 Alessandra Petrelli1
Alessandra Petrelli1 Sarah K. Popp2
Sarah K. Popp2 Riho Fukuda2,3
Riho Fukuda2,3 Christopher R. Parish4
Christopher R. Parish4 Emanuele Bosi1,5
Emanuele Bosi1,5 Charmaine J. Simeonovic2*
Charmaine J. Simeonovic2*- 1San Raffaele Diabetes Research Institute, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Ospedale San Raffaele, Milan, Italy
- 2Immunology and Infectious Disease Division, The John Curtin School of Medical Research, The Australian National University, Canberra, ACT, Australia
- 3Department of Medicine, Tokyo Medical and Dental University, Bunkyo City, Tokyo, Japan
- 4Genome Sciences and Cancer Division, The John Curtin School of Medical Research, The Australian National University, Canberra, ACT, Australia
- 5Department of Medicine, San Raffaele Vita Salute University, Milan, Italy
Type 1 diabetes (T1D) is an autoimmune disease resulting from the destruction of insulin-producing beta cells in pancreatic islets. T lymphocytes are the claimed pathogenic effectors but abnormalities of other immune cell types, including neutrophils, also characterize T1D development. During human T1D natural history, neutrophils are reduced in the circulation, while accumulate in the pancreas where release of neutrophil extracellular traps (NETs), or NETosis, is manifest. Recent-onset T1D patients also demonstrate activated circulating neutrophils, associated with a unique neutrophil gene signature. Neutrophils can bind to platelets, leading to the formation of platelet-neutrophil aggregates (PNAs). PNAs increase in the circulation during the development of human T1D and provide a mechanism for neutrophil activation and mobilization/recruitment to the pancreas. In non-obese diabetic or NOD mice, T1D autoimmunity is accompanied by dynamic changes in neutrophil numbers, activation state, PNAs and/or NETosis/NET proteins in the circulation, pancreas and/or islets. Such properties differ between stages of T1D disease and underpin potentially indirect and direct impacts of the innate immune system in T1D pathogenesis. Supporting the potential for a pathogenic role in T1D, NETs and extracellular histones can directly damage isolated islets in vitro, a toxicity that can be prevented by small polyanions. In human T1D, NET-related damage can target the whole pancreas, including both the endocrine and exocrine components, and contribute to beta cell destruction, providing evidence for a neutrophil-associated T1D endotype. Future intervention in T1D could therefore benefit from combined strategies targeting T cells and accessory destructive elements of activated neutrophils.
Introduction to NETs in T1D
Type 1 diabetes is an autoimmune disease in which insulin-producing beta cells in pancreatic islets undergo immune destruction, ultimately resulting in hyperglycemia. The detection of beta cell autoantigen-reactive T cells in peripheral blood (1, 2) and islet inflammation (insulitis) (3–5) together with the absence of autoantibody-mediated beta cell damage (6, 7), have supported the concept that T1D in humans is T cell-mediated. However, recent evidence has suggested that cells of the innate immune system, particularly neutrophils, also participate in T1D development in at least a subset of individuals. Similarly, differences in islet- infiltrating immune cell populations have been identified in children who develop clinical T1D at different ages (8). These and other studies support the concept that T1D disease is heterogeneous and that disparities between the disease process in different individuals relate to distinct endotypes (9–12).
Studies including the NOD mouse model of T1D, have confirmed multiple roles for activated/NETosing neutrophils in T1D pathogenesis, with the mechanism of neutrophil activation appearing to differ between disease initiation and progression. Although circulating NETs are elevated in recent-onset human T1D (13), the detection of NETs in the pancreas is infrequent, possibly due to their rapid degradation by locally produced exocrine DNAse (14). As a consequence, NET products, e.g., myeloperoxidase or MPO, neutrophil elastase (NE) and citrullinated histones (CitH3), are commonly used as surrogate markers of NETosis and provide a focus for elucidating the function(s) of neutrophils in T1D. Strongly supporting a pathogenic role for neutrophils/NETs in T1D, circulating levels of indirect markers of neutrophil activation (e.g., PNAs) (15) and NET proteins (e.g., NE) (13) follow a dynamic pattern throughout T1D development, largely mirroring levels in islets in NOD mice at different stages of disease progression (15) or negatively correlating with glycemic control/beta cell function in humans (13). Surprisingly, studies of T1D human pancreas samples have also revealed neutrophil infiltration and NETosis in exocrine pancreatic tissue (16, 17), exposing T1D as a complex immune disease of the pancreas which extends beyond autoimmune damage of islet beta cells (18).
Role for Neutrophils, NETs and PNAs in the Development of T1D
Human T1D
Both innate and adaptive immunity participate in T1D pathogenesis in humans. Neutrophils, the most abundant phagocytic cell in human blood (19), display heterogeneous functions and flexibility, developing different profiles in response to different disorders, including autoimmune diseases (20). A role for neutrophils has been consistently shown in human T1D (15–17, 21). Circulating neutrophil counts are reduced to the lower limits of the normal range in patients with recent-onset T1D; first‐degree relatives of T1D patients with 2 or more autoantibodies (i.e., pre-symptomatic or pre-T1D) also exhibit a lower circulating neutrophil count which parallels the development of beta cell dysfunction (16). This neutrophil abnormality is not attributed to neutrophil cell death, impaired differentiation or targeting by anti-neutrophil antibodies (16), suggesting that the fate of neutrophils in pre-T1D may be to exit the circulation and infiltrate the pancreas. The decrease in circulating neutrophil counts is stable for up to 1 year after the onset of T1D (22), with normal levels returning thereafter (13).
Neutrophils from T1D and pre‐symptomatic subjects display an interferon (IFN) pro‐inflammatory signature (17), resembling other autoimmune diseases such as rheumatoid arthritis (23) and systemic lupus erythematosus (SLE) (24). Neutrophils from T1D patients are altered in their phenotype and function. Post-T1D onset, they display reduced migration and chemotaxis (25), paralleling the sharp decline in circulating PNA levels (15). They also exhibit impaired phagocytic capacity, leading to reduced clearance of bacteria (26) and to hyperglycemia-dependent increased susceptibility to infections (27).
Activated neutrophils release a variety of serine proteases from intracellular granules, including NE and proteinase 3 (PR‐3), which help to eliminate microorganisms and regulate immune responses during inflammation (28). Circulating levels of NE and PR3 are increased in patients with T1D and correlate with numbers and titres of autoantibodies (13). Furthermore, MPO, an enzyme involved in microbial killing (29), is similarly increased, regardless of T1D duration (30). Despite the uncertain mechanism underlying neutrophil-mediated development of islet autoimmunity in humans, NE, PR‐3 and MPO may each contribute to T1D pathogenesis, possibly targeting beta cells, resident pancreatic cells, or have other roles, as revealed in NOD mice.
Besides phagocytosis and degranulation, neutrophils can damage host tissues by the release of NETs. NETs are extracellular web-like structures composed of cytosolic and granule proteins assembled on decondensed chromatin (31). Normally the function of NETs is to neutralize and kill microorganisms. If dysregulated, NETs can contribute to the pathogenesis of immune-mediated diseases (32). Enhanced NETs have been described in the circulation of T1D patients (13), where they display an altered composition compared to healthy individuals and induce Th1 polarization (33). Notably, peptidyl arginine deiminase‐4 (PAD4), the enzyme responsible for histone citrullination and NET formation (34), is increased in neutrophils of patients with T1D and T2D, leading to increased NETosis upon stimulation (35). While increased NET formation is triggered by hyperglycemia after T1D-onset, NETosis also occurs in pre-T1D individuals, as suggested by deposition of NET products (CitH3, MPO) in the pancreas (17).
Altered numbers and function of neutrophils have been described not only in the circulation, but also in the pancreas of patients with T1D. Indeed we have identified neutrophils and decondensed DNA decorated with MPO and citrullinated histones, indicating the presence of pancreas-residing neutrophils releasing NETs, in both pre-T1D and T1D individuals (16, 17). Furthermore, we have recently demonstrated that extracellular histones damage human islets in vitro (15), providing a putative mechanism for neutrophil-mediated islet cell damage. Of note, methyl cellobiose sulfate (mCBS), a small polyanionic drug that neutralizes the high positive charge of histones, prevented islet cell death (15, 36). Thus, mCBS could offer potential, in combination with other agents, for delaying T1D progression. These findings corroborate the hypothesis that neutrophils play an ancillary role in the development of islet autoimmunity and beta cell damage during the pathogenesis of T1D.
How neutrophils are activated to release NETs in T1D is not completely understood. Activated platelets can bind neutrophils through an interaction between platelet cell surface CD62P (P-selectin) and neutrophil P-selectin binding glycoprotein ligand 1 (PSGL1) (32, 37), which triggers the release of NETs (32, 38). In this setting, initial platelet activation could occur via the binding of citrullinated histones to platelet TLR2/TLR4 (39), supporting a vicious cycle of platelet and neutrophil activation, or potentially by autoantigen-IgG immune complexes binding to platelet FcγRIIA (40). Platelets have been shown to direct neutrophil trafficking (41) and P-selectin interactions with PSGL-1 at the vessel wall guide neutrophil extravasation (42). We have recently shown that PNAs are elevated in some pre-T1D individuals and new-onset T1D patients, and that a lower circulating neutrophil count correlates with a higher proportion of PNAs (15). Furthermore, we revealed platelet hyperreactivity in PNAs preceding T1D onset (15). These data support the hypothesis that neutrophils are reduced in the circulation prior to the development of T1D because they are bound to activated platelets, with this interaction promoting neutrophil activation, trafficking to the pancreas, subsequent NET release and islet damage/beta cell impairment. Altogether, these data provide evidence for the potential contribution of innate immune responses to T1D disease progression in humans.
NOD Mouse Model of T1D
Studies in the NOD mouse model of autoimmune T1D have revealed possible ancillary and effector roles for neutrophils in the initiation and progression of T1D disease. During the initiation of T1D autoimmunity, a transient elevation in neutrophil numbers in the islets of 3-4 week old NOD mice (43, 44) was accompanied by the activation of neutrophils by antibodies against beta cell DNA (anti-DNA Ig), CRAMP (anti-microbial peptide) release and occasional peri-islet NETosis (43). The in vivo depletion of neutrophils and other leukocyte populations in young NOD mice prevented later T1D-onset and were considered to define a local accessory role for neutrophils in activating plasmacytoid dendritic cells (DCs) and the local production of IFNα. Such neutrophil interactions may indirectly promote autoantigen presentation by conventional DCs in pancreatic lymph nodes, activating autoreactive T cells and initiating early autoimmune damage of beta cells (43).
In contrast, evidence for prolonged systemic neutrophil activation was found in longitudinal studies of NOD mice from 2-30 weeks of age. NE and PR3 activities were elevated in the circulation for > 10 weeks in only NOD mice which progressed to T1D (13). These findings suggested that circulating neutrophils may contribute to both the initiation and progression of T1D disease, potentially either by degranulation in the bloodstream and release of granule-derived enzymes or from the expulsion of such enzymes during NETosis. By treating young NOD mice with pharmacological or genetically engineered inhibitors of NE, Shu et al. reported NE-induced islet inflammation throughout T1D development, inferring an indirect role for activated neutrophils in autoimmune damage of beta cells (45). NOD mouse models have further identified an inter-relationship between certain gut microbiota, increased gut permeability, neutrophil numbers/NETosis and T1D (46, 47). In particular, gut leakage of abnormal microbiota/lipopolysaccharide (LPS) has been reported to stimulate local NETosis in NOD mice, resulting in NET-assisted activation of enteric CD4 T cells which, upon migration, can enhance T1D autoimmunity (47). Such studies provide a possible indirect link between neutrophils/NETs and adaptive immunity in T1D.
Similar to studies of human pancreata (16, 17) and young NOD mice (44), we recently reported a significant increase in the immunohistochemical localization of islet-associated NET products (MPO and CitH3) in the pancreas of adult pre-T1D (10-12 weeks of age) and T1D-onset NOD mice (15) (Figure 1). In parallel, calf thymus histones and NETs were found to be toxic for isolated mouse islets, with NET- as well as histone-induced islet damage being prevented by short-term culture with mCBS (15, 36). Collectively, these findings suggested that citrullinated histones released from NETosing neutrophils could directly contribute to islet/beta cell damage during T1D development, most likely by inducing pores in the plasma membrane of islet/beta cells (15, 36, 49, 50).
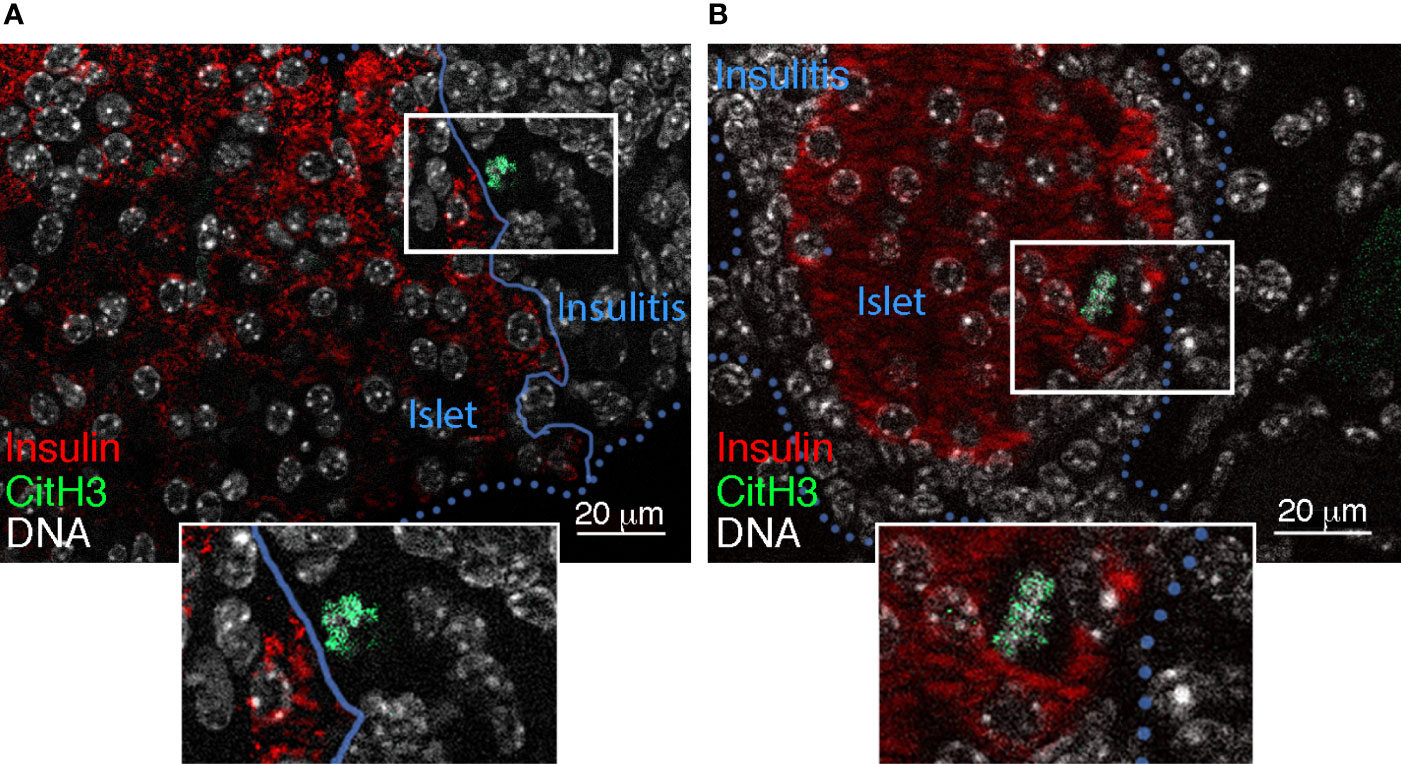
Figure 1 Islet-associated CitH3 in NOD pancreas. Immunofluorescence detection of insulin (red) (48) and CitH3 (green) (15) (using AF488 donkey anti-rabbit Ig) in pancreas sections from (A) 4 week-old and (B) 10-12 week-old female NOD mice. Blue unbroken and dotted lines define the islet border in (A) and insulitis, respectively. DNA is labelled using Hoechst 33342 (white).
The reason underlying the recruitment of neutrophils to the pancreas during the initiation of T1D autoimmunity in very young NOD mice has been attributed to a physiological wave of beta cell apoptosis that occurs during pancreas differentiation (51). This process is envisaged to prompt an acute inflammatory response to islets, triggered especially by chemokines (CXCL1, CXCL2) released from damaged beta cells and/or intra-islet macrophages (43, 52). Endothelial cells inadvertently injured during this early inflammatory response and neutrophil (PSGL1)-endothelial (P-Selectin or CD62P) interactions could facilitate neutrophil influx into the pancreas (44) and the appearance of activated neutrophils in the islet microenvironment (43).
Levels of PNAs are elevated in the circulation of young NOD mice at 4 weeks as well as adult NOD mice at 10-12 weeks (pre-T1D) and at T1D-onset, largely mimicking increased circulating PNAs in at-risk Aab-positive and T1D-onset children (15). PNA levels in NOD islets were similarly increased at 10-12 weeks and T1D-onset, correlating with a significant rise in the proportion of islets that were MPO- or CitH3-positive. High expression of CD62P on platelets within NOD mouse PNAs suggested that CD62P (platelet)-PSGL1 (neutrophil) interactions are key to PNA formation and neutrophil activation, similar to human PNAs. Subsequently, neutrophil mobilization to pancreatic islets, most likely occurs via platelet-derived proplatelet binding protein (PPBP) which undergoes cleavage to form CXCL7/NAP2 (a neutrophil -activating and - recruiting chemokine) (53–55) and has been found elevated in T1D human blood by serum proteome analysis (56). Ultimately this pathway results in islet-associated NETosis (15). Thus, stimulation of neutrophils by activated platelets could provide a supplementary mechanism driving the contribution of neutrophils to the initiation of T1D disease. Thereafter, a pivotal role has been proposed for platelet-activated neutrophils (via PNAs) in islet-associated NETosis (in NOD mice) and disease progression to T1D-onset (in both NOD mice and humans) (15).
From a therapeutic perspective, T1D was prevented by treatment of adult NOD mice with a broad-acting inhibitor of PADs, which citrullinate proteins. Of these PADs, PAD4 citrullinates nuclear histones, a critical step in NETosis (57). This report clearly established the importance of citrullination in T1D autoimmunity, albeit without defining a role for NETs. In contrast, inhibitors that specifically target enzymatic products of neutrophils/NETosis (NE, MPO) or that neutralize NET/histone toxicity have had variable outcomes on T1D incidence (15, 44, 45), with most approaches failing to prevent T1D onset (15, 44). However, treatment of NOD mice from a very young age with NE inhibitors significantly reduced T1D incidence by dampening islet inflammation (45). Such interventions at the very onset of autoimmunity in humans are inevitably unrealistic. Therapies targeting NETs/neutrophil products in combination with short-term protocols targeting T cells (58, 59) may therefore provide more robust prevention of T1D in NOD mice and a basis for an improved clinical strategy for impeding T1D pathogenesis.
T1D Is a Chronic Disease of Both the Endocrine and Exocrine Pancreas
Involvement of the exocrine pancreas in the pathogenesis of T1D, hypothesized long ago (60), is currently supported by documented reduction of pancreatic volume (61–63) and weight (64, 65), tissue damage and infiltration (16, 17, 66, 67) and subclinical impaired exocrine function (68–71). Interestingly, many of these exocrine abnormalities arise during pre-symptomatic T1D stages (17, 61, 64–67, 69–71), in parallel with the silent progression of endocrine dysfunction. These properties underpin the novel concept that T1D is a chronic disease of both the endocrine and exocrine pancreas and is thus an autoimmune or immune -mediated organ-specific disease, seemingly driven by a unique pathogenic process.
Role of Neutrophils and NETosis in the Pathogenesis of T1D-Associated Pancreas Exocrine Disease
In parallel with the mild neutropenia and IFN-associated gene signature in neutrophils that accompanies T1D pathogenesis (16, 17), neutrophils infiltrate the exocrine tissue of the pancreas (16, 17, 67). This neutrophil influx appears very early, during the pre-symptomatic stages of T1D, at the time when islet autoantibodies are detectable, and persists afterwards (17). Notably, a fraction of pancreas-infiltrating neutrophils has been shown to extrude NETs (17), a known mechanism of tissue injury (72). Neutrophils may therefore act as innate immune effectors of exocrine and endocrine pancreas tissue injury in pre-T1D, with the formation of PNAs contributing to neutrophil activation, neutrophil recruitment to the pancreas and NETosis (15, 17).
Innate and Adaptive Immunity in the Course of T1D Endocrine and Exocrine Pancreatic Disease
How does the accumulating evidence for neutrophils in the exocrine pancreas during T1D pathogenesis fit with the current knowledge of the natural history of the disease? The conventional paradigm for T1D identifies a predisposed genetic background, where some yet unidentified environmental factors induce an autoimmune response, marked by the appearance of islet-specific autoantibodies (single, then multiple), the progressive decline of beta cell mass due to autoreactive T cells (3, 73), diminishing beta cell function, finally ensuing in hyperglycemia and clinical T1D (74). Interestingly, in this model, the measurable immune markers, autoantibodies and autoreactive T cells, represent an expression of adaptive immunity towards endocrine pancreas. In contrast, there are no signs of an adaptive immune response against the exocrine pancreas.
Conversely, neutrophil abnormalities are detectable very early in the course of T1D: neutropenia, pancreatic infiltration and elevated circulating PNAs are detectable in pre-symptomatic islet autoantibody-positive individuals (15, 17), while the IFN-gene oriented signature in peripheral neutrophils is found even prior to seroconversion (17). These observations are consistent with a precocious activation of innate immunity during T1D natural history, possibly before the activation of adaptive (auto)-immunity. In such a scenario, exocrine pancreas seems to be damaged by native immunity (i.e., neutrophils) rather than by adaptive immunity (e.g., possible bystander T cells (67)), while the endocrine pancreas is affected by both innate immunity (neutrophils) and adaptive immunity (autoreactive T cells). This may account for the different functional outcomes for the two pancreatic compartments in T1D. While endocrine dysfunction progresses to beta cell failure and clinical diabetes, the impairment of exocrine function remains subclinical, measurable in terms of reduced trypsinogen (71), fecal elastase, pancreatic amylase and lipase (68–70), but never reaching the degree of clinical insufficiency requiring pancreatic enzyme replacement therapy (summarized in Figure 2). How these two distinct processes influence each other is unknown, and remains a matter for future investigation of endotype interactions within T1D (9).
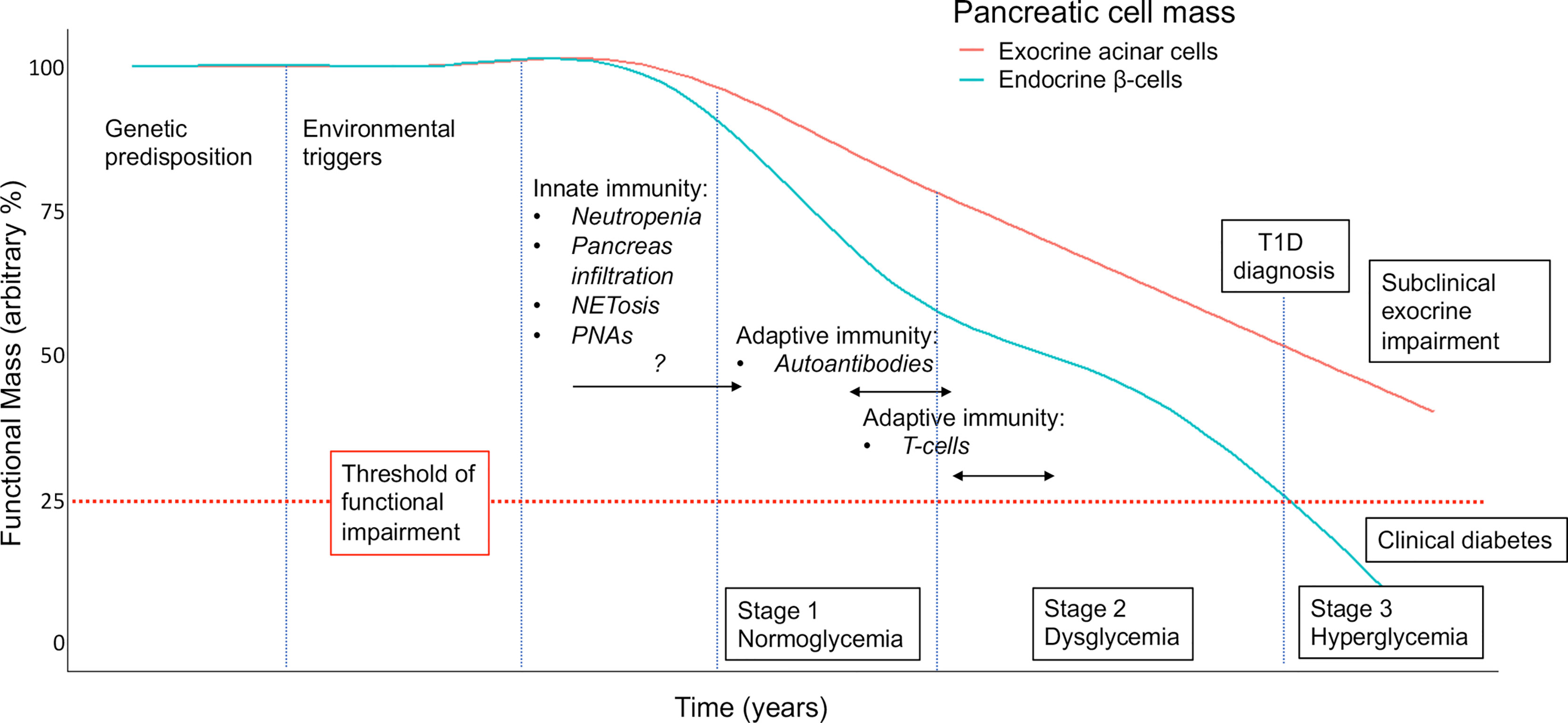
Figure 2 Proposed model for the contribution of neutrophils/NETs to the natural history of the decline in pancreatic endocrine (i.e., beta cell) and exocrine (acinar) function in T1D. Staging, as defined in (74), with modifications and incorporating assumptions based on available clinical and preclinical data. An arbitrary threshold is set at 25% to indicate functional pancreatic insufficiency. NETosis, neutrophils releasing extracellular traps; PNAs, platelet-neutrophil aggregates.
Discussion
Evidence from T1D studies in humans and NOD mice suggests that neutrophils potentially have multiple roles in the pathogenesis of T1D: (i) indirectly aiding the initiation and expansion of autoreactive T cells and promoting islet inflammation by degranulation and/or NETosis, (ii) directly participating as effectors of islet/beta cell damage via NETs/NET proteins (extracellular histones) (15) and (iii) directly injuring pancreatic exocrine tissue. During T1D progression, neutrophils are rarely identified in islets/insulitis, possibly due to NETosis, and are more prevalent throughout the exocrine pancreas (15–17). NET proteins are more readily identified than NETs in both the endocrine and exocrine pancreas (15–17). These findings may in part be due to the rapid degradation of NET DNA by pancreatic DNAse (14) and/or to phagocytosis by macrophages (75), an activity which may be exacerbated by islet-associated inflammation (insulitis). Furthermore, NET proteins are more frequently detected in human exocrine pancreatic tissue than in islets during T1D progression (16, 17), consistent with the accompanying atrophy of exocrine tissue and function (18, 61, 63–71). Finally, heterogeneity in circulating neutrophil/PNA profiles accompanies both pre-symptomatic stages of T1D autoimmunity and T1D-onset in humans and NOD mice. Thus, the neutrophil signature in a subset of T1D and T1D-prone humans lends support to a neutrophil-associated T1D endotype.
Author Contributions
CJS, AP, and EB wrote the manuscript; CRP provided intellectual input and edited the manuscript; SKP reviewed the manuscript; RF and SKP contributed the data in Figure 1. EB contributed Figure 2. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by an Innovation Research Grant to CJS from the Juvenile Diabetes Research Foundation (JDRF)Australian T1D Clinical Research Network (3-SRA-2018-602-M-B). AP was supported by a Juvenile Diabetes ResearchFoundation Advanced Postdoctoral Fellowship (3-APF-2019-744-AN). EB was supported in part by a Fondazione ItalianaDiabete (FID) research grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Claessens LA, Wesselius J, van Lummel M, Laban S, Mulder F, Mul D, et al. Clinical and Genetic Correlates of Islet-Autoimmune Signatures in Juvenile-Onset Type 1 Diabetes. Diabetologia (2020) 63(2):351–61. doi: 10.1007/s00125-019-05032-3
2. Velthuis JH, Unger WW, Abreu JR, Duinkerken G, Franken K, Peakman M, et al. Simultaneous Detection of Circulating Autoreactive CD8+ T-Cells Specific for Different Islet Cell-Associated Epitopes Using Combinatorial MHC Multimers. Diabetes (2010) 59(7):1721–30. doi: 10.2337/db09-1486
3. Anderson AM, Landry LG, Alkanani AA, Pyle L, Powers AC, Atkinson MA, et al. Human Islet T Cells Are Highly Reactive to Preproinsulin in Type 1 Diabetes. Proc Natl Acad Sci U.S.A. (2021) 118(41). doi: 10.1073/pnas.2107208118
4. Pugliese A. Autoreactive T Cells in Type 1 Diabetes. J Clin Invest (2017) 127(8):2881–91. doi: 10.1172/JCI94549
5. So M, Elso CM, Tresoldi E, Pakusch M, Pathiraja V, Wentworth JM, et al. Proinsulin C-Peptide Is an Autoantigen in People With Type 1 Diabetes. Proc Natl Acad Sci U.S.A. (2018) 115(42):10732–7. doi: 10.1073/pnas.1809208115
6. Mannering SI, Pathiraja V, Kay TW. The Case for an Autoimmune Aetiology of Type 1 Diabetes. Clin Exp Immunol (2016) 183(1):8–15. doi: 10.1111/cei.12699
7. Pietropaolo M, Towns R, Eisenbarth GS. Humoral Autoimmunity in Type 1 Diabetes: Prediction, Significance, and Detection of Distinct Disease Subtypes. Cold Spring Harb Perspect Med (2012) 2(10). doi: 10.1101/cshperspect.a012831
8. Leete P, Willcox A, Krogvold L, Dahl-Jorgensen K, Foulis AK, Richardson SJ, et al. Differential Insulitic Profiles Determine the Extent of Beta-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes (2016) 65(5):1362–9. doi: 10.2337/db15-1615
9. Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, et al. Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes Care (2020) 43(1):5–12. doi: 10.2337/dc19-0880
10. Dufort MJ, Greenbaum CJ, Speake C, Linsley PS. Cell Type-Specific Immune Phenotypes Predict Loss of Insulin Secretion in New-Onset Type 1 Diabetes. JCI Insight (2019) 4(4). doi: 10.1172/jci.insight.125556
11. Petrelli A, Atkinson MA, Pietropaolo M, Giannoukakis N. Modulation of Leukocytes of the Innate Arm of the Immune System as a Potential Approach to Prevent the Onset and Progression of Type 1 Diabetes. Diabetes (2021) 70(2):313–22. doi: 10.2337/dbi20-0026
12. Powers AC. Type 1 Diabetes Mellitus: Much Progress, Many Opportunities. J Clin Invest 305 (2021) 131(8). Epub2021/03/25. doi: 10.1172/JCI142242
13. Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, et al. Increased Neutrophil Elastase and Proteinase 3 and Augmented Netosis Are Closely Associated With Beta-Cell Autoimmunity in Patients With Type 1 Diabetes. Diabetes (2014) 63(12):4239–48. doi: 10.2337/db14-0480
14. Takeshita H, Mogi K, Yasuda T, Nakajima T, Nakashima Y, Mori S, et al. Mammalian Deoxyribonucleases I Are Classified Into Three Types: Pancreas, Parotid, and Pancreas-Parotid (Mixed), Based on Differences in Their Tissue Concentrations. Biochem Biophys Res Commun (2000) 269(2):481–4. doi: 10.1006/bbrc.2000.2300
15. Popp SK, Vecchio F, Brown DJ, Fukuda R, Suzuki Y, Takeda Y, et al. Circulating Platelet-Neutrophil Aggregates Characterize the Development of Type 1 Diabetes in Humans and NOD Mice. JCI Insight (2022) 7(2). doi: 10.1172/jci.insight.153993
16. Valle A, Giamporcaro GM, Scavini M, Stabilini A, Grogan P, Bianconi E, et al. Reduction of Circulating Neutrophils Precedes and Accompanies Type 1 Diabetes. Diabetes (2013) 62(6):2072–7. doi: 10.2337/db12-1345
17. Vecchio F, Lo Buono N, Stabilini A, Nigi L, Dufort MJ, Geyer S, et al. Abnormal Neutrophil Signature in the Blood and Pancreas of Presymptomatic and Symptomatic Type 1 Diabetes. JCI Insight (2018) 3(18). doi: 10.1172/jci.insight.122146
18. Campbell-Thompson M, Rodriguez-Calvo T, Battaglia M. Abnormalities of the Exocrine Pancreas in Type 1 Diabetes. Curr Diabetes Rep (2015) 15(10):79. doi: 10.1007/s11892-015-0653-y
19. Rosales C. Neutrophil: A Cell With Many Roles in Inflammation or Several Cell Types? Front Physiol (2018) 9. doi: 10.3389/fphys.2018.00113
20. Zhao Y, Marion TN, Wang Q. Multifaceted Roles of Neutrophils in Autoimmune Diseases. J Immunol Res (2019) 2019:7896738. doi: 10.1155/2019/7896738
21. Harsunen MH, Puff R, D’Orlando O, Giannopoulou E, Lachmann L, Beyerlein A, et al. Reduced Blood Leukocyte and Neutrophil Numbers in the Pathogenesis of Type 1 Diabetes. Horm Metab Res (2013) 45(6):467–70. doi: 10.1055/s-0032-1331226
22. Klocperk A, Petruzelkova L, Pavlikova M, Rataj M, Kayserova J, Pruhova S, et al. Changes in Innate and Adaptive Immunity Over the First Year After the Onset of Type 1 Diabetes. Acta Diabetol (2020) 57(3):297–307. doi: 10.1007/s00592-019-01427-1
23. Lubbers J, Brink M, van de Stadt LA, Vosslamber S, Wesseling JG, van Schaardenburg D, et al. The Type I IFN Signature as a Biomarker of Preclinical Rheumatoid Arthritis. Ann Rheum Dis (2013) 72(5):776–80. doi: 10.1136/annrheumdis-2012-202753
24. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting Neutrophils Are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci Transl Med (2011) 3(73):73ra20. doi: 10.1126/scitranslmed.3001201
25. Huang J, Xiao Y, Zheng P, Zhou W, Wang Y, Huang G, et al. Distinct Neutrophil Counts and Functions in Newly Diagnosed Type 1 Diabetes, Latent Autoimmune Diabetes in Adults, and Type 2 Diabetes. Diabetes Metab Res Rev (2019) 35(1):e3064. doi: 10.1002/dmrr.3064
26. Marhoffer W, Stein M, Schleinkofer L, Federlin K. Evidence of Ex Vivo and in Vitro Impaired Neutrophil Oxidative Burst and Phagocytic Capacity in Type 1 Diabetes Mellitus. Diabetes Res Clin Pract (1993) 19(3):183–8. doi: 10.1016/0168-8227(93)90112-i
27. Chavez-Reyes J, Escarcega-Gonzalez CE, Chavira-Suarez E, Leon-Buitimea A, Vazquez-Leon P, Morones-Ramirez JR, et al. Susceptibility for Some Infectious Diseases in Patients With Diabetes: The Key Role of Glycemia. Front Public Health (2021) 9. doi: 10.3389/fpubh.2021.559595
28. Meyer-Hoffert U, Wiedow O. Neutrophil Serine Proteases: Mediators of Innate Immune Responses. Curr Opin Hematol (2011) 18(1):19–24. doi: 10.1097/MOH.0b013e32834115d1
29. Hirche TO, Gaut JP, Heinecke JW, Belaaouaj A. Myeloperoxidase Plays Critical Roles in Killing Klebsiella Pneumoniae and Inactivating Neutrophil Elastase: Effects on Host Defense. J Immunol (2005) 174(3):1557–65. doi: 10.4049/jimmunol.174.3.1557
30. Rosa JS, Oliver SR, Flores RL, Ngo J, Milne GL, Zaldivar FP, et al. Altered Inflammatory, Oxidative, and Metabolic Responses to Exercise in Pediatric Obesity and Type 1 Diabetes. Pediatr Diabetes (2011) 12(5):464–72. doi: 10.1111/j.1399-5448.2010.00724.x
31. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
32. Papayannopoulos V. Neutrophil Extracellular Traps in Immunity and Disease. Nat Rev Immunol (2018) 18(2):134–47. doi: 10.1038/nri.2017.105
33. Parackova Z, Zentsova I, Vrabcova P, Klocperk A, Sumnik Z, Pruhova S, et al. Neutrophil Extracellular Trap Induced Dendritic Cell Activation Leads to Th1 Polarization in Type 1 Diabetes. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00661
34. Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET Formation. Front Immunol (2012) 3. doi: 10.3389/fimmu.2012.00360
35. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes Primes Neutrophils to Undergo NETosis, Which Impairs Wound Healing. Nat Med (2015) 21(7):815–9. doi: 10.1038/nm.3887
36. Meara CHO, Coupland LA, Kordbacheh F, Quah BJC, Chang CW, Simon Davis DA, et al. Neutralizing the Pathological Effects of Extracellular Histones With Small Polyanions. Nat Commun (2020) 11(1):6408. doi: 10.1038/s41467-020-20231-y
37. Herster F, Bittner Z, Codrea MC, Archer NK, Heister M, Loffler MW, et al. Platelets Aggregate With Neutrophils and Promote Skin Pathology in Psoriasis. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01867
38. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated Platelets Present High Mobility Group Box 1 to Neutrophils, Inducing Autophagy and Promoting the Extrusion of Neutrophil Extracellular Traps. J Thromb Haemost (2014) 12(12):2074–88. doi: 10.1111/jth.12710
39. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular Histones Promote Thrombin Generation Through Platelet-Dependent Mechanisms: Involvement of Platelet TLR2 and TLR4. Blood (2011) 118(7):1952–61. doi: 10.1182/blood-2011-03-343061
40. Zhi H, Dai J, Liu J, Zhu J, Newman DK, Gao C, et al. Platelet Activation and Thrombus Formation Over IgG Immune Complexes Requires Integrin AlphaIIbbeta3 and Lyn Kinase. PloS One (2015) 10(8):e0135738. doi: 10.1371/journal.pone.0135738
41. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils Scan for Activated Platelets to Initiate Inflammation. Science (2014) 346(6214):1234–8. doi: 10.1126/science.1256478
42. Zuchtriegel G, Uhl B, Puhr-Westerheide D, Pornbacher M, Lauber K, Krombach F, et al. Platelets Guide Leukocytes to Their Sites of Extravasation. PloS Biol (2016) 14(5):e1002459. doi: 10.1371/journal.pbio.1002459
43. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk Between Neutrophils, B-1a Cells and Plasmacytoid Dendritic Cells Initiates Autoimmune Diabetes. Nat Med (2013) 19(1):65–73. doi: 10.1038/nm.3042
44. Garciafigueroa Y, Phillips BE, Engman C, Trucco M, Giannoukakis N. Neutrophil-Associated Inflammatory Changes in the Pre-Diabetic Pancreas of Early-Age NOD Mice. Front Endocrinol (Lausanne) (2021) 12. doi: 10.3389/fendo.2021.565981
45. Shu L, Zhong L, Xiao Y, Wu X, Liu Y, Jiang X, et al. Neutrophil Elastase Triggers the Development of Autoimmune Diabetes by Exacerbating Innate Immune Responses in Pancreatic Islets of Non-Obese Diabetic Mice. Clin Sci (Lond) (2020) 134(13):1679–96. doi: 10.1042/CS20200021
46. Huang J, Tan Q, Tai N, Pearson JA, Li Y, Chao C, et al. IL-10 Deficiency Accelerates Type 1 Diabetes Development Via Modulation of Innate and Adaptive Immune Cells and Gut Microbiota in BDC2.5 NOD Mice. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.702955
47. You Q, Shen Y, Wu Y, Li Y, Liu C, Huang F, et al. Neutrophil Extracellular Traps Caused by Gut Leakage Trigger the Autoimmune Response in Nonobese Diabetic Mice. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.711423
48. Simeonovic CJ, Popp SK, Starrs LM, Brown DJ, Ziolkowski AF, Ludwig B, et al. Loss of Intra-Islet Heparan Sulfate Is a Highly Sensitive Marker of Type 1 Diabetes Progression in Humans. PloS One (2018) 13(2):e0191360. doi: 10.1371/journal.pone.0191360
49. Kleine TJ, Lewis PN, Lewis SA. Histone-Induced Damage of a Mammalian Epithelium: The Role of Protein and Membrane Structure. Am J Physiol (1997) 273(6):C1925–36. doi: 10.1152/ajpcell.1997.273.6.C1925
50. Mena HA, Carestia A, Scotti L, Parborell F, Schattner M, Negrotto S. Extracellular Histones Reduce Survival and Angiogenic Responses of Late Outgrowth Progenitor and Mature Endothelial Cells. J Thromb Haemost (2016) 14(2):397–410. doi: 10.1111/jth.13223
51. Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Neonatal Beta-Cell Apoptosis: A Trigger for Autoimmune Diabetes? Diabetes (2000) 49(1):1–7. doi: 10.2337/diabetes.49.1.1
52. Diana J, Lehuen A. Macrophages and Beta-Cells Are Responsible for CXCR2-Mediated Neutrophil Infiltration of the Pancreas During Autoimmune Diabetes. EMBO Mol Med (2014) 6(8):1090–104. doi: 10.15252/emmm.201404144
53. Bdeir K, Gollomp K, Stasiak M, Mei J, Papiewska-Pajak I, Zhao G, et al. Platelet-Specific Chemokines Contribute to the Pathogenesis of Acute Lung Injury. Am J Respir Cell Mol Biol (2017) 56(2):261–70. doi: 10.1165/rcmb.2015-0245OC
54. Gleissner CA, von Hundelshausen P, Ley K. Platelet Chemokines in Vascular Disease. Arterioscler Thromb Vasc Biol (2008) 28(11):1920–7. doi: 10.1161/ATVBAHA.108.169417
55. Ludwig A, Petersen F, Zahn S, Gotze O, Schroder JM, Flad HD, et al. The CXC-Chemokine Neutrophil-Activating Peptide-2 Induces Two Distinct Optima of Neutrophil Chemotaxis by Differential Interaction With Interleukin-8 Receptors CXCR-1 and CXCR-2. Blood (1997) 90(11):4588–97. doi: 10.1182/blood.V90.11.4588
56. Zhang Q, Fillmore TL, Schepmoes AA, Clauss TR, Gritsenko MA, Mueller PW, et al. Serum Proteomics Reveals Systemic Dysregulation of Innate Immunity in Type 1 Diabetes. J Exp Med (2013) 210(1):191–203. doi: 10.1084/jem.20111843
57. Sodre FMC, Bissenova S, Bruggeman Y, Tilvawala R, Cook DP, Berthault C, et al. Peptidylarginine Deiminase Inhibition Prevents Diabetes Development in NOD Mice. Diabetes (2021) 70(2):516–28. doi: 10.2337/db20-0421
58. Hutchings P, O’Reilly L, Parish NM, Waldmann H, Cooke A. The Use of a Non-Depleting Anti-CD4 Monoclonal Antibody to Re-Establish Tolerance to Beta Cells in NOD Mice. Eur J Immunol (1992) 22(7):1913–8. doi: 10.1002/eji.1830220735
59. Yi Z, Diz R, Martin AJ, Morillon YM, Kline DE, Li L, et al. Long-Term Remission of Diabetes in NOD Mice Is Induced by Nondepleting Anti-CD4 and Anti-CD8 Antibodies. Diabetes (2012) 61(11):2871–80. doi: 10.2337/db12-0098
60. Pollard HM, Miller L, Brewer WA. The External Secretion of the Pancreas and Diabetes Mellitus. Am J Dig Dis (1943) 10(1):20–3. doi: 10.1007/BF02997405
61. Campbell-Thompson ML, Filipp SL, Grajo JR, Nambam B, Beegle R, Middlebrooks EH, et al. Relative Pancreas Volume Is Reduced in First-Degree Relatives of Patients With Type 1 Diabetes. Diabetes Care (2019) 42(2):281–7. doi: 10.2337/dc18-1512
62. Gaglia JL, Guimaraes AR, Harisinghani M, Turvey SE, Jackson R, Benoist C, et al. Noninvasive Imaging of Pancreatic Islet Inflammation in Type 1a Diabetes Patients. J Clin Invest (2011) 121(1):442–5. doi: 10.1172/JCI44339
63. Williams AJ, Thrower SL, Sequeiros IM, Ward A, Bickerton AS, Triay JM, et al. Pancreatic Volume Is Reduced in Adult Patients With Recently Diagnosed Type 1 Diabetes. J Clin Endocrinol Metab (2012) 97(11):E2109–13. doi: 10.1210/jc.2012-1815
64. Campbell-Thompson M, Wasserfall C, Montgomery EL, Atkinson MA, Kaddis JS. Pancreas Organ Weight in Individuals With Disease-Associated Autoantibodies at Risk for Type 1 Diabetes. JAMA (2012) 308(22):2337–9. doi: 10.1001/jama.2012.15008
65. Campbell-Thompson ML, Kaddis JS, Wasserfall C, Haller MJ, Pugliese A, Schatz DA, et al. The Influence of Type 1 Diabetes on Pancreatic Weight. Diabetologia (2016) 59(1):217–21. doi: 10.1007/s00125-015-3752-z
66. Lohr M, Kloppel G. Residual Insulin Positivity and Pancreatic Atrophy in Relation to Duration of Chronic Type 1 (Insulin-Dependent) Diabetes Mellitus and Microangiopathy. Diabetologia (1987) 30(10):757–62. doi: 10.1007/BF00275740
67. Rodriguez-Calvo T, Ekwall O, Amirian N, Zapardiel-Gonzalo J, von Herrath MG. Increased Immune Cell Infiltration of the Exocrine Pancreas: A Possible Contribution to the Pathogenesis of Type 1 Diabetes. Diabetes (2014) 63(11):3880–90. doi: 10.2337/db14-0549
68. Dozio N, Indirli R, Giamporcaro GM, Frosio L, Mandelli A, Laurenzi A, et al. Impaired Exocrine Pancreatic Function in Different Stages of Type 1 Diabetes. BMJ Open Diabetes Res Care (2021) 9(1). doi: 10.1136/bmjdrc-2019-001158
69. Giovenzana A, Vecchio F, Cugnata F, Nonis A, Mandelli A, Stabilini A, et al. Exocrine Pancreas Function Is Impaired in Adult Relatives of Patients With Type 1 Diabetes. Acta Diabetol (2022) 59(4):473–9. doi: 10.1007/s00592-021-01819-2
70. Kusmartseva I, Beery M, Hiller H, Padilla M, Selman S, Posgai A, et al. Temporal Analysis of Amylase Expression in Control, Autoantibody-Positive, and Type 1 Diabetes Pancreatic Tissues. Diabetes (2020) 69(1):60–6. doi: 10.2337/db19-0554
71. Li X, Campbell-Thompson M, Wasserfall CH, McGrail K, Posgai A, Schultz AR, et al. Serum Trypsinogen Levels in Type 1 Diabetes. Diabetes Care (2017) 40(4):577–82. doi: 10.2337/dc16-1774
72. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of Neutrophil Extracellular Trap Degradation Is Associated With Lupus Nephritis. Proc Natl Acad Sci U.S.A. (2010) 107(21):9813–8. doi: 10.1073/pnas.0909927107
73. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of Self-Antigen Specificity of Islet-Infiltrating T Cells From Human Donors With Type 1 Diabetes. Nat Med (2016) 22(12):1482–7. doi: 10.1038/nm.4203
74. Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, et al. Staging Presymptomatic Type 1 Diabetes: A Scientific Statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care (2015) 38(10):1964–74. doi: 10.2337/dc15-1419
Keywords: neutrophils, NETs, platelet-neutrophil aggregates, Type 1 diabetes, islets, exocrine dysfunction
Citation: Petrelli A, Popp SK, Fukuda R, Parish CR, Bosi E and Simeonovic CJ (2022) The Contribution of Neutrophils and NETs to the Development of Type 1 Diabetes. Front. Immunol. 13:930553. doi: 10.3389/fimmu.2022.930553
Received: 28 April 2022; Accepted: 13 June 2022;
Published: 06 July 2022.
Edited by:
Daniel (Söderberg) Appelgren, Linköping University, SwedenReviewed by:
Oscar J. Cordero, University of Santiago de Compostela, SpainCopyright © 2022 Petrelli, Popp, Fukuda, Parish, Bosi and Simeonovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charmaine J. Simeonovic, Q2hhcm1haW5lLlNpbWVvbm92aWNAYW51LmVkdS5hdQ==