 Dariusz Szukiewicz
Dariusz Szukiewicz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 22 July 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.943839
This article is part of the Research TopicT cells - Immune Suppression or Polarization in Patients with Autoimmune DiseasesView all 9 articles
Endometriosis is defined as the presence of endometrial-like glands and stroma located outside the uterine cavity. This common, estrogen dependent, inflammatory condition affects up to 15% of reproductive-aged women and is a well-recognized cause of chronic pelvic pain and infertility. Despite the still unknown etiology of endometriosis, much evidence suggests the participation of epigenetic mechanisms in the disease etiopathogenesis. The main rationale is based on the fact that heritable phenotype changes that do not involve alterations in the DNA sequence are common triggers for hormonal, immunological, and inflammatory disorders, which play a key role in the formation of endometriotic foci. Epigenetic mechanisms regulating T-cell responses, including DNA methylation and posttranslational histone modifications, deserve attention because tissue-resident T lymphocytes work in concert with organ structural cells to generate appropriate immune responses and are functionally shaped by organ-specific environmental conditions. Thus, a failure to precisely regulate immune cell transcription may result in compromised immunological integrity of the organ with an increased risk of inflammatory disorders. The coexistence of endometriosis and autoimmunity is a well-known occurrence. Recent research results indicate regulatory T-cell (Treg) alterations in endometriosis, and an increased number of highly active Tregs and macrophages have been found in peritoneal fluid from women with endometriosis. Elimination of the regulatory function of T cells and an imbalance between T helper cells of the Th1 and Th2 types have been reported in the endometria of women with endometriosis-associated infertility. This review aims to present the state of the art in recognition epigenetic reprogramming of T cells as the key factor in the pathophysiology of endometriosis in the context of T-cell-related autoimmunity. The new potential therapeutic approaches based on epigenetic modulation and/or adoptive transfer of T cells will also be outlined.
Epigenetics is focused on studying changes in gene expression, including mitotically and/or meiotically heritable phenotype modifications that arise from changes in chromosomes but do not involve alterations in the DNA sequence (1). Among the better-known epigenetic mechanisms of histone protein posttranslational modifications, higher-order chromatin reorganization, DNA methylation and hydroxymethylation, nucleosome remodeling/repositioning, RNA editing, and noncoding RNA regulation should be mentioned (2, 3) (Figure 1). A variety of these mechanisms to the differentiation of environmental stimuli that trigger the specified epigenetic modification (4). Changes in gene expression without modification of DNA sequence can be induced by several factors, including age, sex, diet, smoking, and other stimulants, exposure to viruses and bacteria, stress, disease state, and chronic alcohol abuse (5, 6). For example, chronic exposure to ethanol modifies DNA and histone methylation, histone acetylation, and microRNA expression (7). Undoubtedly, epigenetic mechanisms play a key role in ensuring homeostasis or maintenance of a constant internal environment. On the other hand, abnormal epigenetic regulation of the human body systems, including the immune system, may predispose to certain diseases or contribute to the development of both rare syndromes and diseases of high prevalence in human populations (8, 9). All epigenetic changes are reversible. This may explain the fact that these modifications are rarely maintained in future generations in humans, even if they have been repeated in numerous cell cycles (10). Pathoepigenetics is an emerging new field dealing with the description of pathologic changes elicited by epigenetic defective reprogramming (11). Considering the reversibility of these changes, elucidation of the epigenetic consequences of environmental-host interactions may have important therapeutic implications (12).
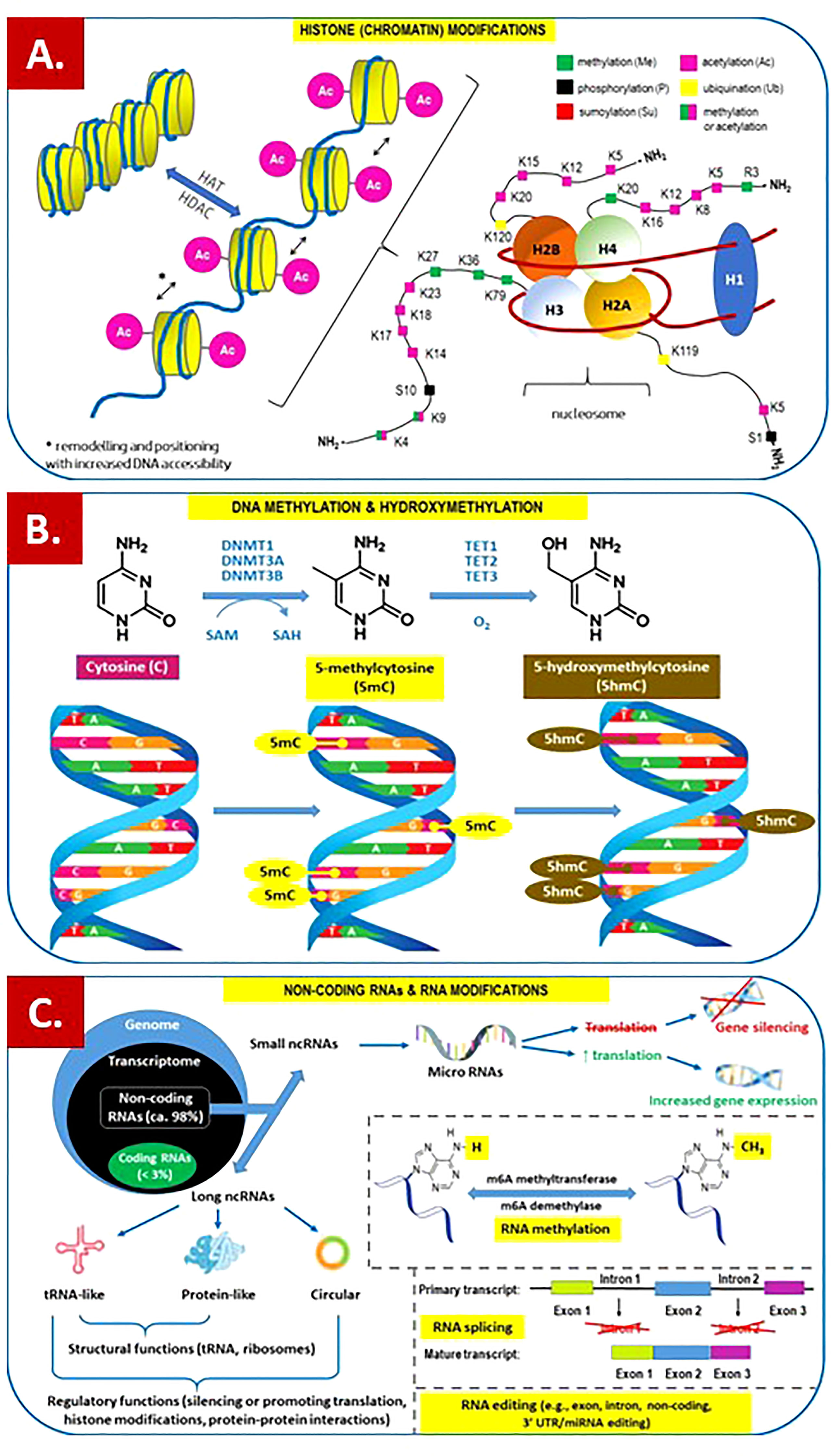
Figure 1 Overview of the main epigenetic mechanisms that regulate gene expression and may establish potentially heritable changes in gene expression without altering the underlying DNA nucleotide sequence. (A) Histone (chromatin) modifications. On the left. Chromatin remodeling is the dynamic modification of chromatin architecture to allow access of condensed genomic DNA to the regulatory transcription machinery proteins, and thereby control gene expression. For example, histone acetylation by HAT (histone acetyl transferase) increases DNA (chromatin) accessibility because acetylated histones cannot pack as well together as deacetylated histones. HDAC – histone deacetylase; On the right. Each nucleosome consists of two subunits, both made of histones H2A, H2B, H3 and H4, also known as core histones, with the linker histone H1 acting as a stabilizer. Histone post-translational modifications are covalent modifications of histones by phosphorylation on serine or threonine residues, methylation on lysine or arginine, acetylation and deacetylation of lysines, ubiquitylation of lysines and sumoylation of lysines. Histone modifications affect chromosome structure and function, especially during transcription and chromatin remodelling processes. (B) DNA methylation and hydroxymethylation. DNA can be modified at cytosine and adenine residues by the addition of chemical groups. Cytosines can be modified by methylation (5mc) or hydroxymethylation (5hmC), while adenines are modified by methylation. CpG islands (regions of the genome that contain a large number of CpG dunucleotide repeats) are DNA methylations regions in promoters known to regulate gene expression through transcriptional silencing of the corresponding gene. DNA methylation at CpG islands is crucial for gene expression and tissue-specific processes. DNMT – DNA methyltransferase; SAM – S-adenosylmethionine; SAH – S-adenosylhomocysteine; TET – ten-eleven-translocation (methylcytosine dioxygenase). (C) Non-coding RNAs (ncRNAs) and RNA modifications. - ncRNAs play an important role in transcription regulation by epigenetic machinery. Within RNA-induced silencing complexes (RISCs), miRNAs mediate the recognition and binding of RNAs that become targeted for degradation. lncRNAs are associated with other complexes and can activate or repress transcription. - RNA methylation is a post-transcriptional level of regulation. At present, more than 150 kinds of RNA modifications have been identified. They are widely distributed in messenger RNA (mRNA), transfer RNA (tRNA), ribosomal RNA (rRNA), noncoding small RNA (sncRNA) and long-chain non-coding RNA (lncRNA). - Alternative splicing (AS) of pre-mRNAs serves as an additional regulatory process for gene expression after transcription, and it generates distinct mRNA species, and even noncoding RNAs (ncRNAs), from one primary transcript. AS contributes to the diversity of proteins in eukaryotes as cells respond to signals from the environment. AS may lead to generation of ncRNAs, especially long noncoding RNAs (lncRNAs). RNA modifications, such as the RNA N6-methyladenosine (m6A) modification, have been found to regulate AS. - RNA editing is an important mechanism of genetic regulation that amplifies genetic plasticity by allowing the production of alternative protein products from a single gene. RNA editing involves the post-transcriptional insertion and deletion of nucleotides (e.g., uridylate – UMP) within nascent transcripts. RNA editing has been observed in mRNAs, tRNAs, and rRNAs, in mitochondrial and chloroplast encoded RNAs, as well as in nuclear encoded RNAs.
In autoimmune diseases, the body’s immune system mistakenly identifies its own healthy tissues as foreign and attacks them. There are over 100 different autoimmune diseases, and the symptoms and effects vary from case to case (13, 14). Most of these diseases run with a more or less evident inflammatory response that can affect many parts of the body. It is estimated that autoimmune diseases affect 3-5% of the population, and widespread diseases include systemic lupus erythematosus (SLE), Hashimoto’s autoimmune thyroiditis, diabetes mellitus type 1, rheumatoid arthritis (RA), Graves’ disease, vitiligo, and celiac disease (CD) (15). There is an upward trend in the prevalence of autoimmunity in developed countries, as was recently found by analysis of the prevalence of the most common biomarker of immunization, antinuclear antibodies (ANA), in the US population (16).
The immune effectors involved in the autoimmune response and related disorders include cells such as natural killer cells (NKs), cytotoxic lymphocytes (CTLs), macrophages, mast cells, and molecules such as antibodies, the complement system, and cytokines (including chemokines). The role of T cells in the pathological immune response, resulting in self-injury, still arouses considerable interest (17). T cells are T lymphocytes, one of the important white blood cells of the immune system. Having the T-cell receptor (TCR) on its surface, a protein complex that is responsible for recognizing fragments of antigens as peptides bound to major histocompatibility complex (MHC) molecules, T cells play a central role in the adaptive immune response. The pathogenesis of autoimmune diseases encompasses both T-cell-induced abnormal (cytotoxic) activation of lymphocytes and macrophages and T-cell-mediated maturation of B cells (B lymphocytes) into memory B cells and plasma cells that produce antibody molecules closely modeled after the receptors of the precursor B cell. The above functions are provided by the T-cell subpopulation named T helper cells (Th cells), also known as CD4+ cells (CD4-positive cells), as they express the CD4 glycoprotein on their surface (17, 18). On the other hand, a T-cell subpopulation named regulatory T cells (Tregs) is essential to maintain immune homeostasis and prevent autoimmune disorders (19, 20). It has been proven that the entire population of T cells is subjected to epigenetic mechanisms that largely govern its maturation and differentiation (21, 22). Thus, precise identification of functional epigenetic pathways in T-cell maturation/differentiation and the modifiers responsible for dysregulation of these processes may provide further insight into the nature of autoimmunity in the context of therapeutic methods.
Endometriosis is a common gynecological disorder affecting approximately 10% (range of 5 to 15%) of reproductive-aged women, whereas significantly higher percentages of endometriosis-related treatments (25 to 50%) have been administered among infertile female patients (23, 24). The term “endometriosis” refers to a condition in which endometrial tissue appears outside the uterine cavity (25). Such endometrial foci may be located either endopelvically or extrapelvically. Abnormally implanted endometrial tissue is typically found in the pelvis, including ovaries, ovarian fossa, fallopian tubes, uterine wall (endometriosis interna or adenomyosis), broad ligaments, round ligaments, uterosacral ligaments, appendix, large bowel, ureters, bladder, or rectovaginal septum (26, 27). Extrapelvic locations of endometriosis are rare. The ectopic endometrium is biologically the same as basal intrauterine endometrial tissue and – because endometriosis cells express estrogen receptors (ERα, Erβ, and GPER) and P4 receptors (PR-A and PR-B) – grows and undergoes cyclic proliferation and breakdown like the ectopic endometrium (28, 29). The local inflammatory response within the endometrial foci is accompanied by pain, including significantly compromising quality of life dyspareunia and dysmenorrhea as well as more serious complications related to fibrosis, scar tissue formation, and adhesions during repair processes (25, 30). A vicious circle of disease arises, where it is difficult to determine whether the inflammatory process favors the development of endometriosis foci or whether the endometriosis foci induce the inflammatory process (31, 32). As a result, patients with endometriosis are less likely to become pregnant and have a successful pregnancy outcome (33). Moreover, it has also been reported that women with endometriosis have a higher incidence of cancer and autoimmune diseases (34, 35). Considering the latter, it was reported that women with endometriosis are at greater risk for autoimmune diseases such as RA, multiple sclerosis (MS), SLE, Sjögren’s Syndrome (SS), and inflammatory bowel disease (IBD) (36). Endometriosis shares several similarities with these autoimmune diseases, including elevated levels of cytokines, decreased cell apoptosis, and T- and B-cell abnormalities.
The immune system is responsible for eliminating cells that are in ectopic sites, and the failure of this elimination in endometriosis is due either to resistance of endometriotic cells to be eliminated by immune cells or to a deficit in the immune response (35, 37). The coexistence of endometriosis and autoimmunity is a well-known occurrence. However, endometriosis has not yet been classified as an autoimmune condition. Despite the still unknown etiology of endometriosis, much evidence suggests the participation of epigenetic mechanisms in the etiopathogenesis of the disease, including immune dysfunction (38, 39). The main rationale is based on the fact that heritable phenotype changes that do not involve alterations in the DNA sequence are common triggers for hormonal, immunological, and inflammatory disorders, which play a key role in the formation of endometriotic foci (39).
This review aims to present the state of the art in recognition epigenetic reprogramming of T cells as the key factor in the pathophysiology of endometriosis. The new potential therapeutic approaches based on epigenetic modulation and/or adoptive transfer of T cells will also be outlined.
The hallmark of the adaptive immune system is clonal expansion of lymphocytes, including a rapid increase in T cells from one or a few cells to millions. T cells play a crucial role in the regulation of the immune system, providing a highly specific, long-lasting and – considering T memory cells – long-term defense mechanism against nonself-pathogens (40). Autoantigens are the result of mutation, neoantigen formation, or exposure of previously hidden self-antigens (41). Immunologic tolerance or a state of unresponsiveness in which lymphocytes remain alive but cannot exert effector functions against a particular antigen ensures a lack of reactivity to self-antigens (autoantigens) (42). In the process of central tolerance, self-reactive T cells possessing receptors specific for autoantigens are eliminated via apoptosis at an early stage in lymphoid cell development. Some CD4+ T cells receive signals in the thymus that select them to differentiate into “natural” T regulatory cells (nTregs), which express the FoxP3 transcription factor and suppress the immune response by both direct and indirect mechanisms. Peripheral tolerance ensures that self-reactive T cells from peripheral tissues are deleted (apoptosis), become anergic (functionally unresponsive to antigen), or can differentiate into “induced” Tregs (iTregs, formerly known as suppressor T cells) (43). Thus, the above two major subsets of CD4+ CD25+ Foxp3+ Tregs (nTregs and iTregs) are essential to the balance between pro- and anti-inflammatory responses. Even temporary malfunction of these checkpoints may cause uncontrolled expansion of these defective (self-reactive) T cells with subsequent development of autoimmunity (42, 43). Aiming to learn about the etiopathogenesis, still accumulating knowledge about the pathways and mediators of the T-cell-dependent autoimmune response includes the T-cell receptor (TCR), T-cell-related cytokines, and defective genes responsible for T-cell regulation and function (42–44). TCR signaling at the level of the membrane of T cells plays a key role in regulating T-cell homeostasis, activation, expansion, and effector function upon recognition of cognate foreign or self-antigens. The specificity of action and properties of the TCR repertoire are acquired during selection, a process in the thymus gland (45, 46). The binding between TCRs and antigens, including autoantigens, is of relatively low affinity and may gradually disengage, which leads to situations in which many TCRs recognize the same antigen peptide and many antigen peptides are recognized by the same TCR (47). TCR forms a TCR complex with six chains of cluster of differentiation 3 (CD3), kinases, coreceptors, and ligands (48).
The TCR is a member of the immunoglobulin superfamily, a large protein superfamily of cell surface and soluble proteins that are involved in the recognition, binding, or adhesion processes of cells. This means that the molecules of TCR share structural features with immunoglobulins (antibodies) (49). It is beyond the scope of this review paper to discuss the structure of the TCR or the TCR-CD3 complex. However, for a quick overview of these issues, please refer to Figure 2. with descriptive legend that includes key components of the TCR signaling pathway.
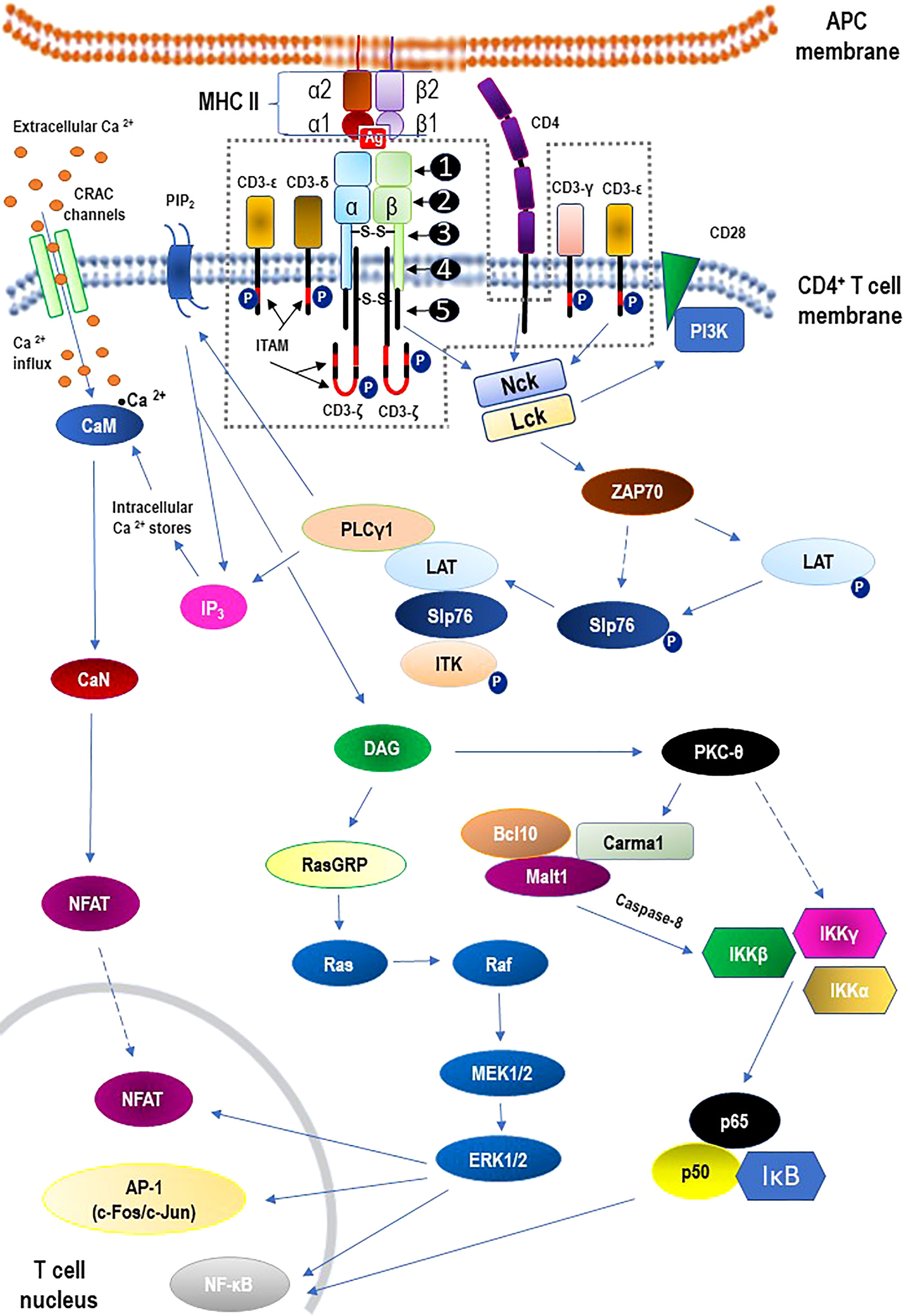
Figure 2 Structure of the αβ T cell receptor (TCR) and the TCR-CD3 complex (the area within the dashed line) including main signaling pathways. - TCR structure: ❶ - variable region; ❷ - constant region; ❸ - hinge region; ❹ - transmembrane region; ❺ - cytoplasmatic tail. - The core TCR signaling complex consists of two TCR chains (αβ heterodimer) that are noncovalently coupled to three dimeric signaling molecules named the cluster of differentiation 3 (CD3): CD3ϵδ, CD3ϵγ, and CD3ζζ. - Signaling via the TCR/CD3 antigen receptor complex is activated after interaction of the TCR with cognate peptide antigen bound to a major histocompatibility complex (MHC) molecule on the surface of antigen-presenting cells (APC), and co-stimulation by co-receptor molecules such as CD28. An early event in the proximal signaling of TCR is the involvement and activation of a set of protein tyrosine kinases (PTKs), such as LCK, FYN, and ZAP-70, that are important components required for activation of TCR signaling through tyrosine phosphorylation on CD3. The proximal TCR signaling is followed by the activation of multiple distal signaling cascades, such as: • Ca2+–calmodulin (CaM) – calcineurin (CaN) – nuclear factor of activated T-cells (NFAT); • diacylglycerol (DAG) – Ras guanyl nucleotide releasing protein (RasGRP) – Ras – proto-oncogene serine/threonine-protein kinase (Raf) – dual-specificity tyrosine/threonine protein kinases (MEK1/2) – extracellular signal-regulated kinases 1/2 (ERK1/2); • protein kinase C-theta (PKCθ) – I kappa B kinases (IKKα, IKKβ, IKKγ) – nuclear factor kappa B (NF-κB). These signaling cascades, regulated largely by epigenetic mechanisms, finally bring out the diverse phenotypic effects, as they control many aspects of T cell biology. For the sake of clarity of the diagram, the presentation of the negative regulation (downregulation) of TCR-mediated signaling has been abandoned. See the main text (2.2. TCR signaling) for details. α1, α2, β1, β2 – domains α1 and α2 and β1 and β2 of the chains (α and β, respectively) that form heterodimeric MHC-II complex; Ag – antigen; AP-1 – activator protein 1; Bcl10 – B cell lymphoma 10; Ca2+ – calcium; CaM – calmodulin; CaN – calcineurin; Carma1 – caspase recruitment domain membrane-associated guanylate kinase protein 1; c-Fos/c-Jun –AP-1-forming dimer of proto-oncogenes; CRAC – calcium release-activated Ca2+; DAG – diacylglycerol; IκB – kinase (IKK) complex containing IKKα, IKKβ, and IKKγ; IKKα – I kappa B kinase α; IKKβ – I kappa B kinase β; IKKγ – I kappa B kinase γ; IP3 – inositol trisphosphate; ITAM –immunoreceptor tyrosine-based activation motif; ITK – interleukin-2 inducible tyrosine kinase; LAT – linker activation of T cells; Lck – leukocyte-specific tyrosine kinase; Malt 1 – mucosa-associated lymphoid tissue protein 1; Nck – adaptor protein non-catalytic region of tyrosine kinase (Nck); NFAT – nuclear factors of activated T cells; NF-κB – nuclear factor kappa B; P –phosphorylated proteins; p50 – regulatory subunit of the NF-kB complex; p65 – subunit of NF-κB; PI3K – phosphatidylinositol-3 kinase; PIP2 – phosphatidylinositol bisphosphate; PKC-theta – protein kinase C-theta; PLCγ1 – phospholipase C gamma 1; RasGRP – Ras guanyl nucleotide releasing protein; Slp76 – SH2-domain containing leukocyte protein of 76 kDa; ZAP70 – zeta-activated protein 70 kDa.
Based on numerous data to date, it can be assumed that TCR signaling is inherently involved in the control of adaptive immune responses but also in the acquisition of immunocompetence by T cells and their development and differentiation (50). As these processes are difficult to separate clearly, they will be discussed together in the context of epigenetic mechanisms, including those leading to autoimmunization. It is worth noting that, as long as histone protein posttranslational modifications, higher order chromatin reorganization, DNA methylation, hydroxymethylation or acetylation, and various noncoding RNA-mediated processes are thought to influence gene expression mainly at the level of transcription, other steps in the process (e.g., translation) may also be regulated epigenetically (51).
During early lymphocyte development, Ig and TCR variable region genes are assembled from germline component variable (V), diversity (D), and joining (J) gene segments (52). Such V(D)J recombination at antigen receptor (AR)-encoding loci (Tcra, Tcrb, Tcrd, and Tcrg) expressed in T cells is initiated by recombination activating proteins 1 (RAG1) and RAG2 via the introduction of DNA double strand breaks (DSBs) between the V, D, and J coding segments and flanking recombination signal (RS) sequences (53). In αβT-cells, this leads to the subsequent expression of TCR β and α chains (54). Such T-cell receptor gene assembly by V(D)J recombination proceeds via successive Dβ-to-Jβ and Vβ-to-DJβ as well as Dα-to-Jα and Vα-to-DJα rearrangements. Basically, V(D)J recombination is strictly controlled at various levels, including these checkpoints that rely on modulation of gene accessibility to the recombination machinery. Biochemical changes in chromatin arrangement and structural modifications of chromosomal organization and positioning form the epigenetic basis for functional development of the TCR antigen (55). Research on the concept of the accessibility model assuming that locus-specific control and temporal ordering of V(D)J recombination primarily involve the modulation of locus and/or gene segment accessibility to a common VDJ recombinase led to groundbreaking findings (56, 57). It was established that both the lineage specificity and temporal ordering of gene rearrangement are reflected in in vitro recombinase cleavage of RSs flanking Ig and TCR gene segments within chromatin in isolated nuclei (58, 59). This means that unlike “compact” chromatin within recombination-inert regions, chromatin at gene segments/loci undergoing V(D)J rearrangement fulfils the criteria of an “open” (noncompacted) configuration (60, 61). Referring to gene expression, the “compact” and “open” regions of chromatin are regions of silent and active transcription, respectively (62). When analyzing, for example, the Tcrb locus, a lightly packed form of chromatin, euchromatin, is linked both locally and in a stage-specific way with Dβ-to-Jβ recombination events taking place with the assistance of germline transcription (GT), accessibility to restriction enzymes, enrichment in histone H3/H4 acetylation and H3K4 methylation, lack of CpG methylation, and diminished nucleosome abundance (54). However, from the double-negative 1-4 to the double positive (DN1-4-to-DP) thymocyte transition onward, lack of GT and decreased histone H3/H4 acetyl lysine (H3/H4ac) predominated along chromosomal regions comprising non-rearranged 5′Vβ genes (54). Expression of a productively rearranged VβDJβ CJ (hereafter VDJ+) and formation of a primary receptor, namely, the pre-TCR, triggers further differentiation into DN4 cells and subsequently CD4/CD8 DP cells. This developmental shift is known as β-selection because it selects for cells that have successfully rearranged their TCR-β chain locus. V(D)J recombination is arrested during this period of cell differentiation. Finally, it resumes in DP cells by selectively targeting the TCRα locus to achieve Vα-to-Jα joining, followed by further selection events involving the completed αβTCR (63). In the studies that involved insertion of a Dβ–Jβ recombination substrate into the endogenous Vβ14 gene segment, accessibility for recombination upon the inserted reporter remained dependent on epigenetically modulated chromatin conformation (64). The significance of this euchromatin-to-heterochromatin transition in health and disease is the subject of intense research aimed at identification of the combination of histone marks (e.g., H3K4ac, H3K4me) that possibly produce restriction at V(D)J rearranging loci (62, 63, 65). It is possible that active epigenetic marks are established through the recombining gene segments and associated RSs before AR V(D)J assembly by interaction with either sense or antisense GT (66, 67). Binding of RAG1 and RAG2 proteins that occurs in a highly focal manner to a small region of active chromatin with high levels of H3K4ac and H3K4me may suggest a close link between RNA polymerase (Pol) II-mediated transcription and epigenetic tagging at these sites of the Tcrb locus, precisely encompassing Tcrb J and proximal D gene segments, in a developmental stage- and lineage-specific manner (68).
Euchromatin and related epigenetic modification to the DNA packaging protein histone H3 at V(D)J rearranging loci exerts effects not limited to the gene accessibility only, but it also has a significant and direct impact on the chromosomal environment in the context of V(D)J recombinase tethering and enhancement of catalytic activity. It was demonstrated that the plant homeodomain (PHD) zinc finger of RAG2 binds much more strongly to histone H3 trimethylated at K4 (H3K4me3) (69–71). In addition, H3K4me3 plays a significant role in the stimulation of purified RAG enzymatic activity at both the nicking (2- to 5-fold) and hairpinning (3- to 11-fold) steps of V(D)J recombination (72). Similarly, the N-terminal part of RAG1 contains a short for Really Interesting New Gene (RING) finger domain preferentially interacting directly with and promoting monoubiquitylation of histone H3. Monoubiquitination of histone H3 (H3ub1) could play a role in regulating the joining phase of chromosomal V(D)J recombination (73). It was established that RAG1 binds specifically to AR gene segments in a cell-type and stage-specific manner, whereas RAG2 has a much broader chromosome binding spectrum because it interacts with H3K4me3-enriched regions genome-wide (68).
Interacting with forkhead Box P3 (Foxp3) transcription factor complexes, TCR signaling plays central roles in Treg differentiation, maintenance, and functional maturation (74). During differentiation, Tregs recognize their cognate antigens and receives TCR signals before initiation of Foxp3 transcription, which is triggered by TCR-induced transcription factors, including nuclear factor of activated T cells (NFAT), activator protein 1 (AP-1), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (75). Naturally, derived Tregs are characterized by stable expression of the transcription factor Foxp3 and characteristic epigenetic imprinting at the Foxp3 gene locus. Foxp3 seizes TCR signal-induced transcriptional and epigenetic mechanisms by interacting with AML1/Runx1 and NFAT. Thus, Foxp3 modifies the gene expression dynamics of TCR-induced genes, which constitute cardinal mechanisms for Treg-mediated immune suppression and related self-tolerance and prevention of autoimmunity (74). It is essential that the NF-κB signaling pathway acts as a versatile regulator of Foxp3 expression during normal T-cell development and enhancing the signal strength of the NF-κB pathway induces Foxp3 expression in T cells, including Tregs (75). Interestingly, recent studies have revealed exciting new roles for NF-κB related to its nontranscriptional activities. It has been proven that NF-κB can also activate diverse epigenetic mechanisms that mediate extensive chromatin remodeling of target genes to regulate T-cell activities. Even epigenetic effects on genes encoding different NF-κB subunits may modulate T-cell inflammatory responses (75–77).
T cells depend on mammalian target of rapamycin kinase (mTOR) signaling to sense and integrate immune signals from dendritic cells (including antigenic signals, costimulatory molecules, and cytokines), environmental cues derived from growth factors and immunoregulatory factors, and nutrients (78). Another manifestation of the epigenetic regulation of TCR signaling in T cells arises from the posttranscriptional modulation of mTOR complex components mTOR and Rictor mRNAs by the microRNAs Let-7 and MiR-16. These results for CD4+ T cells demonstrated that microRNAs regulate the expression of mTOR components in T cells and that this regulation is critical for adjustable mTOR activity. Hence, influencing the interpretation of TCR signaling, microRNAs contribute to the discrimination between T-cell activation and anergy (79). Another important mechanism of miRNA regulation of CD4+ Treg development via modulation of the genes within the mTOR signaling pathway is related to miR-15b/16, miR-24, and miR-29a (80). Suppression of mTOR signaling is essential for induction of iTregs from naïve CD4(+) T cells, and the mTOR complex 2 (TORC2) component, Rictor, contains a functional target site for miR-15b/16. It was confirmed that downregulation of Rictor produces a significant reduction in mTOR signaling as measured by phosphorylation of the downstream target, ribosomal protein S6. In line with the knowledge that CD4+ Tregs are essential for controlling immune responses and preventing autoimmunity, the overexpression of miR-15b/16 in conventional CD4+ T cells adoptively transferred into Rag2(-/-) mice increased the in vivo development of peripheral Tregs and diminished the severity of autoimmune colitis (80).
Signal transduction may also be regulated based on reciprocal allosteric regulation of TCR phosphorylation related to cholesterol and ligand binding to the TCRβ transmembrane region (81). It was reported that cholesterol bound to the TCRβ transmembrane region keeps the TCR in a resting, inactive conformation that cannot be phosphorylated by active kinases (82). This ensures that the αβ T-cell remains quiescent in the absence of antigenic peptide-MHC (the TCR’s ligand) at the variable regions of TCRαβ and decreases the sensitivity of the T-cell toward stimulation. Only TCRs that spontaneously detach from cholesterol can achieve the active conformation (named primed TCRs) that is prone to phosphorylation. On the other hand, cholesterol binding to TCRβ leads to an increased formation of TCR nanoclusters, increasing the avidity of the TCRs toward the antigen and thus increasing the sensitivity of the αβ T-cell (83). The latter mechanism seems to be relevant in autoimmunity, as evidence is building up that cholesterol accumulation in leukocytes is causally associated with the production of autoantibodies (84). In contrast to TCRαβ, TCRγδ does not bind to cholesterol and might be regulated in a different manner (83).
Accumulation of intracellular lipid (cholesterol-containing) droplets in CD4+ T cells, coexisting with elevation of serum triglycerides and cholesterol, was observed in many autoimmune diseases, including rheumatic arthritis, SLE, and psoriasis (85–87). Even if it remains speculative, many authors postulated that lowering blood lipids or normalizing the lipid profile may limit T-cell-dependent autoantibody responses (87–90). It is worth noting that epigenetic modulation of cholesterol binding into TCRs may trigger a specific functional state of TCRs, both the resting and the primed (83, 91–93).
Epigenetic influences on TCR signaling should also be analyzed in the context of counteracting affect, i.e., considering that TCR signaling affects epigenetic modulation (94). T-cell activation induces changes in DNA methylation and acetylation, creating broad and lasting genetic modifications (95, 96). Typical markers of altered access to gene transcription include histone H3 lysine 27 trimethylation (H3K27Me3) and histone H3 lysine 27 acetylation (H3K27Ac), but the repertoire of epigenetic activity also includes phosphorylation, nitrosylation, glycosylation, lipidation, ubiquitination, and (small ubiquitin-related modifier) SUMOylation (97). In the case of histones H3K27Me3 and H3K27Ac, methylation is associated with a closed chromatin conformation that prevents gene transcription, whereas acetylation correlates with an “open” (permissive for transcription) chromatin conformation. Analogously, histone methyltransferases and deacetylases are associated with the silencing of gene expression, and histone demethylases and acetyltransferases promote gene expression (94). In addition, difficult to predict in an individual case, the effects of inadequate methylation/demethylation and acetylation/deacetylation on the chromatin conformation should be considered (21, 98).
For example, the chromatin-modifying enzyme enhancer of zeste homolog 2 (EZH2), the functional unit of polycomb repressive complex 2 (PRC2), is a histone methylase that plays a key role in regulating various aspects of T-cell immunobiology, such as Foxp3+ Treg stability (99, 100). The immune homeostasis associated with normal Treg function requires the induction of EZH2 in response to costimulation with CD28, an extracellular cue intrinsically required for Treg maintenance. Treg-specific deprivation of EZH2 resulted in spontaneous autoimmunity with reduced Foxp3(+) cells in nonlymphoid tissues and impaired resolution of experimental autoimmune encephalomyelitis (100).
Dysregulation of the balance between subsets of CD4+ T cells, Tregs, and Th17 cells may be involved in the pathomechanism of several disorders, including autoimmune disease, cancer, and chronic inflammatory conditions. The Treg/Th17 balance depends on many factors involved in the differentiation of these cells, such as TCR signals, cytokines, and metabolic and epigenetic regulators. The latter or posttranslational modifications modulate the activity of forkhead Box P3 (Foxp3), retinoic acid-related orphan receptor gamma t (RORγt), and signal transducer and activator of transcription (STAT)s. Thus, insufficient posttranslational (epigenetic) modifications of Treg/Th17 differentiation and/or balance may lead to autoimmune diseases (97).
Epigenetic influence applies to all three steps of TCR signaling, i.e., signal reception, transduction, and the response triggered by the signal. Thus, aberrant chromatin landscapes following T-cell activation were demonstrated in various autoimmune diseases, including rheumatoid arthritis, SLE, Grave’s disease, and type 1 diabetes mellitus (T1D) (94, 101). Although TCR signaling defects are associated with mediating pancreatic β cell autoimmunity in T1D, the disease is often complicated with other autoimmune diseases, and anti-islet autoantibodies precede the clinical onset of disease (102). Typically, T1D co-occurs in most cases with other common organ-specific autoimmune diseases, such as autoimmune thyroiditis (predominantly), celiac disease, and gastritis (102, 103). Accordingly, Teffs isolated from nonobese diabetic (NOD) mice display a particular chromatin conformation that allows not only easier access to T1D-associated genetic loci but also access to the genes involved in other autoimmune disorders (104–106).
Expression of foxp3 in naïve T cells during induction of differentiation into induced Foxp3+ regulatory T cells (iTregs) occurs with participation of suboptimal (weaker than maximal) TCR stimulus or TCR stimulus in conjunction with TGF-β signaling. It was demonstrated that optimal (strong) activation of TCR in terms of both ligand affinity and duration results in specific enrichment at the foxp3 locus with the accumulating DNA (cytosine-5)-methyltransferase 1 (DNMT1) and DNMT3b. This in turn leads to increased CpG methylation and inhibits foxp3 transcription (107). Regardless of the transcription factor activation, TCR and TGF-β signals exert epigenetic effects on DNMT1 to modulate the expression of foxp3 by increasing CpG methylation. Augmentation of DNMT1 is regulated through at least two posttranscriptional mechanisms. The first assumes that a strong TCR signal inactivates constitutively active glycogen synthase kinase-3 beta GSK3β to rescue DNMT1 protein from proteasomal degradation. The second mechanism is based on evidence that a strong TCR signal suppresses miR-148a to derepress DNMT1 mRNA translation (107). The opposite effect is related to TGF-β signaling, which antagonizes DNMT1 accumulation via activation of the p38 mitogen-activated protein (p38 MAP) kinase pathway (107, 108). In addition, regulation of foxp3 transcription, which may be important for the induction of self-tolerance and the control of autoimmunity, depends on the production of NF-κB-dependent cytokines (e.g., TNFα, IFN-γ, IL-17 and IL-9) by the T cells themselves (109). In addition to its well-documented transcriptional activity, the NF-κB or NF-κB subunit proto-oncogene RelB (RelB) can also trigger diverse epigenetic mechanisms that mediate extensive chromatin remodeling and histone modifications of target genes to regulate T-cell fate decisions (76).
Vitamin C (l-ascorbic acid), a multifunctional water-soluble antioxidant substance, serves as an essential cofactor for many enzymes, including those influencing epigenetic modulation of gene expression (110, 111). In addition, vitamin C can significantly affect T-cell differentiation and may interfere with T-cell signaling (112). Ascorbic acid was discovered as a cofactor for ten-eleven translocation (TET) methylcytosine dioxygenases that use Fe(II) and 2-oxoglutarate as cosubstrates and are responsible for DNA demethylation. Vitamin C also serves as a likely cofactor for some Jumonji C (JmjC) domain-containing histone demethylases that catalyze histone demethylation (113). Thus, vitamin C deficiency can influence demethylation of both DNA and histones, further leading to different phenotypic presentations with an increased possibility of autoimmune disorders. DNA hypomethylation was demonstrated in T cells from patients with SLE, suggesting the development of autoimmunity by decreasing DNA methyltransferase expression, modifying DNA methylation patterns, and altering gene expression (114, 115). DNA methylation is also regulated in part by the extracellular signal-regulated kinase (ERK) pathway, which is influenced by vitamin C, and ERK pathway signaling is diminished in lupus T cells (116). Interestingly, a lack of vitamin C in scurvy may mimic SLE (117).
Finally, signals beside the TCR receptor may modulate the epigenetic landscape of T-cell subpopulations. For example, Treg epigenetic and functional identity is modulated by interleukin-2 (IL-2) in a TCR-independent manner by regulating the positioning of the pioneer factor special AT-rich sequence binding protein 1 (Satb1) in CD4+ thymocytes and subsequently controlling the genome-wide chromatin accessibility of thymic-derived Tregs (118). Thus, in addition to TCR triggering, the immunomodulatory action of IL-2 contributes to the selection of Foxp3+CD4+ Tregs, the functionally stable cell lineage indispensable for the maintenance of immunological self-tolerance and safeguarding immune homeostasis in vivo (118–120).
The above-mentioned mechanisms are summarized in Figure 3.
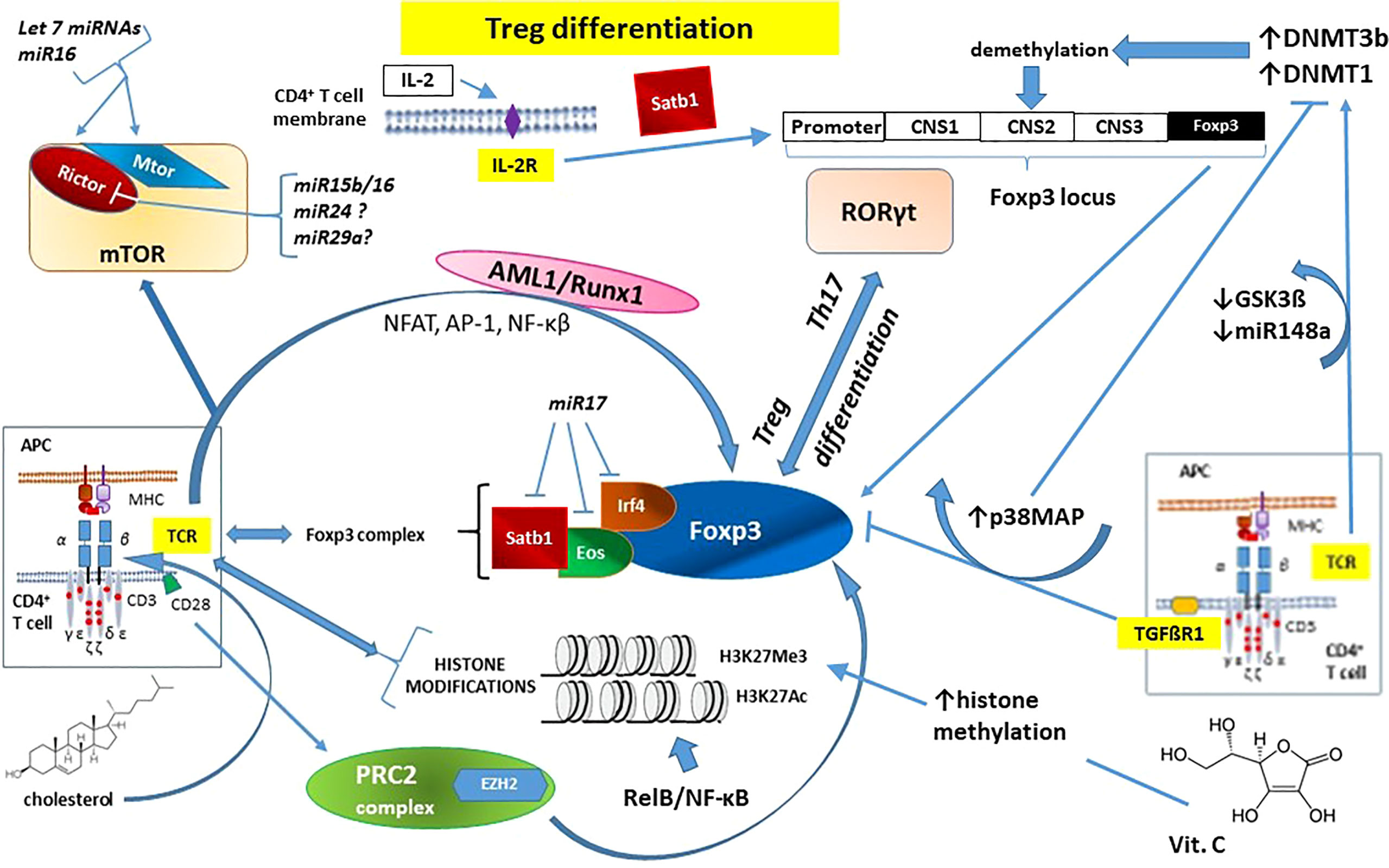
Figure 3 The essential role of epigenetic changes in regulatory T cell (Treg) development and function on the example of forkhead box P3 (Foxp3) - the master regulatory protein involved in Treg-mediated immune system responses. Noticeable that IL-2 action does not involve TCR signaling pathway. See the main text (Chapter 2.2.1. Epigenetic mechanisms influencing TCR signaling and autoimmunity) for details. AML1/Runx1 – Acute myeloid leukemia 1 protein or Runt-related transcription factor 1; AP-1 – activator protein 1; CD – cluster of differentiation (cell surface marker); CNS1-3 – conserved non-coding sequences; APC – antigen-presenting cells; DNMT – DNA methyl-transferase; Eos – transcription factor, member of the Ikaros Zinc Finger (IkZF) family of transcription factors; EZH2 – Enhancer of Zeste Homolog 2; Foxp3 – forkhead box P3 protein; GSK3β – glycogen synthase kinase-3 beta; H3K27Ac – acetylation of the lysine residue at N-terminal position 27 of the histone H3; H3K27Me3 – tri-methylation of lysine residue at N-terminal position 27 on the histone H3; IL-2 – interleukin 2; IL-2R – interleukin 2 receptor; Irf4 – Interferon regulatory factor 4; miRNA/miRNAs –microRNA/microRNAs; mTOR – mammalian target of rapamycin kinase; mTORC2 – mTOR Complex 2; NF-κB – nuclear factor kappa-light-chain-enhancer of activated B cells; NFAT – nuclear factor of activated T cells; p38MAP – p38 mitogen-activated protein kinase; PRC2 – polycomb repressive complex 2; Rictor – Rapamycin-insensitive companion of mammalian target of rapamycin; RelB – NF-κB subunit proto-oncogene RelB; RORγt – Retinoic acid-related orphan receptor gamma t; Satb1 – Special AT-rich sequence-binding protein 1; STAT – signal transducer and activator of transcription; TCR – T-cell receptor; TGFβR1 – transforming growth factor beta-receptor 1; Th17 – T helper 17 cells.
To sum up as simply as possible, the thymus period of T-cell development and maturation includes both positive and negative selection, in which TCR signaling is the major checkpoint (46, 121). T cells expressing TCRs with a low affinity for self-peptide MHC complexes are subjected to differentiation into mature immunocompetent T cells (positive selection), whereas T cells expressing TCRs with a high affinity for self-antigens (self-reactive T cells) undergo negative selection via apoptosis (42, 45). As a result of such selection, only T-cells demonstrating autotolerance survive. Next, these naïve mature T cells, between maturity and activation, move to secondary lymphoid organs, such as the spleen and lymph nodes, including the tonsils and gut-associated lymphoid tissue. This is where they become activated after interaction with foreign peptides presented by the MHC molecules of antigen-presenting cells (APCs), such as macrophages, dendritic cells, and B cells. Thus, to participate in an adaptive immune response, a naïve T-cell must first encounter an antigen and then be induced to proliferate and differentiate into cells capable of contributing to the removal of the antigen (45, 47). Understandably, TCR signaling dysregulation can lead both to a state of near total/total immunologic unresponsiveness named anergy and autoimmunity in the case of impaired negative selection and intolerance of autoantigens. The tissue specificity and progression of T-cell-mediated autoimmunity are dictated in part by the repertoire of TCRs (46).
Although aberrant TCR signaling underlies autoimmunity, it is worth looking at epigenetic cell reprogramming, not only in terms of TCR function but also in a broader sense, considering other properties of T cells.
Histone modification and DNA methylation play important roles in the activation of naïve CD4+ and CD8+ T cells in the presence of specific cytokines with subsequent differentiation into effector or memory cells, and in the case of CD4+ T cells, adoption of distinct T helper fates. Activated naïve CD4+ T cells are highly plastic cells that can differentiate into various Th-cell fates characterized by the expression of effector cytokines such as IFN-γ (Th1), IL-4 (Th2) or IL-17A (Th17). Thus, epigenetic modifications greatly influence the functional differentiation of T-cell subsets, including linage commitment to short-lived effectors, long-term memory T cells, T regulatory cells, and other specific T-cell populations (21). The pattern of DNA methylation at key cytokine (IL-2, IL-4) loci influences the control of CD4+ T-cell differentiation and postthymic T-cell maturation (122). It was demonstrated that CD4+ Th-cell differentiation is modulated by lysine methyltransferase (KMT) Dot1 l-dependent dimethylation of lysine 79 of histone H3 (H3K79me2), which assures lineage-specific gene expression (123). Loss of Dot1 l (mediated by transgene Cd4-Cre, which becomes active in thymocytes at the DP stage) produces augmented expression of Th1-specific genes and excessive production of IFNγ at the expense of Th2 cell development. These events may confirm a central role of Dot1 l in Th1-cell lineage engagement and stability (123). Th1 and Th17 cells are involved in the pathogenesis of organ-specific autoimmune disorders, Crohn’s disease, Helicobacter pylori-induced peptic ulcer, acute kidney allograft rejection, and unexplained recurrent abortions (124, 125).. Moreover, numerous studies have found that the single type II IFN, IFN-γ, plays an essential role in the development and severity of systemic autoimmunity, particularly SLE (126). Dysregulation of KMT Dotl1 (KMT4), resulting in the shift of the Th1/Th2 balance paradigm toward Th1 and IFNγ overproduction, may promote autoimmune responses (127, 128). The role of DNA methylation is also significant in the plasticity of the Th17 subset, and under normal conditions, Th17 and naïve CD4 T cells had a similar methylation profile (129, 130). In addition, multiple studies have confirmed the ability of Th17 cells to convert into other CD4+ T cells in the presence of IL-12, both in vitro and in vivo, including conversion into a functional Th1-cell-like phenotype producing IFN-γ and lacking IL-17A secretion (131, 132).
Maintaining homeostasis and self-tolerance is inherently related to the function of Tregs (formerly known as suppressor T cells) because this specialized subpopulation of T cells can inhibit T-cell proliferation and cytokine production, playing a critical role in preventing autoimmunity (133). The transcription factor forkhead box protein 3 (Foxp3) is an essential molecular marker of Treg development in different microenvironments, and Foxp3 upregulation takes place either in the thymus (tTregs) or periphery (iTregs). Sustained expression of Foxp3 that assures balanced phenotypic plasticity and stability in Tregs requires both fine-tuned transcriptional and epigenetic events (133, 134). Recent reports have suggested that specific modifications of DNA and histones in the regulatory regions of the Foxp3 locus are key determinants for the establishment of the chromatin structure in conventional CD4+ T (Tconv) cells for their future differentiation into the Treg cell lineage (135, 136). In addition to the Foxp3 promoter, the three conserved noncoding DNA sequence (CNS) elements within the Foxp3 locus, i.e., CNS1, CNS2, and CNS3, are also targets of several modifying enzymes and are epigenetically regulated at different stages of Treg development (137). Defective Foxp3 expression involving abnormal Treg development and function may predispose patients to several autoimmune diseases (138). Decreased Foxp3 expression counteracts the suppressive effects, which are tightly regulated by Foxp3 itself and by its cooperation with several cofactors (139, 140). Foxp3 interaction with other transcription factors (e.g., GATA3 – member of the GATA family of conserved zinc-finger transcription factors, NFAT – nuclear factor of activated T cells, Runx – Runt-domain transcription factors, and STAT-3 – signal transducer and activator of transcription 3) may trigger either silenced or augmented gene expression (74, 139, 141). Therefore, epigenetic events that impair Foxp3 expression lead to disinhibition of the immune system with subsequent T-cell mediated autoimmunity (142). In other words, depending on the environment, Tregs gain effector functions upon loss of Foxp3 expression (143).
Posttranscriptional modulation of Foxp3 may be mediated by microRNAs (miRNAs), small single-stranded noncoding RNA molecules (containing approximately 22 nucleotides), which thus take part in epigenetically establishing Treg biological properties in health and disease (144, 145). After their posttranscriptional maturation, miRNAs are loaded into the ribonucleoprotein complex, i.e., RNA-induced silencing complex (RISC) modulates gene expression by binding to the 3’ untranslated region (UTR) of their target mRNAs through base-pairing, which in turn triggers mRNA degradation or translational inhibition (146). Computational estimates suggest that each human miRNA targets between 100 and 200 messages, usually in the 3′- UTR of the mRNA. Over 700 miRNAs are encoded in the human genome, and approximately one-third of all human genes are believed to be under the regulatory control of a miRNA (147). In relation to Tregs, there is mounting evidence that miRNAs regulate the proliferation, differentiation, and apoptosis of these T-cell subpopulations. Blockade of miRNA maturation in mice caused a lymphoproliferative phenotype similar to that observed in the absence of Foxp3 (148). Similarly, selective miRNA disruption in Tregs leads to uncontrolled autoimmunity (145, 149). For example, deletion of miR-146a-5p results in a breakdown of immune tolerance and the development of a fatal spontaneous autoimmune disorder due to inadequate inhibitory function of Tregs (150, 151). Suppression of the specific genes by Foxp3 may involve both direct binding to genetic regulatory elements and induction of miRNAs that specifically target the 3′-UTR of the same genes. Such coordinated action was demonstrated between Foxp3 and miR-155-5p in blocking the special AT-rich sequence binding protein 1 (Satb1) gene and zinc finger E-box-binding homeobox 2 (Zeb2) gene (152–154). With reference to miR-31, miR-24, and miR-210, there are grounds for assuming the possibility of direct action on the Foxp3 3′- UTR with subsequent reduction of Foxp3 expression levels and Treg phenotypic adjustment (155, 156). Another possibility of miRNA action on Tregs that leads to indirect reduction of its transcriptional activity includes interfering with the expression of proteins that cooperate with Foxp3, e.g., Eos (lkzf4), a member of the Ikaros family of transcription factors, interferon regulatory factor (Irf), or Satb1. This type of action is shown by miR-17, an individual mature miRNA of the miR-17-92 cluster (157). In turn, overexpression of miR-15a/16 contributes to the modulation of methylation/demethylation dynamics within the Foxp3 locus that influence Foxp3 expression (158).
Epigenetic modulation of Foxp3 expression also occurs at the protein level via covalent posttranslational modifications, including ubiquitination, acetylation, and phosphorylation of different amino acids (159–161). These processes influence Foxp3 subcellular localization, functional activity, and interaction with other proteins, mainly transcriptional activators or repressors. Thus, the resulting reduction in Foxp3 expression and corresponding reduction in suppressive Treg activity may promote autoimmune disorders (134, 135, 162).
Epigenetic alterations such as DNA methylation and histone modifications can regulate gene expression in mature T cells, with the possibility of dysregulation in autoimmune diseases. For example, in patients with SLE, numerous genes, such as CD11a (ITGAL), perforin (PRF1), CD70 (TNFSF7), and CD40LG (TNFSF5), in T lymphocytes were hypomethylated (163, 164). Several studies on the role of miRNAs in SLE revealed that decreased expression of DNMTs in CD4+ T cells of SLE shows correlation with three microRNAs (miR-21, miR-148a, and miR126) regulated by methylation (165). In addition to miRNAs, long noncoding RNAs (lncRNAs), defined as ≥200 base pairs in length with little or no translation potential, play a key role in imprinting control, immune cell differentiation, apoptosis, and immune responses. Many potential lncRNAs have been revealed to contribute to a new layer of molecular regulation of systemic lupus erythematosus (SLE) (166). LncRNAs play an indispensable role in SLE by interacting with proteins, DNA, and even RNA. Aberrant levels of NEAT1, Gas5, Lnc-DC, Linc0949, linc0597, MALAT1, and TUG1 are involved in the pathogenesis of SLE (167, 168). It was recently demonstrated that the novel lncRNA lincRNA00892 activates CD4+ T cells in SLE by regulating CD40 L, a 261-amino-acid membrane glycoprotein expressed on activated CD4 lymphocytes (169). Variation in the expression of noncoding RNAs (ncRNAs), both miRNAs and lncRNAs, interacting with the immune function of T cells influences susceptibility to SLE and the clinical course of this disorder (170).
Epigenetic reprogramming of T cells caused by ncRNAs is under intense scrutiny in relation to other autoimmune diseases, such as rheumatoid arthritis, systemic sclerosis, Sjogren’s syndrome, and organ-specific autoimmune diseases (e.g., autoimmune thyroid diseases and type 1 diabetes). The results of such investigation resemble those for SLE, however, with other ncRNAs (171–174).
The definition and basic characteristics of endometriosis with reference to its autoimmune linkages are briefly presented in the introduction (Chapter 1.). The reason for these links is unclear, but it might exist because mature endometriotic foci initiate inflammation, which may contribute to an imbalanced immune response inducing autoimmunity (175, 176). On the other hand, the abnormal immune response that occurs in endometriosis may be due to an already existing autoimmune disorder with a disturbed cytokine profile, altered cell apoptosis, and imbalances in immune cell function (177). The evidence is not clear as to which condition causes the other. Thus, there is still no conclusive cause of endometriosis, and researchers do not yet know what triggers the condition (178, 179). As already mentioned in the introduction, this chronic, progressive condition is not currently classified as an autoimmune disease (180). This may be because - at least initially - the immune system fails to recognize and target endometrial tissue growing elsewhere (ectopically) in the body. Endometrial foci themselves may have some ability to evade the immune response – similar to some cancers – by tricking or confusing immune cells that would otherwise attack those cells that form the lesions. Such a failure to recognize and target endometriotic foci may indicate that endometriosis is an immune disease with a deficit of immune recognition but not necessarily an autoimmune condition (181–183). Possible autoimmune pathogenesis of this proliferative disease may be supported by the fact that women with endometriosis may also have an increased risk of autoimmune comorbidities (e.g., SLE, RA, SS, MS, IBD) compared to healthy controls (36). In addition, endometriosis is more severe in patients who are also affected by autoimmune disease (184). As the pathogenesis of endometriosis continues to reveal itself, more autoantibodies are being discovered, and they may offer useful noninvasive tools for early diagnosis of endometriosis. This is important because diagnosis of ectopic dissemination of endometrial cells is usually delayed by an average of 8 to 11 years, leading to significant consequences in terms of disease progression (185). Various anti-endometrial antibodies may be used for early diagnosis in minimal to mild endometriosis, especially anti-SLP2, anti-TMOD3, anti-TPM3, and anti-PDIK1 L. Other nonanti-endometrial antibodies, such as anti-IMP1, anti-CA, aCL, and anti-STX5, may be used as additional noninvasive diagnostic tools (180, 185).
However, one should not forget that in the etiopathogenesis of endometriosis, hormonal disorders play an important, if not the most important, role. The disease is known as an estrogen-dependent and progesterone (P4)-resistant process (186, 187). In contrast to endometriosis tissue, estrogens are not locally produced in the endometrium. Several causes of P4 resistance in the endometrium have been postulated, including congenital “preconditioning”, whereby the in-utero environment renders infants susceptible to neonatal uterine bleeding and endometriosis (188). P4 action is crucial to decreasing inflammation in the endometrium, and deviant progesterone signaling results in a proinflammatory phenotype. Interestingly, chronic inflammation itself can induce a P4-resistant state (32, 189). The importance of excess estrogen exposure and P4 resistance in epigenetic homeostasis failure in endometrial/endometriotic tissue is crucial. Epigenetic alterations regarding transcription factors of estrogen and P4 signaling pathways in mesenchymal stromal cells (MSCs) are robust in endometriotic tissue (38). It is therefore logical that, unlike in autoimmune disorders where targeted immunosuppression is a priority, the treatment in endometriosis concerns hormonal imbalances and is primarily aimed at decreasing the endogenous ovarian production of estrogens (190, 191). In general, it is now appreciated that estrogens, and in particular E2, can control proinflammatory signals/pathways (192). The anti-inflammatory effects of estrogens are associated mostly with signaling via ERα and GPER, whereas even if not without controversy, an increased ratio of ERβ is associated with proinflammatory signatures (193–195). Variations in the expression of different estrogen receptor types may lead to some discrepancies in understanding the effects of estrogen on the immune system in health and endometriosis (192, 196). Markedly higher levels of ERβ and lower levels of ERα in human endometriotic stromal cells corresponds to EnSCs compared with EnSCs within eutopic endometrial tissues were reported (197, 198). Such overexpression of ERβ in endometriosis was associated with abnormally lowered methylation of a CpG island in the promoter region of the ERβ gene (ESR2) (199). High levels of ERβ, in turn, suppress ERα expression and the response to E2 in endometriotic stromal cells by binding to nonclassical DNA motifs in alternatively used ERα promoters (28). Lowered expression of ERα demonstrated in endometriosis may cause insufficient responsiveness to E2 with respect to progesterone receptor (PR) expression, leading to secondary P4 receptor deficiency and P4 resistance, which is commonly observed in women with this disorder (28, 188). In addition to DNA methylation, the epigenetic nature of the interaction between immune and hormonal systems that significantly impact endometriosis pathogenesis and development by modulating the immune response via estrogen and P4 receptors also encompasses noncoding RNAs: miRNAs (e.g., miR-148a, miR-30b-5p, miR-487a-5p, miR-4710, miR-501-3p, miR-378 h, and miR-1244) and lncRNAs (e.g., HOX antisense intergenic RNA - HOTAIR) (200–206). In the spectrum of consequences of hormonal profile modulation by epigenetic factors in endometriosis, the altered function of immune cells, including T cells, deserves attention (177, 207).
Naturally, T cells do not function in isolation from the environment and changes in their environment may play a key role in the etiopathogenesis and course of endometriosis. This applies primarily to the endometriotic lesions and eutopic endometrium but also – although to a different extent – may be the result of an interaction with all of the cells in the body within reach of T cells. Moreover, the immune landscape in endometriosis is largely modulated by epigenetic factors (208). For example, aberrant DNA methylation patterns were demonstrated in the eutopic endometrium in endometriosis patients compared to the eutopic endometrium of endometriosis-free women (209). The level of DNA methylation in the whole genome was also different when comparing endometriotic stromal cells with the endometrium of healthy women. The observed differences in the methylation of the 403 genes examined pertained mainly to those encoding transcription factors, HOXA genes, and genes of nuclear receptors (210). The altered pattern of DNA methylation/demethylation within endometrial stromal cells translates to the upregulation or downregulation of specific proteins. Thus, endometriosis phenotypes are associated with specific proinflammatory and tissue remodeling cytokine profiles influencing the T-cell immune response. In addition, the DNA methylome is responsible for the overexpression of the genes encoding deoxyribonucleic acid methyltransferases DNMT1, DNMT3A, and DNMT3B in ectopic endometrium (209, 211, 212). Their expression levels were positively correlated with each other. Moreover, it was demonstrated that hypermethylation was confirmed only for the DNMT3A transcript but not for DNMT1 and DNMT3B transcripts in endometriotic stromal cells (213). Conversely, a significant reduction in the expression of DNMTs was found in other studies comparing the tissue obtained from endometriotic foci with endometriosis-free control specimens (213, 214). An estrogen-dependent and progesterone (P4)-resistant environment was created at the level of endometriotic stromal cells by the altered expression of estrogen receptor-ß (ERß) and P4 receptors (PRs) concomitantly with an epigenetic switch in GATA transcription factor isoform expression. This switch replaces GATA2, which is expressed in normal healthy endometrium, with GATA6 in endometriotic cells and appears to strongly contribute to the endometriotic phenotype (210, 215). Altered endometrial DNA methylation in endometriosis was most evident in the mid-secretory phase (P4 peak), where a bias toward methylation of CpG islands may lead to the disruption of the normal pattern of cycle-dependent DNA methylation modifications. Considering this, wide-range abnormalities of the chromatin remodeling machinery in endometriosis should become a logical consequence (38, 216).
Modulation of gene expression through histone modifications has been confirmed recently in endometriosis in relation to both eutopic and ectopic endometrial/endometriotic tissues. Profiles of normal and aberrant histone lysine methylation or acetylation patterns were analyzed intensively in animal and human endometrial tissue models (217, 218). In human samples, endometriotic foci are more hypoacetylated at H3 (but not at H4) compared to eutopic endometrium from healthy women. The endometriotic tissue was characterized by significantly lower levels of H3K9ac and H4K16ac compared to eutopic endometrium from patients and controls (219). The use of chromatin immunoprecipitation (ChIP)-polymerase chain reaction made it possible to demonstrate hypoacetylation of H3/H4 within the promoter regions of candidate genes that are recognized as downregulated in endometriosis (e.g., HOXA10, ESR1, CDH1, and p21WAF1/Cip1) when comparing endometriotic lesions and control endometrium (219, 220). The stereoidogenic factor 1 (SF1) promoter region was enriched for acetylated H3 and H4 in endometriotic vs. control endometrial tissues. This acetylation was correlated with the increased expression of SF1 in the lesions. In addition to altered activities of HDACs, hypermethylation at H3K4, H3K9, and H3K27 was demonstrated in endometriosis (218, 219).
Differences in the expression of over 100 miRNAs have been reported in endometriotic cells. Such miRNA profiling may play a pivotal role in the development of miRNA signatures for endometriosis and expand our knowledge on the roles of individual miRNAs in the pathomechanism of this disease (220).
Thus, when looking at a slightly more general perspective of the role of T cells in endometriosis, local endometrial function/dysfunction, including cell proliferation, inflammation, immunogenicity of endometriotic stromal cells, angiogenesis, and sex steroid hormone responsiveness, may be relevant. Regardless of whether the observed disorders are caused by epigenetic factors or DNA sequence changes, the immune landscape in endometriosis interacts with T cells through overproduction of prostaglandins (PGE2), metalloproteinases (MMP-2, -3, -9), cytokines (TNFα, IL-1β, IL-8, IFNγ, MCP-1, and MIF) and adhesive molecules (VCAM-1, ICAM-1) (221). Furthermore, reactive oxygen and nitrogen species (ROS/RNS) production induced by pathological conditions influences aerobic glycolysis in activated CD4+ T cells and has an immunomodulatory effect on the mechanisms of antigen presentation and T-cell receptor signaling (222, 223).
The use of whole-tissue deconvolution with single-cell transcriptomic (scRNAseq) analysis made it possible to create an atlas of the human endometrium during the menstrual cycle (224, 225). Such a high-resolution molecular and cellular characterization of the human endometrium as a dynamic tissue that undergoes cyclic changes provides new opportunities to study the pathophysiology of endometriosis, including the cellular complexity of disease development. scRNAseq analysis can provide insights into the phenotypes of endometrial/endometriotic cell populations (226). Moreover, the full complement of immune and nonimmune cell types contributing to a proinflammatory background can be precisely identified across the menstrual cycle (227, 228). For example, subpopulations of fibroblasts related to endometriosis development were identified (228).
The coexistence of some diseases, including autoi-mmunological ones, may significantly change the influence of environmental factors on T cells. It was recently established that the presence of concomitant autoimmunity is associated with an advanced stage of disease in women with endometriosis. Although without a known explanation, this does indicate the possibility of a more aggressive course of the disease in patients who are also affected by autoimmune disturbances (184).
There is substantial evidence of aberrant function of almost all types of immune cells in women with endometriosis, including altered T-cell reactivity and NK cytotoxicity, polyclonal activation of B cells and increased antibody production, increased number and activation of peritoneal macrophages, and changes in inflammatory mediators (175, 229–232). As in the title of this chapter, T-cell disorders in endometriosis are discussed. In contrast to macrophages, dendritic cells, and toll-like receptors (TLRs), which are integral components of the innate immune system, Th (Th1/Th2/Th17) cells and Tregs are the main components of the adaptive immune system. The rationale for a potential role of T cells, especially Tregs, acting either alone or in combination in the initiation, maintenance, and progression of endometriosis is because the growth and progression of endometriosis continues even in ovariectomized animals. Thus, in addition to ovarian steroid hormones, the establishment and growth of endometriotic foci outside the uterine cavity can be regulated by the innate/adaptive immune system (175, 233, 234). Interpretation of the results of many comparative T-cell studies in women with endometriosis vs. normal (control) women is difficult because most of the research was carried out under different (incompatible) conditions, using small and not precisely defined groups/subgroups of patients. Identification of T-cell subtypes typically took place in the blood, peritoneal fluid, or endometrial/endometriotic foci (235). In connection with the pursuit of greater selectivity T-cell markers are also changing. For example, currently, in both mice and humans, the markers of Tregs are often presented as CD4+CD25 high CD127 – Foxp3+, where CD25 and Foxp3 are constitutive markers appropriate for isolation of Tregs, and CD127 expression is inversely correlated with both Foxp3 expression and related CD4+Tregs immunosuppressive function (236, 237). Previously, such a set of markers was not obvious, which makes the interpretation of the results over many years difficult. After considering the above reservations, which may explain some discrepancies and controversies, it is assumed that there are quantitative and qualitative changes in the T-cell population in endometriosis. Loss of balance between Th1/Th2/Th17 and Tregs leads to inappropriate secretion of T-cell-related cytokines (abnormal cytokine profile) and inflammation that induces progression of endometriotic lesions (238). Regarding T-cell subpopulations, it was demonstrated that the CD4+ T-cell profile in lesions and peripheral blood is altered in women with endometriosis. The proportion of Th1 lymphocytes was significantly lower in endometriotic lesions than in eutopic endometrium, and the Th17 lymphocyte fraction was significantly higher in the lesions than in eutopic endometrium. In addition, in peripheral blood, the Th1-cell fraction was significantly higher in patients with endometriosis than in women without the disease (231, 239). Posttranslational modifications (PTMs) are key molecules involved in Th17/Treg differentiation and function (Foxp3, RORγt, and STATs), regulate the Th17/Treg balance, and initiate autoimmune diseases caused by dysregulation of the Th17/Treg balance. An epigenetic toolkit contains modulators of genome architecture such as phosphorylation, methylation, nitrosylation, acetylation, glycosylation, lipidation, ubiquitination, and SUMOylation. Phosphorylation is the most common PTM contributing to Th17/Treg cell functions, whereas interactions between multiple PTMs influence Th17/Treg differentiation (97, 240, 241).. An increased number of Tregs has been reported in specimens (e.g., eutopic endometrium, peripheral blood, and peritoneal fluid) obtained from women with endometriosis compared to endometriosis-free control women (242–244). It was postulated that such an increased amount of Tregs may modulate the inflammatory response toward the establishment of an anti-inflammatory environment by suppressing activation of the immune system evoked by the endometriotic foci. Consequently, a reduced immune response enables ectopic endometrial implantation and propagation, resembling immune tolerance in allogeneic grafts and pregnancy (245). It can also be the opposite: Tregs could be moving toward the ectopic endometrial focus to reduce the severe inflammatory reaction (246). Thus, a higher frequency of circulating Tregs in patients with endometriosis compared with controls may be considered a compensatory mechanism to regulate the inflammatory condition in this disease (247).
There is also no doubt that, in addition to their immunoregulatory role, Tregs are involved in both normal and pathological angiogenesis. The association between angiogenesis and Tregs can be viewed in terms of either relation to the vascular endothelial growth factor (VEGF) signaling pathway or mediation via modulation of other immune cells and their release of cytokines and growth factors that influence angiogenesis (248). Interestingly, the role of Tregs in angiogenesis has been shown to be highly tissue- and context-specific and, as a result, can yield either pro- or antiangiogenic effects. This also pertains to different stages of endometriosis; however, it makes prediction unreliable (249–251).
CD4+ CD25+ Tregs (or even more precisely characterized in the current papers as CD4+CD25 high CD127 – Foxp3+ cells) are mainly produced in the thymus from where they migrate to the circulation as natural Tregs (nTregs), and a much smaller subpopulation differentiates in the periphery from naïve T cells into induced Tregs (iTregs) (252, 253). It has been shown that concurrent induction of Treg-specific epigenetic changes and the expression of transcription factor Foxp3 controlling a substantial part of Treg development and function is crucial for lineage specification and functional stability of Treg cells (254). Treg deficiency or dysfunction exaggerates local inflammation and angiogenesis and simultaneously facilitates the attachment and growth of endometrial implants (250).
The altered immune response in endometriosis may be attributed to defective apoptotic processes. Increased concentrations of cytotoxic (CD8+) T lymphocytes (CTLs) and HLA-DR- activated T cells were observed in peripheral blood during the luteal phase compared to the follicular phase of the menstrual cycle in healthy women, whereas women with endometriosis did not exhibit fluctuations in the concentrations of cytotoxic and activated peripheral blood lymphocytes during the menstrual cycle. In addition, a marked increase in Treg concentration, which was positively correlated with the serum levels of cortisol, was detected in the peripheral blood of women with endometriosis only (255). The cytoplasmic granules of CTLs contain perforin, a cytolytic mediator that may induce apoptosis, because they form pores when inserted into the target cell membrane (256). A significant reduction in the cytotoxic potential of CTLs was demonstrated in endometriosis, where the number of perforin+ CTLs among CD8+ T cells in the menstrual effluent was decreased compared to healthy controls. Perforin mRNA levels correlate with the methylation status and accessibility of the promoter at the 5′ flanking region of its gene. Thus, the defective apoptotic process may be caused by DNA hypermethylation and changed chromatin structure affecting negatively perforin gene expression in T cells (257).
At the same time, a decreased concentration of intercellular adhesion molecule-1 (ICAM-1) was observed in the serum of endometriosis patients. The transmembrane glycoprotein ICAM-1 plays a role in inflammatory processes and in the T-cell-mediated host defense system. ICAM-1 is constitutively expressed over the cell surface and its expression can be modulated by transcription and epigenetic factors related to cellular stress, proinflammatory cytokines, and viral infection (258). It functions as a costimulatory molecule on antigen-presenting cells to activate MHC class II restricted T cells and on other cell types in association with MHC class I to activate cytotoxic T cells. Deletion of the 5′ flanking region of ICAM-1 gene at positions -329 and -485 upregulates the basal level expression of ICAM-1, suggesting the presence of a regulatory silencer within this region (259).
Both fewer perforin+ CTLs and a reduced concentration of ICAM-1 may reflect a reduced capacity to remove endometrial cells from ectopic locations (260–262). ICAM-1 alone or together with soluble vascular cell adhesion molecule 1 (VCAM-1) may be a promising biomarker for diagnosing endometriosis. However, according to the results of a meta-analysis, ICAM-1 used alone has moderate diagnostic accuracy, while for unknown reasons, the diagnostic accuracy is higher in patients of Asian ethnicity than in those of Caucasian ethnicity (263, 264).
Because of the opposite effect on the immune response, proinflammatory Th1 and Th17 cells should be balanced by Treg subsets with anti-inflammatory capacity. An important element of such a balance is a specific cytokine profile with IFNγ and IL-2 produced by Th1, IL-17A synthetized by Th17 and IL-10 and transforming growth factor (TGF)-β secreted by Tregs (265–268). It was documented that increases in the level of IL-17A and the presence of Th17 in peritoneal fluid correlate positively with the severity of endometriosis and infertility associated with this disorder (269, 270). The number of Th17 cells in peritoneal fluid was higher in severe endometriosis (stages III and IV) than in early/not advanced (stages I and II) endometriosis (270, 271). IL-17A may play a role in the development of endometriosis by stimulating inflammatory responses, angiogenesis, and proliferation of endometriotic stromal cells (272, 273).
An imbalance between the cytokine profile related to Th1 and Th2 responses was suggested in the etiopathology of endometriosis. The shift toward the Th2 immune response component (a reduced Th1/Th2 ratio among T cells in the peritoneal fluid) with the relative increase in cytokines, characteristic of this pattern of immune response (IL-4, IL-5, IL-10, and IL-13) should be considered. These cytokines are associated with the promotion of IgE and eosinophilic responses in atopy and interleukin-10, which has more of an anti-inflammatory response. In excess, Th2 responses counteract the Th1-mediated perpetuating autoimmune responses (274). Indeed, in endometriotic lesions, the levels of IFN-γ and IL10 and the ratios of IL4/IFN-γ, IL4/IL2 IL10/IFN-γ, and IL10/IL2 are significantly elevated in the peritoneal fluid of endometriosis patients compared to healthy controls (238, 275). For example, the release of IL-4 from Th2 cells may lead to a dose-dependent increase in the expression of 3β-hydroxysteroid dehydrogenase (HSD3B2) mRNA, a pivotal enzyme for estrogen production (276). Therefore, endometriosis progression may be stimulated by an IL-4 dependent, local increase in estrogen concentration. Moreover, IL-4 promotes the proliferation of endometriotic stromal cells (ESCs) and endometriosis progression by activating p38 mitogen-activated kinases (p38 MAPKs), stress-activated protein kinase/c-Jun kinase and p42/44 MAPK. All these enzymes are strongly induced in vivo by environmental stresses and inflammatory cytokines (277). Activity of the transcription factor GATA binding protein 3 (GATA3) is regulated by estrogen, and their synergistic action regulates Th2 cytokine (e.g., IL6, IL8, and IL10) expression in endometrial cells (both eutopic and ectopic). Therefore, GATA3 integrates estrogen signaling to induce Th2 cytokine expression in endometriotic lesions, thereby promoting endometriosis progression (278). Interestingly, eutopic endometrial tissues from patients with endometriosis have higher mRNA levels of GATA-binding protein 3 (GATA3) compared to normal endometrial tissue (279). Because the development and maintenance of endometriosis highly depends on the estrogen pathway, overexpression of the two proteins that control key steps of 17β‐estradiol biosynthesis, steroidogenic acute regulatory protein (StAR) and aromatase (CYP19), may contribute to formation of ectopic lesions (280). Hypomethylation of the promoter and/or intronic regions of StAR and CYP19 was shown to cause their incorrect expression in ectopic foci (281, 282). Hypomethylation was also detected within the promoter and/or intronic regions of several aberrantly expressed nuclear receptors that mediate the effect of steroid hormones or modulate steroidogenic activity (e.g., estrogen receptor β, steroidogenic Factor 1 (SF-1)) (283, 284). These data indicate that DNA methylation is coordinately regulated to facilitate production or to enhance activity of 17β‐estradiol (E2) in endometriosis (285). The central role of epigenetic regulation on the steroid hormone pathway manifests itself in two directions. This means that inactivation of E2 is also regulated by DNA methylation. In addition, conversion of E2 to less potent estrone (E1) is suppressed in endometriosis because the converting enzyme 17β‐hydroxysteroid dehydrogenase type II is inactivated in ectopic stromal cells due to hypermethylation (286).
Interleukin 6 (IL-6) promotes CD4+ Th2 differentiation by activating transcription mediated by nuclear factor of activated T cells (NFAT) and – at the same time – inhibits Th1 differentiation by interfering with IFN-γ signaling and expression of suppressor of cytokine signaling 1 (SOCS1) (287, 288). Increased IL-6 levels were demonstrated in ESCs isolated from women with endometriosis compared to healthy controls (289). IL-6 expression in endometriotic cells may be induced by IL-1β and TNF-α (290). According to recent studies, IL-6 pathway gene expression can be affected by DNA methylation, microRNAs, and posttranslational modifications (291, 292).
Interleukin 23 (IL-23) is a proinflammatory cytokine composed of two subunits, IL-23A (p19) and IL-12/23B (p40), produced primarily by activated macrophages and dendritic cells. It drives the differentiation and activation of T helper 17 (Th17) cells and maintains their phenotype, such as their cytokine production, including IL-17A, which is their major effector molecule (293). Well-established experimental data support the concept that defective IL-23/IL-17 axis activation contributes to the development of several autoimmune (e.g., IBD, RA, SS, MS) and inflammatory diseases, including endometriosis (294). Levels of IL23 were significantly higher in the peritoneal fluid of women with endometriosis than in normal controls (295). Activated naïve T cells produce IL23 and consequently increase production of IL10 and IL17, both of which are factors promoting endometriosis progression (296). It was recently documented that environmental factors may significantly contribute to activation, modulation, or dysregulation of the IL-23/IL-17 axis (297).
Despite the fact that the etiology of endometriosis is complex and multifactorial, an abnormal immune response in this disease with evident changes in T-cell activities clearly indicates the possibility of treating these heterogeneous cells as therapeutic targets (32, 133, 175, 235). The suggestions for potential therapeutic measures presented below are limited to epigenetically modulated T-cell disorders.
Unlike autoimmune diseases, in endometriosis, we are dealing with a deficit of immune recognition by T cells (176). Therefore, an augmented T-cell-dependent immune response may improve the elimination of ectopic cells within endometriotic foci (177). The cells can be influenced directly or indirectly. In the latter situation, and generally in the absence of a selective effect, one should consider the effects of epigenetic modulation with respect to other cells and organs, that is, systemic action. It is therefore important to be aware of the possibility of both the synergistic effects and the side effects when acting on other cells (e.g., ectopic and eutopic endometrial tissue). Increased activity of T cells may also adversely affect the course of autoimmune diseases frequently cooccurring with endometriosis (176).
There are three families of epigenetic proteins considered as susceptible to disease modifying drugs: readers, writers, and erasers (298). Initially, the readers recognize and bind to specific covalent DNA modifications, as well as histones, and non-histone proteins. Next, the writers introduce various chemical modifications on DNA and histones. Finally, the erasers are responsible for enzymatic removal of these biochemical tags. Therefore, all these stages of epigenetic modulation are druggable targets using small molecular-inhibitors (SMIs) including approved by US Food and Drug Administration (FDA) azanucleosides, vorinistat, fedratinib. These drugs targeting DNMTs, HDACs, and JAK2, respectively (299, 300). The significant advancement of work on other SMIs, including on-going clinical trials, causes the implementation of new drugs is a matter of time. However, at present the use of SMIs in clinical settings is mostly limited to hematological malignancies (300–302). Safety issues related mostly to the lack of selectivity produce significant limitations with implementation of SMIs. Cardiovascular, CNS, stem cell homeostatic, developmental and reproductive, transgenerational, and carcinogenic effects are among the potential consequences of targeting epigenetic mechanisms (303).
Naturally, at this stage of advancement of research on epigenetically targeting T cells in endometriosis, the rationale and their main directions are signaled. For example, to identify the epigenetic changes involved in endometriosis, a genome-wide analysis of DNA methylation and enrichment of H3K4me3 and H3K27ac histone marks in sorted CD4+ and CD8+ T cells may be performed (304). However, it is important to realize that only full knowledge about the etiopathology of endometriosis, and in this case, the importance of epigenetic interactions, will make it possible to increase the effectiveness of these activities oriented on therapy.
The drugs (SMIs) available on the market today carry too great a risk to women of childbearing age in regards to fertility and embryo-fetal development, leading to a pregnancy category D warning on the label (303). As it was mentioned elsewhere, up to 50% of endometriosis-related treatments have been performed in order to restore the ability to become pregnant (23, 24).
Considering that endometriosis is an estrogen-dependent and P4-resistant disorder, alignment of hormonal dysregulation is a widely used symptomatic treatment. Estrogen has been shown to modulate all subsets of T cells, including CD4+ (Th1, Th2, Th17, and Tregs) and CD8+ cells (305, 306). Human CD4+ T cells and CD8+ T cells express both estrogen receptors, ERα and Erβ, and are involved in the nongenomic G protein-coupled estrogen receptor (GPER) pathway (307–309). It was suggested that targeting abnormally lowered methylation of a CpG island in the promoter region of the ERβ gene (Esr2) may have promising therapeutic effects, reducing proinflammatory signatures of T cells. Moreover, the resulting decrease/normalization in ERβ expression may restore the normal ERα expression required for sufficient responsiveness to E2 with respect to progesterone receptor (PR) expression, which counteracts P4 receptor deficiency and P4 resistance. Alternatively, induction of hypomethylation in the respective CpG islands of the promoter regions of ERα and GPER may be used, as the anti-inflammatory effects of estrogens are associated mostly with signaling via ERα and GPER (164, 310).
In contrast to the mouse, most studies did not confirm the classical P4 receptors (PR-A and PR-B) in human T cells. However, P4 effects may be mediated via the two families of membrane PRs belonging to the progestin and adipoQ receptor (PAQR) family (also known as membrane progestin receptors (mPRs)) and the progesterone receptor membrane component (PGRMC) receptors. In humans, three members of the PAQR family (PAQR5, PAQR7, and PAQR8) and two members of the PGRMC family (PGRMC1 and PGRMC2) were identified. Progesterone modulates the pattern of T-cell cytokine production in a dose-dependent manner (311, 312). Hence, reverting relative P4 resistance may augment the potentially beneficial influence of these cytokines. Downregulation of PR-B due to promoter hypermethylation of PR-B was reported in the endometrium of women with endometriosis (313). Accordingly, with regard to T cells, P4 increases the number of CD4+ CD25+ FoxP3+ Tregs in the maternal-fetal interface of pregnant mice (314). It was demonstrated that Treg deficiency intensifies the course of endometriosis primarily regarding the intensity of inflammation (198, 315). Nevertheless, the recommendation of a particular course of action (i.e., methylation, demethylation) in relation to estrogen and P4 receptors in T cells must be preceded by comparative analysis with endometrial/endometriotic cells because the effects of interactions with these receptors may differ significantly.
In addition to methylation, noncoding RNAs: miRNAs (e.g., miR-148a, miR-30b-5p, miR-487a-5p, miR-4710, miR-501-3p, miR-378 h, and miR-1244) and lncRNAs (e.g., HOX antisense intergenic RNA - HOTAIR) are involved in the epigenetic spectrum of interactions between immune and hormonal systems in endometriosis (316, 317). Future studies are needed to determine whether estrogen and P4 receptors in T cells may be modulated efficiently by noncoding RNAs to restore the environment with normal responsiveness to estrogen and P4.
Another promising treatment option is based on the demonstration that the estrogen – indoleamine 2,3-dioxygenase-1 (IDO1) – mannose receptor C, type 2 (MRC2) axis participates in the differentiation and function of Tregs and is involved in development of endometriosis (318). In cocultured naïve T-cell-macrophage-endometrial stromal cells (ESCs), a specific blocker of IDO1,1-methyl-tryptophan (1-MT) produced a significant decrease in Treg differentiation, particularly in the IL-10+ Treg subpopulation. Therefore, 1-MT-pretreated ESC-educated Tregs exhibited impaired suppressive function. Moreover, estrogen promoted the differentiation of Tregs by elevating IDO1 expression in ectopic lesions (319). At the same time, expression of MRC2, which is an upstream molecule of IL-10 required for Treg differentiation in ectopic lesions (especially CD4high Tregs), was significantly lowered. Thus, blockage of IDO1 in ectopic lesions, which does not influence the physiological functions of estrogen, may be considered a potential therapy for endometriosis (318).
Increasing data show that epigenetic reprogramming of T-cell development and differentiation can contribute to the development of new, breakthrough treatments in endometriosis (320). Quantitative and qualitative disorders in the T-cell population in endometriosis (vs. endometriosis-free T cells) that can now be compensated for under laboratory conditions by using epigenetic mechanisms include the Th1:Th17:Treg lymphocyte ratio and number, influencing T-cell-dependent apoptosis and angiogenesis, modulating Th2 cytokine expression and influencing Th17 activation (156, 242, 321–323).
The discovery of Foxp3 was a turning point in understanding the molecular determinants leading to the generation and maintenance of Tregs (82). Maintenance of Foxp3 protein expression in regulatory Tregs is crucial for a balanced immune response. Recent reports suggest that specific modifications of DNA and histones are necessary for establishing the chromatin structure in conventional CD4+ T cells (T conv) as a prerequisite for their future differentiation into the Treg cell lineage. Thus, Tregs support a distinct DNA methylation pattern compared to Tconv, and specific epigenetic mechanisms critically influence Foxp3 stability (324). Moreover, several studies have demonstrated that during inflammation (e.g., endometriosis-related), Treg cells may lose their phenotypic properties and be converted into effector T cells secondary to the alteration of Foxp3 expression and stability (325–327).
The results of animal studies have shown that Foxp3 expression is regulated by miRNA (96, 97). For example, deletion of miR-146a-5p results in a breakdown of immune tolerance and development of a fatal spontaneous autoimmune disorder, whereas Foxp3 acting together with miR-155-5p blocks key inducers of effector lineage commitment, such as Satb1 and zinc finger E-box binding homeobox (98–100, 328). For future therapeutic purposes, the most important thing is therefore that Foxp3 imposes a multilayered suppression of specific genes in Treg cells by both direct binding to genetic regulatory elements and by induction of miRNAs that specifically target the 3′ UTR of the same genes. Epigenetic machinery adjusting the Treg phenotype for medicinal purposes could be based on the action of miR-31, miR-24, and miR-210, which directly target the Foxp3 3′ UTR (103, 104). Regulation of Foxp3 expression and function may also take place at the protein level in the form of covalent posttranslational modifications, such as ubiquitination, acetylation, and phosphorylation of different amino acids. Following these processes, changes in the subcellular localization and activity of Foxp3 should be expected. Such effects resulting from different interactions with other proteins, mainly transcriptional activators or repressors, deserve attention from a therapeutic point of view.
Sirtuin-1 (SIRT1)-mediated deacetylation of Foxp3 leads to its decreased expression as a result of increased ubiquitination and subsequent degradation in the proteasome (329). Conversely, application of nicotinamide, a SIRT1 inhibitor, reduces Foxp3 degradation together with increased Treg cell number and suppressive activity. These findings may indicate the central role for SIRT1 in the regulation of Foxp3 protein levels and thereby in the regulation of Treg suppressive capacity. Pharmacological modulation of SIRT1 activity in Tregs may therefore provide a novel therapeutic approach for controlling immune responses in endometriosis. This can be done by regulating mammalian sterile 20-like kinase 1 (Mst1), which increases Foxp3 acetylation and promotes its activity both indirectly, by inhibiting the activity of SIRT1, and directly, by interacting with Foxp3 and preventing its binding to SIRT1 (330). Interestingly, influencing the Foxp3 acetylation level deserves attention as a potential therapy aimed at restoring the normal balance between Treg and Th17-cell lineage differentiation (331). This is because the transcriptional coactivator with PDZ-binding motif (TAZ) that promotes differentiation toward Th17 and inhibits Treg development, regulates Foxp3 acetylation by competing with it for binding to TIP60, a nuclear histone acetyltransferase (HAT) that mediates Foxp3 acetylation and inhibits its proteasomal degradation (331). Thus, a decrease in Foxp3 acetylation constrains differentiation toward Tregs. Sirtuin-targeted treatment in altered immune response (autoimmune disease vs. endometriosis) aimed to restore optimal Foxp3 expression in Tregs is presented in Figure 4.
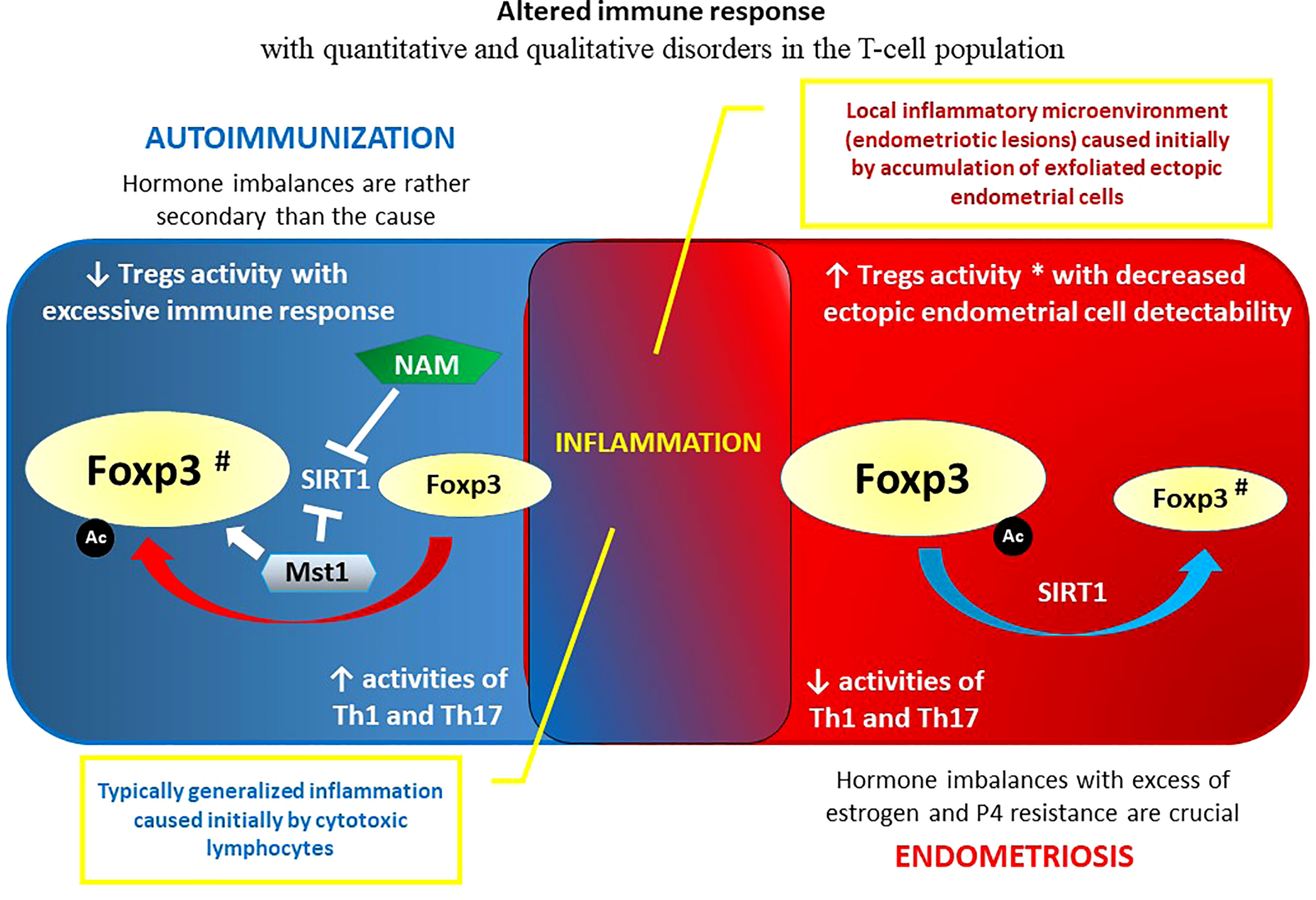
Figure 4 Regulatory T cells and altered immune response: autoimmune disease vs. endometriosis. Maintenance of Foxp3 protein expression in regulatory Tregs is crucial for a balanced immune response. The rationales for pharmacological modulation of SIRT1 activity in Tregs as an example of novel therapeutic approach for controlling immune responses in endometriosis and autoimmunity are also showed. # Restored to normal Foxp3 expression after the treatment; * It can also be opposite: increased number and activity of Tregs may be considered as an insufficient compensatory mechanism to overcome inflammation (194, 195). Epigenetics of Tregs appears to be a common denominator for autoimmunity and endometriosis. See the main text (Chapter 3. The association between endometriosis and autoimmune diseases) for details. Ac – acetylation; Foxp3 – forkhead box P3 protein; Mst1 – mammalian sterile 20-like kinase 1; NAM – nicotinamide; SIRT1 – sirtuin 1 (silent mating type information regulation 2 homolog 1); Th1 – T helper 1 cells; Th17 – T helper 17 cells; Tregs – regulatory T cells.
T-cell reprogramming should be considered an effective measure to overcome the altered immune response in endometriosis, including that caused by epigenetic factors (332). Naturally, the crucial issue is comprehensive understanding of the etiopathogenesis of these disorders that present unclear association with autoimmunization. The question to be answered is as follows: should we potentiate or suppress T cells in endometriosis? More precisely: action of which T-cell subpopulations should be strengthened, and which should be inhibited? Only then can the course of action be correctly determined. Research on T-cell reprogramming in endometriosis is very intense and advanced; however, as opposed to T-cell-based immunotherapy in cancer, it has not yet been translated into clinical practice.
The TCR is sufficient to direct antigen-specific T-cell differentiation and redirect their cytotoxicity. Custom TCR reprogramming may revert the condition in which the immune system fails to recognize and target endometrial tissue growing elsewhere in the body. Now, TCRs can be engineered to possess more specificity, affinity, reactivity, and broad-spectrum binding ability. For instance, TCRs can be engineered to potentially recognize all peptides processed and presented in the context of MHC molecules, thus allowing TCRs to target both surface and intracellular antigens. Exodomains of the α and β subunits of the TCR can be modified by replacing their variable domains with antibody domains that can recognize endometriosis-associated antigens (333). T-cell receptor gene rearrangement may also provide a lower or higher sensitivity to gene silencing (e.g., in human CD8+ memory T cells).
Chimeric antigen receptors (CARs) are a class of synthetic TCR receptors that reprogram lymphocyte specificity and function. Designed to bind to certain proteins (e.g., expressed within endometriotic foci), CARs constitute an effective genetic optimization of T cells to redirect specificity. The use of CARs in the treatment of epigenetic modifications or environmental endometriosis-causing agents is only a matter of time.
Discovery of immune checkpoint inhibitors about ten years ago, and then the development of CARs, created new options for treating hematological malignancies (334, 335). Clinical trials employing CARs are also conducted in solid tumors, including gynecological cancers (336). There are undoubted parallels in the development between cancer and endometriosis. The way of mutagenesis, pelvic spreading, immunological adaptation, and difficulties in eradication justify to consider endometriosis as “a cancer of no kill” (337). Ultra-mutated phenotype of ectopic endometriotic cells requires an escape form the immune surveillance during the development of endometriosis. As in malignant tumors, the interaction TCR/antigen/MHC self-recognition complex and the PD-1/PD-L1 immune checkpoint must be abolished to avoid T cell-mediated cytotoxicity. This is achieved by upregulated expression of PD-1/PD-L1 in endometriosis (338). Accordingly, serum PD-1 level is positively correlated with endometriosis-related infertility (339). Such immune escape in endometriosis makes it possible to apply an immune checkpoint blockade therapy using CARs that has revolutionized cancer treatment. If in a wide range of cancers that are characterized by genomic hypermutation and a high replication error rate, targeting PD-1/PD-L1 was effective, endometriotic cells, with identical phenotype characteristics, may very likely respond similarly (340).
Both custom TCR reprogramming and CAR generation can be performed using in vitro-transcribed (IVT) mRNA. After selection of the target of the CAR T cells, IVT-mRNA is precisely positioned within the created CAR IVT-mRNA molecule. The transfection of the mRNA transcript into T cell must ensure optimal/efficient expression the CAR molecule on T cell surface (341). This method is characterized by rapid and facile production and an acceptable safety profile. Unlike DNA, IVT mRNA has no risk of causing insertional mutagenesis and no long-term concern for side effects because of its labile nature (342).
The inhibition of epigenetic regulators may also be skillfully used to obtain the desired functional profile of T cells suitable for treatment of endometriosis. Inhibitors of DNA methyltransferase (e.g., 5′-azacytidine), histone deacetylase (e.g., valproic acid), histone methyltransferase (e.g., BIX-01294), and histone demethylase (e.g., tranylcypromine) should be considered (343, 344). Remarkable developments in the basic understanding and tools for reprogramming have begun to show the clinical impact of cellular reprogramming. Thus, genetically, and epigenetically reprogrammed T cells hold great promise in the areas of immunotherapy, including endometriosis, offering great hope for curative responses in women with ectopic endometriotic lesions. Adoptive transfer of profiled Tregs is a promising new therapeutic option to treat detrimental inflammatory conditions after transplantation during autoimmune disease and endometriosis, including disorders caused by epigenetic factors (345).
Dysfunction of the immune system is the essence of both autoimmune diseases and endometriosis (346, 347). Therefore, the altered function of T cells in these disorders is the subject of intense research. Understanding the role of epigenetic factors and determining which T-cell functions are controlled epigenetically could lead to a breakthrough in the final elucidation of endometriosis etiopathogenesis (348). The prevailing view thus far is that, in contrast to autoimmune diseases, silencing the immune response prevents detection and destruction of the endometriotic foci (349). Changes in T-cell count and/or T-cell subtype proportion together with modulation of T-cell activity may be a therapeutic target in endometriosis that involves epigenetic mechanisms. However, except for observational, cross-sectional studies, it is difficult to perform reliable investigations on endometriosis patients. Most of the immunological endometriosis research is performed on animals. In nature, spontaneous endometriosis affects only those mammalian species that menstruate, including primates, some bat species, spiny mice, and elephant shrews (350, 351). Thus, many animal models are not fully reliable, since endometriosis is induced artificially and does not represent all the phenomena present in the disorder. Such artificial endometriosis behaves differently than spontaneous endometriosis, even in the same experimental animal model or in the same animal (352, 353). T-cell reprogramming creates a new therapeutic option that may be tested to a large extent without the exploitation of animal models. Since epigenetics appears to be a common denominator for hormonal and immunological aberrations in endometriosis, adjustment of hormonal imbalances and influencing T-cell development, differentiation, and activation should be considered (12, 21, 38, 354, 355). Epigenetic, reversible regulation of Tregs toward higher ectopic endometrial cell detectability, and elimination can be a significant step in human treatment. Further studies are needed to investigate how such therapy influences coexisting autoimmune diseases (36, 356).
As the only author of this review paper, I acknowledge my full contribution and responsibility for the content of the manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor APG declared a past co-authorship with the author.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev (2009) 23(7):781–3. doi: 10.1101/gad.1787609
2. Tollervey JR, Lunyak VV. Epigenetics: judge, jury and executioner of stem cell fate. Epigenetics (2012) 7(8):823–40. doi: 10.4161/epi.21141
3. Boland MJ, Nazor KL, Loring JF. Epigenetic regulation of pluripotency and differentiation. Circ Res (2014) 115(2):311–24. doi: 10.1161/CIRCRESAHA.115.301517
4. Barrere-Cain R, Allard P. An understudied dimension: Why age needs to be considered when studying epigenetic-environment interactions. Epigenet Insights (2020) 13:2516865720947014. doi: 10.1177/2516865720947014
5. Polverino A, Sorrentino P, Pesoli M, Mandolesi L. Nutrition and cognition across the lifetime: an overview on epigenetic mechanisms. AIMS Neurosci (2021) 8(4):448–76. doi: 10.3934/Neuroscience.2021024
6. Mahmoud AM. An overview of epigenetics in obesity: The role of lifestyle and therapeutic interventions. Int J Mol Sci (2022) 23(3):1341. doi: 10.3390/ijms23031341
7. Ajoolabady A, Aslkhodapasandhokmabad H, Zhou Y, Ren J. Epigenetic modification in alcohol-related liver diseases. Med Res Rev (2022) 21:1463–91. doi: 10.1002/med.21881
8. Rastegar M, Yasui DH. Editorial: Epigenetic mechanisms and their involvement in rare diseases. Front Genet (2021) 12. doi: 10.3389/fgene.2021.755076
9. Cañas JA, Núñez R, Cruz-Amaya A, Gómez F, Torres MJ, Palomares F, et al. Epigenetics in food allergy and immunomodulation. Nutrients (2021) 13(12):4345. doi: 10.3390/nu13124345
10. Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell (2014) 157(1):95–109. doi: 10.1016/j.cell.2014.02.045
12. Conway SJ, Woster PM, Greenlee WJ, Georg G, Wang S. Epigenetics: Novel therapeutics targeting epigenetics. J Med Chem (2016) 59(4):1247–8. doi: 10.1021/acs.jmedchem.6b00098
13. Kayser C, Dutra LA, Dos Reis-Neto ET, Castro CHM, Fritzler MJ, Andrade LEC. The role of autoantibody testing in modern personalized medicine. Clin Rev Allergy Immunol. doi: 10.1007/s12016-021-08918-6
14. Autoimmune Registry, Inc. Available at: https://www.autoimmuneregistry.org.
15. Mariette X, Gottenberg JE, Ravaud P, Combe B. Registries in rheumatoid arthritis and autoimmune diseases: data from the French registries. Rheumatol (Oxford) (2011) 50(1):222–9. doi: 10.1093/rheumatology/keq368
16. Dinse GE, Parks CG, Weinberg CR, Co CA, Wilkerson J, Zeldin DC, et al. Increasing prevalence of antinuclear antibodies in the united states. Arthritis Rheumatol (2020) 72(6):1026–35. doi: 10.1002/art.41214
17. Zhang XM, Liu CY, Shao ZH. Advances in the role of helper T cells in autoimmune diseases. Chin Med J (Engl) (2020) 133(8):968–74. doi: 10.1097/CM9.0000000000000748
18. Singh VK, Mehrotra S, Agarwal SS. The paradigm of Th1 and Th2 cytokines: its relevance to autoimmunity and allergy. Immunol Res (1999) 20(2):147–61. doi: 10.1007/BF02786470
19. Mishra S, Srinivasan S, Ma C, Zhang N. CD8+ regulatory T cell - a mystery to be revealed. Front Immunol (2021) 12:708874. doi: 10.3389/fimmu.2021.708874
20. Selck C, Dominguez-Villar M. Antigen-specific regulatory T cell therapy in autoimmune diseases and transplantation. Front Immunol (2021) 12:661875. doi: 10.3389/fimmu.2021.661875
21. Dutta A, Venkataganesh H, Love PE. New insights into epigenetic regulation of T cell differentiation. Cells (2021) 10(12):3459. doi: 10.3390/cells10123459
22. Rodriguez RM, Lopez-Larrea C, Suarez-Alvarez B. Epigenetic dynamics during CD4(+) T cells lineage commitment. Int J Biochem Cell Biol (2015) 67:75–85. doi: 10.1016/j.biocel.2015.04.020
23. Parazzini F, Esposito G, Tozzi L, Noli S, Bianchi S. Epidemiology of endometriosis and its comorbidities. Eur J Obstet Gynecol Reprod Biol (2017) 209:3–7. doi: 10.1016/j.ejogrb.2016.04.021
24. Shafrir AL, Farland LV, Shah DK, Harris HR, Kvaskoff M, Zondervan K, et al. Risk for and consequences of endometriosis: A critical epidemiologic review. Best Pract Res Clin Obstet Gynaecol (2018) 51:1–15. doi: 10.1016/j.bpobgyn.2018.06.001
25. Kim JH, Han E. Endometriosis and female pelvic pain. Semin Reprod Med (2018) 36(2):143–51. doi: 10.1055/s-0038-1676103
26. Bourgioti C, Preza O, Panourgias E, Chatoupis K, Antoniou A, Nikolaidou ME, et al. MR imaging of endometriosis: Spectrum of disease. Diagn Interventional Imaging (2017) 98(11):751–67. doi: 10.1016/j.diii.2017.05.009
27. Charatsi D, Koukoura O, Ntavela IG, Chintziou F, Gkorila G, Tsagkoulis M, et al. Gastrointestinal and urinary tract endometriosis: A review on the commonest locations of extrapelvic endometriosis. Adv Med (2018) 2018:3461209. doi: 10.1155/2018/3461209
28. Bulun SE, Cheng YH, Pavone ME, Xue Q, Attar E, Trukhacheva E, et al. Estrogen receptor-beta, estrogen receptor-alpha, and progesterone resistance in endometriosis. Semin Reprod Med (2010) 28(1):36–43. doi: 10.1055/s-0029-1242991
29. Plante BJ, Lessey BA, Taylor RN, Wang W, Bagchi MK, Yuan L, et al. G Protein-coupled estrogen receptor (GPER) expression in normal and abnormal endometrium. Reprod Sci (Thousand Oaks Calif) (2012) 19(7):684–93. doi: 10.1177/1933719111431000
30. Tanbo T, Fedorcsak P. Endometriosis-associated infertility: aspects of pathophysiological mechanisms and treatment options. Acta Obstetricia Gynecol Scand (2017) 96(6):659–67. doi: 10.1111/aogs.13082
31. Burney RO, Giudice LC. Pathogenesis and pathophysiology of endometriosis. Fertil Steril (2012) 98(3):511–9. doi: 10.1016/j.fertnstert.2012.06.029
32. Patel BG, Lenk EE, Lebovic DI, Shu Y, Yu J, Taylor RN. Pathogenesis of endometriosis: Interaction between endocrine and inflammatory pathways. Best Pract Res Clin Obstet Gynaecol (2018) 50:50–60. doi: 10.1016/j.bpobgyn.2018.01.006
33. Tomassetti C, D'Hooghe T. Endometriosis and infertility: Insights into the causal link and management strategies. Best Pract Res Clin Obstet Gynaecol (2018) 51:25–33. doi: 10.1016/j.bpobgyn.2018.06.002
34. Kajiyama H, Suzuki S, Yoshihara M, Tamauchi S, Yoshikawa N, Niimi K, et al. Endometriosis and cancer. Free Radical Biol Med (2019) 133:186–92. doi: 10.1016/j.freeradbiomed.2018.12.015
35. Zondervan KT, Becker CM, Koga K, Missmer SA, Taylor RN, Viganò P. Endometriosis. Nat Rev Dis Primers (2018) 4(1):9. doi: 10.1038/s41572-018-0008-5
36. Shigesi N, Kvaskoff M, Kirtley S, Feng Q, Fang H, Knight JC, et al. The association between endometriosis and autoimmune diseases: a systematic review and meta-analysis. Hum Reprod Update (2019) 25(4):486–503. doi: 10.1093/humupd/dmz014
37. Klemmt P, Starzinski-Powitz A. Molecular and cellular pathogenesis of endometriosis. Curr Women's Health Rev (2018) 14(2):106–16. doi: 10.2174/1573404813666170306163448
38. Szukiewicz D, Stangret A, Ruiz-Ruiz C, Olivares EG, Soriţău O, Suşman S, et al. Estrogen- and progesterone (P4)-mediated epigenetic modifications of endometrial stromal cells (EnSCs) and/or mesenchymal Stem/Stromal cells (MSCs) in the etiopathogenesis of endometriosis. Stem Cell Rev Rep (2021) 17(4):1174–93. doi: 10.1007/s12015-020-10115-5
39. Smolarz B, Szyłło K, Romanowicz H. Endometriosis: Epidemiology, classification, pathogenesis, treatment and genetics (Review of literature). Int J Mol Sci (2021) 22(19):10554. doi: 10.3390/ijms221910554
40. Whitacre JM, Lin J, Harding A. T Cell adaptive immunity proceeds through environment-induced adaptation from the exposure of cryptic genetic variation. Front Genet (2012) 3:5. doi: 10.3389/fgene.2012.00005
41. Rosen A, Casciola-Rosen L. Autoantigens as partners in initiation and propagation of autoimmune rheumatic diseases. Annu Rev Immunol (2016) 34:395–420. doi: 10.1146/annurev-immunol-032414-112205
42. Xing Y, Hogquist KA. T-Cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol (2012) 4(6):a006957. doi: 10.1101/cshperspect.a006957
43. Khan U, Ghazanfar H. T Lymphocytes and autoimmunity. Int Rev Cell Mol Biol (2018) 341:125–68. doi: 10.1016/bs.ircmb.2018.05.008
44. Fu J, Khosravi-Maharlooei M, Sykes M. High throughput human T cell receptor sequencing: A new window into repertoire establishment and alloreactivity. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.777756
45. Kisielow P. How does the immune system learn to distinguish between good and evil? the first definitive studies of T cell central tolerance and positive selection. Immunogenetics (2019) 71(8-9):513–8. doi: 10.1007/s00251-019-01127-8
46. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol (2014) 14(6):377–91. doi: 10.1038/nri3667
47. Sewell AK. Why must T cells be cross-reactive? Nat Rev Immunol (2012) 12(9):669–77. doi: 10.1038/nri3279
48. Shah K, Al-Haidari A, Sun J, Kazi JU. T Cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther (2021) 6(1):412. doi: 10.1038/s41392-021-00823-w
49. Hawke NA, Yoder JA, Litman GW. Expanding our understanding of immunoglobulin, T-cell antigen receptor, and novel immune-type receptor genes: a subset of the immunoglobulin gene superfamily. Immunogenetics (1999) 50(3-4):124–33. doi: 10.1007/s002510050588
50. Muro R, Takayanagi H, Nitta T. T Cell receptor signaling for γδT cell development. Inflammation Regen (2019) 39:6. doi: 10.1186/s41232-019-0095-z
51. Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity (Edinb) (2010) 105(1):4–13. doi: 10.1038/hdy.2010.54
52. Dudley DD, Sekiguchi J, Zhu C, Sadofsky MJ, Whitlow S, DeVido J, et al. Impaired V(D)J recombination and lymphocyte development in core RAG1-expressing mice. J Exp Med (2003) 198(9):1439–50. doi: 10.1084/jem.20030627
53. Dong Y, Guo H, Wang D, Tu R, Qing G, Liu H. Genome-wide analysis identifies Rag1 and Rag2 as novel Notch1 transcriptional targets in thymocytes. Front Cell Dev Biol (2021) 9:703338. doi: 10.3389/fcell.2021.703338
54. Spicuglia S, Pekowska A, Zacarias-Cabeza J, Ferrier P. Epigenetic control of tcrb gene rearrangement. Semin Immunol (2010) 22(6):330–6. doi: 10.1016/j.smim.2010.07.002
55. Ebert PJ, Li QJ, Huppa JB, Davis MM. Functional development of the T cell receptor for antigen. Prog Mol Biol Transl Sci (2010) 92:65–100. doi: 10.1016/S1877-1173(10)92004-8
56. Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol (2011) 11(4):251–63. doi: 10.1038/nri2941
57. Krangel MS. T Cell development: better living through chromatin. Nat Immunol (2007) 8(7):687–94. doi: 10.1038/ni1484
58. Yancopoulos GD, Blackwell TK, Suh H, Hood L, Alt FW. Introduced T cell receptor variable region gene segments recombine in pre-b cells: evidence that b and T cells use a common recombinase. Cell (1986) 44(2):251–9. doi: 10.1016/0092-8674(86)90759-2
59. Stanhope-Baker P, Hudson KM, Shaffer AL, Constantinescu A, Schlissel MS. Cell type-specific chromatin structure determines the targeting of V(D)J recombinase activity in vitro. Cell (1996) 85(6):887–97. doi: 10.1016/s0092-8674(00)81272-6
60. McMurry MT, Krangel MS. A role for histone acetylation in the developmental regulation of VDJ recombination. Science (2000) 287(5452):495–8. doi: 10.1126/science.287.5452.495
61. Jhunjhunwala S, van Zelm MC, Peak MM, Murre C. Chromatin architecture and the generation of antigen receptor diversity. Cell (2009) 138(3):435–48. doi: 10.1016/j.cell.2009.07.016
62. Ishihara S, Sasagawa Y, Kameda T, Yamashita H, Umeda M, Kotomura N, et al. Local states of chromatin compaction at transcription start sites control transcription levels. Nucleic Acids Res (2021) 49(14):8007–23. doi: 10.1093/nar/gkab587
63. Hilbert L, Sato Y, Kuznetsova K, Bianucci T, Kimura H, Jülicher F, et al. Transcription organizes euchromatin via microphase separation. Nat Commun (2021) 12(1):1360. doi: 10.1038/s41467-021-21589-3
64. Ranganath S, Carpenter AC, Gleason M, Shaw AC, Bassing CH, Alt FW. Productive coupling of accessible Vbeta14 segments and DJbeta complexes determines the frequency of Vbeta14 rearrangement. J Immunol (2008) 180(4):2339–46. doi: 10.4049/jimmunol.180.4.2339
65. Jaeger S, Lima R, Meyroneinc A, Bonnet M, Ugalde E, Ferrier P. A dynamical model of TCRβ gene recombination: Coupling the initiation of dβ-jβ rearrangement to TCRβ allelic exclusion. bioRxiv. doi: 10.1101/200444
66. Abarrategui I, Krangel MS. Noncoding transcription controls downstream promoters to regulate T-cell receptor alpha recombination. EMBO J (2007) 26(20):4380–90. doi: 10.1038/sj.emboj.7601866
67. Bolland DJ, Wood AL, Corcoran AE. Large-Scale chromatin remodeling at the immunoglobulin heavy chain locus: a paradigm for multigene regulation. Adv Exp Med Biol (2009) 650:59–72. doi: 10.1007/978-1-4419-0296-2_5
68. Ji Y, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell (2010) 141(3):419–31. doi: 10.1016/j.cell.2010.03.010
69. Liu Y, Subrahmanyam R, Chakraborty T, Sen R, Desiderio S. A plant homeodomain in RAG-2 that binds hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity (2007) 27(4):561–71. doi: 10.1016/j.immuni.2007.09.005
70. Matthews AG, Kuo AJ, Ramón-Maiques S, Han S, Champagne KS, Ivanov D, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature (2007) 450(7172):1106–10. doi: 10.1038/nature06431
71. Ramón-Maiques S, Kuo AJ, Carney D, Matthews AG, Oettinger MA, Gozani O, et al. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc Natl Acad Sci U S A (2007) 104(48):18993–8. doi: 10.1073/pnas.0709170104
72. Shimazaki N, Tsai AG, Lieber MR. H3K4me3 stimulates the V(D)J RAG complex for both nicking and hairpinning in trans in addition to tethering in cis: implications for translocations. Mol Cell (2009) 34(5):535–44. doi: 10.1016/j.molcel.2009.05.011
73. Grazini U, Zanardi F, Citterio E, Casola S, Goding CR, McBlane F. The RING domain of RAG1 ubiquitylates histone H3: a novel activity in chromatin-mediated regulation of V(D)J joining. J Mol Cell (2010) 37(2):282–93. doi: 10.1016/j.molcel.2009.12.035
74. Ono M. Control of regulatory T-cell differentiation and function by T-cell receptor signalling and Foxp3 transcription factor complexes. Immunology (2020) 160(1):24–37. doi: 10.1111/imm.13178
75. Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity (2009) 31(6):921–31. doi: 10.1016/j.immuni.2009.09.022
76. Zhang L, Xiao X, Arnold PR, Li XC. Transcriptional and epigenetic regulation of immune tolerance: roles of the NF-κB family members. Cell Mol Immunol (2019) 16(4):315–23. doi: 10.1038/s41423-019-0202-8
77. Papoutsopoulou S, Campbell BJ. Epigenetic modifications of the nuclear factor kappa b signalling pathway and its impact on inflammatory bowel disease. Curr Pharm Des (2021) 27(35):3702–13. doi: 10.2174/1381612827666210218141847
78. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol (2012) 12(5):325–38. doi: 10.1038/nri3198
79. Marcais A, Blevins R, Graumann J, Feytout A, Dharmalingam G, Carroll T, et al. microRNA-mediated regulation of mTOR complex components facilitates discrimination between activation and anergy in CD4 T cells. J Exp Med (2014) 211(11):2281–95. doi: 10.1084/jem.20132059
80. Singh Y, Garden OA, Lang F, Cobb BS. MicroRNA-15b/16 enhances the induction of regulatory T cells by regulating the expression of rictor and mTOR. J Immunol (2015) 195(12):5667–77. doi: 10.4049/jimmunol.1401875
81. Schamel WW, Alarcon B, Höfer T, Minguet S. The allostery model of TCR regulation. J Immunol (2017) 198(1):47–52. doi: 10.4049/jimmunol.1601661
82. Swamy M, Beck-Garcia K, Beck-Garcia E, Hartl FA, Morath A, Yousefi OS, et al. A cholesterol-based allostery model of T cell receptor phosphorylation. Immunity (2016) 44(5):1091–101. doi: 10.1016/j.immuni.2016.04.011
83. Pathan-Chhatbar S, Drechsler C, Richter K, Morath A, Wu W, OuYang B, et al. Direct regulation of the T cell antigen receptor's activity by cholesterol. Front Cell Dev Biol (2021) 8. doi: 10.3389/fcell.2020.615996
84. Zhou X, Paulsson G, Stemme S, Hansson GK. Hypercholesterolemia is associated with a T helper (Th) 1/Th2 switch of the autoimmune response in atherosclerotic apo e-knockout mice. J Clin Invest (1998) 101(8):1717–25. doi: 10.1172/JCI1216
85. Steiner G, Urowitz MB. Lipid profiles in patients with rheumatoid arthritis: mechanisms and the impact of treatment. Semin Arthritis Rheum (2009) 38(5):372–81. doi: 10.1016/j.semarthrit.2008.01.015
86. Yuan J, Li LI, Wang Z, Song W, Zhang Z. Dyslipidemia in patients with systemic lupus erythematosus: Association with disease activity and b-type natriuretic peptide levels. BioMed Rep (2016) 4(1):68–72. doi: 10.3892/br.2015.544
87. Ghazizadeh R, Tosa M, Ghazizadeh M. Clinical improvement in psoriasis with treatment of associated hyperlipidemia. Am J Med Sci (2011) 341(5):394–8. doi: 10.1097/MAJ.0b013e3181ff8eeb
88. Qiu J, Wu B, Goodman SB, Berry GJ, Goronzy JJ, Weyand CM. Metabolic control of autoimmunity and tissue inflammation in rheumatoid arthritis. Front Immunol (2021) 12:652771. doi: 10.3389/fimmu.2021.652771
89. Giera M, Ioan-Facsinay A, Toes R, Gao F, Dalli J, Deelder AM, et al. Lipid and lipid mediator profiling of human synovial fluid in rheumatoid arthritis patients by means of LC-MS/MS. Biochim Biophys Acta (2012) 1821(11):1415–24. doi: 10.1016/j.bbalip.2012.07.011
90. Baardman J, Lutgens E. Regulatory T cell metabolism in atherosclerosis. Metabolites (2020) 10(7):279. doi: 10.3390/metabo10070279
91. Li X, Xiao H, Jian X, Zhang X, Zhang H, Mu Y, et al. Epigenetic regulation of key enzymes CYP7a1 and HMGCR affect hepatic cholesterol metabolism in different breeds of piglets. Front Vet Sci (2020) 7. doi: 10.3389/fvets.2020.00231
92. Singh AK, Aryal B, Zhang X, Fan Y, Price NL, Suárez Y, et al. Posttranscriptional regulation of lipid metabolism by non-coding RNAs and RNA binding proteins. Semin Cell Dev Biol (2018) 81:81:129–140. doi: 10.1016/j.semcdb.2017.11.026
93. Lee KH, Hwang HJ, Cho JY. Long non-coding RNA associated with cholesterol homeostasis and its involvement in metabolic diseases. Int J Mol Sci (2020) 21(21):8337. doi: 10.3390/ijms21218337
94. Clark M, Kroger CJ, Ke Q, Tisch RM. The role of T cell receptor signaling in the development of type 1 diabetes. Front Immunol (2021) 11:615371. doi: 10.3389/fimmu.2020.615371
95. Weng NP, Araki Y, Subedi K. The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation. Nat Rev Immunol (2012) 12(4):306–15. doi: 10.1038/nri3173
96. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8+ T cell differentiation. Nat Rev Immunol (2018) 18(5):340–56. doi: 10.1038/nri.2017.146
97. Le Menn G, Jabłońska A, Chen Z. The effects of post-translational modifications on Th17/Treg cell differentiation. Biochim Biophys Acta Mol Cell Res (2022) 1869(6):119223. doi: 10.1016/j.bbamcr.2022.119223
98. Montacchiesi G, Pace L. Epigenetics and CD8+ T cell memory. Immunol Rev (2022) 305(1):77–89. doi: 10.1111/imr.13057
99. He S, Liu Y, Meng L, Sun H, Wang Y, Ji Y, et al. Ezh2 phosphorylation state determines its capacity to maintain CD8+ T memory precursors for antitumor immunity. Nat Commun (2017) 8(1):2125. doi: 10.1038/s41467-017-02187-8
100. DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity (2015) 42(2):227–38. doi: 10.1016/j.immuni.2015.01.007
101. Jerram ST, Dang MN, Leslie RD. The role of epigenetics in type 1 diabetes. Curr Diabetes Rep (2017) 17(10):89. doi: 10.1007/s11892-017-0916-x
102. Kawasaki E. Type 1 diabetes and autoimmunity. Clin Pediatr Endocrinol (2014) 23(4):99–105. doi: 10.1297/cpe.23.99
103. Karavanaki K, Karayianni C, Vassiliou I, Tzanela M, Sdogou T, Kakleas K, et al. Multiple autoimmunity, type 1 diabetes (T1DM), autoimmune thyroiditis and thyroid cancer: is there an association? a case report and literature review. J Pediatr Endocrinol Metab (2014) 27(9-10):1011–6. doi: 10.1515/jpem-2013-0370
104. Fasolino M, Goldman N, Wang W, Cattau B, Zhou Y, Petrovic J, et al. Genetic variation in type 1 diabetes reconfigures the 3D chromatin organization of T cells and alters gene expression. Immunity (2020) 52(2):257–274.e11. doi: 10.1016/j.immuni.2020.01.003
105. Orozco G. Fine mapping with epigenetic information and 3D structure. Semin Immunopathol (2022) 44(1):115–25. doi: 10.1007/s00281-021-00906-4
106. Caliskan M, Brown CD, Maranville JC. A catalog of GWAS fine-mapping efforts in autoimmune disease. Am J Hum Genet (2021) 108(4):549–63. doi: 10.1016/j.ajhg.2021.03.009
107. Li C, Ebert PJ, Li QJ. T Cell receptor (TCR) and transforming growth factor β (TGF-β) signaling converge on DNA (cytosine-5)-methyltransferase to control forkhead box protein 3 (foxp3) locus methylation and inducible regulatory T cell differentiation. J Biol Chem (2013) 288(26):19127–39. doi: 10.1074/jbc.M113.453357
108. Sun X, Cui Y, Feng H, Liu H, Liu X. TGF-β signaling controls Foxp3 methylation and T reg cell differentiation by modulating Uhrf1 activity. J Exp Med (2019) 216(12):2819–37. doi: 10.1084/jem.20190550
109. Molinero LL, Miller ML, Evaristo C, Alegre ML. High TCR stimuli prevent induced regulatory T cell differentiation in a NF-κB-dependent manner. J Immunol (2011) 186(8):4609–17. doi: 10.4049/jimmunol.1002361
110. Guz J, Oliński R. The role of vitamin c in epigenetic regulation. Postepy Hig Med Dosw (Online) (2017) 71(1):747–60. doi: 10.5604/01.3001.0010.3853
111. Camarena V, Wang G. The epigenetic role of vitamin c in health and disease. Cell Mol Life Sci (2016) 73(8):1645–58. doi: 10.1007/s00018-016-2145-x
112. Peters C, Kouakanou L, Kabelitz D. A comparative view on vitamin c effects on αβ- versus γδ T-cell activation and differentiation. J Leukoc Biol (2020) 107(6):1009–22. doi: 10.1002/JLB.1MR1219-245R
113. Pehlivan FE. Vitamin c: An epigenetic regulator. In: LeBlanc JG, editor. Vitamin c - an update on current uses and functions [Internet]. London: IntechOpen (2018). Available at: https://www.intechopen.com/chapters/64868.
114. Oelke K, Richardson B. Decreased T cell ERK pathway signaling may contribute to the development of lupus through effects on DNA methylation and gene expression. Int Rev Immunol (2004) 23(3-4):315–31. doi: 10.1080/08830180490452567
115. Wang Z, Chang C, Lu Q. Epigenetics of CD4+ T cells in autoimmune diseases. Curr Opin Rheumatol (2017) 29(4):361–8. doi: 10.1097/BOR.0000000000000393
116. Ulrich-Merzenich G, Zeitler H, Panek D, Bokemeyer D, Vetter H. Vitamin c promotes human endothelial cell growth via the ERK-signaling pathway. Eur J Nutr (2007) 46(2):87–94. doi: 10.1007/s00394-006-0636-5
117. Likhitweerawong N, Boonchooduang N, Morakote W, Louthrenoo O. Scurvy mimicking as systemic lupus erythematosus. BMJ Case Rep (2021) 14(6):e242958. doi: 10.1136/bcr-2021-242958
118. Li Z, Li D, Tsun A, Li B. FOXP3+ regulatory T cells and their functional regulation. Cell Mol Immunol (2015) 12(5):558–65. doi: 10.1038/cmi.2015.10
119. Chorro L, Suzuki M, Chin SS, Williams TM, Snapp EL, Odagiu L, et al. Interleukin 2 modulates thymic-derived regulatory T cell epigenetic landscape. Nat Commun (2018) 9(1):5368. doi: 10.1038/s41467-018-07806-6
120. Kitagawa Y, Ohkura N, Sakaguchi S. Epigenetic control of thymic treg-cell development. Eur J Immunol (2015) 45(1):11–6. doi: 10.1002/eji.201444577
121. Kondo K, Ohigashi I, Takahama Y. Thymus machinery for T-cell selection. Int Immunol (2019) 31(3):119–25. doi: 10.1093/intimm/dxy081
122. Berkley AM, Hendricks DW, Simmons KB, Fink PJ. Recent thymic emigrants and mature naive T cells exhibit differential DNA methylation at key cytokine loci. J Immunol (2013) 190(12):6180–6. doi: 10.4049/jimmunol.1300181
123. Scheer S, Runting J, Bramhall M, Russ B, Zaini A, Ellemor J, et al. The methyltransferase DOT1L controls activation and lineage integrity in CD4+ T cells during infection and inflammation. Cell Rep (2020) 33(11):108505. doi: 10.1016/j.celrep.2020.108505
124. Romagnani S. Th1/Th2 cells. Inflammation Bowel Dis (1999) 5(4):285–94. doi: 10.1097/00054725-199911000-00009
125. Wang G, Su Z, Li H, Xiao L, Li C, Lian G. The role of metabolism in Th17 cell differentiation and autoimmune diseases. Int Immunopharmacol (2022) 103:108450. doi: 10.1016/j.intimp.2021.108450
126. Pollard KM, Cauvi DM, Toomey CB, Morris KV, Kono DH. Interferon-γ and systemic autoimmunity. Discovery Med (2013) 16(87):123–31.
127. Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun (2008) 31(3):252–6. doi: 10.1016/j.jaut.2008.04.017
128. Ozdemir C, Akdis M, Akdis CA. T Regulatory cells and their counterparts: masters of immune regulation. Clin Exp Allergy (2009) 39(5):626–39. doi: 10.1111/j.1365-2222.2009.03242.x
129. Renaude E, Kroemer M, Loyon R, Binda D, Borg C, Guittaut M, et al. The fate of Th17 cells is shaped by epigenetic modifications and remodeled by the tumor microenvironment. Int J Mol Sci (2020) 21(5):1673. doi: 10.3390/ijms21051673
130. Yang BH, Floess S, Hagemann S, Deyneko IV, Groebe L, Pezoldt J, et al. Development of a unique epigenetic signature during in vivo Th17 differentiation. Nucleic Acids Res (2015) 43(3):1537–48. doi: 10.1093/nar/gkv014
131. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun (2018) 87:1–15. doi: 10.1016/j.jaut.2017.12.007
132. Shin BY, Lee SH, Kim Y, An J, Park TY, Lee SK. Interactomic inhibition of eomes in the nucleus alleviates EAE via blocking the conversion of Th17 cells into non-classic Th1 cells. Immunol Med (2022) 7:1–9. doi: 10.1080/25785826.2022.2031812
133. Bellanti JA, Li D. Treg cells and epigenetic regulation. Adv Exp Med Biol (2021) 1278:95–114. doi: 10.1007/978-981-15-6407-9_6
134. Colamatteo A, Carbone F, Bruzzaniti S, Galgani M, Fusco C, Maniscalco GT, et al. Molecular mechanisms controlling Foxp3 expression in health and autoimmunity: From epigenetic to post-translational regulation. Front Immunol (2020) 10:3136. doi: 10.3389/fimmu.2019.03136
135. Huehn J, Beyer M. Epigenetic and transcriptional control of Foxp3+ regulatory T cells. Semin Immunol (2015) 27(1):10–8. doi: 10.1016/j.smim.2015.02.002
136. Morikawa H, Sakaguchi S. Genetic and epigenetic basis of treg cell development and function: from a FoxP3-centered view to an epigenome-defined view of natural treg cells. Immunol Rev (2014) 259(1):192–205. doi: 10.1111/imr.12174
137. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463(7282):808–12. doi: 10.1038/nature08750
138. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol (2018) 19(7):665–73. doi: 10.1038/s41590-018-0120-4
139. Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol (2012) 13(10):1010–9. doi: 10.1038/ni.2402
140. Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol (2017) 17(11):703–17. doi: 10.1038/nri.2017.75
141. Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell (2012) 151(1):153–66. doi: 10.1016/j.cell.2012.06.053
142. Watts D, Janßen M, Jaykar M, Palmucci F, Weigelt M, Petzold C, et al. Transient depletion of Foxp3+ regulatory T cells selectively promotes aggressive β cell autoimmunity in genetically susceptible DEREG mice. Front Immunol (2021) 12:720133. doi: 10.3389/fimmu.2021.720133
143. Göschl L, Scheinecker C, Bonelli M. Treg cells in autoimmunity: from identification to treg-based therapies. Semin Immunopathol (2019) 41(3):301–14. doi: 10.1007/s00281-019-00741-8
145. Di Marco M, Ramassone A, Pagotto S, Anastasiadou E, Veronese A, Visone R. MicroRNAs in autoimmunity and hematological malignancies. Int J Mol Sci (2018) 19(10):3139. doi: 10.3390/ijms19103139
146. Yapijakis C. Regulatory role of MicroRNAs in brain development and function. Adv Exp Med Biol (2020) 1195:237–47. doi: 10.1007/978-3-030-32633-3_32
147. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell (2005) 120(1):15–20. doi: 10.1016/j.cell.2004.12.035
148. Liston A, Lu LF, O'Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med (2008) 205(9):1993–2004. doi: 10.1084/jem.20081062
149. Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, et al. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med (2008) 205(9):1983–91. doi: 10.1084/jem.20080707
150. Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, et al. Function of miR-146a in controlling treg cell-mediated regulation of Th1 responses. Cell (2010) 142:914–29. doi: 10.1016/j.cell.2010.08.012
151. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med (2011) 208:1189–201. doi: 10.1084/jem.20101823
152. Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity (2009) 30:80–91. doi: 10.1016/j.immuni.2008.11.010
153. Beyer M, Thabet Y, Muller RU, Sadlon T, Classen S, Lahl K, et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat Immunol (2011) 12:898–907. doi: 10.1038/ni.2084
154. Sadlon TJ, Wilkinson BG, Pederson S, Brown CY, Bresatz S, Gargett T, et al. Genome-wide identification of human FOXP3 target genes in natural regulatory T cells. J Immunol (2010) 185:1071–81. doi: 10.4049/jimmunol.1000082
155. Rouas R, Fayyad-Kazan H, El Zein N, Lewalle P, Rothé F, Simion A, et al. Human natural treg microRNA signature: role of microRNA-31 and microRNA-21 in FOXP3 expression. Eur J Immunol (2009) 9(6):1608–18. doi: 10.1002/eji.200838509
156. Fayyad-Kazan H, Rouas R, Fayyad-Kazan M, Badran R, El Zein N, Lewalle P, et al. MicroRNA profile of circulating CD4-positive regulatory T cells in human adults and impact of differentially expressed microRNAs on expression of two genes essential to their function. J Biol Chem (2012) 287(13):9910–22. doi: 10.1074/jbc.M111.337154
157. Yang HY, Barbi J, Wu CY, Zheng Y, Vignali PD, Wu X, et al. MicroRNA-17 modulates regulatory T cell function by targeting Co-regulators of the Foxp3 transcription factor. Immunity (2016) 45(1):83–93. doi: 10.1016/j.immuni.2016.06.022
158. Liu X, Robinson SN, Setoyama T, Tung SS, D'Abundo L, Shah MY, et al. FOXP3 is a direct target of miR15a/16 in umbilical cord blood regulatory T cells. Bone Marrow Transpl (2014) 49(6):793–9. doi: 10.1038/bmt.2014.57
159. Pickart CM. Ubiquitin enters the new millennium. Mol Cell (2001) 8(3):499–504. doi: 10.1016/s1097-2765(01)00347-1
160. Yang X, Lun Y, Jiang H, Liu X, Duan Z, Xin S, et al. SIRT1-regulated abnormal acetylation of FOXP3 induces regulatory T-cell function defect in hashimoto's thyroiditis. Thyroid (2018) 28(2):246–56. doi: 10.1089/thy.2017.0286
161. Chen L, Wu J, Pier E, Zhao Y, Shen Z. mTORC2-PKBα/Akt1 serine 473 phosphorylation axis is essential for regulation of FOXP3 stability by chemokine CCL3 in psoriasis. J Invest Dermatol (2013) 133(2):418–28. doi: 10.1038/jid.2012.333
162. Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol (2009) 9(2):83–9. doi: 10.1038/nri2474
163. Richardson B. Primer: epigenetics of autoimmunity. Nat Clin Pract Rheumatol (2007) 3(9):521–7. doi: 10.1038/ncprheum0573
164. Mazzone R, Zwergel C, Artico M, Taurone S, Ralli M, Greco A, et al. The emerging role of epigenetics in human autoimmune disorders. Clin Epigenet (2019) 11(1):34. doi: 10.1186/s13148-019-0632-2
165. Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S, et al. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheumatol (2011) 63(5):1376–86. doi: 10.1002/art.30196
166. Wu H, Chen S, Li A, Shen K, Wang S, Wang S, et al. LncRNA expression profiles in systemic lupus erythematosus and rheumatoid arthritis: Emerging biomarkers and therapeutic targets. Front Immunol (2021) 12:792884. doi: 10.3389/fimmu.2021.792884
167. Zhao CN, Mao YM, Liu LN, Li XM, Wang DG, Pan HF. Emerging role of lncRNAs in systemic lupus erythematosus. BioMed Pharmacother (2018) 106:584–92. doi: 10.1016/j.biopha.2018.06.175
168. Xu W, Wu Q, Huang A. Emerging role of LncRNAs in autoimmune lupus. Inflammation (2022) 45(3):937–948. doi: 10.1007/s10753-021-01607-8
169. Liu X, Lin J, Wu H, Wang Y, Xie L, Wu J, et al. A novel long noncoding RNA lincRNA00892 activates CD4+ T cells in systemic lupus erythematosus by regulating CD40L. Front Pharmacol (2021) 12:733902. doi: 10.3389/fphar.2021.733902
170. Taheri M, Eghtedarian R, Dinger ME, Ghafouri-Fard S. Exploring the role of non-coding RNAs in the pathophysiology of systemic lupus erythematosus. Biomolecules (2020) 10(6):937. doi: 10.3390/biom10060937
171. Yin L, Zeng C, Yao J, Shen J. Emerging roles for noncoding RNAs in autoimmune thyroid disease. Endocrinology (2020) 161(8):bqaa053. doi: 10.1210/endocr/bqaa053
172. Hur K, Kim SH, Kim JM. Potential implications of long noncoding RNAs in autoimmune diseases. Immune Netw (2019) 19(1):e4. doi: 10.4110/in.2019.19.e4
173. Wang J, Yan S, Yang J, Lu H, Xu D, Wang Z. Non-coding RNAs in rheumatoid arthritis: From bench to bedside. Front Immunol (2020) 10:3129. doi: 10.3389/fimmu.2019.03129
174. Xu F, Jin L, Jin Y, Nie Z, Zheng H. Long noncoding RNAs in autoimmune diseases. J BioMed Mater Res A (2019) 107(2):468–75. doi: 10.1002/jbm.a.36562
175. Ahn SH, Monsanto SP, Miller C, Singh SS, Thomas R, Tayade C. Pathophysiology and immune dysfunction in endometriosis. BioMed Res Int (2015) 2015:795976. doi: 10.1155/2015/795976
176. Zhang T, De Carolis C, Man GCW, Wang CC. The link between immunity, autoimmunity and endometriosis: a literature update. Autoimmun Rev (2018) 17(10):945–55. doi: 10.1016/j.autrev.2018.03.017
177. Herington JL, Bruner-Tran KL, Lucas JA, Osteen KG. Immune interactions in endometriosis. Expert Rev Clin Immunol (2011) 7(5):611–26. doi: 10.1586/eci.11.53
178. Laganà AS, Garzon S, Götte M, Viganò P, Franchi M, Ghezzi F, et al. The pathogenesis of endometriosis: Molecular and cell biology insights. Int J Mol Sci (2019) 20(22):5615. doi: 10.3390/ijms20225615
179. Koninckx PR, Ussia A, Adamyan L, Wattiez A, Gomel V, Martin DC. Pathogenesis of endometriosis: the genetic/epigenetic theory. Fertil Steril (2019) 111(2):327–40. doi: 10.1016/j.fertnstert.2018.10.013
180. Greenbaum H, Galper BL, Decter DH, Eisenberg VH. Endometriosis and autoimmunity: Can autoantibodies be used as a non-invasive early diagnostic tool? Autoimmun Rev (2021) 20(5):102795. doi: 10.1016/j.autrev.2021.102795
181. Anderson G. Endometriosis pathoetiology and pathophysiology: Roles of vitamin a, estrogen, immunity, adipocytes, gut microbiome and melatonergic pathway on mitochondria regulation. Biomol Concepts (2019) 10(1):133–49. doi: 10.1515/bmc-2019-0017
182. Moghaddam MZ, Ansariniya H, Seifati SM, Zare F, Fesahat F. Immunopathogenesis of endometriosis: An overview of the role of innate and adaptive immune cells and their mediators. Am J Reprod Immunol (2022) 87(5):e13537. doi: 10.1111/aji.13537. Epub ahead of print.
183. Kong Y, Shao Y, Ren C, Yang G. Endometrial stem/progenitor cells and their roles in immunity, clinical application, and endometriosis. Stem Cell Res Ther (2021) 12(1):474. doi: 10.1186/s13287-021-02526-z
184. Vanni VS, Villanacci R, Salmeri N, Papaleo E, Delprato D, Ottolina J, et al. Concomitant autoimmunity may be a predictor of more severe stages of endometriosis. Sci Rep (2021) 11(1):15372. doi: 10.1038/s41598-021-94877-z
185. Gajbhiye R, Bendigeri T, Ghuge A, Bhusane K, Begum S, Warty N, et al. Panel of autoimmune markers for noninvasive diagnosis of minimal-mild endometriosis. Reprod Sci (2017) 24(3):413–20. doi: 10.1177/1933719116657190
186. Han SJ, Jung SY, Wu SP, Hawkins SM, Park MJ, Kyo S, et al. Estrogen receptor β modulates apoptosis complexes and the inflammasome to drive the pathogenesis of endometriosis. Cell (2015) 163(4):960–74. doi: 10.1016/j.cell.2015.10.034
187. Chantalat E, Valera MC, Vaysse C, Noirrit E, Rusidze M, Weyl A, et al. Estrogen receptors and endometriosis. Int J Mol Sci (2020) 21(8):2815. doi: 10.3390/ijms21082815
188. Patel BG, Rudnicki M, Yu J, Shu Y, Taylor RN. Progesterone resistance in endometriosis: origins, consequences and interventions. Acta Obstet Gynecol Scand (2017) 96(6):623–32. doi: 10.1111/aogs.13156
189. García-Gómez E, Vázquez-Martínez ER, Reyes-Mayoral C, Cruz-Orozco OP, Camacho-Arroyo I, Cerbón M. Regulation of inflammation pathways and inflammasome by sex steroid hormones in endometriosis. Front Endocrinol (Lausanne) (2020) 10:935. doi: 10.3389/fendo.2019.00935
190. Chandrashekara S. The treatment strategies of autoimmune disease may need a different approach from conventional protocol: a review. Indian J Pharmacol (2012) 44(6):665–71. doi: 10.4103/0253-7613.103235
191. Vannuccini S, Clemenza S, Rossi M, Petraglia F. Hormonal treatments for endometriosis: The endocrine background. Rev Endocr Metab Disord (2022) 23(3):333–55. doi: 10.1007/s11154-021-09666-w.
192. Harding AT, Heaton NS. The impact of estrogens and their receptors on immunity and inflammation during infection. Cancers (Basel) (2022) 14(4):909. doi: 10.3390/cancers14040909
193. Harding AT, Goff MA, Froggatt HM, Lim JK, Heaton NS. GPER1 is required to protect fetal health from maternal inflammation. Science (2021) 371(6526):271–6. doi: 10.1126/science.aba9001
194. Pelekanou V, Kampa M, Kiagiadaki F, Deli A, Theodoropoulos P, Agrogiannis G, et al. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J Leukoc Biol (2016) 99(2):333–47. doi: 10.1189/jlb.3A0914-430RR
195. Toniolo A, Fadini GP, Tedesco S, Cappellari R, Vegeto E, Maggi A, et al. Alternative activation of human macrophages is rescued by estrogen treatment in vitro and impaired by menopausal status. J Clin Endocrinol Metab (2015) 100(1):E50–8. doi: 10.1210/jc.2014-2751
196. Rider V, Li X, Peterson G, Dawson J, Kimler BF, Abdou NI. Differential expression of estrogen receptors in women with systemic lupus erythematosus. J Rheumatol (2006) 33(6):1093–101.
197. Brandenberger AW, Lebovic DI, Tee MK, Ryan IP, Tseng JF, Jaffe RB, et al. Oestrogen receptor (ER)-alpha and ER-beta isoforms in normal endometrial and endometriosis-derived stromal cells. Mol Hum Reprod (1999) 5(7):651–5. doi: 10.1093/molehr/5.7.651
198. Simmen RC, Kelley AS. Reversal of fortune: estrogen receptor-β in endometriosis. J Mol Endocrinol (2016) 57(2):F23–7. doi: 10.1530/JME-16-0080
199. Xue Q, Lin Z, Cheng YH, Huang CC, Marsh E, Yin P, et al. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis. Biol Reprod (2007) 77(4):681–7. doi: 10.1095/biolreprod.107.061804
200. Vidal-Gómez X, Pérez-Cremades D, Mompeón A, Dantas AP, Novella S, Hermenegildo C. MicroRNA as crucial regulators of gene expression in estradiol-treated human endothelial cells. Cell Physiol Biochem (2018) 45(5):1878–92. doi: 10.1159/000487910
201. Lin Y, Xiao L, Zhang Y, Li P, Wu Y, Lin Y. MiR-26b-3p regulates osteoblast differentiation via targeting estrogen receptor α. Genomics (2019) 111(5):1089–96. doi: 10.1016/j.ygeno.2018.07.003
202. Pandey DP, Picard D. miR-22 inhibits estrogen signaling by directly targeting the estrogen receptor alpha mRNA. Mol Cell Biol (2009) 29(13):3783–90. doi: 10.1128/MCB.01875-08
203. He SZ, Li J, Bao HC, Wang MM, Wang XR, Huang X, et al. G Protein coupled estrogen receptor/miR 148a/human leukocyte antigen G signaling pathway mediates cell apoptosis of ovarian endometriosis. Mol Med Rep (2018) 18(1):1141–8. doi: 10.3892/mmr.2018.9039
204. Yan W, Hu H, Tang B. Progress in understanding the relationship between long noncoding RNA and endometriosis. Eur J Obstet Gynecol Reprod Biol X. (2019) 5:100067. doi: 10.1016/j.eurox.2019.100067
205. Bhan A, Hussain I, Ansari KI, Kasiri S, Bashyal A, Mandal SS. Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol. J Mol Biol (2013) 425(19):3707–22. doi: 10.1016/j.jmb.2013.01.022
206. Taheri M, Shoorei H, Dinger ME, Ghafouri-Fard S. Perspectives on the role of non-coding RNAs in the regulation of expression and function of the estrogen receptor. Cancers (Basel) (2020) 12(8):2162. doi: 10.3390/cancers12082162
207. Fazleabas AT, Braundmeier A, Parkin K. Endometriosis-induced changes in regulatory T cells - insights towards developing permanent contraception. Contraception (2015) 92(2):116–9. doi: 10.1016/j.contraception.2015.06.006
208. Vargas E, García-Moreno E, Aghajanova L, Salumets A, Horcajadas JA, Esteban FJ, et al. The mid-secretory endometrial transcriptomic landscape in endometriosis: a meta-analysis. Hum Reprod Open (2022) 2022(2):hoac016. doi: 10.1093/hropen/hoac016
209. Houshdaran S, Nezhat CR, Vo KC, Zelenko Z, Irwin JC, Giudice LC. Aberrant endometrial DNA methylome and associated gene expression in women with endometriosis. Biol Reprod (2016) 95(5):93. doi: 10.1095/biolreprod.116.140434
210. Dyson MT, Roqueiro D, Monsivais D, Ercan CM, Pavone ME, Brooks DC, et al. Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PloS Genet (2014) 10(3):e1004158. doi: 10.1371/journal.pgen.1004158
211. Koukoura O, Sifakis S, Spandidos DA. DNA Methylation in endometriosis (Review). Mol Med Rep (2016) 13:2939–48. doi: 10.3892/mmr.2016.4925
212. Wu Y, Strawn E, Basir Z, Halverson G, Guo SW. Aberrant expression of deoxyribonucleic acid methyltransferases DNMT1, DNMT3A, and DNMT3B in women with endometriosis. Fertil Steril (2007) 87(1):24–32. doi: 10.1016/j.fertnstert.2006.05.077
213. Szczepańska M, Wirstlein P, Skrzypczak J, Jagodziński PP. Expression of HOXA11 in the mid-luteal endometrium from women with endometriosis-associated infertility. Reprod Biol Endocrinol (2012) 10:1. doi: 10.1186/1477-7827-10-1
214. van Kaam KJ, Delvoux B, Romano A, D'Hooghe T, Dunselman GA, Groothuis PG. Deoxyribonucleic acid methyltransferases and methyl-CpG-binding domain proteins in human endometrium and endometriosis. Fertil Steril (2011) 95(4):1421–7. doi: 10.1016/j.fertnstert.2011.01.031
215. Izawa M, Taniguchi F, Harada T. GATA6 expression promoted by an active enhancer may become a molecular marker in endometriosis lesions. Am J Reprod Immunol (2019) 81(2):e13078. doi: 10.1111/aji.13078
216. Chen H, Malentacchi F, Fambrini M, Harrath AH, Huang H, Petraglia F. Epigenetics of estrogen and progesterone receptors in endometriosis. Reprod Sci (2020) 27(11):1967–74. doi: 10.1007/s43032-020-00226-2
217. Xiaomeng X, Ming Z, Jiezhi M, Xiaoling F. Aberrant histone acetylation and methylation levels in woman with endometriosis. Arch Gynecol Obstet (2013) 287(3):487–94. doi: 10.1007/s00404-012-2591-0
218. Colón-Caraballo M, Monteiro JB, Flores I. H3K27me3 is an epigenetic mark of relevance in endometriosis. Reprod Sci (2015) 22(9):1134–42. doi: 10.1177/1933719115578924
219. Monteiro JB, Colón-Díaz M, García M, Gutierrez S, Colón M, Seto E, et al. Endometriosis is characterized by a distinct pattern of histone 3 and histone 4 lysine modifications. Reprod Sci (2014) 21(3):305–18. doi: 10.1177/1933719113497267
220. Nasu K, Kawano Y, Kai K, Aoyagi Y, Abe W, Okamoto M, et al. Aberrant histone modification in endometriosis. Front Biosci (Landmark Ed) (2014) 19(8):1202–14. doi: 10.2741/4276
221. Chopyak VV, Koval HD, Havrylyuk AM, Lishchuk-Yakymovych KA, Potomkina HA, Kurpisz MK. Immunopathogenesis of endometriosis - a novel look at an old problem. Cent Eur J Immunol (2022) 47(1):109–16. doi: 10.5114/ceji.2022.113830
222. Schwenck J, Mehling R, Thaiss WM, Kramer D, Menendez IG, Öz HH, et al. Temporal dynamics of reactive oxygen and nitrogen species and NF-κB activation during acute and chronic T cell-driven inflammation. Mol Imaging Biol (2020) 22(3):504–14. doi: 10.1007/s11307-019-01412-8
223. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res (2018) 122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401
224. Wang W, Vilella F, Alama P, Moreno I, Mignardi M, Isakova A, et al. Single-cell transcriptomic atlas of the human endometrium during the menstrual cycle. Nat Med (2020) 26(10):1644–53. doi: 10.1038/s41591-020-1040-z
225. Zhang X, Li Y, Chen X, Jin B, Shu C, Ni W, et al. Single-cell transcriptome analysis uncovers the molecular and cellular characteristics of thin endometrium. FASEB J (2022) 36(3):e22193. doi: 10.1096/fj.202101579R
226. Bunis DG, Wang W, Vallvé-Juanico J, Houshdaran S, Sen S, Ben Soltane I, et al. Whole-tissue deconvolution and scRNAseq analysis identify altered endometrial cellular compositions and functionality associated with endometriosis. Front Immunol (2022) 12:788315. doi: 10.3389/fimmu.2021.788315
227. Garcia-Alonso L, Handfield LF, Roberts K, Nikolakopoulou K, Fernando RC, Gardner L, et al. Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro. Nat Genet (2021) 53(12):1698–711. doi: 10.1038/s41588-021-00972-2
228. Ma J, Zhang L, Zhan H, Mo Y, Ren Z, Shao A, et al. Single-cell transcriptomic analysis of endometriosis provides insights into fibroblast fates and immune cell heterogeneity. Cell Biosci (2021) 11(1):125. doi: 10.1186/s13578-021-00637-x
229. Riccio LDGC, Santulli P, Marcellin L, Abrão MS, Batteux F, Chapron C. Immunology of endometriosis. Best Pract Res Clin Obstet Gynaecol (2018) 50:39–49. doi: 10.1016/j.bpobgyn.2018.01.010
230. Agostinis C, Balduit A, Mangogna A, Zito G, Romano F, Ricci G, et al. Immunological basis of the endometriosis: The complement system as a potential therapeutic target. Front Immunol (2021) 11:599117. doi: 10.3389/fimmu.2020.599117
231. de Barros IBL, Malvezzi H, Gueuvoghlanian-Silva BY, Piccinato CA, Rizzo LV, Podgaec S. "What do we know about regulatory T cells and endometriosis? a systematic review". J Reprod Immunol (2017) 120:48–55. doi: 10.1016/j.jri.2017.04.003
232. Osuga Y, Koga K, Hirota Y, Hirata T, Yoshino O, Taketani Y. Lymphocytes in endometriosis. Am J Reprod Immunol (2011) 65(1):1–10. doi: 10.1111/j.1600-0897.2010.00887.x
233. Symons LK, Miller JE, Kay VR, Marks RM, Liblik K, Koti M, et al. The immunopathophysiology of endometriosis. Trends Mol Med (2018) 24(9):748–62. doi: 10.1016/j.molmed.2018.07.004
234. Sbracia M, McKinnon B, Scarpellini F, Marconi D, Rossi G, Simmilion C, et al. PreImplantation factor in endometriosis: A potential role in inducing immune privilege for ectopic endometrium. PloS One (2017) 12(9):e0184399. doi: 10.1371/journal.pone.0184399
235. Witz CA, Montoya IA, Dey TD, Schenken RS. Characterization of lymphocyte subpopulations and T cell activation in endometriosis. Am J Reprod Immunol (1994) 32(3):173–9. doi: 10.1111/j.1600-0897.1994.tb01110.x
236. Haddadi MH, Negahdari B. Clinical and diagnostic potential of regulatory T cell markers: From bench to bedside. Transpl Immunol (2022) 70:101518. doi: 10.1016/j.trim.2021.101518
237. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203(7):1701–11. doi: 10.1084/jem.20060772
238. Cho YJ, Lee SH, Park JW, Han M, Park MJ, Han SJ. Dysfunctional signaling underlying endometriosis: current state of knowledge. J Mol Endocrinol (2018) 60(3):R97–R113. doi: 10.1530/JME-17-0227
239. Takamura M, Koga K, Izumi G, Hirata T, Harada M, Hirota Y, et al. Simultaneous detection and evaluation of four subsets of CD4+ T lymphocyte in lesions and peripheral blood in endometriosis. Am J Reprod Immunol (2015) 74(6):480–6. doi: 10.1111/aji.12426
240. Maurer-Alcalá XX, Katz LA. An epigenetic toolkit allows for diverse genome architectures in eukaryotes. Curr Opin Genet Dev (2015) 35:93–9. doi: 10.1016/j.gde.2015.10.005
241. Stein GS, Stein JL, Van Wijnen AJ, Lian JB, Montecino M, Croce CM, et al. Transcription factor-mediated epigenetic regulation of cell growth and phenotype for biological control and cancer. Adv Enzyme Regul (2010) 50(1):160–7. doi: 10.1016/j.advenzreg.2009.10.026
242. Chen S, Zhang J, Huang C, Lu W, Liang Y, Wan X. Expression of the T regulatory cell transcription factor FoxP3 in peri-implantation phase endometrium in infertile women with endometriosis. Reprod Biol Endocrinol (2012) 10:34. doi: 10.1186/1477-7827-10-34
243. Podgaec S, Rizzo LV, Fernandes LF, Baracat EC, Abrao MS. CD4(+) CD25(high) Foxp3(+) cells increased in the peritoneal fluid of patients with endometriosis. Am J Reprod Immunol (2012) 68(4):301–8. doi: 10.1111/j.1600-0897.2012.01173.x
244. Olkowska-Truchanowicz J, Bocian K, Maksym RB, Białoszewska A, Włodarczyk D, Baranowski W, et al. CD4+ CD25+ FOXP3+ regulatory T cells in peripheral blood and peritoneal fluid of patients with endometriosis. Hum Reprod (2013) 28(1):119–24. doi: 10.1093/humrep/des346
245. Basta P, Majka M, Jozwicki W, Lukaszewska E, Knafel A, Grabiec M, et al. The frequency of CD25+CD4+ and FOXP3+ regulatory T cells in ectopic endometrium and ectopic decidua. Reprod Biol Endocrinol (2010) 8:116. doi: 10.1186/1477-7827-8-116
246. Basta P, Koper K, Kazmierczak W, Wisniewski M, Makarewicz A, Dutsch-Wicherek M, et al. The biological role of treg cells in ectopic endometrium homeostasis. Histol Histopathol (2014) 29(10):1217–33. doi: 10.14670/HH-29.1217
247. Delbandi AA, Mahmoudi M, Shervin A, Moradi Z, Arablou T, Zarnani AH. Higher frequency of circulating, but not tissue regulatory T cells in patients with endometriosis. J Reprod Immunol (2020) 139:103119. doi: 10.1016/j.jri.2020.103119
248. Lužnik Z, Anchouche S, Dana R, Yin J. Regulatory T cells in angiogenesis. J Immunol (2020) 205(10):2557–65. doi: 10.4049/jimmunol.2000574
249. Wang XQ, Zhou WJ, Luo XZ, Tao Y, Li DJ. Synergistic effect of regulatory T cells and proinflammatory cytokines in angiogenesis in the endometriotic milieu. Hum Reprod (2017) 32(6):1304–17. doi: 10.1093/humrep/dex067
250. Tanaka Y, Mori T, Ito F, Koshiba A, Takaoka O, Kataoka H, et al. Exacerbation of endometriosis due to regulatory T-cell dysfunction. J Clin Endocrinol Metab (2017) 102(9):3206–17. doi: 10.1210/jc.2017-00052
251. Chung MS, Han SJ. Endometriosis-associated angiogenesis and anti-angiogenic therapy for endometriosis. Front Glob Womens Health (2022) 3:856316. doi: 10.3389/fgwh.2022.856316
252. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity (2013) 38(3):414–23. doi: 10.1016/j.immuni.2013.03.002
253. Li P, Liu C, Yu Z, Wu M. New insights into regulatory T cells: Exosome- and non-coding RNA-mediated regulation of homeostasis and resident treg cells. Front Immunol (2016) 7:574. doi: 10.3389/fimmu.2016.00574
254. Kitagawa Y, Ohkura N, Sakaguchi S. Molecular determinants of regulatory T cell development: the essential roles of epigenetic changes. Front Immunol (2013) 4:106. doi: 10.3389/fimmu.2013.00106
255. Slabe N, Meden-Vrtovec H, Verdenik I, Kosir-Pogacnik R, Ihan A. Cytotoxic T-cells in peripheral blood in women with endometriosis. Geburtshilfe Frauenheilkd (2013) 73(10):1042–8. doi: 10.1055/s-0033-1350702
256. Bolitho P, Voskoboinik I, Trapani JA, Smyth MJ. Apoptosis induced by the lymphocyte effector molecule perforin. Curr Opin Immunol (2007) 19(3):339–47. doi: 10.1016/j.coi.2007.04.007
257. Lu Q, Wu A, Ray D, Deng C, Attwood J, Hanash S, et al. DNA Methylation and chromatin structure regulate T cell perforin gene expression. J Immunol (2003) 170(10):5124–32. doi: 10.4049/jimmunol.170.10.5124
258. Singh M, Thakur M, Mishra M, Yadav M, Vibhuti R, Menon AM, et al. Gene regulation of intracellular adhesion molecule-1 (ICAM-1): A molecule with multiple functions. Immunol Lett (2021) 240:123–36. doi: 10.1016/j.imlet.2021.10.007
259. Aoudjit F, Bossé M, Stratowa C, Voraberger G, Audette M. Regulation of intercellular adhesion molecule-1 expression by retinoic acid: analysis of the 5' regulatory region of the gene. Int J Cancer (1994) 58(4):543–9. doi: 10.1002/ijc.2910580416
260. Voskoboinik I, Dunstone MA, Baran K, Whisstock JC, Trapani JA. Perforin: structure, function, and role in human immunopathology. Immunol Rev (2010) 235(1):35–54. doi: 10.1111/j.0105-2896.2010.00896.x
261. Schmitz T, Hoffmann V, Olliges E, Bobinger A, Popovici R, Nößner E, et al. Reduced frequency of perforin-positive CD8+ T cells in menstrual effluent of endometriosis patients. J Reprod Immunol (2021) 148:103424. doi: 10.1016/j.jri.2021.103424
262. van de Stolpe A, van der Saag PT. Intercellular adhesion molecule-1. J Mol Med (Berl) (1996) 74(1):13–33. doi: 10.1007/BF00202069
263. Kuessel L, Wenzl R, Proestling K, Balendran S, Pateisky P, Yotova 1st, et al. Soluble VCAM-1/soluble ICAM-1 ratio is a promising biomarker for diagnosing endometriosis. Hum Reprod (2017) 32(4):770–9. doi: 10.1093/humrep/dex028
264. Li R, Qiu Y. Diagnostic value of serum ICAM-1 for endometriosis: A meta-analysis. Med (Baltimore) (2018) 97(31):e11760. doi: 10.1097/MD.0000000000011760
265. Ha DY, Jung JS, Choi GH, Ji S. Polarization of human gingival fibroblasts by Th1-, Th2-, Th17-, and treg-derived cytokines. J Periodontal Res (2022) 7(3):487–501. doi: 10.1111/jre.12978.
266. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med (2009) 361(9):888–98. doi: 10.1056/NEJMra0707449
267. Brinkhoff A, Sieberichs A, Engler H, Dolff S, Benson S, Korth J, et al. Pro-inflammatory Th1 and Th17 cells are suppressed during human experimental endotoxemia whereas anti-inflammatory IL-10 producing T-cells are unaffected. Front Immunol (2018) 9:1133. doi: 10.3389/fimmu.2018.01133
268. Corthay A. How do regulatory T cells work? Scand J Immunol (2009) 70(4):326–36. doi: 10.1111/j.1365-3083.2009.02308.x
269. Zhang X, Xu H, Lin J, Qian Y, Deng L. Peritoneal fluid concentrations of interleukin-17 correlate with the severity of endometriosis and infertility of this disorder. BJOG (2005) 112(8):1153–5. doi: 10.1111/j.1471-0528.2005.00639.x
270. Gogacz M, Winkler I, Bojarska-Junak A, Tabarkiewicz J, Semczuk A, Rechberger T, et al. Increased percentage of Th17 cells in peritoneal fluid is associated with severity of endometriosis. J Reprod Immunol (2016) 117:117:39–44. doi: 10.1016/j.jri.2016.04.289
271. Khan KN, Yamamoto K, Fujishita A, Muto H, Koshiba A, Kuroboshi H, et al. Differential levels of regulatory T cells and T-Helper-17 cells in women with early and advanced endometriosis. J Clin Endocrinol Metab (2019) 104(10):4715–29. doi: 10.1210/jc.2019-00350
272. Hirata T, Osuga Y, Hamasaki K, Yoshino O, Ito M, Hasegawa A, et al. Interleukin (IL)-17A stimulates IL-8 secretion, cyclooxygensase-2 expression, and cell proliferation of endometriotic stromal cells. Endocrinology (2008) 149(3):1260–7. doi: 10.1210/en.2007-0749
273. Ahn SH, Edwards AK, Singh SS, Young SL, Lessey BA, Tayade C. IL-17A contributes to the pathogenesis of endometriosis by triggering proinflammatory cytokines and angiogenic growth factors. J Immunol (2015) 195(6):2591–600. doi: 10.4049/jimmunol.1501138
274. Berger A. Th1 and Th2 responses: what are they? BMJ (2000) 321(7258):424. doi: 10.1136/bmj.321.7258.424
275. Podgaec S, Abrao MS, Dias JA Jr, Rizzo LV, de Oliveira RM, Baracat EC. Endometriosis: an inflammatory disease with a Th2 immune response component. Hum Reprod (2007) 22(5):1373–9. doi: 10.1093/humrep/del516
276. Urata Y, Osuga Y, Akiyama I, Nagai M, Izumi G, Takamura M, et al. Interleukin-4 and prostaglandin E2 synergistically up-regulate 3β-hydroxysteroid dehydrogenase type 2 in endometrioma stromal cells. J Clin Endocrinol Metab (2013) 98(4):1583–90. doi: 10.1210/jc.2012-3475
277. OuYang Z, Hirota Y, Osuga Y, Hamasaki K, Hasegawa A, Tajima T, et al. Interleukin-4 stimulates proliferation of endometriotic stromal cells. Am J Pathol (2008) 173(2):463–9. doi: 10.2353/ajpath.2008.071044
278. Chen P, Wang DB, Liang YM. Evaluation of estrogen in endometriosis patients: Regulation of GATA-3 in endometrial cells and effects on Th2 cytokines. J Obstet Gynaecol Res (2016) 42(6):669–77. doi: 10.1111/jog.12957
279. Chen P, Zhang Z, Chen Q, Ren F, Li T, Zhang C, et al. Expression of Th1 and Th2 cytokine-associated transcription factors, T-bet and GATA-3, in the eutopic endometrium of women with endometriosis. Acta Histochem (2012) 114(8):779–84. doi: 10.1016/j.acthis.2012.01.005
280. Tsai SJ, Wu MH, Lin CC, Sun HS, Chen HM. Regulation of steroidogenic acute regulatory protein expression and progesterone production in endometriotic stromal cells. J Clin Endocrinol Metab (2001) 86(12):5765–73. doi: 10.1210/jcem.86.12.8082
281. Wang D, Chen Q, Zhang C, Ren F, Li T. DNA Hypomethylation of the COX-2 gene promoter is associated with up-regulation of its mRNA expression in eutopic endometrium of endometriosis. Eur J Med Res (2012) 17(1):12. doi: 10.1186/2047-783X-17-12
282. Izawa M, Harada T, Taniguchi F, Ohama Y, Takenaka Y, Terakawa N. An epigenetic disorder may cause aberrant expression of aromatase gene in endometriotic stromal cells. Fertil Steril (2008) 89(5 Suppl):1390–6. doi: 10.1016/j.fertnstert.2007.03.078
283. Xue Q, Lin Z, Yin P, Milad MP, Cheng YH, Confino E, et al. Transcriptional activation of steroidogenic factor-1 by hypomethylation of the 5' CpG island in endometriosis. J Clin Endocrinol Metab (2007) 92(8):3261–7. doi: 10.1210/jc.2007-0494
284. Xue Q, Zhou YF, Zhu SN, Bulun SE. Hypermethylation of the CpG island spanning from exon II to intron III is associated with steroidogenic factor 1 expression in stromal cells of endometriosis. Reprod Sci (2011) 18(11):1080–4. doi: 10.1177/1933719111404614
285. Hsiao KY, Wu MH, Tsai SJ. Epigenetic regulation of the pathological process in endometriosis. Reprod Med Biol (2017) 16(4):314–9. doi: 10.1002/rmb2.12047
286. Yamagata Y, Nishino K, Takaki E, Sato S, Maekawa R, Nakai A, et al. Genome-wide DNA methylation profiling in cultured eutopic and ectopic endometrial stromal cells. PloS One (2014) 9(1):e83612. doi: 10.1371/journal.pone.0083612
287. Diehl S, Anguita J, Hoffmeyer A, Zapton T, Ihle JN, Fikrig E, et al. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity (2000) 13(6):805–15. doi: 10.1016/s1074-7613(00)00078-9
288. Diehl S, Rincón M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol (2002) 39(9):531–6. doi: 10.1016/s0161-5890(02)00210-9
289. Tsudo T, Harada T, Iwabe T, Tanikawa M, Nagano Y, Ito M, et al. Altered gene expression and secretion of interleukin-6 in stromal cells derived from endometriotic tissues. Fertil Steril (2000) 73(2):205–11. doi: 10.1016/s0015-0282(99)00496-3
290. Akoum A, Lemay A, Paradis I, Rheault N, Maheux R. Secretion of interleukin-6 by human endometriotic cells and regulation by proinflammatory cytokines and sex steroids. Hum Reprod (1996) 11(10):2269–75. doi: 10.1093/oxfordjournals.humrep.a019088
291. Candido S, Tomasello BMR, Lavoro A, Falzone L, Gattuso G, Libra M. Novel insights into epigenetic regulation of IL6 pathway: In silico perspective on inflammation and cancer relationship. Int J Mol Sci (2021) 22(18):10172. doi: 10.3390/ijms221810172
292. Lamprianidou E, Kordella C, Kazachenka A, Zoulia E, Bernard E, Filia A, et al. Modulation of IL-6/STAT3 signaling axis in CD4+FOXP3- T cells represents a potential antitumor mechanism of azacitidine. Blood Adv (2021) 5(1):129–42. doi: 10.1182/bloodadvances.2020002351
293. Umezawa N, Kawahata K, Mizoguchi F, Kimura N, Yoshihashi-Nakazato Y, Miyasaka N, et al. Interleukin-23 as a therapeutic target for inflammatory myopathy. Sci Rep (2018) 8(1):5498. doi: 10.1038/s41598-018-23539-4
294. Schinocca C, Rizzo C, Fasano S, Grasso G, La Barbera L, Ciccia F, et al. Role of the IL-23/IL-17 pathway in rheumatic diseases: An overview. Front Immunol (2021) 12:637829. doi: 10.3389/fimmu.2021.637829
295. Andreoli CG, Genro VK, Souza CA, Michelon T, Bilibio JP, Scheffel C. & cunha-filho JS 2011 T helper (Th)1, Th2, and Th17 interleukin pathways in infertile patients with minimal/mild endometriosis. Fertil Steril (2011) 95(8):2477–80. doi: 10.1016/j.fertnstert.2011.02.019
296. Vanden Eijnden S, Goriely S, De Wit D, Willems F, Goldman M. IL-23 up-regulates IL-10 and induces IL-17 synthesis by polyclonally activated naive T cells in human. Eur J Immunol (2005) 35(2):469–75. doi: 10.1002/eji.200425677
297. Liao HT, Tsai CY, Lai CC, Hsieh SC, Sun YS, Li KJ, et al. The potential role of genetics, environmental factors, and gut dysbiosis in the aberrant non-coding RNA expression to mediate inflammation and Osteoclastogenic/Osteogenic differentiation in ankylosing spondylitis. Front Cell Dev Biol (2022) 9:748063. doi: 10.3389/fcell.2021.748063
298. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discovery (2012) 11(5):384–400. doi: 10.1038/nrd3674
299. Dzobo K. Epigenomics-guided drug development: Recent advances in solving the cancer treatment "jigsaw puzzle". OMICS (2019) 23(2):70–85. doi: 10.1089/omi.2018.0206
300. Ganesan A, Arimondo PB, Rots MG, Jeronimo C, Berdasco M. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenet (2019) 11(1):174. doi: 10.1186/s13148-019-0776-0
301. Xiao W, Zhou Q, Wen X, Wang R, Liu R, Wang T, et al. Small-molecule inhibitors overcome epigenetic reprogramming for cancer therapy. Front Pharmacol (2021), 12:702360. doi: 10.3389/fphar.2021.702360
302. Heerboth S, Lapinska K, Snyder N, Leary M, Rollinson S, Sarkar S. Use of epigenetic drugs in disease: an overview. Genet Epigenet (2014) 6:9–19. doi: 10.4137/GEG.S12270
303. Butler P, Lawton M, Chapin RE, Hollingshead BD, Leach K, Nambiar PR, et al. CHAPTER 12: Safety considerations for epigenetic mechanisms as drug targets: Are existing toxicology studies fit for purpose? Rsc Drug Discovery Ser (2016), 288–315. doi: 10.1039/9781782628484-00288
304. Tamura I, Ohkawa Y, Sato T, Suyama M, Jozaki K, Okada M, et al. Genome-wide analysis of histone modifications in human endometrial stromal cells. Mol Endocrinol (2014) 28(10):1656–69. doi: 10.1210/me.2014-1117
305. Lelu K, Laffont S, Delpy L, Paulet PE, Perinat T, Tschanz SA, et al. Estrogen receptor alpha signaling in T lymphocytes is required for estradiol-mediated inhibition of Th1 and Th17 cell differentiation and protection against experimental autoimmune encephalomyelitis. J Immunol (2011) 187(5):2386–93. doi: 10.4049/jimmunol.1101578
306. Priyanka HP, Krishnan HC, Singh RV, Hima L, Thyagarajan S. Estrogen modulates in vitro T cell responses in a concentration- and receptor-dependent manner: effects on intracellular molecular targets and antioxidant enzymes. Mol Immunol (2013) 56(4):328–39. doi: 10.1016/j.molimm.2013.05.226
307. Kim DH, Park HJ, Park HS, Lee JU, Ko C, Gye MC, et al. Estrogen receptor α in T cells suppresses follicular helper T cell responses and prevents autoimmunity. Exp Mol Med (2019) 51(4):1–9. doi: 10.1038/s12276-019-0237-z
308. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol (2015) 294(2):63–9. doi: 10.1016/j.cellimm.2015.01.018
309. Notas G, Kampa M, Castanas E. G Protein-coupled estrogen receptor in immune cells and its role in immune-related diseases. Front Endocrinol (Lausanne) (2020) 11:579420. doi: 10.3389/fendo.2020.579420
310. Mahajan V, Farquhar C, Ponnampalam AP. Could DNA hydroxymethylation be crucial in influencing steroid hormone signaling in endometrial biology and endometriosis? Mol Reprod Dev (2020) 87(1):7–16. doi: 10.1002/mrd.23299
311. Lissauer D, Eldershaw SA, Inman CF, Coomarasamy A, Moss PA, Kilby MD. Progesterone promotes maternal-fetal tolerance by reducing human maternal T-cell polyfunctionality and inducing a specific cytokine profile. Eur J Immunol (2015) 45(10):2858–72. doi: 10.1002/eji.201445404
312. Areia A, Vale-Pereira S, Alves V, Rodrigues-Santos P, Santos-Rosa M, Moura P, et al. Can membrane progesterone receptor α on T regulatory cells explain the ensuing human labour? J Reprod Immunol (2016) 113:113:22–6. doi: 10.1016/j.jri.2015.10.002
313. Wu Y, Strawn E, Basir Z, Halverson G, Guo SW. Promoter hypermethylation of progesterone receptor isoform b (PR-b) in endometriosis. Epigenetics (2006) 1(2):106–11. doi: 10.4161/epi.1.2.2766
314. Yang Q, Li M, Zhao M, Lu F, Yu X, Li L, et al. Progesterone modulates CD4+ CD25+ FoxP3+ regulatory T cells and TGF-β1 in the maternal-fetal interface of the late pregnant mouse. Am J Reprod Immunol (2022) 25:e13541. doi: 10.1111/aji.13541
315. Hierweger AM, Engler JB, Friese MA, Reichardt HM, Lydon J, DeMayo F, et al. Progesterone modulates the T-cell response via glucocorticoid receptor-dependent pathways. Am J Reprod Immunol (2019) 81(2):e13084. doi: 10.1111/aji.13084
316. Ghafouri-Fard S, Shoorei H, Taheri M. Role of non-coding RNAs in the pathogenesis of endometriosis. Front Oncol (2020) 10:1370. doi: 10.3389/fonc.2020.01370
317. Hudson QJ, Proestling K, Perricos A, Kuessel L, Husslein H, Wenzl R, et al. The role of long non-coding RNAs in endometriosis. Int J Mol Sci (2021) 22(21):11425. doi: 10.3390/ijms222111425
318. Wei C, Mei J, Tang L, Liu Y, Li D, Li M, et al. 1-methyl-tryptophan attenuates regulatory T cells differentiation due to the inhibition of estrogen-IDO1-MRC2 axis in endometriosis. Cell Death Dis (2016) 7(12):e2489. doi: 10.1038/cddis.2016.375
319. Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol (2009) 183(4):2475–83. doi: 10.4049/jimmunol.0900986
320. Schmidl C, Delacher M, Huehn J, Feuerer M. Epigenetic mechanisms regulating T-cell responses. J Allergy Clin Immunol (2018) 142(3):728–43. doi: 10.1016/j.jaci.2018.07.014
321. Xiao F, Liu X, Guo SW. Platelets and regulatory T cells may induce a type 2 immunity that is conducive to the progression and fibrogenesis of endometriosis. Front Immunol (2020) 11:610963. doi: 10.3389/fimmu.2020.610963
322. Pashizeh F, Mansouri R, Davari-Tanha F, Hosseini R, Asgari Z, Aghaei H, et al. Alterations of CD4+T cell subsets in blood and peritoneal fluid in different stages of endometriosis. Int J Fertil Steril (2020) 14(3):201–8. doi: 10.22074/ijfs.2020.6127
323. Khoufache K, Michaud N, Harir N, Kibangou Bondza P, Akoum A. Anomalies in the inflammatory response in endometriosis and possible consequences: a review. Minerva Endocrinol (2012) 37(1):75–92.
324. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T Cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for treg cell development. Immunity (2012) 37(5):785–99. doi: 10.1016/j.immuni.2012.09.010
325. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20(1):62–8. doi: 10.1038/nm.3432
326. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
327. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity (2013) 39(5):949–62. doi: 10.1016/j.immuni.2013.10.016
328. Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol (2009) 182(5):2578–82. doi: 10.4049/jimmunol.0803162
329. van Loosdregt J, Brunen D, Fleskens V, Pals CE, Lam EW, Coffer PJ. Rapid temporal control of Foxp3 protein degradation by sirtuin-1. PloS One (2011) 6(4):e19047. doi: 10.1371/journal.pone.0019047
330. Li J, Du X, Shi H, Deng K, Chi H, Tao W. Mammalian sterile 20-like kinase 1 (Mst1) enhances the stability of forkhead box P3 (Foxp3) and the function of regulatory T cells by modulating Foxp3 acetylation. J Biol Chem (2015) 290(52):30762–70. doi: 10.1074/jbc.M115.668442
331. Geng J, Yu S, Zhao H, Sun X, Li X, Wang P, et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and treg cells. Nat Immunol (2017) 18(7):800–12. doi: 10.1038/ni.3748
332. Wu J, Shi H. Unlocking the epigenetic code of T cell exhaustion. Transl Cancer Res (2017) 6(Suppl 2):S384–7. doi: 10.21037/tcr.2017.03.02
333. Mitchell DA, Karikari I, Cui X, Xie W, Schmittling R, Sampson JH. Selective modification of antigen-specific T cells by RNA electroporation. Hum Gene Ther (2008) 19(5):511–21. doi: 10.1089/hum.2007.115
334. Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol (2015) 23:32–8. doi: 10.1016/j.coph.2015.05.011
335. Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun (2020) 11(1):3801. doi: 10.1038/s41467-020-17670-y
336. Schepisi G, Casadei C, Toma I, Poti G, Iaia ML, Farolfi A, et al. Immunotherapy and its development for gynecological (Ovarian, endometrial and cervical) tumors: From immune checkpoint inhibitors to chimeric antigen receptor (CAR)-T cell therapy. Cancers (Basel) (2021) 13(4):840. doi: 10.3390/cancers13040840
337. Hsu C-F, Khine AA, Huang H-S, Chu T-Y. The double engines and single checkpoint theory of endometriosis. Biomedicines (2022) 10:1403. doi: 10.3390/biomedicines10061403
338. Wu L, Lv C, Su Y, Li C, Zhang H, Zhao X, et al. Expression of programmed death-1 (PD-1) and its ligand PD-L1 is upregulated in endometriosis and promoted by 17beta-estradiol. Gynecol Endocrinol (2019) 35(3):251–6. doi: 10.1080/09513590.2018.1519787
339. Santoso B, Sa'adi A, Dwiningsih SR, Tunjungseto A, Widyanugraha MYA, Mufid AF, et al. Soluble immune checkpoints CTLA-4, HLA-G, PD-1, and PD-L1 are associated with endometriosis-related infertility. Am J Reprod Immunol (2020) 84(4):e13296. doi: 10.1111/aji.13296
340. Nishihira H, Kigasawa H. Growth of human erythroid and erythroid-granulocytic colonies in culture without addition of exogeneous erythropoietin. Br J Haematol (1981) 49(4):563–6. doi: 10.1111/j.1365-2141.1981.tb07264.x
341. Miliotou AN, Papadopoulou LC. In vitro-transcribed (IVT)-mRNA CAR therapy development. Methods Mol Biol (2020) 2086:87–117. doi: 10.1007/978-1-0716-0146-4_7
342. Foster JB, Barrett DM, Karikó K. The emerging role of In vitro-transcribed mRNA in adoptive T cell immunotherapy. Mol Ther (2019) 27(4):747–56. doi: 10.1016/j.ymthe.2019.01.018
343. Zhao J, Wei K, Chang C, Xu L, Jiang P, Guo S, et al. DNA Methylation of T lymphocytes as a therapeutic target: Implications for rheumatoid arthritis etiology. Front Immunol (2022) 13:863703. doi: 10.3389/fimmu.2022.863703
344. Tabana Y, Moon TC, Siraki A, Elahi S, Barakat K. Reversing T-cell exhaustion in immunotherapy: a review on current approaches and limitations. Expert Opin Ther Targets (2021) 25(5):347–63. doi: 10.1080/14728222.2021.1937123
345. Bruner-Tran KL, Carvalho-Macedo AC, Duleba AJ, Crispens MA, Osteen KG. Experimental endometriosis in immunocompromised mice after adoptive transfer of human leukocytes. Fertil Steril (2010) 93(8):2519–24. doi: 10.1016/j.fertnstert.2009.05.076
346. Oda JM, Hirata BK, Guembarovski RL, Watanabe MA. Genetic polymorphism in FOXP3 gene: imbalance in regulatory T-cell role and development of human diseases. J Genet (2013) 92(1):163–71. doi: 10.1007/s12041-013-0213-7
347. Matarese G, De Placido G, Nikas Y, Alviggi C. Pathogenesis of endometriosis: natural immunity dysfunction or autoimmune disease? Trends Mol Med (2003) 9(5):223–8. doi: 10.1016/s1471-4914(03)00051-0
348. Kokcu A. A current view of the role of epigenetic changes in the aetiopathogenesis of endometriosis. J Obstet Gynaecol (2016) 36(2):153–9. doi: 10.3109/01443615.2015.1036403
349. Huang N, Chi H, Qiao J. Role of regulatory T cells in regulating fetal-maternal immune tolerance in healthy pregnancies and reproductive diseases. Front Immunol (2020) 11:1023. doi: 10.3389/fimmu.2020.01023
350. Catalini L, Fedder J. Characteristics of the endometrium in menstruating species: lessons learned from the animal kingdom†. Biol Reprod (2020) 102(6):1160–9. doi: 10.1093/biolre/ioaa029
351. Maksym RB, Hoffmann-Młodzianowska M, Skibińska M, Rabijewski M, Mackiewicz A, Kieda C. Immunology and immunotherapy of endometriosis. J Clin Med (2021) 10(24):5879. doi: 10.3390/jcm10245879
352. D'Hooghe TM, Kyama CM, Chai D, Fassbender A, Vodolazkaia A, Bokor A, et al. Nonhuman primate models for translational research in endometriosis. Reprod Sci (2009) 16(2):152–61. doi: 10.1177/1933719108322430
353. Nishimoto-Kakiuchi A, Netsu S, Okabayashi S, Taniguchi K, Tanimura H, Kato A, et al. Spontaneous endometriosis in cynomolgus monkeys as a clinically relevant experimental model. Hum Reprod (2018) 33(7):1228–36. doi: 10.1093/humrep/dey095
354. Sun Y, Shao J, Jiang F, Wang Y, Yan Q, Yu N, et al. CD33+ CD14+ CD11b+ HLA-DR- monocytic myeloid-derived suppressor cells recruited and activated by CCR9/CCL25 are crucial for the pathogenic progression of endometriosis. Am J Reprod Immunol (2019) 81(1):e13067. doi: 10.1111/aji.13067
355. Zebley CC, Gottschalk S, Youngblood B. Rewriting history: Epigenetic reprogramming of CD8+ T cell differentiation to enhance immunotherapy. Trends Immunol (2020) 41(8):665–75. doi: 10.1016/j.it.2020.06.008
Keywords: epigenetic mechanisms, T cells, endometriosis, autoimmunity, T-cell reprogramming
Citation: Szukiewicz D (2022) Epigenetic regulation and T-cell responses in endometriosis – something other than autoimmunity. Front. Immunol. 13:943839. doi: 10.3389/fimmu.2022.943839
Received: 14 May 2022; Accepted: 27 June 2022;
Published: 22 July 2022.
Edited by:
Agnieszka Paradowska-Gorycka, National Institute of Geriatrics, Rheumatology and Rehabilitation, PolandReviewed by:
Frederique Ponchel, University of Leeds, United KingdomCopyright © 2022 Szukiewicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dariusz Szukiewicz, ZGFyaXVzei5zenVraWV3aWN6QHd1bS5lZHUucGw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.