 Martin Kolev
Martin Kolev Madhumita Das
Madhumita Das Monica Gerber2
Monica Gerber2 Scott Baver
Scott Baver Maciej M. Markiewski
Maciej M. Markiewski
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 July 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.931273
This article is part of the Research TopicInsights in Cancer Immunity and Immunotherapy: 2021View all 9 articles
The role of complement in cancer has received increasing attention over the last decade. Recent studies provide compelling evidence that complement accelerates cancer progression. Despite the pivotal role of complement in fighting microbes, complement seems to suppress antitumor immunity via regulation of host cell in the tumor microenvironment. Although most studies link complement in cancer to complement activation in the extracellular space, the discovery of intracellular activation of complement, raises the question: what is the relevance of this process for malignancy? Intracellular activation is pivotal for the survival of immune cells. Therefore, complement can be important for tumor cell survival and growth regardless of the role in immunosuppression. On the other hand, because intracellular complement (the complosome) is indispensable for activation of T cells, these functions will be essential for priming antitumor T cell responses. Here, we review functions of complement in cancer with the consideration of extra and intracellular pathways of complement activation and spatial distribution of complement proteins in tumors and periphery and provide our take on potential significance of complement as biomarker and target for cancer therapy.
The complement system comprises of over 50 soluble, membrane, and intracellular proteins and is an essential component of both innate and adaptive immunity (1–3). Complement proteins can be activated through proteolytic cleavage in extracellular and intracellular spaces. Therefore, we use terms extracellular and intracellular complement thereafter in reference to these different activation pathways (4). We also consider the terms intracellular complement and the complosome interchangeable (5).
Extracellular activation pathways are well described. They include the classical (CP), lectin (LP), or alternative (AP) pathways (6). Extracellular activation by danger signals, such as bacterial surfaces or immune complexes, results in the opsonization of pathogens and subsequent lysis or removal by phagocytes. Complement activation and proteolytic cleavages of large inactive fragments lead to the generation of smaller size fragments such as C3b, iC3b, C3d, C3a, C5a, and the protein complex MAC (7). The central step in extracellular activation is the cleavage of C3 and C5 with the subsequent generation of C3a and C5a complement anaphylatoxins that are potent regulators of inflammation (2). C5 cleavage by C5 convertase initiates the formation of membrane attack complex (MAC), which directly lyses gram negative bacteria (8). Extracellular complement is redundant. Therefore, its activation is tightly controlled to prevent disease (3). Thus, regulatory mechanisms that serve to limit complement activation to prevent harm to host cells are of great importance for homeostasis. These complement regulators can block complement activation in the fluid phase (complement factor H [CFH] in conjunction with factor I [CFI]) or on cell surface, as is a case with the membrane-bound complement regulators: CD46, CD55, and CD59 (8, 10). The activation and regulation of extracellular complement has recently been reviewed elsewhere (2, 7, 9).
Conversely, intracellular complement, originally described in T cells in 2013, is less understood (11). CD4+ T cells were demonstrated to contain intracellular C3 stores that are important for cell survival. Intracellular activation was observed in resting CD4+ T cells, where C3 is continuously cleaved by intracellular cathepsin L to bioactive C3a and C3b fragments. C3a then engages with C3aR located on the lysosomal surface, driving mTOR activation and enhancing cell survival (11). Surprisingly, most of intracellular C3 likely derives from serum C3(H2O), which is a transient form of C3 (12). T cells also increase expression of C3 upon T cell activation, following the interaction with antigen presenting cells or during the transmigration to the tissues mediated by the LFA-1-AP-1 axis (13). This intrinsic C3 expression in T cells is important, as T cells lacking CD11a (part of LFA-1 heterodimer together with CD18), from the patients with leukocyte adhesion deficiency syndrome, do not upregulate C3 and, therefore, have defective TH1responses. During activation, C3 is cleaved intracellularly near the cell membrane and the resulting C3a and C3b fragments are shuttled to the surface where they signal via surface expressed C3aR and CD46 receptors, respectively. CD46 plays a crucial role in metabolic reprogramming during T cell activation and enables cell proliferation and cytokine secretion (14). It is therefore not surprising that individuals lacking CD46 expression have reduced TH1 immunity (14). Conversely, uncontrolled intracellular C3 activation and CD46 signalling leads to the dysregulation of human T cell responses and contributes to pathologically hyperactive TH1 cells in autoimmune disease (e.g., rheumatoid arthritis (RA)) (11, 15). CD46 co-stimulation drives increased expression of the glucose transporter GLUT1 (SLC2A1) and the amino acid transporter LAT1 (SLC7A5). These increases result in the elevated influx of glucose and amino acids required for T cell activation, proliferation, and the induction of TH1 response (14). In addition, CD46 upregulates expression of the Late Endosomal-Lysosomal Adaptor, MAPK and mTOR Activator 5 (LAMTOR5), which is a component of the Ragulator complex sensing amino acid influx. Thus, T cells from CD46-deficient patients do not up-regulate GLUT1, LAT1 and LAMTOR5, do not increase mTOR activity upon activation of T cell receptor, and consequently, do not produce IFN-γ (14). In T cells, CD46 also induces the activation and cleavage of intracellular C5 through yet unknown mechanisms, although intracellular factor B was proposed to play a role in this process in macrophages (16). The resultant C5a engages the intracellular C5aR1 to produce reactive oxygen species and to trigger NLRP3 inflammasome activation, resulting in the expression of intrinsically active IL‐1β, which sustains TH1 responses (17). Surface-expressed C5aR2, on the other hand, is a negative regulator of TH1 induction. In CD8+ T cells, CD46 is required for optimal cytotoxic activity by augmenting nutrient-influx and fatty acid synthesis. However, unlike in CD4+ T cells, canonical C5 and NLRP3 inflammasome activity is not required for normal human CD8+ T cell activity (18). In mice that normally do not express CD46 in somatic tissue (19), activation of anaphylatoxin receptors C3aR and C5aR1 on T cells may be driving the induction of TH1, TH17 T cells (20). Additional details on recent advances in intracellular roles of complement in metabolic processes are included in recently published comprehensive reviews (4, 21, 22). Because CD46 plays a crucial role in metabolic changes during T cell activation and metabolic reprograming is critical for cancer cell survival, it is conceivable that similar mechanisms may operate in cancer cells (Figure 1) (23).
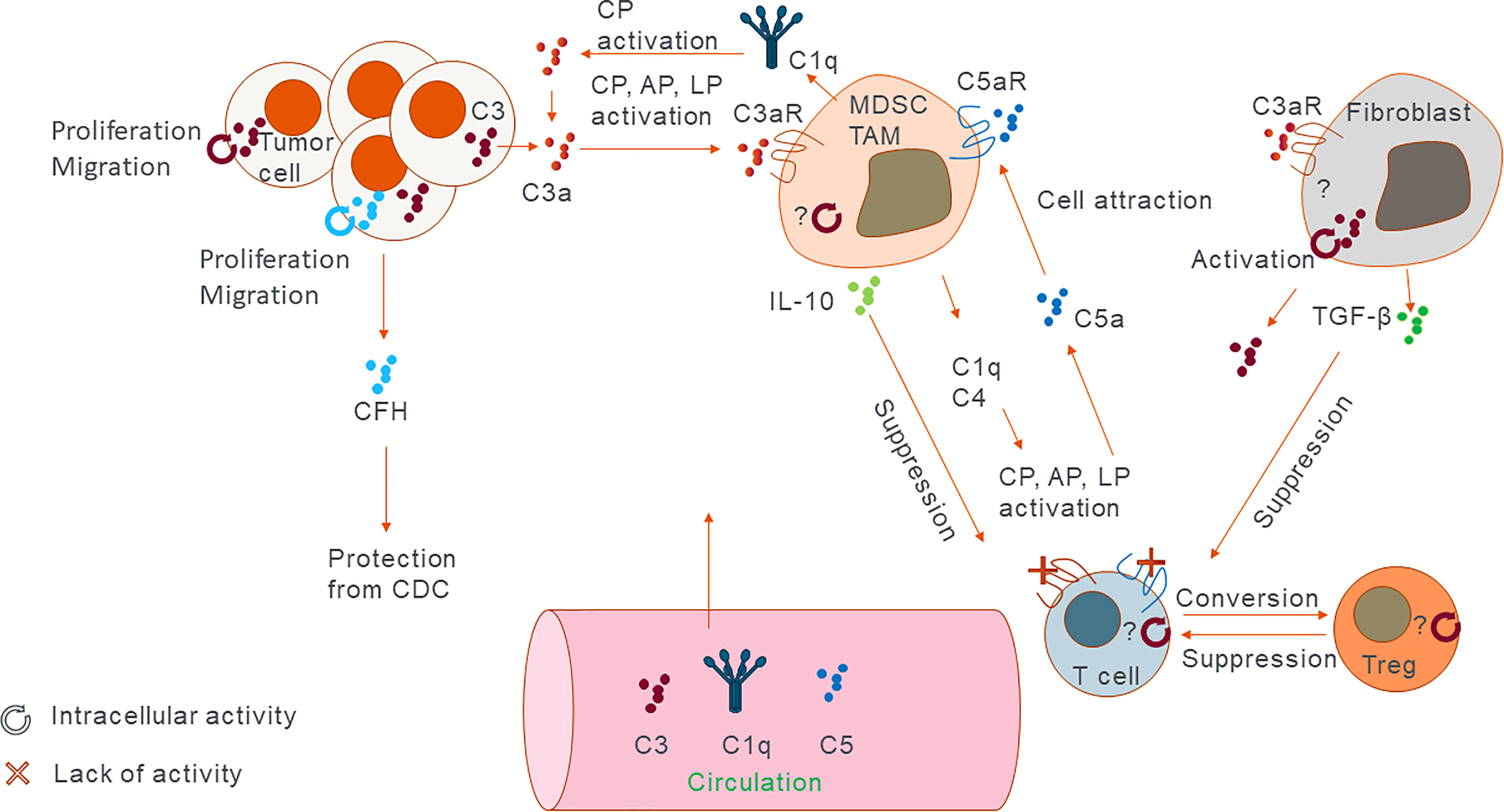
Figure 1 Sites of complement expression and activation. Cartoon representing the main sources of complement in the tumor and their main modes of activation and action. C3 and factor H have shown to exert their function both intra- and extracellularly. Other proteins such as C5 were shown to have extracellular function via its receptors. Generally extracellular complement works by inducing immunosuppressive phenotype. Intracellular complement has the potential to modulate immune cell function in similar fashion or by directly increasing proliferation and migration capacity of the cancer cells themselves. In addition, lack of C3aR and C5aR signalling on T cells can convert them into Tregs which in turn can suppress other T cells’ function.
In summary, the main differences between extra- and intracellular complement include 1) the location of activation process and 2) different overall functions of the complement pathways. Extracellular complement is a danger sensing effector arm of the innate immunity while intracellular complement is probably evolutionary older and is intimately involved in the metabolism and the activation state of innate and adaptive immune cells (24). In addition, intracellular complement activation is not likely to trigger the whole complement cascade until the generation of MAC (C5b-9 complex). While there are suggestions that serum-derived C3 is involved in intracellular activation (12) in T and B cells, recent report suggest that there is a truncated strictly intracellular form of C3 in β-cells in pancreatic islets (25).
An imbalance of activation vs. regulation of both extracellular and intracellular complement results in complement-mediated diseases (1–3, 6, 7). Dysregulation of the extracellular complement system through primary deficiencies in complement regulators results in uncontrolled activation, which leads to archetypical complement-mediated diseases (e.g., paroxysmal nocturnal haemoglobinuria and atypical haemolytic uremic syndrome) (26). Intracellular complement is dysregulated in RA patients where an increase in its activity was observed in synovial CD4+ T cells and (11) and fibroblasts (27). These increases seem to correlate with proinflammatory immune cell phenotypes. Inhibition of cathepsin L using cell permeable inhibitor in T cells from RA synovium leads to the reduction in in vitro activity (11). Importantly, up to 400 diseases including kidney, ocular, neurodegenerative, and autoimmune disorders, and several cancers are associated with complement dysregulation or alterations in complement genes. However, the functional link between these complement-related alterations and the underlying pathology is often unclear and it is likely that other immune and non-immune mechanisms contribute to these diseases (28–30). An interest in targeting complement for treatment of these disorders has significantly increased over the past twenty years, and particularly following the approval of eculizumab in 2007 and the more recent approval of pegcetacoplan for treatment of paroxysmal nocturnal haemoglobinuria in 2021 (31) and is well summarized in recent reviews (26, 32).
Malignancy progression, including growth of primary tumors and cancer metastasis, is regulated by intrinsic mechanisms within cancer cells and crosstalk between tumor cells and elements of the tumor microenvironment, including stromal extracellular matrix proteins, tumor associated fibroblasts, vasculature, and infiltrating immune cells (33). Recent evidence clearly demonstrates a role of complement in orchestrating functions of different components of the tumor microenvironment and that these complement-mediated mechanisms accelerate malignancy progression (8). This notion contrasts the original theory implicating complement in tumor immunosurveillance through complement-mediated cytotoxicity toward cancer cells. While complement activation in solid tumors has been demonstrated (34), there is little evidence that complement can kill a significant number of tumor cells because of evasion mechanisms employed by cancer cells (35). Accordingly, recent preclinical studies demonstrate the role of complement as a tumor promoter (Figure 1). The inhibition of antitumor immunity is a main driver of this inflammation-driven acceleration of cancer progression and complement activation seems to be instrumental for orchestrating immunosuppression (36). This has been illustrated in murine models of cancer with targeted deletion of the CFH. This deletion results in excessive complement activation with concomitant increase in tumor burden (37). Genetic analyses of several human cancers reveal high expression of genes encoding proteins of CP and AP, and high expression of some of complement regulators (29). The magnitude of complement activation corresponds to the generation of different levels of complement. Depending on their abundance, effectors may differently impact tumor progression. For example, low sublytic dose of C5b-9 is pivotal for activation of prosurvival signalling in tumor cells (38), whereas a higher dose of C5b-9 has the potential to lyse cells, although tumor cells appear to be resistant to complement-mediated lysis (35). Thus, progressing tumors likely require optimal levels of complement activation that foster chronic inflammation, immunosuppression, angiogenesis, and prosurvival cancer cell signalling (39). C3a and C5a acting through their cognate receptors C3aR, C5aR1 and C5aR2, respectively, are central mediators of tumor promoting responses in the tumor microenvironment (Figure 1) (40). In cancer, anaphylatoxins disrupt tissue homeostasis and favour altered immune responses accelerating tumor progression. For example, C5a is a positive modulator of T helper 1 (TH1) responses in infection, organ transplantation, and autoimmune diseases (41–43). Conversely, in cancer, C5a inhibits TH1 responses and shifts the balance toward TH2, which are ineffective in promoting cytolytic antitumor CD8+ T cell activity, or toward regulatory T cells (Tregs) (Figure 1). In addition, anaphylatoxins reshape the microenvironment through modulation of myeloid cells. C5a through its interactions with C5aR1 activates and recruits myeloid-derived suppressor cells (MDSC) to tumors and the premetastatic niches and regulates self-renewal of tissue resident pulmonary alveolar macrophages that suppress antitumor immunity (44). The complement system also promotes tumor growth by maintaining stemness of cancer stem cells and promoting tumor proliferation via C3a–C3aR signalling (Figure 1) (45).
Complement-dependent cytotoxicity (CDC) triggered by MAC is an important mechanism utilized by therapeutic anticancer antibody to kill tumor cells (34). In contrast, antitumor antibodies normally found in patients with cancer are unable to trigger complement-mediated tumor cell killing because of their low quantities and low affinity to tumor antigens (34).
Therapeutic antibodies trigger generation of MAC through the activation of CP, initiated by binding of C1q to immune complexes. AP amplifies this activation, thereby, contributing to CDC. Antibodies currently approved for cancer therapy, with confirmed CDC as part of their effector mechanism of action, target CD20 (rituximab), CD38 [daratumumab, (46)], EGFR [cetuximab (47)], GD2 [dinutuximab, (48)], claudin 18 [zolbetuximab, (49)), and HER2 (pertuzumab) (50)].
These antibodies are of IgG1 isotype, as IgG1 is known to trigger strong complement activation (51). Some of these antibodies have been modified to better activate complement and, consequently, more efficaciously induce tumor cell killing. For example, glycosylation and mannosylation of CD20 mAb obinutuzumab triggers the activation of the lectin pathway, which further potentiates CDC (52). Mutations of the Fc fragment (Glu345 to Arg) enables more efficient hexamerization and, consequently, binding of more C1q molecules, which amplifies CP and CDC (53).
The first antibody approved for therapy that utilizes CDC, as part of its therapeutic mechanism, was rituximab (anti-CD20). This antibody is approved for relapsed indolent Non-Hodgkin lymphomas (NHL) and its newer iteration, rituximab with hyaluronidase for subcutaneous injection, was more recently approved for refractory follicular lymphoma, diffuse large B-cell lymphoma (DLBCL), and chronic lymphocytic leukaemia (CLL) (54). Other CD20 antibodies such as ofatumumab and ibritumomab (55), and anti-CD52 (alemtuzumab) were also showed to have complement activating properties and lyse cancer cells through CDC (56).
The therapeutic efficacy of anti-cancer antibodies is, however, hindered by several mechanisms that cancer cells employ to evade complement-mediated lysis. These mechanisms include the removal of MAC from the cell surface (57, 58) and the incorporation of MAC into the cell membrane at sub-lytic concentrations (35). The latter can lead to increased tumorigenesis and faster malignancy progression through increased cancer cell proliferation, enhanced epithelial-to-mesenchymal transition (EMT), and increased motility of cancer cells that all ultimately facilitate metastasis (reviewed in 59). Overexpression of membrane bound complement regulators such as CD46, CD55, and CD 59 and blocking complement activation at the level of C3b also prevent CDC and efficient tumor cell killing. CD46 is overexpressed in breast (60) and ovarian cancers (10), whereas CD59 in sarcomas and melanoma (61). CFH, a fluid phase and cell surface complement regulator, which also reduces CDC, was found in extracellular vesicles (EV) secreted by non-small cell lung carcinoma (NSCLC) (62). In addition, cancer cells evade complement-mediated lysis via modulation of the complement regulators through posttranslational modifications. Lin and colleagues showed that ST3GAL1-mediated sialylation of CD55 renders better inhibition of the complement system at C3 level and that ST3GAL1 silencing results in increased C3 deposition and increased CDC-mediated killing of breast cancer cells (63). Interestingly, tumors can produce analogues of complement regulators that have alike inhibitory functions. FH related protein 5 (FHR5) confers resistance of glioblastoma to complement-mediated killing in vitro (64). In summary, monoclonal antibodies with CDC, as part of their mechanism of action, are efficacious in haematological malignancies but their efficacy in solid tumors is low (65).
The discovery of complement evasion mechanisms that reduce or completely prevent CDC prompted the attempts to disable complement regulators in tumor cells to increase CDC and, consequently, the efficacy of therapeutic antibodies (34, 66). For example, the inhibition of both CD55 and CD59 results in enhanced CDC and better efficacy of trastuzumab (anti-HER2) in killing human lung carcinoma in vitro (67). However, because CD46, CD55, and CD59 are constitutively expressed in human tissues, nonspecific targeting of these molecules can lead to undesired complement activation outside the tumor. In addition to membrane bound complement regulators, targeting soluble complement inhibitors is considered as additional strategy. For instance, the inhibition of CFH, using a recombinant antibody based on the naturally occurring CFH antibodies, was shown to improve rituximab efficacy in vitro and in vivo (68). Interestingly, CFH antibodies were found in patients with early-stage NSCLC (69).
To avoid adverse effects of non-specific targeting complement regulators, bi-specific antibodies targeting a complement regulator and a tumor antigen were developed. Antibodies targeting CD20 and simultaneously CD55 or CD59 increased the survival of mice in a model of Burkitt’s lymphoma. The improved efficacy of the anti-CD20, largely dependent on CDC, was associated with increased deposition of C3 and C9 in the tumor. The combination of bi-specific anti-CD20/anti-CD55 and anti-CD20/anti-CD59 resulted in near full recovery of treated mice while the treatment with either of those resulted in the survival of only 20% of mice (70). Despite these encouraging results, to the best of our knowledge (clinicalstrials.gov and clinicaltrialsregister.eu), bi-specific antibody therapies, involving inhibition of complement regulators on tumor cells, are not being tested in clinical trials at time of writing this review.
Since the discovery of intracellular complement and associated mechanisms referred to as the complosome (5), the question of how and where to target the complement system for cancer therapy has become more complicated. In the past, the objective was to inhibit complement-mediated mechanisms promoting cancer and enhance CDC for better efficacy of therapeutic antibodies. Now we need to consider multifaced functions of complement inside cells vs. functions of complement effectors in the extracellular space. In addition, since tumor growth relies on the interaction of tumor cells with host-derived microenvironment, it is critically important to address functions of complement inside cancer cells vs. inside host-derived components of the tumor microenvironment. The third dimension of a role of complement in cancer is where complement proteins regulating tumor growth are made. The majority of complement proteins are produced in the liver, in their inactive forms, and are secreted to the extracellular space. Given the rich tumor vasculature, these proteins are readily available in the tumor microenvironment. However, many other cells can produce complement fragments, including immune cells, that are an integral part of the tumor microenvironment. In the tumor, cancer cells, stromal cells, and the immune cells all synthesize and secrete complement proteins (8, 29). Given differences between intracellular vs. extracellular complement functions and diversity of complement sources (Table 1 and Figure 1), it is conceivable that outcome of the regulation of cancer by complement depends on these variables. This has potential implications for future immunotherapies targeting complement.

Table 1 Sources, functions, and therapeutic potential of complement proteins in cancer.
Since the discovery of protumor roles of the C5a-C5aR1 axis in a mouse model of human papilloma virus (HPV)-induced cancer, the view on the role of extracellular complement in cancer has shifted from the concept of complement contribution to tumor immune surveillance to complement mediated suppression of antitumor immunity (71). Here, we will discuss most recent examples of these protumor roles in pre-clinical animal models and human cancers. Extracellular complement activation product C3a induces PI3K/PDK1/p-SGK3 signalling in osteoclasts, which results in bone disease in multiple myeloma patients and is associated with poor prognosis (Figure 2) (72). Properdin, which stabilizes the AP C3 convertase, increases the generation of C5a, which via its receptor C5aR1, recruits immunosuppressive MDSCs from the spleen in a mouse model of melanoma (73). Similar mechanism, initiated by the activation of CP by pentraxin 3, leads to the recruitment of immunosuppressive cells in prostate cancer patients (74). The C5a-C5aR1 axis also skews the activation of tumor-associated macrophages (TAM) toward M2 anti-inflammatory phenotypes and enables metastasis in a colon cancer mouse model (Figure 2) (75). Corroborating mouse data, Piao et al. found that in human colon carcinoma patients, C5aR1 expression correlates with tumor grade. C3 was recently showed to play a role in cutaneous squamous cell carcinoma, as C3 deficient mice developed fewer and smaller tumors compared to wild type controls. Conversely, mice deficient in C5aR1 or C5aR2 had higher tumor incidence, suggesting a tumor limiting role of these receptors in this particular model (76). The protumor roles of C3a-C3aR axis was supported by Magrini and colleagues, who showed that C3 and C3aR deficiencies, in a mouse model of 3-methylcholanthrene sarcomagenesis, result in reduced accumulation and activation of immunosuppressive TAM, which in turn leads to the increased activation of T cells and better response to anti-PD-1 therapy (77). In patients with sarcomas, C3 deficiency-associated genetic signatures predicted better clinical outcomes supporting protumorigenic function of C3. C3a-C3aR contribution to cancer progression was also suggested in a mouse model of pancreatic adenocarcinoma. In this model, the microbiome was found to activate mannose binding lectin (MBL) of LP and promote cancer progression via C3 activation because the genetic deletion of either MBL or C3 in the extra tumoral compartment resulted in reduced tumor growth (78). C3, secreted and cleaved extracellularly to C3a and C3b, triggers C3a-C3aR signalling, resulting in increased neutrophil infiltration of tumors in mouse B10 melanoma. These neutrophils were found to promote tumor growth, as their depletion resulted in reduced tumor growth similar to that observed in C3 deficient mice (79). In a mouse renal cell carcinoma (RCC) model, genetic deficiency of C3aR or pharmacological inhibition of either C3aR or C5aR1 resulted in reduced tumor growth. C3aR signalling inhibited antitumor immunity to the greatest extent, as its ablation resulted in highest increase in IFN-γ production by CD8+ T cells. Mechanisms of this inhibition involved T cell exhaustion and dysfunction, as C3aR-deficiency and blockade reduced the expression T cell inhibitory receptors Pdcd1, Ctla4, and Btla. Depletion of CD8+ T cells entirely abrogated beneficial impact of C3aR-deficiency on tumor growth confirming regulation of T cell function by C3aR (80).
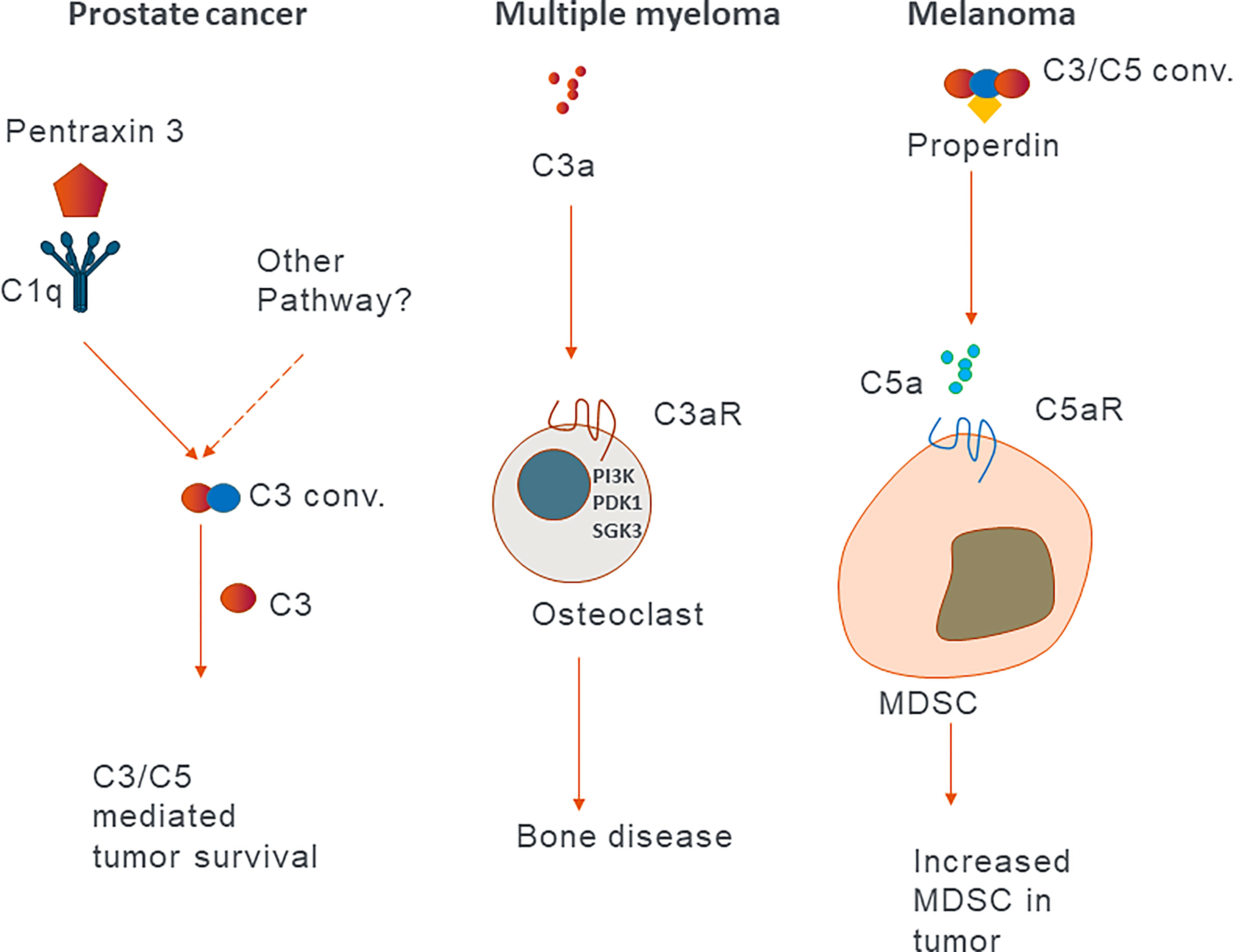
Figure 2 Examples of the effect of extracellular complement in different cancers. Schematic representation of published mechanisms describing different roles of complement proteins in different cancers.
In contrast, the inhibition of C3aR and C5aR1 accelerated tumor growth in a mouse orthotopic head and neck squamous cell carcinoma model. The acceleration of tumor growth associated with the increased number of Tregs in tumors. Mechanistically, locally generated C3a and C5a signalled via their receptors on CD4+ T cells to promote anti-tumor immune responses. The inhibition of this signalling resulted in the differentiation of CD4+ T cells into Tregs. Clearly, Tregs play an important immunosuppressive role in this model, since their depletion resulted in reduced tumor growth (Figure 1) (81). The constitutive C3 deficiency did not result in reduction of tumor growth in this model, which further underscores those functions of complement in cancer are highly context and model dependent. Nevertheless, these findings are consistent with the observations that genetic deficiency or pharmacological blockade of C3aR and/or C5aR signalling augments murine and human non-thymic Treg generation and, consequently, reduces the severity of graft-versus-host disease (82). In summary, C3a, C5a, and their receptors appear to have heterogenous functions in cancer and these functions are highly context dependent. Therefore, additional studies involving human cancer cells and patient data are needed to determine the role of liver-derived complement in human cancers.
Complement proteins secreted by tumor cells in the tumor microenvironment and activated extracellularly appear to promote cancer progression similar to liver-derived complement. This notion seems to be valid across several cancer mouse models and human cell lines. C3 secretion induced by Piwi Like RNA-Mediated Gene Silencing 1 protein (PIWIL1) in human hepatocellular carcinoma (HCC) cells leads to the activation of p38 and MAPK signalling in MDSCs, which, in turn, triggers the expression of immunosuppressive cytokine IL-10 in these cells (83). C3 secreted from liver metastatic breast cancer cells was also found to recruit, in C3aR-dependent manner, immature low-density prometastatic neutrophils to the liver (84).
These immunosuppressive and tumor promoting functions of C3 and its cleavage product C3a are contrasted by findings that CFH, which inhibits extracellular complement activation at the level of C3 convertase, promotes cell growth, migration, invasiveness, enhanced liver tumour formation, and metastasis in a mouse HCC model. In line with these observations, treatment with CFH antibody inhibits metastasis in this model (86). The role of complement in tumor immune surveillance is further corroborated by data showing that FHR5, secreted by primary glioblastoma cells, prevented complement activation and lysis of cancer cells in vitro (64). However, these two reports are in contrast with the observations that more than 50% of aged male CFH-deficient mice develop spontaneous liver carcinoma. Lack of CFH in these mice correlated with increased complement activation in the liver and increased infiltration of livers with CD8+ T cells and macrophages. Accordingly, data from the Cancer Genome Atlas revealed that increased CFH mRNA expression is associated with improved survival in patients with HCC, whereas mutations, reducing CFH function, were associated with worse outcomes. Similar corelative analysis was performed by Daugan et al., who demonstrated that increased CFH mRNA expression is associated with improved survival in patients with RCC (85). While the role of CFH, as an inhibitor of extracellular complement activation, in tumor progression appears to be very much model/context dependent, silencing of intracellular CFH in RCC cell lines results in cell-cycle arrest and death of tumor cells (Figure 1) (Daugan, Revel, Thouenon, et al., 2021). In addition to C3 and CFH, cancer cells have been shown to produce and secrete other complement proteins that suppress anti-tumor immunity. RCC secrete complement proteins such as C1r, C1s, C4, and C3 (87). C1r and C1s, secreted by tumor cells, form a functional C1 complex with C1q, secreted by the TAM, to initiate activation of the CP, leading to the generation of immunosuppressive complement effectors (Figure 1).
Host cells in the tumor microenvironment, including immune and non-immune cells, such as fibroblasts, can express and secrete C3 and other complement proteins. Functional data for complement proteins derived from these cells are scarce, therefore, we will focus on few examples where the connection to the function has been proposed. Single cell RNA sequencing, broadly adopted by cancer research, has yield ample and detailed data on complement protein expression in solid tumors. Using this approach, Davidson et al. demonstrated the highest expression of C3 in fibroblasts with lower levels in T cells and monocytes in mouse melanoma and breast tumors (88). C3 secreted from these cells is cleaved in the extracellular space and C3a, generated through this cleavage, recruits C3aR expressing macrophages that supress antitumor immunity and accelerate tumor growth. Similar pattern of C3 expression in stromal cells and C3aR in macrophages was found in human melanoma and head and neck tumors. These observations are in line with the earlier work demonstrating that cancer-associated fibroblasts in melanoma can produce C3 and other complement proteins including C1S, C1R, C3, C4A, CFB and C1INH (89). C3 is secreted by hepatic stellate cells in HCC and leads to the expansion of MDSCs and induction of T cell apoptosis in vitro (90). Fibroblasts appear to be a major source of C3 and C3a in patients with breast cancer (91). C3a-C3aR signalling in fibroblasts is thought to lead to their activation and subsequent upregulation of pro-tumorigenic cytokines such as transforming growth factor β (TGF-β) and the promotion of lung metastasis via EMT, as C3aR deficiency or pharmacological blockade decrease TGF-β expression, EMT, and thus metastasis in a mouse breast cancer model (Figure 1). C5 cleavage and activation in the tumor can also be accomplished through non-canonical complement pathways, that do not require the engagement of the entire complement cascade. For example, urokinase positive TAMs can convert plasminogen to plasmin, which directly cleaves C5 to C5a and C5b in C3-independent manner. Then C5a triggers the C5aR1 signalling on tumor-associated mast cell and supresses antitumor CD8+ T cells in a mouse model of squamous cell carcinoma (92).
Interestingly, in glioblastoma multiforme (GBM), tumor cells can induce complement protein production in benign host cells through tumor-derived EV. EV contain long noncoding RNA that stimulates C5 and C5a production in microglia. Microglia-derived C5/C5a then promotes the repair of temozolomide (TMZ)-induced DNA damage in C5aR1-dependent manner to confer resistance to TMZ chemotherapy (93). These data were corroborated in vivo in an orthotopic mouse model of GBM where C5aR1 antagonism resulted in increased sensitivity of GBM to TMZ. Using in vitro co-cultures of breast and prostate cancer cells with lymphatic endothelial cells (LEC), Oliveira-Ferrer demonstrated tumor-dependent induction of C3, CFB and C1q secretion as well as that chemokine upregulation (CCL7, CXCL6, CXCL1) by the LECs promotes tumor metastasis (94).
Data on a role of intracellular complement in signalling in tumor cells are limited, recent studies, however, suggest its importance for cancer progression. Cho et al. showed that siRNA downregulation of C3 in ovarian cancer cells SKOV3ip1 leads to reduced tumor cell proliferation and migration, (95). It is conceivable that C3 expressed in tumor cells can be cleaved inside these cells in non-C3 convertase fashion by cathepsins similar to cathepsin L in CD4+ T cells (11). Indeed, intracellular C3 cleavage by cathepsin L and B and subsequent release of C3a was demonstrated in colon carcinoma cells (96). Importantly, reduction of C3a secretion by these cells was observed when they were treated with cathepsin L and B inhibitors. These findings are consistent with data showing that C3 produced by mouse colon carcinoma cells is intracellularly activated. This tumor cell-derived C3a promotes TAM activation via C3a-C3aR-PI3Kγ pathway leading to the subsequent suppression of antitumor CD8+ T cell responses (Figure 1) (97). Importantly, the deletion of C3 from the tumor cells enhances the efficacy of anti-PD-L1 therapy in this model (97). Based on these findings, C3, activated in cancer cells, appears to be directly involved in the regulation of cancer cell survival. C3a, generated through this activation and released from cancer cells, contributes to immunosuppression similar to complement effectors from other sources. Supporting this notion, CFB expression was found to be elevated in pancreatic ductal adenocarcinoma (PDAC) (98). CFB mediated protection from senescence of tumor cells and cell proliferation. Secreted from the tumor cells CFB protein correlated with increases of immunosuppressive cells, including Tregs, MDSCs and TAMs. High CFB expression also correlated with poor survival, prompting authors to propose CFB as a potential therapeutic target for PDAC (Shimazaki et al., 2021).
Given abundance of plasma-derived complement, it is not clear if the effects of tumor expressed complement proteins are exclusively related to intracellular complement activity, therefore, further investigation is warranted. since In our opinion the evidence that intracellular complement has specific role in human cancers is still very limited.
Intracellular complement in CD4+ T cells and monocytes is involved in the regulation of basic metabolic processes and mTOR during T cell activation (14, 16). Although experimental evidence has yet to come, we theorize that intracellular complement in tumor cells may fulfil similar functions as in T cells. Similarities between cancer cells and activated T cells have been noted, particularly, in the utilization of aerobic glycolysis as means to generate energy and intermediate metabolites that are required for cell survival, proliferation, and metastasis (99). Immunohistochemistry of RCC tumors demonstrated that in the majority of tumors staining for complement was found in infiltrating immune, endothelial, and stromal cells whereas tumor cells did not show positive staining (80) These data are consistent with functions of complement in the regulation of the immunosuppressive microenvironment However, there is a portion of high-grade tumors that exhibited strong cytoplasmic staining for C3. Although evidence supporting the functional link between C3 abundance and tumor differentiation is missing, this notion can be further explored.
C1q directly inhibits the proliferation of T cells (100) and reduces mitochondrial metabolism of CD8+ T cells, thereby, restraining their activation (101). Higher expression of C1q and high numbers of the C1q positive macrophages positively correlates with markers of T cell exhaustion (PD-1 and LAG3) and poor clinical outcome for systemic lupus erythematosus (101). Further, C1q+ TAMs might be playing similar role in cancer by promoting T cell exhaustion and their tumor accumulation is often associated with poor prognosis (102). C1q is thought to interact with surface bound receptors on TAMs, in addition to controlling T cell metabolism via mitochondrial receptor engagement as described by Ling et al. (101). Increased expression of C1q is also associated with reduced survival in patients with RCC (80).
Haldar and colleagues discovered that human prostate carcinoma cells secrete mitochondrial DNA, which triggers TLR9 signalling in fibroblasts. Upon this stimulation, fibroblasts secrete C3a in reactive oxygen species (ROS) dependent manner. Then C3a in a paracrine fashion promotes disease progression and docetaxel resistance by increasing tumor cell proliferation and survival (103). In a subcutaneous xenograft of human PC3 prostate cell line, the authors demonstrated superior efficacy of docetaxel and C3aR inhibition over docetaxel alone in decreasing tumor burden. This was mediated via the reduction of AKT and Bcl-2 expression in the tumor cells. However, this paper did not explore in detail if intracellular complement activation occurred, so clearly further studies will be required to elucidate any potential role of intracellular complement in these cells.
Complement has been proposed as prognostic or predictive biomarker for many cancers [reviewed in (97)]. The utility of complement in this context seems to be twofold: (1) Complement proteins are routinely measured in plasma and several studies explore the potential of complement in blood to predict prognosis or the response to therapy. (2) Easy access to publicly available genomic data enables analysis of complement gene expression in tumors and correlations with clinical outcomes.
Recent studies have connected amounts of complement proteins in the blood of cancer patients with clinical outcomes, response to therapy, or diagnosis. The data summarized in Table 2 paints a picture of increasing number of studies usually identifying an elevation of the concentration of activated complement fragments or complement proteins in plasma or serum of cancer pateints. In most cases this increase correlates with the disease progression, outcome for patients or aids diagnosis. There are however some caveats, including relatively low number of patients and controls in some of the studies and lack of systematic approach to further validate these initial findings. Therefore, larger systematic studies are required in order for complement to be used as biomarker in clinical practice (10).
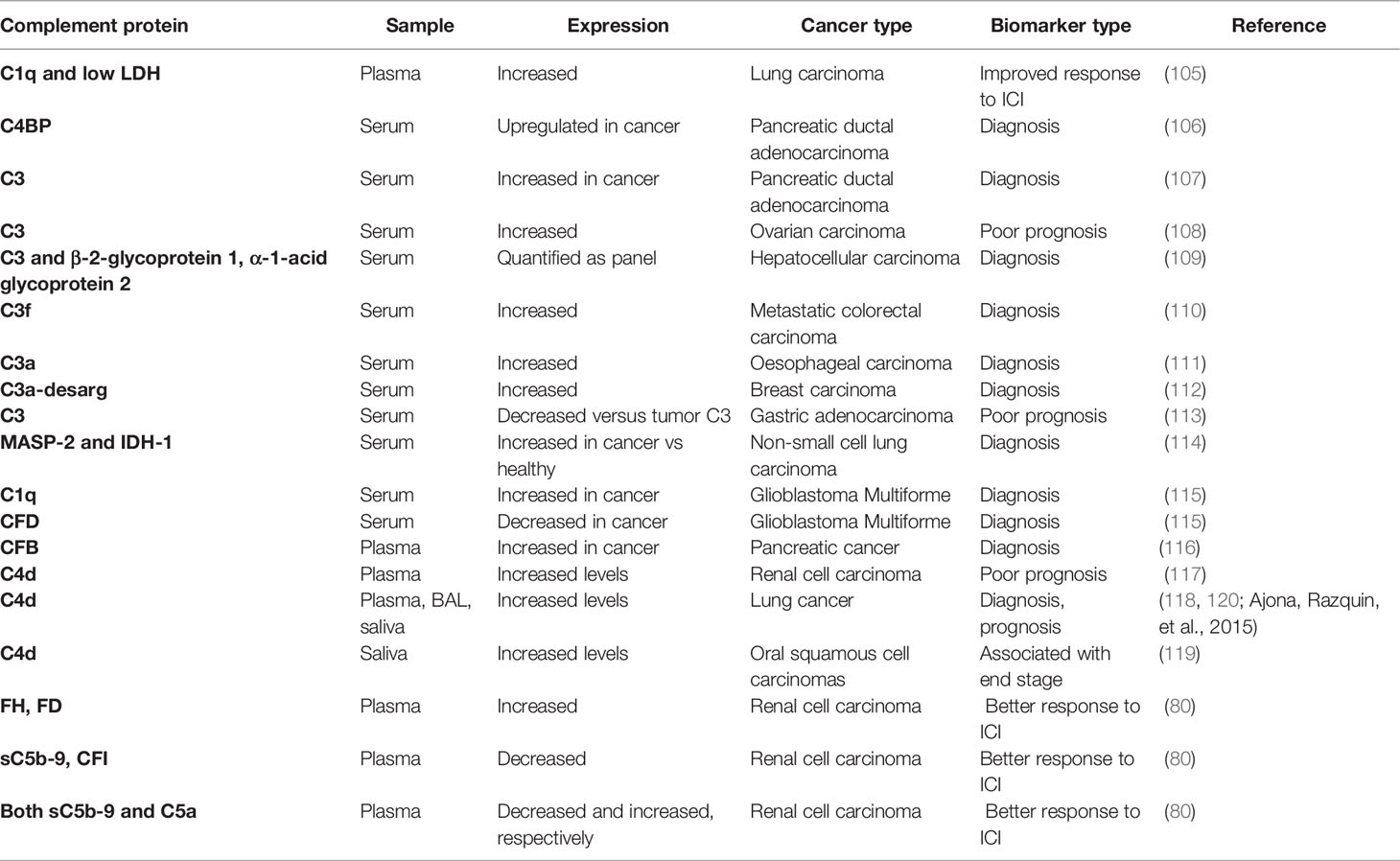
Table 2 Diagnostic, prognostic, and predictive complement biomarkers in blood.
Complement proteins were shown to be overexpressed in several common human malignancies (121), and their expression are linked with clinical outcomes (29). Predictably, prognostic significance of complement gene expression is dependent on malignancy type. Based on genomic analysis, high expression of C1QA, C1QB, C1S, C1R, C2, C3, CFB, CFH, CD55, and C5aR1 is associated with reduced overall survival in RCC. Conversely, high CD59 expression is associated with better outcomes (29, 80, 122). Further support for the role of C3 in RCC comes from Dong and colleagues, who demonstrated that high mRNA and protein expression levels of C3 and fibronectin 1 (FN1) in cancer cells were primarily responsible for RCC tumorigenesis and progression (123). Furthermore, increased expression of C3 was also associated with the advanced clinical stage, high pathological grade, and poor survival. The authors suggest both C3 and FN1 are candidate biomarkers for poor patient survival. Increased expression of C3 was also negatively correlated with survival in ovarian carcinoma (124).
In contrast to RCC and ovarian carcinoma, high expression of C1S, C3, C5, C6, and C7 is associated with favourable prognosis in liver carcinoma. CFB and CR2 are also associated with better prognosis in breast cancer alike high expression of CFD in pancreatic adenocarcinoma. Upregulated C3 in colon adenocarcinoma tumor cells was linked to poor overall survival while its expression was lower in rectal adenocarcinoma and had no prognostic value, suggesting context dependent C3 value as a prognostic biomarker (125). CD59, which is associated with favourable prognosis in RCC, correlates with poor outcomes in pancreatic, head and neck, and cervical cancer. In addition to RCC, C5aR1 is associated with poor prognosis in testis and ovarian cancer but with better outcomes in cervical cancer (80). The functional significance of these associations is poorly understood and appear to contradict some mouse studies. For example, increases in the activation of extracellular complement, which generates complement effectors, is expected to trigger immunosuppressive mechanisms that worsen outcomes. Therefore, high expression of complement regulators, which inhibit this activation, is expected to improve outcomes. However, this “rule of thumb” is not universally applicable, as several complement regulators, including CFH, CD55, and CD59, are associated with poor prognosis in several malignancies (80) For example, increased expression of CD55 in tumor cells correlated with worse survival in stomach and colon adenocarcinomas. Increased C3, complement receptor 4 (CR4), and C5aR1 expression were associated with poor prognosis in gastric cancer patients (125). Finally, C3 expression was found to be higher in primary tumors than lymph node metastases from luminal breast cancer patients (126). The decrease in C3 expression in lymph node metastases was linked to poor prognosis, suggesting that the role of tumor derived C3 might be different in primary tumors vs. metastatic niche and that C3 might play a role in EMT or MET conversion in accordance with earlier studies (127, 128).
Further meta-analyses of the publicly available RNA sequencing or next generation sequencing datasets are required in order to find a link between complement expression in tumor microenvironment and clinical outcomes. Long term this will establish complement as both biomarker and therapeutic target in specific cancers.
Our understanding of the complex role of the complement system in cancer has significantly expanded over the last decade. Initially, complement was viewed as an effector arm to “complement” therapeutic anti-cancer antibodies (such as anti-CD20); however, it is now clear that complement activation, in both the extra- or intracellular space, can directly and indirectly impact tumor growth and metastasis. In addition, complement proteins that influence cancer can be produced not only by ‘traditional’ expressors such as hepatocytes or myeloid cells but also by cancer cells, stromal, and non-myeloid immune cells such as T cells. Correlative analyses linking complement expression in the tumor or blood with clinical outcomes are informative in identifying cancer types where complement may be used as a biomarker, but they still do not address the key question of whether complement functionally drives malignancy progression and poor outcomes. Thus, further pre-clinical mechanistic in vivo and in vitro research linking complement protein expression and activation in cancer and non-cancers cells with tumor progression and metastasis is needed to gain confidence that targeting complement will be beneficial for cancer patients. Currently, one of the biggest weaknesses of available data is that they are mostly derived from animal models and from constitutive complement knockouts. The clinical translation of these findings is not always straightforward (129). Nevertheless, testing of complement inhibition is now underway in the clinic with both C3 and C5aR1 as targets. IPH5401 (anti-C5aR antibody) was tested in a clinical trial together with anti-PD-L1 in patients with advanced solid tumors. However at the time of writing this review no results have been posted (see Table 3). Two other clinical trials are currently active in testing the anti-C5aR monoclonal antibody TJ210001 as monotherapy. C3 extracellular inhibition is also being tested in the clinic in ovarian cancers in combination with Pembrolizumab or Bevacizumab and Pembrolizumab (see Table 3 for further details). The data obtained from these studies will inform further developments for complement-based cancer therapy and could validate extracellular complement as a target in oncology. Emerging data that intracellular complement might have a role in cancer cell proliferation, survival, and metastasis will require further preclinical validation to gain more confidence that this pathway plays a role in cancer and is targetable for therapy. Since this pathway is operative in immune cells as well, a cancer specific intracellular complement targeting may be required to achieve optimal efficacy (outlined in Figure 3).
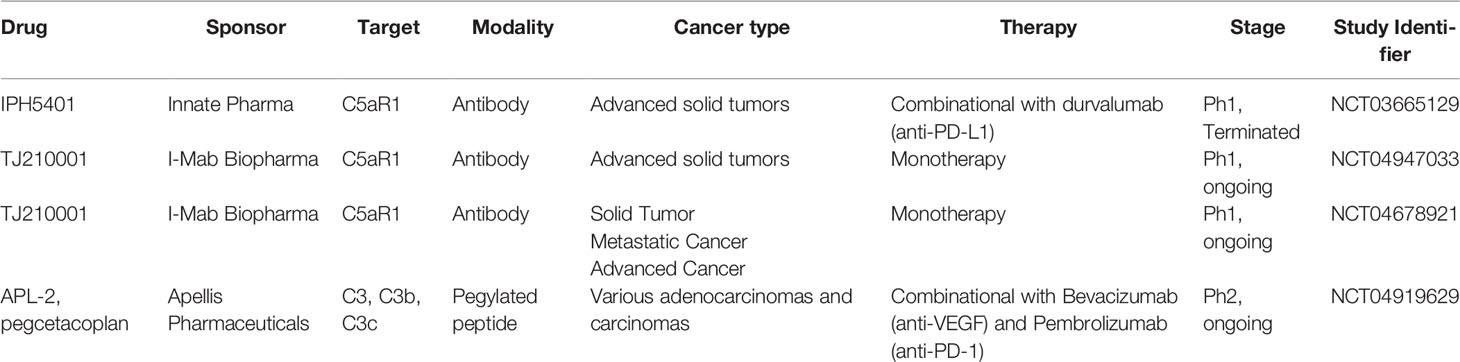
Table 3 Complement therapeutics currently tested in clinical trials.
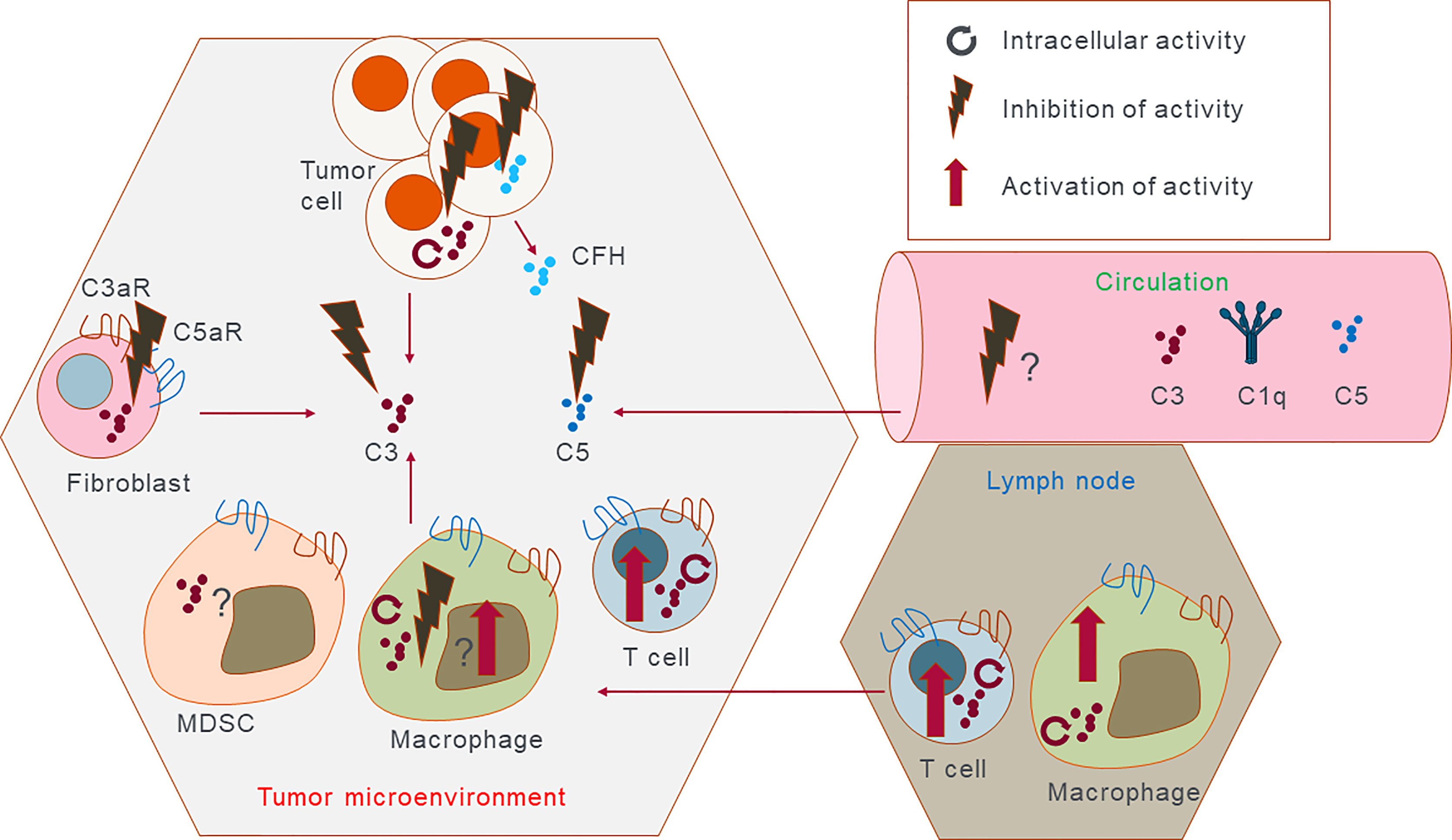
Figure 3 Potential therapeutic intervention points. Complement proteins can be expressed and secreted by almost all cells in the tumor microenvironment, and in the lymph nodes and are present in serum. This cartoon outlines potential targets either intracellularly or extracellularly that could be either inhibited or activated in the case of the immune cells. It is possible that in the future complement inhibiting therapeutics should be able to act extracellularly to promote immune cell antitumor immunity. Reversely intracellular complement in immune cells can be promoted as it is expected to have antitumorigenic effect and promote inflammation. Discreet complement targets present in the tumor cells but not in the immune cells and vice versa would have to be discovered to obtain full clinical efficacy of complement targeting therapy.
Interactions between cancer cells, immune, and non-immune cells in the tumor microenvironment as well as the spatial distribution of complement proteins add complexity to multifaced roles of complement in cancer and present a significant challenge to develop therapy. Ongoing clinical trials and pre-clinical efforts in targeting complement as standalone or combinational therapy will bring a wealth of new information to identify complement targets for tumor specific and cell specific basis and thus enable the design of a new generation of complement therapeutics to harness the full potential of this system for cancer immunotherapy. Furthermore, with the discovery of CRIPSR/Cas9 editing technology and recent advances in AAV and siRNA therapy, options of how-to best target complement in the extra- and intracellular space have never been better.
The idea of the manuscript was conceived by MK and MM. All authors contributed to writing, editing and revising of the paper. All authors contributed to the article and approved the submitted version.
MK, MD, SB, MG and PD are employees and shareholders in Apellis Pharmaceuticals Inc., which actively develops drugs targeting the complement system.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kemper C, Pangburn MK, Fishelson Z. Complement Nomenclature 2014. Mol Immunol (2014) 61(2):56–8. doi: 10.1016/J.MOLIMM.2014.07.004
2. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement System Part II: Role in Immunity. Front Immunol (2015) 6:257(MAY). doi: 10.3389/FIMMU.2015.00257
3. Noris M, Remuzzi G. Overview of Complement Activation and Regulation. Semin Nephrol (2013) 33(6):479. doi: 10.1016/J.SEMNEPHROL.2013.08.001
4. West EE, Kolev M, Kemper C. Complement and the Regulation of T Cell Responses. Annu Rev Immunol (2018) 36:309–38. doi: 10.1146/ANNUREV-IMMUNOL-042617-053245
5. Arbore G, Kemper C, Kolev M. Intracellular Complement – the Complosome – in Immune Cell Regulation. Mol Immunol (2017) 89:2–9. doi: 10.1016/J.MOLIMM.2017.05.012
6. Kolev M, Friec G, Kemper C. Complement-Tapping Into New Sites and Effector Systems. Nat Rev Immunol (2014) 14(12):811–20. doi: 10.1038/NRI3761
7. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I – Molecular Mechanisms of Activation and Regulation. Front Immunol (2015) 2:262. doi: 10.3389/FIMMU.2015.00262
8. Kolev M, Markiewski MM. Targeting Complement-Mediated Immunoregulation for Cancer Immunotherapy. Semin Immunol (2018) 37:85–97. doi: 10.1016/J.SMIM.2018.02.003
9. Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: Untangling an intricate relationship. Nat Rev Immunol (2018) 18(1):5–18. doi: 10.1038/NRI.2017.97
10. Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in Cancer: Untangling an Intricate Relationship. Nat Rev Immunol (2017) 18(1):5–18. doi: 10.1038/nri.2017.97
11. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular Complement Activation Sustains T Cell Homeostasis and Mediates Effector Differentiation. Immunity (2013) 39(6):1143–57. doi: 10.1016/J.IMMUNI.2013.10.018
12. Elvington M, Liszewski MK, Bertram P, Kulkarni HS, Atkinson JP. A C3(H20) Recycling Pathway is a Component of the Intracellular Complement System. J Clin Invest (2017) 127(3):970–81. doi: 10.1172/JCI89412
13. Kolev M, West EE, Kunz N, Chauss D, Moseman EA, Rahman J, et al. Diapedesis-Induced Integrin Signaling via LFA-1 Facilitates Tissue Immunity by Inducing Intrinsic Complement C3 Expression in Immune Cells. Immunity (2020) 52(3):513–527.e8. doi: 10.1016/J.IMMUNI.2020.02.006
14. Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, et al. Complement Regulates Nutrient Influx and Metabolic Reprogramming During Th1 Cell Responses. Immunity (2015) 42(6):1033–47. doi: 10.1016/J.IMMUNI.2015.05.024
15. Cardone J, le Friec G, Vantourout P, Roberts A, Fuchs A, Jackson I, et al. Complement Regulator CD46 Temporally Regulates Cytokine Production by Conventional and Unconventional T Cells. Nat Immunol (2010) 11(9):862–71. doi: 10.1038/ni.1917
16. Niyonzima N, Rahman J, Kunz N, West EE, Freiwald T, Desai J, et al. Mitochondrial C5aR1 Activity in Macrophages Controls IL-1β Production Underlying Sterile Inflammation. Sci Immunol (2021) 6(66):eabf2489. doi: 10.1126/SCIIMMUNOL.ABF2489
17. Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T Helper 1 Immunity Requires Complement-Driven NLRP3 Inflammasome Activity in CD4+ T Cells. Science (2016) 352(6292):aad1210. doi: 10.1126/SCIENCE.AAD1210
18. Arbore G, West EE, Rahman J, le Friec G, Niyonzima N, Pirooznia M, et al. Complement Receptor CD46 Co-Stimulates Optimal Human CD8+ T Cell Effector Function via Fatty Acid Metabolism. Nat Commun (2018) 9(1):1–15. doi: 10.1038/s41467-018-06706-z
19. Tsujimura A, Shida K, Kitamura M, Nomura M, Takeda J, Tanaka H, et al. Molecular Cloning of a Murine Homologue of Membrane Cofactor Protein (CD46): Preferential Expression in Testicular Germ Cells. Biochem J (1998) 330(1):163–8. doi: 10.1042/BJ3300163
20. Grailer JJ, Bosmann M, Ward PA. Regulatory Effects of C5a on IL-17a, IL-17F, and IL-23. Front Immunol (2013) 3:387. doi: 10.3389/FIMMU.2012.00387
21. Kunz N, Kemper C. Complement Has Brains—Do Intracellular Complement and Immunometabolism Cooperate in Tissue Homeostasis and Behavior? Front Immunol (2021) 12:154. doi: 10.3389/FIMMU.2021.629986
22. West EE, Kunz N, Kemper C. Complement and Human T Cell Metabolism: Location, Location, Location. Immunol Rev (2020) 295(1):68–81. doi: 10.1111/IMR.12852
23. Macintyre AN, Rathmell JC. Activated Lymphocytes as a Metabolic Model for Carcinogenesis. Cancer Metab (2013) 1(1):1–12. doi: 10.1186/2049-3002-1-5
24. Kolev M, Kemper C. Keeping It All Going—Complement Meets Metabolism. Front. Immunol (2017). doi: 10.3389/FIMMU.2017.00001
25. King BC, Renström E, Blom AM. Intracellular Cytosolic Complement Component C3 Regulates Cytoprotective Autophagy in Pancreatic Beta Cells by Interaction With ATG16L1. Autophagy (2019) 15(5):919. doi: 10.1080/15548627.2019.1580515
26. Mastellos DC, Ricklin D, Lambris JD. Clinical Promise of Next-Generation Complement Therapeutics. Nat Rev Drug Discov (2019) 18(9):707. doi: 10.1038/S41573-019-0031-6
27. Friščić J, Böttcher M, Reinwald C, Bruns H, Wirth B, Popp S-J, et al. The Complement System Drives Local Inflammatory Tissue Priming by Metabolic Reprogramming of Synovial Fibroblasts. Immunity (2021) 54(5):1002–1021.e10. doi: 10.1016/J.IMMUNI.2021.03.003
28. Ricklin D, Reis ES, Lambris JD. Complement in Disease: A Defence System Turning Offensive. Nat Rev Nephrol (2016) 12(7):383–401. doi: 10.1038/nrneph.2016.70
29. Roumenina LT, Daugan MV, Petitprez F, Sautès-Fridman C, Fridman WH. Context-Dependent Roles of Complement in Cancer. Nat Rev Cancer (2019) 19(12):698–715. doi: 10.1038/s41568-019-0210-0
30. Zelek WM, Xie L, Morgan BP, Harris CL. Compendium of Current Complement Therapeutics. Mol Immunol (2019) 114:341–52. doi: 10.1016/J.MOLIMM.2019.07.030
31. Hillmen P, Szer J, Weitz I, Röth A, Höchsmann B, Panse J, et al. Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. New England Journal of Medicine (2021) 384(11):1028–37. doi: 10.1056/NEJMOA2029073/SUPPL_FILE/NEJMOA2029073_DATA-SHARING.PDF
32. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The Renaissance of Complement Therapeutics. Nat Rev Nephrol (2017) 14(1):26–47. doi: 10.1038/NRNEPH.2017.156
33. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
34. Macor P, Capolla S, Tedesco F. Complement as a Biological Tool to Control Tumor Growth. Front Immunol (2018) 9:2203/BIBTEX(SEP). doi: 10.3389/FIMMU.2018.02203/BIBTEX
35. Fishelson Z, Kirschfink M. Complement C5b-9 and Cancer: Mechanisms of Cell Damage, Cancer Counteractions, and Approaches for Intervention. Front Immunol (2019) 10:752/BIBTEX(APR). doi: 10.3389/FIMMU.2019.00752/BIBTEX
36. Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, et al. PTX3 Is an Extrinsic Oncosuppressor Regulating Complement-Dependent Inflammation in Cancer. Cell (2015) 160(4):700–14. doi: 10.1016/J.CELL.2015.01.004
37. Laskowski J, Renner B, Pickering MC, Serkova NJ, Smith-Jones PM, Clambey ET, et al. Complement Factor H–deficient Mice Develop Spontaneous Hepatic Tumors. J Clin Invest (2020) 130(8):4039–54. doi: 10.1172/JCI135105
38. Vlaicu SI, Tatomir A, Rus V, Rus H. Role of C5b-9 and RGC-32 in Cancer. Front Immunol (2019) 10:1054. doi: 10.3389/FIMMU.2019.01054
39. Pio R, Corrales L, Lambris JD. The Role of Complement in Tumor Growth. Adv Exp Med Biol (2014) 772:229–62. doi: 10.1007/978-1-4614-5915-6_11
40. Ajona D, Ortiz-Espinosa S, Pio R. Complement Anaphylatoxins C3a and C5a: Emerging Roles in Cancer Progression and Treatment. Semin Cell Dev Biol (2019) 85:153–63. doi: 10.1016/J.SEMCDB.2017.11.023
41. Grafals M, Thurman JM. The Role of Complement in Organ Transplantation. Front Immunol (2019) 2380:2380. doi: 10.3389/FIMMU.2019.02380
42. Kim AHJ, Dimitriou ID, Holland MCH, Mastellos D, Mueller YM, Altman JD, et al. Complement C5a Receptor Is Essential for the Optimal Generation of Antiviral CD8+ T Cell Responses. J Immunol (2004) 173(4):2524–9. doi: 10.4049/JIMMUNOL.173.4.2524
43. Thurman JM, Yapa R. Complement Therapeutics in Autoimmune Disease. Front Immunol (2019) 10:672. doi: 10.3389/FIMMU.2019.00672
44. Kochanek DM, Ghouse SM, Karbowniczek MM, Markiewski MM. Complementing Cancer Metastasis. Front Immunol (2018) 9:1629/BIBTEX(JUL). doi: 10.3389/FIMMU.2018.01629/BIBTEX
45. Fan Z, Qin J, Wang D, Geng S. Complement C3a Promotes Proliferation, Migration and Stemness in Cutaneous Squamous Cell Carcinoma. J Cell Mol Med (2019) 23(5):3097–107. doi: 10.1111/JCMM.13959
46. Weers M, Tai Y-T, van der Veer MS, Bakker JM, Vink T, Jacobs DCH, et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J Immunol (2011) 186(3):1840–8. doi: 10.4049/JIMMUNOL.1003032
47. Hsu Y-F, Ajona D, Corrales L, Lopez-Picazo JM, Gurpide A, Montuenga LM, et al. Complement Activation Mediates Cetuximab Inhibition of non-Small Cell Lung Cancer Tumor Growth In Vivo. Mol Cancer (2010) 9(1):1–8. doi: 10.1186/1476-4598-9-139
48. Dhillon S. Dinutuximab: First Global Approval. Drugs (2015) 75(8):923–7. doi: 10.1007/S40265-015-0399-5
49. Sahin U, Schuler M, Richly H, Bauer S, Krilova A, Dechow T, et al. A Phase I Dose-Escalation Study of IMAB362 (Zolbetuximab) in Patients With Advanced Gastric and Gastro-Oesophageal Junction Cancer. Eur J Cancer (2018) 100:17–26. doi: 10.1016/J.EJCA.2018.05.007
50. El-Sahwi K, Bellone S, Cocco E, Cargnelutti M, Casagrande F, Bellone M, et al. In Vitro Activity of Pertuzumab in Combination With Trastuzumab in Uterine Serous Papillary Adenocarcinoma. Br J Cancer (2009) 102(1):134–43. doi: 10.1038/sj.bjc.6605448
51. Vidarsson G, Dekkers G, Rispens T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front Immunol (2014) 5:520(OCT). doi: 10.3389/FIMMU.2014.00520
52. Hayes JM, Wormald MR, Rudd PM, Davey GP. Fc Gamma Receptors: Glycobiology and Therapeutic Prospects. J Inflamm Res (2016) 9:209–19. doi: 10.2147/JIR.S121233
53. Diebolder CA, Beurskens FJ, Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement Is Activated by IgG Hexamers Assembled at the Cell Surface. Science (New York N Y) (2014) 343(6176):1260. doi: 10.1126/SCIENCE.1248943
54. Ioannou N, Jain K, Ramsay AG, Deutsch A, Feichtinger J, Krstic J, et al. Immunomodulatory Drugs for the Treatment of B Cell Malignancies. Int J Mol Sci (2021) 22(16):8572. doi: 10.3390/IJMS22168572
55. Marshall MJE, Stopforth RJ, Cragg MS. Therapeutic Antibodies: What Have We Learnt From Targeting CD20 and Where Are We Going? Front Immunol (2017) 8:1245. doi: 10.3389/FIMMU.2017.01245
56. Zent CS, Secreto CR, LaPlant BR, Bone ND, Call TG, Shanafelt TD, et al. Direct and Complement Dependent Cytotoxicity in CLL Cells From Patients With High-Risk Early–Intermediate Stage Chronic Lymphocytic Leukemia (CLL) Treated With Alemtuzumab and Rituximab. Leuk Res (2008) 32(12):1849–56. doi: 10.1016/J.LEUKRES.2008.05.014
57. Carney DF, Koski CL, Shin ML. Elimination of Terminal Complement Intermediates From the Plasma Membrane of Nucleated Cells: The Rate of Disappearance Differs for Cells Carrying C5b-7 or C5b-8 or a Mixture of C5b-8 With a Limited Number of C5b-9. J Immunol (1985) 134(3).
58. Scolding NJ, Morgan BP, Houston WAJ, Linington C, Campbell AK, Compston DAS. Vesicular Removal by Oligodendrocytes of Membrane Attack Complexes Formed by Activated Complement. Nature (1989) 339(6226):620–2. doi: 10.1038/339620a0
59. Towner LD, Wheat RA, Hughes TR, PaulMorgan B. Complement Membrane Attack and Tumorigenesis: A SYSTEMS BIOLOGY APPROACH *. J Bio Chem (2026) 291(29):14927–38. doi: 10.1074/JBC.M115.708446
60. MacIejczyk A, Szelachowska J, Szynglarewicz B, Szulc R, Szulc A, Wysocka T, et al. CD46 Expression is an Unfavorable Prognostic Factor in Breast Cancer Cases. Appl Immunohistochem Mol Morphol (2011) 19(6):540–6. doi: 10.1097/PAI.0B013E31821A0BE9
61. Zhang R, Liu Q, Liao Q, Zhao Y. CD59: A Promising Target for Tumor Immunotherapy. Future Oncol (2018) 14(8):781–91. doi: 10.2217/FON-2017-0498
62. Campa MJ, Gottlin EB, Bushey RT, Patz EF. Complement Factor H Antibodies From Lung Cancer Patients Induce Complement-Dependent Lysis of Tumor Cells, Suggesting a Novel Immunotherapeutic Strategy. Cancer Immunol Res (2015) 3(12):1325–32. doi: 10.1158/2326-6066.CIR-15-0122
63. Lin W-D, Fan T-C, Hung J, Yeo H-L, Wang S-H, Kuo C-W, et al. Sialylation of CD55 by ST3GAL1 Facilitates Immune Evasion in Cancer. Cancer Immunol Res (2021) 9(1):113–22. doi: 10.1158/2326-6066.CIR-20-0203
64. DeCordova S, Abdelgany A, Murugaiah V, Pathan AA, Nayak A, Walker T, et al. Secretion of Functionally Active Complement Factor H Related Protein 5 (FHR5) by Primary Tumour Cells Derived From Glioblastoma Multiforme Patients. Immunobiology (2019) 224(5):625–31. doi: 10.1016/J.IMBIO.2019.07.006
65. Rogers LM, Veeramani S, Weiner GJ. Complement in Monoclonal Antibody Therapy of Cancer. Immunol Res (2014) 59(0):203. doi: 10.1007/S12026-014-8542-Z
66. Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to Cancer Immunotherapy: Expression of Membrane Complement Regulatory Proteins (mCRPs) in Tumors. Mol Immunol (2003) 40(2–4):109–23. doi: 10.1016/S0161-5890(03)00112-3
67. Zhao W-P, Zhu B, Duan Y-Z, Chen Z-T. Neutralization of Complement Regulatory Proteins CD55 and CD59 Augments Therapeutic Effect of Herceptin Against Lung Carcinoma Cells. Oncol Rep (2009) 21(6):1405–11. doi: 10.3892/OR_00000368
68. Bushey RT, Moody MA, Nicely NL, Haynes BF, Alam SM, Keir ST, et al. A Therapeutic Antibody for Cancer, Derived From Single Human B Cells. Cell Rep (2016) 15(7):1505–13. doi: 10.1016/J.CELREP.2016.04.038
69. Amornsiripanitch N, Hong S, Campa MJ, Frank MM, Gottlin EB, Patz EF. Complement Factor H Autoantibodies Are Associated With Early Stage NSCLC. Clin Cancer Res (2010) 16(12):3226–31. doi: 10.1158/1078-0432.CCR-10-0321
70. Macor P, Secco E, Mezzaroba N, Zorzet S, Durigutto P, Gaiotto T, et al. Bispecific Antibodies Targeting Tumor-Associated Antigens and Neutralizing Complement Regulators Increase the Efficacy of Antibody-Based Immunotherapy in Mice. Leukemia (2014) 29(2):406–14. doi: 10.1038/leu.2014.185
71. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the Antitumor Immune Response by Complement. Nat Immunol (2008) 9(11):1225–35. doi: 10.1038/ni.1655
72. Jiang F, Liu H, Peng F, Liu Z, Ding K, Song J, et al. Complement C3a Activates Osteoclasts by Regulating the PI3K/PDK1/SGK3 Pathway in Patients With Multiple Myeloma. Cancer Biol Med (2021) 18(3):721. doi: 10.20892/J.ISSN.2095-3941.2020.0430
73. Al-Rayahi IAM, Machado LR, Stover CM. Properdin Is a Modulator of Tumour Immunity in a Syngeneic Mouse Melanoma Model. Medicina (2021) 57(2):85. doi: 10.3390/MEDICINA57020085
74. Stallone G, Netti GS, Cormio L, Castellano G, Infante B, Pontrelli P, et al. Modulation of Complement Activation by Pentraxin-3 in Prostate Cancer. Sci Rep (2020) 10(1):18400. doi: 10.1038/S41598-020-75376-Z
75. Piao C, Zhang WM, Li TT, Zhang CC, Qiu S, Liu Y, et al. Complement 5a Stimulates Macrophage Polarization and Contributes to Tumor Metastases of Colon Cancer. Exp Cell Res (2018) 366(2):127–38. doi: 10.1016/J.YEXCR.2018.03.009
76. Jackson WD, Gulino A, Fossati-Jimack L, Seoane RC, Tian K, Best K, et al. C3 Drives Inflammatory Skin Carcinogenesis Independently of C5. J Invest Dermatol (2021) 141(2):404–414.e6. doi: 10.1016/J.JID.2020.06.025
77. Magrini E, di Marco S, Mapelli SN, Perucchini C, Pasqualini F, Donato A, et al. Complement Activation Promoted by the Lectin Pathway Mediates C3aR-Dependent Sarcoma Progression and Immunosuppression. Nat Cancer (2021) 2(2):218. doi: 10.1038/S43018-021-00173-0
78. Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, et al. The Fungal Mycobiome Promotes Pancreatic Oncogenesis via Activation of MBL. Nature (2019) 574(7777):264–7. doi: 10.1038/s41586-019-1608-2
79. Nabizadeh JA, Manthey HD, Steyn FJ, Chen W, Widiapradja A, Akhir FNM, et al. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J Immunol (2016) 196(11):4783–92. doi: 10.4049/JIMMUNOL.1600210
80. Reese B, Silwal A, Daugherity E, Daugherity M, Arabi M, Daly P, et al. Complement as Prognostic Biomarker and Potential Therapeutic Target in Renal Cell Carcinoma. J Immunol (2020) 205(11):3218–29. doi: 10.4049/JIMMUNOL.2000511
81. Gadwa J, Bickett TE, Darragh LB, Knitz MW, Bhatia S, Piper M, et al. Complement C3a and C5a Receptor Blockade Modulates Regulatory T Cell Conversion in Head and Neck Cancer. J Immunother Cancer (2021) 9(3):e002585. doi: 10.1136/JITC-2021-002585
82. Touw van der W, Cravedi P, Kwan W, Paz-Artal E, Merad M, Heeger PS. Cutting Edge: Receptors for C3a and C5a Modulate Stability of Alloantigen-Reactive Induced Regulatory T Cells. J Immunol (2013) 190(12):5921–5. doi: 10.4049/JIMMUNOL.1300847
83. Wang N, Tan H-Y, Lu Y, Chan Y-T, Wang D, Guo W, et al. PIWIL1 Governs the Crosstalk of Cancer Cell Metabolism and Immunosuppressive Microenvironment in Hepatocellular Carcinoma. Signal Transduct Target Ther (2021) 6(1):1–13. doi: 10.1038/s41392-021-00485-8
84. Hsu BE, Roy J, Mouhanna J, Rayes RF, Ramsay L, Tabariès S, et al. C3a Elicits Unique Migratory Responses in Immature Low-Density Neutrophils. Oncogene (2020) 39(12):2612–23. doi: 10.1038/s41388-020-1169-8
85. Daugan MvRevel M, Thouenon R, Dragon-Durey MA, Robe-Rybkine T, Torset C, et al. Intracellular Factor H Drives Tumor Progression Independently of the Complement Cascade. Cancer Immunology Research (2021) 9(8):909–25. doi: 10.1158/2326-6066.CIR-20-0787
86. Mao X, Zhou L, Tey SK, Ma APY, Yeung CLS, Ng TH, et al. Tumour Extracellular Vesicle-Derived Complement Factor H Promotes Tumorigenesis and Metastasis by Inhibiting Complement-Dependent Cytotoxicity of Tumour Cells. J Extracell Vesicles (2020) 10(1):e12031. doi: 10.1002/JEV2.12031
87. Roumenina LT, Daugan MV, Noé R, Petitprez F, Vano YA, Sanchez-Salas R, et al. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol Res (2019) 7(7):1091–105. doi: 10.1158/2326-6066.CIR-18-0891
88. Davidson S, Efremova M, Riedel A, Mahata B, Pramanik J, Huuhtanen J, et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep (2020) 31(7):107628. doi: 10.1016/J.CELREP.2020.107628
89. Tirosh I, Izar B, Prakadan SM, Marc H. Wadsworth I, Treacy D, Trombetta JJ, et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science (2016) 352(6282):189–96. doi: 10.1126/SCIENCE.AAD0501
90. Xu Y, Huang Y, Xu W, Zheng X, Yi X, Huang L, et al. Activated Hepatic Stellate Cells (HSCs) Exert Immunosuppressive Effects in Hepatocellular Carcinoma by Producing Complement C3. Onco Targets Ther (2020) 13:1497–505. doi: 10.2147/OTT.S234920
91. Shu C, Zha H, Long H, Wang X, Yang F, Gao J, et al. C3a-C3aR Signaling Promotes Breast Cancer Lung Metastasis via Modulating Carcinoma Associated Fibroblasts. J Exp Clin Cancer Res (2020) 39(1):1–14. doi: 10.1186/S13046-019-1515-2/FIGURES/6
92. Medler TR, Murugan D, Horton W, Kumar S, Cotechini T, Forsyth AM, et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell (2018) 34(4):561–578.e6. doi: 10.1016/J.CCELL.2018.09.003
93. Li Z, Meng X, Wu P, Zha C, Han B, Li L, et al. Glioblastoma Cell-Derived lncRNA-Containing Exosomes Induce Microglia to Produce Complement C5, Promoting Chemotherapy Resistance. Cancer Immunol Res (2021) 0258:2021. doi: 10.1158/2326-6066.CIR-21-0258
94. Oliveira-Ferrer L, Milde-Langosch K, Eylmann K, Rossberg M, Müller V, Schmalfeldt B, et al. Mechanisms of Tumor-Lymphatic Interactions in Invasive Breast and Prostate Carcinoma. Int J Mol Sci (2020) 21(2):602. doi: 10.3390/IJMS21020602
95. Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, et al. Autocrine Effects of Tumor-Derived Complement. Cell Rep (2014) 6(6):1085–95. doi: 10.1016/J.CELREP.2014.02.014
96. Satyam A, Kannan L, Matsumoto N, Geha M, Lapchak PH, Bosse R, et al. Intracellular Activation of Complement 3 Is Responsible for Intestinal Tissue Damage During Mesenteric Ischemia. J Immunol (2017) 198(2):788–97. doi: 10.4049/JIMMUNOL.1502287
97. Zha H, Wang X, Zhu Y, Chen D, Han X, Yang F, et al. Intracellular Activation of Complement C3 Leads to PD-L1 Antibody Treatment Resistance by Modulating Tumor-Associated Macrophages. Cancer Immunol Res (2019) 7(2):193–207. doi: 10.1158/2326-6066.CIR-18-0272
98. Shimazaki R, Takano S, Satoh M, Takada M, Miyahara Y, Sasaki K, et al. Complement factor B regulates cellular senescence and is associated with poor prognosis in pancreatic cancer. Cell Oncol (2021) 44(4):937–50. doi: 10.1007/S13402-021-00614-Z/FIGURES/6
99. Andrejeva G, Rathmell JC. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab (2017) 26(1):49–70. doi: 10.1016/J.CMET.2017.06.004
100. Ghebrehiwet B, Lu PD, Zhang W, Keilbaugh SA, Leigh LE, Eggleton P, et al. Evidence That the Two C1q Binding Membrane Proteins, Gc1q-R and Cc1q-R, Associate to Form a Complex. J Immunol (1997) 159(3):1429–36.
101. Ling GS, Crawford G, Buang N, Bartok I, Tian K, Thielens NM, et al. C1q Restrains Autoimmunity and Viral Infection by Regulating CD8+ T-Cell Metabolism. Science (New York N Y) (2018) 360(6388):558. doi: 10.1126/SCIENCE.AAO4555
102. Revel M, Sautès-Fridman C, Fridman WH, Roumenina LT. C1q+ macrophages: passengers or drivers of cancer progression. Trends in Cancer (2022) 0(0). doi: 10.1016/J.TRECAN.2022.02.006
103. Haldar S, Mishra R, Billet S, Thiruvalluvan M, Placencio-Hickok VR, Madhav A, et al. Cancer Epithelia-Derived Mitochondrial DNA Is a Targetable Initiator of a Paracrine Signaling Loop That Confers Taxane Resistance. Proc Natl Acad Sci U S A (2020) 117(15):8515–23. doi: 10.1073/PNAS.1910952117/-/DCSUPPLEMENTAL
104. Revel M, Daugan MV, Sautés-Fridman C, Fridman WH, Roumenina LT. Complement System: Promoter or Suppressor of Cancer Progression? Antibodies (2020) 9(4):57. doi: 10.3390/ANTIB9040057
105. Zhang D, Li Y, Li H, Tang T, Zheng Y, Guo X, et al. A Preliminary Study of the Complement Component 1q Levels in Predicting the Efficacy of Combined Immunotherapy in Patients With Lung Cancer. Cancer Manag Res (2021) 13:7131–7. doi: 10.2147/CMAR.S314369
106. Sogawa K, Takano S, Iida F, Satoh M, Tsuchida S, Kawashima Y, et al. Identification of a Novel Serum Biomarker for Pancreatic Cancer, C4b-Binding Protein α-Chain (C4BPA) by Quantitative Proteomic Analysis Using Tandem Mass Tags. Br J Cancer (2016) 115(8):949–56. doi: 10.1038/bjc.2016.295
107. Huang Y, Chen F, Zhang L, Lv Q, Yan J, Cui W. MALDI-TOF-MS Analysis in the Discovery and Identification of the Serum Peptide Pattern of Pancreatic Ductal Adenocarcinoma. Lab Med (2021) 52(6):558–66. doi: 10.1093/LABMED/LMAB024
108. Chen J, Wu W, Chen L, Zhou H, Yang R, Hu L, et al. Profiling the Potential Tumor Markers of Pancreatic Ductal Adenocarcinoma Using 2D-DIGE and MALDI-TOF-MS: Up-Regulation of Complement C3 and Alpha-2-HS-Glycoprotein. Pancreatology (2013) 13(3):290–7. doi: 10.1016/J.PAN.2013.03.010
109. Cao W-Q, Jiang B-Y, Huang J-M, Zhang L, Liu M-Q, Yao J, et al. Straightforward and Highly Efficient Strategy for Hepatocellular Carcinoma Glycoprotein Biomarker Discovery Using a Nonglycopeptide-Based Mass Spectrometry Pipeline. Anal Chem (2019) 91(19):12435–43. doi: 10.1021/ACS.ANALCHEM.9B03074
110. Beitia M, Romano P, Larrinaga G, Solano-Iturri JD, Salis A, Damonte G, et al. The Activation of Prothrombin Seems to Play an Earlier Role Than the Complement System in the Progression of Colorectal Cancer: A Mass Spectrometry Evaluation. Diagnostics (2020) 10(12):1077. doi: 10.3390/DIAGNOSTICS10121077
111. Zhang X, Sun L. Anaphylatoxin C3a: A Potential Biomarker for Esophageal Cancer Diagnosis. Mol Clin Oncol (2018) 8(2):315. doi: 10.3892/MCO.2017.1524
112. Chung L, Moore K, Phillips L, Boyle FM, Marsh DJ, Baxter RC. Novel Serum Protein Biomarker Panel Revealed by Mass Spectrometry and its Prognostic Value in Breast Cancer. Breast Cancer Res (2014) 16(3):R63. doi: 10.1186/BCR3676
113. Yuan K, Ye J, Liu Z, Ren Y, He W, Xu J, et al. Complement C3 Overexpression Activates JAK2/STAT3 Pathway and Correlates With Gastric Cancer Progression. J Exp Clin Cancer Res (2020) 39(1):1–15. doi: 10.1186/S13046-019-1514-3
114. Chen Y, Wu ED, Ma S, Bai WQ, Yin YJ, Shi GL. The Clinical Significance of Serum MASP-2 and IDH1 in the Early Diagnosis of non-Small Cell Lung Cancer. Clin Lab (2020) 66(11):2287–96. doi: 10.7754/CLIN.LAB.2020.200249
115. Bouwens TAM, Trouw LA, Veerhuis R, Dirven CMF, Lamfers MLM, Al-Khawaja H. Complement Activation in Glioblastoma Multiforme Pathophysiology: Evidence From Serum Levels and Presence of Complement Activation Products in Tumor Tissue. J Neuroimmunol (2015) 278:271–6. doi: 10.1016/J.JNEUROIM.2014.11.016
116. Lee MJ, Na K, Jeong SK, Lim JS, Kim SA, Lee MJ, et al. Identification of human complement factor B as a novel biomarker candidate for pancreatic ductal adenocarcinoma. J Proteo Res (2014) 13(11):4878–88. doi: 10.1021/PR5002719/SUPPL_FILE/PR5002719_SI_001.PDF
117. Daugan MV, Revel M, Russick J, Dragon-Durey M-A, Gaboriaud C, Robe-Rybkine T, et al. Complement C1s and C4d as Prognostic Biomarkers in Renal Cancer: Emergence of Noncanonical Functions of C1s. Cancer Immunol Res (2021) 9(8):891–908. doi: 10.1158/2326-6066.CIR-20-0532
118. Ajona D, Pajares MJ, Corrales L, Perez-Gracia JL, Agorreta J, Lozano MD. Investigation of Complement Activation Product C4d as a Diagnostic and Prognostic Biomarker for Lung Cancer. JNCI: J Nat Canc Instit (2013) 105(18):1385–93. doi: 1093/JNCI/DJT205
119. Ajona D, Pajares MJ, Chiara MD, Rodrigo JP, Jantus-Lewintre E, Camps C, et al. Complement activation product C4d in oral and oropharyngeal squamous cell carcinoma. Oral Disease (2015) 21(7):899–904. doi: 10.1111/ODI.12363
120. Ajona D, Ortiz-Espinosa S, Moreno H, Lozano T, Pajares MJ, Agorreta J, et al. A Combined PD-1/C5a Blockade Synergistically Protects Against Lung Cancer Growth and Metastasis. Cancer Discov (2017) 7(7):694–703. doi: 10.1158/2159-8290.CD-16-1184
121. Zhu H, Yu X, Zhang S, Shu K. Targeting the Complement Pathway in Malignant Glioma Microenvironments. Front Cell Dev Biol (2021) 9:758. 10.3389/FCELL.2021.657472
122. Wang T, Lu R, Kapur P, Jaiswal BS, Hannan R, Zhang Z, et al. An Empirical Approach Leveraging Tumorgrafts to Dissect the Tumor Microenvironment in Renal Cell Carcinoma Identifies Missing Link to Prognostic Inflammatory Factors. Cancer Discov (2018) 8(9):1142–55. doi: 10.1158/2159-8290.CD-17-1246
123. Dong Y, Ma W, Yang W, Hao L, Zhang S, Fang K, et al. Identification of C3 and FN1 as Potential Biomarkers Associated With Progression and Prognosis for Clear Cell Renal Cell Carcinoma. BMC Cancer (2021) 21(1):1–17. doi: 10.1186/S12885-021-08818-0
124. Su K-M, Lin T-W, Liu L-C, Yang Y-P, Wang M-L, Tsai P-H, et al. The Potential Role of Complement System in the Progression of Ovarian Clear Cell Carcinoma Inferred From the Gene Ontology-Based Immunofunctionome Analysis. Int J Mol Sci (2020) 21(8):2824. doi: 10.3390/IJMS21082824
125. Liang J-Z, Liang X-L, Zhong L-Y, Wu C-T, Zhang J, Wang Y. Comparative Proteome Identifies Complement Component 3-Mediated Immune Response as Key Difference of Colon Adenocarcinoma and Rectal Adenocarcinoma. Front Oncol (2021) 10:3314. doi: 10.3389/FONC.2020.617890
126. Popeda M, Markiewicz A, Stokowy T, Szade J, Niemira M, Kretowski A, et al. Reduced Expression of Innate Immunity-Related Genes in Lymph Node Metastases of Luminal Breast Cancer Patients. Sci Rep (2021) 11(1):5097. doi: 10.1038/S41598-021-84568-0
127. Cho M, Hu Q, Rupaimoole R, Sood A, Afshar-Kharghan V. Complement Component 3 (C3) Is a Mediator of Epithelial-Mesenchymal Transition (EMT). Blood (2014) 12(21):1421–1421. doi: 10.1182/BLOOD.V124.21.1421.1421
128. Wan J, Zhou X, Cui J, Zou Z, Xu Y, You D. Role of Complement 3 in TNF-α-Induced Mesenchymal Transition of Renal Tubular Epithelial Cells In Vitro. Molecular Biotechnology (2012) 54(1):92–100. doi: 10.1007/S12033-012-9547-2
Keywords: cancer, extracellular complement, intracellular complement, biomarkers, therapy, tumor microenvironment, complosome
Citation: Kolev M, Das M, Gerber M, Baver S, Deschatelets P and Markiewski MM (2022) Inside-Out of Complement in Cancer. Front. Immunol. 13:931273. doi: 10.3389/fimmu.2022.931273
Received: 28 April 2022; Accepted: 06 June 2022;
Published: 01 July 2022.
Edited by:
Katy Rezvani, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Lubka T. Roumenina, U1138 Centre de Recherche des Cordeliers (CRC) (INSERM), FranceCopyright © 2022 Kolev, Das, Gerber, Baver, Deschatelets and Markiewski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Kolev, bWFydGluLmtvbGV2QGFwZWxsaXMuY29t; Maciej M. Markiewski, bWFjaWVqLm1hcmtpZXdza2lAdHR1aHNjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.