 Nagaja Capitani
Nagaja Capitani Cosima T. Baldari
Cosima T. Baldari
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 13 July 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.943344
This article is part of the Research Topic Modulation of the immune system by bacteria: from evasion to therapy View all 17 articles
Similar to other pathogens, bacteria have developed during their evolution a variety of mechanisms to overcome both innate and acquired immunity, accounting for their ability to cause disease or chronic infections. The mechanisms exploited for this critical function act by targeting conserved structures or pathways that regulate the host immune response. A strategic potential target is the immunological synapse (IS), a highly specialized structure that forms at the interface between antigen presenting cells (APC) and T lymphocytes and is required for the establishment of an effective T cell response to the infectious agent and for the development of long-lasting T cell memory. While a variety of bacterial pathogens are known to impair or subvert cellular processes essential for antigen processing and presentation, on which IS assembly depends, it is only recently that the possibility that IS may be a direct target of bacterial virulence factors has been considered. Emerging evidence strongly supports this notion, highlighting IS targeting as a powerful, novel means of immune evasion by bacterial pathogens. In this review we will present a brief overview of the mechanisms used by bacteria to affect IS assembly by targeting APCs. We will then summarize what has emerged from the current handful of studies that have addressed the direct impact of bacterial virulence factors on IS assembly in T cells and, based on the strategic cellular processes targeted by these factors in other cell types, highlight potential IS-related vulnerabilities that could be exploited by these pathogens to evade T cell mediated immunity.
Successful microbial pathogens, such as bacteria, have evolved complex and efficient strategies to evade the host immune response. To establish chronic infection bacteria have to overcome the two powerful arms of the host immune defenses, innate and adaptive immunity. Innate immunity is evolutionarily conserved among higher eukaryotes and represents the first line of defense against infections, with the key role to recognize pathogen components and start the process of microbial clearance. Additionally, innate immune cells are central for the development of adaptive immunity. Hence, not surprisingly, pathogens have evolved a variety of mechanisms to elude this first line of the host immune defenses, from building a protective capsule (e.g. Streptococcus pneumoniae, Haemophilus influenzae, Escherichia coli, Neisseria meningitidis) (1), to interfering with recognition of Pathogen-Associated Molecular Patterns (PAMPs) by host Pattern Recognition Receptors (PRRs) such as Toll-like receptors (TLRs) and C-type lectin receptors (e.g. Helicobacter pylori) (2), to inhibiting phagocytic activity (e.g. H. pylori, Yersinia pestis) (3). Remarkably, evasion of innate immunity is often accompanied by the exploitation of innate immune cells such as macrophages, which have been incapacitated to kill internalized bacteria by specific virulence factors, as a protected niche for replication.
Another strategy deployed by several bacterial pathogens to escape the host immune system is to prevent the development of the exquisitely specific and highly effective adaptive response. Adaptive immunity involves a tightly regulated interplay among B lymphocytes, T lymphocytes and antigen presenting cells (APCs) to activate pathogen-specific and lifelong immunological effector pathways. The development of T cell mediated immunity relies on the assembly of a highly specialized signaling and secretory platform formed by T cells at the interface with cognate APCs, known as the immunological synapse (IS). In this minireview we will briefly review the strategies evolved by bacterial pathogens to suppress T cell activation and discuss emerging evidence that highlights the IS as a key target for pathogens to evade the T cell-mediated host immune response.
T cell activation is initiated in response to the interaction of the T cell antigen receptor (TCR) with antigenic peptides bound to major histocompatibility complex (MHC) molecules (pMHC) expressed on the surface of APCs, which participate in the cellular immune response by processing and presenting antigens for recognition by T lymphocytes. Antigen presentation is a complex multistep process, involving the processing of endogenous or exogenous pathogen-associated antigens, peptide loading on MHC, and localization at the cell surface of pMHC complexes which can interact with T cells expressing a cognate TCR. Bacterial antigen presentation is mainly mediated by MHC class II (MHCII) molecules found on the surface of professional APCs that present antigen-derived peptides to be recognized by CD4+ T cells.
Following TCR interaction with cognate pMHC, a specialized supramolecular structure, defined as immunological synapse (IS), forms at the T cell interface with the APC. IS formation requires not only TCR:pMHC interaction but also the accumulation of coreceptors, adhesion molecules, and signaling and cytoskeletal components at the T cell-APC contact area (4). In its mature configuration the IS features a peculiar “bull’s eye” architecture characterized by concentric domains, known as supramolecular activation clusters (SMAC), that differ in molecular composition and function (5). The central SMAC (cSMAC), mainly enriched in TCRs and TCR-associated proteins, is surrounded by the peripheral (pSMAC), enriched in integrins, such as lymphocyte function-associated antigen (LFA-1), and cytoskeleton-associated proteins. The pSMAC is in turn surrounded by the distal SMAC (dSMAC), which is enriched in F-actin as well as in molecules that are excluded from the IS centre due either to steric hindrance (e.g. CD43) or to their ability to negatively regulate signaling (e.g. CD45) (4). The dSMAC is also the IS domain where signaling starts with the assembly of TCR-CD28 microclusters that move centripetally towards the IS to eventually segregate to the cSMAC (6), where exhausted TCR are internalized to make room to new TCRs microclusters that continuously form at the periphery.
TCRs are associated not only with the plasma membrane, but also with recycling endosomes (7). Delivery to the synaptic membrane of this intracellular TCR pool is essential to replenish the plasma membrane pool as TCRs are internalized at the cSMAC, allowing for the steady inward flow of actively signaling TCR microclusters to sustain signaling for the extended timeframe required for T cell activation (7). This process is dependent on the polarization of the centrosome together with the secretory apparatus to the region beneath the T cell-APC contact (8), which sets the stage for polarized exocytosis. Polarized recycling from an intracellular vesicular pool is a strategy co-opted by a number of molecules that participate in IS architecture and function. These include surface receptors, such as the co-inhibitory receptor cytotoxic T lymphocyte antigen-4 (CTLA-4) (9), and intracellular signaling molecules, such as the lymphocyte-specific protein tyrosine kinase (Lck), the adaptor molecule LAT (10–12), and the small GTPase Rac1 (13).
IS assembly is coordinated by the cytoskeleton (14–16), which plays a key role at different step of IS assembly, from integrin activation (16), to TCR microcluster movement from the periphery to the center of the IS (6), to centrosome translocation toward the IS (14), to the directional vesicular trafficking that ensures the continuous availability of receptors and signaling molecules at the IS (17–19) (Figure 1).
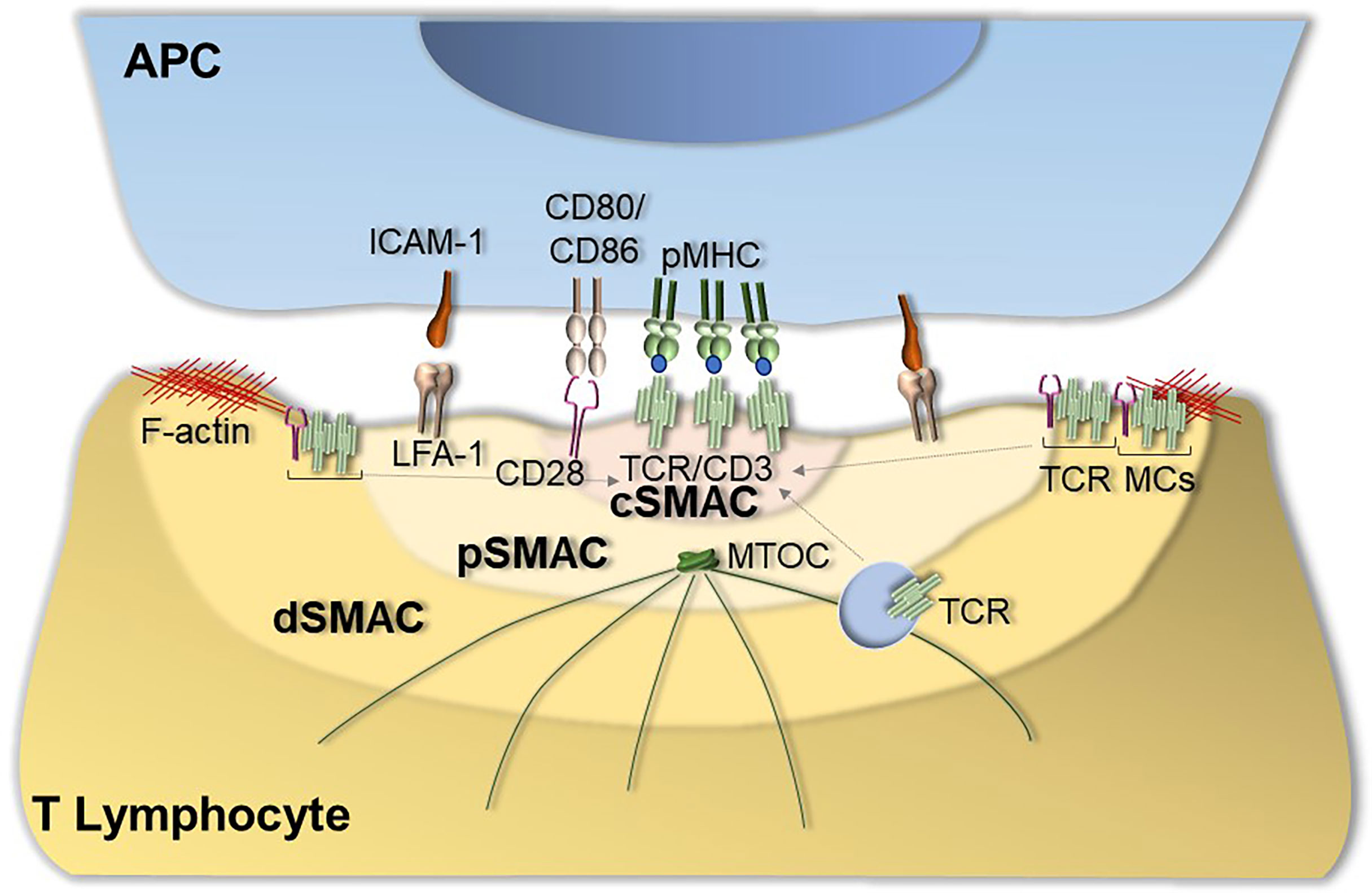
Figure 1 Immunological synapse assembly. The canonical IS shows a well-organized bull’s eye architecture that features the central supramolecular activation cluster (cSMAC) characterized by the presence of TCRs and TCR-associated proteins such as the co-stimulatory receptor CD28, the peripheral SMAC (pSMAC) enriched in the integrin LFA-1 and the distal SMAC (dSMAC) enriched in TCR-CD28 microclusters (TCR MCs) that move centripetally towards the cSMAC driven by F-actin. IS assembly is also coordinated by cytoskeletal dynamics that allow for centrosome translocation toward the IS as well as for the directional vesicular trafficking of receptors and signaling mediators to sustain signaling at the IS.
TCR interaction with pMHC at the IS triggers an intracellular tyrosine phosphorylation cascade, resulting in the activation of multiple signaling pathways. Briefly, the activated TCR recruits the initiating kinases Lck and ζ-associated kinase of 70 kDa (ZAP-70) which phosphorylates LAT, a multifunctional transmembrane adaptor that orchestrates the activation of phospholipase Cγ (PLCγ). By producing key second messengers, PLCγ promotes the activation of the PKC, Ras and Ca2+ pathways which couple TCR triggering to gene expression through the activation of transcription factors such as nuclear factor of activated T cells (NF-AT), nuclear factor-κB (NF-κB) and activating protein 1 (AP-1) (20).
As IS assembly is a key event for the development of T cell-mediated immunity, it is not surprising that many pathogens have developed virulence mechanisms to target IS formation, either indirectly by impairing the ability of APCs to present antigen to the T cell, or, as supported by emerging evidence, by directly inhibiting IS assembly within the T cell.
To initiate adaptive immunity to pathogens, T cells must interact with cognate APCs that have previously taken up antigen at the site of infection and have migrated to the draining lymph node. This role is subserved by dendritic cells (DCs) which are specialized for antigen presentation to naïve T cells, but in the context of bacterial infections it can also be taken over by macrophages. Several steps are required before an APC can acquire the appropriate functional status and be in the appropriate location to form an IS with a cognate T cell. These steps are orchestrated by innate immune receptors, which on recognition of bacterial PAMPs trigger the maturation of DCs, the phagocytic uptake and destruction of the pathogen, and the migration of the phagocyte to the closest lymph node station. As largely documented for viruses (1), also bacterial pathogens have evolved a variety of strategies to interfere with each of these steps, including camouflaging as host components (e.g. GAG proteins of Streptococcus), modifying PAMPs to decrease their potency in innate immune receptor activation (e.g. modified LPS core component lipid A of Salmonella) (21), inhibiting PRR signaling (e.g. Salmonella TIR domain-like TIpA to disrupt TLR4 signaling (22); Yersinia acetyltransferase YopJ to inhibit NF-kB signaling (23); Mycobacterium tuberculosis (M. tubercolosis) ubiquitin ligase PnkG to degrade components of the NF-kB-activating signalosome (24), or exploiting mimicry to activate inhibitory circuits (e.g. sialylated capsular polysaccharides of group B Streptococcus) (25, 26). For details on these upstream steps we refer the reader to excellent reviews (1, 27). Here we will focus on the process that is directly implicated in IS assembly -antigen presentation-, limiting the discussion to MHCII.
Antigen presentation to T cells by APCs plays an essential role in the initiation of adaptive immunity. As such, disruption of the process of antigen presentation is a mechanism co-opted by a number of bacterial pathogens to prevent the generation of specific effector T cells. Bacteria can modulate the MHCII pathway acting at different levels: by inhibiting MHCII gene transcription, by interfering with MHCII loading and trafficking, or by impairing antigen processing. The resulting defects in IS assembly translate into defects in T cell activation and differentiation to pathogen-specific helper T cell effectors. The intracellular pathogen M. tubercolosis is a remarkable example of how an individual pathogen can target the process of antigen presentation at every single level and we will use it as paradigm in the following sections.
M. tubercolosis has the ability to potently downregulate MHCII expression, which occurs as part of the APC activation program triggered by PRR engagement. A well characterized M. tubercolosis factor implicated in this function is the 19-kDa lipoprotein (LpqH) which acts a potent TLR2 agonist. The resulting excessive or prolonged TLR2 activation leads to the expression of isoforms of the transcriptional transactivator C/EBP that inhibit the IFNγ-dependent induction of class II transactivator (CIITA), on which MHCII gene expression crucially depends (28, 29). Preventing MHCII upregulation to disrupt antigen presentation is shared by other M. tubercolosis virulence factors such as the cell envelope-associated serine protease Hip1 (30), and co-opted by a number of pathogenic bacteria (e.g. 31). One such example is H. pylori, which uses ADP183 heptose, an intermediate metabolite in LPS biosynthesis, to promote miR146b expression in macrophages, leading to downmodulation of CIITA expression (32).
Whether the protease activity of Hip1 influences directly CIITA expression is not known. However, this mechanism has been documented for Chlamydia trachomatis, which secretes proteases that promote the degradation of the transcription factor USF-1 that regulates IFN-γ induction of CIITA expression (33). A different mechanism to lower MHCII expression is exploited by Salmonella, which induces surface MHCII internalization by promoting the expression of the E3 ubiquitin ligase MARCH1 and K63-linked MHCII ubiquitination. Internalized ubiquitylated MHCII molecules are subsequently degraded following routing to the endolysosomal system (34) (Figure 2, Table 1).
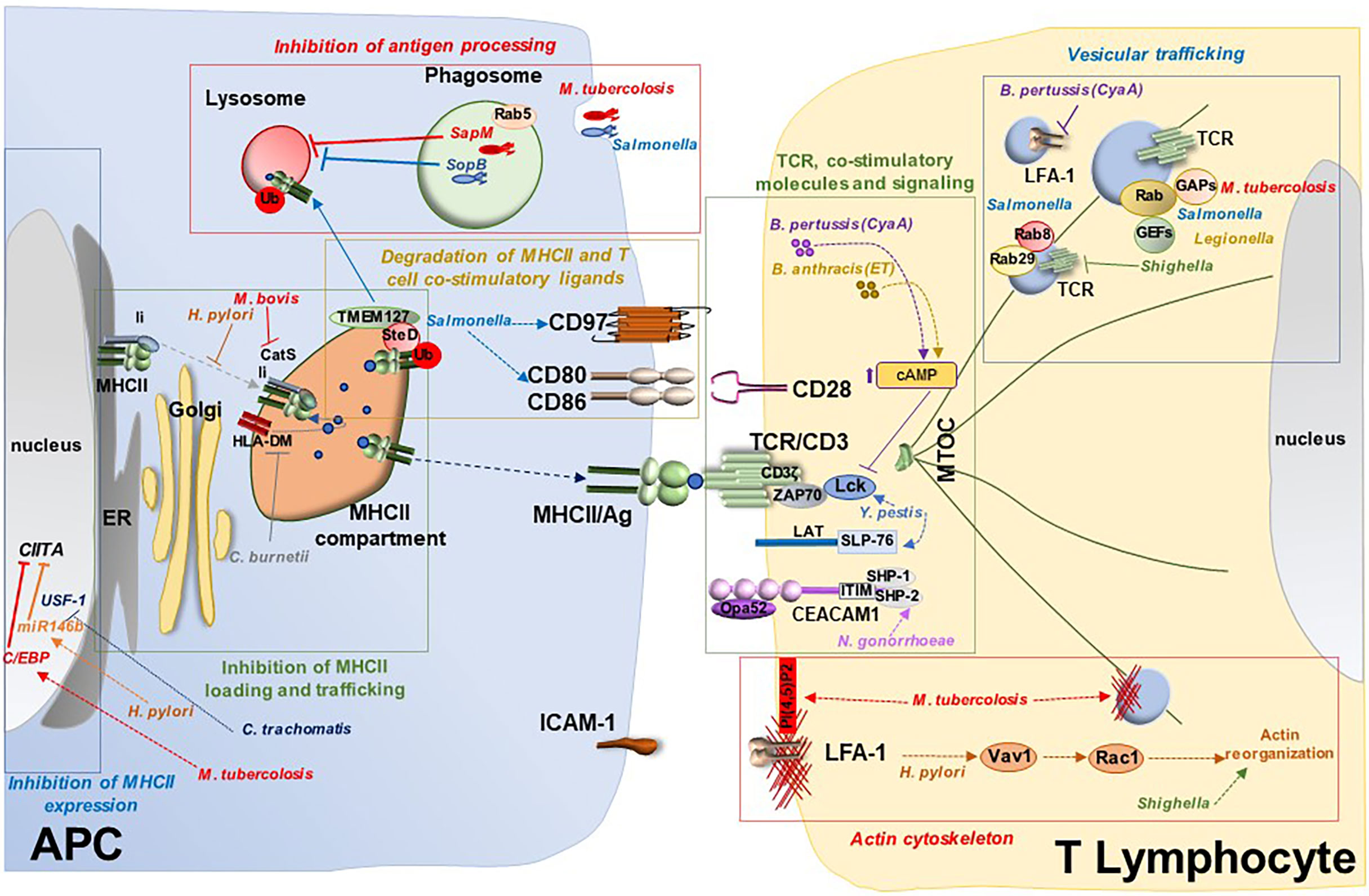
Figure 2 Bacterial targeting of the immunological synapse. Model for suppression of IS assembly by bacterial pathogens. Bacterial pathogens exploit a variety of virulence factors to interfere with IS assembly at different steps, both at the APC side and at the T cell side. Bacteria target APCs and hence indirectly IS assembly by interfering with different mechanisms: i) MHCII inhibition through modulation of transcription factors responsible for its expression (e.g. CIITA regulation by M. tuberculosis, Helicobacter pylori and Chlamydia trachomatis); ii) inhibition of antigen processing through suppression of phagolysosomal fusion (e.g. M. tuberculosis and Salmonella); iii) defective antigen processing and loading onto MHCII in the MHCII compartment (e.g. inhibition of the Ii-dependent pathway by Helicobacter pylori or targeting CatS and HLA-DM by M. tuberculosis and Coxiella burnetii, respectively); iv) degradation of MHCII and T cell co-stimulatory ligands such as CD80/CD86 and CD97 (e.g. Salmonella). Bacteria interfere directly with IS assembly at the T lymphocyte side by i) targeting expression and function of the TCR and co-stimulatory molecules (e.g. CD3ζ degradation by M. tuberculosis, CEACAM1 disabling by Neisseria gonorrhoeae or impairment of TCR signaling by Yersinia pestis, Bordetella pertussis and Bacillus anthracis); ii) subverting the actin cytoskeleton (e.g. Shigella flexneri, Yersinia pestis and Salmonella enterica serovar Typhimurium); and iii) interfering with vesicular trafficking by modulating Rab GTPases (e.g. Salmonella enterica, Legionella pneumophila, Shigella, M. tuberculosis) or by targeting receptor trafficking (e.g the TCR by Shigella or LFA-1 by Bordetella pertussis).

Table 1 Bacterial virulence factors that target directly or indirectly IS as.
Pathogenic bacteria can modulate the MHCII pathway by inhibiting the fusion of the phagosome containing internalized bacteria with the lysosome, which not only allows escape from killing but leads to impaired antigen processing. Again, using M. tubercolosis as paradigm, inhibition of phagolysosomal fusion has been shown to involve retention of the early endosome marker Rab5 at the phagosomal membrane, with concomitant exclusion of the lysosome marker Rab7 (83), which results in a delay in phagosome maturation and defective antigen processing. M. tubercolosis targets this process by using its lipid phosphatase SapM to hydrolyze the phospholipid PI3P, which is essential for phagosome-late endosome fusion (35). Similarly, Salmonella blocks phagosome maturation by modulating the phosphoinositide composition of the Salmonella-containing vacuole through its lipid phosphatase SopB (45). Hence inhibition of phagosome maturation is co-opted by many pathogenic bacteria to prevent antigen processing while escaping killing.
An alternative strategy used by M. tubercolosis for disrupting antigen processing is inhibition of phagosome function. One of the underlying mechanisms involves a M. tubercolosis-derived lipid, the mannose-capped form of lipoarabinomannan (manLAM). manLAM blocks phagosome acidification by reducing the local recruitment of the tethering molecule EEA1, which is essential for delivery of lysosomal hydrolases to the phagosome (36). The failure of EEA1 to associate with the phagosome in M. tubercolosis-infected cells is caused by defective production of PI3P at the phagosome membrane due to defective Ca2+-dependent activation of the PI3K component VPS34 (35, 37, 84). Additionally, the transport of vacuolar ATPase (v-ATPase), which is essential for phagosome acidification and activation of lysosomal hydrolases, is impaired in M. tubercolosis-infected cells due to dephosphorylation of the VPS33B component of the v-ATPase sorting complex by the M. tubercolosis phosphatase PtpA (38) (Figure 2, Table 1).
An alternative strategy exploited by a variety of pathogens to inhibit antigen presentation is to interfere with MHCII loading and trafficking. Macrophage infection with M. bovis leads to the inhibition of both activity and expression of the cystein protease cathepsin S (Cat S) (44), which mediates the late cleavage steps of the invariant chain (Ii) cleavage (85) required for the generation of MHCII molecules that can be efficiently loaded with peptide antigens and delivered to the cell surface. The defect in CatS expression has been ascribed to the M. bovis-dependent induction of the suppressive cytokine IL-10 which blocks Cat S gene expression (44) as well as of M. tubercolosis microRNA miR-106b-5p which downregulates its transcript (39).
Other bacterial pathogens target the key steps of MHC loading and trafficking to suppress the initiation of T cell response. This is the case of H. pylori which, through its major virulence factor Vacuolating cytotoxin A (VacA), interferes with the proteolytic generation of T cell epitopes that are loaded onto newly synthesized MHCII molecules, specifically inhibiting the Ii-dependent pathway (55). In addition, MHCII molecules are retained in the H. pylori-containing vacuoles in H. pylori-infected DCs, such that their trafficking to the cell surface is prevented (56). Coxiella burnetii impairs antigen presentation at a different step -loading of peptide antigen- by altering the interaction of MHCII with HLA-DM, a key step required for displacing from MHCII the Ii CLIP peptide to allow for loading of pathogen-derived peptides and transport to the plasma membrane of functional pMHC complexes. In C. burnetii-infected cells MHCII molecules fail to dissociate form HLA-DM and accumulate in enlarged intracellular compartments (60) (Figure 2, Table 1).
An alternative mechanism for reducing the levels of pMHC complexes at the APC surface has been reported for Salmonella. This function is mediated by the type 3 secretion system effector SteD. This transmembrane protein forms a complex with mature endosome-associated MHCII molecules and the transmembrane host tumor suppressor TMEM127, a Nedd4 family E3 ubiquitin ligase adaptor. TMEM127 recruits the E3 ligase Wwp2 to the complex, inducing ubiquitylation of MHCII for subsequent lysosomal degradation (46). Interestingly, SteD exploits this degradation-promoting activity to reduce the expression of important T cell activating ligands expressed on APCs, including CD86/B7-2 which activated the key co-stimulatory receptor CD28 (47), and the plasma membrane protein CD97 that is required to stabilize the IS formed with T cells (48) (Figure 2, Table 1).
Since the seminal discovery that lymphotropic viruses such as HIV-1 and HTLV-1 not only exploit the IS to evade the T cell response but apply the same building principles to form the virological synapse, a platform for cell-to-cell transmission, the IS has attracted major interest as a target for immune evasion by viral pathogens (86, 87). Whether and how bacterial pathogens can subvert IS assembly to avoid T cell immunity not indirectly by modulating DC activation and function, but directly, are questions that are only beginning to be formulated. DCs are present at the sites of infection where they can readily recognize pathogens through their wide array of PRRs, orchestrating a sophisticated response that not only optimizes their antigen presentation capacity but also provides all the signals that T cells require to differentiate to the most appropriate type of effector. At variance, T cells continuously cycle between blood and lymph and are activated in secondary lymphoid organs, where DCs migrate following pathogen recognition. However, a number of bacterial virulence factors are released as soluble factors that can be transported through the lymph to the closest lymph node station, where they can interact with naïve T cells and even enter them while not establishing a productive infection, as exemplified by the T cell delivery of Shigella T3SS effectors (88). Importantly, following their differentiation, effector T cells, whether CTLs or Th cells, are recruited to the site of infection to coordinate a combined attack with innate immune cells against the invading pathogen. There, effector T cells become a very relevant target for immune evasion.
Examples of IS targeting by bacterial pathogens are as yet very few. However, the substantial body of information acquired on how bacteria subvert pivotal cellular processes in host cells, such as cytoskeletal dynamics and vesicular trafficking, which are essential for IS assembly, suggests that we are looking at the tip of the iceberg. In this section we will present arguments to support this notion, discussing specific instances that provide experimental evidence that the IS is exploited not only by viruses, but also by bacteria, to evade T cell-mediated immunity.
A strategy that mirrors at the T cell side what we described above on the APC side is downregulation of TCR expression, as exemplified in Pneumococcus-related sepsis. Of note, T cells from these patients also coordinately downregulated the expression of the major co-stimulatory receptors CD28, essential for T cell activation, and ICOS and CD40L, required for T cell-dependent B cell maturation (61). A different mechanism to modulate CD3 expression is exploited by M. tubercolosis, involving degradation of its key component CD3ζ. This is achieved through upregulation of the E3 ubiquitin ligase Grail by manLAM (40). Although not tested directly, downregulation of surface TCR is expected to impact on IS assembly and local signaling, as witnessed by primary immunodeficiency disorders with CD3 deficiency (89).
Staphylococcus aureus uses the amply characterized mechanism of forced, antigen-independent TCR binding to MHCII mediated by its toxins SEA, SEB and SEE to promote massive T cell activation and inflammatory cytokine production associated with defective anti-bacterial T cell response. These toxins are able to elicit IS assembly with high efficiency and are in fact used as surrogate antigens to study IS assembly in polyclonal T cells. Interestingly, a different mechanism involving the Staphylococcus superantigens SEA, SEB and TSST-1, has been recently reported, based on cross-linking the co-stimulatory receptor CD28 with its ligand B7.2 on APCs (62). Since CD28 co-localizes with the TCR at the cSMAC, this double locking action is expected to lead to the generation of hyperstable and hyperactive immune synapses.
Another example of co-inhibitory receptor targeting for T cell suppression is CEACAM1 disabling by Neisseria gonorrhoeae. CEACAM1 is expressed as two isoforms differing in the length of its intracellular domain, with the long isoform endowed of two immunoreceptor tyrosine-based inhibitory motifs (ITIM). The gonococcal protein Opa52 interacts with CEACAM1 on CD4+ T cells, leading to phosphorylation of its ITIM motifs and recruitment of the tyrosine phosphatases SHP-1 and SHP-2, which dampen TCR signaling (63). A similar strategy to suppress CD4+ T cell activation is exploited by Fusobacterium nucleatum, Neisseria meningitidis, Moraxella catarrhalis, and Haemophilus influenzae, which also trigger CEACAM1 activation through specific adhesins (90–92). At variance, CEACAM1 has been recently reported to also act as a co-stimulatory receptor essential for the activation and proliferation of CD8+ T cells, preventing their exhaustion and promoting their antiviral activity (93). Interestingly, CEACAM1 engagement leads to the recruitment of Lck to the TCR and stabilizes this key initiating kinase at the IS (93). This finding underscores the IS as a potential important target of bacterial pathogens that produce CEACAM1 ligands (Figure 2, Table 1).
Major bacterial pathogens have the potential to target signaling downstream of the TCR, thereby affecting IS assembly and stability. M. tubercolosis exploits the manLAM-dependent upregulation of Grail mentioned above for CD3ζ downregulation to coordinately promote the degradation of essential mediators of the TCR signaling cascade, including the initiating tyrosine kinases Lck and ZAP-70, and the adaptor LAT required for signal amplification and diversification (40). Again, deficiency of these signaling mediators in experimental systems or primary immunodeficiencies supports the potential negative impact of M. tubercolosis in IS assembly. Another M. tubercolosis-derived molecule, mycolactone, interferes with T cell activation by inhibiting TCR signaling through an as yet unknown mechanism (41), underscoring T cell activation -and by inference IS assembly- as a relevant target for T cell disabling by M. tubercolosis.
Other pathogens have been reported to disrupt specific steps in TCR signaling. One such example is Yersinia pestis, which terminates TCR signaling using one of its outer membrane proteins, the protein tyrosine phosphatase YopH, that dephosphorylates key TCR signalosome components, including Lck, LAT and SLP-76 (64–67). Bordetella pertussis and Bacillus anthracis also suppress TCR signaling from its earliest step -activation of Lck- by elevating the cellular concentration of cAMP through their adenylate cyclase toxins, CyaA and edema toxin, respectively (70, 71). At variance, the H. pylori vacuolating cytotoxin (VacA) inhibits the Ca2+-calcineurin pathway that is responsible for the activation of the key transcription factor NF-AT by inducing plasma membrane depolarization through its anion channel activity (57, 58). Additionally, VacA perturbs TCR signaling through an independent pathway triggered by its receptor-binding moiety, which selectively enhances the activity of the MAP kinase p38 but not Erk, leading to a dysfunctional MAP kinase network (57). That these effects have the potential to target the IS is witnessed by the ability of Bordetella pertussis CyaA to impair IS assembly through local cAMP production (71, 72) (Figure 2, Table 1).
IS assembly is coordinated by the interplay of the actin and tubulin cytoskeletons. F-actin reorganization regulates multiple steps of IS formation, from integrin-mediated T cell adhesion to its cognate APC, to the recruitment of TCR microclusters to the cSMAC, to centrosome polarization beneath the synaptic membrane, to the process of sorting of cargoes, including TCRs, from early endosomes for their recycling to the IS to sustain signaling (94). Bacterial pathogens are masters at exploiting the host cell actin cytoskeleton for engulfment by host cells and intercellular dissemination, as exemplified by Shigella flexneri, Yersinia pestis and Salmonella enterica serovar Typhimurium. This is achieved by a remarkable array of T3SS effectors that promote actin remodeling by targeting directly or indirectly the Rho GTPases. The strategies evolved to modulate the activity of these small GTPases are multifarious, ranging from Rho GEF mimics (e.g. Salmonella SopB and SopE), to GAP mimics (e.g. Salmonella SptP, Yersinia YopE), to direct modulators of the active (GTP-bound) or inactive (GDP-bound) forms of Rho GTPases (e.g. the ADP-ribolysating Clostridium C3 toxin; the Yersinia protease YopT), to the process of F-actin filament assembly (e.g. Shigella IcsA and Listeria ActA mimicking activators of the actin nucleator N-WASP and of the actin adaptor Arp2/3, respectively; M. tubercolosis MtSerB2-mediated dephosphorylation and activation of cofilin) (95, 96). By acting on F-actin remodeling, these bacterial pathogens have the potential to interfere with the highly regulated process of IS assembly.
Direct experimental evidence in support of this hypothesis has been recently generated. Shigella had been previously shown to directly impair T cell chemotaxis through its T3SS effector IpgD, a lipid phosphatase that hydrolyses PI(4,5)P2, thus preventing leading edge formation in which actin dynamics plays a pivotal role (76). Recently Samassa and colleagues demonstrated that Shigella promotes actin polymerization in CD4+ T cell through an as yet unidentified T3SS effector which leads to an increase in cell stiffness, thereby impairing the ability of T cells to scan APCs for the presence of specific pMHC and hence affecting the efficiency of T cell:APC conjugate formation, which is sets the stage for IS assembly (77). Since other bacterial pathogens may exploit their T3SS system to invade, albeit not productively infect, T cells, they might exploit the actin-subverting effectors to similarly affect IS formation. A similar scenario can be hypothesized for the H. pylori vacuolating cytotoxin VacA, which binds T cells by interacting with the integrin LFA-1 (59) and triggers the activation of the Rho family guanine nucleotide exchanger Vav1 and the downstream activation of Rac1, leading to perturbations in the actin cytoskeleton (57).
F-actin reorganization during IS assembly is critically controlled by the dynamic redistribution of lipid kinases and phosphatases that generate local pools of specific phosphoinositides. Actin clearance from the IS center is required to generate the secretory domain where exocytic and endocytic events occur. This is regulated by depletion from the IS center of the lipid kinase PIP5K, which is required to replenish PI(4,5)P2 at the synaptic membrane, thus sustaining actin polymerization (97). Remarkably, modulation of phosphoinositide signaling is a major target shared by a variety of bacterial pathogens (98). An interesting example is the M. tubercolosis lipid phosphatase SapM, which dephosphorylates PI(4,5)P2 and PI3P to regulate the early stages of microbial phagocytosis and phagosome formation (35). Of note, while PI(4,5)P2 is implicated in F-actin polymerization during IS assembly, PI3P plays a crucial role in endosome trafficking, which is also centrally implicated in IS assembly, as detailed in the following section (Figure 2, Table 1).
T cell activation requires TCR signaling to be sustained for several hours (99). This is achieved through the sequential mobilization of two TCR pools associated with the plasma membrane and recycling endosomes, respectively (17–19). Translocation of the centrosome towards the T cell:APC contact sets the stage for the polarized delivery of endosomal TCRs through their dynein-dependent transport along the microtubules. This strategy is co-exploited by a number of other receptors as well as membrane-associated signaling mediators that modulate the TCR signaling cascade (11, 12).
Vesicular trafficking is widely highjacked by bacterial pathogens for infection as well as to disable the bactericidal mechanisms of phagocytes. Major targets in this process are the Rab GTPases, largely through the modulation of their activity by a variety of virulence factors that act as GAPs or GEFs on specific Rab family members (100). Examples of bacterial Rab GAPs are M. tubercolosis Ndk (43), Salmonella enterica SopD2 (53), Legionella pneumophila LepB (80) and Shigella VirA (78), while examples of bacterial Rab GEFs are Legionella pneumophila Lgp0393 (82) and DrrA/SidM (81). Additionally, as mentioned in the previous paragraph, phosphoinositide signaling, which is essential for endosome maturation through recruitment of Rab proteins or their regulators or effectors, is disrupted by phosphoinositide-specific virulence factors, such as the phosphoinositide phosphatases M. tubercolosis SapM and Salmonella enterica SopB (98). Hence, similar to phagocytes, these factors may be expected to interfere with vesicular trafficking in T cells, thereby impacting on IS assembly and function.
Strong support to this hypothesis has been provided by the finding that Shigella impairs IS assembly by disrupting the polarized recycling of TCR-containing endosomes to the IS through two T3SS effectors, the Rab1 GAP VirA and the Arf/Arl targeting cysteine protease IpaJ (77). Additionally, we have shown that forced expression of the Salmonella protease GtgE, which cleaves and inactivates Rab29 and Rab8 (101, 102), similarly impairs IS assembly by inhibiting two sequential steps in the vesicular transport pathway that regulates polarized TCR recycling to the IS (54). Of note, the activity of Rab32 is also modulated by Salmonella SopD2 acting as a GAP (53), highlighting a combined targeting of Rab29 by distinct virulence factors of this pathogen. A different strategy is exploited by Bordetella pertussis, which uses its adenylate cyclase toxin CyaA to impair recycling of the integrin LFA-1, leading to premature IS disassembly (71) (Figure 2, Table 1).
Pathogens are masters in the art of spotting the vulnerabilities of target cells and evolve strategies to either neutralize or subvert these to their own advantage to infect target cells and evade immune mediated destruction. As the platform where the T cell response to antigen recognition is coordinated, the IS represents one of such vulnerabilities. This is witnessed by evidence accumulated over the past several years showing that the processes that regulate IS assembly, from TCR signaling, to cytoskeleton dynamics, to vesicular trafficking, are targeted by lymphotropic viruses to thwart the antiviral T cell response and infect neighboring cells while remaining undetectable (86, 87). Interesting, IS targeting is exploited also by tumor cells to suppress antitumor immunity through both contact-dependent and -independent mechanisms, as amply documented in chronic lymphocytic leukemia (103). Hence, it is not surprising that bacterial pathogens have co-opted this strategy to evade T cell mediated immunity. While the evidence supporting this notion is as yet scant, it is likely to represent only the tip of the iceberg since the cellular processes known to be disrupted or subverted by bacterial virulence factors that coordinate infection of target cells, such as cytoskeletal dynamics, membrane trafficking or phosphoinositide signaling, are also centrally implicated in the process of IS assembly. Hence studies focusing on the IS as target of bacterial virulence factors are expected to provide major insights into the mechanisms of immune evasion by bacterial pathogens. Of note, bacterial pathogens that infect cells that are transported to peripheral lymphoid tissues, such as DCs or macrophages, can interfere with priming pathogen-specific T cells. While pathogens that remain confined in infected tissues may influence T cell priming through soluble factors that can be transported by the lymph, their physical separation prevents them from directly deploying the full array of virulence factors, targeting rather APCs for targeting this process. However, naive T cells differentiated to helper or cytotoxic effectors are recruited to the site of infection to coordinate the fight against the pathogens in concert with the innate immune cells. Since effector T cells assemble immune synapses with target cells for the selective delivery of cytokines and cytotoxic molecules, the potential IS-modulating functions of bacterial virulence factors may be highly effective to evade the effector mechanisms of these cells.
NC and CB wrote the paper. NC prepared the artwork.
CB is supported by grants from EU (ERC Synergy Grant 951329), AIRC (IG-2017 - ID 20148) and 464 Ministero dell’Istruzione, dell’Università e della Ricerca (Grant PRIN bando 2017 - 2017FS5SHL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Finlay BB, McFadden G. Anti-Immunology: Evasion of the Host Immune System by Bacterial and Viral Pathogens. Cell (2006) 124:767–82. doi: 10.1016/j.cell.2006.01.034
2. Karkhah A, Ebrahimpour S, Rostamtabar M, Koppolu V, Darvish S, Vasigala VKR, et al. Helicobacter Pylori Evasion Strategies of the Host Innate and Adaptive Immune Responses to Survive and Develop Gastrointestinal Diseases. Microbiol Res (2019) 218:49–57. doi: 10.1016/j.micres.2018.09.011
3. Celli J, Finlay BB. Bacterial Avoidance of Phagocytosis. Trends Microbiol (2002) 10:232–7. doi: 10.1016/s0966-842x(02)02343-0
4. Dustin ML. The Immunological Synapse. Cancer Immunol Res (2014) 2:1023–33. doi: 10.1158/2326-6066.CIR-14-0161
5. Monks CRF, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-Dimensional Segregation of Supramolecular Activation Clusters in T Cells. Nature (1998) 395:82–6. doi: 10.1038/25764
6. Yokosuka T, Saito T. The Immunological Synapse, TCR Microclusters, and T Cell Activation. Curr Top Microbiol Immunol (2010) 340:81–107. doi: 10.1007/978-3-642-03858-7_5
7. Das V, Nal B, Dujeancourt A, Thoulouze MI, Galli T, Roux P, et al. Activation-Induced Polarized Recycling Targets T Cell Antigen Receptors to the Immunological Synapse; Involvement of SNARE Complexes. Immunity (2004) 20:577–88. doi: 10.1016/s1074-7613(04)00106-2
8. Martín-Cófreces NB, Baixauli F, Sánchez-Madrid F. Immune Synapse: Conductor of Orchestrated Organelle Movement. Trends Cell Biol (2014) 24:61–72. doi: 10.1016/j.tcb.2013.09.005
9. Egen JG, Allison JP. Cytotoxic T Lymphocyte Antigen-4 Accumulation in the Immunological Synapse is Regulated by TCR Signal Strength. Immunity (2002) 16:23–35. doi: 10.1016/s1074-7613(01)00259-x
10. Bunnell SC, Hong DI, Kardon JR, Yamazaki T, McGlade CJ, Barr VA, et al. T Cell Receptor Ligation Induces the Formation of Dynamically Regulated Signaling Assemblies. J Cell Biol (2002) 158:1263–75. doi: 10.1083/jcb.200203043
11. Ehrlich LIR, Ebert PJR, Krummel MF, Weiss A, Davis MM. Dynamics of P56lck Translocation to the T Cell Immunological Synapse Following Agonist and Antagonist Stimulation. Immunity (2002) 17:809–22. doi: 10.1016/s1074-7613(02)00481-8
12. Bonello G, Blanchard N, Montoya MC, Aguado E, Langlet C, He HT, et al. Dynamic Recruitment of the Adaptor Protein LAT: LAT Exists in Two Distinct Intracellular Pools and Controls its Own Recruitment. J Cell Sci (2004) 117:1009–16. doi: 10.1242/jcs.00968
13. Bouchet J, Del Río-Iñiguez I, Lasserre R, Agüera-Gonzalez S, Cuche C, Danckaert A, et al. Rac1-Rab11-FIP3 Regulatory Hub Coordinates Vesicle Traffic With Actin Remodeling and T-Cell Activation. EMBO J (2016) 35:1160–74. doi: 10.15252/embj.201593274
14. Ritter AT, Angus KL, Griffiths GM. The Role of the Cytoskeleton at the Immunological Synapse. Immunol Rev (2013) 256:107–17. doi: 10.1111/imr.12117
15. Martín-Cófreces NB, Sánchez-Madrid F. Sailing to and Docking at the Immune Synapse: Role of Tubulin Dynamics and Molecular Motors. Front Immunol (2018) 9:1174. doi: 10.3389/fimmu.2018.01174
16. Hammer JA, Wang JC, Saeed M, Pedrosa AT. Origin, Organization, Dynamics, and Function of Actin and Actomyosin Networks at the T Cell Immunological Synapse. Annu Rev Immunol (2019) 37:201–24. doi: 10.1146/annurev-immunol-042718-04134
17. Soares H, Lasserre R, Alcover A. Orchestrating Cytoskeleton and Intracellular Vesicle Traffic to Build Functional Immunological Synapses. Immunol Rev (2013) 256:118–32. doi: 10.1111/imr.12110
18. Onnis A, Finetti F, Baldari CT. Vesicular Trafficking to the Immune Synapse: How to Assemble Receptor-Tailored Pathways From a Basic Building Set. Front Immunol (2016) 7:50. doi: 10.3389/fimmu.2016.00050
19. Finetti F, Cassioli C, Baldari CT. Transcellular Communication at the Immunological Synapse: A Vesicular Traffic-Mediated Mutual Exchange. F1000Res (2017 1880) 6. doi: 10.12688/f1000research.11944.1
20. Dustin ML, Chan AC. Signaling Takes Shape in Review the Immune System. Cell (2000) 103:283–94. doi: 10.1016/s0092-8674(00)00120-3
21. Kawasaki K, Ernst RK, Miller SI. 3-O-Deacylation of Lipid A by PagL, a PhoP/PhoQ-Regulated Deacylase of Salmonella Typhimurium, Modulates Signaling Through Toll-Like Receptor 4. J Biol Chem (2004) 279:20044–8. doi: 10.1074/jbc.M401275200
22. Newman RM, Salunkhe P, Godzik A, Reed JC. Identification and Characterization of a Novel Bacterial Virulence Factor That Shares Homology With Mammalian Toll/interleukin-1 Receptor Family Proteins. Infect Immun (2006) 74:594–601. doi: 10.1128/IAI.74.1.594-601.2006
23. Schesser K, Spiik AK, Dukuzumuremyi JM, Neurath MF, Pettersson S, Wolf-Watz H. The yopJ Locus is Required for Yersinia-Mediated Inhibition of NF-kappaB Activation and Cytokine Expression: YopJ Contains a Eukaryotic SH2-Like Domain That is Essential for its Repressive Activity. Mol Microbiol (1998) 28:1067–79. doi: 10.1046/j.1365-2958.1998.00851.x
24. Wang J, Ge P, Lei Z, Lu Z, Qiang L, Chai Q, et al. Mycobacterium Tuberculosis Protein Kinase G Acts as an Unusual Ubiquitinating Enzyme to Impair Host Immunity. EMBO Rep (2021) 22:e52175. doi: 10.15252/embr.202052175
25. Carlin AF, Lewis AL, Varki A, Nizet V. Group B Streptococcal Capsular Sialic Acids Interact With Siglecs (Immunoglobulin-Like Lectins) on Human Leukocytes. J Bacteriol (2007) 189:1231–7. doi: 10.1128/JB.01155-06
26. Carlin AF, Uchiyama S, Chang YC, Lewis AL, Nizet V, Varki A. Molecular Mimicry of Host Sialylated Glycans Allows a Bacterial Pathogen to Engage Neutrophil Siglec-9 and Dampen the Innate Immune Response. Blood (2009) 113:3333–6. doi: 10.1182/blood-2008-11-187302
27. Zhou D, Galán J. Salmonella Entry Into Host Cells: The Work in Concert of Type III Secreted Effector Proteins. Microbes Infect (2001) 3:1293–8. doi: 10.1016/s1286-4579(01)01489-7
28. Pai RK, Convery M, Hamilton TA, Boom WH, Harding CV. Inhibition of IFN-Gamma-Induced Class II Transactivator Expression by a 19-kDa Lipoprotein From Mycobacterium Tuberculosis: A Potential Mechanism for Immune Evasion. J Immunol (2003) 171:175–84. doi: 10.4049/jimmunol.171.1.175
29. Pennini ME, Pai RK, Schultz DC, Boom WH, Harding CV. Mycobacterium Tuberculosis 19-kDa Lipoprotein Inhibits IFN-Gamma-Induced Chromatin Remodeling of MHC2TA by TLR2 and MAPK Signaling. J Immunol (2006) 176:4323–30. doi: 10.4049/jimmunol.176.7.4323
30. Madan-Lala R, Sia JK, King R, Adekambi T, Monin L, Khader SA, et al. Mycobacterium Tuberculosis Impairs Dendritic Cell Functions Through the Serine Hydrolase Hip1. J Immunol (2014) 192:4263–72. doi: 10.4049/jimmunol.1303185
31. Barbaro Ade L, Tosi G, Frumento G, Bruschi E, D'Agostino A, Valle MT, et al. Block of Stat-1 Activation in Macrophages Phagocytosing Bacteria Causes Reduced Transcription of CIITA and Consequent Impaired Antigen Presentation. Eur J Immunol (2002) 32:1309–18. doi: 10.1002/1521-4141(200205)32:5<1309::aid-immu1309>3.0.co;2-4
32. Coletta S, Battaggia G, Della Bella C, Furlani M, Hauke M, Faass L, et al. ADP-Heptose Enables Helicobacter Pylori to Exploit Macrophages as a Survival Niche by Suppressing Antigen-Presenting HLA-II Expression. FEBS Lett (2021) 595:2160–8. doi: 10.1002/1873-3468.14156
33. Zhong G, Fan T, Liu L. Chlamydia Inhibits Interferon Gamma-Inducible Major Histocompatibility Complex Class II Expression by Degradation of Upstream Stimulatory Factor 1. J Exp Med (1999) 189:1931–8. doi: 10.1084/jem.189.12.1931
34. Gogoi M, Ravikumar V, Dixit NM, Chakravortty D. Salmonella Escapes Antigen Presentation Through K63 Ubiquitination Mediated Endosomal Proteolysis of via Modulation of Endosomal Acidification in Dendritic Cells. Pathog Dis (2018) 76. doi: 10.1093/femspd/ftx125
35. Vergne I, Chua J, Lee HH, Lucas M, Belisle J, Deretic V. Mechanism of Phagolysosome Biogenesis Block by Viable Mycobacterium Tuberculosis. Proc Natl Acad Sci USA (2005) 102:4033–8. doi: 10.1073/pnas.0409716102
36. Fratti RA, Chua J, Vergne I, Deretic V. Mycobacterium Tuberculosis Glycosylated Phosphatidylinositol Causes Phagosome Maturation Arrest. Proc Natl Acad Sci USA (2003) 100:5437–42. doi: 10.1073/pnas.0737613100
37. Vergne I, Chua J, Deretic V. Mycobacterium Tuberculosis Phagosome Maturation Arrest: Selective Targeting of PI3P-Dependent Membrane Trafficking. Traffic (2003) 4:600–6. doi: 10.1034/j.1600-0854.2003.00120.x
38. Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium Tuberculosis Protein Tyrosine Phosphatase (PtpA) Excludes Host Vacuolar-H+-ATPase to Inhibit Phagosome Acidification. Proc Natl Acad Sci USA (2011) 108:19371–6. doi: 10.1073/pnas.1109201108
39. Pires D, Bernard EM, Pombo JP, Carmo N, Fialho C, Gutierrez MG, et al. Mycobacterium Tuberculosis Modulates miR-106b-5p to Control Cathepsin S Expression Resulting in Higher Pathogen Survival and Poor T-Cell Activation. Front Immunol (2017) 8:1819. doi: 10.3389/fimmu.2017.01819
40. Sande OJ, Karim AF, Li Q, Ding X, Harding CV, Rojas RE, et al. Mannose-Capped Lipoarabinomannan From Mycobacterium Tuberculosis Induces CD4+ T Cell Anergy via GRAIL. J Immunol (2016) 196:691–702. doi: 10.4049/jimmunol.1500710
41. Boulkroun S, Guenin-Macé L, Thoulouze MI, Monot M, Merckx A, Langsley G, et al. Mycolactone Suppresses T Cell Responsiveness by Altering Both Early Signaling and Posttranslational Events. J Immunol (2010) 184:1436–44. doi: 10.4049/jimmunol.0902854
42. Grant GA. Regulatory Mechanism of Mycobacterium Tuberculosis Phosphoserine Phosphatase Serb2. Biochemistry (2017) 56:6481–90. doi: 10.1021/acs.biochem.7b01082
43. Sun J, Wang X, Lau A, Liao TY, Bucci C, Hmama Z. Mycobacterial Nucleoside Diphosphate Kinase Blocks Phagosome Maturation in Murine RAW 264.7 Macrophages. PloS One (2010) 5:e8769. doi: 10.1371/journal.pone.0008769
44. Sendide K, Deghmane A, Pechkovsky D, Av-Gay Y, Talal A, Hmama Z. Mycobacterium Bovis BCG Attenuates Surface Expression of Mature Class II Molecules Through IL-10-Dependent Inhibition of Cathepsin S. J Immunol (2005) 175:5324–32. doi: 10.4049/jimmunol.175.8.5324
45. Norris FA, Wilson MP, Wallis TS, Galyov EE, Majerus PW. SopB, a Protein Required for Virulence of Salmonella Dublin, is an Inositol Phosphate Phosphatase. Proc Natl Acad Sci USA (1998) 95:14057–9. doi: 10.1073/pnas.95.24.14057
46. Alix E, Godlee C, Cerny O, Blundell S, Tocci R, Matthews S, et al. The Tumour Suppressor TMEM127 is a Nedd4-Family E3 Ligase Adaptor Required by Salmonella SteD to Ubiquitinate and Degrade MHC Class II Molecules. Cell Host Microbe (2020) 28:54–68.e7. doi: 10.1016/j.chom.2020.04.024
47. Bayer-Santos E, Durkin CH, Rigano LA, Kupz A, Alix E, Cerny O, et al. The Salmonella Effector SteD Mediates MARCH8-Dependent Ubiquitination of MHC II Molecules and Inhibits T Cell Activation. Cell Host Microbe (2016) 20:584–95. doi: 10.1016/j.chom.2016.10.007
48. Cerny O, Godlee C, Tocci R, Cross NE, Shi H, Williamson JC, et al. CD97 Stabilises the Immunological Synapse Between Dendritic Cells and T Cells and is Targeted for Degradation by the Salmonella Effector SteD. PloS Pathog (2021) 17:e1009771. doi: 10.1371/journal.ppat.1009771
49. Hardt WD, Chen LM, Schuebel KE, Bustelo XR, Galán JE. S. Typhimurium Encodes an Activator of Rho GTPases That Induces Membrane Ruffling and Nuclear Responses in Host Cells. Cell (1998) 93:815–26. doi: 10.1016/s0092-8674(00)81442-7
50. Fu Y, Galán JE. A Salmonella Protein Antagonizes Rac-1 and Cdc42 to Mediate Host-Cell Recovery After Bacterial Invasion. Nature (1999) 401:293–7. doi: 10.1038/45829
51. Truong D, Boddy KC, Canadien V, Brabant D, Fairn GD, D'Costa VM, et al. Salmonella Exploits Host Rho GTPase Signalling Pathways Through the Phosphatase Activity of SopB. Cell Microbiol (2018) 20:e12938. doi: 10.1111/cmi.12938
52. Hernandez LD, Hueffer K, Wenk MR, Galán JE. Salmonella Modulates Vesicular Traffic by Altering Phosphoinositide Metabolism. Science (2004) 304:1805–7. doi: 10.1126/science.1098188
53. Spanò S, Gao X, Hannemann S, Lara-Tejero M, Galán JE. A Bacterial Pathogen Targets a Host Rab-Family GTPase Defense Pathway With a GAP. Cell Host Microbe (2016) 19:216–26. doi: 10.1016/j.chom.2016.01.004
54. Onnis A, Finetti F, Patrussi L, Gottardo M, Cassioli C, Spanò S, et al. The Small GTPase Rab29 is a Common Regulator of Immune Synapse Assembly and Ciliogenesis. Cell Death Differ (2015) 22:1687–99. doi: 10.1038/cdd.2015.17
55. Molinari M, Salio M, Galli C, Norais N, Rappuoli R, Lanzavecchia A, et al. Selective Inhibition of Ii-Dependent Antigen Presentation by Helicobacter Pylori Toxin VacA. J Exp Med (1998) 187:135–40. doi: 10.1084/jem.187.1.135
56. Wang YH, Gorvel JP, Chu YT, Wu JJ, Lei HY. Helicobacter Pylori Impairs Murine Dendritic Cell Responses to Infection. PloS One (2010) 5:e10844. doi: 10.1371/journal.pone.0010844
57. Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, et al. The Helicobacter Pylori Vacuolating Toxin Inhibits T Cell Activation by Two Independent Mechanisms. J Exp Med (2003) 198:1887–97. doi: 10.1084/jem.20030621
58. Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter Pylori Vacuolating Cytotoxin Inhibits T Lymphocyte Activation. Science (2003) 301:1099–102. doi: 10.1126/science.1086871
59. Sewald X, Gebert-Vogl B, Prassl S, Barwig I, Weiss E, Fabbri M, et al. Integrin Subunit CD18 Is the T-Lymphocyte Receptor for the Helicobacter Pylori Vacuolating Cytotoxin. Cell Host Microbe (2008) 3:20–9. doi: 10.1016/j.chom.2007.11.003
60. Lem L, Riethof DA, Scidmore-Carlson M, Griffiths GM, Hackstadt T, Brodsky FM. Enhanced Interaction of HLA-DM With HLA-DR in Enlarged Vacuoles of Hereditary and Infectious Lysosomal Diseases. J Immunol (1999) 162:523–32.
61. Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, et al. Genomic Landscape of the Individual Host Response and Outcomes in Sepsis: A Prospective Cohort Study. Lancet Respir Med (2016) 4:259–71. doi: 10.1016/S2213-2600(16)00046-1
62. Popugailo A, Rotfogel Z, Supper E, Hillman D, Kaempfer R. Staphylococcal and Streptococcal Superantigens Trigger B7/CD28 Costimulatory Receptor Engagement to Hyperinduce Inflammatory Cytokines. Front Immunol (2019) 10:942. doi: 10.3389/fimmu.2019.00942
63. Boulton IC, Gray-Owen SD. Neisserial Binding to CEACAM1 Arrests the Activation and Proliferation of CD4+ T Lymphocytes. Nat Immunol (2002) 3:229–36. doi: 10.1038/ni769
64. Yao T, Mecsas J, Healy JI, Falkow S, Chien Y. Suppression of T and B Lymphocyte Activation by a Yersinia Pseudotuberculosis Virulence Factor, yopH. J Exp Med (1999) 190:1343–50. doi: 10.1084/jem.190.9.1343
65. Alonso A, Bottini N, Bruckner S, Rahmouni S, Williams S, Schoenberger SP, et al. Lck Dephosphorylation at Tyr-394 and Inhibition of T Cell Antigen Receptor Signaling by Yersinia Phosphatase YopH. J Biol Chem (2004) 279:4922–8. doi: 10.1074/jbc.M308978200
66. Gerke C, Falkow S, Chien YH. The Adaptor Molecules LAT and SLP-76 are Specifically Targeted by Yersinia to Inhibit T Cell Activation. J Exp Med (2005) 201:361–71. doi: 10.1084/jem.20041120
67. de la Puerta ML, Trinidad AG, del Carmen Rodríguez M, Bogetz J, Sánchez Crespo M, Mustelin M, et al. Characterization of New Substrates Targeted by Yersinia Tyrosine Phosphatase YopH. PloS One (2009) 4:e4431. doi: 10.1371/journal.pone.000443
68. Von Pawel-Rammingen U, Telepnev MV, Schmidt G, Aktories K, Wolf-Watz H, Rosqvist R. GAP Activity of the Yersinia YopE Cytotoxin Specifically Targets the Rho Pathway: A Mechanism for Disruption of Actin Microfilament Structure. Mol Microbiol (2000) 36:737–48. doi: 10.1046/j.1365-2958.2000.01898.x
69. Shao F, Vacratsis PO, Bao Z, Bowers KE, Fierke CA, Dixon JE. Biochemical Characterization of the Yersinia YopT Protease: Cleavage Site and Recognition Elements in Rho GTPases. Proc Natl Acad Sci USA (2003) 100:904–9. doi: 10.1073/pnas.252770599
70. Paccani SR, Dal Molin F, Benagiano M, Ladant D, D'Elios MM, Montecucco C, et al. Suppression of T-Lymphocyte Activation and Chemotaxis by the Adenylate Cyclase Toxin of Bordetella Pertussis. Infect Immun (2008) 76:2822–32. doi: 10.1128/IAI.00200-08
71. Paccani SR, Finetti F, Davi M, Patrussi L, D'Elios MM, Ladant D, et al. The Bordetella Pertussis Adenylate Cyclase Toxin Binds to T Cells via LFA-1 and Induces its Disengagement From the Immune Synapse. J Exp Med (2011) 208:1317–30. doi: 10.1084/jem.20101558
72. Arumugham VB, Ulivieri C, Onnis A, Finetti F, Tonello F, Ladant D, et al. Compartmentalized Cyclic AMP Production by the Bordetella Pertussis and Bacillus Anthracis Adenylate Cyclase Toxins Differentially Affects the Immune Synapse in T Lymphocytes. Front Immunol (2018) 9:919. doi: 10.3389/fimmu.2018.00919
73. Paccani SR, Tonello F, Ghittoni R, Natale M, Muraro L, D'Elios MM, et al. Anthrax Toxins Suppress T Lymphocyte Activation by Disrupting Antigen Receptor Signaling. J Exp Med (2005) 201:325–31. doi: 10.1084/jem.20041557
74. Chardin P, Bouquet P, Madaule P, Popoff MR, Rubin EJ, Gill DM. The Mammalian G Protein rhoC is ADP-Ribosylated by Clostridium Botulinum Exoenzyme C3 and Affects Actin Microfilaments in Vero Cells. EMBO J (1989) 8:1087–92. doi: 10.1002/j.1460-2075.1989.tb03477.x
75. Goldberg MB, Theriot JA. Shigella Flexneri Surface Protein IcsA is Sufficient to Direct Actin-Based Motility. Proc Natl Acad Sci USA (1995) 92:6572–6. doi: 10.1073/pnas.92.14.6572
76. Konradt C, Frigimelica E, Nothelfer K, Puhar A, Salgado-Pabon W, di Bartolo V, et al. The Shigella Flexneri Type Three Secretion System Effector IpgD Inhibits T Cell Migration by Manipulating Host Phosphoinositide Metabolism. Cell Host Microbe (2011) 9:263–72. doi: 10.1016/j.chom.2011.03.010
77. Samassa F, Ferrari ML, Husson J, Mikhailova A, Porat Z, Sidaner F, et al. Shigella Impairs Human T Lymphocyte Responsiveness by Hijacking Actin Cytoskeleton Dynamics and T Cell Receptor Vesicular Trafficking. Cell Microbiol (2020) 22:e13166. doi: 10.1111/cmi.13166
78. Ferrari ML, Malardé V, Grassart A, Salavessa L, Nigro G, Descorps-Declere S, et al. Shigella Promotes Major Alteration of Gut Epithelial Physiology and Tissue Invasion by Shutting Off Host Intracellular Transport. Proc Natl Acad Sci USA (2019) 116:13582–91. doi: 10.1073/pnas.1902922116
79. Kocks C, Hellio R, Gounon P, Ohayon H, Cossart P. Polarized Distribution of Listeria Monocytogenes Surface Protein ActA at the Site of Directional Actin Assembly. J Cell Sci (1993) 105:699–710. doi: 10.1242/jcs.105.3.699
80. Ingmundson A, Delprato A, Lambright DG, Roy CR. Legionella Pneumophila Proteins That Regulate Rab1 Membrane Cycling. Nature (2007) 450:365–9. doi: 10.1038/nature06336
81. Machner MP, Isberg RR. Targeting of Host Rab GTPase Function by the Intravacuolar Pathogen Legionella Pneumophila. Dev Cell (2006) 11:47–56. doi: 10.1016/j.devcel.2006.05.013
82. Sohn YS, Shin HC, Park WS, Ge J, Kim CH, Lee BL, et al. Lpg0393 of Legionella Pneumophila is a Guanine-Nucleotide Exchange Factor for Rab5, Rab21 and Rab22. PloS One (2015) 10:e0118683. doi: 10.1371/journal.pone.0118683
83. Via LE, Deretic D, Ulmer RJ, Hibler NS, Huber LA, Deretic V. Arrest of Mycobacterial Phagosome Maturation is Caused by a Block in Vesicle Fusion Between Stages Controlled by Rab5 and Rab7. J Biol Chem (1997) 272:13326–31. doi: 10.1074/jbc.272.20.13326
84. Vergne I, Chua J, Deretic V. Tuberculosis Toxin Blocking Phagosome Maturation Inhibits a Novel Ca2+/Calmodulin-PI3K Hvps34 Cascade. J Exp Med (2003) 198:653–9. doi: 10.1084/jem.20030527
85. Bania J, Gatti E, Lelouard H, David A, Cappello F, Weber E, et al. Human Cathepsin S, But Not Cathepsin L, Degrades Efficiently MHC Class II-Associated Invariant Chain in Nonprofessional APCs. Proc Natl Acad Sci USA (2003) 10:6664–9. doi: 10.1073/pnas.1131604100
86. Fackler OT, Alcover A, Schwartz O. Modulation of the Immunological Synapse: A Key to HIV-1 Pathogenesis? Nat Rev Immunol (2007) 7:310–7. doi: 10.1038/nri2041
87. Bayliss RJ, Piguet V. Masters of Manipulation: Viral Modulation of the Immunological Synapse. Cell Microbiol (2018) 20:e12944. doi: 10.1111/cmi.12944
88. Brunner K, Samassa F, Sansonetti PJ, Phalipon A. Shigella-Mediated Immunosuppression in the Human Gut: Subversion Extends From Innate to Adaptive Immune Responses. Hum Vaccin. Immunother. (2019) 15:1317–25. doi: 10.1080/21645515.2019.1594132
89. Capitani N, Patrussi L, D'Elios MM, Baldari CT. “Dysfunctional Immune Synapses in T Cell Immunodeficiencies”. In: D’Elios MN, Baldari CT, Annunziato F, editors. Cellular Primary Immunodeficiencies. Springer Cham: Springer (2021). p. 43–63.
90. Voges M, Bachmann V, Kammerer R, Gophna U, Hauck CR. CEACAM1 Recognition by Bacterial Pathogens is Species-Specific. BMC Microbiol (2010) 10:117. doi: 10.1186/1471-2180-10-117
91. Brewer ML, Dymock D, Brady RL, Singer BB, Virji M, Hill DJ. Fusobacterium Spp. Target Human CEACAM1 via the Trimeric Autotransporter Adhesin CbpF. J Oral. Microbiol (2019) 11:1565043. doi: 10.1080/20002297.2018.1565043
92. Galaski J, Shhadeh A, Umaña A, Yoo CC, Arpinati L, Isaacson B, et al. Fusobacterium Nucleatum CbpF Mediates Inhibition of T Cell Function Through CEACAM1 Activation. Front. Cell Infect Microbiol (2021) 11:69254. doi: 10.3389/fcimb.2021.69254
93. Khairnar V, Duhan V, Patil AM, Zhou F, Bhat H, Thoens C, et al. CEACAM1 Promotes CD8(+) T Cell Responses and Improves Control of a Chronic Viral Infection. Nat Commun (2018) 9:2561. doi: 10.1038/s41467-018-04832-2
94. Onnis A, Baldari CT. Orchestration of Immunological Synapse Assembly by Vesicular Trafficking. Front Cell Dev Biol (2019) 7:110. doi: 10.3389/fcell.2019.00110
95. Lemichez E, Aktories K. Hijacking of Rho GTPases During Bacterial Infection. Exp Cell Res (2013) 319:2329–36. doi: 10.1016/j.yexcr.2013.04.021
96. Stradal TEB, Schelhaas M. Actin Dynamics in Host-Pathogen Interaction. FEBS Lett (2018) 592:3658–69. doi: 10.1002/1873-3468.13173
97. Gawden-Bone CM, Frazer GL, Richard AC, Ma CY, Strege K, Griffiths GM. PIP5 Kinases Regulate Membrane Phosphoinositide and Actin Composition for Targeted Granule Secretion by Cytotoxic Lymphocytes. Immunity (2018) 49:427–437.e4. doi: 10.1016/j.immuni.2018.08.017
98. Pizarro-Cerdá J, Kühbacher A, Cossart P. Phosphoinositides and Host-Pathogen Interactions. Biochim Biophys Acta (2015) 1851:911–8. doi: 10.1016/j.bbalip.2014.09.011
99. Iezzi G, Karjalainen K, Lanzavecchia A. The Duration of Antigenic Stimulation Determines the Fate of Naive and Effector T Cells. Immunity (1998) 8:89–95. doi: 10.1016/s1074-7613(00)80461-6
100. Spanò S, Galán JE. Taking Control: Hijacking of Rab GTPases by Intracellular Bacterial Pathogens. Small GTPases. (2018) 9:182–91. doi: 10.1080/21541248.2017.1336192
101. Spanò S, Liu X, Galán JE. Proteolytic Targeting of Rab29 by an Effector Protein Distinguishes the Intracellular Compartments of Human-Adapted and Broad-Host Salmonella. Proc Natl Acad Sci USA (2011) 108:18418–23. doi: 10.1073/pnas.1111959108
102. Savitskiy S, Itzen A. SopD From Salmonella Specifically Inactivates Rab8. Biochim Biophys Acta Proteins Proteom. (2021) 186:140661. doi: 10.1016/j.bbapap.2021.140661
103. Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple Inhibitory Ligands Induce Impaired T-Cell Immunologic Synapse Function in Chronic Lymphocytic Leukemia That can be Blocked With Lenalidomide: Establishing a Reversible Immune Evasion Mechanism in Human Cancer. Blood (2012) 120:1412–21. doi: 10.1182/blood-2012-02-411678
Keywords: pathogens, immunological synapse, Antigen Presenting Cell (APC), major histocompatibility complex class II (MHCII), T cell receptor (TCR), actin cytoskeleton
Citation: Capitani N and Baldari CT (2022) The Immunological Synapse: An Emerging Target for Immune Evasion by Bacterial Pathogens. Front. Immunol. 13:943344. doi: 10.3389/fimmu.2022.943344
Received: 13 May 2022; Accepted: 20 June 2022;
Published: 13 July 2022.
Edited by:
Marina De Bernard, University of Padua, ItalyReviewed by:
Yung-Fu Chang, Cornell University, United StatesCopyright © 2022 Capitani and Baldari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nagaja Capitani, Y2FwaXRhbmkyQHVuaXNpLml0; Cosima T. Baldari, Y29zaW1hLmJhbGRhcmlAdW5pc2kuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.