 Chang Wang
Chang Wang Ying Shi
Ying Shi Xiaomei Wang1,3
Xiaomei Wang1,3 Heming Ma
Heming Ma Quan Liu
Quan Liu Yanhang Gao
Yanhang Gao Junqi Niu
Junqi Niu- 1Department of Hepatology, The First Hospital of Jilin University, Changchun, China
- 2Department of Gastroenterology, Henan Provincial People’s Hospital, Zhengzhou, China
- 3Center of Infectious Disease and Pathogen Biology, Key Laboratory of Organ Regeneration and Transplantation of the Ministry of Education, State Key Laboratory of Zoonotic Disease, The First Hospital of Jilin University, Changchun, China
- 4Institute of Translational Medicine, The First Hospital of Jilin University, Changchun, China
Fibrates, which are agonists of peroxisome proliferator-activated receptor alpha, have received increasing attention in the treatment of primary biliary cholangitis. Reduced alkaline phosphatase levels and improved clinical outcomes were observed in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid (UDCA) monotherapy4 when treated with bezafibrate or fenofibrate combined with UDCA. In contrast to obeticholic acid, which exacerbates pruritus in patients, fibrates have been shown to relieve pruritus. Clinical trial outcomes show potential for the treatment of primary biliary cholangitis by targeting peroxisome proliferator-activated receptors. It is currently agreed that primary biliary cholangitis is an autoimmune-mediated cholestatic liver disease, and peroxisome proliferator-activated receptor is a nuclear receptor that regulates the functions of multiple immune cells, thus playing an important role in regulating innate and adaptive immunity. Therefore, this review focuses on the immune disorder of primary biliary cholangitis and summarizes the regulation of hepatic immunity when peroxisome proliferator-activated receptors are targeted for treating primary biliary cholangitis.
Introduction
Primary biliary cholangitis (PBC) is an autoimmune-mediated cholestatic liver disease characterized by progressive destruction of hepatic interlobular bile ducts, which eventually leads to liver cirrhosis (1). The diagnosis of PBC depends on elevated alkaline phosphatase (ALP)/γ-glutamyl transpeptidase (γ-GT) levels; exclusion of other diseases that may cause cholestasis, including drug-induced liver injury, biliary stones, and malignant tumors through patient medical history and imaging examinations; and positive antimitochondrial antibody (AMA) and/or antinuclear antibodies (ANA) tests, including anti-gp210 and anti-sp100. Liver biopsy may be performed when the diagnostic results for these tests are insufficient to determine PBC (2). PBC is the only cholestatic disease which has an established treatment available. Ursodeoxycholic acid (UDCA) has been the only approved treatment for PBC for over 20 years, until 2016, when obeticholic acid (OCA) was licensed for the treatment of primary biliary cholangitis (previously primary biliary cirrhosis) in combination with UDCA in adults showing an inadequate response to UDCA or as monotherapy in adults unable to tolerate UDCA (3). However, there remains a need to develop improved PBC treatments. Up to 40% of patients with PBC respond inadequately to UDCA therapy (4), and OCA aggravates pruritus dose-dependently (5, 6). Recent clinical trials of selective peroxisome proliferator-activated receptor α (SPPARα) agonists for the treatment of PBC have received increasing attention.
PPARs are ligand dependent transcription factors and three isoforms including PPARα (NR1C1), PPARβ/δ (NR1C2) and PPARγ (NR1C3) are found (7). PPAR isoforms heterodimerize with retinoid X receptor. This complex regulates gene expression by binding to specific peroxisome proliferator response elements located in regulatory site of each gene. Due to differences in tissue distribution, ligands sensitivity and target genes, these three PPAR isoforms have distinct but complementary physiological functions (8). PPARα is highly expressed in the liver, skeletal muscle, and mainly regulate lipid and glucose metabolism (9). Fibrates, which are PPARα agonists, are used to treat hyperlipidemia. PPARγ is predominantly expressed in adipose tissue, which plays an important role in insulin sensitivity (10). Thiazolidinediones, the PPARγ agonists, are used to manage type 2 diabetes. PPARβ/δ is ubiquitously expressed and is involved in many physiological processes, including lipid metabolism, wound healing and inflammation (11). PPARβ/δ agonists currently are not approved for clinical use.
The first evidence that fibrates could be used to treat hepatobiliary disease was presented in 1993, when bezafibrate treatment caused a reduction in serum and biliary ALP activities (12). In 1999– 2003, several studies from Japan attempted to treat primary biliary cirrhosis with bezafibrate (13–18), and the results of these preliminary studies indicated that bezafibrate was effective in reducing ALP, γ-GT, and immunoglobulin M (IgM), with or without UDCA. In 2002 and 2004, preliminary clinical trials of fenofibrate in the treatment of primary biliary cirrhosis were conducted in Japan, and the results suggested that combination therapy with UDCA and fenofibrate was useful in reducing ALP, γ-GT, and IgM levels (19, 20). Subsequent clinical trials and retrospective studies have provided new evidence for the use of PPAR agonists in the treatment of PBC. Table 1 summarizes the studies that have been published and registered on the National Institutes of Health clinical trials website (http://clinicaltrials.gov). Systematic reviews and meta-analyses of these clinical trials have confirmed the efficacy of bezafibrate and fenofibrate in improving serological responses and relieving pruritus in patients (58–63). Clinical trials have attempted to treat refractory PBC with triple therapy (UDCA, OCA, and fibrate) (64, 65) with higher risks of adverse events despite a significant reduction in serological markers. Almost all clinical trials take the serological response of patients as the endpoint and lack the detection of immune-related indicators. This is due to a lack of reliable immune-related markers associated with PBC progression and prognosis. One study showed that IgM shows potential as a marker to predict the long-term clinical outcomes of patients with PBC treated with UDCA and bezafibrate (34), but further evidence is needed to confirm this assumption. In a particular clinical trial (NCT02931513), researchers evaluated whether soluble mannose receptor and soluble CD163 (sCD163), a macrophage activation marker, can be used as potential predictors of non-response to UDCA treatment and thus, as predictors of patients needing add-on therapy. Another clinical trial (NCT04514965, in progress) attempts to investigate how treatment with bezafibrate as an add-to therapy to UDCA influences the levels of sCD163, fibrosis markers, and bile acid composition in patients with PBC.
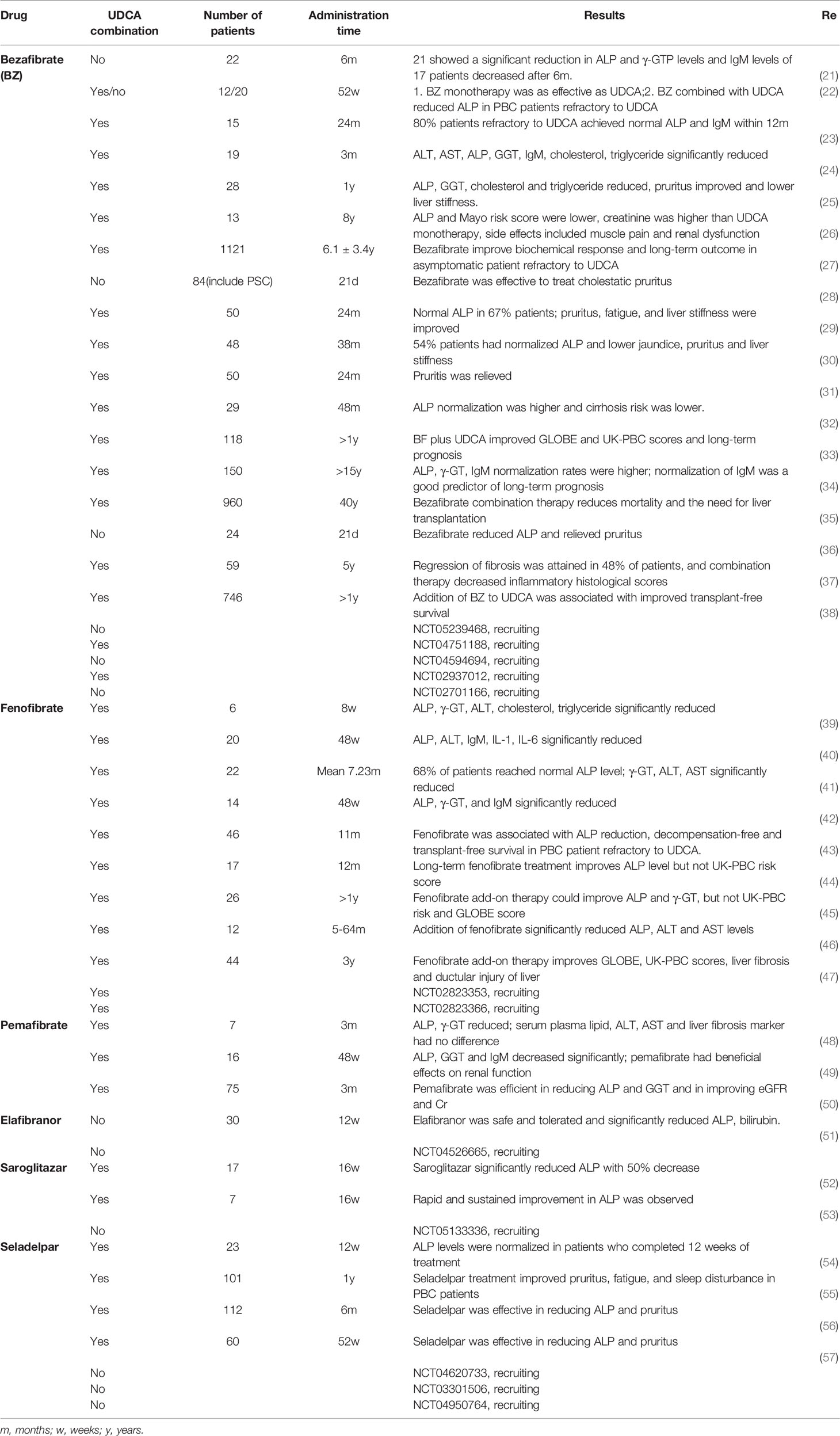
Table 1 clinical studies of PPAR agonists on the treatment of PBC.
Innate and adaptive immune-response abnormalities play an essential role in the occurrence and progression of PBC. Whereas PPAR, an important component of nuclear receptors, regulates the function of multiple innate and adaptive immunity-response cells. However, research on immune regulation related to PPAR in PBC is limited. Previous studies were evaluated for the regulation of PPAR on hepatic immunity in the progression of PBC to identify potentially novel biomarkers and therapeutic drugs that can be further investigated in future studies.
Regulation of Immune Response Abnormality by PPARs in PBC
Genetic susceptibility and exposure to environmental factors are the two main contributors to PBC development. Genome-wide association studies and observations of identical twins have confirmed genetic associations and risk factors for PBC (66–68). Molecular mimicry induced by bacterial infection, especially the pyruvate dehydrogenase complex-E2 (PDC- E2), and xenobiotic exposure are important environmental factors that disrupt hepatic immune tolerance and induce PBC (69, 70). T help 1(Th1)-mediated immunopathological damage to the intrahepatic small bile duct is a characteristic of PBC (71). In fact, innate and adaptive immune-response cells collectively participate in the development of PBC at different stages of the disease, including monocytes and macrophages with hyperreaction, dendritic cells with enhanced antigen presentation, and natural killer (NK)/natural killer T (NKT) cells with enhanced killing properties in the early stage of PBC. Th17 were shown to inhibit Th2/Treg, and B cells were also involved in PBC progression. We describe the specific role of individual immune response cells in PBC progression and the regulatory role of PPARs in these cells.
Th1/Th2
Interleukin-12 (IL-12)-induced Th1 cells produce IFN-γ and IL-2, whereas IL-4 and IL-2-induced Th2 cells secrete a variety of cytokines, including IL-4, IL-10, and IL-13 (66). Excessive Th1 immune response leads to uncontrolled tissue damage. High levels of IFNγ are associated with portal inflammation activity indicating Th1-dominant liver injury in PBC (72). Decreased IL-4 producing CD4+ T cells in patients with advanced PBC also supports this result (73). In addition, a trans-ethnic genome-wide meta-analysis revealed that IL12RB1 is included in the susceptibility loci of PBC, and Th1 differentiation is significant in pathway analysis (66). A decrease in liver-infiltrating CD4+ Th1 cells in patients with PBC indicated an adequate response to UDCA treatment (74). Therefore, reversal of the excessive Th1 immune response is of significance for the treatment of PBC.
Fibrate treatment has also been found to reduce CD4+ T cell migration to the liver. Bezafibrate and fenofibrate have been shown to decrease elevated normal T-cell expressed and secreted (RANTES) levels induced by chenodeoxycholic acid (75). RANTES, a member of the CC chemokine family, mediates the migration of CD4+ T cells to inflamed tissues, and in PBC RANTES expression has been observed to be elevated (75, 76). Research in PBC animal models also indicated that 15d-PGJ2, a PPARγ ligand, effectively attenuated portal inflammation with reduced T cell numbers, which prevented the progression of PBC (77). However, this study did not confirm the reduction in CD4+ T cells because of the limitation of mouse anti-CD4 antibodies.
PPARs activation also promotes Th1/Th2 phenotypic conversion, except for the inhibition of CD4+ T cell migration. Sex differences were found in the expression of PPARα in CD4+ T cells. CD4+ T cells isolated from female peripheral blood produced higher levels of IFNγ than those isolated from male peripheral blood. Knockdown of PPARα by small interfering RNA in male CD4+ T cells contributes to increased IFNγ production (78). Another study indicated that higher PPARα expression was detected in male CD4+ T cells than in females, and deletion of PPARα in male T cells induced increased IFNγ and TNF production (79). One study suggested a possible regulatory mechanism of PPARα in IFNγ production. Interaction of PPARα and nuclear receptor corepressor 1 reduced histone acetylation of sites on cis-regulatory elements in the ifng locus, thereby inhibiting IFNγ production and Th1 dominant immunity (80). Female predisposition characterizes multiple autoimmune diseases, including PBC. Several hypotheses, such as sex hormones, genes, and epigenetic regulation, have attempted to explain the predominance of PBC in females (81), but the reasons remain unclear. The potential relationship between differences in PPARα expression in CD4+ T cells and characteristics of autoimmune diseases remains to be investigated. PPARγ activation also contributes to Th1/Th2 phenotypic conversion (82, 83). Another study indicated that PPARγ binds directly to prospero-related homeobox and inhibits the production of IFNγ (84). PPARδ was also demonstrated to inhibit IFNγ production in other Th1-mediated autoimmune disease (85, 86), but these results need to be confirmed in studies on PBC.
Th17
Th17 cells differentiate from naïve T cells stimulated by IL-1β, IL-6, and TGF-β1 and are characterized by IL-17 production. IL-23 is required to maintain Th17 cellular function (87). Recent studies have confirmed that Th17 cells play an important role in the progression of PBC, although the mechanism has not been fully elucidated. The frequency of Th17 cells in the liver tissues of patients is higher than that in healthy controls (88). Th1 and Th17 differentiation was included in the pathway analysis of the trans-ethnic genome-wide meta-analysis of PBC cohorts. Elevated IL-17 produced by Th17 cells in the liver promotes the proliferation and fibrosis of hepatic stellate cells in PBC (89).
PPARα and PPARγ are involved in the suppression of Th17 differentiation from naïve T cells by inhibiting the expression of the retinoic acid receptor-related orphan receptor (RORγt), an important factor controlling Th17 polarization (90). Fenofibrate inhibits Th17 differentiation through the IL-6/STAT3/RORγt pathway, and this effect could be reversed by MK886, a PPARα antagonist (91). Upregulation of PPARγ also selectively inhibits Th17 differentiation, but not Th1, Th2, or Treg differentiation in CD4+ T cells via inhibition of RORγt (92). PPARγ agonist has also been reported to inhibit Th17 polarization by regulating the expression of cyclin B1 and glutaminase (93; 94). Increased IFNγ and IL-17 levels have been observed in PPARδ-deficient mice, indicating an enhanced Th1/Th17 mediated immune response (95). However, a PPARδ agonist blocks IL-17 production by inhibiting Th17 function (85). These results were obtained from studies on isolated Th17 cells or other autoimmune diseases. More evidence is needed on the effects of PPAR agonists on Th17 cells after PBC treatment.
Dendritic Cell
Dendritic cells (DC) play an essential role in the induction of an adaptive immune response. DCs from patients with PBC have a higher capacity for antigen presentation, and the presence of DCs, especially myeloid DCs, has been confirmed immunohistochemically around the damaged bile ducts (96–98). Bile epithelial cells produce macrophage protein-3α in response to IL-1β, TNFα, and IL-17, which promotes DC infiltration (99). The production of nitric oxide by DCs, which may participate in bile duct injury, was significantly higher in patients with PBC than in healthy controls (100, 101). Cytokines produced by DCs partly determine helper T cell differentiation from naïve T-cells. A study on DC subtypes found that type 2 DCs in patients with PBC were significantly decreased, which is characterized by the expression of CD123 and the promotion of Th2 cell differentiation (102). Therefore, antigen presentation and cytokines of DCs are involved in directing the Th cell response in PBC patients.
PPARα, PPARβ, and PPARγ mRNAs were detected in DCs, but only PPARγ was detected at the protein level (103). Therefore, PPARγ has been extensively studied for its role in the regulation of DC function. Isolation and culture of DCs from peripheral blood of patients with PBC indicated that bezafibrate treatment significantly decreased nitrite production in DCs (104), which was elevated in patients with PBC (100). In monocyte-induced DCs, PPARγ activation reduces DC immunogenicity and increases self-tolerance maintenance by downregulating RelB protein expression (105). Troglitazone and 15d-PGJ2, which are PPARγ ligands, inhibit toll-like receptor-mediated activation of DCs via inhibition of the NF-κB mitogen-activated protein kinase pathway (106). Another study indicated that troglitazone inhibited dectin-1-mediated activation by interfering with curdlan-mediated accumulation of caspase recruitment domain 9, mitogen-activated protein kinase, and the NF-κB pathway (107). Therefore, the upregulation of PPARγ may suppress the immune response in PBC by inhibiting antigen presentation by DCs.
Additionally, PPARγ activation in DCs also inhibits the Th1-dominant immune response via the alteration of cytokines. Activation of PPARγ in DCs maintains the immature status of DCs, which fails to promote the activation and differentiation of CD4+ T cells (108). Rosiglitazone, a PPARγ agonist, can downregulate CD40-induced secretion of IL-12 in DCs, a potent Th1 driving factor (109). Another study also found that PPARγ activation reduces the production of IL-12 in CD1a- monocyte-derived DCs (110). These results were confirmed in another study with unaffected production of IL-1β, IL-6, IL-10, and TNF-α. Reduced Th1 recruiting chemokines, including CXCL10 and CCL5, but not Th2-attracting chemokines including CCL22 and CCL17, were observed in this study (111). It was found through subsequent research that PPARγ directly binds to the PPAR response element in the human IL-10 promoter region, upregulating IL-10 expression of DCs (112).
Treg
Regulatory T (Treg) cells are important in the maintenance of immune tolerance, and the forkhead transcription factor Foxp3 has been shown to be an essential regulator of Treg lineage commitment and function (113). Two subtypes of Tregs, thymus-derived natural Tregs and inducible Tregs from CD4+CD25- T effector cells, have been described (114, 115). The suppressive effects of Treg cells from patients with PBC decreased and differentiated into Th1 cells upon stimulation with low concentrations of IL-12 (116). The relative number of CD4+CD25+ regulatory T cells and Foxp3-expressing Tregs in patients with PBC was significantly reduced compared to that in healthy controls, and the CD8+/Foxp3+ Treg ratio was markedly higher in late-stage patients with PBC than in those with chronic hepatitis C and autoimmune hepatitis (117).
In vitro and in vivo studies confirmed the regulatory effects of PPARs on Tregs. Fenofibrate promotes Foxp3+ regulatory T cell differentiation in vitro by inhibiting Akt and enhancing Smad3 phosphorylation (118). Another study found that the suppressive effect of PPARα-deficient Treg cells on CD4+CD25- and CD8+ T cell proliferation was impaired (119). Bezafibrate and ciglitazone induces stable Foxp3 expression by collaborating with transforming growth factor-β through the downregulation of DNA methyltransferase, which mediates demethylation of Foxp3-conserved noncoding DNA elements (120). Pioglitazone also promotes Foxp3 expression, increasing the percentage of hepatic CD4+CD25+Foxp3+ Treg cells significantly (121). It seems that the upregulation of PPARs contributes to the maintenance of the inhibitory effects and high frequency of Treg cells.
Follicular Helper T Cell
Follicular helper T (Tfh) cells are a subset of CD4+ T cells with the characteristic CXCR5+, whose main function is to regulate humoral immunity. The activation, proliferation, and differentiation to antibody-producing plasma cells depend on Tfh cells (122, 123). It has been demonstrated that the frequency of circulating CD4+CXCR5+ Tfh cells in patients with PBC is significantly higher than that in healthy controls and patients with autoimmune hepatitis (124). In this study, the frequency of Tfh cells was reduced in patients with PBC with an adequate response to UDCA treatment compared to those who showed an inadequate response to UDCA. Another study indicated that elevated Tfh cells were positively correlated with increased plasma B cells, serum AMA, and IgM in patients with PBC (125). Mice with a specific knockout of PPARγ in CD4+ T cells developed an autoimmune phenotype with increased activation of Tfh cells and enhanced autoantibody production of B cells. However, pioglitazone treatment significantly ameliorated the Tfh cell response (126, 127). These results confirm the regulatory effects of PPAR on Tfh cells.
B Cell
The presence of autoantibodies and hyperglobulinemia, particularly IgM, is characteristic of PBC. IgM-producing plasma cells are significantly increased in the serum of patients with PBC (128). Presentation of PDC by cross-reactive B cells may be responsible for the disruption of T cell tolerance to highly conserved self-antigen PDC (129). Whether the levels of autoantibodies and IgM are correlated with the clinical manifestations and outcomes of patients with PBC is still controversial (130, 131). However, decreased IgM levels were observed in patients who responded adequately to UDCA monotherapy or UDCA combined with OCA or fibrates in some clinical trials. One study reported that serum IgM has the potential to be a marker for predicting long-term clinical outcomes of patients with PBC treated with UDCA and bezafibrate (34). The mechanism underlying the high levels of IgM and its role in PBC remains unclear. Genomic and miRNA analyses have indicated that IFNγ and CD40L are central upstream regulators of PBC (132). One study also found that reduced methylation of the CD40L promoter in CD4+ T cells was inversely associated with IgM levels in PBC (133). This provides an epigenetic regulatory explanation for the elevated IgM levels. The depletion of B cells is an immune-related treatment strategy for PBC. Decreased ALP and IgM levels were observed in patients with PBC treated with rituximab, with increased frequency of CD25highCD4+ T cells and increased expression of FoxP3 (134). Although B cell depletion is effective in reducing AMA and IgM levels, serological responses of patients with PBC are not always reproducible in all clinical trials (135).
The regulatory role of PPARs in B cells is limited. B cell-activating factor (BAFF) belongs to the tumor necrosis factor family, which plays an important role in B cell maturation. Overexpression of BAFF is harmful to the immune tolerance of B cells, and increased BAFF is detected in patients with PBC (136). BAFF-activated B cell-mediated Treg cell apoptosis also contributes to impaired immune tolerance, and bezafibrate treatment effectively inhibits BAFF-induced Treg apoptosis (137). In addition, in PPARγ haplo-deficient mice, the proliferation and antigen-specific immune response of B cells are upregulated (138). Research on B cell-specific PPARγ knockout mice demonstrated that reduction of IL-10 producing CD5+CD1dhi regulatory B cells was responsible for exaggerated hypersensitivity (139). PPAR agonist treatment has reduced IgM levels in patients with PBC in numerous clinical trials (Table 1), which may confirm the inhibitory effects of PPARs on B cells.
Macrophage and Monocytes
Resident Kupffer cells and monocyte-derived macrophages from the peripheral blood comprise the hepatic macrophage population, which participates in hepatic inflammation and immune response regulation. Macrophages are roughly divided into classically or alternatively activated phenotypes, which are also called M1 or M2 phenotypes with pro-inflammatory and anti-inflammatory effects, respectively (140). Early research has found that the number of hepatic Kupffer cells is increased in patients with PBC (141). In addition, monocyte chemotactic proteins (MCP), CXCL12, and CX3CL1 in the liver tissue of patients with PBC are significantly increased, which promotes the accumulation of monocytes in the liver (142–144). Monocytes from patients with PBC exhibit higher TLR4 expression, are more sensitive to LPS stimulation, and increase the production of TNFα, IL-1β, IL-6, and IL-8 (145–147). Monocytes and macrophages also influence NK cell function and T-cell differentiation. Increased circulating CD14lowCD16+ monocytes in PBC promote Th1 cell skewing and accelerate liver injury (148). Kupffer cells promote NK cell activation via the direct interaction between NK group 2, member D, and retinoic acid early inducible-1, with increased production of IL-12, TNFα, and IFNγ, which synergistically induces hepatic inflammation in PBC (149). A study of macrophage activation markers demonstrated that an increase in soluble CD163 and mannose receptors is consistent with an increase in ALP, and these can be used as markers to predict disease severity and prognosis of patients with PBC (150).
Previous studies have shown that PPARγ, PPARβ/δ, and PPARα exert regulatory effects on macrophages. Activation of PPARγ in Kupffer cells significantly inhibits the production of nitric oxide and TNFα, resulting in the suppression of inflammation (151). Another study showed that pioglitazone prevents LPS-induced liver injury by inhibiting TNFα production in Kupffer cells (152). Recruitment of monocytes/macrophages is reduced in cholestatic mice treated with 15d-PGJ2 (153). Several studies have confirmed that PPARδ (154) and PPARγ (155–158) activation promotes M2 macrophage polarization, which effectively inhibits hepatic inflammation. However, the mechanisms by which PPARγ and PPARδ inhibits inflammation in classically activated macrophages are distinct. SUMOylated PPARγ inhibits macrophage inflammatory gene expression by blocking the release of the nuclear receptor corepressor complex, and when PPARδ is linked to its ligands, the release of B-cell lymphoma 6 allows it to repress the transcription of inflammatory genes (159). Overexpression or absence of PPARα indicates that PPARα might also promote macrophage polarization from M1 to M2 (160, 161). Macrophages also act as intermediaries for IL-4 to suppress the secretion of IL-2 by T cells because of 12/15-lipoxygenase production in macrophages, whose metabolic product, 13-hydroxyoctadecadienoic acid, is the ligand of PPARγ (162).
Natural Killer Cell
NK cells participate in the innate immune system, mainly through cytotoxic mechanism activation and IFNγ production. Early studies have demonstrated that the number of circulating and liver-infiltrating NK cells is significantly elevated in patients with PBC (163, 164). These increased NK cells had different properties compared with healthy controls, with increased cytotoxic activity and perforin production, but significantly decreased IFN-γ, IL-6, and IL-8 synthesis (164). Recently NK cells of patients with PBC were found to have increased sensitivity to IL-12 stimulation. A minimal amount of IL-12 stimulation can enhance IFN-γ production in NK cells (165).
PPARγ regulates NK cell cytotoxicity and IFN-γ production by interacting with PPARγ ligands. A study found that 15d-PGJ2, a natural ligand of PPARγ, simultaneously inhibited cytotoxicity and IFNγ production in NK cells, regardless of the presence of PPARγ. However, ciglitazone, a synthetic ligand of PPARγ, reduces IFNγ production via PPARγ activation (166).
Natural Killer T Cell
NKT cells are lymphocytes characterized by the simultaneous expression of T-cell receptors and NK cell-related markers (CD56, CD57, and CD161) (167). In the liver, NKT cells reside in hepatic sinuses and secrete a variety of cytokines, including IFN, IL-2, IL-4, and IL-17, to induce Th1, Th2, and Th17 differentiation (168). Studies have demonstrated that NKT cells are involved in immunopathological damage in PBC. The hepatic infiltration of CD1d-αGalCer-restricted NKT cells was significantly higher in patients with PBC than in healthy controls (169). In the dnTGFβRII mouse model of PBC, the lack of CD1d-restricted NKT cells significantly decreased hepatic injury (170). Another study found that the activation of iNKT cells via αGalCer exacerbated hepatic damage, increased AMA production and CD8+ T cell infiltration in 2-OA-BSA, which induced PBC in the animal model (171). One study found that CD57+CD3+NKT accumulation around damaged interlobular bile ducts might be related to an imbalance in Th1/Th2 cytokines (167). CD56+and Fas ligand-positive NKT cells are involved in the death of bile epithelial cells (BECs), which promotes PBC progression. Therefore, the activation of NKT cells promotes the progression of PBC, and inhibition of NKT cells may be a potential therapeutic target for PBC.
There is no direct evidence on whether PPARs regulate NKT cells, and thus affect PBC progression. Studies on other autoimmune diseases and liver inflammation-related diseases have confirmed that PPARα and PPARγ have regulatory effects on NKT cells. One study reported that PPARα activation negatively regulates Ifng gene transcription in NKT cells, whereas PPARα antagonist enhances IFNγ production and induces Th1 dominant immunity (80). Elafibranor, a dual PPARα/δ agonist, ameliorates hepatic inflammation by reducing a variety of immune response cells, including NKT cells (172). PPARγ activation also indirectly enhances iNKT cell expansion via upregulation of CD1d and cathepsin D expression in DCs (89, 173). These two studies only focused on antigen presentation between DC and iNKT cells without evaluating the effects on the Th1/Th2 balance. Another study showed that iNKT cell activation enabled a Th2-dominant immune response upon PPARγ activation (174). Further research is needed to evaluate whether fibrates have the same effects on PBC treatment.
Bile Epithelial Cell
Th-1 mediated damage to hepatic small bile ducts is characteristic of PBC, but bile epithelial cells (BECs) are not just innocent victims. BECs are involved in the maintenance of immune tolerance and immune cells including macrophages are associated with the repair of damaged BECs (175). Bacterial components recognized as pathogen-associated molecular patterns (PAMPs) are detected in bile from patients with PBC and the healthy controls (176). TLR4 in BECs is markedly expressed in patients with PBC and recognizes lipopolysaccharide (177). TLR4 interacts with the adaptor protein myeloid differentiation primary response 88 (MyD88), which recruits IL-1 receptor-associated kinase (IRAK) 1 and subsequently activates the NF-κB and MAPK signaling pathways. Owing to the activation of these pathways, BECs produce more IL-6, IL-8, and MCP-1 (178). PPARγ and IRAK-M, inhibitory kinases of IRAK molecules, strongly inhibit NF-κB pathway activation by inhibiting MyD88 and IRAK1, thus maintaining the immune tolerance of BECs (179). PPARγ expression in cultured human BECs were downregulated in a Th-1-dominant immune environment, which promotes PBC progression. BECs from patients with PBS are more sensitive to LPS stimulation than those from healthy controls. PPARγ activation by 15d-PGJ2 negatively inhibits LPS-induced NF-κB pathway activation (180). Therefore, PPARγ is involved in negative regulation of BECs to maintain immune tolerance.
Therapeutic Strategies for Immune Disorders
Reduction of ALP is currently considered an adequate response to treatment and an endpoint in clinical trials. The mechanism of damage by the immune system in PBC, a disease with strong autoimmune characteristics, has not yet been fully elucidated. Drugs with broad immunosuppressive effects, including glucocorticoids (181), cyclosporine (182), and azathioprine (183), have not produced visible beneficial effects on the clinical outcomes in patients with PBC. In addition, selective depletion of B cells with anti-CD20 monoclonal antibody significantly reduced the titer of autoantibodies in patients with PBC, but the therapeutic effect was nonsignificant (135). Other immunomodulators under development include the IL12/23 monoclonal antibody (ustekinumab), CD40/CD40L antagonist, CX3CL1 antibody, CD80/CD86 antagonist, and selective sphingosine-1-phosphate receptor modulator (184). Current animal models cannot fully reproduce the clinical features and immunological complexity of human PBC (185), which makes it difficult to select suitable models. In addition, the lack of immune-related biomarkers for predicting PBC progression and prognosis also complicates PBC research.
The efficacy of PPAR agonists in the treatment of PBC has been confirmed. Previous studies have primarily focused on the regulation of PPARs in bile acid metabolism. It has been proven that regulation of cytochrome P450 enzymes and bile acid transporters by PPARα contributes to hepatic lipid and bile acid homeostasis, which is involved in alleviating cholestatic liver injury (186). Regulation of immune response and inhibition of disease progression by PPARs have been confirmed in studies of other autoimmune diseases, including colitis (187) and autoimmune encephalomyelitis (188). Although studies on PPAR-regulating immunity in PBC are relatively limited, current research results have preliminarily confirmed that activation of PPARs is involved in the reverse of Th1-dominant immune injury, which may delay the progression of PBC. As shown in Figure 1, PPARs have regulatory effects on multiple immune cells involved in immune disorders. In Table 2, we describe the regulatory effects of different PPAR subtypes on diverse immune cells. In general, PPAR activation promotes the maintenance of immune tolerance by directly or indirectly influencing the differentiation of Th cells. Whether the alterations in immunity are directly related to the decrease in serological indicators or are beneficial to the long-term clinical outcomes of PBC patients requires further evaluation.
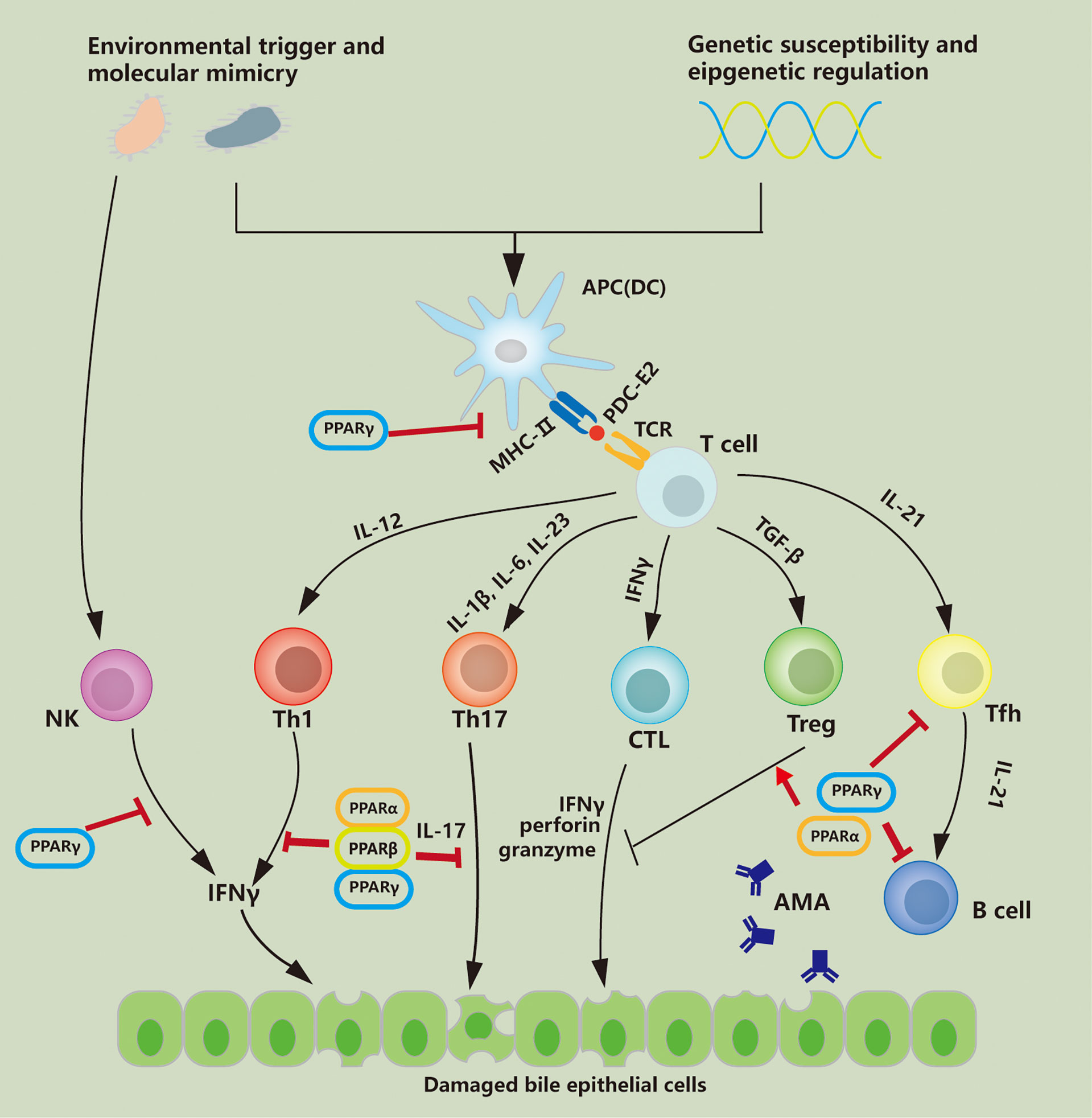
Figure 1 PPAR regulates immune cells involved in PBC pathology. PDC-E2 the E2 component of the mitochondrial pyruvate dehydrogenase complex; APC antigen-presenting cell; DC dendritic cell; IFNγ interferon-γ, TFh follicular helper T cell; AMA anti-mitochondrial autoantibody; TGFβ transforming growth factor-β; Treg regulatory T cell; CTL cytotoxic T lymphocyte; NK natural killer; MHC-II major histocompatibility complex-II; TCR T cell receptor; Th T helper.
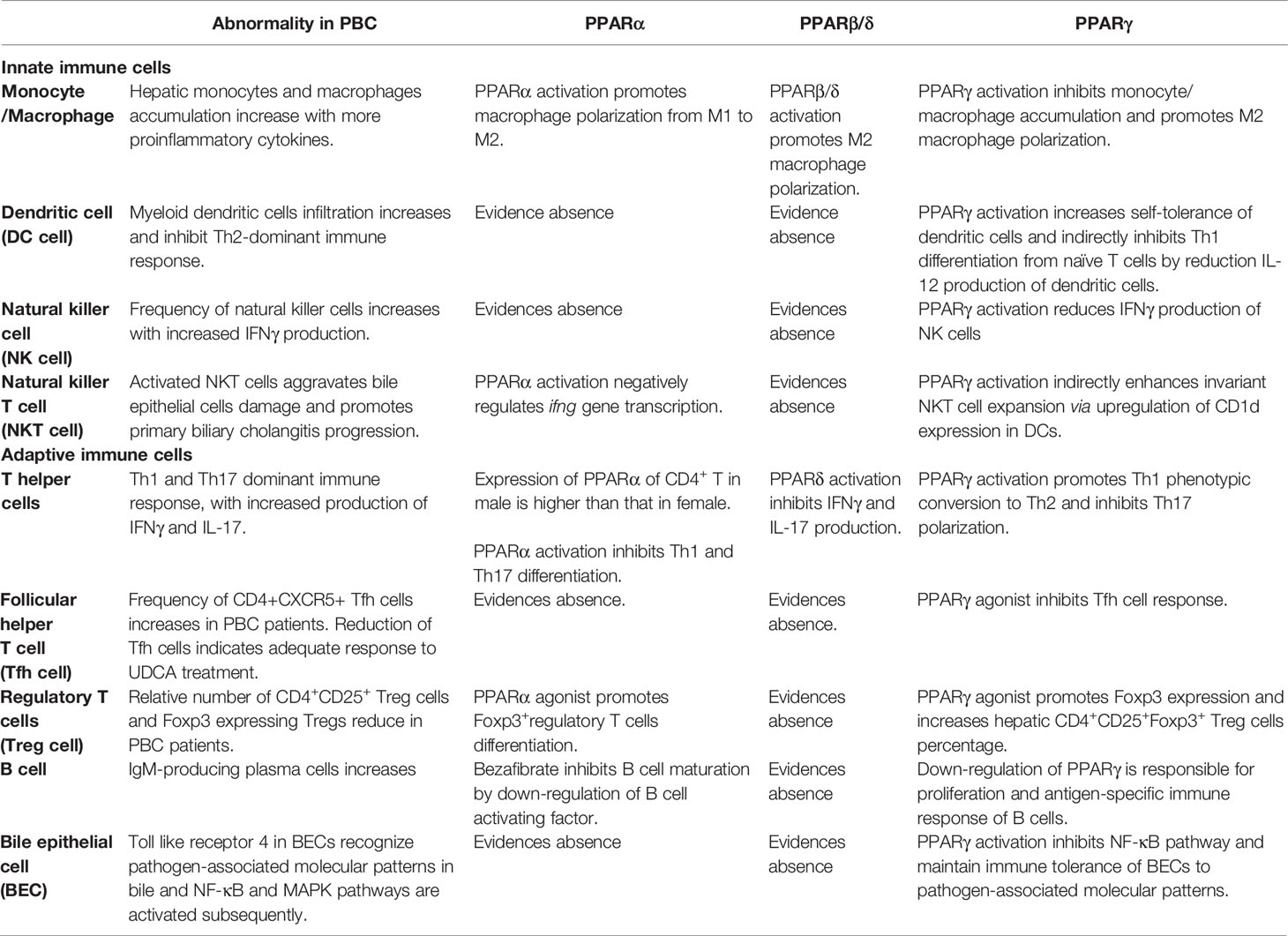
Table 2 Regulatory effects of different PPAR subtypes on diverse immune cells.
Summary and Prospects
The new generation of farnesoid X receptor and PPAR agonists and bile acid uptake inhibitors have effectively expanded the second-line treatment of PBC. UDCA still occupies a dominant position in the treatment of PBC, with its incomparable safety and effectiveness, as confirmed by several clinical trials. Fibrates are currently included in the clinical guidelines for add-on therapy (189). It is not known whether PPAR agonists will be used as monotherapy in the future or in combination with UDCA in patients with PBC, regardless of adequate response to UDCA. PPARα, PPARβ/δ, and PPARγ have distinct, but complementary functions. Dual- or pan-PPAR agonists may have better therapeutic effects than selective agonists. Activation of different subtypes of PPARs has beneficial effects on upstream immune disorders, midstream cholestasis (186), and downstream fibrosis (190) of PBC progression. The side effects may be a barrier to the application of PPAR agonists. Increased creatinine levels and myalgia are common side effects of PBC treatment (29). Cardiotoxicity, hepatotoxicity, and tumorigenesis of PPAR activation also indicate that PPAR agonists should be used circumspectly (191). Agonists with stronger liver targeting and more balanced activation effects may be more competitive in the future.
In this review, we comprehensively summarize the regulation of PPARs on known immune abnormalities of PBC. However, the full picture of the pathogenesis of PBC is not yet understood. In addition, not all the immune cells involved in PBC pathogenesis are associated with PPARs, such as cytotoxic T cells, although regulatory effects have been demonstrated in anti-tumor researches. Therefore, with the deepening of understanding about PBC immunopathogenesis, the regulatory roles of PPARs will be further updated. Interestingly, the expression of PPARα in T cells has gender differences, and whether this difference is related to the female dominance of PBC should be further explored. Comparison of PBC patients who have adequate response to UDCA in combination with fibrates therapy but not to UDCA monotherapy may obtain novel biomarkers which could predict disease progression and treatment response, such as sCD163. Although animal models of PBC are still defective, the effects of PPARs on immune cells in current autoimmunity mice model, such as dnTGF-βRII and IL-2Rα-/- mice models (185), are worthy of further exploration.
Author Contributions
CW and YS completed manuscript writing. XW and HM helped to search literature. QL provided guidance on writing. YG and JN are jointly responsible for the structure of this manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to express their gratitude to professor Zhexiong Lian for his correction of this manuscript, and editage for manuscript polish.
References
1. Lleo A, Leung PSC, Hirschfield GM, Gershwin EM. The Pathogenesis of Primary Biliary Cholangitis: A Comprehensive Review. Semin Liver Dis (2020) 40(1):34–48. doi: 10.1055/s-0039-1697617
2. Younossi ZM, Bernstein D, Shiffman ML, Kwo P, Kim WR, Kowdley KV, et al. Diagnosis and Management of Primary Biliary Cholangitis. Am J Gastroenterol (2019) 114(1):48–63. doi: 10.1038/s41395-018-0390-3
3. Manne V, Kowdley KV. Obeticholic Acid in Primary Biliary Cholangitis: Where We Stand. Curr Opin Gastroenterol (2019) 35(3):191–6. doi: 10.1097/mog.0000000000000525
4. Gochanour EM, Kowdley KV. Investigational Drugs in Early Phase Development for Primary Biliary Cholangitis. Expert Opin Investig Drugs (2021) 30(2):131–41. doi: 10.1080/13543784.2021.1857364
5. Kowdley KV, Luketic V, Chapman R, Hirschfield GM, Poupon R, Schramm C, et al. A Randomized Trial of Obeticholic Acid Monotherapy in Patients With Primary Biliary Cholangitis. Hepatology (2018) 67(5):1890–902. doi: 10.1002/hep.29569
6. Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, et al. Efficacy of Obeticholic Acid in Patients With Primary Biliary Cirrhosis and Inadequate Response to Ursodeoxycholic Acid. Gastroenterology (2015) 148(4):751–U347. doi: 10.1053/j.gastro.2014.12.005
7. Derosa G, Sahebkar A, Maffioli P. The Role of Various Peroxisome Proliferator-Activated Receptors and Their Ligands in Clinical Practice. J Cell Physiol (2018) 233(1):153–61. doi: 10.1002/jcp.25804
8. Dubois V, Eeckhoute J, Lefebvre P, Staels B. Distinct But Complementary Contributions of PPAR Isotypes to Energy Homeostasis. J Clin Invest (2017) 127(4):1202–14. doi: 10.1172/JCI88894
9. Kersten S, Stienstra R. The Role and Regulation of the Peroxisome Proliferator Activated Receptor Alpha in Human Liver. Biochimie (2017) 136:75–84. doi: 10.1016/j.biochi.2016.12.019
10. Kvandova M, Majzunova M, Dovinova I. The Role of PPARgamma in Cardiovascular Diseases. Physiol Res (2016) 65(Suppl 3):S343–63. doi: 10.33549/physiolres.933439
11. Chinetti-Gbaguidi G, Staels B. PPARbeta in Macrophages and Atherosclerosis. Biochimie (2017) 136:59–64. doi: 10.1016/j.biochi.2016.12.008
12. Day AP, Feher MD, Chopra R, Mayne PD. The Effect of Bezafibrate Treatment on Serum Alkaline Phosphatase Isoenzyme Activities. Metabol: Clin Exp (1993) 42(7):839–42. doi: 10.1016/0026-0495(93)90056-t
13. Iwasaki S, Tsuda K, Ueta H, Aono R, Ono M, Saibara T, et al. Bezafibrate may Have a Beneficial Effect in Precirrhotic Primary Biliary Cirrhosis. Hepatol Res (1999) 16:12–8. doi: 10.1016/S1386-6346(99)00033-9
14. Kurihara T, Niimi A, Maeda A, Shigemoto M, Yamashita K. Bezafibrate in the Treatment of Primary Biliary Cirrhosis: Comparison With Ursodeoxycholic Acid. Am J Gastroenterol (2000) 95(10):2990–2. doi: 10.1111/j.1572-0241.2000.03220.x
15. Takeshi K, Furukawa M, Tsuchiya M, Akimoto M, Ishiguro H, Hashimoto H, et al. Effect of Bezafibrate in the Treatment of Primary Biliary Cirrhosis. Curr Ther Res (2000) 61:74–83. doi: 10.1016/S0011-393X(00)88530-6
16. Nakai S, Masaki T, Kurokohchi K, Deguchi A, Nishioka M. Combination Therapy of Bezafibrate and Ursodeoxycholic Acid in Primary Biliary Cirrhosis: A Preliminary Study. Am J Gastroenterol (2000) 95(1):326–7. doi: 10.1111/j.1572-0241.2000.01667.x
17. Ohmoto K, Mitsui Y, Yamamoto S. Effect of Bezafibrate in Primary Biliary Cirrhosis: A Pilot Study. Liver (2001) 21(3):223–4. doi: 10.1034/j.1600-0676.2001.021003223.x
18. Kanda T, Yokosuka O, Imazeki F, Saisho H. Bezafibrate Treatment: A New Medical Approach for PBC Patients? J Gastroenterol (2003) 38(6):573–8. doi: 10.1007/s00535-002-1102-7
19. Hiromasa O, Sato Y, Ueno T, Sata M. Fenofibrate Treatment in Patients With Primary Biliary Cirrhosis. Am J Gastroenterol (2002) 97(8):2147–9. doi: 10.1111/j.1572-0241.2002.05944.x
20. Dohmen K, Mizuta T, Nakamuta M, Shimohashi N, Ishibashi H, Yamamoto K. Fenofibrate for Patients With Asymptomatic Primary Biliary Cirrhosis. World J Gastroenterol (2004) 10(6):894–8. doi: 10.3748/wjg.v10.i6.894
21. Kita R, Takamatsu S, Kimura T, Kokuryu H, Osaki Y, Tomono N. Bezafibrate may Attenuate Biliary Damage Associated With Chronic Liver Diseases Accompanied by High Serum Biliary Enzyme Levels. J Gastroenterol (2006) 41(7):686–92. doi: 10.1007/s00535-006-1831-0
22. Iwasaki S, Ohira H, Nishiguchi S, Zeniya M, Kaneko S, Onji M, et al. The Efficacy of Ursodeoxycholic Acid and Bezafibrate Combination Therapy for Primary Biliary Cirrhosis: A Prospective, Multicenter Study. Hepatol Res (2008) 38(6):557–64. doi: 10.1111/j.1872-034X.2007.00305.x
23. Takeuchi Y, Ikeda F, Fujioka S, Takaki T, Osawa T, Yasunaka T, et al. Additive Improvement Induced by Bezafibrate in Patients With Primary Biliary Cirrhosis Showing Refractory Response to Ursodeoxycholic Acid. J Gastroenterol Hepatol (2011) 26(9):1395–401. doi: 10.1111/j.1440-1746.2011.06737.x
24. Honda A, Ikegami T, Nakamuta M, Miyazaki T, Iwamoto J, Hirayama T, et al. Anticholestatic Effects of Bezafibrate in Patients With Primary Biliary Cirrhosis Treated With Ursodeoxycholic Acid. Hepatology (2013) 57(5):1931–41. doi: 10.1002/hep.26018
25. Lens S, Leoz M, Nazal L, Bruguera M, Pares A. Bezafibrate Normalizes Alkaline Phosphatase in Primary Biliary Cirrhosis Patients With Incomplete Response to Ursodeoxycholic Acid. Liver Int (2014) 34(2):197–203. doi: 10.1111/liv.12290
26. Hosonuma K, Sato K, Yamazaki Y, Yanagisawa M, Hashizume H, Horiguchi N, et al. A Prospective Randomized Controlled Study of Long-Term Combination Therapy Using Ursodeoxycholic Acid and Bezafibrate in Patients With Primary Biliary Cirrhosis and Dyslipidemia. Am J Gastroenterol (2015) 110(3):423–31. doi: 10.1038/ajg.2015.20
27. Tanaka A, Hirohara J, Nakanuma Y, Tsubouchi H, Takikawa H. Biochemical Responses to Bezafibrate Improve Long-Term Outcome in Asymptomatic Patients With Primary Biliary Cirrhosis Refractory to UDCA. J Gastroenterol (2015) 50(6):675–82. doi: 10.1007/s00535-014-0998-z
28. Bolier R, de Vries ES, Pares A, Helder J, Kemper EM, Zwinderman K, et al. Fibrates for the Treatment of Cholestatic Itch (FITCH): Study Protocol for a Randomized Controlled Trial. Trials (2017) 18(1):230. doi: 10.1186/s13063-017-1966-8
29. Corpechot C, Chazouilleres O, Rousseau A, Le Gruyer A, Habersetzer F, Mathurin P, et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N Engl J Med (2018) 378(23):2171–81. doi: 10.1056/NEJMoa1714519
30. Reig A, Sese P, Pares A. Effects of Bezafibrate on Outcome and Pruritus in Primary Biliary Cholangitis With Suboptimal Ursodeoxycholic Acid Response. Am J Gastroenterol (2018) 113(1):49–55. doi: 10.1038/ajg.2017.287
31. Kremer AE, Cleac`h AL, Lemoinne S, Wolf K, De Chaisemartin L, Chollet-Martin S, et al. Antipruritic Effect of Bezafibrate and Serum Autotaxin Measures in Patients With Primary Biliary Cholangitis. Gut (2019) 68(10):1902–3. doi: 10.1136/gutjnl-2018-317426
32. Chung SW, Lee JH, Kim MA, Leem G, Kim SW, Chang Y, et al. Additional Fibrate Treatment in UDCA-Refractory PBC Patients. Liver Int (2019) 39(9):1776–85. doi: 10.1111/liv.14165
33. Honda A, Tanaka A, Kaneko T, Komori A, Abe M, Inao M, et al. Bezafibrate Improves GLOBE and UK-PBC Scores and Long-Term Outcomes in Patients With Primary Biliary Cholangitis. Hepatology (2019) 70(6):2035–46. doi: 10.1002/hep.30552
34. Takano K, Saeki C, Oikawa T, Hidaka A, Mizuno Y, Ishida J, et al. IgM Response is a Prognostic Biomarker of Primary Biliary Cholangitis Treated With Ursodeoxycholic Acid and Bezafibrate. J Gastroenterol Hepatol (2020) 35(4):663–72. doi: 10.1002/jgh.14900
35. Bezafibrate Add-On Therapy Improves Liver Transplantation-Free Survival in Patients With Primary Biliary Cholangitis: A Japanese Nationwide Cohort Study. Gastroenterol Hepatol (2021) 17(2 suppl 3):10–1.
36. de Vries E, Bolier R, Goet J, Pares A, Verbeek J, de Vree M, et al. Fibrates for Itch (FITCH) in Fibrosing Cholangiopathies: A Double-Blind, Randomized, Placebo-Controlled Trial. Gastroenterology (2021) 160(3):734–743.e736. doi: 10.1053/j.gastro.2020.10.001
37. Sorda JA, Gonzalez Ballerga E, Barreyro FJ, Avagnina A, Carballo P, Paes de Lima A, et al. Bezafibrate Therapy in Primary Biliary Cholangitis Refractory to Ursodeoxycholic Acid: A Longitudinal Study of Paired Liver Biopsies at 5 Years of Follow Up. Aliment Pharmacol Ther (2021) 54(9):1202–12. doi: 10.1111/apt.16618
38. Tanaka A, Hirohara J, Nakano T, Matsumoto K, Chazouilleres O, Takikawa H, et al. Association of Bezafibrate With Transplant-Free Survival in Patients With Primary Biliary Cholangitis. J Hepatol (2021) 75(3):565–71. doi: 10.1016/j.jhep.2021.04.010
39. Liberopoulos EN, Florentin M, Elisaf MS, Mikhailidis DP, Tsianos E. Fenofibrate in Primary Biliary Cirrhosis: A Pilot Study. Open Cardiovassular Med J (2010) 4:120–6. doi: 10.2174/1874192401004010120
40. Levy C, Peter JA, Nelson DR, Keach J, Petz J, Cabrera R, et al. Pilot Study: Fenofibrate for Patients With Primary Biliary Cirrhosis and an Incomplete Response to Ursodeoxycholic Acid. Aliment Pharmacol Ther (2011) 33(2):235–42. doi: 10.1111/j.1365-2036.2010.04512.x
41. Han XF, Wang QX, Liu Y, You ZR, Bian ZL, Qiu DK, et al. Efficacy of Fenofibrate in Chinese Patients With Primary Biliary Cirrhosis Partially Responding to Ursodeoxycholic Acid Therapy. J Dig Dis (2012) 13(4):219–24. doi: 10.1111/j.1751-2980.2012.00574.x
42. Dohmen K, Tanaka H, Haruno M. Effectiveness of Fenofibrate in Comparison to Bezafibrate for Patients With Asymptomatic Primary Biliary Cirrhosis. Fukuoka Igaku Zasshi (2013) 104(10):350–61. doi: 10.15017/1398604
43. Cheung AC, Lapointe-Shaw L, Kowgier M, Meza-Cardona J, Hirschfield GM, Janssen HL, et al. Combined Ursodeoxycholic Acid (UDCA) and Fenofibrate in Primary Biliary Cholangitis Patients With Incomplete UDCA Response may Improve Outcomes. Aliment Pharmacol Ther (2016) 43(2):283–93. doi: 10.1111/apt.13465
44. Hegade VS, Khanna A, Walker LJ, Wong LL, Dyson JK, Jones DEJ. Long-Term Fenofibrate Treatment in Primary Biliary Cholangitis Improves Biochemistry But Not the UK-PBC Risk Score. Dig Dis Sci (2016) 61(10):3037–44. doi: 10.1007/s10620-016-4250-y
45. Duan W, Ou X, Wang X, Wang Y, Zhao X, Wang Q, et al. Efficacy and Safety of Fenofibrate Add-on Therapy for Patients With Primary Biliary Cholangitis and a Suboptimal Response to UDCA. Rev Esp Enferm Dig (2018) 110(9):557–63. doi: 10.17235/reed.2018.5533/2018
46. Ghonem NS, Auclair AM, Hemme CL, Gallucci GM, de la Rosa Rodriguez R, Boyer JL, et al. Fenofibrate Improves Liver Function and Reduces the Toxicity of the Bile Acid Pool in Patients With Primary Biliary Cholangitis and Primary Sclerosing Cholangitis Who Are Partial Responders to Ursodiol. Clin Pharmacol Ther (2020) 108(6):1213–23. doi: 10.1002/cpt.1930
47. Wang L, Sun K, Tian A, Liu Y, Zhang M, Zhou X, et al. Fenofibrate Improves GLOBE and UK-PBC Scores and Histological Features in Primary Biliary Cholangitis. Minerva Med (2021). doi: 10.23736/s0026-4806.21.07316-x
48. Joshita S, Umemura T, Yamashita Y, Sugiura A, Yamazaki T, Fujimori N, et al. Biochemical and Plasma Lipid Responses to Pemafibrate in Patients With Primary Biliary Cholangitis. Hepatol Res (2019) 49(10):1236–43. doi: 10.1111/hepr.13361
49. Dohmen K, Onohara SY, Harada S. Effects of Switching From Fenofibrate to Pemafibrate for Asymptomatic Primary Biliary Cholangitis. Korean J Gastroenterol (2021) 78(4):227–34. doi: 10.4166/kjg.2021.092
50. Yamaguchi M, Asano T, Arisaka T, Mashima H, Irisawa A, Tamano M. Effects of Pemafibrate on Primary Biliary Cholangitis With Dyslipidemia. Hepatol Res (2022) 52(6):522–31. doi: 10.1111/hepr.13747
51. Schattenberg JM, Pares A, Kowdley KV, Heneghan MA, Caldwell S, Pratt D, et al. A Randomized Placebo-Controlled Trial of Elafibranor in Patients With Primary Biliary Cholangitis and Incomplete Response to UDCA. J Hepatol (2021) 74(6):1344–54. doi: 10.1016/j.jhep.2021.01.013
52. Vuppalanchi R, Caldwell SH, Pyrsopoulos N, deLemos AS, Rossi S, Levy C, et al. Proof-Of-Concept Study to Evaluate the Safety and Efficacy of Saroglitazar in Patients With Primary Biliary Cholangitis. J Hepatol (2022) 76(1):75–85. doi: 10.1016/j.jhep.2021.08.025
53. Vuppalanchi R, Gonzalez-Huezo MS, Payan-Olivas R, Munoz-Espinosa LE, Shaikh F, Pio Cruz-Lopez JL, et al. A Multicenter, Open-Label, Single-Arm Study to Evaluate the Efficacy and Safety of Saroglitazar in Patients With Primary Biliary Cholangitis. Clin Transl Gastroenterol (2021) 12(4):e00327. doi: 10.14309/ctg.0000000000000327
54. Jones D, Boudes PF, Swain MG, Bowlus CL, Galambos MR, Bacon BR, et al. Seladelpar (MBX-8025), a Selective PPAR-δ Agonist, in Patients With Primary Biliary Cholangitis With an Inadequate Response to Ursodeoxycholic Acid: A Double-Blind, Randomised, Placebo-Controlled, Phase 2, Proof-of-Concept Study. Lancet Gastroenterol Hepatol (2017) 2(10):716–26. doi: 10.1016/s2468-1253(17)30246-7
55. Kremer AE, Mayo MJ, Hirschfield G, Levy C, Bowlus CL, Jones DE, et al. Seladelpar Improved Measures of Pruritus, Sleep, and Fatigue and Decreased Serum Bile Acids in Patients With Primary Biliary Cholangitis. Liver Int (2022) 42(1):112–23. doi: 10.1111/liv.15039
56. ENHANCE: Safety and Efficacy of Seladelpar in Patients With Primary Biliary Cholangitis-A Phase 3, International, Randomized, Placebo-Controlled Study. Gastroenterol Hepatol (2021) 17(2 suppl 3):5–6.
57. . CymaBay Therapeutics Announces Oral Late-Breaking Presentation of Positive Results From the ENHANCE Global Phase 3 Study Evaluating Seladelpar for Primary Biliary Cholangitis at The Liver Meeting (2020).
58. Agrawal R, Majeed M, Attar BM, Omar YA, Mbachi C, Wang Y, et al. Effectiveness of Bezafibrate and Ursodeoxycholic Acid in Patients With Primary Biliary Cholangitis: A Meta-Analysis of Randomized Controlled Trials. Ann Gastroenterol (2019) 32(5):489–97. doi: 10.20524/aog.2019.0403
59. Shen N, Pan J, Miao H, Zhang H, Xing L, Yu X. Fibrates for the Treatment of Pruritus in Primary Biliary Cholangitis: A Systematic Review and Meta-Analysis. Ann Palliat Med (2021) 10(7):7697–705. doi: 10.21037/apm-21-1304
60. Yin Q, Li J, Xia Y, Zhang R, Wang J, Lu W, et al. Systematic Review and Meta-Analysis: Bezafibrate in Patients With Primary Biliary Cirrhosis. Drug Des Devel Ther (2015) 9:5407–19. doi: 10.2147/DDDT.S92041
61. Zhang Y, Chen K, Dai W, Xia Y, Wang F, Shen M, et al. Combination Therapy of Bezafibrate and Ursodeoxycholic Acid for Primary Biliary Cirrhosis: A Meta-Analysis. Hepatol Res (2015) 45(1):48–58. doi: 10.1111/hepr.12373
62. Guo C, Zhang Y, He L, Wang F, Chen K, Li J, et al. Combination Therapy of Fenofibrate and Ursodeoxycholic Acid in Patients With Primary Biliary Cirrhosis Who Respond Incompletely to&Nbsp;UDCA Monotherapy: A Meta-Analysis. Drug Design Dev Ther (2015) 2757:2757–66. doi: 10.2147/dddt.s79837
63. Grigorian AY, Mardini HE, Corpechot C, Poupon R, Levy C. Fenofibrate is Effective Adjunctive Therapy in the Treatment of Primary Biliary Cirrhosis: A Meta-Analysis. Clin Res Hepatol Gastroenterol (2015) 39(3):296–306. doi: 10.1016/j.clinre.2015.02.011
64. Soret PA, Lam L, Carrat F, Smets L, Berg T, Carbone M, et al. Combination of Fibrates With Obeticholic Acid is Able to Normalise Biochemical Liver Tests in Patients With Difficult-to-Treat Primary Biliary Cholangitis. Aliment Pharmacol Ther (2021) 53(10):1138–46. doi: 10.1111/apt.16336
65. Smets L, Verbeek J, Korf H, van der Merwe S, Nevens F. Improved Markers of Cholestatic Liver Injury in Patients With Primary Biliary Cholangitis Treated With Obeticholic Acid and Bezafibrate. Hepatology (2021) 73(6):2598–600. doi: 10.1002/hep.31613
66. Cordell HJ, Fryett JJ, Ueno K, Darlay R, Aiba Y, Hitomi Y, et al. An International Genome-Wide Meta-Analysis of Primary Biliary Cholangitis: Novel Risk Loci and Candidate Drugs. J Hepatol (2021) 75(3):572–81. doi: 10.1016/j.jhep.2021.04.055
67. Wang C, Zheng X, Jiang P, Tang R, Gong Y, Dai Y, et al. Genome-Wide Association Studies of Specific Antinuclear Autoantibody Subphenotypes in Primary Biliary Cholangitis. Hepatology (2019) 70(1):294–307. doi: 10.1002/hep.30604
68. Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, et al. Primary Biliary Cirrhosis in Monozygotic and Dizygotic Twins: Genetics, Epigenetics, and Environment. Gastroenterology (2004) 127(2):485–92. doi: 10.1053/j.gastro.2004.05.005
69. Yang Y, Choi J, Chen Y, Invernizzi P, Yang G, Zhang W, et al. E. Coli and the Etiology of Human PBC: Antimitochondrial Antibodies and Spreading Determinants. Hepatology (2022) 75(2):266–79. doi: 10.1002/hep.32172
70. Tanaka A, Leung PS, Gershwin ME. Environmental Basis of Primary Biliary Cholangitis. Exp Biol Med (Maywood) (2018) 243(2):184–9. doi: 10.1177/1535370217748893
71. Harada K, de Water JV, Leung PS, Coppel RL, Ansari A, Nakanuma Y, Gershwin ME. In Situ Nucleic Acid Hybridization of Cytokines in Primary Biliary Cirrhosis: Predominance of the Th1 Subset. Hepatology (1997) 25(4):791–6. doi: 10.1002/hep.510250402
72. Zhu J. T Helper Cell Differentiation, Heterogeneity, and Plasticity. Cold Spring Harb Perspect Biol (2018) 10(10):a030338. doi: 10.1101/cshperspect.a030338
73. Sekiya H, Komatsu T, Isono E, Furukawa M, Matsushima S, Yamaguchi N, et al. Decrease in the Prevalence of IL-4-Producing CD4+ T Cells in Patients With Advanced Stage of Primary Biliary Cirrhosis. Am J Gastroenterol (1999) 94(12):3589–94. doi: 10.1111/j.1572-0241.1999.01547.x
74. Yu K, Li P, Xu T, Xu J, Wang K, Chai J, et al. Decreased Infiltration of CD4(+) Th1 Cells Indicates a Good Response to Ursodeoxycholic Acid (UDCA) in Primary Biliary Cholangitis. Pathol Res Pract (2021) 217:153291. doi: 10.1016/j.prp.2020.153291
75. Hirano Y, Hirano F, Fujii H, Makino I. Fibrates Suppress Chenodeoxycholic Acid-Induced RANTES Expression Through Inhibition of NF-kappaB Activation. Eur J Pharmacol (2002) 448(1):19–26. doi: 10.1016/s0014-2999(02)01902-7
76. Maltby J, Wright S, Bird G, Sheron N. Chemokine Levels in Human Liver Homogenates: Associations Between GRO Alpha and Histopathological Evidence of Alcoholic Hepatitis. Hepatology (1996) 24(5):1156–60. doi: 10.1053/jhep.1996.v24.pm0008903391
77. Nozaki Y, Harada K, Sanzen T, Nakanuma Y. PPARgamma Ligand Attenuates Portal Inflammation in the MRL-Lpr Mouse: A New Strategy to Restrain Cholangiopathy in Primary Biliary Cirrhosis. Med Mol Morphol (2013) 46(3):153–9. doi: 10.1007/s00795-013-0017-0
78. Zhang MA, Rego D, Moshkova M, Kebir H, Chruscinski A, Nguyen H, et al. Peroxisome Proliferator-Activated Receptor (PPAR)alpha and -Gamma Regulate IFNgamma and IL-17A Production by Human T Cells in a Sex-Specific Way. Proc Natl Acad Sci U.S.A. (2012) 109(24):9505–10. doi: 10.1073/pnas.1118458109
79. Dunn SE, Ousman SS, Sobel RA, Zuniga L, Baranzini SE, Youssef S, et al. Peroxisome Proliferator-Activated Receptor (PPAR)alpha Expression in T Cells Mediates Gender Differences in Development of T Cell-Mediated Autoimmunity. J Exp Med (2007) 204(2):321–30. doi: 10.1084/jem.20061839
80. Zhang MA, Ahn JJ, Zhao FL, Selvanantham T, Mallevaey T, Stock N, et al. Antagonizing Peroxisome Proliferator-Activated Receptor Alpha Activity Selectively Enhances Th1 Immunity in Male Mice. J Immunol (2015) 195(11):5189–202. doi: 10.4049/jimmunol.1500449
81. Gerussi A, Cristoferi L, Carbone M, Asselta R, Invernizzi P. The Immunobiology of Female Predominance in Primary Biliary Cholangitis. J Autoimmun (2018) 95:124–32. doi: 10.1016/j.jaut.2018.10.015
82. Yuan Z. Peroxisome Proliferation-Activated Receptor-γ Ligands Ameliorate Experimental Autoimmune Myocarditis. Cardiovasc Res (2003) 59(3):685–94. doi: 10.1016/s0008-6363(03)00457-7
83. Hasegawa H, Takano H, Zou Y, Qin Y, Hizukuri K, Odaka K, et al. Pioglitazone, a Peroxisome Proliferator-Activated Receptor Gamma Activator, Ameliorates Experimental Autoimmune Myocarditis by Modulating Th1/Th2 Balance. J Mol Cell Cardiol (2005) 38(2):257–65. doi: 10.1016/j.yjmcc.2004.11.010
84. Wang L, Zhu J, Shan S, Qin Y, Kong Y, Liu J, et al. Repression of Interferon-Gamma Expression in T Cells by Prospero-Related Homeobox Protein. Cell Res (2008) 18(9):911–20. doi: 10.1038/cr.2008.275
85. Kanakasabai S, Chearwae W, Walline CC, Iams W, Adams SM, Bright JJ. Peroxisome Proliferator-Activated Receptor Delta Agonists Inhibit T Helper Type 1 (Th1) and Th17 Responses in Experimental Allergic Encephalomyelitis. Immunology (2010) 130(4):572–88. doi: 10.1111/j.1365-2567.2010.03261.x
86. Dunn SE, Bhat R, Straus DS, Sobel RA, Axtell R, Johnson A, et al. Peroxisome Proliferator-Activated Receptor Delta Limits the Expansion of Pathogenic Th Cells During Central Nervous System Autoimmunity. J Exp Med (2010) 207(8):1599–608. doi: 10.1084/jem.20091663
87. Harada K, Shimoda S, Sato Y, Isse K, Ikeda H, Nakanuma Y. Periductal Interleukin-17 Production in Association With Biliary Innate Immunity Contributes to the Pathogenesis of Cholangiopathy in Primary Biliary Cirrhosis. Clin Exp Immunol (2009) 157(2):261–70. doi: 10.1111/j.1365-2249.2009.03947.x
88. Lan RYZ, Salunga TL, Tsuneyama K, Lian Z-X, Yang G-X, Hsu W, et al. Hepatic IL-17 Responses in Human and Murine Primary Biliary Cirrhosis. J Autoimmun (2009) 32(1):43–51. doi: 10.1016/j.jaut.2008.11.001
89. Nakken B, Varga T, Szatmari I, Szeles L, Gyongyosi A, Illarionov PA, et al. Peroxisome Proliferator-Activated Receptor Gamma-Regulated Cathepsin D is Required for Lipid Antigen Presentation by Dendritic Cells. J Immunol (2011) 187(1):240–7. doi: 10.4049/jimmunol.1002421
90. Castro G, Liu X, Ngo K, De Leon-Tabaldo A, Zhao S, Luna-Roman R, et al. RORgammat and RORalpha Signature Genes in Human Th17 Cells. PloS One (2017) 12(8):e0181868. doi: 10.1371/journal.pone.0181868
91. Chang H, Zhao F, Xie X, Liao Y, Song Y, Liu C, et al. PPARalpha Suppresses Th17 Cell Differentiation Through IL-6/STAT3/RORgammat Pathway in Experimental Autoimmune Myocarditis. Exp Cell Res (2019) 375(1):22–30. doi: 10.1016/j.yexcr.2018.12.005
92. Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, et al. The Nuclear Receptor PPAR Gamma Selectively Inhibits Th17 Differentiation in a T Cell-Intrinsic Fashion and Suppresses CNS Autoimmunity. J Exp Med (2009) 206(10):2079–89. doi: 10.1084/jem.20082771
93. Kim DH, Ihn HJ, Moon C, Oh SS, Park S, Kim S, et al. Ciglitazone, a Peroxisome Proliferator-Activated Receptor Gamma Ligand, Inhibits Proliferation and Differentiation of Th17 Cells. Biomol Ther (Seoul) (2015) 23(1):71–6. doi: 10.4062/biomolther.2014.042
94. Miao Y, Zheng Y, Geng Y, Yang L, Cao N, Dai Y, et al. The Role of GLS1-Mediated Glutaminolysis/2-HG/H3K4me3 and GSH/ROS Signals in Th17 Responses Counteracted by PPARgamma Agonists. Theranostics (2021) 11(9):4531–48. doi: 10.7150/thno.54803
95. Kanakasabai S, Walline CC, Chakraborty S, Bright JJ. PPARdelta Deficient Mice Develop Elevated Th1/Th17 Responses and Prolonged Experimental Autoimmune Encephalomyelitis. Brain Res (2011) 1376:101–12. doi: 10.1016/j.brainres.2010.12.059
96. Tomiyama T, Yang GX, Zhao M, Zhang W, Tanaka H, Wang J, et al. The Modulation of Co-Stimulatory Molecules by Circulating Exosomes in Primary Biliary Cirrhosis. Cell Mol Immunol (2017) 14(3):276–84. doi: 10.1038/cmi.2015.86
97. Ballardini G, M.Fallani, Bianchi FB, Pisi E. Antigen Presenting Cells in Liver Biopsies From Patients With Primary Biliary Cirrhosis. Autoimmunity (1989) 3(2):135–44. doi: 10.3109/08916938909019962
98. Tanimoto K, Akbar SMF, Michitaka K, Onji M. Immunohistochemical Localization of Antigen Presenting Cells in Liver From Patients With Primary Biliary Cirrhosis; Highly Restricted Distribution of CD83-Positive Activated Dendritic Cells. Pathol - Res Pract (1999) 195(3):157–62. doi: 10.1016/s0344-0338(99)80028-4
99. Harada K, Shimoda S, Ikeda H, Chiba M, Hsu M, Sato Y, et al. Significance of Periductal Langerhans Cells and Biliary Epithelial Cell-Derived Macrophage Inflammatory Protein-3alpha in the Pathogenesis of Primary Biliary Cirrhosis. Liver Int (2011) 31(2):245–53. doi: 10.1111/j.1478-3231.2010.02367.x
100. Yamamoto K, Akbar SM, Masumoto T, Onji M. Increased Nitric Oxide (NO) Production by Antigen-Presenting Dendritic Cells is Responsible for Low Allogeneic Mixed Leucocyte Reaction (MLR) in Primary Biliary Cirrhosis (PBC). Clin Exp Immunol (1998) 114(1):94–101. doi: 10.1046/j.1365-2249.1998.00696.x
101. Hokari A, Zeniya M., Esumi H, Kawabe T, Gershwin ME, Toda G. Detection of Serum Nitrite and Nitrate in Primary Biliary Cirrhosis: Possible Role of Nitric Oxide in Bile Duct Injury. J Gastroenterol Hepatol (2002) 17(3):308–15. doi: 10.1046/j.1440-1746.2002.02689.x
102. Hiasa Y, Akbar SMF, Abe M, Michitaka K, Horiike N, Onji M. Dendritic Cell Subtypes in Autoimmune Liver Diseases; Decreased Expression of HLA DR and CD123 on Type 2 Dendritic Cells. Hepatol Res (2002) 22(4):241–9. doi: 10.1016/s1386-6346(01)00149-8
103. Jakobsen MA, Petersen RK, Kristiansen K, Lange M, Lillevang ST. Peroxisome Proliferator-Activated Receptor Alpha, Delta, Gamma1 and Gamma2 Expressions are Present in Human Monocyte-Derived Dendritic Cells and Modulate Dendritic Cell Maturation by Addition of Subtype-Specific Ligands. Scand J Immunol (2006) 63(5):330–7. doi: 10.1111/j.1365-3083.2006.01745.x
104. Akbar SM, Furukawa S, Nakanishi S, Abe M, Horiike N, Onji M. Therapeutic Efficacy of Decreased Nitrite Production by Bezafibrate in Patients With Primary Biliary Cirrhosis. J Gastroenterol (2005) 40(2):157–63. doi: 10.1007/s00535-004-1518-3
105. Nencioni A, Grunebach F, Zobywlaski A, Denzlinger C, Brugger W, Brossart P. Dendritic Cell Immunogenicity is Regulated by Peroxisome Proliferator-Activated Receptor Gamma. J Immunol (2002) 169(3):1228–35. doi: 10.4049/jimmunol.169.3.1228
106. Appel S, Mirakaj V, Bringmann A, Weck MM, Grunebach F, Brossart P. PPAR-Gamma Agonists Inhibit Toll-Like Receptor-Mediated Activation of Dendritic Cells via the MAP Kinase and NF-kappaB Pathways. Blood (2005) 106(12):3888–94. doi: 10.1182/blood-2004-12-4709
107. Kock G, Bringmann A, Held SA, Daecke S, Heine A, Brossart P. Regulation of Dectin-1-Mediated Dendritic Cell Activation by Peroxisome Proliferator-Activated Receptor-Gamma Ligand Troglitazone. Blood (2011) 117(13):3569–74. doi: 10.1182/blood-2010-08-302224
108. Klotz L, Dani I, Edenhofer F, Nolden L, Evert B, Paul B, et al. Peroxisome Proliferator-Activated Receptor Gamma Control of Dendritic Cell Function Contributes to Development of CD4+ T Cell Anergy. J Immunol (2007) 178(4):2122–31. doi: 10.4049/jimmunol.178.4.2122
109. Faveeuw C, S.Fougeray, Angeli V, Fontaine J, Chinetti G, Gosset P, et al. Peroxisome Proliferator-Activated Receptor Gamma Activators Inhibit Interleukin-12 Production in Murine Dendritic Cells. FEBS Lett (2000) 486(3):261–6. doi: 10.1016/s0014-5793(00)02319-x
110. Gogolak P, Rethi B, Szatmari I, Lanyi A, Dezso B, Nagy L, et al. Differentiation of CD1a- and CD1a+ Monocyte-Derived Dendritic Cells is Biased by Lipid Environment and PPARgamma. Blood (2007) 109(2):643–52. doi: 10.1182/blood-2006-04-016840
111. Gosset P, Charbonnier AS, Delerive P, Fontaine J, Staels B, Pestel J, et al. Peroxisome Proliferator-Activated Receptor Gamma Activators Affect the Maturation of Human Monocyte-Derived Dendritic Cells. Eur J Immunol (2001) 31(10):2857–65. doi: 10.1002/1521-4141(2001010)31:10<2857::aid-immu2857>3.0.co;2-x
112. Thompson PW, Bayliffe AI, Warren AP, Lamb JR. Interleukin-10 is Upregulated by Nanomolar Rosiglitazone Treatment of Mature Dendritic Cells and Human CD4+ T Cells. Cytokine (2007) 39(3):184–91. doi: 10.1016/j.cyto.2007.07.191
113. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
114. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of Peripheral CD4+CD25- Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-Beta Induction of Transcription Factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi: 10.1084/jem.20030152
115. Jeffrey A Bluestone AKA. Natural Versus Adaptive Regulatory T Cells Nature Reviews. Immunology (2003) 3(3):253–7. doi: 10.1038/nri1032
116. Liaskou E, Patel SR, Webb G, Bagkou Dimakou D, Akiror S, Krishna M, et al. Increased Sensitivity of Treg Cells From Patients With PBC to Low Dose IL-12 Drives Their Differentiation Into IFN-Gamma Secreting Cells. J Autoimmun (2018) 94:143–55. doi: 10.1016/j.jaut.2018.07.020
117. Lan RY, Cheng C, Lian ZX, Tsuneyama K, Yang GX, Moritoki Y, et al. Liver-Targeted and Peripheral Blood Alterations of Regulatory T Cells in Primary Biliary Cirrhosis. Hepatology (2006) 43(4):729–37. doi: 10.1002/hep.21123
118. Zhou Z, Liang Y, Gao Y, Kong W, Feng J, Wang X. Fenofibrate Enhances the In Vitro Differentiation of Foxp3(+) Regulatory T Cells in Mice. PPAR Res (2012) 2012:529035. doi: 10.1155/2012/529035
119. Hichami A, Yessoufou A, Ghiringhelli F, Salvadori F, Moutairou K, Zwetyenga N, et al. Peroxisome Proliferator-Activated Receptor Alpha Deficiency Impairs Regulatory T Cell Functions: Possible Application in the Inhibition of Melanoma Tumor Growth in Mice. Biochimie (2016) 131:1–10. doi: 10.1016/j.biochi.2016.09.001
120. Lei J, Hasegawa H, Matsumoto T, Yasukawa M. Peroxisome Proliferator-Activated Receptor Alpha and Gamma Agonists Together With TGF-Beta Convert Human CD4+CD25- T Cells Into Functional Foxp3+ Regulatory T Cells. J Immunol (2010) 185(12):7186–98. doi: 10.4049/jimmunol.1001437
121. Zhu Y, Ni Y, Liu R, Hou M, Yang B, Song J, et al. PPAR-Gamma Agonist Alleviates Liver and Spleen Pathology via Inducing Treg Cells During Schistosoma Japonicum Infection. J Immunol Res (2018) 2018:6398078. doi: 10.1155/2018/6398078
122. Cannons JL, Lu KT, Schwartzberg PL. T Follicular Helper Cell Diversity and Plasticity. Trends Immunol (2013) 34(5):200–7. doi: 10.1016/j.it.2013.01.001
123. Zhang Y, Garcia.-Ibanez L, Toellner K-M. Regulation of Germinal Center B-Cell Differentiation. Immunol Rev (2016) 270(1):8–19. doi: 10.1111/imr.12396
124. Wang L, Sun Y, Zhang Z, Jia Y, Zou Z, Ding J, et al. CXCR5+ CD4+ T Follicular Helper Cells Participate in the Pathogenesis of Primary Biliary Cirrhosis. Hepatology (2015) 61(2):627–38. doi: 10.1002/hep.27306
125. Wang L, Sun X, Qiu J, Cai Y, Ma L, Zhao P, et al. Increased Numbers of Circulating ICOS(+) Follicular Helper T and CD38(+) Plasma Cells in Patients With Newly Diagnosed Primary Biliary Cirrhosis. Dig Dis Sci (2015) 60(2):405–13. doi: 10.1007/s10620-014-3372-3
126. Park HJ, Kim DH, Choi JY, Kim WJ, Kim JY, Senejani AG, et al. PPARgamma Negatively Regulates T Cell Activation to Prevent Follicular Helper T Cells and Germinal Center Formation. PloS One (2014) 9(6):e99127. doi: 10.1371/journal.pone.0099127
127. Park HJ, Park HS, Lee JU, Bothwell AL, Choi JM. Gender-Specific Differences in PPARgamma Regulation of Follicular Helper T Cell Responses With Estrogen. Sci Rep (2016) 6:28495. doi: 10.1038/srep28495
128. Menéndez-Caro JL, Alvarez.-Mon M, Girón JA, Manzano L, Garrido A, Abreu L, et al. Increased IgM B Cell Differentiation Lymphokine Production by T Lymphocytes From Patients With Primary Biliary Cirrhosis. J Hepatol (1994) 20(4):446–53. doi: 10.1016/s0168-8278(05)80488-x
129. Robe AJ, Kirby JA, Jones DE, Palmer JM. A Key Role for Autoreactive B Cells in the Breakdown of T-Cell Tolerance to Pyruvate Dehydrogenase Complex in the Mouse. Hepatology (2005) 41(5):1106–12. doi: 10.1002/hep.20642
130. Gatselis NK, Zachou K, Norman GL, Gabeta S, Papamichalis P, Koukoulis GK, et al. Clinical Significance of the Fluctuation of Primary Biliary Cirrhosis-Related Autoantibodies During the Course of the Disease. Autoimmunity (2013) 46(7):471–9. doi: 10.3109/08916934.2013.801461
131. Benson GD, Kikuchi K, Miyakawa H, Tanaka A, Watnik MR, Gershwin ME. Serial Analysis of Antimitochondrial Antibody in Patients With Primary Biliary Cirrhosis. Clin Dev Immunol (2004) 11(2):129–33. doi: 10.1080/10446670410001722113
132. Ueno K, Aiba Y, Hitomi Y, Shimoda S, Nakamura H, Gervais O, et al. Integrated GWAS and mRNA Microarray Analysis Identified IFNG and CD40L as the Central Upstream Regulators in Primary Biliary Cholangitis. Hepatol Commun (2020) 4(5):724–38. doi: 10.1002/hep4.1497
133. Lleo A, Liao J, Invernizzi P, Zhao M, Bernuzzi F, Ma L, et al. Immunoglobulin M Levels Inversely Correlate With CD40 Ligand Promoter Methylation in Patients With Primary Biliary Cirrhosis. Hepatology (2012) 55(1):153–60. doi: 10.1002/hep.24630
134. Tsuda M, Moritoki Y, Lian ZX, Zhang W, Yoshida K, Wakabayashi K, et al. Biochemical and Immunologic Effects of Rituximab in Patients With Primary Biliary Cirrhosis and an Incomplete Response to Ursodeoxycholic Acid. Hepatology (2012) 55(2):512–21. doi: 10.1002/hep.24748
135. Myers RP, Swain MG, Lee SS, Shaheen AA, Burak KW. B-Cell Depletion With Rituximab in Patients With Primary Biliary Cirrhosis Refractory to Ursodeoxycholic Acid. Am J Gastroenterol (2013) 108(6):933–41. doi: 10.1038/ajg.2013.51
136. Lied GA, Berstad A. Functional and Clinical Aspects of the B-Cell-Activating Factor (BAFF): A Narrative Review. Scand J Immunol (2011) 73(1):1–7. doi: 10.1111/j.1365-3083.2010.02470.x
137. Zhang B, Hu M, Zhang P, Cao H, Wang Y, Wang Z, et al. BAFF Promotes Regulatory T-Cell Apoptosis and Blocks Cytokine Production by Activating B Cells in Primary Biliary Cirrhosis. Braz J Med Biol Res (2013) 46(5):433–9. doi: 10.1590/1414-431X20132665
138. Setoguchi K, Misaki Y, Terauchi Y, Yamauchi T, Kawahata K, Kadowaki T, et al. Peroxisome Proliferator-Activated Receptor-γ Haploinsufficiency Enhances B Cell Proliferative Responses and Exacerbates Experimentally Induced Arthritis. J Clin Invest (2001) 108(11):1667–75. doi: 10.1172/jci200113202
139. Su J, Wang K, Zhou X, Wang Y, Xu J, Tao L, et al. B-Cell-Specific-Peroxisome Proliferator-Activated Receptor Gamma Deficiency Augments Contact Hypersensitivity With Impaired Regulatory B Cells. Immunology (2019) 156(3):282–96. doi: 10.1111/imm.13027
140. Mosser DM, Edwards JP. Exploring the Full Spectrum of Macrophage Activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
141. Mathew J, Hines JE, Toole K, Johnson SJ, James OF, Burt AD. Quantitative Analysis of Macrophages and Perisinusoidal Cells in Primary Biliary Cirrhosis. Histopathology (1994) 25(1):65–70. doi: 10.1111/j.1365-2559.1994.tb00599.x
142. Tsuneyama K, Harada K, Yasoshima M, Hiramatsu K, Mackay CR, Mackay IR, et al. Monocyte Chemotactic Protein-1, -2, and -3 are Distinctively Expressed in Portal Tracts and Granulomata in Primary Biliary Cirrhosis: Implications for Pathogenesis. J Pathol (2001) 193(1):102–9. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH725>3.0.CO;2-P
143. Yang Z, Liang Y, Lin F, Zhang Z, Luo W, Zhang Y, et al. Implication of Increased Serum Stromal Cell-Derived Factor-1 for Primary Biliary Cholangitis. Int Immunopharmacol (2018) 56:285–90. doi: 10.1016/j.intimp.2018.01.046
144. Isse K, Harada K, Zen Y, Kamihira T, Shimoda S, Harada M, et al. Fractalkine and CX3CR1 are Involved in the Recruitment of Intraepithelial Lymphocytes of Intrahepatic Bile Ducts. Hepatology (2005) 41(3):506–16. doi: 10.1002/hep.20582
145. Mao TK, Lian ZX, Selmi C, Ichiki Y, Ashwood P, Ansari AA, et al. Altered Monocyte Responses to Defined TLR Ligands in Patients With Primary Biliary Cirrhosis. Hepatology (2005) 42(4):802–8. doi: 10.1002/hep.20859
146. Honda Y, Yamagiwa S, Matsuda Y, Takamura M, Ichida T, Aoyagi Y. Altered Expression of TLR Homolog RP105 on Monocytes Hypersensitive to LPS in Patients With Primary Biliary Cirrhosis. J Hepatol (2007) 47(3):404–11. doi: 10.1016/j.jhep.2007.03.012
147. Zhao J, Zhao S, Zhou G, Liang L, Guo X, Mao P, et al. Altered Biliary Epithelial Cell and Monocyte Responses to Lipopolysaccharide as a TLR Ligand in Patients With Primary Biliary Cirrhosis. Scand J Gastroenterol (2011) 46(4):485–94. doi: 10.3109/00365521.2010.539624
148. Peng A, Ke P, Zhao R, Lu X, Zhang C, Huang X, et al. Elevated Circulating CD14(low)CD16(+) Monocyte Subset in Primary Biliary Cirrhosis Correlates With Liver Injury and Promotes Th1 Polarization. Clin Exp Med (2016) 16(4):511–21. doi: 10.1007/s10238-015-0381-2
149. Fu HY, Bao WM, Yang CX, Lai WJ, Xu JM, Yu HY, et al. Kupffer Cells Regulate Natural Killer Cells Via the NK Group 2, Member D (NKG2D)/Retinoic Acid Early Inducible-1 (RAE-1) Interaction and Cytokines in a Primary Biliary Cholangitis Mouse Model. Med Sci Monit (2020) 26:e923726. doi: 10.12659/MSM.923726
150. Bossen L, Rebora P, Bernuzzi F, Jepsen P, Gerussi A, Andreone P, et al. Soluble CD163 and Mannose Receptor as Markers of Liver Disease Severity and Prognosis in Patients With Primary Biliary Cholangitis. Liver Int (2020) 40(6):1408–14. doi: 10.1111/liv.14466
151. Uchimura K, Nakamuta M, Enjoji M, Irie T, Sugimoto R, Muta T, et al. Activation of Retinoic X Receptor and Peroxisome Proliferator-Activated Receptor-Gamma Inhibits Nitric Oxide and Tumor Necrosis Factor-Alpha Production in Rat Kupffer Cells. Hepatology (2001) 33(1):91–9. doi: 10.1053/jhep.2001.21145
152. Enomoto N, Takei Y, Yamashima S, Ikejima K, Kitamura T, Sato N. Protective Effect of Pioglitazone Against Endotoxin-Induced Liver Injury Through Prevention of Kupffer Cell Sensitization. Alcohol Clin Exp Res (2005) 29(12 Suppl):216S–9S. doi: 10.1097/01.alc.0000192394.26573.10
153. Han Z, Zhu T, Liu X, Li C, Yue S, Liu X, et al. 15-Deoxy-Delta12,14 -Prostaglandin J2 Reduces Recruitment of Bone Marrow-Derived Monocyte/Macrophages in Chronic Liver Injury in Mice. Hepatology (2012) 56(1):350–60. doi: 10.1002/hep.25672
154. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 Activation of Kupffer Cells by PPARdelta Ameliorates Obesity-Induced Insulin Resistance. Cell Metab (2008) 7(6):496–507. doi: 10.1016/j.cmet.2008.04.003
155. Chang HY, Lee HN, Kim W, Surh YJ. Docosahexaenoic Acid Induces M2 Macrophage Polarization Through Peroxisome Proliferator-Activated Receptor Gamma Activation. Life Sci (2015) 120:39–47. doi: 10.1016/j.lfs.2014.10.014
156. Luo W, Xu Q, Wang Q, Wu H, Hua J. Effect of Modulation of PPAR-Gamma Activity on Kupffer Cells M1/M2 Polarization in the Development of non-Alcoholic Fatty Liver Disease. Sci Rep (2017) 7:44612. doi: 10.1038/srep44612
157. Linares I, Farrokhi K, Echeverri J, Kaths JM, Kollmann D, Hamar M, et al. PPAR-Gamma Activation is Associated With Reduced Liver Ischemia-Reperfusion Injury and Altered Tissue-Resident Macrophages Polarization in a Mouse Model. PloS One (2018) 13(4):e0195212. doi: 10.1371/journal.pone.0195212
158. Liu Y, Zhang W, Cheng Y, Miao C, Gong J, Wang M. Activation of PPARgamma by Curcumin Protects Mice From Ischemia/Reperfusion Injury Induced by Orthotopic Liver Transplantation via Modulating Polarization of Kupffer Cells. Int Immunopharmacol (2018) 62:270–6. doi: 10.1016/j.intimp.2018.07.013
159. Chawla A. Control of Macrophage Activation and Function by PPARs. Circ Res (2010) 106(10):1559–69. doi: 10.1161/CIRCRESAHA.110.216523
160. Penas F, Mirkin GA, Vera M, Cevey A, Gonzalez CD, Gomez MI, et al. Treatment In Vitro With PPARalpha and PPARgamma Ligands Drives M1-To-M2 Polarization of Macrophages From T. Cruzi-Infected Mice. Biochim Biophys Acta (2015) 1852(5):893–904. doi: 10.1016/j.bbadis.2014.12.019
161. David E, Stec DMG, Hipp JA, Hong S, Mitchell ZL, Franco NR, Robison JW, et al. Loss of Hepatic Pparα Promotes Inflammation and Serum Hyperlipidemia in Diet-Induced Obesity. Am J Physiol Regul Integr Comp Physiol (2019) 317(5):R733–45. doi: 10.1152/ajpregu.00153.2019
162. Yang XY, Wang LH, Mihalic K, Xiao W, Chen T, Li P, et al. Interleukin (IL)-4 Indirectly Suppresses IL-2 Production by Human T Lymphocytes via Peroxisome Proliferator-Activated Receptor Gamma Activated by Macrophage-Derived 12/15-Lipoxygenase Ligands. J Biol Chem (2002) 277(6):3973–8. doi: 10.1074/jbc.M105619200
163. Björkland A, Festin R, Mendel-Hartvig I, Nyberg A, Lööf L, Tötterman TH. Blood and Liver-Infiltrating Lymphocytes in Primary Biliary Cirrhosis: Increase in Activated T and Natural Killer Cells and Recruitment of Primed Memory T Cells. Hepatology (1991) 13(6):1106–11. doi: 10.1002/hep.1840130617
164. Chuang YH, Lian ZX, Tsuneyama K, Chiang BL, Ansari AA, Coppel RL, et al. Increased Killing Activity and Decreased Cytokine Production in NK Cells in Patients With Primary Biliary Cirrhosis. J Autoimmun (2006) 26(4):232–40. doi: 10.1016/j.jaut.2006.04.001
165. Hydes TJ, Blunt MD, Naftel J, Vallejo AF, Seumois G, Wang A, et al. Constitutive Activation of Natural Killer Cells in Primary Biliary Cholangitis. Front Immunol (2019) 10:2633. doi: 10.3389/fimmu.2019.02633
166. Zhang X, Rodriguez-Galan MC, Subleski JJ, Ortaldo JR, Hodge DL, Wang JM, et al. Peroxisome Proliferator-Activated Receptor-Gamma and its Ligands Attenuate Biologic Functions of Human Natural Killer Cells. Blood (2004) 104(10):3276–84. doi: 10.1182/blood-2004-02-0664
167. Harada K, Isse K, Tsuneyama K, Ohta H, Nakanuma Y. Accumulating CD57 + CD3 + Natural Killer T Cells are Related to Intrahepatic Bile Duct Lesions in Primary Biliary Cirrhosis. Liver Int (2003) 23(2):94–100. doi: 10.1034/j.1600-0676.2003.00807.x
168. Santodomingo-Garzon T, Swain MG. Role of NKT Cells in Autoimmune Liver Disease. Autoimmun Rev (2011) 10(12):793–800. doi: 10.1016/j.autrev.2011.06.003
169. Kita H, Naidenko OV, Kronenberg M, Ansari AA, Rogers P, He XS, et al. Quantitation and Phenotypic Analysis of Natural Killer T Cells in Primary Biliary Cirrhosis Using a Human CD1d Tetramer. Gastroenterology (2002) 123(4):1031–43. doi: 10.1053/gast.2002.36020
170. Chuang YH, Lian ZX, Yang GX, Shu SA, Moritoki Y, Ridgway WM, et al. Natural Killer T Cells Exacerbate Liver Injury in a Transforming Growth Factor Beta Receptor II Dominant-Negative Mouse Model of Primary Biliary Cirrhosis. Hepatology (2008) 47(2):571–80. doi: 10.1002/hep.22052
171. Wu SJ, Yang YH, Tsuneyama K, Leung PS, Illarionov P, Gershwin ME, et al. Innate Immunity and Primary Biliary Cirrhosis: Activated Invariant Natural Killer T Cells Exacerbate Murine Autoimmune Cholangitis and Fibrosis. Hepatology (2011) 53(3):915–25. doi: 10.1002/hep.24113
172. Briand F, Heymes C, Bonada L, Angles T, Charpentier J, Branchereau M, et al. A 3-Week Nonalcoholic Steatohepatitis Mouse Model Shows Elafibranor Benefits on Hepatic Inflammation and Cell Death. Clin Transl Sci (2020) 13(3):529–38. doi: 10.1111/cts.12735
173. Szatmari I, Gogolak P, Im JS, Dezso B, Rajnavolgyi E, Nagy L. Activation of PPARgamma Specifies a Dendritic Cell Subtype Capable of Enhanced Induction of iNKT Cell Expansion. Immunity (2004) 21(1):95–106. doi: 10.1016/j.immuni.2004.06.003
174. Dahl A. Pollen Lipids Can Play a Role in Allergic Airway Inflammation. Front Immunol (2018) 9:2816. doi: 10.3389/fimmu.2018.02816
175. Lan T, Qian S, Tang C, Gao J. Role of Immune Cells in Biliary Repair. Front Immunol (2022) 13:866040. doi: 10.3389/fimmu.2022.866040
176. Hiramatsu K, Harada K, Tsuneyama K, Sasaki M, Fujita S, Hashimoto T, et al. Amplification and Sequence Analysis of Partial Bacterial 16S Ribosomal RNA Gene in Gallbladder Bile From Patients With Primary Biliary Cirrhosis. J Hepatol (2000) 33(1):9–18. doi: 10.1016/s0168-8278(00)80153-1
177. Wang AP, Migita K, Ito M, Takii Y, Daikoku M, Yokoyama T, et al. Hepatic Expression of Toll-Like Receptor 4 in Primary Biliary Cirrhosis. J Autoimmun (2005) 25(1):85–91. doi: 10.1016/j.jaut.2005.05.003
178. Yokoyama T, Komori A, Nakamura M, Takii Y, Kamihira T, Shimoda S, et al. Human Intrahepatic Biliary Epithelial Cells Function in Innate Immunity by Producing IL-6 and IL-8 via the TLR4-NF-kappaB and -MAPK Signaling Pathways. Liver Int (2006) 26(4):467–76. doi: 10.1111/j.1478-3231.2006.01254.x
179. Zhang H, Leung PSC, Gershwin ME, Ma X. How the Biliary Tree Maintains Immune Tolerance? Biochim Biophys Acta Mol Basis Dis (2018) 1864(4 Pt B):1367–73. doi: 10.1016/j.bbadis.2017.08.019
180. Harada K, Isse K, Kamihira T, Shimoda S, Nakanuma Y. Th1 Cytokine-Induced Downregulation of PPARgamma in Human Biliary Cells Relates to Cholangitis in Primary Biliary Cirrhosis. Hepatology (2005) 41(6):1329–38. doi: 10.1002/hep.20705
181. Hirschfield GM, Beuers U, Kupcinskas L, Ott P, Bergquist A, Farkkila M, et al. A Placebo-Controlled Randomised Trial of Budesonide for PBC Following an Insufficient Response to UDCA. J Hepatol (2021) 74(2):321–9. doi: 10.1016/j.jhep.2020.09.011
182. Lombard M, Portmann B, Neuberger J, Williams R, Tygstrup N, Ranek L, et al. Cyclosporin a Treatment in Primary Biliary Cirrhosis: Results of a Long-Term Placebo Controlled Trial. Gastroenterology (1993) 104(2):519–26. doi: 10.1016/0016-5085(93)90422-9
183. Heathcote J, Ross A, Sherlock S. A Prospective Controlled Trial of Azathioprine in Primary Biliary Cirrhosis. Gastroenterology (1976) 70(5):656–60. doi: 10.1016/s0016-5085(76)80250-8
184. Wagner M, Fickert P. Drug Therapies for Chronic Cholestatic Liver Diseases. Annu Rev Pharmacol Toxicol (2020) 60:503–27. doi: 10.1146/annurev-pharmtox-010818-021059
185. Katsumi T, Tomita K, Leung PS, Yang GX, Gershwin ME, Ueno Y. Animal Models of Primary Biliary Cirrhosis. Clin Rev Allergy Immunol (2015) 48(2-3):142–53. doi: 10.1007/s12016-015-8482-y
186. Ghonem NS, Assis DN, Boyer JL. Fibrates and Cholestasis. Hepatology (2015) 62(2):635–43. doi: 10.1002/hep.27744
187. Bertin B, Dubuquoy L, Colombel JF, Desreumaux P. PPAR-Gamma in Ulcerative Colitis: A Novel Target for Intervention. Curr Drug Targets (2013) 14(12):1501–7. doi: 10.2174/13894501113149990162
188. Racke MK, Gocke AR, Muir M, Diab A, Drew PD, Lovett-Racke AE. Nuclear Receptors and Autoimmune Disease: The Potential of PPAR Agonists to Treat Multiple Sclerosis. J Nutr (2006) 136(3):700–3. doi: 10.1093/jn/136.3.700
189. You H, Ma X, Efe C, Wang G, Jeong SH, Abe K, et al. APASL Clinical Practice Guidance: The Diagnosis and Management of Patients With Primary Biliary Cholangitis. Hepatol Int (2022) 16(1):1–23. doi: 10.1007/s12072-021-10276-6
190. Li J, Guo C, Wu J. The Agonists of Peroxisome Proliferator-Activated Receptor-Gamma for Liver Fibrosis. Drug Des Devel Ther (2021) 15:2619–28. doi: 10.2147/DDDT.S310163
Keywords: primary biliary cholangitis, fibrate, cholestatic liver disease, hepatic immunity, peroxisome proliferator-activated receptor
Citation: Wang C, Shi Y, Wang X, Ma H, Liu Q, Gao Y and Niu J (2022) Peroxisome Proliferator-Activated Receptors Regulate Hepatic Immunity and Assist in the Treatment of Primary Biliary Cholangitis. Front. Immunol. 13:940688. doi: 10.3389/fimmu.2022.940688
Received: 10 May 2022; Accepted: 13 June 2022;
Published: 08 July 2022.
Edited by:
Jinhang Gao, Sichuan University, ChinaCopyright © 2022 Wang, Shi, Wang, Ma, Liu, Gao and Niu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanhang Gao, eWFuaGFuZ0BtYWlsLmpsdS5lZHUuY24=; Junqi Niu, anVucWluaXVAamx1LmVkdS5jbg==
†These authors have contributed equally to this work