 Ruiyi Deng
Ruiyi Deng Huiling Zheng
Huiling Zheng Hongzhen Cai
Hongzhen Cai Man Li
Man Li Yanyan Shi
Yanyan Shi Shigang Ding
Shigang Ding
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 28 July 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.923477
This article is part of the Research Topic Modulation of the immune system by bacteria: from evasion to therapy View all 17 articles
Helicobacter pylori is closely associated with gastric cancer. During persistent infection, Helicobacter pylori can form a microenvironment in gastric mucosa which facilitates the survival and colony formation of Helicobacter pylori. Tumor stromal cells are involved in this process, including tumor-associated macrophages, mesenchymal stem cells, cancer-associated fibroblasts, and myeloid-derived suppressor cells, and so on. The immune checkpoints are also regulated by Helicobacter pylori infection. Helicobacter pylori virulence factors can also act as immunogens or adjuvants to elicit or enhance immune responses, indicating their potential applications in vaccine development and tumor immunotherapy. This review highlights the effects of Helicobacter pylori on the immune microenvironment and its potential roles in tumor immunotherapy responses.
Helicobacter pylori is a gram-negative, helical, microaerophilic, and flagellated bacteria that colonizes the gastric mucosa in approximately 50% of the world population (1, 2). Helicobacter pylori infection is the main cause of gastric mucosal diseases such as gastric cancer (GC), chronic non-atrophic gastritis, atrophic gastritis, intestinal metaplasia, and dysplasia (3). GC is the fifth most common cancer and the fourth leading cause of cancer-related deaths worldwide (4). H. pylori is classified by the WHO as a class I carcinogen associated with the onset of GC, as chronic H. pylori infection leads to at least 75% of GC cases (5–8). 2% of H. pylori infected patients will develop GC (7).
Tumor growth is supported by oncogene-driven metabolic activities as well as by the microenvironment. Infection with H. pylori promotes gastric tumorigenesis, mainly by influencing the microenvironment (9). Virulence factors such as cytotoxin-associated gene A (CagA), vacuolating cytotoxin A (VacA), urease (Ure), arginase (Arg), lipopolysaccharide (LPS), and neutrophil-activating protein (NAP), enable H. pylori to survive and colonize the gastric mucosa, maintain chronic inflammation, and induce malignant changes within the gastric mucosa (1, 10–12). The immune system plays a pivotal role in eliminating H. pylori infection and controlling inflammation. Throughout a long-term co-existence with human hosts, H. pylori has developed several strategies to maintain a balance between the immune response and immune escape (13, 14). Through regulating tumor stromal cells, immune checkpoints, and other regulatory factors, H. pylori constructs a microenvironment that favors persistent colonization and facilitates tumorigenesis.
However, the influence of H. pylori on responses to immunotherapies and the prognosis of GC remains controversial (15–18). Recent studies have presented that H. pylori infection might affect the curative effect of tumor therapy by the induced immuno-regulation (19, 20). Besides, H. pylori virulence factors such as NAP, VacA, and Ure might elicit or enhance immune responses, which indicates the potential application in vaccine development and tumor immunotherapy (21, 22). These virulence factors are immunodominant antigens of H. pylori and might improve patient prognosis as immunogens or adjuvants in immunotherapy (23). Here, this review describes the mechanisms and effects of H. pylori on the immune microenvironment of GC and tumor immunotherapy responses.
The tumor microenvironment (TME) consists of a continuously evolving complex of tumor cells and stroma. Stroma comprises surrounding non-cancerous fibroblasts, epithelial, immune and blood cells, and extracellular components such as cytokines, growth factors, hormones, and extracellular matrix (ECM) (24, 25). Stroma plays a key role during tumor initiation, progression, and metastasis, meanwhile it significantly influences therapeutic responses and clinical outcomes (26). Helicobacter pylori and its virulence factors can form a microenvironment that facilitates its survival and colony formation by regulating the constituents and functions of the TME. This section summarizes the interactions between H. pylori and tumor stromal cells during GC initiation, progression, and metastasis and describes potential strategies to improve the prognosis (Figure 1; Table 1).
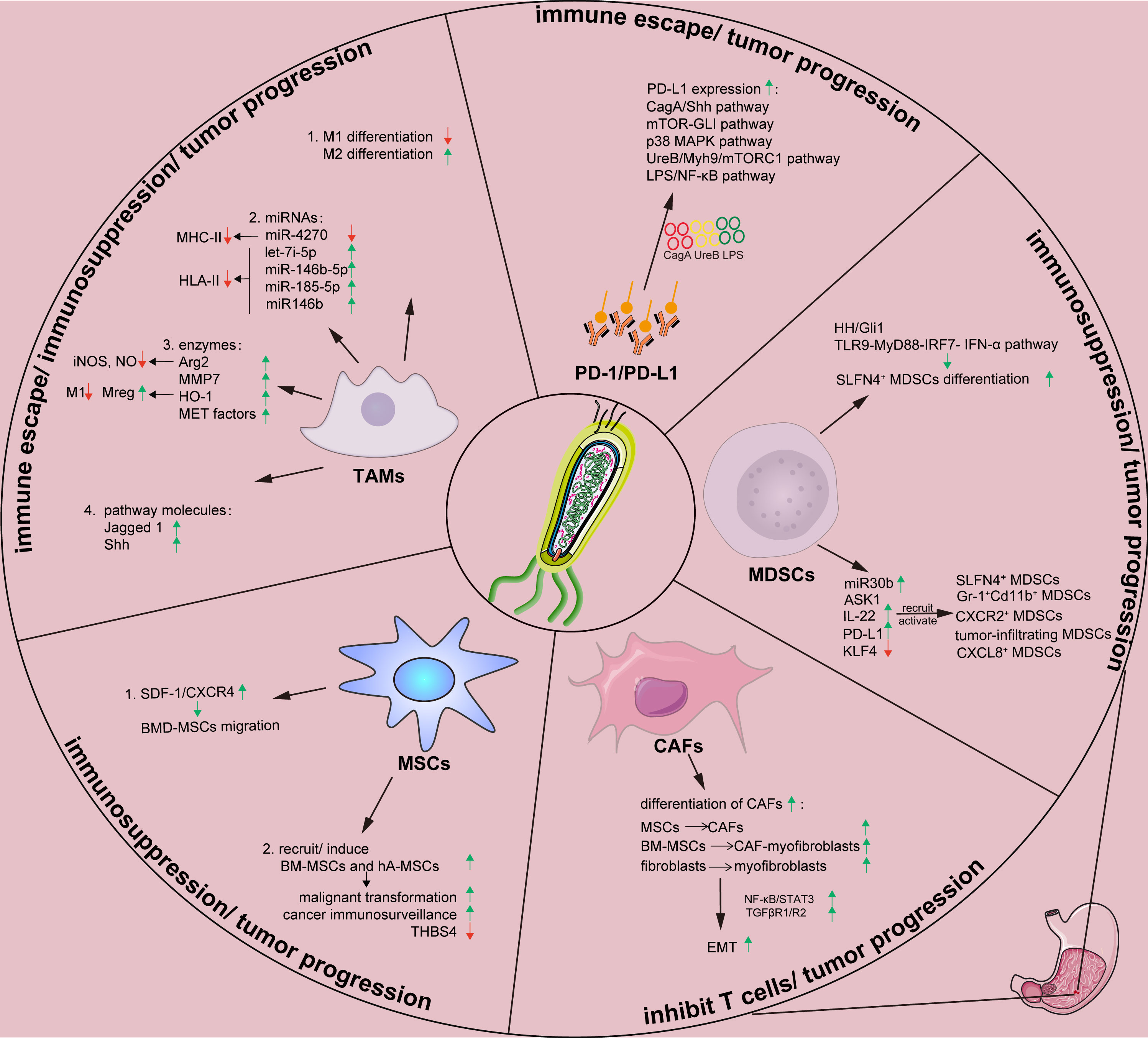
Figure 1 Effects of H. pylori on tumor stromal cells and tumor-related proteins in gastric tumor immune microenvironment. Arg, arginase; ASK1, apoptosis signal-regulating kinase 1; BM-MSC, Bone marrow-derived mesenchymal stem cells; CAF, cancer-associated fibroblast; Cag A, cytotoxin-associated gene A; CXCL8, chemokine (C-X-C motif) ligand 8; EMT, epithelial-mesenchymal transition; hA-MSC, human adipose-derived mesenchymal stem cells; HH, Hedgehog; HO-1, heme oxygenase-1; H.pylori, Helicobacter pylori; IL-22, Interleukin-22; IRF, interferon regulatory factor; IFN, interferon; KLF4, Krüppel-like factor 4; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinases; MDSCs, myeloid-derived suppressor cells; MET, mesenchymal-epithelial transition; MHC-II, major histocompatibility complex class II; MMP, matrix metalloproteinase; mTOR, mammalian target of rapamycin; Myh9, myosin heavy chain 9; NF-κB, nuclear factor kappa B; miR, microRNA; MSCs, mesenchymal stem cells; PD-1, programmed death 1; PD-L1, programmed death-ligand 1; PI3K-AKT, phosphatidylinositol 3 kinase-protein kinase B; ROS, reactive oxygen species; SDF, stromal-derived factor; Shh, Sonic hedgehog; SLFN4, Schlafen 4; STAT3, signal transducer and activator of transcription 3; TAMs, tumor-associated macrophages; TGFβ, transforming growth factor β; TLR, Toll-like receptor; Ure, urease; Vac A, vacuolating cytotoxin A.

Table 1 Effects of H. pylori on tumor cells in gastric tumor immune microenvironment.
Changes in immune responses and the immune escape of H. pylori are closely associated with tumor-associated macrophages (TAMs), which are emerging key players in the TME. Macrophages play crucial roles in host defense against bacterial infections and in the regulation of immune responses during H. pylori infection (68). However, macrophages can also induce angiogenesis and suppress the host immune response during cancer development (37, 69). Generally, TAMs comprise M1 and M2 subtypes (27). Proinflammatory activated M1 macrophages promote the type I T helper (Th1) immune response by producing type I proinflammatory cytokines such as IL-1β, IL-1α, and IL-6 to clear pathogens and inhibit tumor progression, while simultaneously suppressing Th2-type responses (27, 70, 71). Activated M2 macrophages contribute to production of ECM and anti-inflammatory effectors such as IL-4 and IL-10 that are involved in the Th2 immune response, promotion of wound healing, and suppression of Th1 responses (72–75). Additionally, a third type called regulatory macrophages (Mregs) secrete abundant IL-10 that limits inflammation but do not secrete ECM (72). Helicobacter pylori and other pathogens might impair M1 macrophage differentiation while inducing M2 macrophage differentiation or M1 transdifferentiation into M2 macrophages, which can promote tumor progression and invasion by inducing angiogenesis and mediating immunosuppressive signals in solid tumors (27).
Furthermore, H. pylori infection might regulate specific microRNAs (miRNAs) to control macrophage function and affect the TME (28, 76). Infection with H. pylori leads to the downregulated expression of miR-4270 by human monocyte-derived macrophages. This favors upregulation of expression of CD300E immune receptors that enhance the proinflammatory potential of macrophages. However, the expression and exposure of major histocompatibility complex class II (MHC-II) molecules on the plasma membrane are simultaneously compromised. Hence, antigen presentation ability is decreased, leading to persistent H. pylori infection (28). The upregulation of let-7i-5p, miR-146b-5p and miR-185-5p, and miR146b expression in macrophages caused by H. pylori infection can similarly decrease HLA-II expression on the plasma membrane, which ultimately compromises bacterial antigen presentation to Th lymphocytes and impairs immune responses against H. pylori (29, 30). Collectively, H. pylori infection mainly downregulates surface recognition factors at the transcriptional level by rendering macrophages fail to degrade the bacteria. Thus, macrophages become a protective niche for H. pylori.
Helicobacter pylori can induce the production of specific enzymes that regulate macrophage function and affect TME. The production of arginase II (Arg2) in macrophages induced by H. pylori infection results in cell apoptosis and restrained proinflammatory cytokine responses, thus promotes H. pylori immune evasion (31, 32). Matrix metalloproteinase 7 (MMP7) plays a pivotal role in H. pylori-mediated immune escape (33). Heme oxygenase-1 (HO-1) expression in macrophages also be induced, resulting in a polarization switch towards a reduction in the M1 population and an increase in the Mreg profile, causing innate and adaptive immune responses failure (34). Transfer exosomes expressing mesenchymal–epithelial transition (MET) factor, a cell-surface receptor tyrosine kinase from H. pylori‐infected GC cells, can elicit uncontrolled macrophage activation and downstream inflammation and might be associated with tumorigenesis and cancer development (35). These findings shed light on how H. pylori influences the gastric microenvironment by inducing the expression of macrophage-associated enzymes in TAMs.
Moreover, H. pylori upregulates the expression of Jagged 1, a ligand of Notch signaling that plays an important role in M1 macrophage activation and bactericidal activity to prevent H. pylori infection. Upregulated Jagged 1 expression induces an increase in the expression of proinflammatory mediators and phagocytosis and a decrease in the bacterial load, which together impart antibacterial activity in macrophages (36). The hedgehog (HH) signaling pathway also plays an important role in the gastric TME. Sonic hedgehog (SHH) induced by H. pylori infection acts as a macrophage chemoattractant, which is a prerequisite in the gastric immune response (37).
In conclusion, H. pylori infection at the early stage can induce the infiltration of polymorphonuclear leukocytes and mononuclear phagocytes in the gastric mucosa as an innate immune response (77). During the advanced stages of GC, H. pylori can escape immune surveillance by impairing the antigen presentation of TAMs or by disrupting the M1/M2 (or Mreg) balance in favor of an M2 (or Mreg) phenotype (34, 72). Immunosuppressive status eventually promotes tumorigenesis and cancer development (78). These mechanisms also provide the potential for investigating novel targeted drugs (79). Specific miRNAs such as let-7i-5p, miR-146b-5p, and miR-185-5p can be targeted to reduce adverse effects on macrophage antigen presentation (29). Targeting specific enzymes including MMP7 and HO-1 or signaling pathways, such as Notch and HH, to regulate the M1/M2 (or Mreg) balance might also warrant investigation (33, 34).
Multipotent mesenchymal stem cells (MSCs) can self-renew and differentiate into various cell types that play key roles in tissue healing, regeneration, and immune regulation (80). Bone marrow-derived mesenchymal stem cells (BM-MSCs) might play important roles in H. pylori-associated gastric tumorigenesis and immunosuppression. Upon sensing signals indicating gastric mucosa damage, BM-MSCs migrate from bone marrow to stomach via the peripheral circulation. BM-MSCs heal damaged mucosa through a paracrine mechanism and directed differentiation (81, 82). H. pylori-induced persistent inflammation is required for BM-MSC migration and tumorigenesis (43, 83). Upregulated C-X-C chemokine receptor type 4 (CXCR4) interacts with its ligand, stromal-derived factor (SDF-1) and then promote BM-MSC migration to the gastric tissues (38).
Gastric epithelial glands become repopulated with BM-MSCs in mice model one year after H. pylori infection (39). After recruitment to stomach, BM-MSCs can become entrapped in a microenvironment containing H. pylori and malignant cells, 25% of which originate from BM-MSCs. Fusion with epithelial cells might render BM-MSCs more susceptible to malignant transformation or lead to the promotion of cancerous processes (40). BM-MSCs gradually acquire a clonal advantage and undergo stepwise transformation to malignant cells (39). During malignant progression, gastric epithelial glandular units undergo monoclonal transformation, resulting in emerging cancer stem cell (CSC) clones and adenocarcinomas (39, 41). Human adipose-derived mesenchymal stem cells (hA-MSCs) also participate in gastric tumorigenesis by increasing tumor cells invasion and metastasis during H. pylori infection (42).
In addition to malignant transformation, MSCs can promote tumorigenesis locally and systemically by compromising cancer immune surveillance or altering tumor stroma. When transplanting BM-MSCs in H. pylori infected mice model, IL-10 and transforming growth factor-β1 (TGF-β1) can be increased, as well as T cells secreting IL-10 and CD4+ CD25+ Foxp3+ regulatory T (Treg) cells in splenic mononuclear cells (43, 44). BM-MSCs can reduce the fraction of T cells that produce IFN-γ, thus inhibiting CD4+ and CD8+ T cell proliferation. Local and systemic immunosuppression mediated by BM-MSCs contributes to GC development induced by H. pylori (43).
MSCs can also promote tumorigenesis by altering tumor stromal components. Thrombospondin (THBS) promotes tumorigenesis through crosstalk with BM-MSCs. Infection with H. pylori significantly upregulates the expression of THBS4 in BM-MSCs. Overexpressed THBS4 then mediates BM-MSC-induced angiogenesis in GC by activating the THBS4/integrin α2/PI3K/AKT pathway (45). Moreover, BM-MSCs can differentiate into pan-cytokeratin-positive (pan-CK+) epithelial cells and alpha-smooth muscle actin (α-SMA+) cancer-associated fibroblasts (CAFs) by secreting THBS2, thus promoting the development of H. pylori-associated GC (46).
BM-MSCs play pivotal roles in H. pylori-associated GC. The immune regulatory functions of MSCs remain obscure. Shedding light on these functions and their mechanisms will provide clues on therapeutic targets for preventing GC development.
CAFs are activated myofibroblasts that accompany solid tumors and are principal constituents of tumor stroma (84, 85). They play important roles in the TME. They can create a niche for cancer cells and promote cancer progression by stimulating cancer cell proliferation, migration, invasion, and angiogenesis (85–87). Proinflammatory and tumor-associated factors secreted by CAFs might induce persistent inflammation or intervene in tumor immunity, thus mediate tumor immune escape (52, 88). Mainly derived from MSCs, CAFs could induce epithelial-mesenchymal transition (EMT), which enhances the invasive properties of malignant cells (89, 90) that detach from primary tumor site to surrounding tissues (91).
Helicobacter pylori infection can induce MSCs differentiating into CAFs, and upregulate the expression of fibroblast markers, fibroblast activation protein (FAP), CAF activation markers, and aggressive/invasive markers (47). FAP-positive CAFs enhance the survival, proliferation, and migration of GC cell lines and inhibit T cells function (48). H. pylori infection also increases the expression of hepatoma-derived growth factor (HDGF) (49, 50). Exposure to HDGF promotes the recruitment of BM-MSCs, stimulates their differentiation into CAF-myofibroblasts, and enhances tumor cell proliferation, invasiveness, and metastasis (49). Moreover, H. pylori infection can induce fibroblasts transdifferentiating into myofibroblasts, which upregulating the early carcinogenic marker hypoxia-inducible factor 1-alpha (HIF-1α) and downregulating proapoptotic bcl-2-like protein 4 (Bax) expression (51).
CAFs induced by H. pylori propel EMT by nuclear factor kappa B (NF-κB), signal transducer and activator of transcription 3 (STAT3), and TGF-β. Helicobacter pylori might induce the activation or differentiation of rat gastric fibroblasts in vitro, which then activate NF-κB and STAT3 signaling, and upregulate Snail1. This is an EMT-inducing transcription factor (EMT-TF) (52). As a major propeller of EMT in cancer progression and metastasis (53, 54), TGF-β can initiate tumorigenesis by activating EMT-type III initiation in epithelial cell compartments at the early stage of cancer development (55, 92). Gastric fibroblasts activated by H. pylori promote normal gastric epithelial cells to precancerous phenotype, and promote EMT by regulating TGFβ R1/R2-dependent signaling (55). The HH, Wnt, and Notch signaling pathways can interact with TGF-β pathway and induce EMT progression (93–97).
Collectively, persistent H. pylori infection increases the differentiation of CAFs, which propel EMT through NF-κB, STAT3, and TGF-β. As CAFs play key roles in the gastric microenvironment, targeting CAFs might be a potential strategy to improve the prognosis of patients (98, 99).
Immature myeloid (progenitor) cells (IMCs) do not mediate immunosuppression in healthy individuals. However, chronic inflammation, infections, and autoimmune diseases impair IMC differentiation and decrease peripheral myeloid cells numbers, resulting in more myelopoiesis (100–103). This eventually results in myeloid-derived suppressor cells (MDSCs) accumulation and immunosuppression (102, 104). MDSCs mediate immune suppression by inducing immunosuppressive cells (105), blocking lymphocyte homing (106), producing reactive oxygen and nitrogen species (107, 108), exhausting critical metabolites for T cell function (109), expressing negative immune checkpoint molecules (110).
Interactions between H. pylori and MDSCs are important in gastric immune microenvironment. On one hand, H. pylori can induce the differentiation of myeloid cell differentiation factor Schlafen 4 (SLFN4+) MDSCs (56, 58). This factor marks a subset of MDSCs in the stomach during H. pylori-induced spasmolytic polypeptide-expressing metaplasia (SPEM) (57). During chronic H. pylori infection in mice model, a subset of HH-Gli1-dependent immune cells is recruited to the gastric epithelium, and polarizes into SLFN4+ MDSCs. Overexpression of the SHH ligand in infected WT mice accelerates SLFN4+ MDSCs differentiataion in gastric corpus (57). Furthermore, H. pylori can stimulate plasmacytoid dendritic cells to secrete IFN-α through toll-like receptor 9-myeloid differentiation factor 88-interferon regulatory factor 7 (TLR9-MyD88-IRF7 pathway) (58). Differentiated SLFN4+ MDSCs inhibit gastric inflammatory response induced by H. pylori and suppress T cell function (56–59). Persistent immune dysregulation then favors intestinal metaplasia and neoplastic transformation, which leads to immune disorders and tumor progression.
Several markers, such as MiR130b, apoptosis signal-regulating kinase 1 (ASK1), interleukin 22 (IL-22), programmed death-ligand 1 (PD-L1), and Krüppel-like factor 4 (KLF4) play regulatory roles in the interactions between H. pylori and MDSCs. MiR130b produced by SLFN4+ MDSCs suppress T cells function and promote H. pylori-induced metaplasia (59). ASK1 deficiency promotes a Th1-dependent immune response and recruits immature Gr-1+Cd11b+ MDSCs with H. pylori infection. This could lead to the development of gastric atrophy and metaplasia (25, 60). Moreover, IL-22 secreted by polarized Th22 cells induced by H. pylori can stimulate CXCL2 production from gastric epithelial cells. This causes CXCR2+ MDSCs migration to gastric mucosa, where they produce proinflammatory proteins and suppress Th1 cell responses, contributing to the development of H. pylori-associated gastritis (61). PD-L1 upregulation on the surface of gastric epithelial cells at the early stage of H. pylori infection (62) promotes tumor infiltration of MDSCs (63) and then lead to anti-PD-1/PD-L1 treatment resistance (64). KLF4 is an evolutionarily conserved zinc finger transcription factor and key regulator of diverse cellular processes (111–113). Helicobacter pylori and its virulence factor CagA can influence KLF4 expression. The transduction of CagA or infection with H. pylori downregulates KLF4 expression by inducing CXCL8 expression, and low KLF4 expression further upregulates CXCL8 expression (65). Increased CXCL8 expression promotes MDSCs recruitment to tumors as well as tumor growth, and creates an immunosuppressive microenvironment conducive to resistance against immune response (65–67).
A high abundance of MDSCs in patients correlate with more advanced GC and a poor prognosis (114, 115). MDSCs infiltration induced by H. pylori mediates immunosuppression, immune dysfunction, gastric tumorigenesis, and reduces the effect of chemotherapy and immunotherapy (63). The possibility that combining immunotherapy or chemotherapy with MDSC-targeting therapy might overcome drug resistance and improve prognosis warrants investigation (116–118).
In addition to cells in TME, immune checkpoints are involved in regulating H. pylori-associated TME. (Table 2).

Table 2 Effects of H. pylori on tumor-related proteins in gastric tumor immune microenvironment.
The 55 kDa transmembrane protein programmed death 1 (PD-1) is expressed in activated T cells, natural killer (NK) cells, B lymphocytes, macrophages, dendritic cells (DCs), and monocytes. It is abundantly expressed in tumor-specific T cells (126–128). PD-L1 (also known as CD274 or B7-H1) is a 33 kDa type 1 transmembrane glycoprotein that is widely expressed in macrophages, activated T lymphocytes, B cells, DCs, and also expressed in tumor cells (129). Binding of PD-1 and PD-L1 enhances T cell tolerance, inhibits T cell activation and proliferation, increases Th cell transformation to Foxp3+ Treg cell, and prevents T cell cytolysis in tumor cells (130). Thus, interaction between PD-1 and PD-L1 is a double-edged sword. It can inhibit immune responses and promote self-tolerance, while it can also lead to immune escape and tumor progression.
Helicobacter pylori infection could upregulate PD-1/PD-L1 expression in gastric ulcers and GC patients (119), which might be related with poor prognosis (131, 132). Chronic H. pylori infection could cause excessive damage to gastric mucosa. Upregulated PD-1/PD-L1 is launched to avoid such damage, meanwhile this also reduces T cell-mediated cytotoxicity and promotes GC progression (119–121). SHH pathway is involved in PD-L1 upregulating (62). As an HH transcriptional effector, zinc finger protein GL1, mediates mammalian target of rapamycin (mTOR)-induced PD-L1 expression in GC organoids (64). Kinds of H. pylori virulence factors are reported in this process. H. pylori T4SS components activate p38 MAPK pathway and upregulate PD-L1 expression, thus inhibiting T cell proliferation and inducing Treg differentiation from naïve T cells, which lead to immune escape (122, 123). Helicobacter pylori urease B subunit mediates PD-L1 upregulation via myosin heavy chain 9 (Myh9) or mTORC1 signaling in bone marrow-derived macrophages (BMDMs) and, and regulates CD8+ T cells infiltration and activation (124). Helicobacter pylori LPS induces PD-L1 expression via NF‐κB pathway in GC cells and eventually promotes GC progression (125).
Overall, PD-1/PD-L1 play vital roles in H. pylori-infected GC, which presents an opportunity and challenge for treatment. However, numerous unknown mechanisms of PD-1/PD-L1 expression might be the basis for overcoming drug resistance and developing novel immunotherapies (133). The mechanisms and functions of PD1/PD-L1 with H. pylori infection requires further investigation (132, 134–136).
Immunotherapy stimulates the immune system against neoplasms and harnesses the specificity of innate immune to fight cancer, particularly by activating T-cell mediated immunity (137, 138). With the wide application of immune therapy, the immune checkpoint inhibitors (ICIs) targeting immune checkpoint molecules such as PD-1 and CTLA-4, and other immune therapies such as cancer vaccine, the immune cells input, antigen vaccine, oncolytic viruses, and recombinant cytokines, have been receiving worldwide attention and have made a certain progress (139–147). However, as lack of optimal criteria selecting suitable patients until now, the objective response rate of immunotherapy remains low (148, 149). Hence, factors that influence the effectiveness of tumor immunotherapy need to be identified. In this section, we focused on the effects and potential applications of H. pylori infection on tumor immunotherapies (Figure 2; Table 3).
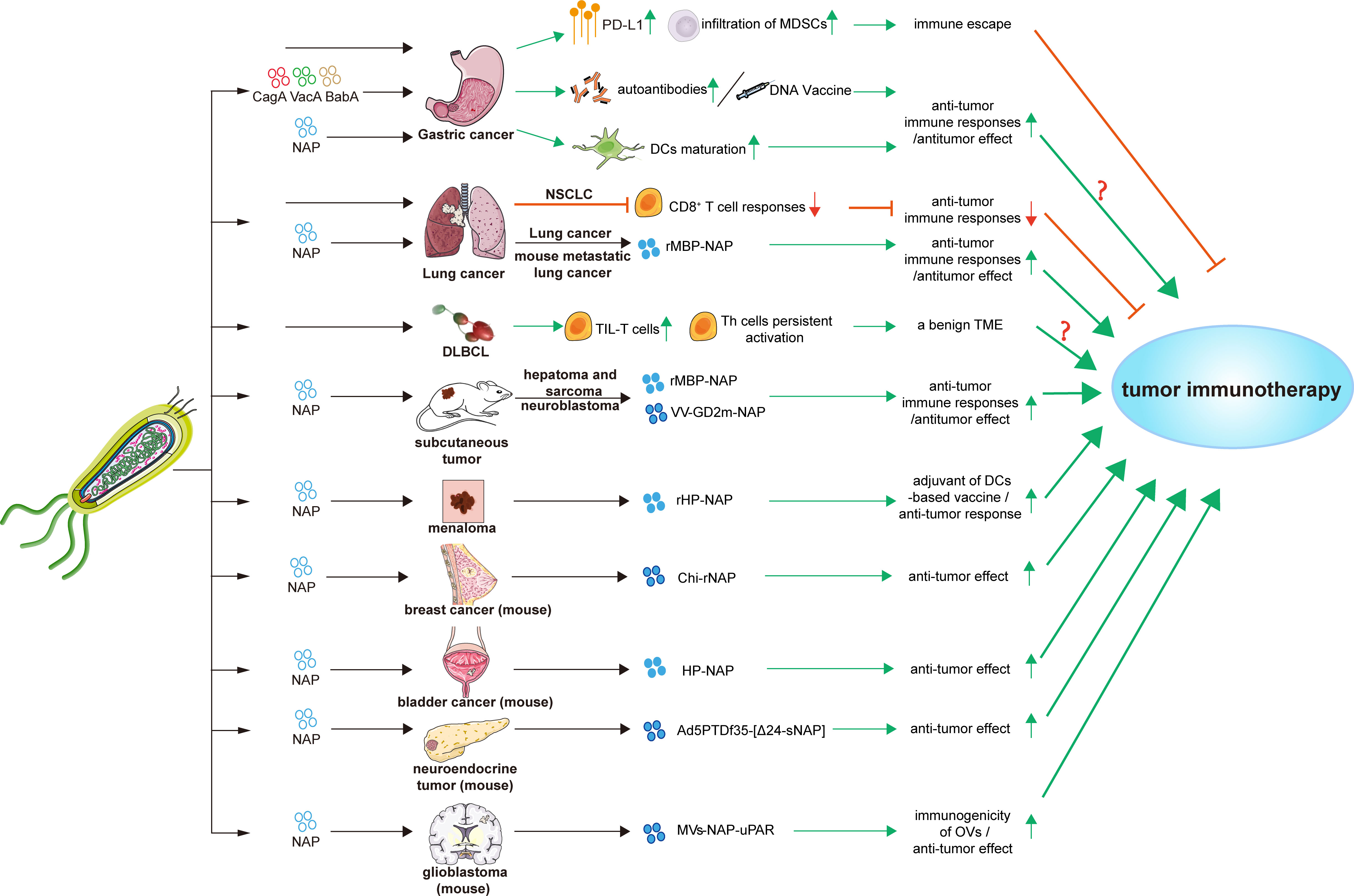
Figure 2 Effects and applications of H. pylori and its factors in tumor immunotherapies. Bab A, blood-group antigen-binding adhesin gene A; Cag A, cytotoxin-associated gene A; Chi-rNap, rNAP coated chitosan nanoparticles; DCs, dendritic cells; DLBCL, diffuse large B-cell lymphoma; HP-NAP, H. pylori neutrophil-activating protein; MDSCs, myeloid-derived suppressor cells; MV-NAP-uPAR, recombinant measles virus-NAP-urokinase-type plasminogen activator receptor; NSCLC, non-small cell lung cancer; OVs, oncolytic viruses; PD-L1, programmed death-ligand 1; rHP-NAP, recombinant H. pylori neutrophil-activating protein; rMBP-NAP, recombinant HP-NAP with the maltose-binding protein of Escherichia coli; Th cells, T helper cells; TIL-T cells, tumor-infiltrating T lymphocytes; TME, tumor microenvironment; Vac A, vacuolating cytotoxin A; VV-GD2m-NAP, vaccinia virus - neuroblastoma-associated antigen disialoganglioside mimotope.
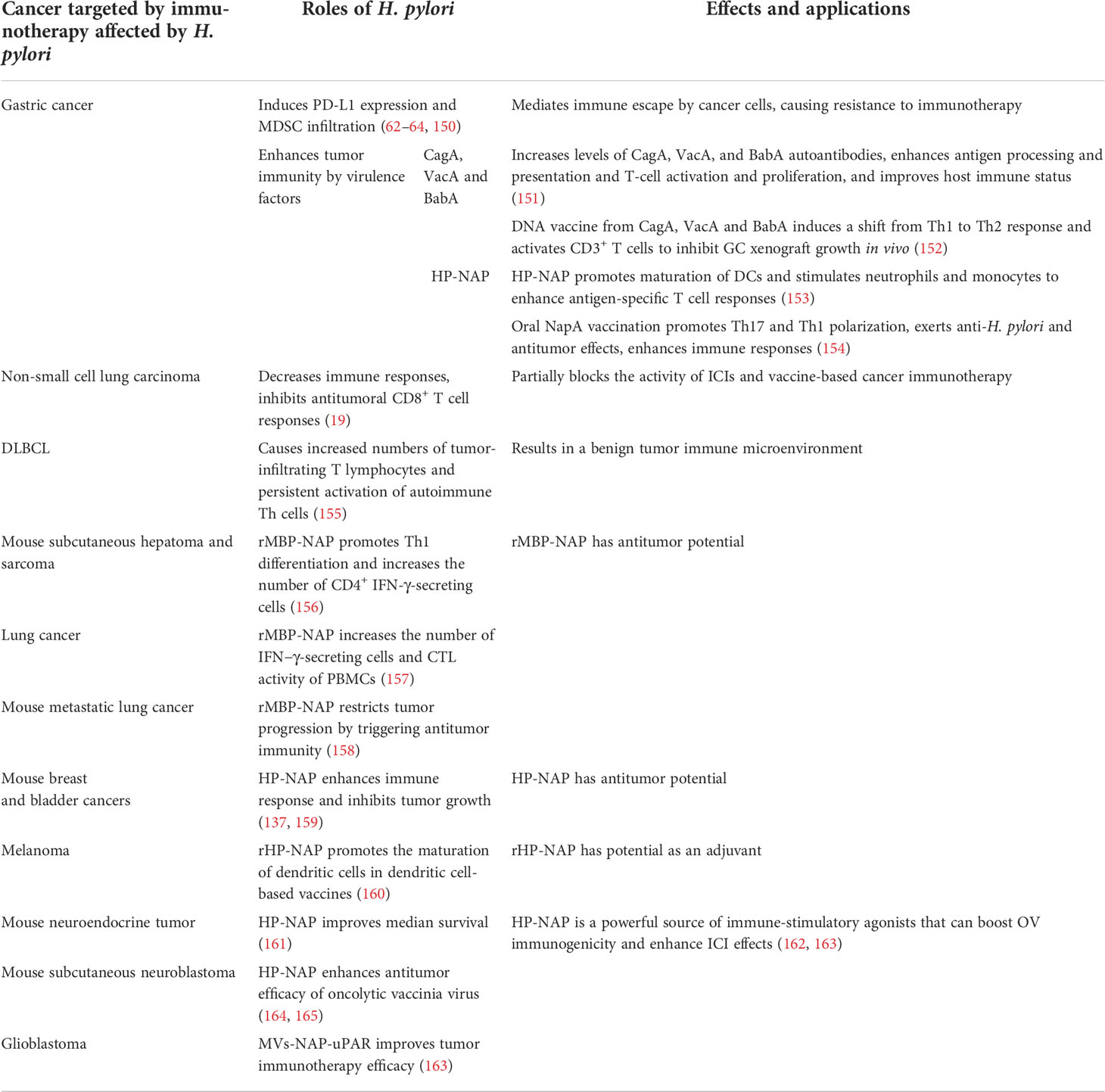
Table 3 Effects of H. pylori on tumor immunotherapy responses.
The 5-year survival rate of advanced GC patients is <30%. Although platinum-fluoropyrimidine combination chemotherapy is the standard first-line treatment for advanced GC, its low complete response rate and severe adverse reactions have limited its application (63, 166). Novel effective therapies are urgently required. For example, PD-1 inhibitor pembrolizumab received accelerated approval from the US Food and Drug Administration (FDA) in 2017 to treat recurrent advanced or metastatic gastric or gastroesophageal junction adenocarcinomas expressing PD-L1 (63, 167–169).
Helicobacter pylori is a class I carcinogen associated with GC (170–172). The overall survival of GC diagnosis is reported to be higher for patients with H. pylori infection (17). Helicobacter pylori infection induces PD-L1 expression and MDSC infiltration that mediate immune escape. HH signaling activated by H. pylori infection induces PD-L1 expression and tumor cell proliferation in GC, resulting in cancer cell resistance to immunotherapy (150). In addition, Helicobacter pylori and its virulence factors can act as antigens or adjuvants to enhance tumor immunity.
Helicobacter pylori virulence factors, such as CagA, VacA, blood-group antigen-binding adhesin gene (BabA), and H. pylori neutrophil-activating protein (HP-NAP), can act as antigens or adjuvants to enhance tumor immunity. The stimulation of autoantibodies during antigen processing and presentation and subsequent T-cell activation and proliferation improves the host immune status, which can kill cancer cells and even suppress metastasis (151). Moreover, H. pylori DNA vaccines encoding fragments of CagA, VacA, and BabA can induce Th1 shift to Th2 response in immunized BALB/c mice, which mimics the immune status of GC patients with chronic H. pylori infection. Stimulated CD3+ T cells inhibit the proliferation of human GC cells in vitro, and the adoptive infusion of CD3+ T cells inhibits the growth of GC xenografts in vivo (152).
HP-NAP is a major virulence factor in H. pylori infection and colony formation, and it can also act as a protective factor (173, 174). As a Toll-like receptor-2 (TLR2) agonist, HP-NAP can bind to TLR2 of neutrophils (161, 175). Furthermore, HP-NAP promotes the maturation of DCs with Th1 polarization and improves migration of mature DCs. Stimulating neutrophils and monocytes by HP-NAP induces IL-12 and IL-23 expression, thus shifting antigen-specific T cell responses from the Th2 to the Th1 phenotype which characterized by abundant IFN-γ and TNF-α expression (153). Vaccination with HP-NAP A subunit (NapA) promotes Th17 and Th1 polarization. Such vaccines have potential effects as an anti-H. pylori oral vaccine candidate and a mucosal immunomodulatory agent, which could be used in antitumor strategies (154).
In addition to GC, the influence of H. pylori on other tumor immunotherapies is also paid much attention recently. Helicobacter pylori infection might disrupt the immune system and exert detrimental effects on the outcomes of cancer immunotherapies (19).
Helicobacter pylori seropositivity could reduce anti-PD-1 immunotherapy effect in non-small cell lung cancer (NSCLC) patients. Helicobacter pylori infection partially blocks the activities of ICIs and vaccine-based cancer immunotherapies. Helicobacter pylori suppresses the innate and adaptive immune responses of infected hosts and inhibits antitumor CD8+ T cell responses by altering the cross-presentation activity of DCs (19). In contrast, a significantly high proportion of tumor-infiltrating T lymphocytes in H. pylori-positive de novo diffuse large B-cell lymphoma (DLBCL) patients preliminarily indicates a benign TME. Inflammation induced by H. pylori confers persistent activation of autoimmune Th cells, which would explain the benign TME (155). More researches are necessary to elucidate how H. pylori infection status influences the effects of tumor immunotherapies.
The immunomodulatory activity and potential applications of NAP in tumor immunotherapy have been investigated. Recombinant HP-NAP with the maltose-binding protein of Escherichia coli (rMBP-NAP) can mediate T helper lymphocytes differentiation into the Th1 phenotype and significantly increase the number of CD4+ IFN-γ-secreting T cells. This induces antitumor effects through a TLR-2-dependent mechanism in subcutaneous hepatoma and sarcoma mice model (156). rMBP-NAP can significantly increase peripheral blood mononuclear cells (PBMCs) that secrete IFN-γ, and prominently increases the cytotoxic activity of PBMCs derived from lung cancer patients (157). Treatment with rMBP-NAP restricts the progression of metastatic lung cancer in mice model by triggering antitumor immunity (158). A therapeutic nanocomplex of HP-NAP altered the production rate of cytokines and increase tumoricidal activities of the immune system, leading to decreased breast tumor growth in mice (137). Local administration of HP-NAP inhibits tumor growth by triggering tumor cell necrosis in bladder cancer mice model (159). Recombinant HP-NAP has potential effects as an adjuvant in DC-based vaccines for treating melanoma (160).
Because of its ideal immunogenicity, NAP has recently been applied as an immune adjuvant to enhance the antitumor immune response. When combined with oncolytic viruses (OVs), HP-NAP can activate the immune response. The intratumoral administration of adenovirus armed with secretory HP-NAP can improve the median survival rate of nude mice xenografted with neuroendocrine tumors (161). A recombinant vaccinia virus (VV) neuroblastoma-associated antigen disialoganglioside mimotope (GD2m)-NAP significantly improved therapeutic efficacy. Helicobacter pylori-NAP might help to overcome virus-mediated suppressive immune responses, resulting in improved anti-GD2 antibody production and a better therapeutic outcome (164, 165). Moreover, recombinant measles virus (MV)-NAP-urokinase-type plasminogen activator receptor (uPAR) can improve immunotherapeutic effects on glioblastoma with a better tumor prognosis and increased susceptibility to CD8+ T cell-mediated lysis. Overall, HP-NAP represents a potential immunostimulatory agonists which can boost the immunogenicity of OVs and enhance ICIs effects (162, 163).
In conclusion, H. pylori and its virulence factors could be closely related with personalized treatment strategies during tumor immunotherapies. The mechanisms of H. pylori infection in tumor immunotherapies requires further elucidation, and the translation of research findings to clinical applications should be accelerated.
This review summarized current knowledge of the effects of H. pylori on the immune microenvironment of GC and tumor immunotherapy responses. Helicobacter pylori elicits powerful immune responses during surviving and colonizing gastric mucosa. Helicobacter pylori has also developed several strategies to evade recognition and disrupt immune function. The constituents and functions of stroma are regulated by H. pylori and its virulence factors to facilitate its survival and colony. Persistent H. pylori infection can induce immune evasion and tumorigenesis.
The stroma provides TME for tumor initiation and development after H. pylori persistent infection. Immunotherapy targeting tumor-associated immune cells is more mature and improved, particularly immunotherapy targeting T cells, such as ICIs. PD-1 inhibitor pembrolizumab has received approval from the US FDA in 2017 to treat recurrent advanced or metastatic gastric or gastroesophageal junction adenocarcinomas (167). While some clinical trials targeting non-immune cells in TME such as CAFs, MSCs, have failed to show promising efficacy in cancer patients (176–178). The main reason might be a lack of deep understanding of the fundamental mechanisms of stromal cells and elements as well as a lack of reliable biomarkers to guide stroma-targeted therapies (176). Of course, because of the important roles of regulating the immune response in TME, targeting TAMs is getting more and more attraction. For example, targeting colony-stimulating factor 1 receptor (CSF1R) signaling and the CCL2-CCR2 axis are developing drugs (179, 180). And there are some developing drugs to reprogram TAMs from a pro-tumor phenotype to an anti-tumor phenotype and interrupt the bad cycle between TAMs and tumor cells (176, 177), such as agonistic anti-CD40 antibodies (181), PI3Kγ inhibitors (182). These ongoing researches show good prospects in immunotherapy. Based on these, it seems that immunotherapy intervening tumor-associated immune cells may be more appropriate currently. However, we should also pay attention to the study of non-immune cells in TME. Further research on these cells may provide clues for developing new therapies in the future.
H. pylori infection might affect the tumor immunotherapy. Although H. pylori infection has been reported as a protective factor in GC immunotherapy while in NSCLC as a negative factor, the mechanisms and effect of H. pylori on GC immunotherapy still remains unclear (19, 183). Helicobacter pylori virulence factors can act as immunogens or adjuvants to elicit or enhance immune responses. Some H. pylori virulence factors such as HP-NAP, have been applied as adjuvants or combined with drugs in pan-tumor treatment to improve immunotherapeutic efficiency. The effects of H. pylori in TME should be further explored, and clinical applications should be performed to select the proper features of population for better immunotherapy benefits.
RD and HZ searched the literature and wrote the manuscript. HC and ML re-checked the literature. YS and SD designed this study and revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was funded by the National Natural Science Foundation of China (Grant No. 81700496 and 81870386), Peking University Medicine Fund of Fostering Young Scholars’ Scientific and Technological Innovation (BMU2021PY002), and Key laboratory for Helicobacter pylori infection and upper gastrointestinal diseases, Beijing Key Laboratory (No.BZ0371).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Baj J, Forma A, Sitarz M, Portincasa P, Garruti G, Krasowska D, et al. Helicobacter pylori virulence factors-mechanisms bacterial pathogenicity gastric microenvironment. Cells (2020) 10(1):27. doi: 10.3390/cells10010027
2. Mentis A, Lehours P, Megraud F. Epidemiology and diagnosis of helicobacter pylori infection. Helicobacter. (2015) 20(Suppl 1):1–7. doi: 10.1111/hel.12250
3. Machlowska J, Baj J, Sitarz M, Maciejewski R, Sitarz R. Gastric cancer: Epidemiology, risk factors, classification, genomic characteristics and treatment strategies. Int J Mol Sci (2020) 21(11):4012. doi: 10.3390/ijms21114012
4. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
5. Plummer M, Franceschi S, Vignat J, Forman D, de Martel C. Global burden of gastric cancer attributable to helicobacter pylori. Int J Cancer (2015) 136(2):487–90. doi: 10.1002/ijc.28999
6. McColl KE. Clinical practice. Helicobacter pylori Infect N Engl J Med (2010) 362(17):1597–604. doi: 10.1056/NEJMcp1001110
7. Ishaq S, Nunn L. Helicobacter pylori and gastric cancer: a state of the art review. Gastroenterol Hepatol Bed Bench. (2015) 8(Suppl 1):S6–S14. doi: 10.22037/ghfbb.v8iSupplement.653
8. Anwar W, Armstrong BK, Correa P, Forman D, Gentile JM, Haswell-Elkins M, et al. Schistosomes, liver flukes and helicobacter pylori. In: IARC working group on the evaluation of carcinogenic risks to humans, vol. 61. . Lyon: IARC Monogr Eval Carcinog Risks Hum. p. 1–241.
9. Seeneevassen L, Bessede E, Megraud F, Lehours P, Dubus P, Varon C. Gastric cancer: Advances in carcinogenesis research and new therapeutic strategies. Int J Mol Sci (2021) 22(7):3418. doi: 10.3390/ijms22073418
10. Liccardi G, Pentimalli F. Cancer, immunity and inflammation. report from the CDD Cambridge conferences 2018 and 2019. Cell Death Dis (2019) 10(11):798. doi: 10.1038/s41419-019-2032-0
11. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
12. Yolanda LV, Sergio PD, Hugo ES, Isabel AF, Rafael BZ, Aldo TD, et al. Gastric cancer progression associated with local humoral immune responses. BMC Cancer (2015) 15:924. doi: 10.1186/s12885-015-1858-9
13. Mejias-Luque R, Gerhard M. Immune evasion strategies and persistence of helicobacter pylori. Curr Top Microbiol Immunol (2017) 400:53–71. doi: 10.1007/978-3-319-50520-6_3
14. Song L, Song M, Rabkin CS, Williams S, Chung Y, Van Duine J, et al. Helicobacter pylori immunoproteomic profiles in gastric cancer. J Proteome Res (2021) 20(1):409–19. doi: 10.1021/acs.jproteome.0c00466
15. Alexander SM, Retnakumar RJ, Chouhan D, Devi TNB, Dharmaseelan S, Devadas K, et al. Helicobacter pylori in human stomach: The inconsistencies in clinical outcomes and the probable causes. Front Microbiol (2021) 12:713955. doi: 10.3389/fmicb.2021.713955
16. Zhang MJ, Chen DS, Li S, Chen L, Qi YX, Zhang CJ. Helicobacter pylori infection as a potential favorable factor for immune checkpoint inhibitor therapy for gastric cancer. Invest New Drugs (2021) 39(5):1436–8. doi: 10.1007/s10637-021-01122-5
17. Fang X, Liu K, Cai J, Luo F, Yuan F, Chen P. Positive helicobacter pylori status is associated with better overall survival for gastric cancer patients: evidence from case-cohort studies. Oncotarget. (2017) 8(45):79604–17. doi: 10.18632/oncotarget.18758
18. Li G, Yu S, Xu J, Zhang X, Ye J, Wang Z, et al. The prognostic role of helicobacter pylori in gastric cancer patients: A meta-analysis. Clin Res Hepatol Gastroenterol (2019) 43(2):216–24. doi: 10.1016/j.clinre.2018.08.012
19. Oster P, Vaillant L, Riva E, McMillan B, Begka C, Truntzer C, et al. Helicobacter pylori infection has a detrimental impact on the efficacy of cancer immunotherapies. Gut. (2021) 71(3):457–66. doi: 10.1136/gutjnl-2020-323392
20. Shi Y, Zheng H, Wang M, Ding S. Influence of helicobacter pylori infection on PD-1/PD-L1 blockade therapy needs more attention. Helicobacter. (2022) 27(2):e12878. doi: 10.1111/hel.12878
21. Mohammadzadeh R, Soleimanpour S, Pishdadian A, Farsiani H. Designing and development of epitope-based vaccines against helicobacter pylori. Crit Rev Microbiol (2021), 1–24. doi: 10.1080/1040841X.2021.1979934
22. Del Giudice G, Malfertheiner P, Rappuoli R. Development of vaccines against helicobacter pylori. Expert Rev Vaccines (2009) 8(8):1037–49. doi: 10.1586/erv.09.62
23. Fu HW. Helicobacter pylori neutrophil-activating protein: from molecular pathogenesis to clinical applications. World J Gastroenterol (2014) 20(18):5294–301. doi: 10.3748/wjg.v20.i18.5294
24. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
25. Navashenaq JG, Shabgah AG, Banach M, Jamialahmadi T, Penson PE, Johnston TP, et al. The interaction of helicobacter pylori with cancer immunomodulatory stromal cells: New insight into gastric cancer pathogenesis. Semin Cancer Biol (2021) S1044-579X(21):00248–0. doi: 10.1016/j.semcancer.2021.09.014
26. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett (2017) 387:61–8. doi: 10.1016/j.canlet.2016.01.043
27. Gambardella V, Castillo J, Tarazona N, Gimeno-Valiente F, Martinez-Ciarpaglini C, Cabeza-Segura M, et al. The role of tumor-associated macrophages in gastric cancer development and their potential as a therapeutic target. Cancer Treat Rev (2020) 86:102015. doi: 10.1016/j.ctrv.2020.102015
28. Pagliari M, Munari F, Toffoletto M, Lonardi S, Chemello F, Codolo G, et al. Helicobacter pylori affects the antigen presentation activity of macrophages modulating the expression of the immune receptor CD300E through miR-4270. Front Immunol (2017) 8:1288. doi: 10.3389/fimmu.2017.01288
29. Codolo G, Toffoletto M, Chemello F, Coletta S, Soler Teixidor G, Battaggia G, et al. Helicobacter pylori dampens HLA-II expression on macrophages via the up-regulation of miRNAs targeting CIITA. Front Immunol (2019) 10:2923. doi: 10.3389/fimmu.2019.02923
30. Coletta S, Battaggia G, Della Bella C, Furlani M, Hauke M, Faass L, et al. ADP-heptose enables helicobacter pylori to exploit macrophages as a survival niche by suppressing antigen-presenting HLA-II expression. FEBS Lett (2021) 595(16):2160–8. doi: 10.1002/1873-3468.14156
31. Lewis ND, Asim M, Barry DP, de Sablet T, Singh K, Piazuelo MB, et al. Immune evasion by helicobacter pylori is mediated by induction of macrophage arginase II. J Immunol (2011) 186(6):3632–41. doi: 10.4049/jimmunol.1003431
32. Hardbower DM, Asim M, Murray-Stewart T, Casero RA Jr., Verriere T, Lewis ND, et al. Arginase 2 deletion leads to enhanced M1 macrophage activation and upregulated polyamine metabolism in response to helicobacter pylori infection. Amino Acids (2016) 48(10):2375–88. doi: 10.1007/s00726-016-2231-2
33. Krakowiak MS, Noto JM, Piazuelo MB, Hardbower DM, Romero-Gallo J, Delgado A, et al. Matrix metalloproteinase 7 restrains helicobacter pylori-induced gastric inflammation and premalignant lesions in the stomach by altering macrophage polarization. Oncogene. (2015) 34(14):1865–71. doi: 10.1038/onc.2014.135
34. Gobert AP, Verriere T, Asim M, Barry DP, Piazuelo MB, de Sablet T, et al. Heme oxygenase-1 dysregulates macrophage polarization and the immune response to helicobacter pylori. J Immunol (2014) 193(6):3013–22. doi: 10.4049/jimmunol.1401075
35. Che Y, Geng B, Xu Y, Miao X, Chen L, Mu X, et al. Helicobacter pylori-induced exosomal MET educates tumour-associated macrophages to promote gastric cancer progression. J Cell Mol Med (2018) 22(11):5708–19. doi: 10.1111/jcmm.13847
36. Wen J, Chen C, Luo M, Liu X, Guo J, Wei T, et al. Notch signaling ligand Jagged1 enhances macrophage-mediated response to helicobacter pylori. Front Microbiol (2021) 12:692832. doi: 10.3389/fmicb.2021.692832
37. Schumacher MA, Donnelly JM, Engevik AC, Xiao C, Yang L, Kenny S, et al. Gastric sonic hedgehog acts as a macrophage chemoattractant during the immune response to helicobacter pylori. Gastroenterology. (2012) 142(5):1150–9.e6. doi: 10.1053/j.gastro.2012.01.029
38. Fakhari S, Kalantar E, Nikzaban M, Hakhamneshi MS, Fathi F, Nikkhoo B, et al. Effect of helicobacter pylori infection on stromal-derived factor-1/CXCR4 axis in bone marrow-derived mesenchymal stem cells. Adv BioMed Res (2014) 3:19. doi: 10.4103/2277-9175.124650
39. Varon C, Dubus P, Mazurier F, Asencio C, Chambonnier L, Ferrand J, et al. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology. (2012) 142(2):281–91. doi: 10.1053/j.gastro.2011.10.036
40. Ferrand J, Lehours P, Schmid-Alliana A, Megraud F, Varon C. Helicobacter pylori infection of gastrointestinal epithelial cells in vitro induces mesenchymal stem cell migration through an NF-kappaB-dependent pathway. PLoS One (2011) 6(12):e29007. doi: 10.1371/journal.pone.0029007
41. Alison MR, Islam S, Wright NA. Stem cells in cancer: instigators and propagators? J Cell Sci (2010) 123(Pt 14):2357–68. doi: 10.1242/jcs.054296
42. Moradi SL, Eslami G, Goudarzi H, Hajishafieeha Z, Soleimani M, Mohammadzadeh A, et al. Role of helicobacter pylori on cancer of human adipose-derived mesenchymal stem cells and metastasis of tumor cells-an in vitro study. Tumou Biol (2016) 37(3):3371–8. doi: 10.1007/s13277-015-4137-0
43. Lin R, Ma H, Ding Z, Shi W, Qian W, Song J, et al. Bone marrow-derived mesenchymal stem cells favor the immunosuppressive T cells skewing in a helicobacter pylori model of gastric cancer. Stem Cells Dev (2013) 22(21):2836–48. doi: 10.1089/scd.2013.0166
44. Yaghoobi M. Bone marrow-derived stem cells in pathogenesis of helicobacter pylori-associated gastric cancer. Clin Transl Gastroenterol (2015) 6:e110. doi: 10.1038/ctg.2015.35
45. He L, Wang W, Shi H, Jiang C, Yao H, Zhang Y, et al. THBS4/integrin alpha2 axis mediates BM-MSCs to promote angiogenesis in gastric cancer associated with chronic helicobacter pylori infection. Aging (Albany NY) (2021) 13(15):19375–96. doi: 10.18632/aging.203334
46. Shi H, Qi C, Meng L, Yao H, Jiang C, Fan M, et al. Bone marrow-derived mesenchymal stem cells promote helicobacter pylori-associated gastric cancer progression by secreting thrombospondin-2. Cell Prolif (2021) 54(10):e13114. doi: 10.1111/cpr.13114
47. Zhang Q, Wang M, Huang F, Yang T, Cai J, Zhang X, et al. H. pylori infection-induced MSC differentiation into CAFs promotes epithelial-mesenchymal transition in gastric epithelial cells. Int J Mol Med (2013) 32(6):1465–73. doi: 10.3892/ijmm.2013.1532
48. Wen X, He X, Jiao F, Wang C, Sun Y, Ren X, et al. Fibroblast activation protein-alpha-Positive fibroblasts promote gastric cancer progression and resistance to immune checkpoint blockade. Oncol Res (2017) 25(4):629–40. doi: 10.3727/096504016X14768383625385
49. Liu CJ, Wang YK, Kuo FC, Hsu WH, Yu FJ, Hsieh S, et al. Helicobacter pylori infection-induced hepatoma-derived growth factor regulates the differentiation of human mesenchymal stem cells to myofibroblast-like cells. Cancers (Basel) (2018) 10(12):479. doi: 10.3390/cancers10120479
50. Lee KH, Choi EY, Kim MK, Lee SH, Jang BI, Kim TN, et al. Hepatoma-derived growth factor regulates the bad-mediated apoptotic pathway and induction of vascular endothelial growth factor in stomach cancer cells. Oncol Res (2010) 19(2):67–76. doi: 10.3727/096504010X12864748215043
51. Krzysiek-Maczka G, Targosz A, Ptak-Belowska A, Korbut E, Szczyrk U, Strzalka M, et al. Molecular alterations in fibroblasts exposed to helicobacter pylori: a missing link in bacterial inflammation progressing into gastric carcinogenesis? J Physiol Pharmacol (2013) 64(1):77–87.
52. Krzysiek-Maczka G, Targosz A, Szczyrk U, Strzalka M, Brzozowski T, Ptak-Belowska A. Involvement of epithelial-mesenchymal transition-inducing transcription factors in the mechanism of helicobacter pylori-induced fibroblasts activation. J Physiol Pharmacol (2019) 70(5):727–36. doi: 10.26402/jpp.2019.5.08
53. Krzysiek-Maczka G, Wrobel T, Targosz A, Szczyrk U, Strzalka M, Ptak-Belowska A, et al. Helicobacter pylori-activated gastric fibroblasts induce epithelial-mesenchymal transition of gastric epithelial cells in vitro in a TGF-beta-dependent manner. Helicobacter. (2019) 24(5):e12653. doi: 10.1111/hel.12653
54. Katsuno Y, Lamouille S, Derynck R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol (2013) 25(1):76–84. doi: 10.1097/CCO.0b013e32835b6371
55. Krzysiek-Maczka G, Targosz A, Szczyrk U, Wrobel T, Strzalka M, Brzozowski T, et al. Long-term helicobacter pylori Infect switches gastric epithelium reprogram towards cancer stem cell-related differ program Hp-activated gastric fibroblast-TGFbeta dependent manner. Microorganisms (2020) 8(10):1519. doi: 10.3390/microorganisms8101519
56. El-Zaatari M, Kao JY, Tessier A, Bai L, Hayes MM, Fontaine C, et al. Gli1 deletion prevents helicobacter-induced gastric metaplasia and expansion of myeloid cell subsets. PLoS One (2013) 8(3):e58935. doi: 10.1371/journal.pone.0058935
57. Ding L, Hayes MM, Photenhauer A, Eaton KA, Li Q, Ocadiz-Ruiz R, et al. Schlafen 4-expressing myeloid-derived suppressor cells are induced during murine gastric metaplasia. J Clin Invest (2016) 126(8):2867–80. doi: 10.1172/JCI82529
58. Xiang X, Wu Y, Li H, Li C, Yan L, Li Q. Plasmacytoid dendritic cell-derived type I interferon is involved in helicobacter pylori infection-induced differentiation of schlafen 4-expressing myeloid-derived suppressor cells. Infect Immun (2021) 89(11):e0040721. doi: 10.1128/IAI.00407-21
59. Ding L, Li Q, Chakrabarti J, Munoz A, Faure-Kumar E, Ocadiz-Ruiz R, et al. MiR130b from Schlafen4(+) MDSCs stimulates epithelial proliferation and correlates with preneoplastic changes prior to gastric cancer. Gut. (2020) 69(10):1750–61. doi: 10.1136/gutjnl-2019-318817
60. Hayakawa Y, Hirata Y, Hata M, Tsuboi M, Oya Y, Kurokawa K, et al. Dysregulated immune responses by ASK1 deficiency alter epithelial progenitor cell fate and accelerate metaplasia development during h. pylori infection. Microorganisms (2020) 8(12):1995. doi: 10.3390/microorganisms8121995
61. Zhuang Y, Cheng P, Liu XF, Peng LS, Li BS, Wang TT, et al. A pro-inflammatory role for Th22 cells in helicobacter pylori-associated gastritis. Gut. (2015) 64(9):1368–78. doi: 10.1136/gutjnl-2014-307020
62. Holokai L, Chakrabarti J, Broda T, Chang J, Hawkins JA, Sundaram N, et al. Increased programmed death-ligand 1 is an early epithelial cell response to helicobacter pylori infection. PLoS Pathog (2019) 15(1):e1007468. doi: 10.1371/journal.ppat.1007468
63. Kim W, Chu TH, Nienhuser H, Jiang Z, Del Portillo A, Remotti HE, et al. PD-1 signaling promotes tumor-infiltrating myeloid-derived suppressor cells and gastric tumorigenesis in mice. Gastroenterology. (2021) 160(3):781–96. doi: 10.1053/j.gastro.2020.10.036
64. Koh V, Chakrabarti J, Torvund M, Steele N, Hawkins JA, Ito Y, et al. Hedgehog transcriptional effector GLI mediates mTOR-induced PD-L1 expression in gastric cancer organoids. Cancer Lett (2021) 518:59–71. doi: 10.1016/j.canlet.2021.06.007
65. Liu Z, Wu X, Tian Y, Zhang W, Qiao S, Xu W, et al. H. pylori infection induces CXCL8 expression and promotes gastric cancer progress through downregulating KLF4. Mol Carcinog (2021) 60(8):524–37. doi: 10.1002/mc.23309
66. Li BH, Garstka MA, Li ZF. Chemokines and their receptors promoting the recruitment of myeloid-derived suppressor cells into the tumor. Mol Immunol (2020) 117:201–15. doi: 10.1016/j.molimm.2019.11.014
67. Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res (2016) 22(15):3924–36. doi: 10.1158/1078-0432.CCR-15-2463
68. Zhang X, Arnold IC, Muller A. Mechanisms of persistence, innate immune activation and immunomodulation by the gastric pathogen helicobacter pylori. Curr Opin Microbiol (2020) 54:1–10. doi: 10.1016/j.mib.2020.01.003
69. Kaparakis M, Walduck AK, Price JD, Pedersen JS, van Rooijen N, Pearse MJ, et al. Macrophages are mediators of gastritis in acute helicobacter pylori infection in C57BL/6 mice. Infect Immun (2008) 76(5):2235–9. doi: 10.1128/IAI.01481-07
70. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
71. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol (2015) 15(5):271–82. doi: 10.1038/nri3831
72. Cassetta L, Cassol E, Poli G. Macrophage polarization in health and disease. ScientificWorldJournal. (2011) 11:2391–402. doi: 10.1100/2011/213962
73. Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol Res (2012) 53(1-3):11–24. doi: 10.1007/s12026-012-8291-9
74. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
75. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3(1):23–35. doi: 10.1038/nri978
76. Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol (2012) 19(6):586–93. doi: 10.1038/nsmb.2296
77. Wilson KT, Crabtree JE. Immunology of helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. (2007) 133(1):288–308. doi: 10.1053/j.gastro.2007.05.008
78. Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, et al. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol (2017) 43:74–89. doi: 10.1016/j.semcancer.2017.03.001
79. Pulendran B, Davis MM. The science and medicine of human immunology. Science. (2020) 369(6511):eaay4014. doi: 10.1126/science.aay4014
80. Baj J, Korona-Glowniak I, Forma A, Maani A, Sitarz E, Rahnama-Hezavah M, et al. Mechanisms of the epithelial-mesenchymal transition and tumor microenvironment in helicobacter pylori-induced gastric cancer. Cells (2020) 9(4):1055. doi: 10.3390/cells9041055
81. Fu X, Liu G, Halim A, Ju Y, Luo Q, Song AG. Mesenchymal stem cell migration and tissue repair. Cells. (2019) 8(8):784. doi: 10.3390/cells8080784
82. Baker N, Boyette LB, Tuan RS. Characterization of bone marrow-derived mesenchymal stem cells in aging. Bone. (2015) 70:37–47. doi: 10.1016/j.bone.2014.10.014
83. Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, et al. Gastric cancer originating from bone marrow-derived cells. Science. (2004) 306(5701):1568–71. doi: 10.1126/science.1099513
84. Arcucci A, Ruocco MR, Granato G, Sacco AM, Montagnani S. Cancer: An oxidative crosstalk between solid tumor cells and cancer associated fibroblasts. BioMed Res Int (2016) 2016:4502846. doi: 10.1155/2016/4502846
85. Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res (2012) 10(11):1403–18. doi: 10.1158/1541-7786.MCR-12-0307
86. Yamaguchi H, Sakai R. Direct interaction between carcinoma cells and cancer associated fibroblasts for the regulation of cancer invasion. Cancers (Basel) (2015) 7(4):2054–62. doi: 10.3390/cancers7040876
87. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer (2006) 6(5):392–401. doi: 10.1038/nrc1877
88. Lim H, Moon A. Inflammatory fibroblasts in cancer. Arch Pharm Res (2016) 39(8):1021–31. doi: 10.1007/s12272-016-0787-8
89. Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res (2010) 70(17):6945–56. doi: 10.1158/0008-5472.CAN-10-0785
90. Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One (2009) 4(4):e4992. doi: 10.1371/journal.pone.0004992
91. Krzysiek-Maczka G, Targosz A, Szczyrk U, Strzalka M, Sliwowski Z, Brzozowski T, et al. Role of helicobacter pylori infection in cancer-associated fibroblast-induced epithelial-mesenchymal transition in vitro. Helicobacter (2018) 23(6):e12538. doi: 10.1111/hel.12538
92. Yoshida K, Murata M, Yamaguchi T, Matsuzaki K, Okazaki K. Reversible human TGF-beta signal shifting between tumor suppression and fibro-carcinogenesis: Implications of smad phospho-isoforms for hepatic epithelial-mesenchymal transitions. J Clin Med (2016) 5(1):7. doi: 10.3390/jcm5010007
93. Nantajit D, Lin D, Li JJ. The network of epithelial-mesenchymal transition: potential new targets for tumor resistance. J Cancer Res Clin Oncol (2015) 141(10):1697–713. doi: 10.1007/s00432-014-1840-y
94. Zhang J, Thorikay M, van der Zon G, van Dinther M, Ten Dijke P. Studying TGF-beta signaling and TGF-beta-induced epithelial-to-mesenchymal transition in breast cancer and normal cells. J Vis Exp (2020) 164:e61830. doi: 10.3791/61830
95. Yoo JY, Ku BJ, Kim TH, Il Ahn J, Ahn JY, Yang WS, et al. Beta-catenin activates TGF-beta-induced epithelial-mesenchymal transition in adenomyosis. Exp Mol Med (2020) 52(10):1754–65. doi: 10.1038/s12276-020-00514-6
96. Zhang J, Fan J, Zeng X, Nie M, Luan J, Wang Y, et al. Hedgehog signaling in gastrointestinal carcinogenesis and the gastrointestinal tumor microenvironment. Acta Pharm Sin B (2021) 11(3):609–20. doi: 10.1016/j.apsb.2020.10.022
97. von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J Hematol Oncol (2017) 10(1):76. doi: 10.1186/s13045-017-0448-5
98. Fujii S, Fujihara A, Natori K, Abe A, Kuboki Y, Higuchi Y, et al. TEM1 expression in cancer-associated fibroblasts is correlated with a poor prognosis in patients with gastric cancer. Cancer Med (2015) 4(11):1667–78. doi: 10.1002/cam4.515
99. Zhai J, Shen J, Xie G, Wu J, He M, Gao L, et al. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett (2019) 454:37–43. doi: 10.1016/j.canlet.2019.04.002
100. Meyer C, Sevko A, Ramacher M, Bazhin AV, Falk CS, Osen W, et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci U S A (2011) 108(41):17111–6. doi: 10.1073/pnas.1108121108
101. Dorhoi A, Du Plessis N. Monocytic myeloid-derived suppressor cells in chronic infections. Front Immunol (2017) 8:1895. doi: 10.3389/fimmu.2017.01895
102. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer (2019) 120(1):16–25. doi: 10.1038/s41416-018-0333-1
103. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
104. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res (2007) 67(1):425; author reply 6. doi: 10.1158/0008-5472.CAN-06-3037
105. Lee CR, Lee W, Cho SK, Park SG. Characterization of multiple cytokine combinations and TGF-beta on differentiation and functions of myeloid-derived suppressor cells. Int J Mol Sci (2018) 19(3):869. doi: 10.3390/ijms19030869
106. Ku AW, Muhitch JB, Powers CA, Diehl M, Kim M, Fisher DT, et al. Tumor-induced MDSC act via remote control to inhibit l-selectin-dependent adaptive immunity in lymph nodes. Elife. (2016) 5:e17375. doi: 10.7554/eLife.17375
107. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182(9):5693–701. doi: 10.4049/jimmunol.0900092
108. Jayaraman P, Parikh F, Lopez-Rivera E, Hailemichael Y, Clark A, Ma G, et al. Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J Immunol (2012) 188(11):5365–76. doi: 10.4049/jimmunol.1103553
109. Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev (2008) 222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x
110. Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. (2016) 5(12):e1247135. doi: 10.1080/2162402X.2016.1247135
111. Ghaleb AM, Yang VW. Kruppel-like factor 4 (KLF4): What we currently know. Gene. (2017) 611:27–37. doi: 10.1016/j.gene.2017.02.025
112. Zhang J, Zhu Z, Wu H, Yu Z, Rong Z, Luo Z, et al. PODXL, negatively regulated by KLF4, promotes the EMT and metastasis and serves as a novel prognostic indicator of gastric cancer. Gastric Cancer (2019) 22(1):48–59. doi: 10.1007/s10120-018-0833-y
113. Ghaleb AM, Nandan MO, Chanchevalap S, Dalton WB, Hisamuddin IM, Yang VW. Kruppel-like factors 4 and 5: the yin and yang regulators of cellular proliferation. Cell Res (2005) 15(2):92–6. doi: 10.1038/sj.cr.7290271
114. Wang L, Chang EW, Wong SC, Ong SM, Chong DQ, Ling KL. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol (2013) 190(2):794–804. doi: 10.4049/jimmunol.1202088
115. Wang PF, Song SY, Wang TJ, Ji WJ, Li SW, Liu N, et al. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: A meta-analysis of 40 studies. Oncoimmunology. (2018) 7(10):e1494113. doi: 10.1080/2162402X.2018.1494113
116. Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. (2017) 543(7647):728–32. doi: 10.1038/nature21676
117. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med (2014) 6(237):237ra67. doi: 10.1126/scitranslmed.3007974
118. Wesolowski R, Markowitz J, Carson WE 3rd. Myeloid derived suppressor cells - a new therapeutic target in the treatment of cancer. J Immunother Cancer (2013) 1:10. doi: 10.1186/2051-1426-1-10
119. Aydin EM, Demir TD, Seymen N, Said SS, Oktem-Okullu S, Tiftikci A, et al. The crosstalk between h. pylori virulence factors and the PD1:PD-L1 immune checkpoint inhibitors in progression to gastric cancer. Immunol Lett (2021) 239:1–11. doi: 10.1016/j.imlet.2021.06.009
120. Shen B, Qian A, Lao W, Li W, Chen X, Zhang B, et al. Relationship between helicobacter pylori and expression of programmed death-1 and its ligand in gastric intraepithelial neoplasia and early-stage gastric cancer. Cancer Manag Res (2019) 11:3909–19. doi: 10.2147/CMAR.S203035
121. Go DM, Lee SH, Lee SH, Woo SH, Kim K, Kim K, et al. Programmed death ligand 1-expressing classical dendritic cells MitigateHelicobacter-induced gastritis. Cell Mol Gastroenterol Hepatol (2021) 12(2):715–39. doi: 10.1016/j.jcmgh.2021.04.007
122. Lina TT, Alzahrani S, House J, Yamaoka Y, Sharpe AH, Rampy BA, et al. Helicobacter pylori cag pathogenicity island's role in B7-H1 induction and immune evasion. PLoS One (2015) 10(3):e0121841. doi: 10.1371/journal.pone.0121841
123. Beswick EJ, Pinchuk IV, Das S, Powell DW, Reyes VE. Expression of the programmed death ligand 1, B7-H1, on gastric epithelial cells after helicobacter pylori exposure promotes development of CD4+ CD25+ FoxP3+ regulatory T cells. Infect Immun (2007) 75(9):4334–41. doi: 10.1128/IAI.00553-07
124. Wu J, Zhu X, Guo X, Yang Z, Cai Q, Gu D, et al. Helicobacter urease suppresses cytotoxic CD8+ T-cell responses through activating Myh9-dependent induction of PD-L1. Int Immunol (2021) 33(9):491–504. doi: 10.1093/intimm/dxab044
125. Li H, Xia JQ, Zhu FS, Xi ZH, Pan CY, Gu LM, et al. LPS promotes the expression of PD-L1 in gastric cancer cells through NF-kappaB activation. J Cell Biochem (2018) 119(12):9997–10004. doi: 10.1002/jcb.27329
126. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res (2020) 10(3):727–42.
127. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol (2016) 34:539–73. doi: 10.1146/annurev-immunol-032414-112049
128. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
129. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med (2012) 4(127):127ra37. doi: 10.1126/scitranslmed.3003689
130. Dong P, Xiong Y, Yue J, Hanley SJB, Watari H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: Beyond immune evasion. Front Oncol (2018) 8:386. doi: 10.3389/fonc.2018.00386
131. Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther (2016) 9:5023–39. doi: 10.2147/OTT.S105862
132. Liu X, Choi MG, Kim K, Kim KM, Kim ST, Park SH, et al. High PD-L1 expression in gastric cancer (GC) patients and correlation with molecular features. Pathol Res Pract (2020) 216(4):152881. doi: 10.1016/j.prp.2020.152881
133. Wu L, Cai S, Deng Y, Zhang Z, Zhou X, Su Y, et al. PD-1/PD-L1 enhanced cisplatin resistance in gastric cancer through PI3K/AKT mediated p-gp expression. Int Immunopharmacol (2021) 94:107443. doi: 10.1016/j.intimp.2021.107443
134. Kawazoe A, Shitara K, Boku N, Yoshikawa T, Terashima M. Current status of immunotherapy for advanced gastric cancer. Jpn J Clin Oncol (2021) 51(1):20–7. doi: 10.1093/jjco/hyaa202
135. Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med (2018) 24(9):1449–58. doi: 10.1038/s41591-018-0101-z
136. Kono K, Nakajima S, Mimura K. Current status of immune checkpoint inhibitors for gastric cancer. Gastric Cancer (2020) 23(4):565–78. doi: 10.1007/s10120-020-01090-4
137. Mohabati Mobarez A, Soleimani N, Esmaeili SA, Farhangi B. Nanoparticle-based immunotherapy of breast cancer using recombinant helicobacter pylori proteins. Eur J Pharm Biopharm (2020) 155:69–76. doi: 10.1016/j.ejpb.2020.08.013
138. Solbrig CM, Saucier-Sawyer JK, Cody V, Saltzman WM, Hanlon DJ. Polymer nanoparticles for immunotherapy from encapsulated tumor-associated antigens and whole tumor cells. Mol Pharm (2007) 4(1):47–57. doi: 10.1021/mp060107e
139. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
140. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med (2018) 50(12):1–11. doi: 10.1038/s12276-018-0191-1
141. Li B, Chan HL, Chen P. Immune checkpoint inhibitors: Basics and challenges. Curr Med Chem (2019) 26(17):3009–25. doi: 10.2174/0929867324666170804143706
142. Taefehshokr S, Parhizkar A, Hayati S, Mousapour M, Mahmoudpour A, Eleid L, et al. Cancer immunotherapy: Challenges and limitations. Pathol Res Pract (2022) 229:153723. doi: 10.1016/j.prp.2021.153723
143. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. (2020) 52(1):17–35. doi: 10.1016/j.immuni.2019.12.011
144. Odunsi K. Immunotherapy in ovarian cancer. Ann Oncol (2017) 28(suppl_8):viii1–7. doi: 10.1093/annonc/mdx444
145. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood. (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
146. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
147. Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol (2017) 47(5):765–79. doi: 10.1002/eji.201646875
148. Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2017) 390(10111):2461–71. doi: 10.1016/S0140-6736(17)31827-5
149. Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: Phase 2 clinical KEYNOTE-059 trial. JAMA Oncol (2018) 4(5):e180013. doi: 10.1001/jamaoncol.2018.0013
150. Chakrabarti J, Holokai L, Syu L, Steele NG, Chang J, Wang J, et al. Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget. (2018) 9(100):37439–57. doi: 10.18632/oncotarget.26473
151. Xue LJ, Su QS, Yang JH, Lin Y. Autoimmune responses induced by helicobacter pylori improve the prognosis of gastric carcinoma. Med Hypotheses (2008) 70(2):273–6. doi: 10.1016/j.mehy.2007.05.045
152. Xue LJ, Mao XB, Liu XB, Gao H, Chen YN, Dai TT, et al. Activation of CD3(+) T cells by helicobacter pylori DNA vaccines in potential immunotherapy of gastric carcinoma. Cancer Biol Ther (2019) 20(6):866–76. doi: 10.1080/15384047.2019.1579957
153. Ramachandran M, Jin C, Yu D, Eriksson F, Essand M. Vector-encoded helicobacter pylori neutrophil-activating protein promotes maturation of dendritic cells with Th1 polarization and improved migration. J Immunol (2014) 193(5):2287–96. doi: 10.4049/jimmunol.1400339
154. Peng X, Zhang R, Duan G, Wang C, Sun N, Zhang L, et al. Production and delivery of helicobacter pylori NapA in lactococcus lactis and its protective efficacy and immune modulatory activity. Sci Rep (2018) 8(1):6435. doi: 10.1038/s41598-018-24879-x
155. Deng Y, Su W, Zhu J, Ji H, Zhou X, Geng J, et al. Helicobacter pylori infection disturbs the tumor immune microenvironment and is associated with a discrepant prognosis in gastric de novo diffuse large b-cell lymphoma. J Immunother Cancer (2021) 9(10):e002947. doi: 10.1136/jitc-2021-002947
156. Wang T, Liu X, Ji Z, Men Y, Du M, Ding C, et al. Antitumor and immunomodulatory effects of recombinant fusion protein rMBP-NAP through TLR-2 dependent mechanism in tumor bearing mice. Int Immunopharmacol (2015) 29(2):876–83. doi: 10.1016/j.intimp.2015.08.027
157. Ding C, Li L, Zhang Y, Ji Z, Zhang C, Liang T, et al. Toll-like receptor agonist rMBP-NAP enhances antitumor cytokines production and CTL activity of peripheral blood mononuclear cells from patients with lung cancer. Oncol Lett (2018) 16(4):4707–12. doi: 10.3892/ol.2018.9182
158. Wang T, Du M, Ji Z, Ding C, Wang C, Men Y, et al. Recombinant protein rMBP-NAP restricts tumor progression by triggering antitumor immunity in mouse metastatic lung cancer. Can J Physiol Pharmacol (2018) 96(2):113–9. doi: 10.1139/cjpp-2017-0186
159. Codolo G, Fassan M, Munari F, Volpe A, Bassi P, Rugge M, et al. HP-NAP inhibits the growth of bladder cancer in mice by activating a cytotoxic Th1 response. Cancer Immunol Immunother (2012) 61(1):31–40. doi: 10.1007/s00262-011-1087-2
160. Hou M, Wang X, Lu J, Guo X, Ding C, Liang T, et al. TLR agonist rHP-NAP as an adjuvant of dendritic cell-based vaccine to enhance anti-melanoma response. Iran J Immunol (2020) 17(1):14–25. doi: 10.22034/iji.2020.80291
161. Ramachandran M, Yu D, Wanders A, Essand M, Eriksson F. An infection-enhanced oncolytic adenovirus secreting h. pylori neutrophil-activating protein with therapeutic effects on neuroendocrine tumors. Mol Ther (2013) 21(11):2008–18. doi: 10.1038/mt.2013.153
162. Yang W, Li Y, Gao R, Xiu Z, Sun T. MHC class I dysfunction of glioma stem cells escapes from CTL-mediated immune response via activation of wnt/beta-catenin signaling pathway. Oncogene. (2020) 39(5):1098–111. doi: 10.1038/s41388-019-1045-6
163. Panagioti E, Kurokawa C, Viker K, Ammayappan A, Anderson SK, Sotiriou S, et al. Immunostimulatory bacterial antigen-armed oncolytic measles virotherapy significantly increases the potency of anti-PD1 checkpoint therapy. J Clin Invest (2021) 131(13):e141614. doi: 10.1172/JCI141614
164. Ma J, Jin C, Cancer M, Wang H, Ramachandran M, Yu D. Concurrent expression of HP-NAP enhances antitumor efficacy of oncolytic vaccinia virus but not for semliki forest virus. Mol Ther Oncolytics (2021) 21:356–66. doi: 10.1016/j.omto.2021.04.016
165. Ma J, Ramachandran M, Jin C, Quijano-Rubio C, Martikainen M, Yu D, et al. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis (2020) 11(1):48. doi: 10.1038/s41419-020-2236-3
166. Smyth EC, Verheij M, Allum W, Cunningham D, Cervantes A, Arnold D, et al. Gastric cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2016) 27(suppl 5):v38–49. doi: 10.1093/annonc/mdw350
167. Fashoyin-Aje L, Donoghue M, Chen H, He K, Veeraraghavan J, Goldberg KB, et al. FDA Approval summary: Pembrolizumab for recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma expressing PD-L1. Oncologist. (2019) 24(1):103–9. doi: 10.1634/theoncologist.2018-0221
168. Fuchs CS, Doi T, Jang RW-J, Muro K, Satoh T, Machado M, et al. KEYNOTE-059 cohort 1: Efficacy and safety of pembrolizumab (pembro) monotherapy in patients with previously treated advanced gastric cancer. J Clin Oncol (2017) 35(15_suppl):4003. doi: 10.1200/JCO.2017.35.15_suppl.4003
169. Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol (2016) 17(6):717–26. doi: 10.1016/S1470-2045(16)00175-3
170. Senchukova MA, Tomchuk O, Shurygina EI. Helicobacter pylori in gastric cancer: Features of infection and their correlations with long-term results of treatment. World J Gastroenterol (2021) 27(37):6290–305. doi: 10.3748/wjg.v27.i37.6290
171. Kayapinar AK, Solakoglu D, Bas K, Oymaci E, Isbilen B, Calik B, et al. Relationship of prognostic factors in stomach cancer with helicobacter pylori: A retrospective study. Acta Gastroenterol Belg (2021) 84(4):607–17. doi: 10.51821/84.4.012
172. Tsai KF, Liou JM, Chen MJ, Chen CC, Kuo SH, Lai IR, et al. Distinct clinicopathological features and prognosis of helicobacter pylori negative gastric cancer. PLoS One (2017) 12(2):e0170942. doi: 10.1371/journal.pone.0170942
173. Tsuruta O, Yokoyama H, Fujii S. A new crystal lattice structure of helicobacter pylori neutrophil-activating protein (HP-NAP). Acta Crystallogr Sect F Struct Biol Cryst Commun (2012) 68(Pt 2):134–40. doi: 10.1107/S1744309111052675
174. Zanotti G, Papinutto E, Dundon W, Battistutta R, Seveso M, Giudice G, et al. Structure of the neutrophil-activating protein from helicobacter pylori. J Mol Biol (2002) 323(1):125–30. doi: 10.1016/S0022-2836(02)00879-3
175. Kottakis F, Papadopoulos G, Pappa EV, Cordopatis P, Pentas S, Choli-Papadopoulou T. Helicobacter pylori neutrophil-activating protein activates neutrophils by its c-terminal region even without dodecamer formation, which is a prerequisite for DNA protection–novel approaches against helicobacter pylori inflammation. FEBS J (2008) 275(2):302–17. doi: 10.1111/j.1742-4658.2007.06201.x
176. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
177. Kozlova N, Grossman JE, Iwanicki MP, Muranen T. The interplay of the extracellular matrix and stromal cells as a drug target in stroma-rich cancers. Trends Pharmacol Sci (2020) 41(3):183–98. doi: 10.1016/j.tips.2020.01.001
178. Ridge SM, Sullivan FJ, Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer (2017) 16(1):31. doi: 10.1186/s12943-017-0597-8
179. Poh AR, Ernst M. Targeting macrophages in cancer: From bench to bedside. Front Oncol (2018) 8:49. doi: 10.3389/fonc.2018.00049
180. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A (2018) 115(17):E4041–E50. doi: 10.1073/pnas.1720948115
181. Li DK, Wang W. Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies. Oncol Lett (2020) 20(5):176. doi: 10.3892/ol.2020.12037
182. Sullivan RJ, Hong DS, Tolcher AW, Patnaik A, Shapiro G, Chmielowski B, et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J Clin Oncol (2018) 36(15):3013. doi: 10.1200/JCO.2018.36.15_suppl.3013
Keywords: Helicobacter pylori, immune evasion, gastric cancer, microenvironment, immunotherapy
Citation: Deng R, Zheng H, Cai H, Li M, Shi Y and Ding S (2022) Effects of helicobacter pylori on tumor microenvironment and immunotherapy responses. Front. Immunol. 13:923477. doi: 10.3389/fimmu.2022.923477
Received: 19 April 2022; Accepted: 04 July 2022;
Published: 28 July 2022.
Edited by:
Maria Kaparakis-Liaskos, La Trobe University, AustraliaReviewed by:
Elba Mónica Vermeulen, Instituto de Biología y Medicina Experimental, ArgentinaCopyright © 2022 Deng, Zheng, Cai, Li, Shi and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanyan Shi, c2hpeWFueWFuQGJqbXUuZWR1LmNu; Shigang Ding, ZGluZ3NoaWdhbmcyMjJAMTYzLmNvbQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.