 (154). The enhancement of the type 2 immune response was detected in fatal SARS-CoV-2 infection (155) and severe COVID-19 cases (156, 157), and has been reported to be associated with adverse effects in other respiratory infections (158, 159). A meta-analysis of more than 50,000 hospitalized COVID-19 patients showed that the incidence of acute respiratory distress syndrome (ARDS) increased to 14.8% (160). In contrast, it has been observed that the prognosis of COVID-19 is not altered in patients with asthma and other eosinophil-related diseases (161, 162). Similarly, although preclinical studies suggest that eosinophils have potential antiviral activity, there is no evidence that eosinopenia induced by anti-eosinophil therapy increases susceptibility to SARS-CoV-2 (163). In addition, it is noteworthy that no eosinophilia has been observed in the lungs of COVID-19 patients (163). Thus, the inflammatory response induced by conventional or pathogenic Th2 cells and the subsequent fibrosis would be expected to have different effects on the pathology of COVID-19. Pulmonary fibrosis, a common signature in patients with virus-associated respiratory diseases, is also observed in the lungs of patients with SARS-CoV infection (164–166) and Middle East respiratory syndrome coronavirus (MERS-CoV) infection (167). In contrast, it has been reported that pulmonary fibrosis is a risk factor for severe COVID-19 infection (168). An international multicenter study reported that the survival rate of patients who were hospitalized with COVID-19 with complication by non-idiopathic or idiopathic pulmonary fibrosis was significantly lower than that of patients without pulmonary fibrosis (169). This means that patients with interstitial lung disease (ILD), especially fibrotic ILD, face a lower survival rate after COVID-19 infection than patients without ILD (169). In addition, risk factors for COVID-19 and idiopathic pulmonary fibrosis share many similarities, including age, sex, obesity, diabetes, smoking history, and hypertension, and both diseases begin with lung injury (170, 171). In short, a better understanding of the cellular and molecular mechanisms underlying tissue fibrosis will help us to control fibrosis in patients with severe diseases induced by viruses, including COVID-19.
Th2 TRM cells have been reported to play an important role in type 2 inflammatory responses in local tissues, but their role in tissue fibrosis induced during chronic inflammation has been unclear. Recently, we reported that tissue-resident CD44hiCD69hi CD4+ T cells showed the higher expression of fibrotic genes in comparison to circulating CD44lo or CD44hiCD69lo CD4+ T cells in the lungs of mice with repeated exposure to Aspergillus fumigatus (13). Furthermore, CD103lo CD4+ TRM cells were defined as pathogenic, while CD103hi CD4+ TRM cells were defined as immunosuppressive Treg cells, and they had a different epigenetic and transcriptional program in the inflamed lung. CD4+CD103lo TRM cells promoted the pathology of allergic inflammation and fibrotic response by increasing Th2 cytokines, such as IL-4, IL-5, and IL-13. In contrast, CD103hi tissue-resident Treg cells expressed Foxp3 and ameliorated the fibrotic response. Thus, tissue-resident CD4+ T cell populations with opposing functionality regulate the pathogenesis of fungal-induced chronic inflammatory response and subsequent fibrotic response in the lung (13) (Figure 4).
While Th2 cells induce tissue fibrosis through type-2 inflammatory responses, Th1 cells inhibit fibrosis through the production of the pro-inflammatory cytokine IFN-γ (Figure 4). First, it was reported that IFN-γ suppresses collagen synthesis by fibroblasts and attenuates fibrosis (172). In addition, Wynn et al. previously used IL-12 for the treatment of S. mansoni infection in mice and found that IL-12 not only increased the Th1 cytokine expression by inducing Th1 differentiation from naïve CD4+ T cells but also suppressed the inflammatory response induced by Th2 cells (173). IFN-γ upregulates the expression of MMPs, such as MMP-2, MMP-7, MMP-9, and MMP-13, by bone marrow-derived cells, resulting in a dramatic improvement in fibrosis (111). This proteolytic activity can alter the remodeling of ECM and help ameliorate fibrosis (174). In contrast, the induction of fibrosis by Th1 cells and IFN-γ has been reported in liver injury (175) and fibrotic diseases (176, 177). Therefore, the function of Th1 cells and IFN-γ in the suppression of fibrosis remains controversial. Another subset of cells that have been reported to suppress fibrosis are Treg cells. First, it has been reported that the function of Treg cells is reduced in cystic fibrosis patients infected with Pseudomonas aeruginosa (178). Second, in hypertensive mice treated with angiotensin II, the adoptive transfer of Treg cells improved cardiac hypertrophy and fibrosis of the heart (179). Furthermore, in Mdr2-deficient mice with sclerosing cholangitis, low-dose IL-2 treatment induced the proliferation of intrahepatic Treg cells and suppressed biliary injury and fibrosis (180). In contrast, there have been several reports on the exacerbation of fibrosis by Treg cells (181. 182). Thus, it is possible that Treg cells may have different functions in different disease models of fibrosis.
IL-13, a major Th2 effector cytokine for the induction of fibrosis, exerts its effects through a receptor complex containing IL-4Rα and IL-13Rα1. There is also a third high-affinity IL-13 receptor, IL-13Rα2, which is an inducible decoy receptor that inhibits the effector function of IL-13 by sequestering the IL-13 from the IL-4Rα/IL-13Rα1 signaling receptor complex (183). Several cytokines that are derived Th1 and Th17 have been reported to increase IL-13Rα2, suggesting a strong reciprocal regulation of the IL-13 function by Th1 and Th17-type immune responses (184). Indeed, combinations of TNF and several cytokines, including IL-4 or IL-17, have been shown to synergistically promote the expression of IL-13Rα2 and inhibit the ability of IL-13 to upregulate downstream targets in mouse and human fibroblasts (184).
A number of studies have reported the continuous activation of signaling via Th2 cytokines in fibrosis. Among these Th2 cytokines, IL-4 and IL-13 particularly promote fibrosis, and indeed, IL-4 and IL-13 levels are increased in many lung diseases (185–188). Therefore, blockade with antagonists and/or antibodies that target various aspects of the IL-4 and IL-13 signaling pathways in combination or alone is being explored as a therapeutic approach (189). However, the results of these trials in various diseases have been contradictory, and some trials have shown even worse outcomes in treated patients than in controls (190–192). These results suggest the complexity of the mechanism underlying the induction of fibrosis by IL-4 and IL-13 signaling.
In the course of experimental murine schistosomiasis and in a pulmonary granuloma model, blocking IL-13 notably reduces fibrosis but simultaneously increases IFN-γ production, followed by increased TNF production and necrosis of inflamed tissue, exacerbating lung and liver damage. However, blocking both IL-13 and IFN-γ markedly suppresses fibrosis and eliminates the simultaneous type 1 inflammation and subsequent tissue damage that is observed with anti-IL-13 alone (193). Similarly, blocking IL-4 or IL-13 in HDM-induced allergy models has been shown to induce considerable neutrophilic inflammation by Th17 cells. However, blocking both IL-13 and IL-17A protects mice from neutrophilic and eosinophilic inflammation and eliminates the associated airway hyperresponsiveness and mucus production, indicating that dual or multiple blockade strategies are effective in combating rebound inflammation (193, 194). Interestingly, some clinical trials in which portions of the IL-4 and/or IL-13 signaling pathways are blocked have shown disappointing results (190–192). This may be due to unintended disruption of important beneficial aspects of IL-4 and IL-13 signaling and/or dysregulation of type 1 and Th17-driven inflammatory responses. In contrast, anti-IL-5 therapy in asthmatic patients is reported to inhibit lung fibrosis without affecting the lung function (195). The result of this clinical trial is expected to be attributed to the recruitment and activation of eosinophils by IL-5 (196, 197). Based on these results, it is expected that targeting IL-5 signaling to specifically inhibit the Tpath2 cell function, rather than targeting IL-4 and IL-13 signaling to inhibit the function of whole Th2 cells, will allow for more precise inhibition of tissue fibrosis. Taken together, these studies suggest that careful targeting of therapeutic agents and consideration of dosage is needed to successfully block the pathological features of persistent type 2-driven inflammation. An ideal treatment would neither sacrifice the beneficial functions (e.g., wound repair and epithelial regeneration) nor induce detrimental relapsing inflammatory responses.
In this review, we provide a systematic overview of the role of conventional Th2 cells in the sequence of tissue inflammation, repair, and fibrosis, and discuss the role of Tpath2 cells (Table 1). As described above, Th2 cells play an important role in type-2 inflammation, tissue repair and tissue fibrosis via Th2 cytokines, which target various immune and non-immune cells. Among Th2 cells, Tpath2 cells have the ability of higher IL-5 production, which induces eosinophilic inflammation. Another subpopulation of Tpath2 cells, which produces amphiregulin, reprograms recruited-eosinophils to produce osteopontin, the fibrosis-inducing immunomodulatory protein. Furthermore, these Tpath2 cells show the signature of tissue residency. In conclusion, Tpath2 cells are expected to induce subsequent tissue fibrosis in addition to the previously reported induction of inflammatory responses.
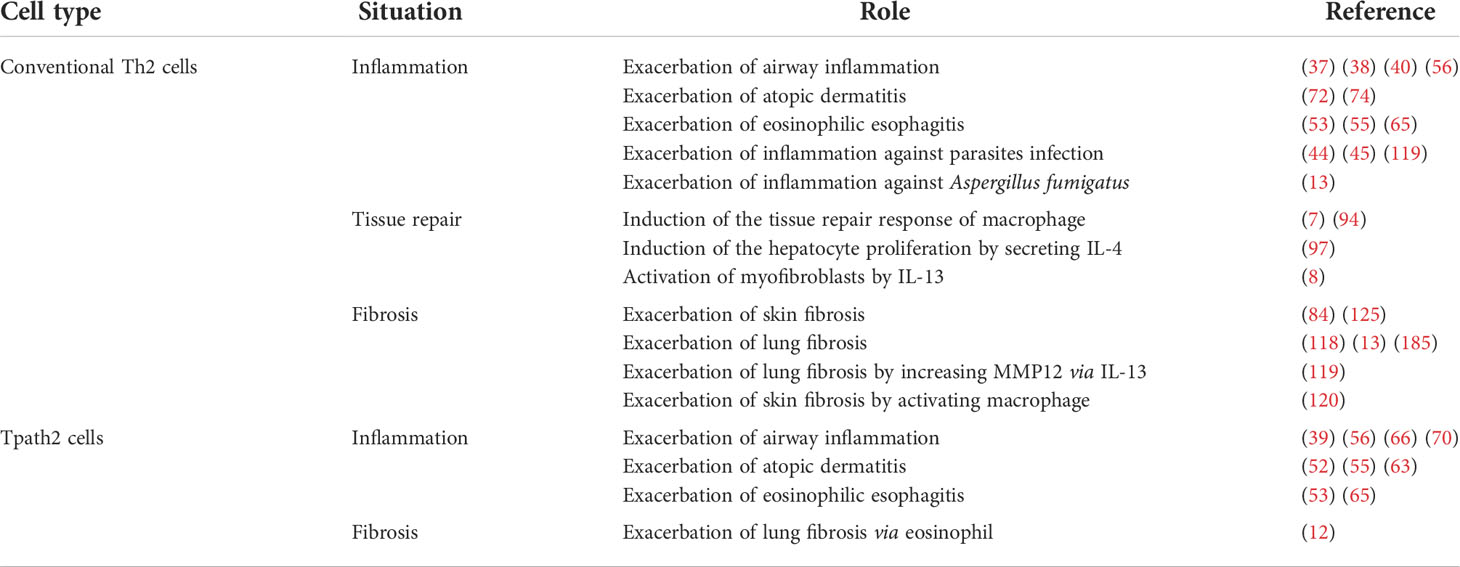
Table 1 The roles of conventional Th2 cells and Tpath2 cells in inflammation, tissue repair, and fibrosis.
Very recently, various novel mechanisms—beyond type-2 inflammation—that induce tissue fibrosis have emerged. For example, it has been reported that altering the gut microbiota may reduce fibrosis (198, 199). Checkpoint inhibitors for cancer treatment have recently received a great deal of attention, but they also show promise in relation to the resolution of fibrosis. It has been reported that checkpoint inhibitors that block costimulatory signals, such as CTLA4 and OX40L, can prevent fibrosis and induce the regression of established fibrosis (200–202). Liver oxidative stress associated with obesity has also been shown to be involved in T cell recruitment and fibrosis (203). Single-cell sequencing technology identifies the transcriptome profiles of individual cells in all cell types in certain tissues, and has allowed us to identify several cell populations that are important for the induction of fibrosis and also novel mechanisms in fibrogenesis (204, 205). In the near the future, this multifaceted approach will hopefully pave the way to the elucidation of the mechanism underlying the induction of fibrosis by Tpath2 cells.
Writing, peer-review, and editing: KK, AO, MK, KT, and KH, TN. All authors contributed to the article and approved the submitted version.
This work was supported by the following grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT Japan) (JP19H05650, JP18H05375, JP20K20383, JP20H03685, JP21H05120, JP21H05121, JP20KK0351, JP19K16683, JP21K20754, JP22H02885, JP22K15485, JP22K15484); the Japan Agency for Medical Research and Development, AMED (JP21gm1210003, JP21ek0410060, JP21ek0410082, JP22ek0410092); JST FOREST Project (JPMJFR200R); MSD Life Science Foundation; Takeda Science Foundation; and Kowa Life Science Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nakayama T, Hirahara K, Onodera A, Endo Y, Hosokawa H, Shinoda K, et al. Th2 cells in health and disease. Annu Rev Immunol (2017) 35:53–84. doi: 10.1146/annurev-immunol-051116-052350
2. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. i. definition according to profiles of lymphokine activities and secreted proteins. J Immunol (1986) 136(7):2348–57.
3. Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature (1996) 383(6603):787–93. doi: 10.1038/383787a0
4. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol (2015) 15(5):271–82. doi: 10.1038/nri3831
5. Endo Y, Hirahara K, Yagi R, Tumes DJ, Nakayama T. Pathogenic memory type Th2 cells in allergic inflammation. Trends Immunol (2014) 35(2):69–78. doi: 10.1016/j.it.2013.11.003
6. Minutti CM, Knipper JA, Allen JE, Zaiss DM. Tissue-specific contribution of macrophages to wound healing. Semin Cell Dev Biol (2017) 61:3–11. doi: 10.1016/j.semcdb.2016.08.006
7. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: Roles in homeostasis and disease. Annu Rev Immunol (2013) 31:317–43. doi: 10.1146/annurev-immunol-032712-095906
8. Gieseck RL 3rd, Ramalingam TR, Hart KM, Vannella KM, Cantu DA, Lu WY, et al. Interleukin-13 activates distinct cellular pathways leading to ductular reaction, steatosis, and fibrosis. Immunity (2016) 45(1):145–58. doi: 10.1016/j.immuni.2016.06.009
9. Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol (2010) 298(6):L715–31. doi: 10.1152/ajplung.00361.2009
10. Pakshir P, Hinz B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol (2018) 68–69:81–93. doi: 10.1016/j.matbio.2018.01.019
11. Nowarski R, Jackson R, Flavell RA. The stromal intervention: Regulation of immunity and inflammation at the epithelial-mesenchymal barrier. Cell (2017) 168(3):362–75. doi: 10.1016/j.cell.2016.11.040
12. Morimoto Y, Hirahara K, Kiuchi M, Wada T, Ichikawa T, Kanno T, et al. Amphiregulin-producing pathogenic memory T helper 2 cells instruct eosinophils to secrete osteopontin and facilitate airway fibrosis. Immunity (2018) 49(1):134–50.e6. doi: 10.1016/j.immuni.2018.04.023
13. Ichikawa T, Hirahara K, Kokubo K, Kiuchi M, Aoki A, Morimoto Y, et al. CD103(hi) treg cells constrain lung fibrosis induced by CD103(lo) tissue-resident pathogenic CD4 T cells. Nat Immunol (2019) 20(11):1469–80. doi: 10.1038/s41590-019-0494-y
14. Mowen KA, Glimcher LH. Signaling pathways in Th2 development. Immunol Rev (2004) 202:203–22. doi: 10.1111/j.0105-2896.2004.00209.x
15. Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol (2010) 10(4):225–35. doi: 10.1038/nri2735
16. Reiner SL. Development in motion: helper T cells at work. Cell (2007) 129(1):33–6. doi: 10.1016/j.cell.2007.03.019
17. Locksley RM. Nine lives: Plasticity among T helper cell subsets. J Exp Med (2009) 206(8):1643–6. doi: 10.1084/jem.20091442
18. O'Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (2010) 327(5969):1098–102. doi: 10.1126/science.1178334
19. Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol (2006) 24:607–56. doi: 10.1146/annurev.immunol.23.021704.115821
20. Onodera A, Nakayama T. Epigenetics of T cells regulated by Polycomb/Trithorax molecules. Trends Mol Med (2015) 21(5):330–40. doi: 10.1016/j.molmed.2015.03.001
21. Kuwahara M, Yamashita M, Shinoda K, Tofukuji S, Onodera A, Shinnakasu R, et al. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-beta and suppresses T(H)2 differentiation. Nat Immunol (2012) 13(8):778–86. doi: 10.1038/ni.2362
22. Horiuchi S, Onodera A, Hosokawa H, Watanabe Y, Tanaka T, Sugano S, et al. Genome-wide analysis reveals unique regulation of transcription of Th2-specific genes by GATA3. J Immunol (2011) 186(11):6378–89. doi: 10.4049/jimmunol.1100179
23. Sasaki T, Onodera A, Hosokawa H, Watanabe Y, Horiuchi S, Yamashita J, et al. Genome-wide gene expression profiling revealed a critical role for GATA3 in the maintenance of the Th2 cell identity. PLoS One (2013) 8(6):e66468. doi: 10.1371/journal.pone.0066468
24. Onodera A, Gonzalez-Avalos E, Lio CJ, Georges RO, Bellacosa A, Nakayama T, et al. Roles of TET and TDG in DNA demethylation in proliferating and non-proliferating immune cells. Genome Biol (2021) 22(1):186. doi: 10.1186/s13059-021-02384-1
25. Onodera A, Kokubo K, Nakayama T. Epigenetic and transcriptional regulation in the induction, maintenance, heterogeneity, and recall-response of effector and memory Th2 cells. Front Immunol (2018) 9:2929. doi: 10.3389/fimmu.2018.02929
26. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity (2013) 39(5):819–32. doi: 10.1016/j.immuni.2013.09.012
27. Nakayama T, Yamashita M. The TCR-mediated signaling pathways that control the direction of helper T cell differentiation. Semin Immunol (2010) 22(5):303–9. doi: 10.1016/j.smim.2010.04.010
28. Kiani A, Rao A, Aramburu J. Manipulating immune responses with immunosuppressive agents that target NFAT. Immunity (2000) 12(4):359–72. doi: 10.1016/S1074-7613(00)80188-0
29. Kiuchi M, Onodera A, Kokubo K, Ichikawa T, Morimoto Y, Kawakami E, et al. The Cxxc1 subunit of the trithorax complex directs epigenetic licensing of CD4+ T cell differentiation. J Exp Med (2021) 218(4):e20201690. doi: 10.1084/jem.20201690
30. Onodera A, Kiuchi M, Kokubo K, Nakayama T. Epigenetic regulation of inflammation by CxxC domain-containing proteins. Immunol Rev (2022) 305(1):137–51. doi: 10.1111/imr.13056
31. Roan F, Obata-Ninomiya K, Ziegler SF. Epithelial cell-derived cytokines: more than just signaling the alarm. J Clin Invest (2019) 129(4):1441–51. doi: 10.1172/JCI124606
32. Iinuma T, Okamoto Y, Yamamoto H, Inamine-Sasaki A, Ohki Y, Sakurai T, et al. Interleukin-25 and mucosal T cells in noneosinophilic and eosinophilic chronic rhinosinusitis. Ann Allergy Asthma Immunol (2015) 114(4):289–98. doi: 10.1016/j.anai.2015.01.013
33. Kotsiou OS, Gourgoulianis KI, Zarogiannis SG. IL-33/ST2 axis in organ fibrosis. Front Immunol (2018) 9:2432. doi: 10.3389/fimmu.2018.02432
34. Chae WJ, Ehrlich AK, Chan PY, Teixeira AM, Henegariu O, Hao L, et al. The wnt antagonist dickkopf-1 promotes pathological type 2 cell-mediated inflammation. Immunity (2016) 44(2):246–58. doi: 10.1016/j.immuni.2016.01.008
35. Onodera A, Kiuchi M, Kokubo K, Kato M, Ogino T, Horiuchi S, et al. Menin controls the memory Th2 cell function by maintaining the epigenetic integrity of Th2 cells. J Immunol (2017) 199(3):1153–62. doi: 10.4049/jimmunol.1602129
36. Ruterbusch M, Pruner KB, Shehata L, Pepper M. In vivo CD4(+) T cell differentiation and function: Revisiting the Th1/Th2 paradigm. Annu Rev Immunol (2020) 38:705–25. doi: 10.1146/annurev-immunol-103019-085803
37. Kimura MY, Hayashizaki K, Tokoyoda K, Takamura S, Motohashi S, Nakayama T. Crucial role for CD69 in allergic inflammatory responses: CD69-Myl9 system in the pathogenesis of airway inflammation. Immunol Rev (2017) 278(1):87–100. doi: 10.1111/imr.12559
38. Hayashizaki K, Kimura MY, Tokoyoda K, Hosokawa H, Shinoda K, Hirahara K, et al. Myosin light chains 9 and 12 are functional ligands for CD69 that regulate airway inflammation. Sci Immunol (2016) 1(3):eaaf9154. doi: 10.1126/sciimmunol.aaf9154
39. Nakayama T, Hirahara K, Kimura MY, Iwamura C, Kiuchi M, Kokubo K, et al. CD4+ T cells in inflammatory diseases: pathogenic T-helper cells and the CD69Myl9 system. Int Immunol (2021) 33(12):699–704. doi: 10.1093/intimm/dxab053
40. Van Dyken SJ, Nussbaum JC, Lee J, Molofsky AB, Liang HE, Pollack JL, et al. A tissue checkpoint regulates type 2 immunity. Nat Immunol (2016) 17(12):1381–7. doi: 10.1038/ni.3582
41. Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol (2018) 18(2):121–33. doi: 10.1038/nri.2017.118
42. Pezzulo AA, Tudas RA, Stewart CG, Buonfiglio LGV, Lindsay BD, Taft PJ, et al. HSP90 inhibitor geldanamycin reverts IL-13- and IL-17-induced airway goblet cell metaplasia. J Clin Invest (2019) 129(2):744–58. doi: 10.1172/JCI123524
43. Kistemaker LE, Hiemstra PS, Bos IS, Bouwman S, van den Berge M, Hylkema MN, et al. Tiotropium attenuates IL-13-induced goblet cell metaplasia of human airway epithelial cells. Thorax (2015) 70(7):668–76. doi: 10.1136/thoraxjnl-2014-205731
44. Dougan M, Dranoff G, Dougan SK. GM-CSF, IL-3, and IL-5 family of cytokines: Regulators of inflammation. Immunity (2019) 50(4):796–811. doi: 10.1016/j.immuni.2019.03.022
45. Ravin KA, Loy M. The eosinophil in infection. Clin Rev Allergy Immunol (2016) 50(2):214–27. doi: 10.1007/s12016-015-8525-4
46. Spencer LA, Weller PF. Eosinophils and Th2 immunity: Contemporary insights. Immunol Cell Biol (2010) 88(3):250–6. doi: 10.1038/icb.2009.115
47. Fahy JV. Type 2 inflammation in asthma–present in most, absent in many. Nat Rev Immunol (2015) 15(1):57–65. doi: 10.1038/nri3786
48. Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O'Neill KR, et al. Defining a link with asthma in mice congenitally deficient in eosinophils. Science (2004) 305(5691):1773–6. doi: 10.1126/science.1099472
49. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature (2010) 464(7293):1367–70. doi: 10.1038/nature08900
50. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature (2013) 502(7470):245–8. doi: 10.1038/nature12526
51. Mikami Y, Takada Y, Hagihara Y, Kanai T. Innate lymphoid cells in organ fibrosis. Cytokine Growth Factor Rev (2018) 42:27–36. doi: 10.1016/j.cytogfr.2018.07.002
52. Islam SA, Chang DS, Colvin RA, Byrne MH, McCully ML, Moser B, et al. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat Immunol (2011) 12(2):167–77. doi: 10.1038/ni.1984
53. Mitson-Salazar A, Yin Y, Wansley DL, Young M, Bolan H, Arceo S, et al. Hematopoietic prostaglandin d synthase defines a proeosinophilic pathogenic effector human T(H)2 cell subpopulation with enhanced function. J Allergy Clin Immunol (2016) 137(3):907–18.e9. doi: 10.1016/j.jaci.2015.08.007
54. Seumois G, Zapardiel-Gonzalo J, White B, Singh D, Schulten V, Dillon M, et al. Transcriptional profiling of Th2 cells identifies pathogenic features associated with asthma. J Immunol (2016) 197(2):655–64. doi: 10.4049/jimmunol.1600397
55. Wambre E, Bajzik V, DeLong JH, O'Brien K, Nguyen QA, Speake C, et al. A phenotypically and functionally distinct human TH2 cell subpopulation is associated with allergic disorders. Sci Transl Med (2017) 9(401):eaam9171. doi: 10.1126/scitranslmed.aam9171
56. Hirahara K, Shinoda K, Morimoto Y, Kiuchi M, Aoki A, Kumagai J, et al. Immune cell-Epithelial/Mesenchymal interaction contributing to allergic airway inflammation associated pathology. Front Immunol (2019) 10:570. doi: 10.3389/fimmu.2019.00570
57. Calise J, Garabatos N, Bajzik V, Farrington M, Robinson D, Jeong D, et al. Optimal human pathogenic TH2 cell effector function requires local epithelial cytokine signaling. J Allergy Clin Immunol (2021) 148(3):867–75.e4. doi: 10.1016/j.jaci.2021.02.019
58. Nagase H, Ueki S, Fujieda S. The roles of IL-5 and anti-IL-5 treatment in eosinophilic diseases: Asthma, eosinophilic granulomatosis with polyangiitis, and eosinophilic chronic rhinosinusitis. Allergol Int (2020) 69(2):178–86. doi: 10.1016/j.alit.2020.02.002
59. Shinoda K, Hirahara K, Iinuma T, Ichikawa T, Suzuki AS, Sugaya K, et al. Thy1+IL-7+ lymphatic endothelial cells in iBALT provide a survival niche for memory T-helper cells in allergic airway inflammation. Proc Natl Acad Sci USA (2016) 113(20):E2842–51. doi: 10.1073/pnas.1512600113
60. Wen T, Rothenberg ME. Cell-by-cell deciphering of T cells in allergic inflammation. J Allergy Clin Immunol (2019) 144(5):1143–8. doi: 10.1016/j.jaci.2019.10.001
61. Hewitt RJ, Lloyd CM. Regulation of immune responses by the airway epithelial cell landscape. Nat Rev Immunol (2021) 21(6):347–62. doi: 10.1038/s41577-020-00477-9
62. Bertschi NL, Bazzini C, Schlapbach C. The concept of pathogenic TH2 cells: Collegium internationale allergologicum update 2021. Int Arch Allergy Immunol (2021) 182(5):365–80. doi: 10.1159/000515144
63. Mitson-Salazar A, Prussin C. Pathogenic effector Th2 cells in allergic eosinophilic inflammatory disease. Front Med (Lausanne) (2017) 4:165. doi: 10.3389/fmed.2017.00165
64. Satkunanathan S, Kumar N, Bajorek M, Purbhoo MA, Culley FJ. Respiratory syncytial virus infection, TLR3 ligands, and proinflammatory cytokines induce CD161 ligand LLT1 expression on the respiratory epithelium. J Virol (2014) 88(5):2366–73. doi: 10.1128/JVI.02789-13
65. Wen T, Aronow BJ, Rochman Y, Rochman M, Kc K, Dexheimer PJ, et al. Single-cell RNA sequencing identifies inflammatory tissue T cells in eosinophilic esophagitis. J Clin Invest (2019) 129(5):2014–28. doi: 10.1172/JCI125917
66. Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med (2019) 25(7):1153–63. doi: 10.1038/s41591-019-0468-5
67. Seumois G, Ramirez-Suastegui C, Schmiedel BJ, Liang S, Peters B, Sette A, et al. Single-cell transcriptomic analysis of allergen-specific T cells in allergy and asthma. Sci Immunol (2020) 5(48):eaba6087. doi: 10.1126/sciimmunol.aba6087
68. Nobs SP, Natali S, Pohlmeier L, Okreglicka K, Schneider C, Kurrer M, et al. PPARgamma in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J Exp Med (2017) 214(10):3015–35. doi: 10.1084/jem.20162069
69. Chen T, Tibbitt CA, Feng X, Stark JM, Rohrbeck L, Rausch L, et al. PPAR-gamma promotes type 2 immune responses in allergy and nematode infection. Sci Immunol (2017) 2(9):eaal5196. doi: 10.1126/sciimmunol.aal5196
70. Hirahara K, Aoki A, Kiuchi M, Nakayama T. Memory-type pathogenic TH2 cells and ILC2s in type 2 allergic inflammation. J Allergy Clin Immunol (2021) 147(6):2063–6. doi: 10.1016/j.jaci.2021.02.006
71. Corren J. New targeted therapies for uncontrolled asthma. J Allergy Clin Immunol Pract (2019) 7(5):1394–403. doi: 10.1016/j.jaip.2019.03.022
72. Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N, et al. Atopic dermatitis is an IL-13-Dominant disease with greater molecular heterogeneity compared to psoriasis. J Invest Dermatol (2019) 139(7):1480–9. doi: 10.1016/j.jid.2018.12.018
73. Oldhoff JM, Darsow U, Werfel T, Katzer K, Wulf A, Laifaoui J, et al. AntiIL-5 recombinant humanized monoclonal antibody (mepolizumab) for the treatment of atopic dermatitis. Allergy (2005) 60(5):693–6. doi: 10.1111/j.1398-9995.2005.00791.x
74. Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers (2018) 4(1):1. doi: 10.1038/s41572-018-0001-z
75. Joost P, Methner A. Phylogenetic analysis of 277 human G-proteincoupled receptors as a tool for the prediction of orphan receptor ligands. Genome Biol (2002) 3(11):RESEARCH0063.
76. Nguyen LP, Pan J, Dinh TT, Hadeiba H, O'Hara E 3rd, Ebtikar A, et al. Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat Immunol (2015) 16(2):207–13. doi: 10.1038/ni.3079
77. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
78. Masopust D, Soerens AG. Tissue-resident T cells and other resident leukocytes. Annu Rev Immunol (2019) 37:521–46. doi: 10.1146/annurev-immunol-042617-053214
79. Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol (2016) 16(2):79–89. doi: 10.1038/nri.2015.3
80. Rahimi RA, Nepal K, Cetinbas M, Sadreyev RI, Luster AD. Distinct functions of tissue-resident and circulating memory Th2 cells in allergic airway disease. J Exp Med (2020) 217(9):e20190865. doi: 10.1084/jem.20190865
81. Hirahara K, Kokubo K, Aoki A, Kiuchi M, Nakayama T. The role of CD4(+) resident memory T cells in local immunity in the mucosal tissue - protection versus pathology. Front Immunol (2021) 12:616309. doi: 10.3389/fimmu.2021.616309
82. Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med (2015) 7(279):279ra39. doi: 10.1126/scitranslmed.3010302
83. Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med (2012) 4(117):117ra7. doi: 10.1126/scitranslmed.3003008
84. Lee EJ, Kim JW, Yoo H, Kwak W, Choi WH, Cho S, et al. Single highdose irradiation aggravates eosinophil-mediated fibrosis through IL-33 secreted from impaired vessels in the skin compared to fractionated irradiation. Biochem Biophys Res Commun (2015) 464(1):20–6. doi: 10.1016/j.bbrc.2015.05.081
85. Martin P. Wound healing–aiming for perfect skin regeneration. Science (1997) 276(5309):75–81. doi: 10.1126/science.276.5309.75
86. Lech M, Anders HJ. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta (2013) 1832(7):989–97. doi: 10.1016/j.bbadis.2012.12.001
87. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
88. Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol (2017) 79:593–617. doi: 10.1146/annurev-physiol-022516-034356
89. Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liverresident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity (2015) 42(1):145–58. doi: 10.1016/j.immuni.2014.12.020
90. Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol (2015) 36(4):229–39. doi: 10.1016/j.it.2015.02.004
91. Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog (2009) 5(4):e1000371. doi: 10.1371/journal.ppat.1000371
92. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci (2013) 16(9):1211–8. doi: 10.1038/nn.3469
93. Busch SA, Hamilton JA, Horn KP, Cuascut FX, Cutrone R, Lehman N, et al. Multipotent adult progenitor cells prevent macrophage-mediated axonal dieback and promote regrowth after spinal cord injury. J Neurosci (2011) 31(3):944–53. doi: 10.1523/JNEUROSCI.3566-10.2011
94. Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest (2016) 126(6):2151–66. doi: 10.1172/JCI85782
95. Villalta SA, Deng B, Rinaldi C, Wehling-Henricks M, Tidball JG. IFNgamma promotes muscle damage in the mdx mouse model of duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J Immunol (2011) 187(10):5419–28. doi: 10.4049/jimmunol.1101267
96. Saclier M, Yacoub-Youssef H, Mackey AL, Arnold L, Ardjoune H, Magnan M, et al. Differentially activated macrophages orchestrate myogenic precursor cell fate during human skeletal muscle regeneration. Stem Cells (2013) 31(2):384–96. doi: 10.1002/stem.1288
97. Goh YP, Henderson NC, Heredia JE, Red Eagle A, Odegaard JI, Lehwald N, et al. Eosinophils secrete IL-4 to facilitate liver regeneration. Proc Natl Acad Sci U S A (2013) 110(24):9914–9. doi: 10.1073/pnas.1304046110
98. Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell (2013) 153(2):376–88. doi: 10.1016/j.cell.2013.02.053
99. Jiang HR, Milovanovic M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-gamma production and inducing alternatively activated macrophages. Eur J Immunol (2012) 42(7):1804–14. doi: 10.1002/eji.201141947
100. Van Dyken SJ, Liang HE, Naikawadi RP, Woodruff PG, Wolters PJ, Erle DJ, et al. Spontaneous chitin accumulation in airways and age-related fibrotic lung disease. Cell (2017) 169(3):497–509.e13. doi: 10.1016/j.cell.2017.03.044
101. Van Dyken SJ, Mohapatra A, Nussbaum JC, Molofsky AB, Thornton EE, Ziegler SF, et al. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gammadelta T cells. Immunity (2014) 40(3):414–24.
102. Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol (2011) 12(11):1071–7. doi: 10.1038/ni.2133
103. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity (2014) 41(2):283–95. doi: 10.1016/j.immuni.2014.06.016
104. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12(11):1045–54. doi: 10.1038/ni.2131
105. Schultz GS, Wysocki A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen (2009) 17(2):153–62. doi: 10.1111/j.1524-475X.2009.00466.x
106. Lindsey ML, Iyer RP, Jung M, DeLeon-Pennell KY, Ma Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J Mol Cell Cardiol (2016) 91:134–40. doi: 10.1016/j.yjmcc.2015.12.018
107. Craig VJ, Zhang L, Hagood JS, Owen CA. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol (2015) 53(5):585–600. doi: 10.1165/rcmb.2015-0020TR
108. Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res (2012) 49(1):35–43. doi: 10.1159/000339613
109. Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Muller W, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol (2010) 184(7):3964–77. doi: 10.4049/jimmunol.0903356
110. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature (2020) 587(7835):555–66. doi: 10.1038/s41586-020-2938-9
111. Zhang M, Zhang S. T Cells in fibrosis and fibrotic diseases. Front Immunol (2020) 11:1142. doi: 10.3389/fimmu.2020.01142
112. Singh B, Kasam RK, Sontake V, Wynn TA, Madala SK. Repetitive intradermal bleomycin injections evoke T-helper cell 2 cytokine-driven pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2017) 313(5):L796–806. doi: 10.1152/ajplung.00184.2017
113. Shoyab M, Plowman GD, McDonald VL, Bradley JG, Todaro GJ. Structure and function of human amphiregulin: A member of the epidermal growth factor family. Science (1989) 243(4894 Pt 1):1074–6. doi: 10.1126/science.2466334
114. Nakayama T, Yoshikawa M, Asaka D, Okushi T, Matsuwaki Y, Otori N, et al. Mucosal eosinophilia and recurrence of nasal polyps - new classification of chronic rhinosinusitis. Rhinology. (2011) 49(4):392–6. doi: 10.4193/Rhino10.261
115. Schafer MJ, Haak AJ, Tschumperlin DJ, LeBrasseur NK. Targeting senescent cells in fibrosis: Pathology, paradox, and practical considerations. Curr Rheumatol Rep (2018) 20(1):3. doi: 10.1007/s11926-018-0712-x
116. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine (2019) 47:446–56. doi: 10.1016/j.ebiom.2019.08.069
117. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17(5):306–21. doi: 10.1038/nri.2017.11
118. Borthwick LA, Barron L, Hart KM, Vannella KM, Thompson RW, Oland S, et al. Macrophages are critical to the maintenance of IL-13-dependent lung inflammation and fibrosis. Mucosal Immunol (2016) 9(1):38–55. doi: 10.1038/mi.2015.34
119. Madala SK, Pesce JT, Ramalingam TR, Wilson MS, Minnicozzi S, Cheever AW, et al. Matrix metalloproteinase 12-deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL-13-dependent fibrosis. J Immunol (2010) 184(7):3955–63. doi: 10.4049/jimmunol.0903008
120. Knipper JA, Willenborg S, Brinckmann J, Bloch W, Maass T, Wagener R, et al. Interleukin-4 receptor alpha signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity (2015) 43(4):803–16. doi: 10.1016/j.immuni.2015.09.005
121. Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol (2003) 24(6):302–6. doi: 10.1016/S1471-4906(03)00132-7
122. Barron L, Smith AM, El Kasmi KC, Qualls JE, Huang X, Cheever A, et al. Role of arginase 1 from myeloid cells in th2-dominated lung inflammation. PLoS One (2013) 8(4):e61961. doi: 10.1371/journal.pone.0061961
123. Aceves SS, Broide DH. Airway fibrosis and angiogenesis due to eosinophil trafficking in chronic asthma. Curr Mol Med (2008) 8(5):350–8. doi: 10.2174/156652408785161023
124. Percopo CM, Qiu Z, Phipps S, Foster PS, Domachowske JB, Rosenberg HF. Pulmonary eosinophils and their role in immunopathologic responses to formalininactivated pneumonia virus of mice. J Immunol (2009) 183(1):604–12. doi: 10.4049/jimmunol.0802270
125. Oyoshi MK, He R, Kanaoka Y, ElKhal A, Kawamoto S, Lewis CN, et al. Eosinophil-derived leukotriene C4 signals via type 2 cysteinyl leukotriene receptor to promote skin fibrosis in a mouse model of atopic dermatitis. Proc Natl Acad Sci USA (2012) 109(13):4992–7. doi: 10.1073/pnas.1203127109
126. Swartz JM, Dyer KD, Cheever AW, Ramalingam T, Pesnicak L, Domachowske JB, et al. Schistosoma mansoni infection in eosinophil lineage-ablated mice. Blood (2006) 108(7):2420–7. doi: 10.1182/blood-2006-04-015933
127. Mesnil C, Raulier S, Paulissen G, Xiao X, Birrell MA, Pirottin D, et al. Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J Clin Invest (2016) 126(9):3279–95. doi: 10.1172/JCI85664
128. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol (2014) 134(6):1422–32.e11. doi: 10.1016/j.jaci.2014.05.011
129. Masterson JC, Capocelli KE, Hosford L, Biette K, McNamee EN, de Zoeten EF, et al. Eosinophils and IL-33 perpetuate chronic inflammation and fibrosis in a pediatric population with stricturing crohn's ileitis. Inflammation Bowel Dis (2015) 21(10):2429–40.
130. Endo Y, Hirahara K, Iinuma T, Shinoda K, Tumes DJ, Asou HK, et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity (2015) 42(2):294–308. doi: 10.1016/j.immuni.2015.01.016
131. Yanaba K, Yoshizaki A, Asano Y, Kadono T, Sato S. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol (2011) 30(6):825–30. doi: 10.1007/s10067-011-1686-5
132. Rankin AL, Mumm JB, Murphy E, Turner S, Yu N, McClanahan TK, et al. IL-33 induces IL-13-dependent cutaneous fibrosis. J Immunol (2010) 184(3):1526–35. doi: 10.4049/jimmunol.0903306
133. Gao Y, Liu Y, Yang M, Guo X, Zhang M, Li H, et al. IL-33 treatment attenuated diet-induced hepatic steatosis but aggravated hepatic fibrosis. Oncotarget (2016) 7(23):33649–61. doi: 10.18632/oncotarget.9259
134. Roussel L, Farias R, Rousseau S. IL-33 is expressed in epithelia from patients with cystic fibrosis and potentiates neutrophil recruitment. J Allergy Clin Immunol (2013) 131(3):913–6. doi: 10.1016/j.jaci.2012.10.019
135. Lopetuso LR, Scaldaferri F, Pizarro TT. Emerging role of the interleukin (IL)-33/ST2 axis in gut mucosal wound healing and fibrosis. Fibrogenesis Tissue Repair (2012) 5(1):18. doi: 10.1186/1755-1536-5-18
136. Son A, Oshio T, Kawamura YI, Hagiwara T, Yamazaki M, Inagaki-Ohara K, et al. TWEAK/Fn14 pathway promotes a T helper 2-type chronic colitis with fibrosis in mice. Mucosal Immunol (2013) 6(6):1131–42. doi: 10.1038/mi.2013.10
137. Truchetet ME, Demoures B, Eduardo Guimaraes J, Bertrand A, Laurent P, Jolivel V, et al. Platelets induce thymic stromal lymphopoietin production by endothelial cells: Contribution to fibrosis in human systemic sclerosis. Arthritis Rheumatol (2016) 68(11):2784–94. doi: 10.1002/art.39817
138. Usategui A, Criado G, Izquierdo E, Del Rey MJ, Carreira PE, Ortiz P, et al. A profibrotic role for thymic stromal lymphopoietin in systemic sclerosis. Ann Rheum Dis (2013) 72(12):2018–23. doi: 10.1136/annrheumdis-2012-202279
139. Christmann RB, Mathes A, Affandi AJ, Padilla C, Nazari B, Bujor AM, et al. Thymic stromal lymphopoietin is up-regulated in the skin of patients with systemic sclerosis and induces profibrotic genes and intracellular signaling that overlap with those induced by interleukin-13 and transforming growth factor beta. Arthritis Rheumatol (2013) 65(5):1335–46. doi: 10.1002/art.37859
140. Lee JU, Chang HS, Lee HJ, Jung CA, Bae DJ, Song HJ, et al. Upregulation of interleukin-33 and thymic stromal lymphopoietin levels in the lungs of idiopathic pulmonary fibrosis. BMC Pulm Med (2017) 17(1):39. doi: 10.1186/s12890-017-0380-z
141. Herro R, Da Silva Antunes R, Aguilera AR, Tamada K, Croft M. Tumor necrosis factor superfamily 14 (LIGHT) controls thymic stromal lymphopoietin to drive pulmonary fibrosis. J Allergy Clin Immunol (2015) 136(3):757–68. doi: 10.1016/j.jaci.2014.12.1936
142. Oh MH, Oh SY, Yu J, Myers AC, Leonard WJ, Liu YJ, et al. IL-13 induces skin fibrosis in atopic dermatitis by thymic stromal lymphopoietin. J Immunol (2011) 186(12):7232–42. doi: 10.4049/jimmunol.1100504
143. Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci USA (2014) 111(1):367–72. doi: 10.1073/pnas.1315854111
144. Yao X, Wang W, Li Y, Lv Z, Guo R, Corrigan CJ, et al. Characteristics of IL-25 and allergen-induced airway fibrosis in a murine model of asthma. Respirology (2015) 20(5):730–8. doi: 10.1111/resp.12546
145. Gharaee-Kermani M, Phan SH. Lung interleukin-5 expression in murine bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol (1997) 16(4):438–47. doi: 10.1165/ajrcmb.16.4.9115755
146. Vannella KM, Ramalingam TR, Borthwick LA, Barron L, Hart KM, Thompson RW, et al. Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci Transl Med (2016) 8(337):337ra65. doi: 10.1126/scitranslmed.aaf1938
147. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med (2020) 382(18):1708–20. doi: 10.1056/NEJMoa2002032
148. Xu YH, Dong JH, An WM, Lv XY, Yin XP, Zhang JZ, et al. Clinical and computed tomographic imaging features of novel coronavirus pneumonia caused by SARS-CoV-2. J Infect (2020) 80(4):394–400. doi: 10.1016/j.jinf.2020.02.017
149. Ojha V, Mani A, Pandey NN, Sharma S, Kumar S. CT in coronavirus disease 2019 (COVID-19): a systematic review of chest CT findings in 4410 adult patients. Eur Radiol (2020) 30(11):6129–38. doi: 10.1007/s00330-020-06975-7
150. Liu Q, Wang RS, Qu GQ, Wang YY, Liu P, Zhu YZ, et al. Gross examination report of a COVID-19 death autopsy. Fa Yi Xue Za Zhi (2020) 36(1):21–3.
151. Yao XH, Li TY, He ZC, Ping YF, Liu HW, Yu SC, et al. [A pathological report of three COVID-19 cases by minimal invasive autopsies]. Zhonghua Bing Li Xue Za Zhi (2020) 49(5):411–7.
152. Deschamp AR, Hatch JE, Slaven JE, Gebregziabher N, Storch G, Hall GL, et al. Early respiratory viral infections in infants with cystic fibrosis. J Cyst Fibros (2019) 18(6):844–50. doi: 10.1016/j.jcf.2019.02.004
153. Notarbartolo S, Ranzani V, Bandera A, Gruarin P, Bevilacqua V, Putignano AR, et al. Integrated longitudinal immunophenotypic, transcriptional and repertoire analyses delineate immune responses in COVID-19 patients. Sci Immunol (2021) 6(62):eabg5021. doi: 10.1126/sciimmunol.abg5021
154. Aydemir MN, Aydemir HB, Korkmaz EM, Budak M, Cekin N, Pinarbasi E, et al. Computationally predicted SARS-COV-2 encoded microRNAs, JAK/STAT and TGFB signaling pathways. Gene Rep (2021) 22:101012. doi: 10.1016/j.genrep.2020.101012
155. Li CK, Wu H, Yan H, Ma S, Wang L, Zhang M, et al. T Cell responses to whole SARS coronavirus in humans. J Immunol (2008) 181(8):5490–500. doi: 10.4049/jimmunol.181.8.5490
156. Lombardi A, Trombetta E, Cattaneo A, Castelli V, Palomba E, Tirone M, et al. Early phases of COVID-19 are characterized by a reduction in lymphocyte populations and the presence of atypical monocytes. Front Immunol (2020) 11:560330. doi: 10.3389/fimmu.2020.560330
157. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature (2020) 584(7821):463–9. doi: 10.1038/s41586-020-2588-y
158. Shah VK, Firmal P, Alam A, Ganguly D, Chattopadhyay S. Overview of immune response during SARS-CoV-2 infection: Lessons from the past. Front Immunol (2020) 11:1949. doi: 10.3389/fimmu.2020.01949
159. Graham MB, Braciale VL, Braciale TJ. Influenza virus-specific CD4+ T helper type 2 T lymphocytes do not promote recovery from experimental virus infection. J Exp Med (1994) 180(4):1273–82. doi: 10.1084/jem.180.4.1273
160. Sun P, Ren J, Li K, Qie S, Liu Z, Xi J. Response to: Sore throat in COVID19: comment on "Clinical characteristics of hospitalized patients with SARS-CoV-2 infection: A single arm meta-analysis". J Med Virol (2020) 92(7):716–8. doi: 10.1002/jmv.25818
161. Adir Y, Saliba W, Beurnier A, Humbert M. Asthma and COVID-19: an update. Eur Respir Rev (2021) 30(162):210152. doi: 10.1183/16000617.0152-2021
162. Rosenberg HF, Foster PS. Eosinophils and COVID-19: diagnosis, prognosis, and vaccination strategies. Semin Immunopathol (2021) 43(3):383–92. doi: 10.1007/s00281-021-00850-3
163. Lindsley AW, Schwartz JT, Rothenberg ME. Eosinophil responses during COVID-19 infections and coronavirus vaccination. J Allergy Clin Immunol (2020) 146(1):1–7. doi: 10.1016/j.jaci.2020.04.021
164. Wendisch D, Dietrich O, Mari T, von Stillfried S, Ibarra IL, Mittermaier M, et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell (2021) 184(26):6243–61.e27. doi: 10.1016/j.cell.2021.11.033
165. Venkataraman T, Coleman CM, Frieman MB. Overactive epidermal growth factor receptor signaling leads to increased fibrosis after severe acute respiratory syndrome coronavirus infection. J Virol (2017) 91(12):e00182–17. doi: 10.1128/JVI.00182-17
166. Venkataraman T, Frieman MB. The role of epidermal growth factor receptor (EGFR) signaling in SARS coronavirus-induced pulmonary fibrosis. Antiviral Res (2017) 143:142–50. doi: 10.1016/j.antiviral.2017.03.022
167. Kim J, Yang YL, Jeong Y, Jang YS. Middle East respiratory SyndromeCoronavirus infection into established hDPP4-transgenic mice accelerates lung damage Via activation of the pro-inflammatory response and pulmonary fibrosis. J Microbiol Biotechnol (2020) 30(3):427–38. doi: 10.4014/jmb.1910.10055
168. Shen H, Zhang N, Liu Y, Yang X, He Y, Li Q, et al. The interaction between pulmonary fibrosis and COVID-19 and the application of related AntiFibrotic drugs. Front Pharmacol (2021) 12:805535. doi: 10.3389/fphar.2021.805535
169. Drake TM, Docherty AB, Harrison EM, Quint JK, Adamali H, Agnew S, et al. Outcome of hospitalization for COVID-19 in patients with interstitial lung disease. an international multicenter study. Am J Respir Crit Care Med (2020) 202(12):1656–65. doi: 10.1164/rccm.202007-2794OC
170. Li HH, Liu CC, Hsu TW, Lin JH, Hsu JW, Li AF, et al. Upregulation of ACE2 and TMPRSS2 by particulate matter and idiopathic pulmonary fibrosis: A potential role in severe COVID-19. Part Fibre Toxicol (2021) 18(1):11. doi: 10.1186/s12989-021-00404-3
171. George PM, Wells AU, Jenkins RG. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir Med (2020) 8(8):807–15. doi: 10.1016/S2213-2600(20)30225-3
172. Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol (2004) 4(8):583–94. doi: 10.1038/nri1412
173. Wynn TA, Cheever AW, Jankovic D, Poindexter RW, Caspar P, Lewis FA, et al. An IL-12-based vaccination method for preventing fibrosis induced by schistosome infection. Nature (1995) 376(6541):594–6. doi: 10.1038/376594a0
174. Roderfeld M, Rath T, Pasupuleti S, Zimmermann M, Neumann C, Churin Y, et al. Bone marrow transplantation improves hepatic fibrosis in Abcb4-/- mice via Th1 response and matrix metalloproteinase activity. Gut (2012) 61(6):907–16. doi: 10.1136/gutjnl-2011-300608
175. Zhang S, Liang R, Luo W, Liu C, Wu X, Gao Y, et al. High susceptibility to liver injury in IL-27 p28 conditional knockout mice involves intrinsic interferongamma dysregulation of CD4+ T cells. Hepatology (2013) 57(4):1620–31. doi: 10.1002/hep.26166
176. Fielding CA, Jones GW, McLoughlin RM, McLeod L, Hammond VJ, Uceda J, et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity (2014) 40(1):40–50. doi: 10.1016/j.immuni.2013.10.022
177. Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, et al. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med (2017) 214(11):3311–29. doi: 10.1084/jem.20161791
178. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
179. Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, et al. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation (2009) 119(22):2904–12. doi: 10.1161/CIRCULATIONAHA.108.832782
180. Taylor AE, Carey AN, Kudira R, Lages CS, Shi T, Lam S, et al. Interleukin 2 promotes hepatic regulatory T cell responses and protects from biliary fibrosis in murine sclerosing cholangitis. Hepatology (2018) 68(5):1905–21. doi: 10.1002/hep.30061
181. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, et al. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation (2019) 139(2):206–21. doi: 10.1161/CIRCULATIONAHA.118.036065
182. do Valle Duraes F, Lafont A, Beibel M, Martin K, Darribat K, Cuttat R, et al. Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight (2020) 5(3):e130651. doi: 10.1172/jci.insight.130651
183. Ulzii D, Kido-Nakahara M, Nakahara T, Tsuji G, Furue K, HashimotoHachiya A, et al. Scratching counteracts IL-13 signaling by upregulating the decoy receptor IL-13Ralpha2 in keratinocytes. Int J Mol Sci (2019) 20(13):3324. doi: 10.3390/ijms20133324
184. Badalyan V, Thompson R, Addo K, Borthwick LA, Fisher AJ, Ort T, et al. TNF-alpha/IL-17 synergy inhibits IL-13 bioactivity via IL-13Ralpha2 induction. J Allergy Clin Immunol (2014) 134(4):975–8.e5. doi: 10.1016/j.jaci.2014.05.019
185. Baurakiades E, Costa VH Jr., Raboni SM, de Almeida VR, Larsen KS, Kohler JN, et al. The roles of ADAM33, ADAM28, IL-13 and IL-4 in the development of lung injuries in children with lethal non-pandemic acute infectious pneumonia. J Clin Virol (2014) 61(4):585–9. doi: 10.1016/j.jcv.2014.10.004
186. Heitmann L, Abad Dar M, Schreiber T, Erdmann H, Behrends J, McKenzie AN, et al. The IL-13/IL-4Ralpha axis is involved in tuberculosis-associated pathology. J Pathol (2014) 234(3):338–50. doi: 10.1002/path.4399
187. Gieseck RL 3rd, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol (2018) 18(1):62–76. doi: 10.1038/nri.2017.90
188. Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, et al. Interleukin-13: Central mediator of allergic asthma. Science (1998) 282(5397):2258–61. doi: 10.1126/science.282.5397.2258
189. Kasaian MT, Miller DK. IL-13 as a therapeutic target for respiratory disease. Biochem Pharmacol (2008) 76(2):147–55. doi: 10.1016/j.bcp.2008.04.002
190. Beck LA, Thaci D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med (2014) 371(2):130–9. doi: 10.1056/NEJMoa1314768
191. Danese S, Rudzinski J, Brandt W, Dupas JL, Peyrin-Biroulet L, Bouhnik Y, et al. Tralokinumab for moderate-to-severe UC: A randomised, double-blind, placebo-controlled, phase IIa study. Gut (2015) 64(2):243–9. doi: 10.1136/gutjnl-2014-308004
192. Reinisch W, Panes J, Khurana S, Toth G, Hua F, Comer GM, et al. Anrukinzumab, an anti-interleukin 13 monoclonal antibody, in active UC: Efficacy and safety from a phase IIa randomised multicentre study. Gut (2015) 64(6):894–900. doi: 10.1136/gutjnl-2014-308337
193. Ramalingam TR, Gieseck RL, Acciani TH, MH K, Cheever AW, MentinkKane MM, et al. Enhanced protection from fibrosis and inflammation in the combined absence of IL-13 and IFN-gamma. J Pathol (2016) 239(3):344–54. doi: 10.1002/path.4733
194. Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med (2015) 7(301):301ra129.
195. Flood-Page P, Menzies-Gow A, Phipps S, Ying S, Wangoo A, Ludwig MS, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest (2003) 112(7):1029–36. doi: 10.1172/JCI17974
196. Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, et al. A critical role for eosinophils in allergic airways remodeling. Science (2004) 305(5691):1776–9. doi: 10.1126/science.1100283
197. Durrani SR, Viswanathan RK, Busse WW. What effect does asthma treatment have on airway remodeling? current perspectives. J Allergy Clin Immunol (2011) 128(3):439–48; quiz 49-50. doi: 10.1016/j.jaci.2011.06.002
198. Tedesco D, Thapa M, Chin CY, Ge Y, Gong M, Li J, et al. Alterations in intestinal microbiota lead to production of interleukin 17 by intrahepatic gammadelta T-cell receptor-positive cells and pathogenesis of cholestatic liver disease. Gastroenterology (2018) 154(8):2178–93. doi: 10.1053/j.gastro.2018.02.019
199. Rai RP, Liu Y, Iyer SS, Liu S, Gupta B, Desai C, et al. Blocking integrin alpha4beta7-mediated CD4 T cell recruitment to the intestine and liver protects mice from western diet-induced non-alcoholic steatohepatitis. J Hepatol (2020) 73(5):1013–22. doi: 10.1016/j.jhep.2020.05.047
200. Elhai M, Avouac J, Hoffmann-Vold AM, Ruzehaji N, Amiar O, Ruiz B, et al. OX40L blockade protects against inflammation-driven fibrosis. Proc Natl Acad Sci U S A (2016) 113(27):E3901–10. doi: 10.1073/pnas.1523512113
201. Boleto G, Guignabert C, Pezet S, Cauvet A, Sadoine J, Tu L, et al. T-Cell costimulation blockade is effective in experimental digestive and lung tissue fibrosis. Arthritis Res Ther (2018) 20(1):197. doi: 10.1186/s13075-018-1694-9
202. Celada LJ, Kropski JA, Herazo-Maya JD, Luo W, Creecy A, Abad AT, et al. PD-1 up-regulation on CD4(+) T cells promotes pulmonary fibrosis through STAT3mediated IL-17A and TGF-beta1 production. Sci Transl Med (2018) 10(460):eaar8356. doi: 10.1126/scitranslmed.aar8356
203. Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, et al. Obesity drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell (2018) 175(5):1289–306.e20. doi: 10.1016/j.cell.2018.09.053
204. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature (2019) 575(7783):512–8. doi: 10.1038/s41586-019-1631-3
Keywords: type 2 helper T (Th2) cell, interleukin-4 (IL-4), interleukin-5 (IL-5), interleukin-13 (IL-13), inflammation, tissue repair, tissue fibrosis, pathogenic Th2 (Tpath2) cell
Citation: Kokubo K, Onodera A, Kiuchi M, Tsuji K, Hirahara K and Nakayama T (2022) Conventional and pathogenic Th2 cells in inflammation, tissue repair, and fibrosis. Front. Immunol. 13:945063. doi: 10.3389/fimmu.2022.945063
Received: 16 May 2022; Accepted: 18 July 2022;
Published: 09 August 2022.
Edited by:
Nicholas Van Panhuys, Sidra Medical and Research Center, QatarReviewed by:
Norman Nausch, Deutsche Gesellschaft für Internationale Zusammenarbeit, GermanyCopyright © 2022 Kokubo, Onodera, Kiuchi, Tsuji, Hirahara and Nakayama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kiyoshi Hirahara, aGlyYWhhcmFrQGNoaWJhLXUuanA=; Toshinori Nakayama, dG5ha2F5YW1hQGZhY3VsdHkuY2hpYmEtdS5qcA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.