 Sarah Vollmers1
Sarah Vollmers1 Annabelle Lobermeyer1
Annabelle Lobermeyer1 Annika Niehrs1Pia Fittje1Daniela Indenbirken1Jacqueline Nakel1Sanamjeet Virdi1Sebastien Brias1,2Timo Trenkner1Gabriel Sauer3Sven Peine4
Annika Niehrs1Pia Fittje1Daniela Indenbirken1Jacqueline Nakel1Sanamjeet Virdi1Sebastien Brias1,2Timo Trenkner1Gabriel Sauer3Sven Peine4 Georg M.N. Behrens5Clara Lehmann6,7,8Anja Meurer9Ramona Pauli10Nils Postel11
Georg M.N. Behrens5Clara Lehmann6,7,8Anja Meurer9Ramona Pauli10Nils Postel11 Julia Roider12,13Stefan Scholten14Christoph D. Spinner13,15Christoph Stephan16Eva Wolf17Christoph Wyen6,18Laura Richert19
Julia Roider12,13Stefan Scholten14Christoph D. Spinner13,15Christoph Stephan16Eva Wolf17Christoph Wyen6,18Laura Richert19 Paul J. Norman20,21
Paul J. Norman20,21 Jürgen Sauter22
Jürgen Sauter22 Alexander H. Schmidt22,23Angelique Hoelzemer1,2,24
Alexander H. Schmidt22,23Angelique Hoelzemer1,2,24 Marcus Altfeld1,24
Marcus Altfeld1,24 Christian Körner1*
Christian Körner1*- 1Leibniz Institute of Virology, Hamburg, Germany
- 2First Department of Medicine, Division of Infectious Diseases, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 3Department I for Internal Medicine, University Hospital of Cologne, Cologne, Germany
- 4Institute for Transfusion Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 5Department for Rheumatology and Clinical Immunology, Hannover Medical School, Hannover, Germany
- 6Department I for Internal Medicine, Division of Infectious Diseases, University Hospital Cologne, Cologne, Germany
- 7German Center for Infection Research (DZIF), Partner Site Bonn-Cologne, Cologne, Germany
- 8Center for Molecular Medicine Cologne, University of Cologne, Cologne, Germany
- 9Center for Internal Medicine and Infectiology, Munich, Germany
- 10Medizinisches Versorgungszentrum (MVZ) am Isartor, Munich, Germany
- 11Prinzmed, Practice for Infectious Diseases, Munich, Germany
- 12Department of Internal Medicine IV, Department of Infectious Diseases, Ludwig-Maximilians University Munich, Munich, Germany
- 13German Center for Infection Research (DZIF), Partner Site Munich, Munich, Germany
- 14Praxis Hohenstaufenring, Cologne, Germany
- 15Technical University of Munich, School of Medicine, University Hospital rechts der Isar, Department of Internal Medicine II, Munich, Germany
- 16Infectious Diseases Unit, Goethe-University Hospital Frankfurt, Frankfurt, Germany
- 17MUC Research, Munich, Germany
- 18Praxis am Ebertplatz, Cologne, Germany
- 19University of Bordeaux, Inserm U1219 Bordeaux Population Health, Inria Sistm, Bordeaux, France
- 20Division of Biomedical Informatics and Personalized Medicine, University of Colorado, Aurora, CO, United States
- 21Department of Immunology and Microbiology, University of Colorado, Aurora, CO, United States
- 22DKMS, Tübingen, Germany
- 23DKMS Life Science Lab, Dresden, Germany
- 24German Center for Infection Research (DZIF), Partner Site Hamburg-Lübeck-Borstel-Riems, Hamburg, Germany
NK cells play a pivotal role in viral immunity, utilizing a large array of activating and inhibitory receptors to identify and eliminate virus-infected cells. Killer-cell immunoglobulin-like receptors (KIRs) represent a highly polymorphic receptor family, regulating NK cell activity and determining the ability to recognize target cells. Human leukocyte antigen (HLA) class I molecules serve as the primary ligand for KIRs. Herein, HLA-C stands out as being the dominant ligand for the majority of KIRs. Accumulating evidence indicated that interactions between HLA-C and its inhibitory KIR2DL receptors (KIR2DL1/L2/L3) can drive HIV-1-mediated immune evasion and thus may contribute to the intrinsic control of HIV-1 infection. Of particular interest in this context is the recent observation that HIV-1 is able to adapt to host HLA-C genotypes through Vpu-mediated downmodulation of HLA-C. However, our understanding of the complex interplay between KIR/HLA immunogenetics, NK cell-mediated immune pressure and HIV-1 immune escape is still limited. Therefore, we investigated the impact of specific KIR/HLA-C combinations on the NK cell receptor repertoire and HIV-1 Vpu protein sequence variations of 122 viremic, untreated HIV-1+ individuals. Compared to 60 HIV-1- controls, HIV-1 infection was associated with significant changes within the NK cell receptor repertoire, including reduced percentages of NK cells expressing NKG2A, CD8, and KIR2DS4. In contrast, the NKG2C+ and KIR3DL2+ NK cell sub-populations from HIV-1+ individuals was enlarged compared to HIV-1- controls. Stratification along KIR/HLA-C genotypes revealed a genotype-dependent expansion of KIR2DL1+ NK cells that was ultimately associated with increased binding affinities between KIR2DL1 and HLA-C allotypes. Lastly, our data hinted to a preferential selection of Vpu sequence variants that were associated with HLA-C downmodulation in individuals with high KIR2DL/HLA-C binding affinities. Altogether, our study provides evidence that HIV-1-associated changes in the KIR repertoire of NK cells are to some extent predetermined by host KIR2DL/HLA-C genotypes. Furthermore, analysis of Vpu sequence polymorphisms indicates that differential KIR2DL/HLA-C binding affinities may serve as an additional mechanism how host genetics impact immune evasion by HIV-1.
Introduction
Natural killer (NK) cells are innate lymphocytes crucially involved in antiviral immunity and tumor surveillance (1, 2). Their main effector functions comprise production of pro-inflammatory cytokines and elimination of virus-infected and transformed cells through direct cytotoxicity (3, 4). NK cells utilize a large number of activating and inhibitory receptors to distinguish between healthy (“self”) and aberrant cells (“non-self”) (5). Inhibitory receptors mainly interact with various human leukocyte antigen (HLA) class I molecules, maintaining self-tolerance, whereas receptors with activating properties are able to recognize stress ligands on potential target cells. An important group of NK cell receptors are killer-cell immunoglobulin-like receptors (KIRs) that predominantly recognize HLA class I molecules. The most recently evolved HLA class I molecule is HLA-C, which is only present in humans and great apes (6). Contrary to HLA-A and HLA-B, virtually all HLA-C allotypes are recognized by KIRs, making HLA-C a dominant ligand for the regulation of NK cell activity (7, 8). Co-evolution of HLA class I and KIRs resulted in a remarkable diversity with distinct binding specificities between their members (8, 9). The inhibitory receptor KIR3DL2 binds only to HLA-A3/11 (10, 11), while KIR3DL1 recognition is limited to a subset of HLA-A and -B molecules that contain the serological motif Bw4 (12). In contrast, HLA-C is recognized by a number of inhibitory and activating KIRs. HLA-C molecules can be distinguished into two groups, defined by a dimorphism at position 80 in the α1 domain (7). HLA-C group 1 (HLA-C1) allotypes are characterized by an asparagine at position 80 and are predominantly recognized by the inhibitory receptors KIR2DL2 and KIR2DL3 as well as some allotypes of the activating receptor KIR2DS2 (13). HLA-C alleles encoding for group 2 (HLA-C2) molecules contain a lysine at position 80 and interact with the inhibitory KIR2DL1, the activating KIR2DS1 and a few KIR2DS5 allotypes (14–16). Additionally, certain KIR2DL2 and KIR2DL3 allotypes are cross-reactive with HLA-C2 molecules. Finally, KIR2DS4 is able to bind HLA-C1 and -C2 molecules with variegated affinities (17, 18).
Both, HLA-C and KIRs, are highly polymorphic, resulting in a large number of allotype combinations within a population that are further characterized by variegating binding affinities (8, 19). Accumulating evidence strongly indicates that interactions between KIRs and HLA-C impact the course of pathologic conditions and infectious diseases, such as preeclampsia (20), arthritis (21) and HCV infection (22). Most prominently, several studies have demonstrated a major impact of HLA class I genes in the outcome of HIV-1 infection, alone or in combination with certain KIRs (23–27). In recent years accumulating evidence suggests a role for HLA-C in immune control as well as HIV-1-associated immune escape. For one, increased HLA-C expression levels were associated with protection against HIV-1 progression (28). The ability of the accessory HIV-1 protein Vpu to downregulate HLA-C expression level on infected cells (29) and its adaption to HLA-C genotypes through sequence variations in Vpu was indicative of a novel HLA-C-associated immune evasion strategy (30). Further epidemiological and experimental evidence demonstrated that KIR+ NK cells can recognize HIV-1-mediated alterations of HLA-C expression (31) or changes in the HLA-C/peptide complex (32–37). However, the complex interplay between host genetics, NK cell-mediated immune pressure and HIV-1 immune escape is still only partially understood. In particular, the high allelic diversity of HLA-C and KIR2DL molecules and previous limitations in the resolution of KIR alleles have complicated the generation of models that would allow a clearer view of the mechanisms underlying the intrinsic control of HIV-1 and the contribution of KIR2DL and their HLA-C ligands.
In this study, we investigated the impact of KIR/HLA-C interactions on the NK cell repertoire and HIV-1 sequence polymorphisms in the context of HIV-1 infection. For this, we determined the binding affinities of various KIR2DL/HLA-C allotype combinations and conducted a comprehensive phenotypical characterization of NK cells from a cohort of viremic, untreated HIV-1+ individuals compared to HIV-1- controls. Finally, we performed next-generation sequencing (NGS) of Vpu from matched plasma and PBMC samples from HIV-1+ individuals. Herein, our results indicated that the underlying host genetics influences the NK cell receptor repertoire in HIV-1+ individuals as well as the selection of Vpu sequence variants.
Materials and Methods
Human Subjects
Peripheral blood samples were obtained from healthy blood donors (n = 45) recruited at the University Medical Center Hamburg-Eppendorf, Hamburg, Germany. In addition, peripheral blood was obtained from anonymized healthy human donors (n = 15) at the Institute for Transfusion Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany. Information on age and sex was not available for all subjects, however was not relevant for the purpose of the study. Cryopreserved peripheral blood mononuclear cells (PBMC) and plasma samples from untreated HIV-1-infected individuals (n = 122) were obtained from the Translational Platform HIV (TP-HIV) Cohort by the German Center for Infection Research (DZIF).
Sample Processing
Peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood from healthy human donors by density gradient centrifugation. Isolated PBMCs were cryopreserved for upcoming experiments in liquid nitrogen tanks in heat-inactivated fetal bovine serum (Sigma-Aldrich) supplemented with 10% (v/v) DMSO (Sigma-Aldrich). Cryopreserved PBMCs were thawed by adding PBMCs dropwise to complete RPMI-1640 medium (Life Technologies) supplemented with 10% (v/v) heat-inactivated FBS (Sigma-Aldrich), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich) and 25 U/ml Benzonase Nuclease (Merck Milipore Novagen). Thawed PBMCs were incubated for 30 min at 37°C and washed with PBS (Sigma-Aldrich) and then used for antibody staining and genomic DNA isolation.
Cell Lines
HEK293T/17 cell line (ATCC, Cat#CRL-11268) was cultured in Dulbecco’s Modified Eagle Medium (DMEM, Life Technologies) supplemented with 10% (v/v) heat-inactivated FBS (Sigma-Aldrich), 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma-Aldrich) and used for the generation of lentivirus. Sf9 insect cell line (CVCL_0549) was used to produce high titer of baculovirus stocks cultured in Sf-900 II medium (Life Technologies) supplemented with 10% (v/v) heat-inactivated FBS, (fetal bovine serum Sigma-Aldrich), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich) and 1% (v/v) L-glutamine (Life Technologies). Hi5 insect cell line (CVCL_C190) was used for KIR2DL-Fc fusion protein production and cultured in Express Five serum free medium (Life Technologies) supplemented with 1% (v/v) L-glutamine (Life Technologies). The HLA class I deficient B cell line 721.221 (RRID: CVCL_6263) (38) was used for expressing HLA-C1 and -C2 allotypes (Table 1). For this, the extracellular domains of HLA-C allotypes were obtained from GeneSynthesis (Thermo Fisher) and cloned into a lentiviral transfer vector (pSIP-ZsGreen) with a puromycin resistance. For the production of lentivirus, Lipofectamine 3000 (Life Technology) was used to transfect HEK293T/17 cells with the lentiviral transfer vector with the gene of interest, a VSV-G envelope vector (pHEF-VSVF; NIH HIV Reagent Program) and a HIV-1 Gag-Pol packaging vector (psPAX2; NIH HIV Reagent Program). After 48 h, the supernatant containing the lentivirus was harvested and used for the transduction of the 721.221 cells. 3 days post-transduction, HLA-C+ 721.221 cells were selected with 1 μg/ml puromycin (Sigma-Aldrich) and later sorted for high HLA-C expression by fluorescence-activated cell sorting. HLA-C-721.221 cell lines were cultured in complete RPMI-1640 medium (Life Technology) supplemented with 10% (v/v) heat-inactivated FBS (Sigma-Aldrich), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich) and 1 μg/ml puromcyin at 37°C and 5% CO2.

Table 1 Overview of generated HLA-C expressing 721.221 cell lines.
Generation of KIR2DL-Fc Fusion Proteins
KIR2DL1*001-Fc, KIR2DL2*003-Fc and KIR2DL3*001-Fc fusion constructs were produced as described in Hilton et al. (39). Further KIR2DL-Fc fusion constructs (Table 2) were generated by site-directed mutagenesis (Agilent Technologies). KIR2DL-Fc fusion constructs were co-transfected with linearized baculovirus (Expression Systems) into Sf9 insect cells with Cellfectin II (Invitrogen). Sf9 cells were cultured for 7 days in Sf-900 II medium (Life Technologies) at 115 rpm, 27°C and 5% CO2. To produce high titer of baculovirus, 3 more rounds of amplification were performed by culturing Sf9 cells in Sf-900 II medium supplemented with 10% (v/v) heat-inactivated FBS (Sigma-Aldrich), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich) and 1% (v/v) L-glutamine (Life Technologies) for 4-5 days with the respective virus stock. Hi5 insect cells were cultured in Express Five serum free medium (Life Technologies) supplemented with 1% (v/v) L-glutamine (Life Technologies). KIR2DL-Fc fusion proteins were produced by infecting Hi5 insect cells with P3 viral stock for 72 h at 115 rpm, 27°C and 5% CO2. The supernatant was collected by centrifugation and filtration. To isolate the KIR2DL-Fc fusion proteins, the supernatant was neutralized with HEPES buffer (Thermo Fisher Scientific) and incubated with protein A Sepharose beads (Thermo Fisher Scientific) rotating overnight. The KIR2DL-Fc fusion proteins were washed with PBS and eluted with 100 mM glycine (pH 2.7) and immediately neutralized with 1 M Tris (pH 9). The eluted proteins were desalted by using a Sephadex G-25 desalting column (GE Healthycare). Protein concentration was measured with a BCA protein assay (Thermo Fisher Scientific).

Table 2 Overview of generated KIR2DL-Fc fusion proteins.
KIR2DL-Fc Binding Assay
HLA-C-721.221 cell lines and untransduced 721.221 were incubated with 25 μg/ml KIR2DL-Fc fusion protein for 15 min at 4°C and then washed and stained with a LIVE/DEAD fixable Near-IR Dead Cell staining kit (Invitrogen) and with a secondary F(ab)2 goat anti-human IgG-PE antibody (Invitrogen). After additional washing steps, the cells were fixed in 1x CellFIX (BD Bioscience) and the binding of the KIR2DL-Fc fusion protein was analyzed via flow cytometry. As a negative control, cells were only stained with anti-human IgG-PE without any KIR2DL-Fc fusion protein. HLA-C expression of all transfected and untransfected 721.221 cell lines was assessed by flow cytometry using the HLA-ABC antibody W6/32 (Supplementary Figure 1). Binding of KIR2DL-Fc fusion proteins was normalized to the negative control and adjusted for the HLA-ABC expression of the respective 721.221 cell lines.
Phenotypical Characterization of PBMCs From Healthy and HIV-1-Infected Individuals
PBMCs from healthy and untreated HIV-1-infected individuals were gently thawed by adding dropwise complete medium supplemented with 25 U/ml Benzonase Nuclease (Merck Milipore Novagen) followed by a 30 min incubation at 37°C and 5% CO2. After counting and washing with PBS, cells were incubated with LIVE/DEAD fixable Near-IR Dead Cell staining kit (Invitrogen) and the following antibodies at 4°C: anti-CD3-PerCP-Cy5.5 (clone UCHT1, BioLegend), anti-CD4-BV650 (clone RPA-T4, BioLegend), anti-CD8-AF700 (clone EB6B, BioLegend), anti-CD14-APC-Cy7 (clone HCD14, BioLegend), anti-CD19-APC-Cy7 (clone HIB19, BioLegend), anti-CD56-BUV395 (clone NCAM16.2, BD Optibuild), anti-CD16-BV785 (clone 3G8, BioLegend), anti-CD57-BV510 (clone QA17A04, BioLegend), anti-NKG2A-PE-Vio615 (clone REA110, Miltenyi), anti-NKG2C-BUV563 (clone 134591, BD Optibuild), anti-KIR2DL1/S1-APC (clone EB6B, Beckman Coulter), anti-KIR2DL1/S5-PE (clone 134211, R&D Systems), anti-KIR2DL2/L3/S2-BV711 (clone DX27, BD Optibuild), anti-KIR2DL3-AF488 (clone 180701, R&D Systems), anti-KIR3DL1-AF700 (clone DX9, BioLegend), anti-KIR3DL1/L2-PE-Vio770 (clone 5.133, Miltenyi) and anti-KIR2DS4-Biotin (clone JJC11.6, Miltenyi) with secondary Strepdavidin-BV421 (BioLegend). Cells were washed and then fixed with FluoroFix Buffer (BioLegend). Cells were analyzed by flow cytometry.
Viral RNA Isolation, cDNA Synthesis and Genomic DNA Isolation
Viral RNA from plasma samples of HIV-1-infected individuals was isolated using the High Pure Viral RNA Kit (Roche) and then used for a reverse transcription reaction (SuperScript III One-Step RT-PCR, Invitrogen) with specific Vpu outer-revers primer (sense primer: 5’-CCT AGA CTA GAG CCC TGG AAG CAT-3’, anti-sense primer: 5’-TTC TTG TGG GTT GGG GTC TGT-3’) described by Pickering et al., 2014 (38). Genomic DNA from PBMCs was isolated using the DNeasy Blood & Tissue Kit (QIAGEN).
Vpu Sequencing
cDNA generated from viral RNA and genomic DNA isolated from PBMC of HIV-1-infected individuals was amplified and prepared for Vpu sequencing via PCR (Platinum SuperFi DNA Polymerase, Invitrogen) using gene specific Vpu inner-primers (38) and overhangs of Illumina-sequencing compatible adapter sequences (highlighted in bold) (sense primer: 5’-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG TAA TAC GAC TCA CTA TAG GCA GGA AGA AGC GGA GAC A-3’, anti-sense primer: 5´-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GCA GGA AAC AGC TAT GAC CCC ATA ATA GAC TGT GAC-3´). In a second PCR using the KAPA HiFi HotStart ReadyMix PCR Kit (Roche) the Illumina-sequencing compatible adapter overhangs served as a template to add dual indices (Nextera XT Index Kit, Illumina) for multiplexing and to complete the Illumina sequencing adapters. Prior to sequencing samples were pooled according to their concentration measured via Qubit dsDNA High Sensitivity (Invitrogen). The amplicon size of each pool was assessed on a TapeStation 4200 High Sensitivity D1000 Screen Tape (Agilent Technologies) to dilute the samples to a concentration of 2 nM. Paired-end sequencing of the generated libraries was performed on an Illumina MiSeq platform (2x250 bp) aiming for 0.1 - 0.2 million reads per sample.
Vpu Sequence Analysis
Raw paired-end reads were aligned to indexed reference sequence AF324493.2 from NCBI GenBank using bwa mem command (40). Resulting SAM alignment files were converted to BAM files, sorted and indexed using samtools (41). Variants were detected using GATK HaplotypeCaller (42) using “–dont-use-soft-clipped-bases true” option, and to identify variant with maximum frequency “–max-alternate-alleles 1” option was used. In downstream process low quality variants were filtered out using following parameters from GATK Best Practices recommendations: “DP < 25 || QD < 2.0 || MQ < 30.0 || FS > 60.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0” (43). Filtered high quality variants meeting the criteria were then taken as input to create mutation VPU nucleotide sequence taken 6061:6306 as reference coordinates using GATK FastaAlternateReferenceMaker command (44).
HLA Class I and KIR Genotyping
Genomic DNA was isolated from cryopreserved PBMCs with the DNeasy Blood and Tissue Kit (QIAGEN). HLA class I and KIR genotyping was performed by the DKMS Life Science Lab, Dresden, Germany as described in (45, 46).
Data Analysis and Statistics
The acquisition of flow cytometric data was performed with a BD LSRFortessa (BD Bioscience) in the core facility Fluorescence Cytometry at the Leibniz Institute of Virology and analyzed using FlowJo software 10.7.1 (BD Life Sciences). Graphical and statistical analyses were performed using GraphPad Prism 9.0.1 software (GraphPad Software, La Jolla, CA, USA). Multiple linear regression analysis was performed to assess the impact of HIV-1 status, age and sex on the expression of NK cell receptors (Figure 2C). One-way ANOVA, test for linear trend was used to test for gene-dose effects (Figure 2A, B, D, E). A non-parametric statistical test (Mann-Whitney) was applied to test for differences between two groups. Adjustment for multiplicity was applied to comparisons of interest using false discovery rate (FDR) with Q = 5% (Benjamini/Krieger/Yekutieli). Bonferroni correction was used in one instance (Figures 3A, B). Variation in the frequency of amino acid residues between individuals of different HLA-C genotypes was analyzed by chi square tests. Statistical parameters are stated in the results section as well as in the figure legends.
Results
KIR2DL/HLA-C Allotype Combinations Display Differential Binding Affinities
Previous assessments of KIR binding affinities to HLA class I have repeatedly confirmed the overall specificities of KIRs to different groups of HLA class I molecules (7, 10, 12). Further stratification of HLA class I and KIR molecules into specific allotypes revealed that binding affinities are exhibited as a continuum, ranging from strong affinities to no binding at all (8, 47, 48). In order to assess the specificity and affinity of the receptors KIR2DL1, KIR2DL2, and KIR2DL3 (henceforth KIR2DL) to various HLA-C allotypes in a cellular system, we assessed binding of various KIR2DL-Fc fusion proteins to HLA-C expressing cell lines using flow cytometry (Figure 1A). Representative and cumulative data in Figure 1B display the overall binding specificities of the three KIR2DL types. All generated KIR2DL-Fc constructs did not bind to untransduced 721.221, while KIR2DL1-Fc allotypes were predominantly associated with binding of HLA-C group 2 (HLA-C2), but did not recognize HLA-C group 1 (HLA-C1) on 721.221s with the exception of KIR2DL1*022. KIR2DL1*022 is characterized by an amino acid substitution at position 44 which leads to a switch in its binding specificity towards HLA-C1 ligands (49). Conversely, KIR2DL2-Fc and KIR2DL3-Fc displayed binding affinities to both HLA-C1 and -C2. For comparison of the binding affinities of all tested KIR2DL/HLA-C allotype combinations, the highest value for KIR2DL-Fc binding was set to 100% and all other values were calculated accordingly (Figure 1C). The displayed affinity matrix indicated hierarchies that applied to KIR2DL1 as well as HLA-C2 allotypes. E.g. 2DL1*003 showed the highest affinities to HLA-C2 cell lines of all tested KIR2DL1 allotypes independent of the HLA-C allotype follwes by 2DL1*004, 2DL1*020 and 2DL1*001. In turn, highest KIR2DL1 binding was observed for the HLA-C2 cell line expressing C*05:01 irrespective of the KIR2DL1 allotype. This was followed by C*02:02, C*04:01 and C*06:02. Overall KIR2DL2-Fc and KIR2DL3-Fc constructs displayed a lower binding affinity to HLA-C expressing cell lines in general and showed a more ambiguous affinity pattern. E.g. the KIR2DL3*016/C*07:01 combination showed the strongest binding of all KIR2DL2/L3 allotypes to HLA-C1 cell lines. But for HLA-C2 cell lines KIR2DL2*009 showed the highest affinity, i.e. C*05:01. Of note, the HLA-C1 cell lines C*01:02, C*03:03, C*12:03 and C*14:02 did not allow for any KIR2DL-Fc binding in our experimental setting, whereas C*03:04, C*07:02 and C*16:01 demonstrated only low binding affinities to the KIR2DL2/L3-Fc fusion proteins. C*07:01 allowed for stronger binding of KIR2DL2/L3 allotypes than any other tested C1 cell line. Altogether, the performed KIR2DL-Fc fusion protein assays showed high binding affinities of KIR2DL1/HLA-C2 combinations but no binding of KIR2DL1 to HLA-C1. Furthermore, KIR2DL2/L3-Fc fusion proteins had only low binding affinities for both, HLA-C1 and -C2 expressing 721.221 cell lines.
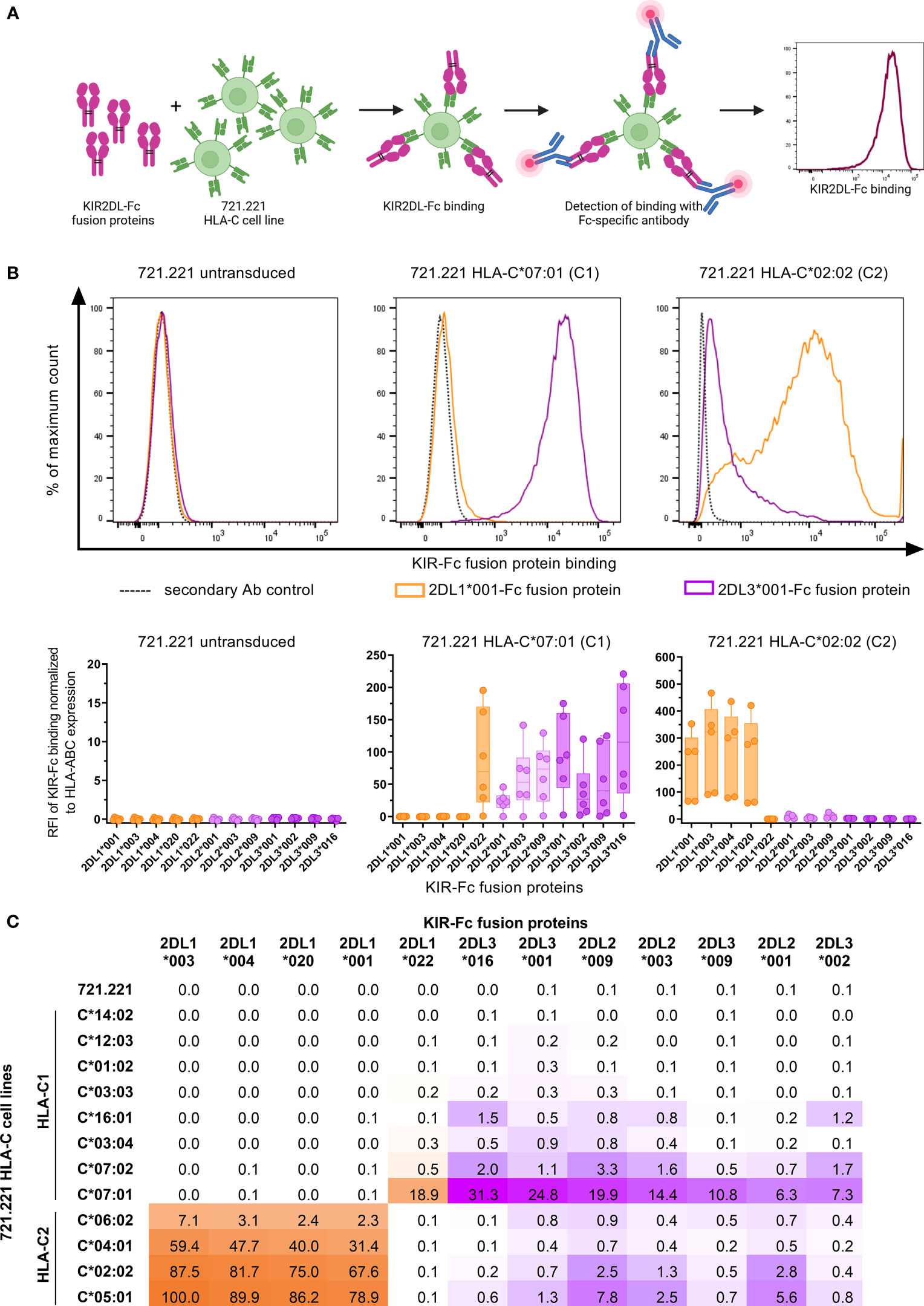
Figure 1 KIR2DL-Fc fusion proteins display differential binding affinities to HLA-C expressing 721.221 cell lines. (A) Overview of the assessment of binding affinities between HLA-C expressing 721.221 cell lines and KIR2DL-Fc fusion proteins using flow cytometry. Created with BioRender.com. (B) Upper panel: Representative histograms depicting binding of KIR2DL1*001-Fc (orange), KIR2DL3*001-Fc (purple) and the respective secondary antibody (Ab) control (black, dotted) to 721.221 (.221),.221-C*07:01 (C1) and .221-C*02:02 (C2) cell lines. Expression was quantified as fluorescence intensity (x-axis). Lower panel: Cumulative data showing binding of KIR2DL-Fc fusion proteins to .221, .221-C*07:01 (C1) and .221-C*02:02 (C2) cell lines. Binding of KIR2DL1-Fc (orange), KIR2DL2-Fc (light purple) and KIR2DL3-Fc (dark purple) allotypes is displayed as relative fluorescence intensity (RFI) normalized to the secondary antibody only control and adjusted for HLA-ABC expression of the respective 721.221 cell lines. Box plots show median and 25%/75% percentile. Data points represent at least five technical replicates (n ≤ 6). (C) Summarizing results of binding assays between 12 KIR2DL-Fc allotypes and .221s expressing 12 different HLA-C allotypes. Highest binding value was set to 100% and all other values were calculated in relation to the 100% value.
HIV-1 Infection Is Associated With an Altered NK Cell Receptor Repertoire
Next, we investigated the impact of HIV-1 infection on the expression patterns of NK cell receptors. For this, we assessed the expression of eleven NK cell receptors within 60 HIV-1- and 122 untreated HIV-1+ individuals (Table 3; Figure 2A). Exemplary expression patterns for all analyzed NK cell receptors and the applied gating are shown in Figure 2B. Initial comparative analyses of bulk NK cells, including adjustment for age and sex, suggested significant changes in the receptor repertoire between HIV-1+ individuals and the HIV-1- control group (Figure 2C and Supplementary Table 1). We observed a lower percentage of NK cells expressing the inhibitory receptor NKG2A in HIV-1+ individuals (median: 42.5%) compared to our control group (54.4%, p < 0.0001 vs. HIV-1-), whereas the percentage of NK cells expressing the activating counterpart NKG2C was considerably increased (23.5% vs. 3.5%, p < 0.0001 vs. HIV-1-). The relative frequency of NK cells expressing CD57, a marker for terminal differentiation, was slightly higher in HIV-1+ individuals (36.7%) compared to the HIV-1- control group (31.5%) but did not reach statistical significance after adjustment for age and sex (p = 0.2). Percentage of CD57+ NK cells was positively associated with the percentage of NKG2C+ NK cells (rs = 0.5, p < 0.0001) and negatively with the percentage of NKG2A+ NK cells (rs = -0.57, p < 0.0001; Figure 2D). The relative frequency of CD8+ NK cells was reduced in HIV-1+ individuals (18.9%) in comparison to HIV-1- donors (32.3%, p < 0.0001) but did not correlate with any other investigated NK cell receptor. For most KIRs expression patterns seemed to be unaffected by HIV-1 infection. No differences in the relative frequency of KIR2DL1+ cells (p = 0.12) and KIR2DL2/L3+ cells (p = 0.41) were observed, even after stratification into KIR2DL2 (p = 0.2) and KIR2DL3 (p = 0.12). The activating KIRs, KIR2DS1 (p = 0.2) remained unaltered as well but KIR2DS4 showed lower relative frequencies in HIV-1+ individuals (26.5% vs. 41.9%, p = 0.049). The proportion of KIR3DL1+ NK cells was not significantly altered (p = 0.20), even when adjusted for its ligand Bw4 or stratified by KIR3DL1 allotype groups (Supplementary Figure 2) in contrast to KIR3DL2, which exhibited increased relative frequencies in HIV-1+ individuals (19.7% vs. 13.4%, p = 0.07). Analysis of relative frequencies and absolute cell numbers of the respective receptor+ NK cells revealed a significant positive correlation for all receptors (Supplementary Figure 3), indicating that the observed changes represent an actual expansion or contraction of NK cell subsets.
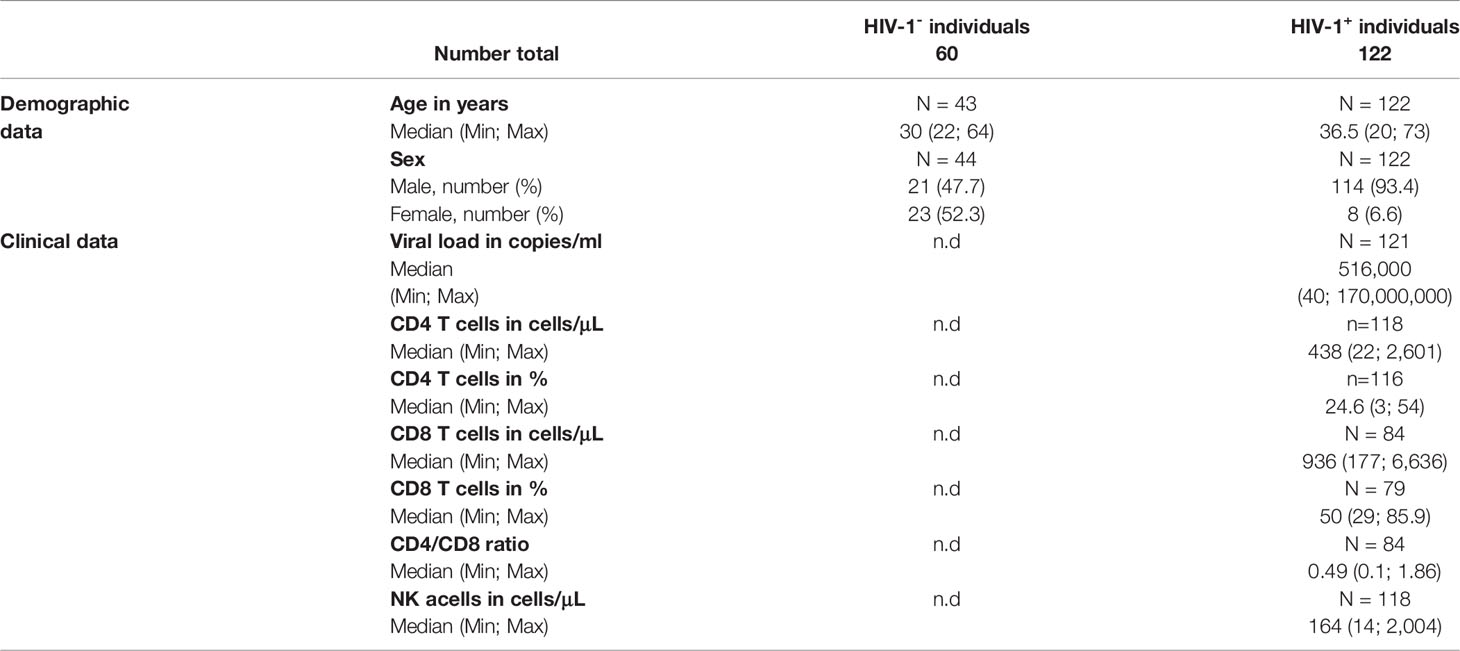
Table 3 Demographic and clinical profile of HIV-1- and HIV-1+ individuals.
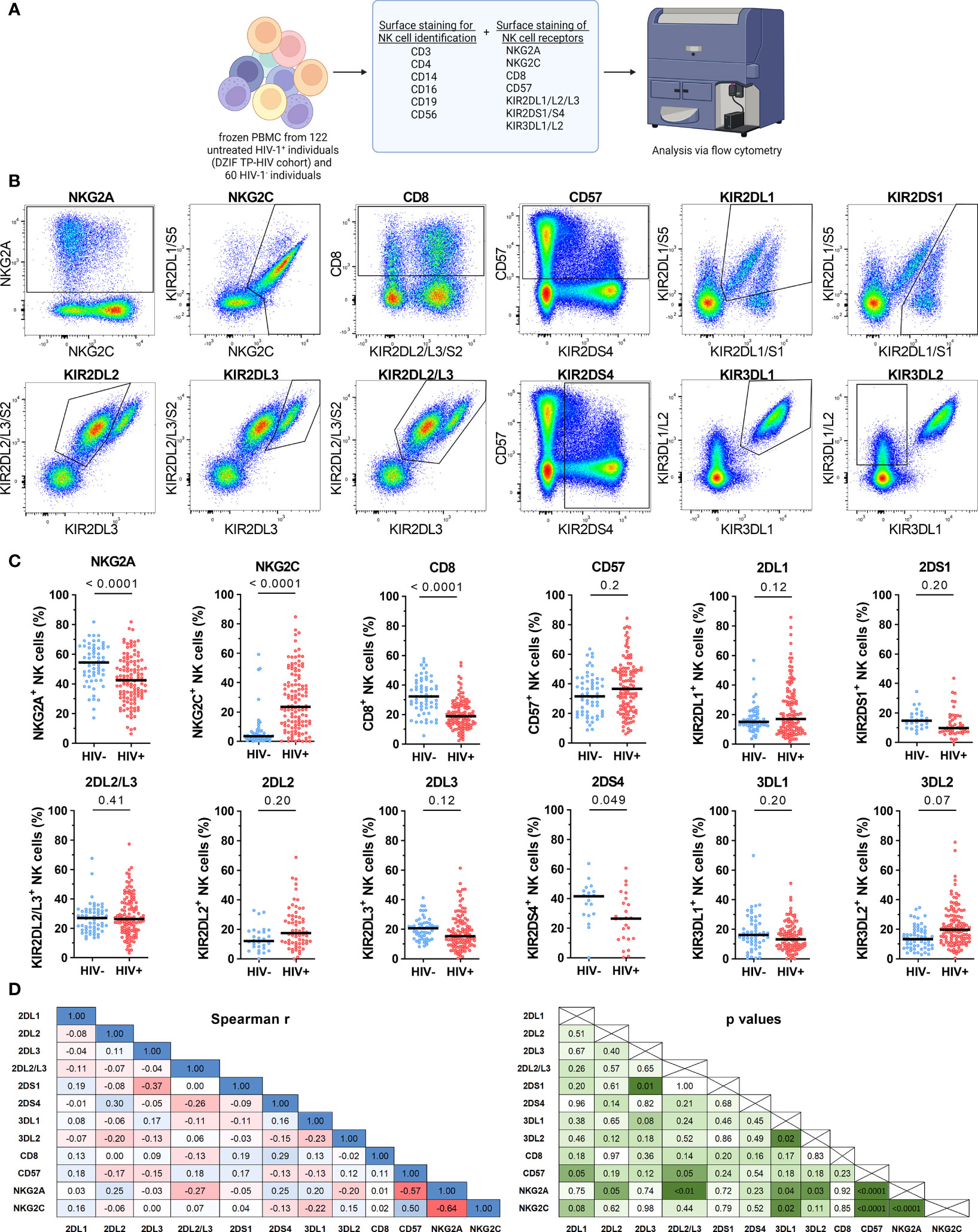
Figure 2 Cell surface expression of NK cell receptors of HIV-1- and untreated HIV-1+ individuals. (A) Overview of the flow cytometric assessment of NK cell receptor expression on NK cells from HIV-1- (n = 60) and untreated HIV-1+ (n = 122) individuals. Created with BioRender.com (B) Respective expression patterns of the NK cell receptors NKG2A, NKG2C, CD8, CD57, KIR2DL1, KIR2DS1, KIR2DL2, KIR2DL3, KIR2DL2/L3, KIR2DS4, KIR3DL1 and KIR3DL2 on bulk NK cells as well as the respective gating. (C) Scatter plots displaying the percentage of NKG2A+, NKG2C+, CD8+, CD57+, KIR2DL1+, KIR2DS1+, KIR2DL2/L3+, KIR2DL2+, KIR2DL3+, KIR2DS4+, KIR3DL1+ and KIR3DL2+ cells within bulk NK cells in HIV-1- (blue) and HIV-1+ (red) individuals. Each data point represents one donor. Donors lacking the respective gene, containing a gene deletion (KIR2DS4-del) or a null allele (KIR3DL1*004) were excluded (NKG2A/NKG2C/CD8/CD57/KIR2DL2/L3: HIV-1-: n = 60, HIV-1+: n = 122; KIR2DL1: n = 59, n = 118; KIR2DS1: n = 26, n = 47; KIR2DL2: n = 28, n = 64; KIR2DL3: n = 55, n = 111; KIR2DS4: n = 19, n = 25; KIR3DL1: n = 55, n = 108). (D) Correlation analyses between the frequencies of all tested receptor+ NK cell subsets from HIV-1+ individuals. Left panel: rs values; right panel: p values. Data information: (C) Bars indicate the median for each group. Multiple linear regression analysis was used to determine differences between HIV-1- and HIV-1+ individuals. (D) Spearman rank analysis. (C, D) p values were adjusted for multiple comparisons (Benjamini/Krieger/Yekutieli).
Subsequently, we investigated whether the altered NK cell receptor repertoire was attributed to changes of the distribution of the three major CD56 NK cell subsets. Based on the expression levels of CD56, NK cells can be distinguished into CD56Bright (CD16-/dim), CD56Dim (CD16+) and CD56Negative sub-populations (CD16+), each displaying specific receptor profiles on their own (50). Compared to the HIV-1- control group, the frequency of CD56Bright NK cells was significantly decreased within the NK cell compartment of HIV-1- infected individuals (p < 0.0001) (Supplementary Figure 4). Conversely, the proportion of CD56Negative NK cells was increased (p < 0.0001). Subsequently, the three CD56 NK cell subsets were analyzed for the expression of the eleven NK cell receptors (Table 4). Changes of NK cell receptor profiles were observed in all CD56 NK cell subsets. In the CD56Bright NK cell subset, eight of the investigated NK cell receptors, namely KIR2DL1, KIR2DL2, KIR2DL3, KIR2DS4, KIR2DS1, KIR3DL1, CD57 and NKG2C exhibited an increased frequency in HIV-1+ individuals, although it should be noted that percentage point differences were rather marginal for all tested KIRS. In contrast, NKG2A and CD8 were significantly decreased. In the CD56Dim subset, CD8+ NK cells showed decreased frequencies, while the KIR3DL2+ NK cell subset was enlarged. CD56Negative NK cells showed similar changes in the expression patterns as the CD56Dim subset in both HIV-1- and HIV-1+ groups, although CD56Negative NK cells displayed an overall lower percentage of receptor+ cells compared to CD56Dim NK cells. Taken together, we observed reduced percentages of CD8+ NK cells in all CD56 NK cell subsets of HIV-1+ individuals compared to HIV-1- individuals, whereas the NKG2C+ subset was enlarged. In contrast, changes in the KIR profiles between HIV-1+ and HIV-1- individuals were rather marginal in the CD56Dim subset in which KIRs are predominantly expressed, with the exception of KIR3DL2.
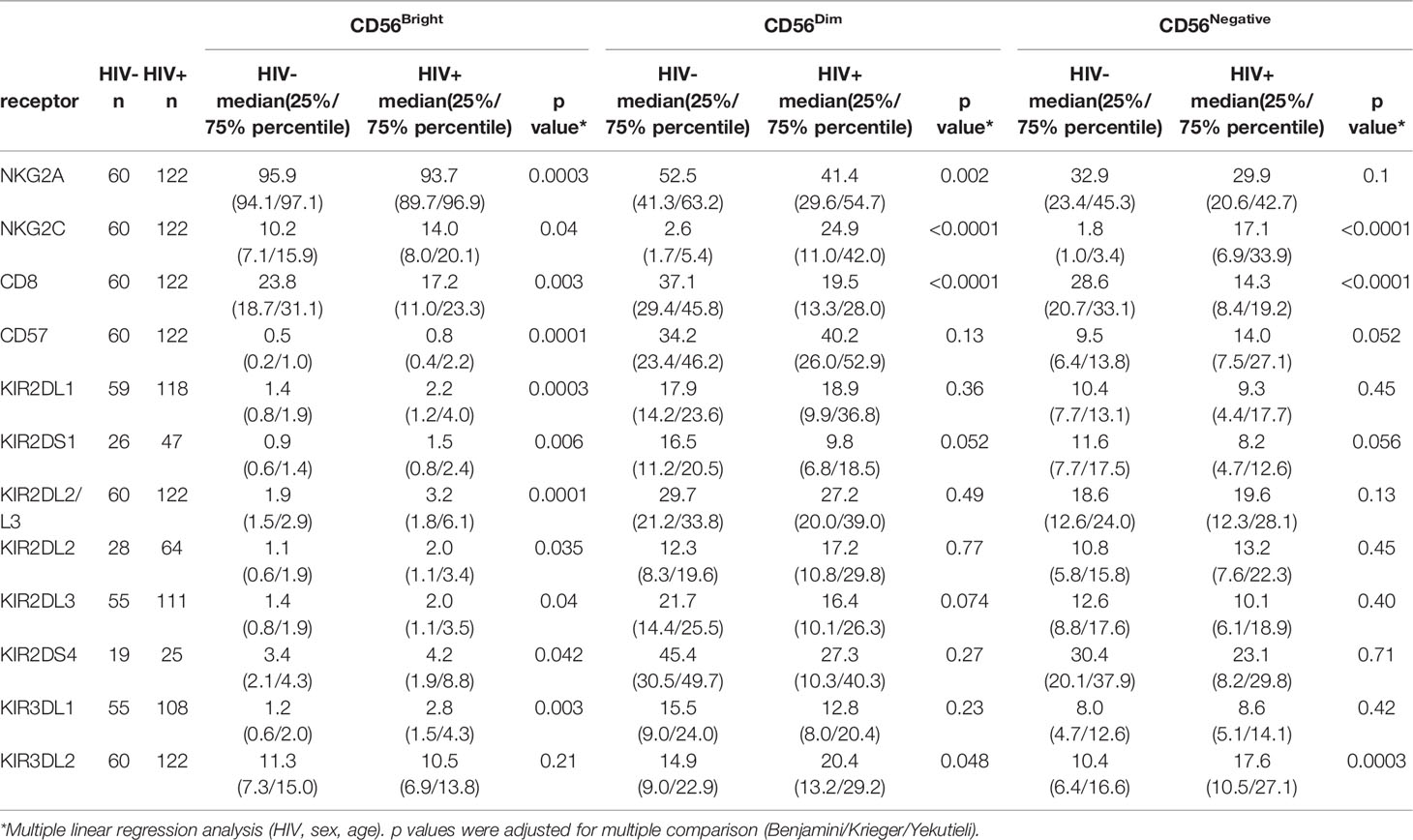
Table 4 NK cell receptor expression in NK cell subsets.
HLA-C and KIR2DL Genotypes Impact Frequency of KIR2DL+ NK Cells
To investigate the influence of KIR2DL/HLA-C genotype combinations on KIR2DL expression on NK cells, HIV-1+ and HIV-1- individuals were stratified according to their HLA-C allotypes. HIV-1+ individuals showed significant differences in the frequency of KIR2DL1+ NK cells between the generated groups, while no differences were detected in the HIV-1- control group (Figure 3A). HIV-1+ individuals carrying the cognate HLA-C2 ligand (C1/C2, C2/C2) showed an increased frequency of KIR2DL1+ NK cells, whereas individuals without HLA-C2 alleles (C1/C1) exhibited a generally lower percentage of KIR2DL1+ NK cells (left panel, linear trend: p < 0.0001). This effect was not due to a skewed distribution of KIR2DL1-C245 and KIR2DL1-R245 allotypes between HIV-1+ and HIV-1- individuals or between HLA-C2+ or HLA-C2- donors (data not shown), which was previously shown to affect KIR2DL1 expression (51). The observed gene-dose dependent effect of HLA-C2 alleles within the HIV-1+ group was also observed when using absolute cell numbers as a readout (right panel, p = 0.006). In contrast, presence of the HLA-C1 ligand did not affect the proportion or cell number of NK cells expressing the respective receptors KIR2DL2/L3 in either HIV-1+ or HIV-1- individuals (Figure 3B, percentage HIV-1-: p > 0. 99, HIV-1+: p = 0.97, cell number: p = 0.69).
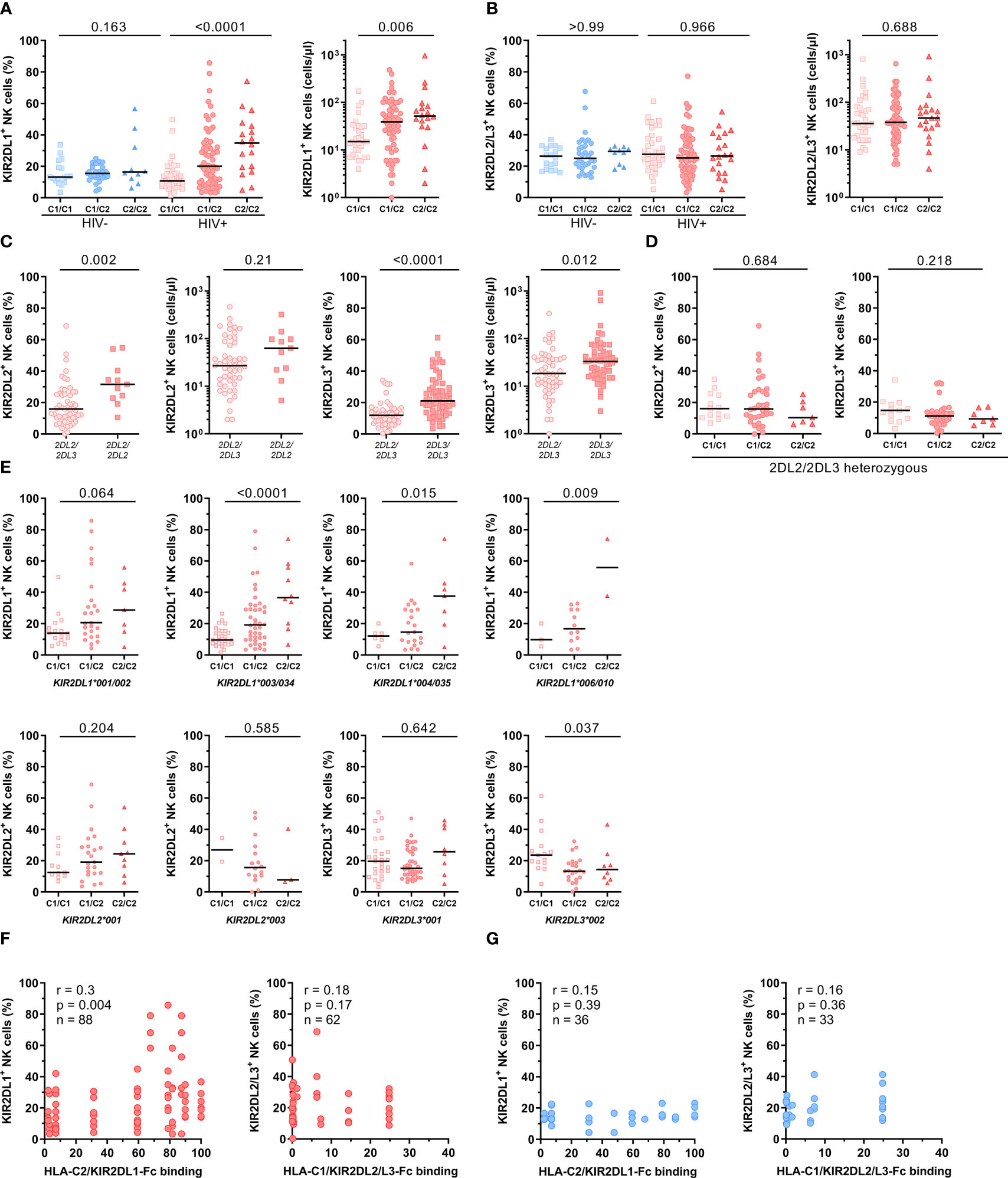
Figure 3 Impact of HLA-C and KIR genotypes on the frequency of NK cells expressing HLA-C-binding KIRs. (A) Percentage of KIR2DL1+ cells within bulk NK cells in HIV-1- and HIV-1+ individuals (left panel). Absolute numbers of KIR2DL1+ NK cells in HIV-1+ individuals (right panel). (B) Percentage of KIR2DL2/L3+ cells within bulk NK cells in HIV-1- and HIV-1+ individuals (left panel). Absolute numbers of KIR2DL2/L3+ NK cells in HIV-1+ individuals (right panel). (A/B) Donors were stratified by HLA-C group genotypes. Genotypes were defined by the presence of HLA-C allotypes carrying either a C1 or C2 epitope (C1/C1 = C1homozygous, C1/C1 = C1/C2heterozygous, C2/C2 = C2homozygous). (C) Percentage and absolute cell numbers of KIR2DL2+ NK cells and KIR2DL3+ NK cells in HIV-1+ individuals stratified by the presence of KIR2DL2 or KIR2DL3 alleles. (D) Percentage of KIR2DL2+ and KIR2DL3+ NK cells in KIR2DL2/L3heterozygous HIV-1+ individuals stratified by HLA-C group genotypes. (E) Percentage of KIR2DL1+, KIR2DL2+ and KIR2DL3+ NK cells in HIV-1+ individuals stratified by HLA-C group genotypes and KIR2DL alleles. (F) Correlation between percentage of KIR2DL1+ NK cells and HLA-C2/KIR2DL1-Fc binding, KIR2DL2/L3+ NK cells and HLA-C1/KIR2DL2/L3-Fc binding in HIV-1+ individuals (red) stratified by KIR/HLA binding affinities determined in Figure 1. (G) Correlation between percentage of KIR2DL1+ NK cells and HLA-C2/KIR2DL1-Fc binding, KIR2DL2/L3+ NK cells and HLA-C1/KIR2DL2/L3-Fc binding in HIV-1- individuals (blue) stratified by KIR/HLA binding affinities determined in Figure 1. Data information: Black bars display the median. (A, B, D, E) One-way ANOVA, test for linear trend. (C) Mann-Whitney test. P values were adjusted for multiple comparisons (A, B, D: Bonferroni; E: Benjamini/Krieger/Yekutieli). (F, G) Spearman rank correlation.
Given that potential changes of KIR2DL2/L3 frequencies between HIV-1+ and HIV-1- groups were masked by the previously observed opposing directions of KIR2DL2+ and KIR2DL3+ NK cell frequencies, we stratified KIR2DL2/3 frequencies based on their KIR2DL2/L3 genotype (Figure 3C). NK cells from homozygous KIR2DL2 individuals (KIR2DL2/L2) showed a significantly increased frequency of KIR2DL2+ NK cells compared to heterozygous (KIR2DL2/L3) individuals independent of the underlying HLA-C genotype (p = 0.002). This effect was also present in KIR2DL3+ NK cells when comparing KIR2DL3 homozygous (KIR2DL3/L3) and heterozygous (KIR2DL2/L3) individuals (p < 0.0001). To exclude the skewing effect of KIR2DL2 and KIR2DL3 homozygosity, stratification of HLA-C allotypes was performed only for KIR2DL2/L3 heterozygous HIV-1+ individuals (Figure 3D). Increased frequencies of KIR2DL2+ and KIR2DL3+ NK cells in donors with two HLA-C1 alleles (C1/C1) were observed compared to individuals lacking HLA-C1 alleles (C2/C2), however the overall trend did not reach statistical significance (KIR2DL2+: p = 0.68, KIR2DL3+: p = 0.22).
Based on the observed effects of homo- or heterozygosity of the KIR2DL/L3 gene, we sought to further stratify donors based on the specific KIR2DL alleles (Figure 3E). In line with the results for KIR2DL1+ NK cells from all HIV-1+ individuals stratified by their HLA-C allotypes, HIV-1+ individuals with KIR2DL1*001/002, *003/*034, *004/*035 or *006/010 alleles showed the same distribution of KIR2DL1+ NK cells with the highest frequency of KIR2DL1+ NK cells in HLA-C2 homozygous individuals and the lowest frequency in HLA-C1 homozygous donors. Moreover, individuals positive for KIR2DL2*001 and with at least one HLA-C2 allele (C1/C2, C2/C2) showed an increased frequency of NK cells expressing KIR2DL2 compared to individuals without HLA-C2. In contrast, individuals carrying a KIR2DL2*003 allele showed reverse KIR2DL2 frequencies with the highest percentages for HLA-C1/C1 homozygous and the lowest percentages for HLA-C2/C2 homozygous individuals. Although, it should be noted that the sample size for this particular allele was rather low. Stratification of HLA-C allotypes for different KIR2DL3 alleles showed a slightly increased frequency of KIR2DL3+ NK cells in HLA-C2/C2 individuals positive for KIR2DL3*001, whereas individuals carrying a KIR2DL3*002 allele had an increased frequency of KIR2DL3 in the group of HLA-C1/C1 genotypes. In addition, the observed distribution of KIR2DL1 alleles in HLA-C genotypes was also present but less pronounced when using absolute cell numbers, whereas KIR2DL2 and KIR2DL3 alleles showed no differences in HLA-C genotypes when using absolute cell numbers a s readout (Supplementary Figure 5). To examine a possible relationship between KIR/HLA-C genotype combinations and the frequency of KIR2DL+ NK cells, correlation analyses between the frequencies of KIR2DL+ NK cells and HLA-C/KIR2DL-Fc binding affinities were performed for HIV-1- and HIV-1+ individuals (Figures 3F, G). For this, we included individuals heterozygous for C1/C2 and carrying HLA-C/KIR combinations with corresponding binding data. Frequency of KIR2DL1+ NK cells showed a significant positive correlation with the assessed binding affinities for HLA-C2/KIR2DL1 combinations (rs = 0.3, p = 0.004) in HIV-1+ individuals. On the other hand, KIR2DL1+ NK cell frequency and HLA-C2/KIR2DL1 binding affinities for HIV-1- individuals, as well as KIR2DL2/L3+ NK cell frequency and HLA-C1/KIR2DL2/L3 binding affinities for HIV-1+ and HIV-1- individuals showed no significant correlation. Overall, these results suggest that both, the underlying HLA-C and KIR genotype of an individual has a direct impact on the expression levels of the corresponding KIR in the respective NK cell pool.
Host Genetics Impact Vpu Sequence Polymorphisms in Plasma and PBMC Samples
Lab-adapted HIV-1 strains and primary isolates display differential abilities to modulate HLA-C expression on the surface of infected CD4+ T cells mediated by the accessory HIV-1 protein Vpu (29, 31). Previously, five amino acid (AA) positions have been identified at which specific residues were independently associated with HLA-C downregulation (30): Proline (P) at position 3, glutamic acid (E) at position 5, glycine (G) or threonine (T) at position 16 and serine (S) at position 24. Conversely, alanine (A) at position 15 negatively affected the ability to downregulate HLA-C. To investigate the impact of HLA-C/KIR2DL allotype combinations on Vpu sequence variations, genomic DNA and viral RNA from matching PBMC and plasma samples from 122 HIV-1+ individuals were isolated and sequenced with next-generation sequencing (NGS) for Vpu (Figure 4A). Sequences of Vpu from 93 plasma and 67 PBMC samples passed quality control, were translated to amino acid sequences and then aligned to the Vpu protein from the lab-adapted HIV-1 strains NL4-3 and JR-CSF (Figure 4B). Comparison of sequence similarity for the five analyzed AA positions between matching PBMC and plasma samples showed a frequency of matching AA residues between 0.64 and 0.92, with the lowest frequency for position 16 and the highest frequency for position 5. In addition, the frequency of all observed residues for the AA position was determined for PBMC and plasma samples. For position 3, the most common AA was proline (PBMC: 0.69, plasma: 0.61) followed by serine and leucine. Position 5 showed the highest variations in AA residues with the highest frequency for isoleucine (PBMC: 0.63, plasma: 0.45). The most common AA at position 15 was alanine (PBMC: 0.92, plasma: 0.73) followed by valine. Moreover, position 16 had comparable frequencies for alanine (PBMC: 0.29, plasma: 0.36), isoleucine (PBMC: 0.28, plasma: 0.14) and glycine (PBMC: 0.33, plasma: 0.36). Position 24 showed the lowest variations in AA residues with a serine as the most common AA (PBMC: 0.61, plasma: 0.63) followed by a threonine.
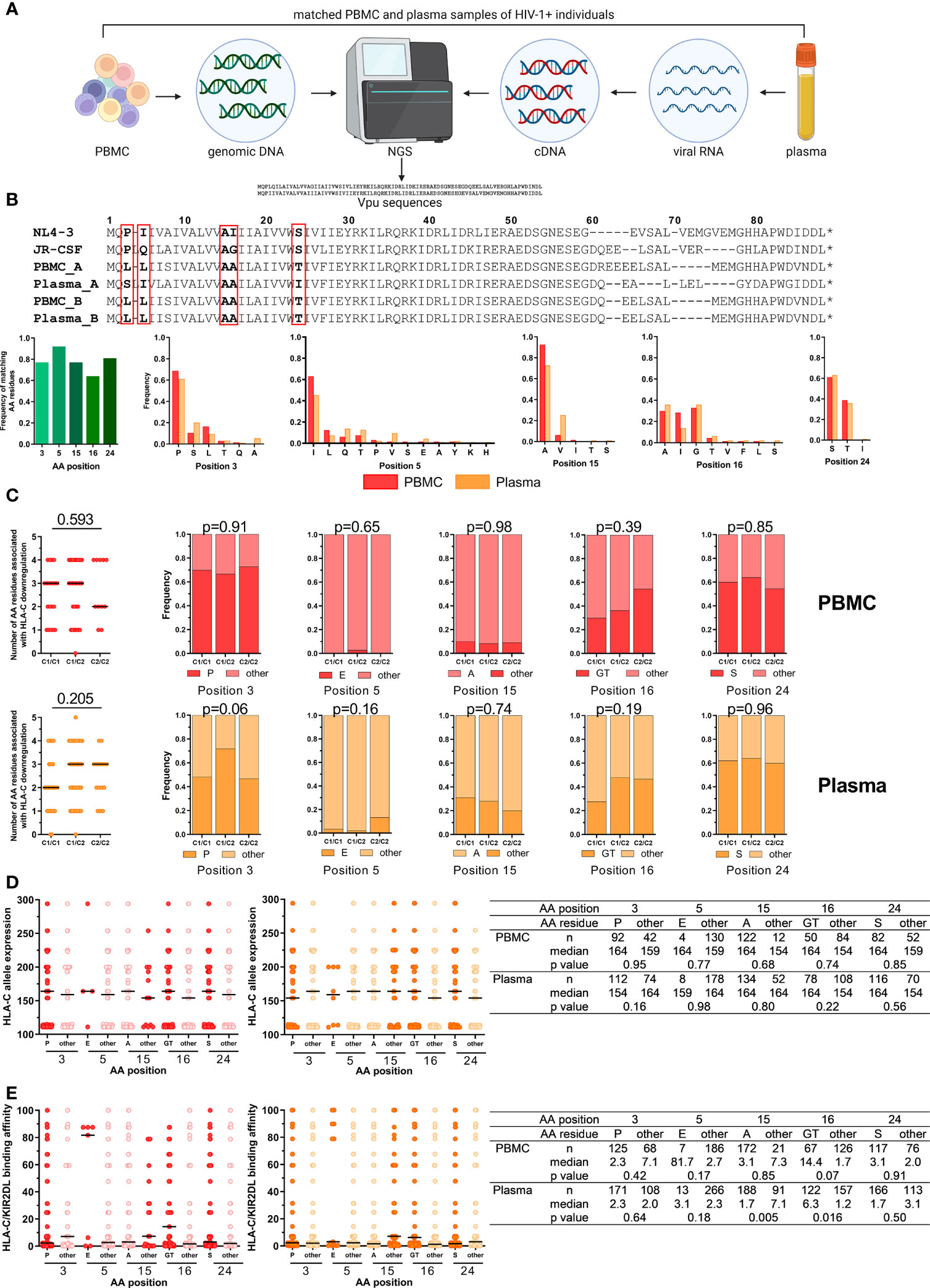
Figure 4 Impact of HLA-C/KIR2DL genotypes on Vpu sequence polymorphisms. (A) Overview of the work flow for Vpu sequencing from matched PBMC (n = 122) and plasma samples (n = 122) of untreated HIV-1+ individuals. Created with BioRender.com (B) Upper panel: Representative alignment of amino acid (AA) sequences of HIV-1 Vpu passing quality control. Lower panels: Frequency of matching AA residues at position 3, 5, 15, 16 and 24 between matched PBMC and plasma samples (n = 53). Frequency of individual AA residues at 3, 5, 15, 16 and 24 in PBMC (n = 67) and plasma (n = 93) samples. (C) Left: Dot plots displaying the cumulative number of AA residues associated with Vpu-mediated HLA-C downregulation per sequence. Right: Frequency of AA residues at position 3, 5, 15, 16 and 24. Individuals were stratified by HLA-C group genotypes (x-axis). Upper panel: PBMC, lower panel: Plasma. (D) Expression levels of HLA-C alleles of HIV-1+ donors. Data points are displayed for each position (3, 5, 15, 16 and 24) and AA residues. AA residues associated with HLA-C downregulation are displayed in red (PBMC, left panel) or dark orange (plasma, right panel). Summary table displaying descriptive and comparative statistics. (E) Binding affinities of KIR/HLA-C combinations of HIV-1+ donors. Data points are displayed in red (PBMC, left panel) or dark orange (plasma, right panel). Summary table displaying descriptive and comparative statistics. Data information: Black bars display the median. (C) Left panel: One-way ANOVA, test for linear trend. Right panels: Chi square test. (D, E) Mann-Whitney test.
Stratification of HLA-C allotypes was performed for Vpu sequences for PBMC and plasma samples and analyzed for the five respective AA previously identified to influence HLA-C expression levels (Figure 4C). Vpu sequences isolated from PBMC samples from HLA-C1+ individuals contained a median of three of the five AA residues, whereas HLA-C2 homozygous individuals had a median of two AA residues that were associated with HLA-C downregulation (linear trend, p = 0.593). In comparison, Vpu sequenced from viral RNA from plasma samples had three of the five AA residues in individuals with at least one HLA-C2 allele and HLA-C1 homozygous individuals had two (p = 0.205). However, there were no differences detectable in the frequency of AA residues associated with HLA-C downregulation when individuals were stratified by their HLA-C genotype.
HLA-C surface expression levels vary and correlate with the HLA-C allotype (28). To identify a potential influence of HLA-C surface expression levels on Vpu sequence variations, Vpu sequences from PBMC and plasma samples were analyzed for the five AA positions that are involved in HLA-C downregulation based on the average MFI HLA-C allele expression (Figure 4D). Our analysis showed no significant influence of HLA-C allele expression on individual AA residuces.
Finally, differences in AA residues for the five positions were analyzed based on the assessed binding affinities of HLA-C1/KIR2DL2/3 and HLA-C2/KIR2DL1 allotype combinations (Figure 4E). HLA-C/KIR2DL combinations with stronger binding affinities were more common in Vpu sequences from plasma samples with AA other than an alanine at position 15 (15other) (p = 0.005) or a glycine or threonine at position 16 (16GT) (p = 0.016) whereas HLA-C/KIR2DL binding affinities were not associated with specific amino acid residues in Vpu sequences from PBMC samples. Overall, these results indicated that HLA-C expression level and HLA-C/KIR2DL binding may have an influence on Vpu sequence polymorphisms.
Discussion
In recent years multiple studies have drawn attention to the potential role of HLA-C and its cognate inhibitory receptors, KIR2DL1, KIR2DL2, and KIR2DL3, in the intrinsic control of HIV-1 infection. Most notably were two studies showing the protective effect of high HLA-C expression levels on HIV-1 progression (28) and the differential ability of HIV-1 to modulate HLA-C expression through the accessory protein Vpu (29). In contrast to Nef-mediated downregulation of HLA-A/B, the magnitude of HLA-C modulation by Vpu varies extensively across viral strains. Further investigations revealed that HIV-1 adapts its ability for downmodulation to the HLA-C genotype of the host (30). However, the majority of the investigated viral strains exhibited only a marginal ability to reduce HLA-C expression on infected cells (30) indicating that other factors than escape from CTL recognition through HLA-C downmodulation may contribute to Vpu sequence polymorphisms. Previous reports by our group demonstrated that KIR2DL+ NK cells are able to sense HIV-1-mediated alterations of HLA-C (31). Based on these studies, we hypothesized that differential binding affinities across KIR and HLA-C allotypes in part pre-determine KIR2DL+ NK cell induced immune pressure and Vpu-associated viral escape.
Our results demonstrated a hierarchy of affinities along the various combinations and confirmed the binding specificities of KIR2DL1 (HLA-C2), KIR2DL3 (HLA-C1) and KIR2DL2 (HLA-C1/-C2) (52). These results are largely consistent with those from the cell-free system of Hilton et al. who used KIR-Fc fusion constructs and microbeads, each coated with a different HLA class I allotype (39). While specificities between KIR2DL and HLA-C are defined by position 44 of the KIR protein (15) and position 80 of the α1 domain of HLA-C (7), other amino acid positions within both molecules fine-tune avidity (19, 53). Data on the functional consequences of the variegated affinities in the context of HIV-1 infection have been limited to KIR3DL1 (54, 55). However, multiple studies have shown that changes in the HLA class I presented peptidome can modulate binding to KIRs and subsequently impact NK cell functions (33, 56, 57). Moreover, HIV-1 sequence polymorphisms have been associated with KIR2DL genes alone and in combination with HLA-C indicating NK cell-mediated immune pressure involving KIR2DL+ NK cells (36, 37). It is therefore conceivable that binding affinities predetermined by KIR2DL/HLA-C genotypes may have a similar impact.
In this context, expansion of KIR+ NK cell subsets can be an indicator for the involvement of specific KIR+ NK cell subsets in the antiviral immune response against HIV-1 (58–60). HIV-1 infection has been associated with significant changes in the NK cell receptor repertoire (61–63). In our phenotypical characterization we investigated the expression of NK cell receptors interacting with various HLA class I molecules in a cohort of untreated, viremic HIV-1+ individuals. Noteworthy, decreased numbers of CD8+ NK cells in HIV-1-infected individuals have been observed in a previous study, where it correlated positively with HIV loads and inversely with CD4+ T cell counts (64). We also observed an overall contraction of the NKG2A+ NK cell subset which was closely tied to an expansion of NKG2C+ and CD57+ NK cells in the same donors. The inhibitory NKG2A receptor interacts with the non-classical HLA class I molecule HLA-E on target cells, preventing healthy cells from NK cell-mediated lysis. Additionally, HLA-E is recognized by NKG2C, which delivers an activating signal to the NK cell (65). NKG2A+ NK cells have been implicated to possess superior anti-HIV activity compared to other subsets, whereas KIR2DL+ NK cells had a diminished ability to degranulate in response to HIV-1-infected CD4+ T cells even in the absence of NKG2A (66). In line with our results, Mavilio et al., showed a decreased expression of NKG2A in HIV-1-infected viremic individuals, which was associated with reduced inhibitory function in a redirected killing assay (61). Loss of NKG2A+ NK cells corresponded with a high expansion of NK cells expressing NKG2C (67). In contrast, higher NKG2A expression levels in cytotoxic NK cell subsets have been reported in individuals with late stage HIV-1 infection (68). Expansion of NKG2C+ and CD57+ NK cells is not unique to HIV-1 infection (69). Similar changes have been observed for CMV and Hantavirus infection (70–72). It is still under investigation if these CD57+NKG2C+ NK cells have memory-like features specific for viral infections (73). Data on CMV seropositivity was not available for this cohort and therefore we cannot rule out a CMV-associated expansion in the HIV-1+ cohort. However, an earlier study of our group indicated a CMV-independent, HIV-1-driven expansion of KIR2DL+ NK cells (32).
Analysis of KIR+ NK cells recognizing HLA-C initially showed a decrease of cells expressing the activating receptor KIR2DS4, while percentage of most inhibitory receptors remained unchanged. KIR2DS4 binds a subset of HLA-C1 and -C2 allotypes and HLA-A*11 (18) however its role in disease progression and NK cells regulation is not fully understood. KIR2DS4 has been associated with high viral loads and promotion of HIV-1 pathogenesis in chronic HIV-1 infection, probably through excessive NK cell activation (74, 75). HIV-1+ individuals showed a higher frequency of the inhibitory KIR3DL2 receptor, which recognizes only HLA-A3 and -A11 in a peptide specific manner (11) and HLA-B27 (76). So far, not much is known about the role of KIR3DL2 in HIV-1 infection but other studies showed also an increased expression of KIR3DL2 in chronic HCV patients (77) and on activated NK cells from patients with spondylarthritis (78). Further stratification of our data based on HLA-C and KIR2DL genotypes indicated that the HIV-1-associated changes in the proportion of KIR2DL+ NK cells within the NK cell pool were predetermined by host genetics. The frequency of KIR2DL1+ NK cells was linked to the number of the cognate HLA-C2 alleles, which we showed previously for individuals with primary HIV-1 infection (32). The increased frequency is potentially related to an increased surface expression of the respective HLA-C2 ligand. In addition, differentiation of KIR2DL1 alleles displayed a similar “gene-dose” effect for the analyzed KIR2DL1 alleles with the exception of KIR2DL1*001/002. Further analysis showed a positive correlation between KIR2DL/HLA-C binding affinities and the frequency of the respective KIR2DL+ NK cell subset, thus further corroborating our hypothesis. However, we cannot rule out that the differences in the relative frequency of NK cells expressing various KIR2DL allotypes is due to changes in non-coding regions that may impact expression on an individual cell and within an individual NK cell pool (79–81).
Lastly, we investigated the impact of KIR/HLA-C genotypes on Vpu sequence polymorphisms. Molecular characterization of Vpu sequences of primary viruses identified five amino acid positions (3, 5, 15, 16 and 24) in the transmembrane and extracellular domain of Vpu that are associated with HLA-C downregulation (30). Grouping the amino acid position and residues based on their HLA-C genotype showed no differences in the frequency of amino acid positions/residues that downregulate HLA-C. We further tested whether differential HLA-C allele expression or HLA-C/KIR2DL binding were associated with AA residues that are involved in HLA-C downmodulation (28, 30). We observed an increased frequency of glycine or threonine at AA position 16 in individuals with stronger HLA-C/KIR2DL binding combinations compared to other amino acid residues at position 16, hinting that the binding affinities between KIR2DL and HLA-C molecules might be an additional factor impacting Vpu sequence polymorphisms. It should be noted that the observed signals contain a certain level of uncertainty that are attributed to the numerous allotype combinations in the cohort and the ambiguity of KIR allele typing. In general, the enormous diversity in HLA-C/KIR2DL binding combinations and different affinities, influencing the activation of NK cells and immune evasion mechanisms of HIV-1, make it challenging to develop a prediction model for specific Vpu sequence polymorphisms and consequences for NK cell activation based on HLA-C/KIR2DL genotypes.
Altogether, this study demonstrates the significant effects of HIV-1 infection on the NK cell pool in viremic, untreated HIV-1+ individuals and provides evidence that the specific changes in the KIR2DL repertoire are predetermined by the underlying KIR2DL/HLA-C genotypes. The results also make a case that high resolution KIR typing and the generation of KIR/HLA binding models may be warranted to improve disease models and to potentially predict NK cell-associated immune responses and disease progression in various pathological settings and subsequently NK cell-based immune therapies.
Data Availability Statement
Sequence data presented in the study are deposited in the European Nucleotide Archive (ENA), accession number #PRJEB53333. Storage of other raw data is performed by the Leibniz Institute of Virology on an internal server. Raw data will be made available upon request and can be shared after confirming that data will be used within the scope of the originally provided informed consent.
Ethics Statement
For this study, residual amounts of anonymized peripheral blood samples were used which were routinely taken from healthy blood donors at the the Institute for Transfusion Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany, and would have been discarded otherwise. All blood donors gave their general written consent to usage of their blood samples for scientific studies in an anonymized form. The anonymized use of human material complies with a vote by the ethics committee of the German Medical Association. Healthy blood donors recruited at the University Medical Center Hamburg-Eppendorf, Hamburg, Germany, provided written informed consent and studies were approved by the ethics committee of the Ärztekammer Hamburg (PV4780). Donors recruited through the Translational Platform HIV (TP-HIV) Cohort by the German Center for Infection Research (DZIF) provided written informed consent and the study was approved by the ethics committee of the Ärztekammer Hamburg (MC-316/14).
Author Contributions
Conceptualization: CK; Funding acquisition: CK, MA, and AH; Methodology: CK, SVo, AN, and AH. Validation: SVo and AL; Formal Analysis: CK, SVo, SVi, and LR; Investigation: SVo, AL, TT, SB, DI, and JN; Resources: SVo, AN, PF, MA, GS, SP, GMNB, CL, AM, RP, NP, JR, SS, CS, CDS, EW, CW, PJN, JS, and AHS; Writing – original draft: CK and SVo; Writing - review and editing: all authors; Visualization: CK and SVo; Supervision: CK. All authors contributed to the article and approved the submitted version.
Funding
The study was supported by the German Research foundation (DFG) (KO5139/3-1) and the German Center for Infection Research (DZIF) (TTU 04.820_01). TT and SB received funding by the state of Hamburg, Germany (LFF-FV74 and LFF-FV78). JR received funding by the German Center for Infection Research (DZIF) (TTU HIV 04.817, 04.819, 04.919) and the Else-Kröner-Fresenius Stiftung (EKFS) (2021_EKEA.25). AH was supported through the Clinician Scientist Program of the Faculty of Medicine, University Medical Centre Hamburg-Eppendorf.
Conflict of Interest
CDS reports grants and personal fees from AbbVie, grants, fees and non-financial support from Gilead Sciences, grants and personal fees from Janssen-Cilag, grants and personal fees from MSD, grants from Cepheid, personal fees from GSK, grants and personal fees from ViiV Healthcare, during the conduct of the study; fees from AstraZeneca, other from Apeiron, grants, personal fees and non-financial support from BBraun Melsungen, grants, personal fees from BioNtech, personal fees from Eli Lilly, personal fees from Formycon, personal fees from Molecular partners, grants and personal fees from Eli Lilly, personal fees from Roche, personal fees from SOBI.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the blood donors and the coordinator of the Healthy Cohort Hamburg and the German Center for Infection Research (DZIF) for the PBMC and plasma samples from untreated HIV+ individuals. We also want to thank the Flow Cytometry core facility of the Leibniz Institute of Virology for their technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.922252/full#supplementary-material
References
1. Jost S, Altfeld M. Control of Human Viral Infections by Natural Killer Cells. Annu Rev Immunol (2013) 31:163–94. doi: 10.1146/annurev-immunol-032712-100001
2. Pahl J, Cerwenka A. Tricking the Balance: NK Cells in Anti-Cancer Immunity. Immunobiology (2017) 222(1):11–20. doi: 10.1016/j.imbio.2015.07.012
3. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of Natural Killer Cells. Nat Immunol (2008) 9(5):503–10. doi: 10.1038/ni1582
4. Cooper MA, Fehniger TA, Caligiuri MA. The Biology of Human Natural Killer-Cell Subsets. Trends Immunol (2001) 22(11):633–40. doi: 10.1016/S1471-4906(01)02060-9
5. Lanier LL. Up on the Tightrope: Natural Killer Cell Activation and Inhibition. Nat Immunol (2008) 9(5):495–502. doi: 10.1038/ni1581
6. Guethlein LA, Older Aguilar AM, Abi-Rached L, Parham P. Evolution of Killer Cell Ig-Like Receptor (KIR) Genes: Definition of an Orangutan KIR Haplotype Reveals Expansion of Lineage III KIR Associated With the Emergence of MHC-C. J Immunol (2007) 179(1):491–504. doi: 10.4049/jimmunol.179.1.491
7. Colonna M, Borsellino G, Falco M, Ferrara GB, Strominger JL. HLA-C is the Inhibitory Ligand That Determines Dominant Resistance to Lysis by NK1- and NK2-Specific Natural Killer Cells. Proc Natl Acad Sci (1993) 90(24):12000–4. doi: 10.1073/pnas.90.24.12000
8. Hilton HG, Guethlein LA, Goyos A, Nemat-Gorgani N, Bushnell DA, Norman PJ, et al. Polymorphic HLA-C Receptors Balance the Functional Characteristics of KIR Haplotypes. J Immunol (2015) 195(7):3160–70. doi: 10.4049/jimmunol.1501358
9. Older Aguilar AM, Guethlein LA, Adams EJ, Abi-Rached L, Moesta AK, Parham P. Coevolution of Killer Cell Ig-Like Receptors With HLA-C To Become the Major Variable Regulators of Human NK Cells. J Immunol (2010) 185(7):4238–51. doi: 10.4049/jimmunol.1001494
10. Pende D, Biassoni R, Cantoni C, Verdiani S, Falco M, Di Donato C, et al. The Natural Killer Cell Receptor Specific for HLA-A Allotypes: A Novel Member of the P58/P70 Family of Inhibitory Receptors That is Characterized by Three Immunoglobulin-Like Domains and Is Expressed as a 140-kD Disulphide-Linked Dimer. J Exp Med (1996) 184(2):505. doi: 10.1084/jem.184.2.505
11. Hansasuta P, Dong T, Thananchai H, Weekes M, Willberg C, Aldemir H, et al. Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is Peptide-Specific. Eur J Immunol (2004) 34(6):1673–9. doi: 10.1002/eji.200425089
12. Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 Public Epitope of HLA-B Molecules Confers Reactivity With Natural Killer Cell Clones That Express NKB1, a Putative HLA Receptor. J Exp Med (1995) 181(3):1133–44. doi: 10.1084/jem.181.3.1133
13. Moesta AK, Parham P. Diverse Functionality Among Human NK Cell Receptors for the C1 Epitope of HLA-C: KIR2DS2, KIR2DL2, and KIR2DL3. Front Immunol (2012) 3:336. doi: 10.3389/fimmu.2012.00336
14. Mandelboim O, Reyburn HT, Valés-Gómez M, Pazmany L, Colonna M, Borsellino G, et al. Protection From Lysis by Natural Killer Cells of Group 1 and 2 Specificity is Mediated by Residue 80 in Human Histocompatibility Leukocyte Antigen C Alleles and Also Occurs With Empty Major Histocompatibility Complex Molecules. J Exp Med (1996) 184(3):913–22. doi: 10.1084/jem.184.3.913
15. Winter CC, Long EO. A Single Amino Acid in the P58 Killer Cell Inhibitory Receptor Controls the Ability of Natural Killer Cells to Discriminate Between the Two Groups of HLA-C Allotypes. J Immunol (1997) 158(9):4026–8.
16. Blokhuis JH, Hilton HG, Guethlein LA, Norman PJ, Nemat-Gorgani N, Nakimuli A, et al. KIR2DS5 Allotypes That Recognize the C2 Epitope of HLA-C Are Common Among Africans and Absent From Europeans. Immunity Inflamm Dis (2017) 5(4):461–8. doi: 10.1002/iid3.178
17. Moesta AK, Norman PJ, Yawata M, Yawata N, Gleimer M, Parham P. Synergistic Polymorphism at Two Positions Distal to the Ligand-Binding Site Makes KIR2DL2 a Stronger Receptor for HLA-C Than Kir2dl3. J Immunol (2008) 180(6):3969–79. doi: 10.4049/jimmunol.180.6.3969
18. Graef T, Moesta AK, Norman PJ, Abi-Rached L, Vago L, Older Aguilar AM, et al. KIR2DS4 Is a Product of Gene Conversion With KIR3DL2 That Introduced Specificity for HLA-A*11 While Diminishing Avidity for HLA-C. J Exp Med (2009) 206(11):2557–72. doi: 10.1084/jem.20091010
19. Moradi S, Stankovic S, O’Connor GM, Pymm P, MacLachlan BJ, Faoro C, et al. Structural Plasticity of KIR2DL2 and KIR2DL3 Enables Altered Docking Geometries Atop HLA-C. Nat Commun (2021) 12(1). doi: 10.1038/s41467-021-22359-x
20. Hiby SE, Walker JJ, O’shaughnessy KM, Redman CWG, Carrington M, Trowsdale J, et al. Combinations of Maternal KIR and Fetal HLA-C Genes Influence the Risk of Preeclampsia and Reproductive Success. J Exp Med (2004) 200(8):957–65. doi: 10.1084/jem.20041214
21. Nelson GW, Martin MP, Gladman D, Wade J, Trowsdale J, Carrington M. Cutting Edge: Heterozygote Advantage in Autoimmune Disease: Hierarchy of Protection/Susceptibility Conferred by HLA and Killer Ig-Like Receptor Combinations in Psoriatic Arthritis. J Immunol (2004) 173(7):4273–6. doi: 10.4049/jimmunol.173.7.4273
22. Khakoo SI. HLA and NK Cell Inhibitory Receptor Genes in Resolving Hepatitis C Virus Infection. Science (2004) 305(5685):872–4. doi: 10.1126/science.1097670
23. Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, et al. Epistatic Interaction Between KIR3DS1 and HLA-B Delays the Progression to AIDS. Nat Genet (2002) 31(4):429–34. doi: 10.1038/ng934
24. Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F, et al. Innate Partnership of HLA-B and KIR3DL1 Subtypes Against HIV-1. Nat Genet (2007) 39(6):733–40. doi: 10.1038/ng2035
25. Bashirova AA, Thomas R, Carrington M. HLA/KIR Restraint of HIV: Surviving the Fittest. Annu Rev Immunol (2011) 29:295–317. doi: 10.1146/annurev-immunol-031210-101332
26. International HIV Controllers Study, Pereyra F, Jia X, PJ M, Telenti A, de Bakker PIW, et al. The Major Genetic Determinants of HIV-1 Control Affect HLA Class I Peptide Presentation. Science (2010) 330(6010):1551–7. doi: 10.1126/science.1195271
27. Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, et al. A Whole-Genome Association Study of Major Determinants for Host Control of HIV-1. Science (2007) 317(5840):944–7. doi: 10.1126/science.1143767
28. Apps R, Qi Y, Carlson JM, Chen H, Gao X, Thomas R, et al. Influence of HLA-C Expression Level on HIV Control. Science (2013) 340(6128):87–91. doi: 10.1126/science.1232685
29. Apps R, Del Prete GQ, Chatterjee P, Lara A, Brumme ZL, Brockman MA, et al. HIV-1 Vpu Mediates HLA-C Downregulation. Cell Host Microbe (2016) 19(5):686–95. doi: 10.1016/j.chom.2016.04.005
30. Bachtel ND, Umviligihozo G, Pickering S, Mota TM, Liang H, Del Prete GQ, et al. HLA-C Downregulation by HIV-1 Adapts to Host HLA Genotype. PloS Pathog (2018) 14(9):e1007257. doi: 10.1371/journal.ppat.1007257
31. Körner C, Simoneau CR, Schommers P, Granoff M, Ziegler M, Hölzemer A, et al. HIV-1-Mediated Downmodulation of HLA-C Impacts Target Cell Recognition and Antiviral Activity of NK Cells. Cell Host Microbe (2017) 22(1):111–119.e4. doi: 10.1016/j.chom.2017.06.008
32. Körner C, Granoff ME, Amero MA, Sirignano MN, Vaidya SA, Jost S, et al. Increased Frequency and Function of KIR2DL1-3+ NK Cells in Primary HIV-1 Infection are Determined by HLA-C Group Haplotypes. Eur J Immunol (2014) 44(10):2938–48. doi: 10.1002/eji.201444751
33. Fadda L, Körner C, Kumar S, van Teijlingen NH, Piechocka-Trocha A, Carrington M, et al. HLA-Cw*0102-Restricted HIV-1 P24 Epitope Variants can Modulate the Binding of the Inhibitory KIR2DL2 Receptor and Primary NK Cell Function. PloS Pathog (2012) 8(7):40. doi: 10.1371/journal.ppat.1002805
34. Mori M, Wichukchinda N, Miyahara R, Rojanawiwat A, Pathipvanich P, Tsuchiya N, et al. The Effect of KIR2D-HLA-C Receptor-Ligand Interactions on Clinical Outcome in a HIV-1 CRF01_AE-Infected Thai Population. AIDS (2015) 29(13):1607–15. doi: 10.1097/QAD.0000000000000747
35. Lin Z, Kuroki K, Kuse N, Sun X, Akahoshi T, Qi Y, et al. HIV-1 Control by NK Cells via Reduced Interaction Between KIR2DL2 and HLA-C∗12:02/C∗14:03. Cell Rep (2016) 17(9):2210–20. doi: 10.1016/j.celrep.2016.10.075
36. Hölzemer A, Thobakgale CF, Jimenez Cruz CA, Garcia-Beltran WF, Carlson JM, van Teijlingen NH, et al. Selection of an HLA-C*03:04-Restricted HIV-1 P24 Gag Sequence Variant Is Associated With Viral Escape From KIR2DL3+ Natural Killer Cells: Data From an Observational Cohort in South Africa. PloS Med (2015) 12(11):1–27. doi: 10.1371/journal.pmed.1001900
37. Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, et al. HIV-1 Adaptation to NK-Cell-Mediated Immune Pressure. Nature. (2011) 476(7358):96–101. doi: 10.1038/nature10237
38. Pickering S, Hué S, Kim E-Y, Reddy S, Wolinsky SM, Neil SJD. Preservation of Tetherin and CD4 Counter-Activities in Circulating Vpu Alleles Despite Extensive Sequence Variation Within HIV-1 Infected Individuals. PloS Pathog (2014) 10(1):e1003895. doi: 10.1371/journal.ppat.1003895
39. Hilton HG, Moesta AK, Guethlein LA, Blokhuis J, Parham P, Norman PJ. The Production of KIR-Fc Fusion Proteins and Their Use in a Multiplex HLA Class I Binding Assay. J Immunol Methods (2015) 425:79–87. doi: 10.1016/j.jim.2015.06.012
40. Li H, Durbin R. Fast and Accurate Short Read Alignment With Burrows-Wheeler Transform. Bioinformatics (2009) 25(14):1754–60. doi: 10.1093/bioinformatics/btp324
41. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map Format and SAMtools. Bioinformatics (2009) 25(16):2078–9. doi: 10.1093/bioinformatics/btp352
42. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res (2010) 20(9):1297–303. doi: 10.1101/gr.107524.110
43. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr Protoc Bioinforma (2013) 43(1110). doi: 10.1002/0471250953.bi1110s43
44. Van der Auwera G, O’Connor B, Safari an OMC. Genomics in the Cloud (2020). Available at: https://www.oreilly.com/library/view/genomics-in-the/9781491975183/.
45. Schöfl G, Lang K, Quenzel P, Böhme I, Sauter J, Hofmann JA, et al. 2.7 Million Samples Genotyped for HLA by Next Generation Sequencing: Lessons Learned. BMC Genomics (2017) 18(1). doi: 10.1186/s12864-017-3575-z
46. Wagner I, Schefzyk D, Pruschke J, Schöfl G, Schöne B, Gruber N, et al. Allele-Level KIR Genotyping of More Than a Million Samples: Workflow, Algorithm, and Observations. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02843
47. Boudreau JE, Mulrooney TJ, Le Luduec J-B, Barker E, Hsu KC. KIR3DL1 and HLA-B Density and Binding Calibrate NK Education and Response to HIV. J Immunol (2016) 196(8):3398–410. doi: 10.4049/jimmunol.1502469
48. Saunders PM, Pymm P, Pietra G, Hughes VA, Hitchen C, O’Connor GM, et al. Killer Cell Immunoglobulin-Like Receptor 3dl1 Polymorphism Defines Distinct Hierarchies of HLA Class I Recognition. J Exp Med (2016) 213(5):791–807. doi: 10.1084/jem.20152023
49. Hilton HG, Norman PJ, Nemat-Gorgani N, Goyos A, Hollenbach JA, Henn BM, et al. Loss and Gain of Natural Killer Cell Receptor Function in an African Hunter-Gatherer Population. PloS Genet (2015) 11(8):e1005439. doi: 10.1371/journal.pgen.1005439
50. Björkström NK, Ljunggren HG, Sandberg JK. CD56 Negative NK Cells: Origin, Function, and Role in Chronic Viral Disease. Trends Immunol (2010) 31(11):401–6. doi: 10.1016/j.it.2010.08.003
51. Le Luduec JB, Boudreau JE, Freiberg JC, Hsu KC. Novel Approach to Cell Surface Discrimination Between KIR2DL1 Subtypes and KIR2DS1 Identifies Hierarchies in NK Repertoire, Education, and Tolerance. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00734
52. Winter CC, Gumperz JE, Parham P, Long EO, Wagtmann N. Direct Binding and Functional Transfer of NK Cell Inhibitory Receptors Reveal Novel Patterns of HLA-C Allotype Recognition. J Immunol (1998) 161(2):571–7.
53. Kaur G, Gras S, Mobbs JI, Vivian JP, Cortes A, Barber T, et al. Structural and Regulatory Diversity Shape HLA-C Protein Expression Levels. Nat Commun (2017) 8(1):15924. doi: 10.1038/ncomms15924
54. Boudreau JE, Hsu KC. Natural Killer Cell Education and the Response to Infection and Cancer Therapy: Stay Tuned. Trends Immunol (2018) 39(3):222–39. doi: 10.1016/j.it.2017.12.001
55. Martin MP, Naranbhai V, Shea PR, Qi Y, Ramsuran V, Vince N, et al. Killer Cell Immunoglobulin-Like Receptor 3dl1 Variation Modifies HLA-B*57 Protection Against HIV-1. J Clin Invest (2018) 128(5):1903–12. doi: 10.1172/JCI98463
56. Van Teijlingen NH, Hölzemer A, Körner C, García-Beltrán WF, Schafer JL, Fadda L, et al. Sequence Variations in HIV-1 P24 Gag-Derived Epitopes can Alter Binding of KIR2DL2 to HLA-C*03:04 and Modulate Primary Natural Killer Cell Function. AIDS (2014) 28(10):1399–408. doi: 10.1097/QAD.0000000000000284
57. Ziegler MC, Nelde A, Weber JK, Schreitmüller CM, Martrus G, Huynh T, et al. HIV-1 Induced Changes in HLA-C*03: 04-Presented Peptide Repertoires Lead to Reduced Engagement of Inhibitory Natural Killer Cell Receptors. AIDS (2020) 34(12):1713–23. doi: 10.1097/QAD.0000000000002596
58. Alter G, Martin MP, Teigen N, Carr WH, Suscovich TJ, Schneidewind A, et al. Differential Natural Killer Cell-Mediated Inhibition of HIV-1 Replication Based on Distinct KIR/HLA Subtypes. J Exp Med (2007) 204(12):3027–36. doi: 10.1084/jem.20070695
59. Alter G, Rihn S, Walter K, Nolting A, Martin M, Rosenberg ES, et al. HLA Class I Subtype-Dependent Expansion of KIR3DS1+ and KIR3DL1+ NK Cells During Acute Human Immunodeficiency Virus Type 1 Infection. J Virol (2009) 83(13):6798–805. doi: 10.1128/JVI.00256-09
60. Pelak K, Need AC, Fellay J, Shianna KV, Feng S, Urban TJ, et al. Copy Number Variation of KIR Genes Influences HIV-1 Control. PloS Biol (2011) 9(11):e1001208. doi: 10.1371/journal.pbio.1001208
61. Mavilio D, Benjamin J, Daucher M, Lombardo G, Kottilil S, Planta MA, et al. Natural Killer Cells in HIV-1 Infection: Dichotomous Effects of Viremia on Inhibitory and Activating Receptors and Their Functional Correlates. Proc Natl Acad Sci U.S.A. (2003) 100(25):15011–6. doi: 10.1073/pnas.2336091100
62. Ahmad R, Sindhu STAK, Tran P, Toma E, Morisset R, Menezes J, et al. Modulation of Expression of the MHC Class I-Binding Natural Killer Cell Receptors, and NK Activity in Relation to Viral Load in HIV-Infected/AIDS Patients. J Med Virol (2001) 65(3):431–40. doi: 10.1002/jmv.2053
63. De Maria A, Fogli M, Costa P, Murdaca G, Puppo F, Mavilio D, et al. The Impaired NK Cell Cytolytic Function in Viremic HIV-1 Infection is Associated With a Reduced Surface Expression of Natural Cytotoxicity Receptors (NKp46, NKp30 and Nkp44). Eur J Immunol (2003) 33(9):2410–8. doi: 10.1002/eji.200324141
64. Ahmad F, Hong HS, Jäckel M, Jablonka A, Lu I-N, Bhatnagar N, et al. High Frequencies of Polyfunctional CD8 + NK Cells in Chronic HIV-1 Infection Are Associated With Slower Disease Progression. J Virol (2014) 88(21):12397–408. doi: 10.1128/JVI.01420-14
65. Braud VM, Allan DSJ, O’Callaghan CA, Söderström K, D’Andrea A, Ogg GS, et al. HLA-E Binds to Natural Killer Cell Receptors CD94/NKG2A, B and C. Nature (1998) 391(6669):795–9. doi: 10.1038/35869
66. Davis ZB, Cogswell A, Scott H, Mertsching A, Boucau J, Wambua D, et al. A Conserved HIV-1-Derived Peptide Presented by HLA-E Renders Infected T-Cells Highly Susceptible to Attack by NKG2A/CD94-Bearing Natural Killer Cells. PloS Pathog (2016) 12(2):e1005421. doi: 10.1371/journal.ppat.1005421
67. Mela CM, Burton CT, Imami N, Nelson M, Steel A, Gazzard BG, et al. Switch From Inhibitory to Activating NKG2 Receptor Expression in HIV-1 Infection: Lack of Reversion With Highly Active Antiretroviral Therapy. AIDS (2005) 19(16):1761–9. doi: 10.1097/01.aids.0000183632.12418.33
68. Zhang R, Xu J, Hong K, Yuan L, Peng H, Tang H, et al. Increased NKG2A Found in Cytotoxic Natural Killer Subset in HIV-1 Patients With Advanced Clinical Status. AIDS (2007) 21(Suppl 8):S9–17. doi: 10.1097/01.aids.0000304691.32014.19
69. Gumá M, Cabrera C, Erkizia I, Bofill M, Clotet B, Ruiz L, et al. Human Cytomegalovirus Infection is Associated With Increased Proportions of NK Cells That Express the CD94/NKG2C Receptor in Aviremic HIV-1-Positive Patients. J Infect Dis (2006) 194(1):38–41. doi: 10.1086/504719
70. Gumá M, Angulo A, Vilches C, Gómez-Lozano N, Malats N, López-Botet M. Imprint of Human Cytomegalovirus Infection on the NK Cell Receptor Repertoire. Blood. (2004) 104(12):3664–71. doi: 10.1182/blood-2004-05-2058
71. Gumá M, Budt M, Sáez A, Brckalo T, Hengel H, Angulo A, et al. Expansion of CD94/NKG2C+ NK Cells in Response to Human Cytomegalovirus-Infected Fibroblasts. Blood (2006) 107(9):3624–31. doi: 10.1182/blood-2005-09-3682
72. Björkström NK, Lindgren T, Stoltz M, Fauriat C, Braun M, Evander M, et al. Rapid Expansion and Long-Term Persistence of Elevated NK Cell Numbers in Humans Infected With Hantavirus. J Exp Med (2011) 208(1):13–21. doi: 10.1084/jem.20100762
73. Kovalenko EI, Streltsova MA. Adaptive Features of Natural Killer Cells, Lymphocytes of Innate Immunity. Orig Russ Text © EI Kovalenko MA Streltsova (2016) 42(6):649–67. doi: 10.1134/S1068162016060066
74. Merino AM, Dugast A-S, Wilson CM, Goepfert PA, Alter G, Kaslow RA, et al. KIR2DS4 Promotes HIV-1 Pathogenesis: New Evidence From Analyses of Immunogenetic Data and Natural Killer Cell Function. PloS One (2014) 9(6):e99353. doi: 10.1371/journal.pone.0099353
75. Olvera A, Pérez-Álvarez S, Ibarrondo J, Ganoza C, Lama JR, Lucchetti A, et al. The HLA-C*04: 01/KIR2DS4 Gene Combination and Human Leukocyte Antigen Alleles With High Population Frequency Drive Rate of HIV Disease Progression. AIDS (2015) 29(5):507–17. doi: 10.1097/QAD.0000000000000574
76. Kollnberger S, Bird L, Sun MY, Retiere C, Braud VM, McMichael A, et al. Cell-Surface Expression and Immune Receptor Recognition of HLA-B27 Homodimers. Arthritis Rheum (2002) 46(11):2972–82. doi: 10.1002/art.10605
77. Podhorzer A, Dirchwolf M, Machicote A, Belen S, Montal S, Paz S, et al. The Clinical Features of Patients With Chronic Hepatitis C Virus Infections Are Associated With Killer Cell Immunoglobulin-Like Receptor Genes and Their Expression on the Surface of Natural Killer Cells. Front Immunol (2018) 8. doi: 10.3389/fimmu.2017.01912
78. Chan AT, Kollnberger SD, Wedderburn LR, Bowness P. Expansion and Enhanced Survival of Natural Killer Cells Expressing the Killer Immunoglobulin-Like Receptor KIR3DL2 in Spondylarthritis. Arthritis Rheum (2005) 52(11):3586–95. doi: 10.1002/art.21395
79. Li H, Wright PW, McCullen M, Anderson SK. Characterization of KIR Intermediate Promoters Reveals Four Promoter Types Associated With Distinct Expression Patterns of KIR Subtypes. Genes Immun (2016) 17(1):66–74. doi: 10.1038/gene.2015.56
80. Wright PW, Li H, Huehn A, O’connor GM, Cooley S, Miller JS, et al. Characterization of a Weakly Expressed KIR2DL1 Variant Reveals a Novel Upstream Promoter That Controls KIR Expression HHS Public Access. Genes Immun (2014) 15(7):440–8. doi: 10.1038/gene.2014.34
Keywords: NK cell, KIR, HLA-C, HIV-1, Vpu
Citation: Vollmers S, Lobermeyer A, Niehrs A, Fittje P, Indenbirken D, Nakel J, Virdi S, Brias S, Trenkner T, Sauer G, Peine S, Behrens GMN, Lehmann C, Meurer A, Pauli R, Postel N, Roider J, Scholten S, Spinner CD, Stephan C, Wolf E, Wyen C, Richert L, Norman PJ, Sauter J, Schmidt AH, Hoelzemer A, Altfeld M and Körner C (2022) Host KIR/HLA-C Genotypes Determine HIV-Mediated Changes of the NK Cell Repertoire and Are Associated With Vpu Sequence Variations Impacting Downmodulation of HLA-C. Front. Immunol. 13:922252. doi: 10.3389/fimmu.2022.922252
Received: 17 April 2022; Accepted: 13 June 2022;
Published: 15 July 2022.
Edited by:
Hui Peng, University of Science and Technology of China, ChinaReviewed by:
Edward Barker, Rush University, United StatesJeanette E. Boudreau, Dalhousie University, Canada
Copyright © 2022 Vollmers, Lobermeyer, Niehrs, Fittje, Indenbirken, Nakel, Virdi, Brias, Trenkner, Sauer, Peine, Behrens, Lehmann, Meurer, Pauli, Postel, Roider, Scholten, Spinner, Stephan, Wolf, Wyen, Richert, Norman, Sauter, Schmidt, Hoelzemer, Altfeld and Körner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Körner, Y2hyaXN0aWFuLmtvZXJuZXJAbGVpYm5pei1saXYuZGU=