 Like Zhao
Like Zhao Xianda Hu
Xianda Hu Fei Xiao
Fei Xiao Xuan Zhang1
Xuan Zhang1 Min Wang
Min Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 July 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.929520
This article is part of the Research Topic Gut Microbiome, Metabolites, and Immune Response in Lupus, Rheumatoid Arthritis, and Sjogren Syndrome View all 10 articles
Nucleic acid autoantibodies, increase type I interferon (IFN-α) levels, and immune cell hyperactivation are hallmarks of systemic lupus erythematosus (SLE). Notably, immune cell activation requires high level of cellular energy that is predominately generated by the mitochondria. Mitochondrial reactive oxygen species (mROS), the byproduct of mitochondrial energy generation, serves as an essential mediator to control the activation and differentiation of cells and regulate the antigenicity of oxidized nucleoids within the mitochondria. Recently, clinical trials on normalization of mitochondrial redox imbalance by mROS scavengers and those investigating the recovery of defective mitophagy have provided novel insights into SLE prophylaxis and therapy. However, the precise mechanism underlying the role of oxidative stress-related mitochondrial molecules in skewing the cell fate at the molecular level remains unclear. This review outlines distinctive mitochondrial functions and pathways that are involved in immune responses and systematically delineates how mitochondrial dysfunction contributes to SLE pathogenesis. In addition, we provide a comprehensive overview of damaged mitochondrial function and impaired metabolic pathways in adaptive and innate immune cells and lupus-induced organ tissues. Furthermore, we summarize the potential of current mitochondria-targeting drugs for SLE treatment. Developing novel therapeutic approaches to regulate mitochondrial oxidative stress is a promising endeavor in the search for effective treatments for systemic autoimmune diseases, particularly SLE.
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by self-tolerance breakdown, excessive production of autoantibodies and impaired clearance of immune complexes, which incites sustained inflammatory response and multi-organ damage (1). As the signature of SLE, excessive production of type I IFN (especially IFN-α) substantially contributes to SLE pathogenesis by triggering the activation and expansion of autoreactive immune cells, promoting autoantibodies production and inflammatory cytokines release, hence leading to the formation of nucleic acid-protein immune complex and perpetuating the autoreactive immune response. Despite great improvement has been achieved in SLE survival, the morbidity from both the disease and the medications makes the prognosis still far from satisfactory (2). Therefore, deeper understanding and further elucidation of the cellular and molecular immune aberrations in SLE pathogenesis are still in desire.
Cellular bioactivity cannot be accomplished without energy and mitochondria is the critical organelle in initiating and maintaining cellular energy metabolism. By generating adenosine triphosphate (ATP) through oxidative phosphorylation, mitochondria provide energy for multiple cell activities and determine the cell fate like activation and differentiation (3). In addition, mitochondria are also the major producer of reactive oxygen species (ROS), which are involved in redox balance and oxidative damage, and regulate iron homeostasis, calcium efflux, and metabolic pathways (4). Abnormal mitochondrial function participate in numerous diseases, such as neurological disorders, endocrinopathy, inflammatory bowel disease, and autoimmune diseases (5). Moreover, under oxidative stress conditions, immune complexes and IFN-α stimulate excessive mitochondrial ROS (mROS) production that consequently induces highly immunogenic oxidative mitochondrial DNA (Ox-mtDNA). This Ox-mtDNA activates inflammatory signaling pathways and promotes cell death progression (6, 7). Notably, several studies have reported that mitochondrial dysfunction is closely associated with SLE (8, 9). Furthermore, regulation of excessive levels of mROS and alleviation of deleterious consequences of mitochondrial dysfunction have emerged as promising therapeutic approaches for SLE treatment (9, 10).
This review aimed to summarize the current research advances on physiological and pathological roles of mitochondria., Mitophagy defects of antioxidant defense and mitochondrial dysfunction contributing to SLE immunopathogenesis were systematically reviewed. The drugs based on the normalization of mitochondria function and mitophagy metabolic pathways were briefly introduced and their potential therapeutic values in SLE were discussed.
Mitochondria are energy-producing cellular organelles that generate energy in the form of ATP via oxidative phosphorylation (OXPHOS) performed by the electron transport chain (ETC) complexes. These complexes consist of five enzymes, wherein complexes I and III are the primary enzymes that catalyze the generation of low concentrations of superoxide anions. These enzymes maintain the mitochondrial antioxidant defenses (11). However, increased mROS levels during oxidative stress cause mitochondrial DNA (mtDNA) impairment and ETC defects, resulting in mitochondrial dysfunction (12).
The tricarboxylic acid (TCA) cycle occurs in the mitochondria is essential for ATP energy supplement and participates in multiple mitochondria metabolic pathways (13, 14). This review mainly discusses defective mROS clearance for nutrient energy signaling activation (e.g., activation of AMP-activated protein kinase (AMPK) and mammalian target of rapamycin complex 1 (mTORC1)) by regulating the mitochondrial dynamic loop and releasing the antigenicity of the mtDNA to induce the immune responses. Furthermore, preclinical safety therapeutics for the clearance of excessive mROS and mtDNA production were collectively recapitulated in autoimmune diseases, particularly SLE (Figure 1).
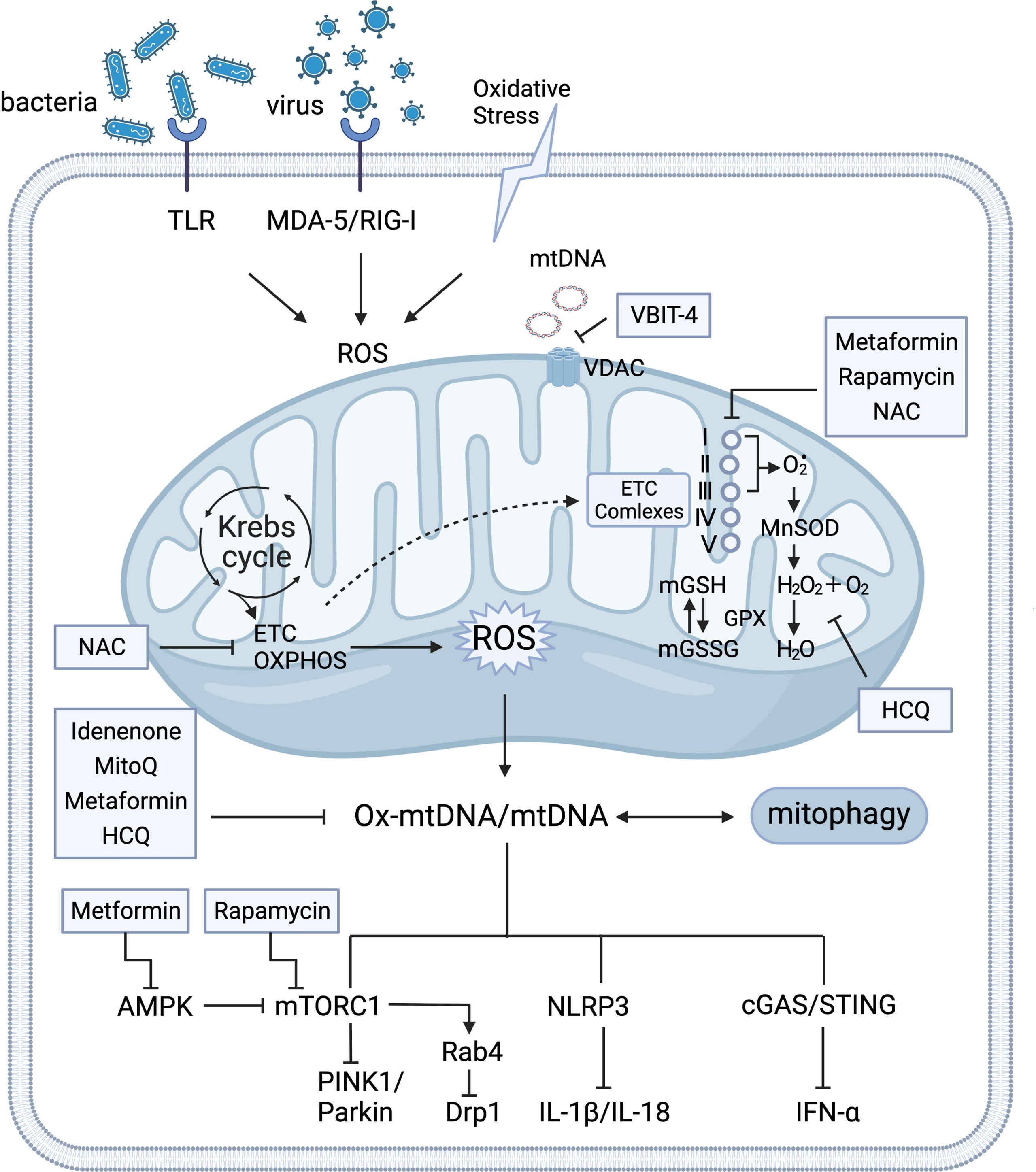
Figure 1 Crosstalk of mitochondria homeostasis and immune signaling. Mitochondria support the major generation of reactive oxygen species(mROS) for modulating various signaling and cellular metabolism. Mitochondria produce ATP through the tricarboxylic acid cycle (TCA) oxidative phosphorylation (OXPHOS) at electron transport chain (ETC) complexes. Detoxify free radicals from ETC complexes reduced by manganese superoxide dismutase (MnSOD) and glutathione peroxidase (GPX) into water (H2O). GPX, as a catalase, also regulates the conversion balance of oxidized glutathione (GSH) and glutathione disulfide (GSSG) in mitochondria. Under the circumstance of oxidative stress that is stimulated by insults and exogenous pathogens, various DNA sensors trigger aberrant mitochondrial function by activation of pattern recognition receptors in the cytosol and mitochondria (e.g. retinoic-acid-inducible protein I (RIG-I), melanoma differentiation-associated protein 5 (MDA-5), and Toll-like receptor(TLR)). Expulsion of highly immunogenic Ox-mtDNA out of mitochondria stimulates AMP-activated protein kinase (AMPK), mammalian target of rapamycin complex 1 (mTORC1), Nucleotide-binding oligomerization domain (NOD)-like receptor pyrin domain-containing protein 3 (NLRP3)-mediated IL-1β and IL-18 production, and cyclic GMP-AMP (cGAS) and the cyclic GMP-AMP receptor stimulator of interferon genes (STING)-dependent IFN-α. Under conditions of nutrient deprivation, AMPK inhibits energy-consuming pathways by mTORC1 for mitophagy activation. The HRES-1/Rab4-mediated dynamin-related protein 1 (Drp1) depletion reverses mitochondrial accumulation via mTOR-independent pathway. Mitochondrial respiration (ETC), mTORC1, and cGAS-STING signaling pathway serve as potential therapy targets. Several medications that targeting mitochondria metabolism process has been observed the therapeutic clinical effectiveness in SLE, including N-acetylcysteine (NAC), Rapamycin, Metformin, Mito Q, Hydroxychloroquine (HCQ), and Idenenone. The voltage-dependent anion selective channel (VDAC) oligomer inhibitor VBIT 4 can inhibit the release of mtDNA. I, II, III, IV, and V represent the electron transport chain complexes I–V. Glutathione reductase (GR).
Mitochondria continuously undergo a cycle of production (biogenesis), morphology (fusion/fission), and degradation (mitophagy); hence, they are crucial for maintaining cellular homeostasis (15) (Figure 2).
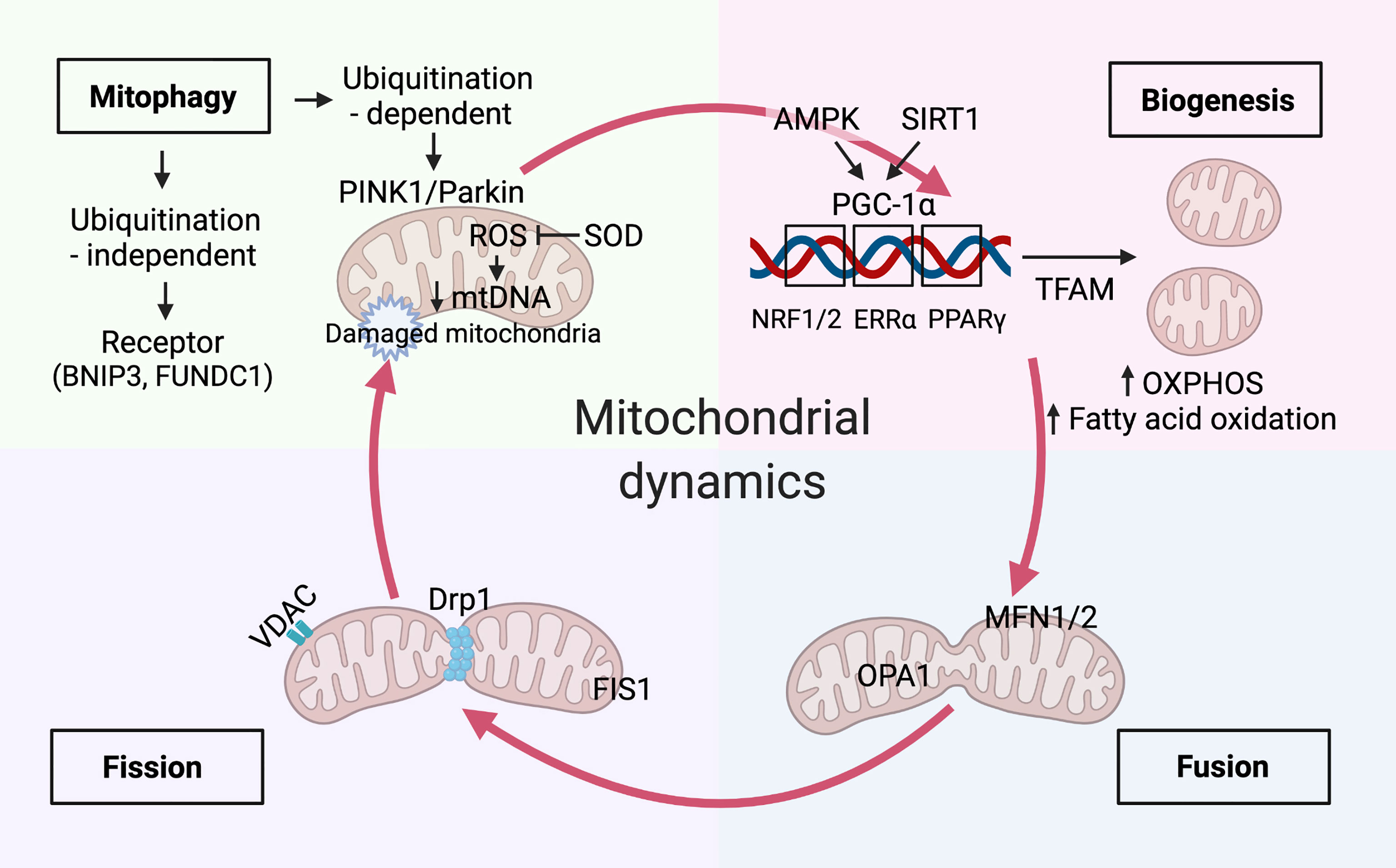
Figure 2 Mitochondria dynamics balanced with biogenesis and mitophagy. Mitochondrial biogenesis. Immune signals of AMP-activated protein kinase (AMPK) and sirtuin 1 (SIRT1) have been reported to increase mitochondrial biogenesis by peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α). The master regulator, PGC-1α, drives mitochondrial biogenesis, oxidative phosphorylation(OXPHOS), and fatty acid oxidation by co-activating transcription factors, including Estrogen Related Receptor Alpha (ERRα), peroxisome proliferator-activated receptor (PPAR)-γ, nuclear respiratory factor (NRF)-1 and-2. Mitochondrial transcription factor A(TFAM) aids in the transcription of genes that is essential for balancing mtDNA transcription and clearance. Mitochondrial dynamics. Mitochondrial dynamics of fission and fusion are regulated by dynamin-related protein-1 (Drp1), fission 1 (Fis1) and mitofusin-1 (Mfn1), mitofusin-2 (Mfn2), and optic atrophy 1 (Opa1) respectively. mtDNA extrudes from the mitochondria through pores formed by voltage-dependent anion-selective channel (VDAC) oligomers. Mitophagy activation. The augment of fission promotes mitophagy activation by the PTEN-induced kinase 1 (PINK1)/Parkin-mediated ubiquitination pathway and receptor-mediated pathways, such as BCL2 interacting protein 3 (BNIP3) and FUN14 domain-containing protein 1(FUNDC1). Timely elimination of damaged and aged mitochondria reduces the excessive accumulation of mitochondria reactive oxygen species (mROS), which can be inhibited by mitochondrial enzymes such as superoxide dismutase 2 (SOD2). Damaged mitochondria with decreased mtDNA undergo mitophagy.
The mitochondrial biogenesis and function require peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1 (PGC) family of transcriptional coactivators, including PGC-1α, PGC-1β, and PGC-1–related coactivator(PRC) (16). PGC-1α is a primary mitochondrial biogenesis core regulator that recruits several transcription factors, such as nuclear respiratory factor (NRF)-1, NRF-2, and estrogen-related receptors. It also promotes the assembly fatty acid β-oxidation, OXPHOS and co-activate the mitochondrial biogenesis. Remarkably, NRF-1 and NRF-2 significantly induce the transcription of mitochondrial proteins in the nucleus; among these proteins, the expression of the mitochondrial transcription factor A (TFAM) is essential for balancing mtDNA transcription and clearance (17). PGC-1α plays a complex role in regulating the expression of ROS-scavenging proteins, such as superoxide dismutase (SOD2) (18). Furthermore, in response to the cellular energy demands, it activates the key energy sensors AMPK and sirtuin 1 under differential nutritional conditions. Owing to its critical role in cellular energy homeostasis, AMPK participates in various catabolic processes. Indeed, its phosphorylation stimulates mitochondrial biogenesis, autophagy, mitophagy, fatty acid oxidation (FAO), and glycolysis, and induces ATP generation during cellular metabolism (19). We discussed AMPK-mediated regulation of PGC-1α expression in kidney podocytes in the SLE organ damage section (4.1 Mitochondria and Lupus Nephritis). However, AMPK inhibits energy-consuming pathways via mTORC1 and ULK1 phosphorylation under nutrient deprivation conditions, consequently activating mitophagy (20). The mammalian target of rapamycin (mTOR) pathway is another nutrient-sensing pathway that regulates mitochondrial biogenesis and activates PGC-1α in response to external stimuli (21). Furthermore, PGC-1α interacts with various transcription factors, such as hypoxia-inducible factor 1 subunit alpha (HIF-1α) that mediates mitochondrial biogenesis and mitophagy (22).
Distinct mitochondrial molecular mechanisms and signaling pathways have been proven to regulate the pathways mentioned above (23). For instance, mitochondrial fusion is primarily mediated by mitofusin 1/2 (Mfn1/2), optic atrophy protein 1 (Opa1), and three GTPases. Mfn1/2 localizes to the outer mitochondrial membrane and facilitates the fusion of adjacent mitochondria by forming homo- or heterodimers. In contrast, Opa1 predominately induces the fusion of the inner mitochondrial membrane in a synchronized manner. Additionally, dynamin-related protein (Drp1) and other receptors play essential roles in regulating mitochondrial fission through phosphorylation and ubiquitination (24). Notably, Drp1 mutation affects mitochondrial fission, ROS generation, and kidney-specific cell damage and effacement, eventually initiating cell apoptosis (25). Mitochondrial fission-mediated fragmentation, concomitant with other mitochondrial damage, initiates mitophagy and mediates mROS production. On the contrary, mitochondrial fusion promotes the exchange and restoration of mitochondrial content by tethering adjacent mitochondria (26). The mitochondrial fission and fusion dynamics modulate a metabolic shift towards cell differentiation. For example, memory T cells have fused mitochondria that favor OXPHOS and FAO, whereas effector T cells enforce fission of punctate mitochondria, promoting the catabolic aerobic glycolysis pathway (27). This suggests that manipulating the mitochondrial structure can determine cell differentiation and, eventually, cell fate.
Mitophagy is closely associated with mitochondrial fission and selectively removes damaged or depolarized mitochondria during lysosomal degradation (28). Similar to autophagy, mitophagy is a catabolic process that is initiated by the recruitment of autophagy machinery proteins to activate the mitochondrial surface. Thereafter, autophagosomes enclose mitochondria and transport them to lysosomes for degradation (29). Two major degradation processes enable the removal of defective mitochondria to maintain the homeostasis of cell function and metabolism. For instance, ATPases specifically degrade senescent and damaged mitochondria into small peptides; however, these mitochondria can also be eliminated in lysosomes through mitophagic degradation (30). Under cellular stress stimuli, mitophagy selectivity initiates multiple cell death signals through the PTEN-induced kinase 1 (PINK1)/Parkin-mediated ubiquitination pathway and receptor-mediated pathways, such as BCL2 interacting protein 3 and FUN14 domain-containing protein 1 (31, 32). PINK1 and Parkin-mediate ubiquitination signaling play crucial roles in the interplay between mitophagy and mitochondrial dynamics. Hence, timely elimination of damaged and aged mitochondria reduces the excessive accumulation of mROS, avoiding triggering the cytoplasmic DNA sensor by releasing the mtDNA (33). Mitochondria are endosymbionts evolved from bacteria. The mtDNA and N-formyl peptides are two mitochondrial components that represent mitochondrial damage-associated molecular patterns (DAMPs), serve as the innate immune response to produce inflammatory cytokines and chemokines via Toll-like receptors (TLR), absent in melanoma 2 (AIM2), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and retinoic acid-inducible protein I (RIG-I)-like receptors (RLRs) (33). Beyond the clearance capability of mitophagy, damaged mitochondria trigger the innate immune response by releasing mitochondrial DAMPs to recognize pattern recognition receptors and G-protein-coupled formyl peptide receptors, which explains the stimulation of a pro-inflammatory response in the innate immune system under microbiology-free condition (34).
Under physiological conditions, mitochondria generate low levels of mROS to modulate various signaling pathways and cellular metabolisms. However, mROS levels beyond a threshold can be pathogenic, causing mitochondrial dysfunction and further cellular damage (35). Glutathione peroxidase (GPX), as one of the antioxidant enzymes to balance between glutathione and glutathione disulfide, detoxifies lipid peroxides and scavenges ROS from the mitochondria (36). Under the oxidative stress condition, mtDNA can be extruded from the mitochondria into the cytoplasm (4). A recent study has discovered that the formation of 8-hydroxy-2-deoxyguanosine (8-OHdG), which is recognized as an indicator of oxidant-induced DNA damage, is greatly eliminated upon the decline of the mtDNA copy number in SLE patients (37). On the other side, highly immunogenic Ox-mtDNA induces mROS overproduction and stimulates an inflammatory response (38). In addition, defective mitochondrial antioxidant enzymes in the immune cells of SLE patients can further aggravate oxidative stress in a vicious cycle (38). Therefore, oxidative stress-mediated mitophagy has been recognized to play a critical role in genetic predisposition and immune cell activation in SLE.
Genetic predisposition along with many other etiological factors contributes to the pathogenesis of SLE. Multiple genome-wide association studies have identified several mitochondrial single nucleotide polymorphisms as responsible for conferring susceptibility towards SLE (39). For example, ATP synthase 5/6, displacement-loop, D310, NADH dehydrogenase subunit (ND) 1, and ND2 have been demonstrated to be involved in mitochondrial biogenesis and mitophagy, proving that mtDNA polymorphisms confer susceptibility to SLE (39–42). As known, pathogenic autoantibodies and the deposition of immune complexes in tissues cause multiple organs damage in SLE. Autoantibodies against various mitochondrial components indicate that mitochondria are key antigenic stimulants that incite an immune response in SLE. Various autoantibodies, such as anti-mitochondrial, anti-whole mitochondrial (43), anti-mtDNA (44), mitochondrial-RNA (45), and anticardiolipin, have been generated by targeting distinct mitochondrial components, including the mitochondrial surface, mtDNA, mitochondrial RNA, and mitochondrial inner membrane, respectively. These findings suggest that mitochondria are an important source of autoantigens that contribute to SLE pathogenesis. Furthermore, accumulating evidence suggests that mitochondrial dysfunction and abnormal mitophagy result in redox disturbance of mtDNA, overproduction of mROS, an imbalance of oxidative stress, and activation of inflammatory pathways. All these factors are involved in the initiation and development of SLE (46).
Mitochondria are hypothesized to originate from endosymbiotic bacteria. Additionally, self-energy modulation in the mitochondria is essential for mitochondrial metabolism and host immune response signaling cascade (47). Mitochondrial dysfunction-induced redox disturbance differentially activates various inflammatory pathways in adaptive and innate immune cells. Studies have investigated multiple cellular signaling pathways that regulate mitophagy in SLE, including the mTORC1, AMPK, NLR family pyrin domain-containing 3 (NLRP3) inflammasome, and cGAS–STING pathways (48–51) (Figure 1).
Mitochondrial dysfunction has been detected in lupus T cells and is characterized by excessive mROS production, elevated transmembrane potential, downregulated mitophagy, and reduced glutathione levels (52–54). Moreover, oxidative stress impairs proximal T cell receptor (TCR) signaling, pathway activation, and exhaustion-associated gene expression program activation (55). Furthermore, chronic antigen stimulation induces irreversible T cell exhaustion and impairs mitophagy, causing mROS overproduction and autoantigen release (56).
Multiple signaling pathways enable the sequestration and successful clearance of damaged mitochondria by mitophagy, suppressing mtROS accumulation (57). Redox-dependent activation of the mTOR pathway plays an essential role in altering TCR signal transduction and T cell differentiation in SLE patients (48, 58). Additionally, it is believed that mitochondrial localization to an immune synapse is required for T cell activation. However, mTOR activation decreases T cell surface receptor/CD3ζ chain levels and in compensation augments tyrosine-protein kinase and Fcgamma chain receptor (FcγR) levels. Consequently, this increases the calcium flux in lupus T cells through HRES-1/Rab4-dependent lysosomal degradation (48). The aberrant Ca2+ alteration may account for the inappropriate activation in lupus T cells. Notably, upregulated expression of the small GTPase HRES-1/Rab4 depletes the Drp1 levels, leading to decreased mitophagy in CD4+ T cells in lupus. In contrast, Rab4 inhibited with 3-pyridinyl ethylidene hydroxyl phosphonocarboxylate (3-PEHPC) restores Drp1 function via the mTOR-independent pathway, reversing mitochondrial accumulation in lupus T cells (59).
Increased calcium flux also promotes oxidative stress-dependent T cell activation. For instance, the calcium-mediated phosphatase calcineurin dephosphorylates nuclear factor of activated T cell (NFAT). Intranuclear NFAT, accompanied by other transcription factors such as AP-1, NF-κB, and Oct-1, promotes the expression of the inflammatory cytokine interleukin (IL)-2 (54). Of note, Rab4A inhibition improves impaired mitophagy of lupus T cells, activation of T cells, and production of autoantibodies in SLE. This demonstrates that recovery of mitochondrial dysfunction caused by insufficient mitophagy and mROS overproduction normalizes the phenotypes of T cells in SLE. Furthermore, few clinical trials have demonstrated that either inhibiting mTORC1 by rapamycin (sirolimus) (60) or supplementing N-acetylcysteine (NAC), a glutathione precursor and an antioxidant (58), is therapeutically effective and improved systemic inflammation in patients with SLE. Interestingly, mitochondrial accumulation depletes memory T cells and regulatory T (Tregs) cells in SLE (60). Indeed, decreased number of Treg cells fail to maintain self-tolerance and immunosuppressive function in SLE, whereas supplementation of Treg cells normalizes the inflammatory response and SLE pathology in lupus mice (61). With regard to mitochondrial energy metabolism, these cells have a higher requirement for OXPHOS via AMPK activation than the T helper 17 (Th17) cell subset (62). Metformin, by targeting AMPK activation, can optimize the immunoregulatory effect of Treg cells by enhancing STAT1 expression in an AMPK-dependent manner in SLE (63).
Little is known about the mitochondrial immune metabolism of B cells in SLE. Nonetheless, induced differentiation of CD27+IgD+ unswitched memory B cells into D27hiCD38hi plasmablasts is accompanied by activation of mTORC1 (64). In fact, increased activation of mTORC1 has a positive correlation with the overproduction of autoantibodies and cytokines in the B cells in SLE (64, 65). Remarkably, suppression of hyper-responsiveness in B cells was observed upon rapamycin treatment in SLE (66). with consistency, suppressing mTOR activity by deletion of RAPTOR, an essential signal adaptor for mTORC1, blocks plasma cell differentiation in a mouse model of lupus (67). Previous evidence has also revealed that long-lived plasma cells require quiescent and slow ATP generation through AMPK signaling and mitochondrial OXPHOS (68). Metformin has been demonstrated to ameliorate SLE manifestations by preventing B cell differentiation into plasma cells and germinal center expansion by altering AMPK–mTOR–STAT3 signaling (69).
Neutrophils and plasmacytoid dendritic cells (pDCs) produce IFN-α under the stimulation of nucleic acid–protein immune complexes (70). Of note, excessive IFN-α production occurring in more than half of all SLE patients stimulates the release of interferongenic DNA (including mtDNA) from neutrophils, forming an autoantigen/antibody immune complex (71). However, exposure to the IFNα/RNP immune complex disrupts mitochondrial degradation, leading to the accumulation of abundant Ox-mtDNA within mitochondria and eventually out of mitochondria, which would be a potent pDC stimulator (49). Owing to the presence of the Ox-mtDNA autoantibody, mtDNA is potent antigen that stimulates autoantibody production in SLE (45). Furthermore, the common oxidative DNA damage marker 8-OHdG is elevated in the blood cells of SLE patients (37). mtDNA served as a DAMP induces the production of IFN-α, which activates the cytosolic DNA-sensing cGAS-STING (72), and NLRP3 signaling pathway (51). Defective mitophagy and consequent mtDNA-dependent DNA sensor activation might conceivably be a fundamental event in SLE pathogenesis (3).
The formation of neutrophil extracellular traps (NETs), which are extrusions of genomic DNA and chromatin to kill invading pathogens (73). The ribonucleoprotein immune complex and IFN-α induce spontaneous NETosis through cGAS–STING pathway activation, mitochondrial hyperpolarization, and mROS overproduction in SLE (74). Notably, mROS-mediated Ox-mtDNA enclosed within NETs that are expelled to the neutrophil cell surface triggers IFN production. Thus, mROS inhibition reduces NETosis by inhibiting mitochondrial respiration in lupus mice. This reinforces the notion that mitochondrial functioning and ROS productivity drive SLE pathogenesis (75).
In addition, high mROS levels and increased oxidative stress have been detected in SLE monocytes exhibiting IFN-α signature. Defection of IFN-α-mediated mtDNA degradation leads to a high mitochondrial membrane potential in SLE monocytes, promoting their autoreactivity in a STING-dependent manner (76). Moreover, the differentiation of circulating monocytes into autoinflammatory dendritic cells in the presence of IFN-α promotes the activation and expansion of autoreactive lymphocytes, partially mediating an adaptive immune response (77). Notably, pDCs are the major producers of IFN-α in SLE, and IFN-α production from pDC mediated by TLR9 is reduced by mTOR inhibitor, which regulates pDC differentiation and activation by modulating mitochondrial biogenesis and energy metabolism (78). Therefore, normalization of mtDNA and repairment of mitophagy by an antioxidant treatment are promising therapeutic strategies for SLE treatment (76).
NLRP3 and AIM2 are DNA sensors that along with apoptosis-associated speck-like protein and pro-caspase-1 (component of the NLRP3 inflammasome) trigger the production of several downstream pro-inflammatory cytokines (e.g., IL-1β and IL-18) and promote pyroptosis (79). Although multiple models of NLRP3 inflammasome activation have been identified in autoimmune diseases, the most common model comprises NLRP3 inflammasome activation with ROS production (80, 81). Clearance of damaged mitochondria by decreasing oxidative stress signals over time and increasing mitophagy physiology are necessary for limited NLRP3 activation. In addition, mROS and mtDNA are crucial for optimal NLRP3 inflammasome activation and concomitant calcium influx (82). Mitochondria are essential for intracellular calcium storage and are key modulators of mitochondrial homeostasis. Of note, calcium influx promotes the release of mROS and mtDNA to amplify NLRP3 activation in a feedback loop (83). NOD2/RIPK2-dependent mitophagy and LC3B/Beclin 1-mediated autophagy contribute to the clearance of damaged mitochondria and thus negatively regulate NLRP3 inflammasome activation (82, 84).
Several studies have also indicated that NETs-mediate the activation of the NLRP3 inflammasome in macrophages of lupus patients, subsequently promoting IL-1β production via ROS and K+ efflux (85). Importantly, insufficient clearance of damaged mitochondria causes excessive mtDNA, mROS, and cardiolipin externalization and that of other stimulators (e.g., K+ efflux), thereby activating the NLRP3 inflammasome (86). However, administration of mROS scavengers significantly decreases inflammasome-related gene expression and mature IL-18 expression in lupus mice (75). Conversely, studies reported that loss-of-function mutations of inflammasomes are correlated with disease development in SLE patients (87). The possible explanation is that homeostatic inflammasome function is required for the equilibrium of protective and destructive immunity. In addition, defective lysosomal degradation of autophagosomes and their cargo in macrophages fail to clear mtDNA and lysosomal TLR7 activation. Notably, lack of mitochondria-related GTPases, e.g. the IRGM1, could lead to sustained activation of IFN, consequently induce cGAS–STING axis activation and autophagy inhibition in various autoimmune diseases (88).
Target organs of SLE, especially kidney, nerves, and gut, require high level of energy supply to drive their function and are more susceptible to oxidative stress and subject to chronic inflammatory injuries (1). Moreover, it is being increasingly evident that impaired mitochondria are central mediators of injury in different tissues and organs in SLE (46), which will be discussed in-depth in the subsequent section and summarized in Figure 3.
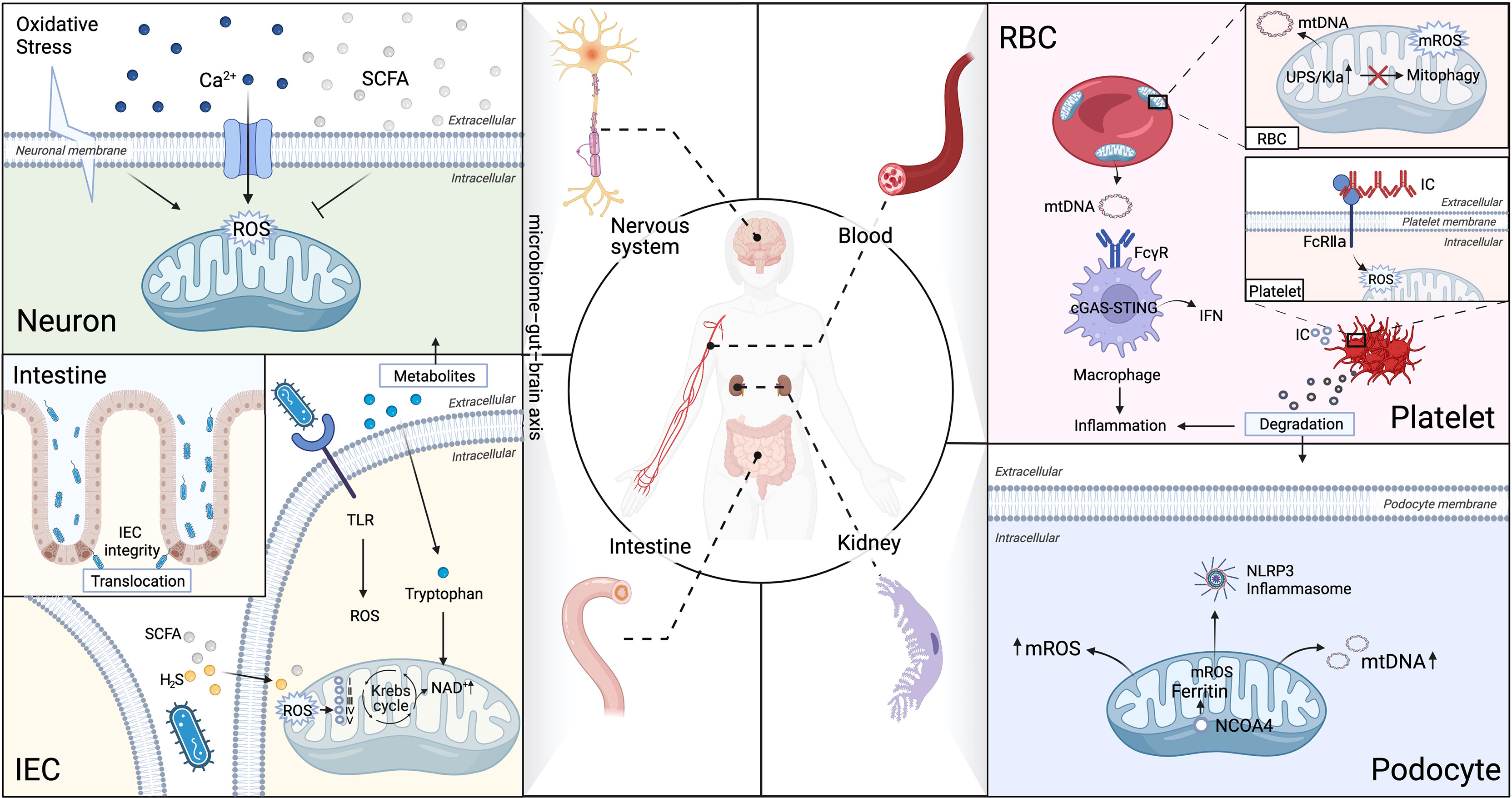
Figure 3 Aberrant mitophagy and redox disturbance cause multiple organs damage in SLE. SLE is a heterogeneous systemic autoimmune disease, affected multiple organs, such as kidney, nerves, gut and hematologic (non-immune cells). (A) Brain-Gut axis is novel therapeutic strategy on modulation of mitochondrial dyshomeostasis by neutralizing excessive ROS in microglial cells in neuropsychiatric SLE (NPSLE). (B) Extracellular mitochondria (specifically platelets mitochondria and erythrocytes) serve as an immunogenic DNA, activated the cGAS–STING pathway, consequently trigger IFN-α production through binding to FcγRIIA in SLE. (C) The microbiota metabolites can directly interfere with the mitochondrial redox status in the host cells. Microbiota metabolites, such as hydrogen sulfide (H2S), short-chain fatty acids (SCFAs), and tryptophan, play an essential role in regulation of intestinal epithelial cells (IEC) integrity in SLE. (D) Impaired mitochondrial degradation in glomerular and tubular cells causes proteinuria and renal failure in LN. Iron loss-induced mitophagy is associated with the mitochondrial ferritin by nuclear receptor coactivator 4(NCOA4). Iron-induced injury of glomerular and tubular cells has been attributed to excessive mROS production, which contributes to drive NLRP3 inflammasome activation in SLE podocytes. Ubiquitin-proteasome system (UPS), Five respiratory complexes (complexes I–V), tricarboxylic acid (TCA) cycle.
The kidney is one of the most energy-consuming organs in human body and the mostly attacked organ in lupus (89). Indeed, glomerular podocytes and tubular epithelial cells require a bundle of mitochondria to provide sufficient energy to remove waste from the blood and maintain the electrolyte balance (90). Current evidence suggests that impaired mitochondrial degradation in glomerular and tubular cells are involved in LN pathogenesis and renal damage (91). Targeted therapies against impaired mitophagy have been designed to prevent LN (91–93).
Dysfunction of podocytes with abnormal permeability and viability and aggravated podocytes apoptosis by pathogenic IgG and IFN-α have been found in LN patients and lupus mice, which could be partially restored by activation of autophagy via mTORC1 inhibition (91). Recent evidence demonstrates that lupus mice with severe proteinuria had low autophagosomes in the podocytes with significant foot process effacement and fusion, while lupus mice with mild proteinuria had a high number of autophagosomes observed with slight podocyte foot fusion. Podocyte function is regulated by an imbalance in mitochondrial division and fusion and activates mitophagy to remove damaged mitochondria via Mfn1 and Drp1 expression. However, once decompensated, mitochondrial dysfunction leads to a vicious cycle of defective mitophagy in LN (94). Therefore, further investigation of systemic and local mitophagy mechanisms is required to elucidate LN.
Since glomerular and tubular cells are metabolically active, they are particularly susceptible to oxidative stress-mediated mitophagy. The uptake of filtered iron and the high mitochondrial content are essential for iron metabolism, which is an under-investigated driver of LN (95). Podocytes serve as the primary components of iron uptake in glomerular epithelial cells. Indeed, they take up hemoglobin and transferrin via megalin–cubilin complex-mediated endocytosis and store the absorbed iron as ferritin (96). Notably, recent evidence has identified novel mechanisms of mitophagy independent of the PINK1/Parkin pathway (97). For instance, mitophagy induced by iron chelator-mediated iron loss is associated with increased expression of mitochondrial ferritin. In fact, the mitochondrial ferritin interacts with nuclear receptor coactivator 4, an autophagic cargo receptor, triggers mitophagy and regulates the expression of antioxidants and detoxifying enzymes via HIF-1α specific protein 1 axis (92). Furthermore, studies have demonstrated that excessive dietary iron intake may aggravates human lupus, whereas iron infusion increases disease progression and worsens symptoms (98). Renal iron accumulation have been reported in different strains of lupus mice, and hepcidin could ameliorates LN development in MRL/lpr mice by regulating iron metabolism (99). Treatment with deferiprone, a U. S. Food and Drug Administration-approved iron chelator, significantly ameliorates pathogenic anti-dsDNA IgG expression and renal injury. Although deferiprone has been authorized for only limited diseases, its ability to interfere with iron loss‐induced mitophagy highlights its therapeutic potential in treating SLE and thus deserves further investigation (95). In lupus-prone mice with neutrophil-specific glutathione peroxidase-4 (GPX4) haploinsufficient, treatment with a specific ferroptosis inhibitor via GPX4, the component most downstream to the ferroptosis pathway, significantly ameliorated disease severity. This further proves the feasibility of manipulating iron metabolism in SLE treatment (100).
Remarkably, the molecular mechanism of iron-induced injury in glomerular and tubular cells has been attributed to mROS production (93). Indeed, excess mROS activates the NLRP3 inflammasome by upregulating IL-1β and IL-18 levels, further exacerbating inflammatory reactions within these cells (51, 93). Studies have also determined the activation of the podocyte NLRP3 inflammasome in both lupus-prone mice and LN patients (101, 102). Mitochondrial ROS inhibitors can suppress the activation of the NLRP3 inflammasomes alleviate the podocyte lesions and proteinuria (101, 103).
To date, there has been a lack of mitochondrial antioxidant therapeutics developed in preclinical studies. Notably, supplementation of coenzyme Q10 (CoQ10), a potent antioxidant, significantly reduces tacrolimus -induced oxidative stress and mitochondrial membrane potential in proximal tubular cells. Thus, this can be a promising approach to reduce nephrotoxicity via regulation of mitochondrial function (104). More potential targets treatment beneficial for mitochondrial function and structure, and preventing oxidative injuries are expected.
It has been widely proposed that mitochondria and the gut microbiome have a shared phylogenetic history, and their crosstalk regulates cellular homeostasis and metabolism (47). Furthermore, increasing evidence suggests that microbiota and their metabolites directly interfere with the mitochondrial respiratory chain and maintain the redox status in host cells (105).
Emerging evidence suggests that a disturbed gut microbiota is an essential pathogenic mechanism in SLE development (106, 107). Owing to their tight junctions and coated mucus layers, host intestinal epithelial cells (IEC) act as frontline machinery that partition the host inner organs from the gut microbiota in the lumen (108). Gut microbiome regulates mitochondrial energy production via biosynthesis and mitophagy, whereas mitochondrial homeostasis in IECs maintains intestinal barrier integrity against pathogens (109, 110). Of note, excessive mROS levels induce a redox imbalance and modify phosphorylation of tight junction proteins (e.g. occludin and ZO-1), and disrupt the tight junctions of the intestinal epithelial barrier (111). However, studies have noted that treatment with the mROS scavenger mitoquinone (MitoQ) and the antioxidant NAC restores mitochondrial ATP synthesis and enhances epithelial cell mitophagy in vivo and in vitro (112, 113).
The interaction between epithelial cell mitophagy and disturbed microbiota in the gut of SLE patients has attracted increasing attention (114). For example, studies have discovered that microbial metabolites, such as hydrogen sulfide (H2S), short-chain fatty acids (SCFAs), and tryptophan, in particular, considerably regulate mitophagy in SLE (114–116). Indeed, a previous study has demonstrated that several intestinal pathogens (e.g., Escherichia coli and Salmonella spp.) exhibiting degradation of sulfur amino acids produce a large amount of H2S (117). Recently, a study has detected detrimental responses to excessive intestinal H2S, which inhibits complex IV of the mitochondrial ETC during inflammation, reduced SCFA production and damage gut epithelial barrier integrity (115, 118). Increased intestinal permeability and gut microbes translocation have been eventually proposed contributing to the pathogenesis (119). Indeed, translocation of Enterococcus gallinarum and Lactobacillus reuteri into various organs has been independently implicated in different mouse models of lupus (120, 121).
Microbial fermentation of indigestible dietary fiber produces SCFAs that are primarily composed of acetate, propionate, and butyrate. These compounds are capable of regulating mitochondrial activity (118). Generally, Firmicutes are the principle producers of butyrate, whereas Bacteroidetes primarily produce acetate and propionates (122). Accumulating evidence has revealed that gut dysbiosis causes a reduced ratio of Firmicutes/Bacteroidetes (F/B) in SLE patients (123). Nevertheless, butyrate treatment has been demonstrated to significantly alter the microbiome diversity and normalizes the F/B ratio in lupus-prone mice (114). A recent study has reported that activated mitochondrial Drp1 reduces the production of SCFAs and perturbs the relevant microbes in a ROS-specific manner, thereby substantially disturbing intestinal homeostasis (124). Intriguingly, butyrate serves as an important fuel for intestinal cells and participates in the TCA cycle by modulating the mitochondrial ETC and mROS clearance (125). Moreover, sodium butyrate alleviates H2O2-induced oxidative stress and intestinal epithelium injury via the AMPK-mitophagy-dependent pathway. However, treatment with an AMPK inhibitor weakens this positive effect of sodium butyrate on mitophagy, highlighting the importance of sodium butyrate in maintaining mitophagy homeostasis (126). Frontier research on the mitochondria and gut microbiome nexus further illustrates that the AMPK activator metformin can enhance SCFA production (125). These previous data suggest that SCFA modulates gut microbiome diversity and is beneficial for mitochondrial homeostasis in intestinal epithelial cells, making it a complex yet fascinating target for lupus studies.
An abundance of the microbial metabolite tryptophan has been observed in the gut of SLE patients and lupus-prone mice and is considered to shift the composition of the gut microbiome (123, 127, 128). Furthermore, fecal transfer from lupus mice fed with a high-tryptophan diet compared to that from mice fed with a low-tryptophan diet is more likely to induce autoimmunity in Germ-free mice. Consequently, this strengthens the link between gut microbiota and tryptophan metabolism in the lupus phenotype (128). Remarkably, tryptophan is metabolically processed through the kynurenine pathway to produce nicotinamide adenine dinucleotide, which is essential for the mitochondrial redox balance (116). However, the complex link microbial metabolites and mitochondrial functions is just at its beginning to be recognized (118). Owing to the evidence provided by current studies on the direct association between mitochondria and gut microbiota in SLE pathogenesis, increasing focus is being directed toward normalizing defective mitophagy by influencing the gut microbiome diversity and microbial metabolites. Notably, probiotics, diet, and/or fecal transplantation are emerging strategies for maintaining redox balance and mitophagy in SLE (129). Nonetheless, multiple interactions of gut microbiota metabolites and mitochondria in SLE require further investigation.
Mitophagy mediates the elimination of organelles that are essential for regulating erythroid maturation. Previously, a study has demonstrated that a deficiency of Nix, a Bcl-2 family member, reduces mature erythrocyte levels and downregulates compensatory expansion of erythroid precursors by regulating the mitochondrial membrane potential (90). A recent study has indicated that over one-third of mature erythrocytes in SLE patients contain mitochondria (130). In this regard, certain SLE patients exhibit a dysfunctional ubiquitin-proteasome system (UPS) that fails to eliminate mitochondria in the mature erythroid cells. It has been proposed that defects in the HIF-1α-mediated metabolic regulation pathway prevent UPS activation and mitophagy initiation, leading to the accumulation of erythrocytes containing mitochondria (131). These mitochondria-containing erythrocytes produce interferongenic mtDNA that serve as immunogenic DNA in SLE, inducing IFN-α production through the activation of the cGAS–STING pathway in macrophages (132).
Presence of antiphospholipid antibodies is prothrombotic and associates with risk of thrombosis. Emerging studies have reported that increased anti-dsDNA autoantibodies or dsDNA immune complexes strongly promote platelet activation and thrombosis events by via binding to the FcγRIIA (133). Despite the sources of circulating autoantigens in SLE have not been completely elucidated. New evidence has indicated that mtDNA and mitochondrial extrusion via platelet activation through FcγRIIA stimulation induces a circulatory autoantigenic load that ultimately leads to SLE pathogenesis (134, 135). Given their abundance in the blood, platelets are a primary source of mitochondria. In addition, platelet-derived IgG-containing microparticles, mitochondria, mtDNA, and cytokines are targets of autoantibodies to form immune complexes in SLE. The DNA-containing immune complexes captured by Fcγ receptor stimulate multiple intracellular signaling pathways for IFN-α production in SLE, including the TLR9, RLRs, and cGAS–STING pathways (70, 134, 136). Since platelets have crucial roles in an immune response, preventing platelet activation by releasing microparticles and mitochondrial antigens has emerged as a therapeutic strategy for SLE treatment (137).
Neuropsychiatric (NP) involvement and cognitive symptoms are some of the most severe manifestations of SLE. These symptoms are induced by multiple immune complexes deposition and by pro-inflammatory cytokines/chemokines (138). However, the mechanism underlying brain abnormalities in NPSLE remains largely unknown, and effective strategies to ameliorate neuropsychiatric disorders are limited.
Since neurons require high energy supplied by healthy mitochondria to get excited, the brain is very sensitive to dyshomeostasis of mitochondrial redox-sensitive signaling (57). Positron emission tomography implemented by a pilot study has revealed that altering the distribution of a mitochondrial translocator protein in the brain significantly affects the cognitive functions of SLE patients (139). The microglia cells are stimulated by pro-inflammatory cytokines to cause impairment in axonal transmission and mitochondrial dysfunction (140, 141). The swollen and vacuolated mitochondria cause Ca2+ dysregulation and caspases signal activation (140, 142). However, a therapeutic strategy of neutralizing excessive ROS in microglial cells to modulate mitochondrial dyshomeostasis requires further investigation in NPSLE patients.
Inspired by the gut-brain axis concept, potential neuromodulatory mechanisms of gut microbial metabolites have been extensively studied. Evidence indicates that metabolites influence the neuro microenvironment by modulating mitophagy and microglial activation (143). Notably, SLE patients exhibit lower levels of butyrate, the mostly studied microbial metabolites. Studies have demonstrated that butyrate induces transforming growth factor (TGF)-β production and is essential for microglial maturation and function in vivo (144, 145). Additionally, butyrate suppresses the side effects of LPS and restraining the mitochondrial function of oligodendrocytes in a mitochondrial redox-dependent manner. Thus, it is considered to be a key mediator of NP-SLE (146, 147). Moreover, gut dysbiosis and neurotoxic substance production (e.g., amyloid proteins and lipopolysaccharides) triggers microglial activation and inflammation. Several studies have also reported that increased kynurenine serum levels are positively correlated with neurological manifestations of SLE (128, 148, 149). Indeed, a kynurenine-derived metabolite, quinolinic acid, influences neuronal activities and causes neurotoxicity (150, 151). More intensive investigation into the mechanism underlying the mitochondrial redox balance in the pathogenesis of NPSLE is expected.
Given the plenty of studies elucidating the essential role of mitophagy, mitochondria have emerged as important pharmacological targets. Thus, novel intervention strategies, such as treatment using antioxidants, metabolic rescue of autolysosomal degradation, and repurposing of conventional drugs, are possible options to counteract or fine-tune the current treatment for SLE (152).
Sirolimus is an mTOR inhibitor that naturally occurs as an antifungal agent and was first discovered in Streptomyces hygroscopicus. It exhibits anti-tumor, anti-proliferative, anti-fibrotic, and immunosuppressive effects (153–155). Sirolimus blocks both mTORC1 activation and T-cell hyperactivity, and its effectiveness has been demonstrated in lupus mice by normalizing proteinuria, protecting renal function, reducing anti-dsDNA titers and inhibiting antiphospholipid antibody production (156, 157). In 2018, a single-arm, open-label trial further verified the role of mTOR inhibition by sirolimus in treating SLE to recover mitochondrial dysfunction (60). Both the off-label drug use and subsequent open-label phase 1/2 clinical trial revealed that sirolimus effectively reduced SLE disease activity (60, 158), and could be an effective therapeutic medication for SLE. Nonetheless, further randomized controlled trials of sirolimus in SLE are warranted.
The glutathione precursor NAC is an antioxidant and ROS scavenger that inhibits mTORC1 and suppresses T cell overactivation (53). By disrupting the mTOR pathway and decreasing ROS levels, NAC displayed beneficial efficacy in disease activity of lupus patients (58). As was aforementioned, impaired mitochondrial function, accompanied by increased mitochondrial transmembrane potential and sustained hyperpolarization, potentiates the death of pro-inflammatory T cells in SLE (53). Importantly, a randomized, double-blind clinical trial has proven NAC to be an effective inhibitor of mTOR activity and a regulator of the mitochondrial transmembrane potential in SLE (58). As it is an ROS scavenger and a glutathione precursor, NAC decreases ROS levels while increasing glutathione levels in peripheral lymphocytes (159). Moreover, NAC inhibits mitochondrial ETC complex I, reduce the oxidative stress (159, 160), diminish serum anti-dsDNA antibodies and improves lupus nephritis as well as prevent disease flares in lupus mice (161).
CoQ10 is a potent antioxidant that prevents the oxidation of cell membranes and cellular components by scavenging free radicals in the cytoplasm. Furthermore, it plays a key role in electron transport from complexes I and II to complex III in the mitochondria (162). It is well recognized that mROS production triggers immune responses against SLE through NET formation (74). A synthetic analog of CoQ10, MitoQ, effectively suppresses autoimmune inflammation in SLE by inhibiting mROS generation. This further downregulates granulocyte NETosis, reduces disease activity, inhibits IFN-α responses, and suppresses immune complex formation in the kidneys in a lupus mouse model (74, 163). Furthermore, another synthetic quinone analog of CoQ10, idebenone, has shown effectiveness in improving immune dysregulation, preventing organ impairment, and restoring mitochondrial abnormalities (75).
Metformin is a first-line antihyperglycemic medication for type II diabetes mellitus; however, it is also a promising therapeutic agent for SLE with immunomodulatory properties. Studies have observed the normalization of T-cell mitochondrial metabolism (including mitochondrial oxidative stress and redox balance) in SLE patients with metformin treatment (164–166). Remarkably, add-on metformin treatment for mild or moderate SLE decreases clinical flares, and reduces glucocorticoid dose (167). Furthermore, metformin selectively inhibits mitochondrial complex I and reduces NADPH oxidase activity. While it significantly downregulates DNA release from NETs by suppressing ROS production (168), it also activates AMPK and suppresses OXPHOS in different immune cells (169). Additionally, it has been proven to be effective in inhibiting B cell differentiation into plasma cells in the germinal center of lupus-prone mice, reduction of NLRP3 inflammasome activation mediated by AMPK, and blockade of pyroptosis of tubular epithelial cells in pristane-induced lupus mice (69, 170). Importantly, the beneficial effects of metformin have been confirmed via a post-hoc pooled analysis, demonstrating its potential in improving SLE disease activity and decreasing flare risks, particularly in serologically quiescent SLE patients (171).
Hydroxychloroquine (HCQ) and chloroquine (CQ) have clinical applications for prevention and treatment of malaria and have been repurposed to treat milder manifestations of SLE, such as skin rash and arthritis (172). Although their precise mechanisms of action remain to be completely elucidated, it has been reported that HCQ inhibits the mitochondrial antioxidant system induced by TCR crosslinking, consequently increasing mROS levels and reducing CD4+ T cell proliferation through oxidative stress (173). Furthermore, HCQ exhibits immunomodulatory properties that downregulate the constitutive activation of TLR7 and TLR9 in antigen-presenting cells, and inhibited the cytokine production thereby (174). Another potential mechanism of HCQ and CQ is to regulate cGAS activity by interfering with its binding to cytosolic DNA, eventually reducing pro-inflammatory cytokine production (175).
A few potential mitochondria-targeting chemicals have emerged as beneficial therapeutics in lupus mice based on the normalization of mitophagy metabolic pathways. For instance, studies have shown that treatment with 3-PEHPC, an inhibitor of Rab geranylgeranyl transferase, restores Drp1 expression, enables mitochondrial accumulation, and prevents Antinuclear antibody production and nephritis in MRL/lpr mice. Additionally, these 3-PEHPC-treated mice exhibit altered inflammatory cytokine profiles and low IL-10 expression post-treatment (59). Another potential mitochondria-targeting agent is pioglitazone, a PPARγ agonist, induces the disassembly of complexes I and III in the mitochondria, depleting cellular ATP production by upregulating the mitochondrial respiratory chain subunit gene (176). Interestingly, murine lupus models treated with pioglitazone or rosiglitazone have displayed positive responses regarding their inflammatory pathways and disease activity (177, 178). Furthermore, KN-93 is another potential therapeutic agent that inhibits CaMK4 via the AKT/mTOR signaling pathway (179). Notably, it can increase Treg cells and decrease the differentiation of Th17 cells in lupus-prone mice, which contribute to suppresses the development of glomerulonephritis and skin diseases (180, 181). cGAS–STING pathway is another possible strategy to develop for SLE patients. Moreover, a potent small-molecule inhibitor of STING, identified as H-151, has been proven to inhibit both murine and human STING activation. Therefore, it is under significant clinical consideration (182).
Only limited mitochondria-targeted drugs have been translated in the clinical application of SLE. With the further understanding of the unique energy metabolism feature of mitochondria in a distinct population of immunocytes, tissue or organ-specific therapeutics of mitochondria-targeting intervention may provide promising strategies for SLE treatment. Due to the critical role of mROS in many pathophysiological processes, the majority of mitochondria-targeted drug focus on antioxidants (183). The antioxidant lipophilic cations have great potential for the treatment of a wide range of pathologies. However, there are still some other prospects for new mitochondrial modulation drugs, depending on increasing lipophilic cation moieties, targeting cardiolipin, and aiming on mitochondrial targeting signal peptides (184). Besides to mitochondria-targeting design strategy, renew and expansion of clinical applicability on the conventional mitochondria-targeting drug is another potential translation
Accumulating evidence has highlighted the importance of mitochondria homeostasis in maintaining immune cell differentiation and activation (33). However, alterations in mitochondrial functions, such as excessive mROS and mtDNA production, impaired mitophagy, altered mitochondrial dynamics, and disrupted redox homeostasis, initiate an immune cascade preceding the onset and progression of SLE (127). Critically, mitophagy downregulation results in the release of autoimmunogenic mtDNA that promotes the production of inflammatory cytokines, such as IL-1β and IFN-α (46). Targeting on mROS scavengers and mitophagy agonists are advantageous in SLE clinical treatment (185). In this review, mitophagy defects of antioxidant defense and mitochondrial dysfunction in SLE were mainly discussed. Nevertheless, certain questions and limitations remain to be addressed. During metabolic processes, the TCA cycle occurs in the mitochondria and involves in not only energy supplement glucose and lipid metabolism pathways. As the sophisticated complexity of mitochondria function, the effect of TCA cycle on immunity is worth to be discussed elsewhere. Moreover, the potential pre-clinical mitochondria-targeting chemicals have been only briefly summarized according to the limited space.
Mitochondria-targeting are promising therapeutics due to the clinical benefits. However, several challenges remain to be addressed. One of these is an intensively debated mechanism involved in mitochondria directing the cell fate in SLE (186). During the pathogenic conditions of systemic and chronic SLE, mitophagy and mitochondrial metabolism pathways form extremely complex networks that mutually affect immune signaling activation (7). Specific mROS scavenger and mitophagy agonist/antagonist are required for their function assessment in cellular fate decision and deepening the understanding of the mitochondria-associated immunometabolism (184). With the aid of the latest single-cell, multi-omics, and visualization technologies, identification of key mitochondrial metabolites that determine cell fate may shed light on SLE pathogenesis.
Furthermore, interdisciplinary research in pharmaceutics and materials engineering is required to develop a novel drug-targeting delivery platform that portrays great potential for SLE treatment, facilitating the release of drug payload in an organelle-specific and controllable manner for manipulating the cell differentiation (152). In addition, drug repurposing, which involves investigation of existing drugs for new therapeutic purposes, particularly uses biophysics-based molecular docking approaches and has been proven to be effective and efficient and of immense value in biomedicine (184, 187). The effect of repurposed drugs, such as metformin and CoQ10 in SLE treatment has been well investigated (75, 171). Therefore, our accumulated data help elucidate the mitochondrial metabolite landscape and in identifying mitochondria-targeting therapeutic strategies that precisely modulate abnormal cell differentiation in SLE. Nonetheless, the safety and efficacy of these strategies still require further investigation.
All authors were involved in drafting the article or revising for important intellectual content, and all authors approved the final version to be published. LDZ and MW designed the literature search and wrote the article with input from all authors. LKZ, XH and MW drafted the manuscript. FX, LDZ and XZ revised the manuscript.
This work was supported by grants from the National Natural Science Foundation of China (81788101, 82171798, 81801635, 81803848, and 32141004), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-040,2021-I2M-1-017,2021-I2M-1-047,2021-I2M-1-016 and 2021-I2M-1-026).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JX declared a shared parent affiliation with the authors LKZ, FX, XZ, LDZ and MW to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol (2016) 12(12):716–30. doi: 10.1038/nrrheum.2016.186
2. Pisetsky DS, Lipsky PE. New insights into the role of antinuclear antibodies in systemic lupus erythematosus. Nat Rev Rheumatol (2020) 16(10):565–79. doi: 10.1038/s41584-020-0480-7
3. Mills EL, Kelly B, O'Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol (2017) 18(5):488–98. doi: 10.1038/ni.3704
4. Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science (2019) 366(6472):1531–6. doi: 10.1126/science.aav4011
5. Javadov S, Kozlov AV, Camara AKS. Mitochondria in health and diseases. Cells (2020) 9(5):1177. doi: 10.3390/cells9051177
6. Postal M, Vivaldo JF, Fernandez-Ruiz R, Paredes JL, Appenzeller S, Niewold TB. Type I interferon in the pathogenesis of systemic lupus erythematosus. Curr Opin Immunol (2020) 67:87–94. doi: 10.1016/j.coi.2020.10.014
7. Huang N, Perl A. Metabolism as a target for modulation in autoimmune diseases. Trends Immunol (2018) 39(7):562–76. doi: 10.1016/j.it.2018.04.006
8. Chen PM, Tsokos GC. Mitochondria in the pathogenesis of systemic lupus erythematosus. Curr Rheumatol Rep (2022) 24(4):88–95. doi: 10.1007/s11926-022-01063-9
9. Yang SK, Zhang HR, Shi SP, Zhu YQ, Song N, Dai Q, et al. The role of mitochondria in systemic lupus erythematosus: A glimpse of various pathogenetic mechanisms. Curr Med Chem (2020) 27(20):3346–61. doi: 10.2174/0929867326666181126165139
10. Schuiveling M, Vazirpanah N, Radstake T, Zimmermann M, Broen JCA. Metformin, a new era for an old drug in the treatment of immune mediated disease? Curr Drug Targets (2018) 19(8):945–59. doi: 10.2174/1389450118666170613081730
11. Pearce SF, Rebelo-Guiomar P, D'Souza AR, Powell CA, Van Haute L, Minczuk M. Regulation of mammalian mitochondrial gene expression: recent advances. Trends Biochem Sci (2017) 42(8):625–39. doi: 10.1016/j.tibs.2017.02.003
12. Leishangthem BD, Sharma A, Bhatnagar A. Role of altered mitochondria functions in the pathogenesis of systemic lupus erythematosus. Lupus (2016) 25(3):272–81. doi: 10.1177/0961203315605370
13. Wojcik P, Gegotek A, Zarkovic N, Skrzydlewska E. Oxidative stress and lipid mediators modulate immune cell functions in autoimmune diseases. Int J Mol Sci (2021) 22(2):723. doi: 10.3390/ijms22020723
14. Martinez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
15. Fu W, Liu Y, Yin H. Mitochondrial dynamics: Biogenesis, fission, fusion, and mitophagy in the regulation of stem cell behaviors. Stem Cells Int (2019) 2019:9757201. doi: 10.1155/2019/9757201
16. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell (1998) 92(6):829–39. doi: 10.1016/S0092-8674(00)81410-5
17. Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev (2006) 27(7):728–35. doi: 10.1210/er.2006-0037
18. Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest (2013) 123(11):4888–99. doi: 10.1172/JCI66218
19. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol (2011) 13(9):1016–23. doi: 10.1038/ncb2329
20. Melser S, Chatelain EH, Lavie J, Mahfouf W, Jose C, Obre E, et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab (2013) 17(5):719–30. doi: 10.1016/j.cmet.2013.03.014
21. Summer R, Shaghaghi H, Schriner D, Roque W, Sales D, Cuevas-Mora K, et al. Activation of the mTORC1/PGC-1 axis promotes mitochondrial biogenesis and induces cellular senescence in the lung epithelium. Am J Physiol Lung Cell Mol Physiol (2019) 316(6):L1049–L60. doi: 10.1152/ajplung.00244.2018
22. LaGory EL, Wu C, Taniguchi CM, Ding CC, Chi JT, von Eyben R, et al. Suppression of PGC-1alpha is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep (2015) 12(1):116–27. doi: 10.1016/j.celrep.2015.06.006
23. Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol (2014) 15(10):634–46. doi: 10.1038/nrm3877
24. Ma K, Chen G, Li W, Kepp O, Zhu Y, Chen Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front Cell Dev Biol (2020) 8:467. doi: 10.3389/fcell.2020.00467
25. Galvan DL, Long J, Green N, Chang BH, Lin JS, Schumacker P, et al. Drp1S600 phosphorylation regulates mitochondrial fission and progression of nephropathy in diabetic mice. J Clin Invest (2019) 129(7):2807–23. doi: 10.1172/JCI127277
26. Lee H, Yoon Y. Mitochondrial fission and fusion. Biochem Soc Trans (2016) 44(6):1725–35. doi: 10.1042/BST20160129
27. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell (2016) 166(1):63–76. doi: 10.1016/j.cell.2016.05.035
28. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol (2018) 20(9):1013–22. doi: 10.1038/s41556-018-0176-2
29. Jin SM, Youle RJ. PINK1- and parkin-mediated mitophagy at a glance. J Cell Sci (2012) 125(Pt 4):795–9. doi: 10.1242/jcs.093849
30. Lazarou M. Keeping the immune system in check: A role for mitophagy. Immunol Cell Biol (2015) 93(1):3–10. doi: 10.1038/icb.2014.75
31. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol (2012) 14(2):177–85. doi: 10.1038/ncb2422
32. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. PloS Biol (2010) 8(1):e1000298. doi: 10.1371/journal.pbio.1000298
33. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity (2015) 42(3):406–17. doi: 10.1016/j.immuni.2015.02.002
34. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi: 10.1038/nature08780
35. Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discovery (2018) 17(12):865–86. doi: 10.1038/nrd.2018.174
36. Ribas V, Garcia-Ruiz C, Fernandez-Checa JC. Glutathione and mitochondria. Front Pharmacol (2014) 5:151. doi: 10.3389/fphar.2014.00151
37. Lee HT, Lin CS, Lee CS, Tsai CY, Wei YH. Increased 8-hydroxy-2'-deoxyguanosine in plasma and decreased mRNA expression of human 8-oxoguanine DNA glycosylase 1, anti-oxidant enzymes, mitochondrial biogenesis-related proteins and glycolytic enzymes in leucocytes in patients with systemic lupus erythematosus. Clin Exp Immunol (2014) 176(1):66–77. doi: 10.1111/cei.12256
38. Lee HT, Wu TH, Lin CS, Lee CS, Pan SC, Chang DM, et al. Oxidative DNA and mitochondrial DNA change in patients with SLE. Front Biosci (Landmark Ed) (2017) 22:493–503. doi: 10.2741/4497
39. Jonsen A, Yu X, Truedsson L, Nived O, Sturfelt G, Ibrahim S, et al. Mitochondrial DNA polymorphisms are associated with susceptibility and phenotype of systemic lupus erythematosus. Lupus (2009) 18(4):309–12. doi: 10.1177/0961203308097477
40. Lee HT, Lin CS, Chen WS, Liao HT, Tsai CY, Wei YH. Leukocyte mitochondrial DNA alteration in systemic lupus erythematosus and its relevance to the susceptibility to lupus nephritis. Int J Mol Sci (2012) 13(7):8853–68. doi: 10.3390/ijms13078853
41. Zhou XJ, Lu XL, Lv JC, Yang HZ, Qin LX, Zhao MH, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis (2011) 70(7):1330–7. doi: 10.1136/ard.2010.140111
42. Lai R, Zhang X, Qiao K, Gao X, Li S, Zhang R, et al. Identification of sequence polymorphisms in the mitochondrial deoxyribonucleic acid displacement-loop region as risk factors for systemic lupus erythematosus. Arch Rheumatol (2021) 36(3):375–80. doi: 10.46497/ArchRheumatol.2021.8101
43. Pisetsky DS, Spencer DM, Mobarrez F, Fuzzi E, Gunnarsson I, Svenungsson E. The binding of SLE autoantibodies to mitochondria. Clin Immunol (2020) 212:108349. doi: 10.1016/j.clim.2020.108349
44. Becker Y, Loignon RC, Julien AS, Marcoux G, Allaeys I, Levesque T, et al. Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci Rep (2019) 9(1):4530. doi: 10.1038/s41598-019-40900-3
45. Becker Y, Marcoux G, Allaeys I, Julien AS, Loignon RC, Benk-Fortin H, et al. Autoantibodies in systemic lupus erythematosus target mitochondrial RNA. Front Immunol (2019) 10:1026. doi: 10.3389/fimmu.2019.01026
46. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy (2020) 16(1):3–17. doi: 10.1080/15548627.2019.1603547
47. Franco-Obregon A, Gilbert JA. The microbiome-mitochondrion connection: common ancestries, common mechanisms, common goals. mSystems (2017) 2(3):e00018-17. doi: 10.1128/mSystems.00018-17
48. Fernandez DR, Telarico T, Bonilla E, Li Q, Banerjee S, Middleton FA, et al. Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol (2009) 182(4):2063–73. doi: 10.4049/jimmunol.0803600
49. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med (2016) 213(5):697–713. doi: 10.1084/jem.20151876
50. Ding L, Dong G, Zhang D, Ni Y, Hou Y. The regional function of cGAS/STING signal in multiple organs: One of culprit behind systemic lupus erythematosus? Med Hypotheses (2015) 85(6):846–9. doi: 10.1016/j.mehy.2015.09.026
51. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
52. Caza TN, Talaber G, Perl A. Metabolic regulation of organelle homeostasis in lupus T cells. Clin Immunol (2012) 144(3):200–13. doi: 10.1016/j.clim.2012.07.001
53. Gergely P Jr., Grossman C, Niland B, Puskas F, Neupane H, Allam F, et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheumatol (2002) 46(1):175–90. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
54. Perl A, Gergely P Jr., Nagy G, Koncz A, Banki K. Mitochondrial hyperpolarization: a checkpoint of T-cell life, death and autoimmunity. Trends Immunol (2004) 25(7):360–7. doi: 10.1016/j.it.2004.05.001
55. Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat Immunol (2020) 21(9):1022–33. doi: 10.1038/s41590-020-0725-2
56. Li W, Cheng H, Li G, Zhang L. Mitochondrial damage and the road to exhaustion. Cell Metab (2020) 32(6):905–7. doi: 10.1016/j.cmet.2020.11.004
57. Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol (2013) 9(11):674–86. doi: 10.1038/nrrheum.2013.147
58. Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol (2012) 64(9):2937–46. doi: 10.1002/art.34502
59. Caza TN, Fernandez DR, Talaber G, Oaks Z, Haas M, Madaio MP, et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann Rheum Dis (2014) 73(10):1888–97. doi: 10.1136/annrheumdis-2013-203794
60. Lai ZW, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet (2018) 391(10126):1186–96. doi: 10.1016/S0140-6736(18)30485-9
61. Scalapino KJ, Daikh DI. Suppression of glomerulonephritis in NZB/NZW lupus prone mice by adoptive transfer of ex vivo expanded regulatory T cells. PloS One (2009) 4(6):e6031. doi: 10.1371/journal.pone.0006031
62. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
63. Jang SG, Lee J, Hong SM, Kwok SK, Cho ML, Park SH. Metformin enhances the immunomodulatory potential of adipose-derived mesenchymal stem cells through STAT1 in an animal model of lupus. Rheumatol (Oxford) (2020) 59(6):1426–38. doi: 10.1093/rheumatology/kez631
64. Torigoe M, Iwata S, Nakayamada S, Sakata K, Zhang M, Hajime M, et al. Metabolic reprogramming commits differentiation of human CD27(+)IgD(+) b cells to plasmablasts or CD27(-)IgD(-) cells. J Immunol (2017) 199(2):425–34. doi: 10.4049/jimmunol.1601908
65. Wang M, Chen H, Qiu J, Yang HX, Zhang CY, Fei YY, et al. Antagonizing miR-7 suppresses b cell hyperresponsiveness and inhibits lupus development. J Autoimmun (2020) 109:102440. doi: 10.1016/j.jaut.2020.102440
66. Wu T, Qin X, Kurepa Z, Kumar KR, Liu K, Kanta H, et al. Shared signaling networks active in b cells isolated from genetically distinct mouse models of lupus. J Clin Invest (2007) 117(8):2186–96. doi: 10.1172/JCI30398
67. Jones DD, Gaudette BT, Wilmore JR, Chernova I, Bortnick A, Weiss BM, et al. mTOR has distinct functions in generating versus sustaining humoral immunity. J Clin Invest (2016) 126(11):4250–61. doi: 10.1172/JCI86504
68. Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, et al. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity (2016) 45(1):60–73. doi: 10.1016/j.immuni.2016.06.011
69. Lee SY, Moon SJ, Kim EK, Seo HB, Yang EJ, Son HJ, et al. Metformin suppresses systemic autoimmunity in roquin(san/san) mice through inhibiting b cell differentiation into plasma cells via regulation of AMPK/mTOR/STAT3. J Immunol (2017) 198(7):2661–70. doi: 10.4049/jimmunol.1403088
70. Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest (2005) 115(2):407–17. doi: 10.1172/JCI23025
71. Fu X, Liu H, Huang G, Dai SS. The emerging role of neutrophils in autoimmune-associated disorders: effector, predictor, and therapeutic targets. MedComm (2020) (2021) 2(3):402–13. doi: 10.1002/mco2.69
72. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature (2018) 561(7722):258–62. doi: 10.1038/s41586-018-0448-9
73. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res (2011) 21(2):290–304. doi: 10.1038/cr.2010.150
74. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22(2):146–53. doi: 10.1038/nm.4027
75. Blanco LP, Pedersen HL, Wang X, Lightfoot YL, Seto N, Carmona-Rivera C, et al. Improved mitochondrial metabolism and reduced inflammation following attenuation of murine lupus with coenzyme Q10 analog idebenone. Arthritis Rheumatol (2020) 72(3):454–64. doi: 10.1002/art.41128
76. Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, et al. IFNalpha impairs autophagic degradation of mtDNA promoting autoreactivity of SLE monocytes in a STING-dependent fashion. Cell Rep (2018) 25(4):921–33 e5. doi: 10.1016/j.celrep.2018.09.001
77. Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science (2001) 294(5546):1540–3. doi: 10.1126/science.1064890
78. Fernandez D, Perl A. mTOR signaling: A central pathway to pathogenesis in systemic lupus erythematosus? Discovery Med (2010) 9(46):173–8.
79. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol (2015) 6:262. doi: 10.3389/fphar.2015.00262
80. Kahlenberg JM, Kaplan MJ. The inflammasome and lupus: Another innate immune mechanism contributing to disease pathogenesis? Curr Opin Rheumatol (2014) 26(5):475–81. doi: 10.1097/BOR.0000000000000088
81. Kim HW, Kwon YJ, Park BW, Song JJ, Park YB, Park MC. Differential expressions of NOD-like receptors and their associations with inflammatory responses in rheumatoid arthritis. Clin Exp Rheumatol (2017) 35(4):630–7.
82. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
83. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature (2012) 492(7427):123–7. doi: 10.1038/nature11588
84. Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol (2013) 14(5):480–8. doi: 10.1038/ni.2563
85. Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol (2013) 190(3):1217–26. doi: 10.4049/jimmunol.1202388
86. Yang CA, Huang ST, Chiang BL. Sex-dependent differential activation of NLRP3 and AIM2 inflammasomes in SLE macrophages. Rheumatol (Oxford) (2015) 54(2):324–31. doi: 10.1093/rheumatology/keu318
87. Lech M, Lorenz G, Kulkarni OP, Grosser MO, Stigrot N, Darisipudi MN, et al. NLRP3 and ASC suppress lupus-like autoimmunity by driving the immunosuppressive effects of TGF-beta receptor signalling. Ann Rheum Dis (2015) 74(12):2224–35. doi: 10.1136/annrheumdis-2014-205496
88. Rai P, Janardhan KS, Meacham J, Madenspacher JH, Lin WC, Karmaus PWF, et al. IRGM1 links mitochondrial quality control to autoimmunity. Nat Immunol (2021) 22(3):312–21. doi: 10.1038/s41590-020-00859-0
89. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol (2017) 13(10):629–46. doi: 10.1038/nrneph.2017.107
90. Zuo Z, Jing K, Wu H, Wang S, Ye L, Li Z, et al. Mechanisms and functions of mitophagy and potential roles in renal disease. Front Physiol (2020) 11:935. doi: 10.3389/fphys.2020.00935
91. Qi YY, Zhou XJ, Cheng FJ, Hou P, Ren YL, Wang SX, et al. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-alpha in lupus nephritis. Ann Rheum Dis (2018) 77(12):1799–809. doi: 10.1136/annrheumdis-2018-213028
92. Hara Y, Yanatori I, Tanaka A, Kishi F, Lemasters JJ, Nishina S, et al. Iron loss triggers mitophagy through induction of mitochondrial ferritin. EMBO Rep (2020) 21(11):e50202. doi: 10.15252/embr.202050202
93. Chun J, Chung H, Wang X, Barry R, Taheri ZM, Platnich JM, et al. NLRP3 localizes to the tubular epithelium in human kidney and correlates with outcome in IgA nephropathy. Sci Rep (2016) 6:24667. doi: 10.1038/srep24667
94. Tian Y, Guo H, Miao X, Xu J, Yang R, Zhao L, et al. Nestin protects podocyte from injury in lupus nephritis by mitophagy and oxidative stress. Cell Death Dis (2020) 11(5):319. doi: 10.1038/s41419-020-2547-4
95. Marks ES, Bonnemaison ML, Brusnahan SK, Zhang W, Fan W, Garrison JC, et al. Renal iron accumulation occurs in lupus nephritis and iron chelation delays the onset of albuminuria. Sci Rep (2017) 7(1):12821. doi: 10.1038/s41598-017-13029-4
96. Wlazlo E, Mehrad B, Morel L, Scindia Y. Iron metabolism: An under investigated driver of renal pathology in lupus nephritis. Front Med (Lausanne) (2021) 8:643686. doi: 10.3389/fmed.2021.643686
97. Bona N, Pezzarini E, Balbi B, Daniele SM, Rossi MF, Monje AL, et al. Oxidative stress, inflammation and disease activity biomarkers in lupus nephropathy. Lupus (2020) 29(3):311–23. doi: 10.1177/0961203320904784
98. Brown AC. Lupus erythematosus and nutrition: a review of the literature. J Ren Nutr (2000) 10(4):170–83. doi: 10.1053/jren.2000.16323
99. Scindia Y, Wlazlo E, Ghias E, Cechova S, Loi V, Leeds J, et al. Modulation of iron homeostasis with hepcidin ameliorates spontaneous murine lupus nephritis. Kidney Int (2020) 98(1):100–15. doi: 10.1016/j.kint.2020.01.025
100. Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol (2021) 22(9):1107–17. doi: 10.1038/s41590-021-00993-3
101. Fu R, Guo C, Wang S, Huang Y, Jin O, Hu H, et al. Podocyte activation of NLRP3 inflammasomes contributes to the development of proteinuria in lupus nephritis. Arthritis Rheumatol (2017) 69(8):1636–46. doi: 10.1002/art.40155
102. Nakamura K, Kawakami T, Yamamoto N, Tomizawa M, Fujiwara T, Ishii T, et al. Activation of the NLRP3 inflammasome by cellular labile iron. Exp Hematol (2016) 44(2):116–24. doi: 10.1016/j.exphem.2015.11.002
103. Ichinose K, Ushigusa T, Nishino A, Nakashima Y, Suzuki T, Horai Y, et al. Lupus nephritis IgG induction of Calcium/Calmodulin-dependent protein kinase IV expression in podocytes and alteration of their function. Arthritis Rheumatol (2016) 68(4):944–52. doi: 10.1002/art.39499
104. Yu JH, Lim SW, Luo K, Cui S, Quan Y, Shin YJ, et al. Coenzyme Q10 alleviates tacrolimus-induced mitochondrial dysfunction in kidney. FASEB J (2019) 33(11):12288–98. doi: 10.1096/fj.201900386RR
105. Jackson DN, Theiss AL. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes (2020) 11(3):285–304. doi: 10.1080/19490976.2019.1592421
106. Zhang X, Chen BD, Zhao LD, Li H. The gut microbiota: Emerging evidence in autoimmune diseases. Trends Mol Med (2020) 26(9):862–73. doi: 10.1016/j.molmed.2020.04.001
107. Xu D, Yang H, Lai CC, Li P, Zhang X, Yang XO, et al. Clinical analysis of systemic lupus erythematosus with gastrointestinal manifestations. Lupus (2010) 19(7):866–9. doi: 10.1177/0961203310365883
108. Li R, Meng X, Chen B, Zhao L, Zhang X. Gut microbiota in lupus: A butterfly effect? Curr Rheumatol Rep (2021) 23(4):27. doi: 10.1007/s11926-021-00986-z
109. Gruber J, Kennedy BK. Microbiome and longevity: Gut microbes send signals to host mitochondria. Cell (2017) 169(7):1168–9. doi: 10.1016/j.cell.2017.05.048
110. Mimoun S, Andriamihaja M, Chaumontet C, Atanasiu C, Benamouzig R, Blouin JM, et al. Detoxification of H(2)S by differentiated colonic epithelial cells: implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid Redox Signal (2012) 17(1):1–10. doi: 10.1089/ars.2011.4186
111. Rao R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci (2008) 13:7210–26. doi: 10.2741/3223
112. Lopes F, Keita AV, Saxena A, Reyes JL, Mancini NL, Al Rajabi A, et al. ER-stress mobilization of death-associated protein kinase-1-dependent xenophagy counteracts mitochondria stress-induced epithelial barrier dysfunction. J Biol Chem (2018) 293(9):3073–87. doi: 10.1074/jbc.RA117.000809
113. Asano J, Sato T, Ichinose S, Kajita M, Onai N, Shimizu S, et al. Intrinsic autophagy is required for the maintenance of intestinal stem cells and for irradiation-induced intestinal regeneration. Cell Rep (2017) 20(5):1050–60. doi: 10.1016/j.celrep.2017.07.019
114. He H, Xu H, Xu J, Zhao H, Lin Q, Zhou Y, et al. Sodium butyrate ameliorates gut microbiota dysbiosis in lupus-like mice. Front Nutr (2020) 7:604283. doi: 10.3389/fnut.2020.604283
115. Ijssennagger N, van der Meer R, van Mil SWC. Sulfide as a mucus barrier-breaker in inflammatory bowel disease? Trends Mol Med (2016) 22(3):190–9. doi: 10.1016/j.molmed.2016.01.002
116. Castro-Portuguez R, Sutphin GL. Kynurenine pathway, NAD(+) synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp Gerontol (2020) 132:110841. doi: 10.1016/j.exger.2020.110841
117. Wallace JL, Blackler RW, Chan MV, Da Silva GJ, Elsheikh W, Flannigan KL, et al. Anti-inflammatory and cytoprotective actions of hydrogen sulfide: translation to therapeutics. Antioxid Redox Signal (2015) 22(5):398–410. doi: 10.1089/ars.2014.5901
118. Saint-Georges-Chaumet Y, Edeas M. Microbiota-mitochondria inter-talk: consequence for microbiota-host interaction. Pathog Dis (2016) 74(1):ftv096. doi: 10.1093/femspd/ftv096
119. Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis (2019) 78(7):947–56. doi: 10.1136/annrheumdis-2018-214856
120. Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science (2018) 359(6380):1156–61. doi: 10.1126/science.aar7201
121. Zegarra-Ruiz DF, El Beidaq A, Iniguez AJ, Lubrano Di Ricco M, Manfredo Vieira S, Ruff WE, et al. A diet-sensitive commensal lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe (2019) 25(1):113–27 e6. doi: 10.1016/j.chom.2018.11.009
122. Kim M, Qie Y, Park J, Kim CH. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe (2016) 20(2):202–14. doi: 10.1016/j.chom.2016.07.001
123. Chen BD, Jia XM, Xu JY, Zhao LD, Ji JY, Wu BX, et al. An autoimmunogenic and proinflammatory profile defined by the gut microbiota of patients with untreated systemic lupus erythematosus. Arthritis Rheumatol (2021) 73(2):232–43. doi: 10.1002/art.41511
124. Duan C, Kuang L, Xiang X, Zhang J, Zhu Y, Wu Y, et al. Activated Drp1-mediated mitochondrial ROS influence the gut microbiome and intestinal barrier after hemorrhagic shock. Aging (Albany NY). (2020) 12(2):1397–416. doi: 10.18632/aging.102690
125. Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab (2011) 13(5):517–26. doi: 10.1016/j.cmet.2011.02.018
126. Li X, Wang C, Zhu J, Lin Q, Yu M, Wen J, et al. Sodium butyrate ameliorates oxidative stress-induced intestinal epithelium barrier injury and mitochondrial damage through AMPK-mitophagy pathway. Oxid Med Cell Longev (2022) 2022:3745135. doi: 10.1155/2022/3745135
127. Tzeng HT, Chyuan IT. Immunometabolism in systemic lupus erythematosus: Relevant pathogenetic mechanisms and potential clinical applications. J Formos Med Assoc (2021) 120(9):1667–75. doi: 10.1016/j.jfma.2021.03.019
128. Choi SC, Brown J, Gong M, Ge Y, Zadeh M, Li W, et al. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Sci Transl Med (2020) 12(551):eaax2220. doi: 10.1126/scitranslmed.aax2220
129. Chen B, Sun L, Zhang X. Integration of microbiome and epigenome to decipher the pathogenesis of autoimmune diseases. J Autoimmun (2017) 83:31–42. doi: 10.1016/j.jaut.2017.03.009
130. Caielli S, Cardenas J, de Jesus AA, Baisch J, Walters L, Blanck JP, et al. Erythroid mitochondrial retention triggers myeloid-dependent type I interferon in human SLE. Cell (2021) 184(17):4464–79 e19. doi: 10.1016/j.cell.2021.07.021
131. Sulkshane P, Ram J, Thakur A, Reis N, Kleifeld O, Glickman MH. Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol (2021) 45:102047. doi: 10.1016/j.redox.2021.102047
132. Morel L. Erythrocyte-derived mitochondria: an unexpected interferon inducer in lupus. Trends Immunol (2021) 42(12):1054–6. doi: 10.1016/j.it.2021.10.010
133. Andrianova IA, Ponomareva AA, Mordakhanova ER, Le Minh G, Daminova AG, Nevzorova TA, et al. In systemic lupus erythematosus anti-dsDNA antibodies can promote thrombosis through direct platelet activation. J Autoimmun (2020) 107:102355. doi: 10.1016/j.jaut.2019.102355
134. Melki I, Allaeys I, Tessandier N, Levesque T, Cloutier N, Laroche A, et al. Platelets release mitochondrial antigens in systemic lupus erythematosus. Sci Transl Med (2021) 13(581):eaav5928. doi: 10.1126/scitranslmed.aav5928
135. Melki I, Allaeys I, Tessandier N, Mailhot B, Cloutier N, Campbell RA, et al. FcgammaRIIA expression accelerates nephritis and increases platelet activation in systemic lupus erythematosus. Blood (2020) 136(25):2933–45. doi: 10.1182/blood.2020004974
136. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339(6121):786–91. doi: 10.1126/science.1232458
137. Linge P, Fortin PR, Lood C, Bengtsson AA, Boilard E. The non-haemostatic role of platelets in systemic lupus erythematosus. Nat Rev Rheumatol (2018) 14(4):195–213. doi: 10.1038/nrrheum.2018.38
138. Schwartz N, Stock AD, Putterman C. Neuropsychiatric lupus: new mechanistic insights and future treatment directions. Nat Rev Rheumatol (2019) 15(3):137–52. doi: 10.1038/s41584-018-0156-8
139. Wang Y, Coughlin JM, Ma S, Endres CJ, Kassiou M, Sawa A, et al. Neuroimaging of translocator protein in patients with systemic lupus erythematosus: a pilot study using [(11)C]DPA-713 positron emission tomography. Lupus (2017) 26(2):170–8. doi: 10.1177/0961203316657432
140. Makinde HM, Winter DR, Procissi D, Mike EV, Stock AD, Kando MJ, et al. A novel microglia-specific transcriptional signature correlates with behavioral deficits in neuropsychiatric lupus. Front Immunol (2020) 11:230. doi: 10.3389/fimmu.2020.00230
141. Qu M, Ni Y, Guo B, Feng X, Jiang Z. Lycopene antagonizes lead toxicity by reducing mitochondrial oxidative damage and mitochondria-mediated apoptosis in cultured hippocampal neurons. MedComm (2020) (2020) 1(2):228–39. doi: 10.1002/mco2.17
142. Rani L, Mondal AC. Emerging concepts of mitochondrial dysfunction in parkinson's disease progression: Pathogenic and therapeutic implications. Mitochondrion (2020) 50:25–34. doi: 10.1016/j.mito.2019.09.010
143. Areti A, Yerra VG, Naidu V, Kumar A. Oxidative stress and nerve damage: role in chemotherapy induced peripheral neuropathy. Redox Biol (2014) 2:289–95. doi: 10.1016/j.redox.2014.01.006
144. Anderson G, Rodriguez M, Reiter RJ. Multiple sclerosis: Melatonin, orexin, and ceramide interact with platelet activation coagulation factors and gut-Microbiome-Derived butyrate in the circadian dysregulation of mitochondria in glia and immune cells. Int J Mol Sci (2019) 20(21):5500. doi: 10.3390/ijms20215500
145. Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci (2015) 18(7):965–77. doi: 10.1038/nn.4030
146. Anderson G, Maes M. Gut dysbiosis dysregulates central and systemic homeostasis via suboptimal mitochondrial function: Assessment, treatment and classification implications. Curr Top Med Chem (2020) 20(7):524–39. doi: 10.2174/1568026620666200131094445
147. Yamawaki Y, Yoshioka N, Nozaki K, Ito H, Oda K, Harada K, et al. Sodium butyrate abolishes lipopolysaccharide-induced depression-like behaviors and hippocampal microglial activation in mice. Brain Res (2018) 1680:13–38. doi: 10.1016/j.brainres.2017.12.004
148. Dehhaghi M, Kazemi Shariat Panahi H, Guillemin GJ. Microorganisms' footprint in neurodegenerative diseases. Front Cell Neurosci (2018) 12:466. doi: 10.3389/fncel.2018.00466
149. Widner B, Sepp N, Kowald E, Kind S, Schmuth M, Fuchs D. Degradation of tryptophan in patients with systemic lupus erythematosus. Adv Exp Med Biol (1999) 467:571–7. doi: 10.1007/978-1-4615-4709-9_71
150. Dehhaghi M, Kazemi Shariat Panahi H, Heng B, Guillemin GJ. The gut microbiota, kynurenine pathway, and immune system interaction in the development of brain cancer. Front Cell Dev Biol (2020) 8:562812. doi: 10.3389/fcell.2020.562812
151. Venkatesan D, Iyer M, Narayanasamy A, Siva K, Vellingiri B. Kynurenine pathway in parkinson's disease-an update. eNeurologicalSci (2020) 21:100270. doi: 10.1016/j.ensci.2020.100270
152. Wincup C, Radziszewska A. Abnormal mitochondrial physiology in the pathogenesis of systemic lupus erythematosus. Rheum Dis Clin North Am (2021) 47(3):427–39. doi: 10.1016/j.rdc.2021.05.001
153. Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): Mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem (1998) 31(5):335–40. doi: 10.1016/S0009-9120(98)00045-9
154. Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab (2014) 19(3):373–9. doi: 10.1016/j.cmet.2014.01.001
155. Ji L, Xie W, Zhang Z. Efficacy and safety of sirolimus in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Semin Arthritis Rheumatol (2020) 50(5):1073–80. doi: 10.1016/j.semarthrit.2020.07.006
156. Oaks Z, Winans T, Caza T, Fernandez D, Liu Y, Landas SK, et al. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis Rheumatol (2016) 68(11):2728–39. doi: 10.1002/art.39791
157. Lui SL, Yung S, Tsang R, Zhang F, Chan KW, Tam S, et al. Rapamycin prevents the development of nephritis in lupus-prone NZB/W F1 mice. Lupus (2008) 17(4):305–13. doi: 10.1177/0961203307088289
158. Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheumatol (2006) 54(9):2983–8. doi: 10.1002/art.22085
159. Garcia RJ, Francis L, Dawood M, Lai ZW, Faraone SV, Perl A. Attention deficit and hyperactivity disorder scores are elevated and respond to n-acetylcysteine treatment in patients with systemic lupus erythematosus. Arthritis Rheumatol (2013) 65(5):1313–8. doi: 10.1002/art.37893
160. Doherty E, Oaks Z, Perl A. Increased mitochondrial electron transport chain activity at complex I is regulated by n-acetylcysteine in lymphocytes of patients with systemic lupus erythematosus. Antioxid Redox Signal (2014) 21(1):56–65. doi: 10.1089/ars.2013.5702
161. Suwannaroj S, Lagoo A, Keisler D, McMurray RW. Antioxidants suppress mortality in the female NZB x NZW F1 mouse model of systemic lupus erythematosus (SLE). Lupus (2001) 10(4):258–65. doi: 10.1191/096120301680416940
162. Procaccio V, Bris C, Chao de la Barca JM, Oca F, Chevrollier A, Amati-Bonneau P, et al. Perspectives of drug-based neuroprotection targeting mitochondria. Rev Neurol (Paris) (2014) 170(5):390–400. doi: 10.1016/j.neurol.2014.03.005
163. Buskiewicz IA, Montgomery T, Yasewicz EC, Huber SA, Murphy MP, Hartley RC, et al. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci Signal (2016) 9(456):ra115. doi: 10.1126/scisignal.aaf1933
164. He L. Metformin and systemic metabolism. Trends Pharmacol Sci (2020) 41(11):868–81. doi: 10.1016/j.tips.2020.09.001
165. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med (2015) 7(274):274ra18. doi: 10.1126/scitranslmed.aaa0835
166. Teng X, Brown J, Morel L. Redox homeostasis involvement in the pharmacological effects of metformin in systemic lupus erythematosus. Antioxid Redox Signal (2022) 36(7-9):462–479. doi: 10.1089/ars.2021.0070
167. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-Concept trial of metformin. Arthritis Rheumatol (2015) 67(12):3190–200. doi: 10.1002/art.39296
168. Yin Y, Choi SC, Xu Z, Zeumer L, Kanda N, Croker BP, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol (2016) 196(1):80–90. doi: 10.4049/jimmunol.1501537
169. Quinn BJ, Kitagawa H, Memmott RM, Gills JJ, Dennis PA. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab (2013) 24(9):469–80. doi: 10.1016/j.tem.2013.05.004
170. Peng X, Yang T, Liu G, Liu H, Peng Y, He L. Piperine ameliorated lupus nephritis by targeting AMPK-mediated activation of NLRP3 inflammasome. Int Immunopharmacol (2018) 65:448–57. doi: 10.1016/j.intimp.2018.10.025
171. Sun F, Geng S, Wang H, Wang H, Liu Z, Wang X, et al. Effects of metformin on disease flares in patients with systemic lupus erythematosus: post hoc analyses from two randomised trials. Lupus Sci Med (2020) 7(1):e000429v. doi: 10.1136/lupus-2020-000429
172. Rainsford KD, Parke AL, Clifford-Rashotte M, Kean WF. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology (2015) 23(5):231–69. doi: 10.1007/s10787-015-0239-y
173. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi: 10.1016/j.immuni.2012.10.020
174. Kim ML, Hardy MY, Edgington-Mitchell LE, Ramarathinam SH, Chung SZ, Russell AK, et al. Hydroxychloroquine inhibits the mitochondrial antioxidant system in activated T cells. iScience (2021) 24(12):103509. doi: 10.1016/j.isci.2021.103509
175. Schrezenmeier E, Dorner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol (2020) 16(3):155–66. doi: 10.1038/s41584-020-0372-x
176. Garcia-Ruiz I, Solis-Munoz P, Fernandez-Moreira D, Munoz-Yague T, Solis-Herruzo JA. Pioglitazone leads to an inactivation and disassembly of complex I of the mitochondrial respiratory chain. BMC Biol (2013) 11:88. doi: 10.1186/1741-7007-11-88
177. Zhao W, Thacker SG, Hodgin JB, Zhang H, Wang JH, Park JL, et al. The peroxisome proliferator-activated receptor gamma agonist pioglitazone improves cardiometabolic risk and renal inflammation in murine lupus. J Immunol (2009) 183(4):2729–40. doi: 10.4049/jimmunol.0804341
178. Zhao W, Berthier CC, Lewis EE, McCune WJ, Kretzler M, Kaplan MJ. The peroxisome-proliferator activated receptor-gamma agonist pioglitazone modulates aberrant T cell responses in systemic lupus erythematosus. Clin Immunol (2013) 149(1):119–32. doi: 10.1016/j.clim.2013.07.002
179. Chang S, Yin T, He F, Ding J, Shang Y, Yang J. CaMK4 promotes abortion-related Th17 cell imbalance by activating AKT/mTOR signaling pathway. Am J Reprod Immunol (2020) 84(6):e13315. doi: 10.1111/aji.13315
180. Koga T, Mizui M, Yoshida N, Otomo K, Lieberman LA, Crispin JC, et al. KN-93, an inhibitor of calcium/calmodulin-dependent protein kinase IV, promotes generation and function of Foxp3(+) regulatory T cells in MRL/lpr mice. Autoimmunity (2014) 47(7):445–50. doi: 10.3109/08916934.2014.915954
181. Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, et al. CaMK4-dependent activation of AKT/mTOR and CREM-alpha underlies autoimmunity-associated Th17 imbalance. J Clin Invest (2014) 124(5):2234–45. doi: 10.1172/JCI73411
182. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING with covalent small-molecule inhibitors. Nature (2018) 559(7713):269–73. doi: 10.1038/s41586-018-0287-8
183. Lopez-Pedrera C, Villalba JM, Patino-Trives AM, Luque-Tevar M, Barbarroja N, Aguirre MA, et al. Therapeutic potential and immunomodulatory role of coenzyme Q10 and its analogues in systemic autoimmune diseases. Antioxid (Basel) (2021) 10(4):600. doi: 10.3390/antiox10040600
184. Zinovkin RA, Zamyatnin AA. Mitochondria-targeted drugs. Curr Mol Pharmacol (2019) 12(3):202–14. doi: 10.2174/1874467212666181127151059
185. McHugh J. Targeting mitochondrial dysfunction in SLE. Nat Rev Rheumatol (2019) 15(12):700. doi: 10.1038/s41584-019-0332-5
186. Sharabi A, Tsokos GC. T Cell metabolism: New insights in systemic lupus erythematosus pathogenesis and therapy. Nat Rev Rheumatol (2020) 16(2):100–12. doi: 10.1038/s41584-019-0356-x
Keywords: systemic lupus erythematosus, mitochondrial reactive oxygen species, mitochondrial dysfunction, mitophagy, oxidative stress, mitochondria targeting therapeutics
Citation: Zhao L, Hu X, Xiao F, Zhang X, Zhao L and Wang M (2022) Mitochondrial impairment and repair in the pathogenesis of systemic lupus erythematosus. Front. Immunol. 13:929520. doi: 10.3389/fimmu.2022.929520
Received: 27 April 2022; Accepted: 28 June 2022;
Published: 25 July 2022.
Edited by:
Ryu Watanabe, Osaka Metropolitan University, JapanReviewed by:
Lele Zhu, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2022 Zhao, Hu, Xiao, Zhang, Zhao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Wang, dml2aWFuMDgxNTIwMDNAMTYzLmNvbQ==; Lidan Zhao, emhhb2xpZGFucHVtY0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.