 Tao Yu
Tao Yu Shao-kun Yu1†
Shao-kun Yu1† Ming Sun
Ming Sun- 1Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China
- 2Suzhou Cancer Center Core Laboratory, Suzhou Municipal Hospital, Gusu School, The Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou, China
Chimeric antigen receptor (CAR)-T cells have enormous potentials for clinical therapies. The CAR-T therapy has been approved for treating hematological malignancies. However, their application is limited in solid tumors owing to antigen loss and mutation, physical barriers, and an immunosuppressive tumor microenvironment. To overcome the challenges of CAR-T, increasing efforts are put into developing CAR-T to expand its applied ranges. Varied receptors are utilized for recognizing tumor-associated antigens and relieving immunosuppression. Emerging co-stimulatory signaling is employed for CAR-T activation. Furthermore, other immune cells such as NK cells and macrophages have manifested potential for delivering CAR. Hence, we collected and summarized the last advancements of CAR engineering from three aspects, namely, the ectodomains, endogenous domains, and immune cells, aiming to inspire the design of next-generation adoptive immunotherapy for treating solid tumors.
Introduction
Over the past decades, the breakthrough of gene engineering and editing technologies has led to the fast improvement of chimeric antigen receptor (CAR)-T therapy. CAR-T cells combine the antitumor abilities of T cells with the recognition abilities of single-chain variable fragments (scFvs) (1). The extracellular scFv can recognize tumors expressing specific tumor-associated antigens (TAAs) in a major histocompatibility complex (MHC)-independent manner. Subsequently, the intracellular domain CD3ζ activates CAR-T to secrete cytokines and eradicate cancer cells regardless of their genomic mutations (2). CAR-T cells targeting CD19 have been approved for treating refractory B-cell malignancies. The designs of CARs have experienced four generations. The first-generation (1G) CAR only carries scFvs and immunoreceptor tyrosine-based activation motifs (ITAMs) of CD3ζ (3). The second- and third-generation (2G/3G) CARs are innovated by adding co-stimulatory molecules from the B7 family and tumor necrosis factor receptor (TNFR) subfamily, such as CD28, DAP10, CD134 (OX40), and CD137 (4-1BB) (4). Co-stimulatory fragments enable T cells to secrete cytokines and differentiate upon repeated contact with antigens (5, 6). CAR-T therapy has been widely utilized for hematological tumors; however, the bulk of difficulties constrain the application of CAR-T in solid tumors. Clinical trials of CAR-T targeting different TAAs in solid tumors have been thoroughly listed elsewhere. Here, we review and summarize the advancements of CAR engineering, T cells, or other immune cells that express CARs, with a particular focus on sophisticated engineering strategies of CAR design to improve the next-generation adoptive immune cell therapy for treating solid tumors (Figure 1).
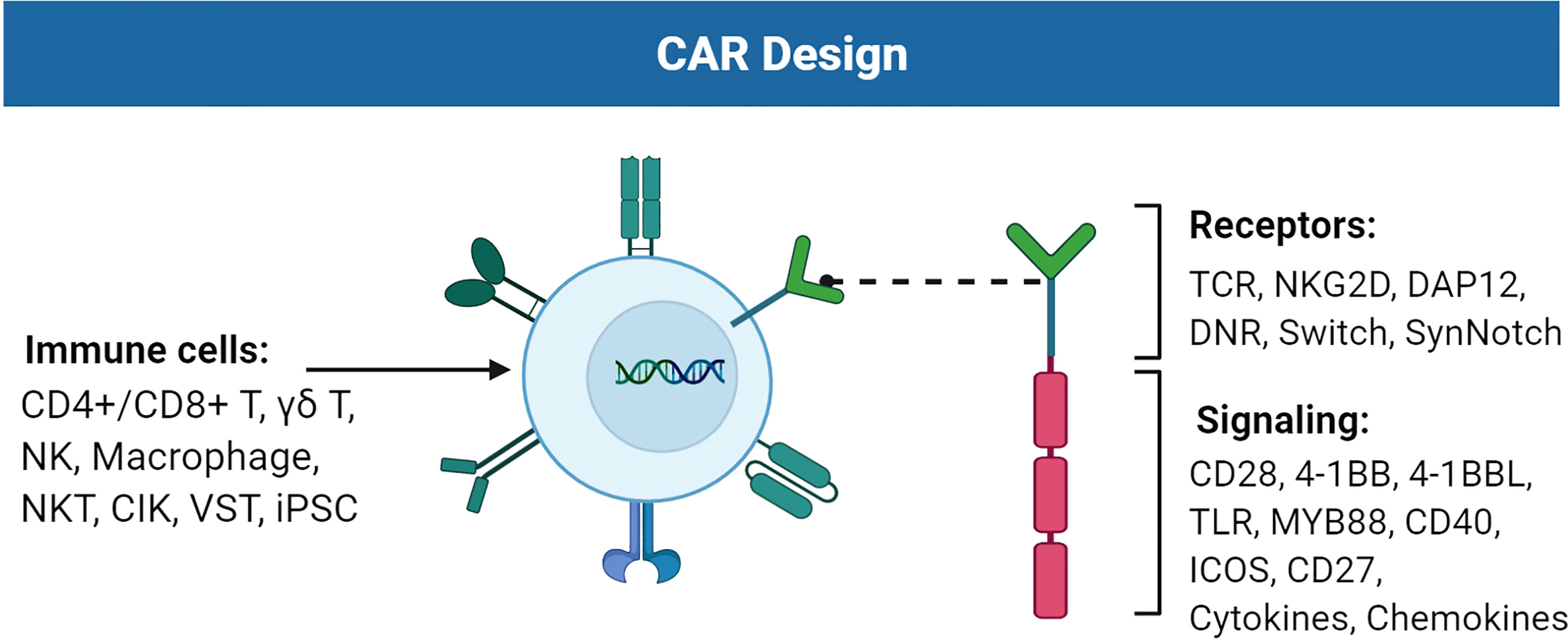
Figure 1 Improvements of the chimeric antigen receptor (CAR) design. CAR consists of receptors and signaling suitable for TAA recognition and cell activation. Immune cells such as T cells, NK cells, and macrophages can carry CAR to perform cytotoxic activities against tumor cells. Figure created using BioRender.com.
CAR-T challenges and improvements in solid tumors
The challenges of CAR-T therapy for solid tumors include trafficking, tumor recognition and killing, proliferation and persistence, immunosuppressive tumor microenvironment (TME), and control of CAR-T (7). One of the major difficulties is that heterogeneous solid tumors rarely express tumor-specific antigens, which prevents the recognition of CAR-T and induces on-target/off-tumor effects (8). Moreover, the conventional CAR structure has shortcomings. Sustained weak CD3ζ motif activation can raise CAR tonic signaling, leading to clustering of CAR molecules and causing T-cell exhaustion (9). Impaired T cells highly express inhibitory markers such as programmed cell death 1 (PD-1), TIM-3, and LAG-3 and are prone to apoptosis (10). Excessive activation of CAR-T following interleukin (IL)-6 overproduction may induce cytokine release syndrome (CRS), which is the most significant treatment-related toxicity (11). In addition, some patients lacking sufficient T cells require allo-CAR-T therapy (12). Endogenous T-cell receptors (TCRs) on allogeneic CAR-T may cause the graft-versus-host disease (GvHD), and immune rejection of T cells would reduce CAR-T efficacy. Although disruption of human leukocyte antigen (HLA) loci on CAR-T might avoid the rejection, it would reciprocally expose CAR-T to the rejection of natural killer (NK) cells.
To overcome the above problems, countermeasures based on optimal targets or novel CAR construct have been developed (13). Firstly, increasing research is conducted to identify recognizable and ideal TAAs for solid tumors, such as mesothelin, GD2, and MUC1 (14). Sequencing and proteomics have also been used for identifying targetable neoantigens (15). At the same time, the newly designed CAR can target multiple antigens with specific antibodies. For instance, camelids use a variable heavy chain domain (VHH), also known as nanobody (Nb), for combining antigens (16, 17). Nbs have the potential to serve as antigen recognition domains in CAR-T due to their small size, high affinity, optimal stability, and manufacturing feasibility (18). Nbs display a large convex paratope, which enables them to approach epitopes that are inaccessible for classical antibodies (19). Nbs are specifically suitable for bi-specific CARs containing two tandem antigen recognition domains (20). VH and VL in two independent scFvs may mispair and aggregate, while using Nbs can avoid domain swapping between variable and constant domains. Multiple antigens of solid tumors such as HER2, programmed cell death 1 ligand 1 (PD-L1), and EGFR could be chosen as targets of Nb-based CAR-T (21–24). Secondly, activation and co-stimulation domains can be optimized. For example, CD19 CAR-T with mutated first and third CD3ζ ITAMs was resistant to apoptosis and presented better antilymphoma efficacy than that with normal ITAMs (25, 26). In addition, improving co-stimulatory molecules seems promising. Merits and demerits of CD28 and 4-1BB have been deeply discussed (27). Substituting CD28 with 4-1BB can avert the CAR tonic signaling (28). A broad range of molecules, such as OX40, CD27, and inducible T-cell co-stimulator (ICOS), are also possible co-stimulators (29). Furthermore, distinctive immune cells beyond T cells delivering CAR might achieve unexpected success (30–32). Interestingly, recent studies have used other immune-activating receptors instead of CD3ζ for CAR design and optimization and have gotten surprising results. Hence, the structural characteristics of CAR-T significantly impact the functionality of adoptive immunotherapy in solid tumors (8, 13). Specifically, improvements in extracellular receptors, intracellular signaling, and carrier immune cells are summarized to enlighten investigators.
Developments of extracellular receptors
Many studies have reported the developments of scFvs (33). Here, we mainly focused on distinctive receptors in extracellular domains (Figures 2–4). They could recognize multiple targets, induce downstream activation, and even reverse inhibitory signaling into the activation pathway (Tables 1, 2).
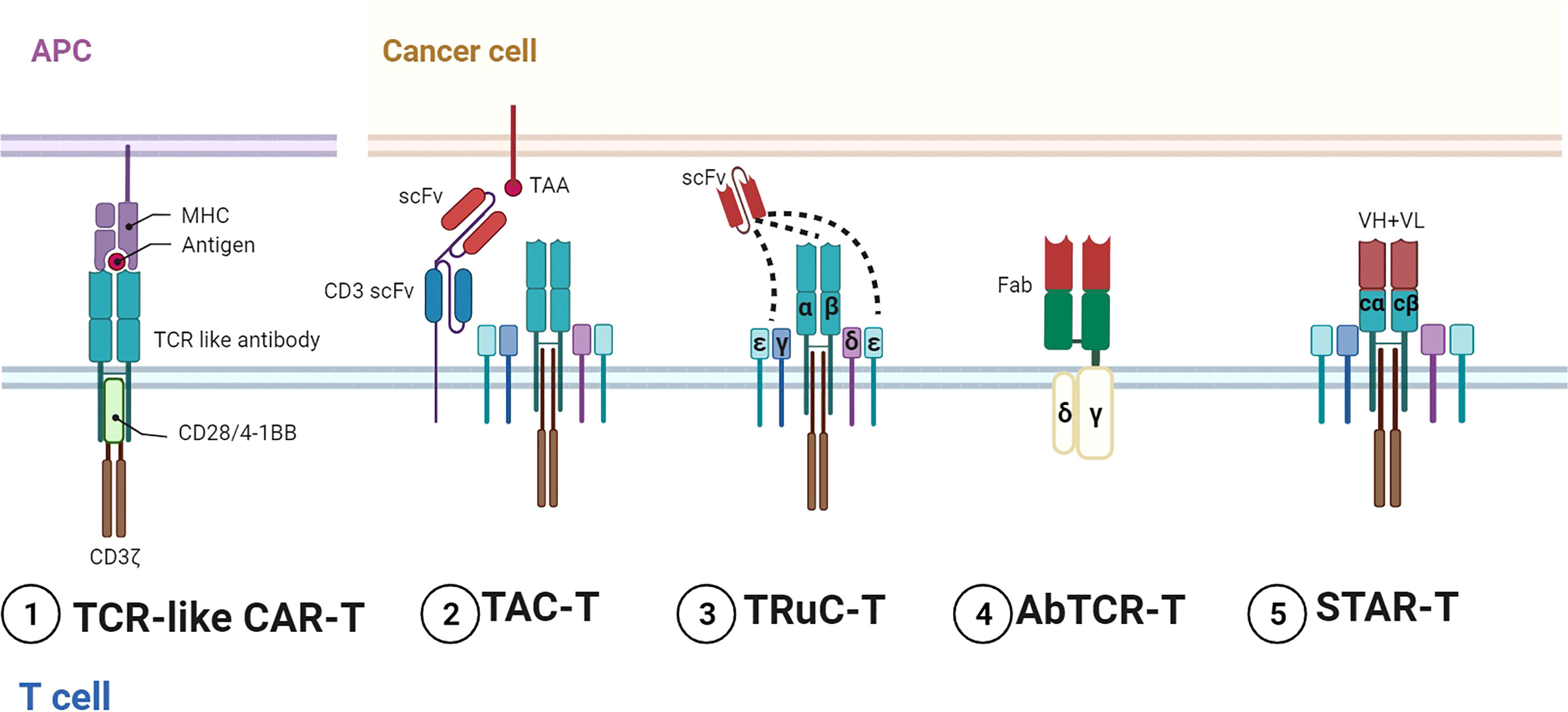
Figure 2 Representative designs of TCR–CARs. Diagrams of TCR–CARs with different configurations for tumor antigen recognition and cell activation. 1) TCR-like CAR-T utilized TCR-like antibodies for recognition and conventional stimulatory domains for activation. 2) TAC-T combined TCR with specific TAAs by two scFvs. 3) TRuC T bound scFvs to different parts of TCR and CD3. 4) AbTCR-T incorporated Fabs and γδ TCR. 5) STAR-T contained VH and VL with TCR-Cα/β. Figure created using BioRender.com.
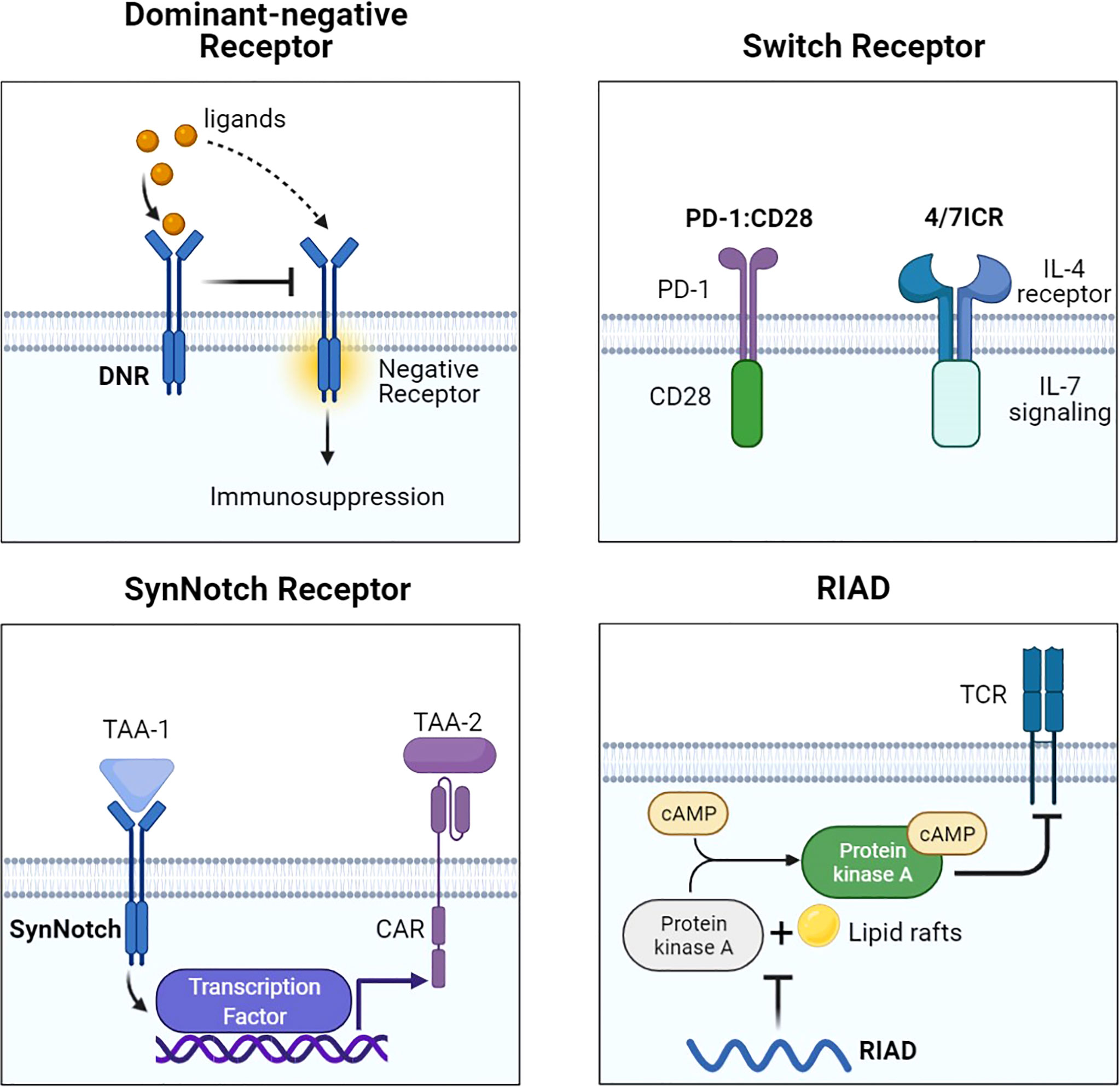
Figure 3 Representative designs of CARs with different receptors and modifications. Diagrams of CAR with different receptors and modifications. DNR avoided the suppressive signal by competing with negative receptors such as PD-1 and TGF-βR. Switch receptors converted the suppressive signal into activation signaling. SynNotch could increase antigen specificity by recognizing another TAA. RIAD inhibited the combination of PKA and cAMP to protect TCR. Figure created using BioRender.com.
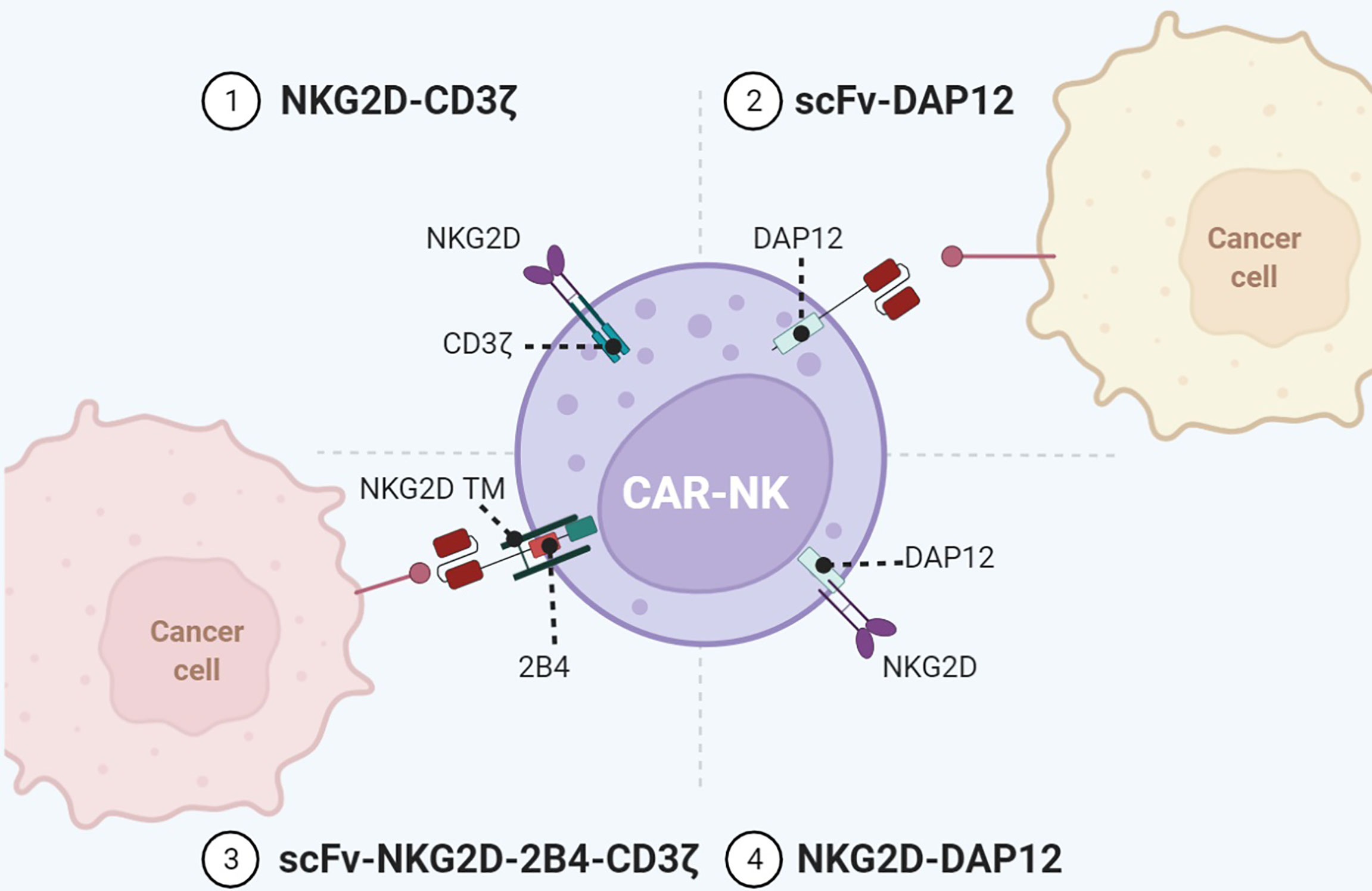
Figure 4 Representative designs of CARs with NK cell-associated repertoires. Diagrams of CAR-NK using NKG2D or DAP12 for immune activity. Co-expression of NK cell-associated receptors/adaptors such as NKG2D, KIR, TREM, DAP12, and 2B4 in the configuration of CARs enables a wider range of targets and enhanced antitumor immunity. Figure created using BioRender.com.
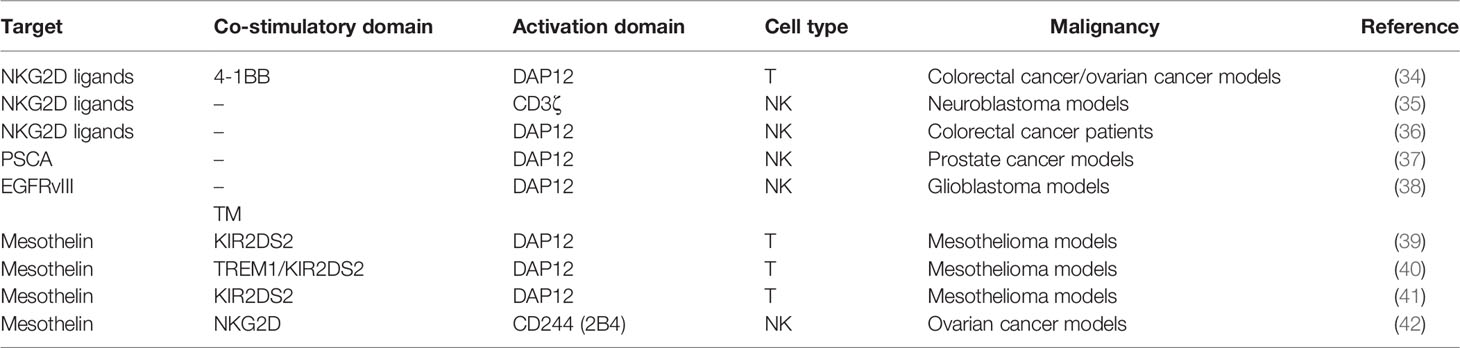
Table 1 CARs modified by NK cell receptors in solid tumors.
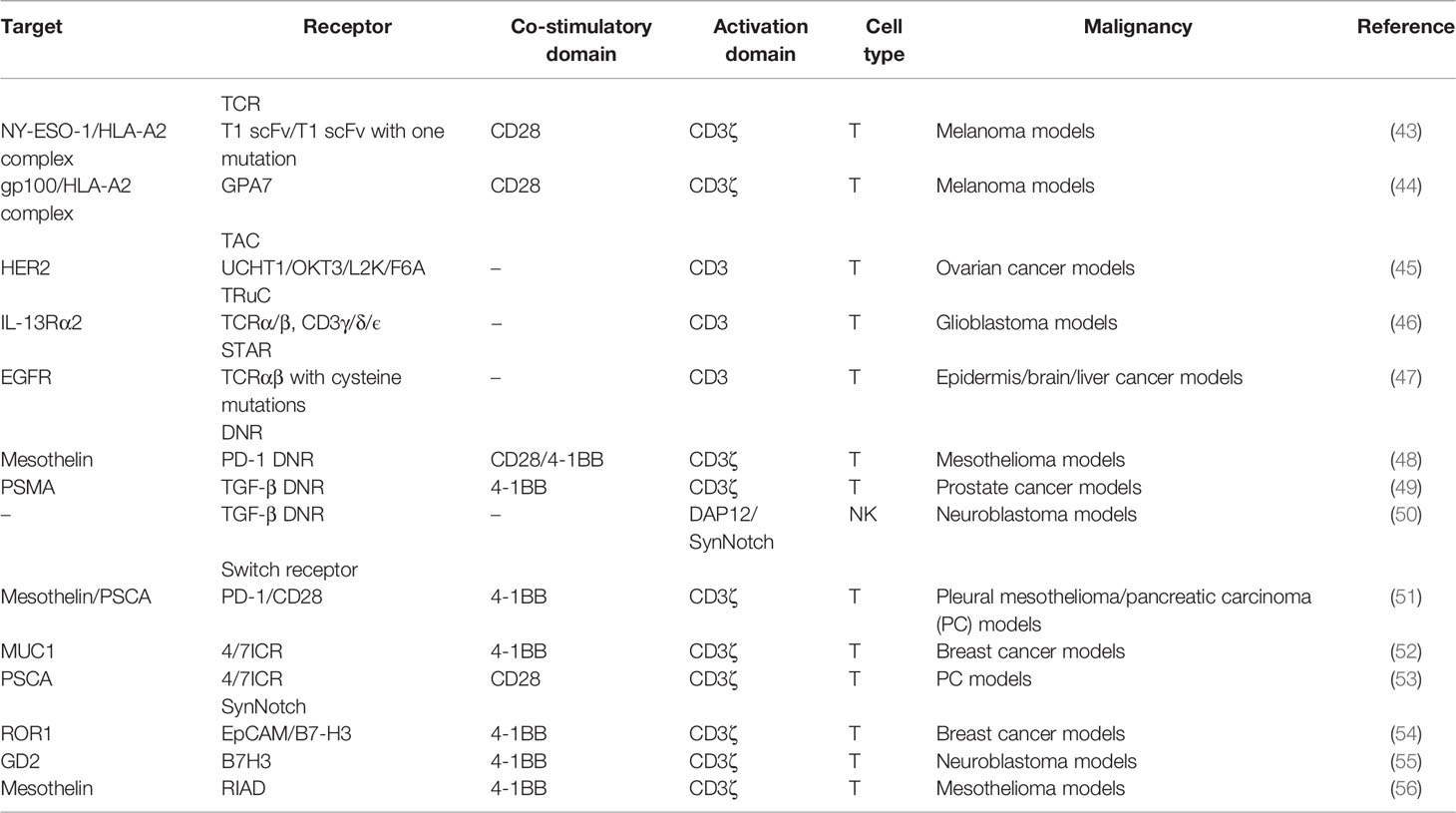
Table 2 CARs with different receptors and modifications in solid tumors.
T-Cell Receptor
Conventional TCR consists of α and β chains that combine with peptide-MHC ligands and cooperate with CD3 signaling, which has three dimers: ϵ/γ, ϵ/δ, and ζ/ζ (57). CD3 subunits have a total of 10 ITAMs that accept up to 20 tyrosine phosphates upon activation (58). On the one hand, TCR can be applied for antigen recognition. Compared with scFv mainly targeting membrane proteins, which account for about 30% of whole proteins (59), TCR could target almost all peptides via the MHC of antigen-presenting cells (APCs) (60). Transducing distinctive TCRs is a promising strategy for recognizing intracellular antigens. TCR–CAR composed of TCR α/β, CD28, and CD3ζ was designed and showed antitumor efficacy (61). Furthermore, antibodies with structural similarities to TCRs can also bind to HLA/peptide complexes. TCR-like CAR could guide T cells to eradicate xenograft melanoma (44). However, the excessive affinity of TCR may reversely reduce the specificity of CAR. Maus et al. developed a 2G CAR with the scFv of the high-affinity antibody (T1) and found that mutations of T1 scFv downregulated the avidity but upregulated the specificity and infiltration (43).
On the other hand, TCR has been exploited for CD3 signaling activation. Compared with CD3ζ containing three ITAMs, the TCR–CD3 complex can activate 10 ITAMs in total. Representative designs in this field include the T-cell antigen coupler (TAC), the TCR fusion constructs (TRuCs), the antibody-TCR (AbTCR), and the synthetic TCR antigen receptor (STAR) (Figure 2). TAC contained three domains, namely, an antigen-binding scFv, an anti-CD3 scFv, and a co-receptor domain (45). TAC could recruit the TCR–CD3 complex via the scFv targeting CD3. TAC-T cells manifested greater infiltration in solid tumors and less expansion in healthy tissues than 1G/2G CAR-T. The TAC platform without exogenous ITAMs could circumvent the tonic signaling. By fusing scFvs to extracellular N-termini of TCR subunits (TCRα/β, CD3γ/δ/ϵ), TRuCs were developed to activate CD3 in an MHC-independent manner (46). TRuC-T demonstrated either superior or equivalent tumor killing to CAR-T and released significantly less cytokines. AbTCR linked the Fab domain to γ and δ chains of TCR and avoided the mispairing with the endogenous αβTCR (62). AbTCR-T showed similar cytotoxicity with 2G CAR-T but less exhausted phenotype in CD19-expressing models. STAR is a double-chain TCR-based receptor with VH and VL, constant regions of TCR (TCR-Cα/β), and endogenous CD3 signaling machinery (47). In multiple tumor models, STAR-T exhibited similar persistence with 4-1BB-CAR-T and superior or equal antitumor effects compared with CD28-CAR-T.
NK Cell Receptor
In addition to TCR, activation receptors on NK cells have also been incorporated into CAR to activate T cells. Among them, the type II transmembrane-anchored C-type lectin-like protein NKG2D is a key activation receptor of NK cells. NKG2D is not only located on NK cells but also presented on CD8+ αβT cells, γδ T cells, and NKT cells (63). NKG2D recognizes eight stress-induced ligands within two families: MHC class I chain-related proteins (MICA and MICB) and UL16-binding proteins (ULBP1–6) (64). These NKG2D ligands are rarely present on the normal cell surface but are upregulated upon DNA damage, hypoxia, infection, and transformation of cells (40). Since NKG2D ligands are widely distributed in various malignancies, NKG2D-CARs are capable of targeting a broad range of tumors. NKG2D-based CARs comprise either the full-length NKG2D receptor (aa 1–216) or the NKG2D ectodomain (aa 83–216) and contain the cytoplasmic tail of CD3ζ. By recognizing antigens via NKG2D, these CAR-T cells eradicate tumors in preclinical animal models (34). In a neuroblastoma xenograft model, NKG2D/CD3ζ-NK cells not only eliminated intratumoral myeloid-derived suppressor cells (MDSCs) with excessive NKG2D ligands but also guided GD2-CAR-T cells to tumor sites (35). It is noteworthy that activated T cells themselves also expressed ligands of NKG2D. NKG2D-CD3ζ-T cells without physiologic inhibitory receptors might encounter fratricide or on-target/off-tumor effects. In contrast, NKG2D-CD3ζ-NK cells merely targeted autologous MDSCs and did not attack NKG2D ligand-expressing T cells.
NKG2D merely has a short cytoplasmic tail, hardly initiating intracellular signaling. Specialized adaptors are indispensable for transmitting activation signaling received by NKG2D, especially the DAP10 in human and DAP12 in mouse. DAP10 can contribute to DAP12-dependent PI3K activation (25). In addition to CD3ζ, DAP12 can be another choice for activation upon NKG2D recognizing ligands. Despite that the NKG2D/DAP12 axis is absent in human immune cells, the artificial NKG2D/DAP12 structure exhibits considerable immunocompetence (Figure 4). DAP12 is a short 12-kDa transmembrane (TM) protein of 113 aa expressed on the surface of a broad range of hematopoietic cells, harboring a single ITAM in the 48-aa cytoplasmic domain (65). The phosphorylation of ITAM activates the downstream cytoplasmic ZAP70 tyrosine kinases to eventually produce cytokine (66). DAP12 ITAM also contains an immunoreceptor tyrosine-based inhibitory motif (ITIM), which is not present in CD3ζ (67). This property makes DAP12 adjust its reaction in response to stimuli with differential strength to avoid CAR tonic signaling. Producing NKG2D/DAP12-CAR-NK by mRNA electroporation could treat patients with metastatic colorectal cancer (36). In two patients with malignant ascites, the number of tumor cells in ascites was significantly reduced after infusion of CAR-NK. The patient treated with ultrasound-guided percutaneous injection of CAR-NK in the liver acquired a complete metabolic response at the injection site. A phase 1 trial of NKG2D/DAP12-CAR-NK in metastatic colorectal cancer patients is ongoing (NCT05213195). Temme’s lab has developed two DAP12-CAR-NK cells with scFvs targeting PSCA or EGFRvIII (37, 38). They reported that co-stimulatory domains are unnecessary for DAP12-mediated NK cell activation.
Moreover, T cells can employ NKG2D/DAP12 as well. Chronic CD3ζ activation may induce tonic signaling and cause impaired expansion of CAR-T, while DAP12 enables non-impaired production of CAR-T because of the ITIM motif. Compared with NKG2D/CD3ζ-CAR-T, NKG2D/DAP12-CAR-T secreted less interferon (IFN)-γ, TNF-α, and IL-2 during tumor cell lysis and mitigated the risk of CRS (34). Considering that the ITIM motif can preclude tonic signaling (25), Epstein et al. designed CAR with DAP signaling domains to prevent CAR-T exhaustion (68). Excluding NKG2D, DAP12 can associate with other receptors to initiate activation. In the lymphoid lineage like NK cells, DAP12 binds to receptors such as NKG2C, NKp44, the short-tailed KIR3DS1, and KIR2DS1/2/5 (37). Killer immunoglobulin-like receptors (KIRs) are transmembrane glycoproteins detecting MHC class I molecules. KIR genes can be translated into inhibitory receptors (2DL and 3DL) or activating receptors (2DS, 3DS, and 2DL4) with two or three C2-type Ig-like extracellular domains. In the myeloid lineage, like macrophages and granulocytes, DAP12 is coupled with a triggering receptor expressed on myeloid cell members (TREM) or myeloid DAP12-associating lectin 1 (MDL1) (69, 70). Investigators have attempted to fuse scFv to KIR2DS2/TREM-1 TM and use DAP12 for activation (40). Compared with CD3ζ-CAR-T, KIR2DS2/TREM-1-CAR-T exhibited superior antitumor activity despite lower IL-2 production. Their enhanced antitumor activity was associated with better maintenance of CAR expression in TILs (39). Our team further designed KIR2DS2/DAP12-CAR-T cells with the presence of 4-1BB (41). DAP12 accompanied by 4-1BB made T cells express more central memory phenotype and less PD-1. Our phase I clinical trial found that CD19-KIRS2/DAP12-4-1BB CAR-T cells were safe and efficient in treating relapsed/refractory B-ALL patients.
DNR
To counteract the PD-1/PD-L1 axis, PD-1 dominant-negative receptor (DNR) saturating PD-L1 was designed to compete with endogenous PD-1. DNR only contained an extracellular receptor and a CD8 TM. CAR-T with PD-1 DNR demonstrated improved efficacy (48). In addition to PD-1, transforming growth factor β (TGF-β) secreted by solid tumors and received by the TGF-βRII/TGF-βRI heterodimer complex can induce an immunosuppressive milieu. The dominant-negative TGF-βRII, which lacks the intracellular domain, can be employed for CAR. TGF-β-insensitive CAR-T had a striking proliferative advantage over wild-type CAR-T in prostate cancer models (49). TGF-β DNR CAR-T showed a gene profile associated with cell cycle progression and cell division and a cytokine profile including the T helper (Th) 2 phenotype (IL-4, IL-5, and IL-13) and the Th1 phenotype (IL-2 and IFN-γ). TGF-β can phosphorylate Smads 2 and 3 to impair the expression of NK receptors. TGF-β DNR was also tailored with DAP12 or a synthetic Notch-like receptor to protect NK cells in neuroblastoma (50).
Switch receptor
External inhibitory receptors with cytoplasmic activation domains are called “switch receptors” (51). For example, a PD-1/CD28 switch receptor was incorporated into mesothelin or PSCA CAR-T and enhanced tumor eradication. The PD-1 switch receptor was more helpful for CAR-T cells than current PD-1 inhibitors. Among the six diffuse large B-cell lymphoma patients treated with CD19-PD-1/CD28-CAR-T, half achieved complete remissions (71). Some cytokines in the TME, such as IL-4, IL-10, and TGF-β, also induce suppressive signals. The MUC1-CAR with the cytokine switch receptor (4/7ICR), which consists of the IL-4 receptor extracellular domain and the IL-7 intracellular signaling domain, efficiently suppressed tumor growth in breast cancer models and did not show markers of exhaustion (52). In the IL-4-rich pancreatic tumor milieu, PSCA-4/7ICR-CAR-T also significantly repressed the tumor (53).
SynNotch receptor
The synthetic notch (SynNotch) system is a gating strategy to minimize the off-tumor toxicity of CAR-T. The SynNotch receptor comprises an extracellular antigen-recognition domain. Matching ligations lead to the release of intracellular transcription factors into the nucleus to activate the secondary CAR gene (72). Intermittent gate-dependent expression of the second CAR relieves tonic signaling and improves metabolic fitness. SynNotch receptors targeting B7-H3 or EpCAM were used to activate ROR1-CAR (54). Choosing GD2 as the gatekeeper for B7H3-CAR also showed improved metabolic plasticity, high specificity, and low toxicity in neuroblastoma (55).
RIAD
Another choice to overcome CAR-T limitations in solid tumors is to maintain TCR-induced T-cell proliferation and cytotoxic ability (73). Protein kinase A (PKA) on the immune synapse phosphorylates key molecules to inhibit the TCR signaling cascade. PKA is activated through a cyclic AMP (cAMP)-dependent manner. It is necessary for PKA to be tethered to lipid rafts in close proximity to the cAMP-generating enzyme adenylyl cyclase. One peptide, called “regulatory subunit I anchoring disruptor” (RIAD), could displace PKA from lipid rafts (74). Newick and colleagues designed RIAD-CAR to protect TCR from cAMP/PKA-mediated immunosuppression (56). Mesothelin-RIAD-CAR-T showed increased TCR signaling and infiltrated into the mesothelioma to secrete more cytokines.
Developments of intracellular signaling
Physiological T-cell activation requires three signals (75). Signal 1 (activation) is regulated by CD3ζ upon antigen recognition. Signal 2 (co-stimulation) originates from co-stimulatory molecules supporting CD3ζ. Signal 3 is secreted by cytokines, promoting T-cell proliferation, differentiation, and development. Transducing signals suitable for tumoricidal cytotoxicity could expand advantages for CAR-T in solid tumors (Table 3).
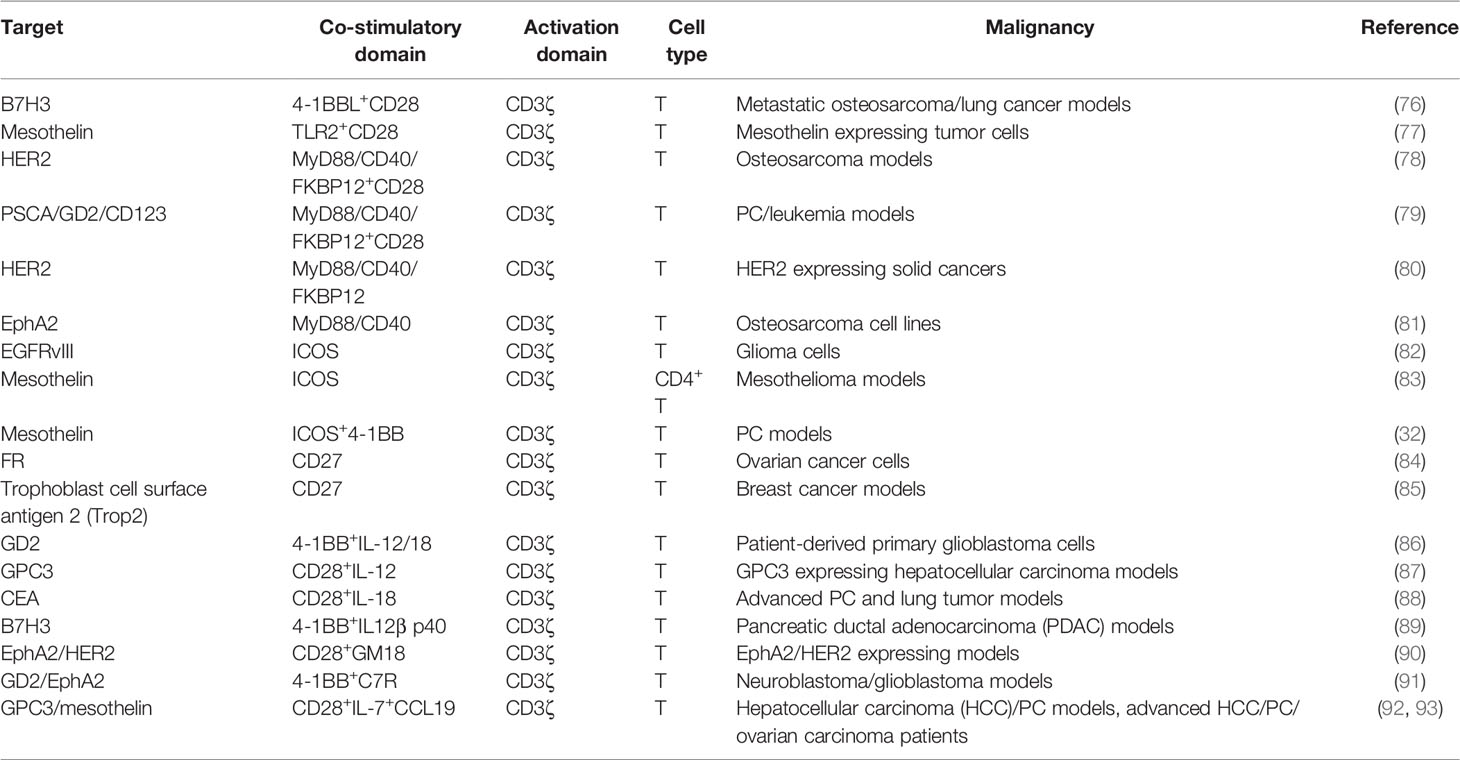
Table 3 CAR with different stimulation domains in solid tumors.
Modification of CD3ζ
The location and number of ITAMs are tightly associated with CAR-T functions. James and colleagues demonstrated that deleting a CD3ζ ITAM helped CD19-CAR-T to focus on CD19 highly expressing cells and ignore normal cells (94). Sadelain and colleagues found that a single functional ITAM was sufficient for antitumor efficacy and superior to the natural triple-ITAM-containing CD3ζ chain in a CD28-based CAR-T (26). The single ITAM configuration prolonged the lifetime of CAR-T and balanced the replicative capacity of long-lived memory cells. Furthermore, ITAM showed a stronger signal transduction ability when closer to TM. However, increasing the number of ITAMs in 4-1BB-based CAR-T was beneficial for recognizing low antigen density-expressing target cells (95). Thus, the function of each ITAM might be qualitatively different. Selecting the optimal ITAM(s) for different CAR signaling domains is a feasible strategy to promote solid tumor eradication.
Selection of Co-Stimulatory Domains
Co-stimulatory molecules can be divided into two groups. The first is basically expressed on resting antigen-naive T cells including CD27 and CD28. The other, including ICOS, 4-1BB, OX40, and GITR, is detectable after the formation of immune synapses (29). CD28 and/or TNFR family members (4-1BB and OX40) are usually chosen as co-stimulatory domains (96). Details of CD28 and 4-1BB have been reviewed elsewhere (97). Here, we presented other co-stimulators and their combinations (Figure 5).
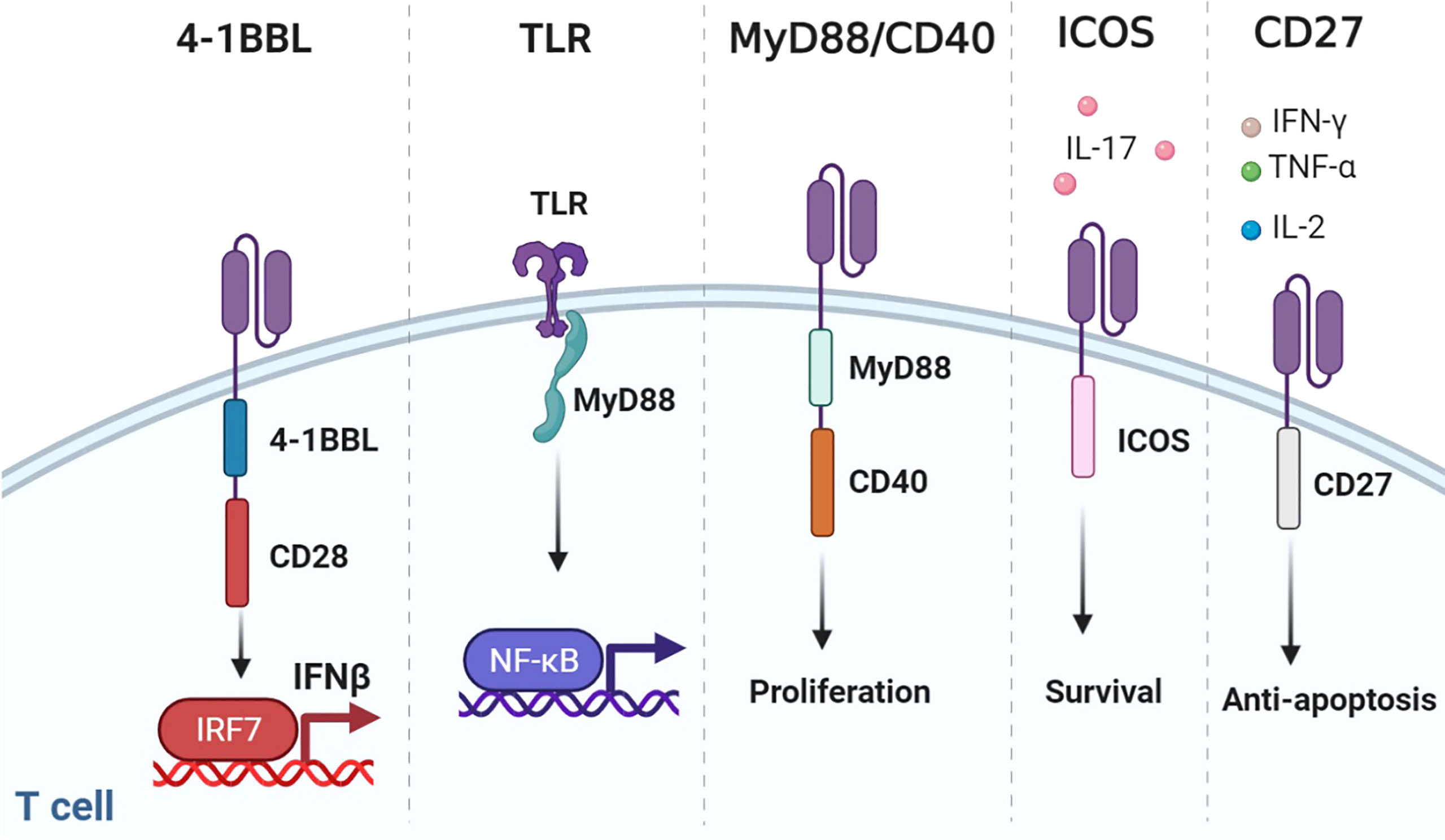
Figure 5 Representative designs of CARs with co-stimulatory domains. Diagrams of CARs with co-stimulatory domains. 1) Co-expressing 4-1BBL and CD28 could recruit the host immune response against the tumor through the IRF7/IFN-β pathway. 2) TLR could combine MyD88 to activate NF-κB. 3) MyD88/CD40 promoted T-cell proliferation. 4) ICOS increased the production of IL-17 and enhanced the survival of T cells. 5) CD27-based CAR-T cells secreted Th1 cytokines (IFN-γ, TNF-α, and IL-2) and were resistant to apoptosis. Figure created using BioRender.com.
4-BBL
Although CD28 and 4-1BB are the most common co-stimulatory molecules, changing their location or construct results in spatial and temporal differences. Considering that CD28 far away from the cell membrane would weaken the function of CAR, multiple attempts have been made to figure out the optimal design of 3G CARs (98, 99). Surprisingly, compared with 3G CAR incorporating 4-1BB, placing 4-1BB ligand (4-1BBL) on the surface endowed CAR-T with superior function in solid tumors (76). This may have resulted from the trimeric conformation of activated 4-1BB distinct from the dimeric CAR design. Co-expressing 4-1BBL and CD28 could recruit the host immune response against the tumor through the IRF7/IFN-β pathway (100). The safety and efficacy of CD28-CAR-T with surface 4-1BBL were evaluated in a clinical trial for adults with CD19-positive malignancies (NCT03085173).
TLR
Toll-like receptors (TLRs) are innate immune receptors on activated T cells for co-stimulation (101). TLR2 signaling can induce the secretion of T-bet protein, IFN-γ, perforin, and granzyme B through the mTOR pathway (102). TLR2 potentiated the antitumor efficacy of CAR-T without increased CRS (77). Genes upregulated by TLR2 co-stimulation were mainly associated with cell adhesion, synaptic transmission, and migration. TLR2 can form weak homodimers to combine MyD88 (103), which activates NF-κB and PI3K/AKT signaling and provides anti-apoptotic signals in T cells (104).
MyD88/CD40
The MyD88/IRF7 complex is critical for IFN production (105). Therefore, investigators exploited MyD88 as the co-stimulatory domain. An artificial design consisting of MyD88, CD40, and FKBP12 could be used for CAR-T (78, 79). CD40 is a cell-surface receptor for T-cell intrinsic activation, differentiation, memory formation, and exhaustion resistance (106). Researchers utilized the small molecule ligand rimiducid (Rim) to dimerize MyD88/CD40 by binding to FKBP12. Dropping Rim induced IL-2 secretion and promoted proliferation. On the contrary, withholding Rim effectively eliminated residual CAR-T. Moreover, CAR-T directly transduced with MyD88/CD40 expressed higher levels of MYB and FOXM1 at baseline and remained at a less differentiated state after stimulation than CD28- or 4-1BB-CAR-T (81).
ICOS
ICOS is a member of the CD28 family. ICOS activates PI3K/AKT signaling more potently than CD28. ICOS-based CAR-CD4+ T cells demonstrated a Th1/T17 polarization, producing more IL-17A and less IL-2, and enhanced the survival of CD28 or 4-1BB-based CAR-CD8+ T cells (32, 83). The combination of ICOS and 4-1BB as the 3G CAR showed synergistic effects on antitumor activity. Placing co-stimulatory domains proximal to the cell membrane could reduce CAR expression on the cell surface to avoid tonic signaling.
CD27
CD27 is a member of the tumor necrosis factor receptor superfamily, constitutively expressed on immune cells (84). CD27-based CAR-T cells secreted Th1 cytokines (IFN-γ, TNF-α, and IL-2) and exhibited stronger toxicity and more Bcl-XL, an anti-apoptotic protein of the Bcl-2 family (107). Increased IL-7Rα and decreased PD-1 were presented on CD27-based CAR-T compared with CD28-based CAR-T, which indicated that CD27 could prevent T-cell exhaustion (85).
Transduction of Signal of Cytokines and Chemokines
4G CAR-T is characterized by using signals derived from immunostimulatory cytokines, known as T cells redirected for universal cytokine-mediated killing (TRUCK) (6). Forced expression of cytokine genes, such as IL-12 and IL-15, can improve the persistence and antitumor effects of CAR-T but increase the risks of serious adverse events (108, 109). To control cytokine signaling only upon antigen engagement, different strategies were used to regulate the cytokine expression (Figure 6). Firstly, scientists transduced T cells with CAR and inducible cytokine expression through nuclear factor of activated T cells (NFAT)-responsive promoters (86, 87). IL-12 or IL-18 expression was ideally induced upon tumor antigen recognition. Armored inducible expression of cytokines could facilitate CAR-T function with fewer potential side effects. Inducible IL-18 could further accumulate FoxO1 transcription factor and alleviate T-bet to sustain a cytolytic phenotype. IL-18 CAR-T could recruit M1 macrophages and NKG2D+ NK cells and drive away immune-suppressive cells (88). Moreover, IL-23 is a STAT3-activating cytokine and consists of IL-23α p19 and IL-12β p40 subunits. Upon TCR stimulation, T cells upregulate the expression of IL-23 receptors and the IL-23α p19 excluding the p40 subunit. Therefore, CAR-T with p40 (p40-Td CAR-T) could couple the release and activity of IL-23 with T-cell activation (89). IL-23 produced by p40-Td cells worked mainly through an autocrine mechanism with limited effects on bystander cells. Additionally, GM-CSF, a cytokine invariably expressed after CAR-T activation, provides another choice for inducible cytokine expression (90). A chimeric cytokine receptor (GM18) was constructed by combining the extracellular domains of the GM-CSF receptor with the TM and signaling domains of the IL-18 receptor. GM18 created a functional loop to secrete IL-18 upon antigen exposure and activated the MyD88 signaling. On the other side, CAR-T with cytokine receptors was developed. Enlightened by the report that constitutively active IL-7 receptors without ligands are sufficient for STAT5 activation, Shum et al. fused a constitutively active IL-7 receptor variant with ectodomains derived from CD34, which blocked the external IL-7, to construct C7R (91). C7R co-expression in CAR-T exerted a broad anti-apoptotic influence by upregulating BCL2 and downregulating FAS and CASP8, consistent with the role of IL-7. C7R-CAR-T showed superior antitumor activities in multiple models. Moreover, IL-2 induces STAT5 activation through tyrosine residues within the IL-2 receptor β-chain (IL-2Rβ), while IL-21 preferentially activates STAT3 through its association motif YXXQ within the IL-21 receptor (110). Investigators have incorporated the truncated cytoplasmic domain of IL-2Rβ and a STAT3-binding YXXQ motif into CAR-T targeting CD19 (111). Compared with a CD28 or 4-1BB domain alone, this kind of CAR-T cells showed antigen-dependent JAK–STAT3/5 pathway activation and gene expression profiles analogous to be triggered by IL-21, which promoted their proliferation and prevented terminal differentiation.
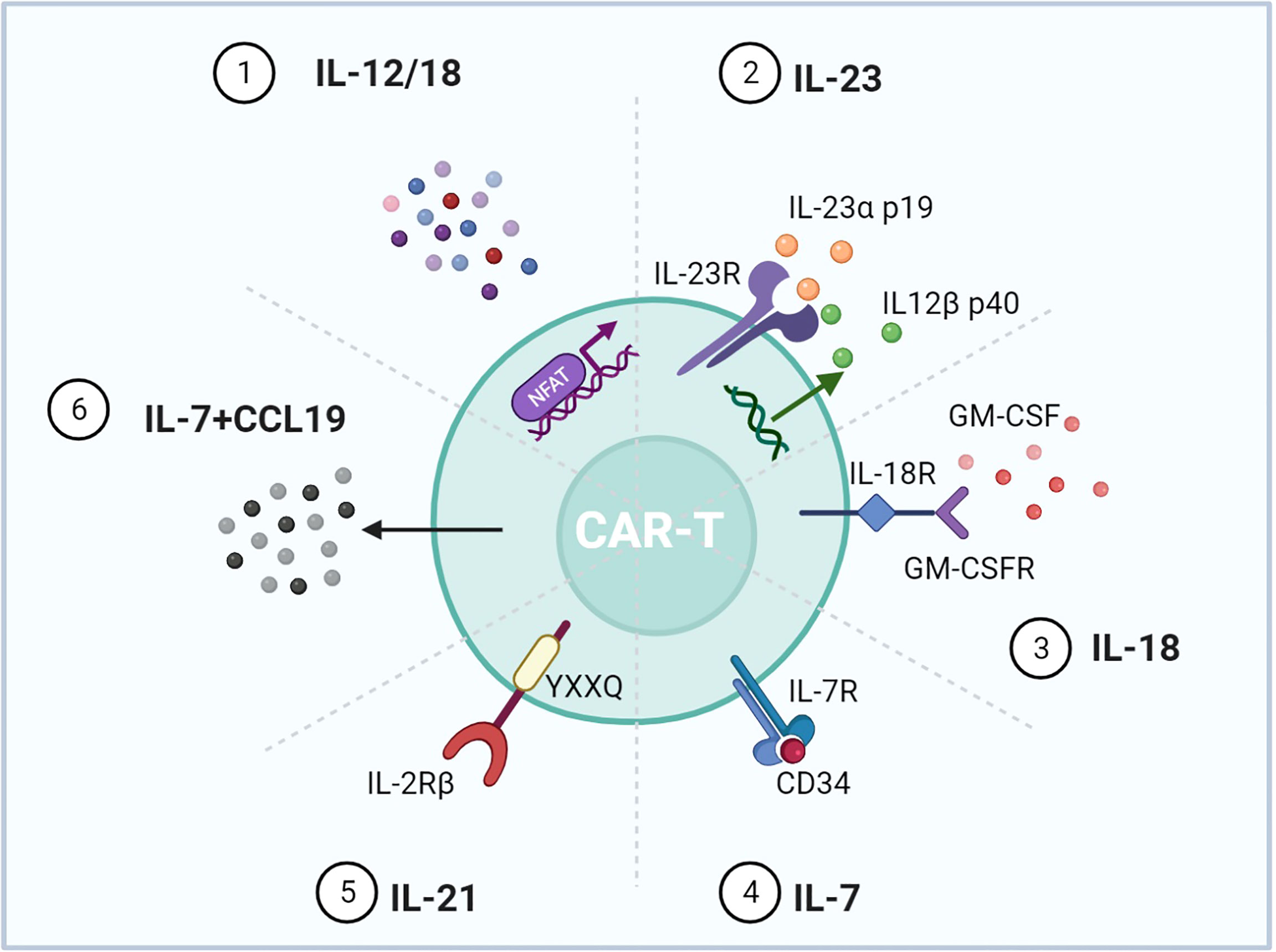
Figure 6 Representative designs of CARs with cytokines and chemokines. Diagrams of CAR mediating cytokine and chemokine expression upon T-cell activation. 1) NFAT activated upon antigen recognition was transduced into T cells to regulate IL-12/18. 2) IL-12β p40 was overexpressed for IL-23 production accompanied by intrinsic IL-23R and IL-23α p19. 3) The GM-CSF receptor sensing GM-CSF upon T-cell activation was incorporated with the IL-18 receptor. 4) The IL-7 receptor was enough for the downstream STAT5 activation, and CD34 blocked the effect of exogenous IL-7. 5) IL-2Rβ with the YXXQ motif could mimic the effect of IL-21. 6) Expressing IL-7 and CCL19 could promote CAR-T migration and recruit DCs and T cells into the tumor. Figure created using BioRender.com.
In addition, chemokines are a kind of small molecule cytokine that can induce chemotaxis. Since solid malignancies are difficult to penetrate, expressing chemokines can attract immune cells to traffic into the tumor. IL-7 and CCL19 are essential for the formation and maintenance of the T-cell zone in lymphoid organs, where both T cells and dendritic cells (DCs) are recruited from the periphery. CAR-T cells expressing IL-7 and CCL19 were designed (7 × 19 CAR-T) (92). IL-7 and CCL19 expression has been identified to improve T-cell infiltration and CAR-T survival in mouse tumors. Compared to conventional CAR-T, 7 × 19 CAR-T showed superior antitumor activity and recruited DCs and T cells. A phase 1 clinical trial was conducted to evaluate the clinical potential of 7 × 19 CAR-T (NCT03198546) (93).
Developments of carrier immune cells
Beyond conventional CD4+/CD8+ T cells, immune cells involved in the innate immune system can carry CAR. Based on their specific constructs, they are with favorable factors to circumvent GvHD and show potent cell-killing abilities (Table 4).
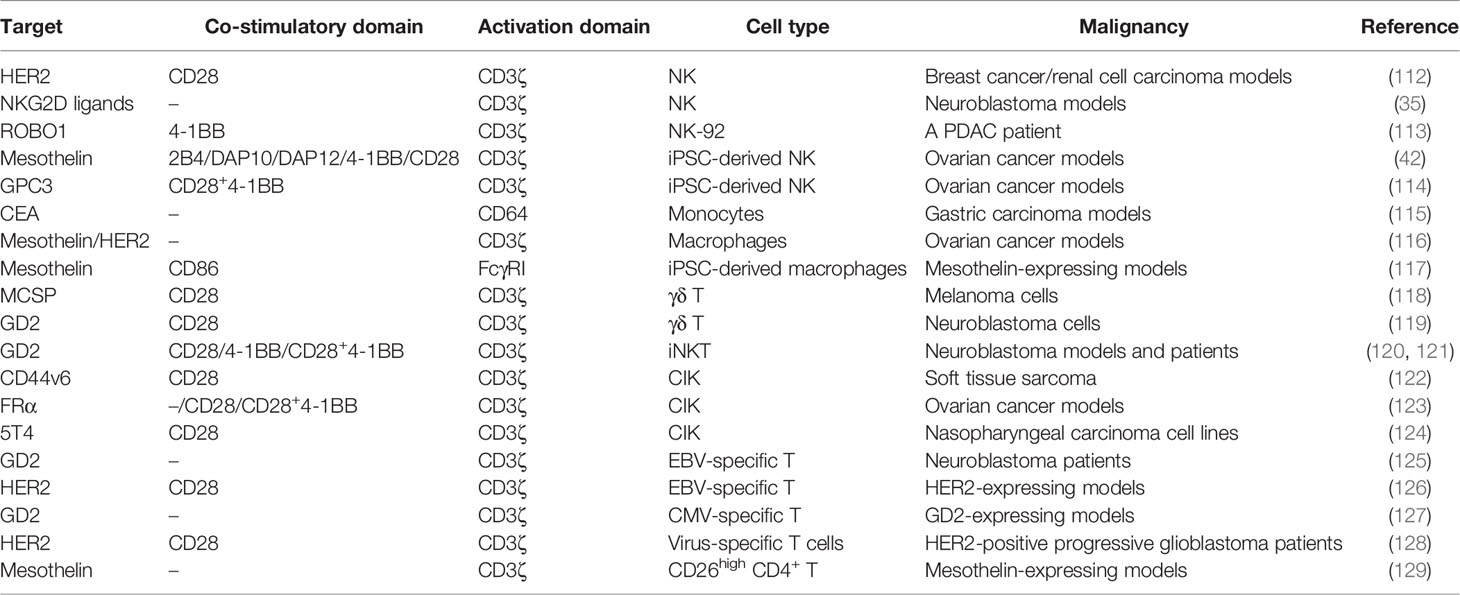
Table 4 CAR carried by different immune cells in solid tumors.
NK Cells
NK cells without TCR-like molecules are distinct from T cells and of paramount importance in the innate immune system by performing MHC-unrestricted cell killing. NK cells express a repertoire of inhibitory and activating receptors and related adaptors, including natural cytotoxic receptors such as NKG2D, CD16 (FcγRIIIa), FasL, and TRAIL, and co-stimulatory receptors such as LFA-1, 4-1BB, and 2B4 (130). When KIRs on the NK cell surface fail to engage with their cognate HLA, cell lysis is activated. This phenomenon is summarized as the “missing self” mechanism (131). The NK cell-based therapy has superiority in the microenvironment with downregulated HLA. Inhibitory receptors of NK cells can combine HLA class I molecules on autologous normal cells to relieve cytotoxic activity, avoiding CRS or neurotoxicity (132). Furthermore, NK cells as allogeneic effectors do not need to be collected from a patient or a specific HLA-matched donor. NK cells can be acquired from umbilical cord blood (UCB) to generate “off-the-shelf” products. With over 500,000 validated banked UCB units worldwide, the source of adoptive NK cells is available (133). NK cells show cytotoxicity advantages over T cells in CAR-driven immunotherapy (134). CD3ζ modification made CAR-NK cells resistant to TGF-β, which inhibits endogenous NK cells by decreasing DAP10 transcription (35, 135). Incorporating CD19-CAR-NK cells with IL-15 could further promote NK cell expansion and persistence. Of the 11 patients with relapsed/refractory CD19-positive cancers, 7 had a complete remission after the treatment of CD19-CAR-NK cells with IL-15 (136).
In solid tumors, a series of trials employing CAR-NK are ongoing (NCT03941457, NCT05194709, NCT03940820) (113, 137). Intriguingly, CAR-expressing lymphoid progenitors were prone to differentiate into cells with NK cell receptors that are called CAR-induced killer cells (CARiK) by inhibiting the transcription factor B-cell CLL/lymphoma 11B (BCL11B) and NOTCH1. Allogeneic CARiK showed increased survival without GvHD, suggesting the potential of CARiK for anticancer immunotherapy (138). Moreover, CAR-expressing induced pluripotent stem cell (iPSC)-derived NK cells with the TM domain of NKG2D showed significant antitumor cytotoxicity. Moreover, the co-stimulatory domain 2B4 instead of DAP10, DAP12, 4-1BB, or CD28 gave the greatest antitumor activity (42). Since 2B4 signaling is necessary for optimal NK cell function and beneficial for activation triggered by other NK cell receptors (139), 2B4 might be the most suitable candidate for the co-stimulatory domain of CAR-NK cells and more investigation is needed.
Macrophages
Macrophages can serve as the platform for CAR (116). CAR macrophages (CAR-Ms) exhibited versatilities including tumor-specific phagocytosis, inflammatory cytokine production, polarization of bystander macrophages to the immunostimulatory M1 phenotype, and cross-presentation of the TAAs to bystander T cells. Resultantly, CAR-Ms could create a pro-inflammatory TME and promote antitumor T-cell activity. Additionally, iPSC−derived macrophages with the CAR consisting of the macrophage receptor FcγRI and CD19 or mesothelin scFvs demonstrated enhanced phagocytosis and immune activities (117).
γδ T Cells
γδ T cells are T cells with γ/δ TCRs, constituting only 1%–5% of circulating lymphocytes but predominant in some epithelial sites, such as the intestine, reproductive organs, tongue, and skin (140). Similarly, γδ T cells could accumulate in the solid TME. αβ T cells exert antitumor effects in an MHC-dependent manner. In contrast, γδ T cells find their targets through innate-like pathways without the restriction of MHC. They sense small, non-peptide antigens called “phosphoantigens.” With the capability of migration toward tumors and cross-presentation of TAAs, γδ T cells are more beneficial for the adoptive T-cell therapy in solid tumors than αβ T cells. Furthermore, γδ T cells are less prone to exhaust or induce GvHD than αβ T cells (141, 142). Consequently, γδ T cells have the potential to become “off-the-shelf” cellular immunotherapy (143). Compared with genetically engineered conventional T cells, γδ T cells with the expression of TCR or CAR released reduced cytokines but presented the equivalent cytotoxicity in the melanoma (118). CAR-γδ T cells with co-stimulation and antigen-presentation molecules (CD86 and HLA-DR) were able to cross-present TAAs to other T cells in the TME (119). The abovementioned AbTCR combined the Fab domain with γ and δ chains. CD19-AbTCR-T cells showed similar cytotoxic activity with CD19–CD28/4-1BB CAR-T cells but with less cytokine and exhaustion (62). More specifically, there are two main subtypes of γδ T cells (Table 5) (151). The first is Vγ9Vδ2 T cells, whose TCR heterodimer is built by a γ chain with the Vγ9 segment and a δ chain with the variable segment Vδ2. Another is Vδ1+ T cells containing the Vδ1 segment. CAR-Vδ1+ cells maintained a T-naive phenotype, and CAR-Vδ2+ cells predominantly adopted an effector memory phenotype. CAR-Vδ1+ cells showed less exhausted phenotypes than their Vδ2+ counterparts (119).

Table 5 Differences between Vδ1+ and Vδ2+ T cells.
iNKT Cells
NKT cells are a subset of T lymphocytes with NK cell surface markers. They are rare but powerful effector T cells. Chemokines secreted by tumor cells and tumor-associated macrophages (TAMs) can recruit NKT cells into solid tumors (152). The non-polymorphic, glycolipid-presenting HLA I-like molecule CD1d on B cells presents antigens for recognition of NKT cells (153). Reciprocally, NKT cells can eliminate TAMs (154). The monomorphic nature of CD1d and its interspecies conservation reduces the risk of GvHD. CD1d-restricted Vα24-invariant (type-I) NKT, also termed iNKT, whose infiltration in colon cancer is associated with a better prognosis, is one of the ideal candidates for CAR carrier (155). Compared with CAR-T cells, CAR-iNKT cells are superior as they can easily arrive at tumor sites, kill CD1d-positive TAMs, and avoid GvHD. In B-cell malignancies, CD19-CAR-iNKT cells exhibited superior proliferation and therapeutic effects than CAR-T cells. Upregulating CD1d expression could enhance the antitumor efficacy of CD19-CAR-iNKT cells (156). In particular, given that the co-stimulatory OX40L–OX40 axis is conducive for iNKT cell-mediated antitumor responses, the inclusion of OX40 in a 3G CAR configuration might be advantageous. In neuroblastoma, GD2-based CAR rendered iNKT cells with stronger cytotoxicity and persistence bypassing CD1d. Moreover, 4-1BB induced T helper 1-like polarization of NKT cells (120). Researchers further engineered GD2-CAR-iNKT cells with IL-15 and initiated a first-in-human CAR-NKT clinical trial (NCT03294954) (121). Co-expression of IL-15 could reduce exhaustion markers and improve tumor control. In addition, they reported using CD28 instead of 4-1BB as the co-stimulatory domain could avoid excessive activation-induced cell death. The interim analysis showed that the GD2-targeted CAR-NKT cells were well tolerated in three patients. One of the three patients achieved an objective response with regressing bone metastatic lesions (157).
CIKs
Cytokine-induced killer cells (CIKs) are ex-vivo-expanded T lymphocytes with T-NK phenotype and potent MHC-independent antitumor ability regulated by the NKG2D receptor. Researchers aimed at combining the CAR specificity with the intrinsic tumor-killing ability of CIK cells to generate bipotential killers. CAR-CIKs have been investigated in solid tumor settings. CAR-CIK produced higher amounts of IL-6 and IFN-γ and enhanced killing activity compared to control CIK (122). The presence of 4-1BB or CD28 in CAR increased the production of cytokines, and the 3G CAR-CIKs showed significant proliferation and long-term inhibition on tumor growth (124, 158).
Virus-Specific T Cells
Cytotoxic T lymphocytes (CTLs) specific for endogenous viral antigens such as Epstein–Barr virus (EBV) and cytomegalovirus (CMV), known as virus-specific T cells (VSTs), receive superior co-stimulation provided by professional APCs. Repeated stimulation with viral antigen gradually increases viral specificity and concomitantly depletes alloreactivity. Clinical application of donor virus-specific T cells indicates a low rate of GvHD (159). EBV-specific T-cell lines contain CD8+ cytotoxic T cells and antigen-specific CD4+ helper T cells, which control EBV latency by providing growth factors (160). The rapid expansion of EBV-specific T cells in vivo and their persistence in a lifelong functional state without further immunization make them attractive candidates for CAR vehicles. CD8+ T cells with a GD2-CAR were activated by EBV antigen and co-stimulated through the B7/CD28 axis to convert into CAR-EBV-specific T cells. Normal levels of EBV DNA instead of evident EBV-positive malignancy were sufficient for their expansion and maintenance (161). In a long-term clinical trial with 19 neuroblastoma patients, CAR-EBV-specific T cells and CAR-T cells demonstrated similar fates. Their duration of persistence was highly concordant with the proportion of helper and T central memory cells (125). Strikingly, researchers have designed a whole-cell vaccine promoting the cross-presentation of viral epitopes to the native virus-specific TCRs aiming at expanding CAR-redirected CTLs (127). They further engineered the vaccine with CD40L and OX40L to promote the overall antitumor activity of CAR-CMV-specific T cells. A phase 1 clinical trial indicated that VSTs expressing HER2-CAR were safe and beneficial for progressive glioblastoma patients (128).
iPSCs
T cells derived from iPSCs can also be a source of CAR-T cells (162). A bank of iPSCs with common HLA haplotypes can be generated to minimize the risk of allo-rejection. CAR-T cells are generated from one clonal engineered pluripotent cell line and are therefore homogeneous. Although iPSC-T cells expressed their rearranged endogenous αβ-TCR on the surface, they acquired an innate-like phenotype instead of that of natural naive or memory CD8αβ+ T lymphocytes (163). Surprisingly, iPSC-derived, CAR-expressing T cells displayed an mRNA expression profile similar to that of γδ T cells (162). Under the stimulation of CD19, expanding CAR-iPSC-T cells expressed NK receptors such as NKp44, NKp46, and NKG2D and switched to the Th1 phenotype. The rate of CAR expression on iPSC-T cells was relatively lower than that on γδ or αβ T cells, which might reduce tonic signaling. A phase 1 study of iPSC-derived CAR-T cells targeting CD19 has been conducted for leukemia patients (164).
CD26high T Cells
It is suggested that some subsets of T cells like CD26high T cells may be more efficacious than others (165). CD26 is an enzymatically active, multifunctional protein beneficial for T-cell co-stimulation as well as the binding of extracellular molecules (166). CD26 might augment antitumor immunity in an MHC-independent manner (167). CD26high CD4+ T cells generated excessive IL-17 showing a phenotype resembling Th17, which exhibited stem cell-like qualities (168). CD26high CAR-T cells with enhanced stemness and apoptosis resistance produced increased cytokines (IL-17A, IFN-γ, IL-2, TNF, and IL-22) and elevated CCR2 and CCR5 (129). Intriguingly, CD26high T cells also expressed multiple co-stimulatory and co-inhibitory markers (PD-1, CD40L, OX40, and GITR). GITR is a co-stimulatory receptor enhancing effector T-cell function and alleviating T regulatory cell (Treg)-mediated immunosuppression (169), suggesting its potential to provide a co-stimulatory signal for CAR.
Conclusion
To overcome the challenges of CAR-T cells in solid tumors and promote their commercial application, we presented the achievements of receptors, signaling, and immune cells beneficial for CAR construction. Either of the stimulation domains such as CD28/4-1BB/CD3ζ or CD4+/CD8+ T cells can be replaced for better efficacy. We reanalyzed the construct of CARs from 1G to 4G and focused on unconventional designs including specific receptors or novel co-stimulatory domains. Except for scFvs, different receptors with high affinities have emerged. Artificial switch receptors can reverse inhibitory signaling and SynNotch receptors can reduce “on-target/off-tumor” effects. Furthermore, incorporating natural TCR and NKG2D into CAR can expand the range of targets. Developments of stimulation domains have been investigated as well. Intriguingly, changing the number or location of ITAMs can promote an activation signal. In addition to CD28 and 4-1BB, more molecules, such as ICOS, CD27, and MyD88, are participating in the CAR machinery and their interaction might achieve synergistic effects. We further compared the characteristics of diverse immune cells including NK cells, NKT cells, macrophages, and several subtypes of T cells and evaluated their antitumor potential. Using virus-specific T cells or iPSC as drivers of CAR can decrease the risk of GvHD. Integrating CAR into NK cells or CIKs with NKG2D can avert off-target effects. In a word, a growing number of candidates of the optimal CAR construct without the conventional framework are appearing. Their effects need to be objectively compared and evaluated to inspire researchers.
The application of NK cells and associated receptors or co-stimulatory molecules in CAR is of great interest. As a pivotal component of the innate immune system, NK cells can show potent antitumor function without severe side effects. Their receptor NKG2D is also expressed on the surface of CIK, γδ T cells, and NKT cells. CAR that contained NKG2D can exhibit superior recognition capacity. Meanwhile, DAP12 as the adaptor of NKG2D shows vigorous activation ability. The ingenious combination of NKG2D and DAP12 or CD3ζ can maximumly activate immune cells and minimize immune suppression. For instance, CD3ζ modification brings CAR-NK cell resistance to TGF-β, which inhibits DAP10 transcription to suppress endogenous NK cells (35). Moreover, strategies of 4G CAR can be applied to CAR-NK cells. Adding IL-15 or its receptor probably extends the persistence of CAR-NK/NKT cells.
It is noteworthy that the design of CAR is undergoing changes from a single chain to double chains or from a series model to a parallel model. The recognition domain and activation domain may be separated into two chains, like KIR/TREM and DAP12, to improve responsiveness and stability. Another strategy is replacing the single chain with only one CD3ζ by two chains similar to TCR, such as STAR, AbTCR, and TRuC, which have advantages in fully stimulating immune cells. These findings suggest that using two chains with different or synergistic functions can improve CAR-T efficacy and avoid tonic signaling. Furthermore, there is still a wide research field awaiting investigation. For example, even Treg cells can express CAR and different co-stimulators might influence their functions (170). Various co-stimulatory molecules like GITR and dectin-1 deserve attention as well (171, 172).
In conclusion, assembling distinct receptors, stimulation domains, and immune cells might realize unexpected achievements. To acquire optimal CAR, multiple novel designs and collocations need to be developed.
Author Contributions
MS planned the manuscript. TY and S-KY wrote the draft and designed the figures. YX corrected the grammar errors and notation mistakes. K-HL reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (82172708).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rafiq S, Hackett CS, Brentjens RJ. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat Rev Clin Oncol (2020) 17(3):147–67. doi: 10.1038/s41571-019-0297-y
2. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma After Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
3. Moritz D, Wels W, Mattern J, Groner B. Cytotoxic T Lymphocytes With a Grafted Recognition Specificity for ERBB2-Expressing Tumor Cells. Proc Natl Acad Sci USA (1994) 91(10):4318–22. doi: 10.1073/pnas.91.10.4318
4. Cartellieri M, Bachmann M, Feldmann A, Bippes C, Stamova S, Wehner R, et al. Chimeric Antigen Receptor-Engineered T Cells for Immunotherapy of Cancer. J BioMed Biotechnol (2010) 2010:956304. doi: 10.1155/2010/956304
5. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T Cells With Chimeric Antigen Receptors Have Potent Antitumor Effects and can Establish Memory in Patients With Advanced Leukemia. Sci Transl Med (2011) 3(95):95ra73. doi: 10.1126/scitranslmed.3002842
6. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: Chimeric Antigen Receptor (CAR) T Cells Engineered With an Inducible Cytokine to Modulate the Tumor Stroma. Immunol Rev (2014) 257(1):83–90. doi: 10.1111/imr.12125
7. Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell (2017) 168(4):724–40. doi: 10.1016/j.cell.2017.01.016
8. Martinez M, Moon EK. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front Immunol (2019) 10:128. doi: 10.3389/fimmu.2019.00128
9. Ajina A, Maher J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol Cancer Ther (2018) 17(9):1795–815. doi: 10.1158/1535-7163.MCT-17-1097
11. Frey N, Porter D. Cytokine Release Syndrome With Chimeric Antigen Receptor T Cell Therapy. Biol Blood Marrow Transplant (2019) 25(4):e123–7. doi: 10.1016/j.bbmt.2018.12.756
12. MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al. Integration of a CD19 CAR Into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol Ther (2017) 25(4):949–61. doi: 10.1016/j.ymthe.2017.02.005
13. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther (2020) 28(11):2320–39. doi: 10.1016/j.ymthe.2020.09.015
14. Newick K, O'Brien S, Moon E, Albelda SM. CAR T Cell Therapy for Solid Tumors. Annu Rev Med (2017) 68:139–52. doi: 10.1146/annurev-med-062315-120245
15. Grunert C, Willimsky G, Peuker CA, Rhein S, Hansmann L, Blankenstein T, et al. Isolation of Neoantigen-Specific Human T Cell Receptors From Different Human and Murine Repertoires. Cancers (Basel) (2022) 14(7):1842. doi: 10.3390/cancers14071842
16. Muyldermans S. Nanobodies: Natural Single-Domain Antibodies. Annu Rev Biochem (2013) 82:775–97. doi: 10.1146/annurev-biochem-063011-092449
17. Muyldermans S. A Guide to: Generation and Design of Nanobodies. FEBS J (2021) 288(7):2084–102. doi: 10.1111/febs.15515
18. Bao C, Gao Q, Li LL, Han L, Zhang B, Ding Y, et al. The Application of Nanobody in CAR-T Therapy. Biomolecules (2021) 11(2):238. doi: 10.3390/biom11020238
19. De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J, et al. Molecular Basis for the Preferential Cleft Recognition by Dromedary Heavy-Chain Antibodies. Proc Natl Acad Sci USA (2006) 103(12):4586–91. doi: 10.1073/pnas.0505379103
20. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T Cells Targeting HER2 and IL13Rα2 Mitigate Tumor Antigen Escape. J Clin Invest (2016) 126(8):3036–52. doi: 10.1172/jci83416
21. Khaleghi S, Rahbarizadeh F, Ahmadvand D, Rasaee MJ, Pognonec P. A Caspase 8-Based Suicide Switch Induces Apoptosis in Nanobody-Directed Chimeric Receptor Expressing T Cells. Int J Hematol (2012) 95(4):434–44. doi: 10.1007/s12185-012-1037-6
22. Jamnani FR, Rahbarizadeh F, Shokrgozar MA, Mahboudi F, Ahmadvand D, Sharifzadeh Z, et al. T Cells Expressing VHH-Directed Oligoclonal Chimeric HER2 Antigen Receptors: Towards Tumor-Directed Oligoclonal T Cell Therapy. Biochim Biophys Acta (2014) 1840(1):378–86. doi: 10.1016/j.bbagen.2013.09.029
23. Xie YJ, Dougan M, Ingram JR, Pishesha N, Fang T, Momin N, et al. Improved Antitumor Efficacy of Chimeric Antigen Receptor T Cells That Secrete Single-Domain Antibody Fragments. Cancer Immunol Res (2020) 8(4):518–29. doi: 10.1158/2326-6066.CIR-19-0734
24. Albert S, Arndt C, Koristka S, Berndt N, Bergmann R, Feldmann A, et al. From Mono- to Bivalent: Improving Theranostic Properties of Target Modules for Redirection of UniCAR T Cells Against EGFR-Expressing Tumor Cells In Vitro and In Vivo. Oncotarget (2018) 9(39):25597–616. doi: 10.18632/oncotarget.25390
25. Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-Dependent Activation of PI3K Requires DAP10 and is Inhibited by SHIP1. Sci Signal (2010) 3(122):ra38. doi: 10.1126/scisignal.2000500
26. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR Activation Potential Directs Alternative T Cell Fates and Therapeutic Potency. Nat Med (2019) 25(1):82–8. doi: 10.1038/s41591-018-0290-5
27. Cappell KM, Kochenderfer JN. A Comparison of Chimeric Antigen Receptors Containing CD28 Versus 4-1BB Costimulatory Domains. Nat Rev Clin Oncol (2021) 18(11):715–27. doi: 10.1038/s41571-021-00530-z
28. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat Med (2015) 21(6):581–90. doi: 10.1038/nm.3838
29. Pourakbari R, Hajizadeh F, Parhizkar F, Aghebati-Maleki A, Mansouri S, Aghebati-Maleki L. Co-Stimulatory Agonists: An Insight Into the Immunotherapy of Cancer. Excli J (2021) 20:1055–85. doi: 10.17179/excli2021-3522
30. Klebanoff CA, Gattinoni L, Restifo NP. Sorting Through Subsets: Which T-Cell Populations Mediate Highly Effective Adoptive Immunotherapy? J Immunother (2012) 35(9):651–60. doi: 10.1097/CJI.0b013e31827806e6
31. Schmueck-Henneresse M, Omer B, Shum T, Tashiro H, Mamonkin M, Lapteva N, et al. Comprehensive Approach for Identifying the T Cell Subset Origin of CD3 and CD28 Antibody-Activated Chimeric Antigen Receptor-Modified T Cells. J Immunol (2017) 199(1):348–62. doi: 10.4049/jimmunol.1601494
32. Guedan S, Posey AD Jr., Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T Cell Persistence Through ICOS and 4-1BB Costimulation. JCI Insight (2018) 3(1):e96976. doi: 10.1172/jci.insight.96976
33. Hong M, Clubb JD, Chen YY. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell (2020) 38(4):473–88. doi: 10.1016/j.ccell.2020.07.005
34. Ng YY, Tay JCK, Li Z, Wang J, Zhu J, Wang S. T Cells Expressing NKG2D CAR With a DAP12 Signaling Domain Stimulate Lower Cytokine Production While Effective in Tumor Eradication. Mol Ther (2021) 29(1):75–85. doi: 10.1016/j.ymthe.2020.08.016
35. Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity Against Solid Tumors. Cancer Immunol Res (2019) 7(3):363–75. doi: 10.1158/2326-6066.CIR-18-0572
36. Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol Ther (2019) 27(6):1114–25. doi: 10.1016/j.ymthe.2019.03.011
37. Topfer K, Cartellieri M, Michen S, Wiedemuth R, Muller N, Lindemann D, et al. DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy. J Immunol (2015) 194(7):3201–12. doi: 10.4049/jimmunol.1400330
38. Muller N, Michen S, Tietze S, Topfer K, Schulte A, Lamszus K, et al. Engineering NK Cells Modified With an EGFRvIII-Specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1alpha-Secreting Glioblastoma. J Immunother (2015) 38(5):197–210. doi: 10.1097/CJI.0000000000000082
39. Wang E, Wang LC, Tsai CY, Bhoj V, Gershenson Z, Moon E, et al. Generation of Potent T-Cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol Res (2015) 3(7):815–26. doi: 10.1158/2326-6066.CIR-15-0054
40. Chen B, Zhou M, Zhang H, Wang C, Hu X, Wang B, et al. TREM1/Dap12-Based CAR-T Cells Show Potent Antitumor Activity. Immunotherapy (2019) 11(12):1043–55. doi: 10.2217/imt-2019-0017
41. Sun M, Xu P, Wang E, Zhou M, Xu T, Wang J, et al. Novel Two-Chain Structure Utilizing KIRS2/DAP12 Domain Improves the Safety and Efficacy of CAR-T Cells in Adults With R/R B-ALL. Mol Ther Oncolytics (2021) 23:96–106. doi: 10.1016/j.omto.2021.08.014
42. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered With Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell (2018) 23(2):181–92.e5. doi: 10.1016/j.stem.2018.06.002
43. Maus MV, Plotkin J, Jakka G, Stewart-Jones G, Riviere I, Merghoub T, et al. An MHC-Restricted Antibody-Based Chimeric Antigen Receptor Requires TCR-Like Affinity to Maintain Antigen Specificity. Mol Ther Oncolytics (2016) 3:1–9. doi: 10.1038/mto.2016.23
44. Zhang G, Wang L, Cui H, Wang X, Zhang G, Ma J, et al. Anti-Melanoma Activity of T Cells Redirected With a TCR-Like Chimeric Antigen Receptor. Sci Rep (2014) 4:3571. doi: 10.1038/srep03571
45. Helsen CW, Hammill JA, Lau VWC, Mwawasi KA, Afsahi A, Bezverbnaya K, et al. The Chimeric TAC Receptor Co-Opts the T Cell Receptor Yielding Robust Anti-Tumor Activity Without Toxicity. Nat Commun (2018) 9(1):3049. doi: 10.1038/s41467-018-05395-y
46. Baeuerle PA, Ding J, Patel E, Thorausch N, Horton H, Gierut J, et al. Synthetic TRuC Receptors Engaging the Complete T Cell Receptor for Potent Anti-Tumor Response. Nat Commun (2019) 10(1):2087. doi: 10.1038/s41467-019-10097-0
47. Liu Y, Liu G, Wang J, Zheng ZY, Jia L, Rui W, et al. Chimeric STAR Receptors Using TCR Machinery Mediate Robust Responses Against Solid Tumors. Sci Transl Med (2021) 13(586):eabb5191. doi: 10.1126/scitranslmed.abb5191
48. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T Cells With Cell-Intrinsic PD-1 Checkpoint Blockade Resist Tumor-Mediated Inhibition. J Clin Invest (2016) 126(8):3130–44. doi: 10.1172/JCI83092
49. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-Negative TGF-Beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther (2018) 26(7):1855–66. doi: 10.1016/j.ymthe.2018.05.003
50. Burga RA, Yvon E, Chorvinsky E, Fernandes R, Cruz CRY, Bollard CM. Engineering the TGFbeta Receptor to Enhance the Therapeutic Potential of Natural Killer Cells as an Immunotherapy for Neuroblastoma. Clin Cancer Res (2019) 25(14):4400–12. doi: 10.1158/1078-0432.CCR-18-3183
51. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res (2016) 76(6):1578–90. doi: 10.1158/0008-5472.CAN-15-2524
52. Bajgain P, Tawinwung S, D'Elia L, Sukumaran S, Watanabe N, Hoyos V, et al. CAR T Cell Therapy for Breast Cancer: Harnessing the Tumor Milieu to Drive T Cell Activation. J Immunother Cancer (2018) 6(1):34. doi: 10.1186/s40425-018-0347-5
53. Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, et al. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol Ther (2017) 25(1):249–58. doi: 10.1016/j.ymthe.2016.10.016
54. Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell (2019) 35(3):489–503.e8. doi: 10.1016/j.ccell.2019.02.003
55. Moghimi B, Muthugounder S, Jambon S, Tibbetts R, Hung L, Bassiri H, et al. Preclinical Assessment of the Efficacy and Specificity of GD2-B7H3 SynNotch CAR-T in Metastatic Neuroblastoma. Nat Commun (2021) 12(1):511. doi: 10.1038/s41467-020-20785-x
56. Newick K, O'Brien S, Sun J, Kapoor V, Maceyko S, Lo A, et al. Augmentation of CAR T-Cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol Res (2016) 4(6):541–51. doi: 10.1158/2326-6066.CIR-15-0263
57. Schamel WW, Arechaga I, Risueno RM, van Santen HM, Cabezas P, Risco C, et al. Coexistence of Multivalent and Monovalent TCRs Explains High Sensitivity and Wide Range of Response. J Exp Med (2005) 202(4):493–503. doi: 10.1084/jem.20042155
59. Almen MS, Nordstrom KJ, Fredriksson R, Schioth HB. Mapping the Human Membrane Proteome: A Majority of the Human Membrane Proteins can be Classified According to Function and Evolutionary Origin. BMC Biol (2009) 7:50. doi: 10.1186/1741-7007-7-50
60. Yin L, Scott-Browne J, Kappler JW, Gapin L, Marrack P. T Cells and Their Eons-Old Obsession With MHC. Immunol Rev (2012) 250(1):49–60. doi: 10.1111/imr.12004
61. Walseng E, Koksal H, Sektioglu IM, Fane A, Skorstad G, Kvalheim G, et al. A TCR-Based Chimeric Antigen Receptor. Sci Rep (2017) 7(1):10713. doi: 10.1038/s41598-017-11126-y
62. Xu Y, Yang Z, Horan LH, Zhang P, Liu L, Zimdahl B, et al. A Novel Antibody-TCR (AbTCR) Platform Combines Fab-Based Antigen Recognition With Gamma/Delta-TCR Signaling to Facilitate T-Cell Cytotoxicity With Low Cytokine Release. Cell Discov (2018) 4:62. doi: 10.1038/s41421-018-0066-6
63. Wensveen FM, Jelencic V, Polic B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front Immunol (2018) 9:441. doi: 10.3389/fimmu.2018.00441
64. Gonzalez S, Lopez-Soto A, Suarez-Alvarez B, Lopez-Vazquez A, Lopez-Larrea C. NKG2D Ligands: Key Targets of the Immune Response. Trends Immunol (2008) 29(8):397–403. doi: 10.1016/j.it.2008.04.007
65. Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature (1998) 391(6668):703–7. doi: 10.1038/35642
66. Lanier LL. DAP10- and DAP12-Associated Receptors in Innate Immunity. Immunol Rev (2009) 227(1):150–60. doi: 10.1111/j.1600-065X.2008.00720.x
67. Barrow AD, Trowsdale J. You Say ITAM and I Say ITIM, Let's Call the Whole Thing Off: The Ambiguity of Immunoreceptor Signalling. Eur J Immunol (2006) 36(7):1646–53. doi: 10.1002/eji.200636195
68. Zheng L, Ren L, Kouhi A, Khawli LA, Hu P, Kaslow HR, et al. A Humanized Lym-1 CAR With Novel DAP10/DAP12 Signaling Domains Demonstrates Reduced Tonic Signaling and Increased Antitumor Activity in B-Cell Lymphoma Models. Clin Cancer Res (2020) 26(14):3694–706. doi: 10.1158/1078-0432.CCR-19-3417
69. Ford JW, McVicar DW. TREM and TREM-Like Receptors in Inflammation and Disease. Curr Opin Immunol (2009) 21(1):38–46. doi: 10.1016/j.coi.2009.01.009
70. Rosen DB, Araki M, Hamerman JA, Chen T, Yamamura T, Lanier LL. A Structural Basis for the Association of DAP12 With Mouse, But Not Human, NKG2D. J Immunol (2004) 173(4):2470–8. doi: 10.4049/jimmunol.173.4.2470
71. Liang Y, Liu H, Lu Z, Lei W, Zhang C, Li P, et al. CD19 CAR-T Expressing PD-1/CD28 Chimeric Switch Receptor as a Salvage Therapy for DLBCL Patients Treated With Different CD19-Directed CAR T-Cell Therapies. J Hematol Oncol (2021) 14(1):26. doi: 10.1186/s13045-021-01044-y
72. Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T Cells With Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell (2016) 167(2):419–32.e16. doi: 10.1016/j.cell.2016.09.011
73. Manz BN, Tan YX, Courtney AH, Rutaganira F, Palmer E, Shokat KM, et al. Small Molecule Inhibition of Csk Alters Affinity Recognition by T Cells. Elife (2015) 4:e08088. doi: 10.7554/eLife.08088
74. Mosenden R, Singh P, Cornez I, Heglind M, Ruppelt A, Moutschen M, et al. Mice With Disrupted Type I Protein Kinase A Anchoring in T Cells Resist Retrovirus-Induced Immunodeficiency. J Immunol (2011) 186(9):5119–30. doi: 10.4049/jimmunol.1100003
75. Bell M, Gottschalk S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front Immunol (2021) 12:684642. doi: 10.3389/fimmu.2021.684642
76. Nguyen P, Okeke E, Clay M, Haydar D, Justice J, O'Reilly C, et al. Route of 41BB/41BBL Costimulation Determines Effector Function of B7-H3-CAR.CD28zeta T Cells. Mol Ther Oncolytics (2020) 18:202–14. doi: 10.1016/j.omto.2020.06.018
77. Lai Y, Weng J, Wei X, Qin L, Lai P, Zhao R, et al. Toll-Like Receptor 2 Costimulation Potentiates the Antitumor Efficacy of CAR T Cells. Leukemia (2018) 32(3):801–8. doi: 10.1038/leu.2017.249
78. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov (2017) 7(11):1306–19. doi: 10.1158/2159-8290.CD-17-0263
79. Foster AE, Mahendravada A, Shinners NP, Chang WC, Crisostomo J, Lu A, et al. Regulated Expansion and Survival of Chimeric Antigen Receptor-Modified T Cells Using Small Molecule-Dependent Inducible Myd88/CD40. Mol Ther (2017) 25(9):2176–88. doi: 10.1016/j.ymthe.2017.06.014
80. Duong MT, Collinson-Pautz MR, Morschl E, Lu A, Szymanski SP, Zhang M, et al. Two-Dimensional Regulation of CAR-T Cell Therapy With Orthogonal Switches. Mol Ther Oncolytics (2019) 12:124–37. doi: 10.1016/j.omto.2018.12.009
81. Prinzing B, Schreiner P, Bell M, Fan Y, Krenciute G, Gottschalk S. MyD88/CD40 Signaling Retains CAR T Cells in a Less Differentiated State. JCI Insight (2020) 5(21):e136093. doi: 10.1172/jci.insight.136093
82. Shen CJ, Yang YX, Han EQ, Cao N, Wang YF, Wang Y, et al. Chimeric Antigen Receptor Containing ICOS Signaling Domain Mediates Specific and Efficient Antitumor Effect of T Cells Against EGFRvIII Expressing Glioma. J Hematol Oncol (2013) 6:33. doi: 10.1186/1756-8722-6-33
83. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-Based Chimeric Antigen Receptors Program Bipolar TH17/TH1 Cells. Blood (2014) 124(7):1070–80. doi: 10.1182/blood-2013-10-535245
84. Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ Jr. CD27 Costimulation Augments the Survival and Antitumor Activity of Redirected Human T Cells In Vivo. Blood (2012) 119(3):696–706. doi: 10.1182/blood-2011-03-344275
85. Chen H, Wei F, Yin M, Zhao Q, Liu Z, Yu B, et al. CD27 Enhances the Killing Effect of CAR T Cells Targeting Trophoblast Cell Surface Antigen 2 in the Treatment of Solid Tumors. Cancer Immunol Immunother (2021) 70(7):2059–71. doi: 10.1007/s00262-020-02838-8
86. Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, et al. Design and Characterization of an "All-In-One" Lentiviral Vector System Combining Constitutive Anti-GD2 CAR Expression and Inducible Cytokines. Cancers (Basel) (2020) 12(2):375. doi: 10.3390/cancers12020375
87. Liu Y, Di S, Shi B, Zhang H, Wang Y, Wu X, et al. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J Immunol (2019) 203(1):198–207. doi: 10.4049/jimmunol.1800033
88. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors That Exhibit Augmented Activity Against Advanced Solid Tumors. Cell Rep (2017) 21(11):3205–19. doi: 10.1016/j.celrep.2017.11.063
89. Ma X, Shou P, Smith C, Chen Y, Du H, Sun C, et al. Interleukin-23 Engineering Improves CAR T Cell Function in Solid Tumors. Nat Biotechnol (2020) 38(4):448–59. doi: 10.1038/s41587-019-0398-2
90. Lange S, Sand LGL, Bell M, Patil SL, Langfitt D, Gottschalk S. A Chimeric GM-CSF/IL18 Receptor to Sustain CAR T-Cell Function. Cancer Discov (2021) 11(7):1661–71. doi: 10.1158/2159-8290.CD-20-0896
91. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive Signaling From an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov (2017) 7(11):1238–47. doi: 10.1158/2159-8290.CD-17-0538
92. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 Expression in CAR-T Cells Improves Immune Cell Infiltration and CAR-T Cell Survival in the Tumor. Nat Biotechnol (2018) 36(4):346–51. doi: 10.1038/nbt.4086
93. Pang N, Shi J, Qin L, Chen A, Tang Y, Yang H, et al. IL-7 and CCL19-Secreting CAR-T Cell Therapy for Tumors With Positive Glypican-3 or Mesothelin. J Hematol Oncol (2021) 14(1):118. doi: 10.1186/s13045-021-01128-9
94. James JR. Tuning ITAM Multiplicity on T Cell Receptors can Control Potency and Selectivity to Ligand Density. Sci Signal (2018) 11(531):eaan1088. doi: 10.1126/scisignal.aan1088
95. Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the Antigen Density Requirement for CAR T-Cell Activity. Cancer Discov (2020) 10(5):702–23. doi: 10.1158/2159-8290.CD-19-0945
96. Sadelain M, Brentjens R, Riviere I. The Promise and Potential Pitfalls of Chimeric Antigen Receptors. Curr Opin Immunol (2009) 21(2):215–23. doi: 10.1016/j.coi.2009.02.009
97. van der Stegen SJ, Hamieh M, Sadelain M. The Pharmacology of Second-Generation Chimeric Antigen Receptors. Nat Rev Drug Discov (2015) 14(7):499–509. doi: 10.1038/nrd4597
98. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell-Mediated Tumor Eradication. Mol Ther (2010) 18(2):413–20. doi: 10.1038/mt.2009.210
99. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of Large, Established Tumor Xenografts With Genetically Retargeted Human T Cells Containing CD28 and CD137 Domains. Proc Natl Acad Sci USA (2009) 106(9):3360–5. doi: 10.1073/pnas.0813101106
100. Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell (2015) 28(4):415–28. doi: 10.1016/j.ccell.2015.09.004
101. Reynolds JM, Dong C. Toll-Like Receptor Regulation of Effector T Lymphocyte Function. Trends Immunol (2013) 34(10):511–9. doi: 10.1016/j.it.2013.06.003
102. Geng D, Zheng L, Srivastava R, Asprodites N, Velasco-Gonzalez C, Davila E. When Toll-Like Receptor and T-Cell Receptor Signals Collide: A Mechanism for Enhanced CD8 T-Cell Effector Function. Blood (2010) 116(18):3494–504. doi: 10.1182/blood-2010-02-268169
103. Brown V, Brown RA, Ozinsky A, Hesselberth JR, Fields S. Binding Specificity of Toll-Like Receptor Cytoplasmic Domains. Eur J Immunol (2006) 36(3):742–53. doi: 10.1002/eji.200535158
104. Rahman AH, Cui W, Larosa DF, Taylor DK, Zhang J, Goldstein DR, et al. MyD88 Plays a Critical T Cell-Intrinsic Role in Supporting CD8 T Cell Expansion During Acute Lymphocytic Choriomeningitis Virus Infection. J Immunol (2008) 181(6):3804–10. doi: 10.4049/jimmunol.181.6.3804
105. Ning S, Pagano JS, Barber GN. IRF7: Activation, Regulation, Modification and Function. Genes Immun (2011) 12(6):399–414. doi: 10.1038/gene.2011.21
106. Bhadra R, Gigley JP, Khan IA. Cutting Edge: CD40-CD40 Ligand Pathway Plays a Critical CD8-Intrinsic and -Extrinsic Role During Rescue of Exhausted CD8 T Cells. J Immunol (2011) 187(9):4421–5. doi: 10.4049/jimmunol.1102319
107. Song DG, Powell DJ. Pro-Survival Signaling via CD27 Costimulation Drives Effective CAR T-Cell Therapy. Oncoimmunology (2012) 1(4):547–9. doi: 10.4161/onci.19458
108. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-Targeted T Cells Modified to Secrete IL-12 Eradicate Systemic Tumors Without Need for Prior Conditioning. Blood (2012) 119(18):4133–41. doi: 10.1182/blood-2011-12-400044
109. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-Specific T Lymphocytes With Interleukin-15 and a Suicide Gene to Enhance Their Anti-Lymphoma/Leukemia Effects and Safety. Leukemia (2010) 24(6):1160–70. doi: 10.1038/leu.2010.75
110. Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ. The Molecular Basis of IL-21-Mediated Proliferation. Blood (2007) 109(10):4135–42. doi: 10.1182/blood-2006-10-054973
111. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A Novel Chimeric Antigen Receptor Containing a JAK-STAT Signaling Domain Mediates Superior Antitumor Effects. Nat Med (2018) 24(3):352–9. doi: 10.1038/nm.4478
112. Schonfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, et al. Selective Inhibition of Tumor Growth by Clonal NK Cells Expressing an ErbB2/HER2-Specific Chimeric Antigen Receptor. Mol Ther (2015) 23(2):330–8. doi: 10.1038/mt.2014.219
113. Li C, Yang N, Li H, Wang Z. Robo1-Specific Chimeric Antigen Receptor Natural Killer Cell Therapy for Pancreatic Ductal Adenocarcinoma With Liver Metastasis. J Cancer Res Ther (2020) 16(2):393–6. doi: 10.4103/jcrt.JCRT_190_20
114. Ueda T, Kumagai A, Iriguchi S, Yasui Y, Miyasaka T, Nakagoshi K, et al. Non-Clinical Efficacy, Safety and Stable Clinical Cell Processing of Induced Pluripotent Stem Cell-Derived Anti-Glypican-3 Chimeric Antigen Receptor-Expressing Natural Killer/Innate Lymphoid Cells. Cancer Sci (2020) 111(5):1478–90. doi: 10.1111/cas.14374
115. Biglari A, Southgate TD, Fairbairn LJ, Gilham DE. Human Monocytes Expressing a CEA-Specific Chimeric CD64 Receptor Specifically Target CEA-Expressing Tumour Cells In Vitro and In Vivo. Gene Ther (2006) 13(7):602–10. doi: 10.1038/sj.gt.3302706
116. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat Biotechnol (2020) 38(8):947–53. doi: 10.1038/s41587-020-0462-y
117. Zhang L, Tian L, Dai X, Yu H, Wang J, Lei A, et al. Pluripotent Stem Cell-Derived CAR-Macrophage Cells With Antigen-Dependent Anti-Cancer Cell Functions. J Hematol Oncol (2020) 13(1):153. doi: 10.1186/s13045-020-00983-2
118. Harrer DC, Simon B, Fujii SI, Shimizu K, Uslu U, Schuler G, et al. RNA-Transfection of Gamma/Delta T Cells With a Chimeric Antigen Receptor or an Alpha/Beta T-Cell Receptor: A Safer Alternative to Genetically Engineered Alpha/Beta T Cells for the Immunotherapy of Melanoma. BMC Cancer (2017) 17(1):551. doi: 10.1186/s12885-017-3539-3
119. Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity With Retention of Cross Presentation. Mol Ther (2018) 26(2):354–65. doi: 10.1016/j.ymthe.2017.12.001
120. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT Cells With Chimeric Antigen Receptor Provide a Novel Platform for Safe and Effective Cancer Immunotherapy. Blood (2014) 124(18):2824–33. doi: 10.1182/blood-2013-11-541235
121. Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced In Vivo Persistence and Antitumor Activity Against Neuroblastoma. Clin Cancer Res (2019) 25(23):7126–38. doi: 10.1158/1078-0432.CCR-19-0421
122. Leuci V, Casucci GM, Grignani G, Rotolo R, Rossotti U, Vigna E, et al. CD44v6 as Innovative Sarcoma Target for CAR-Redirected CIK Cells. Oncoimmunology (2018) 7(5):e1423167. doi: 10.1080/2162402X.2017.1423167
123. Zuo S, Wen Y, Panha H, Dai G, Wang L, Ren X, et al. Modification of Cytokine-Induced Killer Cells With Folate Receptor Alpha (FRalpha)-Specific Chimeric Antigen Receptors Enhances Their Antitumor Immunity Toward FRalpha-Positive Ovarian Cancers. Mol Immunol (2017) 85:293–304. doi: 10.1016/j.molimm.2017.03.017
124. Guo X, Zheng H, Luo W, Zhang Q, Liu J, Yao K. 5T4-Specific Chimeric Antigen Receptor Modification Promotes the Immune Efficacy of Cytokine-Induced Killer Cells Against Nasopharyngeal Carcinoma Stem Cell-Like Cells. Sci Rep (2017) 7(1):4859. doi: 10.1038/s41598-017-04756-9
125. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor Activity and Long-Term Fate of Chimeric Antigen Receptor-Positive T Cells in Patients With Neuroblastoma. Blood (2011) 118(23):6050–6. doi: 10.1182/blood-2011-05-354449
126. Nakazawa Y, Huye LE, Salsman VS, Leen AM, Ahmed N, Rollins L, et al. PiggyBac-Mediated Cancer Immunotherapy Using EBV-Specific Cytotoxic T-Cells Expressing HER2-Specific Chimeric Antigen Receptor. Mol Ther (2011) 19(12):2133–43. doi: 10.1038/mt.2011.131
127. Caruana I, Weber G, Ballard BC, Wood MS, Savoldo B, Dotti G. K562-Derived Whole-Cell Vaccine Enhances Antitumor Responses of CAR-Redirected Virus-Specific Cytotoxic T Lymphocytes In Vivo. Clin Cancer Res (2015) 21(13):2952–62. doi: 10.1158/1078-0432.CCR-14-2998
128. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol (2017) 3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184
129. Bailey SR, Nelson MH, Majchrzak K, Bowers JS, Wyatt MM, Smith AS, et al. Human CD26(high) T Cells Elicit Tumor Immunity Against Multiple Malignancies via Enhanced Migration and Persistence. Nat Commun (2017) 8(1):1961. doi: 10.1038/s41467-017-01867-9
130. Lanier LL. Up on the Tightrope: Natural Killer Cell Activation and Inhibition. Nat Immunol (2008) 9(5):495–502. doi: 10.1038/ni1581
131. Hoerster K, Uhrberg M, Wiek C, Horn PA, Hanenberg H, Heinrichs S. HLA Class I Knockout Converts Allogeneic Primary NK Cells Into Suitable Effectors for "Off-the-Shelf" Immunotherapy. Front Immunol (2020) 11:586168. doi: 10.3389/fimmu.2020.586168
132. Moretta L, Moretta A. Unravelling Natural Killer Cell Function: Triggering and Inhibitory Human NK Receptors. EMBO J (2004) 23(2):255–9. doi: 10.1038/sj.emboj.7600019
133. Lin SJ, Kuo ML. Cytotoxic Function of Umbilical Cord Blood Natural Killer Cells: Relevance to Adoptive Immunotherapy. Pediatr Hematol Oncol (2011) 28(8):640–6. doi: 10.3109/08880018.2011.613092
134. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord Blood NK Cells Engineered to Express IL-15 and a CD19-Targeted CAR Show Long-Term Persistence and Potent Antitumor Activity. Leukemia (2018) 32(2):520–31. doi: 10.1038/leu.2017.226
135. Park YP, Choi SC, Kiesler P, Gil-Krzewska A, Borrego F, Weck J, et al. Complex Regulation of Human NKG2D-DAP10 Cell Surface Expression: Opposing Roles of the Gammac Cytokines and TGF-Beta1. Blood (2011) 118(11):3019–27. doi: 10.1182/blood-2011-04-346825
136. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
137. Mylod E, Lysaght J, Conroy MJ. Natural Killer Cell Therapy: A New Frontier for Obesity-Associated Cancer. Cancer Lett (2022) 535:215620. doi: 10.1016/j.canlet.2022.215620
138. Maluski M, Ghosh A, Herbst J, Scholl V, Baumann R, Huehn J, et al. Chimeric Antigen Receptor-Induced BCL11B Suppression Propagates NK-Like Cell Development. J Clin Invest (2019) 129(12):5108–22. doi: 10.1172/JCI126350
139. Sivori S, Parolini S, Falco M, Marcenaro E, Biassoni R, Bottino C, et al. 2B4 Functions as a Co-Receptor in Human NK Cell Activation. Eur J Immunol (2000) 30(3):787–93. doi: 10.1002/1521-4141(200003)30:3<787::AID-IMMU787>3.0.CO;2-I
140. Kato Y, Tanaka Y, Miyagawa F, Yamashita S, Minato N. Targeting of Tumor Cells for Human Gammadelta T Cells by Nonpeptide Antigens. J Immunol (2001) 167(9):5092–8. doi: 10.4049/jimmunol.167.9.5092
141. Deniger DC, Switzer K, Mi T, Maiti S, Hurton L, Singh H, et al. Bispecific T-Cells Expressing Polyclonal Repertoire of Endogenous Gammadelta T-Cell Receptors and Introduced CD19-Specific Chimeric Antigen Receptor. Mol Ther (2013) 21(3):638–47. doi: 10.1038/mt.2012.267
142. Hoeres T, Smetak M, Pretscher D, Wilhelm M. Improving the Efficiency of Vgamma9Vdelta2 T-Cell Immunotherapy in Cancer. Front Immunol (2018) 9:800. doi: 10.3389/fimmu.2018.00800
143. Airoldi I, Bertaina A, Prigione I, Zorzoli A, Pagliara D, Cocco C, et al. Gammadelta T-Cell Reconstitution After HLA-Haploidentical Hematopoietic Transplantation Depleted of TCR-Alphabeta+/CD19+ Lymphocytes. Blood (2015) 125(15):2349–58. doi: 10.1182/blood-2014-09-599423
144. Almeida AR, Correia DV, Fernandes-Platzgummer A, da Silva CL, da Silva MG, Anjos DR, et al. Delta One T Cells for Immunotherapy of Chronic Lymphocytic Leukemia: Clinical-Grade Expansion/Differentiation and Preclinical Proof of Concept. Clin Cancer Res (2016) 22(23):5795–804. doi: 10.1158/1078-0432.CCR-16-0597
145. Silva-Santos B, Serre K, Norell H. Gammadelta T Cells in Cancer. Nat Rev Immunol (2015) 15(11):683–91. doi: 10.1038/nri3904
146. De Rosa SC, Andrus JP, Perfetto SP, Mantovani JJ, Herzenberg LA, Herzenberg LA, et al. Ontogeny of Gamma Delta T Cells in Humans. J Immunol (2004) 172(3):1637–45. doi: 10.4049/jimmunol.172.3.1637
147. Fisher JP, Yan M, Heuijerjans J, Carter L, Abolhassani A, Frosch J, et al. Neuroblastoma Killing Properties of Vdelta2 and Vdelta2-Negative gammadeltaT Cells Following Expansion by Artificial Antigen-Presenting Cells. Clin Cancer Res (2014) 20(22):5720–32. doi: 10.1158/1078-0432.CCR-13-3464
148. Siegers GM, Lamb LS Jr. Cytotoxic and Regulatory Properties of Circulating Vdelta1+ Gammadelta T Cells: A New Player on the Cell Therapy Field? Mol Ther (2014) 22(8):1416–22. doi: 10.1038/mt.2014.104
149. Silva-Santos B, Strid J. Working in "NK Mode": Natural Killer Group 2 Member D and Natural Cytotoxicity Receptors in Stress-Surveillance by Gammadelta T Cells. Front Immunol (2018) 9:851. doi: 10.3389/fimmu.2018.00851
150. Raverdeau M, Cunningham SP, Harmon C, Lynch L. Gammadelta T Cells in Cancer: A Small Population of Lymphocytes With Big Implications. Clin Transl Immunol (2019) 8(10):e01080. doi: 10.1002/cti2.1080
151. Sebestyen Z, Prinz I, Dechanet-Merville J, Silva-Santos B, Kuball J. Translating Gammadelta (Gammadelta) T Cells and Their Receptors Into Cancer Cell Therapies. Nat Rev Drug Discov (2020) 19(3):169–84. doi: 10.1038/s41573-019-0038-z
152. Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, et al. IL-15 Protects NKT Cells From Inhibition by Tumor-Associated Macrophages and Enhances Antimetastatic Activity. J Clin Invest (2012) 122(6):2221–33. doi: 10.1172/JCI59535
153. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-Shelf' Allogeneic CAR T Cells: Development and Challenges. Nat Rev Drug Discov (2020) 19(3):185–99. doi: 10.1038/s41573-019-0051-2
154. Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The Inflammatory Micro-Environment in Tumor Progression: The Role of Tumor-Associated Macrophages. Crit Rev Oncol Hematol (2008) 66(1):1–9. doi: 10.1016/j.critrevonc.2007.07.004
155. Tachibana T, Onodera H, Tsuruyama T, Mori A, Nagayama S, Hiai H, et al. Increased Intratumor Valpha24-Positive Natural Killer T Cells: A Prognostic Factor for Primary Colorectal Carcinomas. Clin Cancer Res (2005) 11(20):7322–7. doi: 10.1158/1078-0432.CCR-05-0877
156. Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, et al. Enhanced Anti-Lymphoma Activity of CAR19-iNKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell (2018) 34(4):596–610.e11. doi: 10.1016/j.ccell.2018.08.017
157. Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti-GD2 CAR-NKT Cells in Patients With Relapsed or Refractory Neuroblastoma: An Interim Analysis. Nat Med (2020) 26(11):1686–90. doi: 10.1038/s41591-020-1074-2
158. Marin V, Kakuda H, Dander E, Imai C, Campana D, Biondi A, et al. Enhancement of the Anti-Leukemic Activity of Cytokine Induced Killer Cells With an Anti-CD19 Chimeric Receptor Delivering a 4-1BB-Zeta Activating Signal. Exp Hematol (2007) 35(9):1388–97. doi: 10.1016/j.exphem.2007.05.018
159. Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive Immunotherapy With Unselected or EBV-Specific T Cells for Biopsy-Proven EBV+ Lymphomas After Allogeneic Hematopoietic Cell Transplantation. Blood (2012) 119(11):2644–56. doi: 10.1182/blood-2011-08-371971
160. Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. Epstein-Barr Virus-Specific Human T Lymphocytes Expressing Antitumor Chimeric T-Cell Receptors: Potential for Improved Immunotherapy. Blood (2002) 99(6):2009–16. doi: 10.1182/blood.v99.6.2009
161. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-Specific T Cells Engineered to Coexpress Tumor-Specific Receptors: Persistence and Antitumor Activity in Individuals With Neuroblastoma. Nat Med (2008) 14(11):1264–70. doi: 10.1038/nm.1882
162. Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of Tumor-Targeted Human T Lymphocytes From Induced Pluripotent Stem Cells for Cancer Therapy. Nat Biotechnol (2013) 31(10):928–33. doi: 10.1038/nbt.2678
163. Wakao H, Yoshikiyo K, Koshimizu U, Furukawa T, Enomoto K, Matsunaga T, et al. Expansion of Functional Human Mucosal-Associated Invariant T Cells via Reprogramming to Pluripotency and Redifferentiation. Cell Stem Cell (2013) 12(5):546–58. doi: 10.1016/j.stem.2013.03.001
164. Mazza R, Maher J. Prospects for Development of Induced Pluripotent Stem Cell-Derived CAR-Targeted Immunotherapies. Arch Immunol Ther Exp (Warsz) (2021) 70(1):2. doi: 10.1007/s00005-021-00640-7
165. Larson RC, Maus MV. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat Rev Cancer (2021) 21(3):145–61. doi: 10.1038/s41568-020-00323-z
166. Ohnuma K, Takahashi N, Yamochi T, Hosono O, Dang NH, Morimoto C. Role of CD26/dipeptidyl Peptidase IV in Human T Cell Activation and Function. Front Biosci (2008) 13:2299–310. doi: 10.2741/2844
167. Fan H, Yan S, Stehling S, Marguet D, Schuppaw D, Reutter W. Dipeptidyl Peptidase IV/CD26 in T Cell Activation, Cytokine Secretion and Immunoglobulin Production. Adv Exp Med Biol (2003) 524:165–74. doi: 10.1007/0-306-47920-6_20
168. Bengsch B, Seigel B, Flecken T, Wolanski J, Blum HE, Thimme R. Human Th17 Cells Express High Levels of Enzymatically Active Dipeptidylpeptidase IV (Cd26). J Immunol (2012) 188(11):5438–47. doi: 10.4049/jimmunol.1103801
169. Suvas S, Kim B, Sarangi PP, Tone M, Waldmann H, Rouse BT. In Vivo Kinetics of GITR and GITR Ligand Expression and Their Functional Significance in Regulating Viral Immunopathology. J Virol (2005) 79(18):11935–42. doi: 10.1128/JVI.79.18.11935-11942.2005
170. Boroughs AC, Larson RC, Choi BD, Bouffard AA, Riley LS, Schiferle E, et al. Chimeric Antigen Receptor Costimulation Domains Modulate Human Regulatory T Cell Function. JCI Insight (2019) 5:e126194. doi: 10.1172/jci.insight.126194
171. French RR, Taraban VY, Crowther GR, Rowley TF, Gray JC, Johnson PW, et al. Eradication of Lymphoma by CD8 T Cells Following Anti-CD40 Monoclonal Antibody Therapy is Critically Dependent on CD27 Costimulation. Blood (2007) 109(11):4810–5. doi: 10.1182/blood-2006-11-057216
Keywords: CAR-T cells, solid tumor, receptors, co-stimulatory domains, cytokine, immune cells
Citation: Yu T, Yu S-k, Xiang Y, Lu K-H and Sun M (2022) Revolution of CAR Engineering For Next-Generation Immunotherapy In Solid Tumors. Front. Immunol. 13:936496. doi: 10.3389/fimmu.2022.936496
Received: 05 May 2022; Accepted: 16 June 2022;
Published: 12 July 2022.
Edited by:
John - Maher, King’s College London, United KingdomReviewed by:
Yangqiu Li, Jinan University, ChinaYozo Nakazawa, Shinshu University School of Medicine, Japan
Demin Li, University of Oxford, United Kingdom
Copyright © 2022 Yu, Yu, Xiang, Lu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai-Hua Lu, bHVrYWlodWFAbmptdS5lZHUuY24=; Ming Sun, c3VubWluZzM0OEBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work