 Albert J. Czaja
Albert J. Czaja- Retired, Rochester, MN, United States
The observed risk of autoimmune hepatitis exceeds its genetic risk, and epigenetic factors that alter gene expression without changing nucleotide sequence may help explain the disparity. Key objectives of this review are to describe the epigenetic modifications that affect gene expression, discuss how they can affect autoimmune hepatitis, and indicate prospects for improved management. Multiple hypo-methylated genes have been described in the CD4+ and CD19+ T lymphocytes of patients with autoimmune hepatitis, and the circulating micro-ribonucleic acids, miR-21 and miR-122, have correlated with laboratory and histological features of liver inflammation. Both epigenetic agents have also correlated inversely with the stage of liver fibrosis. The reduced hepatic concentration of miR-122 in cirrhosis suggests that its deficiency may de-repress the pro-fibrotic prolyl-4-hydroxylase subunit alpha-1 gene. Conversely, miR-155 is over-expressed in the liver tissue of patients with autoimmune hepatitis, and it may signify active immune-mediated liver injury. Different epigenetic findings have been described in diverse autoimmune and non-autoimmune liver diseases, and these changes may have disease-specificity. They may also be responses to environmental cues or heritable adaptations that distinguish the diseases. Advances in epigenetic editing and methods for blocking micro-ribonucleic acids have improved opportunities to prove causality and develop site-specific, therapeutic interventions. In conclusion, the role of epigenetics in affecting the risk, clinical phenotype, and outcome of autoimmune hepatitis is under-evaluated. Full definition of the epigenome of autoimmune hepatitis promises to enhance understanding of pathogenic mechanisms and satisfy the unmet clinical need to improve therapy for refractory disease.
1 Introduction
Autoimmune hepatitis has genetic risk factors within and outside the major histocompatibility complex (MHC) (1, 2). The genetic risk factors within the MHC affect mainly the predisposition for autoimmune hepatitis. The susceptibility alleles reside on the HLA-DRB1 gene where they can vary in association with ethnicity and age (3–9). The genetic risk factors outside the MHC are less established. They are mainly polymorphisms or point mutations that may affect individual pathways within the immune response (cytokine milieu, lymphocyte activation, and cell migration) (1, 2, 10–18). The major risk-laden loci are present in approximately 50% of patients with autoimmune hepatitis (19), and they do not explain the observed risk of the disease (19–21).
Epigenetics is a burgeoning science that describes molecular modifications and mechanisms that can modulate gene activity without altering the nucleotide sequence of deoxyribonucleic acid (DNA) (22–26). The epigenetic changes have cell type specificity and stability through cell replication (27), and they have been heritable in diverse experimental models (25, 28). Key epigenetic modifications have been described in the nuclear chromatin that can affect gene transcription (29–31), and small non-coding ribonucleic acids are epigenetic agents that can affect translation of the gene product (32, 33). The epigenetic modifications may be induced by environmental cues (34–37), and they have a durability that may contribute to a transgenerational inheritance through the germline (25, 28, 37, 38). Furthermore, the epigenetic changes are modifiable, reversible, and amenable to therapeutic intervention (19, 39–43).
Epigenetics may explain the difference between the genetic risk and observed risk of autoimmune hepatitis, and it may account for individual variations in clinical phenotype and outcome that cannot be explained by the MHC, genetic polymorphisms, or point mutations (39, 44–47). Chromosomal regions may undergo structural adaptations in response to environmental cues that alter DNA transcription (22, 38), and non-coding ribonucleic acids, especially micro-ribonucleic acids (miRNAs), may induce degradation or translational repression of messenger ribonucleic acids (mRNAs) (48–53).
Salient epigenetic effects have already been identified in experimental models and patients with diverse liver diseases, including alcoholic steatohepatitis (54, 55), non-alcoholic fatty liver disease (NAFLD) (56–58), primary biliary cholangitis (PBC) (59–61), primary sclerosing cholangitis (PSC) (62–64), cholangiocarcinoma (62, 65–67), hepatocellular cancer (68, 69), and autoimmune hepatitis (21, 70, 71). They have also been implicated in various non-liver diseases, including systemic lupus erythematosus (SLE) (72, 73), rheumatoid arthritis (74, 75), systemic sclerosis (76, 77), diverse neuro-degenerative diseases (78), and various cancers (79–82). Investigations of the epigenetic modifications affecting gene expression in autoimmune hepatitis may improve its management and satisfy an unmet clinical need for more effective therapy of refractory disease (83–85).
The goals of this review are to describe the epigenetic modifications that affect gene expression, examine transgenerational inheritance of epigenetic marks, present the key epigenetic changes in autoimmune hepatitis and other liver diseases, and indicate the prospects that epigenetics will enhance understanding of pathogenic pathways and treatment options in autoimmune hepatitis.
2 Methods
Abstracts were identified in PubMed using the search words “Epigenetic changes in liver disease,” “Epigenetic changes in autoimmune hepatitis”, “microRNAs in liver disease”, and “microRNAs in autoimmune hepatitis”. Selected full-length articles constituted the primary bibliography. Selected references cited in the primary sources constituted a secondary bibliography, and a tertiary bibliography was developed from references cited in the secondary bibliography. Several hundred abstracts were reviewed, and the number of full-length articles that were examined was 205.
3 Epigenetic Modulation of Gene Transcription
The transcriptional activity of genes occurs within chromatin (86). Chromatin is composed of histones arranged in octamers and double-stranded DNA that makes 1.65 turns around each octamer (38, 39, 86, 87) (Figure 1). Two copies of four core histones (H2A, H2B, H3, and H4) comprise the octamer (38, 86–88), and each DNA-enwrapped octamer constitutes a nucleosome (89). The nucleosomes are linked by a short DNA sequence of 60 base pairs, and the beaded filament is condensed and packaged in the nucleus as chromatin (86). A histone linker molecule maintains proper packaging of the DNA by binding to the site of DNA entry and exit from each nucleosome (86, 87, 90).
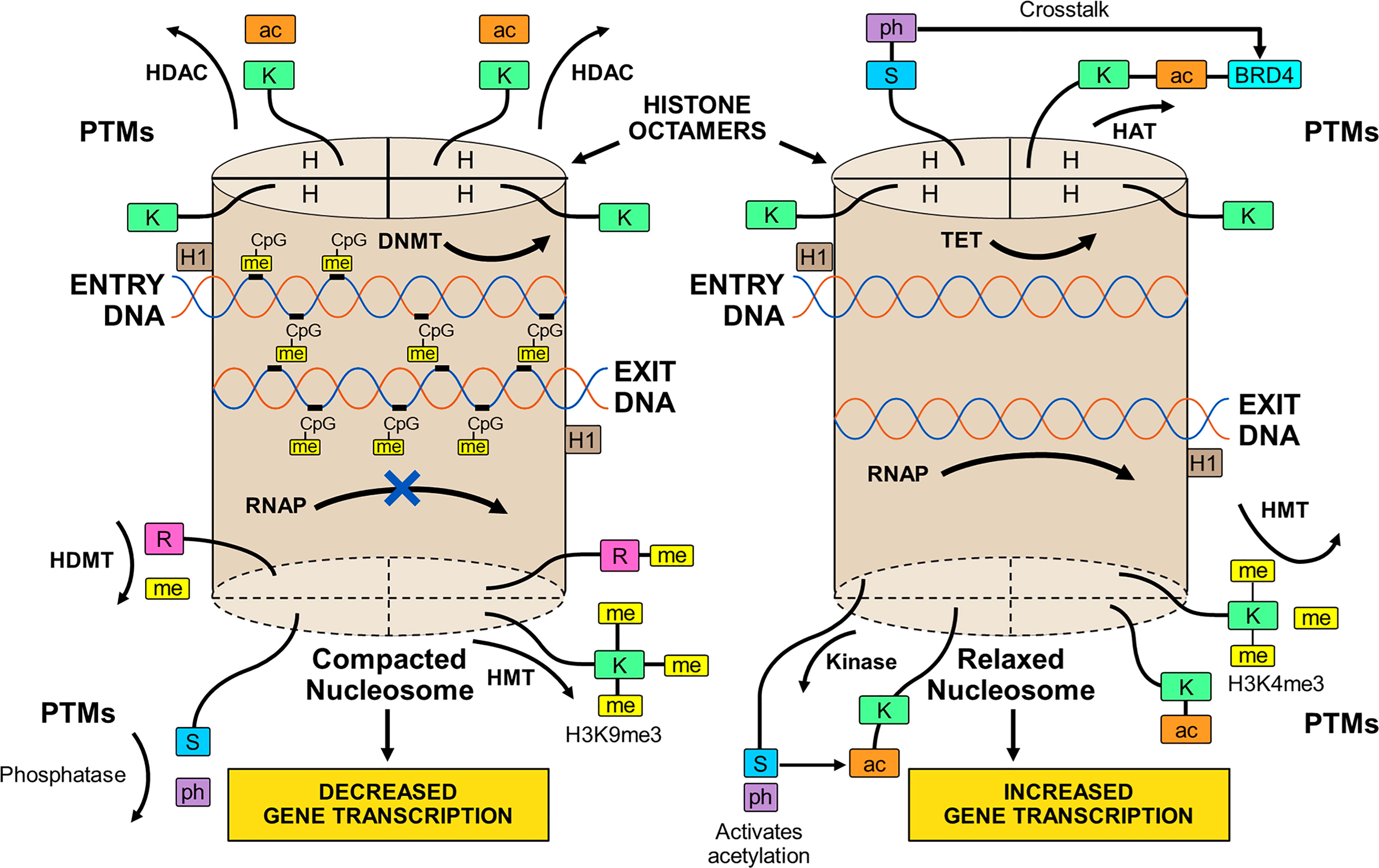
Figure 1 Compacted and relaxed nucleosomes. Nucleosomes consist of two copies of four different histones (H) arranged as a histone octamer and double-stranded deoxyribonucleic acid (DNA) wrapped 1.65 times around each octamer. The entry and exit of the DNA from the nucleosome is secured by a linker histone (H1). Each core histone within the octamer has an N-terminal tail that can undergo post-translational modifications (PTMs) by the attachment of methyl (me), acetyl (ac), or phosphate (ph) groups to a particular amino acid in the histone tail. Lysine (K), serine (S), or arginine (R) are among other amino acids that can serve as attachment sites. The PTMs are orchestrated by various enzymes. Methylation of the histone tail is catalyzed by histone methyltransferase (HMT); acetylation is catalyzed by histone acetyltransferase (HAT); and phosphorylation is catalyzed by kinases. The PTMs can be reversed by enzymes that dissociate the appended groups from the amino acid residues. Acetylation is reversed by histone deacetylase (HDAC); methylation is reversed by histone demethylase (HDMT); and phosphorylation is reversed by phosphatases. Histone acetylation relaxes the nucleosome and promotes gene transcription, and histone de-acetylation compacts the nucleosome (heterochromatin) and represses gene transcription. Histone methylation can decrease (H3K9me3) or increase (H3K4me3) transcription depending on the methylation site and other variables. Histone phosphorylation can recruit other molecules, such as bromo-domain-containing protein 4 (BRD4), to the acetylation site (crosstalk) and promote gene transcription. DNA can be methylated by DNA methyltransferase (DNMT) or de-methylated by ten-eleven translocation methylcytosine dioxygenase (TET). DNA methylation is restricted to sites in which cytosine (C) is separated from guanine (G) by a phosphate (p). Methylated DNA is compacted and transcription factors have limited access to transcription sites. Ribonucleic acid polymerase (RNAP) is prevented (X) from copying the nucleotide sequence, and gene transcription is decreased. De-methylated DNA is relaxed; RNAP can open the double-stranded DNA; and gene transcription is increased.
3.1 Impact of DNA Methylation on Gene Transcription
The methylated state of the DNA (39, 91–93) influences transcriptional activity within the nucleosome. Modifications in the chromatin structure can alter access and binding of transcription factors to the enhancer/promoter sequences of the DNA that are pivotal for transcription (94, 95) (Figure 1). The inability of RNA polymerase (RNAP) to access the DNA binding site can prevent opening of the double-stranded DNA and copying of the nucleotide sequence (39, 92).
DNA methylation occurs at a site in which a cytosine nucleotide (C) is separated from a guanine nucleotide (G) by a phosphate molecule (p) (46) (Table 1). Methylation of the cytosine in the CpG dinucleotide to 5-methylcytosine is mediated by DNA methyltransferases (DNMTs), and the methylation inhibits the binding of transcription factors to the DNA (91, 92) (Figure 1). It can also alter chromatin structure by attracting proteins that bind to the methylated cytosine (46, 92). The net effect of DNA methylation is to repress transcriptional activity and silence gene expression (39).
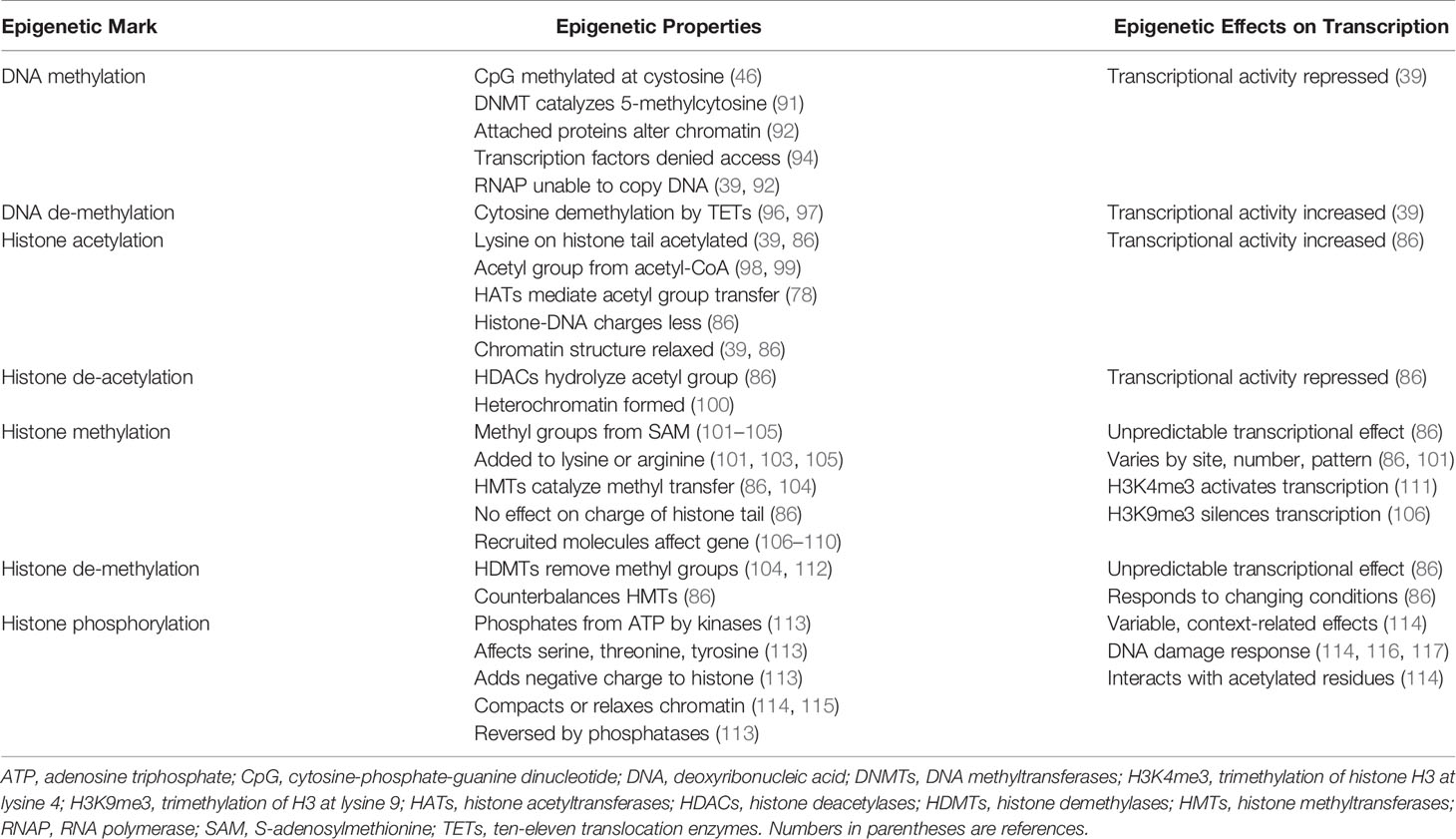
Table 1 Epigenetic properties and effects on gene transcription.
Ten-eleven translocation methylcytosine dioxygenase (TET) enzymes mediate the oxidation of the methylated cytosine to 5-hydroxymethylcytosine (39, 96, 97, 118, 119) (Table 1). This product can then undergo additional processing and demethylation by thymine-DNA-glycosylase and excision repair (118, 120, 121). The restoration of cytosine to its unmodified state can de-repress transcriptional activity and promote gene expression (Figure 1). The counter effects of DNMTs and TET enzymes on DNA methylation constitute a homeostatic mechanism that can respond to diverse stimuli, be disrupted in disease states, and be manipulated by therapeutic interventions (39, 122).
3.2 Impact of Histone Modifications on Gene Transcription
The N-terminal tail of the core histones can undergo multiple post-translational modifications (PTMs) that include acetylation, methylation, and phosphorylation (39, 86, 113, 123–126) (Table 1). The PTMs can alter the chemical structure, charge, and configuration of the histones, and the cumulative effect of multiple histone modifications can determine the transcriptional activity of the DNA (127) (Figure 1). PTMs also influence the cellular repair response to DNA injury (128). The modification of histones is a dynamic process that can preserve the integrity of the genome (129) and modulate transcriptional activity to maintain biological homeostasis (39, 86).
3.2.1 Histone Acetylation
The transfer of an acetyl group from acetyl-coenzyme A (acetyl-CoA) to a lysine residue on the histone tail constitutes histone acetylation, and the process is mediated by the histone acetyltransferases (HATs) (39, 78, 86, 98, 99) (Table 1). Histone acetylation can promote transcriptional activity by neutralizing differences in charge between the positively charged histones and the negatively charged DNA. The relaxed chromatin can promote transcriptional activity (39, 86) (Figure 1). Histone deacetylases (HDACs) can reverse the acetylation process by hydrolyzing the acetyl group on the lysine residue, compacting the chromatin into heterochromatin, and repressing transcriptional activity of the DNA (78, 86, 99, 100).
3.2.2 Histone Methylation
The transfer of methyl groups from S-adenosylmethionine (SAM) to lysine or arginine residues on the histone tail constitutes histone methylation (101–105), and the methylation process is mediated by histone methyltransferases (HMTs) (86, 104, 128, 130) (Table 1). The impact of histone methylation on DNA transcription is less predictable than histone acetylation, and it varies by methylation site (lysine versus arginine), number of methylations (mono-, di-, or tri-methylation) and pattern of methylation (symmetric versus asymmetric) (101, 103, 104, 128). Trimethylation of histone H3 at lysine 4 (H3K4me3) is the start site of transcription for most active genes (111, 128, 131, 132) (Figure 1), whereas trimethylation of H3 at lysine 9 (H3K9me3) is associated with heterochromatin and gene silencing (106, 128, 132).
Histone methylation does not affect the charge of the histone tail, and the impact of histone methylation on transcriptional activity relates mainly to the effects of molecules recruited to the methylated state and the sequence of adjacent amino acids (86, 107). Lysine methylation attracts diverse proteins mainly with chromo-domains that can modify chromatin structure and affect DNA transcription (108–110). Histone demethylases (HDMTs) can reverse the methylated PTM by removing methyl groups from the histone tails (86, 104, 112). The balance between HMTs and HDMTs is another homeostatic mechanism by which the genome can respond to changing conditions.
3.2.3 Histone Phosphorylation
Histone phosphorylation is a dynamic process affecting serine, threonine, and tyrosine residues in the N-terminal tail of the core histones (113) (Table 1). Kinases transfer phosphate groups from adenosine triphosphate (ATP) to the amino acid residues. Phosphorylation adds a negative charge to the histone, and the change in charge can remodel the chromatin. The phosphorylation process can be reversed by phosphatases that catalyze the hydrolysis and removal of the phosphate group (113) (Figure 1).
Histone phosphorylation occurs rapidly after DNA damage, and it is involved in the DNA damage response (DDR) (114, 116, 117) (Table 1). Phosphorylated histone residues are also associated with gene expression, including proto-oncogenes (133–135), and they can interrelate with histone residues that are acetylated to activate DNA transcription (114, 136, 137) (Figure 1). Phosphorylation of serine 10 at histone H3 (H3S10ph) activates DNA transcription by triggering acetylation of lysine 16 at histone 4 (H4K16ac) (138). The crosstalk between histone PTMs recruits bromo-domain-containing protein 4 (BRD4) to the nucleosome where it can bind to the acetylated lysine residue and promote DNA transcription (138–140).
The phosphorylation of histone is a rapidly changing process that can have contradictory effects depending on the context of the microenvironment (114). Histone phosphorylation is associated with chromatin compaction during mitosis and meiosis, but it can also be associated with chromatin relaxation under other circumstances (114, 115, 141). Therapeutic efforts to modulate histone phosphorylation must recognize the dynamic, interactive, labile, and context-dependent nature of the PTM.
4 Epigenetic Modulation of Gene Translation
MiRNAs are a subgroup of non-coding RNAs that by definition do not encode protein (142) (Table 2). They constitute a functional minority of non-coding RNAs (143), and they are members of a class that includes small interfering RNAs (siRNAs) (144) and Piwi-interacting RNAs (piRNAs) (145, 146). MiRNAs are small (21-25 nucleotides), natural, genomic products that have multiple functions within their cell of origin (48, 50). They are present in the nucleus, nucleolus, and mitochondria where they can influence the intracellular processes of DNA transcription, repair, and splicing (147, 148, 170, 171). They can also silence the expression of genes that encode protein by preventing the translation of mRNA into a protein product (48, 50, 51) (Figure 2). MiRNAs are key epigenetic agents that act primarily outside the chromatin to degrade mRNA within the cytoplasm or otherwise repress its translation.

Table 2 Epigenetic Properties and Effects of Micro-Ribonucleic Acids.
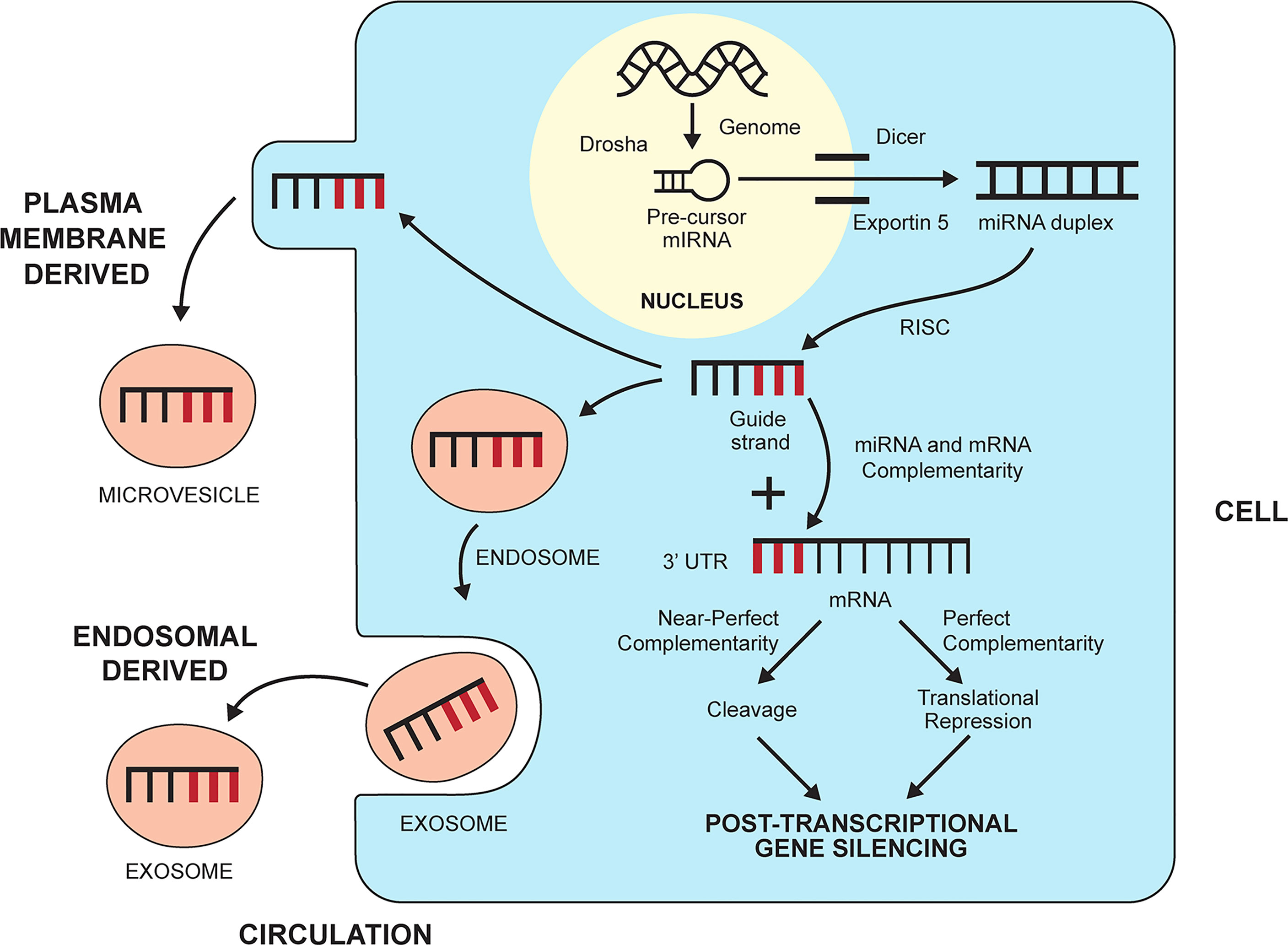
Figure 2 Biogenesis and gene silencing action of micro-ribonucleic acids (miRNAs). MiRNAs are derived from the cell genome and processed within the nucleus by the ribonuclease III enzyme, Drosha, into pre-cursor miRNA. The precursor miRNA is transported to the cytoplasm by exportin 5 and processed further by the ribonuclease II enzyme, Dicer, to a miRNA duplex. The duplex is processed in a RNA-induced silencing complex (RISC), and the strand with less stable 5’ end is selected as the guide strand. The guide strand probes for complementary base pairs (bold lines) in the 3’ untranslated region (3’UTR) of messenger RNA (mRNA). The degree of complementarity between the guide strand and the mRNA determines if the mRNA will undergo cleavage by endonucleases (perfect complementarity) or translational repression (near perfect complementarity). Either fate induces post-transcriptional gene silencing. MiRNAs can leave the cell and enter the circulation by forming a plasma membrane-derived microvesicle or an endosomal-derived exosome.
MiRNAs can enter the circulation within vesicles that develop from the endosomal compartment (exosomes) or separate from the plasma membrane (microvesicles, apoptotic bodies) (150, 151) (Figure 2). Circulating miRNAs have the potential to engage in cell-to-cell communication and affect the function of other cells, albeit their role in this capacity remains obscure (149, 152) (Table 2). Despite this limitation, circulating levels of miRNAs have been measured and correlated with the inflammatory activity of diverse diseases, including autoimmune hepatitis (70).
The number of miRNAs in humans has been estimated by a manually curated miRNA database as over 500 (154) (Table 2). Multiple miRNAs can regulate the expression of a single gene, and a single miRNA can influence multiple genes (155, 156). Diverse cell types produce miRNAs, and tissue and disease specificity can be difficult to demonstrate (153). Critical physiological and pathological effects have been ascribed to single miRNA-mRNA interactions that are context-dependent (157, 158), and certain miRNAs have been highly specific for individual cell lines (159). Preferential expression of particular miRNAs has been recognized in diverse diseases, including chronic liver disease (70, 71, 160, 161).
4.1 Biogenesis and Regulatory Actions of MiRNAs
MiRNAs originate in the nucleus as double-stranded RNA molecules that are encoded by the genome as primary miRNAs (149, 162) (Table 2). The primary miRNAs are then modified in the nucleus by a microprocessor complex containing the ribonuclease III enzyme, Drosha, to precursor miRNAs (149, 163, 172, 173) (Figure 2). The precursor miRNAs are exported to the cytoplasm by exportin 5 where the ribonuclease II enzyme, Dicer, modifies the precursor molecules further to form mature miRNA duplexes (164, 174). The duplexes are processed in a RNA-induced silencing complex (RISC) within the cytoplasm (51), and the strand with the less stable 5’ end is selected for incorporation in the RISC-loading complex (RLC) as the guide strand (165). The other strand (passenger strand) is degraded by endonucleases (175).
The guide strand probes for complementary base pairs in the 3’ untranslated region (3’ UTR) of mRNAs in the cytoplasm (Figure 2). The “seed region” that identifies complementarity in the mRNA may consist of only 2-7 bases (51, 155). Near perfect complementarity between the miRNA and the mRNA triggers degradation of the mRNA by endonucleases and complete gene silencing (169) (Table 2). More commonly, the complementarity is less complete, and the miRNA mainly disrupts the translation of mRNA without triggering its degradation (translational repression) (51, 149). MiRNAs can also develop along non-canonical pathways that do not involve Drosha or Dicer (166–168). The biological functions of these miRNAs are uncertain in humans.
5 Transgenerational Inheritance of Epigenetic Marks
The DNA sequence and the epigenome are replicated during cell mitosis (25, 27), and DNA methylation (176), histone PTMs (38), and miRNAs (177) can be transmitted in the germline of mammals. Extensive re-programming of the epigenetic information occurs during gametogenesis and after fertilization, and transgenerational inheritance requires re-assembly or reconstruction of the epigenetic marks. DNA methylation and histone modifications can be re-assembled after mitosis (replicative transmission) or the epigenetic changes can be reconstructed in the germline by another inherited signal (reconstructive transmission) (28). Non-coding RNAs are templates that are pivotal to the reconstructive process, and they can be transmitted to the next generation in oocytes and sperm (28, 178, 179). Transgenerational inheritance requires proof that the original epigenetic signal is successfully transmitted and that heritability extends beyond the second generation.
The transmitted epigenetic changes may reflect environmental adaptations made by the parent and transmitted to the offspring through the germline (28, 35, 180, 181). The offspring of male mice who have been fed a low-protein diet inherit epigenetic marks that affect the peroxisome proliferator-activated receptor alpha (PPARA) gene which regulates lipid and cholesterol metabolism (180). The heritable epigenetic changes may also re-program responses to disease (182). Two generations of offspring from male rats with a history of liver fibrosis have inherited a resistance to hepatic fibrosis manifested by impaired differentiation of hepatic myofibroblasts, increased expression of the anti-fibrotic peroxisome proliferator-activated receptor-gamma (PPAR-γ) protein, and decreased production of the pro-fibrotic transforming growth factor beta 1 (TGF-β1) cytokine (182).
The demonstration of heritable epigenetic marks has been difficult to establish in humans because of confounding genetic, cultural, and environmental factors (37, 183), and heritability has been eliminated from the definition of epigenetics (22). Epigenetic changes within an individual may be acquired by external pressures (diet, lifestyle, toxic exposures) (184–187) or by intrinsic instability of the epigenome through successive cell divisions (“epigenetic drift”) (188–192). Shared changes in the somatic epigenome of individuals in the same environment does not connote heritability unless expressed in the germline (sperm or egg) (25). Furthermore, the epigenetic marks in individuals with genetic identity cannot be assumed to be inherited. Genetically identical monozygotic twins may acquire epigenetic changes that are distributed throughout the genome and related to the commonality or diversity of their environment (187).
Family studies assessing discordant and concordant phenotypes have demonstrated the complexity of distinguishing inherited and acquired determinants. Fatty liver occurs in 17% of siblings and 37% of parents of overweight children (193). The severity of hepatic steatosis in the family members strongly correlates with body mass index (BMI) (193). Complete hereditability for fatty liver is evident after adjustments for age, gender, race, and BMI, but the phenotypic expression of the inherited risk probably relates to family attitudes about diet and exercise (193). Similarly, heritable miRNAs for NAFLD (miR-331-3p and miR-30c) have been demonstrated in monozygotic and dizygotic twins, but most of the 21 miRNAs that have distinguished the twins with and without NAFLD have not been inherited (194). Although transgenerational inheritance of epigenetic marks has been demonstrated in experimental models (28, 180, 182) and humans (26, 37, 194), its impact on the occurrence of an individual disease is unsettled (35). Large longitudinal studies over several generations are necessary to establish the heritability of epigenetic changes in particular human diseases, and they would require concurrent analyses of the genome and epigenome (37, 183, 195).
6 Epigenetic Changes in Autoimmune Hepatitis
Investigations of the epigenetic changes in patients with autoimmune hepatitis have been limited, and they have focused mainly on DNA methylation patterns in circulating and liver-infiltrating lymphocytes (21) and on the profile of circulating miRNAs (70).
6.1 DNA Methylation Patterns
Most genes in the circulating CD4+ and CD19+ T lymphocytes of untreated patients with autoimmune hepatitis have been hypo-methylated, and this pattern has contrasted with the hyper-methylated pattern in PBC (21) (Table 3). The predominant hypo-methylated pattern has also been recognized in liver-infiltrating, periportal lymphocytes, and it has been reversible after glucocorticoid-induced, laboratory remission (21). The shift in the pre-treatment pattern of DNA hypo-methylation to the post-treatment pattern of DNA hyper-methylation has occurred in most genes, and it suggests that DNA hypo-methylation promotes disease activity by broadly enhancing the transcriptional activity of multiple genes. The cues that trigger the hypo-methylated state, the hypo-methylated genes that account for active disease, and the glucocorticoid actions that shift the methylation status and achieve remission are unclear.
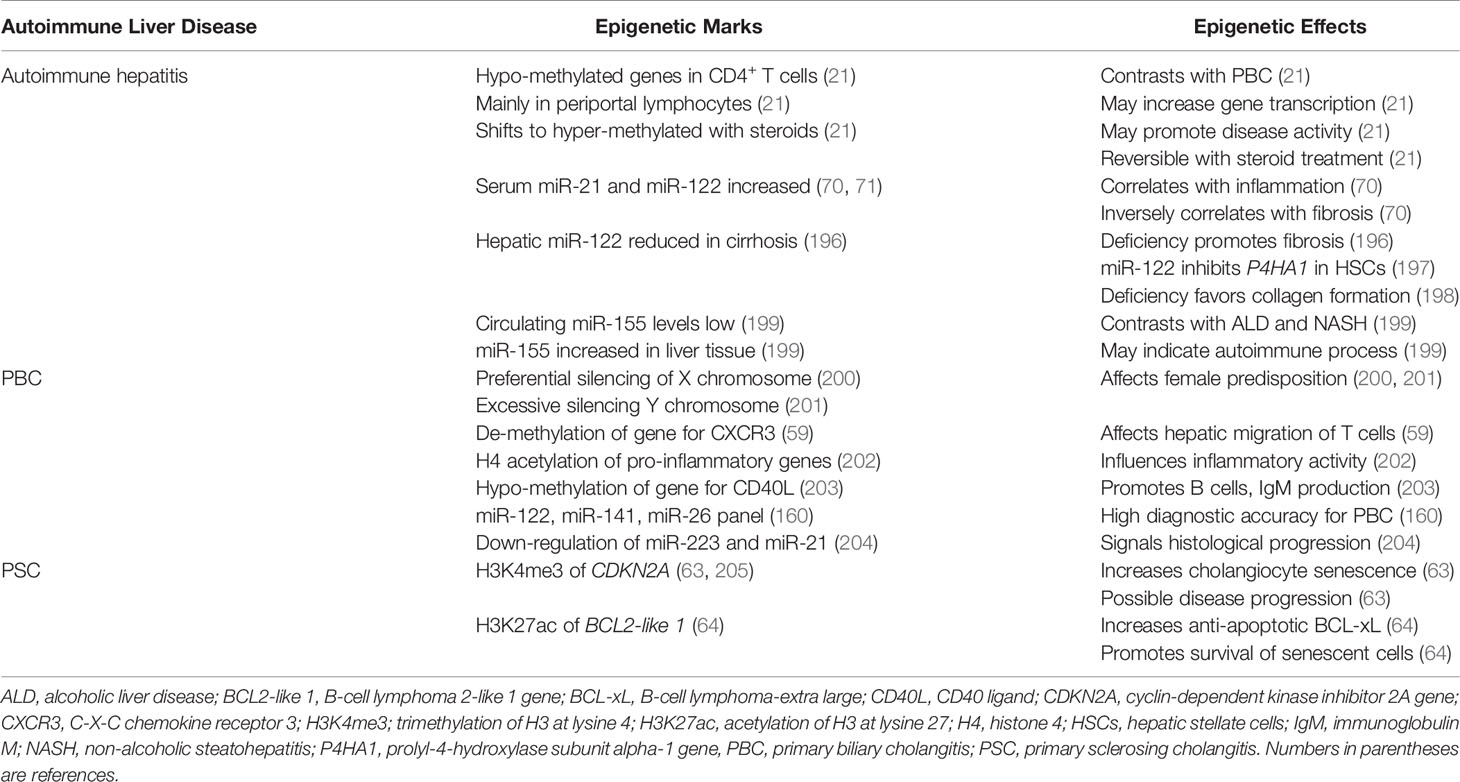
Table 3 Epigenetic marks in autoimmune hepatitis and other autoimmune liver diseases.
6.2 MiRNA Profiles
Circulating levels of miR-21 and miR-122 have been increased in untreated patients with type 1 autoimmune hepatitis (70, 71, 196), and miR-155 has been increased in hepatic tissue (71, 196, 199) (Table 3). The serum miR-21 and miR-122 levels have correlated with serum alanine aminotransferase (ALT) levels, and the serum miR-21 level has correlated with the histological grade of liver inflammation (70). The histological expression of miR-21 in liver tissue has also correlated with serum ALT levels (196).
In contrast, the serum levels of both miR-21 and miR-122 have correlated inversely with the stage of hepatic fibrosis (70), and reduced hepatic concentrations of miR-122 have been associated with cirrhosis (196) (Table 3). MiR-122 markedly attenuates the expression of the gene for prolyl-4-hydroxylase subunit alpha-1 (P4HA1) in hepatic stellate cells (197), thereby preventing the hydroxylation and maturation of stable collagen (198). The findings in autoimmune hepatitis suggest that serum miR-21 and miR-122 levels are biomarkers of inflammatory activity (206) and that a pathological deficiency of miR-122 may promote hepatic fibrosis by de-repressing P4HA1 (196, 197).
Circulating levels of miR-155 have been significantly lower in patients with autoimmune hepatitis regardless of glucocorticoid therapy than in normal individuals (199) (Table 3). In contrast, miR-155 concentrations in liver tissue from patients with autoimmune hepatitis have been 7.6 ± 5.6-fold higher than in liver tissue obtained from normal control subjects (P< 0.01) and significantly higher than in liver tissue from patients with alcoholic liver disease or non-alcoholic steatohepatitis (NASH) (199). The findings suggest that the hepatic expression of miR-155 in autoimmune hepatitis is particularly associated with immune-mediated liver injury. This possibility is supported by the implication of miR-155 in the pathogenesis of other autoimmune diseases (207). The discrepancy between serum and tissue levels may reflect active mobilization of miR-155 from the circulation to the liver.
6.3 Familial Occurrence
The heritability of autoimmune hepatitis through epigenetic traits is unexplored. The familial occurrence of autoimmune hepatitis in Sweden has been mainly among siblings (208, 209) and spouses (208). Among 6269 patients with autoimmune hepatitis in a Swedish database, only siblings have had a significantly increased risk [standardized incidence ratio (SIR), 3.83, 95% confidence interval (CI), 2.09-6.45] (208). Furthermore, the risk for autoimmune hepatitis has been greater among spouses than among siblings (SIR for husbands, 5.91, 95% CI, 2.53-11.7; SIR for wives, 6.07 (95% CI, 2.59-12.02) (208). The risk of autoimmune hepatitis among siblings and spouses in Sweden suggests that epigenetic changes induced by environmental factors may be contributory.
The SIR of autoimmune hepatitis among first-degree relatives has been 4.9 (95% confidence interval [CI], 1.8-10.7) in a Danish database, and the 10-year cumulative risk of autoimmune hepatitis in this group has been 0.10% (95% CI, 0.04-0.23) (210). Among second-degree relatives, there has been no increased risk, whereas among monozygotic twins, the concordance rate for autoimmune hepatitis has been 8.7% (95% CI, 1.1-28) (210). In the composite experience of 32 medical centers in the Netherlands, familial occurrence has been recognized in 0.3% of 564 patients with autoimmune hepatitis, and the disease has occurred in monozygotic twins, the mother of a patient, and the cousin of another patient (211). In each of these experiences, the overall risk of autoimmune hepatitis in family members has been low; heritability has rarely extended beyond the first generation; shared environmental exposures have not been assessed; and the contribution of shared genetic factors has not been evaluated. Differences in the community occurrence of autoimmune hepatitis might also be valuable in assessing non-genetic factors for the disease.
7 Epigenetic Changes in Other Autoimmune Liver Diseases
Histone modifications, DNA methylation status, and miRNAs in blood and liver tissue have been evaluated in experimental models and patients with diverse autoimmune and non-autoimmune liver diseases (19, 39, 86). The investigations have been driven by efforts to catalogue the disease-associated findings and identify associations with pivotal pathogenic mechanisms. Key insights have been derived from studies of PBC (212–214) and PSC (63, 64, 212), and they may prompt and direct future investigations of autoimmune hepatitis (19, 21, 70). The epigenetic changes have not been evaluated for disease-specificity nor have they been fully translated into clinical phenotypes.
7.1 Epigenetic Findings in PBC
The epigenetic changes described in PBC have been discovered mainly by assessing factors influencing its clinical phenotype (212–214). The importance of epigenetic changes has been demonstrated in monozygotic twins concordant (215) and discordant (216) for PBC, and the female predisposition for PBC has guided investigations of the epigenetic influence on the X-chromosome. A preferential, parent-specific, silencing of the X chromosome has been described in women with PBC (200), and an excessive epigenetic silencing of alleles of the Y chromosome has been demonstrated in men with PBC (201). Furthermore, an aberrant DNA methylation pattern of the promoter region of CXCR3 on the X chromosome of CD4+, CD8+, and CD14+ T cells may affect their differentiation and hepatic migration (59) (Table 3). The acetylation of histone 4 in the promoter region of diverse pro-inflammatory genes can enhance their expression in PBC (202), and DNA hypo-methylation of the gene expressing the CD40 ligand (CD40L, also called CD154) in CD4+ T cells may promote B cell maturation and immunoglobulin class switching. The epigenetic effect may contribute to the increased serum levels of immunoglobulin M (IgM) in PBC (203).
A panel of miRNAs, including miR-122-5p, miR-141-3p, and miR-26b-5p, has had high diagnostic accuracy for PBC and a sensitivity that has exceeded that of the serum alkaline phosphatase level (160) (Table 3). Step-down expression of miR-223-3p and miR-21-5p in peripheral blood B cells has signaled histological progression of PBC from stage I to stage III (204), and decreased levels of the molecules involved in the biogenesis of miRNAs (prolyl 4-hydroxylase subunit alpha 1 and Argonaute 2) have suggested a widespread disruption of the homeostatic network in a murine model of PBC (61). This hypothesis has been supported by experimental evidence that non-selective stimulation of miRNA biogenesis with enoxacin can up-regulate miRNA production in CD8+ T cells, decrease T cell proliferation, and reduce interferon-gamma (IFN-γ) production (61).
7.2 Epigenetic Findings in PSC
The epigenetic factors contributing to the progression of PSC have focused mainly on factors influencing the phenotype of the cholangiocytes. Senescent cholangiocytes, defined as cells that have been irreversibly arrested in the G1 or G2 phase of the cell cycle (217, 218), are abundant in the liver of patients with PSC (205) (Table 3). The cholangiocytes exhibit features of a senescence-associated secretory phenotype (SASP) that is characterized by the hypersecretion of pro-inflammatory cytokines, chemokines and growth factors (205, 219). The cyclin-dependent kinase inhibitor 2A (CDKN2A) gene has been associated with cholangiocyte senescence (205), and histone methylation (H3K4me3) increases its transcriptional activity and the possibility of disease progression (63). Histone acetylation (H3K27ac) of the promoter of the B-cell lymphoma 2-like 1 gene (BCL2-like 1) increases expression of the anti-apoptotic protein, B-cell lymphoma-extra large (BCL-xL). This epigenetic change may promote the resistance of senescent cholangiocytes to apoptosis and prolong their survival (64). Both sites of histone modification have been proposed as potential therapeutic targets (63, 64).
The studies in PBC and PSC affirm the strong association of epigenetic modifications in immune-mediated chronic liver disease, and they suggest that the epigenetic modifications can impact on the clinical phenotype, reflect disease-specificity, aid in diagnosis, and direct future therapeutic interventions. They also identify key areas in autoimmune hepatitis that have been unassessed or under-evaluated. Investigations of the epigenetic effects on the X and Y chromosomes, familial predisposition, and heritability of autoimmune hepatitis are wanting.
8 Epigenetic Findings in Non-Autoimmune Liver Diseases
Studies in alcoholic liver disease (54, 55) and non-alcoholic fatty liver disease (NAFLD) (220–229) have emphasized the pervasive, interactive, and composite effects of epigenetic modifications in each disease. They have also indicated the needs to associate changes in disease expression to clinically relevant features and to explore the heritable and adaptive nature of the epigenetic modifications. These insights are foundational for future studies in autoimmune hepatitis since they may clarify the mechanisms of occurrence, recurrence and progression.
8.1 Epigenetic Findings in Alcoholic Liver Disease
A plethora of epigenetic changes involving DNA methylation, histone modification, and circulating miRNA levels have been described in experimental models and patients with alcoholic liver disease and alcoholic steatohepatitis (54, 55). Epigenetic modifications have been demonstrated in genes that may influence the metabolism of ethanol (230–234), the activity of enzymes that mediate histone acetylation (HATs, sirtuins) (55, 235), the vigor of the inflammatory response (236), and the generation of hepatic fibrosis (224, 237) (Table 4). Furthermore, increased circulating levels of several miRNAs have been described that may be biomarkers of alcohol-related liver injury (miR-155) (55, 238, 241, 242), indicators of a disrupted intestinal mucosal barrier (miR-212) (239), or mediators of lipid and cholesterol metabolism (miR-122) (240). The abundance of epigenetic changes and interactions has indicated a complexity that must be edited for clinical relevance. A similar complexity of epigenetic interactions can be anticipated in autoimmune hepatitis, and future investigations must be directed by the pivotal clinical needs to understand and control the severity, progression, and recurrence of the disease.
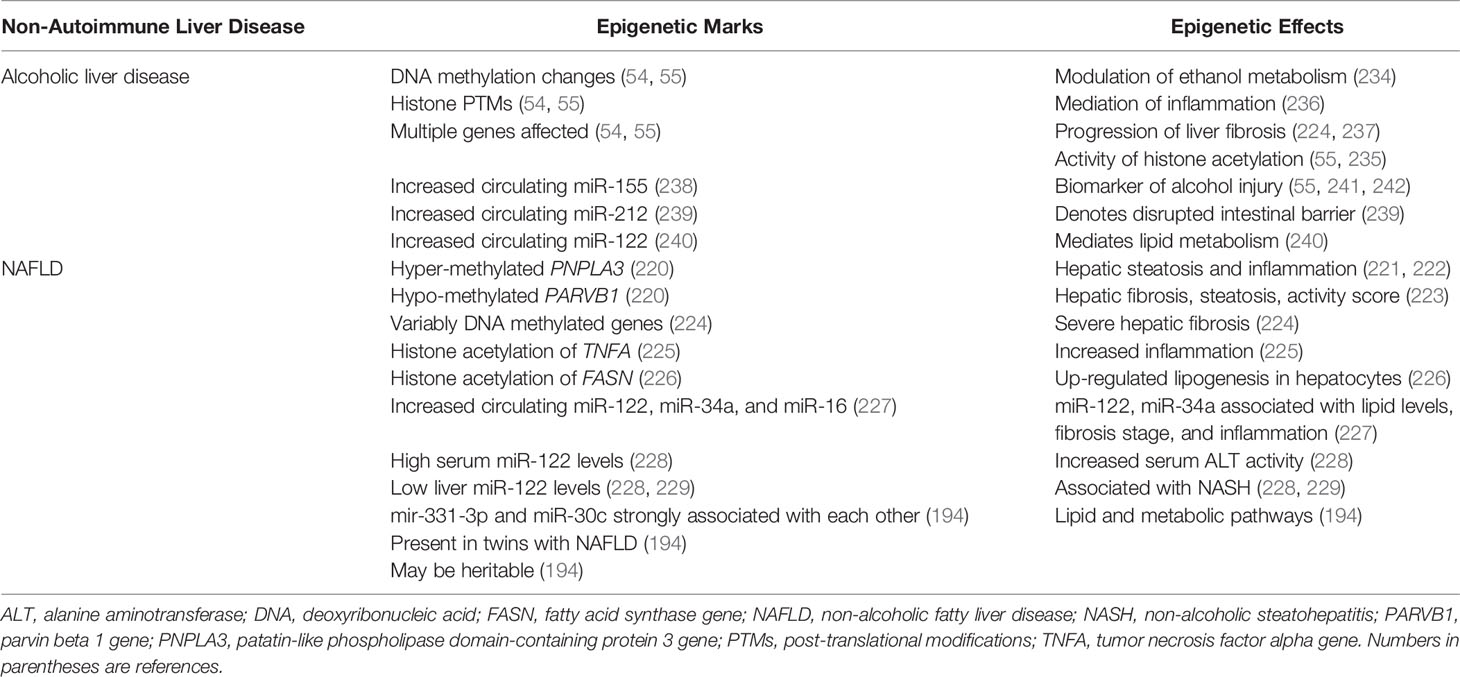
Table 4 Epigenetic marks in non-autoimmune liver diseases.
8.2 Epigenetic Findings in NAFLD
Hypermethylation of CpG99 in the regulatory region of the patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene and hypomethylation of CpG26 in the regulatory region of the parvin beta 1 (PARVB1) gene have been associated with advanced hepatic fibrosis in patients with NAFLD (220) (Table 4). The rs738409 (G-allele) polymorphism of PNPLAS has been associated with hepatic steatosis and inflammation in patients with NAFLD (221, 222), and the PARVB variant has been associated with steatosis grade, NAFLD activity score, and hepatic fibrosis (223). Differential methylation of CpG sites within other genes known to affect hepatic fibrosis have also distinguished patients with severe fibrosis (224).
Histone acetylation of the pro-inflammatory genes, tumor necrosis factor alpha (TNFA) and monocyte chemotactic protein 1 (MCP1; also called CD2) have been up-regulated in a murine model of obesity (225), and the histone acetylation of the gene stimulating transcription of fatty acid synthase (FASN) has been associated with de novo lipogenesis in human hepatocytes (226) (Table 4). Serum levels of miR-122, miR-34a and miR-16 have also been increased in patients with NAFLD compared to patients with chronic hepatitis C, and the serum levels of miR-122 and miR-34a have correlated with biochemical tests and histological assessments of fibrosis stage and inflammatory activity (227).
Serum levels of miR-122 have also been 7.2-fold higher in patients with non-alcoholic steatohepatitis (NASH) than in healthy control subjects and 3.1-fold higher in patients with NASH than in patients with simple steatosis (228) (Table 4). Hepatic expression of miR-122 has been down-regulated in NASH compared to patients with simple steatosis (228) or normal liver (229), and the hepatic expression of miR-122 has been mostly near lipid-laden hepatocytes (228). The physiological significance of miR-122 in the development of NASH has been postulated, but not evident in all investigations (243) or validated as a pivotal pathogenic factor (194).
Studies of monozygotic and dizygotic twins have demonstrated that discordance for NAFLD has been associated with 21 miRNAs, including miR-122 (P=0.002) and miR-34a (P=0.04) (194) (Table 4). MiR-331-3p (P=0.0007) and miR-30c (P=0.011) have been preferentially expressed in the twins with NAFLD, and the strong correlation of miR-331-3p and miR-30c with each other (R=0.90, P=2.2 x 10-16) has suggested their shared involvement in NAFLD (194). This hypothesis has been supported by evidence that the seven gene targets shared by miR-331-3p and miR-30c have included genes affecting lipid and metabolic pathways (194).
The multiplicity of epigenetic changes associated with NAFLD may reflect differences in environmental cues (lifestyle, diet, age-related exposures, surgeries) (184, 187, 193) and transgenerational inheritance of gene modifiers (25, 38, 194). The profiling of the epigenome of sperm from lean and obese men has disclosed marked differences in the expression of small non-coding RNA and DNA methylation patterns which may have reflected inherited and acquired changes (184) (Table 4). The rapid remodeling of DNA methylation in the sperm of morbidly obese men who have undergone bariatric surgery has indicated the dynamic plasticity of epigenetic changes under environmental pressure (184). The challenge has been to identify the key factor or combination of factors that can be moderated in a particular clinical situation. The plasticity of the epigenetic changes in response to environmental cues or therapeutic intervention and the expression of these epigenetic responses in the germline are key features that warrant investigation in autoimmune hepatitis.
9 Epigenetic Manipulations
Therapeutic manipulation of disease-associated epigenetic changes is possible by interventions that affect the enzymes that modify the chromatin structure, the targets recognized by circulating miRNAs, and the environmental factors that promote instability of the epigenome (39, 86, 244). Interventions that affect enzymatic modulation of the chromatin structure have been directed at DNA methylation (245), histone methylation (246, 247), and histone acetylation (248–250). Interventions that affect target recognition by pivotal miRNAs have involved engineered molecules that mask the chosen gene product or substitute a decoy (251–256). Interventions that stabilize the epigenome have included risk-reduction, lifestyle modifications (19, 34, 257) and dietary supplementation with S-adenosylmethionine (258, 259), methyl group donors (260, 261), vitamin C (122), or vitamin D (262–267). The major concerns have been the lack of target selectivity and the uncertain risk of deleterious off-target consequences (19, 43).
9.1 Therapeutic Modulation of Chromatin Structure
DNMT inhibitors, HDAC inhibitors, HDAC activators, and HMT inhibitors have been the principal interventions directed at the enzymatic bases for disease-associated epigenetic changes in chromatin. These interventions have been studied mainly in experimental models of liver disease and patients with malignancy (39, 86, 244) (Table 5).
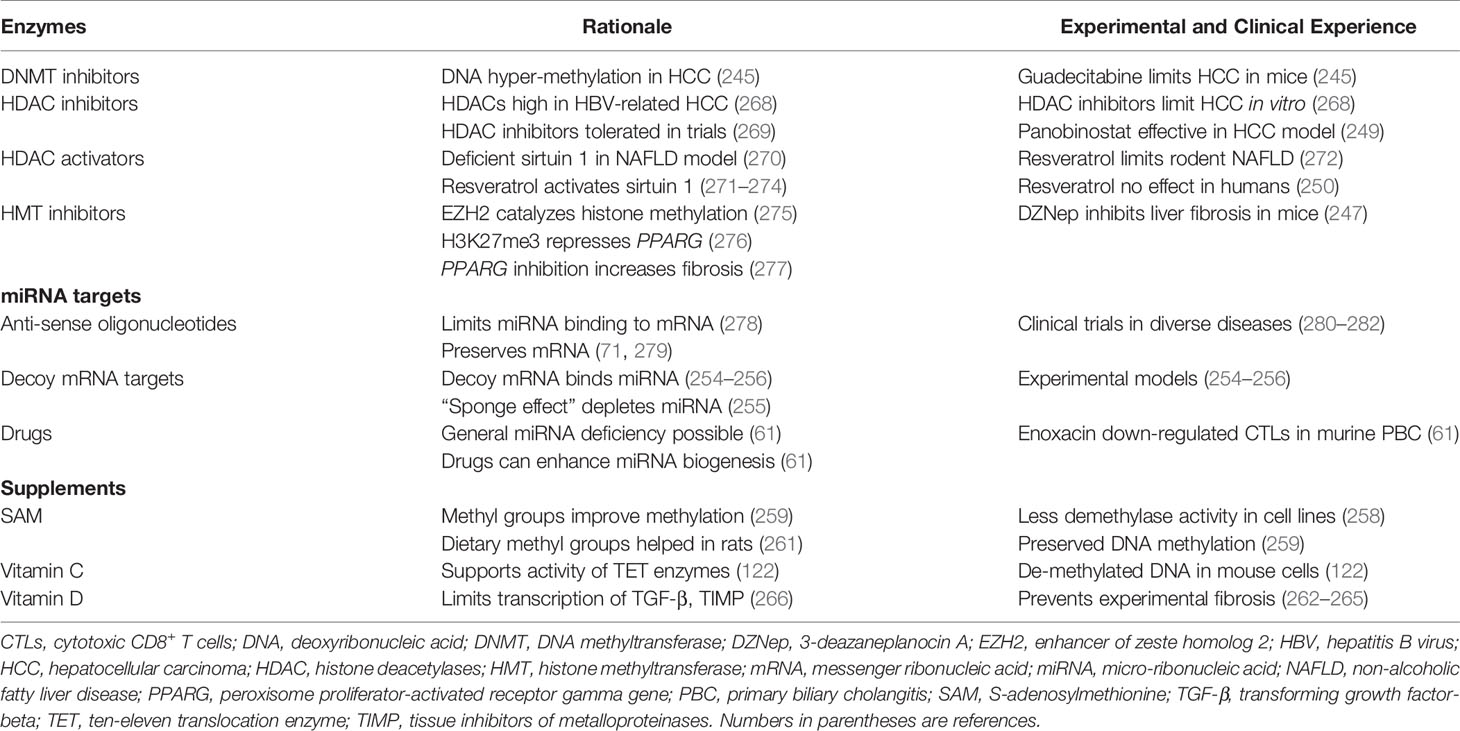
Table 5 Therapeutic manipulations of epigenome.
9.1.1 DNA Methyltransferase Inhibition
DNA hyper-methylation has been a strong feature of hepatocellular carcinoma (HCC), and guadecitabine (also called SGI-110) is a DNMT inhibitor. Guadecitabine has sensitized HCC cells to oxaliplatin by inhibiting signaling pathways that have promoted HCC growth in mice (245).
9.1.2 Histone Deacetylase Inhibition
HDACs have been highly expressed in patients with HCC related to chronic hepatitis B virus infection, and they have been a prognostic biomarker associated with tumor growth and reduced survival (268). HDAC inhibition has suppressed proliferation of HCC cells in vitro (268), and the pan-HDCA inhibitor, panobinostat, has been effective in experimental models of HCC when combined with sorafenib (249). HDAC inhibitors have been well-tolerated in clinical protocols, and trials have been extended to non-tumorous diseases, including neurodegenerative diseases and inflammatory disorders (269) (Table 5).
9.1.3 Histone Deacetylase Activation
Sirtuin 1 (SIRT1) promotes the deacetylation of histones and regulates glucose and fat metabolism (270, 283). Deficient hepatic expression of SIRT1 has been accompanied by metabolic dysfunction in a murine model (270). The polyphenol, resveratrol, activates the deacetylase, SIRT1 (248, 284, 285), and it has improved the survival of mice on a high calorie diet (286) (Table 5). Resveratrol has also protected rodents from diet-induced steatohepatitis through a variety of signaling pathways (271–274). Resveratrol has not had a therapeutic benefit in overweight and obese men with established NAFLD (250), and the role of HDAC activation as a protective or therapeutic intervention for NAFLD remains unclear in humans.
9.1.4 Histone Methyltransferase Inhibition
Epigenetic modifications of chromatin have been implicated in the trans-differentiation of hepatic stellate cells into myofibroblasts (39), and the enzymes that regulate the methylation of DNA (287, 288) and histone (246) have been prime therapeutic targets (247, 277). Hepatic fibrosis is regulated by a series of epigenetic relays that include down-regulation of miR-132, binding of the methyl-CpG binding protein 2 (MeCP2) to the 5’ end of the PPAR-γ-producing gene (PPARG), and activation of the enhancer of zeste homolog 2 (EZH2) (277) (Table 5).
EZH2 is an epigenetic regulator that represses gene transcription by catalyzing the trimethylation of histone 3 at lysine 27 (H3K27me3) (275, 289, 290). The formation of H3K27me3 in the 3’ exon of PPARG represses the anti-fibrotic effect of this gene (276) and promotes hepatic fibrosis (277) (Table 5). Therapeutic disruption of the pro-fibrotic epigenetic pathway is possible at multiple sites, but the pivotal epigenetic step for myofibroblast differentiation is trimethylation of PPARG at H3K27 (276). 3-Deazaneplanocin A (DZNep) is a pan-inhibitor of histone methyltransferase, and its use in a murine model of toxin-induced liver injury has inhibited the histological progression of hepatic fibrosis (247).
9.2 Therapeutic Modulation of MiRNAs
MiRNAs are prime targets for therapeutic manipulation because circulating miRNA levels have distinguished certain diseases and the gene silencing action of miRNAs can disrupt pivotal homeostatic pathways that regulate immune and inflammatory responses (40, 42, 43). The principal method of targeting miRNAs in experimental models and patients in clinical trials has been the use of anti-sense oligonucleotides (antimirs) (71, 278, 279) (Table 5). These molecules are engineered to block the binding of a selected miRNA to its targeted mRNA, and they prevent the miRNA from silencing the gene product. The binding affinity, stability, and potency of antimirs can be enhanced by diverse modifications of the core molecule. The modified molecules have been designated antagomirs (252, 253, 291). Anti-sense obligonucleotides have been evaluated in clinical treatment trials for Alport syndrome (280), chronic hepatitis C (281), and chronic lymphocytic leukemia (282).
RNA transcripts have also been designed to mimic the selected natural mRNA and protect it from degradation or translational repression by miRNAs. The decoy mRNA binds with the natural miRNA and prevents it from silencing the natural mRNA (254–256) (Table 5). Drugs have also been used to non-selectively stimulate the biogenesis of miRNAs (61). Widespread deficiency of miRNAs may allow the expression of genes that promote disease activity, and drug-induced, non-selective stimulation of miRNA biogenesis may silence the expression of these deleterious genes (61). These interventions await rigorous preclinical evaluations and clarification of their safety profile (43).
9.3 Therapeutic Modulation of Environmental Factors and Use of Dietary Supplements
Multiple environmental factors have been associated with diverse epigenetic changes, and lifestyle modifications may reduce the risk of disease-provoking epigenetic changes (19, 34, 58, 257, 292, 293) (Table 5). Medications (procainamide, hydralazine, and 5-azacytidine) (294–297), pollutants (tobacco smoke, aerosolized contaminants, and heavy metals) (298–301), and infection (302) have been associated with changes in DNA methylation that may affect gene expression. Furthermore, environmentally-induced epigenetic changes have been associated with the occurrence or progression of diverse immune-mediated diseases (rheumatoid arthritis, PBC, and SLE) (72, 298, 303–305). Epigenetic changes that are potentially deleterious and heritable have also been associated with nutritional deficiencies, stress, ultraviolet light, radiation, and trauma (19, 34, 257). Lifestyle modifications that avoid excessive, high risk exposures may protect against deleterious epigenetic effects, but their efficacy has been difficult to establish.
Dietary supplements have also been described in experimental animals that enhance the supply of methyl groups (S-adenosylmethionine, diverse methyl donors) (258–261), activate the TET enzymes that de-methylate DNA (vitamin C) (122), and alter the transcription of mRNAs that promote hepatic fibrosis (vitamin D) (262–267) (Table 5). S-adenosylmethionine has inhibited demethylase activity and preserved DNA methylation in cell lines (258, 259). Dietary supplementation with methyl groups has promoted DNA hyper-methylation and prevented transgenerational amplification of obesity in a mouse model (260). It has also modified the methylation profile of the gene expressing fatty acid synthase and reduced hepatic triglyceride accumulation in rats fed a high fat, high sucrose diet (261). Vitamin C has supported the activity of TET enzymes, and it has promoted the de-methylation of DNA in the embryonic stem cells of mice (122). 1, 25-dihydroxyvitamin D has repressed the transcription of mRNAs for TGF-β and tissue inhibitors of metalloproteinases (TIMP). It has also up-regulated the transcription of metalloproteinases and prevented progressive hepatic fibrosis (262–267). These promising pre-clinical experiences await validation in randomized clinical trials that define their utility in specific diseases (259).
10 Epigenetic Prospects in Autoimmune Hepatitis
Findings that have already been made in diverse autoimmune (63, 64, 212–214) and non-autoimmune (220, 223–226, 232) liver diseases support the prospect that multiple, clinically-relevant, epigenetic marks will be identified in autoimmune hepatitis. They also support the prospect that pivotal genes affecting critical pathogenic pathways will be recognized and that interventions will be assessed to modify an aberrant gene expression or pattern (247, 250, 260, 261, 271, 281). The success of these projections in changing the management of autoimmune hepatitis will depend on proofs of causality, confident identification of critical gene expressions or patterns, and precise editing of the epigenetic landscape.
10.1 Proofs of Causality
Progress toward targeted epigenetic management of autoimmune hepatitis ideally requires proof of causality for each epigenetic mark and a hierarchy of candidates based on measured consequences. Methods that disrupt and restore the epigenetic mark in experimental models or cell systems can establish and quantify causality. The clustered, regularly interspaced, short palindromic repeats (CRISPR) of base sequences in segmental DNA and the CRISPR-associated protein 9 (Cas9) system consists of a guide RNA that matches the DNA target site and an endonuclease (Cas9) that performs site-specific DNA cleavage (306, 307) (Table 6). This system has been re-purposed for epigenetic editing by engineering a “deactivated” Cas9 protein (dCas9) that lacks nuclease activity (309–312, 320, 321). The CRISPR-dCas9 system can target specific DNA loci without changing the DNA sequence, and dCas9 can deliver sequence-specific motifs to a desired location in the epigenome (308). Site-specific epigenetic editing that can block or restore gene expression in experimental models or cell lines can prove causality and develop a hierarchy of candidates for therapeutic targeting.
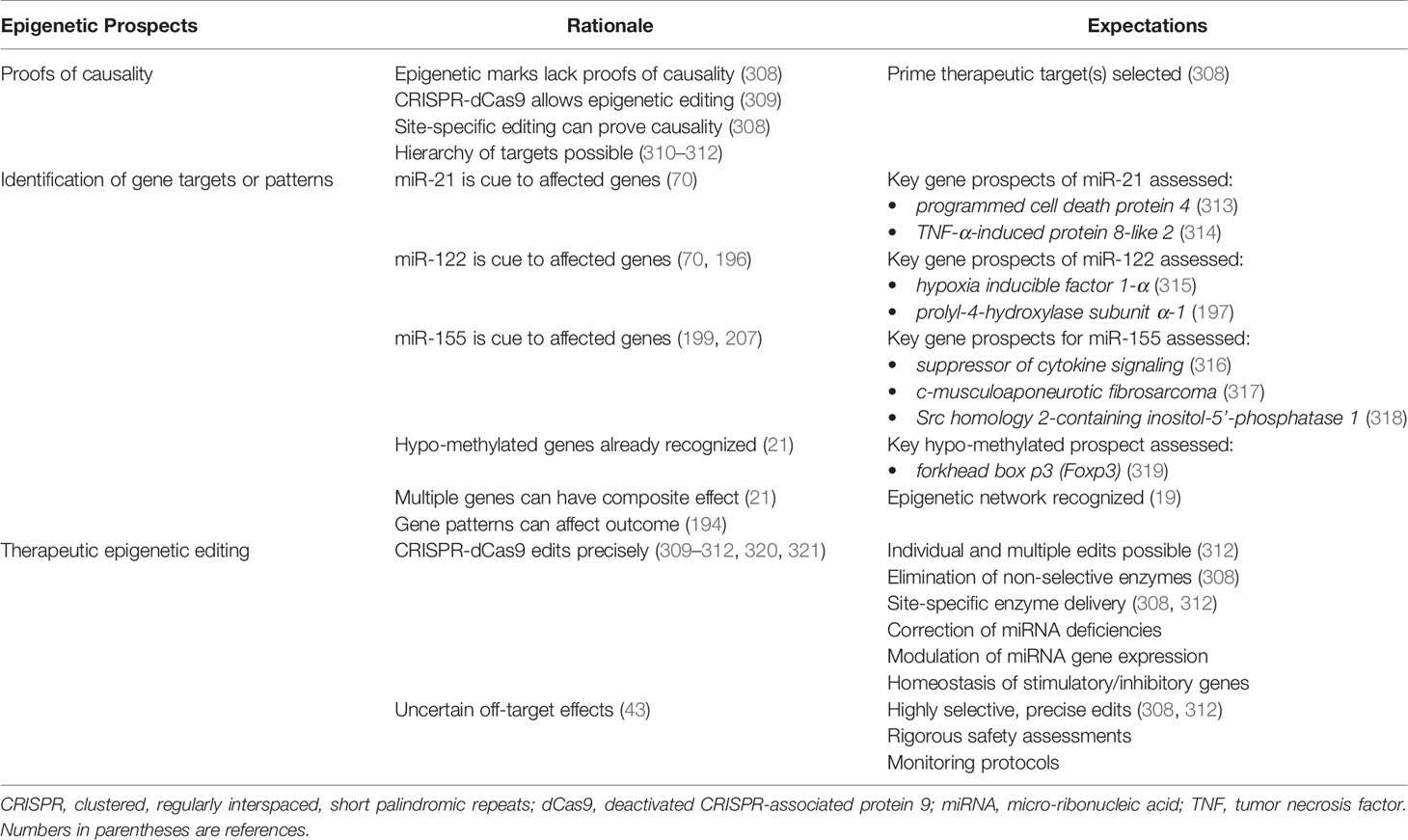
Table 6 Epigenetic prospects in autoimmune hepatitis.
10.2 Identification of Critical Gene Targets or Patterns
A distinctive profile of circulating miRNAs (70, 71, 196, 199) and the hypo-methylation of multiple genes (21) have already been described in autoimmune hepatitis. Future investigations must identify the genes whose expressions are affected by these miRNAs (miR-21, miR-122, and miR-155) and the hypo-methylation (Table 6). The hypo-methylated forkhead box p3 (Foxp3) gene stabilizes the expression of Foxp3 on regulatory T cells (Tregs) and maintains their integrity (319). Preservation of this hypo-methylated state may constitute a mechanism by which to achieve and maintain quiescent disease (322, 323). Hypo-methylation may also stimulate genes with deleterious actions, and treatments that hyper-methylate genes non-selectively may compromise Treg function (322). Clarification of the genes implicated in autoimmune hepatitis by the circulating miRNAs and their hypo-methylated status will be essential in understanding the complexity and interactivity of potential epigenetic targets.
Additional characterization of the epigenome of autoimmune hepatitis can be anticipated, and it may identify multiple up- and down-regulated genes that have a composite effect. Multiple gene expressions have distinguished patients with NAFLD (194), and multiple hypo-methylated genes have been described in autoimmune hepatitis (21). The multiplicity of implicated genes may reveal a pattern that distinguishes autoimmune hepatitis and influences its phenotype and outcome. The pattern may also reveal a common basis for autoimmunity or have disease-specificity.
10.3 Editing the Epigenetic Landscape
The CRISPR-dCas9 system promises to replace the use of enzymes that non-selectively alter DNA methylation and PTMs (309–312, 320, 321) (Table 6). It may also limit or eliminate the need to target miRNAs with anti-sense oligonucleotides (71, 252, 278, 279) or mRNA mimics (254–256). HATs, acetyl groups, DNMTs, and TET enzymes can be tethered to the dCas9 protein and delivered to the chosen epigenetic site by the CRISPR-dCas9 system (312, 320). The effectiveness of the CRISPR-dCas9 system in editing the epigenome of autoimmune diseases is unknown, but future investigations should evaluate its ability to edit multiple epigenetic marks, restore homeostatic balance between immune stimulatory and inhibitory genes, and modulate the genes that generate particular miRNAs. The major safety concern is the uncertainty of unintended off-target effects (34, 43).
11 Conclusions
The epigenome is a largely unstudied domain in autoimmune hepatitis, and its rigorous evaluation may yield results that complement, complete, or change the current knowledge base. The epigenome is dynamic, reactive, adaptable, reversible, and potentially heritable. The epigenetic landscape could influence the predisposition, phenotype, pathogenesis, and outcome of autoimmune hepatitis, and it could reflect environmental factors that can be modified or avoided. The epigenetic landscape could also have diagnostic and prognostic implications that could help direct management. Methods that allow highly selective editing of the epigenome promise to expand treatment options by modulating the expression of pivotal genes or the composite effect of multiple genes. The key challenges are to determine the pivotal epigenetic changes or patterns associated with autoimmune hepatitis, understand the interactive network of genes with opposing actions that promote the disease, and develop interventions that restore homeostatic balance with minimal risk of unintended off-target consequences.
Author Contributions
AC researched, designed, and wrote this article. The tables and figure are original, constructed by the author, and developed solely for this review.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor NK declared a past co-authorship with the author.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Czaja AJ. Genetic Factors Affecting the Occurrence, Clinical Phenotype, and Outcome of Autoimmune Hepatitis. Clin Gastroenterol Hepatol (2008) 6(4):379–88. doi: 10.1016/j.cgh.2007.12.048
2. Czaja AJ. Transitioning From Idiopathic to Explainable Autoimmune Hepatitis. Dig Dis Sci (2015) 60(10):2881–900. doi: 10.1007/s10620-015-3708-7
3. Donaldson PT, Doherty DG, Hayllar KM, McFarlane IG, Johnson PJ, Williams R. Susceptibility to Autoimmune Chronic Active Hepatitis: Human Leukocyte Antigens DR4 and A1-B8-DR3 Are Independent Risk Factors. Hepatology (1991) 13(4):701–6. doi: 10.1002/hep.1840130415
4. Fainboim L, Marcos Y, Pando M, Capucchio M, Reyes GB, Galoppo C, et al. Chronic Active Autoimmune Hepatitis in Children. Strong Association With a Particular HLA-DR6 (DRB1*1301) Haplotype. Hum Immunol (1994) 41(2):146–50. doi: 10.1016/0198-8859(94)90008-6
5. Strettell MD, Donaldson PT, Thomson LJ, Santrach PJ, Moore SB, Czaja AJ, et al. Allelic Basis for HLA-Encoded Susceptibility to Type 1 Autoimmune Hepatitis. Gastroenterology (1997) 112(6):2028–35. doi: 10.1053/gast.1997.v112.pm9178696
6. Bittencourt PL, Goldberg AC, Cancado EL, Porta G, Carrilho FJ, Farias AQ, et al. Genetic Heterogeneity in Susceptibility to Autoimmune Hepatitis Types 1 and 2. Am J Gastroenterol (1999) 94(7):1906–13. doi: 10.1111/j.1572-0241.1999.01229.x
7. Djilali-Saiah I, Fakhfakh A, Louafi H, Caillat-Zucman S, Debray D, Alvarez F. HLA Class II Influences Humoral Autoimmunity in Patients With Type 2 Autoimmune Hepatitis. J Hepatol (2006) 45(6):844–50. doi: 10.1016/j.jhep.2006.07.034
8. Czaja AJ, Carpenter HA. Distinctive Clinical Phenotype and Treatment Outcome of Type 1 Autoimmune Hepatitis in the Elderly. Hepatology (2006) 43(3):532–8. doi: 10.1002/hep.21074
9. van Gerven NM, de Boer YS, Zwiers A, Verwer BJ, Drenth JP, van Hoek B, et al. HLA-DRB1*03:01 and HLA-DRB1*04:01 Modify the Presentation and Outcome in Autoimmune Hepatitis Type-1. Genes Immun (2015) 16(4):247–52. doi: 10.1038/gene.2014.82
10. Cookson S, Constantini PK, Clare M, Underhill JA, Bernal W, Czaja AJ, et al. Frequency and Nature of Cytokine Gene Polymorphisms in Type 1 Autoimmune Hepatitis. Hepatology (1999) 30(4):851–6. doi: 10.1002/hep.510300412
11. Czaja AJ, Cookson S, Constantini PK, Clare M, Underhill JA, Donaldson PT. Cytokine Polymorphisms Associated With Clinical Features and Treatment Outcome in Type 1 Autoimmune Hepatitis. Gastroenterology (1999) 117(3):645–52. doi: 10.1016/S0016-5085(99)70458-0
12. Vogel A, Strassburg CP, Manns MP. Genetic Association of Vitamin D Receptor Polymorphisms With Primary Biliary Cirrhosis and Autoimmune Hepatitis. Hepatology (2002) 35(1):126–31. doi: 10.1053/jhep.2002.30084
13. Vogel A, Strassburg CP, Manns MP. 77 C/G Mutation in the Tyrosine Phosphatase CD45 Gene and Autoimmune Hepatitis: Evidence for a Genetic Link. Genes Immun (2003) 4(1):79–81. doi: 10.1038/sj.gene.6363918
14. Fan LY, Tu XQ, Zhu Y, Pfeiffer T, Feltens R, Stoecker W, et al. Genetic Association of Cytokines Polymorphisms With Autoimmune Hepatitis and Primary Biliary Cirrhosis in the Chinese. World J Gastroenterol (2005) 11(18):2768–72. doi: 10.3748/wjg.v11.i18.2768
15. Fan L, Tu X, Zhu Y, Zhou L, Pfeiffer T, Feltens R, et al. Genetic Association of Vitamin D Receptor Polymorphisms With Autoimmune Hepatitis and Primary Biliary Cirrhosis in the Chinese. J Gastroenterol Hepatol (2005) 20(2):249–55. doi: 10.1111/j.1440-1746.2005.03532.x
16. Agarwal K, Czaja AJ, Donaldson PT. A Functional Fas Promoter Polymorphism Is Associated With a Severe Phenotype in Type 1 Autoimmune Hepatitis Characterized by Early Development of Cirrhosis. Tissue Antigens (2007) 69(3):227–35. doi: 10.1111/j.1399-0039.2006.00794.x
17. de Boer YS, van Gerven NM, Zwiers A, Verwer BJ, van Hoek B, van Erpecum KJ, et al. Genome-Wide Association Study Identifies Variants Associated With Autoimmune Hepatitis Type 1. Gastroenterology (2014) 147(2):443–52. doi: 10.1053/j.gastro.2014.04.022
18. Umemura T, Joshita S, Hamano H, Yoshizawa K, Kawa S, Tanaka E, et al. Association of Autoimmune Hepatitis With Src Homology 2 Adaptor Protein 3 Gene Polymorphisms in Japanese Patients. J Hum Genet (2017) 62(11):963–7. doi: 10.1038/jhg.2017.74
19. Czaja AJ. Epigenetic Changes and Their Implications in Autoimmune Hepatitis. Eur J Clin Invest (2018) 48(4):e12899. doi: 10.1111/eci.12899
20. Webb GJ, Hirschfield GM. Using GWAS to Identify Genetic Predisposition in Hepatic Autoimmunity. J Autoimmun. (2016) 66:25–39. doi: 10.1016/j.jaut.2015.08.016
21. Zachou K, Arvaniti P, Lyberopoulou A, Dalekos GN. Impact of Genetic and Environmental Factors on Autoimmune Hepatitis. J Transl Autoimmun. (2021) 4:100125. doi: 10.1016/j.jtauto.2021.100125
23. Greally JM. A User’s Guide to the Ambiguous Word ‘Epigenetics’. Nat Rev Mol Cell Biol (2018) 19(4):207–8. doi: 10.1038/nrm.2017.135
24. Aristizabal MJ, Anreiter I, Halldorsdottir T, Odgers CL, McDade TW, Goldenberg A, et al. Biological Embedding of Experience: A Primer on Epigenetics. Proc Natl Acad Sci USA (2020) 117(38):23261–9. doi: 10.1073/pnas.1820838116
25. Skinner MK, Nilsson EE. Role of Environmentally Induced Epigenetic Transgenerational Inheritance in Evolutionary Biology: Unified Evolution Theory. Environ Epigenet. (2021) 7(1):dvab012. doi: 10.1093/eep/dvab012
26. Mohajer N, Joloya EM, Seo J, Shioda T, Blumberg B. Epigenetic Transgenerational Inheritance of the Effects of Obesogen Exposure. Front Endocrinol (Lausanne) (2021) 12. doi: 10.3389/fendo.2021.787580
27. Skinner MK. Environmental Epigenetic Transgenerational Inheritance and Somatic Epigenetic Mitotic Stability. Epigenetics (2011) 6(7):838–42. doi: 10.4161/epi.6.7.16537
28. Fitz-James MH, Cavalli G. Molecular Mechanisms of Transgenerational Epigenetic Inheritance. Nat Rev Genet (2022). 23(6):325–41. doi: 10.1038/s41576-021-00438-5
29. Bartova E, Krejci J, Harnicarova A, Galiova G, Kozubek S. Histone Modifications and Nuclear Architecture: A Review. J Histochem. Cytochem. (2008) 56(8):711–21. doi: 10.1369/jhc.2008.951251
30. Cedar H, Bergman Y. Linking DNA Methylation and Histone Modification: Patterns and Paradigms. Nat Rev Genet (2009) 10(5):295–304. doi: 10.1038/nrg2540
31. Taylor BC, Young NL. Combinations of Histone Post-Translational Modifications. Biochem J (2021) 478(3):511–32. doi: 10.1042/BCJ20200170
32. Wei JW, Huang K, Yang C, Kang CS. Non-Coding RNAs as Regulators in Epigenetics (Review). Oncol Rep (2017) 37(1):3–9. doi: 10.3892/or.2016.5236
33. Zeng X, Yuan X, Cai Q, Tang C, Gao J. Circular RNA as an Epigenetic Regulator in Chronic Liver Diseases. Cells (2021) 10(8):1945. doi: 10.3390/cells10081945
34. Skinner MK, Manikkam M, Guerrero-Bosagna C. Epigenetic Transgenerational Actions of Environmental Factors in Disease Etiology. Trends Endocrinol Metab (2010) 21(4):214–22. doi: 10.1016/j.tem.2009.12.007
35. Boyce WT, Kobor MS. Development and the Epigenome: The ‘Synapse’ of Gene-Environment Interplay. Dev Sci (2015) 18(1):1–23. doi: 10.1111/desc.12282
36. Cavalli G, Heard E. Advances in Epigenetics Link Genetics to the Environment and Disease. Nature (2019) 571(7766):489–99. doi: 10.1038/s41586-019-1411-0
37. Ghai M, Kader F. A Review on Epigenetic Inheritance of Experiences in Humans. Biochem Genet (2021). doi: 10.1007/s10528-021-10155-7
38. Margueron R, Reinberg D. Chromatin Structure and the Inheritance of Epigenetic Information. Nat Rev Genet (2010) 11(4):285–96. doi: 10.1038/nrg2752
39. Hardy T, Mann DA. Epigenetics in Liver Disease: From Biology to Therapeutics. Gut (2016) 65(11):1895–905. doi: 10.1136/gutjnl-2015-311292
40. Broderick JA, Zamore PD. MicroRNA Therapeutics. Gene Ther (2011) 18(12):1104–10. doi: 10.1038/gt.2011.50
41. Burnett JC, Rossi JJ, Tiemann K. Current Progress of SiRNA/ShRNA Therapeutics in Clinical Trials. Biotechnol J (2011) 6(9):1130–46. doi: 10.1002/biot.201100054
42. Takahashi K, Yan I, Wen HJ, Patel T. MicroRNAs in Liver Disease: From Diagnostics to Therapeutics. Clin Biochem (2013) 46(10-11):946–52. doi: 10.1016/j.clinbiochem.2013.01.025
43. Wittrup A, Lieberman J. Knocking Down Disease: A Progress Report on SiRNA Therapeutics. Nat Rev Genet (2015) 16(9):543–52. doi: 10.1038/nrg3978
44. Beck S, Trowsdale J. The Human Major Histocompatability Complex: Lessons From the DNA Sequence. Annu Rev Genomics Hum Genet (2000) 1:117–37. doi: 10.1146/annurev.genom.1.1.117
45. Choo SY. The HLA System: Genetics, Immunology, Clinical Testing, and Clinical Implications. Yonsei Med J (2007) 48(1):11–23. doi: 10.3349/ymj.2007.48.1.11
47. Jeffries MA, Sawalha AH. Autoimmune Disease in the Epigenetic Era: How Has Epigenetics Changed Our Understanding of Disease and How Can We Expect the Field to Evolve? Expert Rev Clin Immunol (2015) 11(1):45–58. doi: 10.1586/1744666X.2015.994507
48. Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell (2004) 116(2):281–97. doi: 10.1016/s0092-8674(04)00045-5
49. Kutter C, Svoboda P. MiRNA, SiRNA, PiRNA: Knowns of the Unknown. RNA Biol (2008) 5(4):181–8. doi: 10.4161/rna.7227
50. Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell (2009) 136(2):215–33. doi: 10.1016/j.cell.2009.01.002
51. Pratt AJ, MacRae IJ. The Rna-Induced Silencing Complex: A Versatile Gene-Silencing Machine. J Biol Chem (2009) 284(27):17897–901. doi: 10.1074/jbc.R900012200
52. O’Hara SP, Mott JL, Splinter PL, Gores GJ, LaRusso NF. Micrornas: Key Modulators of Posttranscriptional Gene Expression. Gastroenterology (2009) 136(1):17–25. doi: 10.1053/j.gastro.2008.11.028
53. Loosen SH, Schueller F, Trautwein C, Roy S, Roderburg C. Role of Circulating MicroRNAs in Liver Diseases. World J Hepatol (2017) 9(12):586–94. doi: 10.4254/wjh.v9.i12.586
54. Mandrekar P. Epigenetic Regulation in Alcoholic Liver Disease. World J Gastroenterol (2011) 17(20):2456–64. doi: 10.3748/wjg.v17.i20.2456
55. Kim HG, Cho JH, Kim J, Kim SJ. The Role of Epigenetic Changes in the Progression of Alcoholic Steatohepatitis. Front Physiol (2021) 12. doi: 10.3389/fphys.2021.691738
56. Li YY. Genetic and Epigenetic Variants Influencing the Development of Nonalcoholic Fatty Liver Disease. World J Gastroenterol (2012) 18(45):6546–51. doi: 10.3748/wjg.v18.i45.6546
57. Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, et al. Relationship Between Methylome and Transcriptome in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology (2013) 145(5):1076–87. doi: 10.1053/j.gastro.2013.07.047
58. Sodum N, Kumar G, Bojja SL, Kumar N, Rao CM. Epigenetics in NAFLD/NASH: Targets and Therapy. Pharmacol Res (2021) 167:105484. doi: 10.1016/j.phrs.2021.105484
59. Lleo A, Zhang W, Zhao M, Tan Y, Bernuzzi F, Zhu B, et al. DNA Methylation Profiling of the X Chromosome Reveals an Aberrant Demethylation on CXCR3 Promoter in Primary Biliary Cirrhosis. Clin Epigenet (2015) 7:61. doi: 10.1186/s13148-015-0098-9
60. Tomiyama T, Yang GX, Zhao M, Zhang W, Tanaka H, Wang J, et al. The Modulation of Co-Stimulatory Molecules by Circulating Exosomes in Primary Biliary Cirrhosis. Cell Mol Immunol (2017) 14(3):276–84. doi: 10.1038/cmi.2015.86
61. Itoh A, Adams D, Huang W, Wu Y, Kachapati K, Bednar KJ, et al. Enoxacin Up-Regulates MicroRNA Biogenesis and Down-Regulates Cytotoxic CD8 T-Cell Function in Autoimmune Cholangitis. Hepatology (2021) 74(2):835–46. doi: 10.1002/hep.31724
62. Bernuzzi F, Marabita F, Lleo A, Carbone M, Mirolo M, Marzioni M, et al. Serum MicroRNAs as Novel Biomarkers for Primary Sclerosing Cholangitis and Cholangiocarcinoma. Clin Exp Immunol (2016) 185(1):61–71. doi: 10.1111/cei.12776
63. O’Hara SP, Splinter PL, Trussoni CE, Pisarello MJ, Loarca L, Splinter NS, et al. ETS Proto-Oncogene 1 Transcriptionally Up-Regulates the Cholangiocyte Senescence-Associated Protein Cyclin-Dependent Kinase Inhibitor 2a. J Biol Chem (2017) 292(12):4833–46. doi: 10.1074/jbc.M117.777409
64. O’Hara SP, Splinter PL, Trussoni CE, Guicciardi ME, Splinter NP, Al Suraih MS, et al. The Transcription Factor ETS1 Promotes Apoptosis Resistance of Senescent Cholangiocytes by Epigenetically Up-Regulating the Apoptosis Suppressor Bcl2l1. J Biol Chem (2019) 294(49):18698–713. doi: 10.1074/jbc.RA119.010176
65. Yang B, House MG, Guo M, Herman JG, Clark DP. Promoter Methylation Profiles of Tumor Suppressor Genes in Intrahepatic and Extrahepatic Cholangiocarcinoma. Mod Pathol (2005) 18(3):412–20. doi: 10.1038/modpathol.3800287
66. Andresen K, Boberg KM, Vedeld HM, Honne H, Hektoen M, Wadsworth CA, et al. Novel Target Genes and a Valid Biomarker Panel Identified for Cholangiocarcinoma. Epigenetics (2012) 7(11):1249–57. doi: 10.4161/epi.22191
67. Timmer MR, Beuers U, Fockens P, Ponsioen CY, Rauws EA, Wang KK, et al. Genetic and Epigenetic Abnormalities in Primary Sclerosing Cholangitis-Associated Cholangiocarcinoma. Inflammation Bowel Dis (2013) 19(8):1789–97. doi: 10.1097/MIB.0b013e318281f49a
68. Lambert MP, Paliwal A, Vaissiere T, Chemin I, Zoulim F, Tommasino M, et al. Aberrant DNA Methylation Distinguishes Hepatocellular Carcinoma Associated With HBV and HCV Infection and Alcohol Intake. J Hepatol (2011) 54(4):705–15. doi: 10.1016/j.jhep.2010.07.027
69. Liu N, Chang CW, Steer CJ, Wang XW, Song G. Microrna-15a/16-1 Prevents Hepatocellular Carcinoma by Disrupting the Communication Between Kupffer Cells and Regulatory T Cells. Gastroenterology (2022) 162(2):575–89. doi: 10.1053/j.gastro.2021.10.015
70. Migita K, Komori A, Kozuru H, Jiuchi Y, Nakamura M, Yasunami M, et al. Circulating MicroRNA Profiles in Patients With Type-1 Autoimmune Hepatitis. PLoS One (2015) 10(11):e0136908. doi: 10.1371/journal.pone.0136908
71. Huang C, Xing X, Xiang X, Fan X, Men R, Ye T, et al. MicroRNAs in Autoimmune Liver Diseases: From Diagnosis to Potential Therapeutic Targets. BioMed Pharmacother (2020) 130:110558. doi: 10.1016/j.biopha.2020.110558
72. Somers EC, Richardson BC. Environmental Exposures, Epigenetic Changes and the Risk of Lupus. Lupus (2014) 23(6):568–76. doi: 10.1177/0961203313499419
73. Relle M, Foehr B, Schwarting A. Epigenetic Aspects of Systemic Lupus Erythematosus. Rheumatol. Ther (2015) 2(1):33–46. doi: 10.1007/s40744-015-0014-y
74. Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-Wide Association Data Implicate DNA Methylation as an Intermediary of Genetic Risk in Rheumatoid Arthritis. Nat Biotechnol (2013) 31(2):142–7. doi: 10.1038/nbt.2487
75. Smigielska-Czepiel K, van den Berg A, Jellema P, van der Lei RJ, Bijzet J, Kluiver J, et al. Comprehensive Analysis of MiRNA Expression in T-Cell Subsets of Rheumatoid Arthritis Patients Reveals Defined Signatures of Naive and Memory Tregs. Genes Immun (2014) 15(2):115–25. doi: 10.1038/gene.2013.69
76. Lei W, Luo Y, Lei W, Luo Y, Yan K, Zhao S, et al. Abnormal DNA Methylation in CD4+ T Cells From Patients With Systemic Lupus Erythematosus, Systemic Sclerosis, and Dermatomyositis. Scand J Rheumatol. (2009) 38(5):369–74. doi: 10.1080/03009740902758875
77. Lian X, Xiao R, Hu X, Kanekura T, Jiang H, Li Y, et al. DNA Demethylation of CD40l in CD4+ T Cells From Women With Systemic Sclerosis: A Possible Explanation for Female Susceptibility. Arthritis Rheum (2012) 64(7):2338–45. doi: 10.1002/art.34376
78. Schneider A, Chatterjee S, Bousiges O, Selvi BR, Swaminathan A, Cassel R, et al. Acetyltransferases (HATs) as Targets for Neurological Therapeutics. Neurotherapeutics (2013) 10(4):568–88. doi: 10.1007/s13311-013-0204-7
79. Dawson MA, Kouzarides T. Cancer Epigenetics: From Mechanism to Therapy. Cell (2012) 150(1):12–27. doi: 10.1016/j.cell.2012.06.013
80. Rius M, Lyko F. Epigenetic Cancer Therapy: Rationales, Targets and Drugs. Oncogene (2012) 31(39):4257–65. doi: 10.1038/onc.2011.601
81. You JS, Jones PA. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell (2012) 22(1):9–20. doi: 10.1016/j.ccr.2012.06.008
82. Jha G, Azhar S, Rashid U, Khalaf H, Alhalabi N, Ravindran D, et al. Epigenetics: The Key to Future Diagnostics and Therapeutics of Lung Cancer. Cureus (2021) 13(11):e19770. doi: 10.7759/cureus.19770
83. Jones D, Manns MP, Terracciano L, Torbenson M, Vierling JM. Unmet Needs and New Models for Future Trials in Autoimmune Hepatitis. Lancet Gastroenterol Hepatol (2018) 3(5):363–70. doi: 10.1016/S2468-1253(18)30043-8
84. Mack CL, Adams D, Assis DN, Kerkar N, Manns MP, Mayo MJ, et al. Diagnosis and Management of Autoimmune Hepatitis in Adults and Children: 2019 Practice Guidance and Guidelines From the American Association for the Study of Liver Diseases. Hepatology (2020) 72(2):671–722. doi: 10.1002/hep.31065
85. Vierling JM, Kerkar N, Czaja AJ, Mack CL, Adams D, Assis DN, et al. Immunosuppressive Treatment Regimens in Autoimmune Hepatitis: Systematic Reviews and Meta-Analyses Supporting American Association for the Study of Liver Diseases Guidelines. Hepatology (2020) 72(2):753–69. doi: 10.1002/hep.31407
86. Cai Q, Gan C, Tang C, Wu H, Gao J. Mechanism and Therapeutic Opportunities of Histone Modifications in Chronic Liver Disease. Front Pharmacol (2021) 12. doi: 10.3389/fphar.2021.784591
87. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal Structure of the Nucleosome Core Particle at 2.8A Resolution. Nature (1997) 389(6648):251–60. doi: 10.1038/38444
88. Strahl BD, Allis CD. The Language of Covalent Histone Modifications. Nature (2000) 403(6765):41–5. doi: 10.1038/47412
89. Lawrence M, Daujat S, Schneider R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet (2016) 32(1):42–56. doi: 10.1016/j.tig.2015.10.007
90. Fyodorov DV, Zhou BR, Skoultchi AI, Bai Y. Emerging Roles of Linker Histones in Regulating Chromatin Structure and Function. Nat Rev Mol Cell Biol (2018) 19(3):192–206. doi: 10.1038/nrm.2017.94
91. Renaudineau Y, Youinou P. Epigenetics and Autoimmunity, With Special Emphasis on Methylation. Keio. J Med (2011) 60(1):10–6. doi: 10.2302/kjm.60.10
92. Cedar H, Bergman Y. Programming of DNA Methylation Patterns. Annu Rev Biochem (2012) 81:97–117. doi: 10.1146/annurev-biochem-052610-091920
93. Chen L, Huang W, Wang L, Zhang Z, Zhang F, Zheng S, et al. The Effects of Epigenetic Modification on the Occurrence and Progression of Liver Diseases and the Involved Mechanism. Expert Rev Gastroenterol Hepatol (2020) 14(4):259–70. doi: 10.1080/17474124.2020.1736042
94. Iguchi-Ariga SM, Schaffner W. CpG Methylation of the Camp-Responsive Enhancer/Promoter Sequence TGACGTCA Abolishes Specific Factor Binding as Well as Transcriptional Activation. Genes Dev (1989) 3(5):612–9. doi: 10.1101/gad.3.5.612
95. Campanero MR, Armstrong MI, Flemington EK. Cpg Methylation as a Mechanism for the Regulation of E2F Activity. Proc Natl Acad Sci USA (2000) 97(12):6481–6. doi: 10.1073/pnas.100340697
96. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by Mll Partner Tet1. Science (2009) 324(5929):930–5. doi: 10.1126/science.1170116
97. Wu X, Zhang Y. TET-Mediated Active DNA Demethylation: Mechanism, Function and Beyond. Nat Rev Genet (2017) 18(9):517–34. doi: 10.1038/nrg.2017.33
98. Daskalaki MG, Tsatsanis C, Kampranis SC. Histone Methylation and Acetylation in Macrophages as a Mechanism for Regulation of Inflammatory Responses. J Cell Physiol (2018) 233(9):6495–507. doi: 10.1002/jcp.26497
99. Guo P, Chen W, Li H, Li M, Li L. The Histone Acetylation Modifications of Breast Cancer and Their Therapeutic Implications. Pathol Oncol Res (2018) 24(4):807–13. doi: 10.1007/s12253-018-0433-5
100. Janssen A, Colmenares SU, Karpen GH. Heterochromatin: Guardian of the Genome. Annu Rev Cell Dev Biol (2018) 34:265–88. doi: 10.1146/annurev-cellbio-100617-062653
101. Wysocka J, Allis CD, Coonrod S. Histone Arginine Methylation and Its Dynamic Regulation. Front Biosci (2006) 11. doi: 10.2741/1802
102. Bedford MT, Clarke SG. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol Cell (2009) 33(1):1–13. doi: 10.1016/j.molcel.2008.12.013
103. Black JC, Van Rechem C, Whetstine JR. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol Cell (2012) 48(4):491–507. doi: 10.1016/j.molcel.2012.11.006
104. Greer EL, Shi Y. Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance. Nat Rev Genet (2012) 13(5):343–57. doi: 10.1038/nrg3173
105. Blanc RS, Richard S. Arginine Methylation: The Coming of Age. Mol Cell (2017) 65(1):8–24. doi: 10.1016/j.molcel.2016.11.003
106. Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, et al. Selective Recognition of Methylated Lysine 9 on Histone H3 by the HP1 Chromo Domain. Nature (2001) 410(6824):120–4. doi: 10.1038/35065138
107. Hyun K, Jeon J, Park K, Kim J. Writing, Erasing and Reading Histone Lysine Methylations. Exp Mol Med (2017) 49(4):e324. doi: 10.1038/emm.2017.11
108. Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the Epigenetic Landscape Through Histone Readers. Nat Struct Mol Biol (2012) 19(12):1218–27. doi: 10.1038/nsmb.2436
109. Patel DJ, Wang Z. Readout of Epigenetic Modifications. Annu Rev Biochem (2013) 82:81–118. doi: 10.1146/annurev-biochem-072711-165700
110. Musselman CA, Khorasanizadeh S, Kutateladze TG. Towards Understanding Methyllysine Readout. Biochim Biophys Acta (2014) 1839(8):686–93. doi: 10.1016/j.bbagrm.2014.04.001
111. Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, et al. Active Genes Are Tri-Methylated at K4 of Histone H3. Nature (2002) 419(6905):407–11. doi: 10.1038/nature01080
112. D’Oto A, Tian QW, Davidoff AM, Yang J. Histone Demethylases and Their Roles in Cancer Epigenetics. J Med Oncol Ther (2016) 1(2):34–40. doi: 10.35841/medical-oncology.1.2.34-40
113. Bannister AJ, Kouzarides T. Regulation of Chromatin by Histone Modifications. Cell Res (2011) 21(3):381–95. doi: 10.1038/cr.2011.22
114. Rossetto D, Avvakumov N, Cote J. Histone Phosphorylation: A Chromatin Modification Involved in Diverse Nuclear Events. Epigenetics (2012) 7(10):1098–108. doi: 10.4161/epi.21975
115. de la Barre AE, Gerson V, Gout S, Creaven M, Allis CD, Dimitrov S. Core Histone N-Termini Play an Essential Role in Mitotic Chromosome Condensation. EMBO J (2000) 19(3):379–91. doi: 10.1093/emboj/19.3.379
116. Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2ax: The Histone Guardian of the Genome. DNA Repair (Amst) (2004) 3(8-9):959–67. doi: 10.1016/j.dnarep.2004.03.024
117. Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, et al. Binding of Chromatin-Modifying Activities to Phosphorylated Histone H2a at DNA Damage Sites. Mol Cell (2004) 16(6):979–90. doi: 10.1016/j.molcel.2004.12.003
118. Rasmussen KD, Helin K. Role of TET Enzymes in DNA Methylation, Development, and Cancer. Genes Dev (2016) 30(7):733–50. doi: 10.1101/gad.276568.115
119. Ma C, Seong H, Liu Y, Yu X, Xu S, Li Y. Ten-Eleven Translocation Proteins (TETs): Tumor Suppressors or Tumor Enhancers? Front Biosci (Landmark Ed) (2021) 26(10). doi: 10.52586/4996
120. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. TET-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science (2011) 333(6047):1303–7. doi: 10.1126/science.1210944
121. Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu GL, et al. Thymine DNA Glycosylase Specifically Recognizes 5-Carboxylcytosine-Modified DNA. Nat Chem Biol (2012) 8(4):328–30. doi: 10.1038/nchembio.914
122. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martinez JA, Goyal P, Mahapatra S, et al. Vitamin C Induces TET-Dependent DNA Demethylation and a Blastocyst-Like State in ES Cells. Nature (2013) 500(7461):222–6. doi: 10.1038/nature12362
123. Li B, Carey M, Workman JL. The Role of Chromatin During Transcription. Cell (2007) 128(4):707–19. doi: 10.1016/j.cell.2007.01.015
124. Clapier CR, Cairns BR. The Biology of Chromatin Remodeling Complexes. Annu Rev Biochem (2009) 78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223
125. Suganuma T, Workman JL. Signals and Combinatorial Functions of Histone Modifications. Annu Rev Biochem (2011) 80:473–99. doi: 10.1146/annurev-biochem-061809-175347
126. Allis CD, Jenuwein T. The Molecular Hallmarks of Epigenetic Control. Nat Rev Genet (2016) 17(8):487–500. doi: 10.1038/nrg.2016.59
127. Tarakhovsky A. Tools and Landscapes of Epigenetics. Nat Immunol (2010) 11(7):565–8. doi: 10.1038/ni0710-565
128. Gong F, Miller KM. Histone Methylation and the DNA Damage Response. Mutat Res Rev Mutat Res (2019) 780:37–47. doi: 10.1016/j.mrrev.2017.09.003
129. Lukas J, Lukas C, Bartek J. More Than Just a Focus: The Chromatin Response to DNA Damage and Its Role in Genome Integrity Maintenance. Nat Cell Biol (2011) 13(10):1161–9. doi: 10.1038/ncb2344
130. Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, et al. Regulation of Chromatin Structure by Site-Specific Histone H3 Methyltransferases. Nature (2000) 406(6796):593–9. doi: 10.1038/35020506
131. Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, et al. A Phd Finger of Nurf Couples Histone H3 Lysine 4 Trimethylation With Chromatin Remodelling. Nature (2006) 442(7098):86–90. doi: 10.1038/nature04815
132. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell (2007) 129(4):823–37. doi: 10.1016/j.cell.2007.05.009
133. Chadee DN, Hendzel MJ, Tylipski CP, Allis CD, Bazett-Jones DP, Wright JA, et al. Increased Ser-10 Phosphorylation of Histone H3 in Mitogen-Stimulated and Oncogene-Transformed Mouse Fibroblasts. J Biol Chem (1999) 274(35):24914–20. doi: 10.1074/jbc.274.35.24914
134. Choi HS, Choi BY, Cho YY, Mizuno H, Kang BS, Bode AM, et al. Phosphorylation of Histone H3 at Serine 10 Is Indispensable for Neoplastic Cell Transformation. Cancer Res (2005) 65(13):5818–27. doi: 10.1158/0008-5472.CAN-05-0197
135. Lau AT, Lee SY, Xu YM, Zheng D, Cho YY, Zhu F, et al. Phosphorylation of Histone H2b Serine 32 Is Linked to Cell Transformation. J Biol Chem (2011) 286(30):26628–37. doi: 10.1074/jbc.M110.215590
136. Lau PN, Cheung P. Histone Code Pathway Involving H3 S28 Phosphorylation and K27 Acetylation Activates Transcription and Antagonizes Polycomb Silencing. Proc Natl Acad Sci USA (2011) 108(7):2801–6. doi: 10.1073/pnas.1012798108
137. Clements A, Poux AN, Lo WS, Pillus L, Berger SL, Marmorstein R. Structural Basis for Histone and Phosphohistone Binding by the Gcn5 Histone Acetyltransferase. Mol Cell (2003) 12(2):461–73. doi: 10.1016/s1097-2765(03)00288-0
138. Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, Oliviero S. Histone Crosstalk Between H3S10ph and H4K16ac Generates a Histone Code That Mediates Transcription Elongation. Cell (2009) 138(6):1122–36. doi: 10.1016/j.cell.2009.07.031
139. Zeng L, Zhou MM. Bromodomain: An Acetyl-Lysine Binding Domain. FEBS Lett (2002) 513(1):124–8. doi: 10.1016/s0014-5793(01)03309-9
140. Shi J, Vakoc CR. The Mechanisms Behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol Cell (2014) 54(5):728–36. doi: 10.1016/j.molcel.2014.05.016
141. Wei Y, Mizzen CA, Cook RG, Gorovsky MA, Allis CD. Phosphorylation of Histone H3 at Serine 10 Is Correlated With Chromosome Condensation During Mitosis and Meiosis in Tetrahymena. Proc Natl Acad Sci USA (1998) 95(13):7480–4. doi: 10.1073/pnas.95.13.7480
142. Mattick JS, Makunin IV. Non-Coding RNA. Hum Mol Genet (2006) 15:R17–29. doi: 10.1093/hmg/ddl046
143. Palazzo AF, Lee ES. Non-Coding RNA: What Is Functional and What Is Junk? Front Genet (2015) 6. doi: 10.3389/fgene.2015.00002
144. Choudhuri S. Small Noncoding RNAs: Biogenesis, Function, and Emerging Significance in Toxicology. J Biochem Mol Toxicol (2010) 24(3):195–216. doi: 10.1002/jbt.20325
145. Seto AG, Kingston RE, Lau NC. The Coming of Age for Piwi Proteins. Mol Cell (2007) 26(5):603–9. doi: 10.1016/j.molcel.2007.05.021
146. Siomi MC, Sato K, Pezic D, Aravin AA. Piwi-Interacting Small RNAs: The Vanguard of Genome Defence. Nat Rev Mol Cell Biol (2011) 12(4):246–58. doi: 10.1038/nrm3089
147. Leung AKL. The Whereabouts of MicroRNA Actions: Cytoplasm and Beyond. Trends Cell Biol (2015) 25(10):601–10. doi: 10.1016/j.tcb.2015.07.005
148. Roberts TC. The MicroRNA Biology of the Mammalian Nucleus. Mol Ther Nucleic Acids (2014) 3:e188. doi: 10.1038/mtna.2014.40
149. O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne) (2018) 9. doi: 10.3389/fendo.2018.00402
150. O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA Delivery by Extracellular Vesicles in Mammalian Cells and Its Applications. Nat Rev Mol Cell Biol (2020) 21(10):585–606. doi: 10.1038/s41580-020-0251-y
151. Kalluri R, LeBleu VS. The Biology, Function, and Biomedical Applications of Exosomes. Science (2020) 367(6478):eaau6977. doi: 10.1126/science.aau6977
152. Makarova JA, Shkurnikov MU, Wicklein D, Lange T, Samatov TR, Turchinovich AA, et al. Intracellular and Extracellular MicroRNA: An Update on Localization and Biological Role. Prog Histochem. Cytochem. (2016) 51(3-4):33–49. doi: 10.1016/j.proghi.2016.06.001
153. Haider BA, Baras AS, McCall MN, Hertel JA, Cornish TC, Halushka MK. A Critical Evaluation of MicroRNA Biomarkers in Non-Neoplastic Disease. PloS One (2014) 9(2):e89565. doi: 10.1371/journal.pone.0089565
154. Fromm B, Domanska D, Hoye E, Ovchinnikov V, Kang W, Aparicio-Puerta E, et al. Mirgenedb 2.0: The Metazoan MicroRNA Complement. Nucleic Acids Res (2020) 48(D1):D132–D41. doi: 10.1093/nar/gkz885
155. Lewis BP, Burge CB, Bartel DP. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates That Thousands of Human Genes Are MicroRNA Targets. Cell (2005) 120(1):15–20. doi: 10.1016/j.cell.2004.12.035
156. Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray Analysis Shows That Some MicroRNAs Downregulate Large Numbers of Target mRNAs. Nature (2005) 433(7027):769–73. doi: 10.1038/nature03315
157. Lu LF, Gasteiger G, Yu IS, Chaudhry A, Hsin JP, Lu Y, et al. A Single MiRNA-mRNA Interaction Affects the Immune Response in a Context- and Cell-Type-Specific Manner. Immunity (2015) 43(1):52–64. doi: 10.1016/j.immuni.2015.04.022
158. Erhard F, Haas J, Lieber D, Malterer G, Jaskiewicz L, Zavolan M, et al. Widespread Context Dependency of MicroRNA-Mediated Regulation. Genome Res (2014) 24(6):906–19. doi: 10.1101/gr.166702.113
159. McCall MN, Kent OA, Yu J, Fox-Talbot K, Zaiman AL, Halushka MK. MicroRNA Profiling of Diverse Endothelial Cell Types. BMC Med Genomics (2011) 4:78. doi: 10.1186/1755-8794-4-78
160. Tan Y, Pan T, Ye Y, Ge G, Chen L, Wen D, et al. Serum MicroRNAs as Potential Biomarkers of Primary Biliary Cirrhosis. PLoS One (2014) 9(10):e111424. doi: 10.1371/journal.pone.0111424
161. Yang Z, Zhang T, Kusumanchi P, Tang Q, Sun Z, Radaeva S, et al. Transcriptomic Analysis Reveals the MicroRNAs Responsible for Liver Regeneration Associated With Mortality in Alcohol-Associated Hepatitis. Hepatology (2021) 74(5):2436–51. doi: 10.1002/hep.31994
162. Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of Novel Genes Coding for Small Expressed RNAs. Science (2001) 294(5543):853–8. doi: 10.1126/science.1064921
163. Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 Complex in Primary MicroRNA Processing. Genes Dev (2004) 18(24):3016–27. doi: 10.1101/gad.1262504
164. Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear Export of MicroRNA Precursors. Science (2004) 303(5654):95–8. doi: 10.1126/science.1090599
165. Khvorova A, Reynolds A, Jayasena SD. Functional SiRNAs and MiRNAs Exhibit Strand Bias. Cell (2003) 115(2):209–16. doi: 10.1016/s0092-8674(03)00801-8
166. Ruby JG, Jan CH, Bartel DP. Intronic MicroRNA Precursors That Bypass Drosha Processing. Nature (2007) 448(7149):83–6. doi: 10.1038/nature05983
167. Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse Es Cells Express Endogenous ShRNAs, SiRNAs, and Other Microprocessor-Independent, Dicer-Dependent Small RNAs. Genes Dev (2008) 22(20):2773–85. doi: 10.1101/gad.1705308
168. Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A Dicer-Independent MiRNA Biogenesis Pathway That Requires AGO Catalysis. Nature (2010) 465(7298):584–9. doi: 10.1038/nature09092
169. Tolia NH, Joshua-Tor L. Slicer and the Argonautes. Nat Chem Biol (2007) 3(1):36–43. doi: 10.1038/nchembio848
170. Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, Liu D, et al. Nuclear MiRNA Regulates the Mitochondrial Genome in the Heart. Circ Res (2012) 110(12):1596–603. doi: 10.1161/CIRCRESAHA.112.267732
171. Sripada L, Tomar D, Prajapati P, Singh R, Singh AK, Singh R. Systematic Analysis of Small RNAs Associated With Human Mitochondria by Deep Sequencing: Detailed Analysis of Mitochondrial Associated MiRNA. PLoS One (2012) 7(9):e44873. doi: 10.1371/journal.pone.0044873
172. Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of Primary MicroRNAs by the Microprocessor Complex. Nature (2004) 432(7014):231–5. doi: 10.1038/nature03049
173. Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, et al. The Microprocessor Complex Mediates the Genesis of MicroRNAs. Nature (2004) 432(7014):235–40. doi: 10.1038/nature03120
174. Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single Processing Center Models for Human Dicer and Bacterial RNase III. Cell (2004) 118(1):57–68. doi: 10.1016/j.cell.2004.06.017
175. Ha M, Kim VN. Regulation of MicroRNA Biogenesis. Nat Rev Mol Cell Biol (2014) 15(8):509–24. doi: 10.1038/nrm3838
176. Wigler M, Levy D, Perucho M. The Somatic Replication of DNA Methylation. Cell (1981) 24(1):33–40. doi: 10.1016/0092-8674(81)90498-0
177. Cecere G. Small RNAs in Epigenetic Inheritance: From Mechanisms to Trait Transmission. FEBS Lett (2021) 595(24):2953–77. doi: 10.1002/1873-3468.14210
178. Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, et al. Endogenous SiRNAs From Naturally Formed DsRNAs Regulate Transcripts in Mouse Oocytes. Nature (2008) 453(7194):539–43. doi: 10.1038/nature06908
179. Zhang X, Gao F, Fu J, Zhang P, Wang Y, Zeng X. Systematic Identification and Characterization of Long Non-Coding RNAs in Mouse Mature Sperm. PLoS One (2017) 12(3):e0173402. doi: 10.1371/journal.pone.0173402
180. Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, et al. Paternally Induced Transgenerational Environmental Reprogramming of Metabolic Gene Expression in Mammals. Cell (2010) 143(7):1084–96. doi: 10.1016/j.cell.2010.12.008
181. Tuong ZK, Stewart BJ, Guo SA, Clatworthy MR. Epigenetics and Tissue Immunity-Translating Environmental Cues Into Functional Adaptations. Immunol Rev (2022) 305(1):111–36. doi: 10.1111/imr.13036
182. Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, et al. Multigenerational Epigenetic Adaptation of the Hepatic Wound-Healing Response. Nat Med (2012) 18(9):1369–77. doi: 10.1038/nm.2893
183. Horsthemke B. A Critical View on Transgenerational Epigenetic Inheritance in Humans. Nat Commun (2018) 9(1):2973. doi: 10.1038/s41467-018-05445-5
184. Donkin I, Versteyhe S, Ingerslev LR, Qian K, Mechta M, Nordkap L, et al. Obesity and Bariatric Surgery Drive Epigenetic Variation of Spermatozoa in Humans. Cell Metab (2016) 23(2):369–78. doi: 10.1016/j.cmet.2015.11.004
185. Jaenisch R, Bird A. Epigenetic Regulation of Gene Expression: How the Genome Integrates Intrinsic and Environmental Signals. Nat Genet (2003) 33 Suppl:245–54. doi: 10.1038/ng1089
186. Bjornsson HT, Fallin MD, Feinberg AP. An Integrated Epigenetic and Genetic Approach to Common Human Disease. Trends Genet (2004) 20(8):350–8. doi: 10.1016/j.tig.2004.06.009
187. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic Differences Arise During the Lifetime of Monozygotic Twins. Proc Natl Acad Sci USA (2005) 102(30):10604–9. doi: 10.1073/pnas.0500398102
188. Cooney CA. Are Somatic Cells Inherently Deficient in Methylation Metabolism? A Proposed Mechanism for DNA Methylation Loss, Senescence and Aging. Growth Dev Aging (1993) 57(4):261–73.
189. Bennett-Baker PE, Wilkowski J, Burke DT. Age-Associated Activation of Epigenetically Repressed Genes in the Mouse. Genetics (2003) 165(4):2055–62. doi: 10.1093/genetics/165.4.2055
190. Maegawa S, Gough SM, Watanabe-Okochi N, Lu Y, Zhang N, Castoro RJ, et al. Age-Related Epigenetic Drift in the Pathogenesis of MDS and AML. Genome Res (2014) 24(4):580–91. doi: 10.1101/gr.157529.113
191. Issa JP. Aging and Epigenetic Drift: A Vicious Cycle. J Clin Invest (2014) 124(1):24–9. doi: 10.1172/JCI69735
192. Dozmorov MG, Coit P, Maksimowicz-McKinnon K, Sawalha AH. Age-Associated DNA Methylation Changes in Naive CD4+ T Cells Suggest an Evolving Autoimmune Epigenotype in Aging T Cells. Epigenomics (2017) 9(4):429–45. doi: 10.2217/epi-2016-0143
193. Schwimmer JB, Celedon MA, Lavine JE, Salem R, Campbell N, Schork NJ, et al. Heritability of Nonalcoholic Fatty Liver Disease. Gastroenterology (2009) 136(5):1585–92. doi: 10.1053/j.gastro.2009.01.050
194. Zarrinpar A, Gupta S, Maurya MR, Subramaniam S, Loomba R. Serum MicroRNAs Explain Discordance of Non-Alcoholic Fatty Liver Disease in Monozygotic and Dizygotic Twins: A Prospective Study. Gut (2016) 65(9):1546–54. doi: 10.1136/gutjnl-2015-309456
195. Morkve Knudsen T, Rezwan FI, Jiang Y, Karmaus W, Svanes C, Holloway JW. Transgenerational and Intergenerational Epigenetic Inheritance in Allergic Diseases. J Allergy Clin Immunol (2018) 142(3):765–72. doi: 10.1016/j.jaci.2018.07.007
196. Halasz T, Horvath G, Par G, Werling K, Kiss A, Schaff Z, et al. MiR-122 Negatively Correlates With Liver Fibrosis as Detected by Histology and Fibroscan. World J Gastroenterol (2015) 21(25):7814–23. doi: 10.3748/wjg.v21.i25.7814
197. Li J, Ghazwani M, Zhang Y, Lu J, Li J, Fan J, et al. MiR-122 Regulates Collagen Production Via Targeting Hepatic Stellate Cells and Suppressing P4HA1 Expression. J Hepatol (2013) 58(3):522–8. doi: 10.1016/j.jhep.2012.11.011
198. Myllyharju J. Prolyl 4-Hydroxylases, the Key Enzymes of Collagen Biosynthesis. Matrix Biol (2003) 22(1):15–24. doi: 10.1016/s0945-053x(03)00006-4
199. Blaya D, Aguilar-Bravo B, Hao F, Casacuberta-Serra S, Coll M, Perea L, et al. Expression of MicroRNA-155 in Inflammatory Cells Modulates Liver Injury. Hepatology (2018) 68(2):691–706. doi: 10.1002/hep.29833
200. Miozzo M, Selmi C, Gentilin B, Grati FR, Sirchia S, Oertelt S, et al. Preferential X Chromosome Loss But Random Inactivation Characterize Primary Biliary Cirrhosis. Hepatology (2007) 46(2):456–62. doi: 10.1002/hep.21696
201. Lleo A, Oertelt-Prigione S, Bianchi I, Caliari L, Finelli P, Miozzo M, et al. Y Chromosome Loss in Male Patients With Primary Biliary Cirrhosis. J Autoimmun. (2013) 41:87–91. doi: 10.1016/j.jaut.2012.12.008
202. Hu Z, Huang Y, Liu Y, Sun Y, Zhou Y, Gu M, et al. Beta-Arrestin 1 Modulates Functions of Autoimmune T Cells From Primary Biliary Cirrhosis Patients. J Clin Immunol (2011) 31(3):346–55. doi: 10.1007/s10875-010-9492-4
203. Lleo A, Liao J, Invernizzi P, Zhao M, Bernuzzi F, Ma L, et al. Immunoglobulin M Levels Inversely Correlate With CD40 Ligand Promoter Methylation in Patients With Primary Biliary Cirrhosis. Hepatology (2012) 55(1):153–60. doi: 10.1002/hep.24630
204. Wang X, Wen X, Zhou J, Qi Y, Wu R, Wang Y, et al. MicroRNA-223 and MicroRNA-21 in Peripheral Blood B Cells Associated With Progression of Primary Biliary Cholangitis Patients. PloS One (2017) 12(9):e0184292. doi: 10.1371/journal.pone.0184292
205. Tabibian JH, O’Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte Senescence by Way of N-RAS Activation Is a Characteristic of Primary Sclerosing Cholangitis. Hepatology (2014) 59(6):2263–75. doi: 10.1002/hep.26993
206. Czaja AJ. Emerging Therapeutic Biomarkers of Autoimmune Hepatitis and Their Impact on Current and Future Management. Expert Rev Gastroenterol Hepatol (2018) 12(6):547–64. doi: 10.1080/17474124.2018.1453356
207. Pashangzadeh S, Motallebnezhad M, Vafashoar F, Khalvandi A, Mojtabavi N. Implications the Role of MiR-155 in the Pathogenesis of Autoimmune Diseases. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.669382
208. Thomsen H, Li X, Sundquist K, Sundquist J, Forsti A, Hemminki K. Familial Associations Between Autoimmune Hepatitis and Primary Biliary Cholangitis and Other Autoimmune Diseases. PloS One (2020) 15(10):e0240794. doi: 10.1371/journal.pone.0240794
209. Hodges S, Lobo-Yeo A, Donaldson P, Tanner MS, Vergani D. Autoimmune Chronic Active Hepatitis in a Family. Gut (1991) 32(3):299–302. doi: 10.1136/gut.32.3.299
210. Gronbaek L, Vilstrup H, Pedersen L, Christensen K, Jepsen P. Family Occurrence of Autoimmune Hepatitis: A Danish Nationwide Registry-Based Cohort Study. J Hepatol (2018) 69(4):873–7. doi: 10.1016/j.jhep.2018.05.035
211. van Gerven NM, Verwer BJ, Witte BI, van Erpecum KJ, van Buuren HR, Maijers I, et al. Epidemiology and Clinical Characteristics of Autoimmune Hepatitis in the Netherlands. Scand J Gastroenterol (2014) 49(10):1245–54. doi: 10.3109/00365521.2014.946083
212. Cheung AC, LaRusso NF, Gores GJ, Lazaridis KN. Epigenetics in the Primary Biliary Cholangitis and Primary Sclerosing Cholangitis. Semin Liver Dis (2017) 37(2):159–74. doi: 10.1055/s-0037-1603324
213. Tanaka A, Leung PSC, Gershwin ME. The Genetics and Epigenetics of Primary Biliary Cholangitis. Clin Liver Dis (2018) 22(3):443–55. doi: 10.1016/j.cld.2018.03.002
214. Li Y, Tang R, Ma X. Epigenetics of Primary Biliary Cholangitis. Adv Exp Med Biol (2020) 1253:259–83. doi: 10.1007/978-981-15-3449-2_10
215. Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, et al. Primary Biliary Cirrhosis in Monozygotic and Dizygotic Twins: Genetics, Epigenetics, and Environment. Gastroenterology (2004) 127(2):485–92. doi: 10.1053/j.gastro.2004.05.005
216. Selmi C, Cavaciocchi F, Lleo A, Cheroni C, De Francesco R, Lombardi SA, et al. Genome-Wide Analysis of DNA Methylation, Copy Number Variation, and Gene Expression in Monozygotic Twins Discordant for Primary Biliary Cirrhosis. Front Immunol (2014) 5. doi: 10.3389/fimmu.2014.00128
217. Campisi J, d’Adda di Fagagna F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat Rev Mol Cell Biol (2007) 8(9):729–40. doi: 10.1038/nrm2233
218. Mao Z, Ke Z, Gorbunova V, Seluanov A. Replicatively Senescent Cells Are Arrested in G1 and G2 Phases. Aging (Albany NY) (2012) 4(6):431–5. doi: 10.18632/aging.100467
219. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PloS Biol (2008) 6(12):2853–68. doi: 10.1371/journal.pbio.0060301
220. Kitamoto T, Kitamoto A, Ogawa Y, Honda Y, Imajo K, Saito S, et al. Targeted-Bisulfite Sequence Analysis of the Methylation of CpG Islands in Genes Encoding PNPLA3, SAMM50, and PARVB of Patients With Non-Alcoholic Fatty Liver Disease. J Hepatol (2015) 63(2):494–502. doi: 10.1016/j.jhep.2015.02.049
221. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic Variation in PNPLA3 Confers Susceptibility to Nonalcoholic Fatty Liver Disease. Nat Genet (2008) 40(12):1461–5. doi: 10.1038/ng.257
222. Anstee QM, Day CP. The Genetics of NAFLD. Nat Rev Gastroenterol Hepatol (2013) 10(11):645–55. doi: 10.1038/nrgastro.2013.182
223. Kitamoto T, Kitamoto A, Yoneda M, Hyogo H, Ochi H, Nakamura T, et al. Genome-Wide Scan Revealed That Polymorphisms in the PNPLA3, SAMM50, and PARVB Genes Are Associated With Development and Progression of Nonalcoholic Fatty Liver Disease in Japan. Hum Genet (2013) 132(7):783–92. doi: 10.1007/s00439-013-1294-3
224. Zeybel M, Hardy T, Robinson SM, Fox C, Anstee QM, Ness T, et al. Differential DNA Methylation of Genes Involved in Fibrosis Progression in Non-Alcoholic Fatty Liver Disease and Alcoholic Liver Disease. Clin Epigenet (2015) 7:25. doi: 10.1186/s13148-015-0056-6
225. Mikula M, Majewska A, Ledwon JK, Dzwonek A, Ostrowski J. Obesity Increases Histone H3 Lysine 9 and 18 Acetylation at TNFA and CCl2 Genes in Mouse Liver. Int J Mol Med (2014) 34(6):1647–54. doi: 10.3892/ijmm.2014.1958
226. Cai C, Yu H, Huang G, Du X, Yu X, Zhou Y, et al. Histone Modifications in Fatty Acid Synthase Modulated by Carbohydrate Responsive Element Binding Protein Are Associated With Nonalcoholic Fatty Liver Disease. Int J Mol Med (2018) 42(3):1215–28. doi: 10.3892/ijmm.2018.3702
227. Cermelli S, Ruggieri A, Marrero JA, Ioannou GN, Beretta L. Circulating MicroRNAs in Patients With Chronic Hepatitis C and Non-Alcoholic Fatty Liver Disease. PloS One (2011) 6(8):e23937. doi: 10.1371/journal.pone.0023937
228. Pirola CJ, Fernandez Gianotti T, Castano GO, Mallardi P, San Martino J, Mora Gonzalez Lopez Ledesma M, et al. Circulating MicroRNA Signature in Non-Alcoholic Fatty Liver Disease: From Serum Non-Coding RNAs to Liver Histology and Disease Pathogenesis. Gut (2015) 64(5):800–12. doi: 10.1136/gutjnl-2014-306996
229. Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW, et al. Nonalcoholic Steatohepatitis Is Associated With Altered Hepatic MicroRNA Expression. Hepatology (2008) 48(6):1810–20. doi: 10.1002/hep.22569
230. Park PH, Miller R, Shukla SD. Acetylation of Histone H3 at Lysine 9 by Ethanol in Rat Hepatocytes. Biochem Biophys Res Commun (2003) 306(2):501–4. doi: 10.1016/s0006-291x(03)01040-4
231. Park PH, Lim RW, Shukla SD. Involvement of Histone Acetyltransferase (HAT) in Ethanol-Induced Acetylation of Histone H3 in Hepatocytes: Potential Mechanism for Gene Expression. Am J Physiol Gastrointest. Liver Physiol (2005) 289(6):G1124–36. doi: 10.1152/ajpgi.00091.2005
232. Pal-Bhadra M, Bhadra U, Jackson DE, Mamatha L, Park PH, Shukla SD. Distinct Methylation Patterns in Histone H3 at Lys-4 and Lys-9 Correlate With Up- & Down-Regulation of Genes by Ethanol in Hepatocytes. Life Sci (2007) 81(12):979–87. doi: 10.1016/j.lfs.2007.07.030
233. Park PH, Lim RW, Shukla SD. Gene-Selective Histone H3 Acetylation in the Absence of Increase in Global Histone Acetylation in Liver of Rats Chronically Fed Alcohol. Alcohol Alcohol (2012) 47(3):233–9. doi: 10.1093/alcalc/ags004
234. Kriss CL, Gregory-Lott E, Storey AJ, Tackett AJ, Wahls WP, Stevens SM Jr. In Vivo Metabolic Tracing Demonstrates the Site-Specific Contribution of Hepatic Ethanol Metabolism to Histone Acetylation. Alcohol Clin Exp Res (2018) 42(10):1909–23. doi: 10.1111/acer.13843
235. Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, et al. Deletion of SIRT1 From Hepatocytes in Mice Disrupts Lipin-1 Signaling and Aggravates Alcoholic Fatty Liver. Gastroenterology (2014) 146(3):801–11. doi: 10.1053/j.gastro.2013.11.008
236. Nagy LE. The Role of Innate Immunity in Alcoholic Liver Disease. Alcohol Res (2015) 37(2):237–50.
237. Hardy T, Zeybel M, Day CP, Dipper C, Masson S, McPherson S, et al. Plasma DNA Methylation: A Potential Biomarker for Stratification of Liver Fibrosis in Non-Alcoholic Fatty Liver Disease. Gut (2017) 66(7):1321–8. doi: 10.1136/gutjnl-2016-311526
238. Hartmann P, Tacke F. Tiny RNA With Great Effects: MiR-155 in Alcoholic Liver Disease. J Hepatol (2016) 64(6):1214–6. doi: 10.1016/j.jhep.2016.02.039
239. Tang Y, Zhang L, Forsyth CB, Shaikh M, Song S, Keshavarzian A. The Role of MiR-212 and INOS in Alcohol-Induced Intestinal Barrier Dysfunction and Steatohepatitis. Alcohol Clin Exp Res (2015) 39(9):1632–41. doi: 10.1111/acer.12813
240. Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. MiR-122 Regulation of Lipid Metabolism Revealed by in Vivo Antisense Targeting. Cell Metab (2006) 3(2):87–98. doi: 10.1016/j.cmet.2006.01.005
241. Bala S, Petrasek J, Mundkur S, Catalano D, Levin I, Ward J, et al. Circulating MicroRNAs in Exosomes Indicate Hepatocyte Injury and Inflammation in Alcoholic, Drug-Induced, and Inflammatory Liver Diseases. Hepatology (2012) 56(5):1946–57. doi: 10.1002/hep.25873
242. Bala S, Szabo G. MicroRNA Signature in Alcoholic Liver Disease. Int J Hepatol (2012) 2012:498232. doi: 10.1155/2012/498232
243. Celikbilek M, Baskol M, Taheri S, Deniz K, Dogan S, Zararsiz G, et al. Circulating MicroRNAs in Patients With Non-Alcoholic Fatty Liver Disease. World J Hepatol (2014) 6(8):613–20. doi: 10.4254/wjh.v6.i8.613
244. Bates SE. Epigenetic Therapies for Cancer. N Engl J Med (2020) 383(7):650–63. doi: 10.1056/NEJMra1805035
245. Kuang Y, El-Khoueiry A, Taverna P, Ljungman M, Neamati N. Guadecitabine (Sgi-110) Priming Sensitizes Hepatocellular Carcinoma Cells to Oxaliplatin. Mol Oncol (2015) 9(9):1799–814. doi: 10.1016/j.molonc.2015.06.002
246. Perugorria MJ, Wilson CL, Zeybel M, Walsh M, Amin S, Robinson S, et al. Histone Methyltransferase ASH1 Orchestrates Fibrogenic Gene Transcription During Myofibroblast Transdifferentiation. Hepatology (2012) 56(3):1129–39. doi: 10.1002/hep.25754
247. Zeybel M, Luli S, Sabater L, Hardy T, Oakley F, Leslie J, et al. A Proof-Of-Concept for Epigenetic Therapy of Tissue Fibrosis: Inhibition of Liver Fibrosis Progression by 3-Deazaneplanocin a. Mol Ther (2017) 25(1):218–31. doi: 10.1016/j.ymthe.2016.10.004
248. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol Improves Mitochondrial Function and Protects Against Metabolic Disease by Activating SIRT1 and PGC-1alpha. Cell (2006) 127(6):1109–22. doi: 10.1016/j.cell.2006.11.013
249. Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A, et al. Combination Therapy for Hepatocellular Carcinoma: Additive Preclinical Efficacy of the HDAC Inhibitor Panobinostat With Sorafenib. J Hepatol (2012) 56(6):1343–50. doi: 10.1016/j.jhep.2012.01.009
250. Chachay VS, Macdonald GA, Martin JH, Whitehead JP, O’Moore-Sullivan TM, Lee P, et al. Resveratrol Does Not Benefit Patients With Nonalcoholic Fatty Liver Disease. Clin Gastroenterol Hepatol (2014) 12(12):2092–103 e1-6. doi: 10.1016/j.cgh.2014.02.024
251. Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-Specific Inhibition of MicroRNA- and SiRNA-Induced RNA Silencing. RNA (2004) 10(3):544–50. doi: 10.1261/rna.5235104
252. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of MicroRNAs in Vivo With ‘Antagomirs’. Nature (2005) 438(7068):685–9. doi: 10.1038/nature04303
253. Krutzfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, et al. Specificity, Duplex Degradation and Subcellular Localization of Antagomirs. Nucleic Acids Res (2007) 35(9):2885–92. doi: 10.1093/nar/gkm024
254. Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, et al. Target Mimicry Provides a New Mechanism for Regulation of MicroRNA Activity. Nat Genet (2007) 39(8):1033–7. doi: 10.1038/ng2079
255. Ebert MS, Neilson JR, Sharp PA. Microrna Sponges: Competitive Inhibitors of Small RNAs in Mammalian Cells. Nat Methods (2007) 4(9):721–6. doi: 10.1038/nmeth1079
256. Haraguchi T, Ozaki Y, Iba H. Vectors Expressing Efficient Rna Decoys Achieve the Long-Term Suppression of Specific MicroRNA Activity in Mammalian Cells. Nucleic Acids Res (2009) 37(6):e43. doi: 10.1093/nar/gkp040
257. Nilsson EE, Sadler-Riggleman I, Skinner MK. Environmentally Induced Epigenetic Transgenerational Inheritance of Disease. Environ Epigenet. (2018) 4(2):dvy016. doi: 10.1093/eep/dvy016
258. Detich N, Hamm S, Just G, Knox JD, Szyf M. The Methyl Donor S-Adenosylmethionine Inhibits Active Demethylation of DNA: A Candidate Novel Mechanism for the Pharmacological Effects of S-Adenosylmethionine. J Biol Chem (2003) 278(23):20812–20. doi: 10.1074/jbc.M211813200
259. Anstee QM, Day CP. S-Adenosylmethionine (Same) Therapy in Liver Disease: A Review of Current Evidence and Clinical Utility. J Hepatol (2012) 57(5):1097–109. doi: 10.1016/j.jhep.2012.04.041
260. Waterland RA, Travisano M, Tahiliani KG, Rached MT, Mirza S. Methyl Donor Supplementation Prevents Transgenerational Amplification of Obesity. Int J Obes (Lond) (2008) 32(9):1373–9. doi: 10.1038/ijo.2008.100
261. Cordero P, Gomez-Uriz AM, Campion J, Milagro FI, Martinez JA. Dietary Supplementation With Methyl Donors Reduces Fatty Liver and Modifies the Fatty Acid Synthase DNA Methylation Profile in Rats Fed an Obesogenic Diet. Genes Nutr (2013) 8(1):105–13. doi: 10.1007/s12263-012-0300-z
262. Artaza JN, Norris KC. Vitamin D Reduces the Expression of Collagen and Key Profibrotic Factors by Inducing an Antifibrotic Phenotype in Mesenchymal Multipotent Cells. J Endocrinol (2009) 200(2):207–21. doi: 10.1677/JOE-08-0241
263. Abramovitch S, Dahan-Bachar L, Sharvit E, Weisman Y, Ben Tov A, Brazowski E, et al. Vitamin D Inhibits Proliferation and Profibrotic Marker Expression in Hepatic Stellate Cells and Decreases Thioacetamide-Induced Liver Fibrosis in Rats. Gut (2011) 60(12):1728–37. doi: 10.1136/gut.2010.234666
264. Potter JJ, Liu X, Koteish A, Mezey E. 1,25-Dihydroxyvitamin D3 and Its Nuclear Receptor Repress Human Alpha1 (I) Collagen Expression and Type I Collagen Formation. Liver Int (2013) 33(5):677–86. doi: 10.1111/liv.12122
265. Abramovitch S, Sharvit E, Weisman Y, Bentov A, Brazowski E, Cohen G, et al. Vitamin D Inhibits Development of Liver Fibrosis in an Animal Model But Cannot Ameliorate Established Cirrhosis. Am J Physiol Gastrointest. Liver Physiol (2015) 308(2):G112–20. doi: 10.1152/ajpgi.00132.2013
266. Czaja AJ, Montano-Loza AJ. Evolving Role of Vitamin D in Immune-Mediated Disease and Its Implications in Autoimmune Hepatitis. Dig. Dis Sci (2019) 64(2):324–44. doi: 10.1007/s10620-018-5351-6
267. Snegarova V, Naydenova D. Vitamin D: A Review of Its Effects on Epigenetics and Gene Regulation. Folia Med (Plovdiv) (2020) 62(4):662–7. doi: 10.3897/folmed.62.e50204
268. Wu LM, Yang Z, Zhou L, Zhang F, Xie HY, Feng XW, et al. Identification of Histone Deacetylase 3 as a Biomarker for Tumor Recurrence Following Liver Transplantation in HBV-Associated Hepatocellular Carcinoma. PloS One (2010) 5(12):e14460. doi: 10.1371/journal.pone.0014460
269. da Costa TP, El-Cheikh MC, Carneiro K. Epigenetic Therapy as a Putative Molecular Target to Modulate B Cell Biology and Behavior in the Context of Immunological Disorders. J Immunol Res (2020) 2020:1589191. doi: 10.1155/2020/1589191
270. Jin J, Iakova P, Jiang Y, Medrano EE, Timchenko NA. The Reduction of SIRT1 in Livers of Old Mice Leads to Impaired Body Homeostasis and to Inhibition of Liver Proliferation. Hepatology (2011) 54(3):989–98. doi: 10.1002/hep.24471
271. Ahn J, Cho I, Kim S, Kwon D, Ha T. Dietary Resveratrol Alters Lipid Metabolism-Related Gene Expression of Mice on an Atherogenic Diet. J Hepatol (2008) 49(6):1019–28. doi: 10.1016/j.jhep.2008.08.012
272. Bujanda L, Hijona E, Larzabal M, Beraza M, Aldazabal P, Garcia-Urkia N, et al. Resveratrol Inhibits Nonalcoholic Fatty Liver Disease in Rats. BMC Gastroenterol (2008) 8:40. doi: 10.1186/1471-230X-8-40
273. Shang J, Chen LL, Xiao FX, Sun H, Ding HC, Xiao H. Resveratrol Improves Non-Alcoholic Fatty Liver Disease by Activating AMP-Activated Protein Kinase. Acta Pharmacol Sin (2008) 29(6):698–706. doi: 10.1111/j.1745-7254.2008.00807.x
274. Gomez-Zorita S, Fernandez-Quintela A, Macarulla MT, Aguirre L, Hijona E, Bujanda L, et al. Resveratrol Attenuates Steatosis in Obese Zucker Rats by Decreasing Fatty Acid Availability and Reducing Oxidative Stress. Br J Nutr (2012) 107(2):202–10. doi: 10.1017/S0007114511002753
275. Jiang Y, Xiang C, Zhong F, Zhang Y, Wang L, Zhao Y, et al. Histone H3K27 Methyltransferase EZH2 and Demethylase JMJD3 Regulate Hepatic Stellate Cells Activation and Liver Fibrosis. Theranostics (2021) 11(1):361–78. doi: 10.7150/thno.46360
276. Hazra S, Xiong S, Wang J, Rippe RA, Krishna V, Chatterjee K, et al. Peroxisome Proliferator-Activated Receptor Gamma Induces a Phenotypic Switch From Activated to Quiescent Hepatic Stellate Cells. J Biol Chem (2004) 279(12):11392–401. doi: 10.1074/jbc.M310284200
277. Mann J, Chu DC, Maxwell A, Oakley F, Zhu NL, Tsukamoto H, et al. MECP2 Controls an Epigenetic Pathway That Promotes Myofibroblast Transdifferentiation and Fibrosis. Gastroenterology (2010) 138(2):705–14. doi: 10.1053/j.gastro.2009.10.002
278. Huang CK, Kafert-Kasting S, Thum T. Preclinical and Clinical Development of Noncoding RNA Therapeutics for Cardiovascular Disease. Circ Res (2020) 126(5):663–78. doi: 10.1161/CIRCRESAHA.119.315856
279. Bernardo BC, Ooi JY, Lin RC, McMullen JR. MiRNA Therapeutics: A New Class of Drugs With Potential Therapeutic Applications in the Heart. Future Med Chem (2015) 7(13):1771–92. doi: 10.4155/fmc.15.107
280. Gomez IG, MacKenna DA, Johnson BG, Kaimal V, Roach AM, Ren S, et al. Anti-MicroRNA-21 Oligonucleotides Prevent Alport Nephropathy Progression by Stimulating Metabolic Pathways. J Clin Invest (2015) 125(1):141–56. doi: 10.1172/JCI75852
281. Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV Infection by Targeting MicroRNA. N Engl J Med (2013) 368(18):1685–94. doi: 10.1056/NEJMoa1209026
282. Anastasiadou E, Seto AG, Beatty X, Hermreck M, Gilles ME, Stroopinsky D, et al. Cobomarsen, an Oligonucleotide Inhibitor of MiR-155, Slows DLBCL Tumor Cell Growth in Vitro and in Vivo. Clin Cancer Res (2021) 27(4):1139–49. doi: 10.1158/1078-0432.CCR-20-3139
283. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional Silencing and Longevity Protein SIR2 Is an NAD-Dependent Histone Deacetylase. Nature (2000) 403(6771):795–800. doi: 10.1038/35001622
284. Baur JA, Sinclair DA. Therapeutic Potential of Resveratrol: The in Vivo Evidence. Nat Rev Drug Discov (2006) 5(6):493–506. doi: 10.1038/nrd2060
285. Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, et al. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell Metab (2012) 15(5):675–90. doi: 10.1016/j.cmet.2012.04.003
286. Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol Improves Health and Survival of Mice on a High-Calorie Diet. Nature (2006) 444(7117):337–42. doi: 10.1038/nature05354
287. Page A, Paoli P, Moran Salvador E, White S, French J, Mann J. Hepatic Stellate Cell Transdifferentiation Involves Genome-Wide Remodeling of the DNA Methylation Landscape. J Hepatol (2016) 64(3):661–73. doi: 10.1016/j.jhep.2015.11.024
288. Mann J, Oakley F, Akiboye F, Elsharkawy A, Thorne AW, Mann DA. Regulation of Myofibroblast Transdifferentiation by DNA Methylation and MECP2: Implications for Wound Healing and Fibrogenesis. Cell Death Differ. (2007) 14(2):275–85. doi: 10.1038/sj.cdd.4401979
289. Tan JZ, Yan Y, Wang XX, Jiang Y, Xu HE. EZH2: Biology, Disease, and Structure-Based Drug Discovery. Acta Pharmacol Sin (2014) 35(2):161–74. doi: 10.1038/aps.2013.161
290. Duan R, Du W, Guo W. EZH2: A Novel Target for Cancer Treatment. J Hematol Oncol (2020) 13(1):104. doi: 10.1186/s13045-020-00937-8
291. Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, et al. LNA-Mediated MicroRNA Silencing in Non-Human Primates. Nature (2008) 452(7189):896–9. doi: 10.1038/nature06783
292. Canas CA, Canas F, Bonilla-Abadia F, Ospina FE, Tobon GJ. Epigenetics Changes Associated to Environmental Triggers in Autoimmunity. Autoimmunity (2016) 49(1):1–11. doi: 10.3109/08916934.2015.1086996
293. Javierre BM, Hernando H, Ballestar E. Environmental Triggers and Epigenetic Deregulation in Autoimmune Disease. Discovery Med (2011) 12(67):535–45.
294. Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and Procainamide Inhibit T Cell DNA Methylation and Induce Autoreactivity. J Immunol (1988) 140(7):2197–200.
295. Scheinbart LS, Johnson MA, Gross LA, Edelstein SR, Richardson BC. Procainamide Inhibits DNA Methyltransferase in a Human T Cell Line. J Rheumatol. (1991) 18(4):530–4.
296. Quddus J, Johnson KJ, Gavalchin J, Amento EP, Chrisp CE, Yung RL, et al. Treating Activated CD4+ T Cells With Either of Two Distinct DNA Methyltransferase Inhibitors, 5-Azacytidine or Procainamide, Is Sufficient to Cause a Lupus-Like Disease in Syngeneic Mice. J Clin Invest (1993) 92(1):38–53. doi: 10.1172/JCI116576
297. Christman JK. 5-Azacytidine and 5-Aza-2’-Deoxycytidine as Inhibitors of DNA Methylation: Mechanistic Studies and Their Implications for Cancer Therapy. Oncogene (2002) 21(35):5483–95. doi: 10.1038/sj.onc.1205699
298. Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, et al. A New Model for an Etiology of Rheumatoid Arthritis: Smoking May Trigger HLA-DR (Shared Epitope)-Restricted Immune Reactions to Autoantigens Modified by Citrullination. Arthritis Rheum. (2006) 54(1):38–46. doi: 10.1002/art.21575
299. Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA Methylation Changes After Exposure to Traffic Particles. Am J Respir Crit Care Med (2009) 179(7):572–8. doi: 10.1164/rccm.200807-1097OC
300. Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, et al. Effects of Particulate Matter on Genomic DNA Methylation Content and iNOS Promoter Methylation. Environ Health Perspect (2009) 117(2):217–22. doi: 10.1289/ehp.11898
301. Martinez-Zamudio R, Ha HC. Environmental Epigenetics in Metal Exposure. Epigenetics (2011) 6(7):820–7. doi: 10.4161/epi.6.7.16250
302. Minarovits J, Hu LF, Marcsek Z, Minarovits-Kormuta S, Klein G, Ernberg I. RNA Polymerase III-Transcribed EBER 1 and 2 Transcription Units Are Expressed and Hypomethylated in the Major Epstein-Barr Virus-Carrying Cell Types. J Gen Virol (1992) 73(Pt 7):1687–92. doi: 10.1099/0022-1317-73-7-1687
303. Costenbader KH, Kim DJ, Peerzada J, Lockman S, Nobles-Knight D, Petri M, et al. Cigarette Smoking and the Risk of Systemic Lupus Erythematosus: A Meta-Analysis. Arthritis Rheum. (2004) 50(3):849–57. doi: 10.1002/art.20049
304. Zein CO, Beatty K, Post AB, Logan L, Debanne S, McCullough AJ. Smoking and Increased Severity of Hepatic Fibrosis in Primary Biliary Cirrhosis: A Cross Validated Retrospective Assessment. Hepatology (2006) 44(6):1564–71. doi: 10.1002/hep.21423
305. Corpechot C, Gaouar F, Chretien Y, Johanet C, Chazouilleres O, Poupon R. Smoking as an Independent Risk Factor of Liver Fibrosis in Primary Biliary Cirrhosis. J Hepatol (2012) 56(1):218–24. doi: 10.1016/j.jhep.2011.03.031
306. Hsu PD, Lander ES, Zhang F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell (2014) 157(6):1262–78. doi: 10.1016/j.cell.2014.05.010
307. Redman M, King A, Watson C, King D. What Is CRISPR/Cas9? Arch Dis Child Educ Pract Ed (2016) 101(4):213–5. doi: 10.1136/archdischild-2016-310459
308. Jeffries MA. Epigenetic Editing: How Cutting-Edge Targeted Epigenetic Modification Might Provide Novel Avenues for Autoimmune Disease Therapy. Clin Immunol (2018) 196:49–58. doi: 10.1016/j.clim.2018.02.001
309. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell (2013) 152(5):1173–83. doi: 10.1016/j.cell.2013.02.022
310. Kungulovski G, Jeltsch A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet (2016) 32(2):101–13. doi: 10.1016/j.tig.2015.12.001
311. Thakore PI, Black JB, Hilton IB, Gersbach CA. Editing the Epigenome: Technologies for Programmable Transcription and Epigenetic Modulation. Nat Methods (2016) 13(2):127–37. doi: 10.1038/nmeth.3733
313. Poria DK, Guha A, Nandi I, Ray PS. Rna-Binding Protein Hur Sequesters MicroRNA-21 to Prevent Translation Repression of Proinflammatory Tumor Suppressor Gene Programmed Cell Death 4. Oncogene (2016) 35(13):1703–15. doi: 10.1038/onc.2015.235
314. Ruan Q, Wang P, Wang T, Qi J, Wei M, Wang S, et al. MicroRNA-21 Regulates T-Cell Apoptosis by Directly Targeting the Tumor Suppressor Gene Tipe2. Cell Death Dis (2014) 5:e1095. doi: 10.1038/cddis.2014.47
315. Satishchandran A, Ambade A, Rao S, Hsueh YC, Iracheta-Vellve A, Tornai D, et al. MicroRNA 122, Regulated by GRLH2, Protects Livers of Mice and Patients From Ethanol-Induced Liver Disease. Gastroenterology (2018) 154(1):238–52. doi: 10.1053/j.gastro.2017.09.022
316. Sporri B, Kovanen PE, Sasaki A, Yoshimura A, Leonard WJ. JAB/SOCS1/SSI-1 Is an Interleukin-2-Induced Inhibitor of IL-2 Signaling. Blood (2001) 97(1):221–6. doi: 10.1182/blood.v97.1.221
317. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of BIC/MicroRNA-155 for Normal Immune Function. Science (2007) 316(5824):608–11. doi: 10.1126/science.1139253
318. O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol Phosphatase SHIP1 Is a Primary Target of Mir155. Proc Natl Acad Sci USA (2009) 106(17):7113–8. doi: 10.1073/pnas.0902636106
319. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA Methylation Controls Foxp3 Gene Expression. Eur J Immunol (2008) 38(6):1654–63. doi: 10.1002/eji.200838105
320. Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, et al. Epigenome Editing by a CRISPR-Cas9-Based Acetyltransferase Activates Genes From Promoters and Enhancers. Nat Biotechnol (2015) 33(5):510–7. doi: 10.1038/nbt.3199
321. Pei WD, Zhang Y, Yin TL, Yu Y. Epigenome Editing by CRISPR/Cas9 in Clinical Settings: Possibilities and Challenges. Brief Funct Genomics (2020) 19(3):215–28. doi: 10.1093/bfgp/elz035
322. Taubert R, Hardtke-Wolenski M, Noyan F, Wilms A, Baumann AK, Schlue J, et al. Intrahepatic Regulatory T Cells in Autoimmune Hepatitis Are Associated With Treatment Response and Depleted With Current Therapies. J Hepatol (2014) 61(5):1106–14. doi: 10.1016/j.jhep.2014.05.034
323. Taubert R, Danger R, Londono MC, Christakoudi S, Martinez-Picola M, Rimola A, et al. Hepatic Infiltrates in Operational Tolerant Patients After Liver Transplantation Show Enrichment of Regulatory T Cells Before Proinflammatory Genes Are Downregulated. Am J Transplant (2016) 16(4):1285–93. doi: 10.1111/ajt.13617
Keywords: autoimmune, hepatitis, epigenome, chromatin modifications, micro-ribonucleic acids, treatment
Citation: Czaja AJ (2022) Epigenetic Aspects and Prospects in Autoimmune Hepatitis. Front. Immunol. 13:921765. doi: 10.3389/fimmu.2022.921765
Received: 16 April 2022; Accepted: 12 May 2022;
Published: 30 June 2022.
Edited by:
Nanda Kerkar, University of Rochester, United StatesReviewed by:
Antonia Follenzi, Università degli Studi del Piemonte Orientale, ItalyYanhong Guo, University of Michigan, United States
Luigi Muratori, Università di Bologna, Italy
Copyright © 2022 Czaja. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Albert J. Czaja, Y3phamEuYWxiZXJ0QG1heW8uZWR1; orcid.org/0000-0002-5024-3065