 Shaoying Zhang
Shaoying Zhang Shemin Lu
Shemin Lu Zongfang Li
Zongfang Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 August 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.941721
This article is part of the Research Topic Hepatic Immune Response underlying Liver Cirrhosis and Portal Hypertension View all 19 articles
The liver is a site of complex immune activity. The hepatic immune system tolerates harmless immunogenic loads in homeostasis status, shelters liver function, while maintaining vigilance against possible infectious agents or tissue damage and providing immune surveillance at the same time. Activation of the hepatic immunity is initiated by a diverse repertoire of hepatic resident immune cells as well as non-hematopoietic cells, which can sense “danger signals” and trigger robust immune response. Factors that mediate the regulation of hepatic immunity are elicited not only in liver, but also in other organs, given the dual blood supply of the liver via both portal vein blood and arterial blood. Emerging evidence indicates that inter-organ crosstalk between the liver and other organs such as spleen, gut, lung, adipose tissue, and brain is involved in the pathogenesis of liver diseases. In this review, we present the features of hepatic immune regulation, with particular attention to the correlation with factors from extrahepatic organ. We describe the mechanisms by which other organs establish an immune association with the liver and then modulate the hepatic immune response. We discuss their roles and distinct mechanisms in liver homeostasis and pathological conditions from the cellular and molecular perspective, highlighting their potential for liver disease intervention. Moreover, we review the available animal models and methods for revealing the regulatory mechanisms of these extrahepatic factors. With the increasing understanding of the mechanisms by which extrahepatic factors regulate liver immunity, we believe that this will provide promising targets for liver disease therapy.
The liver is the largest organ engaged in metabolic, nutrient storage and detoxification activities, but has increasingly been recognized as a unique immune organ with its own immune features. Since the dual blood supply of portal venous and systemic blood transport a large number of foreign but harmless molecules, the liver immune cells are largely in an activated state due to continuous exposure to low concentrations of antigens, and the default immune status of the liver is anti-inflammatory or immune-tolerant (1–4). Generally, the hepatic immune system tolerates harmless molecules under healthy condition. In face of immune activation challenge posed by pathogens or tissue damage, however, the liver could mount rapid and robust immune response and attempt to resolve inflammation to maintain liver homeostasis. Otherwise, failure to clear ‘dangerous’ stimuli or appropriately regulate activated immune mechanisms can lead to chronic and pathological inflammation (5–8). Maintenance of liver function requires a balance between immunity and tolerance, therefore, appropriate regulation of the complex hepatic immune activities is necessary.
Chronic liver disease is generally a multi-stage, multi-hit process; it is therefore not surprising that multiple cells within the liver contribute to the immune regulation during disease progression. Active modulation of immune responses in the liver could stem from its unique microenvironment including certain cell types like resident immune cells (9–13), hepatocytes (14), hepatic stellate cells (HSCs) (15) and liver sinusoidal endothelial cells (LSECs) (16, 17). In addition to complications relating to the liver, patients with chronic liver disease also develop concomitant extrahepatic functional disturbances of multiple organ systems (18–20), which in turn affect the progression of liver disease. Given this, the contribution of signals outside the liver to the ‘balance control’ of hepatic immune calls for meticulous exploration. In this review, we briefly describe the changes in hepatic immune status from homeostasis to disease, as well as the cellular, molecular and neural factors that mediate these changes. From the perspective of organ-organ communication, we elaborate on the effects of other organs on hepatic immune regulation and liver disease progression, and discuss the available animal models and methods for revealing the regulatory mechanisms of the extrahepatic factors.
The liver has its special intraparenchymal vascular conduits named hepatic sinusoid. The hepatic sinusoids involve multiple and disparate cell types: LSECs form the walls, HSCs harbor in the Space of Disse between the sinusoidal wall and the adjacent hepatocytes, and various immune cells locate within the sinusoid (21). All these cells play active roles in regulating the hepatic immune. The liver is considered a unique immunological organ for its predominant innate immune role, as it contains an unusually large number of innate immune cells, including myeloid cells like macrophages, dendritic cells (DCs), innate lymphocytes like natural killer (NK) cells, innate lymphoid cells (ILCs), and innate-like T lymphocytes like NKT cells, and γδ T cells (22). Of course, the adaptive immune cells (T and B cells) also count in the hepatic immunity (23). Here, we outline the hepatic immune state in homeostasis and their alterations upon activation, and summarize the mechanisms underlying the alterations.
Hepatic resident immune cells together with non-hematopoietic cell populations maintain hepatic immune tolerance during healthy state. The liver resident macrophages, Kupffer cells (KCs), play a key role by producing anti-inflammatory mediators such as IL-10 and prostaglandins (24, 25), and downregulating expression of co-stimulatory molecules to limit the adaptive immune response (26). Hepatic resident DCs appear phenotypically immature and are less potent activators of T cells, and shown to produce significantly more IL-10 compared with peripheral derived myeloid DCs (27). Non-hematopoietic cells such as LSECs, HSCs and hepatocytes possess the ability to directly present antigen to T cells, but their presentation of antigens in the liver biases T cells towards tolerance for their lack of co-stimulatory molecules (e.g., driving naïve CD4+ T cells differentiation to regulatory T cells rather than to T helper cells) (28–31). Meanwhile, LSECs and hepatocytes constitutively express IL-10 and TGF-β (32, 33). Besides, the healthy liver also has basal expression of pro-inflammatory cytokines (including IL-2, IL-7, IL-12, IL-15 and IFN-γ). The complex cytokine milieu helps orchestrate the homeostasis (34).
Once the hepatic homeostasis sheltered by local and systemic tolerance is disrupted, the innate immune system is first activated, driving the full development of inflammatory hepatocellular injury. Depending on the underlying liver disease, such as viral hepatitis, autoimmune hepatitis, cholestasis, liver ischemia reperfusion or metabolic associated steatohepatitis, various triggers mediate immune-cell activation. The initiative inflammatory activation of HSCs and KCs results in the chemokine-mediated infiltration of monocytes (35), neutrophils (36), NK and NKT cells (37). KCs and the recruited monocyte-derived macrophages (MoMFs), as key cellular components of the liver, adapt their phenotype to local signaling, taking an active part in either inflammation or the subsequent resolution (38, 39). The ultimate outcome of the intrahepatic immune response depends largely on the functional diversity of macrophages. Innate lymphocytes like NK and NKT cells are another source of immune-regulatory cytokines in diseased livers, contributing to the elimination of activated myofibroblasts and infected or injured cells (40). These disease conditions are also closely linked to T cell immunity, in which the heterogeneous pool of hepatic T cells is activated upon shifting to inflammation (41, 42), Th1 and Th17 responses are induced, resulting in the secretion of immune-stimulatory cytokine as well as direct cytopathic function (43). A systematic understanding of the initiation and regulation of the activated hepatic immune regulation is critical for the development of liver disease therapy strategies based on intervention of hepatic immunity.
Mechanisms of hepatic immune regulation involves cell activation, molecule interaction and neural signal transmission. At the cellular level, most types of liver ‘insults’ damage epithelial cells, leading to the release of inflammatory mediators and the initiation of inflammatory cascade. In response to these inflammatory signals, KCs are first activated (44), releasing pro-inflammatory mediators that lead to the recruitment of circulatory-derived immune cells into the inflamed liver (45). MoMFs, as the primary leukocytes being recruited, could further recruit T cells and neutrophils to promote fibrosis by generating pro-inflammatory cytokines (e.g., TNF-α, IL-6, and IL-1β), and secreting pro-inflammatory chemokines (e.g., CCL2, CCL5, and CXCL2), thus further amplifying the inflammatory response (46–48). Also, the behavior of MoMFs themselves is regulated by the complex hepatic microenvironment. Emerging understandings of hepatic macrophage heterogeneity identify a group of CD11bhiF4/80intLY6Clow restorative macrophage as a phenotypical switch subpopulation derived from the pro-inflammatory LY6Chi subset, contributing to inflammation resolution (49). Another important myeloid cells, DCs, exert their role in hepatic immune regulation by forming a bridge between the innate and the adaptive immune system (50). Although T cells are also activated during hepatic inflammation, the mechanisms of their activation and “shift” (to Th1 and Th17) are not fully elucidated, which might require further analysis of the interaction between hepatic DCs and T cells.
Molecularly, pathogen-associated molecular patterns (PAMPs) and danger/death-associated molecular patterns (DAMPs) are the most famous “danger signals”. PAMPs are conserved structures vital to pathogens, presenting in microbes and absent in eukaryotes (51, 52). DAMPs represent damaged cells of the host which are a threat to self (53). PAMPs and DAMPs initiate the immune response via pattern recognition receptors (PRRs), which are present in immune cells, as well as in LSECs and HSCs (51, 54, 55). The recognition of pathogen molecules by PRRs would lead to activation of the complement cascade, cytokines, antimicrobial peptides and antigen-presenting cells, resulting in a complex interplay of pro- and anti-inflammatory responses and immunogenic and suppressive responses in the host (53, 56). Among PRRs, Toll-like receptors (TLRs) are most extensively studied, responding to most DAMPs and PAMPs, and have a major influence in liver diseases (57). Nucleotide binding oligomerization domain-like receptors (NLRs) represent another subtype of PRRs, for example, NLPR3 is one of the NLRs expressed on inflammasome (44). Inflammasomes are intracellular multiprotein complexes that sense intracellular danger signals including DAMPs, PAMPs, and ROS, and the activation of inflammasomes triggers a pro-inflammatory response commonly associated with caspase-1 activation followed by activated secretion of IL-1β and IL-18 (58). In addition to serving as an important mechanism for macrophage activation signal transduction, inflammasome hyperactivation can also result in hepatocyte pyroptosis, a specific form of cell death, leading to increased liver inflammation and fibrosis development in mice (59). Besides, exosomes have also been identified to play important roles in hepatic immune regulation by mediating the intrahepatic cell-cell communication and transmission of information from other organs to the liver (60).
Furthermore, the liver is innervated by both the sympathetic and the parasympathetic nerve systems. These nerves are derived from the splanchnic and vagal nerves that surround the portal vein, hepatic artery, and bile duct. The nervous system and the immune system communicate in response to pathogen invasion, tissue injury, and other “insults” (61). Intrahepatic efferent nerves endings containing catecholaminergic (releasing neurotransmitters like epinephrine and norepinephrine), cholinergic (releasing neurotransmitter acetylcholine) nerves terminate at the Space of Disse (62, 63), and trigger responsiveness in effector cells. Findings in preclinical conditions have indicated the use of vagus nerve stimulation and acetylcholine (Ach) receptor agonists and centrally acting AchE inhibitors as therapies for liver inflammatory diseases. The exact mechanisms of hepatic nervous system regulating the function of hepatic immune cells and secretion of effector factors remain to be further studied.
Hepatic immunity is a complex and adaptable process, which is flexibly regulated by the changing hepatic responses in homeostasis and various disease states. In addition to complications relating to the liver, patients with liver disease often develop concomitant extrahepatic functional disturbances of multiple organ systems; and the liver itself also intensively participates in the acute phase reaction in response to inflammation that occurs in other organs. There have been clues of the liver communicating with other organs through molecular and cellular mediators and the nervous system, but how these extrahepatic factors are involved in hepatic immune regulation is incompletely understood. Below, we discuss in detail the various ways in which other organs regulate hepatic immunity (Figure 1).
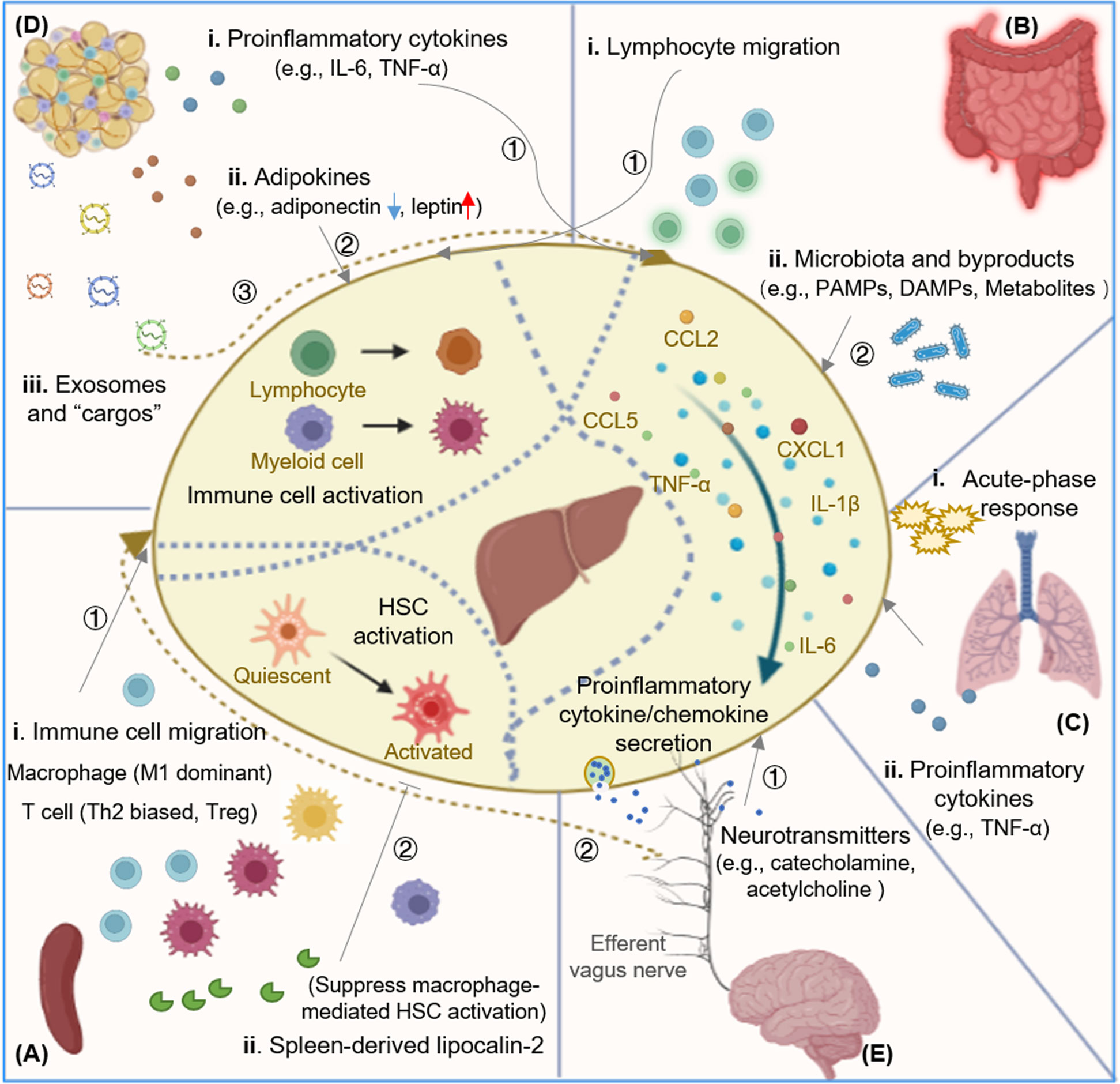
Figure 1 Mechanisms of extrahepatic factors regulating hepatic immunity. Extrahepatic factors in hepatic immune regulation. (A) Spleen and hepatic immune regulation. The spleen exerts its role in hepatic immune regulation by affecting the composition of both innate and adaptive immune cells. Spleen-derived Lcn-2 suppresses macrophage mediated HSC activation. (B) Gut and hepatic immune regulation. Intestinal microbiota and their byproducts (including PAMPs, DAPMs, and metabolites) could be translocated to the liver to active hepatic immune cells and promote the secretion of pro-inflammatory cytokines. Active lymphocytes could also be recruited from the gut into liver to modulate hepatic immunity. (C) Lung and hepatic immune regulation. Hepatic acute phase response is induced by the crosstalk between lung and liver communication, and pro-inflammatory cytokine like TNF-a acts as "shuttle" factor in modulating the lung-liver axis. (D) Adipose tissue and hepatic immune regulation. Adipose tissue-derived proinflammatory cytokines, adipokines, and exosomes modulate hepatic immunity by activating hepatic immune cells and promoting the secretion of pro-inflammatory cytokines. Adipose tissue macrophages are activated and migrate to the diseased liver. (E) Brain and hepatic immune regulation. The hepatic inflammatory signals are transmitted and integrated in the CNS, and then descend via sympathetic and efferent vagus nerve fibers, releasing catecholamine and acetylcholine through hepatic nerve endings and modulating the liver immune response. Lcn-2, lipocalin-2; HSC, hepatic stellate cell; PAMP, pathogen associated molecular pattern; DAMP, danger/death-associated molecular pattern; CNS, central nervous system; KC, Kupffer cell; NKT, natural killer T cell.
The spleen is the largest immune organ and plays a critical role in the production of various immune cells and numerous cytokines. Compared with other tissues and organs, the spleen is more closely linked with the liver in anatomical structure and function, all spleen blood flows into liver through the portal vein, which facilitates transportation of immune mediators such as immune cells and cytokines into the liver. In chronic liver diseases, splenomegaly and hypersplenism are always manifested following the development of portal hypertension. Splenectomy has been reported to have a role of ameliorating patients’ condition or suppressing liver fibrosis in clinical condition (64–66). Moreover, a large number of studies have shown that the abnormal spleen is involved in the modulation of hepatic immunity, and the role of the so-called “liver-spleen axis” is gaining increasing attention in liver diseases (67–69).
The spleen exerts its role in hepatic immune regulation by affecting the composition of both innate and adaptive immune cells in liver (Table 1). For innate immune cells, the spleen serves as a reservoir of monocytes, contributing to the heterogeneity of hepatic macrophages which are indispensable for rapid responses to liver injury (81). In mice with diet-induced NAFLD, macrophages produced increased inflammatory cytokines like TNF-α and IL-6 (M1-type) were increased, but macrophages mainly secreting the anti-inflammatory IL-10 were unchanged (70, 82). The increased hepatic macrophages during the progression of NAFLD was indicated to migrate from BM to liver via the spleen (70). The similar M1 dominant phenotype was also observed in a CCl4-induced rat liver fibrosis model. In this model, the increased monocyte recruitment and the establishment of an M1-dominant hepatic macrophage phenotype was facilitated by up-regulated secretion of hepatic CCL2, which was prompted by splenic macrophages (71). In another TAA-induced liver fibrosis model, splenectomy attenuated murine liver fibrosis with hypersplenism and stimulated accumulation of Ly-6Clo macrophages in the liver (83). Our previous study in a chronic restraint stress prompted hepatocellular carcinoma mice model also found that splenectomy could inhibit tumor growth and prevent the increase of macrophage in tumor tissues (72). Clinically, patients suffered with liver cancer showed improved prognosis upon splenectomy, but this is only viable in the subgroup with an increased neutrophil-lymphocyte ratio (NLR) and increased infiltration of CD163+ tumor associated macrophages (TAMs) in the tumor stroma (84), indicating the crucial role of spleen-derived macrophages in tumor progression.

Table 1 Immune cells migrating from extrahepatic organs to the liver.
As for hepatic adaptive immune cells, their composition can also be altered by the spleen. In a CCl4-induced liver fibrosis mice model, researchers found that splenectomy biased the Th1/Th2 balance in the liver towards Th1 dominance. Upon the transplantation of labelled splenocytes into the spleens of syngeneic wild-type mice, labelled CD4+ lymphocytes appeared in the liver after fibrosis induction, among which the vast majority were Th2 lymphocytes (73). That is, Th2-dominant splenic lymphocytes were recruited to the liver and promoted liver fibrosis by transforming the cytokine balance into Th2 dominance, and splenectomy suppressed the progression of fibrosis at least partly by restoring the Th1/Th2 balance. In the Schistosoma japonicum infection caused liver fibrosis model, dynamic changes of lymphocyte populations in the spleen and concurrent upregulation of chemokines and cell adhesion molecules in the liver also suggested a recruitment of active immune cells from spleen to the liver (74), among which CXCR3+ Tregs were supposed to occupy a considerable proportion of the lymphocytes that migrate from spleen to Th1-infiltrated liver tissues to regulate liver fibrosis (75).
To date, the role of spleen in affecting the composition of hepatic immune cell composition have been well acknowledged, however, these conclusions are mostly based on splenectomy. We have limited information about whether splenic immune cells are directly recruited to the liver, and whether the spleen delivers specific subtypes of immune cells to liver at different stages of liver disease progression. In addition, it is also worth investigating whether and how the diseased liver regulates the composition of splenic immune cells. A better understanding of these issues is crucial to delineate spleen-mediated hepatic immune regulation and lay a foundation for developing novel strategies for liver disease immunotherapy.
Lipocalin-2 (Lcn2) is an antimicrobial protein that regulates macrophage activation. Significant increase of splenic Lcn2 was detected in mice with liver fibrosis, but levels of all other measured cytokines were unchanged. The splenectomy mice showed enhanced liver fibrosis and inflammation, accompanying significantly decreased Lcn2 in portal vein. Upon treatment with recombinant Lcn2 in vitro, LPS-stimulated primary KCs produced less TNF-α and CCL2, and the activation of HSCs was suppressed by co-culture with rLcn2-treated KCs. The mechanism of splenic protection against liver fibrosis development may involve the splenic Lcn2. The splenic Lcn2 might have an important role in regulating hepatic immune tolerance during the development of liver fibrosis (85). The liver has an extraordinary capacity to regenerate upon various injuries (86). Several experimental studies have demonstrated that removal of the spleen accelerates liver regeneration and inhibits the development of liver fibrosis (71, 87–89), indicating a certain role of the spleen in liver regeneration. TGF-β is recognized as the critical factor in the performance of spleen to inhibit liver regeneration in both the thioacetamide-induced liver fibrosis rat model (90) and the partial hepatectomy rat model (91). Upon injury, the liver goes through a process from initiation and proliferation to resolution and repair. These results suggest that the spleen might also plays a role in the resolution and repair of fibrotic liver.
Close anatomical and physiologic connections exist between the gut and liver. These two organs are linked through the portal circulation, and the liver receives 70% of its blood supply from the intestine through the portal vein. Therefore, the liver acts as the first line of defense against gut-derived antigens, and one of the most exposed organs to gut-derived toxic factors, such as bacteria and bacterial byproducts (92). Besides, the gut and liver also communicate through biliary tract and systemic circulation, the bidirectional crosstalk facilitates the formation of gut-liver axis.
The gut microbiota consists of various microorganisms that normally coexist in the gut and have a role of maintaining the homeostasis of the host. A shift in gut microbiota composition can lead to activation of the mucosal immune response, causing homeostasis imbalance. This imbalance results in the translocation of metabolites and components derived from the gut microbiota, and also leads to the transport of active immune cells to the liver, thus inducing pathologic effects in the liver (8, 93). Clinical observations and animal experimental studies reveal that the gut barrier damage seldom leads to liver injury independently but aggravates pre-existing liver diseases, and the circulatory homeostasis is largely intact in patients with early cirrhosis and portal hypertension (94). With the progression of liver fibrosis, regardless of the cause, pathophysiology extends to the intestinal tract with increased intestinal permeability and overgrowth of gut microbiota. The microbiota and their byproducts could then enter the liver through the portal vein, causing inflammation and damage in the liver (95–100). Extra evidence of this process is provided by transplantation of intestinal microbiota from humans with acute alcoholic hepatitis into germ-free and conventionally housed mice (101, 102). Intestinal microbiota entering the liver regulates hepatic immunity via several mechanisms.
The signature and role of gut microbiota in different liver diseases has been reviewed elsewhere (103). Here we emphasize its role in hepatic immune regulation and attempt to disclose the mechanism of its influence on liver disease progression from the perspective of immunity.
In a ConA-induced hepatitis model, ConA treatment failed to activate hepatic NKT cells in germ-free mice, but supplementation with killed intestinal bacteria facilitate NKT cell activation (104). Also, another study with mice transplanted with gut microbiota from a patient with severe ALD found that the mice developed more severe liver inflammation with increased NKT cells (101). Growing evidence suggests that γδ T cells expand in response to invading bacterial pathogens and modulate tissue injuries (105, 106). As the major producers of IL-17A, the production of IL-17A by hepatic γδ T cells was found modulated by the commensal bacterial load (107). Both NKT and γδ T cells are innate lymphocytes enriched in the liver (108). That is, gut-derived microbiota activate innate lymphocytes in the liver, although the mechanism is unclear.
PAMPs, conserved structures vital to microbiota, are one of the main mechanisms of microbiota to activate hepatic immunity. Once gut-derived PAMPs enter the liver through the portal vein, they can activate cells expressing PRRs (e.g., TLRs, NLRs) and induce inflammation (109). Examples of relevant gut-derived PAMPs include LPS, lipoteichoic acid (LTA), and β-glucan (110). LPS is one of the most well-known components of gram‐negative bacteria, and activates hepatic macrophages through interaction with TLR4 (103, 111). Indeed, in NAFLD patients, LPS-induced activation of liver macrophages is associated with inflammation and fibrosis, TLR4 knockout attenuates experimental NASH (112). LTA is a gram-positive microbial component, functions through up-regulating the expression of cyclooxygenase-2 (COX-2), and COX-2-mediated prostaglandin E2 (PGE2) production suppresses antitumor immunity, thereby contributing to the immunosuppressed hepatic microenvironment (93). 1,3-β-glucan, from the overgrowth of fungi, on the one hand binds to the C-type lectin domain family 7 member A (CLEC7A) of KCs and possibly other bone marrow–derived cells and promotes liver inflammation, on the another hand increases PGE2 production in the liver (113, 114). DAMPs, another famous “danger signal”, have also been identified deriving from the intestine and triggering immune response in the liver. In the ASC−/- mice on a high-fat diet (HFD), Chen and colleagues (115) identified a DAMP molecule high-mobility group protein B1 (HMGB1) as a “cargo” transported by exosomes from the intestine to the liver, triggering hepatic steatosis. Recently, injection of intestinal exosomes from ischemia/reperfusion (I/R) mice to healthy mice was also shown able to cause macrophage infiltration, M1 polarization, and liver inflammation in mice (116).
In addition, metabolites derived from the gut microbiota also play roles in hepatic immune regulation. BAs (bile acids) represent one of the classic components that function in the gut-liver axis. BAs including chenodeoxycholic acid and deoxycholic acid (DCA), could upregulate NLRP3 in hepatic macrophages, contributing to cholestatic liver diseases (117). Another important component, D-lactate, could protect against pathogen dissemination by upregulating the phagocytic capability of KCs, thereby generating an intravascular immune firewall (118).
The gut microbiota also shape the hepatic immune milieu by regulating inflammatory cytokines (119–121). In the alcohol-related liver disease (ALD) model, LPS-TLR4 signal in macrophages was delivered by the recruitment of adapter molecules, such as MyD88 and TRIF (122). MyD88-mediated NF-κB activation produced pro-inflammatory cytokines (e.g., TNF-α, IL-6, and IL-1β) and chemokine CCL2, whereas the TRIF pathway induced the production of type-I interferons (123, 124). In murine liver fibrosis, translocation of gut microbiota induced tonic type I IFN expression in the liver, and then conditioned liver myeloid cells to produce high concentrations of IFN in response to intracellular infection with bacteria. Such IFN-receptor signaling also caused myeloid cell IL-10 production that corrupted antibacterial immunity, leading to loss of infection control and to infection-associated mortality (125). The prominent liver IFN signature and myeloid cells with increased IL-10 production after bacterial infection was also found in patients with liver cirrhosis. The augmented IFN and IL-10 expression incapacitated antibacterial immunity of myeloid cells and caused failure to control bacterial infection in severe liver fibrosis and cirrhosis (126–128). HSCs could also response to LPS by releasing pro-inflammatory cytokines (e.g., TNF-α, IL-6, and IL-8) and chemokines (e.g., CCL2, CCL5) and gained expression of adhesion molecules (55, 124). In addition to the TLRs, LPS can also activate inflammasomes by binding to NLRs, which leads to increased release of IL-1β and IL-18 (129, 130). These studies depicted changes in cytokine profile induced by PAMPs, providing potential therapeutic targets for liver diseases based on the gut-liver axis.
In parallel to the ‘leaky gut’ as described above, the ‘gut lymphocyte homing’ is another supposed interaction between the gut and liver immune system. Primary sclerosing cholangitis is strongly linked to inflammatory bowel disease, in which the liver disease develops in the absence of a diseased colon. In this condition, some mucosal lymphocytes generate in the gut during active inflammatory disease and persist as long-lived memory cells are supposed to home to the liver (131). Subsequent studies showed that the CCR9+ gut-homing lymphocytes were recruited by gut-specific chemokine CCL25 expressed by the hepatic endothelium (76, 132). The LSECs also expressed increased levels of mucosal addressin cell adhesion molecule-1 (MAdCAM-1), inter-cellular adhesion molecule-1 (ICAM-1) and vascular-cell adhesion molecule-1 (VCAM-1) for lymphocyte adhesion (77, 132, 133). In addition, there were other studies revealed existence of T cells expressing clonally related TCRβ chain and recognizing the same antigen in the intestine and liver (134), and hepatic B cells that produce IgA deriving from intestinal lymphoid tissue (135). These phenomenon highlight the association of lymphocyte recruitment in gut-liver axis, and call for further exploration of other communication in this axis.
Physiologically, the lung and liver are closely coordinated. When the liver function is perturbed, dysfunctional liver can lead to the abnormal expansion of pulmonary, triggering hypoxemia and a series of other pathophysiological changes and clinical symptoms known as hepato-pulmonary syndrome, which is common in patients with cirrhosis (78). Correspondingly, many hepatic manifests are often secondary to pulmonary disease such as pneumonia, due to the fact that mediators derived from the inflamed lungs can cause liver inflammation. Therefore, the pulmonary-mediated hepatic immune regulation will be reviewed in conditions of both lung disease and liver disease.
The acute-phase response (APR) is a prominent systemic reaction of the organism to local or systemic disturbances in its homeostasis, defined by significant changes in plasma concentrations of inflammation markers. These inflammation markers are acute-phase proteins (APPs). The liver is intensively involved in the APR of the organism in pneumonia and other cases (136). During pneumonia, inflammatory response caused within the airspaces is typified by cytokine production (e.g., IL-6, TNF, and IL-1), leukocyte recruitment and plasma extravasation (136, 137). Plasma extravasation could then induce the APR in the liver. The hepatocyte-derived APPs exert a direct role of curbing injury induced by TNF-α in the liver itself, but also promote survival in association with innate immunity in the lungs (136). The APR is an orchestrated response to tissue injury, infection or inflammation, and the APPs induced during this response act to limit proteolytic and/or fibrogenic activity and tissue damage, thereby contributing to the restoration of homeostasis (138, 139). APR provides novel signaling axis for the immune-mediated lung-liver communication (140, 141).
Insult like chronic alcohol exposure results in both alcohol-related liver disease and alcohol-related susceptibility to acute lung injury. Alcohol-induced injuries to these two organs share a deal of parallel mechanisms, including: damages to both organs are involved to oxidative stress that favors tissue injury (142, 143), inflammatory injuries to both organs are enhanced by alcohol exposure (144, 145), and most importantly, dysregulated cytokine production in the development and progression of both diseases (146, 147). These phenomena indicate that there exists a “shuttle” between the two organs, promoting the pathogenesis of both organs. Study in the mechanically ventilated (MV) lung injury model provides evidence for this assumption: perfusate from injured lungs was able to cause a robust inflammatory response with significantly increased production of pro-inflammatory factors such as G-CSF, IL-6, CXCL1, CXCL2, and CCL2 in LSECs (148); liver tissues obtained from mice subjected to in vivo MV also demonstrated significant increases in hepatic gene transcription of IL-6, CXCL1, and CXCL2 (148).
TNF-α is a common mechanism of alcohol-induced pathology in both lung and liver (146, 149). In the lung, TNF-α led to elevated levels of TNF-α-responsive chemokines, CXCL2 and keratinocyte chemoattractant, all of which were correlated with increased pulmonary neutrophil recruitment (150). Moreover, in a chronic alcohol pre-exposure enhanced endotoxemia-induced acute lung injury model, the lung injury could be prevented by blocking systemic TNF-α with etanercept (147). In the liver, TNF-α activates several intracellular pathways to regulate inflammation, cell death, and proliferation, and is closely associated with liver injury (151). Therefore, although evidence about the predominate source of TNF-α is still lack, it is reasonable of us to speculate that TNF-α may act as one of the mediators that derived from the inflammatory lung to promote the occurrence of hepatic inflammation. Future studies are required to identify more mediators that contribute to the hepato-pulmonary association other than TNF-α.
Alcoholic (ALD) and non-alcoholic fatty liver diseases (NAFLD) are clinical symptoms of hepatocellular injury and inflammation caused by alcohol consumption, high fat diet, obesity and diabetes, among others, and are both characterized by the expandability of adipose tissue. Anatomically, adipose tissue consists of visceral adipose tissue (VAT) and subcutaneous adipose tissue (152). VAT is mainly present within the abdominal cavity, and visceral fat venous blood is drained directly into the liver through the portal vein, and abnormal metabolic pathways and inflammation in VAT are implicated in the pathogenesis of ALD and NAFLD (153). Deregulated adipose tissue has increased lipolysis in adipocyte and activated inflammatory responses in adipose immune cells such as macrophages, which in turn lead to the release of free fatty acids, adipokines, and cytokines into the portal circulation (154–157), and these factors are associated with hepatic immune regulation.
Upregulated expression of pro-inflammatory cytokines (e.g., IL-6 and TNF-α) and chemokines (e.g., CCL2) in adipose tissue are observed in both alcoholic patients (158) and rodent models (159), particularly, the VAT is found to secret large quantities of IL-6 (160–162). The development of NAFLD and insulin resistance is also supposed to be resulted from imbalanced cytokines (increased pro-inflammatory and decreased anti-inflammatory cytokines) (163, 164). These pro-inflammatory cytokines can be delivered into the portal circulation, and directly associate with liver inflammation and fibrosis in hepatic steatosis.
Adipokines are a class of adipose-derived signaling molecules that contribute to the development of ALD and NAFLD. Adiponectin, one of the well-known adipokines, has insulin-sensitization and anti-inflammatory effects in insulin target tissues including liver, and acts as an important regulator for the development of hepatic diseases. Correlation between the onset of hepatic disease and reduced circulating adiponectin levels, decreased expression of adiponectin receptors, and impaired adiponectin-mediated signaling is shown in several animal models of hepatic syndromes (165). Via its cognate receptors, adiponectin receptors 1 and 2, adiponectin potently suppresses hepatic inflammation. KCs constitutively express AdipoR2 (166, 167), suggesting a role of KCs in adiponectin-mediated hepatic anti-inflammation properties. Adiponectin is also found has a role of blocking TNF-α-stimulated CCL2 expression, and thus resulting in reduced macrophage infiltration in the liver (168, 169). However, there is also data showing decreased adiponectin in the plasma of alcohol-fed rodents. This might due to increased TNF-α expression in adipose tissue caused by alcohol administration in rodents (170), and TNF-α could directly inhibit the release of adiponectin from the adipose tissue (171). Leptin, another important adipokine, is able to induce hepatic inflammation and fibrogenic responses by activating HSCs and KCs (172, 173). Increased production of leptin and decreased production of adiponectin were observed in alcoholic patients and mouse models (110). Therefore, different adipokines might have distinct roles in hepatic immune regulation, and their mechanisms might be complicated due to disease conditions.
Extracellular vesicles (EVs) are another important way by which adipose tissue transmits information to other organs, in addition to canonical hormones, growth factors and cytokines. EVs, including microvesicles (MVs) and exosomes or exosome-like vesicles (ELVs), are secreted by donor cells and transferred to the recipient cells, releasing encapsulated nucleic acids, lipids, and proteins to transfer information (134). Roles of adipose-derived exosomes in regulating liver metabolism have been widely documented both clinically and in animal models (174, 175), but their roles in modulating hepatic immune responses are less clear. Deng and colleagues (176) first found that adipose-derived exosomes of obese mice activated monocyte differentiation into adipose tissue macrophages (ATMs), leading to increased production of pro-inflammatory cytokines IL-6 and TNF-α. This process enhanced the migration of ATMs to liver and promoted the development of insulin resistance. ATMs accumulated in the liver also released miRNA-rich exosomes (e.g., miR-155) to regulate hepatic insulin sensitivity and inflammatory response (79). Exosomes from the adipose tissue derived mesenchymal stem cells were later demonstrated capable of promoting NK cells to exert antitumor roles on rat HCC, thereby inhibiting tumor growth (80). Together, these data indicate the possibility of adipose-derived EVs functioning as an intriguing mode for adipose tissues to regulate liver disease progression by modulating hepatic immunity.
The brain and liver bidirectionally communicate via humoral and neural networks (177, 178). The neural axis between brain and liver interacts closely with the central nervous system (CNS) via the autonomic nervous system (ANS). The hepatic sympathetic and parasympathetic nervous systems are collectively known as the ANS, which is part of the peripheral nervous system and plays a key role in the regulation of numerous physiological events (including inflammation) in the liver (179, 180). The hepatic ANS transmits information from the liver to the CNS, and also receive signals from the CNS to regulate liver function, that is, the liver acts as both a sensor and effector affected by neurological signals.
The brain function can be severely affected in severe liver diseases with considerable inflammatory involvement, and these alterations in brain are associated with brain cholinergic dysfunction (181), which is involved with immune regulation. Cholinergic modulation of liver inflammation by the vagus nerve was first reported by Tracey and colleagues (182) more than 20 years ago. In the liver of rodents, they showed that electrical stimulation of the cervical vagus nerve could attenuate LPS-induced TNF production. In rats with hemorrhagic shock, Guarini and colleagues (183) demonstrated that the brain mAChR-mediated activation of efferent vagus nerve signaling to liver also caused significant suppression of hepatic TNF release. Later studies demonstrated a role of KCs in the cholinergic mediated modulation of hepatic immunity in several chronic liver diseases (182, 184, 185), indicating the involvement of immune cells in hepatic neuro-immune regulation. Such involvement was also demonstrated in hepatic NKT cells, which received signals from the catecholamine neurotransmitters, leading to phenotypic transformation (61, 186). Thus, neural signal-expressing cells involved with hepatic immune regulation deserve further study.
Pathogens are also triggers of the intrahepatic neuro-immune responses. On the one hand, immune cells in the liver could detect the presence of pathogen components and release cytokines (e.g., IL-1β and TNF-α) which function as chemical messengers. On the other hand, pathogens can also directly activate the hepatic neurons. These signals are transmitted and integrated in the CNS, and then descend via sympathetic and efferent vagus nerve fibers, releasing catecholamine and acetylcholine through hepatic nerve endings and modulating the liver immune response (181, 187). Particularly, in this brain-liver axis, hypothalamus is recognized to be the critical part for sensing and integrating signals from the periphery tissue and effecting appropriate changes to maintain metabolic and immunologic homeostasis (180, 188). Hypothalamic inflammation is shown an important event in brain-involved hepatic immune regulation and insulin resistance (189–191). As a summary, the nervous system and the immune system communicate in response to pathogen invasion, tissue injury, and other homeostatic threats. A systematic understanding of the mechanism by which dysregulated liver triggers hypothalamic inflammation is critical for realizing the nervous system mediated hepatic immune regulation.
There are also cross-talk between the liver and other organs, such as the BM, the pancreas, and the kidney, but the role of these organs in hepatic immune regulation remains to be further studied.
The BM is an immune-regulatory organ that has a role not only in hematopoiesis but also in immune responses (192). In addition to liver-resident immune cells, most inflammatory cells are derived from the BM (193, 194). MoMFs, key effector cells in the hepatic immune activities, are derived from infiltrated bone marrow-derived CCR2+CX3CR1loLy6Chi monocytes, whose recruitment is dependent on the CCL2-CCR2 axis (195, 196), which has been well-recognized in plenty of liver diseases including liver fibrosis and hepatic carcinoma. The BM is also regarded as source of other leukocytes, however, a detailed depiction of how the BM is involved in hepatic immune regulation in other ways (e.g., cytokines, hormones, and exosomes) is still lacking.
Pancreas is a potential candidate extrahepatic organ to be involved in hepatic immune regulation. Fetuin-A secreted by the inflammatory liver could stimulate chemokines like CCL2 and IL-8, and pro-inflammatory cytokines such as IL-6 and IL-1β expression in the pancreas, and lead to damaged pancreas. In reverse, damaged pancreas may secrete pro-inflammatory cytokines (e.g., TNF-α) to directly attack the liver (197). Besides, there exists a gut-liver-kidney axis during the development and progression of chronic kidney disease associated with chronic fatty liver diseases. Kidney dysfunction led to metabolic acidosis, accumulation of toxins that have serious impacts on various liver functions, for example, changing glucose homeostasis, endothelial dysfunction, enhanced inflammation, and pro-inflammatory cytokines (198). Furthermore, the skin might also play a role in hepatic immune regulation. While clinical studies indicated that psoriasis may be more severe in patients with NAFLD/NASH (199, 200), livers from psoriatic mice were also found enriched for macrophages, polymorphonuclear neutrophils, and T cells (201).
In addition to the organs detailed in our review, correlation of other organs in hepatic immune regulation are also indicated, but the specific connection and exact mechanisms remain to be explored and validated. Nevertheless, the “shuttle” role of cytokines in these processes has been repeatedly mentioned, warranting our high attention to the overall changes of cytokines in the organism during disease development.
To date, studies on the role of spleen in the regulation have provided the most evidence, but these evidence mainly come from splenectomy as an intervention. Novel animal models, including spleen-specific photo-conversion with KikGR transgenic mice (in which KikGreen cells are turned into KikRed by site-specific irradiation) (202, 203) and spleen transplantation (204) between congenic mice strain carrying differential markers, have been proven effective in studying the cell communication between spleen and other organs, employment of these models in studies about spleen-liver crosstalk may help reveal more details. As for other organs, some available models, such as bone marrow chimeras and CD11b-diphtheria toxin receptor mice, have been sophisticatedly used in studying recruitment of liver infiltrating macrophages from the peripheral (205, 206). Recently, Zhou et al. (207, 208) developed a multi-lineage tracing system for in vivo study of hematopoietic cell migration and development (basing on the Cre-loxP and Dre-rox dual recombinase), this could potentially be used to track the movement and differentiation of cells between organs.
Generally, evidence that cytokines or exosomes from other organs influence hepatic immune response is not straightforward, because of the lack of ways to track these factors in vivo. The effects of these factors are always assessed by their corresponding changes in the target organs and liver, and the effect of these factors isolated from the target organs on liver cells during in vitro treatment. Real-time detecting, tracking and quantification of these factors will help assess their effects in vivo. Nanoparticle tracking analysis (NTA) is a technology developed based on the principle of light scattering and Brownian motion of particles in suspension and has been used for quantitative detection of exosomes (209). NTA also has different filters for analyzing fluorescent samples, so that exosomes with different markers on their surface (e.g., CD63, HSP70, and TSG-101) could be distinguished, and the results are more reliable than flow cytometry (210). For cytokine detection, various methods based on the antigen-antibody interaction (e.g., ELISA, ELISpot, bead-based flow assays) have been developed, but there are still many challenges. The impact and function of cytokines is directly linked to their extracellular expression levels, which often drastically vary with time and spatial location. Recent publications have suggested an interesting way forward for cytokine detection by combining molecular target-specific sensors that bind the respective analyte, and detection of successful binding through electric signals (211, 212). These biochips not only allow for fully automated detection of dozens to hundreds of cytokines in parallel, but also allow live and continuous detection of cytokines without the need to obtain any type of sample. However, methods for tracing and identifying the source of specific cytokine are not available yet.
Liver disease is generally a multi-stage, multi-hit process, which may not only be the link between two organs, but also the link between several organs, especially in metabolic-related liver diseases such as ALD, NAFLD, and MAFLD. Hepatic immune alteration from homeostasis to activation is a complex process involving both intrahepatic and extrahepatic factors. The emerging understanding of cross-talk between the liver and other organs complements and completes our knowledge of the role of hepatic immune regulation in liver disease development. Better understanding of the origin specialization and cascade effects of shuttle mediators such as exosomes and cytokines like TNF-α, the trigger factors and recruitment mechanisms of immune cells from other organs to the liver, and the temporal and spatial changes of these events will provide the key to intervening in liver disease progression and other organ complications by modulating hepatic and systemic immunity. These findings will benefit the development of therapeutic strategies for liver diseases that target the cellular and molecular levels to minimize adverse reactions and maximize therapeutic effects. After all, there is an urgent need for more up-to-date models and methods relating to tracking and specific intervention to explore the role of extrahepatic factors in hepatic immune regulation.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was funded by the National Natural Science Foundation of China (# 91842307 and 82173207 to ZL, # 82101915 to SZ), and the Second Affiliated Hospital of Xi'an Jiaotong University Foundation (# 2020YJ (ZYTS) 546-01 to SZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ahmed O, Robinson MW, O'Farrelly C. Inflammatory processes in the liver: Divergent roles in homeostasis and pathology. Cell Mol Immunol (2021) 18:1375–86. doi: 10.1038/s41423-021-00639-2
2. Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol (2016) 13:267–76. doi: 10.1038/cmi.2016.3
3. Kubes P, Jenne C. Immune responses in the liver. Annu Rev Immunol (2018) 36:247–77. doi: 10.1146/annurev-immunol-051116-052415
4. Zheng M, Tian Z. Liver-mediated adaptive immune tolerance. Front Immunol (2019) 10:2525. doi: 10.3389/fimmu.2019.02525
5. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest (2017) 127:55–64. doi: 10.1172/JCI88881
6. Stauffer JK, Scarzello AJ, Jiang Q, Wiltrout RH. Chronic inflammation, immune escape, and oncogenesis in the liver: A unique neighborhood for novel intersections. Hepatology (2012) 56:1567–74. doi: 10.1002/hep.25674
7. Luci C, Bourinet M, Leclere PS, Anty R, Gual P. Chronic inflammation in non-alcoholic steatohepatitis: Molecular mechanisms and therapeutic strategies. Front Endocrinol (Lausanne). (2020) 11:597648. doi: 10.3389/fendo.2020.597648
8. Kronsten VT, Tranah TH, Pariante C, Shawcross DL. Gut-derived systemic inflammation as a driver of depression in chronic liver disease. J Hepatol (2022) 76:665–80. doi: 10.1016/j.jhep.2021.11.008
9. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol (2009) 27:147–63. doi: 10.1146/annurev.immunol.021908.132629
10. O'Farrelly C, Crispe IN. Prometheus Through the looking glass: Reflections on the hepatic immune system. Immunol Today (1999) 20:394–8. doi: 10.1016/S0167-5699(99)01518-2
11. Kelly A, Fahey R, Fletcher JM, Keogh C, Carroll AG, Siddachari R, et al. CD141+ myeloid dendritic cells are enriched in healthy human liver. J Hepatol (2014) 60:135–42. doi: 10.1016/j.jhep.2013.08.007
12. Doherty DG, Norris S, Madrigal-Estebas L, McEntee G, Traynor O, Hegarty JE, et al. The human liver contains multiple populations of NK cells, T cells, and CD3+CD56+ natural T cells with distinct cytotoxic activities and Th1, Th2, and Th0 cytokine secretion patterns. J Immunol (1999) 163:2314–21.
13. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol (2013) 3:785–97. doi: 10.1002/cphy.c120026
14. Burghardt S, Erhardt A, Claass B, Huber S, Adler G, Jacobs T, et al. Hepatocytes contribute to immune regulation in the liver by activation of the notch signaling pathway in T cells. J Immunol (2013) 191:5574–82. doi: 10.4049/jimmunol.1300826
15. Maher JJ. Interactions between hepatic stellate cells and the immune system. Semin Liver Dis (2001) 21:417–26. doi: 10.1055/s-2001-17555
16. Schildberg FA, Sharpe AH, Turley SJ. Hepatic immune regulation by stromal cells. Curr Opin Immunol (2015) 32:1–6. doi: 10.1016/j.coi.2014.10.002
17. Gracia-Sancho J, Caparros E, Fernandez-Iglesias A, Frances R. Role of liver sinusoidal endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol (2021) 18:411–31. doi: 10.1038/s41575-020-00411-3
18. Cacoub P, Comarmond C, Domont F, Savey L, Desbois AC, Saadoun D. Extrahepatic manifestations of chronic hepatitis c virus infection. Ther Adv Infect Dis (2016) 3:3–14. doi: 10.1177/2049936115585942
19. Contreras GV, Marugan MT, Cuervas-Mons V. Autoimmune extrahepatic disorders in patients with autoimmune liver disease. Transplant Proc (2021) 53:2695–7. doi: 10.1016/j.transproceed.2021.06.031
20. Kodama K, Tokushige K, Hashimoto E, Taniai M, Shiratori K. Hepatic and extrahepatic malignancies in cirrhosis caused by nonalcoholic steatohepatitis and alcoholic liver disease. Alcohol Clin Exp Res (2013) 37(Suppl 1):E247–52. doi: 10.1111/j.1530-0277.2012.01900.x
21. Wake K, Sato T. "The sinusoid" in the liver: Lessons learned from the original definition by Charles Sedgwick Minot (1900). Anat Rec (Hoboken) (2015) 298:2071–80. doi: 10.1002/ar.23263
22. Gao B, Jeong WI, Tian Z. Liver: An organ with predominant innate immunity. Hepatology (2008) 47:729–36. doi: 10.1002/hep.22034
23. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. (2006) 43:S54–62. doi: 10.1002/hep.21060
24. Callery MP, Mangino MJ, Flye MW. Kupffer cell prostaglandin-E2 production is amplified during hepatic regeneration. Hepatology (1991) 14:368–72. doi: 10.1002/hep.1840140225
25. Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Büschenfelde KH, Gerken G. Human kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol (1995) 22:226–9. doi: 10.1016/0168-8278(95)80433-1
26. Groux H, Bigler M, de Vries JE, Roncarolo MG. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+ T cells. J Exp Med (1996) 184:19–29. doi: 10.1084/jem.184.1.19
27. Bamboat ZM, Stableford JA, Plitas G, Burt BM, Nguyen HM, Welles AP, et al. Human liver dendritic cells promote T cell hyporesponsiveness. J Immunol (2009) 182:1901–11. doi: 10.4049/jimmunol.0803404
28. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol (2010) 10:753–66. doi: 10.1038/nri2858
29. Wuensch SA, Spahn J, Crispe IN. Direct, help-independent priming of CD8+ T cells by adeno-associated virus-transduced hepatocytes. Hepatology (2010) 52:1068–77. doi: 10.1002/hep.23745
30. Knolle PA, Germann T, Treichel U, Uhrig A, Schmitt E, Hegenbarth S, et al. Endotoxin down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells. J Immunol (1999) 162:1401–7.
31. Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, et al. Ito Cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity (2007) 26:117–29. doi: 10.1016/j.immuni.2006.11.011
32. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol (2018) 15:555–67. doi: 10.1038/s41575-018-0020-y
33. Knolle PA, Gerken G. Local control of the immune response in the liver. Immunol Rev (2000) 174:21–34. doi: 10.1034/j.1600-0528.2002.017408.x
34. Kelly AM, Golden-Mason L, Traynor O, Geoghegan J, McEntee G, Hegarty JE, et al. Changes in hepatic immunoregulatory cytokines in patients with metastatic colorectal carcinoma: Implications for hepatic anti-tumour immunity. Cytokine. (2006) 35:171–9. doi: 10.1016/j.cyto.2006.07.019
35. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut (2012) 61:416–26. doi: 10.1136/gutjnl-2011-300304
36. Su L, Li N, Tang H, Lou Z, Chong X, Zhang C, et al. Kupffer cell-derived TNF-α promotes hepatocytes to produce CXCL1 and mobilize neutrophils in response to necrotic cells. Cell Death Dis (2018) 9:323. doi: 10.1038/s41419-018-0377-4
37. Wehr A, Baeck C, Heymann F, Niemietz PM, Hammerich L, Martin C, et al. Chemokine receptor CXCR6-dependent hepatic NK T cell accumulation promotes inflammation and liver fibrosis. J Immunol (2013) 190:5226–36. doi: 10.4049/jimmunol.1202909
38. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
39. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol (2014) 60:1090–6. doi: 10.1016/j.jhep.2013.12.025
40. Tian Z, Chen Y, Gao B. Natural killer cells in liver disease. Hepatology (2013) 57:1654–62. doi: 10.1002/hep.26115
41. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
42. Libby P. Inflammatory mechanisms: The molecular basis of inflammation and disease. Nutr Rev (2007) 65:S140–146. doi: 10.1301/nr.2007.dec.S140-S146
43. Ficht X, Iannacone M. Immune surveillance of the liver by T cells. Sci Immunol (2020) 5, eaba2351. doi: 10.1126/sciimmunol.aba2351
44. Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol (2015) 12:387–400. doi: 10.1038/nrgastro.2015.94
45. Blazka ME, Wilmer JL, Holladay SD, Wilson RE, Luster MI. Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol (1995) 133:43–52. doi: 10.1006/taap.1995.1125
46. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (2010) 330:362–6. doi: 10.1126/science.1195491
47. Wang X, Sun R, Wei H, Tian Z. High-mobility group box 1 (HMGB1)-toll-like receptor (TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: Interaction of γδ T cells with macrophages. Hepatology (2013) 57:373–84. doi: 10.1002/hep.25982
48. Markose D, Kirkland P, Ramachandran P, Henderson NC. Immune cell regulation of liver regeneration and repair. J Immunol Regen Med (2018) 2:1–10. doi: 10.1016/j.regen.2018.03.003
49. Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol (2014) 193:344–53. doi: 10.4049/jimmunol.1400574
50. Steinman RM. Lasker basic medical research award. dendritic cells: versatile controllers of the immune system. Nat Med (2007) 13:1155–9. doi: 10.1038/nm1643
51. Alisi A, Carsetti R, Nobili V. Pathogen- or damage-associated molecular patterns during nonalcoholic fatty liver disease development. Hepatology (2011) 54:1500–2. doi: 10.1002/hep.24611
52. Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol (2006) 7:131–7. doi: 10.1038/ni1303
53. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol (2012) 4:a006049. doi: 10.1101/cshperspect.a006049
54. Yao Z, Mates JM, Cheplowitz AM, Hammer LP, Maiseyeu A, Phillips GS, et al. Blood-borne lipopolysaccharide is rapidly eliminated by liver sinusoidal endothelial cells via high-density lipoprotein. J Immunol (2016) 197:2390–9. doi: 10.4049/jimmunol.1600702
55. Gupta G, Khadem F, Uzonna JE. Role of hepatic stellate cell (HSC)-derived cytokines in hepatic inflammation and immunity. Cytokine (2019) 124:154542. doi: 10.1016/j.cyto.2018.09.004
56. Sipeki N, Antal-Szalmas P, Lakatos PL, Papp M. Immune dysfunction in cirrhosis. World J Gastroenterol (2014) 20:2564–77. doi: 10.3748/wjg.v20.i10.2564
57. Yang L, Seki E. Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms. Front Physiol (2012) 3:138. doi: 10.3389/fphys.2012.00138
58. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi: 10.1038/nm.3893
59. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology (2014) 59:898–910. doi: 10.1002/hep.26592
60. Xu Z, Zeng S, Gong Z, Yan Y. Exosome-based immunotherapy: A promising approach for cancer treatment. Mol Cancer (2020) 19:160. doi: 10.1186/s12943-020-01278-3
61. Trakhtenberg EF, Goldberg JL. Immunology. Neuroimmune communication. Science (2011) 334:47–8. doi: 10.1126/science.1213099
62. Peinado MA, del Moral ML, Jimenez A, Rodrigo J, Esteban FJ. The nitrergic autonomic innervation of the liver. Auton Neurosci (2002) 99:67–9. doi: 10.1016/S1566-0702(02)00135-2
63. Bioulac-Sage P, Lafon ME, Saric J, Balabaud C. Nerves and perisinusoidal cells in human liver. J Hepatol (1990) 10:105–12. doi: 10.1016/0168-8278(90)90080-B
64. Kedia S, Goyal R, Mangla V, Kumar A, Shalimar S, Das P, et al. Splenectomy in cirrhosis with hypersplenism: improvement in cytopenias, child's status and institution of specific treatment for hepatitis c with success. Ann Hepatol (2012) 11:921–9. doi: 10.1016/S1665-2681(19)31419-X
65. Lv X, Yang F, Guo X, Yang T, Zhou T, Dong X, et al. Hypersplenism is correlated with increased risk of hepatocellular carcinoma in patients with post-hepatitis cirrhosis. Tumour Biol (2016) 37:8889–900. doi: 10.1007/s13277-015-4764-5
66. Nomura Y, Kage M, Ogata T, Kondou R, Kinoshita H, Ohshima K, et al. Influence of splenectomy in patients with liver cirrhosis and hypersplenism. Hepatol Res (2014) 44:E100–109. doi: 10.1111/hepr.12234
67. Barrea L, Di Somma C, Muscogiuri G, Tarantino G, Tenore GC, Orio F, et al. Nutrition, inflammation and liver-spleen axis. Crit Rev Food Sci Nutr (2018) 58:3141–58. doi: 10.1080/10408398.2017.1353479
68. Tarantino G, Scalera A, Finelli C. Liver-spleen axis: Intersection between immunity, infections and metabolism. World J Gastroenterol (2013) 19:3534–42. doi: 10.3748/wjg.v19.i23.3534
69. Li L, Duan M, Chen W, Jiang A, Li X, Yang J, et al. The spleen in liver cirrhosis: revisiting an old enemy with novel targets. J Transl Med (2017) 15:111. doi: 10.1186/s12967-017-1214-8
70. Fukushima H, Kono H, Hirayama K, Akazawa Y, Nakata Y, Wakana H, et al. Changes in function and dynamics in hepatic and splenic macrophages in non-alcoholic fatty liver disease. Clin Exp Gastroenterol (2020) 13:305–14. doi: 10.2147/CEG.S248635
71. Li L, Wei W, Li Z, Chen H, Li Y, Jiang W, et al. The spleen promotes the secretion of CCL2 and supports an M1 dominant phenotype in hepatic macrophages during liver fibrosis. Cell Physiol Biochem (2018) 51:557–74. doi: 10.1159/000495276
72. Jiang W, Li Y, Li ZZ, Sun J, Li JW, Wei W, et al. Chronic restraint stress promotes hepatocellular carcinoma growth by mobilizing splenic myeloid cells through activating β-adrenergic signaling. Brain Behav Immun (2019) 80:825–38. doi: 10.1016/j.bbi.2019.05.031
73. Tanabe K, Taura K, Koyama Y, Yamamoto G, Nishio T, Okuda Y, et al. Migration of splenic lymphocytes promotes liver fibrosis through modification of T helper cytokine balance in mice. J Gastroenterol (2015) 50:1054–68. doi: 10.1007/s00535-015-1054-3
74. Burke ML, McManus DP, Ramm GA, Duke M, Li Y, Jones MK, et al. Co-Ordinated gene expression in the liver and spleen during schistosoma japonicum infection regulates cell migration. PloS Negl Trop Dis (2010) 4:e686. doi: 10.1371/journal.pntd.0000686
75. Romano A, Hou X, Sertorio M, Dessein H, Cabantous S, Oliveira P, et al. FOXP3+ regulatory T cells in hepatic fibrosis and splenomegaly caused by schistosoma japonicum: The spleen may be a major source of tregs in subjects with splenomegaly. PloS Negl Trop Dis (2016) 10:e0004306. doi: 10.1371/journal.pntd.0004306
76. Trivedi PJ, Bruns T, Ward S, Mai M, Schmidt C, Hirschfield GM, et al. Intestinal CCL25 expression is increased in colitis and correlates with inflammatory activity. J Autoimmun (2016) 68:98–104. doi: 10.1016/j.jaut.2016.01.001
77. Lee WY, Kubes P. Leukocyte adhesion in the liver: Distinct adhesion paradigm from other organs. J Hepatol (2008) 48:504–12. doi: 10.1016/j.jhep.2007.12.005
78. Fuhrmann V, Krowka M. Hepatopulmonary syndrome. J Hepatol (2018) 69:744–5. doi: 10.1016/j.jhep.2018.01.002
79. Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB, et al. Adipose tissue macrophage-derived exosomal miRNAs can modulate in vivo and in vitro insulin sensitivity. Cell (2017) 171:372–384 e312. doi: 10.1016/j.cell.2017.08.035
80. Ko SF, Yip HK, Zhen YY, Lee CC, Lee CC, Huang CC, et al. Adipose-derived mesenchymal stem cell exosomes suppress hepatocellular carcinoma growth in a rat model: Apparent diffusion coefficient, natural killer T-cell responses, and histopathological features. Stem Cells Int (2015) 2015:853506. doi: 10.1155/2015/853506
81. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science (2009) 325:612–6. doi: 10.1126/science.1175202
82. Li Z, Soloski MJ, Diehl AM. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology (2005) 42:880–5. doi: 10.1002/hep.20826
83. Yada A, Iimuro Y, Uyama N, Uda Y, Okada T, Fujimoto J. Splenectomy attenuates murine liver fibrosis with hypersplenism stimulating hepatic accumulation of ly-6C(lo) macrophages. J Hepatol (2015) 63:905–16. doi: 10.1016/j.jhep.2015.05.010
84. Motomura T, Shirabe K, Mano Y, Muto J, Toshima T, Umemoto Y, et al. Neutrophil-lymphocyte ratio reflects hepatocellular carcinoma recurrence after liver transplantation via inflammatory microenvironment. J Hepatol (2013) 58:58–64. doi: 10.1016/j.jhep.2012.08.017
85. Aoyama T, Kuwahara-Arai K, Uchiyama A, Kon K, Okubo H, Yamashina S, et al. Spleen-derived lipocalin-2 in the portal vein regulates kupffer cells activation and attenuates the development of liver fibrosis in mice. Lab Invest (2017) 97:890–902. doi: 10.1038/labinvest.2017.44
86. Tanaka M, Miyajima A. Liver regeneration and fibrosis after inflammation. Inflammation Regen (2016) 36:19. doi: 10.1186/s41232-016-0025-2
87. Ueda S, Yamanoi A, Hishikawa Y, Dhar DK, Tachibana M, Nagasue N. Transforming growth factor-beta1 released from the spleen exerts a growth inhibitory effect on liver regeneration in rats. Lab Invest (2003) 83:1595–603. doi: 10.1097/01.LAB.0000095686.10639.C8
88. Murata K, Shiraki K, Sugimoto K, Takase K, Nakano T, Furusaka A, et al. Splenectomy enhances liver regeneration through tumor necrosis factor (TNF)-alpha following dimethylnitrosamine-induced cirrhotic rat model. Hepatogastroenterology (2001) 48:1022–7. doi: 10.1038/s12276-021-00574-2
89. Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology (2010) 52:1390–400. doi: 10.1002/hep.23795
90. Akahoshi T, Hashizume M, Tanoue K, Shimabukuro R, Gotoh N, Tomikawa M, et al. Role of the spleen in liver fibrosis in rats may be mediated by transforming growth factor beta-1. J Gastroenterol Hepatol (2002) 17:59–65. doi: 10.1046/j.1440-1746.2002.02667.x
91. Lee SC, Jeong HJ, Choi BJ, Kim SJ. Role of the spleen in liver regeneration in relation to transforming growth factor-β1 and hepatocyte growth factor. J Surg Res (2015) 196:270–7. doi: 10.1016/j.jss.2015.02.025
92. Adams DH, Eksteen B, Curbishley SM. Immunology of the gut and liver: A love/hate relationship. Gut (2008) 57:838–48. doi: 10.1136/gut.2007.122168
93. Ohtani N, Kawada N. Role of the gut-liver axis in liver inflammation, fibrosis, and cancer: A special focus on the gut microbiota relationship. Hepatol Commun (2019) 3:456–70. doi: 10.1002/hep4.1331
94. Gustot T, Durand F, Lebrec D, Vincent JL, Moreau R. Severe sepsis in cirrhosis. Hepatology (2009) 50:2022–33. doi: 10.1002/hep.23264
95. Garcia-Tsao G, Lee FY, Barden GE, Cartun R, West AB. Bacterial translocation to mesenteric lymph nodes is increased in cirrhotic rats with ascites. Gastroenterology (1995) 108:1835–41. doi: 10.1016/0016-5085(95)90147-7
96. Dunn W, Shah VH. Pathogenesis of alcoholic liver disease. Clin Liver Dis (2016) 20:445–56. doi: 10.1016/j.cld.2016.02.004
97. Aron-Wisnewsky J, Gaborit B, Dutour A, Clement K. Gut microbiota and non-alcoholic fatty liver disease: new insights. Clin Microbiol Infect (2013) 19:338–48. doi: 10.1111/1469-0691.12140
98. Gkolfakis P, Dimitriadis G, Triantafyllou K. Gut microbiota and non-alcoholic fatty liver disease. Hepatobiliary Pancreat Dis Int (2015) 14:572–81. doi: 10.1016/S1499-3872(15)60026-1
99. Cui Y, Wang Q, Chang R, Zhou X, Xu C. Intestinal barrier function-non-alcoholic fatty liver disease interactions and possible role of gut microbiota. J Agric Food Chem (2019) 67:2754–62. doi: 10.1021/acs.jafc.9b00080
100. Safari Z, Gérard P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD). Cell Mol Life Sci (2019) 76:1541–58. doi: 10.1007/s00018-019-03011-w
101. Llopis M, Cassard AM, Wrzosek L, Boschat L, Bruneau A, Ferrere G, et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut. (2016) 65:830–9. doi: 10.1136/gutjnl-2015-310585
102. Tranah TH, Edwards LA, Schnabl B, Shawcross DL. Targeting the gut-liver-immune axis to treat cirrhosis. Gut. (2021) 70:982–94. doi: 10.1136/gutjnl-2020-320786
103. Wang R, Tang R, Li B, Ma X, Schnabl B, Tilg H. Gut microbiome, liver immunology, and liver diseases. Cell Mol Immunol (2021) 18:4–17. doi: 10.1038/s41423-020-00592-6
104. Wei Y, Zeng B, Chen J, Cui G, Lu C, Wu W, et al. Enterogenous bacterial glycolipids are required for the generation of natural killer T cells mediated liver injury. Sci Rep (2016) 6:36365. doi: 10.1038/srep36365
105. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. (2009) 31:321–30. doi: 10.1016/j.immuni.2009.06.020
106. Paget C, Chow MT, Gherardin NA, Beavis PA, Uldrich AP, Duret H, et al. CD3bright signals on γδT cells identify IL-17A-producing Vγ6Vδ1+ T cells. Immunol Cell Biol (2015) 93:198–212. doi: 10.1038/icb.2014.94
107. Li F, Hao X, Chen Y, Bai L, Gao X, Lian Z, et al. The microbiota maintain homeostasis of liver-resident γδT-17 cells in a lipid antigen/CD1d-dependent manner. Nat Commun (2017) 7:13839. doi: 10.1038/ncomms13839
108. Bandyopadhyay K, Marrero I, Kumar V. NKT cell subsets as key participants in liver physiology and pathology. Cell Mol Immunol (2016) 13:337–46. doi: 10.1038/cmi.2015.115
109. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol (2018) 15:397–411. doi: 10.1038/s41575-018-0011-z
110. Shim YR, Jeong WI. Recent advances of sterile inflammation and inter-organ cross-talk in alcoholic liver disease. Exp Mol Med (2020) 52:772–80. doi: 10.1038/s12276-020-0438-5
111. Won SM, Park E, Jeong JJ, Ganesan R, Gupta H, Gebru YA, et al. The gut microbiota-derived immune response in chronic liver disease. Int J Mol Sci (2021) 22:8309. doi: 10.3390/ijms22158309
112. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol (2007) 47:571–9. doi: 10.1016/j.jhep.2007.04.019
113. Yang AM, Inamine T, Hochrath K, Chen P, Wang L, Llorente C, et al. Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest. (2017) 127:2829–41. doi: 10.1172/JCI90562
114. Sun S, Wang K, Sun L, Cheng B, Qiao S, Dai H, et al. Therapeutic manipulation of gut microbiota by polysaccharides of wolfiporia cocos reveals the contribution of the gut fungi-induced PGE(2) to alcoholic hepatic steatosis. Gut Microbes (2020) 12:1830693. doi: 10.1080/19490976.2020.1830693
115. Chen Y, Sun H, Bai Y, Zhi F. Gut dysbiosis-derived exosomes trigger hepatic steatosis by transiting HMGB1 from intestinal to liver in mice. Biochem Biophys Res Commun (2019) 509:767–72. doi: 10.1016/j.bbrc.2018.12.180
116. Zhao J, Chen XD, Yan ZZ, Huang WF, Liu KX, Li C. Gut-derived exosomes induce liver injury after intestinal Ischemia/Reperfusion by promoting hepatic macrophage polarization. Inflammation (2022). doi: 10.1007/s10753-022-01695-0
117. Hao H, Cao L, Jiang C, Che Y, Zhang S, Takahashi S, et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab (2017) 25:856–867 e855. doi: 10.1016/j.cmet.2017.03.007
118. Gong S, Yan Z, Liu Z, Niu M, Fang H, Li N, et al. Intestinal microbiota mediates the susceptibility to polymicrobial sepsis-induced liver injury by granisetron generation in mice. Hepatology (2019) 69:1751–67. doi: 10.1002/hep.30361
119. Abu-Shanab A, Quigley EM. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol (2010) 7:691–701. doi: 10.1038/nrgastro.2010.172
120. Alisi A, Ceccarelli S, Panera N, Nobili V. Causative role of gut microbiota in non-alcoholic fatty liver disease pathogenesis. Front Cell Infect Microbiol (2012) 2:132. doi: 10.3389/fcimb.2012.00132
121. Machado MV, Cortez-Pinto H. A gut microbiota and nonalcoholic fatty liver disease. Ann Hepatol (2012) 11:440–9. doi: 10.1016/S1665-2681(19)31457-7
122. Jarvelainen HA, Orpana A, Perola M, Savolainen VT, Karhunen PJ, Lindros KO. Promoter polymorphism of the CD14 endotoxin receptor gene as a risk factor for alcoholic liver disease. Hepatology (2001) 33:1148–53. doi: 10.1053/jhep.2001.24236
123. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med (2007) 13:1324–32. doi: 10.1038/nm1663
124. Guo J, Friedman SL. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair. (2010) 3:21. doi: 10.1186/1755-1536-3-21
125. Monroe KM, McWhirter SM, Vance RE. Induction of type I interferons by bacteria. Cell Microbiol (2010) 12:881–90. doi: 10.1111/j.1462-5822.2010.01478.x
126. Balmer ML, Slack E, de Gottardi A, Lawson MA, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med (2014) 6:237ra266. doi: 10.1126/scitranslmed.3008618
127. Hackstein CP, Assmus LM, Welz M, Klein S, Schwandt T, Schultze J, et al. Gut microbial translocation corrupts myeloid cell function to control bacterial infection during liver cirrhosis. Gut. (2017) 66:507–18. doi: 10.1136/gutjnl-2015-311224
128. Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol (2014) 60:197–209. doi: 10.1016/j.jhep.2013.07.044
129. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13:325–32. doi: 10.1038/ni.2231
130. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
131. Grant AJ, Lalor PF, Salmi M, Jalkanen S, Adams DH. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet (2002) 359:150–7. doi: 10.1016/S0140-6736(02)07374-9
132. Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hübscher SG, et al. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med (2004) 200:1511–7. doi: 10.1084/jem.20041035
133. Lalor PF, Edwards S, McNab G, Salmi M, Jalkanen S, Adams DH. Vascular adhesion protein-1 mediates adhesion and transmigration of lymphocytes on human hepatic endothelial cells. J Immunol (2002) 169:983–92. doi: 10.4049/jimmunol.169.2.983
134. Henriksen EK, Jorgensen KK, Kaveh F, Holm K, Hamm D, Olweus J, et al. Gut and liver T-cells of common clonal origin in primary sclerosing cholangitis-inflammatory bowel disease. J Hepatol (2017) 66:116–22. doi: 10.1016/j.jhep.2016.09.002
135. Moro-Sibilot L, Blanc P, Taillardet M, Bardel E, Couillault C, Boschetti G, et al. Mouse and human liver contain immunoglobulin a-secreting cells originating from peyer's patches and directed against intestinal antigens. Gastroenterology (2016) 151:311–23. doi: 10.1053/j.gastro.2016.04.014
136. Hilliard KL, Allen E, Traber KE, Yamamoto K, Stauffer NM, Wasserman GA, et al. The lung-liver axis: A requirement for maximal innate immunity and hepatoprotection during pneumonia. Am J Respir Cell Mol Biol (2015) 53:378–90. doi: 10.1165/rcmb.2014-0195OC
137. Young RP, Hopkins RJ, Marsland B. The gut-Liver-Lung axis. modulation of the innate immune response and its possible role in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol (2016) 54:161–9. doi: 10.1165/rcmb.2015-0250PS
138. Hiippala K, Jouhten H, Ronkainen A, Hartikainen A, Kainulainen V, Jalanka J, et al. The potential of gut commensals in reinforcing intestinal barrier function and alleviating inflammation. Nutrients (2018) 10:988. doi: 10.3390/nu10080988
139. Imai Y, Parodo J, Kajikawa O, de Perrot M, Fischer S, Edwards V, et al. Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. JAMA (2003) 289:2104–12. doi: 10.1001/jama.289.16.2104
140. Quinton LJ, Jones MR, Robson BE, Mizgerd JP. Mechanisms of the hepatic acute-phase response during bacterial pneumonia. Infect Immun (2009) 77:2417–26. doi: 10.1128/IAI.01300-08
141. Weber M, Lambeck S, Ding N, Henken S, Kohl M, Deigner HP, et al. Hepatic induction of cholesterol biosynthesis reflects a remote adaptive response to pneumococcal pneumonia. FASEB J (2012) 26:2424–36. doi: 10.1096/fj.11-191957
142. Guidot DM, Roman J. Chronic ethanol ingestion increases susceptibility to acute lung injury: role of oxidative stress and tissue remodeling. Chest. (2002) 122:309S–14S. doi: 10.1378/chest.122.6_suppl.309S
143. Arteel GE. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology (2003) 124:778–90. doi: 10.1053/gast.2003.50087
144. Beier JI, Luyendyk JP, Guo L, von Montfort C, Staunton DE, Arteel GE. Fibrin accumulation plays a critical role in the sensitization to lipopolysaccharide-induced liver injury caused by ethanol in mice. Hepatology (2009) 49:1545–53. doi: 10.1002/hep.22847
145. Deaciuc IV, Nikolova-Karakashian M, Fortunato F, Lee EY, Hill DB, McClain CJ. Apoptosis and dysregulated ceramide metabolism in a murine model of alcohol-enhanced lipopolysaccharide hepatotoxicity. Alcohol Clin Exp Res (2000) 24:1557–65. doi: 10.1111/j.1530-0277.2000.tb04575.x
146. Iimuro Y, Gallucci RM, Luster MI, Kono H, Thurman RG. Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology (1997) 26:1530–7. doi: 10.1002/hep.510260621
147. Massey VL, Poole LG, Siow DL, Torres E, Warner NL, Schmidt RH, et al. Chronic alcohol exposure enhances lipopolysaccharide-induced lung injury in mice: Potential role of systemic tumor necrosis factor-alpha. Alcohol Clin Exp Res (2015) 39:1978–88. doi: 10.1111/acer.12855
148. Patterson EK, Yao LJ, Ramic N, Lewis JF, Cepinskas G, McCaig L, et al. Lung-derived mediators induce cytokine production in downstream organs via an NF-κB-dependent mechanism. Mediators Inflamm (2013) 2013:586895. doi: 10.1155/2013/586895
149. Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology (1999) 117:942–52. doi: 10.1016/S0016-5085(99)70354-9
150. Poole LG, Dolin CE, Arteel GE. Organ-organ crosstalk and alcoholic liver disease. Biomolecules (2017) 7:62. doi: 10.3390/biom7030062
151. Schwabe RF, Brenner DA. Mechanisms of liver injury. i. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol (2006) 290:G583–589. doi: 10.1152/ajpgi.00422.2005
152. Parker R, Kim SJ, Gao B. Alcohol, adipose tissue and liver disease: mechanistic links and clinical considerations. Nat Rev Gastroenterol Hepatol (2018) 15:50–9. doi: 10.1038/nrgastro.2017.116
153. Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discovery (2016) 15:639–60. doi: 10.1038/nrd.2016.75
154. Marcelin G, Silveira ALM, Martins LB, Ferreira AV, Clement K. Deciphering the cellular interplays underlying obesity-induced adipose tissue fibrosis. J Clin Invest (2019) 129:4032–40. doi: 10.1172/JCI129192
155. Wree A, Schlattjan M, Bechmann LP, Claudel T, Sowa JP, Stojakovic T, et al. Adipocyte cell size, free fatty acids and apolipoproteins are associated with non-alcoholic liver injury progression in severely obese patients. Metabolism (2014) 63:1542–52. doi: 10.1016/j.metabol.2014.09.001
156. Steiner JL, Lang CH. Alcohol, adipose tissue and lipid dysregulation. Biomolecules (2017) 7:16. doi: 10.3390/biom7010016
157. Kawai T, Autieri MV, Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol (2021) 320:C375–91. doi: 10.1152/ajpcell.00379.2020
158. Voican CS, Njike-Nakseu M, Boujedidi H, Barri-Ova N, Bouchet-Delbos L, Agostini H, et al. Alcohol withdrawal alleviates adipose tissue inflammation in patients with alcoholic liver disease. Liver Int (2015) 35:967–78. doi: 10.1111/liv.12575
159. Sebastian BM, Roychowdhury S, Tang H, Hillian AD, Feldstein AE, Stahl GL, et al. Identification of a cytochrome P4502E1/Bid/C1q-dependent axis mediating inflammation in adipose tissue after chronic ethanol feeding to mice. J Biol Chem (2011) 286:35989–97. doi: 10.1074/jbc.M111.254201
160. Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes (2007) 56:1010–3. doi: 10.2337/db06-1656
161. van der Poorten D, Milner KL, Hui J, Hodge A, Trenell MI, Kench JG, et al. Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology (2008) 48:449–57. doi: 10.1002/hep.22350
162. Zhang X, Ji X, Wang Q, Li JZ. New insight into inter-organ crosstalk contributing to the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Protein Cell (2018) 9:164–77. doi: 10.1007/s13238-017-0436-0
163. Diehl AM, Li ZP, Lin HZ, Yang SQ. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut (2005) 54:303–6. doi: 10.1136/gut.2003.024935
164. Day CP. From fat to inflammation. Gastroenterology (2006) 130:207–10. doi: 10.1053/j.gastro.2005.11.017
165. Park PH, Sanz-Garcia C, Nagy LE. Adiponectin as an anti-fibrotic and anti-inflammatory adipokine in the liver. Curr Pathobiol Rep (2015) 3:243–52. doi: 10.1007/s40139-015-0094-y
166. Alzahrani B, Iseli T, Ramezani-Moghadam M, Ho V, Wankell M, Sun EJ, et al. The role of AdipoR1 and AdipoR2 in liver fibrosis. Biochim Biophys Acta Mol Basis Dis (2018) 1864:700–8. doi: 10.1016/j.bbadis.2017.12.012
167. Thakur V, Pritchard MT, McMullen MR, Nagy LE. Adiponectin normalizes LPS-stimulated TNF-alpha production by rat kupffer cells after chronic ethanol feeding. Am J Physiol Gastrointest Liver Physiol (2006) 290:G998–1007. doi: 10.1152/ajpgi.00553.2005
168. Ryu J, Hadley JT, Li Z, Dong F, Xu H, Xin X, et al. Adiponectin alleviates diet-induced inflammation in the liver by suppressing MCP-1 expression and macrophage infiltration. Diabetes (2021) 70:1303–16. doi: 10.2337/db20-1073
169. Mandal P, Pritchard MT, Nagy LE. Anti-inflammatory pathways and alcoholic liver disease: Role of an adiponectin/interleukin-10/heme oxygenase-1 pathway. World J Gastroenterol (2010) 16:1330–6. doi: 10.3748/wjg.v16.i11.1330
170. He Z, Li M, Zheng D, Chen Q, Liu W, Feng L. Adipose tissue hypoxia and low-grade inflammation: A possible mechanism for ethanol-related glucose intolerance? Br J Nutr (2015) 113:1355–64. doi: 10.1017/S000711451500077X
171. Zappala G, Rechler MM. IGFBP-3, hypoxia and TNF-alpha inhibit adiponectin transcription. Biochem Biophys Res Commun (2009) 382:785–9. doi: 10.1016/j.bbrc.2009.03.112
172. Ikejima K, Honda H, Yoshikawa M, Hirose M, Kitamura T, Takei Y, et al. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology (2001) 34:288–97. doi: 10.1053/jhep.2001.26518
173. Shen J, Sakaida I, Uchida K, Terai S, Okita K. Leptin enhances TNF-alpha production via p38 and JNK MAPK in LPS-stimulated kupffer cells. Life Sci (2005) 77:1502–15. doi: 10.1016/j.lfs.2005.04.004
174. Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest (2020) 130:1453–60. doi: 10.1172/JCI134165
175. Wu J, Dong T, Chen T, Sun J, Luo J, He J, et al. Hepatic exosome-derived miR-130a-3p attenuates glucose intolerance via suppressing PHLPP2 gene in adipocyte. Metabolism (2020) 103:154006. doi: 10.1016/j.metabol.2019.154006
176. Deng ZB, Poliakov A, Hardy RW, Clements R, Liu C, Liu Y, et al. Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes (2009) 58:2498–505. doi: 10.2337/db09-0216
177. Huang SSY, Makhlouf M, AbouMoussa EH, Ruiz Tejada Segura ML, Mathew LS, Wang K, et al. Differential regulation of the immune system in a brain-liver-fats organ network during short-term fasting. Mol Metab (2020) 40:101038. doi: 10.1016/j.molmet.2020.101038
178. Lee JS, O'Connell EM, Pacher P, Lohoff FW. PCSK9 and the gut-Liver-Brain axis: A novel therapeutic target for immune regulation in alcohol use disorder. J Clin Med (2021) 10:1758. doi: 10.3390/jcm10081758
179. Mizuno K, Ueno Y. Autonomic nervous system and the liver. Hepatol Res (2017) 47:160–5. doi: 10.1111/hepr.12760
180. Kenney MJ, Ganta CK. Autonomic nervous system and immune system interactions. Compr Physiol (2014) 4:1177–200. doi: 10.1002/cphy.c130051
181. Metz CN, Pavlov VA. Vagus nerve cholinergic circuitry to the liver and the gastrointestinal tract in the neuroimmune communicatome. Am J Physiol Gastrointest Liver Physiol (2018) 315:G651–8. doi: 10.1152/ajpgi.00195.2018
182. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature (2000) 405:458–62. doi: 10.1038/35013070
183. Guarini S, Cainazzo MM, Giuliani D, Mioni C, Altavilla D, Marini H, et al. Adrenocorticotropin reverses hemorrhagic shock in anesthetized rats through the rapid activation of a vagal anti-inflammatory pathway. Cardiovasc Res (2004) 63:357–65. doi: 10.1016/j.cardiores.2004.03.029
184. Nishio T, Taura K, Iwaisako K, Koyama Y, Tanabe K, Yamamoto G, et al. Hepatic vagus nerve regulates kupffer cell activation via α7 nicotinic acetylcholine receptor in nonalcoholic steatohepatitis. J Gastroenterol (2017) 52:965–76. doi: 10.1007/s00535-016-1304-z
185. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Susarla S, Czura CJ, et al. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J Vasc Surg (2002) 36:1231–6. doi: 10.1067/mva.2002.129643
186. Wong CH, Jenne CN, Lee WY, Leger C, Kubes P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science (2011) 334:101–5. doi: 10.1126/science.1210301
187. Jensen KJ, Alpini G, Glaser S. Hepatic nervous system and neurobiology of the liver. Compr Physiol (2013) 3:655–65. doi: 10.1002/cphy.c120018
188. Kreier F, Swaab DF. History of hypothalamic research: "The spring of primitive existence". Handb Clin Neurol (2021) 179:7–43. doi: 10.1016/B978-0-12-819975-6.00031-5
189. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology (2005) 146:4192–9. doi: 10.1210/en.2004-1520
190. Milanski M, Arruda AP, Coope A, Ignacio-Souza LM, Nunez CE, Roman EA, et al. Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes (2012) 61:1455–62. doi: 10.2337/db11-0390
191. Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab (2009) 296:E1003–1012. doi: 10.1152/ajpendo.90377.2008
192. Zhao E, Xu H, Wang L, Kryczek I, Wu K, Hu Y, et al. Bone marrow and the control of immunity. Cell Mol Immunol (2012) 9:11–9. doi: 10.1038/cmi.2011.47
193. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11:762–74. doi: 10.1038/nri3070
194. Soehnlein O, Steffens S, Hidalgo A, Weber C. Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol (2017) 17:248–61. doi: 10.1038/nri.2017.10
195. Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Mockel D, et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut (2014) 63:1960–71. doi: 10.1136/gutjnl-2013-306294
196. Brempelis KJ, Crispe IN. Infiltrating monocytes in liver injury and repair. Clin Transl Immunol (2016) 5:e113. doi: 10.1038/cti.2016.62
197. Gerst F, Wagner R, Kaiser G, Panse M, Heni M, Machann J, et al. Metabolic crosstalk between fatty pancreas and fatty liver: Effects on local inflammation and insulin secretion. Diabetologia (2017) 60:2240–51. doi: 10.1007/s00125-017-4385-1
198. Raj D, Tomar B, Lahiri A, Mulay SR. The gut-liver-kidney axis: Novel regulator of fatty liver associated chronic kidney disease. Pharmacol Res (2020) 152:104617. doi: 10.1016/j.phrs.2019.104617
199. Madanagobalane S, Anandan S. The increased prevalence of non-alcoholic fatty liver disease in psoriatic patients: A study from south India. Australas J Dermatol (2012) 53:190–7. doi: 10.1111/j.1440-0960.2012.00905.x
200. Miele L, Vallone S, Cefalo C, La Torre G, Di Stasi C, Vecchio FM, et al. Prevalence, characteristics and severity of non-alcoholic fatty liver disease in patients with chronic plaque psoriasis. J Hepatol (2009) 51:778–86. doi: 10.1016/j.jhep.2009.06.008
201. Vasseur P, Pohin M, Jegou JF, Favot L, Venisse N, McHeik J, et al. Liver fibrosis is associated with cutaneous inflammation in the imiquimod-induced murine model of psoriasiform dermatitis. Br J Dermatol (2018) 179:101–9. doi: 10.1111/bjd.16137
202. Nowotschin S, Hadjantonakis AK. Use of KikGR a photoconvertible green-to-red fluorescent protein for cell labeling and lineage analysis in ES cells and mouse embryos. BMC Dev Biol (2009) 9:49. doi: 10.1186/1471-213X-9-49
203. Shand FH, Ueha S, Otsuji M, Koid SS, Shichino S, Tsukui T, et al. Tracking of intertissue migration reveals the origins of tumor-infiltrating monocytes. Proc Natl Acad Sci U S A (2014) 111:7771–6. doi: 10.1073/pnas.1402914111
204. Wang JJ, Qiu L, Fernandez R, Yeap XY, Lin CX, Zhang ZJ. A mouse model of vascularized heterotopic spleen transplantation for studying spleen cell biology and transplant immunity. J Vis Exp (2019). 148:e59616. doi: 10.3791/59616
205. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest (2005) 115:56–65. doi: 10.1172/JCI200522675
206. Ferreira FM, Palle P, Vom Berg J, Prajwal P, Laman JD, Buch T. Bone marrow chimeras-a vital tool in basic and translational research. J Mol Med (Berl) (2019) 97:889–96. doi: 10.1007/s00109-019-01783-z
207. Jin H, Liu K, Tang J, Huang X, Wang H, Zhang Q, et al. Genetic fate-mapping reveals surface accumulation but not deep organ invasion of pleural and peritoneal cavity macrophages following injury. Nat Commun (2021) 12:2863. doi: 10.1038/s41467-021-23197-7
208. He L, Pu W, Liu X, Zhang Z, Han M, Li Y, et al. Proliferation tracing reveals regional hepatocyte generation in liver homeostasis and repair. Science (2021) 371:eabc4346. doi: 10.1126/science.abc4346
209. Carnell-Morris P, Tannetta D, Siupa A, Hole P, Dragovic R. Analysis of extracellular vesicles using fluorescence nanoparticle tracking analysis. Methods Mol Biol (2017) 1660:153–73. doi: 10.1007/978-1-4939-7253-1_13
210. Bachurski D, Schuldner M, Nguyen PH, Malz A, Reiners KS, Grenzi PC, et al. Extracellular vesicle measurements with nanoparticle tracking analysis - an accuracy and repeatability comparison between NanoSight NS300 and ZetaView. J Extracell Vesicles (2019) 8:1596016. doi: 10.1080/20013078.2019.1596016
211. Ni S, Shen Z, Zhang P, Liu G. Enhanced performance of an electrochemical aptasensor for real-time detection of vascular endothelial growth factor (VEGF) by nanofabrication and ratiometric measurement. Anal Chim Acta (2020) 1121:74–82. doi: 10.1016/j.aca.2020.05.003
Keywords: hepatic immune regulation, extrahepatic factor, liver disease, immune cells, inflammation
Citation: Zhang S, Lu S and Li Z (2022) Extrahepatic factors in hepatic immune regulation. Front. Immunol. 13:941721. doi: 10.3389/fimmu.2022.941721
Received: 11 May 2022; Accepted: 28 July 2022;
Published: 16 August 2022.
Edited by:
Yongzhan Nie, Fourth Military Medical University, ChinaCopyright © 2022 Zhang, Lu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zongfang Li, bGl6b25nZmFuZ0B4anR1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.