 Meng Lv
Meng Lv Yan Liu2†
Yan Liu2† Yabing Xing
Yabing Xing Shengnan Zhang
Shengnan Zhang- 1Department of Pharmacy, Children’s Hospital Affiliated to Zhengzhou University, Zhengzhou, China
- 2Department of Pharmacy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 3Department of Hematology Oncology, Children’s Hospital Affiliated to Zhengzhou University, Zhengzhou, China
Pediatric acute lymphoblastic leukemia (ALL) is the most common subtype of childhood leukemia, which is characterized by the abnormal proliferation and accumulation of immature lymphoid cell in the bone marrow. Although the long-term survival rate for pediatric ALL has made significant progress over years with the development of contemporary therapeutic regimens, patients are still suffered from relapse, leading to an unsatisfactory outcome. Since the immune system played an important role in the progression and relapse of ALL, immunotherapy including bispecific T-cell engagers and chimeric antigen receptor T cells has been demonstrated to be capable of enhancing the immune response in pediatric patients with refractory or relapsed B-cell ALL, and improving the cure rate of the disease and patients’ quality of life, thus receiving the authorization for market. Nevertheless, the resistance and toxicities associated with the current immunotherapy remains a huge challenge. Novel therapeutic options to overcome the above disadvantages should be further explored. In this review, we will thoroughly discuss the emerging immunotherapeutics for the treatment of pediatric ALL, as well as side-effects and new development.
Introduction
Acute lymphoblastic leukemia (ALL), characterized by the abnormal clonal proliferation of the early lymphoid stem cells or progenitor cells and the depletion of the normal hematopoietic cells in the marrow, is the most prevalent subtype of leukemia with a rapidly growing incidence worldwide (1–5). Although ALL occurs in both adults and children, children represent up to 80% of cases (6). Currently, improved long-term survival rates have increased to more than 90% in pediatric ALL thanks to the contemporary therapeutic regimens (7, 8). However, approximately 20% of the patients remain refractory to primary therapy or suffer from relapse after initial complete remission (CR), leading to a poor prognosis (9, 10). Therefore, the exploration of novel therapeutic approaches for pediatric refractory/relapse (R/R) ALL are urgently needed and will eventually benefit this population.
Until now, accumulating evidence has suggested that tumor microenvironment (TME) contributes to the cancer development and progression (11–13). As key members in TME, immune cells consisting of T cells, macrophages, and natural killer cells (NKs) have reduced activity and poise in an immunosuppressive state. Activating the immune system to recognize and eradicate cancer cells rather than remove or directly attack the cancer cells, termed immunotherapy, has been proposed as an alternative to conventional cancer treatment and widely explored over the last decade (14, 15). In particular, immunotherapy promoted rapid development for the treatment of the hematologic malignancies (16–18). The approval for market of blinatumomab, a bispecific CD3/CD19 T-cell engager, and tisagenlecleucel, a CAR T cell therapy, has demonstrated dramatic progress in the treatment of pediatric R/R B-cell precursor ALL (R/R BCP ALL). Combination or replacement of conventional chemotherapy with immunotherapy to further improve the cure rates and life quality of pediatric patients with ALL has become the priority issue for the moment (19–21). Generally, pediatric cancers are not smaller versions of adult cancers (6, 22). The progress and development in pediatric cancers lags behind adult patients (23). Herein, we aim to provide a comprehensive overview of the emerging immunotherapeutic approaches in pediatric ALL, thus guiding the development of novel therapeutic options. The preclinical research and ongoing clinical trials in this field will be extensively summarized in this review (Table 1). Due to T-cell ALL accounts for merely 15% of the pediatric ALL patients and has a different immunophenotype from B-cell ALL, immunotherapy for pediatric T-cell ALL is outside the scope of this article.
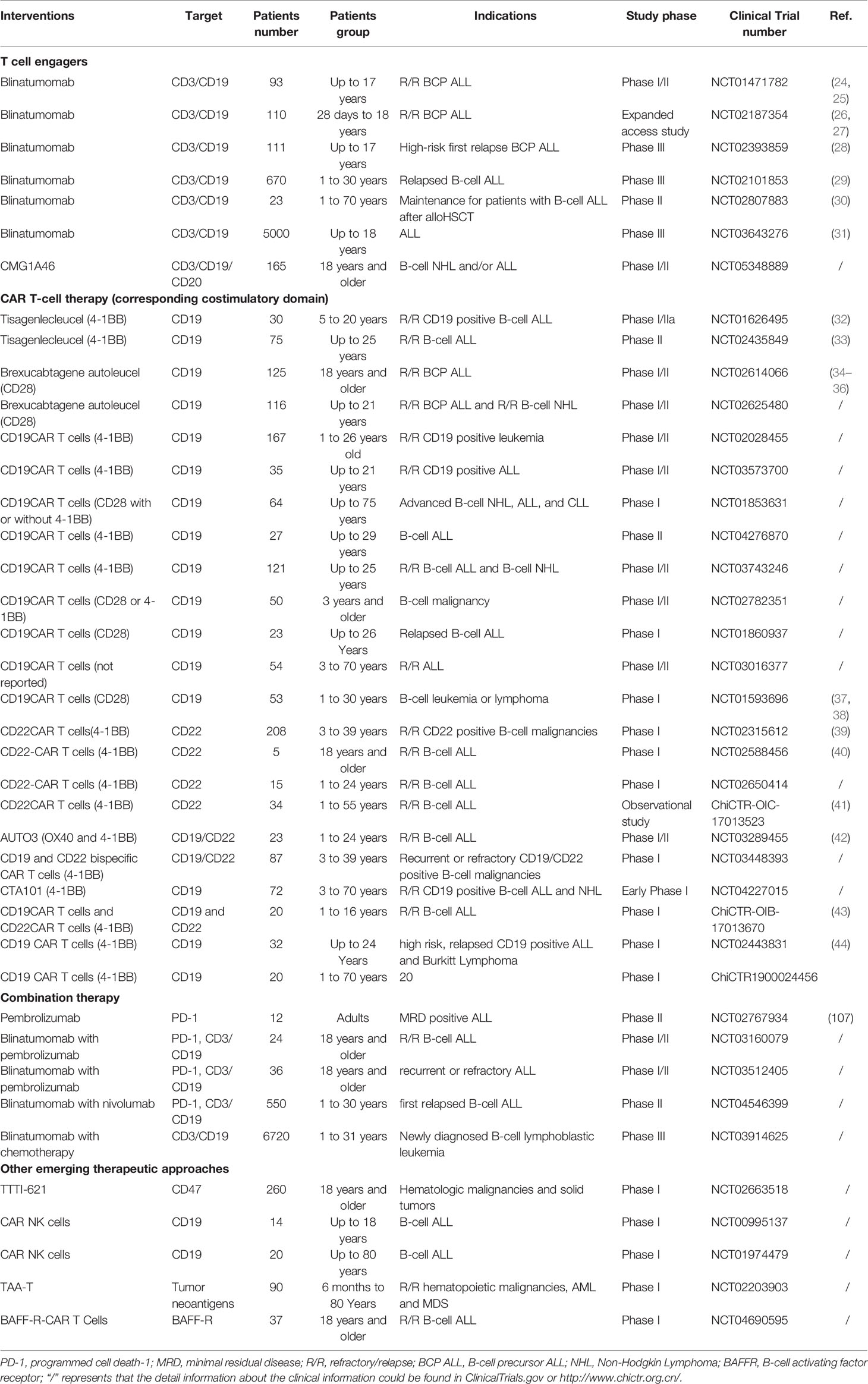
Table 1 Emerging immunotherapeutic approaches for pediatric B-cell ALL.
T Cell-Based Immunotherapy
T cells have become an ideal weapon and attracted great research enthusiasm in cancer immunotherapy due to its capacity for antigen-directed cytotoxicity (45–47). Over the last decade, various T cell-based immunotherapeutic approaches, including blocking programmed cell death-1 (PD-1)/programmed cell death-ligand 1 (PD-L1) axis, bispecific/trispecific T-cell engagers, and chimeric antigen receptor (CAR) T cells have revolutionized the field of cancer therapeutics. The following context will highlight the T cell-based immunotherapeutic strategies available to attenuate pediatric B-cell ALL (Figure 1).
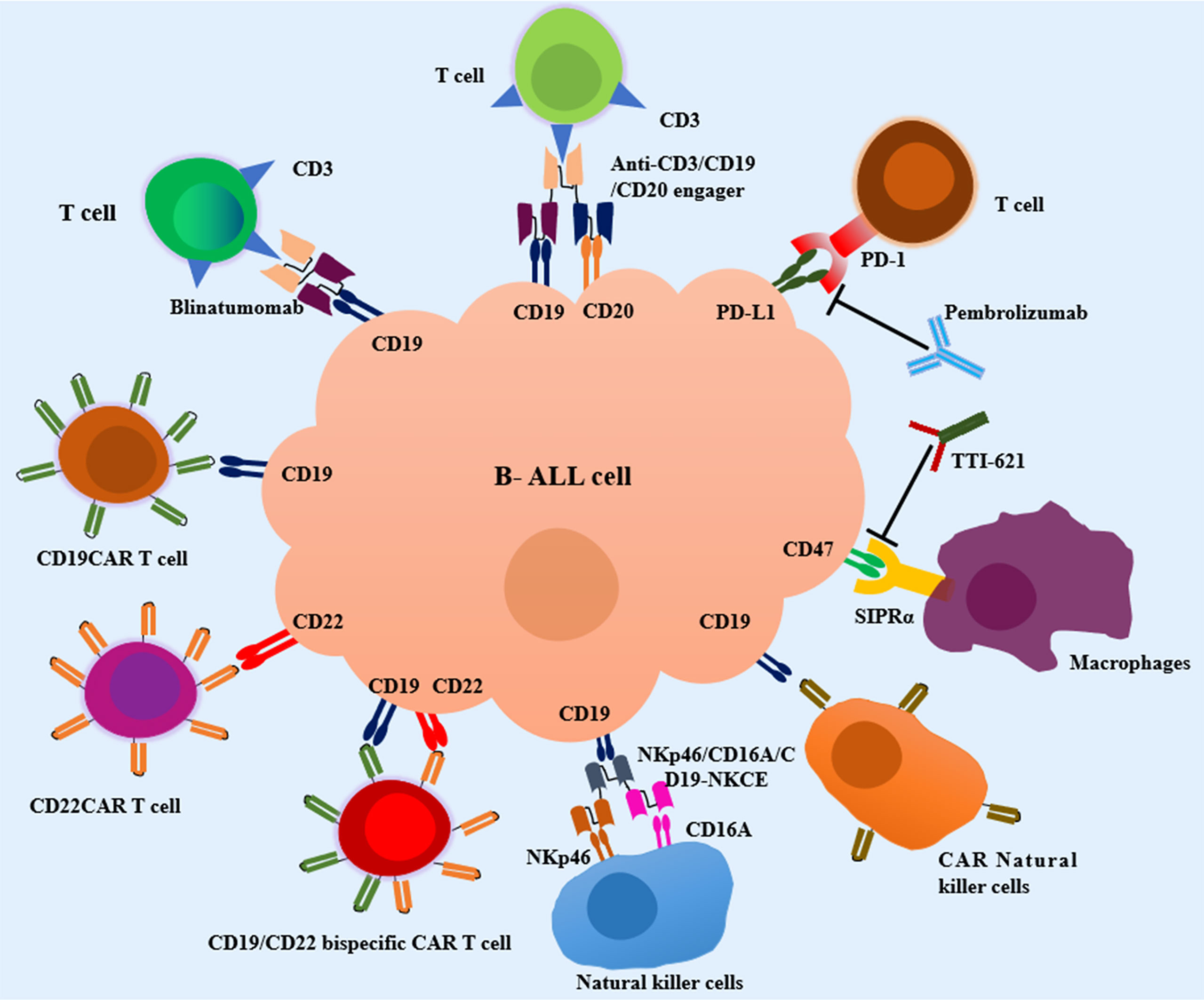
Figure 1 Current immunotherapeutic strategies in pediatric Acute Lymphoblastic Leukemia (ALL). T-cell engagers, CAR T cell therapy, macrophages-based immunotherapy, NK cell-based immunotherapy and other emerging immunotherapies are in development for pediatric B-cell ALL. SIRPα, signal regulatory protein alpha; NKp46/CD16A/CD19-NKCE, a NK cell engager with CD16A on the surface of NKs binding NKp46 and CD19 on the surface of B-cell ALL cells.
Bispecific CD3/CD19 T Cell Engagers
Bispecific antibodies are designated to recognize and bind two distinct epitopes or antigens simultaneously, which have demonstrated great therapeutic potential toward cancer immunotherapy and are in rapid clinical development (48, 49). Blinatumomab, which is comprised of two different single-chain variable fragment regions (scFv) linked via a glycine-serine linker (50), triggers a cytotoxic immune response and shows significant cytotoxic activity at ultra-low concentrations, through binding specifically to antigen CD19 that is overexpressed on the surface of B-cell ALL lymphocytes and antigen CD3 on the surface of T cells (51–53). As the benchmarking case, blinatumomab conveyed good efficiency and safety in a phase I/II study (NCT01471782), which demonstrated that blinatumomab was maximumly tolerated at 15 μg/m2/day in 49 children with R/R BCP-ALL, 39% of whom achieved CR with single-agent blinatumomab treatment (24, 25). In the subsequent multi-center, expanded access study (RIALTO trial, NCT02187354), 63% of children had CR and the MRD negativity was obtained in 83% of responders after the first two cycles of blinatumomab treatment, further confirming the efficiency of blinatumomab (26, 27). Accordingly, blinatumomab was approved to treat R/R B-cell ALL in children by the US Food and Drug Administration (FDA) in 2018. Recently, two randomized trials (NCT02393859 and NCT02101853) exemplify the advantages of blinatumomab as post reinduction consolidation treatment vs chemotherapy before allogeneic hematopoietic stem cell transplant (alloHSCT), resulting in superior in eradicating MRD (28, 29). Therefore, blinatumomab gained accelerated approval by FDA to treat BCP ALL with MRD greater than or equal to 0.1% after the first or second CR. Moreover, a phase II study (NCT02807883) has proved the feasibility of blinatumomab maintenance following alloHSCT for patients with B-cell ALL at high-risk for relapse, with the 1-year overall survival (OS), progression-free survival (PFS), and nonrelapse mortality (NRM) rates of 85%, 71%, and 0%, respectively (30). In addition, to incorporate blinatumomab as part of upfront treatment for pediatric B-cell ALL, several trials are currently ongoing (NCT03643276 and NCT03914625) (31). Of note, the main toxicities including cytokine release syndrome (CRS) and neurotoxicity are tolerable under blinatumomab therapy (54, 55).
Although blinatumomab has obtained an authorization for treating pediatric R/R BCP ALL and eliminating the MRD, several disadvantages, such as the short half-life caused by the low molecular weight, leading to the need to continuous intravenous infusion, limited wide clinical application (56). Besides, blinatumomab resistance caused by the loss of CD19 expression and lineage switch in BCP ALL remains a significant problem (57, 58). For example, in a phase I/II study, four patients harboring CD19 negative B-cell ALL relapsed after prior blinatumomab-induced hematologic remission and one patient with CD19-negative had disease progression (57). In the future, screening tumor markers to predict CD19-negative relapse should be paid more attention. What’s more, overexpression of checkpoint molecules including T-cell immunoglobulin and mucin domain 3 (TIM-3) on T cells and PD-L1 on tumor cells represented an additional potential escape mechanism from immunosurveillance (30, 59, 60). Adding immune checkpoint inhibitors to blinatumomab treatment thus overcoming resistance may be feasible (60) and are under clinical investigation (NCT03160079, NCT03512405 and NCT04546399).
Bispecific CD3/CD20 T-Cell Engagers
In addition to CD19, other antigens on the surface of leukemic blasts are currently under active research and development. CD20 is a signature B cell differentiation antigen and its overexpression is identified as an inferior prognosis marker associated with a worse event-free survival (EFS) in childhood BCP ALL according to the Pediatric Oncology Group treatment protocols (61). Also, CD20 expression was significantly up-regulated in pediatric B-cell leukemias during induction treatment (62), rendering it an appealing target for immunotherapy. Currently, several promising bispecific antibodies of anti-CD3/CD20, such as glofitamab, mosunetuzumab, REGN1979, and epcoritamab, may be of interest for future studies in B-cell Non-Hodgkin Lymphoma (NHL) (63, 64). However, further investigation is warranted to evaluate this strategy in pediatrics with B-cell ALL.
Trispecific CD3/CD19/CD20 T-Cell Engagers
A-2019, a novel trispecific CD3/CD19/CD20 T-cell engagers possessing the anti-CD19 and anti-CD20 scFvs, was designed by Wang lab (65). It mediated autologous B cell depletion ex vivo by inducing the activation and proliferation of T cells, and the production and release of cytokines (65). Furthermore, A-2019 bound to CD19 with a lower affinity compared with blinatumomab (1.06 × 10-8 mol/L vs. 1.49 × 10-9 mol/L, respectively), which potentially reduce the off-target effect on mural cells, thus lowering the risk of neurotoxicity (66). Plus, a general decrease in overall cytokine including IL-6 and IFN-γ induced by A-2019 was observed in preclinical study, reducing the risk of CRS. In short, trispecific T-cell engagers targeting CD3/CD19/CD20 represents a novel strategy for not only preventing and treating CD19 negative relapse, but also has the potential for the treatment of CD20 positive and/or CD19 positive B-cell ALL. In a more recent preclinical study, CMG1A46, another CD3/CD19/CD20 T-cell engager generated from Chimagen’s TRIAD platform, displayed superior potency and safety in comparation with CD3/CD20 bispecific T-cell engagers (67). The phase I/II clinical trial is undergoing to evaluate the safety and efficacy of CMG1A46 in adult patients with advanced CD19 and/or CD20 positive B-cell NHL or ALL (NCT05348889).
CAR T Cell Therapy
CAR T cells that commonly consists of an antigen-binding domain and costimulatory signaling domain such as CD28 and/or 4-1BB (68, 69), is a revolutionary and promising immunotherapy approach in cancer treatment (70). Antigen markers on B-cell ALL cells surface such as CD19 and CD22 can be specifically recognized by CAR that is independent from the major histocompatibility complex receptor, thus activating T cells to kill tumor cells.
CD19 Targeting CAR T Cell Therapy
Tisagenlecleucel, also named CTL019, is autologous T cells engineered ex vivo with a CAR containing a 4-1BB domain (71). In 2012, two children diagnosed with R/R B-cell ALL were infused with tisagenlecleucel and both achieved CR, although one patient had a CD19 negative relapse (72, 73). These encouraging data brought a hope of this therapy for the treatment of R/R B-cell ALL. Subsequently, a trial was expanded to 30 patients with the age of 5 to 22 years with R/R B-cell ALL in a phase I/IIa study (NCT01626495), which demonstrated a 90% rate of CR at the first month, an EFS rate of 67% and overall survival (OS) rate of 78% at 6 months after the single infusion of tisagenlecleucel (32). An international phase II study using tisagenlecleucel in pediatric and young adult patients with R/R B-cell ALL showed a CR rate of 81% at 3 months, EFS rate of 73%, and OS rate of 90% at 6 months (NCT02435849) (33). Given the unprecedented successes in clinical trials, tisagenlecleucel was commercially approved by the FDA and was indicated for R/R B-cell ALL patients up to 25 years old in 2017.
Brexucabtagene autoleucel, also named KTE-X19, is another CD19CAR T cell therapy with a CD28 costimulatory subunit generated from peripheral blood monocular cells by removing CD19 positive malignant cells to avoid T cell exhaustion, and has received FDA approval for mantle cell lymphoma (74). Based on the data from the pivotal phase I/II clinical trial (ZUMA-3, NCT02614066), brexucabtagene autoleucel was successfully manufactured and administered as a single infusion in 55 adult patients with R/R BCP ALL (34–36). Ultimately, 31 (56%) patients achieved CR and 8 (15%) patients achieved CR with incomplete haematological recovery, 38 (97%) of whom had MRD negativity. Due to the striking efficacy of brexucabtagene autoleucel in R/R BCP ALL adult patients, study to evaluate brexucabtagene autoleucel in pediatric and young adult patients with BCP ALL is ongoing (ZUMA-4, NCT02625480) and the results will be anticipated. Furthermore, several other CD19CAR T cell therapies are undergoing clinical research (Table 1) (75–77).
Although CD19CAR T cell therapy has shown great success for pediatric B-cell ALL, some patients displayed no response and a great part of patients suffered from relapse with poor outcomes (NCT01593696) (37, 38). Unfortunately, even a secondary infusion of CD19CAR T cells can’t prevent CD 19-positive relapse, which may be partly due to immune-mediated clearance of CAR T cells (78, 79). Antigen loss caused by mutations or alternate splice variants of CD19 have been elucidated as the major resistance mechanism to CD19CAR T cell immunotherapy, which is widely acknowledged as an urgent problem to be solved (80, 81). Therefore, new therapeutic approaches are required for R/R B-cell ALL patients who have failed previous CD19CAR T cell therapy.
CD22 Targeting CAR T Cell Therapy
CD22 represents an alternative target with a high expression level on most B-cell ALL cells, while has restricted expression on normal B cells, especially in the absence of CD19 expression (82–84). Recently, a phase I dose escalation study of CD22CAR T cells with a 4-1BB costimulatory domain in pediatric and young adults with recurrent or refractory CD22 positive B cell malignancies was conducted sponsored by National Cancer Institute (NCT02315612). The results showed that 70.2% of the patients achieved CR and 87.5% of whom were MRD negative (39, 85). However, an CD22CAR possessing the similar structure with the above mentioned CD22CAR but the heavy and light chains were connected by a standard 20-aminoacid linker instead of a short 5-amino acid sequence, proved surprisingly poor response in pediatric and adult patients with B-cell ALL (NCT02588456) (40). By performing detailed interrogation responsible for the entirely different findings from the two independent clinical trials, mechanisms that short scFv linker and tonic signaling enhanced the antileukemic function of 4-1BB-based CAR T cells induced the phenomena (40). Based on this work, the pilot study (NCT02650414) was amended to determine the feasibility and safety of a single dose administered CD22CAR T cells expressing 4-1BB costimulatory domains in pediatric R/R B-cell ALL patients (40). Moreover, Pan and his colleges constructed an CD22CAR with a 4-1BB costimulatory domain and initiated a clinical trial, which demonstrated a CR rate of 80% on day 30 after infusion in 34 R/R B-cell ALL pediatric and adult patients who have mostly failed from first CD19CAR T cell therapy (ChiCTR-OIC-17013523) (41). However, most patients relapsed with a diminished CD22 site density on B-cell ALL cells (85), which raised the question that single target therapy permit the occurrence of resistant variants. Combinatory or tandem CARs, which contain two or more antigen-recognition moieties, may prevent from relapse due to escape variants but need further validation (86, 87).
Dual CD19 and CD22 Targeting CAR T Cell Therapy
The downregulation or loss of pre-designed antigen on ALL cells leads to the failure of CD19CAR T or CD22CAR T cell monotherapy. Therefore, a dual CD19 and CD22 antigen targeting CAR T cell therapy such as CD19/CD22 bispecific CAR T cell therapy and sequential CD19CAR T and CD19CAR T cell therapy, appears to be a strategy to prevent the escape mechanism given that B cells are unlikely to downregulate both CD19 and CD22 simultaneously in a single cell.
AUTO3 is a representative CD19/CD22 bispecific CAR T cell therapy, which is designed and developed through transduction of autologous T cells expressing two CARs targeting CD19 and CD22 by Autolus Therapeutics. A multi-center, phase I/II study has been completed to explore the safety and efficacy in pediatric and young adult patients with B-cell ALL (AMELIA trial, NCT03289455) (42). In the phase I dose-escalation study, dose-limiting toxicities, severe CRS, and neurotoxicity were not reported, which demonstrated a favorable safety profile for clinical application. 13 of 15 patients achieved CR or CR with incomplete bone marrow recovery after AUTO3 infusion for one month. The OS and EFS rates were 60% and 32%, respectively. Consequently, FDA has granted orphan drug designation of AUTO3 for ALL treatment. Nevertheless, unavailability of long-term persistence of AUTO3 in patients resulted in disease relapse. Hence, prolongation of CAR T cell persistence are needed to fully fulfill the therapeutic potential of dual targeting CAR T cell in B-cell ALL. Another phase I trial (NCT03448393) sponsored by National Cancer Institute to evaluate the safety and efficiency of dual CD19/CD22 targeting CAR T cell therapy is undergoing.
CTA101, also called CRISPR-edited allogeneic off-the-shelf CD19/CD22 bispecific CAR T cells, was composed of scFV targeting CD19 and CD22, 4-1BB costimulatory domain, and CRISPR/Cas9-disrupted TRAC region to avoid host immune-mediated rejection (88). The phase I clinical trial (NCT04227015) to evaluate the safety and efficiency of a single dose of CTA101 in R/R B-cell ALL patients aged from 3 to 70 years is ongoing.
In addition to CD19/CD22 bispecific CAR T cells, sequential infusion of CD19CAR T and CD22CAR T cells are investigated in a phase I trial in pediatric patients with R/R B-cell ALL (ChiCTR-OIB-17013670). 17 of the 20 patients remained CR at the study end point and only 2 patients relapsed caused by loss of CD19, indicating that the risk of relapse associated with antigen escape was greatly reduced (43).
Strategies to Reduce Toxicities of CAR T Cell Therapy
Despite the huge clinical success, CAR T cell therapy-related severe toxicities such as CRS, cannot be neglected and are remained to be resolved (33, 89, 90). For example, around 50% of children and young adult patients treated with tisagenlecleucel for R/R B-cell ALL had ≥ Grade 3 CRS (33, 91), 24% of adult patients infused with brexucabtagene autoleucel had ≥ Grade 3 CRS (36), while lower incidence of ≥ Grade 3 CRS, 8.6% and 2.9% respectively, was observed after CD22-CART cell treatment (39, 41). Moreover, no ≥ Grade 3 CRS was observed in pediatric patients with R/R B-cell ALL receiving AUTO3, which is in concordance with the modest elevation of cytokines production (42). Finally, extensive studies to ameliorate CAR T cell related toxicities are ongoing, such as altering CAR structure, modifying CAR transduced T cells, and inserting CAR “off-switches” (70, 92, 93). Optimizing CAR binding affinity by developing a new CD19 scFV with a lower affinity than FMC63 could be a useful approach to enhance CAR T cell expansion and persistence, and alleviate toxicity in the pediatric R/R B-cell ALL as illustrated in the CARPALL clinical trial (NCT02443831) (44).
Strategies to Increase the Efficacy of CAR T Cell Therapy
Besides reducing toxicities of CAR T cell therapy, attempts have been made to increase the efficacy of CAR T cell therapy. As mentioned above, targeting two or more antigens by dual CD19 and CD22 targeting CAR T cells or sequential infusion of CD19CAR T and CD22CAR T cells broadened the spectrum of targets, decreased the risk of antigen negative relapse and enhanced the potential therapeutic efficiency. Moreover, tisagenlecleucel and other CAR T cell therapies for pediatric B-cell ALL were commonly composed of the costimulatory domains with CD28 and/or 4-1BB (Table 1), which were the second and third generation CARs (94). CAR T cells were further optimized to secrete cytokines or express cytokine receptors to generate the fourth and fifth generation CARs. In a relapsed patient with B-cell ALL after treatment with CD19CAR and CD22CAR T cell therapy, autologous murine CD19CAR T cells expressing membrane-bound IL-15 achieved CR for five months (95). CD19CAR T cells encoded with interleukin 2 receptor β-chain and a STAT3-binding tyrosine-X-X-glutamine motif in the cytoplasmic domain showed grater antitumor effects and superior duration than CAR T cells without this structure (96). Besides, upregulation of TIM-3 increased the risk of relapse in pediatric B-cell ALL (97). Hence, TIM-3-CD28 fusion proteins were combined with the first and second CD19CAR T cells to enhance the proliferation capacity of T cells and improve the functionality of conventional CAR T cells by turning inhibition into activation of T cells (98, 99). Relapse due to the short persistence of CAR T cells and resistance to same murine CAR T cell therapy were attributed to immunogenicity caused by murine scFv (37, 38). 68% of the pediatric and adult patients after failure of murine CD19CAR T cell therapy achieved CR treated with 4-1BB based humanized CD19 CAR-T cells (ChiCT R1900 024456) (100). In brief, various strategies have been developed to increase the efficacy of CAR T cell therapy and warranted to be applied in the treatment of pediatric B-cell ALL.
Combination Therapy
Numerous studies have demonstrated that immune responses by maintaining negative regulatory pathways via PD-1/PD-L1 axis played a significant role in the immune escape, thus leading to growth and spread of malignant tumors (101, 102). In this regard, blocking PD-1/PD-L1 pathway has aroused great success as an effective therapeutic approach in a variety of cancers. Significantly, there is a high expression level of PD-1 on the surface of T cells in B-cell ALL pediatric patients, which is associated with an inferior prognosis (103). Inspired by the encouraging therapeutic outcomes of blocking the PD-1/PD-L1 pathway in the treatment of solid tumors, researchers nowadays have tried to integrate PD-1/PD-L1 inhibitors into the treatment course of hematological malignancy (104, 105). Pembrolizumab is a humanized anti-PD-1 monoclonal antibody with a wide range of indications (106). However, the phase II study of single-agent pembrolizumab in treating minimal residual disease (MRD) in adults with ALL was terminated due to lack of efficacy despite good tolerability (NCT02767934) (107). Immunosuppression due to chemotherapy prior treatment and relatively low mutational rate may lead to the treatment failure of targeting PD-1/PD-L1 pathway (107). That said, monotherapy by blocking PD-1/PD-L1 signaling pathway seems not a viable therapeutic method for the treatment of ALL. Nevertheless, combinatory treatment of immune checkpoint inhibitors including pembrolizumab and nivolumab with blinatumomab for B-cell ALL in adults and pediatrics are undergoing clinical trials (NCT03160079 and NCT03512405 and NCT04546399), which may prove the potential advantage of anti PD-1 antibody as an adjunct or rescue strategy. Besides immune checkpoint inhibitors, blinatumomab in combination of with chemotherapy in patients with newly diagnosed B-lymphoblastic leukemia aged 365 days to 31 years are being investigated (NCT03914625).
Other Emerging T-Cell Based Immunotherapies
Donor-derived tumor associated antigen-specific T cells termed TAA-T (108), reported by Kinoshita and colleges, is capable of targeting three overexpressed and immunogenic tumor associated antigens WT1, PRAME and survivn in most hematologic malignancies (109–111). A phase I clinical is currently ongoing at Children’s National and Johns Hopkins Hospitals and Johns Hopkins University to evaluate the safety of TAA-T for the treatment of very high-risk hematopoietic malignancies (NCT02203903). Preliminary results showed that none of the included acute myeloid leukemia (AML) and ALL patients developed CRS or neurotoxicity, and only one patient developed grade 3 graft-versus-host disease (GVHD), which demonstrated the good safety of TAA-T (108). Moreover, persist remissions were observed in high-risk and relapsed patients (108). Later phase of the study is required to determine long-term clinical disease outcomes.
Adoptive cell therapy using T-cell receptor (TCR)-engineered T cells represents another novel and potential strategy for cancer treatment (112–114). ET190L1-AbTCR is one of TCR-T cell therapies generated by replacing the α and β chains of the TCR in the antigen recognition domain with an anti-CD19 antibody-derived Fab fragment (115). Compared with the clinically commonly used CD19CAR T cells with a CD28 or 4-1BB costimulatory subunit, ET190L1-AbTCR activated cytotoxic T-cell responses, but showed less cytokine release in xenograft mouse models of primary B-cell ALL.
B-cell activating factor receptor (BAFF-R) is a B-lineage marker expressed almost exclusively on B cells (116, 117), making it an ideal target for immunotherapy. BAFF-R-CAR T cells with a 4-1BB costimulatory signaling domain demonstrated therapeutic effects against CD19 negative B-cell ALL in vitro and in vivo (117), and are currently undergoing clinical trials for the treatment of adult ALL (NCT04690595). Moreover, dual CD19/BAFF-R CAR T cells were developed and exhibited anti-ALL activity in vivo, supporting clinical translation of BAFF-R/CD19 dual CAR T cells to treat ALL (118).
Macrophage-Based Immunotherapy
Targeting CD47/SIRPα Pathway
Macrophages are important components of mononuclear phagocytic system, and generates a “don’t eat me” signal to suppresses phagocytosis by expressing a signal regulatory protein alpha (SIRPα) that interacts with CD47 (CD47/SIRPα axis), thus contributing to the development and progression of most cancers (119–121). Therefore, the CD47/SIRPα axis has been identified as an essential and promising immune checkpoint in the homeostatic clearance by macrophages. Generally, CD47 is overexpressed on the surface of B-cell ALL cells and recognized as an inferior prognosis marker associated with worse outcomes in pediatric ALL patients, such as treatment failure and even death (122, 123). Additionally, the anti-CD47 antibody and anti-SIRPα antibody both increased phagocytosis of ALL cells in vitro, suggesting that blockade of the CD47/SIRPα signaling enhanced phagocytosis (123). In vivo, anti-CD47 antibody inhibited tumor engraftment and induced remission in ALL engrafted mice (123). These data provided pre-clinical evidence for disrupting CD47/SIRPα signaling as a potential therapy for ALL.
TTI-621 is a novel soluble fusion protein composed of human SIRPα and IgG1, and exerts its effect by blocking CD47/SIRPα pathway (124) (Figure 1). Recently, a phase Ia/Ib dose escalation and expansion trial of TTI-621 in R/R hematologic malignancies and selected solid tumors is ongoing (NCT02663518). TTI-621 was well-tolerated in the dose escalation phase. The expansion phase conducted in R/R NHL patients has been completed and demonstrated a sound efficacy. The evaluation of clinical efficacy and safety of TTI-621 in ALL patients is still ongoing.
Additionally, several strategies have been proposed and are being intensively explored, for example, anti-CD47 antibody (124–128), anti-SIRPα antibody (129), bi-specific antibodies to CD47 or SIRPα or other molecules (129), SIRPα-related fusion proteins (130), and others (131), to improve therapeutic efficacy during targeting CD47/SIRPα pathway while overcome on-target/off-tumor effects (132, 133). A large number of clinical trials related to cutting off the CD47/SIRPα pathway are currently undergoing at various stages, which are centered on solid tumors and hematological malignancies such as NHL and AML (132). The research on blocking the CD47/SIRPα pathway in pediatric ALL is lagging behind and is undergoing preclinical study.
Notably, targeting CD47 induced hematotoxicity including anemia and thrombocytopenia due to the off-target effect on the blocking of CD47 expressed on surface of platelets and erythrocytes (134). Designing and developing SIRPα fusion proteins could diminish the hematotoxicity, thereby safeguarding the clinical safety. For example, TTI-621 exhibited minimal impact on human erythrocytes, resulting in a lower incidence of anemia than that reported in a phase I study of combinatory treatment of humanized anti-CD47 antibody and rituximab (13% vs 41%) (124, 135). Moreover, thrombocytopenia associated with TTI-621 was transient and reversible, and no bleeding events were observed in the clinical trials (136).
NK Cell-Based Immunotherapy
NKs derived from lymphocyte cell lineage were discovered to be critical to the innate immunity (137). NKs are composed of two subtypes, i.e., CD3-/CD56dim/CD16+ NKs and CD3-/CD56bright/CD16- NKs (138). CD3-/CD56dim/CD16+ NKs display strong cytotoxic activity on targeted cells by perforin and granzyme B in peripheral blood, and CD3-/CD56bright/CD16- NKs produce and release cytokines such as IFN-γ and TNF-α in response cytokines stimulation in lymphoid tissues (139, 140). Mounting studies have demonstrated the innate lymphoid cells as the first line of defense by exerting cytotoxicity against diverse tumor cell types (141, 142), especially in the field of hematologic malignancies (143). The presence of NK cells in bone marrow conferred a better response to chemotherapy and prognosis, and has a high chance of efficacy in pediatric patients with ALL (144, 145). Hence, different artificial engagers have been equipped to selectively redirect NK cells towards tumor cells. In vitro, NKp46/CD16A/CD19-NKCE, a NK cell engager with CD16A on the surface of NKs binding NKp46 and CD19 highly expressed on the surface of B-cell ALL cells, enhanced the activation of NKs and promoted NK cell-mediated lytic effects against pediatric BCP ALL (Figure 1) (146).
Moreover, immunotherapy based on NKs displayed several advantages (147, 148). As the important immune system components, NKs exerted the cytotoxicity upon recognizing specific patterns without prior antigen sensitization. CAR modified NK cells (CAR NKs) are qualified for simultaneously improving efficacy and controlling adverse effects including CRS, neurotoxicity, and GVHD, which offer an alternative option to CAR-T cells (149, 150). To date, two phase I studies to determine the maximum tolerated dose of genetically modified haploidentical NKs infusions targeting CD19 to treat B-cell ALL have been completed in St. Jude Children’s Research Hospital (NCT00995137 and NCT01974479). However, the results have not been disclosed.
Conclusion and Future Perspectives
Immunotherapy represents a novel and promising therapeutic weapon against pediatric B-cell ALL. As alluded to above, T cell engagers and CAR T cell therapies were successful in treating pediatric R/R B-cell ALL. Currently, blinatumomab has been approved for treating R/R BCP ALL in pediatrics and is further investigated in removing MRD as post-reinduction consolidation treatment, which is believed to be more efficacious and less toxic than chemotherapy. In addition, the approval of tisagenlecleucel, a typical representative of CAR T cell therapies, provided us an alteration when none was available for children and young adults with R/R B-cell ALL, although a large proportion of patients suffered relapse with poor outcomes. However, monotherapy by immune checkpoint inhibitors targeting PD-1/PD-L1 pathway achieved poor clinical efficiency. Immune check inhibitors including anti-PD-1 and anti-CD47 antibodies as an adjunct strategy in pediatric B-cell ALL required to be verified in the further study and may be worth awaiting. Moreover, strategies targeting CD47/SIRPα pathway and CAR-NKs demonstrated potential for treating pediatric ALL and were evaluated in clinical trials.
Since native macrophages are critical effectors and regulators of the innate immune system, CAR macrophages can directly target the desired cancer cells and has been developed to phagocytose human solid tumor cells (151, 152). Nevertheless, treatment of pediatric ALL by using this promising immunotherapy is absent (153). Neoantigen, a new mutation-derived protein in tumor cells, is a non-normal cellular product and has proven to be a breakthrough for therapeutic immune targets (154–157). Immunotherapy by harnessing neoepitope-CD8+ T cells to recognize and respond to the neoantigens in pediatric patients with ALL provided us an alternative treatment option (158).
Despite all these advances, the resistance and toxicity should be taken into consideration and further studies should focus on the development of new agents guided to ameliorate some of the toxicities and prevent the recurrence. Variable mechanisms including the downregulation/loss of antigens and antigen escape through lineage switch influence responses to immunotherapy result in resistance or relapse. The relationship between genetic and cytogenetic alterations and immunotherapy responses are warranted to be further explored in the future. Moreover, novel potential targets such as cell surface antigens, kinases and signaling pathways should be consistently identified and explored. On the other hand, blocking a single cell surface antigen or pathway of B-cell ALL may lead to drug resistance. It is anticipated that immunotherapy targeting multiple antigens such as trispecific CD3/CD19/CD20 T-cell engagers and dual CD19 and CD22 targeting CAR T cell therapy will not only overcome the challenge of antigen modulation but also dramatically enhance therapeutic efficacy.
To sum up, the successful outcome of immunotherapy from clinical trials as described in our context has proved the effectiveness of immunotherapy in the treatment of pediatric B-cell ALL and lead to dramatic improvements in outcome for R/R subtype. We believe that the integration and expansion of these therapeutics into frontline therapy and the discovery of novel anti-multiple antigens modifications will further augment efficacy and reduce toxicity, thus improving long-term outcomes in pediatric B-cell ALL patients.
Author Contributions
ML and YL designed the study, drafted the initial manuscript, critically reviewed and revised the manuscript, and contributed equally to this work. WL provided us professional knowledge in the field of pediatric ALL. YX and SZ reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Dr. Wenhui Liu for the professional guidance during writing the manuscript. The authors would also like to thank Dr. Zutao Yu, a researcher from Cancer Research UK Cambridge Institute (University of Cambridge), for the manuscript proof-reading.
References
1. Liu YF, Wang BY, Zhang WN, Huang JY, Li BS, Zhang M, et al. Genomic Profiling of Adult and Pediatric B-Cell Acute Lymphoblastic Leukemia. EBioMedicine (2016) 8:173–83. doi: 10.1016/j.ebiom.2016.04.038
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin (2021) 71:7–33. doi: 10.3322/caac.21654
3. Malard F, Mohty M. Acute Lymphoblastic Leukaemia. Lancet (2020) 395:1146–62. doi: 10.1016/s0140-6736(19)33018-1
4. Sung H, Ferlay J, Siegel RL. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
5. Boyiadzis MM, Aksentijevich I, Arber DA, Barrett J, Brentjens RJ, Brufsky J, et al. The Society for Immunotherapy of Cancer (SITC) Clinical Practice Guideline on Immunotherapy for the Treatment of Acute Leukemia. J Immunother Cancer (2020) 8:e000810. doi: 10.1136/jitc-2020-000810
6. Brown P, Inaba H, Annesley C, Beck J, Colace S, Dallas M, et al. Pediatric Acute Lymphoblastic Leukemia, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Netw (2020) 18:81–112. doi: 10.6004/jnccn.2020.0001
7. Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J Clin Oncol (2015) 33:2938–48. doi: 10.1200/jco.2014.59.1636
8. Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, et al. Improved Survival for Children and Adolescents With Acute Lymphoblastic Leukemia Between 1990 and 2005: A Report From the Children's Oncology Group. J Clin Oncol (2012) 30:1663–9. doi: 10.1200/jco.2011.37.8018
9. Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med (2015) 373:1541–52. doi: 10.1056/NEJMra1400972
10. Hunger SP, Raetz EA. How I Treat Relapsed Acute Lymphoblastic Leukemia in the Pediatric Population. Blood (2020) 136:1803–12. doi: 10.1182/blood.2019004043
11. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res (2019) 79:4557–66. doi: 10.1158/0008-5472.can-18-3962
12. Anderson NM, Simon MC. The Tumor Microenvironment. Curr Biol (2020) 30:R921–r925. doi: 10.1016/j.cub.2020.06.081
13. Jin MZ, Jin WL. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct Target Ther (2020) 5:166. doi: 10.1038/s41392-020-00280-x
14. Farkona S, Diamandis EP, Blasutig IM. Cancer Immunotherapy: The Beginning of the End of Cancer? BMC Med (2016) 14:73. doi: 10.1186/s12916-016-0623-5
15. Im A, Pavletic SZ. Immunotherapy in Hematologic Malignancies: Past, Present, and Future. J Hematol Oncol (2017) 10:94. doi: 10.1186/s13045-017-0453-8
16. Noh JY, Seo H, Lee J, Jung H. Immunotherapy in Hematologic Malignancies: Emerging Therapies and Novel Approaches. Int J Mol Sci (2020) 21:8000. doi: 10.3390/ijms21218000
17. Craddock C, Friedberg JW. Immunotherapy for Hematologic Malignancies. J Clin Oncol (2021) 39:343–5. doi: 10.1200/jco.20.03106
18. Wang H, Kaur G, Sankin AI, Chen F, Guan F, Zang X. Immune Checkpoint Blockade and CAR-T Cell Therapy in Hematologic Malignancies. J Hematol Oncol (2019) 12:59. doi: 10.1186/s13045-019-0746-1
19. Jeha S, Pei D, Choi J, Cheng C, Sandlund JT, Coustan-Smith E, et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J Clin Oncol (2019) 37:3377–91. doi: 10.1200/JCO.19.01692
20. Teachey DT, Pui CH. Comparative Features and Outcomes Between Paediatric T-Cell and B-Cell Acute Lymphoblastic Leukaemia. Lancet Oncol (2019) 20:e142–54. doi: 10.1016/s1470-2045(19)30031-2
21. Inaba H, Pui CH. Immunotherapy in Pediatric Acute Lymphoblastic Leukemia. Cancer Metastasis Rev (2019) 38:595–610. doi: 10.1007/s10555-019-09834-0
22. Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-Cancer Genome and Transcriptome Analyses of 1,699 Paediatric Leukaemias and Solid Tumours. Nature (2018) 555:371–6. doi: 10.1038/nature25795
23. Laetsch TW, DuBois SG, Bender JG, Macy ME. Opportunities and Challenges in Drug Development for Pediatric Cancers. Cancer Discov (2021) 11:545–59. doi: 10.1158/2159-8290.cd-20-0779
24. von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J Clin Oncol (2016) 34:4381–9. doi: 10.1200/JCO.2016.67.3301
25. Gore L, Locatelli F, Zugmaier G, Handgretinger R, O'Brien MM, Bader P, et al. Survival After Blinatumomab Treatment in Pediatric Patients With Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Blood Cancer J (2018) 8:80. doi: 10.1038/s41408-018-0117-0
26. Locatelli F, Zugmaier G, Mergen N, Bader P, Jeha S, Schlegel PG, et al. Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia: Results of the RIALTO Trial, an Expanded Access Study. Blood Cancer J (2020) 10:77. doi: 10.1038/s41408-020-00342-x
27. Locatelli F, Zugmaier G, Mergen N, Bader P, Jeha S, Schlegel PG, et al. Blinatumomab In Pediatric Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia: RIALTO Expanded Access Study Final Analysis. Blood Adv (2022) 6:1004–14. doi: 10.1182/bloodadvances.2021005579
28. Locatelli F, Zugmaier G, Rizzari C, Morris JD, Gruhn B, Klingebiel T, et al. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children With High-Risk First-Relapse B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA (2021) 325:843–54. doi: 10.1001/jama.2021.0987
29. Brown PA, Ji L, Xu X, Devidas M, Hogan LE, Borowitz MJ, et al. Effect of Postreinduction Therapy Consolidation With Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults With First Relapse of B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA (2021) 325:833–42. doi: 10.1001/jama.2021.0669
30. Gaballa MR, Banerjee P, Milton DR, Jiang X. Blinatumomab Maintenance After Allogeneic Hematopoietic Cell Transplantation for B-Lineage Acute Lymphoblastic Leukemia. Blood (2022) 139:1908–19. doi: 10.1182/blood.2021013290
31. Tüfekçi Ö, Evim MS, Güneş AM, Celkan T, Karapinar DY, Kaya Z, et al. Assessment of Minimal Residual Disease in Childhood Acute Lymphoblastic Leukemia: A Multicenter Study From Turkey. J Pediatr Hematol Oncol (2022) 44:e396–402. doi: 10.1097/mph.0000000000002419
32. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
33. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
34. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for Relapsed or Refractory Adult B-Cell Acute Lymphoblastic Leukaemia: Phase 2 Results of the Single-Arm, Open-Label, Multicentre ZUMA-3 Study. Lancet (2021) 398:491–502. doi: 10.1016/s0140-6736(21)01222-8
35. Shah BD, Bishop MR, Oluwole OO. KTE-X19 Anti-CD19 CAR T-Cell Therapy in Adult Relapsed/Refractory Acute Lymphoblastic Leukemia: ZUMA-3 Phase 1 Results. Blood (2021) 138:11–22. doi: 10.1182/blood.2020009098
36. Killock D. KTE-X19 Efficacious in Adults With B-ALL. Nat Rev Clin Oncol (2021) 18:470. doi: 10.1038/s41571-021-00537-6
37. Shah NN, Lee DW. Long-Term Follow-Up of CD19-CAR T-Cell Therapy in Children and Young Adults With B-ALL. J Clin Oncol (2021) 39:1650–9. doi: 10.1200/jco.20.02262
38. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia in Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet (2015) 385:517–28. doi: 10.1016/s0140-6736(14)61403-3
39. Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J Clin Oncol (2020) 38:1938–50. doi: 10.1200/jco.19.03279
40. Singh N, Frey NV, Engels B, Barrett DM, Shestova O, Ravikumar P, et al. Antigen-Independent Activation Enhances the Efficacy of 4-1BB-Costimulated CD22 CAR T Cells. Nat Med (2021) 27:842–50. doi: 10.1038/s41591-021-01326-5
41. Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. CD22 CAR T-Cell Therapy in Refractory or Relapsed B Acute Lymphoblastic Leukemia. Leukemia (2019) 33:2854–66. doi: 10.1038/s41375-019-0488-7
42. Cordoba S, Onuoha S, Thomas S, Pignataro DS, Hough R, Ghorashian S, et al. CAR T Cells With Dual Targeting of CD19 and CD22 in Pediatric and Young Adult Patients With Relapsed or Refractory B Cell Acute Lymphoblastic Leukemia: A Phase 1 Trial. Nat Med (2021) 27:1797–805. doi: 10.1038/s41591-021-01497-1
43. Pan J, Zuo S, Deng B, Xu X, Li C, Zheng Q, et al. Sequential CD19-22 CAR T Therapy Induces Sustained Remission in Children With R/R B-ALL. Blood (2020) 135:387–91. doi: 10.1182/blood.2019003293
44. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T Cell Expansion and Prolonged Persistence in Pediatric Patients With ALL Treated With a Low-Affinity CD19 CAR. Nat Med (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
45. Raskov H, Orhan A, Christensen JP, Gögenur I. Cytotoxic CD8(+) T Cells in Cancer and Cancer Immunotherapy. Br J Cancer (2021) 124:359–67. doi: 10.1038/s41416-020-01048-4
46. O'Donnell JS, Teng MWL, Smyth MJ. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat Rev Clin Oncol (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
47. Waldman AD, Fritz JM, Lenardo MJ. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
48. Labrijn AF, Janmaat ML. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat Rev Drug Discov (2019) 18:585–608. doi: 10.1038/s41573-019-0028-1
49. Rader C. Bispecific Antibodies in Cancer Immunotherapy. Curr Opin Biotechnol (2020) 65:9–16. doi: 10.1016/j.copbio.2019.11.020
50. May MB, Glode A. Blinatumomab: A Novel, Bispecific, T-Cell Engaging Antibody. Am J Health Syst Pharm (2016) 73:e6–e13. doi: 10.2146/ajhp150134
51. Loüffler A, Kufer P, Lutterbüse R, Zettl F, Daniel PT, Schwenkenbecher JM, et al. A Recombinant Bispecific Single-Chain Antibody, CD19 × CD3, Induces Rapid and High Lymphoma-Directed Cytotoxicity by Unstimulated T Lymphocytes. Blood (2000) 95:2098–103. doi: 10.1182/blood.V95.6.2098
52. Orentas RJ, Yang JJ, Wen X, Wei JS, Mackall CL, Khan J. Identification of Cell Surface Proteins as Potential Immunotherapy Targets in 12 Pediatric Cancers. Front Oncol (2012) 2:194. doi: 10.3389/fonc.2012.00194
53. Wang K, Wei G, Liu D. CD19: A Biomarker for B Cell Development, Lymphoma Diagnosis and Therapy. Exp Hematol Oncol (2012) 1:36. doi: 10.1186/2162-3619-1-36
54. Beneduce G, De Matteo A. Blinatumomab in Children and Adolescents With Relapsed/Refractory B Cell Precursor Acute Lymphoblastic Leukemia: A Real-Life Multicenter Retrospective Study in Seven AIEOP (Associazione Italiana Di Ematologia E Oncologia Pediatrica) Centers. Cancers (Basel) (2022) 14:426. doi: 10.3390/cancers14020426
55. Queudeville M, Ebinger M. Blinatumomab in Pediatric Acute Lymphoblastic Leukemia-From Salvage to First Line Therapy (A Systematic Review). J Clin Med (2021) 10:2544. doi: 10.3390/jcm10122544
56. Zhu M, Wu B, Brandl C, Johnson J, Wolf A, Chow A, et al. Blinatumomab, a Bispecific T-Cell Engager (BiTE((R))) for CD-19 Targeted Cancer Immunotherapy: Clinical Pharmacology and Its Implications. Clin Pharmacokinet (2016) 55:1271–88. doi: 10.1007/s40262-016-0405-4
57. Mejstrikova E, Hrusak O, Borowitz MJ, Whitlock JA, Brethon B, Trippett TM, et al. CD19-Negative Relapse of Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia Following Blinatumomab Treatment. Blood Cancer J (2017) 7:659. doi: 10.1038/s41408-017-0023-x
58. Mikhailova E, Gluhanyuk E, Illarionova O, Zerkalenkova E, Kashpor S, Miakova N, et al. Immunophenotypic Changes of Leukemic Blasts in Children With Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia, Who Have Been Treated With Blinatumomab. Haematologica (2021) 106:2009–12. doi: 10.3324/haematol.2019.241596
59. Köhnke T, Krupka C, Tischer J, Knösel T, Subklewe M. Increase of PD-L1 Expressing B-Precursor ALL Cells in a Patient Resistant to the CD19/CD3-Bispecific T Cell Engager Antibody Blinatumomab. J Hematol Oncol (2015) 8:111. doi: 10.1186/s13045-015-0213-6
60. Feucht J, Kayser S, Gorodezki D, Hamieh M, Döring M, Blaeschke F, et al. T-Cell Responses Against CD19+ Pediatric Acute Lymphoblastic Leukemia Mediated by Bispecific T-Cell Engager (BiTE) are Regulated Contrarily by PD-L1 and CD80/CD86 on Leukemic Blasts. Oncotarget (2016) 7:76902–19. doi: 10.18632/oncotarget.12357
61. Borowitz MJ, Shuster J, Carroll AJ, Nash M, Look AT, Camitta B, et al. Prognostic Significance of Fluorescence Intensity of Surface Marker Expression in Childhood B-Precursor Acute Lymphoblastic Leukemia. A Pediatr Oncol Group Study. Blood (1997) 89:3960–6. doi: 10.1182/blood.V89.11.3960
62. Dworzak MN, Schumich A, Printz D, Pötschger U, Husak Z, Attarbaschi A, et al. CD20 Up-Regulation in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia During Induction Treatment: Setting the Stage for Anti-CD20 Directed Immunotherapy. Blood (2008) 112:3982–8. doi: 10.1182/blood-2008-06-164129
63. Lussana F, Gritti G, Rambaldi A. Immunotherapy of Acute Lymphoblastic Leukemia and Lymphoma With T Cell-Redirected Bispecific Antibodies. J Clin Oncol (2021) 39:444–55. doi: 10.1200/jco.20.01564
64. Hutchings M, Morschhauser F, Iacoboni G. Glofitamab, a Novel, Bivalent CD20-Targeting T-Cell-Engaging Bispecific Antibody, Induces Durable Complete Remissions in Relapsed or Refractory B-Cell Lymphoma: A Phase I Trial. J Clin Oncol (2021) 39:1959–70. doi: 10.1200/jco.20.03175
65. Wang S, Peng L, Xu W, Zhou Y, Zhu Z, Kong Y, et al. Preclinical Characterization and Comparison Between CD3/CD19 Bispecific and Novel CD3/CD19/CD20 Trispecific Antibodies Against B-Cell Acute Lymphoblastic Leukemia: Targeted Immunotherapy for Acute Lymphoblastic Leukemia. Front Med (2021) 16:139–49. doi: 10.1007/s11684-021-0835-8
66. Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell (2020) 183:126–42.e117. doi: 10.1016/j.cell.2020.08.022
67. Zhang J, Zhou Z. Preclinical Study of a Novel Tri-Specific Anti-CD3/CD19/CD20 T Cell-Engaging Antibody As a Potentially Better Treatment for NHL. Blood (2020) 136:22–2. doi: 10.1182/blood-2020-140154
68. Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T Design: Elements and Their Synergistic Function. EBioMedicine (2020) 58:102931. doi: 10.1016/j.ebiom.2020.102931
69. Mao R, Hussein MS, He Y. Chimeric Antigen Receptor Engineered T Cells and Their Application in the Immunotherapy of Solid Tumours. Expert Rev Mol Med (2022) 24:e7. doi: 10.1017/erm.2021.32
70. Sterner RC, Sterner RM. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J (2021) 11:69. doi: 10.1038/s41408-021-00459-7
71. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol Ther (2009) 17:1453–64. doi: 10.1038/mt.2009.83
72. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. N Engl J Med (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
73. Rosenbaum L. Tragedy, Perseverance, and Chance - The Story of CAR-T Therapy. N Engl J Med (2017) 377:1313–5. doi: 10.1056/NEJMp1711886
74. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med (2020) 382:1331–42. doi: 10.1056/NEJMoa1914347
75. Gardner RA, Finney O, Annesley C. Intent-To-Treat Leukemia Remission by CD19 CAR T Cells of Defined Formulation and Dose in Children and Young Adults. Blood (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
76. Curran KJ, Margossian SP, Kernan NA, Silverman LB, Williams DA, Shukla N, et al. Toxicity and Response After CD19-Specific CAR T-Cell Therapy in Pediatric/Young Adult Relapsed/Refractory B-ALL. Blood (2019) 134:2361–8. doi: 10.1182/blood.2019001641
77. Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T Cell Product and Disease Attributes Predict Leukemia Remission Durability. J Clin Invest (2019) 129:2123–32. doi: 10.1172/jci125423
78. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/jci85309
79. Cao J, Wang G, Cheng H, Wei C, Qi K, Sang W, et al. Potent Anti-Leukemia Activities of Humanized CD19-Targeted Chimeric Antigen Receptor T (CAR-T) Cells in Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. Am J Hematol (2018) 93:851–8. doi: 10.1002/ajh.25108
80. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
81. Fischer J, Paret C, El Malki K, Alt F, Wingerter A, Neu MA, et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J Immunother (2017) 40:187–95. doi: 10.1097/cji.0000000000000169
82. Raponi S, De Propris MS, Intoppa S, Milani ML, Vitale A, Elia L, et al. Flow Cytometric Study of Potential Target Antigens (CD19, CD20, CD22, CD33) for Antibody-Based Immunotherapy in Acute Lymphoblastic Leukemia: Analysis of 552 Cases. Leuk Lymphoma (2011) 52:1098–107. doi: 10.3109/10428194.2011.559668
83. Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-Chimeric Antigen Receptors Targeting B-Cell Precursor Acute Lymphoblastic Leukemia. Blood (2013) 121:1165–74. doi: 10.1182/blood-2012-06-438002
84. Shah NN, Stevenson MS, Yuan CM, Richards K, Delbrook C, Kreitman RJ, et al. Characterization of CD22 Expression in Acute Lymphoblastic Leukemia. Pediatr Blood Cancer (2015) 62:964–9. doi: 10.1002/pbc.25410
85. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-Targeted CAR T Cells Induce Remission in B-ALL That is Naive or Resistant to CD19-Targeted CAR Immunotherapy. Nat Med (2018) 24:20–8. doi: 10.1038/nm.4441
86. Qin H, Ramakrishna S, Nguyen S, Fountaine TJ, Ponduri A, Stetler-Stevenson M, et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol Ther Oncol (2018) 11:127–37. doi: 10.1016/j.omto.2018.10.006
87. Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D, et al. Bispecific CAR-T Cells Targeting Both CD19 and CD22 for Therapy of Adults With Relapsed or Refractory B Cell Acute Lymphoblastic Leukemia. J Hematol Oncol (2020) 13:30. doi: 10.1186/s13045-020-00856-8
88. Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia. Clin Cancer Res (2021) 27:2764–72. doi: 10.1158/1078-0432.CCR-20-3863
89. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and Biological Correlates of Neurotoxicity Associated With CAR T-Cell Therapy in Patients With B-Cell Acute Lymphoblastic Leukemia. Cancer Discov (2018) 8:958–71. doi: 10.1158/2159-8290.cd-17-1319
90. Gofshteyn JS, Shaw PA, Teachey DT, Grupp SA, Maude S, Banwell B, et al. Neurotoxicity After CTL019 in a Pediatric and Young Adult Cohort. Ann Neurol (2018) 84:537–46. doi: 10.1002/ana.25315
91. Fitzgerald JC, Weiss SL, Maude SL, Barrett DM, Lacey SF, Melenhorst JJ, et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit Care Med (2017) 45:e124–31. doi: 10.1097/ccm.0000000000002053
92. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A Safe and Potent Anti-CD19 CAR T Cell Therapy. Nat Med (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
93. Sheth VS, Gauthier J. Taming the Beast: CRS and ICANS After CAR T-Cell Therapy for ALL. Bone Marrow Transplant (2021) 56:552–66. doi: 10.1038/s41409-020-01134-4
94. Voynova E, Kovalovsky D. From Hematopoietic Stem Cell Transplantation to Chimeric Antigen Receptor Therapy: Advances, Limitations and Future Perspectives. Cells (2021) 10:2845. doi: 10.3390/cells10112845
95. Sun Y, Su Y, Wang Y, Liu N, Li Y, Chen J, et al. CD19 CAR-T Cells With Membrane-Bound IL-15 for B-Cell Acute Lymphoblastic Leukemia After Failure of CD19 and CD22 CAR-T Cells: Case Report. Front Immunol (2021) 12:728962. doi: 10.3389/fimmu.2021.728962
96. Kagoya Y, Tanaka S, Guo T. A Novel Chimeric Antigen Receptor Containing a JAK-STAT Signaling Domain Mediates Superior Antitumor Effects. Nat Med (2018) 24:352–9. doi: 10.1038/nm.4478
97. Blaeschke F, Willier S, Stenger D, Lepenies M, Horstmann MA, Escherich G, et al. Leukemia-Induced Dysfunctional TIM-3(+)CD4(+) Bone Marrow T Cells Increase Risk of Relapse in Pediatric B-Precursor ALL Patients. Leukemia (2020) 34:2607–20. doi: 10.1038/s41375-020-0793-1
98. Zhao S, Wang C, Lu P, Lou Y, Liu H, Wang T, et al. Switch Receptor T3/28 Improves Long-Term Persistence and Antitumor Efficacy of CAR-T Cells. J Immunother Cancer (2021) 9:e003176. doi: 10.1136/jitc-2021-003176
99. Blaeschke F, Ortner E, Stenger D, Mahdawi J, Apfelbeck A, Habjan N, et al. Design and Evaluation of TIM-3-CD28 Checkpoint Fusion Proteins to Improve Anti-CD19 CAR T-Cell Function. Front Immunol (2022) 13:845499. doi: 10.3389/fimmu.2022.845499
100. An L, Lin Y, Deng B, Yin Z, Zhao D, Ling Z, et al. Humanized CD19 CAR-T Cells in Relapsed/Refractory B-ALL Patients Who Relapsed After or Failed Murine CD19 CAR-T Therapy. BMC Cancer (2022) 22:393. doi: 10.1186/s12885-022-09489-1
101. Chen DS, Mellman I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature (2017) 541:321–30. doi: 10.1038/nature21349
102. Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J, et al. Immune Cells Within the Tumor Microenvironment: Biological Functions and Roles in Cancer Immunotherapy. Cancer Lett (2020) 470:126–33. doi: 10.1016/j.canlet.2019.11.009
103. Kang SH, Hwang HJ, Yoo JW, Kim H, Choi ES, Hwang SH, et al. Expression of Immune Checkpoint Receptors on T-Cells and Their Ligands on Leukemia Blasts in Childhood Acute Leukemia. Anticancer Res (2019) 39:5531–9. doi: 10.21873/anticanres.13746
104. Younes A, Santoro A, Shipp M, Zinzani PL, Timmerman JM, Ansell S, et al. Nivolumab for Classical Hodgkin's Lymphoma After Failure of Both Autologous Stem-Cell Transplantation and Brentuximab Vedotin: A Multicentre, Multicohort, Single-Arm Phase 2 Trial. Lancet Oncol (2016) 17:1283–94. doi: 10.1016/s1470-2045(16)30167-x
105. Armand P, Rodig S, Melnichenko V, Thieblemont C, Bouabdallah K, Tumyan G, et al. Pembrolizumab in Relapsed or Refractory Primary Mediastinal Large B-Cell Lymphoma. J Clin Oncol (2019) 37:3291–9. doi: 10.1200/jco.19.01389
106. Kwok G, Yau TC, Chiu JW, Tse E, Kwong YL. Pembrolizumab (Keytruda). Hum Vaccin Immunother (2016) 12:2777–89. doi: 10.1080/21645515.2016.1199310
107. Cassaday RD, Garcia KA, Fromm JR, Percival MM, Turtle CJ, Nghiem PT, et al. Phase 2 Study of Pembrolizumab for Measurable Residual Disease in Adults With Acute Lymphoblastic Leukemia. Blood Adv (2020) 4:3239–45. doi: 10.1182/bloodadvances.2020002403
108. Kinoshita H, Cooke KR, Grant M. Outcome of Donor-Derived TAA T Cell Therapy in Patients With High-Risk or Relapsed Acute Leukemia Post Allogeneic BMT. Blood Adv (2022) 6:2520–34. doi: 10.1182/bloodadvances.2021006831
109. Candoni A, Tiribelli M, Toffoletti E, Cilloni D, Chiarvesio A, Michelutti A, et al. Quantitative Assessment of WT1 Gene Expression After Allogeneic Stem Cell Transplantation is a Useful Tool for Monitoring Minimal Residual Disease in Acute Myeloid Leukemia. Eur J Haematol (2009) 82:61–8. doi: 10.1111/j.1600-0609.2008.01158.x
110. Rezvani K, Yong AS, Tawab A, Jafarpour B, Eniafe R, Mielke S, et al. Ex Vivo Characterization of Polyclonal Memory CD8+ T-Cell Responses to PRAME-Specific Peptides in Patients With Acute Lymphoblastic Leukemia and Acute and Chronic Myeloid Leukemia. Blood (2009) 113:2245–55. doi: 10.1182/blood-2008-03-144071
111. Park E, Gang EJ, Hsieh YT, Schaefer P, Chae S, Klemm L, et al. Targeting Survivin Overcomes Drug Resistance in Acute Lymphoblastic Leukemia. Blood (2011) 118:2191–9. doi: 10.1182/blood-2011-04-351239
112. Ecsedi M, McAfee MS, Chapuis AG. The Anticancer Potential of T Cell Receptor-Engineered T Cells. Trends Cancer (2021) 7:48–56. doi: 10.1016/j.trecan.2020.09.002
113. Liu H, Pan C, Song W, Liu D, Li Z, Zheng L. Novel Strategies for Immuno-Oncology Breakthroughs With Cell Therapy. Biomark Res (2021) 9:62. doi: 10.1186/s40364-021-00316-6
114. Ping Y, Liu C, Zhang Y. T-Cell Receptor-Engineered T Cells for Cancer Treatment: Current Status and Future Directions. Protein Cell (2018) 9:254–66. doi: 10.1007/s13238-016-0367-1
115. Xu Y, Yang Z, Horan LH, Zhang P, Liu L, Zimdahl B, et al. A Novel Antibody-TCR (AbTCR) Platform Combines Fab-Based Antigen Recognition With Gamma/Delta-TCR Signaling to Facilitate T-Cell Cytotoxicity With Low Cytokine Release. Cell Discov (2018) 4:62. doi: 10.1038/s41421-018-0066-6
116. Rodig SJ, Shahsafaei A, Li B, Mackay CR, Dorfman DM. BAFF-R, the Major B Cell-Activating Factor Receptor, is Expressed on Most Mature B Cells and B-Cell Lymphoproliferative Disorders. Hum Pathol (2005) 36:1113–9. doi: 10.1016/j.humpath.2005.08.005
117. Qin H, Dong Z. CAR T Cells Targeting BAFF-R can Overcome CD19 Antigen Loss in B Cell Malignancies. Sci Transl Med (2019) 11:eaaw9414. doi: 10.1126/scitranslmed.aaw9414
118. Wang X, Dong Z, Awuah D, Chang WC, Cheng WA, Vyas V, et al. CD19/BAFF-R Dual-Targeted CAR T Cells for the Treatment of Mixed Antigen-Negative Variants of Acute Lymphoblastic Leukemia. Leukemia (2022) 36:1015–24. doi: 10.1038/s41375-021-01477-x
119. Boissonnas A, Combadiere C. Modulating the Tumor-Associated Macrophage Landscape. Nat Immunol (2022) 23:481–2. doi: 10.1038/s41590-022-01159-5
120. Tian L, Lei A, Tan T, Zhu M, Zhang L, Mou H, et al. Macrophage-Based Combination Therapies as a New Strategy for Cancer Immunotherapy. Kidney Dis (Basel) (2022) 8:26–43. doi: 10.1159/000518664
121. Russ A, Hua AB, Montfort WR, Rahman B, Riaz IB, Khalid MU, et al. Blocking "Don't Eat Me" Signal of CD47-Sirpα in Hematological Malignancies, an in-Depth Review. Blood Rev (2018) 32:480–9. doi: 10.1016/j.blre.2018.04.005
122. Yang K, Xu J, Liu Q, Li J, Xi Y. Expression and Significance of CD47, PD1 and PDL1 in T-Cell Acute Lymphoblastic Lymphoma/Leukemia. Pathol Res Pract (2019) 215:265–71. doi: 10.1016/j.prp.2018.10.021
123. Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, et al. Therapeutic Antibody Targeting of CD47 Eliminates Human Acute Lymphoblastic Leukemia. Cancer Res (2011) 71:1374–84. doi: 10.1158/0008-5472.CAN-10-2238
124. Petrova PS, Viller NN, Wong M, Pang X, Lin GH, Dodge K, et al. TTI-621 (Sirpαfc): A CD47-Blocking Innate Immune Checkpoint Inhibitor With Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin Cancer Res (2017) 23:1068–79. doi: 10.1158/1078-0432.ccr-16-1700
125. Uno S, Kinoshita Y, Azuma Y, Tsunenari T, Yoshimura Y, Iida S, et al. Antitumor Activity of a Monoclonal Antibody Against CD47 in Xenograft Models of Human Leukemia. Oncol Rep (2007) 17:1189–94. doi: 10.3892/or.17.5.1189
126. Puro RJ, Bouchlaka MN, Hiebsch RR, Capoccia BJ, Donio MJ, Manning PT, et al. Development of AO-176, a Next-Generation Humanized Anti-CD47 Antibody With Novel Anticancer Properties and Negligible Red Blood Cell Binding. Mol Cancer Ther (2020) 19:835–46. doi: 10.1158/1535-7163.mct-19-1079
127. Peluso MO, Adam A, Armet CM, Zhang L, O'Connor RW, Lee BH, et al. The Fully Human Anti-CD47 Antibody SRF231 Exerts Dual-Mechanism Antitumor Activity via Engagement of the Activating Receptor CD32a. J Immunother Cancer (2020) 8:e000413. doi: 10.1136/jitc-2019-000413
128. Ma L, Zhu M, Gai J, Li G, Chang Q, Qiao P, et al. Preclinical Development of a Novel CD47 Nanobody With Less Toxicity and Enhanced Anti-Cancer Therapeutic Potential. J Nanobiotechnol (2020) 18:12. doi: 10.1186/s12951-020-0571-2
129. Ring NG, Herndler-Brandstetter D, Weiskopf K, Shan L, Volkmer JP, George BM, et al. Anti-Sirpα Antibody Immunotherapy Enhances Neutrophil and Macrophage Antitumor Activity. Proc Natl Acad Sci U S A (2017) 114:E10578–e10585. doi: 10.1073/pnas.1710877114
130. Tahk S, Vick B, Hiller B, Schmitt S, Marcinek A, Perini ED, et al. Sirpα-αcd123 Fusion Antibodies Targeting CD123 in Conjunction With CD47 Blockade Enhance the Clearance of AML-Initiating Cells. J Hematol Oncol (2021) 14:155. doi: 10.1186/s13045-021-01163-6
131. Logtenberg MEW, Jansen JHM, Raaben M, Toebes M, Franke K, Brandsma AM, et al. Glutaminyl Cyclase is an Enzymatic Modifier of the CD47- Sirpα Axis and a Target for Cancer Immunotherapy. Nat Med (2019) 25:612–9. doi: 10.1038/s41591-019-0356-z
132. Jiang Z, Sun H, Yu J, Tian W, Song Y. Targeting CD47 for Cancer Immunotherapy. J Hematol Oncol (2021) 14:180. doi: 10.1186/s13045-021-01197-w
133. Narla RK, Modi H, Bauer D, Abbasian M, Leisten J, Piccotti JR, et al. Modulation of CD47-Sirpα Innate Immune Checkpoint Axis With Fc-Function Detuned Anti-CD47 Therapeutic Antibody. Cancer Immunol Immunother (2021) 71:473–89. doi: 10.1007/s00262-021-03010-6
134. Chen YC, Shi W, Shi JJ, Lu JJ. Progress of CD47 Immune Checkpoint Blockade Agents in Anticancer Therapy: A Hematotoxic Perspective. J Cancer Res Clin Oncol (2022) 148:1–14. doi: 10.1007/s00432-021-03815-z
135. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin's Lymphoma. N Engl J Med (2018) 379:1711–21. doi: 10.1056/NEJMoa1807315
136. Ansell SM, Maris MB, Lesokhin AM, Chen RW, Flinn IW. Phase I Study of the CD47 Blocker TTI-621 in Patients With Relapsed or Refractory Hematologic Malignancies. Clin Cancer Res (2021) 27:2190–9. doi: 10.1158/1078-0432.ccr-20-3706
137. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years on. Cell (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
138. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
139. Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright Natural Killer (NK) Cells: An Important NK Cell Subset. Immunology (2009) 126:458–65. doi: 10.1111/j.1365-2567.2008.03027.x
140. Moretta L, Montaldo E, Vacca P, Del Zotto G, Moretta F, Merli P, et al. Human Natural Killer Cells: Origin, Receptors, Function, and Clinical Applications. Int Arch Allergy Immunol (2014) 164:253–64. doi: 10.1159/000365632
141. Chiossone L, Dumas PY. Natural Killer Cells and Other Innate Lymphoid Cells in Cancer. Nat Rev Immunol (2018) 18:671–88. doi: 10.1038/s41577-018-0061-z
142. Ljunggren HG, Malmberg KJ. Prospects for the Use of NK Cells in Immunotherapy of Human Cancer. Nat Rev Immunol (2007) 7:329–39. doi: 10.1038/nri2073
143. Sivori S, Meazza R. NK Cell-Based Immunotherapy for Hematological Malignancies. J Clin Med (2019) 8:1702. doi: 10.3390/jcm8101702
144. Mizia-Malarz A, Sobol-Milejska G. NK Cells as Possible Prognostic Factor in Childhood Acute Lymphoblastic Leukemia. Dis Markers (2019) 2019:1–4. doi: 10.1155/2019/3596983
145. Sullivan EM, Jeha S, Kang G, Cheng C, Rooney B, Holladay M, et al. NK Cell Genotype and Phenotype at Diagnosis of Acute Lymphoblastic Leukemia Correlate With Postinduction Residual Disease. Clin Cancer Res (2014) 20:5986–94. doi: 10.1158/1078-0432.ccr-14-0479
146. Colomar-Carando N, Gauthier L, Merli P, Loiacono F, Canevali P, Falco M, et al. Exploiting Natural Killer Cell Engagers to Control Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. Cancer Immunol Res (2022) 10:291–302. doi: 10.1158/2326-6066.CIR-21-0843
147. Vaněk O, Kalousková B, Abreu C, Nejadebrahim S, Skořepa O. Natural Killer Cell-Based Strategies for Immunotherapy of Cancer. Adv Protein Chem Struct Biol (2022) 129:91–133. doi: 10.1016/bs.apcsb.2022.02.001
148. Zhang L, Meng Y, Feng X, Han Z. CAR-NK Cells for Cancer Immunotherapy: From Bench to Bedside. Biomark Res (2022) 10:12. doi: 10.1186/s40364-022-00364-6
149. Herrera L, Santos S, Vesga MA, Anguita J. Adult Peripheral Blood and Umbilical Cord Blood NK Cells are Good Sources for Effective CAR Therapy Against CD19 Positive Leukemic Cells. Sci Rep (2019) 9:18729. doi: 10.1038/s41598-019-55239-y
150. Müller S, Bexte T, Gebel V, Kalensee F, Stolzenberg E, Hartmann J, et al. High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia. Front Immunol (2020) 10:3123. doi: 10.3389/fimmu.2019.03123
151. DeNardo DG, Ruffell B. Macrophages as Regulators of Tumour Immunity and Immunotherapy. Nat Rev Immunol (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
152. Franken L, Schiwon M, Kurts C. Macrophages: Sentinels and Regulators of the Immune System. Cell Microbiol (2016) 18:475–87. doi: 10.1111/cmi.12580
153. Klichinsky M, Ruella M. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat Biotechnol (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
154. Kast F, Klein C. Advances in Identification and Selection of Personalized Neoantigen/T-Cell Pairs for Autologous Adoptive T Cell Therapies. Oncoimmunology (2021) 10:1869389. doi: 10.1080/2162402x.2020.1869389
155. Lu F, Ma XJ, Jin WL, Luo Y, Li X. Neoantigen Specific T Cells Derived From T Cell-Derived Induced Pluripotent Stem Cells for the Treatment of Hepatocellular Carcinoma: Potential and Challenges. Front Immunol (2021) 12:690565. doi: 10.3389/fimmu.2021.690565
156. Verdon DJ, Jenkins MR. Identification and Targeting of Mutant Peptide Neoantigens in Cancer Immunotherapy. Cancers (Basel) (2021) 13:4245. doi: 10.3390/cancers13164245
157. Borden ES, Buetow KH, Wilson MA, Hastings KT. Cancer Neoantigens: Challenges and Future Directions for Prediction, Prioritization, and Validation. Front Oncol (2022) 12:836821. doi: 10.3389/fonc.2022.836821
Keywords: pediatric ALL, T-cell engagers, CAR T cell therapy, macrophage-based immunotherapy, NK cell-based immunotherapy
Citation: Lv M, Liu Y, Liu W, Xing Y and Zhang S (2022) Immunotherapy for Pediatric Acute Lymphoblastic Leukemia: Recent Advances and Future Perspectives. Front. Immunol. 13:921894. doi: 10.3389/fimmu.2022.921894
Received: 16 April 2022; Accepted: 19 May 2022;
Published: 13 June 2022.
Edited by:
Xuyao Zhang, Fudan University, ChinaReviewed by:
Yan Cao, Sun Yat-sen University Cancer Center (SYSUCC), ChinaBowen Xing, Children’s Hospital of Philadelphia, United States
Longchao Liu, University of Texas Southwestern Medical Center, United States
Copyright © 2022 Lv, Liu, Liu, Xing and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yabing Xing, eHliMDZAbGl2ZS5jb20=; Shengnan Zhang, enNucGhhckAxMjYuY29t
†These authors have contributed equally to this work