 Elena Erausquin
Elena Erausquin Pau Serra
Pau Serra Daniel Parras
Daniel Parras Pere Santamaria4,5*
Pere Santamaria4,5* Jacinto López-Sagaseta
Jacinto López-Sagaseta
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 28 July 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.924311
We recently provided evidence for promiscuous recognition of several different hybrid insulin peptides (HIPs) by the highly diabetogenic, I-Ag7-restricted 4.1-T cell receptor (TCR). To understand the structural determinants of this phenomenon, we solved the structure of an agonistic HIP/I-Ag7 complex, both in isolation as well as bound to the 4.1-TCR. We find that HIP promiscuity of the 4.1-TCR is dictated, on the one hand, by an amino acid sequence pattern that ensures I-Ag7 binding and, on the other hand, by the presence of three acidic residues at positions P5, P7 and P8 that favor an optimal engagement by the 4.1-TCR’s complementary determining regions. Surprisingly, comparison of the TCR-bound and unbound HIP/I-Ag7 structures reveals that 4.1-TCR binding triggers several novel and unique structural motions in both the I-Ag7 molecule and the peptide that are essential for docking. This observation indicates that the type 1 diabetes-associated I-Ag7 molecule is structurally malleable and that this plasticity allows the recognition of multiple peptides by individual TCRs that would otherwise be unable to do so.
Type 1 diabetes (T1D) in nonobese diabetic (NOD) mice results from destruction of pancreatic beta-cells by T-cells targeting a growing list of autoantigens (1, 2). Compelling experimental evidence have shown that T1D onset requires the activation and recruitment of autoreactive CD4+ T-cells, especially those recognizing insulin B-chain epitopes (3). T-cell specificities recognizing hybrid insulin peptides (HIPs), which arise from the fusion of fragments of insulin chains with peptides from other hormones that are exclusively expressed in the insulin secretory granules, have also been implicated in T1D initiation and progression (4). In turn, disease-initiating T-cells orchestrate the activation of downstream effectors of diabetogenic autoimmunity, such as beta cell-cytotoxic CD8+ T-cells (5–11).
Susceptibility to T1D (and other autoimmune diseases) involves a small number of genes with large effects (e.g. Human Leukocyte Antigen (HLA)-coding genes) and a larger number of genes with smaller contributions (12–15). HLA-DQ and its murine I-A counterpart play key roles, albeit through unclear mechanisms. DQβ chains carrying Ser, Ala or Val at position 57 provide risk, whereas those carrying Asp at this position afford protection (16, 17). The nonobese diabetic (NOD) H-2 haplotype (H-2g7) encodes a unique I-Aαd/I-Aβg7 heterodimer in which the Pro and Asp found at positions 56 and 57 in most I-Aβ chains are replaced by His and Ser, respectively. Thus, the I-Aαd/I-Aβg7 pair conforms the murine major histocompatibility complex class II (MHCII) molecule I-Ag7 that is uniquely expressed by NOD mice.
These polymorphisms allow the T1D-associated MHCII molecules to bind to peptides carrying acidic residues at position 9 (P9) (18).
Transgenic NOD mice expressing the I-Ag7-restricted 4.1-TCR, cloned from islet-associated CD4+ T-cells of diabetic NOD mice, spontaneously develop a dramatically accelerated form of T1D (9). However, in H-2 heterozygous NOD mice co-expressing anti-diabetogenic MHCII molecules this TCR undergoes negative selection/Treg cell re-programming by recognizing the latter on hematopoietic APCs in a β56-67-regulated and peptide-dependent manner, indicating that it is MHCII promiscuous (19–24). More recently, we have shown that the 4.1-TCR is also antigenically promiscuous, recognizing seven different I-Ag7-binding HIPs that result from the fusion of various chromogranin A and/or insulin C fragments, including post-translationally modified peptides (25).
The work described herein was undertaken to gain an understanding of the molecular underpinnings underlying the antigenic promiscuity of this TCR. Our crystallographic studies show that the HIP promiscuity of the 4.1-TCR is primarily dictated by (1) the presence of three acidic residues at positions P5, P7 and P8 that help lock a conformationally flexible I-Aβg7 residue away from its position in the TCR-unbound state, which precludes 4.1-TCR docking; and (2) by structural adaptations of several other I-Aαd, I-Aβg7 and peptide residues that promote 4.1-TCR engagement. Collectively, these observations indicate that the pro-diabetogenic I-Ag7 molecule is structurally malleable and suggest that this plasticity might contribute to diabetogenesis by expanding the antigenic repertoire of specific TCRs.
The sequence of the agonist HIP39 antigen lacking its three last amino acids was fused to the I-Aβg7 chain through an Asp>Cys mutation [LQTLALEVEDDPC] to enable covalent linkage through a disulfide bridge with a concomitant Ile74Cys replacement in I-Aαd.
HIP39/I-Ag7 monomers were expressed and produced in lentiviral-transduced CHO-S cells as knob-into-hole (KIH) Fc fusions (26). Briefly, CHO-S cells were transduced sequentially with lentiviral particles encoding a HIP39 peptide-tethered, KIH-based pMHCII heterodimer carrying a 3C cleavage site immediately upstream of each of the two KIH halves and downstream IRES-fluorochrome-encoding cassettes (Figure 1). Sorted CFPhi/GFPhi CHO-S cells were cultured as described (26), and the HIP39/I-Ag7 heterodimers purified from cell culture supernatants via protein G affinity chromatography. The heterodimers were first digested with a 6xHis-tagged human rhinovirus 3C (HRV 3C) protease and then purified away from the KIH tail and the 3C protease on a nickel-affinity column (Figure 1). To express the 4.1-TCR, we engineered expression constructs encoding the 4.1-TCR variable domains fused with human constant regions carrying cysteine-engineered pairs to favor assembly of the TCRαβ as described (27). The TCRα and TCRβ constructs were cloned into the pet28b vector and expressed separately in E.coli. The heterodimeric 4.1-TCRαβ complex was refolded from inclusion bodies as described (28), isolated and purified to homogeneity for crystallization studies (Figure 2).
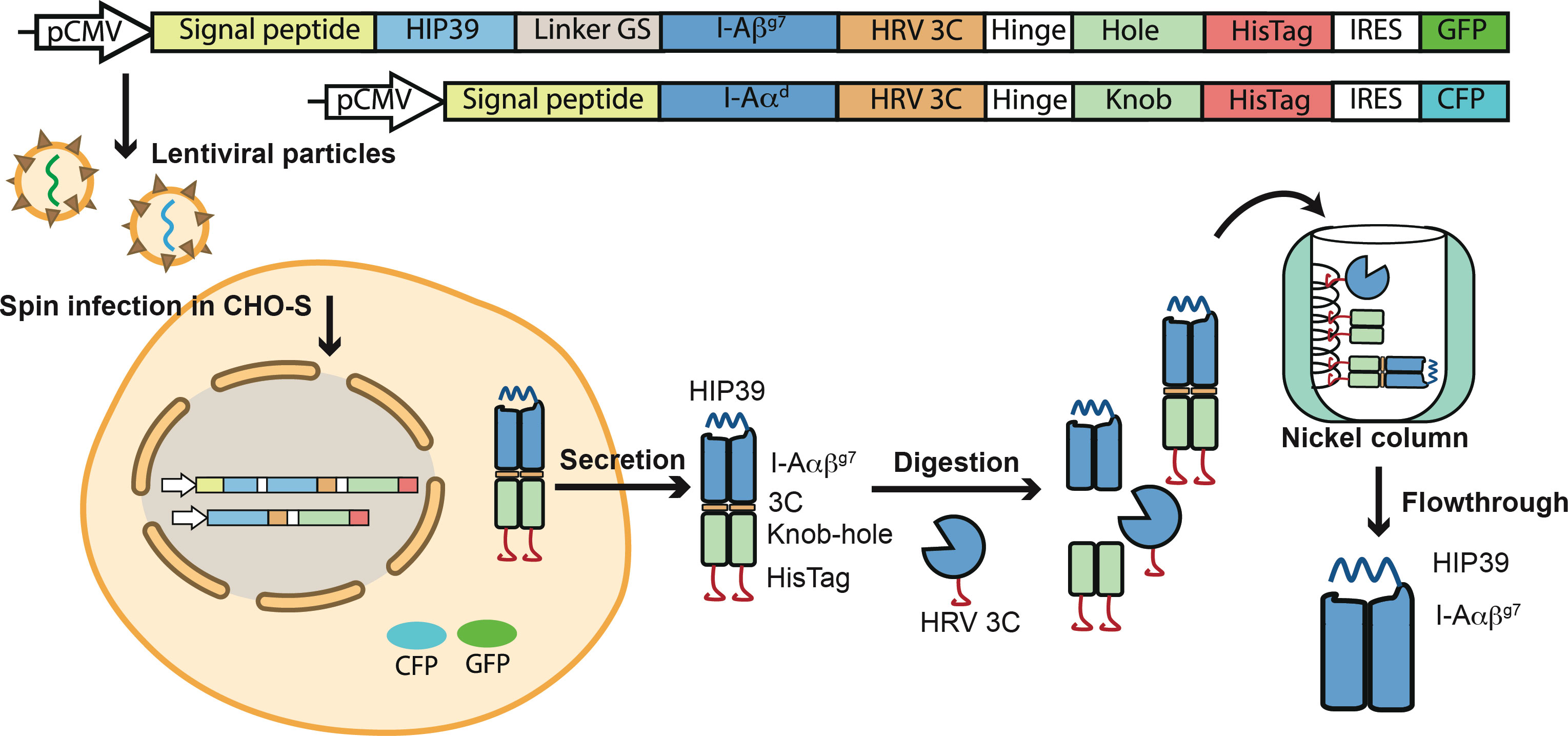
Figure 1 Production of the 4.1-TCR and HIP39/I-Ag7 complexes. Top, lentiviral constructs encoding the HIP39/I-Ag7. The HIP39 peptide sequence is tethered to the I-Aβg7 chain via a flexible GS linker. The HIP39-linker-I-Aβg7 fusion was then fused to an Fc-knob carrying an upstream cleavage site for the HRV 3C enzyme, as well as a downstream 6xHis tag and an IRES-eGFP-coding cassette. The I-Aαd chain was fused to an Fc-hole, also carrying an upstream 3C cleavage site and downstream 6xHis tag and an IRES-CFP-coding cassette. Bottom, transduced CHO-S cells were sorted for high levels of CFP and GFP expression and the corresponding cell culture supernatants used as a source of heterodimeric HIP39-I-Aαβg7-3C-KIH-HisTag molecules. The heterodimers were purified via protein G chromatography, digested with 6xHis-tagged HRV 3C and purified away from the KIH tail and the HRV 3C enzyme via sequential nickel-affinity and protein G chromatography.
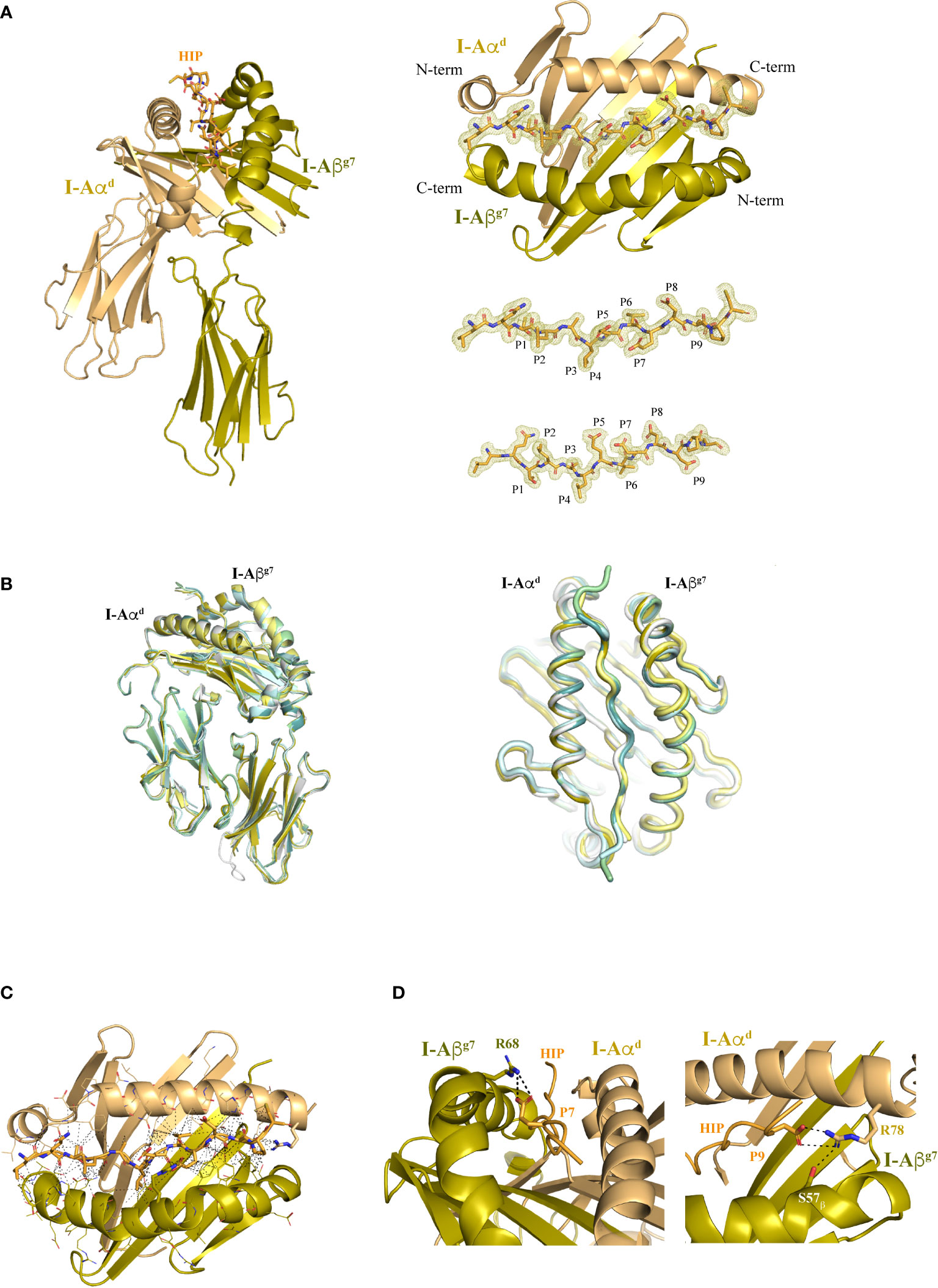
Figure 2 Structure of I-Ag7 with a bound agonistic hybrid insulin peptide (HIP39). (A) Left panel, I-Aαd and I-Aβg7 chains are shown in dun and olive colors, respectively, with the bound HIP39 peptide shown in stick format in the central cavity. Right top panel, top view of the I-Ag7 MHC platform and the HIP antigen inside the 2Fo-Fc map, shown as a light yellow colored mesh. Right lower panel, top and side views of the HIP antigen surrounded by the 2Fo-Fc map. (B) Lateral and top views of HIP39/I-Ag7 overlayed with other I-Ag7 complexes crystallized with their respective peptides (HIP, gray, PDB 7QHP; pale yellow, GAD65, PDB 1ES0; RLGL-WE14, olive, PDB 5DMK; p8E9E, pale green, PDB 6BLQ; p8E9E6SS, pale cyan, PDB 6BLR; p8G9E, light teal, PDB 6BLX). (C) The contacts (≤ 4 Å) between I-Ag7 and the HIP are displayed as dashed lines. (D) Detail of the salt bridge formed between I-Aβg7 Arg68 and Glu (P7) in the HIP and between I-Aαd Arg78 with Asp (P9). The H-bond of Arg78 with I-Aβg7 Ser57 is also shown.
The HIP39/I-Ag7 complex crystallized in space group C2221 with a single pMHCII molecule per asymmetric unit (Supplementary Figure 1). The structure was solved with a resolution of 1.8 Å (Table 1). Difference Fo-Fc map was readily evident for the bound peptide in the MHC’s peptide-binding groove and enabled a precise identification of the HIP39-derived residues in the complex (Figure 2A). Overall, the I-Ag7 molecular structure in this pMHCII is essentially identical to that seen in previously crystallized peptide-I-Ag7 complexes (Figure 2B). The HIP39 epitope is tightly packaged into the peptide-binding groove of the MHCII molecule through a nourished net of interactions with both the I-Aαd and I-Aβg7 chains (Figures 2C, D and Supplementary Tables S1, S2). These include Van der Waals (VDW), hydrogen bonding (H-bonds), ionic interactions (salt bridges) and water bridges. Contacts are not concentrated on a particular region of the antigen, rather, these are distributed longitudinally across the peptide-binding groove, consistent with a strong binding affinity of HIP39 for I-Ag7, as documented previously (25) (Figure 2C). Positions P5, P7 and P8 in the peptide are occupied by acidic residues (Glu/Glu/Asp) that expose side chains outwardly, making them available for potential interactions with cognate TCRs. As expected (18), the negatively charged Asp at the peptide’s P9 position and the Ser at I-Aβg7’s position 57 stabilize the I-Aβg7’s Arg78 side chain through H-bonding (Figure 2D).
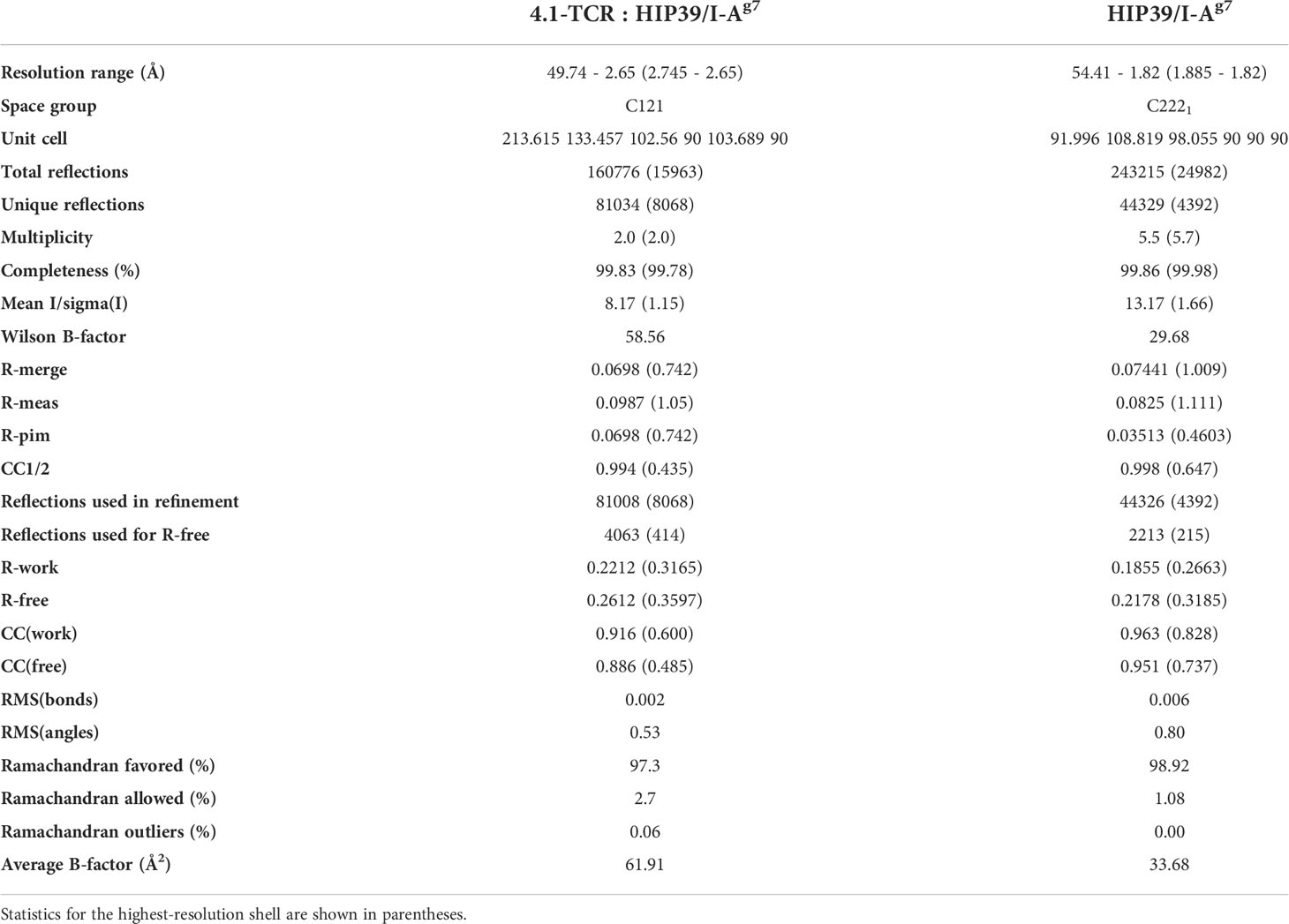
Table 1 Diffraction data collection and refinement statistics.
The I-Ag7-restricted 4.1-TCR was originally isolated from autoreactive CD4+ T lymphocytes infiltrating the pancreatic islets of NOD mice. The α and β chains are encoded by TRAV5D-4*04/TRAJ40*01 and TRBV16*01/TRBJ2-4*01 VDJ rearrangements, respectively. For crystallization studies, the variable regions of the α and β chains were fused with the corresponding human TCR constant regions mutated to encode a pair of heterodimerizing cysteines. TCRα and β chains were folded jointly from inclusion bodies, purified and incubated with recombinant HIP39/I-Ag7 complexes prior to crystallization trials. Plate-like crystals were used to collect a 2.65 Å full native dataset that enabled solution of the ternary complex structure. Crystals belonged to the C121 space group and two equivalent 4.1-TCR : HIP39/I-Ag7 complexes (RMSD 0.216) were obtained in the asymmetric unit (Supplementary Figure 1). Recognition of HIP39/I-Ag7 by the 4.1-TCR follows an overall canonical TCR-pMHCII docking pattern (Figure 3). That is, the 4.1-TCR binds on top of the peptide-binding MHCαβ scaffold. Whereas both CDR3α and CDR3β loops are aligned over the peptide-binding groove, the germline encoded CDR1αβ/CDR2αβ sit on top of the MHC α-helices (Figure 3A). As seen in other TCR-pMHCII structures, the 4.1-TCRα chain interacts primarily with the MHCβ chain, while its 4.1-TCRβ counterpart largely contacts the MHCα chain (Figure 3B). The docking mode presents a geometry often seen in TCR-MHC-II complexes (Figure 3B). The contacts with the TCR are of varied nature, with a predominant weight for non-polar interactions. The side chain-side chain contacts are distributed in a manner such that both alpha and beta chains in the TCR and MHC-II interact with one another (Figure 3C). And a role for water molecules is observed that enables additional intermolecular contacts.
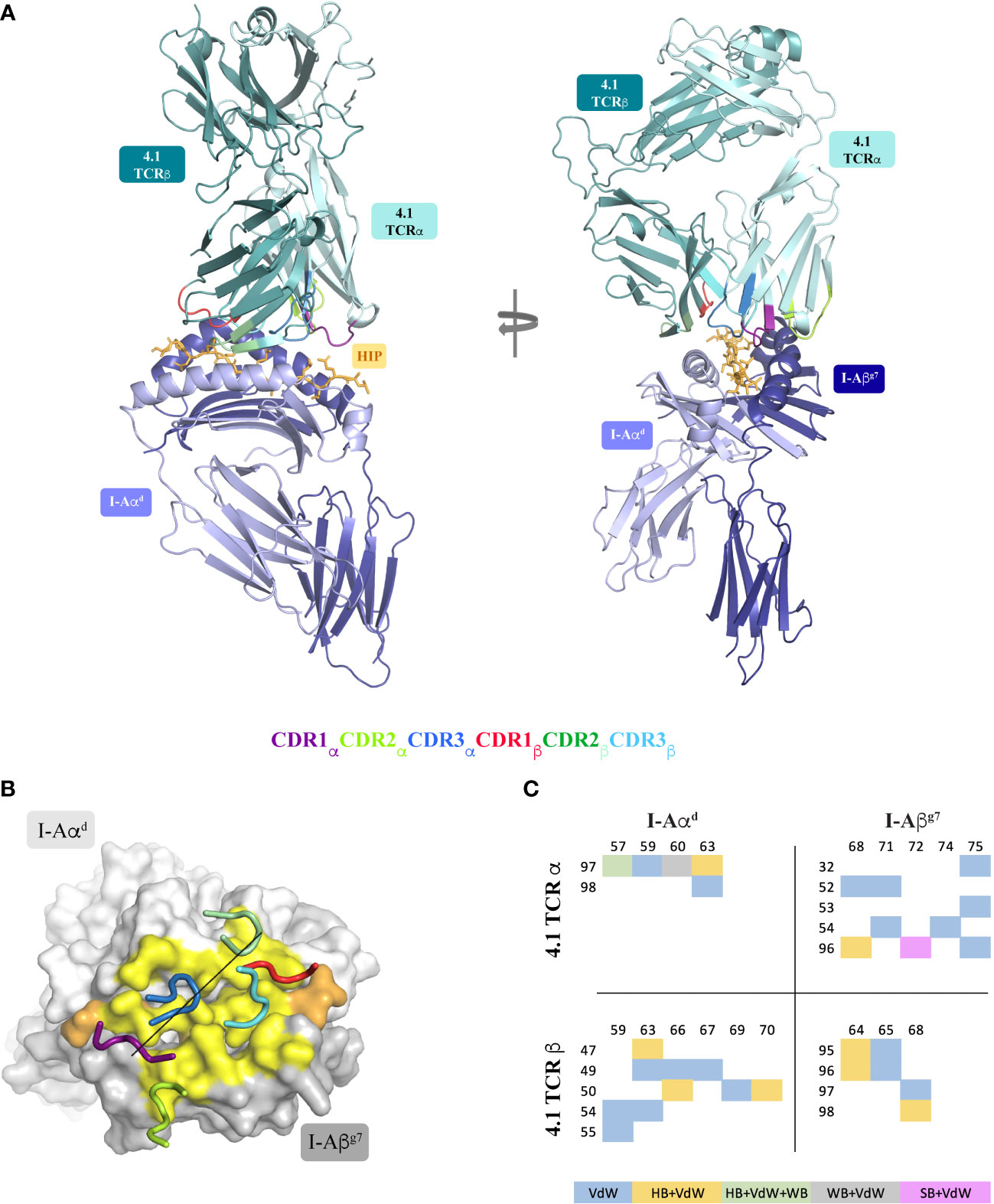
Figure 3 Structure of the 4.1-TCR : HIP39/I-Ag7 complex. (A) Side and front views of the 4.1-TCR bound to HIP39/I-Ag7. The α/β chains of the TCR are colored pale cyan and light teal, respectively, and the CDR loops highlighted as described in the legend underneath. I-Aαd and I-Aβg7 are in light blue and deep blue colors and the HIP antigen shown in stick format in orange color. (B) Surface representation of I-Aαd and I-Aβg7 in light and dark grey colors, respectively, and of the bound HIP antigen, in orange color. The 4.1-TCR footprint on I-Ag7 is colored yellow and highlights residues on the MHC and antigen that establish any type of contact (≤ 4 Å) with the TCR. The CDR loops are displayed in tube format with the color register described in (A). The black straight line connects the conserved disulfide bridges of the TCRα and TCRβ variable regions. (C) Distribution and chemical nature of the intermolecular contacts in the 4.1-TCR : HIP39/I-Ag7 complex. VdW, Van der Waals; HB, Hydrogen bond; WB, Water bridge; SB, Salt bridge.
The TCR-pMHCII interface covers an average buried surface area (BSA) of 1172 A2, a value somewhat lower than that reported for most other TCR-pMHCII interfaces.
Importantly, most of the residues in I-Aαd and I-Aβg7 that make contacts with the TCR are conserved across murine MHCII molecules. The I-Aαd chain contributes to TCR binding with multiple residues and contacts of various types (Figure 4A and Supplementary Tables S3–S6). Glu57, Gln59 and Gln63 play a more significant role. While Glu57 engages both the 4.1-TCR and the HIP39 peptide via H-bonds (Figures 4A, B and Supplementary Tables S7, S8), Gln59 and Gln63 contribute various VDW contacts with CDR3α and CDR2β residues (Figures 4A–C and Supplementary Tables S3–S6). Gln63 participates also in a well-defined polar triad with CDR3α ’s Asn97 and CDR2β’s Tyr47 side chains (Figure 4C).
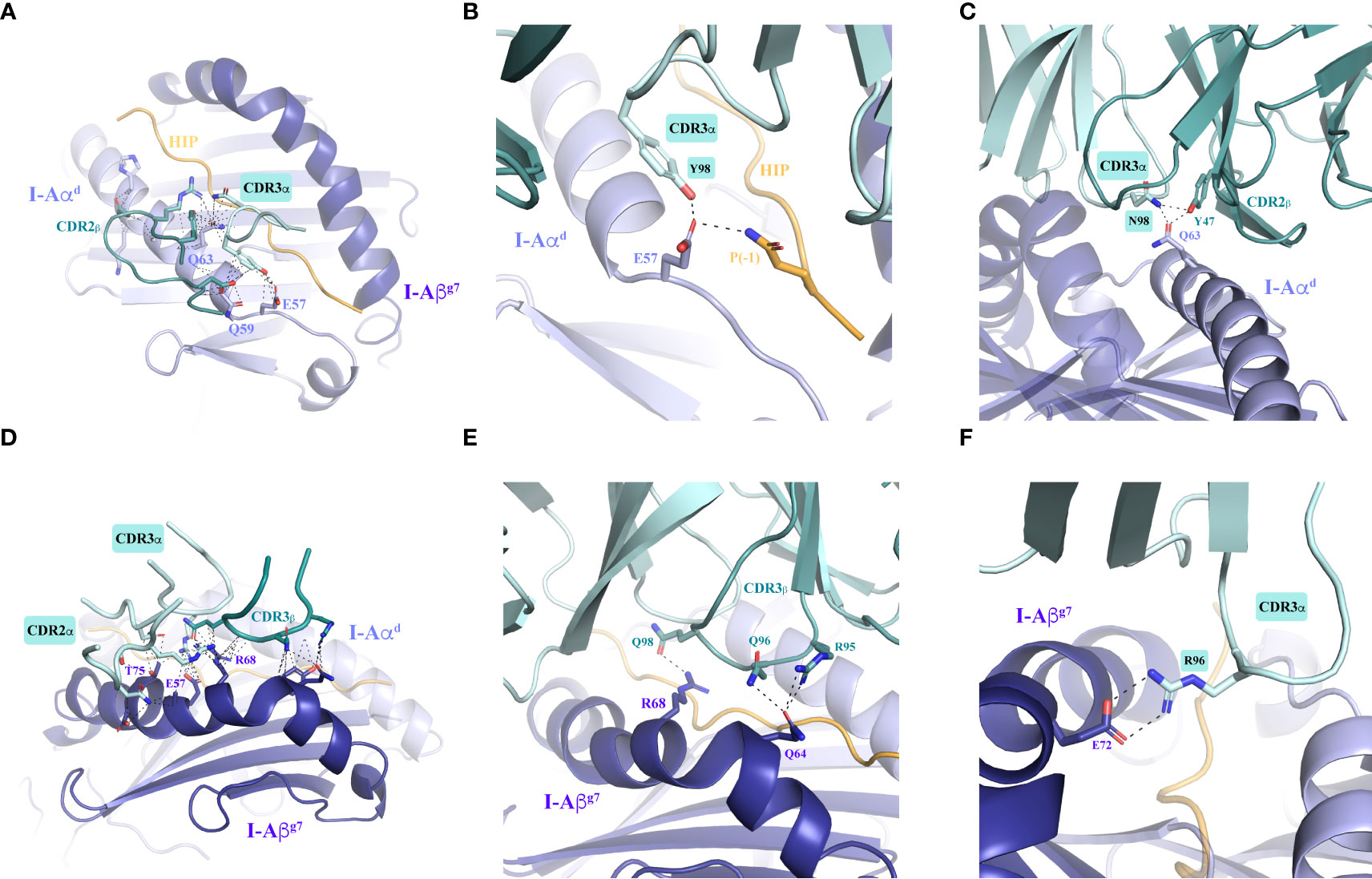
Figure 4 Intermolecular contacts between the 4.1-TCR and HIP39/I-Ag7. (A) Overall contacts (≤ 4 Å) with the TCR mediated by I-Aαd, displayed as dashed lines. (B, C) Polar interactions mediated by I-Aαd are shown in zoomed-in images. The panels show specific interactions established by Glu57 in I-Aαd with Tyr98 in the TCR CDR3α and Gln side chain in the HIP P(-1) position (B) and by Gln63 in I-Aβg7 with Asn98 and Tyr47 in the TCR CDR3α and CDR2β, respectively (C). (D) Overall contacts (≤ 4 Å) with the TCR mediated by I-Aβg7. (E, F) Polar and ionic contacts established by Arg68 and Gln64 (panel E) in I-Aβg7 with the TCR CDR3β loop, and a close salt bridge by Glu72 with CDR3α Arg96 side chain (panel F). In all cases the interacting residues are highlighted as sticks. Contacts are displayed as dashed black lines. The TCRα and TCRβ chains are colored pale cyan and deep teal, respectively.
The I-Aβg7 residues that participate in interactions with the 4.1-TCR (Figures 4D, E and Supplementary Figures S2, S4A and Supplementary Tables S9–S12) are also largely conserved. Arg68 and Glu72 engage both CDR3αβ loops through VDW and polar interactions. Glu72 and Thr75 establish remarkable interactions through a salt bridge with CDR3α’s Arg96 (Figure 4F) and VDW contacts with the three CDRs of the 4.1-TCRα chain (Supplementary Figure 2), respectively. A water bridge provides an additional contact between the 4.1-CDR3β and Gln64 in I-Aβg7 (Supplementary Figure 3A).
We have recently shown that the 4.1-TCR can recognize 6 additional HIPs, in addition to HIP39 (HIPs 15, 18, 30, 30 Q10E, 32 and 32 Q9E), conforming to the following nonamer motif: TLALE(V/A/G)E(D/E)(D/E/P/Q). In all these HIPs, the left arm is donated by the Insulin C-peptide (InsC), truncated at different carboxyterminal residues, whereas the right arm corresponds to different naturally occurring proteolytic products of either Chromogranin A (ChgA) or InsC57-63. HIP30Q10E and HIP32Q9E correspond to the deamidated forms of HIPs 30 and 32, respectively. All these HIPs promoted IFNγ secretion by activated 4.1-CD4+ T-cells, albeit with different functional avidities. Experiments using pMHCII tetramers indicated that these 7 HIPs could be further subclassified into three different subsets: one with high functional avidity and strong tetramer binding avidity (HIP39 and HIP32Q9E); a second subset having a slightly lower functional avidity but only marginal tetramer binding avidity (HIP32 and HIP30Q10E); and a third subset displaying low functional and physical binding avidities (HIP30, HIP15 and HIP18). A fundamental difference between groups 1 and 2 lies in the replacement of a negatively charge amino acid (D or E) at P9, involved in I-Ag7 binding, with P or Q. Unlike tetramer binding, which requires the display of the peptide on the same register on its four monomers, the IFNγ secretion assay can withstand register multiplicity, particularly at high concentration of soluble peptide (25). Of note, additional homologous HIPs carrying E7A or E5V replacements (HIP40 and HIPD1, respectively), were completely devoid of agonistic or tetramer binding activity (25).
Collectively, these data suggested that positions E5, E7 and D8 are key 4.1-TCR contact residues. Close analysis of the ternary structure described herein demonstrates that this is indeed the case. The first noticeable feature is that E5 and D8 are solvent exposed and, therefore, easily accessible for TCR recognition (Figure 5A and Supplementary Tables S13–S16). Thus, whereas E5 makes H-bonds with CDR3α’s backbone nitrogens, D8 engages CDR1β’s Ser29 (via an H-bond) and CDR2β’s Arg49 (via a salt bridge) (Figure 5C). E7, on the other hand, located in the P7 pocket defined by Tyr65 and Arg68 (Figure 5B), interacts with the 4.1-TCR by tethering to the CDR3β’s backbone nitrogen through H-bonding (Figure 5C). Further, a water molecule coordinates a 4-arm interaction that connects the CDR2β and CDR3β with Glu7 and Asp8 in the HIP antigen (Supplementary Figure S3B). These key interactions are further strengthened by the Ala3 backbone oxygen, which interacts with CDR3α’s Arg96 η1/η2 nitrogen, and by additional VDW contacts between HIP and CDR1α residues. As in other TCR:pI-Ag7 structures, the CDR2α does not target residues in the antigen peptide.
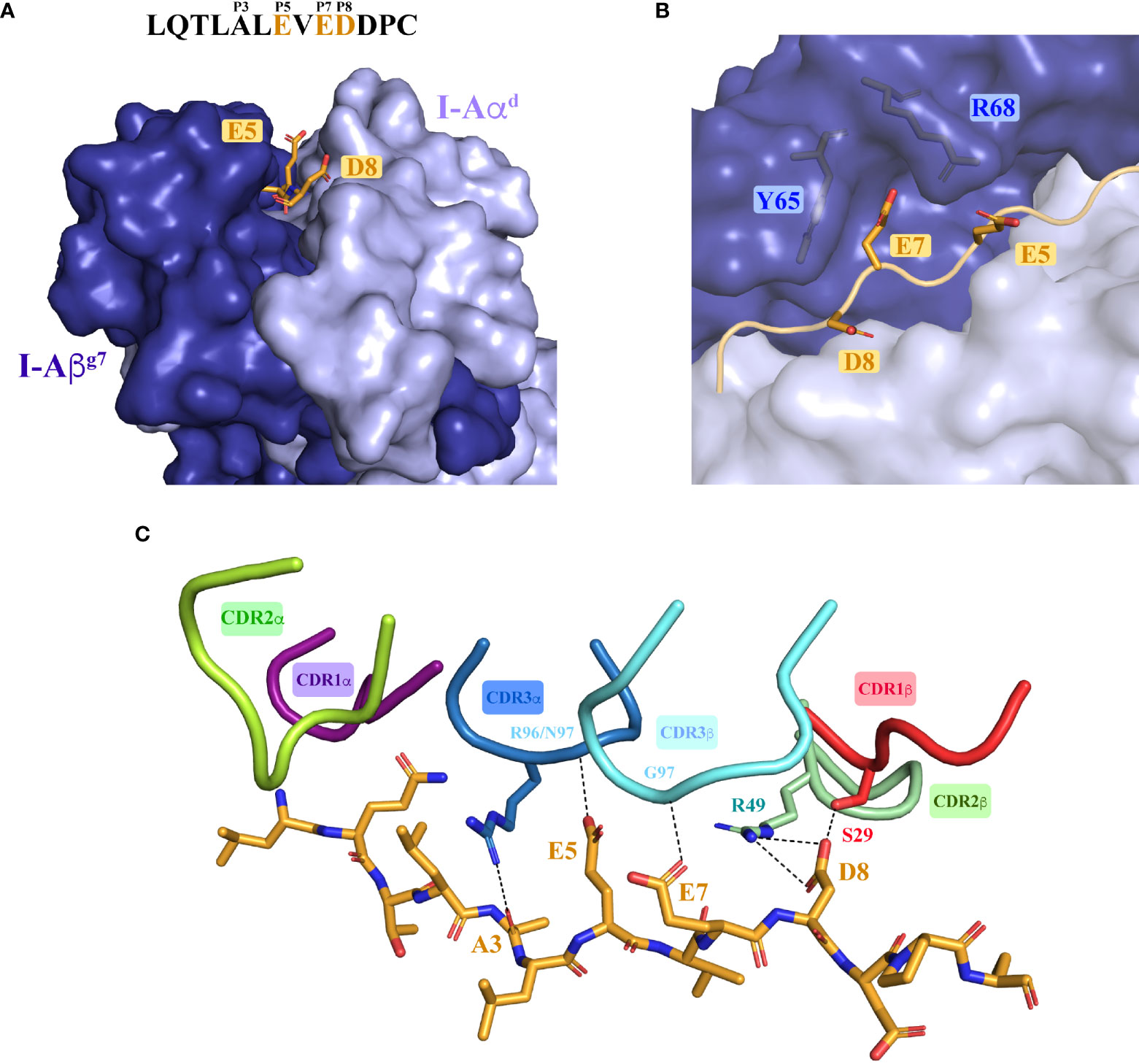
Figure 5 Molecular recognition of the agonistic I-Ag7-presented HIP39 antigen by the diabetogenic 4.1-TCR. (A) Front view of the I-Ag7 molecule, shown as surfaces with the I-Aαd and I-Aβg7 chains colored light and deep blue. The HIP39 (displayed as an orange ribbon in the MHC groove) residues Glu5, Glu7 and Asp8 are highlighted as sticks. The sequence of HIP39 is shown above the molecule with the acidic interacting residues highlighted in orange. (B) Detail of Glu7 in the HIP39 embedded in the P7 pocket between Tyr65 and Arg68 in I-Aβg7. (C) Polar and ionic interactions (dashed lines) established between the 4.1-CDRs and the HIP39 peptide. The CDR colors are consistent with those shown in Figure 2. Note that VDW contacts with CDR1α are not displayed. The CDR2α does not contact the HIP.
Collectively, these observations indicate that the presence of concurrent acidic residues at positions 5, 7 and 8 in peptides anchored onto I-Ag7 enables autorecognition by the 4.1-CD4+ T-cell.
Availability of the TCR-bound and free HIP39/I-Ag7 structures afforded a unique opportunity to investigate whether promiscuous recognition of HIP/I-Ag7 complexes by the 4.1-TCR involved structural adaptations of the pMHCII and/or the TCR. At first sight, no remarkable differences were noticed in the unbound HIP39/I-Ag7 complex as compared to its 4.1-TCR-bound counterpart, consistent with an overall RMSD of 0.383 for the HIP39/I-Ag7 complex. However, comparison of amino acid side chain positioning in the two structures revealed that 4.1-TCR engagement involves subtle, yet critical motions of certain I-Aβg7 residues’ side chains without which TCR engagement would not be possible.
The most obvious conformational change involves I-Aβg7’s Arg68, which switches the positioning of its side chain from an initial arrangement away from I-Ag7’s peptide-binding groove, to a locked and HIP39-oriented conformation in the presence of the 4.1-TCR (Figure 6A and Supplementary Figure S4). The incompatibility of such initial arrangement with docking of the 4.1-TCR’s is readily evident, as steric hindrance caused by the positioning of Arg68 side chain in the TCR-unbound structure is incompatible with CDR3β accommodation (Figure 6A). The TCR-bound coordinates show how Arg68 shifts its side chain towards the HIP39 peptide, creating a novel set of polar interactions that pair Arg68’s ε and η nitrogens with both 4.1-CDR3β’s Gln98 and the peptide’s E5 and E7 residues via salt bridges (Figure 6B and Supplementary Tables S5, S6, S17 and S18).
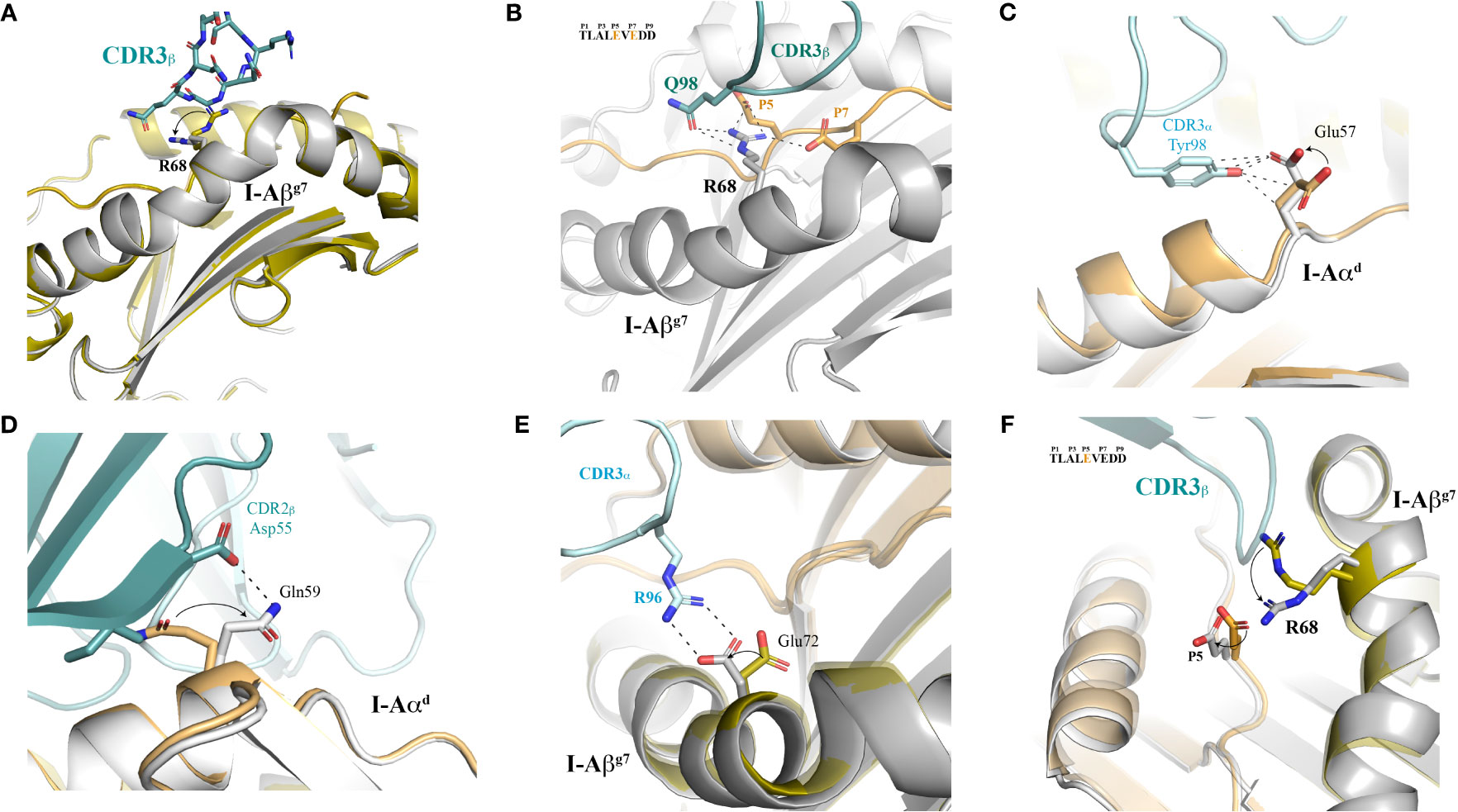
Figure 6 Structural plasticity of both I-Ag7 and peptide contribute to 4.1-TCR docking. (A) The MHC platform is displayed in cartoon mode and both the free and TCR-bound I-Ag7 molecules are aligned and superimposed to one another. The TCR-bound MHC is shown in grey color. The 4.1 CDR3β is shown as sticks. (B) H-bonds and salt bridges between I-Aβg7 Arg68, the 4.1-TCR and the HIP are highlighted with dashed lines. The sequence of the HIP is shown as an inset, with the interacting residues highlighted in orange color. (C–E) Structural motions observed in Glu57 in I-Aαd and contacts established with the CDR3α Tyr98 (C). Structural transition in the I-Aβg7 Gln59 side chain and polar contacts made with the TCR CDR2β Asp55 side chain (D). The presence of the TCR Arg96 in the CDR3α is concomitant with a structural motion of I-Aβg7 Glu72 that enables a closer and stronger salt bridge (panel E) between the two proteins. (F) Sequential transitions of Arg68 and HIP (Glu5) in the presence of 4.1-TCR. In all cases, structural motions are indicated with arrows, and go from the TCR-free to the TCR-bound structures.
This positioning of Arg68’s side chain away from the peptide-binding groove is also seen in other pI-Ag7 crystal structures in the absence of a TCR (Supplementary Figure S4, upper panel), or in TCR-bound pI-Ag7 structures in which the peptide’s P5 and P7 positions are occupied by neutral residues, as is the case for the I.29 and 8F.10 TCR ligands (Supplementary Figure S4, lower panel). In contrast, the Arg68’s side chain is found pointing towards the peptide-binding groove in the structure of a hen egg lysozyme (HEL) peptide/I-Ag7 complex bound to its cognate 21.30 TCR. A major difference between these structures is that P5 and P7 in the HEL peptide are occupied by Glu and Tyr, respectively, which lock Arg68 through a polar set of interactions. In summary, these results indicate that I-Aβg7 Arg68’s plasticity is critical for recognition of peptides with non-neutral residues at P5 and P7 by cognate I-Ag7-restricted TCRs.
Other relevant structural motions are observed in I-Aαd residues Glu57, Gln59 and Glu72 (Figures 6C–E). Glu57 shows a modest but elegant movement of its side chain that facilitates H-bonding with CDR3α’s Tyr98 (Figure 6C). Gln59 undergoes a more significant transition of about 180°, where the side chain rearranges in such a way that enables pairing with CDR2β’s Asp55 via H-bonds (Figure 6D). Likewise, the Glu72 side chain slightly shifts its location to enable stronger salt bridges with CDR3α’s Arg96 (Figure 6E).
We also observed mild, yet pivotal structural transitions in the HIP39 peptide residues that occur in synch with the Arg68 switch discussed above (Figure 6F). Specifically, the structural re-positioning of the Arg68’s side chain (Figure 6A) appears to force a translation of HIP39’s E5 towards I-Aαd. This translation eliminates a steric hindrance between Arg68 and E5, and enables accommodation of Arg68, which is essential for 4.1-CDR3β’s docking (Figure 6F), as described above.
To gain further insights into the driving forces behind HIP recognition, we explored the impact of 4.1-TCR binding in the energy signature of the pMHCII complex. To accomplish this purpose, we examined the energy values in HIP39/I-Ag7 in a per-residue basis both on the absence or presence of the 4.1-TCR using a prediction algorithm (pyDockEneRes, see methods) focused on electrostatics, Van der Waals and third component related with desolvation energy. By following this approach, a severe impact was readily noticeable in the HIP antigen. This effect was particularly relevant for residue Glu5, whose energy gain reached -12.5 kcal mol-1 in the presence of the 4.1-TCR (Supplementary Figure S5). More in detail, the greatest weight of this energy balance falls in the electrostatics term, showing a severe impact due to recognition by the 4.1-TCR: -2.8 kcal mol-1 in the absence of the TCR and -14.8 kcal mol-1 in the ternary complex. Overall, this variation represents by far the largest energy gain in the HIP39/I-Ag7 complex upon binding of the 4.1-TCR. The second greatest variation is in Glu7, with an associated energetic transition of -4.5 kcal mol-1 (-1.1 to -5.6 kcal mol-1). Moreover, the docking of the 4.1-TCR results in an energetically favorable transition for 9 out of the 11 amino acids that conform the peptide (Supplementary Figure S5). As for I-Ag7, as expected, the energetic variations are concentrated at the TCR-binding interface. Here, the calculations suggest an advantageous outcome for the alpha chain while a penalty in the energy term for the beta chain. Altogether, the energy landscape determines that recognition of HIP39/I-Ag7 by the 4.1-TCR is linked to an energetically favorable interaction.
In sum, our data expose a previously unrecognized role for structural malleability of certain pMHCII complexes in TCR binding, long thought to be largely a property of the TCR CDR loops (29–33). Collectively, our data suggest that peptide-tunable conformational flexibility of autoimmune disease-predisposing MHCII molecules such as I-Ag7 along with a HIP-driven energetically favorable recognition may contribute to their pathogenicity, perhaps by broadening the repertoire of peptides that can be recognized by individual TCRs.
Compelling experimental evidence have suggested that T1D initiation/progression involves the initial recognition of insulin/HIP epitopes followed by the recruitment of autoreactive T-cells targeting many other beta cell autoantigens (1, 2, 5–11, 34). It has been established that MHCII polymorphisms, particularly around beta-chain position 57 (β57) afford susceptibility to several autoimmune diseases, including T1D, in both mice and humans. Structural studies have indicated that the positive association between T1D and non-Asp MHCIIβ alleles are in part mediated by the formation of an MHC binding pocket that, in the absence of Asp, allows the binding of peptides carrying an acidic residue at P9 (3, 18). However, the potential contribution of other structural features has not been ruled out and remained elusive.
We have shown that transgenic NOD and NOD.Rag2–/– mice expressing the pancreatic islet-derived I-Ag7-restricted and MHCII-promiscuous 4.1-TCR develop a highly accelerated form of T1D (9, 19–24). More recently, we have established that this TCR is also antigenically promiscuous, recognizing seven different HIPs that result from the fusion of various chromogranin A and/or insulin C fragments, including post-translational modifications (25). Here, by resolving the crystal structures of a HIP/I-Ag7 molecule both in isolation and in a ternary complex with the 4.1-TCR, we show that the HIP promiscuity of the 4.1-TCR is dictated by (1) the presence of acidic amino acid residues at peptide positions P5, P7 and P8 that favor an optimal engagement of the 4.1-TCR’s complementary determining regions; and (2) TCR-induced conformational motions in both the I-Ag7 molecule and the peptide that are likely to stabilize docking of the TCR-pMHC-II complex. Thus, the T1D-associated I-Ag7 molecule is structurally malleable, and this malleability allows both the docking of TCRs that would otherwise be unable to do so, as well as the recognition of different peptides sharing specific features.
Although the overall morphology of the I-Ag7-MHC molecule is preserved in both the free and TCR-bound structures, the side chains of several residues in I-Ag7 dramatically changed their orientation upon TCR binding. Particularly striking were the rearrangements involving Gln59 in I-Aαd and Arg68 in I-Aβg7. These rearrangements not only eliminated intermolecular clashes precluding TCR binding but also enabled the formation of new interactions that contributed to tighter TCR engagement. Of note, the Arg68 rearrangement was accompanied by a subtle yet key movement of the HIP’s E5 residue that resulted in proper positioning of the 4.1-TCR’s CDR3β on the HIP39/I-Ag7 complex. Additional key, albeit less pronounced shifts were noted for I-Aαd’s Glu57 and I-Aβg7’s Glu72, which also enabled stronger interactions with the 4.1-TCR through novel ad hoc contacts. These data strongly suggest that these conformational changes occur upon, and are triggered by, engagement of the 4.1-TCR to enable a stable docking.
An alternative interpretation of the data is that the monomeric pMHCII preparations used for crystallization of ternary pMHCII-TCR complexes correspond to heterogeneous populations of structurally diverse pMHCII monomers, each with alternative conformations in the side chains of specific amino acids. Under this hypothetical scenario, only a fraction of conformationally-compatible and energetically favorable pMHCII complexes would license 4.1-TCR engagement and crystallization. However, in-depth analyses of other pI-Ag7-TCR crystal structures, such as those corresponding to the Insulin B:9-23-specific I.29/8F10 and HEL-specific 21.20 TCRs, respectively, strongly argue against this alternative interpretation of the data. As noted further above, the positioning of Arg68’s side chain away from the peptide-binding groove can also be seen in other pI-Ag7 crystal structures in the absence of a TCR, or in TCR-bound pI-Ag7 structures in which the peptide’s P5 and P7 positions are occupied by neutral residues, as is the case for the I.29 and 8F10 TCR ligands. In contrast, the Arg68’s side chain is found pointing towards the peptide-binding groove in the structure of a HEL peptide/I-Ag7 complex bound to its cognate 21.30 TCR. A key difference between these structures is that P5 and P7 in the HEL peptide are occupied by Glu and Tyr, respectively, which lock Arg68 through a set of polar interactions. Furthermore, limited and isolated displacement of the Arg68 side chain can also be seen in the crystal structures of a pI-Ab or a pI-Au molecules upon engagement of their respective cognate TCRs, in both cases in a peptide-independent manner (35, 36). Thus, our data are not only incompatible with the HIP/I-Ag7 complex and the 4.1-TCR as being rigid molecules that fit one another like two pieces in a jigsaw (29), but rather strongly support the idea that I-Aβg7 Arg68’s ability to make interactions with peptide residues upon TCR engagement promotes the recognition of peptides with non-neutral residues at P5 and P7.
Recent structures of human TCRs bound to a HIP/HLA-DQ8 pMHCII complex (37) enable cross-species comparison for MHCII-presentation of HIPs. Despite the natural divergence in both the TCR repertoire and MHCII sequences, HIP presentation and recognition in the human and murine contexts follow a classical TCR:pMHCII binding pattern. Like other TCRs (38), including those recognizing this HIP/DQ8 pMHCII, the TCRα and TCRβ chains interact, respectively, with the HIP antigen N- and C-terminal residues. The footprint coverage is similar in both scenarios and encompass MHCII α and β chains as well as a large central region of the HIP molecules. One difference stands out, however. In the murine system, the 4.1-TCR : HIP interaction is defined by polar contacts mediated with up to 4 HIP residues. Of these, three are established by solvent accessible side chains that belong to acidic residues located in P5, P7 and P8 via H-bonding and, additionally, via salt bridges with P8. In contrast, in its human counterpart, P5, P7 and P8 positions in the corresponding HIP (derived from splicing of insulin and islet amyloid polypeptide) are occupied by Gly, Ala and Val, that is, neutral amino acids that would not recapitulate the polar contacts seen with the 4.1-TCR. Instead, the scarce polar contacts seen in the three reported human TCR : HIP/DQ8 pairs are mediated by backbone oxygen and nitrogen atoms. Remarkably, however, the peptides bound to HLA-DQ8 molecules in the B-lymphoblastoid 9033 cell line were found to be highly enriched for presence of acidic residues at positions P5, P7 and P8 (37). This observation suggests that recognition of human HIPs carrying acidic residues at these positions by DQ8-restricted TCRs might be dictated by further molecular mechanisms that share common features with the 4.1-TCR : HIP39/I-Ag7 signature.
In addition to the structural analysis, we assessed the energetic yield of the 4.1-TCR : HIP39/I-Ag7 interaction in a per-residue basis. The most outstanding finding were the net energy gain values observed upon binding of the 4.1-TCR, for the glutamic acids in P5 and P7 of the HIP antigen. At the structural level, we observed that these residues acquire conformations whereby the side chains are solvent accesible, i.e., sterically favored for TCR recognition. Indeed, in the 4.1-TCR : HIP39/I-Ag7 complex, the 4.1-TCR CDR3α Asn97 connects with the HIP antigen through an H-bond with Glu5. This interaction is concomitant with a modest shift of Glu5 side chain triggered by a more severe conformational change observed in I-Aβg7 Arg68. Despite the moderate structural rearrangement of Glu5, this motion seems to be key as it brings the glutamic acid side chain closer to the CDR3α, enabling the H-bond. From an energetic point of view, this structural adaptation associates with an energetic gain of -12.5 kcal mol-1. Likewise, the HIP Glu7 is targeted by the 4.1-TCR, in this case through the CDR3β Gly97. Thus, it can be concluded that the 4.1-TCR CDR3 α and β loops recognize the HIP antigen through two acidic residues in a highly favourable manner from an energetic point of view. Moreover, the HIP Asp8, which makes multiple polar contacts with the TCR also presents a high energy gain [-2.4 kcal mol-1].
We also noticed that the conformational changes observed for certain side chains in I-Aαd and I-Aβg7 do not necessarily represent energetically favorable transitions. For instance, the conformation of the side chain of Glu57 in I-Aαd in the presence of the 4.1-TCR shows a penalty of 1.4 kcal mol-1 with respect to that of the TCR-unbound structure. Gln59 in I-Aαd presents a weak energy gain in the presence of the TCR [-0.08 kcal mol-1], while the conformation of Glu72 side chain in I-Aβg7 has a more evident energy benefit [-1.2 kcal mol-1], likely due to the possibility of establishing close salt bridges with CDR3α Arg96.
Collectively, our findings add another level of complexity to previous observations of conformational changes in TCRs, MHC molecules and MHCII-bound peptides. The TCR CDR loops are known to undergo structural rearrangements to recognize peptides and lipids presented by classical (29, 39) and non-classical MHC molecules (40), respectively. Likewise, there is documented evidence for conformational changes in peptide antigens during pMHC-TCR interactions (32, 41, 42). A particularly noteworthy study (43) of a human autoreactive TCR targeting a Multiple Sclerosis-relevant pMHCII identified structural rearrangements in both the MHCII molecule and the peptide in response to TCR engagement. In this case, the peptide-binding cleft opened up due to an outwardly displacement of 1.5 Å of MHCII β chain residues 60-74 in the presence of the TCR. Thus, although conformational flexibility per se is not a unique property of the I-Ag7 molecule, the nature and extent of the molecular shifts described for the HIP/I-Ag7 complex studied herein are unique and provide a structural mechanism for the antigenic promiscuity of the highly diabetogenic 4.1-TCR. Our studies suggest that HIP recognition by the 4.1-TCR is dictated by an energetically favorable interaction that stabilizes the HIP antigen within the MHCII groove, while the conformational plasticity of key I-Ag7 residues at the TCR interface allows the complete settlement of the 4.1-TCR on HIP39/I-Ag7. Most importantly, our studies raise the possibility that these phenomena compound the proclivity to autoimmune disease afforded by the non-Asp β57 residue.
The 4.1-TCR variable domains were fused via gene synthesis (Bio Basic, Canada) to human constant regions for protein production in E. coli and TCRαβ refolding. Both α and β genes were initially delivered in a pUC57 vector carrying flanking NcoI and XhoI restriction sites. The target genes were extracted at 37°C with FastDigest Restriction Enzymes (Fisher Scientific) and subsequently cloned into the pET28 vector (kindly provided by Dr. Lasa) using the Optizyme™ T4 DNA Ligase (Thermo Fisher Scientific). Competent DH5α E. coli cells (Invitrogen) were transformed with the ligation products and positive clones were grown under kanamycin (50 µg/mL, Panreac AppliChem) selection. Plasmid DNA was isolated and purified with the GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. All sequences were validated by Sanger sequencing and the recombinant plasmids were used to transform BL21DE3 competent cells (Agilent). Single colonies were inoculated in LB and left on an orbital shaker at 37°C. Protein expression was induced by addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (Thermo Fisher Scientific), upon which the cultures were further left shaken at 37°C for 4 hours. Bacterial pellets were obtained by centrifugation at 4500xg. Pellets were then resuspended in lysis buffer containing 50 mM Tris, 150mM NaCl, pH 7.4, phenylmethanesulfonyl fluoride 1 mM (Thermo Fisher Scientific), lysozyme 0.25mg/mL (Sigma), DTT 1mM (Thermo Fisher Scientific) and an EDTA free protease inhibitor cocktail (Sigma). The mixture was kept in ice for sonication at 40% amplitude for 5 minutes (30 seconds burst/rest intervals). The sonicated samples were centrifuged at 10,000 x g, the inclusion bodies recovered and washed twice with 10 mM EDTA pH 8.0 in the presence of 1% Triton X100 (Sigma). A final wash step was carried out without Triton X100 and the inclusion bodies were depleted of liquid prior to storage at -20 °C. One hundred mg of each alpha and beta chain inclusion bodies were thawed, solubilized in 8.0 M Urea, 50 mM Tris pH 8.0, 10 mM DTT, and jointly diluted into a volume of 200 ml in the presence of 0.4 M L-arginine, 5 mM reduced glutathione and 0.5 mM oxidized glutathione. The soluble inclusion bodies were then refolded by lowering the urea content via dialysis against 20 mM Tris pH 8.0 for 72 hours. Two liters of fresh 4 °C buffer were used every 12 hours. The dialyzed sample was freed of particulate material by centrifugation at 10.000 x g, and the cleared sample was loaded onto a 5-ml HiTrap Q FF anion exchange chromatography column (Cytiva) to capture the protein. The refolded heterodimeric TCR was isolated with a NaCl gradient (0-0.5 M) elution. The quality of the refolded material was assessed for homogeneity and monodispersity via size exclusion chromatography through a S200 Increase 10/300 chromatography column (Cytiva). All chromatographic procedures were carried out and monitored in an ÄKTA pure 25M station (Cytiva).
HIP39/I-Ag7 monomers were expressed and produced in CHO-S cells (Invitrogen). Briefly, CHO-S cells were transduced sequentially with lentiviral particles encoding a HIP39 peptide-tethered, knob-into-hole (KIH)-based pMHCII heterodimer carrying a 3C cleavage site immediately upstream of each of the two KIH halves (HIP39-linker GS-I-Aβg7–HRV3C-Fc(Hole)-6xHisTag-IRES-GFP and I-Aαd–HRV3C-Fc(Knob)-6xHisTag-IRES-CFP. Sorted CFPhi/GFPhi CHO-S cells were cultured as described (26). Briefly, CHO-S cells were grown in 2L baffled flasks (Nalgene) in a shaker incubator at 125 rpm, 5% CO2 and 37°C. Basal medium was Power-CHO-2 (Lonza) supplemented with 8mM glutamine (Lonza) and gentamicin sulfate (0.05mg/mL) (Lonza). The cultures were started in 400 mL of basal medium at 350,000-400,000 cells/mL and were supplemented with feeds: Cell Boost 7a (Hyclone) at 3% v/v and Cell Boost 7b (Hyclone) at 0.3% v/v on days 0, 3, 4, 5, 6, 8, 9 and 10. A temperature shift to 34°C was done when cell densities reached 5-7x106 cells/mL. Additional glutamine was added on day 7, to 2 mM. Glucose was added to 4.5 g/L when levels dropped below 3.5 g/L. Cells were harvested on Day 14 or when cell viability fell below 60%. Secreted HIP39/I-Ag7 heterodimers were purified from the CHO-S culture supernatants via protein G affinity chromatography. The purified heterodimers were then digested with the human rhinovirus 3C (HRV 3C) protease (ThermoFischer), to cleave the KIH tail. A second round of purification on a nickel-affinity column was done to remove both the 6xHis-tagged enzyme and KIH. The flow through contained the KIH-less HIP39/I-Ag7 heterodimer. Prior to crystallization studies, and to remove any remaining impurities or aggregates, the purified HIP39/I-Ag7 heterodimer was further processed through a size exclusion S200 Increase 10/300 chromatographic column (Cytiva) using 20 mM Tris pH 8.0, 150 mM NaCl as running buffer. The protein was concentrated to 10.0 mg/ml in a 10 molecular weight cut-off Amicon concentrator (Millipore). Protein concentrations were determined using extinction coefficients and absorbance values at 280 nm. The ternary 4.1-TCR : HIP39/I-Ag7 complex was prepared by mixing stoichiometric amounts of the purified 4.1-TCR and HIP39/I-Ag7. The mixture was concentrated in a Nanosep device (Pall) to 7.5 mg/ml and used for crystallization trials.
Crystallization was accomplished by manual screening of over 700 crystallization conditions for each protein sample in 96-well plates using the sitting drop method and 20 °C incubation. Crystals of HIP39/I-Ag7 appeared in 24-48 hours and had needle or rod-shaped morphology. Large optimized three-dimensional crystals were obtained in 0.17 M ammonium sulfate and 25% w/v PEG 4000. Square-shaped thin crystals of 4.1-TCR : HIP39/I-Ag7 appeared after 2-4 weeks at 20 °C and were optimized in 0.1 M MgCl2, 0.1 M Na HEPES pH 7.0, 15% w/v PEG 4000. Crystals were harvested, soaked in their crystallization medium supplemented with 20% glycerol (HIP39/I-Ag7) or 20% ethylenglycol (4.1-TCR : HIP39/I-Ag7) and transferred to liquid nitrogen prior to diffraction analyses.
Crystals were mounted in the diffractometer at the Xaloc beamline of the Alba Synchrothron (Cerdanyola del Vallès, Barcelona, Spain). Diffraction datasets were collected for HIP39/I-Ag7 and 4.1-TCR : HIP39/I-Ag7 crystals. X-ray diffraction data of HIP39/I-Ag7 crystals were processed with autoPROC (44). Diffraction data of the 4.1-TCR : HIP39/I-Ag7 crystals were indexed, reduced and integrated in the XDS Package (45). Data were then merged and scaled using Aimless (46). Phases were obtained via molecular replacement with Phaser (47) for HIP39/I-Ag7 and Molrep (48) for the 4.1-TCR : HIP39/I-Ag7 ternary complex. For HIP39/I-Ag7 the structural model used was obtained from the atomic coordinates of I-Ag7 in the Protein Data Bank (PDB), ID 1ES0. Prior to phasing, the coordinates of the peptide antigen were removed. For the ternary complex, three independent templates were used for phasing. First, the refined coordinates of HIP39/I-Ag7 without the antigen, the human constant domains from PDB ID 4EN3 (49), and lastly the variable domains from PDB 1D9K (50). All molecular replacement solutions were subsequently refined with Refmac5 (51) or phenix.refine (52). A set of 5% random reflections were kept aside for assessment of R-free values during the refinement procedures. Protein building was guided according to the calculated Fo-Fc and 2Fo-Fc electron density maps in Coot (53). Further iterative cycles of refinement and manual building were carried out to optimize the coordinates and geometry prior to data deposition in the Protein Data Bank.
The atomic coordinates of the HIP39/I-Ag7 and 4.1-TCR : HIP39/I-Ag7 complex structures were used to calculate energy values in a per-residue basis with pyDockEneRes (54), deconvoluted into electrostatics, Van der Waals forces and desolvation components. The algorithm calculates the energetic contribution for protein-protein interactions in a systematic way for each of the residues in a set of atomic coordinates
Atomic coordinates and structure factors of the HIP39/I-Ag7 and 4.1-TCR:HIP39/I-Ag7 complexes have been deposited in the Protein Data Bank with accession codes 7QHP and 7Z50, respectively.
Conceptualization: JL-S and PSa. Methodology: JL-S, PSe and PSa. Experimental research: EE, JL-S, DP, PSe. Manuscript writing: JL-S and PSa. All authors contributed to the article and approved the submitted version.
JL-S is a Ramón y Cajal Investigator supported by the Ministerio de Ciencia e Investigación of Spain (RYC‐2017‐21683). DP was supported by a pre-doctoral studentship from FPU (MINECO). This work was supported by the Ministerio de Ciencia e Investigación of Spain (RTI2018-093964-B-I00), Generalitat de Catalunya (SGR and CERCA Programmes), the ISCIII and FEDER (PIE14/00027, PI15/0797), and the Canadian Institutes of Health Research (CIHR-136866). PSe was supported by the Ramon y Cajal program and by a JDRF Career Development Award.
We thank the staff of XALOC beamline at ALBA Synchrotron for their assistance with X-ray diffraction data collection. We thank M.G. Dichiara Rodríguez, A. Ochoa Echeverria, C. Fandos, M. Ortega, S. Thiessen and F. Liu for technical support.
PSa is scientific founder of Parvus Therapeutics and has a financial interest in the company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.924311/full#supplementary-material
Supplementary Figure 1 | Crystal samples and composition of the asymmetric unit in the HIP39/I-Ag7 and 4.1-TCR:HIP39/I-Ag7 ternary complexes. (A) A crystal of the HIP39/I-Ag7 complex is shown mounted in a loop prior to diffraction analysis. Right, the composition of the asymmetric unit in the C2221 unit cell. (B) A single crystal of the 4.1-TCR-HIP39/I-Ag7 complex is ready for diffraction analysis. Two 4.1-TCR:HIP39/I-Ag7 ternary assemblies are displayed inside the C121 unit cell in the right panel.
Supplementary Figure 2 | Multiple contacts mediated by I-Aβg7 Thr75 with the 4.1-TCR. Thr75 engages with all CDR loops in the 4.1-TCRα chain. Residues in the CDRs making contacts are shown as sticks. VDW contacts are displayed as blue dash lines, while the H-bond with Ser53 can be seen in darkgrey color. All contacts are < 4.0 Å.
Supplementary Figure 3 | Additional contacts mediated by water molecules between the 4.1-TCR and the HIP39/I-Ag7 complex. (A) A water molecule, shown as a pale yellow sphere, enables further interactions (blue dashed lines) between the 4.1-TCR β chain and the HIP39 antigen. Interacting residues in the 4.1-TCR CDRs and the peptide are highlighted as sticks. Water bridges displayed in this figure are < 3.4 Å. The salt bridges between HIP39 Asp8 and the side chain of Arg49 in the TCR β are indicated with red dashed lines. (B) Water-mediated contacts between the 4.1-TCR CDR3β and Gln64 in I-Aβg7.
Supplementary Figure 4 | Structural plasticity of I-Aβg7 Arg68. (A) The MHC platforms of I-Ag7 complexed with different peptides are aligned and superposed over that of free HIP39/I-Aβg7 (yellow color) and 4.1-TCR/bound HIP39/I-Aβg7 (green). The I-Ag7-peptide complexes superposed are: PDB 5DMK, RLGL-WE14/ I-Ag7; PDB 6BLQ, p8E9E/ I-Ag7; PDB 6BLR, p8E9E6SS/ I-Ag7; PDB 6BLX, p8G9E/ I-Ag7; the structure of I-Ag7 complexed with GAD65 is not included because we could not appreciate a solid 2Fo-Fc signal for this residue, likely due to its flexibility. The position of Arg68 in the 4.1-TCR:HIP39/I-Aβg7 complex is highlighted with an asterisk. (B) The position of Arg68 in the 4.1-TCR:HIP39/I-Aβg7 complex is compared with those present in three alternative complex structures, as indicated.
Supplementary Figure 5 | Energy balance of the HIP39/I-Aβg7 complex upon recognition by the 4.1-TCR. Electrostatics, Van der Waals forces and desolvation terms were computed for both the HIP antigen and the MHCII in a per-residue basis with pyDockEneRes (see methods). The net energy gain for the HIP39/I-Aβg7 complex was assessed with the difference of the calculated values in the presence and absence of the TCR. The graphic shows the net energy gain values in a per-residue manner for the HIP antigen (residues -1 to 11), I-Aαd and I-Aβg7.
Supplementary Table 1 | I-Aαd chain with HIP39 interactions found in 7QHP. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 2 | I-Aβg7 chain with HIP39 interactions found in 7QHP. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 3 | I-Aαd (chain A) with HIP39 (chain T) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 4 | I-Aαd (chain C) with HIP39 (chain W) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 5 | I-Aβg7 (chain B) with HIP39 (chain T) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 6 | I-Aβg7 (chain D) with HIP39 (chain W) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 7 | 4.1-TCR α (chain H) with HIP39 (chain T) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 8 | 4.1-TCR α (chain G) with HIP39 (chain W) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 9 | 4.1-TCR β (chain E) with HIP39 (chain T) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 10 | 4.1-TCR β (chain F) with HIP39 (chain W) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 11 | I-Aαd (chain A) with 4.1-TCR α (chain H) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 12 | I-Aαd (chain C) with 4.1-TCR α (chain G) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 13 | I-Aαd (chain A) with 4.1-TCR β (chain E) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 14 | I-Aαd (chain C) with 4.1-TCR β (chain F) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 15 | I-Aβg7 (chain B) with 4.1-TCR α (chain H) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 16 | I-Aβg7 (chain D) with 4.1-TCR α (chain G) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 17 | I-Aβg7 (chain B) with 4.1-TCR β (chain E) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
Supplementary Table 18 | I-Aβg7 (chain D) with 4.1-TCR β (chain F) interactions found in 7Z50. All intermolecular contacts are assigned according to the following distance cutoffs: hydrogen bonds ≤ 3.4 Å, salt bridges ≤ 4.5 Å, Van der Waals ≤ 4.0 Å.
1. Babad J, Geliebter A, DiLorenzo TP. T-Cell autoantigens in the non-obese diabetic mouse model of autoimmune diabetes. Immunology (2010) 131:459–65. doi: 10.1111/j.1365-2567.2010.03362.x
2. Purcell AW, Sechi S, DiLorenzo TP. The evolving landscape of autoantigen discovery and characterization in type 1 diabetes. Diabetes (2019) 68:879–86. doi: 10.2337/dbi18-0066
3. Gioia L, Holt M, Costanzo A, Sharma S, Abe B, Kain L, et al. Position β57 of I-A(g7) controls early anti-insulin responses in NOD mice, linking an MHC susceptibility allele to type 1 diabetes onset. Sci Immunol (2019) 4. doi: 10.1126/sciimmunol.aaw6329
4. Baker RL, Jamison BL, Wiles TA, Lindsay RS, Barbour G, Bradley B, et al. CD4 T cells reactive to hybrid insulin peptides are indicators of disease activity in the NOD mouse. Diabetes (2018) 67:1836–46. doi: 10.2337/db18-0200
5. Anderson B, Park BJ, Verdaguer J, Amrani A, Santamaria P. Prevalent CD8(+) T cell response against one peptide/MHC complex in autoimmune diabetes. Proc Natl Acad Sci USA (1999) 96:9311–6. doi: 10.1073/pnas.96.16.9311
6. Han B, Serra P, Amrani A, Yamanouchi J, Marée AFM, Edelstein-Keshet L, et al. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high efficiency of a low-affinity peptide. Nat Med (2005) 11:645–52. doi: 10.1038/nm1250
7. Lieberman SM, Evans AM, Han B, Takaki T, Vinnitskaya Y, Caldwell JA, et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci USA (2003) 100:8384–8. doi: 10.1073/pnas.0932778100
8. Trudeau JD, Chandler T, Soukhatcheva G, Verchere CB, Tan R. Prospective prediction of spontaneous but not recurrent autoimmune diabetes in the non-obese diabetic mouse. Diabetologia (2007) 50:1015–23. doi: 10.1007/s00125-007-0600-9
9. Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med (1997) 186:1663–76. doi: 10.1084/jem.186.10.1663
10. Verdaguer J, Yoon JW, Anderson B, Averill N, Utsugi T, Park BJ, et al. Acceleration of spontaneous diabetes in TCR-beta-transgenic nonobese diabetic mice by beta-cell cytotoxic CD8+ T cells expressing identical endogenous TCR-alpha chains. J Immunol (1996) 157:4726–35.
11. Santamaria P. The long and winding road to understanding and conquering type 1 diabetes. Immunity (2010) 32:437–45. doi: 10.1016/j.immuni.2010.04.003
12. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet (2009) 41:703–7. doi: 10.1038/ng.381
13. Barton NH, Keightley PD. Understanding quantitative genetic variation. Nat Rev Genet (2002) 3:11–21. doi: 10.1038/nrg700
14. Robertson CC, Inshaw JRJ, Onengut-Gumuscu S, Chen W-M, Santa Cruz DF, Yang H, et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat Genet (2021) 53:962–71. doi: 10.1038/s41588-021-00880-5
15. Zhang M, Wang Y, Li X, Meng G, Chen X, Wang L, et al. A single L/D-substitution at Q4 of the mInsA(2-10) epitope prevents type 1 diabetes in humanized NOD mice. Front Immunol (2021) 12:713276. doi: 10.3389/fimmu.2021.713276
16. Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell (1996) 85:291–7. doi: 10.1016/s0092-8674(00)81106-x
17. Vyse TJ, Todd JA. Genetic analysis of autoimmune disease. Cell (1996) 85:311–8. doi: 10.1016/s0092-8674(00)81110-1
18. Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science (2000) 288:505–11. doi: 10.1126/science.288.5465.505
19. Schmidt D, Amrani A, Verdaguer J, Bou S, Santamaria P. Autoantigen-independent deletion of diabetogenic CD4+ thymocytes by protective MHC class II molecules. J Immunol (1999) 162:4627–36.
20. Schmidt D, Verdaguer J, Averill N, Santamaria P. A mechanism for the major histocompatibility complex-linked resistance to autoimmunity. J Exp Med (1997) 186:1059–75. doi: 10.1084/jem.186.7.1059
21. Thiessen S, Serra P, Amrani A, Verdaguer J, Santamaria P. T-Cell tolerance by dendritic cells and macrophages as a mechanism for the major histocompatibility complex-linked resistance to autoimmune diabetes. Diabetes (2002) 51:325–38. doi: 10.2337/diabetes.51.2.325
22. Tsai S, Santamaria P. MHC class II polymorphisms, Autoreactive T-cells, and autoimmunity. Front Immunol (2013) 4:321. doi: 10.3389/fimmu.2013.00321
23. Tsai S, Serra P, Clemente-Casares X, Yamanouchi J, Thiessen S, Slattery RM, et al. Antidiabetogenic MHC class II promotes the differentiation of MHC-promiscuous autoreactive T cells into FOXP3+ regulatory T cells. Proc Natl Acad Sci USA (2013) 110:3471–6. doi: 10.1073/pnas.1211391110
24. Tsai S, Serra P, Clemente-Casares X, Slattery RM, Santamaria P. Dendritic cell-dependent in vivo generation of autoregulatory T cells by antidiabetogenic MHC class II. J Immunol (2013) 191:70–82. doi: 10.4049/jimmunol.1300168
25. Parras D, Solé P, Delong T, Santamaría P, Serra P. Recognition of multiple hybrid insulin peptides by a single highly diabetogenic T-cell receptor. Front Immunol (2021) 12:737428. doi: 10.3389/fimmu.2021.737428
26. Serra P, Garabatos N, Singha S, Fandos C, Garnica J, Solé P, et al. Increased yields and biological potency of knob-into-hole-based soluble MHC class II molecules. Nat Commun (2019) 10:4917. doi: 10.1038/s41467-019-12902-2
27. Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, et al. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng (2003) 16:707–11. doi: 10.1093/protein/gzg087
28. Lopez-Sagaseta J, Dulberger CL, Crooks JE, Parks CD, Luoma AM, McFedries A, et al. The molecular basis for mucosal-associated invariant T cell recognition of MR1 proteins. Proc Natl Acad Sci U S A (2013) 110:E1771–8. doi: 10.1073/pnas.1222678110
29. Armstrong KM, Piepenbrink KH, Baker BM. Conformational changes and flexibility in T-cell receptor recognition of peptide-MHC complexes. Biochem J (2008) 415:183–96. doi: 10.1042/BJ20080850
30. Borbulevych OY, Piepenbrink KH, Baker BM. Conformational melding permits a conserved binding geometry in TCR recognition of foreign and self molecular mimics. J Immunol (2011) 186:2950–8. doi: 10.4049/jimmunol.1003150
31. Borbulevych OY, Piepenbrink KH, Gloor BE, Scott DR, Sommese RF, Cole DK, et al. T Cell receptor cross-reactivity directed by antigen-dependent tuning of peptide-MHC molecular flexibility. Immunity (2009) 31:885–96. doi: 10.1016/j.immuni.2009.11.003
32. Tynan FE, Reid HH, Kjer-Nielsen L, Miles JJ, Wilce MCJ, Kostenko L, et al. A T cell receptor flattens a bulged antigenic peptide presented by a major histocompatibility complex class I molecule. Nat Immunol (2007) 8:268–76. doi: 10.1038/ni1432
33. Bulek AM, Cole DK, Skowera A, Dolton G, Gras S, Madura F, et al. Structural basis for the killing of human beta cells by CD8(+) T cells in type 1 diabetes. Nat Immunol (2012) 13:283–9. doi: 10.1038/ni.2206
34. Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature (2005) 435:220–3. doi: 10.1038/nature03523
35. Dai S, Huseby ES, Rubtsova K, Scott-Browne J, Crawford F, Macdonald WA, et al. Crossreactive T cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity (2008) 28:324–34. doi: 10.1016/j.immuni.2008.01.008
36. Maynard J, Petersson K, Wilson DH, Adams EJ, Blondelle SE, Boulanger MJ, et al. Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity (2005) 22:81–92. doi: 10.1016/j.immuni.2004.11.015
37. Tran MT, Faridi P, Lim JJ, Ting YT, Onwukwe G, Bhattacharjee P, et al. T Cell receptor recognition of hybrid insulin peptides bound to HLA-DQ8. Nat Commun (2021) 12:5110. doi: 10.1038/s41467-021-25404-x
38. Wang Y, Sosinowski T, Novikov A, Crawford F, White J, Jin N, et al. How c-terminal additions to insulin b-chain fragments create superagonists for T cells in mouse and human type 1 diabetes. Sci Immunol (2019) 4. doi: 10.1126/sciimmunol.aav7517
39. Garcia KC, Adams EJ. How the T cell receptor sees antigen–a structural view. Cell (2005) 122:333–6. doi: 10.1016/j.cell.2005.07.015
40. López-Sagaseta J, Sibener LV, Kung JE, Gumperz J, Adams EJ. Lysophospholipid presentation by CD1d and recognition by a human natural killer T-cell receptor (2012). Available at: https://www.scopus.com/inward/record.uri?eid=2-s2.0-84859906269&doi=10.1038%2Femboj.2012.54&partnerID=40&md5=bb36d6c7039bc907381cb4df187a2a16.
41. Garcia KC, Degano M, Pease LR, Huang M, Peterson PA, Teyton L, et al. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science (1998) 279:1166–72. doi: 10.1126/science.279.5354.1166
42. Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature (1996) 384:134–41. doi: 10.1038/384134a0
43. Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. EMBO J (2005) 24:2968–79. doi: 10.1038/sj.emboj.7600771
44. Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr (2011) 67:293–302. doi: 10.1107/S0907444911007773
45. Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr (2010) 66:125–32. doi: 10.1107/s0907444909047337
46. Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr (2013) 69:1204–14. doi: 10.1107/S0907444913000061
47. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr (2007) 40:658–74. doi: 10.1107/S0021889807021206
48. Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr (2010) 66:22–5. doi: 10.1107/S0907444909042589
49. López-Sagaseta J, Kung JE, Savage PB, Gumperz J, Adams EJ. The molecular basis for recognition of CD1d/α-galactosylceramide by a human non-Vα24 T cell receptor. PloS Biol (2012) 10:e1001412. doi: 10.1371/journal.pbio.1001412
50. Reinherz EL, Tan K, Tang L, Kern P, Liu J, Xiong Y, et al. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science (1999) 286:1913–21. doi: 10.1126/science.286.5446.1913
51. Kovalevskiy O, Nicholls RA, Murshudov GN. Automated refinement of macromolecular structures at low resolution using prior information. Acta Crystallogr Sect D Struct Biol (2016) 72:1149–61. doi: 10.1107/S2059798316014534
52. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr (2010) 66:213–21. doi: 10.1107/s0907444909052925
53. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr (2010) 66:486–501. doi: 10.1107/s0907444910007493
Keywords: type I diabetes, hybrid insulin peptides, antigenic promiscuity, autoreactivity, MHC class II, T cell receptor, complex structure, x-ray
Citation: Erausquin E, Serra P, Parras D, Santamaria P and López-Sagaseta J (2022) Structural plasticity in I-Ag7 links autoreactivity to hybrid insulin peptides in type I diabetes. Front. Immunol. 13:924311. doi: 10.3389/fimmu.2022.924311
Received: 20 April 2022; Accepted: 04 July 2022;
Published: 28 July 2022.
Edited by:
Roland Michael Tisch, University of North Carolina at Chapel Hill, United StatesReviewed by:
Gustaf Christoffersson, Uppsala University, SwedenCopyright © 2022 Erausquin, Serra, Parras, Santamaria and López-Sagaseta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacinto López-Sagaseta, amFjaW50by5sb3Blei5zYWdhc2V0YUBuYXZhcnJhLmVz; Pere Santamaria, cHNhbnRhbWFAdWNhbGdhcnkuY2E=
†These authors have contributed equally to this work and share first authorship
‡Lead Author
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.