 Yue Xu
Yue Xu Yongkang Chen
Yongkang Chen Xuan Zhang1
Xuan Zhang1 Jie Ma
Jie Ma Yudong Liu
Yudong Liu Liyan Cui
Liyan Cui- 1Department of Rheumatology, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, China
- 2Department of Laboratory Medicine, Peking University Third Hospital, Beijing, China
- 3Center of Biotherapy, Beijing Hospital, National Center of Gerontology; Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, China
Autoimmune diseases (AIDs) refer to connective tissue inflammation caused by aberrant autoantibodies resulting from dysfunctional immune surveillance. Most of the current treatments for AIDs use non-selective immunosuppressive agents. Although these therapies successfully control the disease process, patients experience significant side effects, particularly an increased risk of infection. There is a great need to study the pathogenesis of AIDs to facilitate the development of selective inhibitors for inflammatory signaling to overcome the limitations of traditional therapies. Immune cells alter their predominant metabolic profile from mitochondrial respiration to glycolysis in AIDs. This metabolic reprogramming, known to occur in adaptive immune cells, i.e., B and T lymphocytes, is critical to the pathogenesis of connective tissue inflammation. At the cellular level, this metabolic switch involves multiple signaling molecules, including serine–threonine protein kinase, mammalian target of rapamycin, and phosphoinositide 3-kinase. Although glycolysis is less efficient than mitochondrial respiration in terms of ATP production, immune cells can promote disease progression by enhancing glycolysis to satisfy cellular functions. Recent studies have shown that active glycolytic metabolism may also account for the cellular physiology of innate immune cells in AIDs. However, the mechanism by which glycolysis affects innate immunity and participates in the pathogenesis of AIDs remains to be elucidated. Therefore, we reviewed the molecular mechanisms, including key enzymes, signaling pathways, and inflammatory factors, that could explain the relationship between glycolysis and the pro-inflammatory phenotype of innate immune cells such as neutrophils, macrophages, and dendritic cells. Additionally, we summarize the impact of glycolysis on the pathophysiological processes of AIDs, including systemic lupus erythematosus, rheumatoid arthritis, vasculitis, and ankylosing spondylitis, and discuss potential therapeutic targets. The discovery that immune cell metabolism characterized by glycolysis may regulate inflammation broadens the avenues for treating AIDs by modulating immune cell metabolism.
Introduction
Autoimmune diseases (AIDs) encompass various chronic disorders involving multiple organs and have various clinical manifestations caused by connective tissue inflammation (1). Most of the current treatments for AIDs use non-selective immunosuppressive agents. Although these therapies successfully control the disease process, patients inevitably suffer from various side effects, particularly an increased risk of infection (2). Investigating the pathogenesis of AIDs is crucial for developing novel selective immunotherapies.
Immune tolerance is established during the maturation of immune cells in the bone marrow and peripheral lymphoid organs (3). Aberrant antigen presentation and differentiation of B cells into autoantibody-secreting plasma cells lead to the development of AIDs (3). Multiple signals that activate the differentiation of CD4+ T lymphocytes are destroyed in AIDs, leading to the breakdown of immune tolerance (4, 5). Hyperactive immune responses triggered by pathogenic autoantibodies are responsible for uncontrolled inflammation in connective tissue (6). Increasing lymphocytes accumulate in lesion locations and even form ectopic germinal centers as the disease progresses (6). However, abnormalities in cell development and function are not limited to T and B cells (7). Recently, it has been suggested that metabolic abnormalities in innate immune cells play a critical role in the pathogenesis of AIDs, such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) (8, 9). However, the comprehensive mechanism of innate immunity involvement in AIDs remains to be elaborated.
Glucose metabolism is an important metabolic pathway that provides energy to cells and consists of multiple enzymes that catalyze the conversion of glucose into metabolized products and energy in the form of ATP (10). As shown in Figure 1, glucose transported into the cytoplasm undergoes glycolysis, and the produced pyruvate is used mainly in oxidative phosphorylation (OXPHOS) after the tricarboxylic acid (TCA) cycle to generate more ATP. Glycolytic intermediates glucose-6-phosphate (G6P) and 3-phosphoglycerate are involved in the pentose phosphate pathway (PPP) and amino acid synthesis, respectively. In addition to providing energy, glycolytic intermediates support immune cells in reprogramming their phenotypes in response to external stimuli (11, 12). Thus, although glycolysis is less efficient than the TCA cycle or OXPHOS in producing ATP, it serves as an important metabolic pathway for activated immune cells (13). During active inflammation, immune cells use glycolysis as the major metabolic pathway to meet the demands of inflammatory activity whereas, they restore OXPHOS during the resolution of inflammation (14). Recently, it has become evident that the glycolytic switch in AIDs determines the fate of immune cells and affects the inflammatory response. More importantly, increasing metabolism has been well characterized in innate immune cells involved in AIDs (15, 16). However, the mechanism by which glycolytic activity in innate immune cells is involved in the pathogenesis of AIDs remains unclear.
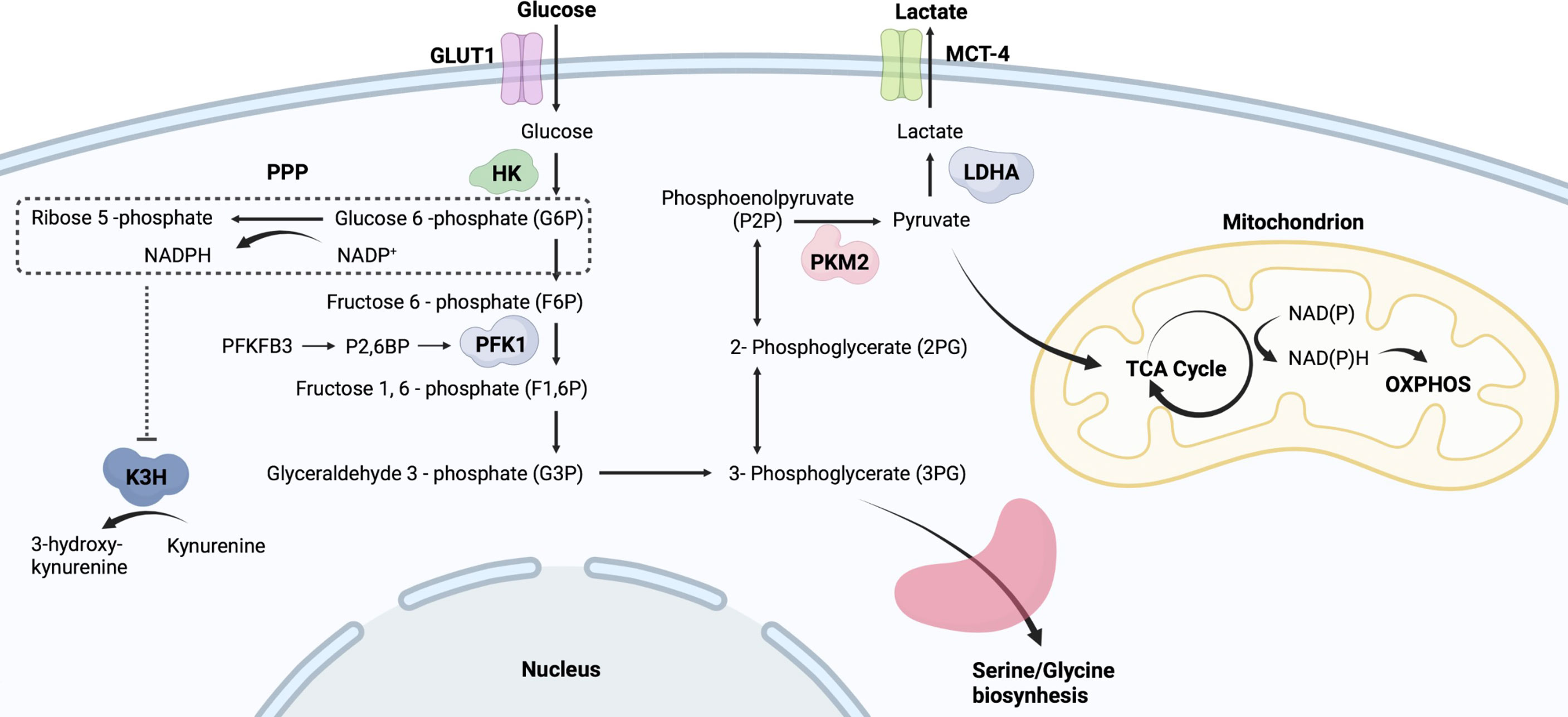
Figure 1 Simplified flowchart of glycolysis. Glucose entering cells is metabolized by HK to G6P, which provides a substrate for PPP. PPP generates ribose 5-phosphate and abundant NADPH. Those NADPH-dependent hydroxylases are manipulated by PPP activity, such as K3H. G6P undergoes a series of oxidative decompositions to generate 3-phosphoglycerate, providing raw materials for serine/glycine biosynthesis. PKM2 controls the final step of glycolysis and generates pyruvate. The produced pyruvate is used mainly in OXPHOS and the tricarboxylic acid TCA cycle to generate more ATP. Monocarboxylate transporter 4, MCT4; Lactic dehydrogenase A, LDHA; Phosphofructokinase-1, PFK-1; Fructose-2,6-bisphosphate, F2,6BP; Kynurenine 3-hydroxylase, K3H.
In this review, we discuss the mechanisms through which glycolysis alters innate immune cells and how metabolic pathways control inflammatory responses in AIDs, thus providing insights for developing new therapeutic targets.
Role of Glycolysis in Innate Immune Cells
The Warburg effect is important for understanding metabolic changes that occur in innate immune cells upon activation (Figure 2). In resting immune cells, pyruvate can enter the TCA cycle for complete oxidation to CO2, generating reduced nicotinamide adenine dinucleotide and reduced flavin adenine dinucleotide (17). This process produces energy efficiently. In contrast, in an inflammatory microenvironment, pyruvate in immune cells undergoes aerobic glycolysis and regenerates nicotinamide adenine dinucleotide (NAD+) to rapidly meet the demands of a pro-inflammatory phenotype (17). The upregulation of glycolytic activity is caused by multiple processes, including the transfer of extracellular glucose into the cell (glucose transporter, GLUT), the breakdown of glucose (hexokinase, HK), and the conversion of glucose-6P to pyruvate (glyceraldehyde-3-phosphate dehydrogenase, GAPDH; pyruvate kinase isoenzyme M2, PKM2) (Figure 1) (18, 19). Besides, G6P undergoes oxidative decomposition in the PPP to generate nicotinamide adenine dinucleotide phosphate (NADPH) and ribose 5-phosphate. This metabolic branch provides the essential metabolite ribose 5-phosphate for nucleotide biosynthesis and cell proliferation (20). The abundantly produced NADPH in PPP supplies reducing power for synthetic reactions, providing antioxidant defenses for cells (20). Thus, those highly NADPH-dependent hydroxylases are manipulated by PPP activity, such as kynurenine 3-hydroxylase (21) (Figure 1). Immune receptors on the cell surface induce a phenotypic switch in immunometabolism. Immune receptors activate various transcription factors to induce the expression of glycolytic genes via kinase signaling pathways, including phosphatidylinositol 3 kinase (PI3K)/serine-threonine protein kinase (Akt), mammalian target of rapamycin (mTOR), and mitogen-activated protein kinase (MAPK) (22, 23). Hypoxia-inducible factor 1α (HIF-1α) nuclear factor kappa B (NF-kB) are major transcription factors mediating immunometabolic and inflammatory activities, inducing glucose uptake, glycolysis, and lipid synthesis (24, 25). mTOR can form different protein complexes, mTOR complexes 1 and 2 (mTORC1 and mTORC2). mTORC2 is responsible for controlling Akt activation through phosphorylation, while PI3K/Akt activates mTORC1 (17). mTORC2 enhances GLUT1 expression and aerobic glycolytic activity. mTORC1 is not only involved in the signaling of glycolysis but also promotes the synthesis of proteins and lipids (17). mTOR signaling is an important regulator of intracellular metabolic activity.
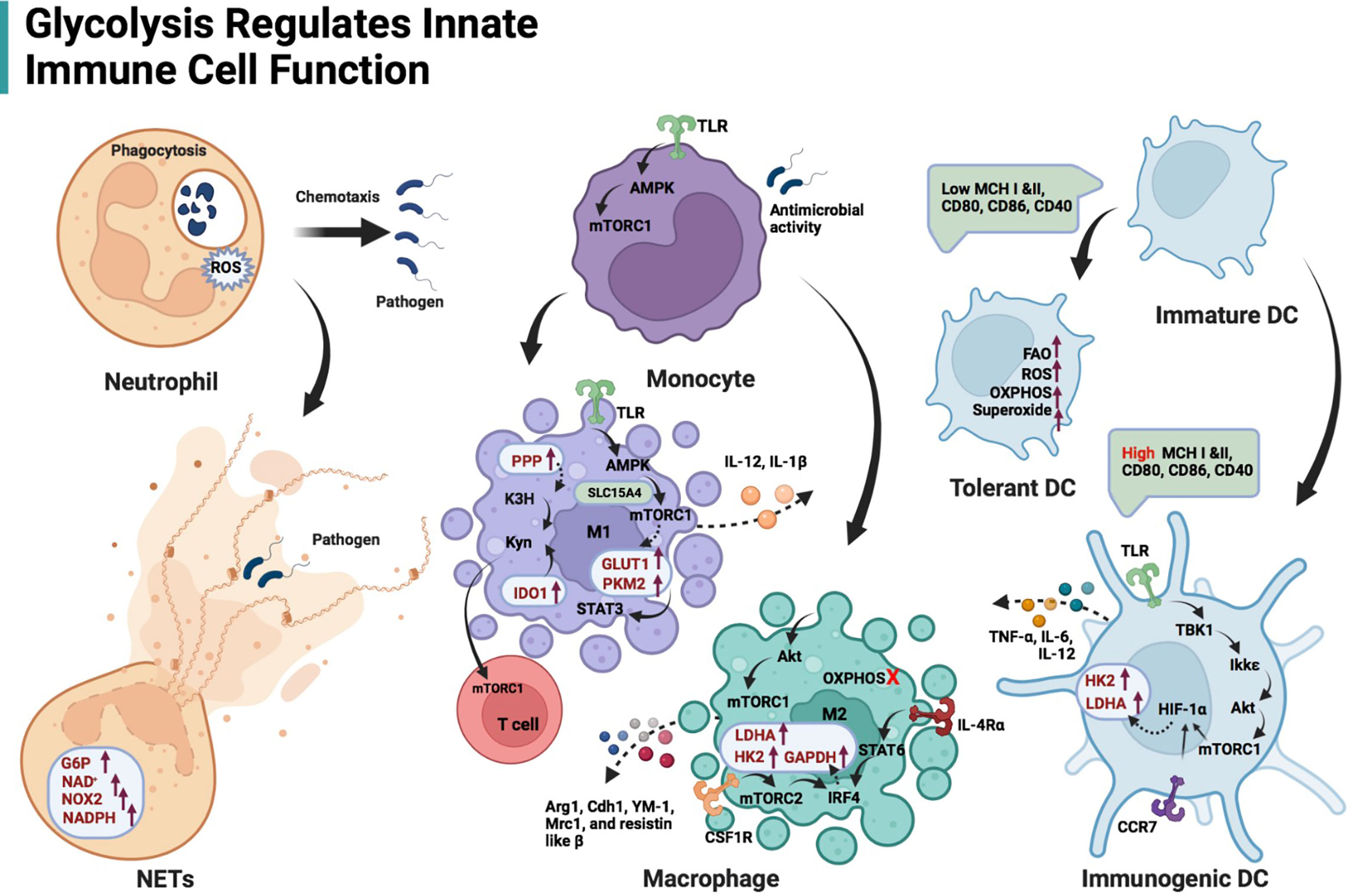
Figure 2 Schematic illustration of glycolysis regulating innate immune cell function. Glycolysis is the main energy production pathway for neutrophils. Impairing glycolysis and PPP can destroy neutrophil function, including chemotaxis and ROS production, even phagocytosis. NETs formation is dependent on adequate glucose flux, G6P, NOX2, and NAD+/NADPH. The TLR/AMPK/mTORC1 regulates glycolysis-dependent antimicrobial activity in monocytes. TLR/AMPK/mTORC1 axis is also responsible for M1-type macrophage induction, expression of glycolytic enzymes (GLUT1 and PKM2) in these cells and their IL-12 secretion. Solute carrier family 15 member A4 (SLC15A4) is likely to maintain the interaction of AMPK and mTORC1 by acting as a scaffold. PKM2 in M1 macrophages contributes to IL-1β transcription via STAT3 signaling. Both enhanced PPP and IDO-1 in M1 macrophages facilitate kynurenine accumulation, stimulating mTORC1 activity in T cells. Akt/mTORC1-mediated glycolysis also affects M2-like macrophage differentiation and gene profile expression (Arg1, Cdh1, YM-1, Mrc1, and resistin-like β) when OXPHOS in macrophages is inhibited. The interferon regulatory factor 4 (IRF4), which is downstream of the IL-4 receptor α/STAT6 and colony-stimulating factor 1 receptor (CSF1R)/mTORC2 signaling axis, promotes glycolysis (enhanced expression of LDHA, GAPDH and HK2) during M2 activation. DCs activated by TLRs depend on glycolysis flux to fulfill metabolic and functional requirements, including secretion of TNF-α, IL-6 and IL-12. TBK1/Ikkϵ-mediated Akt phosphorylation responds to lipopolysaccharide stimulation of TLRs on DCs. p-Akt/mTORC1 immediately promotes the transcription of HK2 and LDHA via HIF-1α. Cxc chemokine receptor 7 (CCR7)-mediated HIF-1α induction contributes to DC migration.
Neutrophils
Glycolysis is the main energy production pathway for neutrophils, although glucose is also metabolized via the hexose monophosphate pathway. Mitochondria in neutrophils are not the main source of ATP. The mitochondria in neutrophils are primarily involved in the initiation of cell death. Therefore, the energy required for inflammatory functions and phagocytic activity originates from glycolysis in these cells (26). Phagocytosis by neutrophils involves the uptake of pathogens into plasma membrane-derived vacuoles and subsequent fusion of lysosomes with pathogen-containing phagosomes. Sodium iodoacetate, which selectively inhibits glycolysis by irreversibly inhibiting GAPDH, and 2-deoxyglucose (2-DG), which inhibits both glycolysis and PPP by competitively inhibiting G6P production, is reported to inhibit neutrophil phagocytosis (27, 28).
Neutrophils that migrate to inflammatory tissues can kill pathogens through phagocytosis, proteolytic enzymes, and reactive oxygen species (29). Neutrophils also function by releasing antimicrobial granules extracellularly (29). Antimicrobial peptides and chromatin released by neutrophils form an extracellular network that binds pathogenic microorganisms and limits the progress of infection. These extracellular fiber-structures are called neutrophil extracellular traps (NETs) (29). Scientists believe that neither NETs nor the chromatin within them originate from cell disintegration. However, mature neutrophils die within a short time after entering the circulation. The formation of NETs is likely an early event in neutrophil death (29). Various microorganisms, bacterial products, and pharmacological stimuli, such as 2-acetoxy-1-methoxypropane, induce NETs formation (30). Stimulation with 2-acetoxy-1-methoxypropane results in increased GLUT levels, glucose uptake, and glycolysis rates. However, when neutrophils are exposed to 2-acetoxy-1-methoxypropane under low glucose conditions, polymorphic nuclei are not maintained, and NETs are not formed (30). This suggests that NETs formation is strictly dependent on glucose levels. G6P, a glycolytic intermediate, enters the PPP. Nicotinamide adenine dinucleotide phosphate (NADPH) is produced during the oxidative phase of the PPP, which maintains NADPH oxidase 2 activity and reactive oxygen species (ROS) production. Both G6P and NADPH are involved in chromatin depolymerization, NADPH oxidase 2-dependent NET formation and NET release (31). NADPH appears to be the core metabolite supporting NETs. The glycolysis inhibitor 2-deoxyglucose (2-DG) affects NET formation (30). Mechanistically, after HK2 is blocked by 2-DG, G6P as the substrate for HK2 decreases immediately. Additionally, the metabolism of glucose to pyruvate in the cytoplasm generates relatively low levels of ATP and NADPH. Neutrophils use aerobic glycolysis to reduce pyruvate to lactate (which is processed by lactic dehydrogenase A, LDHA) and recycle the resultant NAD+ in glycolysis. Therefore, impairing LDHA also restrains the induction of NETs (32). The glucose-6-phosphate transporter/G6Pase-β complex regulates energy metabolism, glycolysis, and the PPP, in neutrophils by controlling G6P levels in the cytoplasm (31). Disturbances in glucose metabolism caused by defective glucose-6-phosphate transporter activity impair neutrophil function, including chemotaxis and ROS production (33). Moreover, NETs formation is, to some extent, dependent on glutamine and, to a lesser extent, affected by the ATP synthase inhibitor oligomycin (30).
Optic atrophy 1 (OPA1) is a mitochondrial structural protein that is essential for mitochondrial integrity and plasticity. Meanwhile, OPA1-dependent ATP is required for the formation and maintenance of NETs (34). Mechanistically, OPA1 in neutrophils can ensure the generation of NAD+ by maintaining the activity of electron transport complex I. Dysfunctional OPA1 reduces the NAD+ levels, which results in a consequent decrease in ATP from glycolysis. This suggests that glycolytic ATP plays an important role in the formation of NETs (34). Additionally, dimethyl malonate acts as a neutrophil succinate dehydrogenase inhibitor and reduces NETs release (35). These results suggest that glycolysis may be involved in NET formation through various biological mechanisms.
Macrophages
The plasticity of macrophages enables them to switch from one phenotype to another in response to stimuli from the microenvironment (36, 37). The balance between different macrophage subtypes maintains health, while phenotype dysregulation of macrophages contributes to the development of diseases, including AIDs (38). Although the M1/M2 classification has been found insufficient to represent the diversity and complexity of macrophage subtypes as two distinct phenotypes with distinct functions, M1/M2 typing can reflect the plasticity of phenotypic transition. Interferon gamma (IFN-γ) secreted by helper T cells and bacterial lipopolysaccharide can induce macrophage differentiation to classically activated M1 (inflammatory). They produce inflammatory cytokines that are involved in pathogen elimination activities and interfere with wound healing and tissue repair (39, 40). These cytokines underlie the pathology of M1 macrophage-mediated AIDs (41). Indoleamine (2,3)-dioxygenase (IDO) is highly expressed in M1 macrophages and orchestrates tryptophan-kynurenine metabolism (42). Kynurenine stimulation alters the phenotype of human T cells via the eukaryotic translation initiation factor 4E binding protein 1/mTORC1 axis (43). Microenvironment-derived interleukin (IL)-21, IL-33, IL-10, and Th2-cell-derived IL-13 and IL-4 all boost macrophage polarization to the M2 type (39). This polarization appears to be a negative feedback regulation of inflammatory responses in the microenvironment, as M2-like macrophages facilitate inflammation resolution and wound healing (38) via secretion of the vascular endothelial growth factor and transforming growth factor-beta.
Alterations in metabolic signature support functional switching. Macrophages use OXPHOS in the resting state. M1 macrophages overexpress GLUT1 and catabolize arginine to produce nitric oxide and ROS (44). PKM2 in these cells is mobilized and phosphorylated (45), contributing to IL-1β transcription via signal transducer and activator of transcription (STAT) 3 signaling (46). M1 macrophages are heavily dependent on glycolysis and undergo two disruptions in the TCA cycle, resulting in an accumulation of citrate acid, succinic acid, and lactate (47). Mechanistically, 1) Downregulated isocitrate dehydrogenase caused by metabolic reprogramming inefficiently converts isocitrate to α-ketoglutarate; and 2) A large amount of itaconic acid in M1 macrophages not only limits the function of succinate dehydrogenase, resulting in substrate accumulation, but also enhances the activity of lactate dehydrogenase. As a signaling hub for toll-like receptors (TLRs), solute carrier family 15 member A4 is likely to maintain the interaction of adenosine monophosphate-activated protein kinase (AMPK) and mTORC1 by acting as a scaffold (48). Activation of TLR/AMPK/mTORC1 signaling is critical for the induction of M1-like metabolic phenotype and IL-12 secretion (48).
In contrast, M2 macrophages, which have an intact TCA cycle that provides substrates for the electron transport chain, are involved in tissue repair and wound healing and use oxidative metabolism to fuel their long-term functions (47). The inflammasome activity in M1 macrophages is regulated by NETs and ATP-binding cassette transporter A1/G1/cholesterol crystal dual signaling in tissues (49–51), followed by upregulation of several glycolytic signals such as Eno3, Aldoc, Bpgm, Pgam1, Pgam2, Pkm, and Hk3 (52). Conversely, M2 macrophages exhibit a similar level of glycolysis as unstimulated cells (53, 54). This may result from the upregulated gene expression of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB) 1 induced by M2-type activating signals. Phospho-fructokinase 2 encoded by PFKFB1 exhibits lower activity than that encoded by PFKFB3, thereby reducing glycolytic flux (53). Inhibiting glycolysis does not affect IL-4-induced macrophage activation without blocking OXPHOS. When OXPHOS and glycolysis were simultaneously inhibited (high doses of 2-DG, 10 mM), M2 differentiation was hindered (55). Based on the results of several recent studies, investigators suggest that glycolysis is also involved in M2 activation. Low doses of 2-DG (1 mM) can downregulate the expression of a series of M2-like gene profiles, including Arg1, Cdh1, YM-1, Mrc1, and resistin-like β through the Akt/mTORC1 signaling pathway (56–58). The IFN regulatory factor 4, which is downstream of the IL-4 receptor α/STAT6 and colony-stimulating factor 1 receptor/mTORC2 signaling axis, promotes glycolysis (enhanced expression of LDHA, GAPDH and HK2) and M2 activation (59). These results suggest that M2 activation is preferentially dependent on OXPHOS, but glycolysis is required if OXPHOS is impaired. Additional studies must elucidate the role of glycolysis in M2 activation.
Recruited by apoptotic cells, the process by which macrophages recognize, engulf, and degrade dying cells is called efferocytosis (60). Macrophages in the injured tissue exhibit exocytosis, and their metabolite load becomes equivalent to that of phagocytes (61). However, the rise in IL-10 levels observed during efferocytosis is regulated by mitochondrial β-oxidation and the electron transport chain and is not dependent on glycolysis (62). Conversely, the chemotactic behavior of macrophages in the inflammatory microenvironment relies on glycolysis, both in vivo and in vitro (63). ATP generated by glycolysis can rapidly replenish the energy required for actin synthesis and pseudopodia extension under mild hypoxia in the inflamed area (63).
Activation of myeloid-derived cells by microbial β-glucan is mediated through metabolic pathways that induce epigenetic reprogramming, also known as “trained immunity” (64). Metabolomic and transcriptomic data suggest that glutaminolysis and glycolysis are involved in β-glucan-induced immune reprogramming of monocytes. Glutamine-induced accumulation of fumarate in the TCA cycle can inhibit histone demethylase lysine-specific demethylase 5A to initiate epigenetic reprogramming in monocytes (65). This is consistent with metabolic changes in glucose mass consumption, lactate production, and inversion of the NAD+/NADH ratio observed in trained monocytes (52).
The Akt/mTOR/HIF-1α signaling axis regulates glycolysis in monocytes (66). Metformin activates AMPK and inhibits the antibacterial activity of monocytes that is induced by mTOR. This is also observed in the mTOR inhibitor rapamycin. Solute carrier family 15 member A4 is an amino acid/oligopeptide transporter in immune cells (67). Additionally, stimulation of the extracellular immune complex (IC) can activate HIF-1α signaling via the Syk/Erk/mTOR and Syk/PI3K/Akt/mTOR pathways, resulting in a switch of macrophage energy metabolism to glycolysis. Fcγ receptor IIb and aFcγ receptor are involved in the transduction of extracellular IC signals (68). This metabolic switch is observed in renal Fcγ receptor IIb-depleted macrophages associated with decreased glycolytic activity, increased mitochondrial respiratory activity, and respiratory reserves in antibody-mediated nephritis (69). Overall, these findings suggest that inhibition of glycolysis in macrophages reduces tissue inflammation, highlighting its potential as a therapeutic strategy for AIDs.
Dendritic Cells (DCs)
As the primary antigen-presenting cells of the peripheral immune system, DCs act as a bridge between innate and adaptive immunity and are also responsible for inducing lymphocyte activation and differentiation (70). Non-activated DCs exhibit oxidative metabolism. Pathogen-associated molecular patterns bind TLRs on DCs and induce chemokines and inflammatory factors. TLRs signaling, upregulation of glycolytic activity, and increased lactate production are within the scope of Warburg physiology (71). Additionally, mitochondrial activity is progressively lost following TLR signaling. Moreover, both glucose deprivation and 2-DG blockade reverse these effects but result in decreased OXPHOS, and glucose restriction prevents DC activation, resulting in premature death of DCs (72).
This reprogramming of glucose metabolism, a switch from oxidative metabolism to glycolysis, is required for DC activation, phenotype maintenance, and migration to lymph nodes (73–75). DCs are rapidly activated by TLR signal heavily depends on a surge in glycolysis flux to fulfill metabolic and functional requirements (73), including the secretion of tumor necrosis factor-α (TNF-α), IL-6, and IL-12. Unlike conventional PI3K-dependent signaling, tumor necrosis receptor-associated factor family member associated NF-κB activator binding kinase 1 (also known as TBK1)/I-kappaB kinase ϵ-mediated Akt phosphorylation responds to lipopolysaccharide stimulation of TLRs in DCs (73). p-Akt/mTORC1 immediately promotes the transcription of HK2 and LDHA via HIF-1α (76). Subsequent HK2 upregulation and enrichment around the ion channels of mitochondria assist DCs to prime T cells in the microenvironment (73). Supplementing 2-DG into the DC culture mixture in vitro significantly weakened the ability to alter shape and remodel the cytoskeleton (74). Even the compensatory supply of ATP by mitochondrial OXPHOS could not reverse the decline in overall distance traveled and velocity (74). It has been proven that cxc chemokine receptor 7-mediated HIF-1α induction contributes to DC migration (74, 75).
Immature DCs exhibit a phenotype with lower cross-expression capacity (histocompatibility complex I and II) and lower expression of co-stimulatory molecules (CD80, CD86, and CD40) than mature DCs, which confers tolerance characteristics (i.e., tolerant DCs) in the peripheral immune system (77). Tolerant DCs exhibit significantly enhanced catabolic pathways, including OXPHOS and fatty acid oxidation, compared with the marked pro-inflammatory activity of activated DCs (also known as immunogenic DCs). Mitochondrial oxidative activity, ROS production, and superoxide production are more pronounced in tolerant DCs (78). The extracellular acidification rate (mpH/min) analysis can indicate the rate, capacity, and reserves of glycolysis. Although tolerant and immunogenic DCs exhibited similar rates of glycolysis, tolerant DCs demonstrate higher glycolytic capacity and reserves, and therefore have higher ATP reserves (78). In contrast, tolerant DCs exhibit more active fatty acid oxidation than immunogenic DCs. Inhibition of fatty acid oxidation inhibits the functions of tolerant DCs and partially restores T-cell stimulation capacity (78).
Glycolysis in Autoimmune Diseases
The mechanism of hyperactivated glycolysis varies for different AIDs. Hallmark lesion sites represent not only differences in clinical symptoms but also differences in metabolic abnormalities. For example, expression signatures in III/IV lupus nephritis tubulointerstitium exhibit down-regulated glycolytic activity (79) while increased glucose uptake and glycolysis are observed in RA-lesioned joints (80). Additionally, glycolysis in diverse stages contributes to the development of AIDs. In the early stage of RA, naive T cells demonstrate reduced phosphofructokinase-1 activity, a deficiency of glycolysis-derived ATP, and increased cell death (81). In the late stage of RA, up-regulated GLUT1 in synovial cells of joint tissue furthers HIF-1α function (82). Therefore, blockade of hypermetabolic states and inhibition of glycolytic mediators may be therapeutically useful for AIDS. In this section, we will review the primary features of glycolysis and innate immune cells in AIDs (as shown in Table 1), referring to the association between glycolysis and innate immune cell function.

Table 1 The primary features of glycolysis and innate immune cells in AIDs.
SLE
Neutrophils contribute to SLE pathogenesis through multiple mechanisms, including the secretion of NETs, which are potent stimulators of type I IFN production. In SLE, neutrophil death and NETs formation are enhanced, leading to an increased debris burden associated with antinuclear autoantibodies (117–119). In healthy individuals, following uptake by macrophages, NETs are shuttled in phagosomes to lysosomes for degradation, and this process is promoted by DNase I and C1q (120). The authors showed that 62% of patients with SLE were serum positive for anti-DNase antibodies as opposed to 8% of healthy volunteers, suggesting that defects in the clearance of aberrant neutrophils by macrophages contribute to SLE pathogenesis (121). Moreover, the antimicrobial peptides and self-DNA composing the extracellular traps activate the plasmacytoid dendritic cells and autoimmune B cells via TLR9 engagement (122). IFN-α, a member of the type I interferon family, is majorly generated from plasmacytoid DCs in SLE (122).
The glycolytic key enzymes and metabolites are involved in the propensity to form NETs. Compared with that in neutrophils from healthy volunteers, the expression of GLUT-3 and GLUT-6 is reported to be decreased in the cell membranes of neutrophils from patients with SLE, along with a concomitant decrease in intracellular glucose concentration (83, 84). When intracellular glucose and glycolytic fluxes are declined to levels where neutrophil viability is difficult to maintain, B-cell lymphoma 2 apoptosis regulator-dependent apoptosis is activated and neutrophil numbers reduce (85). Besides, Perner et al. found that a deficiency of PPP-derived NAPDH was directly associated with reduced NADPH oxidase 2 activity and ROS production (86). Neutrophils may compensate for PPP-related ROS using mitochondrial-originated ROS. But all these changes result in reduced cellular redox capacity and oxidation of mitochondrial DNA (87). Oxidized DNA is subsequently expelled through the mechanism of NETs (87). Some scientists have revealed that neutrophils suffer from a distinct form of cell death, named NETosis, after releasing NETs (88, 89). NETosis has been indicated as an important cause of neutropenia in SLE (89). Although a recent study has revealed that ferroptosis (a novel kind of cell death related to iron overload and abnormal lipid metabolism) (123) may be the major contributor to neutropenia in SLE (124), it is undeniable that alteration of glycolytic activity is involved in the progression of this disease.
Similarly, single-cell RNA sequencing analysis of metabolism-related genes revealed decreased glycolysis and TCA cycling but increased OXPHOS in lupus nephritis renal epithelial clusters (79). The proximal tubule cells have more mitochondria than other renal epithelial cells and are therefore dependent on oxidative metabolism, which prompts the renal epithelium in lupus nephritis to develop such a metabolic switch (79). Additionally, peroxisome biogenesis signatures were markedly upregulated in proximal tubule cells, suggesting that disease progression results in altered mitochondrial and peroxisomal metabolism (79). The simultaneous administration of the mitochondrial metabolism inhibitor metformin and glycolysis inhibitor 2-DG significantly restored immune tolerance of lupus mice, dampening autoimmune inflammation. These findings indicate that immunometabolic regulation may be a practical strategy for SLE therapy (125).
Conversely, activated glycolysis regulates the functions of T cells involved in SLE pathogenesis. Glutaminase Gls1 promotes HIF-1α expression and glycolytic activity, enabling the differentiation of CD4+ T cells into T helper (Th) 17 cells to promote SLE disease activity (126). Aberrant tryptophan metabolites are the mediators that manipulate CD4+ T cells in SLE (127, 128). IDO, which is activated by IFN-γ, is primarily responsible for converting tryptophan into kynurenine and tryptamine. In an in vitro assay, kynurenine induces mTORC1 activity in double negative T cells but not in CD4+ or CD8+ T cells derived from human (43). Interestingly, kynurenine enhances CD4+ T cells to produce IFN-γ in lupus-prone mice, whereas tryptamine stimulates mTORC1 signaling and glycolytic activity in CD4+ T cells (129). It has been proven that gut microbiota predominantly growing in SLE contributes to this aberrant tryptophan metabolism (128, 129). IDO levels are higher in SLE patients (130) and are associated with the active phenotype in the sunny season (131). In response to inflammation, IDO is often highly expressed by antigen-presenting cells (such as macrophages and DCs) that appear specialized for rapid response (132). IL-1β alone with IDO-1 is identified as hyperactivated in autoimmune macrophages (42, 133). M1-like macrophages may be involved in generating kynurenine and modulating T-cell immunity. Besides, inhibition of kynurenine 3-hydroxylase results in decreased intracellular catabolism of kynurenine, which is driven by enhanced PPP (43). Importantly, increased PPP transcription is closely associated with CD68+ macrophages in the lupus kidney and is to blame for the reduced glomerular filtration rate (134). All of these show that oxidative stress in macrophages promotes kynurenine accumulation.
Recently, the glycolytic propensity in macrophages may have facilitated the inflammatory features of SLE. A study has found that human and mouse macrophages undergo a glycolytic switch in response to IgG IC stimulation, reflecting changes in macrophage metabolism in inflamed tissues in vivo (68). This metabolic reprogramming contributes to the production of many pro-inflammatory mediators, including IL-1β, which is further regulated by mTOR and HIF-1α. Inhibition of glycolysis or knockdown of HIF-1α attenuates IgG IC-induced macrophage activation in vitro, including in primary human kidney macrophage cell lines (68). Additionally, inhibition of glycolysis in a mouse model of antibody-mediated nephritis resulted in decreased renal macrophage IL-1β levels and decreased neutrophil recruitment (68).
PKM2 regulates the final step of glycolysis. PKM2 expression is reported to be higher in monocytes, DCs, and B cells derived from patients with SLE than in those derived from healthy volunteers (90). Additionally, a PKM2-MAPK/NF-κB-PKM2 feedback loop was reported in spontaneous lupus MRL/lpr mice and imiquimod-induced lupus mice (90). PKM2 inhibitors suppress proline-rich tyrosine kinase 2, preventing TLR4/TLR7/TLR9 from activating the MAPK/NF-κB pathway (90). PKM2 and lactate levels were reported to be abnormally increased in the hippocampus of MRL/lpr lupus mice (91). PKM2-β-catenin signaling leads to neuronal synapse loss by promoting microglial hyperactivation, hypersecretion of IL-6 and IL-1β, and hyperphagocytosis. In vivo application of AAV9-shPKM2 in a neuropsychiatric SLE mouse model was shown to delay cognitive impairment and brain damage (91). Hdac7, a histone deacetylase, prevents acetylation from limiting PKM2 activity in macrophages (92). Mouse bone marrow-specific deficient hdac7 can disrupt glycolysis-dependent inflammatory responses (92). Hdac7 is considered a susceptibility loci for SLE (93). These pieces of evidence reveal that, differing from neutrophils, controlling the glycolytic mechanism in macrophages may be a novel metabolic regulatory strategy for SLE treatment.
RA
The metabolic microenvironment of RA exhibits different characteristics at different stages and disease sites (81). In the first stage of RA, T cells lose self-tolerance, a critical function, and facilitate autoantibody production by B cells (135). A switch in the metabolic profile within immune cells is the primary mechanism at this stage. In the second stage, autoantibodies cause metabolic alterations in synovial cells, causing uncontrolled inflammation in the joint (135). Adaptive autoimmunity initiates abnormal innate immune functions in the third stage (135).
Increasing evidence suggests metabolic feature changes in stromal and immune cells in RA (136). Recent studies have shown that the hexokinase 2 (HK2) level is elevated in Th17, DC, and fibroblast-like synoviocyte (FLS). By inhibiting glycolysis-dependent DCs activation and Th17/Treg imbalance, specific inhibition o-f HK2 (3-bromopyruvate) significantly reduced the degree of joint swelling and histological damage in SKG mice (a RA model) (137). Upregulation of HK2 is associated with hypertrophy of the synovial lining, which is involved in the bone and cartilage damage observed in RA (138). Hyperactivated HK2 promotes the proliferation and secretory function of synovial cells by mediating AMPK to activate NF-κB signaling (139). HK2 inhibitors effectively restrain the production of inflammatory factors (140). Additionally, AMPK can limit the activity of mTORC1 in immune cells. Wen et al. showed that dysfunctional AMPK induces hyperactive glycolysis in helper T cells by mediating aberrant activation of mTORC1, which facilitates the exacerbation of synovitis in RA (141). Citrate synthase levels decrease in RA synovial fluid, which suggests that the anaerobic glycolytic activity in the joint is upregulated under hypoxic conditions (142). Additionally, lactate levels in the blood samples of patients with early RA correlate with the degree of inflammation (143). Synovial fluid metabolomics in patients with RA demonstrated decreased glucose and increased lactate levels, which correlated with disease activity and markers, such as C-reactive protein (143). GLUT1 is the major glucose transporter in RA-FLSs, macrophages, and T cells. Upregulation of GLUT1 has been reported in the lining and sublining of the RA synovium (82). GLUT1 is also associated with increased glucose uptake and a hypoxic microenvironment in the joints with increased HIF-1α activity (144).
A recent study provided new insights into how glycolysis in innate immune cells contributes to RA progression. Using [18F]-fluoro-2-deoxy-d-glucose ([18F]FDG)-positron emission tomography-computed tomography tracing, Kubota et al. reported that increased glucose uptake in patients with RA correlated with disease severity and treatment response (145). Higher levels of [18F]FDG accumulation in swollen joints are associated with pannus rather than periarticular infiltration of inflammatory cells and are positively associated with arthritis progression (146). This observation suggests that activated macrophages contribute to the accumulation of [18F]FDG in the pannus, with hypoxia and cytokine stimulation promoting [18F]FDG uptake by macrophages.
Depending on glucose uptake and NADPH flux, NET induction is strongly correlated with joint damage and the pathogenesis of RA. The formation of anti-cyclic citrullinated peptide antibodies (anti-CCPs) and rheumatoid factor in the peripheral blood is a characteristic phenomenon of RA (94, 95). The citrullinated self-antigens recognized by anti-CCP are produced by peptidyl arginine deiminase (PAD) (94, 95). Neutrophils express aberrant levels of PAD in the synovial fluid of RA patients (94, 96). This is proved by the evidence that the PAD alone with myeloperoxidase (MPO) is located in the necrotic areas of synovial tissue (97). Aggrandized NETs are detected in the neutrophils from circulating, synovial tissue (98) and rheumatoid nodules in RA, which exhibit a correlation with anti-CCP levels (99). NETs formation is triggered by co-engagement of anti-CCP, IL-17A, and TNF-α (99). Subsequently, NETs induce further pro-inflammatory activity (including secretion of IL-6 and IL-8) in FLSs (99). Various proteins in NETs can damage the cartilage matrix and aggravate joint damage, including elastase and matrix metalloproteinase (2, 8, and 9) (100, 101, 147). Among them, elastase can promote the release of PAD2 from FLSs, which can citrullinate cartilage fragments (101). FLS phagocytose modified cartilage fragments (autoantigens), present antigens and induce autoimmune CD4+ T cells (101).
Both undifferentiated monocytes and macrophages in RA are in a hypermetabolic state (148). IL-1β, which is a consequence of metabolic reprogramming, seems to be a vital contributor to the pathogenesis of RA (102–108). Cinnamaldehyde inhibits the expression of the succinate receptor 1 and the accumulation of succinate, leading to restricted HIF-1α activation and ultimately limiting glycolytic activity (102). Inhibitors of TLR7 and IL-1 receptor-associated kinase 4 also interrupt HIF-1α/c-Myc signaling (103). Impairing macrophage glycolysis in RA is associated with inflammasome disassembly, decreased IL-1β production, and arthritis remission (102). The cellular membrane zinc transporter Zip8 is reported to be present at higher levels in the synovial tissue of patients with RA than in healthy volunteers (104). Bioavailable zinc in the monocyte cytoplasm is essential for protein phosphatase 2 phosphatase activator inhibition and mTORC1/p-S6K phosphorylation. Zip8 expression is associated with IL-1β production in monocytes/macrophages in severe arthritis (104). Immunostaining confirmed that glycogen synthase kinase 3b expression is prevalent in macrophages in the RA-injured synovium, and glycogen synthase kinase 3b inactivation is a metabolic switch that induces mitochondria in macrophages to use oxygen inefficiently to produce sufficient ATP (105). The resultant large amount of ATP maintains the activity of collagenase cathepsin K and promotes the production of IL-1β and IL-6 (105).
PKM2 may be a connection between glycolysis and activated macrophages in RA. PKM2 is an active enzyme involved in glycolysis in macrophages, and its dimerization depends on the intracellular ROS concentration (106). Hypermetabolic macrophages in RA accumulate ROS and activate the PKM2/STAT3 signaling pathway and transcription of IL-1β and IL-6 (106). Production of TNF-α and IL-1β is triggered by STAT1 under PKM2 stimulation in the induced RA model (Dark Agouti rats) (107). Although FLSs express higher levels of PKM2 than immune cells, obviously, FLSs extracellularly secrete a small amount. PKM2 in synovial fluid and plasma of RA patients originates from activated macrophages rather than FLSs (108). The released PKM2 facilitates macrophages to differentiate into osteoclasts involved in joint destruction (108). A hypoxic environment, active glycolysis, and massive secretion of inflammatory factors are characteristic of the macrophage-to-osteoclast transition in the RA synovium (109, 110). This process also involves a deficiency of inhibitory signals, including the copper metabolism domain-containing 1/receptor activator of the nuclear factor kappa-B ligand (RANKL) axis (110). Lack of regulation of NF-κB signaling and the transcription factor E2F1 are the primary drivers for the metabolic shift in osteoclasts (110).
Anti-Neutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis (AAV)
AAVs are a heterogeneous group of AIDs characterized by impaired microvasculature (149). Most of autoantibodies in most patients with ANCA-AAV are directed against autoantigens in the primary granules of neutrophils and lysosomes of monocytes, including MPO and protease 3. Patients with different autoantibodies exhibit different clinical manifestations. Patients with anti-protease 3 antibodies present with granulomatous inflammation, while patients with anti-MPO often present with sclerosis (149). Studies have reported that cross-linked ANCA Fab regions may be involved in disease progression by activating neutrophil superoxide production and inducing respiratory bursts (150, 151). Additionally, ANCA can induce NETs formation and simultaneously promote histone citrullination, which triggers disseminated intravascular coagulation, leading to toxic damage to the endothelium (111). This can be prevented by binding recombinant thrombomodulin to neutrophils via the macrophage 1 antigen (111). As mentioned earlier, lactate and G6P-dependent-PPP support the NET formation (31, 33, 34). These results indicate the crucial role of glycolysis in AAV.
Few studies have focused on the pathogenic role of monocytes in AAV. Recently, some evidence has found clues to how glycolysis in monocytes contributes to AAV development. It has also been suggested that ANCA F(ab)2, but not intact ANCA IgG, activates the respiratory burst of monocytes involved in AAV pathogenesis (152). The anti-MPO antibody stimulates monocyte differentiation into macrophages through macrophage-colony stimulating factor 1 (112), downregulates IL-10 secretion (113), and upregulates the secretion of several M1-like cytokines, such as IL-1β, IL-6, and IL-8 (114). Pro-inflammatory macrophages play a significant role in microvascular-targeted inflammation in AAV. O’Brien et al. reported that the metabolic transition of monocytes occurs after ANCA stimulation (115). Anti-MPO-stimulated monocytes exhibited massive uptake of glucose, increased glycolysis and OXPHOS (115). Glycolysis was also activated in anti-protease 3-stimulated monocytes, albeit for a shorter duration. Additionally, OXPHOS induces the subsequent secretion of IL-1β (115).
Ankylosing Spondylitis (AS)
AS refers to a group of diseases called spondyloarthropathies that present with chronic back pain predominantly affecting the spine and sacroiliac joints, the diagnosis of which is often delayed (153, 154). Genetic factors (153) and environmental factors, including smoking and infection (154), are the main risk factors for AS development; however, the exact mechanisms underlying AS pathogenesis are unclear. Ou et al. established a serum metabolism-associated diagnostic panel and found 55 metabolites that were significantly different between patients with AS before and after TNF inhibitor treatment (116). Healthy volunteers and patients with AS could be differentiated using five metabolites: L-glutamic acid, arachidonic acid, L-phenylalanine, phosphocholine [18:1(9Z)/18:1(9Z)], and 1-palmitoylglycerol (area under the curve 0.998, 95% confidence interval: 0.992–1.000) (116). Pathway analysis showed that multiple pathways, including amino acid biosynthesis, glycolysis, glutaminolysis, fatty acid biosynthesis, and choline metabolism, were significantly active in patients with AS (116). This study provided new insights into AS pathogenesis; however, more mechanical studies investigating the role of immunometabolism in AS are required.
Anti-Glycolysis Drugs That Target Innate Immune Cells
One possible use of metabolically targeted therapy is as an “adjuvant” to increase the effectiveness of co-administered biological or conventional antirheumatic drugs. Multiple pathways affect innate immune cells through different mechanisms that alter multiple aspects of the immune system and synergistically reduce the production and release of pro-inflammatory cytokines. Due to their cost-effectiveness and efficacy in metabolic reprogramming, many anti-malarial drugs, such as hydroxychloroquine and chloroquine, are also used to treat rheumatic diseases. While glycolysis plays a key role in activated FLSs, lactate levels in the FLSs of patients with RA are higher than those in the synovial fluid of patients with osteoarthritis (155). Increasing the reliance of immune cells on accelerated glycolysis makes them more vulnerable to apoptosis (155). This suggests that the biological mechanisms of metabolism-targeted drugs and their therapeutic effects deserve attention. In the section below, we review the molecular mechanisms of these drugs and their corresponding clinical applications.
Rapamycin
mTOR is recognized as a central regulator of multiple metabolic pathways that control cell differentiation, death, and inflammatory activity. mTOR activity mediated by various kinase signals upregulates the expression of glycolytic and inflammatory genes, which in turn helps immune cells meet metabolic demands (156). Given the role of mTOR signaling in regulating inflammation, it serves as a critical link between metabolic phenotypes and AIDs.
Notably, as an mTORC1 inhibitor, N-acetylcysteine can safely reduce disease activity in patients with SLE by suppressing T-cell inflammatory factors (157). Lupus nephritis is a comorbidity in late-stage SLE. Rapamycin treatment reduced renal tissue damage and proteinuria, improved renal function, and prolonged survival in NZBW/F1 lupus-prone mice (158). Aberrant elevation in antiphospholipid antibodies is thought to be associated with liver damage in SLE (159). Rapamycin can restore liver mitochondrial function by targeting mTORC1 and effectively reducing anti-β2-glycoprotein I and anticardiolipin in SLE mice (159).
Previous evidence suggests that controlling Th17-triggered inflammation is the mechanism by which rapamycin treats SLE. KN-93 can restrain Th17 differentiation by inhibiting the calmodulin-dependent protein kinase IV/Akt/mTOR pathway and reduce disease damage in SLE mice (160, 161). A recent study found that the HIF1α-dependent glycolytic pathway in macrophages can exacerbate IgG deposition-induced lupus nephritis (68). PI3K- and Erk-mediated mTOR hyperactivation increases HIF1α transcriptional activity (68). MRL-lpr mice treated with rapamycin exhibited a decline in neutrophil recruitment, IL-1β, prostaglandin E2, and ROS secreted from macrophages (68). These results suggest a potential therapeutic application of rapamycin in targeting macrophages for SLE therapy.
In a single-arm, open-label, phase 1/2 trial of sirolimus (rapamycin) in patients with active SLE, both the British Isles Lupus Assessment Group score and the SLE Disease Activity Index decreased significantly after 12 months of treatment (162). The drug had no adverse effects on liver function or lymphocyte count. In another trial of pediatric patients with SLE, most achieved durable remission with sirolimus treatment (163). Moreover, sirolimus has greater advantages for serological reduction and glucocorticoid tapering, compared to the classic immunosuppressant tacrolimus, in treating SLE (164).
Everolimus is a 40-O-(2-hydroxyethyl) derivative of rapamycin (165). By inhibiting mTOR signaling to block IL-2 activation in T cells, everolimus prevents T-cell hyperactivation and reduces arthritis activity in patients with RA. Patients with RA treated with everolimus for 12 weeks (36.1%) had significantly higher rates of pain reduction and decreased disease activity than those treated with placebo (16.7%) (165). However, the everolimus-treated group showed more fluctuating liver function and blood lipid levels. Another mTOR inhibitor, sirolimus, relieves RA symptoms while it exhibits no impact on routine blood tests and liver and renal functions (166, 167). Additionally, an ongoing clinical trial is evaluating the efficacy of temsirolimus (a mTORC1 inhibitor; ClinicalTrials.gov identifier: NCT00076206) in patients with active RA on concomitant methotrexate therapy. These data demonstrate the potential use of rapamycin for RA treatment.
Dimethyl fumarate
Dimethyl fumarate (DMF), an electrophilic, cell-permeable derivative of the TCA cycle metabolite fumarate, has been clinically approved as an immunomodulatory drug for treating multiple sclerosis (168). An increasing number of studies have reported that DMF mitigates the progression of AIDs by exerting effects on innate immune cells. DMF can succinylate the glycolytic enzyme GAPDH by covalently modifying cysteine residues. DMF can reportedly reduce GAPDH activity, inhibit aerobic glycolysis in bone marrow-derived cells and lymphocytes, and exert anti-inflammatory effects, both in vitro and in vivo (169). DMF reciprocally inhibits the survival, differentiation, and effector functions of Th1 and Th17 cells while promoting the development of regulatory T cells. Therefore, DMF selectively targets effector cells while sparing regulatory and naive T cells (170).
DMF induces nuclear translocation of nuclear factor E2-related factor 2 (Nrf2) and enhances NRF2 promoter activity while attenuating RANKL-mediated intracellular ROS generation (171). The latter is an osteoclast effector that inhibits RANKL-induced bone destruction (171). Activation of Nrf2 in osteoclasts/macrophages by DMF inhibits bone and joint destruction in patients with RA (171). Additionally, DMF inhibits MAPK signaling, thereby downregulating RANKL-induced expression of c-Fos, calcineurin-dependent 1, and nuclear factor of activated T cell cytoplasmic-1 (172). DMF disrupts actin ring formation by inhibiting the abovementioned signaling pathways, ultimately inhibiting the pit-forming activity of osteoclasts (172). DMF inhibits the extracellular release of high-mobility group box 1 by activating Nrf2. Simultaneously, DMF reduces the phosphorylation of p38 MAPK and extracellular signal-regulated kinase in osteoclasts (173). Additionally, DMF succinylates IL-1 receptor-associated kinase 4 in plasmacytoid DCs, preventing the binding of IL-1 receptor-associated kinase 4 to the adaptor protein MyD88 and decreasing the inflammatory cytokines IL-1, IL-18, INF-α, and TNF-α (174). Plasmacytoid DCs are activated in SLE and accumulate in the skin and produce interferons (175). DMF is potentially an important regulator of the innate immune response and may be a novel treatment strategy for SLE.
Hexokinase Inhibitors
Hexokinase can catalyze the phosphorylation of glucose, initiating the glycolysis process. Owing to its structural similarity to glucose, 2-DG can competitively bind to HK2, inhibiting HK2 production by accumulation of phosphorylated 2-DG (176). In pre-clinical experiments, 2-DG significantly attenuated arthritis progression and reduced adaptive and innate immune cell activation in K/BxN mice (176). Cai et al. reported that inducible-nitric oxide synthase expressing M1 macrophages are significantly increased in arthritic joints and that 2-DG can effectively induce arginase 1 expressing M2 macrophages via the AMPK/NF-κB axis (177). Lonidamine, an inhibitor of HK1 and HK2, attenuates joint destruction in collagen-induced arthritis mice (178). The suppression of HK1 and HK2 downregulates the IL-1β and TNF-α expression and restores the anti-inflammatory activity of macrophages in the RA model. Currently, lonidamine and 2-DG are used in phase I/II trials for treating advanced cancers (179). The investigators observed only minor adverse effects, including nausea and blood glucose reduction (179).
Metformin
Metformin was initially used in the first-line treatment of type 2 diabetes owing to its safe glucose-lowering effect (180). Increasing studies have taken advantage of the metabolism modulation property of metformin for treating various diseases, including cancer, cardiovascular disease, and AIDs (180). Metformin can modulate immunometabolism by mediating AMPK signaling. AMPK/mTOR is involved in the differentiation of CD4+ T cells (181). The expression of transcription factors, especially STAT, mediates mTORC1-dependent Th17 differentiation, which contributes to the development of RA (182). Additionally, metformin-activated AMPK suppresses the function of STAT3 and NF-κB, and boosts macrophages to exhibit an anti-inflammatory phenotype (183). In an Israeli cohort, patients on high-dose (2,550 mg/day) metformin had a lower risk of developing RA than those on low-dose (850 mg/day) metformin (adjusted hazard ratio of 0.62, 95% confidence interval 0.46–0.84), especially in women (184). A randomized controlled trial shows that RA patients treated with metformin for 12 weeks (80.8%) had significantly higher rates of pain reduction and decreased disease activity than those treated with placebo (54.7%) (185). No serious adverse effects were reported in these two groups.
Since glycolysis contributes to NETs formation, metformin reduced phorbol 12-myristate 13-acetate-induced NET formation via the AMPK/mTOR axis (186). NET-derived mitochondrial DNA induces IFNα production in pDCs, which is an important pathogenesis of SLE (186). Metformin with concomitant hydroxychloroquine in patients with mild or moderate SLE can reduce clinical flares and disease progression (186, 187).
Summary and Prospect
Uncontrolled inflammatory bursts are a common feature of AIDs, including SLE, RA, AS, and ANCA-AAV. Various studies have demonstrated that innate immune cells adapt their metabolism to maintain or change their inflammatory phenotype. Additionally, a growing body of evidence supports the immunomodulatory properties of glycolytic metabolites in AIDs. Molecule machines, such as mTOR, AMPK, and HK2, which were initially thought to be simply regulators of cellular metabolism, are now regarded as therapeutic targets for modulating inflammatory responses. Hence, the discovery that an immune metabolism characterized by glycolysis may regulate inflammation broadens the avenues for treating AIDs. Strategies to target the abovementioned signaling molecules can be developed to modulate immune cell metabolism. Therefore, additional studies are needed to increase our understanding of the metabolic pathways in innate immune cells that are involved in the pathogenesis of AIDs, could potentially be exploited therapeutically to weaken exacerbated inflammatory responses.
Author Contributions
LC and FW contributed to support the conception of the review. YX and YC wrote the manuscript and prepared the figures and the table. XZ, JM, and YL read, discussed, and revised the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (62071011, 81788101), the Chinese Academy of Medical Science Innovation Fund for Medical Sciences (CIFMS) (2021-1-I2M-017, 2021-1-I2M-047, 2021-1-I2M-040, 2021-1-I2M-016, and 2021-1-I2M-026), the Capital’s Funds for Health Improvement and Research (2020-2-4019), and the Key Clinical Specialty Funding Project of Beijing.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Stathopoulou C, Nikoleri D, Bertsias G. Immunometabolism: An Overview and Therapeutic Prospects in Autoimmune Diseases. Immunotherapy (2019) 11(9):813–29. doi: 10.2217/imt-2019-0002
2. Jung SM, Kim WU. Targeted Immunotherapy for Autoimmune Disease. Immune Netw (2022) 22(1):e9. doi: 10.4110/in.2022.22.e9
3. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant Autoantibody Production by Early Human B Cell Precursors. Science (2003) 301(5638):1374–7. doi: 10.1126/science.1086907
4. Anaya JM, Restrepo-Jimenez P, Ramirez-Santana C. The Autoimmune Ecology: An Update. Curr Opin Rheumatol (2018) 30(4):350–60. doi: 10.1097/BOR.0000000000000498
5. Yang Z, Matteson EL, Goronzy JJ, Weyand CM. T-Cell Metabolism in Autoimmune Disease. Arthritis Res Ther (2015) 17:29. doi: 10.1186/s13075-015-0542-4
6. Lin X, Lu L. B Cell-Mediated Autoimmune Diseases. Adv Exp Med Biol (2020) 1254:145–60. doi: 10.1007/978-981-15-3532-1_11
7. Wang L, Wang FS, Gershwin ME. Human Autoimmune Diseases: A Comprehensive Update. J Intern Med (2015) 278(4):369–95. doi: 10.1111/joim.12395
8. Chen J, Liu C, Ye S, Lu R, Zhu H, Xu J. UPLC-MS/MS-Based Plasma Lipidomics Reveal a Distinctive Signature in Systemic Lupus Erythematosus Patients. MedComm (2020) (2021) 2(2):269–78. doi: 10.1002/mco2.67
9. Fu X, Liu H, Huang G, Dai SS. The Emerging Role of Neutrophils in Autoimmune-Associated Disorders: Effector, Predictor, and Therapeutic Targets. MedComm (2020) (2021) 2(3):402–13. doi: 10.1002/mco2.69
10. Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol (1927) 8(6):519–30. doi: 10.1085/jgp.8.6.519
11. Warburg O, Gawehn K, Geissler AW. Metabolism of Leukocytes. Z Naturforsch B (1958) 13B(8):515–6. doi: 10.1515/znb-1958-0806
12. Finlay DK. Regulation of Glucose Metabolism in T Cells: New Insight Into the Role of Phosphoinositide 3-Kinases. Front Immunol (2012) 3:247. doi: 10.3389/fimmu.2012.00247
13. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T Cell Metabolism Reverses Lupus. Sci Transl Med (2015) 7(274):274ra18. doi: 10.1126/scitranslmed.aaa0835
14. Puleston DJ, Villa M, Pearce EL. Ancillary Activity: Beyond Core Metabolism in Immune Cells. Cell Metab (2017) 26(1):131–41. doi: 10.1016/j.cmet.2017.06.019
15. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling Immunity: Insights Into Metabolism and Lymphocyte Function. Science (2013) 342(6155):1242454. doi: 10.1126/science.1242454
16. Mills EL, Kelly B, O'Neill LAJ. Mitochondria Are the Powerhouses of Immunity. Nat Immunol (2017) 18(5):488–98. doi: 10.1038/ni.3704
17. Chou WC, Rampanelli E, Li X, Ting JP. Impact of Intracellular Innate Immune Receptors on Immunometabolism. Cell Mol Immunol (2022) 19(3):337–51. doi: 10.1038/s41423-021-00780-y
18. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate Kinase M2 Regulates Hif-1alpha Activity and IL-1beta Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab (2015) 21(1):65–80. doi: 10.1016/j.cmet.2014.12.005
19. Wang T, Jiao Y, Zhang X. Immunometabolic Pathways and Its Therapeutic Implication in Autoimmune Diseases. Clin Rev Allergy Immunol (2021) 60(1):55–67. doi: 10.1007/s12016-020-08821-6
20. Ryan K, Tekwani BL. Current Investigations on Clinical Pharmacology and Therapeutics of Glucose-6-Phosphate Dehydrogenase Deficiency. Pharmacol Ther (2021) 222:107788. doi: 10.1016/j.pharmthera.2020.107788
21. Breton J, Avanzi N, Magagnin S, Covini N, Magistrelli G, Cozzi L, et al. Functional Characterization and Mechanism of Action of Recombinant Human Kynurenine 3-Hydroxylase. Eur J Biochem (2000) 267(4):1092–9. doi: 10.1046/j.1432-1327.2000.01104.x
22. Saravia J, Raynor JL, Chapman NM, Lim SA, Chi H. Signaling Networks in Immunometabolism. Cell Res (2020) 30(4):328–42. doi: 10.1038/s41422-020-0301-1
23. Huang H, Long L, Zhou P, Chapman NM, Chi H. mTOR Signaling at the Crossroads of Environmental Signals and T-Cell Fate Decisions. Immunol Rev (2020) 295(1):15–38. doi: 10.1111/imr.12845
24. Zhang T, Li H, Shi J, Li S, Li M, Zhang L, et al. P53 Predominantly Regulates IL-6 Production and Suppresses Synovial Inflammation in Fibroblast-Like Synoviocytes and Adjuvant-Induced Arthritis. Arthritis Res Ther (2016) 18(1):271. doi: 10.1186/s13075-016-1161-4
25. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-Dependent Glycolytic Pathway Orchestrates a Metabolic Checkpoint for the Differentiation of TH17 and Treg Cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
26. Kumar S, Dikshit M. Metabolic Insight of Neutrophils in Health and Disease. Front Immunol (2019) 10:2099. doi: 10.3389/fimmu.2019.02099
27. Sbarra AJ, Karnovsky ML. The Biochemical Basis of Phagocytosis. I. Metabolic Changes During the Ingestion of Particles by Polymorphonuclear Leukocytes. J Biol Chem (1959) 234(6):1355–62. doi: 10.1016/S0021-9258(18)70011-2
28. Borregaard N, Herlin T. Energy Metabolism of Human Neutrophils During Phagocytosis. J Clin Invest (1982) 70(3):550–7. doi: 10.1172/JCI110647
29. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
30. Rodriguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MM, Lopez-Villegas EO, Sanchez-Garcia FJ. Metabolic Requirements for Neutrophil Extracellular Traps Formation. Immunology (2015) 145(2):213–24. doi: 10.1111/imm.12437
31. Azevedo EP, Rochael NC, Guimaraes-Costa AB, de Souza-Vieira TS, Ganilho J, Saraiva EM, et al. A Metabolic Shift Toward Pentose Phosphate Pathway Is Necessary for Amyloid Fibril- and Phorbol 12-Myristate 13-Acetate-Induced Neutrophil Extracellular Trap (NET) Formation. J Biol Chem (2015) 290(36):22174–83. doi: 10.1074/jbc.M115.640094
32. Awasthi D, Nagarkoti S, Sadaf S, Chandra T, Kumar S, Dikshit M. Glycolysis Dependent Lactate Formation in Neutrophils: A Metabolic Link Between NOX-Dependent and Independent NETosis. Biochim Biophys Acta Mol Basis Dis (2019) 1865(12):165542. doi: 10.1016/j.bbadis.2019.165542
33. Cooper MR, DeChatelet LR, McCall CE, LaVia MF, Spurr CL, Baehner RL. Complete Deficiency of Leukocyte Glucose-6-Phosphate Dehydrogenase With Defective Bactericidal Activity. J Clin Invest (1972) 51(4):769–78. doi: 10.1172/JCI106871
34. Amini P, Stojkov D, Felser A, Jackson CB, Courage C, Schaller A, et al. Neutrophil Extracellular Trap Formation Requires OPA1-Dependent Glycolytic ATP Production. Nat Commun (2018) 9(1):2958. doi: 10.1038/s41467-018-05387-y
35. Miranda FJB, Rocha BC, Pereira MCA, Pereira LMN, de Souza EHM, Marino AP, et al. Toxoplasma Gondii-Induced Neutrophil Extracellular Traps Amplify the Innate and Adaptive Response. mBio (2021) 12(5):e0130721. doi: 10.1128/mBio.01307-21
36. Rana AK, Li Y, Dang Q, Yang F. Monocytes in Rheumatoid Arthritis: Circulating Precursors of Macrophages and Osteoclasts and, Their Heterogeneity and Plasticity Role in RA Pathogenesis. Int Immunopharmacol (2018) 65:348–59. doi: 10.1016/j.intimp.2018.10.016
37. Jain N, Moeller J, Vogel V. Mechanobiology of Macrophages: How Physical Factors Coregulate Macrophage Plasticity and Phagocytosis. Annu Rev BioMed Eng (2019) 21:267–97. doi: 10.1146/annurev-bioeng-062117-121224
38. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of Macrophage Polarization in Autoimmunity. Immunology (2018) 154(2):186–95. doi: 10.1111/imm.12910
39. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage Plasticity, Polarization, and Function in Health and Disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
40. Atri C, Guerfali FZ, Laouini D. Role of Human Macrophage Polarization in Inflammation During Infectious Diseases. Int J Mol Sci (2018) 19(6):1801. doi: 10.3390/ijms19061801
41. Cuda CM, Pope RM, Perlman H. The Inflammatory Role of Phagocyte Apoptotic Pathways in Rheumatic Diseases. Nat Rev Rheumatol (2016) 12(9):543–58. doi: 10.1038/nrrheum.2016.132
42. Mohammadi S, Saghaeian-Jazi M, Sedighi S, Memarian A. Sodium Valproate Modulates Immune Response by Alternative Activation of Monocyte-Derived Macrophages in Systemic Lupus Erythematosus. Clin Rheumatol (2018) 37(3):719–27. doi: 10.1007/s10067-017-3922-0
43. Perl A, Hanczko R, Lai ZW, Oaks Z, Kelly R, Borsuk R, et al. Comprehensive Metabolome Analyses Reveal N-Acetylcysteine-Responsive Accumulation of Kynurenine in Systemic Lupus Erythematosus: Implications for Activation of the Mechanistic Target of Rapamycin. Metabolomics (2015) 11(5):1157–74. doi: 10.1007/s11306-015-0772-0
44. Liu Y, Xu R, Gu H, Zhang E, Qu J, Cao W, et al. Metabolic Reprogramming in Macrophage Responses. Biomark Res (2021) 9(1):1. doi: 10.1186/s40364-020-00251-y
45. Rao J, Wang H, Ni M, Wang Z, Wang Z, Wei S, et al. FSTL1 Promotes Liver Fibrosis by Reprogramming Macrophage Function Through Modulating the Intracellular Function of PKM2. Gut (2022):gutjnl-2021-325150. doi: 10.1136/gutjnl-2021-325150
46. Timmons GA, Carroll RG, O'Siorain JR, Cervantes-Silva MP, Fagan LE, Cox SL, et al. The Circadian Clock Protein BMAL1 Acts as a Metabolic Sensor in Macrophages to Control the Production of Pro IL-1beta. Front Immunol (2021) 12:700431. doi: 10.3389/fimmu.2021.700431
47. Russo S, Kwiatkowski M, Govorukhina N, Bischoff R, Melgert BN. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Front Immunol (2021) 12:746151. doi: 10.3389/fimmu.2021.746151
48. Kobayashi T, Nguyen-Tien D, Sorimachi Y, Sugiura Y, Suzuki T, Karyu H, et al. SLC15A4 Mediates M1-Prone Metabolic Shifts in Macrophages and Guards Immune Cells From Metabolic Stress. Proc Natl Acad Sci USA (2021) 118(33):e2100295118. doi: 10.1073/pnas.2100295118
49. Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation (2018) 138(9):898–912. doi: 10.1161/CIRCULATIONAHA.117.032636
50. Van Avondt K, Maegdefessel L, Soehnlein O. Therapeutic Targeting of Neutrophil Extracellular Traps in Atherogenic Inflammation. Thromb Haemost (2019) 119(4):542–52. doi: 10.1055/s-0039-1678664
51. Josefs T, Barrett TJ, Brown EJ, Quezada A, Wu X, Voisin M, et al. Neutrophil Extracellular Traps Promote Macrophage Inflammation and Impair Atherosclerosis Resolution in Diabetic Mice. JCI Insight (2020) 5(7):e134796. doi: 10.1172/jci.insight.134796
52. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell (2017) 169(4):570–86. doi: 10.1016/j.cell.2017.04.004
53. Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, et al. Substrate Fate in Activated Macrophages: A Comparison Between Innate, Classic, and Alternative Activation. J Immunol (2010) 185(1):605–14. doi: 10.4049/jimmunol.0901698
54. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative Metabolism and PGC-1beta Attenuate Macrophage-Mediated Inflammation. Cell Metab (2006) 4(1):13–24. doi: 10.1016/j.cmet.2006.05.011
55. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab (2018) 28(3):463–75 e4. doi: 10.1016/j.cmet.2018.08.012
56. Tan Z, Xie N, Cui H, Moellering DR, Abraham E, Thannickal VJ, et al. Pyruvate Dehydrogenase Kinase 1 Participates in Macrophage Polarization via Regulating Glucose Metabolism. J Immunol (2015) 194(12):6082–9. doi: 10.4049/jimmunol.1402469
57. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep (2016) 17(3):684–96. doi: 10.1016/j.celrep.2016.09.008
58. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 Signaling Regulates Acly to Integrate Metabolic Input to Control of Macrophage Activation. Elife (2016) 5:e11612. doi: 10.7554/eLife.11612
59. Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity (2016) 45(4):817–30. doi: 10.1016/j.immuni.2016.09.016
60. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The Clearance of Dead Cells by Efferocytosis. Nat Rev Mol Cell Biol (2020) 21(7):398–414. doi: 10.1038/s41580-020-0232-1
61. Doran AC, Yurdagul A Jr., Tabas I. Efferocytosis in Health and Disease. Nat Rev Immunol (2020) 20(4):254–67. doi: 10.1038/s41577-019-0240-6
62. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab (2019) 29(2):443–56.e5. doi: 10.1016/j.cmet.2018.12.004
63. Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, et al. HIF-1alpha-PDK1 Axis-Induced Active Glycolysis Plays an Essential Role in Macrophage Migratory Capacity. Nat Commun (2016) 7:11635. doi: 10.1038/ncomms11635
64. Arts RJW, Joosten LAB, Netea MG. The Potential Role of Trained Immunity in Autoimmune and Autoinflammatory Disorders. Front Immunol (2018) 9:298. doi: 10.3389/fimmu.2018.00298
65. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab (2016) 24(6):807–19. doi: 10.1016/j.cmet.2016.10.008
66. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science (2014) 345(6204):1250684. doi: 10.1126/science.1250684
67. Kobayashi T, Shimabukuro-Demoto S, Yoshida-Sugitani R, Furuyama-Tanaka K, Karyu H, Sugiura Y, et al. The Histidine Transporter SLC15A4 Coordinates mTOR-Dependent Inflammatory Responses and Pathogenic Antibody Production. Immunity (2014) 41(3):375–88. doi: 10.1016/j.immuni.2014.08.011
68. Jing C, Castro-Dopico T, Richoz N, Tuong ZK, Ferdinand JR, Lok LSC, et al. Macrophage Metabolic Reprogramming Presents a Therapeutic Target in Lupus Nephritis. Proc Natl Acad Sci USA (2020) 117(26):15160–71. doi: 10.1073/pnas.2000943117
69. Bhunyakarnjanarat T, Udompornpitak K, Saisorn W, Chantraprapawat B, Visitchanakun P, Dang CP, et al. Prominent Indomethacin-Induced Enteropathy in Fcgriib Defi-Cient Lupus Mice: An Impact of Macrophage Responses and Immune Deposition in Gut. Int J Mol Sci (2021) 22(3):1377. doi: 10.3390/ijms22031377
70. Collin M, Bigley V. Human Dendritic Cell Subsets: An Update. Immunology (2018) 154(1):3–20. doi: 10.1111/imm.12888
71. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-Like Receptor-Induced Changes in Glycolytic Metabolism Regulate Dendritic Cell Activation. Blood (2010) 115(23):4742–9. doi: 10.1182/blood-2009-10-249540
72. Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to Glycolysis Sustains Survival of NO-Producing Inflammatory Dendritic Cells. Blood (2012) 120(7):1422–31. doi: 10.1182/blood-2012-03-419747
73. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-Driven Early Glycolytic Reprogramming via the Kinases TBK1-IKKvarepsilon Supports the Anabolic Demands of Dendritic Cell Activation. Nat Immunol (2014) 15(4):323–32. doi: 10.1038/ni.2833
74. Guak H, Al Habyan S, Ma EH, Aldossary H, Al-Masri M, Won SY, et al. Glycolytic Metabolism is Essential for CCR7 Oligomerization and Dendritic Cell Migration. Nat Commun (2018) 9(1):2463. doi: 10.1038/s41467-018-04804-6
75. Liu J, Zhang X, Chen K, Cheng Y, Liu S, Xia M, et al. CCR7 Chemokine Receptor-Inducible lnc-Dpf3 Restrains Dendritic Cell Migration by Inhibiting HIF-1alpha-Mediated Glycolysis. Immunity (2019) 50(3):600–15.e15. doi: 10.1016/j.immuni.2019.01.021
76. Lawless SJ, Kedia-Mehta N, Walls JF, McGarrigle R, Convery O, Sinclair LV, et al. Glucose Represses Dendritic Cell-Induced T Cell Responses. Nat Commun (2017) 8:15620. doi: 10.1038/ncomms15620
77. Sim WJ, Ahl PJ, Connolly JE. Metabolism Is Central to Tolerogenic Dendritic Cell Function. Mediators Inflamm (2016) 2016:2636701. doi: 10.1155/2016/2636701
78. Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, et al. High Mitochondrial Respiration and Glycolytic Capacity Represent a Metabolic Phenotype of Human Tolerogenic Dendritic Cells. J Immunol (2015) 194(11):5174–86. doi: 10.4049/jimmunol.1303316
79. Kingsmore KM, Bachali P, Catalina MD, Daamen AR, Heuer SE, Robl RD, et al. Altered Expression of Genes Controlling Metabolism Characterizes the Tissue Response to Immune Injury in Lupus. Sci Rep (2021) 11(1):14789. doi: 10.1038/s41598-021-93034-w
80. Wang H, Zhang N, Fang K, Chang X. 2-Deoxy-D-Glucose Alleviates Collagen-Induced Arthritis of Rats and Is Accompanied by Metabolic Regulation of the Spleen and Liver. Front Immunol (2021) 12:713799. doi: 10.3389/fimmu.2021.713799
81. Weyand CM, Goronzy JJ. Immunometabolism in Early and Late Stages of Rheumatoid Arthritis. Nat Rev Rheumatol (2017) 13(5):291–301. doi: 10.1038/nrrheum.2017.49
82. Biniecka M, Canavan M, McGarry T, Gao W, McCormick J, Cregan S, et al. Dysregulated Bioenergetics: A Key Regulator of Joint Inflammation. Ann Rheum Dis (2016) 75(12):2192–200. doi: 10.1136/annrheumdis-2015-208476
83. Maratou E, Dimitriadis G, Kollias A, Boutati E, Lambadiari V, Mitrou P, et al. Glucose Transporter Expression on the Plasma Membrane of Resting and Activated White Blood Cells. Eur J Clin Invest (2007) 37(4):282–90. doi: 10.1111/j.1365-2362.2007.01786.x
84. Li KJ, Wu CH, Hsieh SC, Lu MC, Tsai CY, Yu CL. Deranged Bioenergetics and Defective Redox Capacity in T Lymphocytes and Neutrophils are Related to Cellular Dysfunction and Increased Oxidative Stress in Patients With Active Systemic Lupus Erythematosus. Clin Dev Immunol (2012) 2012:548516. doi: 10.1155/2012/548516
85. Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose Metabolism in Lymphocytes Is a Regulated Process With Significant Effects on Immune Cell Function and Survival. J Leukoc Biol (2008) 84(4):949–57. doi: 10.1189/jlb.0108024
86. Perner A, Nielsen SE, Rask-Madsen J. High Glucose Impairs Superoxide Production From Isolated Blood Neutrophils. Intensive Care Med (2003) 29(4):642–5. doi: 10.1007/s00134-002-1628-4
87. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA are Interferogenic and Contribute to Lupus-Like Disease. Nat Med (2016) 22(2):146–53. doi: 10.1038/nm.4027
88. Frangou E, Vassilopoulos D, Boletis J, Boumpas DT. An Emerging Role of Neutrophils and NETosis in Chronic Inflammation and Fibrosis in Systemic Lupus Erythematosus (SLE) and ANCA-Associated Vasculitides (AAV): Implications for the Pathogenesis and Treatment. Autoimmun Rev (2019) 18(8):751–60. doi: 10.1016/j.autrev.2019.06.011
89. Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, et al. Neutrophil Extracellular Traps (NETs) in Autoimmune Diseases: A Comprehensive Review. Autoimmun Rev (2017) 16(11):1160–73. doi: 10.1016/j.autrev.2017.09.012
90. Zhang X, Yang Y, Jing L, Zhai W, Zhang H, Ma Q, et al. Pyruvate Kinase M2 Contributes to TLR-Mediated Inflammation and Autoimmunity by Promoting Pyk2 Activation. Front Immunol (2021) 12:680068. doi: 10.3389/fimmu.2021.680068
91. Lu L, Wang H, Liu X, Tan L, Qiao X, Ni J, et al. Pyruvate Kinase Isoform M2 Impairs Cognition in Systemic Lupus Erythematosus by Promoting Microglial Synaptic Pruning via the Beta-Catenin Signaling Pathway. J Neuroinflammation (2021) 18(1):229. doi: 10.1186/s12974-021-02279-9
92. Das Gupta K, Shakespear MR, Curson JEB, Murthy AMV, Iyer A, Hodson MP, et al. Class IIa Histone Deacetylases Drive Toll-Like Receptor-Inducible Glycolysis and Macrophage Inflammatory Responses via Pyruvate Kinase M2. Cell Rep (2020) 30(8):2712–28.e8. doi: 10.1016/j.celrep.2020.02.007
93. Batu ED, Kosukcu C, Taskiran E, Sahin S, Akman S, Sozeri B, et al. Whole Exome Sequencing in Early-Onset Systemic Lupus Erythematosus. J Rheumatol (2018) 45(12):1671–9. doi: 10.3899/jrheum.171358
94. Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of Active Peptidyl Arginine Deiminases by Neutrophils Can Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol (2015) 67(12):3135–45. doi: 10.1002/art.39313
95. Hemon MF, Lambert NC, Arnoux F, Roudier J, Auger I. PAD4 Immunization Triggers Anti-Citrullinated Peptide Antibodies in Normal Mice: Analysis With Peptide Arrays. Front Immunol (2022) 13:840035. doi: 10.3389/fimmu.2022.840035
96. Chapman EA, Lyon M, Simpson D, Mason D, Beynon RJ, Moots RJ, et al. Caught in a Trap? Proteomic Analysis of Neutrophil Extracellular Traps in Rheumatoid Arthritis and Systemic Lupus Erythematosus. Front Immunol (2019) 10:423. doi: 10.3389/fimmu.2019.00423
97. Turunen S, Huhtakangas J, Nousiainen T, Valkealahti M, Melkko J, Risteli J, et al. Rheumatoid Arthritis Antigens Homocitrulline and Citrulline are Generated by Local Myeloperoxidase and Peptidyl Arginine Deiminases 2, 3 and 4 in Rheumatoid Nodule and Synovial Tissue. Arthritis Res Ther (2016) 18(1):239. doi: 10.1186/s13075-016-1140-9
98. Birkelund S, Bennike TB, Kastaniegaard K, Lausen M, Poulsen TBG, Kragstrup TW, et al. Proteomic Analysis of Synovial Fluid From Rheumatic Arthritis and Spondyloarthritis Patients. Clin Proteomics (2020) 17:29. doi: 10.1186/s12014-020-09292-9
99. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci Transl Med (2013) 5(178):178ra40. doi: 10.1126/scitranslmed.3005580
100. Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial Fibroblast-Neutrophil Interactions Promote Pathogenic Adaptive Immunity in Rheumatoid Arthritis. Sci Immunol (2017) 2(10):eaag3358. doi: 10.1126/sciimmunol.aag3358
101. Carmona-Rivera C, Carlucci PM, Goel RR, James E, Brooks SR, Rims C, et al. Neutrophil Extracellular Traps Mediate Articular Cartilage Damage and Enhance Cartilage Component Immunogenicity in Rheumatoid Arthritis. JCI Insight (2020) 5(13):e139388. doi: 10.1172/jci.insight.139388
102. Liu P, Wang J, Wen W, Pan T, Chen H, Fu Y, et al. Cinnamaldehyde Suppresses NLRP3 Derived IL-1beta via Activating Succinate/HIF-1 in Rheumatoid Arthritis Rats. Int Immunopharmacol (2020) 84:106570. doi: 10.1016/j.intimp.2020.106570
103. Umar S, Palasiewicz K, Volin MV, Zanotti B, Al-Awqati M, Sweiss N, et al. IRAK4 Inhibitor Mitigates Joint Inflammation by Rebalancing Metabolism Malfunction in RA Macrophages and Fibroblasts. Life Sci (2021) 287:120114. doi: 10.1016/j.lfs.2021.120114
104. Kim B, Kim HY, Yoon BR, Yeo J, In Jung J, Yu KS, et al. Cytoplasmic Zinc Promotes IL-1beta Production by Monocytes and Macrophages Through mTORC1-Induced Glycolysis in Rheumatoid Arthritis. Sci Signal (2022) 15(716):eabi7400. doi: 10.1126/scisignal.abi7400
105. Zeisbrich M, Yanes RE, Zhang H, Watanabe R, Li Y, Brosig L, et al. Hypermetabolic Macrophages in Rheumatoid Arthritis and Coronary Artery Disease Due to Glycogen Synthase Kinase 3b Inactivation. Ann Rheum Dis (2018) 77(7):1053–62. doi: 10.1136/annrheumdis-2017-212647
106. Alquraishi M, Puckett DL, Alani DS, Humidat AS, Frankel VD, Donohoe DR, et al. Pyruvate Kinase M2: A Simple Molecule With Complex Functions. Free Radic Biol Med (2019) 143:176–92. doi: 10.1016/j.freeradbiomed.2019.08.007
107. Xu J, Jiang C, Wang X, Geng M, Peng Y, Guo Y, et al. Upregulated PKM2 in Macrophages Exacerbates Experimental Arthritis via STAT1 Signaling. J Immunol (2020) 205(1):181–92. doi: 10.4049/jimmunol.1901021
108. Han DW, Choi YS, Kim HW, Shin S, Ha YJ, Kang EH, et al. Extracellular Pyruvate Kinase M2 Promotes Osteoclastogenesis and is Associated With Radiographic Progression in Early Rheumatoid Arthritis. Sci Rep (2022) 12(1):4024. doi: 10.1038/s41598-022-07667-6
109. Van Raemdonck K, Umar S, Palasiewicz K, Volin MV, Elshabrawy HA, Romay B, et al. Interleukin-34 Reprograms Glycolytic and Osteoclastic Rheumatoid Arthritis Macrophages via Syndecan 1 and Macrophage Colony-Stimulating Factor Receptor. Arthritis Rheumatol (2021) 73(11):2003–14. doi: 10.1002/art.41792
110. Murata K, Fang C, Terao C, Giannopoulou EG, Lee YJ, Lee MJ, et al. Hypoxia-Sensitive COMMD1 Integrates Signaling and Cellular Metabolism in Human Macrophages and Suppresses Osteoclastogenesis. Immunity (2017) 47(1):66–79.e5. doi: 10.1016/j.immuni.2017.06.018
111. Watanabe-Kusunoki K, Nakazawa D, Kusunoki Y, Kudo T, Hattanda F, Nishio S, et al. Recombinant Thrombomodulin Ameliorates Autoimmune Vasculitis via Immune Response Regulation and Tissue Injury Protection. J Autoimmun (2020) 108:102390. doi: 10.1016/j.jaut.2019.102390
112. Popat RJ, Hakki S, Thakker A, Coughlan AM, Watson J, Little MA, et al. Anti-Myeloperoxidase Antibodies Attenuate the Monocyte Response to LPS and Shape Macrophage Development. JCI Insight (2017) 2(2):e87379. doi: 10.1172/jci.insight.87379
113. Rousselle A, Kettritz R, Schreiber A. Monocytes Promote Crescent Formation in Anti-Myeloperoxidase Antibody-Induced Glomerulonephritis. Am J Pathol (2017) 187(9):1908–15. doi: 10.1016/j.ajpath.2017.05.003
114. O'Brien EC, Abdulahad WH, Rutgers A, Huitema MG, O'Reilly VP, Coughlan AM, et al. Intermediate Monocytes in ANCA Vasculitis: Increased Surface Expression of ANCA Autoantigens and IL-1beta Secretion in Response to Anti-MPO Antibodies. Sci Rep (2015) 5:11888. doi: 10.1038/srep11888
115. O'Brien EC, White CA, Wyse J, Leacy E, Porter RK, Little MA, et al. Pro-Inflammatory Stimulation of Monocytes by ANCA Is Linked to Changes in Cellular Metabolism. Front Med (Lausanne) (2020) 7:553. doi: 10.3389/fmed.2020.00553
116. Ou J, Xiao M, Huang Y, Tu L, Chen Z, Cao S, et al. Serum Metabolomics Signatures Associated With Ankylosing Spondylitis and TNF Inhibitor Therapy. Front Immunol (2021) 12:630791. doi: 10.3389/fimmu.2021.630791
117. Kono M, Yoshida N, Maeda K, Suarez-Fueyo A, Kyttaris VC, Tsokos GC. Glutaminase 1 Inhibition Reduces Glycolysis and Ameliorates Lupus-Like Disease in MRL/lpr Mice and Experimental Autoimmune Encephalomyelitis. Arthritis Rheumatol (2019) 71(11):1869–78. doi: 10.1002/art.41019
118. Fresneda Alarcon M, McLaren Z, Wright HL. Neutrophils in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Same Foe Different M.O. Front Immunol (2021) 12:649693. doi: 10.3389/fimmu.2021.649693
119. Chiewchengchol D, Midgley A, Sodsai P, Deekajorndech T, Hirankarn N, Beresford MW, et al. The Protective Effect of GM-CSF on Serum-Induced Neutrophil Apoptosis in Juvenile Systemic Lupus Erythematosus Patients. Clin Rheumatol (2015) 34(1):85–91. doi: 10.1007/s10067-014-2800-2
120. Courtney PA, Crockard AD, Williamson K, Irvine AE, Kennedy RJ, Bell AL. Increased Apoptotic Peripheral Blood Neutrophils in Systemic Lupus Erythematosus: Relations With Disease Activity, Antibodies to Double Stranded DNA, and Neutropenia. Ann Rheum Dis (1999) 58(5):309–14. doi: 10.1136/ard.58.5.309
121. Farrera C, Fadeel B. Macrophage Clearance of Neutrophil Extracellular Traps is a Silent Process. J Immunol (2013) 191(5):2647–56. doi: 10.4049/jimmunol.1300436
122. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA-Peptide Complexes in Systemic Lupus Erythematosus. Sci Transl Med (2011) 3(73):73ra19. doi: 10.1126/scitranslmed.3001180
123. Xu S, Min J, Wang F. Ferroptosis: An Emerging Player in Immune Cells. Science Bulletin (2021) 66:2257–60. doi: 10.1016/j.scib.2021.02.026
124. Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, et al. Glutathione Peroxidase 4-Regulated Neutrophil Ferroptosis Induces Systemic Autoimmunity. Nat Immunol (2021) 22(9):1107–17. doi: 10.1038/s41590-021-00993-3
125. Wilson CS, Stocks BT, Hoopes EM, Rhoads JP, McNew KL, Major AS, et al. Metabolic Preconditioning in CD4+ T Cells Restores Inducible Immune Tolerance in Lupus-Prone Mice. JCI Insight (2021) 6(19):e143245. doi: 10.1172/jci.insight.143245
126. Mahajan A, Herrmann M, Munoz LE. Clearance Deficiency and Cell Death Pathways: A Model for the Pathogenesis of SLE. Front Immunol (2016) 7:35. doi: 10.3389/fimmu.2016.00035
127. Widner B, Sepp N, Kowald E, Ortner U, Wirleitner B, Fritsch P, et al. Enhanced Tryptophan Degradation in Systemic Lupus Erythematosus. Immunobiology (2000) 201(5):621–30. doi: 10.1016/S0171-2985(00)80079-0
128. Choi SC, Brown J, Gong M, Ge Y, Zadeh M, Li W, et al. Gut Microbiota Dysbiosis and Altered Tryptophan Catabolism Contribute to Autoimmunity in Lupus-Susceptible Mice. Sci Transl Med (2020) 12(551):eaax2220. doi: 10.1126/scitranslmed.aax2220
129. Brown J, Abboud G, Ma L, Choi SC, Kanda N, Zeumer-Spataro L, et al. Microbiota-Mediated Skewing of Tryptophan Catabolism Modulates CD4(+) T Cells in Lupus-Prone Mice. iScience (2022) 25(5):104241. doi: 10.1016/j.isci.2022.104241
130. Akesson K, Pettersson S, Stahl S, Surowiec I, Hedenstrom M, Eketjall S, et al. Kynurenine Pathway is Altered in Patients With SLE and Associated With Severe Fatigue. Lupus Sci Med (2018) 5(1):e000254. doi: 10.1136/lupus-2017-000254
131. Pertovaara M, Hasan T, Raitala A, Oja SS, Yli-Kerttula U, Korpela M, et al. Indoleamine 2,3-Dioxygenase Activity Is Increased in Patients With Systemic Lupus Erythematosus and Predicts Disease Activation in the Sunny Season. Clin Exp Immunol (2007) 150(2):274–8. doi: 10.1111/j.1365-2249.2007.03480.x
132. Munn DH, Mellor AL. Indoleamine 2,3 Dioxygenase and Metabolic Control of Immune Responses. Trends Immunol (2013) 34(3):137–43. doi: 10.1016/j.it.2012.10.001
133. Mohammadi S, Memarian A, Sedighi S, Behnampour N, Yazdani Y. Immunoregulatory Effects of Indole-3-Carbinol on Monocyte-Derived Macrophages in Systemic Lupus Erythematosus: A Crucial Role for Aryl Hydrocarbon Receptor. Autoimmunity (2018) 51(5):199–209. doi: 10.1080/08916934.2018.1494161
134. Grayson PC, Eddy S, Taroni JN, Lightfoot YL, Mariani L, Parikh H, et al. Metabolic Pathways and Immunometabolism in Rare Kidney Diseases. Ann Rheum Dis (2018) 77(8):1226–33. doi: 10.1136/annrheumdis-2017-212935
135. Qiu J, Wu B, Goodman SB, Berry GJ, Goronzy JJ, Weyand CM. Metabolic Control of Autoimmunity and Tissue Inflammation in Rheumatoid Arthritis. Front Immunol (2021) 12:652771. doi: 10.3389/fimmu.2021.652771
136. Garcia-Carbonell R, Divakaruni AS, Lodi A, Vicente-Suarez I, Saha A, Cheroutre H, et al. Critical Role of Glucose Metabolism in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Arthritis Rheumatol (2016) 68(7):1614–26. doi: 10.1002/art.39608
137. Okano T, Saegusa J, Nishimura K, Takahashi S, Sendo S, Ueda Y, et al. 3-Bromopyruvate Ameliorate Autoimmune Arthritis by Modulating Th17/Treg Cell Differentiation and Suppressing Dendritic Cell Activation. Sci Rep (2017) 7:42412. doi: 10.1038/srep42412
138. Bustamante MF, Oliveira PG, Garcia-Carbonell R, Croft AP, Smith JM, Serrano RL, et al. Hexokinase 2 as a Novel Selective Metabolic Target for Rheumatoid Arthritis. Ann Rheum Dis (2018) 77(11):1636–43. doi: 10.1136/annrheumdis-2018-213103
139. Wang Y, Xian H, Qi J, Wei F, Cheng X, Li S, et al. Inhibition of Glycolysis Ameliorate Arthritis in Adjuvant Arthritis Rats by Inhibiting Synoviocyte Activation Through AMPK/NF-Small Ka, CyrillicB Pathway. Inflamm Res (2020) 69(6):569–78. doi: 10.1007/s00011-020-01332-2
140. Kishimoto K, Terabe K, Takahashi N, Yokota Y, Ohashi Y, Hattori K, et al. Metabolic Changes in Synovial Cells in Early Inflammation: Involvement of CREB Phosphorylation in the Anti-Inflammatory Effect of 2-Deoxyglucose. Arch Biochem Biophys (2021) 708:108962. doi: 10.1016/j.abb.2021.108962
141. Wen Z, Jin K, Shen Y, Yang Z, Li Y, Wu B, et al. N-Myristoyltransferase Deficiency Impairs Activation of Kinase AMPK and Promotes Synovial Tissue Inflammation. Nat Immunol (2019) 20(3):313–25. doi: 10.1038/s41590-018-0296-7
142. Yang XY, Zheng KD, Lin K, Zheng G, Zou H, Wang JM, et al. Energy Metabolism Disorder as a Contributing Factor of Rheumatoid Arthritis: A Comparative Proteomic and Metabolomic Study. PloS One (2015) 10(7):e0132695. doi: 10.1371/journal.pone.0132695
143. Young SP, Kapoor SR, Viant MR, Byrne JJ, Filer A, Buckley CD, et al. The Impact of Inflammation on Metabolomic Profiles in Patients With Arthritis. Arthritis Rheumatol (2013) 65(8):2015–23. doi: 10.1002/art.38021
144. Koedderitzsch K, Zezina E, Li L, Herrmann M, Biesemann N. TNF Induces Glycolytic Shift in Fibroblast Like Synoviocytes via GLUT1 and HIF1A. Sci Rep (2021) 11(1):19385. doi: 10.1038/s41598-021-98651-z
145. Kubota K, Yamashita H, Mimori A. Clinical Value of FDG-PET/CT for the Evaluation of Rheumatic Diseases: Rheumatoid Arthritis, Polymyalgia Rheumatica, and Relapsing Polychondritis. Semin Nucl Med (2017) 47(4):408–24. doi: 10.1053/j.semnuclmed.2017.02.005
146. Matsui T, Nakata N, Nagai S, Nakatani A, Takahashi M, Momose T, et al. Inflammatory Cytokines and Hypoxia Contribute to 18F-FDG Uptake by Cells Involved in Pannus Formation in Rheumatoid Arthritis. J Nucl Med (2009) 50(6):920–6. doi: 10.2967/jnumed.108.060103
147. Mattey DL, Nixon NB, Dawes PT. Association of Circulating Levels of MMP-8 With Mortality From Respiratory Disease in Patients With Rheumatoid Arthritis. Arthritis Res Ther (2012) 14(5):R204. doi: 10.1186/ar4042
148. Weyand CM, Zeisbrich M, Goronzy JJ. Metabolic Signatures of T-Cells and Macrophages in Rheumatoid Arthritis. Curr Opin Immunol (2017) 46:112–20. doi: 10.1016/j.coi.2017.04.010
149. van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, et al. Autoantibodies Against Neutrophils and Monocytes: Tool for Diagnosis and Marker of Disease Activity in Wegener's Granulomatosis. Lancet (1985) 1(8426):425–9. doi: 10.1016/S0140-6736(85)91147-X
150. Kettritz R, Jennette JC, Falk RJ. Crosslinking of ANCA-Antigens Stimulates Superoxide Release by Human Neutrophils. J Am Soc Nephrol (1997) 8(3):386–94. doi: 10.1681/ASN.V83386
151. Keogan MT, Esnault VL, Green AJ, Lockwood CM, Brown DL. Activation of Normal Neutrophils by Anti-Neutrophil Cytoplasm Antibodies. Clin Exp Immunol (1992) 90(2):228–34. doi: 10.1111/j.1365-2249.1992.tb07934.x
152. Weidner S, Neupert W, Goppelt-Struebe M, Rupprecht HD. Antineutrophil Cytoplasmic Antibodies Induce Human Monocytes to Produce Oxygen Radicals In Vitro. Arthritis Rheum (2001) 44(7):1698–706. doi: 10.1002/1529-0131(200107)44:7<1698::AID-ART294>3.0.CO;2-J
153. Brown MA, Kenna T, Wordsworth BP. Genetics of Ankylosing Spondylitis–Insights Into Pathogenesis. Nat Rev Rheumatol (2016) 12(2):81–91. doi: 10.1038/nrrheum.2015.133
154. Whyte JM, Ellis JJ, Brown MA, Kenna TJ. Best Practices in DNA Methylation: Lessons From Inflammatory Bowel Disease, Psoriasis and Ankylosing Spondylitis. Arthritis Res Ther (2019) 21(1):133. doi: 10.1186/s13075-019-1922-y
155. Sanchez-Lopez E, Cheng A, Guma M. Can Metabolic Pathways Be Therapeutic Targets in Rheumatoid Arthritis? J Clin Med (2019) 8(5):753. doi: 10.3390/jcm8050753
156. Geier C, Perl A. Therapeutic mTOR Blockade in Systemic Autoimmunity: Implications for Antiviral Immunity and Extension of Lifespan. Autoimmun Rev (2021) 20(12):102984. doi: 10.1016/j.autrev.2021.102984
157. Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-Acetylcysteine Reduces Disease Activity by Blocking Mammalian Target of Rapamycin in T Cells From Systemic Lupus Erythematosus Patients: A Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol (2012) 64(9):2937–46. doi: 10.1002/art.34502
158. Stylianou K, Petrakis I, Mavroeidi V, Stratakis S, Vardaki E, Perakis K, et al. The PI3K/Akt/mTOR Pathway is Activated in Murine Lupus Nephritis and Downregulated by Rapamycin. Nephrol Dial Transplant (2011) 26(2):498–508. doi: 10.1093/ndt/gfq496
159. Oaks Z, Winans T, Caza T, Fernandez D, Liu Y, Landas SK, et al. Mitochondrial Dysfunction in the Liver and Antiphospholipid Antibody Production Precede Disease Onset and Respond to Rapamycin in Lupus-Prone Mice. Arthritis Rheumatol (2016) 68(11):2728–39. doi: 10.1002/art.39791
160. Koga T, Mizui M, Yoshida N, Otomo K, Lieberman LA, Crispin JC, et al. KN-93, an Inhibitor of Calcium/Calmodulin-Dependent Protein Kinase IV, Promotes Generation and Function of Foxp3(+) Regulatory T Cells in MRL/lpr Mice. Autoimmunity (2014) 47(7):445–50. doi: 10.3109/08916934.2014.915954
161. Ichinose K, Ushigusa T, Nishino A, Nakashima Y, Suzuki T, Horai Y, et al. Lupus Nephritis IgG Induction of Calcium/Calmodulin-Dependent Protein Kinase IV Expression in Podocytes and Alteration of Their Function. Arthritis Rheumatol (2016) 68(4):944–52. doi: 10.1002/art.39499
162. Lai ZW, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in Patients With Clinically Active Systemic Lupus Erythematosus Resistant to, or Intolerant of, Conventional Medications: A Single-Arm, Open-Label, Phase 1/2 Trial. Lancet (2018) 391(10126):1186–96. doi: 10.1016/S0140-6736(18)30485-9
163. Bride KL, Vincent T, Smith-Whitley K, Lambert MP, Bleesing JJ, Seif AE, et al. Sirolimus is Effective in Relapsed/Refractory Autoimmune Cytopenias: Results of a Prospective Multi-Institutional Trial. Blood (2016) 127(1):17–28. doi: 10.1182/blood-2015-07-657981
164. Jiang N, Li M, Zhang H, Duan X, Li X, Fang Y, et al. Sirolimus Versus Tacrolimus for Systemic Lupus Erythematosus Treatment: Results From a Real-World CSTAR Cohort Study. Lupus Sci Med (2022) 9(1):e000617. doi: 10.1136/lupus-2021-000617
165. Bruyn GA, Tate G, Caeiro F, Maldonado-Cocco J, Westhovens R, Tannenbaum H, et al. Everolimus in Patients With Rheumatoid Arthritis Receiving Concomitant Methotrexate: A 3-Month, Double-Blind, Randomised, Placebo-Controlled, Parallel-Group, Proof-of-Concept Study. Ann Rheum Dis (2008) 67(8):1090–5. doi: 10.1136/ard.2007.078808
166. Niu HQ, Li ZH, Zhao WP, Zhao XC, Zhang C, Luo J, et al. Sirolimus Selectively Increases Circulating Treg Cell Numbers and Restores the Th17/Treg Balance in Rheumatoid Arthritis Patients With Low Disease Activity or in DAS28 Remission Who Previously Received Conventional Disease-Modifying Anti-Rheumatic Drugs. Clin Exp Rheumatol (2020) 38(1):58–66.
167. Wen HY, Wang J, Zhang SX, Luo J, Zhao XC, Zhang C, et al. Low-Dose Sirolimus Immunoregulation Therapy in Patients With Active Rheumatoid Arthritis: A 24-Week Follow-Up of the Randomized, Open-Label, Parallel-Controlled Trial. J Immunol Res (2019) 2019:7684352. doi: 10.1155/2019/7684352
168. von Glehn F, Dias-Carneiro RPC, Moraes AS, Farias AS, Silva V, Oliveira FTM, et al. Dimethyl Fumarate Downregulates the Immune Response Through the HCA2/GPR109A Pathway: Implications for the Treatment of Multiple Sclerosis. Mult Scler Relat Disord (2018) 23:46–50. doi: 10.1016/j.msard.2018.04.016
169. Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl Fumarate Targets GAPDH and Aerobic Glycolysis to Modulate Immunity. Science (2018) 360(6387):449–53. doi: 10.1126/science.aan4665
170. Liao ST, Han C, Xu DQ, Fu XW, Wang JS, Kong LY. 4-Octyl Itaconate Inhibits Aerobic Glycolysis by Targeting GAPDH to Exert Anti-Inflammatory Effects. Nat Commun (2019) 10(1):5091. doi: 10.1038/s41467-019-13078-5
171. Yamaguchi Y, Kanzaki H, Katsumata Y, Itohiya K, Fukaya S, Miyamoto Y, et al. Dimethyl Fumarate Inhibits Osteoclasts via Attenuation of Reactive Oxygen Species Signalling by Augmented Antioxidation. J Cell Mol Med (2018) 22(2):1138–47. doi: 10.1111/jcmm.13367
172. Im NK, Choi JY, Oh H, Kim YC, Jeong GS. 6,4'-Dihydroxy-7-Methoxyflavanone Inhibits Osteoclast Differentiation and Function. Biol Pharm Bull (2013) 36(5):796–801. doi: 10.1248/bpb.b12-00964
173. Nishioku T, Kawamoto M, Okizono R, Sakai E, Okamoto K, Tsukuba T. Dimethyl Fumarate Prevents Osteoclastogenesis by Decreasing NFATc1 Expression, Inhibiting of Erk and P38 MAPK Phosphorylation, and Suppressing of HMGB1 Release. Biochem Biophys Res Commun (2020) 530(2):455–61. doi: 10.1016/j.bbrc.2020.05.088
174. Zaro BW, Vinogradova EV, Lazar DC, Blewett MM, Suciu RM, Takaya J, et al. Dimethyl Fumarate Disrupts Human Innate Immune Signaling by Targeting the IRAK4-MyD88 Complex. J Immunol (2019) 202(9):2737–46. doi: 10.4049/jimmunol.1801627
175. Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid Dendritic Cells (Natural Interferon- Alpha/Beta-Producing Cells) Accumulate in Cutaneous Lupus Erythematosus Lesions. Am J Pathol (2001) 159(1):237–43. doi: 10.1016/S0002-9440(10)61689-6
176. Abboud G, Choi SC, Kanda N, Zeumer-Spataro L, Roopenian DC, Morel L. Inhibition of Glycolysis Reduces Disease Severity in an Autoimmune Model of Rheumatoid Arthritis. Front Immunol (2018) 9:1973. doi: 10.3389/fimmu.2018.01973
177. Cai W, Cheng J, Zong S, Yu Y, Wang Y, Song Y, et al. The Glycolysis Inhibitor 2-Deoxyglucose Ameliorates Adjuvant-Induced Arthritis by Regulating Macrophage Polarization in an AMPK-Dependent Manner. Mol Immunol (2021) 140:186–95. doi: 10.1016/j.molimm.2021.10.007
178. Song G, Lu Q, Fan H, Zhang X, Ge L, Tian R, et al. Inhibition of Hexokinases Holds Potential as Treatment Strategy for Rheumatoid Arthritis. Arthritis Res Ther (2019) 21(1):87. doi: 10.1186/s13075-019-1865-3
179. Teng X, Cornaby C, Li W, Morel L. Metabolic Regulation of Pathogenic Autoimmunity: Therapeutic Targeting. Curr Opin Immunol (2019) 61:10–6. doi: 10.1016/j.coi.2019.07.001
180. Kim JW, Choe JY, Park SH. Metformin and its Therapeutic Applications in Autoimmune Inflammatory Rheumatic Disease. Korean J Intern Med (2022) 37(1):13–26. doi: 10.3904/kjim.2021.363
181. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The Kinase mTOR Regulates the Differentiation of Helper T Cells Through the Selective Activation of Signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12(4):295–303. doi: 10.1038/ni.2005
182. El-Sayyad SM, Ali MA, Kandil LS, Ragab GM, Abdelhamid Ibrahim SS. Metformin and Omega-3 Fish Oil Elicit Anti-Inflammatory Effects via Modulation of Some Dysregulated Micro RNAs Expression and Signaling Pathways in Experimental Induced Arthritis. Int Immunopharmacol (2021) 92:107362. doi: 10.1016/j.intimp.2020.107362
183. Zhou Z, Tang Y, Jin X, Chen C, Lu Y, Liu L, et al. Metformin Inhibits Advanced Glycation End Products-Induced Inflammatory Response in Murine Macrophages Partly Through AMPK Activation and RAGE/NFkappaB Pathway Suppression. J Diabetes Res (2016) 2016:4847812. doi: 10.1155/2016/4847812
184. Naffaa ME, Rosenberg V, Watad A, Tiosano S, Yavne Y, Chodick G, et al. Adherence to Metformin and the Onset of Rheumatoid Arthritis: A Population-Based Cohort Study. Scand J Rheumatol (2020) 49(3):173–80. doi: 10.1080/03009742.2019.1695928
185. Abdallah MS, Alarfaj SJ, Saif DS, El-Naggar ME, Elsokary MA, Elsawah HK, et al. The AMPK Modulator Metformin as Adjunct to Methotrexate in Patients With Rheumatoid Arthritis: A Proof-of-Concept, Randomized, Double-Blind, Placebo-Controlled Trial. Int Immunopharmacol (2021) 95:107575. doi: 10.1016/j.intimp.2021.107575
186. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-Of-Concept Trial of Metformin. Arthritis Rheumatol (2015) 67(12):3190–200. doi: 10.1002/art.39296
Keywords: autoimmune diseases, innate immune cells, immunometabolism, glycolysis, therapeutic target
Citation: Xu Y, Chen Y, Zhang X, Ma J, Liu Y, Cui L and Wang F (2022) Glycolysis in Innate Immune Cells Contributes to Autoimmunity. Front. Immunol. 13:920029. doi: 10.3389/fimmu.2022.920029
Received: 14 April 2022; Accepted: 31 May 2022;
Published: 01 July 2022.
Edited by:
Andras Perl, Upstate Medical University, United StatesReviewed by:
Jaya Talreja, Wayne State University, United StatesFang Wei, Bengbu Medical College, China
Copyright © 2022 Xu, Chen, Zhang, Ma, Liu, Cui and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liyan Cui, Y2xpeWFuQDE2My5jb20=; Fang Wang, eXV5aW5nemhvdXlpZmFuQDEyNi5jb20=
†These authors have contributed equally to this work