 Xiaoqian Yan1†
Xiaoqian Yan1† Qi Xiao
Qi Xiao Xiaoyan Zhu
Xiaoyan Zhu- 1Department of Critical Care Medicine, The Affiliated Hospital of Qingdao University, Qingdao, China
- 2Department of Neurology, The Affiliated Hospital of Qingdao University, Qingdao, China
- 3Department of Neurology, The Affiliated Hiser Hospital of Qingdao University, Qingdao, China
Sepsis-associated encephalopathy (SAE) is a cognitive impairment associated with sepsis that occurs in the absence of direct infection in the central nervous system or structural brain damage. Microglia are thought to be macrophages of the central nervous system, devouring bits of neuronal cells and dead cells in the brain. They are activated in various ways, and microglia-mediated neuroinflammation is characteristic of central nervous system diseases, including SAE. Here, we systematically described the pathogenesis of SAE and demonstrated that microglia are closely related to the occurrence and development of SAE. Furthermore, we comprehensively discussed the function and phenotype of microglia and summarized their activation mechanism and role in SAE pathogenesis. Finally, this review summarizes recent studies on treating cognitive impairment in SAE by blocking microglial activation and toxic factors produced after activation. We suggest that targeting microglial activation may be a putative treatment for SAE.
Introduction
According to expert consensus guidelines in 2016, sepsis is defined as a life-threatening organ dysfunction caused by the dysfunctional response of the body to a pathogenic infection (1). Brain dysfunction caused by sepsis is not associated with direct brain infection (2, 3) and occurs in approximately 70% of patients with sepsis (4, 5). Sepsis-associated encephalopathy (SAE) is a cognitive impairment associated with sepsis resulting in diffuse brain dysfunction without direct central nervous system infection or structural brain damage. Sepsis - associated encephalopathy may be the first symptom of sepsis (6), which is likely to have occurred before patients with sepsis were admitted to general wards and intensive care units (ICU) (6, 7). In an established murine model of sepsis, acute encephalopathy followed by long-term cognitive impairment, could be observed in the surviving mice (8). This long-term cognitive impairment was observed in more than half of the survivors, and their quality of life was significantly decreased (9).
Although attention has been brought to the harm caused by SAE, its pathogenesis remains unclear. There is a lack of clear diagnostic criteria and effective treatment measures. The SAE diagnostic criteria are based on the detection of electroencephalogram (EEG) abnormalities (10), cognitive impairment, and neuroimaging evaluation (6, 7). SAE onset is characterized by changes in the mental state that may range from delirium to coma. A wide range of neurological changes can be observed, such as impaired cognitive function and consciousness, inattention, personality changes, and the onset of depressive mood. Some patients have occasional tremors, stiffness, and EEG deviations (10, 11). Abnormal EEG waveform is related to the presence and severity of encephalopathy (12). Neuroinflammatory processes involve damage to the blood-brain barrier, pathways of inflammatory mediators, and activation of microglias, which amplify this process by releasing more inflammatory factors (13). Sedation is not a viable treatment owing to the complex pathophysiology of SAE (4, 14).
Therefore, the pathogenesis and pathophysiology of SAE must be comprehensively investigated to develop effective treatment measures to reduce the incidence of SAE and improve the quality of life of survivors. In this review, we describe the pathogenesis of SAE and demonstrate that microglia are closely related to the pathogenesis of SAE. Simultaneously, we systematically investigated the critical role of microglia in SAE, focusing on the phenotype, state, and function of microglia. Furthermore, we summarized the effects of inhibiting microglial activation or toxic factors after activation to alleviate cognitive impairment in SAE in recent years. Thus, it is reasonable to envisage SAE treatment by targeting microglia.
Mechanism of sepsis-associated encephalopathy
The pathophysiological mechanisms of SAE, including various factors such as endothelial injury, inflammation, cerebral ischemia, blood-brain barrier (BBB) injury, and excitatory toxicity, remain unclear. Neuroinflammatory reactions, cerebral ischemic changes, and excitatory toxicity are common manifestations of severe sepsis (15). The comprehensive summary of several pathogenic mechanisms underlying SAE underscored the crucial role of microglia in these processes.
Neuroinflammation
During sepsis, inflammatory factors and signals reach different regions of the brain through various means, such as body fluids and nerves (16). Neuroinflammation plays a vital role in the pathogenesis of SAE, as uncontrolled inflammatory responses are the main manifestations of sepsis. Additionally, neuroinflammation is the primary cause of brain dysfunction and apoptosis in brain cells (17).
When pro-inflammatory cytokines enter the brain, microglia are activated to release nitric oxide (NO), active nitrogen, and glutamate, further causing structural damage and inflammation of the cell membranes. Increased peroxynitrite production in the brain under the influence of NO and free radicals may affect brain cell function, further affecting glial cells, neurons, and the blood-brain barrier, leading to SAE-induced brain dysfunction (18–21). In vivo pro-inflammatory mediators promote the expression of brain endothelial cell adhesion molecules and active transport across the BBB through specific receptors, further promoting the entry of neurotoxic and inflammatory factors into the brain tissue (22–24). Pro-inflammatory cytokines that affect the brain include interleukin-1α (IL-1α), IL-1β, IL-6 (25, 26), and high mobility group box-1 protein (HMGB1). Tumor necrosis factor (TNF) passes through the BBB via tumor necrosis factor receptor 1 (TNF-R1) and TNF-R2 (27). As TNF is directly associated with BBB destruction, brain edema, neutrophil infiltration, astrocyte proliferation, and brain cell apoptosis, TNF may be a crucial mediator of SAE, and these manifestations do not occur in mice lacking the TNFR gene (28). In animal models, TNFR, IL-6, and IL-1 receptors antagonists (IL-1RA) are inversely associated with memory, suggesting that inflammatory factors are closely associated with cognitive impairment (29). In the later stages of sepsis, HMGB1 levels significantly increase in different brain regions (30). Antagonistic HMGB1 in the blood and brain regions can improve SAE by preventing damage to the brain cells and restoring neural cognitive function. This suggests that inflammatory cytokines are vital for the pathogenesis of SAE (30–32).
Changes in cerebral ischemia and perfusion
In the pathological mechanism of sepsis, changes in blood flow and inflammatory responses may be critical steps in SAE development (33, 34). Impaired cerebral circulation during sepsis can lead to insufficient cerebral blood flow, which may be associated with electrophysiological and neurological changes (35, 36). Inadequate cerebral blood flow can lead to a cascade of cerebral ischemia which is controlled by three main processes: the reduction of oxygen and energy delivery (37), enhancement of stress signals (38), and activation of microglia (39). A decrease in energy supply can cause mitochondrial dysfunction, resulting in neuronal apoptosis and the release of pro-inflammatory cytokines (40). The increase of stress signals leads to the expression of adhesive molecules, which enhance the expression of matrix metalloproteinases (MMPs) signaling, which is related to an increase BBB permeability (41). Peripheral immune cells migrate to the brain and promote neuroinflammation (42, 43). Microglia can protect neurons to a certain extent as well as produce pro-inflammatory factors that cause neuronal apoptosis. Hemodynamic changes precede cognitive impairment and structural changes in the brain (14, 44, 45). Studies have shown that a continuous decrease in cerebral blood flow in patients with septic shock leads to impaired self-regulation and is associated with the onset of delirium (46). Neurovascular dysfunction is highly associated with decreased language and memory (47, 48). Maintaining the integrity of blood vessels in the brain is vital for cognitive function (34).
Neurotransmitter dysfunction
During sepsis, the dopaminergic, β-adrenergic, GBAB receptors, and cholinergic nervous systems are impaired to a certain extent (15). An imbalance between the dopaminergic and cholinergic nerves is associated with cognitive impairment (49). This is related to the onset of SAE. Increased release of neurotransmitters, such as glutamate and acetylcholine, and reduced reuptake is one of the causes of neurotoxicity (50). Glutamate plays a role in neuronal apoptosis via excitatory toxicity (51). NO production may be related to neurotransmission disorders (52) or the excessive release of neurotoxic amino acids, such as ammonia, tyrosine, and tryptophan, into circulation by the liver and muscles during sepsis (53). Microglia express multiple neurotransmitter receptors, including glutamate, tyrosine, and acetylcholine. They also release glutamate, which induce neuronal apoptosis. Microglia communicate with each other and work together to regulate neuronal function.
Disruption of the blood-brain barrier
The entry of aromatic amino acids into the brain through the damaged BBB leads to increased uptake of these amino acids by the brain (54), which further causes SAE, leading to an altered mental state (55). The BBB plays a vital role in stabilizing the brain milieu and maintaining adequate neural function by regulating the movement of ions and fluids between the blood and brain, thereby providing certain nutrients to the brain (56–58). The barrier also prevents external white blood cells from entering the central nervous system and playing an immunogenic role in the brain (59). Increased expression of complement activation, inflammatory cytokines (60, 61), and adhesion molecules further increase BBB destruction and helps white blood cells enter the brain, enhancing neuroinflammation.
During sepsis, inflammatory cytokines enter the central nervous system through various pathways, including receptor-mediated transcellular action, transcellular diffusion, and carrier proteins (62, 63).Proinflammatory cytokines such as IL-1β, IL-6, and lipopolysaccharide, reactive oxygen species, and NO act on BBB to alter brain function, resulting in the disruption of brain homeostasis and changes in BBB permeability (64–66). These inflammatory factors enter the brain tissue and activate microglia (49). Prior to the change in BBB permeability, microglia begin to migrate to the cerebrovascular site and respond to the surrounding inflammatory factors, which play a certain protective role in the BBB in the early stage (67). However, further inflammation leads to a more active microglial phenotype, increased phagocytosis of astrocyte terminal feet, and increased BBB permeability. Microglia can decrease paracellular connexins expression and further enhance BBB permeability.
Overall, the pathogenesis of SAE is not caused by a single factor but by the joint action of multiple factors. (Figure 1) Microglial activation is essential in the pathogenesis of SAE. It is involved in almost every stage of SAE pathogenesis. Moreover, it interacts with various central nervous system components and plays a vital role in maintaining brain function and integrity. Furthermore, it is closely associated with cognitive dysfunction in central nervous system diseases; therefore, we will focus on the relationship between microglia and cognitive dysfunction in different nervous system diseases.
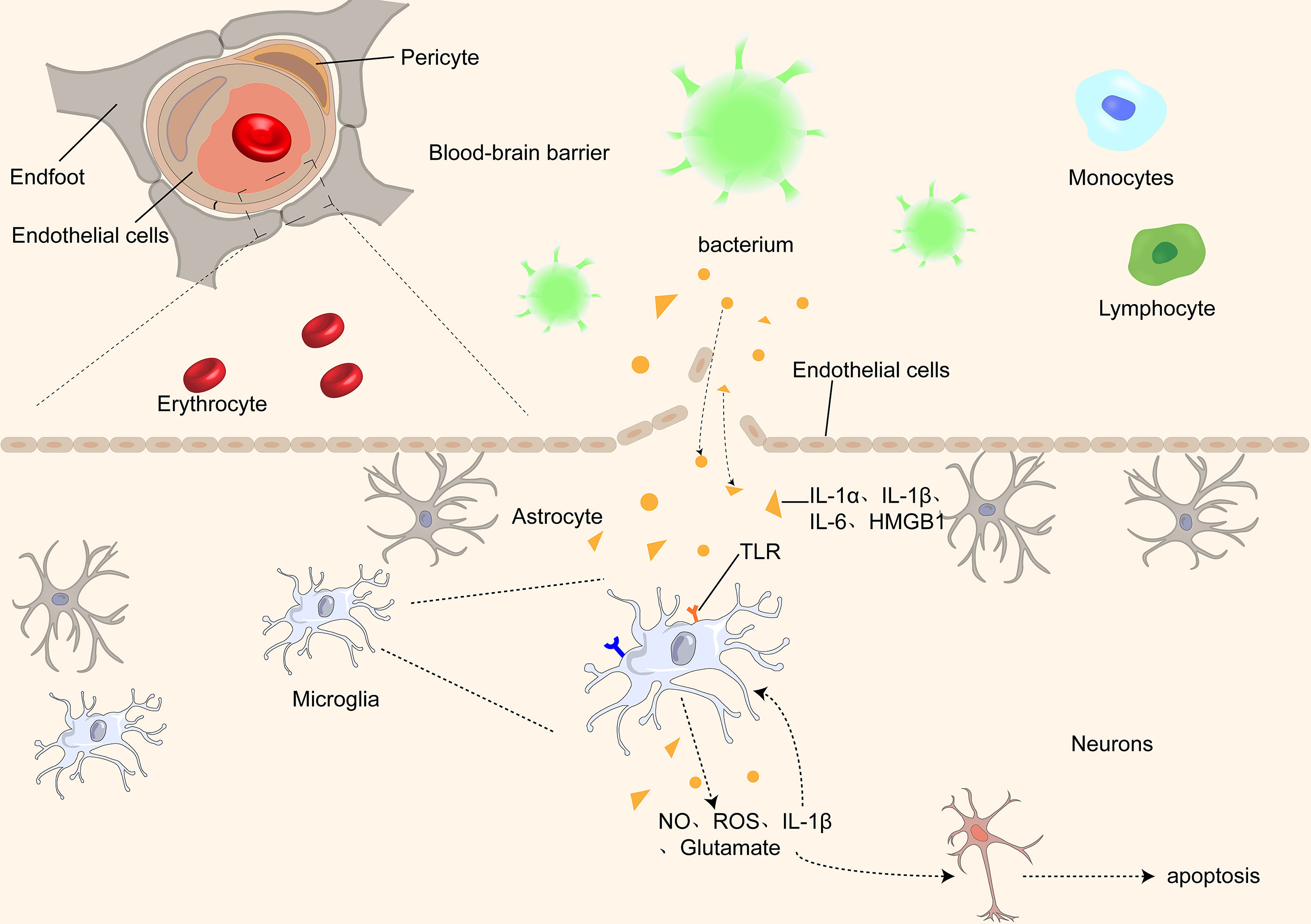
Figure 1 Proinflammatory cytokines, such as IL-1α, IL-1β, IL-1, and HMGB1, pass through the BBB through different receptors and activate microglia, which further damage the BBB by releasing inflammatory factors, causing an inflammatory cascade. At the same time, microglia release ROS and NO, causing damage and even apoptosis of neurons in the brain.
Function, phenotype, and the role of microglia in SAE
Microglia play a vital role and are closely related to SAE pathogenesis. Therefore, we focused on the function, phenotype, and phenotypic transformation of microglia. Finally, the activation of microglia and their role in SAE development are highlighted in this section.
Function of microglia
Microglia are the primary immune cells in the brain parenchyma and differ from other macrophages in the brain (68). Studies of ApoE4 alleles showed that microglia play an important role in neurodegenerative diseases. E4-expressing microglia showed higher innate immune reactivity after LPS treatment, significantly reduced neuronal activity, and secreted more elevated levels of TNF when co-cultured with neurons (69). Microglia perform some critical functions in the brain (70) synaptic genesis, modification, and plasticity changes (71); (2) detection of local steady state (72); (3) immune function, including phagocytosis, antigen presentation, secretion of anti-inflammatory (such as IL-10, IL-4) and pro-inflammatory (such as IL-1β, IL-6) cytokines, and regulation of neuronal apoptosis (73); (4) regulation of myelin sheath (74); (5) neurotrophic support (75); and (6) communicate with astrocytes to regulate these functions (76). (Figure 2) Furthermore, microglia are involved in synapses and neurogenesis, as well as in the removal of unwanted neuronal and other cellular waste. Monitoring the changes in the brain microenvironment suggested that microglia alter both in shape and function through microglial activation (77).
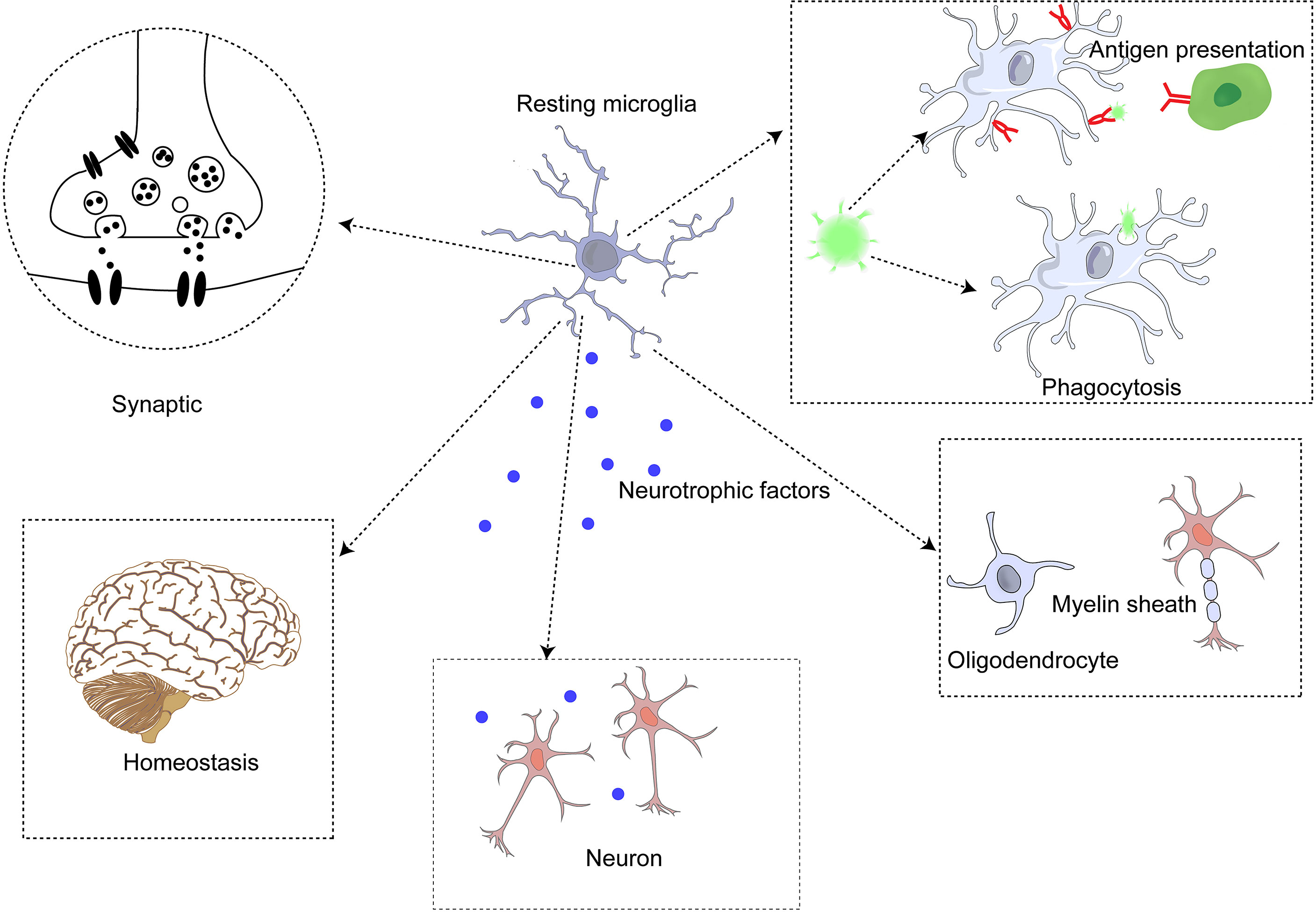
Figure 2 Microglia perform functions in the central nervous system.
Microglia exist in resting and active states (78). In dynamic homeostasis, microglia secrete neurotrophic factors and monitor the microenvironment with scavenger receptors (SR) to remove unnecessary cell debris and apoptotic neurons, thus further maintaining the homeostasis and connection of neuronal functions (72, 79). Microglia in dynamic equilibrium are rod-shaped with many forked processes (73). Although they are in equilibrium, they still monitor the state of their microenvironment and surrounding tissues to acutely alert them to abnormal signals. The morphology of microglia changes significantly after activation, the cell body is enlarged, and the process is shortened (68). The status of microglia can be assessed by their movement and morphology in different environments (77).
Microglial phenotypes
Under different microenvironments, microglia undergo produce phenotypic changes, including the M1 pro-inflammatory, M2 anti-inflammatory and other phenotypes (75, 80). M1 microglia can lead to neuroinflammation and neuronal apoptosis, whereas M2 microglia can protect neurons and repair brain tissues. M1 is generally activated by interferon-γ (IFN-γ) and lipopolysaccharide (LPS) to produce IL-6, and CC-chemokine ligand 2 (CCL2), which ultimately leads to neuronal damage and even apoptosis (68, 81). M2 microglia are generally induced by anti-inflammatory cell mediators, including IL-13 and IL-3. It produces IL-10 and neurotrophic factors to repair brain tissue and neurons (68, 75). (Figure 3)
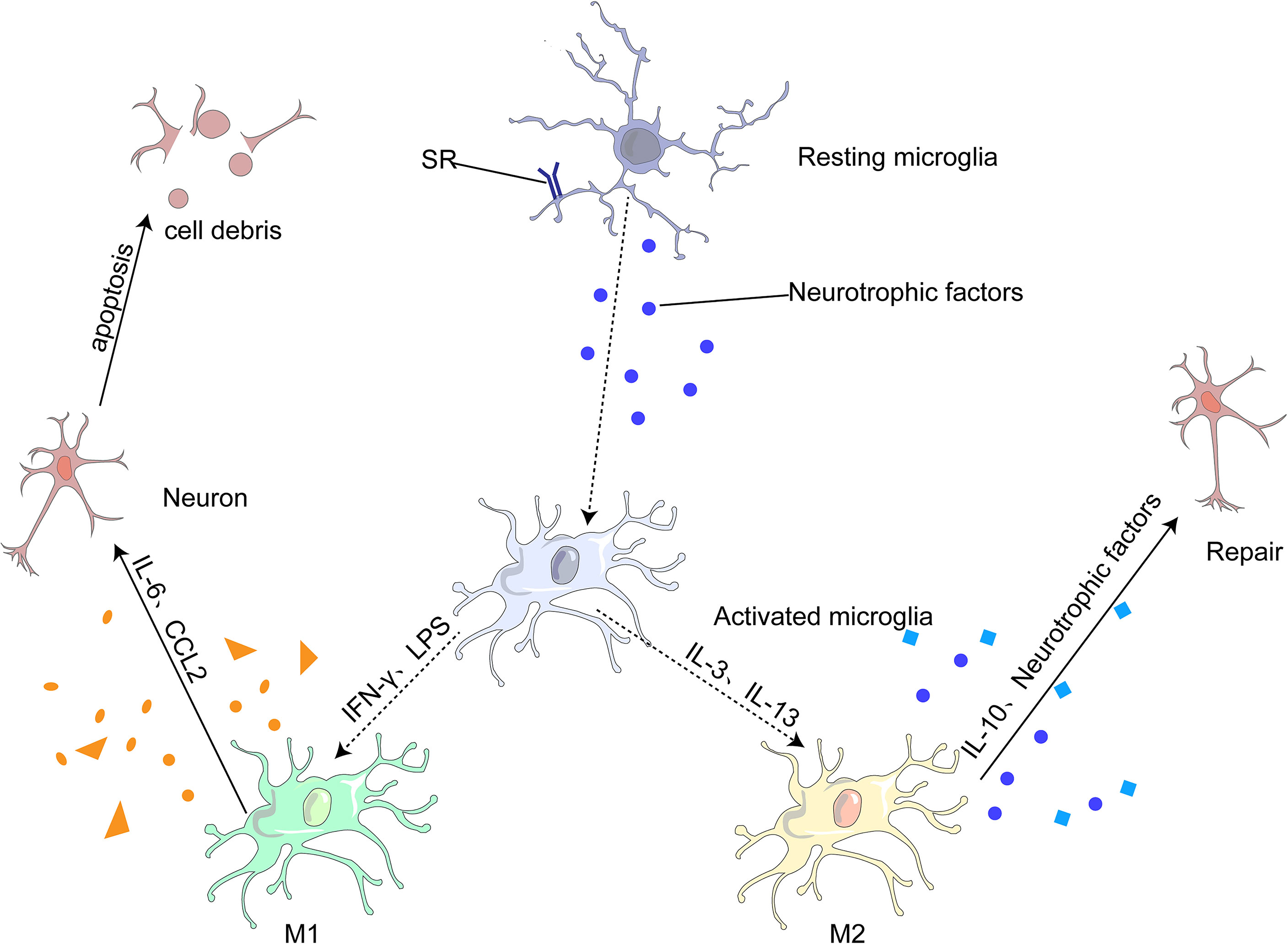
Figure 3 Most microglia can be divided into two opposite types: classical (M1) or alternative (M2). M1 microglia release inflammatory mediators, such as IL-6 and CCL2, which induce inflammation and neurotoxicity. M2 microglia release anti-inflammatory mediators, such as IL-10 and neurotrophic factor, which induce anti-inflammatory and neuroprotective effects.
Microglia can change their phenotype from M1 to M2 in the following ways (82): (1) different signaling pathways, such as the Toll-like receptor signaling pathway (83), Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway (84, 85), NF-κβ signaling pathway (86, 87), and mitogen-activated protein kinase signaling pathway (88); and (2) regulatory transcription factors, such as PPAR-γ (89), which exert anti-inflammatory effects by inhibiting NF-κβ, STAT, and other transcription factors. These transcription factors are closely associated with microglial polarization into type M1, a transcription pro-inflammatory factor. Inhibition of these factors increased the polarization of microglia from M1 to M2; (3) regulation of microglial surface receptors, such as TREM2 (90)and α7 nicotinic acetylcholine receptor (α7nAChR) (91); (3) regulation of different cytokines, such as IL-4 (92), IL-10, TGF-β (75), neurotrophic factor; (5; change in channels (93); (6) bioactive compounds and certain drugs (94–96). The transformation of microglia from the M1 to M2 phenotype is not caused by a single process described in this section but by a combination of several different mechanisms.
Microglial activation damages nearby healthy brain tissue, while the affected nerve tissue may secrete substances that, in turn, promote microglial activation. The activation of microglia by chronic inflammation in the human body increases the occurrence of synapses, resulting in enhanced phagocytosis and neuronal apoptosis (97). Microglia, supported by neurotrophic factors, contribute to the formation of synapses associated with learning and memory, which are implicated in cognitive function (98).
Microglia in CNS-related cognitive impairment
Microglial dysfunction, which affects the structure and function of the brain, is associated with almost all brain diseases, including neurodegenerative diseases (such as Alzheimer’s disease [AD], stroke, and Parkinson’s disease), as well as inflammatory brain diseases (99, 100), that can cause long-term cognitive impairment (97).
Microglia are carriers of amyloid precursor protein and promote Aβ production in rat brain tissue, which is closely related to AD (101). Microglia can bind to Aβ by expressing the corresponding receptors CD36, TLR2, and TLR4, induce the release of IL-1β, and trigger neuroinflammation (102). In AD mouse models, TLR and IL-1β deficiency can reduce Aβ deposition and prevent cognitive impairment (102). Datta et al. believed that in neurodegeneration after stroke, an increase in misfolded proteins and microglial activation can be found in the thalamus, leading to neuronal loss and further deterioration of cognitive function (103). Activation of microglia and increased pro-inflammatory factors are also important mechanisms in Parkinson’s disease (PD) (104, 105). Microglia were significantly active in the substantia nigra pars compacta in the PD murine model, and the secretion of inflammatory cytokines in this region was also significantly enhanced. These inflammatory cytokines can cause neuronal damage and even apoptosis (106) and lead to the degeneration of dopaminergic neurons (107), resulting in cognitive dysfunction. Simultaneously, the function of microglia declines with the increase age (68). The immune receptor expression is increased along with the release of more neurotoxic substances, which goes hand to hand with neurodegenerative diseases (108–110). These studies suggest that the activation of microglia is one of the crucial links in the pathogenesis of AD, stroke, and PD (105, 106, 111). Notably, we also observed that microglial activation was strongly associated with cognitive impairment in patients.
Microglia play a vital role in the pathogenesis of neurodegenerative diseases and in infectious encephalopathy. In studies of viral encephalitis diseases (77) (such as Japanese encephalitis, West Nile and Zika viruses), INF-γ induced microglial activation, then produced different inflammatory signals such as IL-1, and IL-6, which can directly cause neurotoxic lesions. It can also lead to cognitive impairment and neuronal over-firing (112–114).
Treatment of intracranial malignant tumors targets cancer cells and leads to the activation of microglia, which changes from a neurotrophic to neurotoxic state (115). In a glioma mouse model, cognitive impairment is associated with microglial activation induced by repeated cranial irradiation rather than the tumor itself (116). These findings suggest that microglial activation can lead to cognitive dysfunction in central nervous system diseases.
Microglia in SAE
After activation, microglia can cause neuron injury or even apoptosis by releasing inflammatory mediators, reactive oxygen species, neurotransmitters and other substances, which play an important role in the pathogenesis of SAE (Fig 1). Chemokines and inflammatory cytokines secreted by microglia can help the brain defend against inflammatory responses, regulate the migration of white blood cells, and facilitate the repair of neurons in brain tissue (117). However, long-term microglia activation has a minimal protective effect on neurons and further worsen the inflammatory response in the brain. The most common SAE models are divided into two types: intraperitoneal injection of LPS and caecal ligation perforation (CLP) (118, 119). An increase in the number of ED-1-positive microglia was observed 24 h after establishing an SAE mouse model by LPS injection. They are distributed around the cerebrovascular system and around the parenchyma. Most microglia are reportedly distributed around cerebral vessels 4h after LPS injection. The extent of microglial activation was time-dependent, and the highest microglia numbers were observed at 8h in all brain regions (20, 120). The production of TNF was induced by LPS stimulation in vitro (121). LPS strongly stimulates microglia activation, and disordered activation of microglia during SAE may lead to further deterioration of already damaged brain tissue (119, 122). The establishment of CLP model showed a significant increase in the size and number of microglial processes by immunofluorescence (121). Simultaneously, the SAE model established by CLP can also cause microglial overactivation and neuronal pyroptosis, aggravating brain tissue destruction and cognitive dysfunction (118).
Peripheral circulating inflammatory factors induce immune-related responses in the central nervous system through various pathways, including inflammatory mediators (42, 49), adjacent cells (123, 124), and neurotransmitters (49, 125), which may be strongly linked to the role of microglia in sepsis. In particular, microglia are activated through the pathways discussed earlier, resulting in neuronal damage and even apoptosis, further leading to SAE.
As described above, microglial activation is strongly associated with the occurrence of SAE. Microglia can recognize various damage signals, including microorganisms, complements, and cytokines. They are thought to injury to the central nervous system (126). Microglia are activated by bacteria and other substances through Toll-like receptors (TLR-2, TLR-4, and TLR-9) and nucleotide-binding oligonucleotide 2 (NOD2) (42). Simultaneously, inflammatory cytokines such as IL-1β and IL-6 activate microglia through the damaged BBB, leading to brain cell destruction and even apoptosis during sepsis (49). IL-17A/IL-17R signaling pathway forms a vicious inflammation cycle and amplifies the role of inflammation in the brain by promoting the secretion of inflammatory factors by microglia and intensifying IL-17A secretion by immune cells (42, 125). Adjacent cells, such as astrocytes, endothelial cells, and Th1/Th17 cells, have regulatory effects on microglia. Astrocytes attach to vascular endothelial cells or via meningeal cells in peripheral blood vessels and express multiple cytokine receptors that enable astrocytes to respond to inflammation (123). Microglia are activated by bacteria and other substances through cytokines secreted by astrocytes, such as granulocyte colony-stimulating factor (G-CSF) and CCL11. G-CSF is a microglial growth factor, and CCL11 can promote microglial migration to inflammatory sites, causing microglia to produce reactive oxygen species, resulting in the destruction and even apoptosis of brain cells (123). Astrocytes activate microglia and induce apoptosis of brain cells as well as produce anti-inflammatory substances that inhibit inflammation in the central nervous system (127). CX3CR1 is a chemokine and transmembrane protein that promotes leukocyte migration in monocytes, dendritic cells, and microglia (128). CX3CR1 is a receptor for CX3CL1; the interaction between CX3CL1 in neurons and CX3CR1 in microglia mediates the functional phenotype of microglia and its overactivation under inflammatory conditions. Increased CX3CL1 expression on endothelial cells activates endothelial cells and promote increased leukocyte adhesion, microthrombus formation, coagulation disorders, and metastasis of microglia to inflammatory sites (124, 125). Th1/Th17 cells produce large amounts of IL-17A in the brain, inducing microglial activation and prolonging inflammatory processes (43). Microglia express receptors for various neurotransmitters, including glutamate and acetylcholine, and communicate with each other to maintain normal neuronal function (129). During sepsis, an imbalance in the expression of different types of neurotransmitters, such as glutamate and acetylcholine, affects the function of microglia and neurological function. Activated microglia can produce several inflammatory factors around the cerebrovascular or cerebral solid, leading to an enhanced brain immune response, further causing neuronal damage, loss of function, and even apoptosis (130, 131).
Previous studies have shown that in case-control studies of patients who died of sepsis, CD68 expression was significantly elevated in the cortex of the experimental group compared to that in the control group, and deformed microglial cells were also observed (132, 133). Activated microglia can induce neurological dysfunction and memory loss in patients with sepsis by releasing pro-inflammatory cytokines and including the expression of related enzymes (134). Microglia regulate neuronal function through neurotransmitters levels. In a postmortem case-control study of patients with delirium, the expression of microglial markers CD68 and HLA-DR were significantly increased compared to that in the controls, suggesting that microglial activation may be associated with delirium (135). Microglial activation has also been detected during sepsis (136–138). During sepsis, intraventricular injections of minocycline inhibit microglia and reduce acute brain injury, inflammation, and long-term cognitive impairment in survivors (139).
Therefore, when activated microglia sense surrounding injury signals, the cells may be more prone to release several inflammatory cytokines, resulting in cognitive dysfunction and exacerbation of SAE. Further exploration of the role of microglial activation in SAE cognitive dysfunction can deepen our understanding of the pathogenesis of SAE and may provide evidence for the treatment of SAE.
Targeting microglia to treat SAE cognitive impairment
The activation of microglia, as a central link in the development of cognitive deficits in sepsis-associated encephalopathy, could represent an effective therapeutic target (140). Several studies have shown that blocking microglia activation or alleviating a series of neurotoxic reactions after microglia activation can improve neurological symptoms and long-term cognitive dysfunction to a certain extent (42, 139, 141). Based on this concept, we have summarized the potential value of targeting microglia in diagnosing and treating of SAE cognitive impairment.
Prevents microglial activation
Inflammatory mediators, neurotransmitters, and intercellular interactions with surrounding cells accelerate microglial activation (125). Once activated, inflammatory factors, reactive oxygen species, NO, prostaglandins, and neurotoxic glutamate continue to act on the neighboring neurons, causing neuronal damage and, ultimately, cognitive impairment (15, 142). Therefore, early identification and prevention of microglial activation are crucial.
Aseptic neuroinflammation caused by circulating inflammatory mediators in the brain has long been accepted as the pathogenesis of SAE. However, a study on the intestinal flora in septic encephalopathy found that in the absence of evident sepsis, the bacteria temporarily translocate to the brain and cause microglial activation and neuroinflammatory responses (121). Activation of the host immune mechanism may cause persistent cognitive dysfunction. Therefore, early attention to specific microbiota may later improve cognitive dysfunction (121, 143). A recent study by Zhang et al. (143) demonstrated for the first time the exact relationship between gut microbiota and its metabolite butyric acid and SAE. SAE mouse models of different severity were constructed by CLP and fecal microbiota transplantation(FMT) was performed on sterile mice, confirming the significant role of the gut-brain axis in SAE. In particular, butyrate has been found to reduce oxidative stress response and nerve damage through the GPR109A receptor on microglia and the Nrf2/HO-1 signaling pathway. Furthermore, recent sequencing analysis of microglia and brain endothelial cells revealed endotheliitis as the earliest microglial activation event. Microglia are activated by cerebral endothelial cells (CECs)-derived inflammatory mediators. Therefore, early recognition and blocking of CECs activation can also reduce microglial activation and subsequent reactions (144).
Sirtuins have been widely studied as long-lived proteins (145, 146). Sirt3 is a mitochondrial enzyme that plays a vital role in the metabolic cycle and participates in the regulation of apoptosis. High levels of Sirt3 were detected in LPS-induced mouse microglia. In contrast, the Sirt3 levels decreased after treatment with Single-wall carbon nano horns (SWNHs), which delayed the mitosis of microglia and promoted their apoptosis. Therefore, SWNHs may be a therapeutic approach to inhibit microglial activation by blocking Sirt3 (147). Another study used resveratrol (a SIRT1 activator) to induce SIRT1 overexpression, which plays an important role in inflammatory regulation, inhibiting microglial activation and proliferation, as well as inflammatory processes in SAE mice (148). Shi et al. (149) observed that SIRT1 regulates oxidative stress in hypoxic and glucose-deficient hippocampal neurons and has a protective effect on nerve cells after oxygen and glucose deprivation (OGD). These studies have revealed the crucial role of SIRT1 in microglial activation and neuronal protection.
TLR4, an immune recognition receptor, is highly expressed in LPS-induced microglia and is closely associated with neuroinflammation through a cascade of downstream pathways after activation (150, 151). The exposure TLR4 to G+ bacterial LPS activates a series of downstream proteins, one of which ultimately activates NF-κβ, initiating transcription and producing a pro-inflammatory effect. In septic mice treated with sodium butyrate (NaB), activation of hippocampal microglia and secretion of inflammatory factors were reduced, and improvements in neuroinflammation and anxiety were observed. The mechanism underlying these benign results is due to NaB antagonization of TLR4 activation, consequently inhibiting subsequent nuclear transcription (152). Protein kinase C-interacting protein (PICK1) is the most abundant protein in the brain and plays a unique role in the progression of many neurological diseases. In addition, PICK1 is involved in several inflammatory pathological processes (153, 154). Wang et al. observed overactivated microglia, TLR4 pathway, and PICK1/TLR complex in an SAE mouse model with a PICK1 knockout. However, PICK1 levels were not significantly altered in LPS-induced sepsis mice. They demonstrated for the first time that PICK1 plays a protective role in SAE by forming a complex with TLR4 (144). Another study used electroacupuncture to improve neural function, possibly by increasing the PICK1/TLR4 complex in microglia to provide protection (155).
IL-17A is reportedly involved in this acute cycle of microglial activation. Adjacent cells, such as CD4+T cells and Th17 cells, secrete IL-17A, and act on the surface receptors of microglia, thereby activating them. Activated microglia secrete several inflammatory factors, including IL-17A, which undoubtedly aggravate microglial activation (156, 157). By injecting recombinant IL-17A, anti-IL-17A antibody, and anti-IL-17R antibody into CLP mouse models, Ye et al. (42) revealed the potential role of IL-17A/IL-17R blockade in preventing SAE.
Blocking the activation of SAE microglia by blocking inflammatory factors, signaling pathways, and other pathways is of great significance for the early prevention of SAE cognitive impairment. Further studies are warranted in this regard.
Reduce neuronal injury after activation of microglia
Once microglia are activated, their neurotoxic effects accelerate the progression of SAE and are strongly associated with long-term cognitive impairment (140). Survivors of sepsis-associated encephalopathy are at a higher risk of developing dementia, and long-term cognitive impairment is considered a transitional state before the onset of dementia (158). Therefore, improving cognitive impairment is essential for the outcome of patients with SAE, and this process can be achieved by reducing the neurotoxicity of activated microglia. Based on this pathogenesis, we believe that inflammation, oxidative stress, apoptosis, and immune response can reduce microglial neurotoxicity and improve cognitive impairment. Memory disorders are mainly dominated by neuroinflammation in the hippocampus, and IL-1 β levels are negatively correlated with the severity of memory disorders (159). Activated microglia can release many cytokines including IL-1β. Therefore, regulating the inflammatory response after activation of microglia may play a role in improving a range of cognitive disorders such as memory impairment.
Water maze and fear conditioning tests were performed on SAE mice constructed by cecal ligation and perforation (CLP). Impaired learning and memory functions were observed, whereas C-X-C chemokine receptor type (CXCR) 5 expression was upregulated. When CXCR5 was knocked out, cognitive deficits and M1 polarization were reversed, and similar results were observed in primary microglia in vitro. Downregulation of CXCR5 reduces the pro-inflammatory microenvironment in the hippocampus, which may be a potential therapeutic target (160). Previous studies have shown that inflammasome activation is essential in SAE, with NOD, LRR and pyrin domain-containing protein 3 (NLRP3) inflammasome being the most representative. Resveratrol inhibits the NLRP3/IL-1β axis of microglia, reduces hippocampal inflammation, and improves spatial memory in SAE mice (161). In another study, treatment with ethyl pyruvate significantly reduced cognitive impairment in CLP mice by inhibiting NLRP3 and inducing IL-1β cleavage (162). In conclusion, inhibition of NLRP3 can potentially improve cognitive impairment in SAE.
Stanniocalcin-1 (STC-1), a neuroprotective protein, plays an anti-inflammatory and antioxidant role by inducing the uncoupling proteins (UCPs). Injection of recombinant human STC-1 (rhSTC1) inhibited microglia production of pro-inflammatory factors and improves cognitive impairment (163). Moreover, the positive effects of NOS2 gene deletion and propofol inhibition of NMDA receptors on cognitive impairment in sepsis-associated encephalopathy are achieved by inhibiting microglial inflammation (134, 164).
Activating the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway is beneficial to SAE. In addition to mediating the inactivation of NLRP3 and playing an anti-inflammatory role, Nrf2 acts as an endogenous antioxidant and plays a neuroprotective role (165). H2 protects neurons from activated microglia by upregulating the Nrf2 pathway and antagonizing oxidative stress (166). In an experiment on the effect of ginsenoside on SAE, it was found that ginsenoside inhibited oxidative stress and apoptosis, and the mechanism was related to the upregulation of Nrf2 and heme oxygenase-1(HO-1) (167).
Collectively, reducing the toxic effects of microglial activation is of great significance in treating cognitive impairment in sepsis-associated encephalopathy. It provides a new idea to design a treatment for SAE cognitive impairment by targeting various pathways in microglia.
Conclusion
SAE is associated with increased mortality in patients with sepsis and reduced quality of life in survivors; therefore, further research is required to treat cognitive impairment in SAE. In this review, we have provided a comprehensive overview of the different functions and phenotypes of microglia, and their role in SAE pathogenesis. Notably, we summarized recent advances in treating of cognitive impairment in SAE based on microglial activation and the associated toxic effects of microglia activation. We are confident that further research on microglia will provide novel insights into the treatment of SAE.
Author contributions
XY, KY and QX were involved in reading and editing the manuscript and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315(8):801–10. doi: 10.1001/jama.2016.0287
2. Opal SM. Endotoxins and Other Sepsis Triggers. Contrib to Nephrol (2010) 167:14–24. doi: 10.1159/000315915
3. Kikuchi DS, Campos ACP, Qu H, Forrester SJ, Pagano RL, Lassegue B, et al. Poldip2 Mediates Blood-Brain Barrier Disruption in a Model of Sepsis-Associated Encephalopathy. J Neuroinflammation (2019) 16(1):241. doi: 10.1186/s12974-019-1575-4
4. Andonegui G, Zelinski EL, Schubert CL, Knight D, Craig LA, Winston BW, et al. Targeting Inflammatory Monocytes in Sepsis-Associated Encephalopathy and Long-Term Cognitive Impairment. JCI Insight (2018) 3(9):e99364. doi: 10.1172/jci.insight.99364
5. Young GB. Encephalopathy of Infection and Systemic Inflammation. J Clin Neurophys (2013) 30(5):454–61. doi: 10.1097/WNP.0b013e3182a73d83
6. Helbing DL, Bohm L, Witte OW. Sepsis-Associated Encephalopathy. CMAJ (2018) 190(36):E1083. doi: 10.1503/cmaj.180454
7. Gofton TE, Young GB. Sepsis-Associated Encephalopathy. Nat Rev Neurol (2012) 8(10):557–66. doi: 10.1038/nrneurol.2012.183
8. Mina F, Comim CM, Dominguini D, Cassol OJ Jr., Dall Igna DM, Ferreira GK, et al. Il1-β Involvement in Cognitive Impairment After Sepsis. Mol Neurobiol (2014) 49(2):1069–76. doi: 10.1007/s12035-013-8581-9
9. Chung HY, Wickel J, Brunkhorst FM, Geis C. Sepsis-Associated Encephalopathy: From Delirium to Dementia? J Clin Med (2020) 9(3):703. doi: 10.3390/jcm9030703
10. Mazeraud A, Righy C, Bouchereau E, Benghanem S, Bozza FA, Sharshar T. Septic-Associated Encephalopathy: A Comprehensive Review. Neurotherapeutics (2020) 17(2):392–403. doi: 10.1007/s13311-020-00862-1
11. Hosokawa K, Gaspard N, Su F, Oddo M, Vincent JL, Taccone FS. Clinical Neurophysiological Assessment of Sepsis-Associated Brain Dysfunction: A Systematic Review. Crit Care (2014) 18(6):674. doi: 10.1186/s13054-014-0674-y
12. Sutter R, Stevens RD, Kaplan PW. Clinical and Imaging Correlates of EEG Patterns in Hospitalized Patients With Encephalopathy. J Neurol (2013) 260(4):1087–98. doi: 10.1007/s00415-012-6766-1
13. Mazeraud A, Bozza FA, Sharshar T. Sepsis-Associated Encephalopathy is Septic. Am J Respir Crit Care Med (2018) 197(6):698–9. doi: 10.1164/rccm.201712-2593ED
14. Catarina AV, Branchini G, Bettoni L, De Oliveira JR, Nunes FB. Sepsis-Associated Encephalopathy: From Pathophysiology to Progress in Experimental Studies. Mol Neurobiol (2021) 58(6):2770–9. doi: 10.1007/s12035-021-02303-2
15. Heming N, Mazeraud A, Verdonk F, Bozza FA, Chretien F, Sharshar T. Neuroanatomy of Sepsis-Associated Encephalopathy. Crit Care (2017) 21(1):65. doi: 10.1186/s13054-017-1643-z
16. Castro LVG, Goncalves-de-Albuquerque CF, Silva AR. Polarization of Microglia and its Therapeutic Potential in Sepsis. Int J Mol Sci (2022) 23(9):4925. doi: 10.3390/ijms23094925
17. Schwalm MT, Pasquali M, Miguel SP, Dos Santos JP, Vuolo F, Comim CM, et al. Acute Brain Inflammation and Oxidative Damage are Related to Long-Term Cognitive Deficits and Markers of Neurodegeneration in Sepsis-Survivor Rats. Mol Neurobiol (2014) 49(1):380–5. doi: 10.1007/s12035-013-8526-3
18. Danielski LG, Giustina AD, Goldim MP, Florentino D, Mathias K, Garbossa L, et al. Vitamin B(6) Reduces Neurochemical and Long-Term Cognitive Alterations After Polymicrobial Sepsis: Involvement of the Kynurenine Pathway Modulation. Mol Neurobiol (2018) 55(6):5255–68. doi: 10.1007/s12035-017-0706-0
19. Zhu SZ, Huang WP, Huang LQ, Han YL, Han QP, Zhu GF, et al. Huperzine A Protects Sepsis Associated Encephalopathy by Promoting the Deficient Cholinergic Nervous Function. Neurosci Letters (2016) 631:70–8. doi: 10.1016/j.neulet.2016.07.009
20. Semmler A, Okulla T, Sastre M, Dumitrescu-Ozimek L, Heneka MT. Systemic Inflammation Induces Apoptosis With Variable Vulnerability of Different Brain Regions. J Chem Neuroanatomy (2005) 30(2-3):144–57. doi: 10.1016/j.jchemneu.2005.07.003
21. Sharshar T, Gray F, Lorin de la Grandmaison G, Hopkinson NS, Ross E, Dorandeu A, et al. Apoptosis of Neurons in Cardiovascular Autonomic Centres Triggered by Inducible Nitric Oxide Synthase After Death From Septic Shock. Lancet (London England) (2003) 362(9398):1799–805. doi: 10.1016/S0140-6736(03)14899-4
22. He H, Geng T, Chen P, Wang M, Hu J, Kang L, et al. NK Cells Promote Neutrophil Recruitment in the Brain During Sepsis-Induced Neuroinflammation. Sci Rep (2016) 6:27711. doi: 10.1038/srep27711
23. Moinuddin A, Morley JE, Banks WA. Regional Variations in the Transport of Interleukin-1alpha Across the Blood-Brain Barrier in ICR and Aging SAMP8 Mice. Neuroimmunomodulation (2000) 8(4):165–70. doi: 10.1159/000054814
24. Ding H, Cao XY, Ma XG, Zhou WJ. Endothelial Cell Injury With Inflammatory Cytokine and Coagulation in Patients With Sepsis. World J Emergency Med (2013) 4(4):285–9. doi: 10.5847/wjem.j.issn.1920-8642.2013.04.008
25. Tiegs G, Horst AK. TNF in the Liver: Targeting a Central Player in Inflammation. Semin Immunopathol (2022). doi: 10.1007/s00281-022-00910-2
26. Rock CS, Lowry SF. Tumor Necrosis Factor-Alpha. J Surg Res (1991) 51(5):434–45. doi: 10.1016/0022-4804(91)90146-D
27. Rothwell NJ, Luheshi G. Pharmacology of Interleukin-1 Actions in the Brain. Adv Pharmacol (1994) 25:1–20. doi: 10.1016/S1054-3589(08)60428-7
28. Alexander JJ, Jacob A, Cunningham P, Hensley L, Quigg RJ. TNF is a Key Mediator of Septic Encephalopathy Acting Through its Receptor, TNF Receptor-1. Neurochem Int (2008) 52(3):447–56. doi: 10.1016/j.neuint.2007.08.006
29. Krabbe KS, Reichenberg A, Yirmiya R, Smed A, Pedersen BK, Bruunsgaard H. Low-Dose Endotoxemia and Human Neuropsychological Functions. Brain Behav Immun (2005) 19(5):453–60. doi: 10.1016/j.bbi.2005.04.010
30. Ren C, Tong YL, Li JC, Dong N, Hao JW, Zhang QH, et al. Early Antagonism of Cerebral High Mobility Group Box-1 Protein is Benefit for Sepsis Induced Brain Injury. Oncotarget (2017) 8(54):92578–88. doi: 10.18632/oncotarget.21502
31. Chavan SS, Huerta PT, Robbiati S, Valdes-Ferrer SI, Ochani M, Dancho M, et al. HMGB1 Mediates Cognitive Impairment in Sepsis Survivors. Mol Med (Cambridge Mass) (2012) 18(1):930–7. doi: 10.2119/molmed.2012.00195
32. Zhang QH, Sheng ZY, Yao YM. Septic Encephalopathy: When Cytokines Interact With Acetylcholine in the Brain. Military Med Res (2014) 1:20. doi: 10.1186/2054-9369-1-20
33. Taccone FS, Scolletta S, Franchi F, Donadello K, Oddo M. Brain Perfusion in Sepsis. Curr Vasc Pharmacol (2013) 11(2):170–86. doi: 10.2174/1570161111311020007
34. Lin AL, Parikh I, Hoffman JD, Ma D. Neuroimaging Biomarkers of Caloric Restriction on Brain Metabolic and Vascular Functions. Curr Nutr Rep (2017) 6(1):41–8. doi: 10.1007/s13668-017-0187-9
35. Semmler A, Hermann S, Mormann F, Weberpals M, Paxian SA, Okulla T, et al. Sepsis Causes Neuroinflammation and Concomitant Decrease of Cerebral Metabolism. J Neuroinflammation (2008) 5:38. doi: 10.1186/1742-2094-5-38
36. Everson-Rose SA, Ryan JP. Diabetes, Obesity, and the Brain: New Developments in Biobehavioral Medicine. Psychosomatic Med (2015) 77(6):612–5. doi: 10.1097/PSY.0000000000000223
37. Kinoshita H, Maki T, Yasuda K, Kishida N, Sasaoka N, Takagi Y, et al. KUS121, a Valosin-Containing Protein Modulator, Attenuates Ischemic Stroke via Preventing ATP Depletion. Sci Rep (2019) 9(1):11519. doi: 10.1038/s41598-019-47993-w
38. Yang C, Hawkins KE, Doré S, Candelario-Jalil E. Neuroinflammatory Mechanisms of Blood-Brain Barrier Damage in Ischemic Stroke. Am J Physiol Cell Physiol (2019) 316(2):C135–c53. doi: 10.1152/ajpcell.00136.2018
39. Song D, Zhang X, Chen J, Liu X, Xue J, Zhang L, et al. Wnt Canonical Pathway Activator TWS119 Drives Microglial Anti-Inflammatory Activation and Facilitates Neurological Recovery Following Experimental Stroke. J Neuroinflammation (2019) 16(1):256. doi: 10.1186/s12974-019-1660-8
40. Liu F, Lu J, Manaenko A, Tang J, Hu Q. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Disease (2018) 9(5):924–37. doi: 10.14336/AD.2017.1126
41. Ludewig P, Sedlacik J, Gelderblom M, Bernreuther C, Korkusuz Y, Wagener C, et al. Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 Inhibits MMP-9-Mediated Blood-Brain-Barrier Breakdown in a Mouse Model for Ischemic Stroke. Circ Res (2013) 113(8):1013–22. doi: 10.1161/CIRCRESAHA.113.301207
42. Ye B, Tao T, Zhao A, Wen L, He X, Liu Y, et al. Blockade of IL-17a/IL-17R Pathway Protected Mice From Sepsis-Associated Encephalopathy by Inhibition of Microglia Activation. Mediators Inflamm (2019) 2019:8461725. doi: 10.1155/2019/8461725
43. Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 Cells and Activation of Microglia in the CNS During the Course of Experimental Autoimmune Encephalomyelitis. Brain Behav Immun (2010) 24(4):641–51. doi: 10.1016/j.bbi.2010.01.014
44. Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, et al. Patterns of Brain Activation in People at Risk for Alzheimer's Disease. N Engl J Med (2000) 343(7):450–6. doi: 10.1056/NEJM200008173430701
45. Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S, et al. Brain Fuel Metabolism, Aging, and Alzheimer's Disease. Nutr (Burbank Los Angeles County Calif) (2011) 27(1):3–20. doi: 10.1016/j.nut.2010.07.021
46. Taccone FS, Su F, Pierrakos C, He X, James S, Dewitte O, et al. Cerebral Microcirculation is Impaired During Sepsis: An Experimental Study. Crit Care (2010) 14(4):R140. doi: 10.1186/cc9205
47. Keaney J, Campbell M. The Dynamic Blood-Brain Barrier. FEBS J (2015) 282(21):4067–79. doi: 10.1111/febs.13412
48. Engelhardt B, Sorokin L. The Blood-Brain and the Blood-Cerebrospinal Fluid Barriers: Function and Dysfunction. Semin Immunopathol (2009) 31(4):497–511. doi: 10.1007/s00281-009-0177-0
49. van Gool WA, van de Beek D, Eikelenboom P. Systemic Infection and Delirium: When Cytokines and Acetylcholine Collide. Lancet (London England). (2010) 375(9716):773–5. doi: 10.1016/S0140-6736(09)61158-2
50. Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, et al. CXCR4-Activated Astrocyte Glutamate Release via TNFalpha: Amplification by Microglia Triggers Neurotoxicity. Nat Neurosci (2001) 4(7):702–10. doi: 10.1038/89490
51. Viviani B, Boraso M, Marchetti N, Marinovich M. Perspectives on Neuroinflammation and Excitotoxicity: A Neurotoxic Conspiracy? Neurotoxicology (2014) 43:10–20. doi: 10.1016/j.neuro.2014.03.004
52. Jacob A, Brorson JR, Alexander JJ. Septic Encephalopathy: Inflammation in Man and Mouse. Neurochem Int (2011) 58(4):472–6. doi: 10.1016/j.neuint.2011.01.004
53. Basler T, Meier-Hellmann A, Bredle D, Reinhart K. Amino Acid Imbalance Early in Septic Encephalopathy. Intensive Care Med (2002) 28(3):293–8. doi: 10.1007/s00134-002-1217-6
54. Sprung CL, Cerra FB, Freund HR, Schein RM, Konstantinides FN, Marcial EH, et al. Amino Acid Alterations and Encephalopathy in the Sepsis Syndrome. Crit Care Med (1991) 19(6):753–7. doi: 10.1097/00003246-199106000-00004
55. Berg RM, Taudorf S, Bailey DM, Lundby C, Larsen FS, Pedersen BK, et al. Cerebral Net Exchange of Large Neutral Amino Acids After Lipopolysaccharide Infusion in Healthy Humans. Crit Care (2010) 14(1):R16. doi: 10.1186/cc8873
56. Danielski LG, Giustina AD, Badawy M, Barichello T, Quevedo J, Dal-Pizzol F, et al. Brain Barrier Breakdown as a Cause and Consequence of Neuroinflammation in Sepsis. Mol Neurobiol (2018) 55(2):1045–53. doi: 10.1007/s12035-016-0356-7
57. Weighardt H, Holzmann B. Role of Toll-Like Receptor Responses for Sepsis Pathogenesis. Immunobiology (2007) 212(9-10):715–22. doi: 10.1016/j.imbio.2007.09.010
58. Janeway CA Jr., Medzhitov R. Innate Immune Recognition. Annu Rev Immunol (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
59. Daneman R, Prat A. The Blood-Brain Barrier. Cold Spring Harb Perspect Biol (2015) 7(1):a020412. doi: 10.1101/cshperspect.a020412
60. Ward PA. Role of the Complement in Experimental Sepsis. J Leukoc Biol (2008) 83(3):467–70. doi: 10.1189/jlb.0607376
61. Ward PA. Sepsis, Apoptosis and Complement. Biochem Pharmacol (2008) 76(11):1383–8. doi: 10.1016/j.bcp.2008.09.017
62. Erickson MA, Banks WA. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol Rev (2018) 70(2):278–314. doi: 10.1124/pr.117.014647
63. Gao Q, Hernandes MS. Sepsis-Associated Encephalopathy and Blood-Brain Barrier Dysfunction. Inflammation (2021) 44(6):2143–50. doi: 10.1007/s10753-021-01501-3
64. Janeway CA Jr. How the Immune System Protects the Host From Infection. Microbes Infection (2001) 3(13):1167–71. doi: 10.1016/S1286-4579(01)01477-0
65. Chapouly C, Tadesse Argaw A, Horng S, Castro K, Zhang J, Asp L, et al. Astrocytic TYMP and VEGFA Drive Blood-Brain Barrier Opening in Inflammatory Central Nervous System Lesions. Brain J Neurol (2015) 138(Pt6):1548–67. doi: 10.1093/brain/awv077
66. Vincent VA, Tilders FJ, Van Dam AM. Inhibition of Endotoxin-Induced Nitric Oxide Synthase Production in Microglial Cells by the Presence of Astroglial Cells: A Role for Transforming Growth Factor Beta. Glia (1997) 19(3):190–8. doi: 10.1002/(SICI)1098-1136(199703)19:3<190::AID-GLIA2>3.0.CO;2-3
67. Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A, et al. Dual Microglia Effects on Blood Brain Barrier Permeability Induced by Systemic Inflammation. Nat Commun (2019) 10(1):5816. doi: 10.1038/s41467-019-13812-z
68. Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
69. Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature (2017) 549(7673):523–7. doi: 10.1038/nature24016
70. Paasila PJ, Aramideh JA, Sutherland GT, Graeber MB. Synapses, Microglia, and Lipids in Alzheimer's Disease. Front Neurosci (2021) 15:778822. doi: 10.3389/fnins.2021.778822
71. Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, et al. Microglia Contact Induces Synapse Formation in Developing Somatosensory Cortex. Nat Commun (2016) 7:12540. doi: 10.1038/ncomms12540
72. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting Microglial Cells are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Sci (New York NY) (2005) 308(5726):1314–8. doi: 10.1126/science.1110647
73. Kofler J, Wiley CA. Microglia: Key Innate Immune Cells of the Brain. Toxicol pathol (2011) 39(1):103–14. doi: 10.1177/0192623310387619
74. Healy LM, Perron G, Won SY, Michell-Robinson MA, Rezk A, Ludwin SK, et al. MerTK is a Functional Regulator of Myelin Phagocytosis by Human Myeloid Cells. J Immunol (Baltimore Md 1950). (2016) 196(8):3375–84. doi: 10.4049/jimmunol.1502562
75. Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol (2016) 53(2):1181–94. doi: 10.1007/s12035-014-9070-5
76. Matejuk A, Ransohoff RM. Crosstalk Between Astrocytes and Microglia: An Overview. Front Immunol (2020) 11:1416. doi: 10.3389/fimmu.2020.01416
77. Waltl I, Kalinke U. Beneficial and Detrimental Functions of Microglia During Viral Encephalitis. Trends Neurosci (2022) 45(2):158–70. doi: 10.1016/j.tins.2021.11.004
78. Cherry JD, Olschowka JA, O'Banion MK. Neuroinflammation and M2 Microglia: The Good, the Bad, and the Inflamed. J Neuroinflammation (2014) 11:98. doi: 10.1186/1742-2094-11-98
79. Cherry JD, Olschowka JA, O'Banion MK. Are "Resting" Microglia More "M2"? Front Immunol (2014) 5:594. doi: 10.3389/fimmu.2014.00594
80. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 Polarization and Metabolic States. Br J Pharmacol (2016) 173(4):649–65. doi: 10.1111/bph.13139
81. Nguyen HM, Grössinger EM, Horiuchi M, Davis KW, Jin LW, Maezawa I, et al. Differential Kv1.3, KCa3.1, and Kir2.1 Expression in "Classically" and "Alternatively" Activated Microglia. Glia (2017) 65(1):106–21. doi: 10.1002/glia.23078
82. Correale J. The Role of Microglial Activation in Disease Progression. Multiple Sclerosis (Houndmills Basingstoke England). (2014) 20(10):1288–95. doi: 10.1177/1352458514533230
83. Ma K, Guo J, Wang G, Ni Q, Liu X. Toll-Like Receptor 2-Mediated Autophagy Promotes Microglial Cell Death by Modulating the Microglial M1/M2 Phenotype. Inflammation (2020) 43(2):701–11. doi: 10.1007/s10753-019-01152-5
84. Ruganzu JB, Zheng Q, Wu X, He Y, Peng X, Jin H, et al. TREM2 Overexpression Rescues Cognitive Deficits in APP/PS1 Transgenic Mice by Reducing Neuroinflammation via the JAK/STAT/SOCS Signaling Pathway. Exp Neurol (2021) 336:113506. doi: 10.1016/j.expneurol.2020.113506
85. Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of Microglial Activation and Polarization After Intracerebral Haemorrhage. Nat Rev Neurol (2017) 13(7):420–33. doi: 10.1038/nrneurol.2017.69
86. Kawai T, Akira S. Signaling to NF-kappaB by Toll-Like Receptors. Trends Mol Med (2007) 13(11):460–9. doi: 10.1016/j.molmed.2007.09.002
87. Zhang H, Li Y, Yu J, Guo M, Meng J, Liu C, et al. Rho Kinase Inhibitor Fasudil Regulates Microglia Polarization and Function. Neuroimmunomodulation (2013) 20(6):313–22. doi: 10.1159/000351221
88. Zhang B, Wei YZ, Wang GQ, Li DD, Shi JS, Zhang F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front Cell Neurosci (2018) 12:531. doi: 10.3389/fncel.2018.00531
89. Carta AR, Pisanu A. Modulating Microglia Activity With PPAR-γ Agonists: A Promising Therapy for Parkinson's Disease? Neurotoxicity Res (2013) 23(2):112–23. doi: 10.1007/s12640-012-9342-7
90. Sekiyama K, Sugama S, Fujita M, Sekigawa A, Takamatsu Y, Waragai M, et al. Neuroinflammation in Parkinson's Disease and Related Disorders: A Lesson From Genetically Manipulated Mouse Models of α-Synucleinopathies. Parkinson's Disease (2012) 2012:271732. doi: 10.1155/2012/271732
91. Zhang Q, Lu Y, Bian H, Guo L, Zhu H. Activation of the α7 Nicotinic Receptor Promotes Lipopolysaccharide-Induced Conversion of M1 Microglia to M2. Am J Trans Res (2017) 9(3):971–85.
92. Tang RH, Qi RQ, Liu HY. Interleukin-4 Affects Microglial Autophagic Flux. Neural Regeneration Res (2019) 14(9):1594–602. doi: 10.4103/1673-5374.255975
93. Jiang CT, Wu WF, Deng YH, Ge JW. Modulators of Microglia Activation and Polarization in Ischemic Stroke (Review). Mol Med Rep (2020) 21(5):2006–18. doi: 10.3892/mmr.2020.11003
94. Xiang B, Xiao C, Shen T, Li X. Anti-Inflammatory Effects of Anisalcohol on Lipopolysaccharide-Stimulated BV2 Microglia via Selective Modulation of Microglia Polarization and Down-Regulation of NF-κb P65 and JNK Activation. Mol Immunol (2018) 95:39–46. doi: 10.1016/j.molimm.2018.01.011
95. Qiao P, Ma J, Wang Y, Huang Z, Zou Q, Cai Z, et al. Curcumin Prevents Neuroinflammation by Inducing Microglia to Transform Into the M2-Phenotype via Camkkβ-Dependent Activation of the AMP-Activated Protein Kinase Signal Pathway. Curr Alzheimer Res (2020) 17(8):735–52. doi: 10.2174/1567205017666201111120919
96. Qiu Z, Lu P, Wang K, Zhao X, Li Q, Wen J, et al. Dexmedetomidine Inhibits Neuroinflammation by Altering Microglial M1/M2 Polarization Through MAPK/ERK Pathway. Neurochem Res (2020) 45(2):345–53. doi: 10.1007/s11064-019-02922-1
97. Wang XL, Li L. Microglia Regulate Neuronal Circuits in Homeostatic and High-Fat Diet-Induced Inflammatory Conditions. Front Cell Neurosci (2021) 15:722028. doi: 10.3389/fncel.2021.722028
98. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, et al. Microglia Promote Learning-Dependent Synapse Formation Through Brain-Derived Neurotrophic Factor. Cell (2013) 155(7):1596–609. doi: 10.1016/j.cell.2013.11.030
99. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of Microglia. Physiol Rev (2011) 91(2):461–553. doi: 10.1152/physrev.00011.2010
100. Cunningham C. Microglia and Neurodegeneration: The Role of Systemic Inflammation. Glia (2013) 61(1):71–90. doi: 10.1002/glia.22350
101. d'Errico P, Ziegler-Waldkirch S, Aires V, Hoffmann P, Mezö C, Erny D, et al. Microglia Contribute to the Propagation of Aβ Into Unaffected Brain Tissue. Nat Neurosci (2022) 25(1):20–5. doi: 10.1038/s41593-021-00951-0
102. Heneka MT, Golenbock DT, Latz E. Innate Immunity in Alzheimer's Disease. Nat Immunol (2015) 16(3):229–36. doi: 10.1038/ni.3102
103. Datta A, Sarmah D, Kalia K, Borah A, Wang X, Dave KR, et al. Advances in Studies on Stroke-Induced Secondary Neurodegeneration (SND) and its Treatment. Curr Top Med Chem (2020) 20(13):1154–68. doi: 10.2174/1568026620666200416090820
104. Liu WW, Wei SZ, Huang GD, Liu LB, Gu C, Shen Y, et al. BMAL1 Regulation of Microglia-Mediated Neuroinflammation in MPTP-Induced Parkinson's Disease Mouse Model. FASEB J (2020) 34(5):6570–81. doi: 10.1096/fj.201901565RR
105. Li S, Bi G, Han S, Huang R. MicroRNAs Play a Role in Parkinson's Disease by Regulating Microglia Function: From Pathogenetic Involvement to Therapeutic Potential. Front Mol Neurosci (2021) 14:744942. doi: 10.3389/fnmol.2021.744942
106. Bartels T, De Schepper S, Hong S. Microglia Modulate Neurodegeneration in Alzheimer's and Parkinson's Diseases. Sci (New York NY). (2020) 370(6512):66–9. doi: 10.1126/science.abb8587
107. Brudek T. Inflammatory Bowel Diseases and Parkinson's Disease. J Parkinson's Disease (2019) 9(s2):S331–s44. doi: 10.3233/JPD-191729
108. Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, et al. Microglial Brain Region-Dependent Diversity and Selective Regional Sensitivities to Aging. Nat Neurosci (2016) 19(3):504–16. doi: 10.1038/nn.4222
109. Yoo HJ, Kwon MS. Aged Microglia in Neurodegenerative Diseases: Microglia Lifespan and Culture Methods. Front Aging Neurosci (2021) 13:766267. doi: 10.3389/fnagi.2021.766267
110. Galloway DA, Phillips AEM, Owen DRJ, Moore CS. Phagocytosis in the Brain: Homeostasis and Disease. Front Immunol (2019) 10:790. doi: 10.3389/fimmu.2019.00790
111. Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism Against Stroke. Int J Mol Sci (2017) 18(10):2135. doi: 10.3390/ijms18102135
112. Garber C, Soung A, Vollmer LL, Kanmogne M, Last A, Brown J, et al. T Cells Promote Microglia-Mediated Synaptic Elimination and Cognitive Dysfunction During Recovery From Neuropathogenic Flaviviruses. Nat Neurosci (2019) 22(8):1276–88. doi: 10.1038/s41593-019-0427-y
113. Chen CJ, Ou YC, Chang CY, Pan HC, Liao SL, Raung SL, et al. TNF-α and IL-1β Mediate Japanese Encephalitis Virus-Induced RANTES Gene Expression in Astrocytes. Neurochem Int (2011) 58(2):234–42. doi: 10.1016/j.neuint.2010.12.009
114. Manocha GD, Mishra R, Sharma N, Kumawat KL, Basu A, Singh SK. Regulatory Role of TRIM21 in the Type-I Interferon Pathway in Japanese Encephalitis Virus-Infected Human Microglial Cells. J Neuroinflammation (2014) 11:24. doi: 10.1186/1742-2094-11-24
115. Gibson EM, Monje M. Microglia in Cancer Therapy-Related Cognitive Impairment. Trends Neurosci (2021) 44(6):441–51. doi: 10.1016/j.tins.2021.02.003
116. Feng X, Liu S, Chen D, Rosi S, Gupta N. Rescue of Cognitive Function Following Fractionated Brain Irradiation in a Novel Preclinical Glioma Model. Elife (2018) 7:e38865. doi: 10.7554/eLife.38865
117. Hanisch UK. Microglia as a Source and Target of Cytokines. Glia (2002) 40(2):140–55. doi: 10.1002/glia.10161
118. Yang L, Li Z, Xu Z, Zhang B, Liu A, He Q, et al. Protective Effects of Cannabinoid Type 2 Receptor Activation Against Microglia Overactivation and Neuronal Pyroptosis in Sepsis-Associated Encephalopathy. Neuroscience (2022) 493:99–108. doi: 10.1016/j.neuroscience.2022.04.011
119. Song G, Liang H, Song H, Ding X, Wang D, Zhang X, et al. Metformin Improves the Prognosis of Adult Mice With Sepsis-Associated Encephalopathy Better Than That of Aged Mice. J Immunol Res (2022) 2022:3218452. doi: 10.1155/2022/3218452
120. Deng YY, Fang M, Zhu GF, Zhou Y, Zeng HK. Role of Microglia in the Pathogenesis of Sepsis-Associated Encephalopathy. CNS Neurolo Disord Drug Targets (2013) 12(6):720–5. doi: 10.2174/18715273113126660178
121. Singer BH, Dickson RP, Denstaedt SJ, Newstead MW, Kim K, Falkowski NR, et al. Bacterial Dissemination to the Brain in Sepsis. Am J Respir Crit Care Med (2018) 197(6):747–56. doi: 10.1164/rccm.201708-1559OC
122. Lee JS, Jeon YJ, Kang JY, Lee SK, Lee HD, Son CG. Aquilariae Lignum Methylene Chloride Fraction Attenuates IL-1β-Driven Neuroinflammation in BV2 Microglial Cells. Int J Mol Sci (2020) 21(15):5465. doi: 10.3390/ijms21155465
123. Shimada A, Hasegawa-Ishii S. Histological Architecture Underlying Brain-Immune Cell-Cell Interactions and the Cerebral Response to Systemic Inflammation. Front Immunol (2017) 8:17. doi: 10.3389/fimmu.2017.00017
124. Wang H, Hong LJ, Huang JY, Jiang Q, Tao RR, Tan C, et al. P2RX7 Sensitizes Mac-1/ICAM-1-Dependent Leukocyte-Endothelial Adhesion and Promotes Neurovascular Injury During Septic Encephalopathy. Cell Res (2015) 25(6):674–90. doi: 10.1038/cr.2015.61
125. Li Y, Yin L, Fan Z, Su B, Chen Y, Ma Y, et al. Microglia: A Potential Therapeutic Target for Sepsis-Associated Encephalopathy and Sepsis-Associated Chronic Pain. Front Pharmacol (2020) 11:600421. doi: 10.3389/fphar.2020.600421
126. Maccioni RB, Gonzalez A, Andrade V, Cortes N, Tapia JP, Guzman-Martinez L. Alzheimer’s Disease in the Perspective of Neuroimmunology. Open Neurol J (2018) 12:50–6. doi: 10.2174/1874205X01812010050
127. Michels M, Sonai B, Dal-Pizzol F. Polarization of Microglia and its Role in Bacterial Sepsis. J Neuroimmunol (2017) 303:90–8. doi: 10.1016/j.jneuroim.2016.12.015
128. Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, et al. Role for Neuronally Derived Fractalkine in Mediating Interactions Between Neurons and CX3CR1-Expressing Microglia. Proc Natl Acad Sci USA (1998) 95(18):10896–901. doi: 10.1073/pnas.95.18.10896
129. Wolf SA, Boddeke HW, Kettenmann H. Microglia in Physiology and Disease. Annu Rev Physiol (2017) 79:619–43. doi: 10.1146/annurev-physiol-022516-034406
130. Mayer AM, Oh S, Ramsey KH, Jacobson PB, Glaser KB, Romanic AM. Escherichia Coli Lipopolysaccharide Potentiation and Inhibition of Rat Neonatal Microglia Superoxide Anion Generation: Correlation With Prior Lactic Dehydrogenase, Nitric Oxide, Tumor Necrosis Factor-Alpha, Thromboxane B2, and Metalloprotease Release. Shock (1999) 11(3):180–6. doi: 10.1097/00024382-199903000-00005
131. Gimenez MA, Sim JE, Russell JH. TNFR1-Dependent VCAM-1 Expression by Astrocytes Exposes the CNS to Destructive Inflammation. J Neuroimmunol (2004) 151(1-2):116–25. doi: 10.1016/j.jneuroim.2004.02.012
132. Lemstra AW, Groen in't Woud JC, Hoozemans JJ, van Haastert ES, Rozemuller AJ, Eikelenboom P, et al. Microglia Activation in Sepsis: A Case-Control Study. J Neuroinflammation (2007) 4:4. doi: 10.1186/1742-2094-4-4
133. Moraes CA, Zaverucha-do-Valle C, Fleurance R, Sharshar T, Bozza FA, d'Avila JC. Neuroinflammation in Sepsis: Molecular Pathways of Microglia Activation. Pharm (Basel). (2021) 14(5). doi: 10.3390/ph14050416
134. Wu Q, Zhao Y, Chen X, Zhu M, Miao C. Propofol Attenuates BV2 Microglia Inflammation via NMDA Receptor Inhibition. Can J Physiol Pharmacol (2018) 96(3):241–8. doi: 10.1139/cjpp-2017-0243
135. Munster BC, Aronica E, Zwinderman AH, Eikelenboom P, Cunningham C, Rooij SE. Neuroinflammation in Delirium: A Postmortem Case-Control Study. Rejuvenation Res (2011) 14(6):615–22. doi: 10.1089/rej.2011.1185
136. Bozza FA, D'Avila JC, Ritter C, Sonneville R, Sharshar T, Dal-Pizzol F. Bioenergetics, Mitochondrial Dysfunction, and Oxidative Stress in the Pathophysiology of Septic Encephalopathy. Shock (2013) 39 Suppl 1:10–6. doi: 10.1097/SHK.0b013e31828fade1
137. Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic Inflammation and Microglial Activation: Systematic Review of Animal Experiments. J Neuroinflammation (2015) 12:114. doi: 10.1186/s12974-015-0332-6
138. Towner RA, Garteiser P, Bozza F, Smith N, Saunders D, d'Avila JCP, et al. In Vivo Detection of Free Radicals in Mouse Septic Encephalopathy Using Molecular MRI and Immuno-Spin Trapping. Free Radic Biol Med (2013) 65:828–37. doi: 10.1016/j.freeradbiomed.2013.08.172
139. Michels M, Vieira AS, Vuolo F, Zapelini HG, Mendonça B, Mina F, et al. The Role of Microglia Activation in the Development of Sepsis-Induced Long-Term Cognitive Impairment. Brain Behavior Immunity (2015) 43:54–9. doi: 10.1016/j.bbi.2014.07.002
140. Michels M, Danielski LG, Dal-Pizzol F, Petronilho F. Neuroinflammation: Microglial Activation During Sepsis. Curr Neurovascular Res (2014) 11(3):262–70. doi: 10.2174/1567202611666140520122744
141. Wang L, Lin F, Ren M, Liu X, Xie W, Zhang A, et al. The PICK1/TLR4 Complex on Microglia is Involved in the Regulation of LPS-Induced Sepsis-Associated Encephalopathy. Int Immunopharmacol (2021) 100:108116. doi: 10.1016/j.intimp.2021.108116
142. Sharshar T, Polito A, Checinski A, Stevens RD. Septic-Associated Encephalopathy–Everything Starts at a Microlevel. Crit Care (London England) (2010) 14(5):199. doi: 10.1186/cc9254
143. Zhang H, Xu J, Wu Q, Fang H, Shao X, Ouyang X, et al. Gut Microbiota Mediates the Susceptibility of Mice to Sepsis-Associated Encephalopathy by Butyric Acid. J Inflammation Res (2022) 15:2103–19. doi: 10.2147/JIR.S350566
144. Kodali MC, Chen H, Liao FF. Temporal Unsnarling of Brain's Acute Neuroinflammatory Transcriptional Profiles Reveals Panendothelitis as the Earliest Event Preceding Microgliosis. Mol Psychiatry (2021) 26(8):3905–19. doi: 10.1038/s41380-020-00955-5
145. Schiedel M, Robaa D, Rumpf T, Sippl W, Jung M. The Current State of NAD(+) -Dependent Histone Deacetylases (Sirtuins) as Novel Therapeutic Targets. Medicinal Res Rev (2018) 38(1):147–200. doi: 10.1002/med.21436
146. Chang HC, Guarente L. SIRT1 and Other Sirtuins in Metabolism. Trends Endocrinol Metabolism: TEM (2014) 25(3):138–45. doi: 10.1016/j.tem.2013.12.001
147. Li L, Zhang J, Yang Y, Wang Q, Gao L, Yang Y, et al. Single-Wall Carbon Nanohorns Inhibited Activation of Microglia Induced by Lipopolysaccharide Through Blocking of Sirt3. Nanoscale Res Letters (2013) 8(1):100. doi: 10.1186/1556-276X-8-100
148. Li L, Sun Q, Li Y, Yang Y, Yang Y, Chang T, et al. Overexpression of SIRT1 Induced by Resveratrol and Inhibitor of miR-204 Suppresses Activation and Proliferation of Microglia. J Mol Neurosci MN. (2015) 56(4):858–67. doi: 10.1007/s12031-015-0526-5
149. Shi L, Zhang J, Wang Y, Hao Q, Chen H, Cheng X. Sirt1 Regulates Oxidative Stress in Oxygen-Glucose Deprived Hippocampal Neurons. Front Pediatrics (2020) 8:455. doi: 10.3389/fped.2020.00455
150. Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and Toll-Like Receptors 2 and 4 are Required for Fibrillar A{beta}-Stimulated Microglial Activation. J Neurosci (2009) 29(38):11982–92. doi: 10.1523/JNEUROSCI.3158-09.2009
151. Hansson E, Björklund U, Skiöldebrand E, Rönnbäck L. Anti-Inflammatory Effects Induced by Pharmaceutical Substances on Inflammatory Active Brain Astrocytes-Promising Treatment of Neuroinflammation. J Neuroinflammation (2018) 15(1):321. doi: 10.1186/s12974-018-1361-8
152. Zhang H, Fang H, Wang Y, Xu J, Chen C. [Mechanisms of Sodium Butyrate Inhibition of Microglia Inflammatory Activation in Hippocampus via Toll-Like Receptor 4/Nuclear Factor-κb P65 Pathway]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue (2021) 33(12):1471–8.
153. Staudinger J, Zhou J, Burgess R, Elledge SJ, Olson EN. PICK1: A Perinuclear Binding Protein and Substrate for Protein Kinase C Isolated by the Yeast Two-Hybrid System. J Cell Biol (1995) 128(3):263–71. doi: 10.1083/jcb.128.3.263
154. Mo Y, Lou Y, Zhang A, Zhang J, Zhu C, Zheng B, et al. PICK1 Deficiency Induces Autophagy Dysfunction via Lysosomal Impairment and Amplifies Sepsis-Induced Acute Lung Injury. Mediators Inflamm (2018) 2018:6757368. doi: 10.1155/2018/6757368
155. Mo Y, Wang L, Ren M, Xie W, Ye X, Zhou B, et al. Electroacupuncture Prevents LPS- Induced Neuroinflammation via Upregulation of PICK-TLR4 Complexes in the Microglia of Hippocampus. Brain Res Bulletin (2021) 177:295–304. doi: 10.1016/j.brainresbull.2021.10.010
156. Waisman A, Hauptmann J, Regen T. The Role of IL-17 in CNS Diseases. Acta Neuropathol (2015) 129(5):625–37. doi: 10.1007/s00401-015-1402-7
157. Lv M, Liu Y, Zhang J, Sun L, Liu Z, Zhang S, et al. Roles of Inflammation Response in Microglia Cell Through Toll-Like Receptors 2/Interleukin-23/Interleukin-17 Pathway in Cerebral Ischemia/Reperfusion Injury. Neuroscience (2011) 176:162–72. doi: 10.1016/j.neuroscience.2010.11.066
158. Widmann CN, Heneka MT. Long-Term Cerebral Consequences of Sepsis. Lancet Neurology (2014) 13(6):630–6. doi: 10.1016/S1474-4422(14)70017-1
159. Tang H, Ji M, Zong M, Jia M, Luo D, Zhou Z, et al. Individual Differences in the Brain are Associated With Resilience Versus Susceptibility to Lipopolysaccharide-Induced Memory Impairment. Neurosci Letters (2018) 662:361–7. doi: 10.1016/j.neulet.2017.10.064
160. Shen Y, Zhang Y, Du J, Jiang B, Shan T, Li H, et al. CXCR5 Down-Regulation Alleviates Cognitive Dysfunction in a Mouse Model of Sepsis-Associated Encephalopathy: Potential Role of Microglial Autophagy and the P38mapk/NF-κb/STAT3 Signaling Pathway. J Neuroinflammation (2021) 18(1):246. doi: 10.1186/s12974-021-02300-1
161. Sui DM, Xie Q, Yi WJ, Gupta S, Yu XY, Li JB, et al. Resveratrol Protects Against Sepsis-Associated Encephalopathy and Inhibits the NLRP3/IL-1β Axis in Microglia. Mediators Inflammation (2016) 2016:1045657. doi: 10.1155/2016/1045657
162. Zhong X, Xie L, Yang X, Liang F, Yang Y, Tong J, et al. Ethyl Pyruvate Protects Against Sepsis-Associated Encephalopathy Through Inhibiting the NLRP3 Inflammasome. Mol Med (Cambridge Mass) (2020) 26(1):55. doi: 10.1186/s10020-020-00181-3
163. Bonfante S, Joaquim L, Fileti ME, Giustina AD, de Souza Goldim MP, Danielski LG, et al. Stanniocalcin 1 Inhibits the Inflammatory Response in Microglia and Protects Against Sepsis-Associated Encephalopathy. Neurotoxicity Res (2021) 39(2):119–32. doi: 10.1007/s12640-020-00293-y
164. Weberpals M, Hermes M, Hermann S, Kummer MP, Terwel D, Semmler A, et al. NOS2 Gene Deficiency Protects From Sepsis-Induced Long-Term Cognitive Deficits. J Neurosci (2009) 29(45):14177–84. doi: 10.1523/JNEUROSCI.3238-09.2009
165. Xie K, Zhang Y, Wang Y, Meng X, Wang Y, Yu Y, et al. Hydrogen Attenuates Sepsis-Associated Encephalopathy by NRF2 Mediated NLRP3 Pathway Inactivation. Inflammation Res (2020) 69(7):697–710. doi: 10.1007/s00011-020-01347-9
166. Chen H, Dong B, Shi Y, Yu Y, Xie K. Hydrogen Alleviates Neuronal Injury and Neuroinflammation Induced by Microglial Activation via the Nuclear Factor Erythroid 2-Related Factor 2 Pathway in Sepsis-Associated Encephalopathy. Neuroscience (2021) 466:87–100. doi: 10.1016/j.neuroscience.2021.05.003
Keywords: sepsis-associated encephalopathy (SAE), sepsis, microglia, cognitive impairment, inflammatory factors
Citation: Yan X, Yang K, Xiao Q, Hou R, Pan X and Zhu X (2022) Central role of microglia in sepsis-associated encephalopathy: From mechanism to therapy. Front. Immunol. 13:929316. doi: 10.3389/fimmu.2022.929316
Received: 26 April 2022; Accepted: 22 June 2022;
Published: 26 July 2022.
Edited by:
Yi Tang, Capital Medical University, ChinaReviewed by:
Andreia Barroso, IQVIA, BrazilRoland Nau, University Medical Center Göttingen, Germany
Yunchang Mo, Wenzhou Medical University, China
Copyright © 2022 Yan, Yang, Xiao, Hou, Pan and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rongyao Hou, MTM3MzA5ODU2MTBAMTM5LmNvbQ==; Xudong Pan, ZHJwYW4wMjJAcWR1LmVkdS5jbg==; Xiaoyan Zhu, enh5c2RqbUBxZHUuZWR1LmNu
†These authors have contributed equally to this work