 Hasan Hashem
Hasan Hashem Dimana Dimitrova
Dimana Dimitrova Isabelle Meyts
Isabelle Meyts
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 07 July 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.932385
This article is part of the Research Topic DADA2 and other Monogenic Vasculitides View all 9 articles
Deficiency of adenosine deaminase 2 (DADA2) is an inherited autosomal recessive disease characterized by autoinflammation (recurrent fever), vasculopathy (livedo racemosa, polyarteritis nodosa, lacunar ischemic strokes, and intracranial hemorrhages, end organ vasculitis), immunodeficiency, lymphoproliferation, immune cytopenias, and bone marrow failure. Allogeneic hematopoietic cell transplantation (HCT) is curative for DADA2 as it reverses the hematological, immune and vascular phenotype of DADA2. The primary goal of HCT in DADA2, like in other non-malignant diseases, is engraftment with the establishment of normal hematopoiesis and normal immune function. Strategies in selecting a preparative regimen should take into consideration the specific vulnerabilities to endothelial dysfunction and liver toxicity in DADA2 patients. Overcoming an increased risk of graft rejection while minimizing organ toxicity, graft-versus-host disease, and infections can be particularly challenging in DADA2 patients. This review will discuss approaches to HCT in DADA2 patients including disease-specific considerations, barriers to successful engraftment, post-HCT complications, and clinical outcomes of published patients with DADA2 who have undergone HCT to date.
The deficiency of adenosine deaminase 2 (DADA2), initially described in 2014, caused by biallelic deleterious mutations in the cat eye chromosome region 1 gene (CECR1, subsequently renamed ADA2), is a rare autosomal recessive inborn error of immunity disease (1, 2). DADA2 diagnosis is based on an absence or low levels of plasma ADA2 enzymatic activity, and the demonstration of biallelic loss-of-function mutations of ADA2 (3, 4). DADA2 was initially recognized as a syndrome that manifests with fevers, mild immunodeficiency, polyarteritis nodosa, and early-onset stroke (1, 2, 5, 6). The clinical phenotype has expanded significantly since it was first described in 2014 to also include bone marrow failure (e.g. pure red cell aplasia), autoimmune cytopenias, and liver disease (7–9).
Treatment of DADA2 is challenging and case mortality is estimated to be around 8%, mostly in childhood and related to vasculopathy-associated complications and infections (10–13). Immunosuppressants such as corticosteroids are often the choice for initial treatment efforts, albeit with variable success. Other agents such as azathioprine, cyclosporine, tacrolimus, mycophenolate mofetil, cyclophosphamide, and methotrexate have all been used depending on phenotype specifics, but with little or transient effect. At present, the mainstay of treatment of vasculopathy and autoinflammation phenotypes consists of anti-TNF-agents (etanercept, infliximab, adalimumab) with impressive reductions in stroke incidence (14, 15). Nevertheless, anti-TNF-agents were not effective or have shown only modest control of the hematological and severe immunodeficiency phenotypes (14, 16). Moreover, breakthrough inflammation has also been described (14). In these patients, HCT is the only curative treatment until present (17, 18).
The pathophysiology of DADA2 remains unclear but increased neutrophil extracellular trap formation (NETosis) and reduced M2 macrophage differentiation have been proposed (1, 19, 20). ADA2 is predominantly expressed in monocytes, and the immunological profile of DADA2 patients indeed shows a polarization toward proinflammatory M1 macrophages (21–23). Activation of multiple inflammatory responses, including type I and II interferon (IFN) pathways and production of the proinflammatory cytokines IL-1B, IL-6, and TNF-alpha, in turn results in tissue damage and reduced endothelial integrity (20, 21, 24–28).
At present time, more than 240 patients have been reported in the literature (10, 11, 29–31). The onset of disease is usually in childhood but adult-onset has also been described with the oldest patients reported at 59 years of age (27, 31, 32). Presentations are highly heterogenous, rendering early diagnosis difficult. Even within single kindreds, there is tremendous variability in DADA2 phenotypes and sometimes incomplete or variable penetrance (4). Some insight into a potential genotype-phenotype correlation was provided by Lee et al. who suggested that the phenotype is dependent on the residual ADA2 enzyme activity function, as measured in the supernatant of a HEK293T overexpression model (16).
Since DADA2 was first described in 2014, the phenotype has been extended significantly to include not only vasculopathy and autoinflammation but also immunodeficiency and hematologic manifestations. Vasculopathy of small- and medium-sized arteries is a major clinical feature of DADA2 with skin and central nervous system most commonly involved, ranging from livedo racemosa and polyarteritis nodosa (PAN) to intracranial vasculopathy with lacunar ischemic strokes and hemorrhages. Autoinflammation with recurrent fevers and elevated CRP and ESR are also reported in more than half of patients (31). Recurrent bacterial and viral infections have been described, with exceptional susceptibility to herpes virus infection, especially CMV and EBV (33–36). Laboratory evaluations may show lymphopenia, hypogammaglobulinemia, and decreased memory B-cells (32, 37). Hematological manifestations, particularly as an indication for HCT, have been increasingly appreciated in recent years. Pure red cell aplasia (PRCA) was described initially in three patients and further confirmed by additional reports (7, 8, 38). Other patients present with neutropenia, thrombocytopenia, pancytopenia, and autoimmune cytopenias (8, 17, 18, 39). Rarely, some DADA2 patients can present with hemophagocytic lymphohistiocytosis (HLH). Bone marrow biopsies show reticulin fibrosis and characteristic lymphoid aggregates (13, 31). Lymphoproliferation is another important feature; autoimmune lymphoproliferative syndrome-like presentation, clonal lymphoproliferation, T-large granular lymphocytosis, and lymphoma have all been described (40–42).
Currently, there are 36 patients who underwent 45 HCT for DADA2 in 15 countries and 24 centers (18, 31, 43, 44). Indications for HCT included PRCA, neutropenia, combined RCA and immune-mediated neutropenia, pancytopenia, autoimmune hemolytic anemia (AIHA), diffuse large B-cell lymphoma (DLBL), immune dysregulation, severe aplastic anemia, and severe lymphopenia (Table 1).
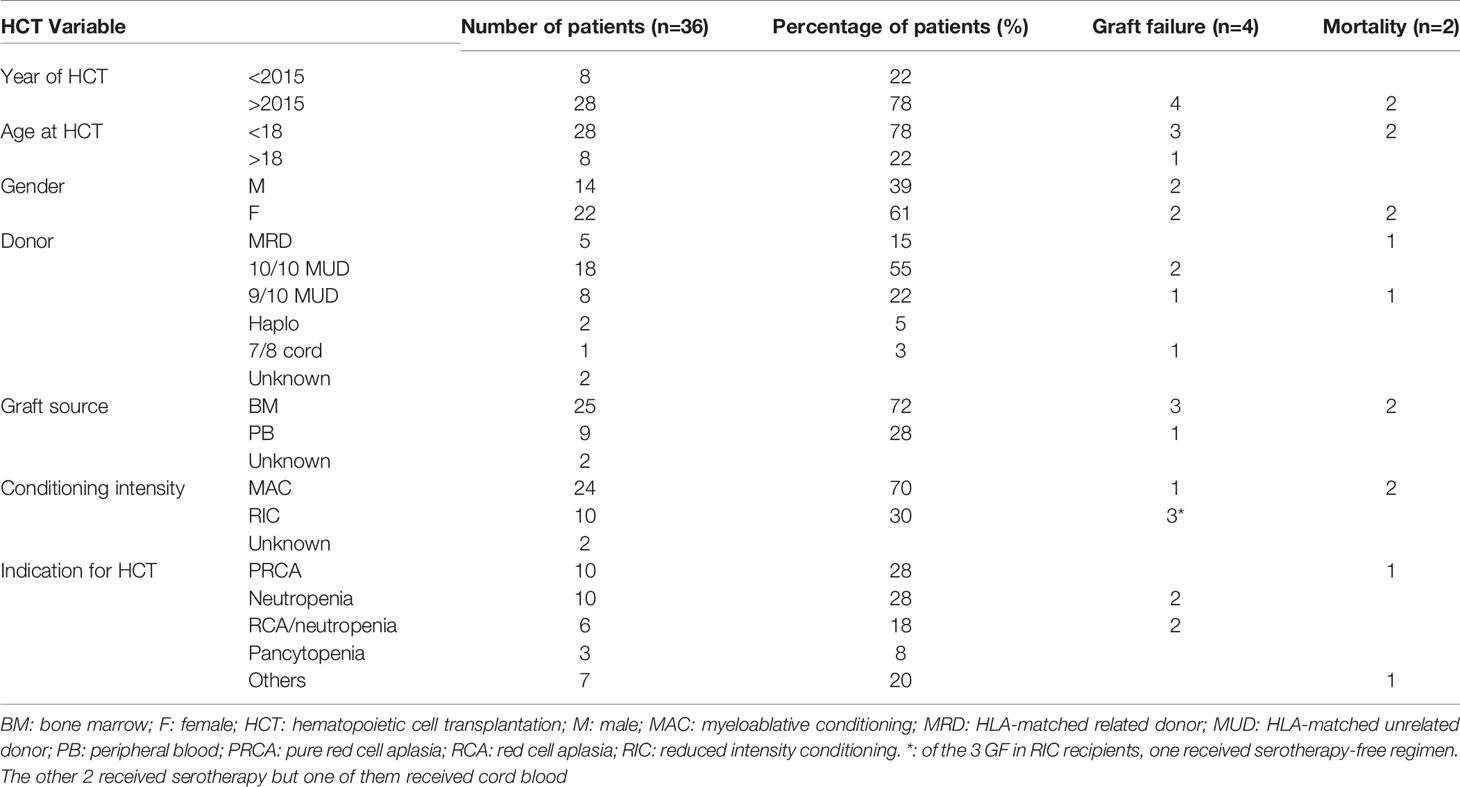
Table 1 HCT characteristics and outcomes for published DADA2 patients .
All but 2 of the patients are alive and well and are cured at a median follow-up of two years with an overall survival of >95%. The majority of patients were transplanted after 2015 and were children. Almost half of HCTs were from HLA-matched unrelated donors. Myeloablative conditioning and bone marrow as a graft source constituted more than two thirds of HCTs (Table 1).
Anti-TNF agents are the mainstay of treatment of vasculopathy and autoinflammation phenotypes and it is recommended that patients already on these agents, who show some response, continue therapy until conditioning for HCT starts, even until engraftment. This might help decrease inflammation associated with DADA2 and decrease peri-transplant complications. Moreover, some patients withDADA2 especially those with liver disease and/or iron overload due to frequent blood transfusions, might benefit from iron chelation prior to HCT.
The choice of preparative regimen should first be guided by the patient’s disease phenotype and end-organ status (Table 2). Previous cohort reports have pointed to a specific hepatic vulnerability in DADA2 patients, with some patients developing chronic liver disease post-HCT even in the absence of clinically overt liver disease prior to HCT especially in patients with PRCA who tend to have iron overload due to previous frequent blood transfusions (17, 18, 31). A careful work-up of the patient, including at least transient elastography, imaging, or preferably, liver biopsy, seems mandatory to adequately assess the status of the liver, going into HCT. Depending on the availability of specific drugs, a less toxic regiment may be used, such as treosulfan instead of busulfan. Also the combination of sirolimus as a GvHD prophylaxis after busulfan conditioning should be avoided (45, 46). Center-specific sinusoidal obstruction syndrome prophylaxis should be adhered to (ursodiol, relative fluid restriction and defibrotide if available). For DADA2 patients who present with HLH, specific conditioning regimens similar to the ones used for HLH might be advised. It is also important to perform a careful assessment of other end organs, including renal imaging and function assessment (microalbuminuria, proteinuria) esp. given the added renal toxicity of most immunosuppressants and of anti-viral agents.
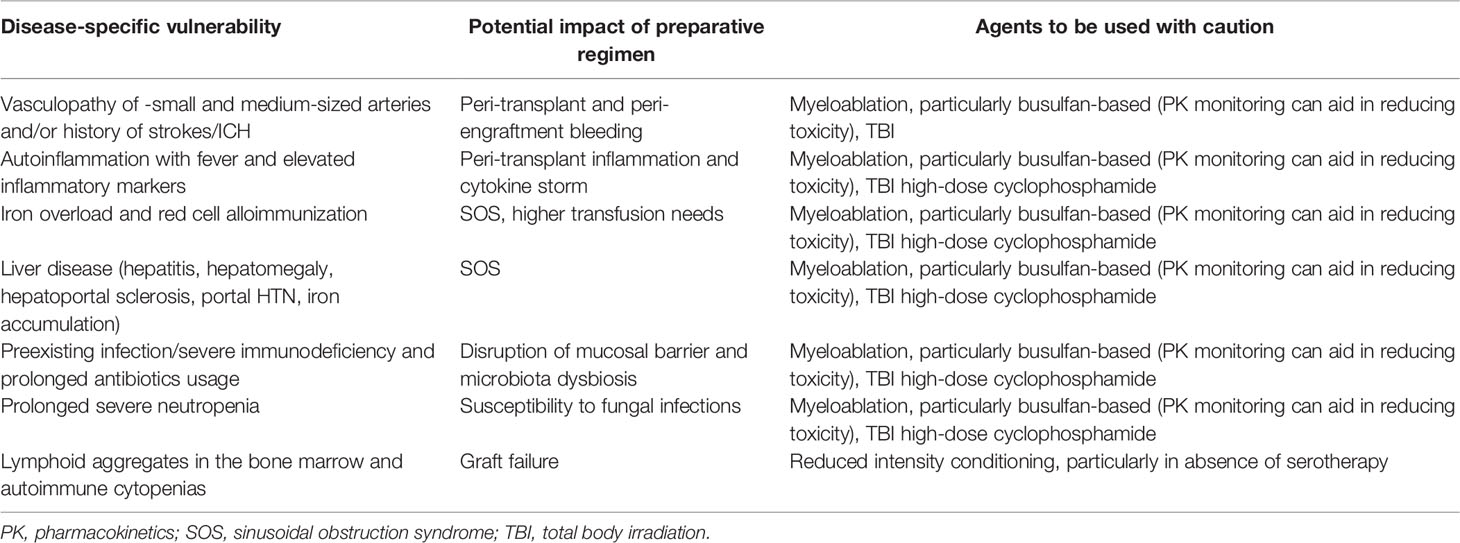
Table 2 DADA2 specific-disease vulnerabilities.
From the reports thus far, myeloablative conditioning regimens have been most commonly used although reduced intensity conditioning has also been successfully used. The most commonly used regimen was treosulfan/fludarabine +/- thiotepa with rabbit ATG or alemtuzumab, reflecting protocols A, B, C, D from a recent IEWP-EBMT update (47). Most patients transplanted received serotherapy, with the choice for either rabbit ATG or alemtuzumab mostly dependent on the center preference. In the context of serotherapy and given the specific vulnerability to viral infections esp. herpesvirus infections, in DADA2, careful monitoring of viremia (EBV, CMV, Adv, HHV6) and prophylaxis when available are advised.
Potentially more important than any other consideration is that these HCTs should be performed by an experienced HCT team, in a center with the necessary HCT expertise available, as well as the necessary logistics such as quick turnaround chimerism analysis etc. An equally important consideration is the timing of HCT –given the risk of life-threatening mortality and morbidity associated with this condition, we suggest that, as with other inherited disorders of immunity, HCT early in life for patients with severe phenotypes is preferred before organ damage has evolved, notwithstanding successes of adolescent and adult HCTs.
All related donors under consideration must be screened for ADA2 mutations, even if they appear clinically healthy. Prior to elucidation of disease genetics, at least two known HCT procedures were performed using donors subsequently found to have biallelic pathogenic mutations (17, 18). Ideally, plasma ADA2 levels should also be checked (3, 48).
Because DADA2 is inherited in an autosomal recessive fashion, mutation-negative related donor options are often scarce. Thus, the question of whether to prioritize a matched unrelated donor over a carrier matched sibling, or whether to use a heterozygous related donor at all in the absence of unrelated donor options, arises frequently. Evaluations of subjects with monoallelic mutations have shown significant variability. Most are healthy with a plasma ADA2 concentration intermediate between a healthy control and a patient with biallelic ADA2 defects, but clinically affected carriers with both detectable ADA2 and undetectable ADA2 levels have been described (1, 48–50). Furthermore, adult onset or mild disease manifestations have been described in carriers, along with in vitro abnormalities such as enhanced NET formation, suggesting the potential for an immunological phenotype that may not be readily apparent in a young prospective donor (20, 50).
Utilizing a heterozygous donor therefore raises theoretical concerns regarding the possibility of developing disease manifestations over time, even in the context of full donor chimerism. Furthermore, because the donor chimerism sufficient for phenotype reversal is unknown, recipients of a graft from a carrier donor could be more prone to developing disease manifestations in the setting of mixed chimerism. Even using an unaffected donor, mixed whole blood chimerism up to 30% donor was associated with a decline in plasma ADA2 enzyme activity and new hematological DADA2 disease manifestations in one patient (17, 18, 51).
Four HCTs have been performed using a clinically healthy donor with a confirmed monoallelic ADA2 mutation. In one, bone marrow collection yielded insufficient CD34+ cell counts (0.5x10^6/kg), requiring a subsequent stem cell boost, while bone marrow collection yields were as expected in the other case. These two patients are clinically well with full donor chimerism and without major complications at 3 and 6 years post-HCT. A third patient died, with further details not available (17, 18). A fourth patient received a salvage HCT from her carrier mother after prior graft failure and has continued reversal of disease phenotype in the setting of full donor chimerism >2 years later (31). Given a risk of graft instability, particularly in DADA2 patients with immune-mediated neutropenia, donor selection must also take into consideration factors related to donor availability and logistics, as unplanned donor cell infusions may be necessary to prevent graft rejection (17, 18, 31).
Of 36 known patients who received HCT for DADA2 to date, graft failure occurred in four patients (primary, n=1; secondary, n=3), while declining chimerism was observed in two patients despite myeloablative conditioning (18, 31). All three patients with secondary graft failure had neutropenia as an indication for HCT, associated with increased T cell infiltration in the bone marrow along with a myeloid maturation arrest at baseline. Remarkably, two of these patients had continued graft instability even after subsequent HCT, manifested by repeated rapid declines in donor T cell chimerism (0% and 9% with myeloid chimerism of 60% and 78%, respectively), coinciding with development of dense neutropenia and necessitating prompt immunosuppression as well as multiple donor lymphocyte infusions. In all three cases, graft stabilization was tied to development of graft-versus-host disease with presumed concurrent graft-versus-marrow effect (31). Interestingly, a fourth patient, who experienced declining donor chimerism (30% whole blood, 0% CD3) rescued with two stem cell boosts, developed new agranulocytosis as ADA2 levels declined in the context of a viral reactivation. Further details to confirm whether host T cells were implicated as in the other patients are not available. A fifth patient, who was transplanted for pure red cell aplasia without neutropenia, tolerated a decline in donor chimerism to 55% without disease manifestations or need for subsequent cell infusion (18).
Thus, a subset of DADA2 patients with T-cell-mediated neutropenia may be more prone to graft rejection and should be counseled accordingly, taking this risk into consideration in planning the most appropriate HCT platform, graft, and post-HCT monitoring. In clinical practice, these HCT candidates may have a history of neutropenia responsive to T lymphocyte-targeted immunosuppressive methods, such as high-dose corticosteroids or cyclosporine. Serotherapy-containing conditioning platforms resulting in robust lymphodepletion are most effective for such patients, while myeloablation is not protective and likely not necessary, and may be particularly harmful in a patient population that is already vulnerable to endothelial damage and hepatic complications. Other strategies aimed at reducing graft rejection may include utilizing T-cell replete grafts, peripheral blood stem cell grafts, and/or higher graft doses. Given the potential for extremely rapid graft rejection, donor chimerism including T cell chimerism should be monitored closely and frequently post-HCT. Unexpected declines in neutrophil count should raise concern, and augmenting immunosuppression may be considered to stall resurgence of host T cell-mediated destruction. Ideally, HCT planning should include a contingency plan to ensure prompt collection and infusion of additional donor cells can be readily arranged if threatened graft failure is identified, although certain patients may benefit from additional donor cell infusion (lymphocytes or stem cell boost, depending on the clinical scenario) or even require a second HCT using a different donor.
HCT is a potentially life-saving and definitive treatment for DADA2 as it reverses the hematological, immunological and vascular phenotype of the disease. Most HCT procedures reported thus far were performed in children with a bone marrow failure phenotype. Overall survival is excellent, but graft failure is a frequent complication, and optimizing immunoablation through use of serotherapy containing regimens may be beneficial. Careful attention to the presence of end-organ disease, particularly liver dysfunction, is warranted in all DADA2 patients but especially for those undergoing HCT. Continued follow-up of DADA2 patients receiving HCT will aid us in defining the most appropriate HCT regimens in DADA2.
HH, DD, and IM designed the study, reviewed the literature, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
IM is a Senior Clinical Investigator at the FWO – Flanders, and is supported by the KU Leuven C1 grant C16/18/007, by a VIB GC PID Grant, by the FWO Grants G0B5120N (DADA2) and G0E8420N and by the Jeffrey Modell Foundation. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 948959).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the DADA2 foundation (www.dada2.org) for organizing the Inaugural International Conference on Deficiency of ADA2, with special thanks to Chip Chambers, MD for forming the foundation. We thank the patients and their parents for confiding in us.
1. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-Onset Stroke and Vasculopathy Associated With Mutations in ADA2. N Engl J Med (2014) 370(10):911–20. doi: 10.1056/NEJMoa1307361
2. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant Adenosine Deaminase 2 in a Polyarteritis Nodosa Vasculopathy. N Engl J Med (2014) 370(10):921–31. doi: 10.1056/NEJMoa1307362
3. Hashem H, Kelly SJ, Ganson NJ, Hershfield MS. Deficiency of Adenosine Deaminase 2 (DADA2), an Inherited Cause of Polyarteritis Nodosa and a Mimic of Other Systemic Rheumatologic Disorders. Curr Rheumatol Rep Curr Rheumatol Reports (2017) 19(11):70. doi: 10.1007/s11926-017-0699-8
4. Schnappauf O, Zhou Q, Moura NS, Ombrello AK, Michael DG, Deuitch N, et al. Deficiency of Adenosine Deaminase 2 (DADA2): Hidden Variants, Reduced Penetrance, and Unusual Inheritance. J Clin Immunol J Clin Immunol (2020) 40(6):917–26. doi: 10.1007/s10875-020-00817-3
5. Caorsi R, Penco F, Schena F, Gattorno M. Monogenic Polyarteritis: The Lesson of ADA2 Deficiency. Pediatr Rheumatol (2016) 14(1):51. doi: 10.1186/s12969-016-0111-7
6. Özen S, Batu ED, Taşkiran EZ, Özkara HA, Ünal ŞVerifytat, Güleray N, et al. A Monogenic Disease With a Variety of Phenotypes: Deficiency of Adenosine Deaminase 2. J Rheumatol (2020) 47(1):117–25. doi: 10.3899/jrheum.181384
7. Hashem H, Egler R, Dalal J. Refractory Pure Red Cell Aplasia Manifesting as Deficiency of Adenosine Deaminase 2. J Pediatr Hematol Oncol (2017) 39(5):e293–6. doi: 10.1097/MPH.0000000000000805
8. Ben-Ami T, Revel-Vilk S, Brooks R, Shaag A, Hershfield MS, Kelly SJ, et al. Extending the Clinical Phenotype of Adenosine Deaminase 2 Deficiency. J Pediatr (2016) 177:316–20. doi: 10.1016/j.jpeds.2016.06.058
9. Van Eyck L, Liston A, Wouters C. Mutant ADA2 in Vasculopathies. N Engl J Med (2014) 371(5):480. doi: 10.1056/NEJMc1405506
10. Meyts I, Aksentijevich I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. J Clin Immunol (2018) 38(5):569–78. doi: 10.1007/s10875-018-0525-8
11. Sahin S, Adrovic A, Barut K, Ugurlu S, Turanli ET, Ozdogan H, et al. Clinical, Imaging and Genotypical Features of Three Deceased and Five Surviving Cases With ADA2 Deficiency. Rheumatol Int (2017) 1:129–36. doi: 10.1007/s00296-017-3740-3
12. Sharma A, Naidu G, Sharma V, Jha S, Dhooria A, Dhir V, et al. Deficiency of Adenosine Deaminase 2 (DADA2) in Adults and Children: Experience From India. Arthritis Rheumatol (2020) 73(2):276–85. doi: 10.1002/art.41500
13. Batu ED, Karadag O, Taskiran EZ, Kalyoncu U, Aksentijevich I, Alikasifoglu M, et al. A Case Series of Adenosine Deaminase 2-Deficient Patients Emphasizing Treatment and Genotype-Phenotype Correlations in Rheumatology and Related Fields. J Rheumatol (2015) 42(8):1532–4. doi: 10.3899/jrheum.150024
14. Cooray S, Omyinmi E, Hong Y, Papadopoulou C, Harper L, Al-Abadi E, et al. Anti-Tumour Necrosis Factor Treatment for the Prevention of Ischaemic Events in Patients With Deficiency of Adenosine Deaminase 2 (DADA2). Rheumatol. (2021) 60(9):4373–8. doi: 10.1093/rheumatology/keaa837
15. Ombrello AK, Qin J, Bethesda ID, Hoffmann PM, Kumar P, Stone D, et al. Treatment Strategies for Deficiency of Adenosine Deaminase 2. HHS Public Access (2020) 380(16):1582–4. doi: 10.1056/NEJMc1801927
16. Lee PY, Kellner ES, Huang Y, Furutani E, Huang Z, Bainter W, et al. Genotype and Functional Correlates of Disease Phenotype in Deficiency of Adenosine Deaminase 2 (DADA2). J Allergy Clin Immunol (2020) 145(6):1664–72.e10. doi: 10.1016/j.jaci.2019.12.908
17. Hashem H, Kumar AR, Müller I, Babor F, Bredius R, Dalal J, et al. Hematopoietic Stem Cell Transplantation Rescues the Hematological, Immunological, and Vascular Phenotype in DADA2. Blood (2017) 130(24):2682–8. doi: 10.1182/blood-2017-07-798660
18. Hashem H, Bucciol G, Ozen S, Unal S, Bozkaya IO, Akarsu N, et al. Hematopoietic Cell Transplantation Cures Adenosine Deaminase 2 Deficiency: Report on 30 Patients. J Clin Immunol (2021) 41(7):1633–47. doi: 10.1007/s10875-021-01098-0
19. Belot A, Wassmer E, Twilt M, Lega J-C, Zeef LA, Oojageer A, et al. Mutations in CECR1 Associated With a Neutrophil Signature in Peripheral Blood. Pediatr Rheumatol Online J (2014) 12(1):44. doi: 10.1186/1546-0096-12-44
20. Carmona-Rivera C, Khaznadar SS, Shwin KW, Irizarry-Caro JA, O’Neil LJ, Liu Y, et al. Deficiency of Adenosine Deaminase 2 Triggers Adenosine-Mediated NETosis and TNF Production in Patients With DADA2. Blood (2019) 134(4):395–406. doi: 10.1182/blood.2018892752
21. Martinon F, Aksentijevich I. New Players Driving Inflammation in Monogenic Autoinflammatory Diseases. Nat Rev Rheumatol Nat Publ Group (2014) 11(1):11–20. doi: 10.1038/nrrheum.2014.158
22. Zavialov AV, Gracia E, Glaichenhaus N, Franco R, Zavialov AV. Human Adenosine Deaminase 2 Induces Differentiation of Monocytes Into Macrophages and Stimulates Proliferation of T Helper Cells and Macrophages. J Leukoc Biol (2010) 88:279–90. doi: 10.1189/jlb.1109764
23. Kaljas Y, Liu C, Skaldin M, Wu C, Zhou Q, Lu Y, et al. Human Adenosine Deaminases ADA1 and ADA2 Bind to Different Subsets of Immune Cells. Cell Mol Life Sci (2017) 74(3):555–70. doi: 10.1007/s00018-016-2357-0
24. Dhanwani R, Takahashi M, Mathews IT, Lenzi C, Romanov A, Watrous JD, et al. Cellular Sensing of Extracellular Purine Nucleosides Triggers an Innate IFN-β Response. Sci Adv (2020) 6(30):eaba3688. doi: 10.1126/sciadv.aba3688
25. Nihira H, Izawa K, Ito M, Umebayashi H, Okano T, Kajikawa S, et al. Detailed Analysis of Japanese Patients With Adenosine Deaminase 2 Deficiency Reveals Characteristic Elevation of Type II Interferon Signature and STAT1 Hyperactivation. J Allergy Clin Immunol Elsevier Inc.; (2021) 148(2):550–62. doi: 10.1016/j.jaci.2021.01.018
26. Insalaco A, Moneta GM, Pardeo M, Caiello I, Messia V, Bracaglia C, et al. Variable Clinical Phenotypes and Relation of Interferon Signature With Disease Activity in ADA2 Deficiency. J Rheumatol (2019) 46(5):523–6. doi: 10.3899/jrheum.180045
27. Betrains A, Staels F, Moens L, Delafontaine S, Hershfield MS, Blockmans D, et al. Diagnosis of Deficiency of Adenosine Deaminase Type 2 in Adulthood. Scand J Rheumatol (2021) 50(6):493–6. doi: 10.1080/03009742.2021.1881156
28. Lee PY, Aksentijevich I, Zhou Q. Mechanisms of Vascular Inflammation in Deficiency of Adenosine Deaminase 2 (DADA2). Semin Immunopathol (2022) 44(3):269–80. doi: 10.1007/s00281-022-00918-8
29. Kendall JL, Springer JM. The Many Faces of a Monogenic Autoinflammatory Disease: Adenosine Deaminase 2 Deficiency. Curr Rheumatol Rep (2020) 22(10):64. doi: 10.1007/s11926-020-00944-1
30. Zhang B, Xu N, Chen J, Zhang S, Huang X, Shen M, et al. Treatment and Outcome in Deficiency of Adenosine Deaminase 2: A Literature Review. J Investig Allergol Clin Immunol (2022) 32(1):13–22. doi: 10.18176/jiaci.0748
31. Barron KS, Aksentijevich I, Deuitch NT, Stone DL, Hoffmann P, Videgar-Laird R, et al. The Spectrum of the Deficiency of Adenosine Deaminase 2: An Observational Analysis of a 60 Patient Cohort. Front Immunol (2022) 12:1–18. doi: 10.3389/fimmu.2021.811473
32. Yap JY, Moens L, Lin MW, Kane A, Kelleher A, Toong C, et al. Intrinsic Defects in B Cell Development and Differentiation, T Cell Exhaustion and Altered Unconventional T Cell Generation Characterize Human Adenosine Deaminase Type 2 Deficiency. J Clin Immunol Springer US (2021) 41(8):1915–35. doi: 10.1007/s10875-021-01141-0
33. Schepp J, Bulashevska A, Mannhardt-Laakmann W, Cao H, Yang F, Seidl M, et al. Deficiency of Adenosine Deaminase 2 Causes Antibody Deficiency. J Clin Immunol (2016) 36(3):179–86. doi: 10.1007/s10875-016-0245-x
34. Schepp J, Proietti M, Frede N, Buchta M, Hübscher K, Rojas Restrepo J, et al. Screening of 181 Patients With Antibody Deficiency for Deficiency of Adenosine Deaminase 2 Sheds New Light on the Disease in Adulthood. Arthritis Rheumatol (2017) 69(8):1689–700. doi: 10.1002/art.40147
35. Le Voyer T, Boutboul D, Ledoux-Pilon A, de Fontbrune FS, Boursier G, Latour S, et al. Late-Onset EBV Susceptibility and Refractory Pure Red Cell Aplasia Revealing Dada2. J Clin Immunol (2020) 40(6):948–53. doi: 10.1007/s10875-020-00812-8
36. Brooks JP, Rice AJ, Ji W, Lanahan SM, Konstantino M, Dara J, et al. Uncontrolled Epstein-Barr Virus as an Atypical Presentation of Deficiency in ADA2 (Dada2). J Clin Immunol (2021) 41(3):680–3. doi: 10.1007/s10875-020-00940-1
37. Schena F, Penco F, Volpi S, Pastorino C, Caorsi R, Kalli F, et al. Dysregulation in B-Cell Responses and T Follicular Helper Cell Function in ADA2 Deficiency Patients. Eur J Immunol (2020) 57(1):206–19. doi: 10.1002/eji.202048549
38. Hashem H, Vatsayan A, Gupta A, Nagle K, Hershfield M, Dalal J. Successful Reduced Intensity Hematopoietic Cell Transplant in a Patient With Deficiency of Adenosine Deaminase 2. Bone Marrow Transpl (2017) 52(11):1575–6. doi: 10.1038/bmt.2017.173
39. Van Eyck L, Hershfield MS, Pombal D, Kelly SJ, Ganson NJ, Moens L, et al. Hematopoietic Stem Cell Transplantation Rescues the Immunologic Phenotype and Prevents Vasculopathy in Patients With Adenosine Deaminase 2 Deficiency. J Allergy Clin Immunol (2015) 135(1):283–7. doi: 10.1016/j.jaci.2014.10.010
40. Alabbas F, Elyamany G, Alsharif O, Hershfield M, Meyts I. Childhood Hodgkin Lymphoma: Think Dada2. J Clin Immunol J Clin Immunol (2019) 39(1):26–9. doi: 10.1007/s10875-019-0590-7
41. Barzaghi F, Minniti F, Mauro M, De Bortoli M, Balter R, Bonetti E, et al. ALPS-Like Phenotype Caused by ADA2 Deficiency Rescued by Allogeneic Hematopoietic Stem Cell Transplantation. Front Immunol (2019) 9:2767. doi: 10.3389/fimmu.2018.02767
42. Trotta L, Martelius T, Siitonen T, Hautala T, Hämäläinen S, Juntti H, et al. ADA2 Deficiency: Clonal Lymphoproliferation in a Subset of Patients. J Allergy Clin Immunol (2018) 141(4):1534–7.e8. doi: 10.1016/j.jaci.2018.01.012
43. Liu L, Wang W, Wang Y, Hou J, Ying W, Hui X, et al. A Chinese DADA2 Patient: Report of Two Novel Mutations and Successful HSCT. Immunogenetics (2019) 71(4):299–305. doi: 10.1007/s00251-018-01101-w
44. Gibson KM, Morishita KA, Dancey P, Moorehead P, Drögemöller B, Han X, et al. Identification of Novel Adenosine Deaminase 2 Gene Variants and Varied Clinical Phenotype in Pediatric Vasculitis. Arthritis Rheumatol (2019) 71(10):1747–55. doi: 10.1002/art.40913
45. Cutler C, Stevenson K, Kim HT, Richardson P, Ho VT, Linden E, et al. Sirolimus is Associated With Veno-Occlusive Disease of the Liver After Myeloablative Allogeneic Stem Cell Transplantation. Blood (2008) 112(12):4425–31. doi: 10.1182/blood-2008-07-169342
46. Mohty M, Malard F, Abecasis M, Aerts E, Alaskar AS, Aljurf M, et al. Prophylactic, Preemptive, and Curative Treatment for Sinusoidal Obstruction Syndrome/Veno-Occlusive Disease in Adult Patients: A Position Statement From an International Expert Group. Bone Marrow Transpl Springer US (2020) 55(3):485–95. doi: 10.1038/s41409-019-0705-z
47. Lankester AC, Albert MH, Booth C, Gennery AR, Güngör T, Hönig M, et al. EBMT/ESID Inborn Errors Working Party Guidelines for Hematopoietic Stem Cell Transplantation for Inborn Errors of Immunity. Bone Marrow Transpl Springer US (2021) 56(9):2052–62. doi: 10.1038/s41409-021-01378-8
48. Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 Deficiency ( DADA2 ) as an Unrecognised Cause of Early Onset Polyarteritis Nodosa and Stroke : A Multicentre National Study. Ann Rheum Dis (2017) 76(10):1648–56. doi: 10.1136/annrheumdis-2016-210802
49. Schnappauf O, Ombrello AK, Kastner DL. Deficiency of Adenosine Deaminase 2: Is it an Elephant After All? J Allergy Clin Immunol (2020) 145(6):1560–1. doi: 10.1016/j.jaci.2020.04.023
50. Moi L, Schnider C, Riccio O, Hershfield MS, Candotti F. Common Variable Immunodeficiency in a Carrier of the ADA2 R169Q Variant: Coincidence or Causality? J Clin Immunol (2022), 3–5. doi: 10.1007/s10875-022-01271-z
51. Bucciol G, Delafontaine S, Segers H, Bossuyt X, Hershfield MS, Moens L, et al. Hematopoietic Stem Cell Transplantation in ADA2 Deficiency: Early Restoration of ADA2 Enzyme Activity and Disease Relapse Upon Drop of Donor Chimerism. J Clin Immunol (2017) 37(8):746–50. doi: 10.1007/s10875-017-0449-8
Keywords: hematopoietic cell transplantation, HCT, deficiency of adenosine deaminase 2, DADA2, inborn error of immunity, bone marrow failure, immunodeficiency, immune dysregulation
Citation: Hashem H, Dimitrova D and Meyts I (2022) Allogeneic Hematopoietic Cell Transplantation for Patients With Deficiency of Adenosine Deaminase 2 (DADA2): Approaches, Obstacles and Special Considerations. Front. Immunol. 13:932385. doi: 10.3389/fimmu.2022.932385
Received: 29 April 2022; Accepted: 20 June 2022;
Published: 07 July 2022.
Edited by:
Pui Y. Lee, Boston Children’s Hospital and Harvard Medical School, United StatesReviewed by:
Maria Pia Cicalese, San Raffaele Scientific Institute (IRCCS), ItalyCopyright © 2022 Hashem, Dimitrova and Meyts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hasan Hashem, SEguMDg4NDdAa2hjYy5qbw==; Isabelle Meyts, SXNhYmVsbGUubWV5dHNAdXpsZXV2ZW4uYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.