 Wenlin Qiu
Wenlin Qiu Tong Yu
Tong Yu Guo-Min Deng
Guo-Min Deng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 13 October 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.924766
This article is part of the Research Topic Systemic Lupus Erythematosus - Predisposition Factors, Pathogenesis, Diagnosis, Treatment and Disease Models View all 21 articles
Systemic lupus erythematosus (SLE), often known simply as lupus, is a severe chronic autoimmune disease that is characterized by multi-organ and tissue damage and high levels of autoantibodies in serum. We have recently investigated, using animal models, the role of organ-deposited IgG autoantibodies in the pathogenesis of organ and tissue damage in SLE. We found that intra-organ injection of serum from mice with lupus (i.e., lupus mice) into healthy mice triggered inflammation in tissue and organs but that serum from other healthy mice did not, and that the severity of inflammation was related to the dose of serum injected. Immunohistochemistry showed that a large number of IgG molecules are deposited at the site of organ and tissue damage in lupus mice, and that IgG is a major contributor to the development of tissue inflammation triggered by serum from lupus mice or patients. The development of tissue inflammation induced by IgG in serum from lupus mice requires the presence of monocytes/macrophages, but not of lymphocytes or neutrophils; tumor necrosis factor (TNF)/tumor necrosis factor receptor 1 (TNFR1) and interleukin 1 (IL-1) also play essential roles in the development of tissue inflammation triggered by IgG. In addition, it has been found that TNFR1 inhibitors can suppress skin injury in lupus mice and that spleen tyrosine kinase (Syk) inhibitors, which can block the signaling transduction of IgG/Fc gamma receptors (FcγRs), can prevent and treat skin injury and kidney damage in lupus mice. We have also observed that lupus IgG might protect against bone erosion. Based on these results, we conclude that IgG plays a crucial role in the development of organ and tissue damage in SLE and in protecting bone erosion and arthritis, and we suggest that the IgG/FcγR signaling pathway is an important therapeutic target in SLE.
Systemic lupus erythematosus (SLE), which affects mostly women of childbearing age, is a chronic severe autoimmune disease characterized by damage to multiple organs and tissues and a high level of autoantibodies in serum (1–3). The incidence and prevalence of SLE vary considerably between different regions of the world (1, 4). In adults, the prevalence of SLE worldwide ranges from 30 to 150 per 100,000, and the incidence ranges from 2.2 to 23.1 per 100,000 per year (4). The typical initial symptoms of lupus are fever, erythema, and arthritis (5). Renal involvement is the most common manifestation: the prevalence of nephritis among lupus patients is 29–82% (6). Skin injury is the second most common symptom, and occurs in 70–85% of all lupus patients (7). Arthralgia is also common, and lupus patients with arthralgia often suffer from varying degrees of synovitis, but without bone erosion (8). Only 2.8–4.3% of patients have Jaccoud’s arthropathy, which may result in joint deformities (8). With advancements in therapies, 5-, 10-, 15-, and 20-year survival rates have risen to 95%, 91%, 85%, and 78%, respectively (4). The main causes of death in SLE patients are infection, cardiovascular events, and active disease (4, 9). Glucocorticoids and antimalarial agents (especially hydroxychloroquine) are the most important and most common first-line agents for the treatment of SLE, but B-cell-targeting drugs have shown promising results (4, 5). Belimumab, a humanized monoclonal antibody targeting soluble B-cell activating factor (BAFF), a B-cell stimulator, can significantly and sustainedly reduce the level of IgG autoantibodies, including anti-double-stranded DNA (anti-dsDNA), anti-Smith (anti-Sm), anti-cardiolipin, and anti-ribosomal P autoantibodies (10). In addition, the immature dendritic cell (iDC) vaccine, an underdeveloped therapy, has been shown to have a protective effect in lupus-like nephritis induced by pathogenic autoantibodies (11). These indicates that SLE IgG is involved in the therapy of SLE.
The organ and tissue damage that occurs in the region of IgG deposition, is due to infiltration by inflammatory cells, leading to the destruction of tissue organization. Severe damage to multiple organs is one of the leading causes of death in patients with SLE (3, 12). Commonly damaged organs include the kidneys, skin, joints, liver, spleen, lungs, and brain (1–3).
The immune system is damaged in SLE patients, and B cells are abnormally over activated and produce multiple types of autoantibodies. Patients with SLE produce IgA, IgE, IgG, and IgM autoantibodies (12), but IgG autoantibodies predominate. Inflammation is the pathological basis of organ and tissue damage caused by the local deposition of IgG and immune complexes (ICs) (12–17). The pathogenesis of SLE is closely associated with a high level of autoreactive IgG (2, 3, 18, 19). However, the mechanism by which IgG causes organ and tissue damage is still unclear.
Tissue-deposited ICs formed by autoantibodies and autoantigens activate immune cells to produce inflammatory cytokines by binding to Fc gamma receptors (FcγRs) (20–24). Recently, we have demonstrated that organ-deposited IgG induces local inflammation by binding to FcγRs on the surface of monocytes/macrophages, resulting in multi-organ and tissue damage (20–28).
In this review, we summarize recent studies of the role of organ-deposited IgG in the pathogenesis of inflammation and organ and tissue damage in SLE, and potential therapeutic targets in the IgG/FcγRs signaling pathways, including studies investigating animal models, immune cells, cytokines (Figure 1)
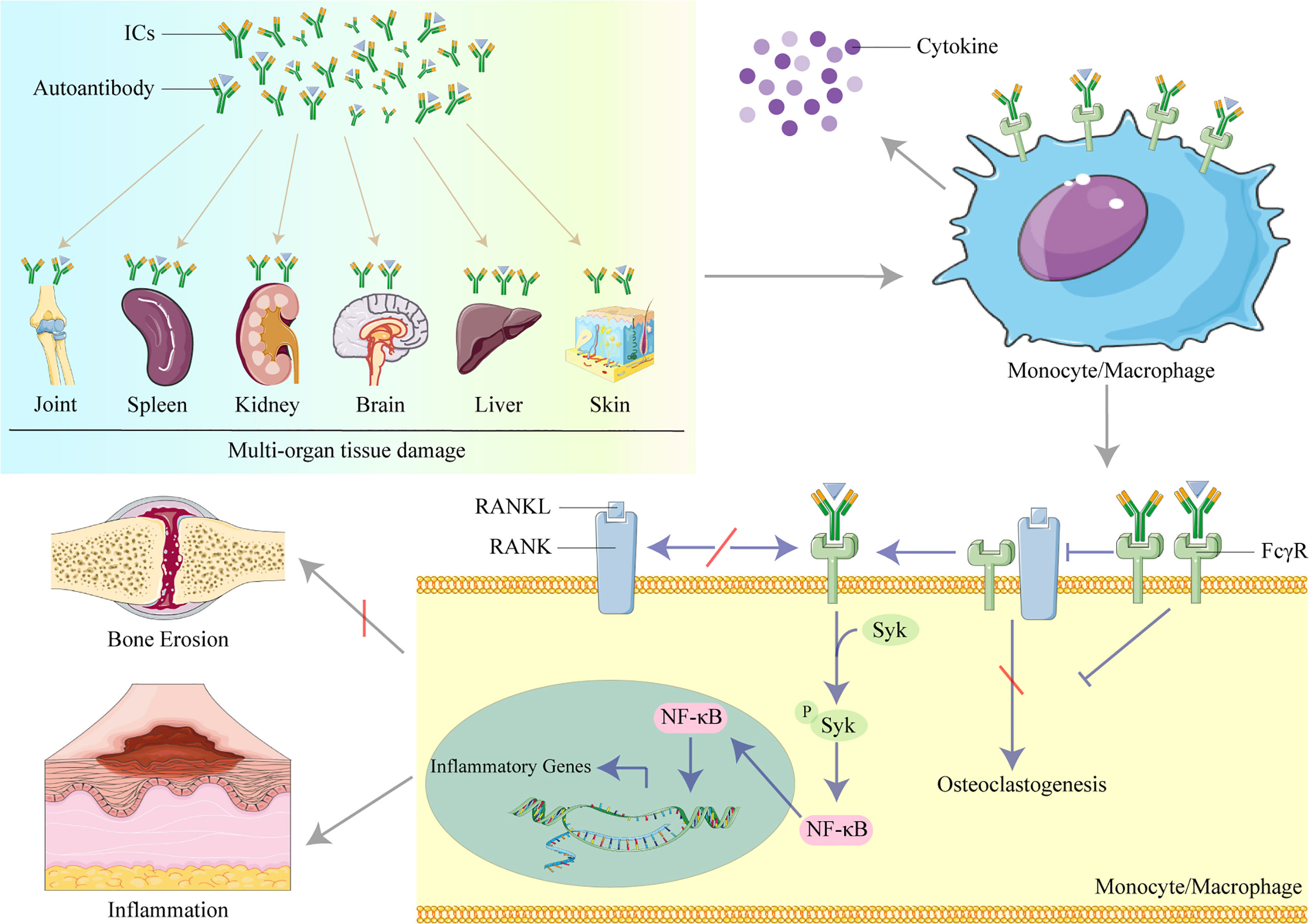
Figure 1 The role of IgG in organ and tissue damage in systemic lupus erythematosus (SLE). Lupus IgG and immune complexes (ICs) deposited in several organs and tissues cause multi-organ and tissue damage. IgG and ICs binding to Fc gamma receptors (FcγRs) activate monocytes/macrophages to secret cytokines. IgG inhibits osteoclastogenesis induced by the receptor activator of nuclear factor kappa B ligand (RANKL) to protect against bone erosion by binding to FcγRI and induces inflammation by activating the spleen tyrosine kinase (Syk)/nuclear factor kappa B (NF-κB) signal pathway to upregulate the transcription of inflammatory genes in monocytes/macrophages.
IgG and ICs have been found in the kidneys of SLE patients, and the most common autoantibodies identified are anti-dsDNA antibodies (18). IgM and IgG autoantibodies are deposited at the dermis–epidermis junction (29), forming so-called “lupus bands,” which can be detected using direct immunofluorescence (30, 31). Some autoantibodies, including anti-galectin 3 antibody, anti-acidic ribosomal protein P0 antibody (anti-RPLP0), and anti-Ro52 antibody, have been reported to be deposited in the skin and to be involved in the development of skin damage (32–34). We have demonstrated that IgG is deposited in the skin, liver, spleen, and joints of MRL/lpr mice (17, 23, 27, 28), and shown that the level of IgG deposited is correlated with the severity of tissue damage (17, 23, 27, 28). These findings suggest that tissue-deposited IgG may have an essential role in the pathogenesis of multi-organ and tissue damage in SLE.
We investigated whether or not serum containing a high level of IgG induces tissue inflammation in order to determine the role of tissue-deposited IgG in the multi-organ and tissue damage that occurs in SLE. Through intra-organ injection of serum from lupus mice and SLE patients (lupus serum), we have established an animal model of SLE organ and tissue damage (17, 20–22, 24, 27, 28). We found that intra-organ injection of serum from SLE patients and lupus mice, but not from healthy humans and mice, induced tissue inflammation in the skin, liver, spleen, and joints, and that the severity of the inflammation was dose-dependent (17, 23, 27, 28). Using toll-like receptor 4 (TLR4)-deficient mice that were lipopolysaccharide (LPS) resistant, we established that organ and tissue inflammation could be induced by serum from SLE patients and lupus-prone mice, but not by LPS contamination (17, 27). We found that tissue inflammation caused by SLE serum appeared in the skin, liver, spleen, and joints a few hours after intra-organ injection, reached its peak after 3 days, and lasted for up to 14 days (17, 23, 27, 28).
Inflammatory skin reactions were induced by injecting serum from SLE patients into six different mouse strains: C57BL/6, SWISS, BALB/c, C3H/HeN, C3H/HeJ, and B-17 mice (23). Skin inflammation was induced by injection of serum only if the patients from which the serum was derived also exhibited skin inflammation and the severity of skin inflammation was not correlated with the severity of systemic disease (23); lupus serum had a synergistic effect with CpG DNA in inducing skin inflammation (23, 35).
Other studies have also shown that intracerebroventricular injection of lupus serum induces an inflammatory response (36–38). These studies suggest that lupus serum can cause organ and tissue inflammation.
SLE is characterized by a high level of autoantibodies, particularly IgG. Many studies have shown that IgG is involved in the pathogenesis of organ and tissue inflammation and organ damage (17, 20–22, 24, 27, 28, 39, 40). We removed IgG from lupus serum to evaluate the role of IgG in the development of tissue inflammation. We found that the inflammation was significantly less severe in mice that received intra-organ injection of IgG-depleted lupus serum than in those injected with IgG-containing lupus serum (17, 23, 27, 28). The skin, liver, spleen, and joints could be directly inflamed by IgG isolated from lupus serum (17, 23, 27, 28). Furthermore, IgG extracted from lupus serum caused more severe inflammation than the same dose of IgG extracted from healthy serum (17, 23, 27, 28). The severity of inflammation was also related to the dose of lupus IgG (17, 23, 27, 28).
Recently, antibody glycosylation in autoimmune diseases has attracted attention, and the importance of Fc regions for inflammation development has been demonstrated (41, 42). In addition to the antigen-binding site, the level of antibody glycosylation may differ in serum from healthy individuals and SLE patients. In vitro experiments have revealed that microglia are activated by phagocytosis of IgG (36). In a study in which mice received intrasplenic injection of lupus serum or of IgG-depleted lupus serum, the numbers of germinal centers(GCs) and IgG-secreting plasmacytes incresased after 8 days in the whole lupus serum group. (28). IgG has also been shown to induce monocyte differentiation into dendritic cells (DCs) (24). In addition, intrahepatic injection of IgG has also been shown to increase levels of ALT alanine transaminase (ALT) and aspartate aminotransferase (AST) in serum and to cause accumulation of apoptotic hepatocytes around the region of inflammation (17).
In another study, the severity of IgG-induced skin inflammation was found not to be related to the titer of the anti-dsDNA antibody and anti-Ro antibody (23). In addition, it has been reported that injection of anti-DNA antibodies causes proteinuria and promotes lupus nephritis in mice only when the antibodies were bound to the basement membrane (43). These findings demonstrate that IgG plays a key role in tissue inflammation induced by lupus serum.
The effect of IgG is mediated by FcγR (44). FcγRI and FcγRIII are activating receptors, and FcγRII is the inhibitory receptor. The only inhibitory receptor, FcγRIIB, transmits its inhibitory signal through the immunoreceptor tyrosine-based inhibitory motif (ITIM) (45). Polymorphism of the genes encoding FcγRs has been reported and confirmed to be a heritable susceptibility factor for SLE (2, 3, 46). It has been reported that the FCGR2B-I232T genotype suppresses ligand binding, leading to low affinity for IgG (47). Because FcγRIIB is an inhibitory receptor, the low affinity for IgG contributes to a reduction in inhibitory signals, thus reducing inflammation, and alters the balance between inhibitory and activatory signals, which is associated with susceptibility to SLE (45, 47).
Similarly, polymorphism of the genes encoding activating receptors also influences susceptibility to SLE (2, 3, 47). FcγRI is the only high-affinity FcγR for IgG, and is usually expressed on the surface of monocytes/macrophages (48). The immunoreceptor tyrosine-based activation motif (ITAM) embedded in the intracellular structure of FcγR is responsible for signal transduction of IgG and ICs (44, 48). The expression of FcγRI, but not of FcγRII and FcγRIII, has been found to be increased in monocytes from SLE patients, and the level of FcγRI expression was found to be related to the SLE disease activity index (SLEDAI) (49).
We used FcγRI-deficient mice with IgG-induced inflammation to investigate the role of FcγRI in the inflammation induced by SLE (49). The inflammation induced by IgG was reduced in FcγRI-deficient mice compared with wild-type mice (49). In addition, the expression level of FcγRI was associated with IgG deposition and skin inflammation in MRL/lpr mice (37). Activation of the inflammatory signal pathway in monocytes/macrophages required FcγR expression (49). However, in vitro experiments have shown that IgG reduces the surface expression of FcγRI, but not of FcγRII and FcγRIII, on monocytes/macrophages (27). N-glycosylation of antibodies and IgG subclasses affects the binding affinity of antibodies to different Fc receptors (50). Abnormal glycosylation of autoreactive IgG may have a higher affinity to FcγRI, suggesting that the molecular modification of autoreactive IgG may influence its pathogenicity. Further work is required to identify the Fc region provided by which state of the SLE IgG.
Using various strains of cell-deficient and cell-depleted mice, we investigated the role of different types of immune cells in inflammation induced by IgG. Immunohistochemistry demonstrated that regions of tissue inflammation in the skin, kidneys, liver, and spleen are characterized by infiltration of a large number of monocytes/macrophages (17, 23, 27, 28, 51). For example, F4/80+ macrophages were identified in the red pulp of the spleen (28), liver macrophages (Kupffer cells) were seen in areas of hepatitis induced by SLE IgG (17), and large numbers of CD11b+ monocytes/macrophage and CD11c+ DCs were detected in sites of skin inflammation; however, CD3+ T cells and CD20+ B cells were absent (23, 52). The injection of lupus serum and IgG significantly reduced the inflammation caused by the depletion of monocytes/macrophages (17, 23, 27, 28).
Furthermore, skin inflammation induced by lupus IgG did not develop in Csf1-deficient mice lacking mature monocytes (21). Rag 1-deficient mice (which lack mature T and B cells but whose monocytes/macrophages are intact) were used to identify the role of T and B cells in IgG-induced inflammation (17, 24, 27). The lack of mature T and B cells did not affect the development of organ tissue inflammation induced by IgG (17, 24, 27). Neutrophil depletion by anti-Ly6G antibody injection did not affect the development of IgG-induced arthritis and dermatitis (20, 27). Another study showed that IgG induced lupus nephritis by activating macrophages (51).
In yet another study, IgG activated microglia to produce proinflammatory cytokines (53). In addition, polymorphism of the ITGAM (integrin subunit alpha M) gene (a gene associated with the activation of monocytes/macrophages) is associated with SLE susceptibility (11), and inflammation and organ damage in lupus-prone mice were suppressed, and splenic macrophages decreased, after treatment with the agonist of ITGAM (54). These findings indicated that monocytes/macrophages but not T cells, B cells, or neutrophils, play a crucial role in IgG-induced inflammation.
Many cytokines are involved in inflammation induced by IgG. Tumor necrosis factor alpha (TNF-α) is an important proinflammatory cytokine that is mainly secreted by macrophages and associated with SLE activity (55, 56). TNF-α has been found to be highly expressed in the kidneys and skin of SLE patients (57), and in the joints, skin, and kidneys of MRL/lpr mice (21, 27, 58). In vitro stimulation of macrophages with IgG has been found to cause IC to produce TNF-α (17, 28, 51),, and the severity of inflammation induced by IgG in skin, liver, spleen, and joints has been shown to be significantly reduced in mice with a TNF-α deficiency (17, 22, 27, 28). TNF exerts its effect through its receptors, such as TNFR1 and TNFR2. The skin inflammation induced by IgG was found to be reduced in mice with a TNFR1 deficiency, but not in those mice with a TNFR2 deficiency (24). Furthermore, we found that a deficiency of interleukin (IL) 1 (IL1) relieved inflammation induced by IgG by reducing the production of TNF-α (22). These results suggest that TNF-α/TNFR1 is crucial for inflammation induced by IgG in lupus serum. In addition, BAFF, produced by activated macrophages, was found in the region of inflammation to prolong the survival of macrophages in lupus nephritis (59, 60). In addition, IL-10, interferon alpha (IFN-α), and interferon gamma (IFN-γ) have been shown to increase CD64 expression in monocytes/macrophages (49, 61). In one study, a lack of macrophage migration inhibitory factor (MIF) attenuated infiltration of macrophages and alleviated lupus nephritis (62).
Based on the findings from animal models of tissue inflammation induced by lupus IgG, the IgG/FcγRs signaling pathway is an important therapeutic target. Thus, we investigated whether an IgG/FcγRs signaling molecule inhibitor prevents and alleviates multi-organ and tissue damage in SLE. Spleen tyrosine kinase (Syk), a non-receptor tyrosine kinase of the Src family, plays a fundamental role in IgG/FcγRs signaling pathway conduction in autoimmune diseases (27, 63–65). FcγRI was required to activate Syk induced by SLE serum in macrophages (49). Syk inhibitors were used to treat lupus MRL/lpr mice, and have been found to prevent and treat damaged skin, kidneys, and spleen in MRL/lpr mice (28, 63). In addition, Syk inhibitors reduced inflammation and damage in the spleen, liver, and skin induced by injection of IgG from lupus serum (17, 23, 28). Intravenous immunoglobin (IVIG) is an anti-inflammation therapy used to treat various acute and chronic autoimmune diseases (66). Its anti-inflammation effect is due to the blocking of Fc receptors, which cannot bind with pathological autoantibodies (66). Glycosylation of the Fc region of IgG affects its function of inflammation modulation. The sialic acid-rich IgG fraction of IVIG has an improved anti-inflammatory activity (66). These results suggest that IgG/FcγRs signaling molecules may be a potential therapeutic target in multi-organ tissue damage of SLE.
Bone erosion is an important feature in inflammatory arthritis, such as rheumatoid arthritis; however, it does not occur in SLE arthritis. The receptor activator of nuclear factor kappa B ligand (RANKL) induces monocytes/macrophages to separate into osteoclasts that mitigate bone erosion (26). FcγRI is a costimulatory molecule of RANK, the receptor for RANKL that activates macrophage differentiation into osteoclasts (14). We investigated the role of IgG in macrophage differentiation to understand the mechanism by which IgG from lupus serum induces arthritis without bone erosion (27). We found that IgG inhibited osteoclastogenesis induced by RANKL in a dose-dependent manner, and deficiency of FcγRII and FcγRIII did not influence the inhibitory effect of IgG in the formation of osteoclasts; both IgG from lupus serum and RANKL reduced the level of FcγRI (27). The inhibitory effect on osteoclastogenesis was greater at high doses than in at doses of IgG, and increasing doses of RANKL gradually blocked the inhibitory effects of lupus IgG. The stronger inhibitory effects of lupus IgG on RANKL-mediated osteoclastogenesis were presented in cells pre-treated for 24 hours when compared with cells treated with both RANKL and lupus IgG at the same time; at 24 hours after RANKL stimulation, the inhibitory effect of lupus IgG on osteoclastogenesis was eliminated (27). Based on these data, it appears that the relationship between IgG and RANKL is competition for FcγRI. IgG inhibits RANKL-induced osteoclastogenesis through competition for FcγRI binding. The binding of IgG to FcγRI may lead to a functional deficiency of FcγRI on the cellular membrane, which is required for RANKL-induced osteoclastogenesis. The competitive occupation of FcγRI by IgG may be exploited to develop therapeutic approaches to prevent bone destruction in autoimmune/inflammatory arthritis.
Based on these results, we conclude that IgG plays a crucial role in developing organ and tissue damage in SLE and in protecting against bone erosion in SLE arthritis, and that the IgG/FcγRs signaling pathway is an important therapeutic target in SLE.
Conception and design, G-MD; manuscript writing, WQ and TY; critical revision of the manuscript, G-MD; and, final approval of manuscript, all authors.
This study was supported by the National Natural Science Foundation of China (NSFC) (G-MD, 82171786).
Figure 1 was modified from Servier Medical Art (http://smart.servier.com/).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Barber MRW, Drenkard C, Falasinnu T, Hoi A, Mak A, Kow NY, et al. Global epidemiology of systemic lupus erythematosus. Nat Rev Rheumatol (2021) 17(9):515–32. doi: 10.1038/s41584-021-00668-1
2. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med (2008) 358(9):929–39. doi: 10.1056/NEJMra071297
3. Tsokos GC. Systemic lupus erythematosus. N Engl J Med (2011) 365(22):2110–21. doi: 10.1056/NEJMra1100359
4. Durcan L, O'Dwyer T, Petri M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet (2019) 393(10188):2332–43. doi: 10.1016/s0140-6736(19)30237-5
5. Kiriakidou M, Ching CL. Systemic lupus erythematosus. Ann Intern Med (2020) 172(11):ITC81–96. doi: 10.7326/AITC202006020
6. Almaani S, Meara A, Rovin BH. Update on lupus nephritis. Clin J Am Soc Nephrol (2017) 12(5):825–35. doi: 10.2215/CJN.05780616
7. Vera-Recabarren MA, Garcia-Carrasco M, Ramos-Casals M, Herrero C. Comparative analysis of subacute cutaneous lupus erythematosus and chronic cutaneous lupus erythematosus: Clinical and immunological study of 270 patients. Br J Dermatol (2010) 162(1):91–101. doi: 10.1111/j.1365-2133.2009.09472.x
8. Ball EM, Bell AL. Lupus arthritis–do we have a clinically useful classification? Rheumatol (Oxford) (2012) 51(5):771–9. doi: 10.1093/rheumatology/ker381
9. Fors Nieves CE, Izmirly PM. Mortality in systemic lupus erythematosus: An updated review. Curr Rheumatol Rep (2016) 18(4):21. doi: 10.1007/s11926-016-0571-2
10. Stohl W, Hiepe F, Latinis KM, Thomas M, Scheinberg MA, Clarke A, et al. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select b cell populations in patients with systemic lupus erythematosus. Arthritis Rheum (2012) 64(7):2328–37. doi: 10.1002/art.34400
11. Xia Y, Jiang S, Weng S, Lv X, Cheng H, Fang C. Antigen-specific immature dendritic cell vaccine ameliorates anti-dsdna antibody-induced renal damage in a mouse model. Rheumatol (Oxford) (2011) 50(12):2187–96. doi: 10.1093/rheumatology/ker231
12. Olsen NJ, Karp DR. Autoantibodies and sle: The threshold for disease. Nat Rev Rheumatol (2014) 10(3):181–6. doi: 10.1038/nrrheum.2013.184
13. Suurmond J, Zou YR, Kim SJ, Diamond B. Therapeutics to block autoantibody initiation and propagation in systemic lupus erythematosus and rheumatoid arthritis. Sci Transl Med (2015) 7(280):280ps5. doi: 10.1126/scitranslmed.aaa3809
14. Deng GM. Autoantibody igg protects bone erosion and destruction in sle arthritis. Am J Biomed Sci Res (2020) 11(2):136–7. doi: 10.34297/ajbsr.2020.11.001605
16. Deng GM. Lupus igg induces synovial inflammation but inhibits bone erosions in sle arthritis. Int J Orthopaedics Res (2020) 3(4):157–9. doi: 10.33140/IJOR.03.04.05
17. Fang X, Zaman MH, Guo X, Ding H, Xie C, Zhang X, et al. Role of hepatic deposited immunoglobulin G in the pathogenesis of liver damage in systemic lupus erythematosus. Front Immunol (2018) 9:1457. doi: 10.3389/fimmu.2018.01457
18. Pisetsky DS. Anti-DNA antibodies–quintessential biomarkers of sle. Nat Rev Rheumatol (2016) 12(2):102–10. doi: 10.1038/nrrheum.2015.151
19. Deng GM. Pathogenesis of skin injury of systemic lupus erythematosus. Curr Rheumatol Rep (2018) 20(2):5. doi: 10.1007/s11926-018-0713-9
20. Guo X, Fang X, He G, Zaman MH, Fei X, Qiao W, et al. The role of neutrophils in skin damage induced by tissue-deposited lupus igg. Immunology (2018) 154(4):604–12. doi: 10.1111/imm.12908
21. Cai Z, Xie C, Qiao W, Fei X, Guo X, Liu H, et al. The role of estrogen membrane receptor (G protein-coupled estrogen receptor 1) in skin inflammation induced by systemic lupus erythematosus serum igg. Front Immunol (2017) 8:1723. doi: 10.3389/fimmu.2017.01723
22. Li X, Guo X, Liu H, Gao G, Xu G, Fei X, et al. Skin inflammation induced by lupus serum was inhibited in il-1r deficient mice. Clin Immunol (2017) 180:63–8. doi: 10.1016/j.clim.2017.03.015
23. Liu L, Xu G, Dou H, Deng GM. The features of skin inflammation induced by lupus serum. Clin Immunol (2016) 165:4–11. doi: 10.1016/j.clim.2016.02.007
24. Deng GM, Liu L, Kyttaris VC, Tsokos GC. Lupus serum igg induces skin inflammation through the Tnfr1 signaling pathway. J Immunol (2010) 184(12):7154–61. doi: 10.4049/jimmunol.0902514
25. Deng GM, Tsokos GC. Pathogenesis and targeted treatment of skin injury in sle. Nat Rev Rheumatol (2015) 11(11):663–9. doi: 10.1038/nrrheum.2015.106
26. Zuo Y, Deng GM. Fc gamma receptors as regulators of bone destruction in inflammatory arthritis. Front Immunol (2021) 12:688201. doi: 10.3389/fimmu.2021.688201
27. Qiao W, Ding H, Zuo Y, Jiang L, Zhou J, Han X, et al. Lupus igg deposition causes arthritis but inhibits bone destruction through competitive occupation of fcgammari and reduced rankl signalling. Clin Transl Immunol (2020) 9(9):e1174. doi: 10.1002/cti2.1174
28. Zhang Q, Xiang L, Zaman MH, Dong W, He G, Deng GM. Predominant role of immunoglobulin G in the pathogenesis of splenomegaly in murine lupus. Front Immunol (2019) 10:3020. doi: 10.3389/fimmu.2019.03020
29. Kuhn A, Landmann A. The classification and diagnosis of cutaneous lupus erythematosus. J Autoimmun (2014) 48-49:14–9. doi: 10.1016/j.jaut.2014.01.021
30. Dahl MV. Usefulness of direct immunofluorescence in patients with lupus erythematosus. Arch Dermatol (1983) 119(12):1010–7. doi: 10.1001/archderm.119.12.1010
31. Provost TT. Lupus band test. Int J Dermatol (1981) 20(7):475–81. doi: 10.1111/j.1365-4362.1981.tb04906.x
32. Lee LA, Gaither KK, Coulter SN, Norris DA, Harley JB. Pattern of cutaneous immunoglobulin G deposition in subacute cutaneous lupus erythematosus is reproduced by infusing purified anti-ro (Ssa) autoantibodies into human skin-grafted mice. J Clin Invest (1989) 83(5):1556–62. doi: 10.1172/JCI114052
33. Shi ZR, Tan GZ, Meng Z, Yu M, Li KW, Yin J, et al. Association of anti-acidic ribosomal protein P0 and anti-galectin 3 antibodies with the development of skin lesions in systemic lupus erythematosus. Arthritis Rheumatol (2015) 67(1):193–203. doi: 10.1002/art.38891
34. Oke V, Vassilaki I, Espinosa A, Strandberg L, Kuchroo VK, Nyberg F, et al. High Ro52 expression in spontaneous and uv-induced cutaneous inflammation. J Invest Dermatol (2009) 129(8):2000–10. doi: 10.1038/jid.2008.453
35. Zhou J, Deng GM. The role of bacterial DNA containing cpg motifs in diseases. J Leukoc Biol (2021) 109(5):991–8. doi: 10.1002/JLB.3MR1220-748RRRRR
36. Wang X, Li Y, Wang Y, Feng Q, Yang P, Qin L. Intracerebroventricular administration of lupus serum induces microglia activation and leukocyte adhesion in the cerebromicrovasculature of mice. J Neuroimmunol (2019) 334:576994. doi: 10.1016/j.jneuroim.2019.576994
37. Yang C, Hou X, Feng Q, Li Y, Wang X, Qin L, et al. Lupus serum igg induces microglia activation through fc fragment dependent way and modulated by b-cell activating factor. J Transl Med (2019) 17(1):426. doi: 10.1186/s12967-019-02175-0
38. Kowal C, Degiorgio LA, Lee JY, Edgar MA, Huerta PT, Volpe BT, et al. Human lupus autoantibodies against nmda receptors mediate cognitive impairment. Proc Natl Acad Sci U S A (2006) 103(52):19854–9. doi: 10.1073/pnas.0608397104
39. ter Borg EJ, Horst G, Hummel EJ, Limburg PC, Kallenberg CG. Measurement of increases in anti-Double-Stranded DNA antibody levels as a predictor of disease exacerbation in systemic lupus erythematosus. A Long-Term Prospective Study. Arthritis Rheum (1990) 33(5):634–43. doi: 10.1002/art.1780330505
40. Forger F, Matthias T, Oppermann M, Becker H, Helmke K. Clinical significance of anti-dsdna antibody isotypes: Igg/Igm ratio of anti-dsdna antibodies as a prognostic marker for lupus nephritis. Lupus (2004) 13(1):36–44. doi: 10.1191/0961203304lu485oa
41. Flevaris K, Kontoravdi C. Immunoglobulin G n-glycan biomarkers for autoimmune diseases: Current state and a glycoinformatics perspective. Int J Mol Sci (2022) 23(9). doi: 10.3390/ijms23095180
42. Vletter EM, Koning MT, Scherer HU, Veelken H, Toes REM. A comparison of immunoglobulin variable region n-linked glycosylation in healthy donors, autoimmune disease and lymphoma. Front Immunol (2020) 11:241. doi: 10.3389/fimmu.2020.00241
43. Krishnan MR, Wang C, Marion TN. Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney Int (2012) 82(2):184–92. doi: 10.1038/ki.2011.484
44. Bruhns P, Jonsson F. Mouse and human fcr effector functions. Immunol Rev (2015) 268(1):25–51. doi: 10.1111/imr.12350
45. Wang J, Li Z, Xu L, Yang H, Liu W. Transmembrane domain dependent inhibitory function of fcgammariib. Protein Cell (2018) 9(12):1004–12. doi: 10.1007/s13238-018-0509-8
46. Brown EE, Edberg JC, Kimberly RP. Fc receptor genes and the systemic lupus erythematosus diathesis. Autoimmunity (2007) 40(8):567–81. doi: 10.1080/08916930701763710
47. Hu W, Zhang Y, Sun X, Zhang T, Xu L, Xie H, et al. Fcgammariib-I232t polymorphic change allosterically suppresses ligand binding. Elife (2019) 8. doi: 10.7554/eLife.46689
48. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol (2008) 8(1):34–47. doi: 10.1038/nri2206
49. Jiang L, Han X, Qiu W, Yu T, Feng R, Wang X, et al. Amelioration of lupus serum-induced skin inflammation in Cd64-deficient mice. Front Immunol (2022) 13:824008. doi: 10.3389/fimmu.2022.824008
50. Zhou X, Motta F, Selmi C, Ridgway WM, Gershwin ME, Zhang W. Antibody glycosylation in autoimmune diseases. Autoimmun Rev (2021) 20(5):102804. doi: 10.1016/j.autrev.2021.102804
51. Jing C, Castro-Dopico T, Richoz N, Tuong ZK, Ferdinand JR, Lok LSC, et al. Macrophage metabolic reprogramming presents a therapeutic target in lupus nephritis. Proc Natl Acad Sci U S A (2020) 117(26):15160–71. doi: 10.1073/pnas.2000943117
52. Deng GM, Liu L, Tsokos GC. Targeted tumor necrosis factor receptor I preligand assembly domain improves skin lesions in Mrl/Lpr mice. Arthritis Rheum (2010) 62(8):2424–31. doi: 10.1002/art.27534
53. Wang J, Yang C, Zhao Q, Zhu Z, Li Y, Yang P. Microglia activation induced by serum of sle patients. J Neuroimmunol (2017) 310:135–42. doi: 10.1016/j.jneuroim.2017.07.010
54. Faridi MH, Khan SQ, Zhao W, Lee HW, Altintas MM, Zhang K, et al. Cd11b activation suppresses tlr-dependent inflammation and autoimmunity in systemic lupus erythematosus. J Clin Invest (2017) 127(4):1271–83. doi: 10.1172/JCI88442
55. Idborg H, Eketjall S, Pettersson S, Gustafsson JT, Zickert A, Kvarnstrom M, et al. Tnf-alpha and plasma albumin as biomarkers of disease activity in systemic lupus erythematosus. Lupus Sci Med (2018) 5(1):e000260. doi: 10.1136/lupus-2018-000260
56. Aringer M. Inflammatory markers in systemic lupus erythematosus. J Autoimmun (2020) 110:102374. doi: 10.1016/j.jaut.2019.102374
57. Zampieri S, Alaibac M, Iaccarino L, Rondinone R, Ghirardello A, Sarzi-Puttini P, et al. Tumour necrosis factor alpha is expressed in refractory skin lesions from patients with subacute cutaneous lupus erythematosus. Ann Rheum Dis (2006) 65(4):545–8. doi: 10.1136/ard.2005.039362
58. Postal M, Appenzeller S. The role of tumor necrosis factor-alpha (Tnf-alpha) in the pathogenesis of systemic lupus erythematosus. Cytokine (2011) 56(3):537–43. doi: 10.1016/j.cyto.2011.08.026
59. Schiffer L, Bethunaickan R, Ramanujam M, Huang W, Schiffer M, Tao H, et al. Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. J Immunol (2008) 180(3):1938–47. doi: 10.4049/jimmunol.180.3.1938
60. Ramanujam M, Davidson A. Targeting of the immune system in systemic lupus erythematosus. Expert Rev Mol Med (2008) 10:e2. doi: 10.1017/S1462399408000562
61. Li Y, Lee PY, Kellner ES, Paulus M, Switanek J, Xu Y, et al. Monocyte surface expression of fcgamma receptor ri (Cd64), a biomarker reflecting type-I interferon levels in systemic lupus erythematosus. Arthritis Res Ther (2010) 12(3):R90. doi: 10.1186/ar3017
62. Hoi AY, Hickey MJ, Hall P, Yamana J, O'Sullivan KM, Santos LL, et al. Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in Mrl/Lpr mice. J Immunol (2006) 177(8):5687–96. doi: 10.4049/jimmunol.177.8.5687
63. Deng GM, Liu L, Bahjat FR, Pine PR, Tsokos GC. Suppression of skin and kidney disease by inhibition of spleen tyrosine kinase in lupus-prone mice. Arthritis Rheum (2010) 62(7):2086–92. doi: 10.1002/art.27452
64. Deng GM, Kyttaris VC, Tsokos GC. Targeting syk in autoimmune rheumatic diseases. Front Immunol (2016) 7:78. doi: 10.3389/fimmu.2016.00078
65. Deng GM, Tsokos GC. The role of syk in cutaneous lupus erythematosus. Exp Dermatol (2016) 25(9):674–5. doi: 10.1111/exd.13018
Keywords: SLE, organ damage, autoantibody, macrophage, IgG, FcγR
Citation: Qiu W, Yu T and Deng G-M (2022) The role of organ-deposited IgG in the pathogenesis of multi-organ and tissue damage in systemic lupus erythematosus. Front. Immunol. 13:924766. doi: 10.3389/fimmu.2022.924766
Received: 20 April 2022; Accepted: 14 September 2022;
Published: 13 October 2022.
Edited by:
Kerstin Nündel, University of Massachusetts Medical School, United StatesReviewed by:
Anna Ghirardello, University of Padua, ItalyCopyright © 2022 Qiu, Yu and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guo-Min Deng, Z21kZW5nQGh1c3QuZWR1LmNu
†These authors contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.