 Atefe Ghamar Talepoor
Atefe Ghamar Talepoor Mehrnoosh Doroudchi
Mehrnoosh Doroudchi- Department of Immunology, School of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
Immune system is a versatile and dynamic body organ which offers survival and endurance of human beings in their hostile living environment. However, similar to other cells, immune cells are hijacked by senescence. The ageing immune cells lose their beneficial functions but continue to produce inflammatory mediators which draw other immune and non-immune cells to the senescence loop. Immunosenescence has been shown to be associated with different pathological conditions and diseases, among which atherosclerosis has recently come to light. There are common drivers of both immunosenescence and atherosclerosis; e.g. inflammation, reactive oxygen species (ROS), chronic viral infections, genomic damage, oxidized-LDL, hypertension, cigarette smoke, hyperglycaemia, and mitochondrial failure. Chronic viral infections induce inflammaging, sustained cytokine signaling, ROS generation and DNA damage which are associated with atherogenesis. Accumulating evidence shows that several DNA and RNA viruses are stimulators of immunosenescence and atherosclerosis in an interrelated network. DNA viruses such as CMV, EBV and HBV upregulate p16, p21 and p53 senescence-associated molecules; induce inflammaging, metabolic reprogramming of infected cells, replicative senescence and telomere shortening. RNA viruses such as HCV and HIV induce ROS generation, DNA damage, induction of senescence-associated secretory phenotype (SASP), metabolic reprogramming of infected cells, G1 cell cycle arrest, telomere shortening, as well as epigenetic modifications of DNA and histones. The newly emerged SARS-CoV-2 virus is also a potent inducer of cytokine storm and SASP. The spike protein of SARS-CoV-2 promotes senescence phenotype in endothelial cells by augmenting p16, p21, senescence-associated β-galactosidase (SA-β-Gal) and adhesion molecules expression. The impact of SARS-CoV-2 mega-inflammation on atherogenesis, however, remains to be investigated. In this review we focus on the common processes in immunosenescence and atherogenesis caused by chronic viral infections and discuss the current knowledge on this topic.
Immunosenescence is a widespread phenomenon
Ageing is defined as progressive physiological changes along with decline of biological functions in a cell, organ, or the total organism which is associated with increased risk of debility, disease, and death (1). Inflammaging is a key underlying mechanism in processes leading to aging and ageing-related diseases. The term “inflammaging” refers to a chronic, low level systemic inflammation that results in overstimulation of the immune system and elevated mortality and morbidity in elderly individuals (2). Growing evidence suggests the existence of interplay between ageing and immune system alterations, which is called immunosenescence (3). Therefore, immunosenescence is referred to the gradual deterioration of immune parameters as a consequence of ageing process. The important hallmarks of immunosenescence are the followings: [1] reduced frequencies of naïve T cells, [2] alternation in CD4:CD8 ratio, [3] defective response to new antigens, [4] accumulation of short-lived memory T cells, [5] impaired calcium-mediated signaling, and [6] inflammaging (4–6). Immunosenescence affects both the ability to respond to infectious agents as well as development of appropriate and long-term immune responses (7). It has also been shown that immunosenescence affects adaptive immune system more than the innate immune system (8). The main characteristics of immunosenescence with respect to the innate immunity include reduced number of circulating CD14+ CD16−classical monocytes, macrophages and dendritic cells (DCs), reduced chemotaxis and phagocytosis of macrophages and neutrophils, defective reactive oxygen species (ROS) production by neutrophils, increased neutrophil susceptibility to apoptosis, elevated levels of inflammatory cytokines and chemokines, lower expression level of major histocompatibility complex (MHC) class II by DCs and macrophages, increased number of natural killer (NK) cells, decreased NK cells cytotoxicity and impaired antigen presentation by DCs (9–17). In terms of adaptive immunity, immunosenescence can result in reduced naive CD4+ and CD8+T cells pools, restricted T cell receptor (TCR) repertoire, loss of CD28 expression by T cells, increased frequencies of effector (TEff) and effector memory T (TEM) cells, decreased accumulation of central memory T (TCM) cells, elevated accumulation of memory B cells, restricted B cell receptor (BCR) repertoire, impaired class-switching and somatic recombination of B cells and defective antibody responses to antigens (17–19). It has been demonstrated that immunosenescence, through stimulating proinflammatory factors and inflammaging, can drive cellular senescence in other tissues and development of age-related diseases, such as infections, cancer, autoimmune disorders and chronic inflammatory and metabolic diseases (20, 21) in the elderly. Although immunosenescence and cellular senescence are slightly different in mechanism, they share several similarities. In the following sections we will describe different properties, drivers and regulators of cellular senescence and immunosenescence with the focus on atherosclerosis.
Overview of cellular senescence
Different types of somatic cells isolated from mammalian tissues undergo multiple proliferations before they stop growing. For the first time in 1961, Hayflick and Moorhead discovered that human fibroblast cells proliferation potency is limited upon serial culture, and they called this phenomenon “cellular senescence” (22).
Cellular senescence is an irreversible cell cycle arrest in the G1 phase, in which the senescent cells remain metabolically active but they hold a growth arrested status (23). Indeed, upregulation of the p16, p21 and p53 cell cycle inhibitors lead to the irreversible cell cycle arrest in the senescent cells (2, 24). Hence, cellular senescence is different from other non-dividing processes such as quiescence, exhaustion or terminally differentiation by several markers and morphological changes (24–26). The replicative senescence (RS), oncogene-induced senescence (OIS), genotoxicity-induced senescence (GIS), developmental senescence and tissue repair senescence are considered different types of cellular senescence in human (25, 27, 28).
In general, senescence in any cell type is accompanied by phenotypic alterations including but not limited to cytoplasm enlargement, flattened, vacuolated, and multinucleated cell morphologies, depending on cell type, genetic background, and the type of senescence inducing stimuli (29). In addition, senescent immune cells show reduced DNA replication, chromatin remodeling, metabolic reprogramming, increased resistance to apoptosis, and increased expression of senescence-associated β-galactosidase (SA-β-Gal) as well as producing a set of inflammatory secretome (30–32). Moreover, immunoscenesent cells downregulate surface markers like CD27, CD28, CCR7 and CD45RO, while they upregulate the Killer cell lectin-like receptor subfamily G (KLRG-1), CD57, PD-1 and CD153 (29, 33–35).
Senescent immune and non-immune cells produce a combination of inflammatory factors, called senescence-associated secretory phenotype (SASP) (36). SASP factors consist of several interleukins (ILs) including IL-6 and IL-1, and chemokines such as CXCL-4,-5, -6, -12 and CCL-2, -3, -7, -8, -13, -16, -20 and -26, proteases, growth factors, bioactive lipids and extracellular vesicles which are involved in immune cell migration, innate and adaptive immune responses, modification of the extracellular matrix (ECM), cell cycle arrest and phagocytosis (24, 37, 38). Particularly, secreted SASP factors act as paracrine and autocrine mediators that promote inflammatory responses as well as defective tissue remodeling which ultimately may lead to chronic autoimmune disorders and different cancers (39–41). A variety of transcription factors and signaling pathways like nuclear factor kappa B (NF-κB) and beta CCAAT/enhancer– binding protein (42, 43), DNA damage (44), p38α mitogen-activated protein kinase (MAPK) (45), mammalian target of rapamycin (mTOR) (46, 47), and mixed lineage leukemia 1 (48) and GATA4 (49) regulate the key effectors of the SASP.
Two major tumor suppressor pathways, p16INK4A/pRB and p53/p21WAF1/CIP1, through upstream regulators and downstream effectors along with extensive inter-pathway crosstalk, are responsible for the stable growth arrest in the senescence (50, 51). The p16INK4A/pRB pathway mainly acts as a negative regulator of cell cycle progression by downregulating cyclin-dependent kinase (CDK) 4/6 activity. As a result, hypo-phosphorylated Rb binds to E2F complex and leads to repression of E2F target gene transcription required for cell cycle progression (52). P53 is activated in response to DNA damage and induces transcription of cyclin-dependent kinase inhibitors (CDKIs) and p21CIP1 which triggers growth arrest and cellular senescence (31, 53). Therefore, overexpression of these four components including p53, p21CIP1, p16INK4A and PRB are sufficient to induce and maintain cellular senescence.
Senescence in physiological and pathological processes
Several studies have shown that cellular senescence playsphysiological roles in the embryonic and later life development, wound healing, tissue repair and protective response to stress (54–57). In addition, as senescent immune and non-immune cells accumulate in multiple tissues, they may contribute to regulate the non-pathological and pathological senescence-related states (39). In some events, acute senescence exhibits protective effects and thus prevents the progression of the diseases. For instance, during early stages of tumor, the activation of oncogenes stimulates cellular senescence in an attempt to inhibit cell growth and tumor progression (58). In other circumstances, chronic and aberrant accumulation of senescent cells in tissues generates a pro-inflammatory environment that affects the onset, development or progression of several senescence-associated diseases, such as atherosclerosis, cardiovascular diseases (CVDs), cancer, hepatic steatosis, Alzheimer’s disease, fibrotic pulmonary disease, osteoarthritis, glaucoma, type 2 diabetes and renal dysfunction (59–64).
Senescence and immunosenescence in atherosclerosis
Atherosclerosis is a chronic inflammatory and age-associated condition of medium to large arteries which is linked to the progression of CVDs, such as abdominal aortic aneurysm (AAA), coronary artery disease (CAD), peripheral artery disease (PAD), heart failure (HF) and ischemic strokes (65). Endothelial abnormalities, vascular inflammation, adhesion molecules expression, cytokines and chemokines secretion, immune cells infiltration, monocytes entry into intima, engulfment of oxidized low density lipoprotein (Ox-LDL) and foam cell formation result in atherosclerotic plaque formation. Subsequently, aberrant efferocytosis, foam cells apoptosis, migration of vascular smooth muscle cells (VSMCs) from media to intima, degradation of ECM and extensive coagulation lead to the rupture and erosion of atherosclerotic plaques, vessel occlusion and the risk of death (66–68).
Several findings showed that cellular senescence in the immune and non-immune cells are involved in the early and advanced stages of atherosclerosis (69). Certainly, accumulation of the senescent endothelial cells (ECs) (70, 71), VSMCs (72, 73), monocytes (74), macrophages and foam cells (71, 75), fibroblasts (74) and T cells (76, 77) influence atherosclerosis process by necrotic core enlargement, ECM destruction, atherosclerotic plaque calcification, intra- plaque angiogenesis and its rupture (78). ECs, as the lining of the intima layer, play major roles in the maintenance of the integrity of arteries. Thus, EC dysfunction and senescence, which can cause elevated blood pressure, increased coagulation and angiogenesis as well as elevated inflammatory response, contribute in progression of atherosclerosis (79, 80). It is not clear, however, if they take part in onset of atherosclerosis, as well. Senescent ECs are frequently found in atherosclerotic plaques of elderly patients and senescent cells have been proposed to be a major contributor to development of vascular diseases (81, 82). ECs in advanced human plaques show evidence of senescence, including telomere shortening, increased p53/p21 signaling pathways and elevated SA-β-GAL activity (83, 84). Senescent ECs are associated with increased levels of ROS, inflammatory cytokines and extracellular vesicles secretion as well as vascular calcification (85, 86). Furthermore, it has been shown that senescent ECs produce lower levels of nitric oxide (NO) and prostacyclin and thereby affect vascular homeostasis (87). On the other hand, secretion of inflammatory mediators and ROS by senescent ECs, retention of Ox-LDL in the arterial intima, adhesion of monocytes to endothelium, activation of NF-κB signaling and production of extracellular vesicles are potent promoters of SASP-mediated senescence and possibly play a role in the atherosclerotic plaque development (88–92). Current studies, however, do not provide information on the senescence of ECs or immune cells in early onset atherosclerosis and cardiovascular diseases.
VSMCs are located in the arterial media, synthesizing the ECM and are responsible for the vessel contraction (72). A number of documents have provided evidence for the occurrence of senescence in VSMCs during atherosclerosis, such as lower level of proliferation capacity (93), higher expression levels of p16 and p21, and decreased telomere length (94) as well as larger and flattened morphology in comparison to cells separated from healthy arterial media (95). Senescent VSMCs in atherosclerotic plaques reduce the expression of proteins required for contraction, including α-smooth muscle actin (α-SMA), smooth muscle myosin heavy chain (SM-MHC) and calponin, but they increase production of inflammatory cytokines (96, 97). Furthermore, senescent human VSMCs in atherosclerotic lesions exhibit reduced expression of anti-inflammatory factors and secrete SASP mediators which induce chemotaxis of monocytes, expression of adhesion molecules and cytokines by ECs and promote atherosclerosis progression. Moreover, senescent human VSMCs can increase plaque vulnerability by producing lower amounts of collagen (98). In addition to senescent ECs and VSMCs, senescent immune cells, like macrophages and T cells, mainly contribute to the development of atheroma and progression of atherosclerotic plaques (99).
Senescent macrophages characterized by increased SA-β-GAL activity as well as p53 and p16 expression, display impaired cholesterol efflux and enhanced senescent-related atherosclerosis (100, 101). On the other hand Ox-LDL inhibits macrophages proliferation and migration, induces cellular senescence and promotes the secretion of inflammatory factors, such as TNF-α, monocyte chemoattractant protein-1(MCP-1), and IL-1β, which may create a positive feedback loop (102).
In addition to macrophages, senescent T cells with CD8+CD57+CD27-CD28null phenotype, through producing large amounts of IFN-γ and TNF-α, promote inflammation and development of atherosclerosis (103). Furthermore, telomere shortening in T cells has been observed in patients with atherosclerosis. Terminal restriction fragments (TRF) analysis has shown that the mean length of TRF in leukocytes of CAD patients is shorter than control individuals who had no family history of CAD (104). It has been found that IFN-γ-producing CD28nullCD4+ T cells accumulate in the heart-draining lymph nodes of aged mice and adoptive transfer of these cells results in proinflammatory responses in young mice (105, 106). It is also shown that senescent CD4+ T cells can infiltrate to heart and promote myocardial inflammation and stress response leading to age-related cardiac dysfunction (107). Additionally, the importance of senescent T cells has been reported in human hypertension. A higher frequency of CD57+CD28−CD8+ T cells as well as increased expression of CXCL11 has been reported in the patients with hypertension compared to healthy controls, suggesting a role for immunosenescent proinflammatory cytotoxic CD8+ T cells in hypertension (108). Interestingly, a higher frequency of senescent CD57+CD8+ T cells has been observed in patients with acute myocardial infarction (MI), which correlated with cardiovascular mortality 6 months after acute MI (109). However, it is still unclear whether accumulation of senescent T cells is the cause or the result of atherosclerosis. In this regard, it has been hypothesized that senescent T cells, through secreting large amounts of IFN-γ, are directly involved in the macrophages activation, metalloproteinases production, ECM destruction and thereby pathophysiology of atherosclerosis (110). In addition, senescent T cells may participate in VSMC and ECs lysis by producing large amounts of perforin and granzyme and lead to atherosclerosis progression (98).
Drivers of immunosenescence and atherosclerosis
Both immunosenescence and atherosclerosis are multifactorial conditions which share common stimulators, including Ox-LDL (111, 112), inflammation (113, 114), ROS (115, 116), cigarette smoke (117, 118), hypertension (119, 120), hyperglycaemia (121, 122), viral infection (123, 124), mitochondrial failure (125, 126) and genomic damage (127, 128) (Figure 1). Certainly, various drivers of senescence induce SASP secretion and thereby stimulate chronic and low-grade inflammation that participates in atherosclerosis development, and in turn, diverse stimulators of atherosclerosis induce cellular senescence and SASP production.
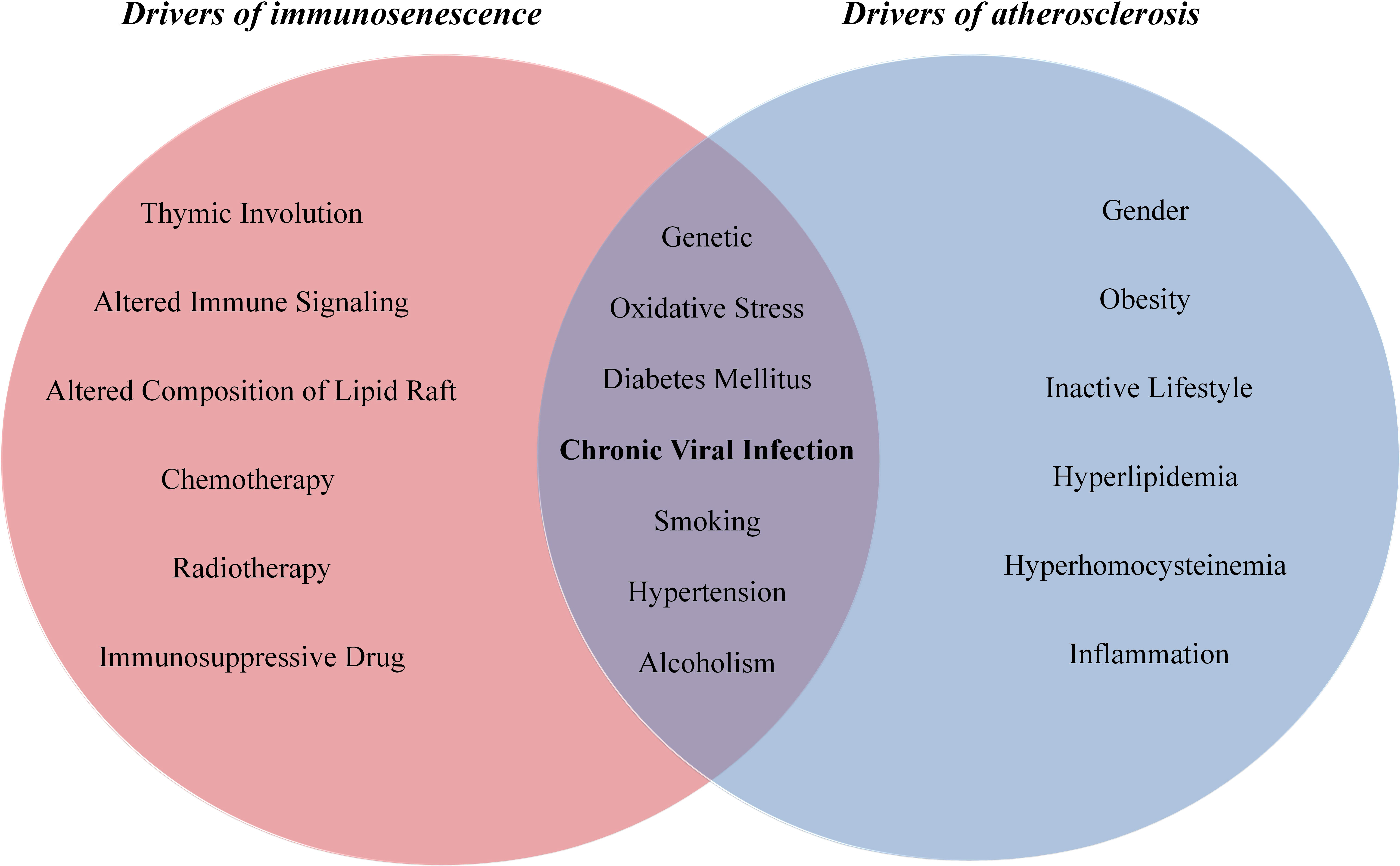
Figure 1 The drivers of immunosenescence and atherosclerosis.
One of the most important causes of immunosenescence, which also may participate in atherosclerosis development/progression, is viral infection. Chronic viral infections can trigger cellular senescence directly and indirectly mainly via inducing DNA damage and prolonged cytokine signaling, respectively (129, 130). It has been shown that viruses, through induction of senescence affect proliferation capacity and function of T cells, quality, and quantity of innate and adaptive immune responses, recruitment of immune cells and secretion of proinflammatory mediators. Therefore, accumulation of viral-induced senescent cells in blood and tissues may contribute to senescence-associated inflammatory onditions, such as atherosclerosis (4, 131–134). Indeed, virus infections through different mechanisms can induce immunosenescence and atherosclerosis (Table 1). In the following sections, we will discuss different chronic human infections with DNA and RNA viruses which play parallel roles in the immunosenescence and atherosclerosis processes.

Table 1 Induction of atherosclerosis and immunosenescence by different viruses.
Cytomegalovirus
CMV is an enveloped, double stranded DNA β-herpesvirus which establishes lifelong latent infection in the population with sporadic reactivation in immunocompromised patients (161). The CMV infection/reactivation is mainly controlled but not eradicated by IFN-γ-producing CD4+ and CD8+ T cells in healthy immunocompetent individuals (162). Between 10-30% of memory T cells in the peripheral circulation of infected individuals are found specific to CMV epitopes (163, 164). CMV-specific CD8+ and CD4+ T lymphocytes display hallmarks of senescence defined by CD57 expression that is associated with decreased proliferation capacity and function of these cells (165). It is assumed that continued antigen exposure during CMV infection results in telomere erosion and replicative senescence generally in CD8+ cytotoxic T lymphocytes (CTL) (166–168). CMV-specific T lymphocytes also exhibit higher levels of proinflammatory cytokines, such as IFN-γ and TNF-α and lower proliferative responses upon stimulation, possibly due to the their telomeres shortening (169).
Recent cohort studies revealed that the excess mortality rate in CMV-seropositive elderly subjects is primarily related to senescence-associated vascular disease (135, 136). Several studies found relationships between increased inflammatory mediators and CVD-related deaths in CMV-infected elderly individuals (136, 170). It has been shown that CMV infection is correlated with increased risk of multiple senescence-associated comorbidities, especially diabetes, atherosclerosis, and CVDs (171). Furthermore, DNA and proteins of CMV have been found in human atherosclerotic plaques (137). It is also suggested that CMV can participate in atherosclerosis by inducing vascular endothelial dysfunction and apoptosis, reduced matrix metalloproteinase 9 (MMP9) activity, increased coagulation and thrombosis, elevated release of proatherosclerotic or proinflammatory molecules, increased intimal thickening and lipid deposition (138). Therefore, systemic inflammation as well as increased circulating senescent and proinflammatory CD8+ T cells during CMV infection may underlie proper condition for initiation and progression of atherosclerosis.
Epstein-Barr virus
EBV is an enveloped double stranded DNA, B lymphotropic γ-herpesvirus that causes an acute infection known as infectious mononucleosis. However, EBV establishes a latent infection in human hosts that is associated with a variety of malignancies, including Extranodal NK/T-cell lymphoma, Burkitt lymphoma, Angioimmunoblast T-cell lymphoma, Hodgkin’s lymphoma and Post-transplant lymphoproliferative disease (172, 173). EBV specific humoral and cellular immune responses are particularly important for controlling and treating EBV-induced acute lymphoproliferative diseases (174). It has been found that EBV infection induces B cell hyper-proliferation via upregulating the viral latency proteins, EBNA2 and EBNA-LP, which results in replicative stress, DNA damage, activation of the DNA damage response (DDR) pathway and eventually, cellular senescence (139, 140, 175–177). A previous study reported that EBV-infected B cells trigger G1 phase cellular arrest (141). Even early EBV-infected B cells exhibit increased markers of OIS, including H3K9me3 senescence-associated heterochromatic foci as well as higher levels of p16, p21 and p53 (178). EBV-infected cells also undergo metabolic reprogramming such as decreased oxidative phosphorylation and purine nucleotide pools, which contribute to increased replication stress and establishment of persistent DNA damage (178). Additionally, antigen-specific T cells isolated from the peripheral blood of EBV and CMV positive patients have higher expression levels of several senescence-associated markers; including KLRG-1 while exhibit reduced antigen-specific T cell repertoire diversity (179). These data suggest that chronic EBV infection may participate in replicative and oncogene-induced cellular senescence.
The effect of EBV to increase the risk of atherosclerosis CAD has been studied in detail. Some studies showed higher levels of EBV-specific antibodies in patients with atherosclerosis (142, 180). Moreover, it has been found that following reactivation of latent virus, EBV-encoded deoxyuridine-5’-triphosphate nucleotidohydrolase (dUTPase) can induce proinflammatory cytokines secretion from peripheral blood monocytes which is associated with atherosclerosis progression (181). EBV may also be involved in the atherosclerosis by triggering IL-6 and TNF-α secretion by macrophages, intercellular adhesion molecule-1 (ICAM-1) expression on ECs and lipid profile alternations in blood (143).
Hepatitis B virus
HBV is an enveloped, double stranded DNA virus of the Hepadnaviridae family that attacks the liver, triggering both acute disease and chronic disease such as cirrhosis, liver cancer and liver failure (182). Recently it has been found that 60% of hepatocellular carcinomas (HCCs) and more than 80% of liver cirrhosis cases exhibited features of replicative senescence as compared to 10% in normal liver (183, 184). In addition, there is higher level of SA-β-Gal activity in cirrhotic hepatocytes as compared with large-cell dysplasia (144, 184). Elevated expression levels of p21, reduced telomere length and lower levels of S, G2 and M phase markers in hepatocytes of chronic HBV-infected patients is also reported which correlated with liver fibrosis (185). Furthermore, in chronic human infection, HBV inhibits Top 1 protein and leads to topological DNA damage and telomere attrition in CD4+ T cells (145). Therefore, Top 1 inhibition by the virus is correlated with premature T cell immunosenescence (145). Several studies indicated that HBx protein of HBV, a transcriptional transactivator for virus replication, can carry out pro-senescent roles by increasing the expression levels of p16 (INK4a) and p21 (Waf1/Cip1) and reducing phosphorylation of Rb (146, 186). Additionally, increased secretion of SASP components, including IL-6 and angiogenin-2, has been reported in chronic HBV infection (187).
The association between HBV infection and CVDs remains controversial. Recent reports revealed that patients with HBV infection exhibited more subclinical atherosclerosis and carotid plaques as compared with non-infected controls (188, 189). A significant association between liver damage, as an independent factor, and development of subclinical atherosclerosis has also been shown (190). A previous study showed that HBV surface antigen (HBsAg) is a major contributor of atherosclerosis (147), but two other studies found no significant association between chronic HBV infection and development of carotid atherosclerotic plaques (191, 192). Moreover, a significant negative correlation between serum levels of triglycerides and HBV infection is also reported (148). In contrast, results of a meta-analysis study represented that exposure to HBV led to increase atherosclerosis-associated morbidity rate (193).
Hepatitis C virus
HCV is an enveloped, positive sense single-stranded RNA virus that causes liver inflammation, leading to serious liver damage and hepatitis (194). Chronic HCV is a long-lasting infection, ranging in severity from a mild illness to a serious disease including liver cirrhosis and cancer (195). Several studies reported that chronic HCV infection can trigger cellular senescence by inducing higher concentrations of ROS, secretion of proinflammatory cytokines and growth factors, G1 cell cycle arrest, DNA damage, as well as epigenetic modifications of DNA and histones (149, 150, 196–200). In addition, it has been found that HCV-associated liver inflammation can promote the telomere shortening process and finally replicative senescence and HCC (184). Isolated memory T cells from HCV-positive patients exhibit shorter telomeres compared to healthy subjects (201). Additionally, CD8+ T cells isolated from peripheral blood of HCV-infected subjects show higher levels of DNA damage and hypophosphorylated signal transducer and activator of transcription 1 (STAT1) and STAT5 in response to IL-6 or IL-2 stimulation, respectively (202). Altogether, these data suggest that chronic HCV infection results in cellular senescence, and since these senescent cells are non-functional, they may predispose HCV-infected individuals to HCC (203).
Growing evidence suggests metabolic reprogramming and chronic hepatic and systemic inflammation induced by HCV can be involved in the development of atherosclerosis (204–207). It has been shown that HCV can live and replicate in the carotid plaques and is associated with arterial inflammation through increased levels of proatherogenic chemokines and cytokines as well as triggering proatherogenic metabolic factors (151, 208, 209). Furthermore, structural and non-structural proteins of HCV induce ROS generation and interfere with glucose and lipid metabolism leading to chronic inflammation, insulin resistance (IR), diabetes and fatty liver, which are known as major atherosclerosis risk factors (152). A higher TNF-α/adiponectin ratio in HCV-infected subjects is shown to be correlated with the development of IR and atherosclerosis (210). A recent multicenter study reported that direct-acting antiviral agents (DAAs) therapies in HCV-infected patients led to eradication of HCV as well as improvement in carotid atherosclerosis, reduction in carotid thickness and alternation in patients’ plaques value (211). The most surprising results of cohort studies have been published recently in which DAA treatments significantly reduced the risk of CVD outcomes in HCV-infected patients (212, 213). Therefore, a strong association between HCV infection and the atherosclerosis process exists.
Human immunodeficiency virus
HIV is an enveloped, diploid positive sense single-stranded RNA retrovirus from Retroviridae family that mainly attacks CD4+ T cells and results in progressive loss of T cell subsets. HIV infection results in immunodeficiency, increased susceptibility to opportunistic infections as well as certain types of cancer such as Kaposi sarcoma and B cell lymphoma, and ultimately a syndrome defined as acquired immune deficiency syndrome (AIDS) (214). Recently, it has been found that HIV-infected individuals display T cell properties similar to those identified in elderly; i.e. decreased expression of CD28, lower naive/memory T cell ratios and hyporesponsiveness to vaccine (215). Growing evidence revealed higher expression level of p16 in active HIV-infected subjects as compared with healthy controls that is not correlated with the age of patients, suggesting that HIV infection is associated with cellular senescence. Also, anti-retroviral therapy (ART) decreased the expression levels of p16 in the CD4+ T cells population compared to healthy controls (153, 154). Moreover, results of a cohort study elucidated that corneal ECs of HIV-infected patients exhibit features of senescence, including decreased cell density as well as variation in cell size and shape as compared with the uninfected subjects (216). In a cohort in sub-Saharan Africa, higher expression levels of p16 and decreased telomere length in peripheral blood leukocytes of HIV infected patients is found (217).
In view of persistent inflammation and immune activation, HIV-positive individuals may have increased risk of HIV-related comorbidities and atherosclerosis-related risk factors such as hypertension, diabetes, and dyslipidemia (218, 219). The higher levels of IL-6 protein and mRNA in HIV-infected individuals can stimulate the production of several acute phase proteins, including C-reactive protein (CRP), serum amyloid A (SSA) and fibrinogen. The elevated levels of these inflammatory mediators have been associated with increased cardiovascular mortality in patients with HIV (155, 220, 221). Moreover, it has been reported that the HIV Tat protein induces endothelial dysfunction and MCP-1 secretion in porcine coronary arteries (156, 222). The gp120 protein of HIV has also been found to increase the levels of TNF-α and subsequently retention and oxidation of LDL in the arterial intima (223). The HIV Nef protein is also identified as an activator of macrophages by increasing CD36 expression, and it promotes macrophages transformation to foam cells through decreasing cholesterol efflux from these cells (224, 225).
Severe acute respiratory syndrome coronavirus 2
SARS-CoV-2 is an enveloped, positive-sense, single-stranded RNA, highly infectious beta-coronavirus, which has been described as the causative agent of upper respiratory tract infections and subsequently coronavirus disease 2019 (COVID-19) (157). A recent study found that COVID-19 patients exhibited features of senescence in their airway mucosa (157). Increased secretion levels of SASP factors in patients with COVID-19 resulted in complement mediated lysis, SASP-induced paracrine senescence of ECs, macrophage and neutrophil infiltration, endothelial damage and extensive thrombosis in SARS-CoV-2- infected lung tissues (157). It has been found that SARS-CoV-2 leads to increased expression levels of senescence markers such as p16, p21 and Lamin B1(LMNB1) as well as SASP factors in cultured human bronchial epithelial cells 14 days after infection when the virus was undetectable (158). These results suggest that SARS-CoV-2 can provoke paracrine senescence through sustained production of virus-induced inflammatory mediators, even after SARS-CoV-2 is no longer detectable (158). Furthermore, other studies indicated that administration of senolytic drugs, including Navitoclax, Dasatinib and Quercetin can inhibit cellular senescence and therefore alleviated COVID-19-related lung disease and reduce inflammation in SARS-CoV-2-infected hamsters and mice (157, 158, 226). Additionally, it has been indicated that the spike protein of SARS-CoV-2 stimulates senescence phenotype in ECs by increasing p16, p21, SA-β-Gal (158).
COVID-19 infection is primarily considered as a destructive disease of the respiratory system, but its complications also lead to the cardiovascular system damage and occurrence of a variety of CVDs, including myocarditis, myocardial damage, heart failure and myocardial infarction (227, 228). It has been found that angiotensin-converting enzyme 2 (ACE2), one of the most important receptors for SARS-CoV-2 entry, is expressed on the cardiomyocytes (229). Furthermore, higher serum levels of troponin and N-terminal pro-brain-natriuretic peptide (NT-proBNP) were reported in severe COVID-19 patients, which were associated with more disease manifestation and mortality rate (159). Another observational study revealed the effect of SARS-CoV-2 infection on lipid metabolism and atherogenesis (22). In this regard, higher serum levels of free fatty acids, lysophosphatidylcholine, lysophosphatidylethanolamine, phosphatidylglycerol and subsequently increased presence of CVDs in recovered SARS infected patients were found (230). Moreover, two other studies represented that SARS-CoV-2 infection, through activating the coagulation pathway, secreting inflammatory cytokines and chemokines by ECs, inducing production of fibrinogen, antithrombin and D-dimers and eventually triggering disseminated intravascular coagulopathy (DIC), may affect the onset or development of atherosclerosis in severe COVID-19 individuals (231, 232). The induction of senescence in immune cells, especially in patients with Long COVID, however, remains to be investigated.
The role of viral co-infections in the immunosenescence and atherosclerosis
The interplay between different viral co-infections and their synergistic effect on immunosenescence has been under investigation, recently. Viral co-infections such as in the case of CMV or HCV infection with HIV synergistically increase immunosenescence progression in patients (233–235). A previous study showed decreased telomere length within CD4+, CD8+ and immature (CD27+CD57+) T cell subsets in HIV/HCV co-infected Injecting Drug Users (IDU) as compared to HCV mono-infected and healthy subjects (233). This report is in line with the higher level of IL-6 in the same group which independently correlated with HIV/HCV co-infection and increasing age (236). Moreover, higher expression levels of immunosenescence markers, CD57 and P16INK4a, were observed in the liver tissue of HIV/HCV co-infected individuals compared to healthy controls (237). In addition, HIV co-infection increases DNA methylation and accelerated epigenetic ageing in chronic HCV patients compared to mono-infected patients (238). Similarly, higher levels of inflammatory markers such as IP-10, TNF-RII, and D-dimer are elevated in HIV/CMV co-infected individuals compared to HIV-mono-infected subjects or healthy controls (235).
In a similar manner, mounting evidence suggest that chronic viral co-infections may accelerate atherosclerosis. A cross sectional study showed higher prevalence of subclinical carotid plaque in relatively young (aged 46 years) HIV/HCV co-infected patientscompared to HIV mono-infected patients (239). Moreover, higher soluble vascular CAM-1 (sVCAM-1) and intercellular CAM-1 (sICAM-1) levels, as pro-atherosclerotic inflammatory biomarkers, have been reported in HIV/HCV co-infected patients as compared to HIV mono-infected patients (240). Increased morbidity and mortality rate and subclinical carotid artery diseasein HIV/HCV co-infected women were also associated with higher CMV IgG levels (241). However, another study on women found that HIV/HCV co-infection was not associated with greater carotid artery intima-media thickness (242).
Chronic viral infections, immunosenescence, CVDs and premature biological ageing
The premature biological ageing process is defined as age-related changes in function and composition of the human body. This process is characterized by chronic systemic immune activation, elevated inflammatory and coagulation markers (243, 244), decreased leukocyte telomere length (217), mitochondrial DNA mutations (245), cell senescence, impaired autophagy (246), epigenetic alterations and higher DNA methylation levels (247), which are correlated with onset of age-related comorbidities such as diabetes, CVDs, or obesity (248). Chronic viral infections are probably one of the most important drivers of premature biological ageing. Previous studies found increased age acceleration and higher DNA methylation in both blood cells and brain tissues of chronic HIV-infected patients compared to HIV-negative individuals (247, 249, 250). Another study on PBMCs of 122 nonagenarians and 21 young controls represented increased DNA methylation in chronically CMV infected individuals irrespective of age. Their findings also indicated a significant correlation between CMV seropositivity and higher epigenetic age in both groups (251). Another study confirmed a positive correlation between the host cell methylation state and CMV seropositivity, as well (252).
Such ageing related epigenetic alterations were also reported in COVID-19 survivors under 60 years of age (253). These individuals represented accelerated telomere shortening and increased DNA methylation in CpG islands of leukocytes and lymphocytes (253). This is noteworthy in the sense that SARS-CoV-2 infection is currently considered an acute viral infection.
The accelerated biological ageing induced by viruses can be a risk factor for vascular diseases and the subsequent cardiovascular and cerebrovascular events (254). The associations between age-related telomere shortening and CAD as well as CVD-related mortality have already been investigated (255–257). In addition, structural alterations in biologically ageing arteries such as elastin fragmentation (258), collagen accumulation (259), decreased vascular compliance (260) and increased arterial stiffness (261) as well as elevated blood pressure (262) are reported as the consequence of chronic viral infections, too. Premature ageing may impose similar risks of developing CVDs in younger individuals. In this regard, a previous study investigated the association between biological age acceleration and carotid intima-media thickness, in a 21-year follow-up cohort. Their results showed that 5-years increase in the epigenetic age in the whole blood was related to approximately 0.01 mm greater carotid intima–media thickness suggesting a potential contribution of biological age acceleration to the development of CVD (263). A shorter telomere length of the buccal epithelium in CVD patients is also shown to be associated with biological ageing (264).
Conclusion
Immunosenescence is the hallmark of many inflammatory and autoimmune diseases that is stimulated by multiple factors. Recent data indicate that chronic viral infections manipulate the pathways involved in replicative senescence (RS), oncogene-induced senescence (OIS), and possibly genotoxicity-induced senescence (GIS) in immune and non-immune cells. The senescence pathways induced by infectious agents are shared with other senescence inducing stimuli. The induction of senescence in immune cells is more robust in chronic viral infections due to direct stimulation of the immune system by viral antigens. From early childhood, the immune cells of human-beings are challenged with viral infections and fortunately enough, in most cases the virus is contained and even eradicated by immune system. However, continuous encounter with viruses and especially establishment of chronic viral infections in the body results in a state of more inflammatory and less protective immune response. In atherosclerosis, as one of the old inflammatory conditions and the mother of cardiovascular and cerebral stroke, immunosenescence is induced both in immune and non-immune cells. Therefore, chronic viral infections, through induction of immunosenescence, may directly or indirectly play a role in development or progression of atherosclerosis (Figure 2). The premature ageing as a result of viral co-infections may also accelerate immunosenescence and inflammatory diseases. In this article, we reviewed evidence of the possible role of chronic viral infections and co-infections in the induction of immunosenescence as a contributor in atherosclerosis.
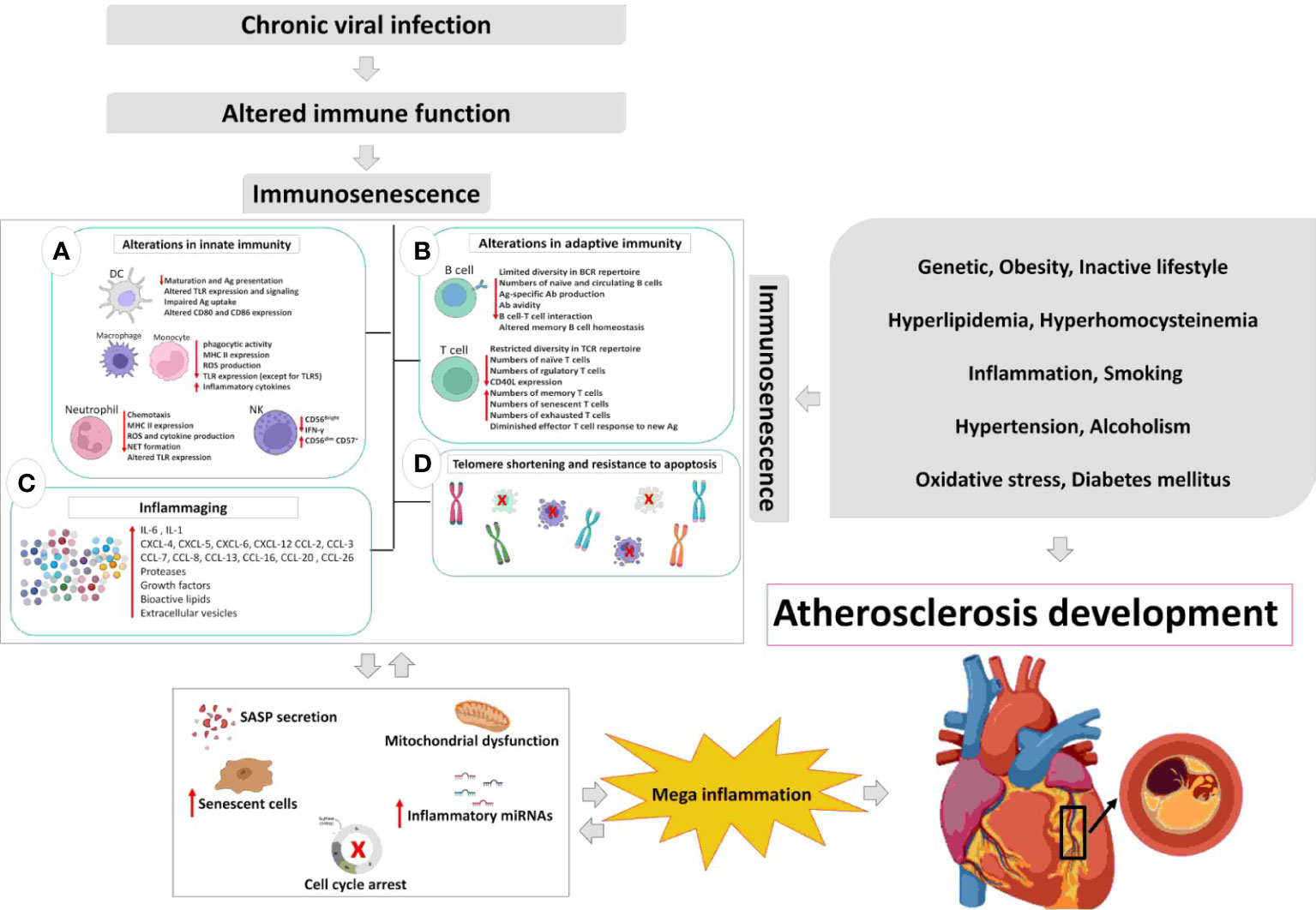
Figure 2 The relation between chronic viral infection-induced immunosenescence and atherosclerosis development. Chronic viral infections modify immune responses and immune functions over the time course of infection. The hallmark of most chronic viral infections is inflammaging, which triggers cell cycle arrest, SASP secretion, mitochondrial dysfunction, telomere shortening, and immunosenescence. Such alterations are accompanied by acceleration of atherosclerosis even in younger adults, thereby; in a positive feedback loop inflammaging accelerates atherosclerosis and ageing of the immune system. DC, dendritic cell; Ag, antigen; TLR, toll-like receptor; MHC, major histocompatibility complex; ROS, reactive oxygen species; NET, neutrophil extracellular traps; NK, natural killer; BCR, B cell receptor; Ab, antibody; TCR, T cell receptor; SASP, senescence-associated secretory phenotype.
Author contributions
AGT wrote the first draft of the manuscript. MD conceptualized, designed the study and critically revised the manuscript. All authors reviewed and approved the final version of the manuscript.
Funding
This study was supported by grants no. 93-7072, and 97-17106 from Shiraz University of Medical Sciences, Shiraz Iran.
Acknowledgments
The authors would like to thank all the individuals who participated in the mentioned studies by donating blood, tissue, etc. without whom these knowledge would have not been available.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lu RJ, Wang EK, Benayoun BA. Functional genomics of inflamm-aging and immunosenescence. Briefings Funct Genomics (2022) 21(1):43–55. doi: 10.1093/bfgp/elab009
2. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153(6):1194–217. doi: 10.1016/j.cell.2013.05.039
3. Aw D, Silva AB, Palmer DB. Immunosenescence: emerging challenges for an ageing population. Immunology (2007) 120(4):435–46. doi: 10.1111/j.1365-2567.2007.02555.x
4. Aiello A, Farzaneh F, Candore G, Caruso C, Davinelli S, Gambino CM, et al. Immunosenescence and its hallmarks: How to oppose aging strategically? a review of potential options for therapeutic intervention. Front Immunol (2019) 10:2247. doi: 10.3389/fimmu.2019.02247
5. Pawelec G. Hallmarks of human "immunosenescence": adaptation or dysregulation? Immun Ageing I A (2012) 9(1):15. doi: 10.1186/1742-4933-9-15
6. Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, et al. An aged immune system drives senescence and ageing of solid organs. Nature (2021) 594(7861):100–5. doi: 10.1038/s41586-021-03547-7
7. Teissier T, Boulanger E, Cox LS. Interconnections between inflammageing and immunosenescence during ageing. Cells (2022) 11(3). doi: 10.3390/cells11030359
8. Pangrazzi L, Weinberger B. T Cells, aging and senescence. Exp Gerontol (2020) 134:110887. doi: 10.1016/j.exger.2020.110887
9. Sadighi Akha AA. Aging and the immune system: An overview. J Immunol Methods (2018) 463:21–6. doi: 10.1016/j.jim.2018.08.005
10. Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood (2014) 123(2):239–48. doi: 10.1182/blood-2013-08-519520
11. Butcher SK, Chahal H, Nayak L, Sinclair A, Henriquez NV, Sapey E, et al. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukocyte Biol (2001) 70(6):881–6.
12. Hearps AC, Martin GE, Angelovich TA, Cheng WJ, Maisa A, Landay AL, et al. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell (2012) 11(5):867–75. doi: 10.1111/j.1474-9726.2012.00851.x
13. Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J, Sambhara S. Innate immunity in aging: impact on macrophage function. Aging Cell (2004) 3(4):161–7. doi: 10.1111/j.1474-9728.2004.00102.x
14. Panda A, Arjona A, Sapey E, Bai F, Fikrig E, Montgomery RR, et al. Human innate immunosenescence: causes and consequences for immunity in old age. Trends Immunol (2009) 30(7):325–33. doi: 10.1016/j.it.2009.05.004
15. Zhang Y, Wallace DL, de Lara CM, Ghattas H, Asquith B, Worth A, et al. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology (2007) 121(2):258–65. doi: 10.1111/j.1365-2567.2007.02573.x
16. Ogata K, An E, Shioi Y, Nakamura K, Luo S, Yokose N, et al. Association between natural killer cell activity and infection in immunologically normal elderly people. Clin Exp Immunol (2001) 124(3):392–7. doi: 10.1046/j.1365-2249.2001.01571.x
17. Bulut O, Kilic G, Domínguez-Andrés J, Netea MG. Overcoming immune dysfunction in the elderly: trained immunity as a novel approach. Int Immunol (2020) 32(12):741–53. doi: 10.1093/intimm/dxaa052
18. Rodriguez IJ, Lalinde Ruiz N, Llano León M, Martínez Enríquez L, Montilla Velásquez MDP, Ortiz Aguirre JP, et al. Immunosenescence study of T cells: A systematic review. Front Immunol (2020) 11:604591. doi: 10.3389/fimmu.2020.604591
19. Tu W, Rao S. Mechanisms underlying T cell immunosenescence: Aging and cytomegalovirus infection. Front Microbiol (2016) 7:2111. doi: 10.3389/fmicb.2016.02111
20. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: Friends or foes? Front Immunol (2017) 8:1960. doi: 10.3389/fimmu.2017.01960
21. Rodrigues LP, Teixeira VR, Alencar-Silva T, Simonassi-Paiva B, Pereira RW, Pogue R, et al. Hallmarks of aging and immunosenescence: Connecting the dots. Cytokine Growth Factor Rev (2021) 59:9–21. doi: 10.1016/j.cytogfr.2021.01.006
22. Kumari R, Jat P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol (2021) 9:645593. doi: 10.3389/fcell.2021.645593
23. McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol (2018) 217(1):65–77. doi: 10.1083/jcb.201708092
24. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol (2014) 15(7):482–96. doi: 10.1038/nrm3823
25. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: Defining a path forward. Cell (2019) 179(4):813–27. doi: 10.1016/j.cell.2019.10.005
26. Mohamad Kamal NS, Safuan S, Shamsuddin S, Foroozandeh P. Aging of the cells: Insight into cellular senescence and detection methods. Eur J Cell Biol (2020) 99(6):151108.
27. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell (2013) 155(5):1104–18. doi: 10.1016/j.cell.2013.10.019
28. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell (2014) 31(6):722–33. doi: 10.1016/j.devcel.2014.11.012
29. Xu W, Larbi A. Markers of T cell senescence in humans. Int J Mol Sci (2017) 18(8). doi: 10.3390/ijms18081742
30. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev (2010) 24(22):2463–79. doi: 10.1101/gad.1971610
31. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev (2014) 28(2):99–114. doi: 10.1101/gad.235184.113
32. Pawelec G. The human immunosenescence phenotype: does it exist? Semin Immunopathology (2020) 42(5):537–44. doi: 10.1007/s00281-020-00810-3
33. Larbi A, Fulop T. From "truly naïve" to "exhausted senescent" T cells: when markers predict functionality. Cytometry Part A J Int Soc Anal Cytol (2014) 85(1):25–35. doi: 10.1002/cyto.a.22351
34. Stubbe M, Vanderheyde N, Goldman M, Marchant A. Antigen-specific central memory CD4+ T lymphocytes produce multiple cytokines and proliferate in vivo in humans. J Immunol (Baltimore Md 1950) (2006) 177(11):8185–90. doi: 10.4049/jimmunol.177.11.8185
35. Ghamar Talepoor A, Khosropanah S, Doroudchi M. Partial recovery of senescence in circulating follicular helper T cells after dasatinib treatment. Int Immunopharmacol (2021) 94:107465. doi: 10.1016/j.intimp.2021.107465
36. Lian J, Yue Y, Yu W, Zhang Y. Immunosenescence: a key player in cancer development. J Hematol Oncol (2020) 13(1):151. doi: 10.1186/s13045-020-00986-z
37. Moreno-Blas D, Gorostieta-Salas E, Pommer-Alba A, Muciño-Hernández G, Gerónimo-Olvera C, Maciel-Barón LA, et al. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging (2019) 11(16):6175–98. doi: 10.18632/aging.102181
38. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathology (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
39. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol (2018) 28(6):436–53. doi: 10.1016/j.tcb.2018.02.001
40. Byun HO, Lee YK, Kim JM, Yoon G. From cell senescence to age-related diseases: differential mechanisms of action of senescence-associated secretory phenotypes. BMB Rep (2015) 48(10):549–58. doi: 10.5483/BMBRep.2015.48.10.122
41. Tchkonia T, Kirkland JL. Aging, cell senescence, and chronic disease: Emerging therapeutic strategies. JAMA (2018) 320(13):1319–20. doi: 10.1001/jama.2018.12440
42. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell (2008) 133(6):1019–31. doi: 10.1016/j.cell.2008.03.039
43. Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell (2008) 133(6):1006–18. doi: 10.1016/j.cell.2008.03.038
44. Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol (2009) 11(8):973–9. doi: 10.1038/ncb1909
45. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J (2011) 30(8):1536–48. doi: 10.1038/emboj.2011.69
46. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol (2015) 17(9):1205–17. doi: 10.1038/ncb3225
47. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol (2015) 17(8):1049–61. doi: 10.1038/ncb3195
48. Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev (2016) 30(3):321–36. doi: 10.1101/gad.271882.115
49. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science (2015) 349(6255):aaa5612. doi: 10.1126/science.aaa5612
50. Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer (2003) 3(2):130–8. doi: 10.1038/nrc993
51. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer (2009) 9(10):749–58. doi: 10.1038/nrc2723
52. Fischer M, Müller GA. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit Rev Biochem Mol Biol (2017) 52(6):638–62. doi: 10.1080/10409238.2017.1360836
53. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol (2015) 16(7):393–405. doi: 10.1038/nrm4007
54. Cox LS, Redman C. The role of cellular senescence in ageing of the placenta. Placenta (2017) 52:139–45. doi: 10.1016/j.placenta.2017.01.116
55. Rhinn M, Ritschka B, Keyes WM. Cellular senescence in development, regeneration and disease. Development (2019) 146(20). doi: 10.1242/dev.151837
56. Pratsinis H, Mavrogonatou E, Kletsas D. Scarless wound healing: From development to senescence. Adv Drug Deliv Rev (2019) 146:325–43. doi: 10.1016/j.addr.2018.04.011
57. Hasan MR, Ho SH, Owen DA, Tai IT. Inhibition of VEGF induces cellular senescence in colorectal cancer cells. Int J cancer (2011) 129(9):2115–23. doi: 10.1002/ijc.26179
58. Krizhanovsky V, Xue W, Zender L, Yon M, Hernando E, Lowe SW. Implications of cellular senescence in tissue damage response, tumor suppression, and stem cell biology. Cold Spring Harbor Symp Quant Biol (2008) 73:513–22. doi: 10.1101/sqb.2008.73.048
59. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature (2016) 530(7589):184–9. doi: 10.1038/nature16932
60. Cohen J, Torres C. Astrocyte senescence: Evidence and significance. Aging Cell (2019) 18(3):e12937. doi: 10.1111/acel.12937
61. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun (2017) 8:15691. doi: 10.1038/ncomms15691
62. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun (2017) 8:14532. doi: 10.1038/ncomms14532
63. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular senescence: Aging, cancer, and injury. Physiol Rev (2019) 99(2):1047–78. doi: 10.1152/physrev.00020.2018
64. Naylor RM, Baker DJ, van Deursen JM. Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin Pharmacol Ther (2013) 93(1):105–16. doi: 10.1038/clpt.2012.193
65. Robinson JG, Fox KM, Bullano MF, Grandy S. Atherosclerosis profile and incidence of cardiovascular events: a population-based survey. BMC Cardiovasc Disord (2009) 9:46. doi: 10.1186/1471-2261-9-46
66. Rahman MS, Woollard K. Atherosclerosis. Adv Exp Med Biol (2017) 1003:121–44. doi: 10.1007/978-3-319-57613-8_7
67. Lu H, Daugherty A. Atherosclerosis. Arteriosclerosis thrombosis Vasc Biol (2015) 35(3):485–91. doi: 10.1161/ATVBAHA.115.305380
68. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature (2011) 473(7347):317–25. doi: 10.1038/nature10146
69. Centner AM, Bhide PG. Nicotine in senescence and atherosclerosis. Cells (2020) 9(4). doi: 10.3390/cells9041035
70. Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation (2002) 105(13):1541–4. doi: 10.1161/01.CIR.0000013836.85741.17
71. Pham LM, Kim EC, Ou W, Phung CD, Nguyen TT, Pham TT, et al. Targeting and clearance of senescent foamy macrophages and senescent endothelial cells by antibody-functionalized mesoporous silica nanoparticles for alleviating aorta atherosclerosis. Biomaterials (2021) 269:120677. doi: 10.1016/j.biomaterials.2021.120677
72. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res (2016) 118(4):692–702. doi: 10.1161/CIRCRESAHA.115.306361
73. Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res (2018) 114(4):622–34. doi: 10.1093/cvr/cvy007
74. Wu CM, Zheng L, Wang Q, Hu YW. The emerging role of cell senescence in atherosclerosis. Clin Chem Lab Med (2020) 59(1):27–38.
75. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (2016) 354(6311):472–7. doi: 10.1126/science.aaf6659
76. Bernal E, Martinez M, Torres A, Guillamón CF, Alcaraz A, Alcaraz MJ, et al. T Cell senescence predicts subclinical atherosclerosis in HIV-infected patients similarly to traditional cardiovascular risk factors. Antiviral Res (2019) 162:163–70. doi: 10.1016/j.antiviral.2018.12.014
77. Yu HT, Park S, Shin EC, Lee WW. T Cell senescence and cardiovascular diseases. Clin Exp Med (2016) 16(3):257–63. doi: 10.1007/s10238-015-0376-z
78. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res (2014) 114(12):1852–66. doi: 10.1161/CIRCRESAHA.114.302721
79. Lee DY, Chiu JJ. Atherosclerosis and flow: roles of epigenetic modulation in vascular endothelium. J BioMed Sci (2019) 26(1):56. doi: 10.1186/s12929-019-0551-8
80. de Yébenes VG, Briones AM, Martos-Folgado I, Mur SM, Oller J, Bilal F, et al. Aging-associated miR-217 aggravates atherosclerosis and promotes cardiovascular dysfunction. Arterioscler Thromb Vasc Biol (2020) 40(10):2408–24. doi: 10.1161/ATVBAHA.120.314333
81. Grootaert MOJ, Roth L, Schrijvers DM, De Meyer GRY, Martinet W. Defective autophagy in atherosclerosis: To die or to senesce? Oxid Med Cell Longev (2018) 2018:7687083. doi: 10.1155/2018/7687083
82. Jia G, Aroor AR, Jia C, Sowers JR. Endothelial cell senescence in aging-related vascular dysfunction. Biochim Biophys Acta Mol Basis Dis (2019) 1865(7):1802–9. doi: 10.1016/j.bbadis.2018.08.008
83. Lowe D, Raj K. Premature aging induced by radiation exhibits pro-atherosclerotic effects mediated by epigenetic activation of CD44 expression. Aging Cell (2014) 13(5):900–10. doi: 10.1111/acel.12253
84. Warboys CM, de Luca A, Amini N, Luong L, Duckles H, Hsiao S, et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler Thromb Vasc Biol (2014) 34(5):985–95. doi: 10.1161/ATVBAHA.114.303415
85. Venkat P, Cui C, Chopp M, Zacharek A, Wang F, Landschoot-Ward J, et al. MiR-126 mediates brain endothelial cell exosome treatment-induced neurorestorative effects after stroke in type 2 diabetes mellitus mice. Stroke (2019) 50(10):2865–74. doi: 10.1161/STROKEAHA.119.025371
86. Yang HH, Chen Y, Gao CY, Cui ZT, Yao JM. Protective effects of MicroRNA-126 on human cardiac microvascular endothelial cells against Hypoxia/Reoxygenation-induced injury and inflammatory response by activating PI3K/Akt/eNOS signaling pathway. Cell Physiol Biochem Int J Exp Cell Physiology Biochem Pharmacol (2017) 42(2):506–18. doi: 10.1159/000477597
87. Kida Y, Goligorsky MS. Sirtuins, cell senescence, and vascular aging. Can J Cardiol (2016) 32(5):634–41. doi: 10.1016/j.cjca.2015.11.022
88. Zhang J, Patel JM, Block ER. Enhanced apoptosis in prolonged cultures of senescent porcine pulmonary artery endothelial cells. Mech Ageing Dev (2002) 123(6):613–25. doi: 10.1016/S0047-6374(01)00412-2
89. Krouwer VJ, Hekking LH, Langelaar-Makkinje M, Regan-Klapisz E, Post JA. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc Cell (2012) 4(1):12. doi: 10.1186/2045-824X-4-12
90. Carracedo J, Ramírez-Carracedo R. Protein carbamylation: A marker reflecting increased age-related cell oxidation. Int J Mol Sci (2018) 19(5). doi: 10.3390/ijms19051495
91. Carmona A, Guerrero F, Buendia P, Obrero T, Aljama P, Carracedo J. Microvesicles derived from indoxyl sulfate treated endothelial cells induce endothelial progenitor cells dysfunction. Front Physiol (2017) 8:666. doi: 10.3389/fphys.2017.00666
92. Alique M, Ruíz-Torres MP, Bodega G, Noci MV, Troyano N, Bohórquez L, et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging (2017) 9(3):778–89. doi: 10.18632/aging.101191
93. Bennett MR, Macdonald K, Chan SW, Boyle JJ, Weissberg PL. Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ Res (1998) 82(6):704–12. doi: 10.1161/01.RES.82.6.704
94. Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res (2006) 99(2):156–64. doi: 10.1161/01.RES.0000233315.38086.bc
95. Gorenne I, Kavurma M, Scott S, Bennett M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res (2006) 72(1):9–17. doi: 10.1016/j.cardiores.2006.06.004
96. Lacolley P, Regnault V, Avolio AP. Smooth muscle cell and arterial aging: basic and clinical aspects. Cardiovasc Res (2018) 114(4):513–28. doi: 10.1093/cvr/cvy009
97. Schurgers LJ, Akbulut AC, Kaczor DM, Halder M, Koenen RR, Kramann R. Initiation and propagation of vascular calcification is regulated by a concert of platelet- and smooth muscle cell-derived extracellular vesicles. Front Cardiovasc Med (2018) 5:36. doi: 10.3389/fcvm.2018.00036
98. Gardner SE, Humphry M, Bennett MR, Clarke MC. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α-Dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol (2015) 35(9):1963–74. doi: 10.1161/ATVBAHA.115.305896
99. Nakajima T, Schulte S, Warrington KJ, Kopecky SL, Frye RL, Goronzy JJ, et al. T-Cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation (2002) 105(5):570–5. doi: 10.1161/hc0502.103348
100. Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res (2006) 99(10):1044–59. doi: 10.1161/01.RES.0000249379.55535.21
101. Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res (2018) 114(4):540–50. doi: 10.1093/cvr/cvy022
102. Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation (2015) 132(20):1909–19. doi: 10.1161/CIRCULATIONAHA.115.016457
103. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. an evolutionary perspective on immunosenescence. Ann New York Acad Sci (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
104. Brouilette SW, Whittaker A, Stevens SE, van der Harst P, Goodall AH, Samani NJ. Telomere length is shorter in healthy offspring of subjects with coronary artery disease: support for the telomere hypothesis. Heart (British Cardiac Society) (2008) 94(4):422–5.
105. Liuzzo G, Biasucci LM, Trotta G, Brugaletta S, Pinnelli M, Digianuario G, et al. Unusual CD4+CD28null T lymphocytes and recurrence of acute coronary events. J Am Coll Cardiol (2007) 50(15):1450–8. doi: 10.1016/j.jacc.2007.06.040
106. Ramos GC, van den Berg A, Nunes-Silva V, Weirather J, Peters L, Burkard M, et al. Myocardial aging as a T-cell-mediated phenomenon. Proc Natl Acad Sci USA (2017) 114(12):E2420–e9. doi: 10.1073/pnas.1621047114
107. Delgobo M, Heinrichs M, Hapke N, Ashour D, Appel M, Srivastava M, et al. Terminally differentiated CD4(+) T cells promote myocardial inflammaging. Front Immunol (2021) 12:584538. doi: 10.3389/fimmu.2021.584538
108. Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, et al. Immunosenescent CD8+ T cells and c-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension (Dallas Tex 1979) (2013) 62(1):126–33. doi: 10.1161/HYPERTENSIONAHA.113.00689
109. Tae Yu H, Youn JC, Lee J, Park S, Chi HS, Lee J, et al. Characterization of CD8(+)CD57(+) T cells in patients with acute myocardial infarction. Cell Mol Immunol (2015) 12(4):466–73.
110. Varricchi G, Bencivenga L, Poto R, Pecoraro A, Shamji MH, Rengo G. The emerging role of T follicular helper (T(FH)) cells in aging: Influence on the immune frailty. Ageing Res Rev (2020) 61:101071. doi: 10.1016/j.arr.2020.101071
111. Wang J, Bai Y, Zhao X, Ru J, Kang N, Tian T, et al. oxLDL-mediated cellular senescence is associated with increased NADPH oxidase p47phox recruitment to caveolae. Bioscience Rep (2018) 38(3). doi: 10.1042/BSR20180283
112. Talepoor AG, Kalani M, Dahaghani AS, Doroudchi M. Hydrogen peroxide and lipopolysaccharide differentially affect the expression of MicroRNAs 10a, 33a, 21, 221 in endothelial cells before and after coculture with monocytes. Int J toxicology (2017) 36(2):133–41. doi: 10.1177/1091581817695270
113. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis - from experimental insights to the clinic. Nat Rev Drug Discovery (2021) 20(8):589–610. doi: 10.1038/s41573-021-00198-1
114. Neves J, Sousa-Victor P. Regulation of inflammation as an anti-aging intervention. FEBS J (2020) 287(1):43–52. doi: 10.1111/febs.15061
115. Nowak WN, Deng J, Ruan XZ, Xu Q. Reactive oxygen species generation and atherosclerosis. Arterioscler Thromb Vasc Biol (2017) 37(5):e41–52. doi: 10.1161/ATVBAHA.117.309228
116. Davalli P, Mitic T, Caporali A, Lauriola A, D'Arca D. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid Med Cell Longevity (2016) 2016:3565127. doi: 10.1155/2016/3565127
117. Cottage CT, Peterson N, Kearley J, Berlin A, Xiong X, Huntley A, et al. Targeting p16-induced senescence prevents cigarette smoke-induced emphysema by promoting IGF1/Akt1 signaling in mice. Commun Biol (2019) 2:307. doi: 10.1038/s42003-019-0532-1
118. Wang W, Zhao T, Geng K, Yuan G, Chen Y, Xu Y. Smoking and the pathophysiology of peripheral artery disease. Front Cardiovasc Med (2021) 8:704106. doi: 10.3389/fcvm.2021.704106
119. McCarthy CG, Wenceslau CF, Webb RC, Joe B. Novel contributors and mechanisms of cellular senescence in hypertension-associated premature vascular aging. Am J Hypertension (2019) 32(8):709–19. doi: 10.1093/ajh/hpz052
120. Westhoff JH, Hilgers KF, Steinbach MP, Hartner A, Klanke B, Amann K, et al. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension (Dallas Tex 1979) (2008) 52(1):123–9. doi: 10.1161/HYPERTENSIONAHA.107.099432
121. Maeda M, Hayashi T, Mizuno N, Hattori Y, Kuzuya M. Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: role of superoxide production by NADPH oxidase. PloS One (2015) 10(4):e0123169. doi: 10.1371/journal.pone.0123169
122. Poznyak A, Grechko AV, Poggio P. The diabetes mellitus-atherosclerosis connection: The role of lipid and glucose metabolism and chronic inflammation. Int J Mol Sci (2020) 21(5). doi: 10.3390/ijms21051835
123. Seoane R, Vidal S, Bouzaher YH, El Motiam A. The interaction of viruses with the cellular senescence response. Biol (Basel) (2020) 9(12). doi: 10.3390/biology9120455
124. Fouladseresht H, Ghamar Talepoor A, Farjadian S, Khosropanah S, Doroudchi M. Anti-varicella zoster virus IgG and hsCRP levels correlate with progression of coronary artery atherosclerosis. Iranian J Allergy Asthma Immunol (2019) 18(5):543–53. doi: 10.18502/ijaai.v18i5.1924
125. Peng W, Cai G, Xia Y, Chen J, Wu P, Wang Z, et al. Mitochondrial dysfunction in atherosclerosis. DNA Cell Biol (2019) 38(7):597–606. doi: 10.1089/dna.2018.4552
126. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab (2016) 23(2):303–14. doi: 10.1016/j.cmet.2015.11.011
127. Busuttil RA, Dollé M, Campisi J, Vijga J. Genomic instability, aging, and cellular senescence. Ann New York Acad Sci (2004) 1019:245–55. doi: 10.1196/annals.1297.041
128. Shah NR, Mahmoudi M. The role of DNA damage and repair in atherosclerosis: A review. J Mol Cell Cardiol (2015) 86:147–57. doi: 10.1016/j.yjmcc.2015.07.005
129. Martínez I, García-Carpizo V, Guijarro T, García-Gomez A, Navarro D, Aranda A, et al. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence (2016) 7(4):427–42. doi: 10.1080/21505594.2016.1144001
130. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol (2013) 15(8):978–90. doi: 10.1038/ncb2784
131. Chou JP, Effros RB. T Cell replicative senescence in human aging. Curr Pharm Design (2013) 19(9):1680–98.
132. Nikolich-Žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol (2018) 19(1):10–9. doi: 10.1038/s41590-017-0006-x
133. Barbé-Tuana F, Funchal G, Schmitz CRR, Maurmann RM, Bauer ME. The interplay between immunosenescence and age-related diseases. J Semin Immunopathol (2020) 42(5):545–57. doi: 10.1007/s00281-020-00806-z
134. Simpson RJ. Aging, persistent viral infections, and immunosenescence: can exercise "make space"? Exercise Sport Sci Rev (2011) 39(1):23–33. doi: 10.1097/JES.0b013e318201f39d
135. Heath JJ, Fudge NJ, Gallant ME, Grant MD. Proximity of cytomegalovirus-specific CD8(+) T cells to replicative senescence in human immunodeficiency virus-infected individuals. Front Immunol (2018) 9:201. doi: 10.3389/fimmu.2018.00201
136. Spyridopoulos I, Martin-Ruiz C, Hilkens C, Yadegarfar ME, Isaacs J, Jagger C, et al. CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: results from the Newcastle 85+ study. Aging Cell (2016) 15(2):389–92. doi: 10.1111/acel.12430
137. Tanaka S, Toh Y, Mori R, Komori K, Okadome K, Sugimachi K. Possible role of cytomegalovirus in the pathogenesis of inflammatory aortic diseases: a preliminary report. J Vasc Surg (1992) 16(2):274–9. doi: 10.1016/0741-5214(92)90118-R
138. Zhu W, Liu S. The role of human cytomegalovirus in atherosclerosis: a systematic review. Acta Biochim Biophys Sinica (2020) 52(4):339–53. doi: 10.1093/abbs/gmaa005
139. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Sci (New York NY) (2008) 319(5868):1352–5. doi: 10.1126/science.1140735
140. Hafez AY, Luftig MA. Characterization of the EBV-induced persistent DNA damage response. Viruses (2017) 9(12). doi: 10.3390/v9120366
141. Nikitin PA, Price AM, McFadden K, Yan CM, Luftig MA. Mitogen-induced b-cell proliferation activates Chk2-dependent G1/S cell cycle arrest. PloS One (2014) 9(1):e87299. doi: 10.1371/journal.pone.0087299
142. Al-Ghamdi A. Role of herpes simplex virus-1, cytomegalovirus and Epstein-Barr virus in atherosclerosis. Pakistan J Pharm Sci (2012) 25(1):89–97.
143. Waldman WJ, Williams MV Jr., Lemeshow S, Binkley P, Guttridge D, Kiecolt-Glaser JK, et al. Epstein-Barr Virus-encoded dUTPase enhances proinflammatory cytokine production by macrophages in contact with endothelial cells: evidence for depression-induced atherosclerotic risk. Brain Behav Immun (2008) 22(2):215–23. doi: 10.1016/j.bbi.2007.07.007
144. Wege H, Le HT, Chui MS, Liu L, Wu J, Giri R, et al. Telomerase reconstitution immortalizes human fetal hepatocytes without disrupting their differentiation potential. Gastroenterology (2003) 124(2):432–44. doi: 10.1053/gast.2003.50064
145. Ji Y, Dang X, Nguyen LNT, Nguyen LN, Zhao J, Cao D, et al. Topological DNA damage, telomere attrition and T cell senescence during chronic viral infections. Immun Ageing I A (2019) 16:12. doi: 10.1186/s12979-019-0153-z
146. Madden CR, Slagle BL. Stimulation of cellular proliferation by hepatitis b virus X protein. Dis Markers (2001) 17(3):153–7. doi: 10.1155/2001/571254
147. Ishizaka N, Ishizaka Y, Takahashi E, Toda Ei E, Hashimoto H, Ohno M, et al. Increased prevalence of carotid atherosclerosis in hepatitis b virus carriers. Circulation (2002) 105(9):1028–30. doi: 10.1161/hc0902.105718
148. Jabeen S, Rasheed A, Jabeen N, Naz SA, Raza A. Prevalence and association of HBV and HCV infection with cardiovascular disease risk factors in a peri-urban population. JPMA J Pakistan Med Assoc (2020) 70(1):58–63.
149. Valgimigli M, Valgimigli L, Trerè D, Gaiani S, Pedulli GF, Gramantieri L, et al. Oxidative stress EPR measurement in human liver by radical-probe technique. correlation with etiology, histology and cell proliferation. Free Radical Res (2002) 36(9):939–48.
150. Pal S, Polyak SJ, Bano N, Qiu WC, Carithers RL, Shuhart M, et al. Hepatitis c virus induces oxidative stress, DNA damage and modulates the DNA repair enzyme NEIL1. J Gastroenterol Hepatology (2010) 25(3):627–34. doi: 10.1111/j.1440-1746.2009.06128.x
151. Adinolfi LE, Zampino R, Restivo L, Lonardo A, Guerrera B, Marrone A, et al. Chronic hepatitis c virus infection and atherosclerosis: clinical impact and mechanisms. World J Gastroenterology (2014) 20(13):3410–7. doi: 10.3748/wjg.v20.i13.3410
152. Adinolfi LE, Restivo L, Zampino R, Guerrera B, Lonardo A, Ruggiero L, et al. Chronic HCV infection is a risk of atherosclerosis. role of HCV and HCV-related steatosis. Atherosclerosis (2012) 221(2):496–502.
153. Nelson JA, Krishnamurthy J, Menezes P, Liu Y, Hudgens MG, Sharpless NE, et al. Expression of p16(INK4a) as a biomarker of T-cell aging in HIV-infected patients prior to and during antiretroviral therapy. Aging Cell (2012) 11(5):916–8. doi: 10.1111/j.1474-9726.2012.00856.x
154. Ribeiro SP, Milush JM, Cunha-Neto E, Kallas EG, Kalil J, Passero LFD, et al. p16INK4a expression and immunologic aging in chronic HIV infection. PloS One (2016) 11(11):e0166759.
155. Reingold J, Wanke C, Kotler D, Lewis C, Tracy R, Heymsfield S, et al. Association of HIV infection and HIV/HCV coinfection with c-reactive protein levels: the fat redistribution and metabolic change in HIV infection (FRAM) study. J Acquir Immune Defic Syndr (1999) (2008) 48(2):142–8. doi: 10.1097/QAI.0b013e3181685727
156. Park IW, Wang JF, Groopman JE. HIV-1 tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood (2001) 97(2):352–8. doi: 10.1182/blood.V97.2.352
157. Lee S, Yu Y, Trimpert J, Benthani F, Mairhofer M, Richter-Pechanska P, et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature (2021) 599(7884):283–9. doi: 10.1038/s41586-021-03995-1
158. Meyer K, Patra T, Vijayamahantesh, Ray R. SARS-CoV-2 spike protein induces paracrine senescence and leukocyte adhesion in endothelial cells. J virology (2021) 95(17):e0079421. doi: 10.1128/JVI.00794-21
159. Gao L, Jiang D, Wen XS, Cheng XC, Sun M, He B, et al. Prognostic value of NT-proBNP in patients with severe COVID-19. Respir Res (2020) 21(1):83. doi: 10.1186/s12931-020-01352-w
160. Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res (2020) 116(6):1097–100. doi: 10.1093/cvr/cvaa078
161. Forte E, Zhang Z, Thorp EB, Hummel M. Cytomegalovirus latency and reactivation: An intricate interplay with the host immune response. Front Cell Infect Microbiol (2020) 10:130. doi: 10.3389/fcimb.2020.00130
162. van den Berg SPH, Pardieck IN. The hallmarks of CMV-specific CD8 T-cell differentiation. Med Microbiol Immunol (2019) 208(3-4):365–73. doi: 10.1007/s00430-019-00608-7
163. Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med (2005) 202(5):673–85. doi: 10.1084/jem.20050882
164. Hadrup SR, Strindhall J, Køllgaard T, Seremet T, Johansson B, Pawelec G, et al. Longitudinal studies of clonally expanded CD8 T cells reveal a repertoire shrinkage predicting mortality and an increased number of dysfunctional cytomegalovirus-specific T cells in the very elderly. J Immunol (Baltimore Md 1950) (2006) 176(4):2645–53. doi: 10.4049/jimmunol.176.4.2645
165. Lopez-Vergès S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, et al. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood (2010) 116(19):3865–74. doi: 10.1182/blood-2010-04-282301
166. Effros RB, Boucher N, Porter V, Zhu X, Spaulding C, Walford RL, et al. Decline in CD28+ T cells in centenarians and in long-term T cell cultures: a possible cause for both in vivo and in vitro immunosenescence. Exp Gerontology (1994) 29(6):601–9. doi: 10.1016/0531-5565(94)90073-6
167. Czesnikiewicz-Guzik M, Lee WW, Cui D, Hiruma Y, Lamar DL, Yang ZZ, et al. T Cell subset-specific susceptibility to aging. Clin Immunol (Orlando Fla) (2008) 127(1):107–18. doi: 10.1016/j.clim.2007.12.002
168. Bellon M, Nicot C. Telomere dynamics in immune senescence and exhaustion triggered by chronic viral infection. (2017) 9(10):. doi: 10.3390/v9100289
169. J Heath J, D Grant M. The immune response against human cytomegalovirus links cellular to systemic senescence. Cells (2020) 9(3). doi: 10.3390/cells9030766
170. Roberts ET, Haan MN, Dowd JB, Aiello AE. Cytomegalovirus antibody levels, inflammation, and mortality among elderly latinos over 9 years of follow-up. Am J Epidemiol (2010) 172(4):363–71. doi: 10.1093/aje/kwq177
171. Savva GM, Pachnio A, Kaul B, Morgan K, Huppert FA, Brayne C, et al. Cytomegalovirus infection is associated with increased mortality in the older population. Aging Cell (2013) 12(3):381–7. doi: 10.1111/acel.12059
172. Williams H, Crawford DH. Epstein-Barr Virus: the impact of scientific advances on clinical practice. Blood (2006) 107(3):862–9. doi: 10.1182/blood-2005-07-2702
173. Dierickx D, Habermann TM. Post-transplantation lymphoproliferative disorders in adults. New Engl J Med (2018) 378(6):549–62. doi: 10.1056/NEJMra1702693
174. Hue SS, Oon ML, Wang S, Tan SY, Ng SB. Epstein-Barr Virus-associated T- and NK-cell lymphoproliferative diseases: an update and diagnostic approach. Pathology (2020) 52(1):111–27. doi: 10.1016/j.pathol.2019.09.011
175. Nikitin PA, Yan CM, Forte E, Bocedi A, Tourigny JP, White RE, et al. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human b cells. Cell Host Microbe (2010) 8(6):510–22. doi: 10.1016/j.chom.2010.11.004
176. Hafez AY, Messinger JE, McFadden K, Fenyofalvi G, Shepard CN, Lenzi GM, et al. Limited nucleotide pools restrict Epstein-Barr virus-mediated b-cell immortalization. Oncogenesis (2017) 6(6):e349. doi: 10.1038/oncsis.2017.46
177. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature (2006) 444(7119):633–7. doi: 10.1038/nature05268
178. McFadden K, Hafez AY, Kishton R, Messinger JE, Nikitin PA, Rathmell JC, et al. Metabolic stress is a barrier to Epstein-Barr virus-mediated b-cell immortalization. Proc Natl Acad Sci U S A (2016) 113(6):E782–90. doi: 10.1073/pnas.1517141113
179. Lanfermeijer J, de Greef PC, Hendriks M, Vos M, van Beek J, Borghans JA, et al. Age and CMV-infection jointly affect the EBV-specific CD8+ T-cell repertoire. Front Aging (2021) 2:11. doi: 10.3389/fragi.2021.665637
180. Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Victor A, et al. Impact of infectious burden on progression of carotid atherosclerosis. Stroke (2002) 33(11):2581–6. doi: 10.1161/01.STR.0000034789.82859.A4
181. Priyanka S, Kaarthikeyan G, Nadathur JD, Mohanraj A, Kavarthapu A. Detection of cytomegalovirus, Epstein-Barr virus, and torque teno virus in subgingival and atheromatous plaques of cardiac patients with chronic periodontitis. J Indian Soc Periodontol (2017) 21(6):456–60.
182. Suhail M, Abdel-Hafiz H, Ali A, Fatima K, Damanhouri GA, Azhar E, et al. Potential mechanisms of hepatitis b virus induced liver injury. World J gastroenterology (2014) 20(35):12462–72. doi: 10.3748/wjg.v20.i35.12462
183. Oura K, Morishita A. Molecular and functional roles of MicroRNAs in the progression of hepatocellular carcinoma-a review. Int J Mol Sci (2020) 21(21). doi: 10.3390/ijms21218362
184. Paradis V, Youssef N, Dargère D, Bâ N, Bonvoust F, Deschatrette J, et al. Replicative senescence in normal liver, chronic hepatitis c, and hepatocellular carcinomas. Hum Pathol (2001) 32(3):327–32. doi: 10.1053/hupa.2001.22747
185. Tachtatzis PM, Marshall A, Arvinthan A, Verma S, Penrhyn-Lowe S, Mela M, et al. Chronic hepatitis b virus infection: The relation between hepatitis b antigen expression, telomere length, senescence, inflammation and fibrosis. PloS One (2015) 10(5):e0127511.
186. Idrissi ME, Hachem H, Koering C, Merle P, Thénoz M, Mortreux F, et al. HBx triggers either cellular senescence or cell proliferation depending on cellular phenotype. J Viral Hepatitis (2016) 23(2):130–8. doi: 10.1111/jvh.12450
187. Zhou X, Yang F, Yang Y, Hu Y, Liu W, Huang C, et al. HBV facilitated hepatocellular carcinoma cells proliferation by up-regulating angiogenin expression through IL-6. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2018) 46(2):461–70. doi: 10.1159/000488614
188. Wijarnpreecha K, Thongprayoon C, Panjawatanan P, Ungprasert P. Hepatitis b virus infection and risk of coronary artery disease: a meta-analysis. Ann Trans Med (2016) 4(21):423. doi: 10.21037/atm.2016.11.12
189. Kumar A, Shariff M, Doshi R. Association between past hepatitis b infection and ischemic heart disease: An analysis from the 2007-2016 NHANES data. Am J Med Sci (2020) 360(4):372–7. doi: 10.1016/j.amjms.2020.05.034
190. Riveiro-Barciela M, Marcos-Fosch C, Martinez-Valle F, Bronte F, Orozco O, Sanz-Pérez I, et al. Naïve hepatitis b e antigen-negative chronic hepatitis b patients are at risk of carotid atherosclerosis: A prospective study. World J Gastroenterol (2021) 27(30):5112–25. doi: 10.3748/wjg.v27.i30.5112
191. Tong DY, Wang XH, Xu CF, Yang YZ, Xiong SD. Hepatitis b virus infection and coronary atherosclerosis: results from a population with relatively high prevalence of hepatitis b virus. World J Gastroenterol (2005) 11(9):1292–6. doi: 10.3748/wjg.v11.i9.1292
192. Kiechl S, Egger G, Mayr M, Wiedermann CJ, Bonora E, Oberhollenzer F, et al. Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation (2001) 103(8):1064–70. doi: 10.1161/01.CIR.103.8.1064
193. Li CG, Liao SJ, Que JL, Liu KJ, Wang HY, Yu J. The relationship between exposure to hepatitis b virus and increased atherosclerosis-associated morbidity - a meta-analysis. Kardiologia polska (2018) 76(1):119–24. doi: 10.5603/KP.a2017.0165
194. Malikova AZ, Shcherbakova AS, Konduktorov KA, Zemskaya AS, Dalina AA, Popenko VI, et al. Pre-senescence induction in hepatoma cells favors hepatitis c virus replication and can be used in exploring antiviral potential of histone deacetylase inhibitors. Int J Mol Sci (2021) 22(9). doi: 10.3390/ijms22094559
195. Bukh J. The history of hepatitis c virus (HCV): Basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J Hepatol (2016) 65(1 Suppl):S2–s21. doi: 10.1016/j.jhep.2016.07.035
196. de Souza-Cruz S, Victória MB, Tarragô AM, da Costa AG, Pimentel JP, Pires EF, et al. Liver and blood cytokine microenvironment in HCV patients is associated to liver fibrosis score: a proinflammatory cytokine ensemble orchestrated by TNF and tuned by IL-10. BMC Microbiol (2016) 16:3. doi: 10.1186/s12866-015-0610-6
197. Gomaa WM, Ibrahim MA, Shatat ME. Overexpression of cyclooxygenase-2 and transforming growth factor-beta 1 is an independent predictor of poor virological response to interferon therapy in chronic HCV genotype 4 patients. Saudi J Gastroenterol (2014) 20(1):59–65. doi: 10.4103/1319-3767.126324
198. Salum GM, Bader El Din NG, Ibrahim MK, Anany MA, Dawood RM, Khairy A, et al. Vascular endothelial growth factor expression in hepatitis c virus-induced liver fibrosis: A potential biomarker. J Interferon Cytokine Res (2017) 37(7):310–6. doi: 10.1089/jir.2016.0127
199. Wijetunga NA, Pascual M, Tozour J, Delahaye F, Alani M, Adeyeye M, et al. A pre-neoplastic epigenetic field defect in HCV-infected liver at transcription factor binding sites and polycomb targets. Oncogene (2017) 36(14):2030–44. doi: 10.1038/onc.2016.340
200. Hamdane N, Jühling F, Crouchet E, El Saghire H, Thumann C, Oudot MA, et al. HCV-induced epigenetic changes associated with liver cancer risk persist after sustained virologic response. Gastroenterology (2019) 156(8):2313–29.e7. doi: 10.1053/j.gastro.2019.02.038
201. Hoare M, Gelson WT, Das A, Fletcher JM, Davies SE, Curran MD, et al. CD4+ T-lymphocyte telomere length is related to fibrosis stage, clinical outcome and treatment response in chronic hepatitis c virus infection. J Hepatol (2010) 53(2):252–60. doi: 10.1016/j.jhep.2010.03.005
202. Hoare M, Shankar A, Shah M, Rushbrook S, Gelson W, Davies S, et al. γ-H2AX+CD8+ T lymphocytes cannot respond to IFN-α, IL-2 or IL-6 in chronic hepatitis c virus infection. J Hepatol (2013) 58(5):868–74. doi: 10.1016/j.jhep.2012.12.009
203. Giannakoulis VG, Dubovan P. Senescence in HBV-, HCV- and NAFLD- mediated hepatocellular carcinoma and senotherapeutics: Current evidence and future perspective. J Cancers (Basel) (2021) 13(18). doi: 10.3390/cancers13184732
204. Babiker A, Jeudy J, Kligerman S, Khambaty M, Shah A, Bagchi S. Risk of cardiovascular disease due to chronic hepatitis c infection: A review. J Clin Trans hepatology (2017) 5(4):343–62. doi: 10.14218/JCTH.2017.00021
205. Mazzaro C, Quartuccio L. A review on extrahepatic manifestations of chronic hepatitis c virus infection and the impact of direct-acting antiviral therapy. J Viruses (2021) 13(11). doi: 10.3390/v13112249
206. Goossens N, Negro F. Cardiovascular manifestations of hepatitis c virus. Clin Liver Dis (2017) 21(3):465–73. doi: 10.1016/j.cld.2017.03.003
207. Negro F. Facts and fictions of HCV and comorbidities: steatosis, diabetes mellitus, and cardiovascular diseases. J Hepatol (2014) 61(1 Suppl):S69–78. doi: 10.1016/j.jhep.2014.08.003
208. Zampino R, Marrone A, Restivo L, Guerrera B, Sellitto A, Rinaldi L, et al. Chronic HCV infection and inflammation: Clinical impact on hepatic and extra-hepatic manifestations. World J Hepatol (2013) 5(10):528–40. doi: 10.4254/wjh.v5.i10.528
209. Boddi M, Abbate R, Chellini B, Giusti B, Giannini C, Pratesi G, et al. Hepatitis c virus RNA localization in human carotid plaques. J Clin Virol (2010) 47(1):72–5. doi: 10.1016/j.jcv.2009.10.005
210. Durante-Mangoni E, Zampino R, Marrone A, Tripodi MF, Rinaldi L, Restivo L, et al. Hepatic steatosis and insulin resistance are associated with serum imbalance of adiponectin/tumour necrosis factor-alpha in chronic hepatitis c patients. Alimentary Pharmacol Ther (2006) 24(9):1349–57. doi: 10.1111/j.1365-2036.2006.03114.x
211. Petta S, Adinolfi LE, Fracanzani AL, Rini F, Caldarella R, Calvaruso V, et al. Hepatitis c virus eradication by direct-acting antiviral agents improves carotid atherosclerosis in patients with severe liver fibrosis. J Hepatol (2018) 69(1):18–24. doi: 10.1016/j.jhep.2018.02.015
212. Cacoub P, Nahon P, Layese R, Blaise L, Desbois AC, Bourcier V, et al. Prognostic value of viral eradication for major adverse cardiovascular events in hepatitis c cirrhotic patients. Am Heart J (2018) 198:4–17. doi: 10.1016/j.ahj.2017.10.024
213. Singer A, Osinusi A, Brainard D, Chokkalingam A. Risk of cardiovascular and cerebrovascular events in hepatitis c patients following completion of direct-acting antiviral therapy: a retrospective cohort study. J Hepatol (2017) 1(66):S282–S3. doi: 10.1016/S0168-8278(17)30882-6
214. Sabin CA, Lundgren JD. The natural history of HIV infection. Curr Opin HIV AIDS (2013) 8(4):311–7. doi: 10.1097/COH.0b013e328361fa66
215. Deeks SG. HIV Infection, inflammation, immunosenescence, and aging. Annu Rev Med (2011) 62:141–55. doi: 10.1146/annurev-med-042909-093756
216. Pathai S, Lawn SD, Shiels PG, Weiss HA, Cook C, Wood R, et al. Corneal endothelial cells provide evidence of accelerated cellular senescence associated with HIV infection: a case-control study. PloS One (2013) 8(2):e57422. doi: 10.1371/journal.pone.0057422
217. Pathai S, Lawn SD, Gilbert CE, McGuinness D, McGlynn L, Weiss HA, et al. Accelerated biological ageing in HIV-infected individuals in south Africa: a case-control study. AIDS (London England) (2013) 27(15):2375–84. doi: 10.1097/QAD.0b013e328363bf7f
218. Shrestha S, Irvin MR, Grunfeld C, Arnett DK. HIV, Inflammation, and calcium in atherosclerosis. Arterioscler Thromb Vasc Biol (2014) 34(2):244–50. doi: 10.1161/ATVBAHA.113.302191
219. Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab (2007) 92(7):2506–12. doi: 10.1210/jc.2006-2190
220. Stone SF, Price P, Keane NM, Murray RJ, French MA. Levels of IL-6 and soluble IL-6 receptor are increased in HIV patients with a history of immune restoration disease after HAART. HIV Med (2002) 3(1):21–7. doi: 10.1046/j.1464-2662.2001.00096.x
221. Yudkin JS, Juhan-Vague I, Hawe E, Humphries SE, di Minno G, Margaglione M, et al. Low-grade inflammation may play a role in the etiology of the metabolic syndrome in patients with coronary heart disease: the HIFMECH study. Metabolism: Clin Exp (2004) 53(7):852–7. doi: 10.1016/j.metabol.2004.02.004
222. Paladugu R, Fu W, Conklin BS, Lin PH, Lumsden AB, Yao Q, et al. Hiv tat protein causes endothelial dysfunction in porcine coronary arteries. J Vasc Surg (2003) 38(3):549–55. doi: 10.1016/S0741-5214(03)00770-5
223. Rieckmann P, Poli G, Fox CH, Kehrl JH, Fauci AS. Recombinant gp120 specifically enhances tumor necrosis factor-alpha production and ig secretion in b lymphocytes from HIV-infected individuals but not from seronegative donors. J Immunol (Baltimore Md 1950) (1991) 147(9):2922–7.
224. Maniar A, Ellis C, Asmuth D, Pollard R, Rutledge J. HIV Infection and atherosclerosis: evaluating the drivers of inflammation. Eur J Prev Cardiol (2013) 20(5):720–8. doi: 10.1177/2047487312447843
225. Swingler S, Mann A, Jacqué J, Brichacek B, Sasseville VG, Williams K, et al. HIV-1 nef mediates lymphocyte chemotaxis and activation by infected macrophages. Nat Med (1999) 5(9):997–103. doi: 10.1038/12433
226. Camell CD, Yousefzadeh MJ. Senolytics reduce coronavirus-related mortality in old mice. Science (2021) 373(6552). doi: 10.1126/science.abe4832
227. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020) 579(7798):270–3. doi: 10.1038/s41586-020-2012-7
228. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol (2020) 5(7):811–8. doi: 10.1001/jamacardio.2020.1017
229. Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res (2020) 116(6):1097–100. doi: 10.1093/cvr/cvaa078
230. Wu Q, Zhou L, Sun X, Yan Z, Hu C, Wu J, et al. Altered lipid metabolism in recovered SARS patients twelve years after infection. Sci Rep (2017) 7(1):9110. doi: 10.1038/s41598-017-09536-z
231. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet (2020) 395(10234):1417–8. doi: 10.1016/S0140-6736(20)30937-5
232. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood (2020) 135(23):2033–40. doi: 10.1182/blood.2020006000
233. Grady BP, Nanlohy NM, van Baarle D. HCV monoinfection and HIV/HCV coinfection enhance T-cell immune senescence in injecting drug users early during infection. Immun Ageing I A (2016) 13:10. doi: 10.1186/s12979-016-0065-0
234. Effros RB. The silent war of CMV in aging and HIV infection. Mech Ageing Dev (2016) 158:46–52. doi: 10.1016/j.mad.2015.09.003
235. Freeman ML, Mudd JC, Shive CL, Younes SA, Panigrahi S, Sieg SF, et al. CD8 T-cell expansion and inflammation linked to CMV coinfection in ART-treated HIV infection. Clin Infect Disc (2016) 62(3):392–6. doi: 10.1093/cid/civ840
236. Salter ML, Lau B, Mehta SH, Go VF, Leng S, Kirk GD. Correlates of elevated interleukin-6 and c-reactive protein in persons with or at high risk for HCV and HIV infections. Acquir Immune Defic Syndr (1999) (2013) 64(5):488–95. doi: 10.1097/QAI.0b013e3182a7ee2e
237. Naggie S, Swiderska-Syn M, Choi S, Lusk S, Lan A, Ferrari G eds. Markers of tissue repair and cellular aging are increased in the liver tissue of patients with HIV infection regardless of presence of HCV coinfection. In: Open forum infectious diseases. Oxford University Press US.
238. Gindin Y, Gaggar A, Lok AS, Janssen HLA, Ferrari C, Subramanian GM, et al. DNA Methylation and immune cell markers demonstrate evidence of accelerated aging in patients with chronic hepatitis b virus or hepatitis c virus, with or without human immunodeficienct virus Co-infection. Clin Infect Dis (2021) 73(1):e184–e90. doi: 10.1093/cid/ciaa1371
239. Sosner P, Wangermez M, Chagneau-Derrode C, Le Moal G, Silvain C. Atherosclerosis risk in HIV-infected patients: the influence of hepatitis c virus co-infection. Atherosclerosis (2012) 222(1):274–7. doi: 10.1016/j.atherosclerosis.2012.02.027
240. Masiá M, Padilla S, Robledano C, Ramos JM, Gutiérrez F. Evaluation of endothelial function and subclinical atherosclerosis in association with hepatitis c virus in HIV-infected patients: a cross-sectional study. BMC Infect Dis (2011) 11:265. doi: 10.1186/1471-2334-11-265
241. Kuniholm MH, Parrinello CM, Anastos K, Augenbraun M, Plankey M, Nowicki M, et al. Hepatitis c viremia is associated with cytomegalovirus IgG antibody levels in HIV-infected women. PloS One (2013) 8(4):e61973. doi: 10.1371/journal.pone.0061973
242. Tien PC, Schneider MF, Cole SR, Cohen MH, Glesby MJ, Lazar J, et al. Association of hepatitis c virus and HIV infection with subclinical atherosclerosis in the women's interagency HIV study. AIDS (London England) (2009) 23(13):1781–4. doi: 10.1097/QAD.0b013e32832d7aa8
243. Dolan SE, Hadigan C, Killilea KM, Sullivan MP, Hemphill L, Lees RS, et al. Increased cardiovascular disease risk indices in HIV-infected women. J Acquir Immune Defic Syndr (1999) (2005) 39(1):44–54. doi: 10.1097/01.qai.0000159323.59250.83
244. Duprez DA, Neuhaus J, Kuller LH, Tracy R, Belloso W, De Wit S, et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PloS One (2012) 7(9):e44454. doi: 10.1371/journal.pone.0044454
245. Payne BA, Wilson IJ, Hateley CA, Horvath R, Santibanez-Koref M, Samuels DC, et al. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nat Genet (2011) 43(8):806–10. doi: 10.1038/ng.863
246. Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol (2021) 18(1):58–68. doi: 10.1038/s41569-020-0431-7
247. Horvath S, Levine AJ. HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis (2015) 212(10):1563–73. doi: 10.1093/infdis/jiv277
248. Cao X, Li W, Wang T, Ran D, Davalos V, Planas-Serra L, et al. Accelerated biological aging in COVID-19 patients. Nat Commun (2022) 13(1):2135. doi: 10.1038/s41467-022-29801-8
249. Levine AJ, Quach A, Moore DJ, Achim CL, Soontornniyomkij V, Masliah E, et al. Accelerated epigenetic aging in brain is associated with pre-mortem HIV-associated neurocognitive disorders. J Neurovirol (2016) 22(3):366–75. doi: 10.1007/s13365-015-0406-3
250. Gross AM, Jaeger PA, Kreisberg JF, Licon K, Jepsen KL, Khosroheidari M, et al. Methylome-wide analysis of chronic HIV infection reveals five-year increase in biological age and epigenetic targeting of HLA. Mol Cell (2016) 62(2):157–68. doi: 10.1016/j.molcel.2016.03.019
251. Kananen L, Nevalainen T, Jylhävä J, Marttila S, Hervonen A, Jylhä M, et al. Cytomegalovirus infection accelerates epigenetic aging. Exp Gerontology (2015) 72:227–9. doi: 10.1016/j.exger.2015.10.008
252. Esteki-Zadeh A, Karimi M, Strååt K, Ammerpohl O, Zeitelhofer M, Jagodic M, et al. Human cytomegalovirus infection is sensitive to the host cell DNA methylation state and alters global DNA methylation capacity. Epigenetics (2012) 7(6):585–93. doi: 10.4161/epi.20075
253. Mongelli A, Barbi V. Evidence for biological age acceleration and telomere shortening in COVID-19 survivors. Int J Mol Sci (2021) 22(11). doi: 10.3390/ijms22116151
254. Hamczyk MR, del Campo L, Andrés V. Aging in the cardiovascular system: Lessons from Hutchinson-gilford progeria syndrome. Annu Rev Physiol (2018) 80:27–48. doi: 10.1146/annurev-physiol-021317-121454
255. Jylhävä J, Pedersen NL, Hägg S. Biological age predictors. EBioMedicine (2017) 21:29–36. doi: 10.1016/j.ebiom.2017.03.046
256. Blackburn EH, Epel ES, Lin J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science (2015) 350(6265):1193–8. doi: 10.1126/science.aab3389
257. De Meyer T, Nawrot T, Bekaert S, De Buyzere ML, Rietzschel ER, Andrés V. Telomere length as cardiovascular aging biomarker: JACC review topic of the week. J Am Coll Cardiol (2018) 72(7):805–13. doi: 10.1016/j.jacc.2018.06.014
258. Reungoat E, Grigorov B. Molecular crosstalk between the hepatitis c virus and the extracellular matrix in liver fibrogenesis and early carcinogenesis. Cancers (Basel) (2021) 13(9). doi: 10.3390/cancers13092270
259. Hillmann A, Crane M, Ruskin HJ. Assessing the impact of HIV treatment interruptions using stochastic cellular automata. J Theor Biol (2020) 502:110376. doi: 10.1016/j.jtbi.2020.110376
260. De Alwis PM, Smith BR, Wu T, Artrip C, Steinbach S, Morse C, et al. In-vivo MRI reveals changes to intracerebral vasculature caliber in HIV infection. Front Neuro (2019) 10:687. doi: 10.3389/fneur.2019.00687
261. Wall NA, Chue CD, Edwards NC, Pankhurst T, Harper L, Steeds RP, et al. Cytomegalovirus seropositivity is associated with increased arterial stiffness in patients with chronic kidney disease. PloS One (2013) 8(2):e55686. doi: 10.1371/journal.pone.0055686
262. Ballegaard V, Pedersen KK, Brændstrup P, Kirkby N, Stryhn A, Ryder LP, et al. Cytomegalovirus-specific CD8+ T-cell responses are associated with arterial blood pressure in people living with HIV. Plose One (2020) 15(1):e0226182. doi: 10.1371/journal.pone.0226182
263. Roetker NS, Pankow JS, Bressler J, Morrison AC, Boerwinkle E. Prospective study of epigenetic age acceleration and incidence of cardiovascular disease outcomes in the ARIC study (Atherosclerosis risk in communities). Circ Genomic Precis Med (2018) 11(3):e001937. doi: 10.1161/CIRCGEN.117.001937
Keywords: immunosenescence, cell senescence, atherosclerosis, inflammaging, viral infections
Citation: Ghamar Talepoor A and Doroudchi M (2022) Immunosenescence in atherosclerosis: A role for chronic viral infections. Front. Immunol. 13:945016. doi: 10.3389/fimmu.2022.945016
Received: 16 May 2022; Accepted: 26 July 2022;
Published: 17 August 2022.
Edited by:
Elias A. Said, Sultan Qaboos University, OmanReviewed by:
Mohamed El-Far, University of Montreal Hospital Centre (CRCHUM), CanadaMohammed Albalushi, Sultan Qaboos University, Oman
Copyright © 2022 Ghamar Talepoor and Doroudchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mehrnoosh Doroudchi, bWRvcm91ZEBzdW1zLmFjLmly