 Linlin Xu1†
Linlin Xu1† Zhaodi Che
Zhaodi Che Bin Wu
Bin Wu Cunchuan Wang
Cunchuan Wang Jia Xiao
Jia Xiao- 1Department of Obstetrics and Gynecology, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou, China
- 2Clinical Medicine Research Institute, Department of Metabolic and Bariatric Surgery, The First Affiliated Hospital of Jinan University, Guangzhou, China
- 3Department of Gastroenterology, The Third Affiliated Hospital of Sun Yat-Sen University, Guangzhou, China
Most liver diseases, including acute liver injury, drug-induced liver injury, viral hepatitis, metabolic liver diseases, and end-stage liver diseases, are strongly linked with hormonal influences. Thus, delineating the clinical manifestation and underlying mechanisms of the “sexual dimorphism” is critical for providing hints for the prevention, management, and treatment of those diseases. Whether the sex hormones (androgen, estrogen, and progesterone) and sex-related hormones (gonadotrophin-releasing hormone, luteinizing hormone, follicle-stimulating hormone, and prolactin) play protective or toxic roles in the liver depends on the biological sex, disease stage, precipitating factor, and even the psychiatric status. Lifestyle factors, such as obesity, alcohol drinking, and smoking, also drastically affect the involving mechanisms of those hormones in liver diseases. Hormones deliver their hepatic regulatory signals primarily via classical and non-classical receptors in different liver cell types. Exogenous sex/sex-related hormone therapy may serve as a novel strategy for metabolic liver disease, cirrhosis, and liver cancer. However, the undesired hormone-induced liver injury should be carefully studied in pre-clinical models and monitored in clinical applications. This issue is particularly important for menopause females with hormone replacement therapy (HRT) and transgender populations who want to receive gender-affirming hormone therapy (GAHT). In conclusion, basic and clinical studies are warranted to depict the detailed hepatoprotective and hepatotoxic mechanisms of sex/sex-related hormones in liver disease. Prolactin holds a promising perspective in treating metabolic and advanced liver diseases.
Introduction
Chronic liver diseases refer to a progressive deterioration of liver functions over six months. The major etiologies of chronic liver disease are genetic defect, toxin ingestion, excessive alcohol consumption, infection, autoimmune reaction, and metabolic syndromes (1, 2). Long-term inflammatory, lipid peroxidation, and necrotic insults lead to liver parenchyma destruction and scar formation (liver fibrosis). A minority of patients will progress to end-stage cirrhosis and/or hepatocellular carcinoma (3, 4). Cirrhosis, characterized by evident fibrosis and nodule formation after chronic injury, is the 11th leading cause of death and the 15th leading cause of morbidity around the world (5). Currently, hepatitis B virus (HBV; 31.5% in males and 24.0% in females), hepatitis C virus (HCV; 25.5% in males and 26.7% in females), and alcohol abuse (27.3% in males and 20.6% in females) are the major etiologies of cirrhosis-induced death. Obesity-related non-alcoholic fatty liver disease (NAFLD; currently 7.7% in males and 11.3% in females), which has overweight and obvious hepatic fat accumulation without a history of alcohol abuse, is also anticipated to account for increasing proportions of death in the future because of its strikingly high prevalence in the world (6). A relatively rare but important cause of cirrhosis is drug-induced liver injury (DILI), which rapidly provokes cell death, severe inflammation, oxidative stress, lipid peroxidation, and alterations in bile acid dysfunction of the liver. Some cases even have loss of immune tolerance, as well as the abnormalities of both innate and adaptive immunity (7). Even after drug cessation, a non-negligible proportion of DILI patients may progress into chronic DILI. Subsequently, more than 40% of the long-term unsolved DILI patients will develop cirrhosis (8). In the United States, the most common drugs that can induce DILI include acetaminophen, antibiotics, herbal/dietary supplements, and immunomodulatory agents (9). A recent epidemiological study indicated that in China, the leading drugs responsible for DILI are traditional Chinese medicines, herbal supplements, and antituberculosis medications (10). Since the incidence of cirrhosis and acute-on-chronic liver failure (ACLF; characterized by acute hepatic decompensation, hepatic and other key organ failure, and high short-term mortality) in DILI patients remains high, efficacious drugs that can control the progression of chronicity are urgently needed.
Sex hormones (include androgen, estrogen, and progesterone), also called sex steroids, are steroid hormones having critical functions such as reproduction, sexual development, puberty, lipid metabolism, body fat distribution, neuronal transmission, and hair growth, in the reproductive and non-reproductive systems. They are mainly produced by the gonads and adrenal glands, including the adrenal cortex, gonads (testes and ovaries), and placenta (11). The circulating and tissue levels of sex hormones are under sophisticated regulations to maintain the body homeostasis and avoid health issues such as infertility, obesity, and hair/bone loss. Several well-documented factors are able to affect the fluctuations of sex hormone levels, including aging, menstruation, stress, menopause, and medications (12). The involving roles of sex hormones in metabolic diseases received mass attention in the past decades since both basic and clinical studies found that those hormones could substantially influence the pathogenesis of or applied as novel therapies for obesity (13), type 2 diabetes (14), cardiovascular diseases (15), and NAFLD (16). Genes regulated by sex hormones are especially important for liver metabolism since the liver expresses receptors for all three sex hormones in males and females (17). In addition, there are “sexual dimorphism” for several common chronic liver diseases. For instance, females exhibit severer liver injury in alcoholic liver disease (ALD; with similar liver pathological phenotypes with NAFLD but has a history of acute or chronic alcohol abuse) and an increased risk of autoimmune liver disease than males. In comparison, hepatocellular carcinoma (HCC; the most common primary malignancy of the liver and strongly associated with cirrhosis caused by alcohol abuse and viral hepatitis) is more common in males (18). Although hormone replacement therapy (HRT) is well-tolerated by the liver, whether the therapy will change liver function and worsen the precipitated liver diseases is in debate, which is probably influenced by hormone dose, duration of therapy, alcohol drinking, smoking, genetic susceptibility, and age (19). Thus, the current review will introduce the hepatoprotective and hepatotoxic roles and mechanisms of sex/sex-related hormones and focus on the advances in elucidating the biological functions of hormone receptors. Since other sex-related hormones, including luteinizing hormone, follicle-stimulating hormone, gonadotrophin-releasing hormone, and prolactin closely coordinate with sex steroid hormones in both physiological and pathological conditions of the liver, we also summarized their involving mechanisms in chronic liver diseases.
Sex Hormones in Liver Diseases
Androgen
Androgens play essential roles in both sexes’ reproductive health and body metabolism. In males, testosterone is the most common androgen, which is produced by the Leydig cells of the testes, or to a lower extent, by the adrenal glands. Dihydrotestosterone (DHT), dehydroepiandrosterone (DHEA), androstenedione (A4), androstenediol (A5), and androsterone are other common types of androgens (20). In females, androgens are produced by the ovaries (testosterone), the adrenal glands (androgen precursors such as DHEA and A4), and the placenta (testosterone) during pregnancy. Androgens play an important role in female reproduction and pregnancy. Although excessive androgen clearly impairs female fertility, physiological level of androgen plays a positive role (21). Pathological conditions of females such as polycystic ovary syndrome (PCOS), obesity, and endocrinopathies (e.g. Cushing’s disease) are associated with pathologically high levels of androgens (22).
Ectopic androgen production directly induces hepatic fat accumulation, indicating a lipid regulatory role of this hormone (23–26). A low level of testosterone increases lipoprotein lipase activity, which in turn promotes triglyceride uptake into the adipocytes and subsequent visceral adiposity. Moreover, low serum DHEA levels are associated with male metabolic syndrome, possibly via the exacerbation of insulin resistance (27). In male rodents, androgen deficiency due to orchiectomized (ORX) also leads to hepatic steatosis (28). Mechanistically, liver lipid deposition is primarily attributed to the up-regulation of genes for de novo lipogenesis (DNL) (e.g. Srebp-1c and Fasn) (23). A prospective follow-up study of 942 Boston males with a median follow-up of 8.9 years reported that adiposity might influence testosterone production via the hypothalamus-pituitary-gonadal axis and confirmed the inverse associations between obesity and total/free testosterones (29). Therefore, in males, obesity is directly associated with low testosterone levels. Contrarily, in females, excessive androgen promotes hepatic steatosis. An important example is that androgen suppression and/or blockade significantly improves hepatic steatosis in female patients with PCOS (30). Moreover, several studies have demonstrated that excessive androgen promoted female food intake to cause obesity and metabolic dysfunction (31–33). A meta-analysis of 5,840 females (including both pre-and postmenopausal women) reported that females with higher circulating testosterone had a higher odds ratio of overweight prevalence (34). Underlying mechanisms probably include hepatic inhibition of phosphoenolpyruvate carboxykinase or up-regulation of proinflammatory mitogen-activated protein kinase 4 (MAP2K4) (35).
The contributing roles of androgens in NALFD seem to be controversial and sex-dependent (36–39). In a longitudinal analysis of 1,944 Korean men (median follow-up of 4.2 years) with repeated liver ultrasonography checks, baseline testosterone concentrations do not predict subsequent NAFLD development (40). A study investigating a cohort of 117 males shows that raising serum testosterone concentrations to normal levels by parenteral testosterone treatments reduces the serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), body weight, body mass index (BMI), waist size, and improved lipid profiles (41). In comparison to males, one study of 22 postmenopausal females with biopsy-proven NAFLD indicates no significant difference in total testosterone levels with 18 matched controls (42). A large cohort study of 1,052 females in the United States identifies a novel association between free testosterone and risk of prevalent NAFLD in midlife. Importantly, this association is present even among females without androgen excess, suggesting a role of testosterone on NAFLD risk in a broader spectrum of females (43). Indeed, low androgen levels in males and high androgen levels in females facilitate NAFLD occurrence and progression. Therefore, testosterone may act as a potential new target for NAFLD treatment. In a clinical study of advanced hepatitis C-related liver disease in males, it is reported that higher serum testosterone is associated with increased risk of liver inflammation and fibrosis (44). For end-stage liver diseases, Sinclair et al. measure serum testosterone levels in 268 patients with cirrhosis and identify low testosterone is associated with adverse outcomes and mortality. The result significantly worsened below a total testosterone threshold of 8.3 nmol/L or a free testosterone threshold of 139 pmol/L (45). This conclusion is consistent with an observational study of 171 male cirrhotic patients in which low testosterone is found to be an independent but reliable predictor of mortality (46). Androgen signaling seems to be a potential therapeutic target of HCC since surgical castration and liver-specific androgen receptor knockout retard hepatocarcinogenesis (47). A recent study also demonstrates that pharmacologic androgen receptor antagonism with enzalutamide inhibits hepatocellular carcinogenesis in a diethylnitrosamine- (DEN-) induced HCC mouse model. More important, the upregulation of androgen receptor is only observed in portal fibroblasts and leukocytes, but not hepatocytes, implying that hepatocyte-autonomous androgen receptor signaling is not required for DEN-induced HCC (48). PD-L1 expression is negatively regulated by androgen receptor, leading to a transcriptional repression of PD-L1 and enhancement of CD8+T function. Thus, inhibition of androgen receptor might improve the efficacy of HCC immune-therapy to PD-L1 inhibitor (49).
Overall, decreased androgen in males or increased androgen in females may lead to metabolic disorders and end-stage liver diseases. Moderate reduction of testosterone in male is a marker of NAFLD. However, for male patients with established hypogonadism, there is no evidence that testosterone replacement therapy can induce hepatotoxicity. In females, the same focus should be on the association between ectopic androgen production and NAFLD-related risk factors/complications. Further clinical trials are needed to determine whether the reduction of physiological androgen levels will have a beneficial effect on the liver.
Estrogen
Estrogen is one of the major female hormones, mainly secreted by the ovaries, small amounts by the liver, adrenal cortex, and breast. Estradiol is the most important form of estrogen, responsible for the regulation of female characteristics, the maturation of accessory sex organs, the menstruation-ovulation cycle, and the production of the mammary duct system (50). Serum concentrations of estrogens such as estradiol vary periodically throughout the menstrual cycle, with estradiol being the most abundant estrogen in females of childbearing age, except in the early follicular phase (51).
It is well known that the liver is a vital target tissue for estrogen signaling (52). Estrogen has a wide range of protective effects on hepatocytes. Estradiol reduces hepatic susceptibility to steatosis by strengthening cellular mitochondrial function in a substrate-specific manner (53). Moreover, 17β-estradiol (E2) enhances hepatocytes mitochondrial content and oxidative capacity to alleviate hepatic lipid accumulation and oxidative stress. Mechanistically, peroxisome proliferator-activated receptor gamma coactivator 1B (PGC1B), but not PGC1A, functions as a modulator of E2 to promote the mitochondrial biogenesis (54). Since activation of c-Jun N-terminal kinase (JNK) is a key pathological event during the development of obesity and NAFLD and E2 can inhibit the activation of JNK, E2 has been considered in NAFLD treatment in a very cautious way (55, 56).
The pathways responsible for estrogen-mediated hepatic lipid metabolism could be quite complicated and are not fully understood. Hepatocyte estrogen receptor alpha (Erα) promotes hepatic absorption of cholesterol and systemic reverse cholesterol transport, a process that is particularly important in females (57). In particular, the ability of high-density lipoprotein (HDL) to initiate reverse cholesterol transport during the female reproductive cycle is related to plasma estrogen content and hepatic ERα activity, which effectively induces cholesterol efflux from macrophages. Moreover, there is a physiologically functional cross-coupling between ERα and liver X receptor alpha (LXRα) which is another important regulator of hepatic lipid metabolism (58). Estrogen is negatively correlated with serum triglyceride (TG) level, achieved by regulating the expression of apolipoprotein A5 in the liver. Estrogen and the G protein-coupled receptor 30 coactivate protein kinase A (PKA) to enhance the expression of hepatic peroxisome proliferator-activated receptor alpha (PPARα) and hepatocyte nuclear factor 4alpha (HNF4α), thereby increasing the expression of hepatic apolipoprotein (59). Estrogen is found to directly inhibit liver inflammation since (1) postmenopause females often exhibit accumulated lipid peroxidation and inflammation in the liver (60); (2) estrogen signaling suppresses pro-inflammatory cytokine release and reactive oxygen species (ROS) production in hepatocytes (55, 61); (3) estrogen supplementation restores depressed Kupffer cell phagocytic capacity via the activation of Akt (62).
Estrogen is also critical in the amelioration of liver fibrosis and cirrhosis. It is reported that in a carbon tetrachloride-induced mouse liver fibrosis model, exogenous E2 significantly alleviated fibrosis and other liver injuries, partly via a restoration of miR-29a and miR-29b expression (63). Another study of male dimethyl nitrosamine (DMN) model found that estradiol treatment decreased the deposition of type I and III collagen protein, the total hepatic collagen content, and malondialdehyde (MDA), a product of lipid peroxidation (64). In high fructose diet-induced NASH-fibrosis mice models, E2 supplementation reversed liver cell destruction, macrophage accumulation, and hepatic stellate cell activation (65). Clinical features of a disrupted gonadal function (e.g. libido loss and reduced potency) and feminization (e.g. gynecomastia and female habitus) can be found in two-thirds of males with alcoholic cirrhosis. After transjugular intrahepatic portosystemic stent shunt (TIPS), serum E2 is significantly increased (with aggravated sex hormone dysbalance) in males but remain persistent in females with cirrhosis (66). In DMN-induced rat cirrhosis models, administration with E2 significant decreases portal pressure and increases hepatic blood flow, which are abolished by the co-treatment with an estrogen receptor antagonist (ICI-182.780) (67). In addition, estrogen stimulates the expression of nitric oxide synthase 3 (endothelial NOS, eNOS) in sinusoidal endothelial cells to provoke nitric oxide production, contributing toward a reduction in portal pressure (67, 68). Inactivated estrogen sulfates are converted to activated estrogen by the action of steroid sulfatase (STS), which is elevated in patients with chronic inflammatory liver diseases and accompanied by increased circulating estrogen levels. STS serves as a novel nuclear factor kappa-B (NF-κB) target gene to alleviate liver inflammation, partly via the provoked estrogen signaling (69). Estrogen is generally thought to be an anti-HCC hormone (70). Possible mechanisms include: (1) to inhibit inflammasome activation through estrogen receptor (71); (2) to repress HCC growth via inhibiting alternative activation of tumor-associated macrophages (72); (3) to inhibit HCC progression because of transition from pro-inflammatory to anti-inflammatory phenotype of Kupffer cell via the physical interaction between estrogen receptor alpha and NF-κB (73).
Progesterone
Progesterone is a steroid hormone secreted by the granulosa luteal cells of the ovary. Ovulation, reproduction, mammary gland growth, and pregnancy maintenance are the main functions of progesterone. The primary target tissues of progesterone are the endometrium, breast, and central nervous system. In the liver, progesterone is inactivated to estradiol and excreted into the urine in combination with glucuronic acid. Progesterone can be divided into two groups according to its chemical structure: 17α-hydroxyprogesterone and 19-nortestosterone. Biological responses to progesterone are mediated by both genomic (e.g. progesterone receptor acts through specific progesterone response elements within the promoter region of target genes) and non-genomic mechanisms (e.g. non-classical progesterone receptor is activated to elicit the activation of downstream signaling) (74, 75).
Studies have shown that sex hormones have complex and variable effects on NAFLD (76). Increased progesterone level is associated with the development of systemic insulin resistance (77). This hormone is also an independent predictor of insulin resistance in adolescent girls (78). In NASH patients, progesterone use, but not estrogen use, will induce observable hepatic lobular inflammation (79). The mechanisms responsible for progesterone-induced metabolic liver injury are not characterized. A recent study indicates that deficiency of progesterone receptor membrane component 1 induces hepatic steatosis through de novo lipogenesis in the liver (80). Another metabolism-related study suggests that progesterone increases hepatic glucose production via the modulation of gluconeogenesis by progesterone receptor membrane component 1 (PGRMC1), which may exacerbate hyperglycemia in diabetes where insulin action is limited (81).
An interesting clinical phenomenon is that females usually have worse outcomes from DILI than males. It could be partly explained by progesterone-induced immune toxic responses via Kupffer cells and the extracellular-signal-regulated kinase (ERK) pathway (82). Moreover, progesterone itself is reported to induce DILI in females (83). Hepatitis E is usually a self-limited liver disease with relatively good prognosis. However, during pregnancy, fulminant hepatic failure with high mortality rate is commonly observed in clinical hepatitis E virus (HEV)-infected patients. Elevated progesterone and HEV RNA levels have been observed in pregnant females with fulminant hepatic failure. Because progesterone is essential for the maintenance of pregnancy, studies on the potential role of progesterone in HEV replication and disease pathogenesis have demonstrated that in human hepatocytes, progesterone could enhance HEV replication but could not modulate HEV-induced interferon response. Loss of the progesterone noncanonical receptor, PGRMC1/2, was associated with decreased levels of HEV replication and increased levels of HEV-induced type III interferon (IFN-λ1) mRNA expression via the ERK pathway (84). In addition, there is a significant association between vaginal progesterone level and intrahepatic cholestasis of pregnancy (ICP). ICP is accompanied by unique maternal pruritus, abnormal liver function tests, elevated serum total bile acids, and an increased incidence of adverse fetal outcomes (e.g. intrauterine fetal death). Pregnant females who receive long-term daily vaginal progesterone treatment to prevent preterm birth are at increased risk of ICP. Progesterone metabolites (PM2DiS, PM3S, PM3DiS) are abundant during pregnancy of genetically susceptible females, leading to supersaturation of the hepatic transport system for biliary excretion of these compounds (85, 86). In animal models, progesterone induces proliferation and abnormal mitotic processes in rat liver cells. Of note, treatment with progesterone, even at pharmacologically-relevant doses, shows an increase in the percentage of binucleated hepatocytes (87). Progesterone may serve as an autocrine/paracrine mediator of cholangiocyte proliferation. Cholangiocytes express progesterone nuclear receptor (PR-B) and progesterone membrane receptors (PRGMC1, PRGMC2, and mPRα). Moreover, progesterone increases the number of bile ducts in normal rats both in vivo and in vitro, while anti-progesterone antibodies inhibit bile duct ligation-stimulated cholangiocyte growth. Thus, antiprogesterone therapy may therefore benefit patients with cholangiocyte proliferation, such as those with extrahepatic cholestasis (88). In terms of end-stage liver diseases, progesterone is reported to stimulate the production of ROS through progesterone receptor, leading to transforming growth factor (TGF)-β1 expression, hepatic stellate cells (HSCs) activation, and extracellular collagen formation (89). This phenomenon increases the possibility that progesterone can establish a favorable microenvironment for tumors and thus contribute to the development of liver cancer. PGRMC1 is considered to be a biomarker of tumor cell proliferation (90) and is strongly expressed in different kinds of cancers (91). Hepatic PGRMC1 and progesterone receptor are continuously active in the presence of high serum progesterone levels and may facilitate the chemoresistance of HCC (91, 92). Known protective and toxic mechanisms of sex hormones in the liver are summarized in Figure 1 and Table 1.
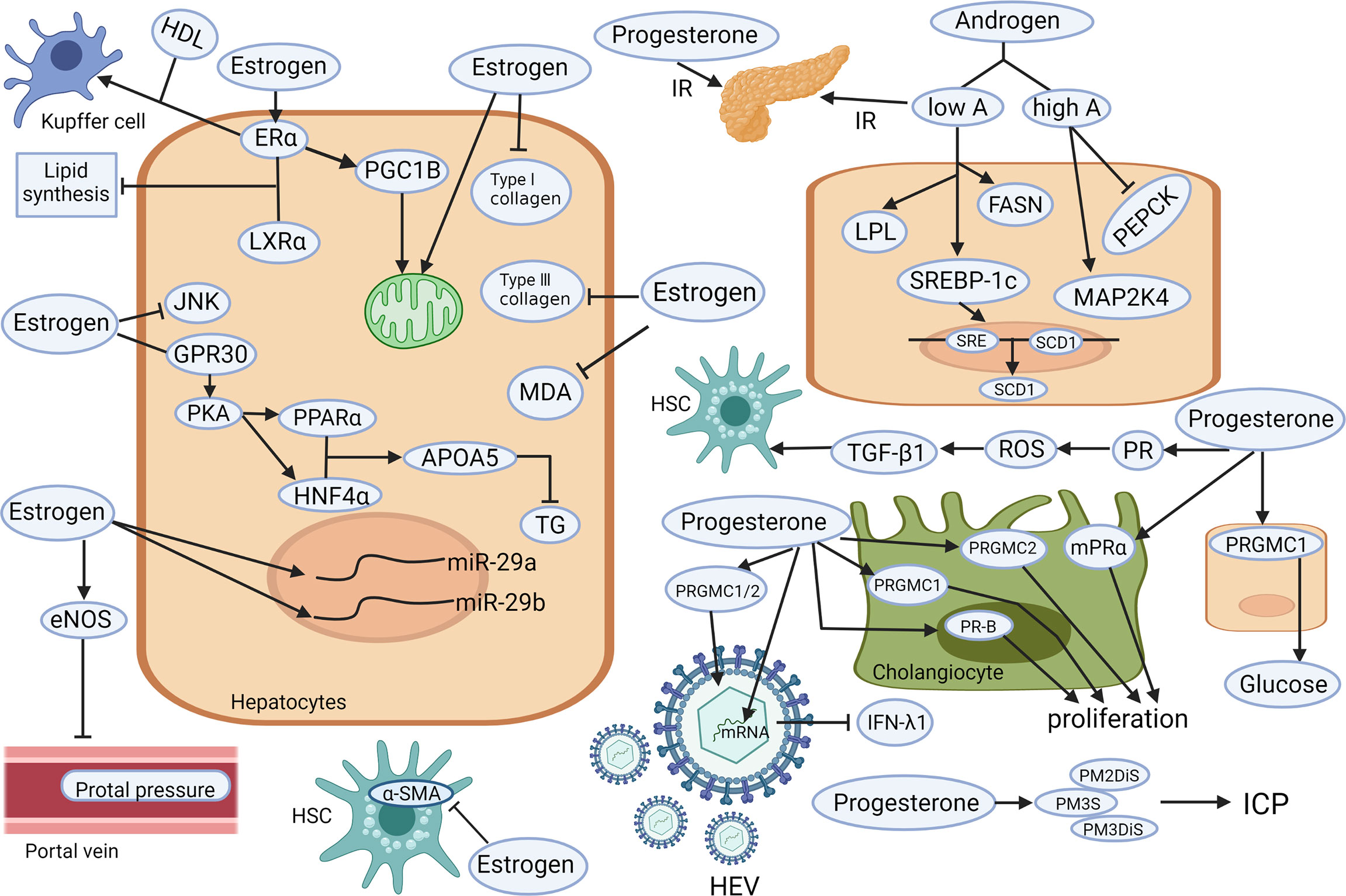
Figure 1 Known mechanisms of the hepatoprotective and hepatotoxic effects of sex hormones (androgen, estrogen, and progesterone) on different liver cell types. Low level of testosterone increases the activity of LPL. The up-regulation of SREBP-1c and FASN lead to liver lipid deposition and aggravated insulin resistance. Those changes, together with the down-regulation of PEPCK and up-regulation of MAPK may lead to steatosis. Estrogen increases the content and oxidation capacity of mitochondria in hepatocytes. PGC1B promotes mitochondrial biogenesis. Estrogen also inhibits the activation of JNK and GPR30 to co-activate PKA and enhance liver PPARα and HNF4α to increase APOA5 expression and reduce TG. Estrogen (via its receptor alpha) induce cholesterol efflux from Kupffer cells with HDL. Estrogen can restore the expression of miR-29a/b to reduce the deposition of type I and III collagen, MDA, and α-SMA, to reduce liver fibrosis and other types of liver damage. Estrogen can also significantly reduce portal vein pressure by stimulating eNOS expression. Elevated progesterone leads to insulin resistance, stimulates PGRMC1 to increase hepatic glucose production, stimulates PGRMC1/2 to promote HEV replication, and inhibits IFN- λ1 expression. In addition, the accumulation of progesterone metabolites (PM2DiS, PM3S, PM3DiS) will increase the risk of ICP. Progesterone stimulates PR-B, PRGMC1, and PRGMC2 to facilitate bile duct cell proliferation. It also causes ROS environment via its receptor signaling, resulting in TGF-β1-activated HSC. APOA5, apolipoprotein A5; α-SMA, alpha-smooth muscle actin; eNOS, endothelial nitric oxide synthase 3; ERα, estrogen receptor alpha; FASN, fatty acid synthase; HDL, high-density lipoprotein; HSC, hepatic stellate cell; GPR30, G protein-coupled receptor 30; HNF4α, hepatocyte nuclear factor 4-alpha; ICP, Intrahepatic cholestasis of pregnancy; IFN-λ1, type III interferon-λ1; JNK, Jun N-terminal kinase; LPL, lipoprotein lipase; IR, insulin resistance; LXRα, liver X receptor alpha; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; mPRα, membrane progestin receptor alpha; PEPCK, phosphoenolpyruvate carboxykinase; PGC1B, proliferator-activated receptor gamma coactivator 1B; PGRMC, progesterone receptor membrane component; PKA, protein kinase A; PM2DiS/PM3DiS/PM3S, progesterone metabolites; PPARα, peroxisome proliferator-activated receptor alpha; PR, progesterone receptor; ROS, reactive oxygen species; SCD1, stearoyl-CoA desaturase1; SRE, sterol regulatory element; SREBP-1c, sterol regulatory element-binding protein-1c; TG, triglyceride; TGF, transforming growth factor (Created with Biorender.com with a publication license).
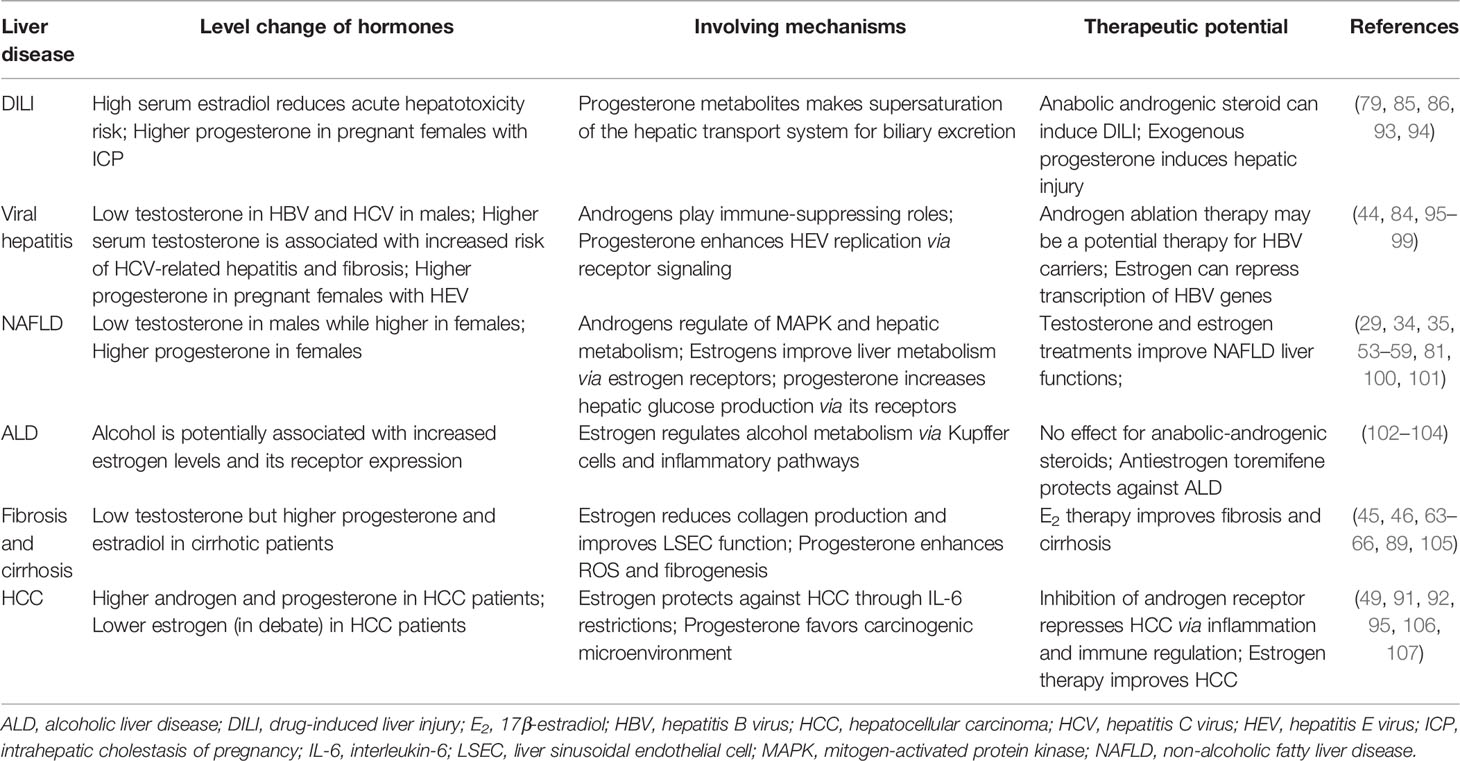
Table 1 The involving roles and therapeutic potentials of sex hormones in liver diseases.
Sex-Related Hormones in Liver Diseases
Gonadotrophin-Releasing Hormone
Gonadotropin-releasing hormone (GnRH) is a decapeptide produced in the hypothalamus and secreted by scattered hypothalamic GnRH neurons in a pulsatile manner. GnRH acts on its receptor (GnRHR) on the surface of gonadotropin cells in the pituitary gland to stimulate the release of LH and FSH, which, in turn, enhance the production and release of testosterone (male testes) and estrogen (female ovaries and placenta) (108).
Experiments from murine models demonstrate that increased GnRH causes obesity after ovariectomy. As the upstream regulator of the gonad axis, GnRH stimulates fat accumulation by directly promoting the cell cycle of preadipocytes via the protein kinase A- cAMP-response element binding protein (PKA-CREB) pathway and increasing the FSH secretion to accelerate adipocyte differentiation in adipose tissue of female mice (109, 110). In humans, the pulsatility of serum LH levels is accepted as a GnRH pulse generator activity marker due to its short half-life (111, 112). Therefore, the exact role of GnRH in human obesity cannot be determined at present. In cirrhotic patients, disturbance in gonadotrophin secretion with inappropriately low levels of LH and FSH has been observed in amenorrheic females with alcoholic or non-alcoholic cirrhosis (113). However, these GnRH responses can only indicate the hypothalamus rather than the pituitary as the site of gonadotropin secretion disorder. Knockdown of hepatic GnRH alleviates liver fibrosis in a murine primary sclerosing cholangitis model by the downregulation of miR-200b (108). Another study confirms above findings by showing that GnRH stimulates fibrosis gene expression in HSCs in a bile duct-ligated-induced liver fibrosis rat model (114). Few studies have reported the interaction between GnRH and metabolic liver diseases, which clearly warrants further epidemiological, observational, and mechanistic investigations.
Luteinizing Hormone
Luteinizing hormone (LH) is a glycoprotein gonadotropin secreted by adenohypophysis cells under the control of GnRH. LH can promote the conversion of cholesterol into sex hormones in gonadal cells. In females, LH stimulates the ovaries to release eggs, and its periodic surge leads to monthly ovulation. Moreover, LH stimulates the production of progesterone and estrogen, as well as the growth of the corpus luteum. In males, LH facilitates the production and release of testosterone from testicular interstitial cells in the testes.
A cross-sectional study of obese male patients in Belgium reports that NAFLD is associated with lower levels of LH, FSH, and total testosterone than controls (115). Another study with Chinese exhibits a slight but not significantly decreased LH level in NAFLD patients than that in healthy controls (116). In Chinese postmenopausal female patients, a significantly reduced LH is only observed in severe steatosis subgroup, but not in mild/moderate steatosis subgroups, when compared with that in controls (117). The direct regulatory mechanism of LH in hepatic lipid accumulation, inflammation, and cell death is largely unknown.
The LH level in seminal fluid of HCV patient is slightly, although not significantly, higher than that in healthy controls. Moreover, anti-HCV therapy does not significantly influence such level of LH in patients (118). Another similar study high larger cohort finds a slightly but not significantly lower semen LH level in HCV patients than healthy controls (119). LH and FSH levels are commonly decreased in males with advanced liver disease (120). In a Meta-analysis Of Observational Studies in Epidemiology (MOOSE) guideline report (including 21 studies with 1,274 patients), results indicate that liver transplantation (LT) improves hormonal disturbances associated with chronic liver disease by restoring circulating physiological levels of growth hormone (GH), insulin-like growth factor-1 (IGF-1), testosterone, estradiol, prolactin, FSH, and LH (121).
Follicle-Stimulating Hormone
FSH is a glycosylated protein hormone secreted by basophils in the pituitary, because of its stimulating capacity of female follicles maturation. FSH function is mediated primarily by the FSH receptor (FSHR), which is located on the plasma membrane of hepatocytes (122). It is one of the most important hormones for development, growth, puberty, sexual maturation, and reproduction in both males and females. The level of FSH is usually low in childhood and becomes high after menopause in females. Its secretion is also in pulses with body weight change and the menstrual cycle. The determination of serum FSH is of great significance in understanding the endocrine function of the pituitary, hypothalamus, and ovary, as well as the diagnosing and treating infertility and endocrine diseases (123).
Endocrine changes during menopause, especially the dramatic increase in serum FSH levels, have a negative impact on blood lipid levels. FSH interacts with its hepatocyte receptor to decrease LDL receptor (LDLR) levels, which in turn attenuates low-density-lipoprotein cholesterol (LDL-C) endocytosis (124). After HRT, postmenopausal females with high baseline FSH levels have more significant improvement in LDL-C levels than those with low baseline FSH levels (124). Thus, HRT might be a preventive therapy in postmenopausal patients with higher basal FSH levels, and these females are encouraged to take HRT for several years after menopause. Epidemiological findings suggest that serum FSH levels are positively correlated with serum total cholesterol levels. Mechanistically, in the liver, FSH activates the Gi2α/β-arrestin-2/Akt pathway by binding to the hepatic FSHR and subsequently inhibits the binding between forkhead box protein O1 (FoxO1) and the sterol regulatory element binding protein (SREBP)-2 promoter, thereby driving 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) transcription and de novo cholesterol biosynthesis, resulting in increased cholesterol accumulation. Therefore, blocking FSH signaling could be a novel strategy for the treatment of menopausal hypercholesterolemia, especially in perimenopausal females characterized only by elevated FSH (122).
There is an established association between FSH and NAFLD in postmenopausal females. However, it is not known whether FSH affects the risk of NAFLD in males. A community-based study of males aged 20 - 69 years observes a gradual increase in FSH with age (125). Another cross-sectional study in 444 Chinese elderly males aged 80-98 years demonstrates that high FSH levels might enhance the risk of NAFLD. Elevated FSH may be one of the possible mechanisms explaining the greater number of NAFLD subjects found in elderly males (126). Considering the age-related changes in circulating FSH levels and the prevalence of NAFLD, FSH might have a novel extragonadal role in the regulation of hepatic gluconeogenesis via FSHR in the liver. Moreover, there is a positive correlation between FSH and fasting blood glucose (127). FSH enhances cyclic AMP-regulated transcriptional coactivator 2-mediated gluconeogenesis via adenosine monophosphate-activated protein kinase (AMPK) phosphorylation regulation in the liver, leading to the pathogenesis of fasting hyperglycemia (128). In a cohort of postmenopausal females with HCV infection, there is a progressive decline in FSH from Child-Turcotte-Pugh class A to C subgroups (129). However, in male HBV-induced cirrhotic patients, such difference is not observed (130). Menotrophin is a female infertility gonadotropin treatment contains purified FSH and LH. It is reported to induce autoimmune hepatitis in a female patient after several cycles of treatment (131).
Prolactin
Prolactin, also known as lactotropin, is a polypeptide hormone produced and secreted from the pituitary gland. The main functions of prolactin include milk production and the development of the mammary gland within breast tissues. During pregnancy, elevated prolactin promotes the growth of mammary alveoli and stimulates the breast alveolar epithelial cells to produce milk components, such as lactose, casein, and lipids (132). Notably, the level of prolactin receptor (PRLR) is suppressed on mammary glandular tissue during periods of elevated progesterone levels and is enhanced to enable lactogenesis when the serum progesterone level drops (133).
Prolactin has a major role in determining the deposition and mobilization of fat. Thus, it is suggested that in both adults and children, increased body weight alters the secretion of prolactin, possibly due to hyperinsulinemia-induced hypothalamic-pituitary dysfunction. In obese females, enhanced prolactin release is in proportion to the size of the visceral fat mass. A possible explanation is reduced dopamine D2 receptor (D2R) availability in the brain, since prolactin is inhibited by D2R activation (134). After the loss of 50% of overweight, such elevated prolactin secretion rate in obese females is significantly blunted, along with increased dopaminergic signaling (135). However, a large cross-sectional study assessing serum prolactin levels in 344 males and females obese subjects’ samples one year after gastric bypass surgery finds no significant association between basal prolactin levels and the degree of obesity or between the change of systematic prolactin level and weight loss. Thus, there does not seem to be a significant role of prolactin in the pathophysiology of obesity (136). Since obesity is associated with higher NAFLD incidence, whether prolactin has hepatoprotective roles in NAFLD received mass attention. A recent clinical study with 859 adults (456 patients with NAFLD and 403 controls without NAFLD) identifies that circulating prolactin levels and hepatic Prlr gene expression levels are lower in NAFLD patients than those of healthy controls (in both sexes). Moreover, in cell models, prolactin ameliorates hepatic steatosis via PRLR and fatty acid translocase (FAT)/CD36, an important hepatic transporter of free fatty acid (137). Thus, prolactin level, body mass index, alanine aminotransferase, HDL cholesterol, and HbA1cA are included in a new noninvasive model for the prediction of NAFLD presence (138). In terms of ALD, since acute and repeated alcohol ingestions sharply rise plasma prolactin levels and decrease plasma testosterone levels in male volunteers (139) and ethanol induces hyperprolactinemia lactotrope growth in female rats (140), it is speculated that increased prolactin is an endogenous protective mechanism to alleviated injury of the liver, and possibly other ethanol-targeted tissues, via unknown pathways. Another study finds a drastic increase in serum prolactin in cirrhotic patients than in healthy control, regardless of hepatic encephalopathy presence, and a cut-off value (50 ng/ml) is capable to predict the mortality (141). This result is consistent with a clinical report including 114 male cirrhotic patients whose increased prolactin level is parallel to growing cirrhosis severity (142). In HCC, prolactin prevents cancer growth by restricting innate immune activation of c-Myc in mice (143). A recent study reports that prolactin upregulated female-predominant cytochrome P450 genes in female mice and downregulated male-predominant 450 genes in male mice, which may explain the abnormal drug metabolism and DILI during pregnancy and lactation (144). In conclusion, increased secretion of prolactin from the pituitary seems to be beneficial for the development of both metabolic and end-stage liver diseases. Compared to other sex/sex-related hormones, prolactin holds a promising perspective in exogenous hormone therapy for those diseases. However, molecular mechanisms and well-designed RCTs are still warranted to be investigated. Known protective and toxic mechanisms of sex-related hormones in the liver are summarized in Figure 2 and Table 2.
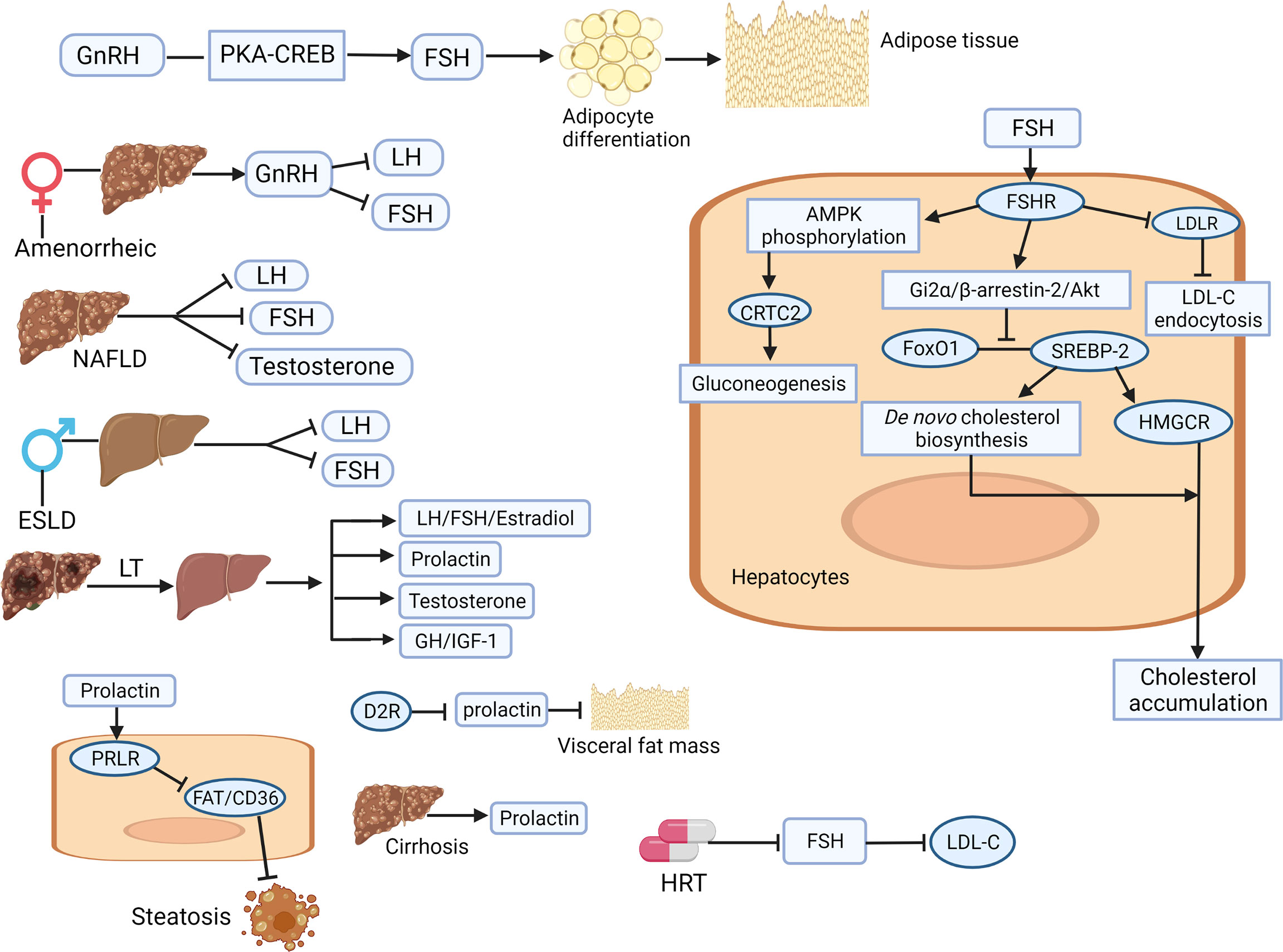
Figure 2 Illustration of the involving mechanisms of sex-related hormones (gonadotrophin-releasing hormone, luteinizing hormone, follicle-stimulating hormone, and prolactin) in liver physiology and pathology. GnRH increases the secretion of FSH through the PKA/CREB pathway, accelerates the differentiation of adipocytes in adipose tissue, which finally lead to hepatic fat accumulation. In amenorrhoea females with cirrhosis, abnormal GnRH secretion leads to low LH and FSH levels. FSH interacts with FSHR to reduce the level of LDLR, weaken the endocytosis of LDL-C, and lead to the increase of circulating LDL-C. After HRT treatment, FSH is inhibited and LDL-C content is improved. FSH activates Gi2 by binding to liver FSHRα/β-Arrestin-2/Akt pathway, which subsequently inhibits the binding between FoxO1 and SREBP-2, drives HMGCR transcription and de novo cholesterol biosynthesis, resulting in increased cholesterol accumulation. Liver transplantation can improve the hormone disorder related to chronic liver disease by restoring the circulating physiological levels of estradiol, FSH, LH, prolactin, testosterone, GH and IGF-1. Inhibition of prolactin by D2R activation leads to reduction of visceral adipose tissue. Prolactin improves hepatic steatosis through PRLR down regulation of FAT/CD36. Prolactin levels are significantly increased in patients with liver cirrhosis. Akt, protein kinase B; AMPK, adenosine monophosphate-activated protein kinase; CREB, cAMP response element binding protein; ESLD, End-stage liver disease; FAT, fatty acid translocase; FoxO1, forkhead box protein O1; FSH, follicle-stimulating hormone; FSHR, FSH receptor; GH, growth hormone; GnRH, gonadotrophin-releasing hormone; HMGCR, 3-hydroxy-3-methylglutaryl coenzyme A reductase; HRT, hormone-replacement therapy; IGF, insulin growth factor; LDL, low-density lipoprotein; LDL-C, low-density-lipoprotein cholesterol; LDLR, LDL receptor; LH, luteinizing hormone; LT, liver transplantation; PKA, protein kinase A; PRLR, prolactin receptor (Created with Biorender.com with a publication license).

Table 2 The involving roles and therapeutic potentials of sex-related hormones in liver diseases.
Gut-Liver Axis Regulation by Sex/Sex-Related Hormones
The gut microbiome is a microbial ecosystem involved in nutrient acquisition and energy metabolism of the host (154). Sex plays an important role in the composition diversity of the gut microbiota (155, 156). The alpha diversity of the gut microbiota is higher in females than in males (157, 158), with differences occurring at the onset of puberty, suggesting that sex hormones cause important composition changes of the gut microbiome (159, 160). Hyperandrogenism is a key factor in impaired follicular development and metabolic disorders in PCOS. In the animal model, intestinal dysbacteriosis is reproduced in DHEA-induced PCOS-like rats. Antibiotic mixtures can be used to eliminate the gut microbiota during DHEA treatment. However, depletion of the gut microbiota does not prevent the development of the PCOS phenotype in DHEA-treated rats. The DHEA type intestinal microflora transplanted into a pseudo-sterile recipient cause disorders of hepatic glycolipid metabolism and reproductive hormone imbalance. These findings suggest that androgen-induced dysbacteriosis may exacerbate metabolic and endocrine dysfunction in PCOS (161). Males are generally more vulnerable to glucose imbalance and diabetes than females. It is revealed that the depletion of the mice gut microbiome largely eliminates sexual dimorphism in glucose metabolism. Glucose tolerance in male mice is more evidently influenced by the gut microbiome than in female mice. Androgen treatment improves glucose tolerance and insulin sensitivity, in part by modulating the gut microbiome, leading to sexual dimorphism in glucose metabolism. Androgens also regulate circulating glutamine and glutamine/glutamate (Gln/Glu) ratios partly via the actions of gut microbiome. Exogenous glutamine supplementation may increase insulin sensitivity in vitro (162). In terms of estrogen, its related receptor alpha (ESRRA) acts as a key regulator of gut homeostasis by activating autophagic flux and controlling host gut microbiota to improve colonic inflammation. In animal models, ESRRA-deficient mice exhibit a distinct gut microbiota composition and significantly higher microbial diversity compared to wild-type mice. ESRRA promotes gut homeostasis through autophagy activation and gut microbiota control to protect the host from harmful inflammation and mitochondrial dysfunction (163). One of the major regulators of circulating estrogens is the gut microbiome, which modulates estrogen by secreting β-glucuronidase (GUS), an enzyme that breaks down estrogen into its active form. When this process is impaired by dysbacteriosis in the gut, the reduction in deconjugation results in a decreased level of circulating estrogens (164). Changes in circulating estrogens may lead to the development of several diseases (obesity, metabolic syndrome, cancer, endometrial hyperplasia, endometriosis, PCOS, infertility, cardiovascular disease, and cognitive function). Modulation of microbiome composition has been shown to alleviate many estrogen-regulated disease progressions (164). Pregnancy is accompanied by changes in the microbiome, and progesterone, the main pregnancy hormone, is found to directly regulate the intestinal microbial composition during pregnancy, such as promoting the growth of bifidobacteria species (probiotics that live in the intestines) in late pregnancy, in order to transmit them to newborns (165). In the serum of ICP patients, the level of a progesterone metabolite, epiallopregnanolone sulfate is significantly elevated, which can inhibit farnesoid X receptor (FXR)-mediated bile acid export and synthesis. Administration with probiotic Lactobacillus rhamnosus GG prevents epiallopregnanolone sulfate-induced hepatic bile acid accumulation and liver injury, possibly mediated by hepatic FXR activation (166). GnRH is associated with gut motility through GnRH receptors signaling, primarily in cells of parasympathetic ganglion and myenteric plexus of the enteric nervous (167). There is a bidirectional relationship between intestinal flora and GnRH/GnRH receptor signaling axis (168). The potential interaction between GnRH and the gut microbiota has been suggested through a lipopolysaccharide (LPS)-induced proinflammatory pathway (169). Disruption of gut microbiota or large bacterial translocations may lead to greater circulation alterations in LPS, inflammatory responses, and GnRH production (170). A study monitoring the effects of the probiotic Bifidobacterium lactis V9 on the gut microbiome, gut-brain mediators, and sex hormones in 14 PCOS patients shows significant higher levels of prolactin, LH and LH/FSH ratio when compared with 9 volunteers. The levels of sex hormones, brain-gut mediators (e.g. ghrelin) and intestinal short-chain fatty acids (SCFAs) are conversely regulated (171). In a rat model, dietary flaxseed oil (FO) intake improves the disturbance of estrous cycle and ovarian morphology, as well as the disorder of sex/sex-related hormones, including testosterone, estrogen, progesterone, and LH/FSH, body weight, dyslipidemia, and insulin resistance. One of the major mechanisms is through the sex steroid hormone-microbiota-inflammatory axis (172). Possible involving mechanisms of sex/sex-related hormones in gut-liver axis regulation are illustrated in Figure 3.
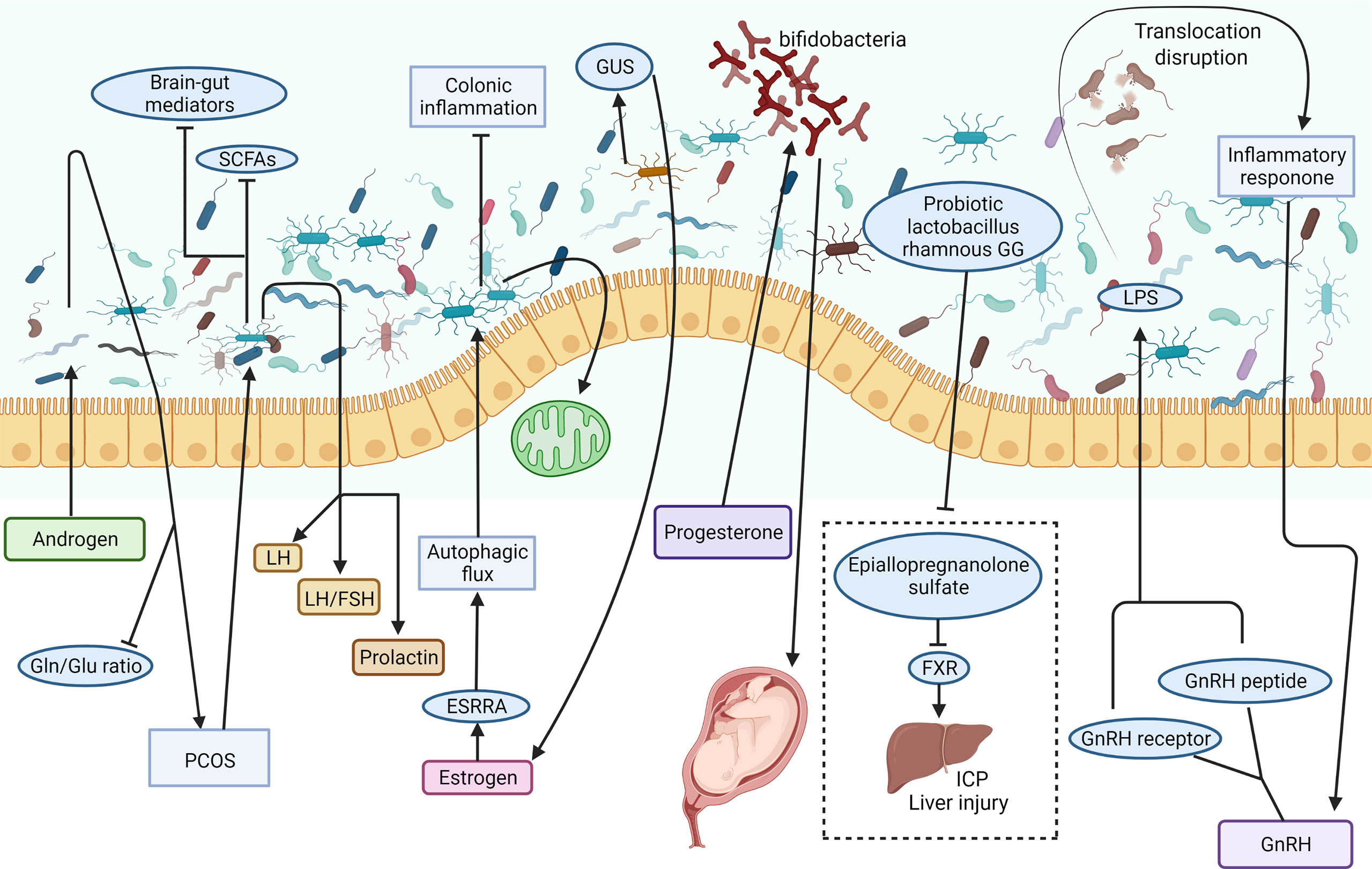
Figure 3 Possible involving mechanisms of sex/sex-related hormones in the regulation of the gut-liver axis. The intestinal microbiome is a complex microbial ecosystem. Androgen induced dysbacteriosis may aggravate PCOS and reduce the circulating Gln/Glu ratio. The study on the effects of intestinal microorganisms on gut brain mediators and sex hormones in patients with PCOS showed that prolactin, LH and LH/FSH ratio increased significantly, while brain-gut mediators and SCFAs decreased. Estrogen can improve colitis and protect mitochondrial function by ESRRA-mediated autophagy. One of the main regulators of circulating estrogen is the intestinal microbiome via the secretion of GUS. Progesterone promotes the growth of bifidobacteria in the third trimester of pregnancy and transmits it to newborns. Probiotic Lactobacillus rhamnosus GG can prevent epicallopregnanolone sulfate-mediated FXR activation and bile acid synthesis, so as to reduce liver bile acid accumulation and liver injury in ICP patients. Dysfunction of intestinal microbiota may lead to LPS leakage, inflammatory response, and GnRH secretion abnormality. ESRRA, estrogen related receptor alpha; FSH, follicle-stimulating hormone; FXR, farnesoid X receptor; GnRH, gonadotrophin-releasing hormone; GUS, β-glucuronidase; ICP, intrahepatic cholestasis of pregnancy; LH, luteinizing hormone; LPS, lipopolysaccharide; PCOS, polycystic ovary syndrome; SCFA, short chain fatty acid (Created with Biorender.com with a publication license).
Hepatic Safety Issue of Gender-Affirming Hormone Therapy in Transgender Populations
Transgender people are a diverse population whose assigned sex at birth are different from their current gender identity. The global prevalence of people who identify as transgender is estimated as 0.3-0.5%, which depends on the definition of transgender used (173). Many transgender people are suffering from health inequities such social marginalization, discrimination, stigma, and violence (174). In the past decades, increasing numbers of people with gender dysphoria have sought medical treatments. According to the clinical practice guidelines from World Professional Association for Transgender Health, those treatments consist of puberty suppression, masculinizing or feminizing hormone treatment, and gender-affirming surgery (175). Application of gonadotropin-releasing hormone analog (GnRHa) or estrogen for puberty suppression in adults and adolescents is recommended by the guidelines. However, data on the efficacy and safety, including the possible metabolic dysfunction and hepatotoxicity, are scarce. A study monitoring triptorelin treatment in gender dysphoric adolescents reports that this agent suppresses puberty in most participated gender dysphoric adolescents. No sustained elevations of liver enzymes or creatinine are observed (176). Another study of 28 transgirl adolescents treated with oral estrogen for more than one year reveals that modest breast development can be found in most participants. The BMI, lean body mass percentage, fat percentage, and liver function do not change during two years of estrogen treatment (177). In a European cohort study of 155 transwomen and 233 transmen, testosterone and estradiol levels are not significantly correlated with amenorrhoea in transmen and breast development in transwomen, respectively. Elevations of liver values are rare (< 4%) and transient in most cases (178). Thus, it seems that GAHT with a safe and effective hormone regimen recommended by the guidelines will not induce liver injury. However, a very recent longitudinal cohort study, which incorporates follow up of over 10 years of 624 transwomen and 438 transmen indicates that, transwomen are likely to experience a moderate elevation of ALT and AST following testosterone initiation, while feminizing GAHT is unlikely to induce such changes. Importantly, alcohol abuse and obesity are strongly associated with liver function abnormalities in transgender populations (179). Thus, more clinical trials and basic studies are needed to delineate the molecular pathways that mediate the sex difference in the liver. Although long-term GAHT under the supervision of clinicians and mental health professionals is not likely to induce evident liver injury, we cannot ignore that many transpeople commonly use sex hormones without any medical supervision and the aware of the potential risks, particularly in the developing world (180). Since irregular and high dosages of sex hormones are common in those transpeople, it is important to test the possible hepatotoxicity and hepatoprotection of those hormones in animal models and, if available, from medical records (Figure 4).
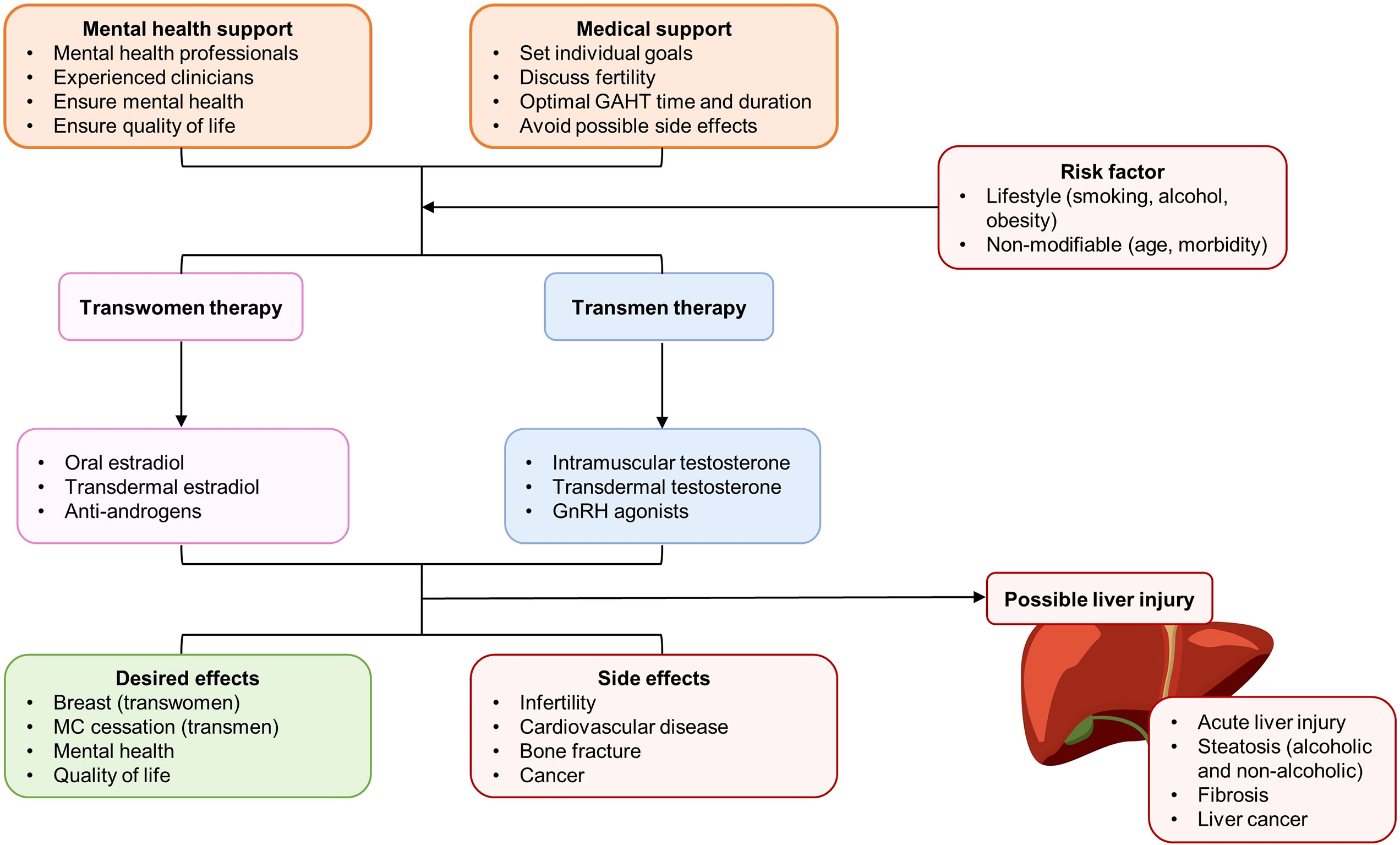
Figure 4 Patient care, therapy selection, and balance between desired effects and potential side effects (with special emphasis in the liver) of gender-affirming hormone therapy (GAHT) for transgender populations. AMPK, D2R, dopamine D2 receptor; GnRH, gonadotropin-releasing hormone; MC, menstrual cycle.
Conclusion
Virtually all kinds of liver diseases are strongly linked with hormonal influences (181, 182). Identifying the clinical manifestation and underlying mechanisms of the “sexual dimorphism” is critical for providing hints for the prevention, management, and treatment of those diseases (76). HRT is used to alleviate vasomotor (e.g. hot flushes and night sweats) and vaginal (e.g. dryness and itching) symptoms of females during menopause because of the reduction in estrogen levels. Previously, the use of HRT is cautioned for patients with basic liver disease since it may provoke or worsen cholestasis (183). Other studies, however, have proved that HRT in patients with chronic liver disease was quite safe and efficacious (184–186). In particular, for patients with chronic liver disease and osteoporosis, transdermal HRT and oral calcium/vitamin D supplementation have been the first-line therapy (187, 188). Results of exogenous sex hormone therapy in liver diseases, both in humans and animals, are controversial. As identified by a Cochrane Review, there is no significant beneficial effect of anabolic-androgenic steroids on clinical outcomes (e.g. liver histology, mortality, and liver-related mortality) of patients with ALD (102). Several small clinical trials tried to examine the efficacy of testosterone therapy in males with cirrhosis, but none of them found beneficial outcomes (189, 190). Since androgen receptor signaling has been shown to suppress metastasis of HCC, combined therapy of Sorafenib and agents that enhance the functional expression of androgen receptor may suppress the HCC progression (106). Similar results are reported in HBV-induced HCC because a small chemical compound that can degrade androgen receptor (ASC-J9) successfully reduce tumor foci and volume in a mice model (95). Estrogen therapy and hormone treatment are generally considered to protect against fatty liver, insulin resistance, and diabetes, although this beneficial effect is not equal in males and females (100). Nuclear receptor proteins (e.g. peroxisome proliferator-activated receptors) are possibly the main targets mediating such protection in the liver (191). Importantly, active estrogen metabolites and derivatives, which have limited affinity for ERs, may play fibrosuppressive roles in the liver (192). Thus, this provides novel therapeutic options for patients with cirrhosis and portal hypertension. An unexplored but promising therapy is the clinical use of prolactin since administration with prolactin or prolactin-releasing peptide evidently improves steatosis in mice obesity models (137), and ablation of prolactin receptor increases hepatic triglyceride accumulation (193). More pre-clinical studies and well-designed RCTs are needed to establish the possible therapeutic effects of prolactin on NAFLD or other chronic liver diseases. Perspectives and side-effects of sex/sex-related hormones or their agonists/antagonists in liver diseases therapy are summarized in Table 3.
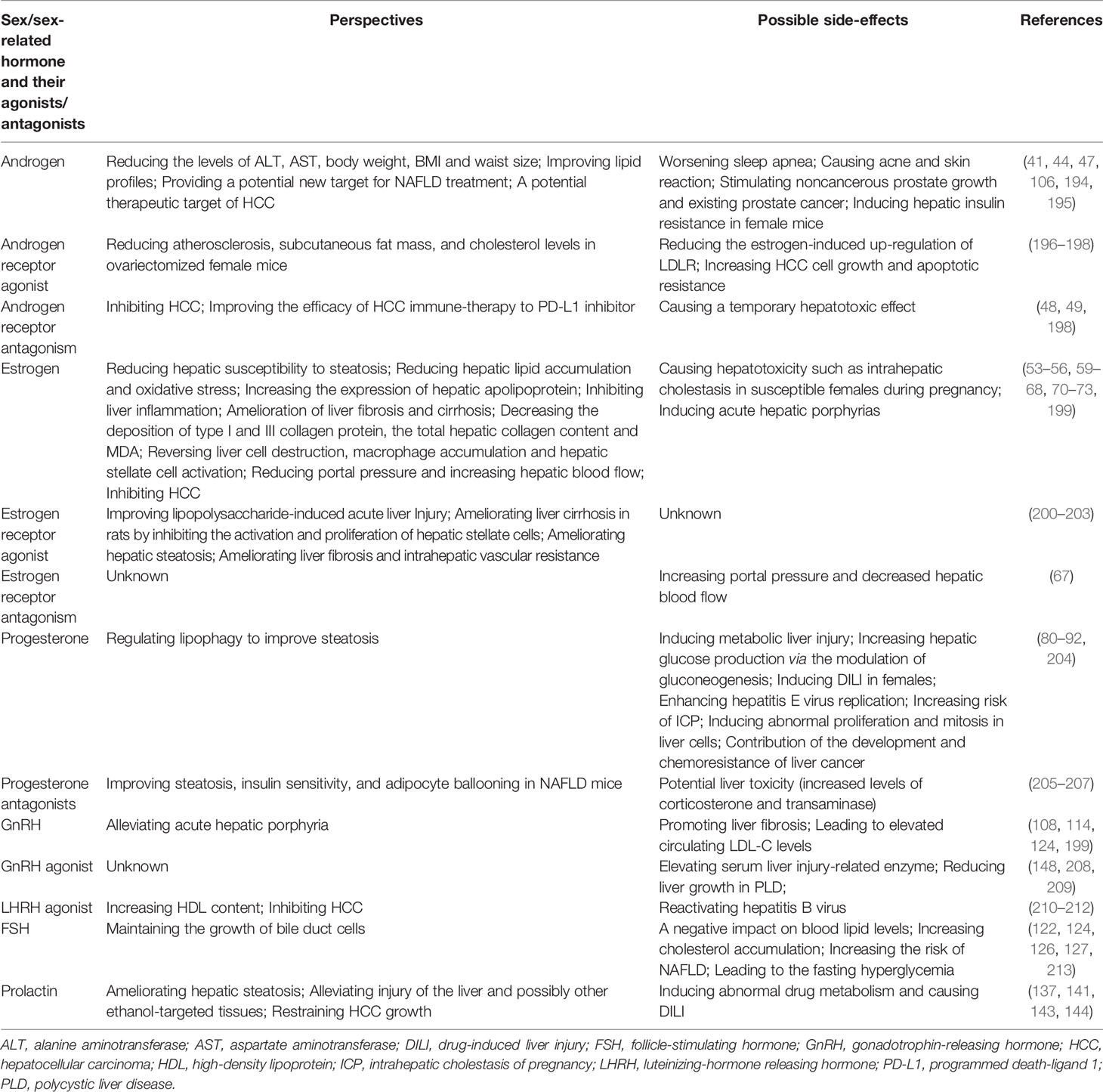
Table 3 Perspectives and side-effects of sex/sex-related hormones or their agonists/antagonists in liver diseases therapy.
Several problems hinder the development of sex and sex-related hormone-based therapy in liver diseases: (1) lack of mechanistic study, particularly the roles of canonical receptor pathway and non-canonical receptor pathway, which provides the detailed information of drug design and adverse effect; (2) lack of study investigating the complicated interplay between sex/sex-related hormones and other hormones, because several source glands do not only secrete sex/sex-related hormones; (3) the involving roles of precipitating factors of liver diseases, such as alcohol abuse, smoking, and obesity, in sex and sex-related hormone-based therapy need further investigation, both in pre-clinical experiments and clinical trials; (4) maximize the alleviative effects and minimize the side effects of synthesized hormones or their derivatives in clinical application are necessary (e.g. ethynyleestradiol has greater side effects than estradiol valerate); (5) well-designed RCT studies are warranted to ensure the efficacy and safety of novel sex and sex-related hormone-based therapy of liver diseases, with special emphasis in the difference caused by biological sex, age, psychiatric status, and menopause.
We cannot ignore the urgent need for clinical study of possible liver injury after GAHT. Although the standards for optimal individual clinical protocols pf GAHT are generally consistent around the world, the implementation of such service is unequal because of health system infrastructure and socio-cultural contexts (214). The large number of transgender populations that meet difficulty in seeking professional medical help for sex hormone recipes must not be overlooked. Developing novel therapeutic agents for over-dose hormone-induced liver injury is critically urgent for those populations.
In conclusion, both clinical and basic studies provide evidence of sexual dimorphism in liver diseases, from acute liver injury to cirrhosis and HCC. Delineating these observations requires a deep understanding of the characters of sex/sex-related hormones in disease initiation and progression. Whether supplementation of a specific hormone can ameliorate liver injury with acceptable side effects require further basic and clinical studies, particularly for transgender people needing GAHT.
Author Contributions
LX and YY contributed equally to this paper as co-first authors. LX, YY, CX, and JX conceived and drafted the paper. LX, YY, ZC, and XT prepared the tables and figures. BW instructed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China under grant 82122009 and 82070606.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hirode G, Saab S, Wong RJ. Trends in the Burden of Chronic Liver Disease Among Hospitalized US Adults. JAMA Netw Open (2020) 3:e201997. doi: 10.1001/jamanetworkopen.2020.1997
2. Lan T, Qian S, Tang C, Gao J. Role of Immune Cells in Biliary Repair. Front Immunol (2022) 13:866040. doi: 10.3389/fimmu.2022.866040
3. Ellis EL, Mann DA. Clinical Evidence for the Regression of Liver Fibrosis. J Hepatol (2012) 56:1171–80. doi: 10.1016/j.jhep.2011.09.024
4. Cai Q, Gan C, Tang C, Wu H, Gao J. Mechanism and Therapeutic Opportunities of Histone Modifications in Chronic Liver Disease. Front Pharmacol (2021) 12:784591. doi: 10.3389/fphar.2021.784591
5. Cheemerla S, Balakrishnan M. Global Epidemiology of Chronic Liver Disease. Clin Liver Dis (Hoboken) (2021) 17:365–70. doi: 10.1002/cld.1061
6. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD Disease Burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the Period 2016-2030. J Hepatol (2018) 69:896–904. doi: 10.1016/j.jhep.2018.05.036
7. Liu W, Zeng X, Liu Y, Liu J, Li C, Chen L, et al. The Immunological Mechanisms and Immune-Based Biomarkers of Drug-Induced Liver Injury. Front Pharmacol (2021) 12:723940. doi: 10.3389/fphar.2021.723940
8. Medina-Caliz I, Robles-Diaz M, Garcia-Muñoz B, Stephens C, Ortega-Alonso A, Garcia-Cortes M, et al. Definition and Risk Factors for Chronicity Following Acute Idiosyncratic Drug-Induced Liver Injury. J Hepatol (2016) 65:532–42. doi: 10.1016/j.jhep.2016.05.003
9. Sandhu N, Navarro V. Drug-Induced Liver Injury in GI Practice. Hepatol Commun (2020) 4:631–45. doi: 10.1002/hep4.1503
10. Shen T, Liu Y, Shang J, Xie Q, Li J, Yan M, et al. Incidence and Etiology of Drug-Induced Liver Injury in Mainland China. Gastroenterology (2019) 156:2230–2241.e2211. doi: 10.1053/j.gastro.2019.02.002
11. Kadel S, Kovats S. Sex Hormones Regulate Innate Immune Cells and Promote Sex Differences in Respiratory Virus Infection. Front Immunol (2018) 9:1653. doi: 10.3389/fimmu.2018.01653
12. Ortona E, Pierdominici M, Rider V. Editorial: Sex Hormones and Gender Differences in Immune Responses. Front Immunol (2019) 10:1076. doi: 10.3389/fimmu.2019.01076
13. Gambineri A, Pelusi C. Sex Hormones, Obesity and Type 2 Diabetes: Is There a Link? Endocr Connect (2019) 8:R1–r9. doi: 10.1530/EC-18-0450
14. Ding EL, Song Y, Malik VS, Liu S. Sex Differences of Endogenous Sex Hormones and Risk of Type 2 Diabetes: A Systematic Review and Meta-Analysis. Jama (2006) 295:1288–99. doi: 10.1001/jama.295.11.1288
15. Zhao D, Guallar E, Ouyang P, Subramanya V, Vaidya D, Ndumele CE, et al. Endogenous Sex Hormones and Incident Cardiovascular Disease in Post-Menopausal Women. J Am Coll Cardiol (2018) 71:2555–66. doi: 10.1016/j.jacc.2018.01.083
16. Phan H, Richard A, Lazo M, Nelson WG, Denmeade SR, Groopman J, et al. The Association of Sex Steroid Hormone Concentrations With non-Alcoholic Fatty Liver Disease and Liver Enzymes in US Men. Liver Int (2021) 41:300–10. doi: 10.1111/liv.14652
17. Kur P, Kolasa-Wołosiuk A, Misiakiewicz-Has K, Wiszniewska B. Sex Hormone-Dependent Physiology and Diseases of Liver. Int J Environ Res Public Health (2020) 17(8):2620. doi: 10.3390/ijerph17082620
18. Lonardo A, Nascimbeni F, Ballestri S, Fairweather D, Win S, Than TA, et al. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology (2019) 70:1457–69. doi: 10.1002/hep.30626
19. McGlynn KA, Hagberg K, Chen J, Braunlin M, Graubard BI, Suneja N, et al. Menopausal Hormone Therapy Use and Risk of Primary Liver Cancer in the Clinical Practice Research Datalink. Int J Cancer (2016) 138:2146–53. doi: 10.1002/ijc.29960
20. Schiffer L, Arlt W, Storbeck KH. Intracrine Androgen Biosynthesis, Metabolism and Action Revisited. Mol Cell Endocrinol (2018) 465:4–26. doi: 10.1016/j.mce.2017.08.016
21. Hammes SR, Levin ER. Impact of Estrogens in Males and Androgens in Females. J Clin Invest (2019) 129:1818–26. doi: 10.1172/JCI125755
22. Kostakis EK, Gkioni LN, Macut D, Mastorakos G. Androgens in Menopausal Women: Not Only Polycystic Ovary Syndrome. Front Horm Res (2019) 53:135–61. doi: 10.1159/000494909
23. Kelly DM, Nettleship JE, Akhtar S, Muraleedharan V, Sellers DJ, Brooke JC, et al. Testosterone Suppresses the Expression of Regulatory Enzymes of Fatty Acid Synthesis and Protects Against Hepatic Steatosis in Cholesterol-Fed Androgen Deficient Mice. Life Sci (2014) 109:95–103. doi: 10.1016/j.lfs.2014.06.007
24. Abruzzese GA, Heber MF, Ferreira SR, Velez LM, Reynoso R, Pignataro OP, et al. Prenatal Hyperandrogenism Induces Alterations That Affect Liver Lipid Metabolism. J Endocrinol (2016) 230:67–79. doi: 10.1530/JOE-15-0471
25. Grossmann M, Wierman ME, Angus P, Handelsman DJ. Reproductive Endocrinology of Nonalcoholic Fatty Liver Disease. Endocr Rev (2019) 40:417–46. doi: 10.1210/er.2018-00158
26. Seidu T, McWhorter P, Myer J, Alamgir R, Eregha N, Bogle D, et al. DHT Causes Liver Steatosis via Transcriptional Regulation of SCAP in Normal Weight Female Mice. J Endocrinol (2021) 250:49–65. doi: 10.1530/JOE-21-0040
27. Haring R, Völzke H, Felix SB, Schipf S, Dörr M, Rosskopf D, et al. Prediction of Metabolic Syndrome by Low Serum Testosterone Levels in Men: Results From the Study of Health in Pomerania. Diabetes (2009) 58:2027–31. doi: 10.2337/db09-0031
28. Nikolaenko L, Jia Y, Wang C, Diaz-Arjonilla M, Yee JK, French SW, et al. Testosterone Replacement Ameliorates Nonalcoholic Fatty Liver Disease in Castrated Male Rats. Endocrinology (2014) 155:417–28. doi: 10.1210/en.2013-1648
29. Derby CA, Zilber S, Brambilla D, Morales KH, McKinlay JB. Body Mass Index, Waist Circumference and Waist to Hip Ratio and Change in Sex Steroid Hormones: The Massachusetts Male Ageing Study. Clin Endocrinol (Oxf) (2006) 65:125–31. doi: 10.1111/j.1365-2265.2006.02560.x
30. Azziz R, Carmina E, Chen Z, Dunaif A, Laven JS, Legro RS, et al. Polycystic Ovary Syndrome. Nat Rev Dis Primers (2016) 2:16057. doi: 10.1038/nrdp.2016.57
31. Sowers MF, Beebe JL, McConnell D, Randolph J, Jannausch M. Testosterone Concentrations in Women Aged 25-50 Years: Associations With Lifestyle, Body Composition, and Ovarian Status. Am J Epidemiol (2001) 153:256–64. doi: 10.1093/aje/153.3.256
32. Naessén S, Carlström K, Byström B, Pierre Y, Hirschberg AL. Effects of an Antiandrogenic Oral Contraceptive on Appetite and Eating Behavior in Bulimic Women. Psychoneuroendocrinology (2007) 32:548–54. doi: 10.1016/j.psyneuen.2007.03.008
33. Hirschberg AL. Sex Hormones, Appetite and Eating Behaviour in Women. Maturitas (2012) 71:248–56. doi: 10.1016/j.maturitas.2011.12.016
34. Jaruvongvanich V, Sanguankeo A, Riangwiwat T, Upala S. Testosterone, Sex Hormone-Binding Globulin and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Ann Hepatol (2017) 16:382–94. doi: 10.5604/01.3001.0009.8593
35. She P, Shiota M, Shelton KD, Chalkley R, Postic C, Magnuson MA. Phosphoenolpyruvate Carboxykinase is Necessary for the Integration of Hepatic Energy Metabolism. Mol Cell Biol (2000) 20:6508–17. doi: 10.1128/MCB.20.17.6508-6517.2000
36. Völzke H, Aumann N, Krebs A, Nauck M, Steveling A, Lerch MM, et al. Hepatic Steatosis is Associated With Low Serum Testosterone and High Serum DHEAS Levels in Men. Int J Androl (2010) 33:45–53. doi: 10.1111/j.1365-2605.2009.00953.x
37. Jones H, Sprung VS, Pugh CJ, Daousi C, Irwin A, Aziz N, et al. Polycystic Ovary Syndrome With Hyperandrogenism is Characterized by an Increased Risk of Hepatic Steatosis Compared to Nonhyperandrogenic PCOS Phenotypes and Healthy Controls, Independent of Obesity and Insulin Resistance. J Clin Endocrinol Metab (2012) 97:3709–16. doi: 10.1210/jc.2012-1382
38. Zhang H, Liu Y, Wang L, Li Z, Zhang H, Wu J, et al. Differential Effects of Estrogen/Androgen on the Prevention of Nonalcoholic Fatty Liver Disease in the Male Rat. J Lipid Res (2013) 54:345–57. doi: 10.1194/jlr.M028969
39. Cai J, Wu CH, Zhang Y, Wang YY, Xu WD, Lin TC, et al. High-Free Androgen Index is Associated With Increased Risk of non-Alcoholic Fatty Liver Disease in Women With Polycystic Ovary Syndrome, Independent of Obesity and Insulin Resistance. Int J Obes (Lond) (2017) 41:1341–7. doi: 10.1038/ijo.2017.116
40. Seo NK, Koo HS, Haam JH, Kim HY, Kim MJ, Park KC, et al. Prediction of Prevalent But Not Incident non-Alcoholic Fatty Liver Disease by Levels of Serum Testosterone. J Gastroenterol Hepatol (2015) 30:1211–6. doi: 10.1111/jgh.12935
41. Haider A, Gooren LJ, Padungtod P, Saad F. Improvement of the Metabolic Syndrome and of non-Alcoholic Liver Steatosis Upon Treatment of Hypogonadal Elderly Men With Parenteral Testosterone Undecanoate. Exp Clin Endocrinol Diabetes (2010) 118:167–71. doi: 10.1055/s-0029-1202774
42. Polyzos SA, Kountouras J, Tsatsoulis A, Zafeiriadou E, Katsiki E, Patsiaoura K, et al. Sex Steroids and Sex Hormone-Binding Globulin in Postmenopausal Women With Nonalcoholic Fatty Liver Disease. Hormones (Athens) (2013) 12:405–16. doi: 10.1007/BF03401306
43. Sarkar M, Wellons M, Cedars MI, VanWagner L, Gunderson EP, Ajmera V, et al. Testosterone Levels in Pre-Menopausal Women are Associated With Nonalcoholic Fatty Liver Disease in Midlife. Am J Gastroenterol (2017) 112:755–62. doi: 10.1038/ajg.2017.44
44. White DL, Tavakoli-Tabasi S, Kuzniarek J, Pascua R, Ramsey DJ, El-Serag HB. Higher Serum Testosterone is Associated With Increased Risk of Advanced Hepatitis C-Related Liver Disease in Males. Hepatology (2012) 55:759–68. doi: 10.1002/hep.24618
45. Sinclair M, Gow PJ, Grossmann M, Shannon A, Hoermann R, Angus PW. Low Serum Testosterone is Associated With Adverse Outcome in Men With Cirrhosis Independent of the Model for End-Stage Liver Disease Score. Liver Transpl (2016) 22:1482–90. doi: 10.1002/lt.24607
46. Grossmann M, Hoermann R, Gani L, Chan I, Cheung A, Gow PJ, et al. Low Testosterone Levels as an Independent Predictor of Mortality in Men With Chronic Liver Disease. Clin Endocrinol (Oxf) (2012) 77:323–8. doi: 10.1111/j.1365-2265.2012.04347.x
47. Ma WL, Hsu CL, Wu MH, Wu CT, Wu CC, Lai JJ, et al. Androgen Receptor is a New Potential Therapeutic Target for the Treatment of Hepatocellular Carcinoma. Gastroenterology (2008) 135:947–955, 955.e941-945. doi: 10.1053/j.gastro.2008.05.046
48. Helms TH, Mullins RD, Thomas-Ahner JM, Kulp SK, Campbell MJ, Lucas F, et al. Inhibition of Androgen/AR Signaling Inhibits Diethylnitrosamine (DEN) Induced Tumour Initiation and Remodels Liver Immune Cell Networks. Sci Rep (2021) 11:3646. doi: 10.1038/s41598-021-82252-x
49. Jiang G, Shi L, Zheng X, Zhang X, Wu K, Liu B, et al. Androgen Receptor Affects the Response to Immune Checkpoint Therapy by Suppressing PD-L1 in Hepatocellular Carcinoma. Aging (Albany NY) (2020) 12:11466–84. doi: 10.18632/aging.103231
50. Morselli E, Santos RS, Criollo A, Nelson MD, Palmer BF, Clegg DJ. The Effects of Oestrogens and Their Receptors on Cardiometabolic Health. Nat Rev Endocrinol (2017) 13:352–64. doi: 10.1038/nrendo.2017.12
51. Yu K, Huang ZY, Xu XL, Li J, Fu XW, Deng SL. Estrogen Receptor Function: Impact on the Human Endometrium. Front Endocrinol (Lausanne) (2022) 13:827724. doi: 10.3389/fendo.2022.827724
52. Uebi T, Umeda M, Imai T. Estrogen Induces Estrogen Receptor Alpha Expression and Hepatocyte Proliferation in the Livers of Male Mice. Genes Cells (2015) 20:217–23. doi: 10.1111/gtc.12214
53. Fuller KNZ, McCoin CS, Von Schulze AT, Houchen CJ, Choi MA, Thyfault JP. Estradiol Treatment or Modest Exercise Improves Hepatic Health and Mitochondrial Outcomes in Female Mice Following Ovariectomy. Am J Physiol Endocrinol Metab (2021) 320:E1020–e1031. doi: 10.1152/ajpendo.00013.2021
54. Galmés-Pascual BM, Nadal-Casellas A, Bauza-Thorbrügge M, Sbert-Roig M, García-Palmer FJ, Proenza AM, et al. 17β-Estradiol Improves Hepatic Mitochondrial Biogenesis and Function Through PGC1B. J Endocrinol (2017) 232:297–308. doi: 10.1530/JOE-16-0350
55. Galmés-Pascual BM, Martínez-Cignoni MR, Morán-Costoya A, Bauza-Thorbrügge M, Sbert-Roig M, Valle A, et al. 17β-Estradiol Ameliorates Lipotoxicity-Induced Hepatic Mitochondrial Oxidative Stress and Insulin Resistance. Free Radic Biol Med (2020) 150:148–60. doi: 10.1016/j.freeradbiomed.2020.02.016
56. Okamura T, Hamaguchi M, Bamba R, Nakajima H, Yoshimura Y, Kimura T, et al. Immune Modulating Effects of Additional Supplementation of Estradiol Combined With Testosterone in Murine Testosterone-Deficient NAFLD Model. Am J Physiol Gastrointest Liver Physiol (2020) 318:G989–g999. doi: 10.1152/ajpgi.00310.2019
57. Zhu L, Shi J, Luu TN, Neuman JC, Trefts E, Yu S, et al. Hepatocyte Estrogen Receptor Alpha Mediates Estrogen Action to Promote Reverse Cholesterol Transport During Western-Type Diet Feeding. Mol Metab (2018) 8:106–16. doi: 10.1016/j.molmet.2017.12.012
58. Della Torre S, Mitro N, Fontana R, Gomaraschi M, Favari E, Recordati C, et al. An Essential Role for Liver Erα in Coupling Hepatic Metabolism to the Reproductive Cycle. Cell Rep (2016) 15:360–71. doi: 10.1016/j.celrep.2016.03.019
59. Luo F, Guo Y, Ruan GY, Peng R, Li XP. Estrogen Lowers Triglyceride via Regulating Hepatic APOA5 Expression. Lipids Health Dis (2017) 16:72. doi: 10.1186/s12944-017-0463-0
60. Polyzos SA, Lambrinoudaki I, Goulis DG. Menopausal Hormone Therapy in Women With Dyslipidemia and Nonalcoholic Fatty Liver Disease. Hormones (Athens) (2022). doi: 10.1007/s42000-022-00369-8
61. Kamada Y, Kiso S, Yoshida Y, Chatani N, Kizu T, Hamano M, et al. Estrogen Deficiency Worsens Steatohepatitis in Mice Fed High-Fat and High-Cholesterol Diet. Am J Physiol Gastrointest Liver Physiol (2011) 301:G1031–1043. doi: 10.1152/ajpgi.00211.2011
62. Hsieh CH, Nickel EA, Chen J, Schwacha MG, Choudhry MA, Bland KI, et al. Mechanism of the Salutary Effects of Estrogen on Kupffer Cell Phagocytic Capacity Following Trauma-Hemorrhage: Pivotal Role of Akt Activation. J Immunol (2009) 182:4406–14. doi: 10.4049/jimmunol.0803423
63. Zhang Y, Wu L, Wang Y, Zhang M, Li L, Zhu D, et al. Protective Role of Estrogen-Induced miRNA-29 Expression in Carbon Tetrachloride-Induced Mouse Liver Injury. J Biol Chem (2012) 287:14851–62. doi: 10.1074/jbc.M111.314922
64. Yasuda M, Shimizu I, Shiba M, Ito S. Suppressive Effects of Estradiol on Dimethylnitrosamine-Induced Fibrosis of the Liver in Rats. Hepatology (1999) 29:719–27. doi: 10.1002/hep.510290307
65. Ohashi T, Kato M, Yamasaki A, Kuwano A, Suzuki H, Kohjima M, et al. Effects of High Fructose Intake on Liver Injury Progression in High Fat Diet Induced Fatty Liver Disease in Ovariectomized Female Mice. Food Chem Toxicol (2018) 118:190–7. doi: 10.1016/j.fct.2018.05.006
66. Nolte W, Schindler CG, Figulla HR, Wuttke W, Hüfner M, Hartmann H, et al. Increase of Serum Estradiol in Cirrhotic Men Treated by Transjugular Intrahepatic Portosystemic Stent Shunt. J Hepatol (2001) 34:818–24. doi: 10.1016/S0168-8278(01)00052-6
67. Sakamoto M, Ueno T, Nakamura T, Sakata R, Hasimoto O, Torimura T, et al. Improvement of Portal Hypertension and Hepatic Blood Flow in Cirrhotic Rats by Oestrogen. Eur J Clin Invest (2005) 35:220–5. doi: 10.1111/j.1365-2362.2005.01476.x
68. Morgan-Martins MI, Jacques SI, Hartmann RM, Marques CM, Marroni CA, Marroni NP. Protection of Estrogen in Portal Hypertension Gastropathy: An Experimental Model. Arq Gastroenterol (2011) 48:211–6. doi: 10.1590/S0004-28032011000300011
69. Jiang M, Klein M, Zanger UM, Mohammad MK, Cave MC, Gaikwad NW, et al. Inflammatory Regulation of Steroid Sulfatase: A Novel Mechanism to Control Estrogen Homeostasis and Inflammation in Chronic Liver Disease. J Hepatol (2016) 64:44–52. doi: 10.1016/j.jhep.2015.07.022
70. Hassan MM, Botrus G, Abdel-Wahab R, Wolff RA, Li D, Tweardy D, et al. Estrogen Replacement Reduces Risk and Increases Survival Times of Women With Hepatocellular Carcinoma. Clin Gastroenterol Hepatol (2017) 15:1791–9. doi: 10.1016/j.cgh.2017.05.036
71. Wei Q, Guo P, Mu K, Zhang Y, Zhao W, Huai W, et al. Estrogen Suppresses Hepatocellular Carcinoma Cells Through Erβ-Mediated Upregulation of the NLRP3 Inflammasome. Lab Invest (2015) 95:804–16. doi: 10.1038/labinvest.2015.63
72. Yang W, Lu Y, Xu Y, Xu L, Zheng W, Wu Y, et al. Estrogen Represses Hepatocellular Carcinoma (HCC) Growth via Inhibiting Alternative Activation of Tumor-Associated Macrophages (TAMs). J Biol Chem (2012) 287:40140–9. doi: 10.1074/jbc.M112.348763
73. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production. Science (2007) 317:121–4. doi: 10.1126/science.1140485
74. Singhal H, Greene ME, Tarulli G, Zarnke AL, Bourgo RJ, Laine M, et al. Genomic Agonism and Phenotypic Antagonism Between Estrogen and Progesterone Receptors in Breast Cancer. Sci Adv (2016) 2:e1501924. doi: 10.1126/sciadv.1501924
75. Garg D, Ng SSM, Baig KM, Driggers P, Segars J. Progesterone-Mediated Non-Classical Signaling. Trends Endocrinol Metab (2017) 28:656–68. doi: 10.1016/j.tem.2017.05.006
76. Ballestri S, Nascimbeni F, Baldelli E, Marrazzo A, Romagnoli D, Lonardo A. NAFLD as a Sexual Dimorphic Disease: Role of Gender and Reproductive Status in the Development and Progression of Nonalcoholic Fatty Liver Disease and Inherent Cardiovascular Risk. Adv Ther (2017) 34:1291–326. doi: 10.1007/s12325-017-0556-1
77. Yeung EH, Zhang C, Mumford SL, Ye A, Trevisan M, Chen L, et al. Longitudinal Study of Insulin Resistance and Sex Hormones Over the Menstrual Cycle: The BioCycle Study. J Clin Endocrinol Metab (2010) 95:5435–42. doi: 10.1210/jc.2010-0702
78. Aldhoon-Hainerová I, Zamrazilová H, Hill M, Hainer V. Insulin Sensitivity and its Relation to Hormones in Adolescent Boys and Girls. Metabolism (2017) 67:90–8. doi: 10.1016/j.metabol.2016.10.005
79. Yang JD, Abdelmalek MF, Guy CD, Gill RM, Lavine JE, Yates K, et al. Patient Sex, Reproductive Status, and Synthetic Hormone Use Associate With Histologic Severity of Nonalcoholic Steatohepatitis. Clin Gastroenterol Hepatol (2017) 15:127–131.e122. doi: 10.1016/j.cgh.2016.07.034
80. Lee SR, Kwon SW, Kaya P, Lee YH, Lee JG, Kim G, et al. Loss of Progesterone Receptor Membrane Component 1 Promotes Hepatic Steatosis via the Induced De Novo Lipogenesis. Sci Rep (2018) 8:15711. doi: 10.1038/s41598-018-34148-6
81. Lee SR, Choi WY, Heo JH, Huh J, Kim G, Lee KP, et al. Progesterone Increases Blood Glucose via Hepatic Progesterone Receptor Membrane Component 1 Under Limited or Impaired Action of Insulin. Sci Rep (2020) 10:16316. doi: 10.1038/s41598-020-73330-7
82. Toyoda Y, Endo S, Tsuneyama K, Miyashita T, Yano A, Fukami T, et al. Mechanism of Exacerbative Effect of Progesterone on Drug-Induced Liver Injury. Toxicol Sci (2012) 126:16–27. doi: 10.1093/toxsci/kfr326
83. Choudhary NS, Bodh V, Chaudhari S, Saraf N, Saigal S. Norethisterone Related Drug Induced Liver Injury: A Series of 3 Cases. J Clin Exp Hepatol (2017) 7:266–8. doi: 10.1016/j.jceh.2017.01.001
84. Sooryanarain H, Ahmed SA, Meng XJ. Progesterone-Mediated Enhancement of Hepatitis E Virus Replication in Human Liver Cells. mBio (2021) 12:e0143421. doi: 10.1128/mBio.01434-21
85. Vallejo M, Briz O, Serrano MA, Monte MJ, Marin JJ. Potential Role of Trans-Inhibition of the Bile Salt Export Pump by Progesterone Metabolites in the Etiopathogenesis of Intrahepatic Cholestasis of Pregnancy. J Hepatol (2006) 44:1150–7. doi: 10.1016/j.jhep.2005.09.017
86. Abu-Hayyeh S, Ovadia C, Lieu T, Jensen DD, Chambers J, Dixon PH, et al. Prognostic and Mechanistic Potential of Progesterone Sulfates in Intrahepatic Cholestasis of Pregnancy and Pruritus Gravidarum. Hepatology (2016) 63:1287–98. doi: 10.1002/hep.28265
87. Boada LD, Zumbado M, del RI, Blanco A, Torres S, Monterde JG, et al. Steroid Hormone Progesterone Induces Cell Proliferation and Abnormal Mitotic Processes in Rat Liver. Arch Toxicol (2002) 75:707–16. doi: 10.1007/s00204-001-0297-3
88. Glaser S, DeMorrow S, Francis H, Ueno Y, Gaudio E, Vaculin S, et al. Progesterone Stimulates the Proliferation of Female and Male Cholangiocytes via Autocrine/Paracrine Mechanisms. Am J Physiol Gastrointest Liver Physiol (2008) 295:G124–g136. doi: 10.1152/ajpgi.00536.2007
89. Itagaki T, Shimizu I, Cheng X, Yuan Y, Oshio A, Tamaki K, et al. Opposing Effects of Oestradiol and Progesterone on Intracellular Pathways and Activation Processes in the Oxidative Stress Induced Activation of Cultured Rat Hepatic Stellate Cells. Gut (2005) 54:1782–9. doi: 10.1136/gut.2004.053728
90. Wheeler KT, Wang LM, Wallen CA, Childers SR, Cline JM, Keng PC, et al. Sigma-2 Receptors as a Biomarker of Proliferation in Solid Tumours. Br J Cancer (2000) 82:1223–32. doi: 10.1054/bjoc.1999.1067
91. Peluso JJ. Progesterone Signaling Mediated Through Progesterone Receptor Membrane Component-1 in Ovarian Cells With Special Emphasis on Ovarian Cancer. Steroids (2011) 76:903–9. doi: 10.1016/j.steroids.2011.02.011
92. Yeh YT, Chang CW, Wei RJ, Wang SN. Progesterone and Related Compounds in Hepatocellular Carcinoma: Basic and Clinical Aspects. BioMed Res Int (2013) 2013:290575. doi: 10.1155/2013/290575
93. Medina-Caliz I, Garcia-Cortes M, Gonzalez-Jimenez A, Cabello MR, Robles-Diaz M, Sanabria-Cabrera J, et al. Herbal and Dietary Supplement-Induced Liver Injuries in the Spanish DILI Registry. Clin Gastroenterol Hepatol (2018) 16:1495–502. doi: 10.1016/j.cgh.2017.12.051
94. Huang S, Liu M, Fu F, Liu H, He B, Xiao D, et al. High Serum Estradiol Reduces Acute Hepatotoxicity Risk Induced by Epirubicin Plus Cyclophosphamide Chemotherapy in Premenopausal Women With Breast Cancer. Front Pharmacol (2020) 11:572444. doi: 10.3389/fphar.2020.572444
95. Wu MH, Ma WL, Hsu CL, Chen YL, Ou JH, Ryan CK, et al. Androgen Receptor Promotes Hepatitis B Virus-Induced Hepatocarcinogenesis Through Modulation of Hepatitis B Virus RNA Transcription. Sci Transl Med (2010) 2:32ra35. doi: 10.1126/scitranslmed.3001143
96. Wang SH, Yeh SH, Lin WH, Yeh KH, Yuan Q, Xia NS, et al. Estrogen Receptor α Represses Transcription of HBV Genes via Interaction With Hepatocyte Nuclear Factor 4α. Gastroenterology (2012) 142:989–998.e984. doi: 10.1016/S0016-5085(12)60375-8
97. Ruggieri A, Gagliardi MC, Anticoli S. Sex-Dependent Outcome of Hepatitis B and C Viruses Infections: Synergy of Sex Hormones and Immune Responses? Front Immunol (2018) 9:2302. doi: 10.3389/fimmu.2018.02302
98. Chaudhury CS, Mee T, Chairez C, McLaughlin M, Silk R, Gross C, et al. Testosterone in Men With Chronic Hepatitis C Infection and After Hepatitis C Viral Clearance. Clin Infect Dis (2019) 69:571–6. doi: 10.1093/cid/ciy965
99. Huang Y, Yan D, Zhang H, Lou B, Yan R, Yao Y, et al. Lower Testosterone Levels Predict Increasing Severity and Worse Outcomes of Hepatitis B Virus-Related Acute-on-Chronic Liver Failure in Males. BMC Gastroenterol (2021) 21:457. doi: 10.1186/s12876-021-01993-1
100. Palmisano BT, Zhu L, Stafford JM. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv Exp Med Biol (2017) 1043:227–56. doi: 10.1007/978-3-319-70178-3_12
101. Al-Qudimat A, Al-Zoubi RM, Yassin AA, Alwani M, Aboumarzouk OM, AlRumaihi K, et al. Testosterone Treatment Improves Liver Function and Reduces Cardiovascular Risk: A Long-Term Prospective Study. Arab J Urol (2021) 19:376–86. doi: 10.1080/2090598X.2021.1959261
102. Rambaldi A, Iaquinto G, Gluud C. Anabolic-Androgenic Steroids for Alcoholic Liver Disease: A Cochrane Review. Am J Gastroenterol (2002) 97:1674–81. doi: 10.1111/j.1572-0241.2002.05826.x
103. Järveläinen HA, Lukkari TA, Heinaro S, Sippel H, Lindros KO. The Antiestrogen Toremifene Protects Against Alcoholic Liver Injury in Female Rats. J Hepatol (2001) 35:46–52. doi: 10.1016/S0168-8278(01)00050-2
104. Ohashi K, Pimienta M, Seki E. Alcoholic Liver Disease: A Current Molecular and Clinical Perspective. Liver Res (2018) 2:161–72. doi: 10.1016/j.livres.2018.11.002
105. Lustik SJ, Chhibber AK, Kolano JW, Hilmi IA, Henson LC, Morris MC, et al. The Hyperventilation of Cirrhosis: Progesterone and Estradiol Effects. Hepatology (1997) 25:55–8. doi: 10.1002/hep.510250110
106. Ma WL, Lai HC, Yeh S, Cai X, Chang C. Androgen Receptor Roles in Hepatocellular Carcinoma, Fatty Liver, Cirrhosis and Hepatitis. Endocr Relat Cancer (2014) 21:R165–182. doi: 10.1530/ERC-13-0283
107. Shi L, Feng Y, Lin H, Ma R, Cai X. Role of Estrogen in Hepatocellular Carcinoma: Is Inflammation the Key? J Transl Med (2014) 12:93. doi: 10.1186/1479-5876-12-93
108. Kyritsi K, Meng F, Zhou T, Wu N, Venter J, Francis H, et al. Knockdown of Hepatic Gonadotropin-Releasing Hormone by Vivo-Morpholino Decreases Liver Fibrosis in Multidrug Resistance Gene 2 Knockout Mice by Down-Regulation of miR-200b. Am J Pathol (2017) 187:1551–65. doi: 10.1016/j.ajpath.2017.03.013
109. Thompson IR, Ciccone NA, Xu S, Zaytseva S, Carroll RS, Kaiser UB. GnRH Pulse Frequency-Dependent Stimulation of Fshβ Transcription is Mediated via Activation of PKA and CREB. Mol Endocrinol (2013) 27:606–18. doi: 10.1210/me.2012-1281
110. Wang Y, Wang Y, Liu L, Cui H. Ovariectomy Induces Abdominal Fat Accumulation by Improving Gonadotropin-Releasing Hormone Secretion in Mouse. Biochem Biophys Res Commun (2022) 588:111–7. doi: 10.1016/j.bbrc.2021.12.039
111. Conn PM, Crowley WF Jr. Gonadotropin-Releasing Hormone and its Analogues. N Engl J Med (1991) 324:93–103. doi: 10.1056/NEJM199101103240205
112. Hayes FJ, Crowley WF Jr. Gonadotropin Pulsations Across Development. Horm Res (1998) 49:163–8. doi: 10.1159/000023165
113. Bell H, Raknerud N, Falch JA, Haug E. Inappropriately Low Levels of Gonadotrophins in Amenorrhoeic Women With Alcoholic and non-Alcoholic Cirrhosis. Eur J Endocrinol (1995) 132:444–9. doi: 10.1530/eje.0.1320444
114. McMillin M, DeMorrow S, Glaser S, Venter J, Kyritsi K, Zhou T, et al. Melatonin Inhibits Hypothalamic Gonadotropin-Releasing Hormone Release and Reduces Biliary Hyperplasia and Fibrosis in Cholestatic Rats. Am J Physiol Gastrointest Liver Physiol (2017) 313:G410–g418. doi: 10.1152/ajpgi.00421.2016
115. Van de Velde F, Bekaert M, Hoorens A, Geerts A, T’Sjoen G, Fiers T, et al. Histologically Proven Hepatic Steatosis Associates With Lower Testosterone Levels in Men With Obesity. Asian J Androl (2020) 22:252–7. doi: 10.4103/aja.aja_68_19
116. Li Y, Liu L, Wang B, Chen D, Wang J. Nonalcoholic Fatty Liver Disease and Alteration in Semen Quality and Reproductive Hormones. Eur J Gastroenterol Hepatol (2015) 27:1069–73. doi: 10.1097/MEG.0000000000000408
117. Wang N, Li Q, Han B, Chen Y, Zhu C, Chen Y, et al. Follicle-Stimulating Hormone is Associated With non-Alcoholic Fatty Liver Disease in Chinese Women Over 55 Years Old. J Gastroenterol Hepatol (2016) 31:1196–202. doi: 10.1111/jgh.13271
118. Durazzo M, Premoli A, Di Bisceglie C, Bertagna A, Faga E, Biroli G, et al. Alterations of Seminal and Hormonal Parameters: An Extrahepatic Manifestation of HCV Infection? World J Gastroenterol (2006) 12:3073–6. doi: 10.3748/wjg.v12.i19.3073
119. Hofny ER, Ali ME, Taha EA, Nafeh HM, Sayed DS, Abdel-Azeem HG, et al. Semen and Hormonal Parameters in Men With Chronic Hepatitis C Infection. Fertil Steril (2011) 95:2557–9. doi: 10.1016/j.fertnstert.2011.05.014
120. Guéchot J, Chazouillères O, Loria A, Hannoun L, Balladur P, Parc R, et al. Effect of Liver Transplantation on Sex-Hormone Disorders in Male Patients With Alcohol-Induced or Post-Viral Hepatitis Advanced Liver Disease. J Hepatol (1994) 20:426–30. doi: 10.1016/S0168-8278(94)80020-0
121. Gariani K, Toso C, Philippe J, Orci LA. Effects of Liver Transplantation on Endocrine Function: A Systematic Review. Liver Int (2016) 36:1401–11. doi: 10.1111/liv.13158
122. Guo Y, Zhao M, Bo T, Ma S, Yuan Z, Chen W, et al. Blocking FSH Inhibits Hepatic Cholesterol Biosynthesis and Reduces Serum Cholesterol. Cell Res (2019) 29:151–66. doi: 10.1038/s41422-018-0123-6
123. Simoni M, Huhtaniemi I, Santi D, Casarini L. Editorial: Follicle-Stimulating Hormone: Fertility and Beyond. Front Endocrinol (Lausanne) (2019) 10:610. doi: 10.3389/fendo.2019.00610
124. Song Y, Wang ES, Xing LL, Shi S, Qu F, Zhang D, et al. Follicle-Stimulating Hormone Induces Postmenopausal Dyslipidemia Through Inhibiting Hepatic Cholesterol Metabolism. J Clin Endocrinol Metab (2016) 101:254–63. doi: 10.1210/jc.2015-2724
125. Tian GX, Sun Y, Pang CJ, Tan AH, Gao Y, Zhang HY, et al. Oestradiol is a Protective Factor for non-Alcoholic Fatty Liver Disease in Healthy Men. Obes Rev (2012) 13:381–7. doi: 10.1111/j.1467-789X.2011.00978.x
126. Zhu Y, Xu J, Zhang X, Ke Y, Fu G, Guo Q. A Low Follicle-Stimulating Hormone Level is a Protective Factor for non-Alcoholic Fatty Liver Disease in Older Men Aged Over 80. BMC Geriatr (2021) 21:544. doi: 10.1186/s12877-021-02490-6
127. Kulaksizoglu M, Ipekci SH, Kebapcilar L, Kebapcilar AG, Korkmaz H, Akyurek F, et al. Risk Factors for Diabetes Mellitus in Women With Primary Ovarian Insufficiency. Biol Trace Elem Res (2013) 154:313–20. doi: 10.1007/s12011-013-9738-0
128. Qi X, Guo Y, Song Y, Yu C, Zhao L, Fang L, et al. Follicle-Stimulating Hormone Enhances Hepatic Gluconeogenesis by GRK2-Mediated AMPK Hyperphosphorylation at Ser485 in Mice. Diabetologia (2018) 61:1180–92. doi: 10.1007/s00125-018-4562-x
129. Sarkar M, Lai JC, Sawinski D, Zeigler TE, Cedars M, Forde KA. Sex Hormone Levels by Presence and Severity of Cirrhosis in Women With Chronic Hepatitis C Virus Infection. J Viral Hepat (2019) 26:258–62. doi: 10.1111/jvh.13027
130. Shehata R, Abdel Hafez K, Fawzi E, Kamal E, Hassany S, Zakaria M. Impact of Hepatitis B on Male Reproductive Hormones. J Curr Med Res Pract (2016) 1:28–30. doi: 10.4103/2357-0121.192542
131. Alqrinawi SH, Akbar N, AlFaddag H, Akbar S, Akbar L, Butt SA, et al. Menotrophin Induced Autoimmune Hepatitis. Case Rep Gastrointest Med (2019) 2019:7343805. doi: 10.1155/2019/7343805
132. Freeman ME, Kanyicska B, Lerant A, Nagy G. Prolactin: Structure, Function, and Regulation of Secretion. Physiol Rev (2000) 80:1523–631. doi: 10.1152/physrev.2000.80.4.1523
133. Sairenji TJ, Ikezawa J, Kaneko R, Masuda S, Uchida K, Takanashi Y, et al. Maternal Prolactin During Late Pregnancy is Important in Generating Nurturing Behavior in the Offspring. Proc Natl Acad Sci U.S.A. (2017) 114:13042–7. doi: 10.1073/pnas.1621196114
134. Kok P, Roelfsema F, Frölich M, Meinders AE, Pijl H. Prolactin Release is Enhanced in Proportion to Excess Visceral Fat in Obese Women. J Clin Endocrinol Metab (2004) 89:4445–9. doi: 10.1210/jc.2003-032184
135. Kok P, Roelfsema F, Langendonk JG, de Wit CC, Frölich M, Burggraaf J, et al. Increased Circadian Prolactin Release is Blunted After Body Weight Loss in Obese Premenopausal Women. Am J Physiol Endocrinol Metab (2006) 290:E218–224. doi: 10.1152/ajpendo.00156.2005
136. Ernst B, Thurnheer M, Schultes B. Basal Serum Prolactin Levels in Obesity–Unrelated to Parameters of the Metabolic Syndrome and Unchanged After Massive Weight Loss. Obes Surg (2009) 19:1159–62. doi: 10.1007/s11695-009-9856-0
137. Zhang P, Ge Z, Wang H, Feng W, Sun X, Chu X, et al. Prolactin Improves Hepatic Steatosis via CD36 Pathway. J Hepatol (2018) 68:1247–55. doi: 10.1016/j.jhep.2018.01.035
138. Zhang P, Feng W, Chu X, Sun X, Zhu D, Bi Y. A Newly Noninvasive Model for Prediction of non-Alcoholic Fatty Liver Disease: Utility of Serum Prolactin Levels. BMC Gastroenterol (2019) 19:202. doi: 10.1186/s12876-019-1120-z
139. Ida Y, Tsujimaru S, Nakamaura K, Shirao I, Mukasa H, Egami H, et al. Effects of Acute and Repeated Alcohol Ingestion on Hypothalamic-Pituitary-Gonadal and Hypothalamic-Pituitary-Adrenal Functioning in Normal Males. Drug Alcohol Depend (1992) 31:57–64. doi: 10.1016/0376-8716(92)90009-2
140. De A, Boyadjieva N, Oomizu S, Sarkar DK. Ethanol Induces Hyperprolactinemia by Increasing Prolactin Release and Lactotrope Growth in Female Rats. Alcohol Clin Exp Res (2002) 26:1420–9. doi: 10.1111/j.1530-0277.2002.tb02687.x
141. Jha SK, Kannan S. Serum Prolactin in Patients With Liver Disease in Comparison With Healthy Adults: A Preliminary Cross-Sectional Study. Int J Appl Basic Med Res (2016) 6:8–10. doi: 10.4103/2229-516X.173984
142. Paternostro R, Heinisch BB, Reiberger T, Mandorfer M, Bardach C, Lampichler K, et al. Dysbalanced Sex Hormone Status is an Independent Predictor of Decompensation and Mortality in Patients With Liver Cirrhosis. Hepatol Res (2019) 49:201–11. doi: 10.1111/hepr.13253
143. Hartwell HJ, Petrosky KY, Fox JG, Horseman ND, Rogers AB. Prolactin Prevents Hepatocellular Carcinoma by Restricting Innate Immune Activation of C-Myc in Mice. Proc Natl Acad Sci U.S.A. (2014) 111:11455–60. doi: 10.1073/pnas.1404267111
144. Sato Y, Kaneko Y, Cho T, Goto K, Otsuka T, Yamamoto S, et al. Prolactin Upregulates Female-Predominant P450 Gene Expressions and Downregulates Male-Predominant Gene Expressions in Mouse Liver. Drug Metab Dispos (2017) 45:586–92. doi: 10.1124/dmd.116.074658
145. Bauer SR, Fortner RT, Gates MA, Eliassen AH, Hankinson SE, Tworoger SS. Analgesic Use in Relation to Sex Hormone and Prolactin Concentrations in Premenopausal Women. Cancer Causes Control (2013) 24:1087–97. doi: 10.1007/s10552-013-0186-0
146. Moreno-Carranza B, Goya-Arce M, Vega C, Adán N, Triebel J, López-Barrera F, et al. Prolactin Promotes Normal Liver Growth, Survival, and Regeneration in Rodents: Effects on Hepatic IL-6, Suppressor of Cytokine Signaling-3, and Angiogenesis. Am J Physiol Regul Integr Comp Physiol (2013) 305:R720–726. doi: 10.1152/ajpregu.00282.2013
147. Matyas RA, Mumford SL, Schliep KC, Ahrens KA, Sjaarda LA, Perkins NJ, et al. Effects of Over-the-Counter Analgesic Use on Reproductive Hormones and Ovulation in Healthy, Premenopausal Women. Hum Reprod (2015) 30:1714–23. doi: 10.1093/humrep/dev099
148. Yoon EL, Yuk JS. Use of Ulipristal Acetate and Risk of Liver Disease: A Nationwide Cohort Study. J Clin Endocrinol Metab (2021) 106:1773–82. doi: 10.1210/clinem/dgab081
149. Serin A, Akarsu M, Akpinar H, Simsek I. Changes of Some Hormones Levels in Patients With Hepatitis B Virus-Related Chronic Liver Disease. Gastroenterol Res (2013) 6:134–8. doi: 10.4021/gr532w
150. Gordon GG, Southren AL, Lieber CS. The Effects of Alcoholic Liver Disease and Alcohol Ingestion on Sex Hormone Levels. Alcohol Clin Exp Res (1978) 2:259–63. doi: 10.1111/j.1530-0277.1978.tb05809.x
151. Milenković L, D’Angelo G, Kelly PA, Weiner RI. Inhibition of Gonadotropin Hormone-Releasing Hormone Release by Prolactin From GT1 Neuronal Cell Lines Through Prolactin Receptors. Proc Natl Acad Sci U.S.A. (1994) 91:1244–7. doi: 10.1073/pnas.91.4.1244
152. Zhang Y, Xu J, Zhao R, Liu J, Wu J. Inhibition Effects on Liver Tumors of BALB/c Mice Bearing H22 Cells by Immunization With a Recombinant Immunogen of GnRH Linked to Heat Shock Protein 65. Vaccine (2007) 25:6911–21. doi: 10.1016/j.vaccine.2007.07.034
153. El Mahdy Korah T, Abd Elfatah Badr E, Mohamed Emara M, Ahmed Samy Kohla M, Gamal Saad Michael G. Relation Between Sex Hormones and Hepatocellular Carcinoma. Andrologia (2016) 48:948–55. doi: 10.1111/and.12536
154. Zheng D, Liwinski T, Elinav E. Interaction Between Microbiota and Immunity in Health and Disease. Cell Res (2020) 30:492–506. doi: 10.1038/s41422-020-0332-7
155. Bolnick DI, Snowberg LK, Hirsch PE, Lauber CL, Org E, Parks B, et al. Individual Diet has Sex-Dependent Effects on Vertebrate Gut Microbiota. Nat Commun (2014) 5:4500. doi: 10.1038/ncomms5500
156. Ma ZS, Li W. How and Why Men and Women Differ in Their Microbiomes: Medical Ecology and Network Analyses of the Microgenderome. Adv Sci (Weinh) (2019) 6:1902054. doi: 10.1002/advs.201902054
157. Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, et al. Gender Bias in Autoimmunity is Influenced by Microbiota. Immunity (2013) 39:400–12. doi: 10.1016/j.immuni.2013.08.013
158. Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, et al. Population-Level Analysis of Gut Microbiome Variation. Science (2016) 352:560–4. doi: 10.1126/science.aad3503
159. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human Gut Microbiome Viewed Across Age and Geography. Nature (2012) 486:222–7. doi: 10.1038/nature11053
160. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex Differences in the Gut Microbiome Drive Hormone-Dependent Regulation of Autoimmunity. Science (2013) 339:1084–8. doi: 10.1126/science.1233521
161. Han Q, Wang J, Li W, Chen ZJ, Du Y. Androgen-Induced Gut Dysbiosis Disrupts Glucolipid Metabolism and Endocrinal Functions in Polycystic Ovary Syndrome. Microbiome (2021) 9:101. doi: 10.1186/s40168-021-01046-5
162. Gao A, Su J, Liu R, Zhao S, Li W, Xu X, et al. Sexual Dimorphism in Glucose Metabolism is Shaped by Androgen-Driven Gut Microbiome. Nat Commun (2021) 12:7080. doi: 10.1038/s41467-021-27187-7
163. Kim S, Lee JY, Shin SG, Kim JK, Silwal P, Kim YJ, et al. ESRRA (Estrogen Related Receptor Alpha) is a Critical Regulator of Intestinal Homeostasis Through Activation of Autophagic Flux via Gut Microbiota. Autophagy (2021) 17:2856–75. doi: 10.1080/15548627.2020.1847460
164. Baker JM, Al-Nakkash L, Herbst-Kralovetz MM. Estrogen-Gut Microbiome Axis: Physiological and Clinical Implications. Maturitas (2017) 103:45–53. doi: 10.1016/j.maturitas.2017.06.025
165. Nuriel-Ohayon M, Neuman H, Ziv O, Belogolovski A, Barsheshet Y, Bloch N, et al. Progesterone Increases Bifidobacterium Relative Abundance During Late Pregnancy. Cell Rep (2019) 27:730–736.e733. doi: 10.1016/j.celrep.2019.03.075
166. Ren L, Song Q, Liu Y, Zhang L, Hao Z, Feng W. Probiotic Lactobacillus Rhamnosus GG Prevents Progesterone Metabolite Epiallaopregnanolone Sulfate-Induced Hepatic Bile Acid Accumulation and Liver Injury. Biochem Biophys Res Commun (2019) 520:67–72. doi: 10.1016/j.bbrc.2019.09.103
167. Ohlsson B. Gonadotropin-Releasing Hormone and Its Role in the Enteric Nervous System. Front Endocrinol (Lausanne) (2017) 8:110. doi: 10.3389/fendo.2017.00110
168. Sand E, Linninge C, Lozinska L, Egecioglu E, Roth B, Molin G, et al. Buserelin Treatment to Rats Causes Enteric Neurodegeneration With Moderate Effects on CRF-Immunoreactive Neurons and Enterobacteriaceae in Colon, and in Acetylcholine-Mediated Permeability in Ileum. BMC Res Notes (2015) 8:824. doi: 10.1186/s13104-015-1800-x
169. Im E, Riegler FM, Pothoulakis C, Rhee SH. Elevated Lipopolysaccharide in the Colon Evokes Intestinal Inflammation, Aggravated in Immune Modulator-Impaired Mice. Am J Physiol Gastrointest Liver Physiol (2012) 303:G490–497. doi: 10.1152/ajpgi.00120.2012
170. Banks WA, Robinson SM. Minimal Penetration of Lipopolysaccharide Across the Murine Blood-Brain Barrier. Brain Behav Immun (2010) 24:102–9. doi: 10.1016/j.bbi.2009.09.001
171. Zhang J, Sun Z, Jiang S, Bai X, Ma C, Peng Q, et al. Probiotic Bifidobacterium Lactis V9 Regulates the Secretion of Sex Hormones in Polycystic Ovary Syndrome Patients Through the Gut-Brain Axis. mSystems (2019) 4(2):e00017–19. doi: 10.1128/mSystems.00017-19
172. Wang T, Sha L, Li Y, Zhu L, Wang Z, Li K, et al. Dietary α-Linolenic Acid-Rich Flaxseed Oil Exerts Beneficial Effects on Polycystic Ovary Syndrome Through Sex Steroid Hormones-Microbiota-Inflammation Axis in Rats. Front Endocrinol (Lausanne) (2020) 11:284. doi: 10.3389/fendo.2020.00284
173. Winter S, Diamond M, Green J, Karasic D, Reed T, Whittle S, et al. Transgender People: Health at the Margins of Society. Lancet (2016) 388:390–400. doi: 10.1016/S0140-6736(16)00683-8
174. White Hughto JM, Reisner SL, Pachankis JE. Transgender Stigma and Health: A Critical Review of Stigma Determinants, Mechanisms, and Interventions. Soc Sci Med (2015) 147:222–31. doi: 10.1016/j.socscimed.2015.11.010
175. Hembree WC, Cohen-Kettenis PT, Gooren L, Hannema SE, Meyer WJ, Murad MH, et al. Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2017) 102:3869–903. doi: 10.1210/jc.2017-01658
176. Schagen SE, Cohen-Kettenis PT, Delemarre-van de Waal HA, Hannema SE. Efficacy and Safety of Gonadotropin-Releasing Hormone Agonist Treatment to Suppress Puberty in Gender Dysphoric Adolescents. J Sex Med (2016) 13:1125–32. doi: 10.1016/j.jsxm.2016.05.004
177. Hannema SE, Schagen SEE, Cohen-Kettenis PT, Delemarre-van de Waal HA. Efficacy and Safety of Pubertal Induction Using 17β-Estradiol in Transgirls. J Clin Endocrinol Metab (2017) 102:2356–63. doi: 10.1210/jc.2017-00373
178. Meyer G, Mayer M, Mondorf A, Flügel AK, Herrmann E, Bojunga J. Safety and Rapid Efficacy of Guideline-Based Gender-Affirming Hormone Therapy: An Analysis of 388 Individuals Diagnosed With Gender Dysphoria. Eur J Endocrinol (2020) 182:149–56. doi: 10.1530/EJE-19-0463
179. Hashemi L, Zhang Q, Getahun D, Jasuja GK, McCracken C, Pisegna J, et al. Longitudinal Changes in Liver Enzyme Levels Among Transgender People Receiving Gender Affirming Hormone Therapy. J Sex Med (2021) 18:1662–75. doi: 10.1016/j.jsxm.2021.06.011
180. Hana T, Butler K, Young LT, Zamora G, Lam JSH. Transgender Health in Medical Education. Bull World Health Organ (2021) 99:296–303. doi: 10.2471/BLT.19.249086
181. Loria P, Carulli L, Bertolotti M, Lonardo A. Endocrine and Liver Interaction: The Role of Endocrine Pathways in NASH. Nat Rev Gastroenterol Hepatol (2009) 6:236–47. doi: 10.1038/nrgastro.2009.33
182. Burra P. Liver Abnormalities and Endocrine Diseases. Best Pract Res Clin Gastroenterol (2013) 27:553–63. doi: 10.1016/j.bpg.2013.06.014
183. Dourakis SP, Tolis G. Sex Hormonal Preparations and the Liver. Eur J Contracept Reprod Health Care (1998) 3:7–16. doi: 10.3109/13625189809167479
184. Collier JD, Ninkovic M, Compston JE. Guidelines on the Management of Osteoporosis Associated With Chronic Liver Disease. Gut (2002) 50 Suppl 1:i1–9. doi: 10.1136/gut.50.suppl_1.i1
185. Boone RH, Cheung AM, Girlan LM, Heathcote EJ. Osteoporosis in Primary Biliary Cirrhosis: A Randomized Trial of the Efficacy and Feasibility of Estrogen/Progestin. Dig Dis Sci (2006) 51:1103–12. doi: 10.1007/s10620-006-8015-x
186. Shimizu I, Kohno N, Tamaki K, Shono M, Huang HW, He JH, et al. Female Hepatology: Favorable Role of Estrogen in Chronic Liver Disease With Hepatitis B Virus Infection. World J Gastroenterol (2007) 13:4295–305. doi: 10.3748/wjg.v13.i32.4295
187. Villareal DT, Binder EF, Williams DB, Schechtman KB, Yarasheski KE, Kohrt WM. Bone Mineral Density Response to Estrogen Replacement in Frail Elderly Women: A Randomized Controlled Trial. Jama (2001) 286:815–20. doi: 10.1001/jama.286.7.815
188. Lukert B, Satram-Hoang S, Wade S, Anthony M, Gao G, Downs R. Physician Differences in Managing Postmenopausal Osteoporosis: Results From the POSSIBLE US™ Treatment Registry Study. Drugs Aging (2011) 28:713–27. doi: 10.2165/11595190-000000000-00000
189. Testosterone Treatment of Men With Alcoholic Cirrhosis: A Double-Blind Study. The Copenhagen Study Group for Liver Diseases. Hepatology (1986) 6:807–13. doi: 10.1002/hep.1840060502
190. Wells R. Prednisolone and Testosterone Propionate in Cirrhosis of the Liver. A Controlled Trial Lancet (1960) 2:1416–9. doi: 10.1016/s0140-6736(60)92566-6
191. Besse-Patin A, Léveillé M, Oropeza D, Nguyen BN, Prat A, Estall JL. Estrogen Signals Through Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α to Reduce Oxidative Damage Associated With Diet-Induced Fatty Liver Disease. Gastroenterology (2017) 152:243–56. doi: 10.1053/j.gastro.2016.09.017
192. Zhang B, Wu ZY. Estrogen Derivatives: Novel Therapeutic Agents for Liver Cirrhosis and Portal Hypertension. Eur J Gastroenterol Hepatol (2013) 25:263–70. doi: 10.1097/MEG.0b013e32835ab5dc
193. Shao S, Yao Z, Lu J, Song Y, He Z, Yu C, et al. Ablation of Prolactin Receptor Increases Hepatic Triglyceride Accumulation. Biochem Biophys Res Commun (2018) 498:693–9. doi: 10.1016/j.bbrc.2018.03.048
194. Bassil N, Alkaade S, Morley JE. The Benefits and Risks of Testosterone Replacement Therapy: A Review. Ther Clin Risk Manag (2009) 5:427–48. doi: 10.2147/tcrm.s3025
195. Andrisse S, Feng M, Wang Z, Awe O, Yu L, Zhang H, et al. Androgen-Induced Insulin Resistance is Ameliorated by Deletion of Hepatic Androgen Receptor in Females. FASEB J (2021) 35:e21921. doi: 10.1096/fj.202100961R
196. Croston GE, Milan LB, Marschke KB, Reichman M, Briggs MR. Androgen Receptor-Mediated Antagonism of Estrogen-Dependent Low Density Lipoprotein Receptor Transcription in Cultured Hepatocytes. Endocrinology (1997) 138:3779–86. doi: 10.1210/endo.138.9.5404
197. Fagman JB, Wilhelmson AS, Motta BM, Pirazzi C, Alexanderson C, De Gendt K, et al. The Androgen Receptor Confers Protection Against Diet-Induced Atherosclerosis, Obesity, and Dyslipidemia in Female Mice. FASEB J (2015) 29:1540–50. doi: 10.1096/fj.14-259234
198. Rooney JP, Ryan N, Chorley BN, Hester SD, Kenyon EM, Schmid JE, et al. From the Cover: Genomic Effects of Androstenedione and Sex-Specific Liver Cancer Susceptibility in Mice. Toxicol Sci (2017) 160:15–29. doi: 10.1093/toxsci/kfx153
199. Anderson KE. Acute Hepatic Porphyrias: Current Diagnosis & Management. Mol Genet Metab (2019) 128:219–27. doi: 10.1016/j.ymgme.2019.07.002
200. Zhang B, Zhang CG, Ji LH, Zhao G, Wu ZY. Estrogen Receptor β Selective Agonist Ameliorates Liver Cirrhosis in Rats by Inhibiting the Activation and Proliferation of Hepatic Stellate Cells. J Gastroenterol Hepatol (2018) 33:747–55. doi: 10.1111/jgh.13976
201. Ma H, Liu J, Du Y, Zhang S, Cao W, Jia Z, et al. Estrogen-Related Receptor γ Agonist DY131 Ameliorates Lipopolysaccharide-Induced Acute Liver Injury. Front Pharmacol (2021) 12:626166. doi: 10.3389/fphar.2021.626166
202. Chow JD, Jones ME, Prelle K, Simpson ER, Boon WC. A Selective Estrogen Receptor α Agonist Ameliorates Hepatic Steatosis in the Male Aromatase Knockout Mouse. J Endocrinol (2011) 210:323–34. doi: 10.1530/JOE-10-0462
203. Zhang CG, Zhang B, Deng WS, Duan M, Chen W, Wu ZY. Role of Estrogen Receptor β Selective Agonist in Ameliorating Portal Hypertension in Rats With CCl4-Induced Liver Cirrhosis. World J Gastroenterol (2016) 22:4484–500. doi: 10.3748/wjg.v22.i18.4484
204. Kanazawa T. Effect of Ovariectomy and Ovarian Hormone Administration on Hepatic Autophagy in Female Rats. J Nutr Sci Vitaminol (Tokyo) (2019) 65:357–61. doi: 10.3177/jnsv.65.357
205. Klijn JG, Setyono-Han B, Foekens JA. Progesterone Antagonists and Progesterone Receptor Modulators in the Treatment of Breast Cancer. Steroids (2000) 65:825–30. doi: 10.1016/S0039-128X(00)00195-1
206. Hashimoto T, Igarashi J, Hasan AU, Ohmori K, Kohno M, Nagai Y, et al. Mifepristone Promotes Adiponectin Production and Improves Insulin Sensitivity in a Mouse Model of Diet-Induced-Obesity. PloS One (2013) 8:e79724. doi: 10.1371/journal.pone.0079724
207. Pal’chikova NA, Selyatitskaya VG, Kuz’minova OI, Pasechnaya KV. Effects Mifepristone on Aminotransferase Activities in the Liver in Rats With Streptozotocin-Induced Diabetes Mellitus. Bull Exp Biol Med (2018) 165:474–7. doi: 10.1007/s10517-018-4197-4
208. Leuprolide. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD: National Institute of Diabetes and Digestive and Kidney Diseases (2012).
209. Aapkes SE, Bernts LHP, van den Berg AP, van den Berg M, Blokzijl H, Cantineau AEP, et al. Protocol for a Randomized Controlled Multicenter Trial Assessing the Efficacy of Leuprorelin for Severe Polycystic Liver Disease: The AGAINST-PLD Study. BMC Gastroenterol (2022) 22:82. doi: 10.1186/s12876-022-02142-y
210. Cirkel U, Schweppe KW, Ochs H, Hanker JP, Schneider HP. LH-RH Agonist (Buserelin): Treatment of Endometriosis. Clinical, Laparoscopic, Endocrine and Metabolic Evaluation. Arch Gynecol Obstet (1989) 246:139–51. doi: 10.1007/BF00934075
211. Yamauchi R, Takeyama Y, Takata K, Fukunaga A, Sakurai K, Tanaka T, et al. Hepatitis B Virus Reactivation After Receiving Cancer Chemotherapy Under Administration of Leuprorelin Acetate. Intern Med (2020) 59:1163–6. doi: 10.2169/internalmedicine.3805-19
212. Grimaldi C, Bleiberg H, Gay F, Messner M, Rougier P, Kok TC, et al. Evaluation of Antiandrogen Therapy in Unresectable Hepatocellular Carcinoma: Results of a European Organization for Research and Treatment of Cancer Multicentric Double-Blind Trial. J Clin Oncol (1998) 16:411–7. doi: 10.1200/JCO.1998.16.2.411
213. Onori P, Mancinelli R, Franchitto A, Carpino G, Renzi A, Brozzetti S, et al. Role of Follicle-Stimulating Hormone on Biliary Cyst Growth in Autosomal Dominant Polycystic Kidney Disease. Liver Int (2013) 33:914–25. doi: 10.1111/liv.12177
Keywords: sex hormone, chronic liver diseases, cirrhosis, mechanism, therapy
Citation: Xu L, Yuan Y, Che Z, Tan X, Wu B, Wang C, Xu C and Xiao J (2022) The Hepatoprotective and Hepatotoxic Roles of Sex and Sex-Related Hormones. Front. Immunol. 13:939631. doi: 10.3389/fimmu.2022.939631
Received: 09 May 2022; Accepted: 13 June 2022;
Published: 04 July 2022.
Edited by:
Jinhang Gao, Sichuan University, ChinaReviewed by:
Minyi Qian, Nanjing University of Chinese Medicine, ChinaLeandro Fernández-Pérez, University of Las Palmas de Gran Canaria, Spain
Weizhen Zhang, Peking University, China
Copyright © 2022 Xu, Yuan, Che, Tan, Wu, Wang, Xu and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chengfang Xu, bGluZGF4dTIwMTFAcXEuY29t; Jia Xiao, ZWR3aW5zaXVAY29ubmVjdC5oa3UuaGs=
†These authors have contributed equally to this work