 Qingxia Huang
Qingxia Huang Song Gao3
Song Gao3 Yao Yao
Yao Yao Yisa Wang
Yisa Wang Jing Li
Jing Li Jinjin Chen
Jinjin Chen Daqing Zhao
Daqing Zhao Xiangyan Li
Xiangyan Li- 1Research Center of Traditional Chinese Medicine, College of Traditional Chinese Medicine, Changchun University of Chinese Medicine, Changchun, China
- 2Jilin Ginseng Academy, Key Laboratory of Active Substances and Biological Mechanisms of Ginseng Efficacy, Ministry of Education, Jilin Provincial Key Laboratory of Bio-Macromolecules of Chinese Medicine, Changchun University of Chinese Medicine, Changchun, China
- 3Jilin Xiuzheng Pharmaceutical New Drug Development Co., Ltd., Changchun, China
Hemorrhagic shock (HS) is a shock result of hypovolemic injury, in which the innate immune response plays a central role in the pathophysiology ofthe severe complications and organ injury in surviving patients. During the development of HS, innate immunity acts as the first line of defense, mediating a rapid response to pathogens or danger signals through pattern recognition receptors. The early and exaggerated activation of innate immunity, which is widespread in patients with HS, results in systemic inflammation, cytokine storm, and excessive activation of complement factors and innate immune cells, comprised of type II innate lymphoid cells, CD4+ T cells, natural killer cells, eosinophils, basophils, macrophages, neutrophils, and dendritic cells. Recently, compelling evidence focusing on the innate immune regulation in preclinical and clinical studies promises new treatment avenues to reverse or minimize HS-induced tissue injury, organ dysfunction, and ultimately mortality. In this review, we first discuss the innate immune response involved in HS injury, and then systematically detail the cutting-edge therapeutic strategies in the past decade regarding the innate immune regulation in this field; these strategies include the use of mesenchymal stem cells, exosomes, genetic approaches, antibody therapy, small molecule inhibitors, natural medicine, mesenteric lymph drainage, vagus nerve stimulation, hormones, glycoproteins, and others. We also reviewed the available clinical studies on immune regulation for treating HS and assessed the potential of immune regulation concerning a translation from basic research to clinical practice. Combining therapeutic strategies with an improved understanding of how the innate immune system responds to HS could help to identify and develop targeted therapeutic modalities that mitigate severe organ dysfunction, improve patient outcomes, and reduce mortality due to HS injury.
Introduction
Hemorrhagic shock (HS) is a life-threatening condition occurring in various clinical situations, including trauma, childbirth, gastrointestinal hemorrhage, and aneurysmal rupture (1, 2). It is a represents a substantial global problem, which more than 1.9 million deaths per year worldwide, of which 1.5 million results from traumatic injury (3). Although 20% of the fatal cases of HS are considered avoidable, current practice has failed to improve the survival rate (4). Clinically, HS is treated with an expedited anatomic control of bleeding in conjunction with intravenous blood products, deliberate hypotension, antifibrinolytic therapy, and vasodilation (5, 6). Despite advances in clinical treatment aimed at the hypovolemic injury, patients who survive the initial HS insult have severe complications due to organ reperfusion injury, delayed infections, immune dysfunction, and the risk of developing organ, with incidences of 37.2%, or multiple organ failure (MOF), with incidences of 22.1% (7, 8).
Increasing evidence has proved that the modulation of innate immune responses is a promising therapeutic strategy for preventing and treating HS-induced MOF injury and complications (9). Soon after HS insult, exposure to exogenous pathogen-associated molecular pattern molecules (PAMPs) and endogenous damaged-associated molecular pattern molecules (DAMPs) extensively activate the innate immune defense, mainly comprising type II innate lymphoid cells (ILC2), CD4+ T cells, natural killer (NK) cells, eosinophils, dendritic cells (DCs), basophils, macrophages, neutrophils, and the complement cascade (10). The innate immune response initiated and propagated in response to HS triggers inflammatory and anti-inflammatory mechanisms within 30 min post-injury, followed by systemic immune response syndrome (SIRS) and counterbalancing anti-inflammatory response syndrome (CARS), which are related to organ injury and complications (11). The systemic parameters during HS suggest that restoration of innate immunity offers exciting and promising directions for developing novel therapeutics for HS-induced second injuries. Recently, compelling evidence has focused on the innate immune response for the monitoring and therapy of HS (8, 9, 12, 13). The emerging understanding is poised to revolutionize the treatment of HS through targeted immune modulators.
In this article, we firstly summarize recent advances in the pathomechanistic insights associated with the innate immune response following HS injury, then systematically detail the cutting-edge therapeutic strategies used in the past decade regarding the innate immune regulation in this field, such as mesenchymal stem cells (MSCs), MSC-derived exosomes, MSC-derived extracellular vesicles (MSC-EVs), MSC-derived soluble factors (FS-MSC), genetic approaches, antibody therapy, small molecule inhibitors, natural medicine, mesenteric lymph (ML) drainage, vagus nerve stimulation (VNS), hormones, glycoproteins, and others. We also reviewed clinical studies on the regulation of immunity for treating HS and assessed the potential of immune regulation concerning a translation from basic research to clinical practice. Combining these therapeutic strategies with an improved understanding of how the innate immune system responds to HS could help to identify and develop targeted therapeutic modalities that mitigate severe organ dysfunction, improve patient outcomes, and reduce mortality due to HS injury.
Innate immune response to HS
Increasing experimental and clinical evidence indicates that innate immunity is the predominant mediator in the pathophysiology of HS injury that unequivocally leads to organ damage and failure. The innate immune activation and immunosuppression responses to HS injury obtained in clinical trials and preclinical experiments are summarized in Figure 1.
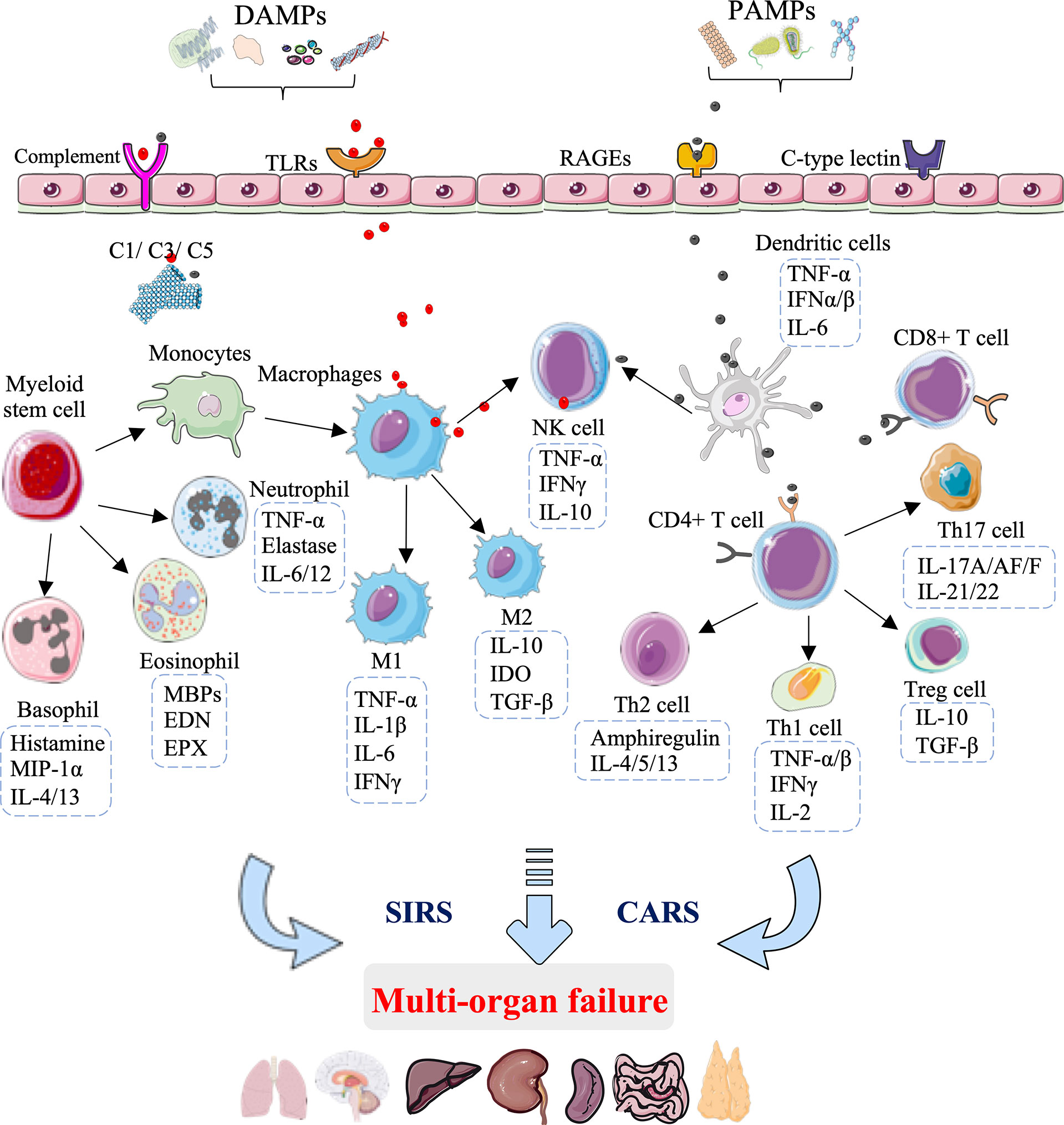
Figure 1 Innate immunity-mediated pathomechanisms in multi-organ failure development after hemorrhagic shock. During the development of hemorrhagic shock, the innate immunity rapid response to damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) is mediated through pattern recognition receptors, including toll-like receptors (TLRs), receptors of advanced glycation end products (RAGEs), C-type lectin receptors, and complement receptors. The early and exaggerated activation of innate immunity results in excessive activation of complement factors(C1/C3/C5) and innate immune cells, comprising macrophages, DC cells, T cells, natural killer (NK) cells, eosinophils, basophils, and neutrophils. The activation of innate immune cells leads to the secretion of cytokines and chemokines, which exaggerates inflammation and subsequent immunosuppression, causing systemic immune response syndrome (SIRS) and a counterbalancing anti-inflammatory response syndrome (CARS), ultimately leading to multi-organ failure.
At the site of hemorrhage, the immune system is challenged with “alarmins”, among which exogenous PAMPs are expressed on invading microorganisms and endogenous DAMPs are released from damaged and host cells, and include mitochondrial DNA, cold-inducible RNA-binding protein (CIRP), high mobility group box 1 (HMGB1), interleukin (IL)-25, IL-33, mitochondrial N-formyl peptides (F-MIT), and F-Actin (14–16). These “alarmins” are recognized by distressed immune cells through groups of pattern-recognition receptors, including toll-like receptors (TLRs), receptors of advanced glycation end products (RAGEs), C-type lectin receptors, and complement receptors (17). The excessive proinflammatory response SIRS and parallel immunosuppression CARS are induced after these damage molecules engage with their corresponding receptors, which is characterized by the release of cytokines, chemokines, complement factors, and coagulation proteins, as well as activation of innate immune cells (18). In terms of negative feedback, the excessive innate immune response can promote the circulation of new DAMPs, thereby amplifying a vicious cycle of cell and tissue injuries (19, 20).
During the development of HS, the innate immune cells act as the first line of defense, providing a rapid response to pathogens or danger signals through pattern recognition receptors (21). One of the first innate immune cell types to the site of injury is antigen-presenting cells, including tissue-resident macrophages, which sense damaging molecules and then differentiate from M1-type macrophages that secrete proinflammatory factors (TNF-α, IL-1β, IL-6, and interferon-γ) to M2-type macrophages that secrete anti-inflammatory factors (IL-10, IGF-1, and TGF-β) (22). In addition to defending against pathogens, macrophages are crucial to the maintenance of tissue homeostasis (23). Indeed, data suggest that the macrophage phenotype can correspondingly switch from M1-type macrophages, polarized by Th1 cytokines (GM-CSF, TNF-α, and interferon-γ), to M2-type macrophages, polarized by Th2 cytokines (IL-4 and IL-13) to deal with tissue repair (24, 25). Interestingly, neutrophils also regulate T cell function, and the M1-to-M2 macrophage switch represents a central element in the clearance of neutrophils by efferocytosis (26–28).
DCs are another type of antigen-presenting cell, which deliver antigens to T and NK cells. DCs show rapid responsiveness to pathogens or danger signals, which is followed by the secretion of TNF-α, interferon (IFN)-α, IFN-β, as well as IL-6 within a few hours after HS (29). The activation of T lymphocytes by DCs or danger signals is essential in exaggerating inflammation and subsequent immunosuppression (30). CD4+ T cells are the primary lymphocytes involved in HS injury and are classically divided into four categories: Th1, Th2, Th17, and T regulatory (Treg) cells (31). Treg cells can suppress T-cell activation and Th1 cytokine production after injury. Data suggest that HMGB1 binding to RAGE, TLR4 and TLR9 can promote the activation of DCs, CD4+ T, CD8+ T, Th17, and Treg cells in response to HS injury (32–34). The imbalance of Th17/Treg has been suggested to be positively correlated with the degree of acute liver injury (35). The balance of Th1/Th2 is attributed to conversion to type 2 responses during HS development (36–38). DCs subject to HS were more inclined to polarize naive CD4+ T cells into Th2 and Treg cells, consistent with the clinically observed immunosuppressive phenomena in severe patients (39). Clinically, the peak of organ damage and MOF occurs within the 3 days of HS, with lung failure being the most common (40, 41).
Taken together, innate immunity is activated early after HS injury, and cross-talk between various immune cells ultimately results in MOF. Many attempts of therapeutic strategies focusing on the innate immune regulation in preclinical and clinical studies have achieved promising results in reducing tissue injury, organ dysfunction, and ultimately mortality due to HS.
New insights into HS therapeutics with innate immune regulation
Stem cell therapy in HS
Stem cells possess a remarkable potential for developing new cell-based treatments in the context of HS by modulating local and systemic deleterious immune responses (42, 43). Table 1 shows the main stem cell-related therapeutic strategies in the HS model in order of stem-cell type: MSC (MSCs, MSC-EVs, FS-MSC, MSC-derived exosomes, and IL-1β primed MSC), adipose-derived stem cells, and neutrophil progenitors. Each of these therapeutic strategies is discussed in detail below.

Table 1 Overview of the applications of stem cell-related therapeutic strategies in a hemorrhagic shock model.
MSCs are multipotent stem cells, which are commonly used as a clinical cell therapeutic strategy for immunomodulation and tissue repair (9, 56). Treg cells, an immunosuppressive T cell subset, are essential for maintaining immune homeostasis and tolerance (57). In a rat model of unilateral lung contusion followed by HS, impaired wound healing and lung structure were improved by MSCs treatment by increasing the Treg cell population (44, 45). Cell-based therapies using MSCs or MSC-EVs are beneficial for improving neurologic outcomes and lung injury in animal models of HS (9). Moreover, in an HS-induced mild lung injury rat model, leukocyte infiltrates (CD68+ and MPO+ cells) were significantly reduced in the lung after treatment with MSCs (46). The latest transcriptome data demonstrated that treatment with MSCs or MSC-EVs was associated with the inactivation of inflammation-chemokine and cytokine pathways in the lung of HS mice (47). In a porcine model of HS, the neuroprotective and neurorestorative properties observed in MSC-EVs treatment were also associated with the attenuation of inflammation-related transcription in the brain (48).
A previous study showed that IL-10, an immunoregulatory cytokine, binds to the IL-10 receptor and inhibits inflammation following HS (58, 59). Yunwei Zhang et al. found that IL-10-deficient MSCs lost the protective function compared to WT MSCs in an HS-induced hepatic injury model (49). Another experiment indicated that MSC-EVs carrying IL-10 as cargo were mainly taken up by macrophages in the liver, mediating M2-type macrophage polarization and consequent immunosuppression in HS-induced hepatic injury (49). Neutrophils are the first innate immune cells against pathogens due to their array of microbicidal activities (60, 61). Clinically, the drop in circulating neutrophils is positively correlated with the occurrence of MOF (62, 63). FS-MSCs have an immunomodulatory action through paracrine activity by secreting anti-inflammatory cytokines and growth factors (64). Recent studies have shown that treatment with FS-MSCs significantly reduced inflammation and lung neutrophil infiltrates in an HS-induced rat model (50).
An early single dose of exosomes derived from MSC treatment has been shown to attenuate neurological injury by decreasing IL-1, IL-6, and IL-18, and increasing granulocyte-macrophage colony-stimulating factor (GM-CSF) levels in the Yorkshire swine model of HS (51). Additionally, the administration of human MSC-derived exosomes induces transcriptomic changes of neuroinflammation after HS injury in swine (52). In several in vitro and in vivo studies, IL-1β priming maximized the immunomodulation effect of MSCs by regulating IL-6 and IL-8 expression and influencing the polarization of peritoneal macrophages (65, 66). Moreover, systemic cytokines (IL-1α, IL-6, and IL-10) and the programmed cell death receptor (PD)-1/PD-L1 axis were decreased by IL-1β-primed MSCs on monocytes and granulocytes in HS-induced kidney and liver injury model (53). Similarly, the IL-6 concentration also decreased with adipose-derived stem cell treatment in HS-induced liver injury (54). In rodent models of HS, it has been demonstrated that, in addition to suppressive proinflammatory cytokines in the lungs, there is an increase in neutrophil migration into the airspace from the bone marrow after neutrophil progenitor transplant, which can be used in the treatment and prevention of secondary infection following HS (55).
Indeed, several studies—mostly in rodents but also in porcine—have concluded that MSCs, MSC-EVs, FS-MSCs, MSC-derived exosomes, IL-1β-primed MSCs, adipose-derived stem cells, and neutrophil progenitors can relieve lung, neurologic, kidney, and liver injury by regulating innate immunity (Figure 2, Table 1). The innate immune processes involved in the above effects include reducing the leukocyte and neutrophil infiltrate, increasing the Treg population, mediating M2-type macrophage polarization and consequent immunosuppression, and inactivating inflammatory chemokines and cytokines (Figure 2, Table 1). These stem cell-related therapeutic strategies represent a potential opportunity for treating HS-induced second injuries.
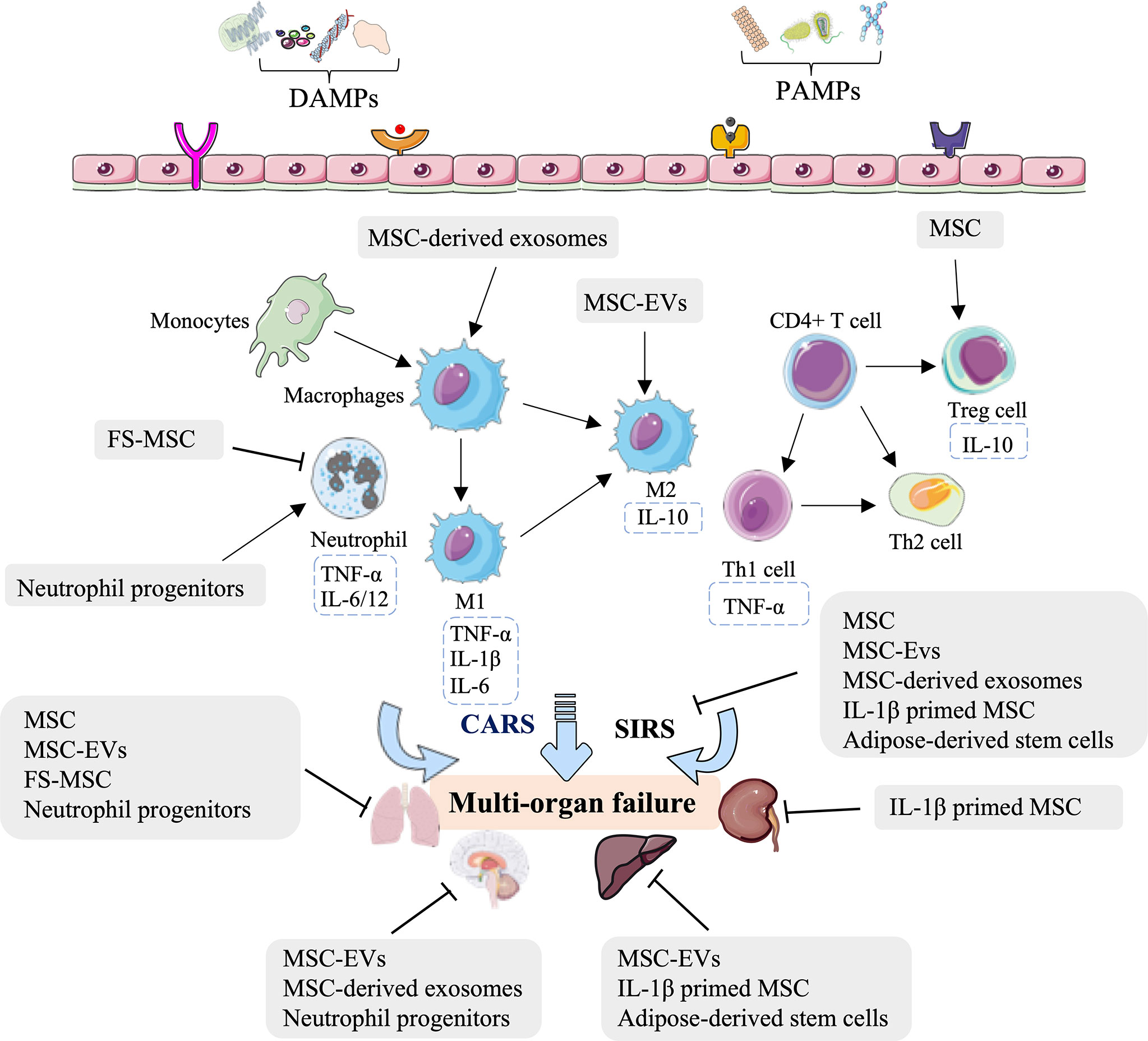
Figure 2 Overview of the immune therapeutic strategies of stem cells in hemorrhagic shock. The mesenchymal stem cell (MSC), MSC-derived extracellular vesicles (MSC-EVs), MSC-derived soluble factors (FS-MSC), MSC-derived exosomes, adipose-derived stem cells, and neutrophil progenitors can relieve the lung, neurologic, kidney, and liver injury by regulating neutrophil infiltration, increasing the Treg population, mediating M2-type macrophage polarization and consequent immunosuppression, and inactivation of inflammatory chemokine and cytokines.
Antibody therapy and genetic approaches in HS
IL-6 plays a prominent role in the differentiation from Th1 to Th2 in the development of HS (67). The secretion of IL-6 is positively correlated with the prognosis patients with shock and organ dysfunction (68, 69). Zhang Yong et al. reported that treatment with anti-mouse IL-6 monoclonal antibody immediately before resuscitation can prevent Th2 cytokine production, suppress the lymphocyte response, reduce the level of IL-10, keratinocyte-derived chemokine (KDC), monocyte chemoattractant protein 1(MCP-1), and macrophage inhibitory protein 1 (MIP-1) in a mouse model combining HS and lower-extremity injury (67). The mucosal address in cell adhesion molecule-1 (MAdCAM-1), a critical mediator of the early innate immune response to HS, mainly mediates lymphocyte recruitment to the gut during the inflammatory storm phase (70–72). This observation is supported by a finding that antibody blockade of MAdCAM-1 can decrease the secretion of IL-1β, IL-6, and TNF-α, reduce lymphocyte infiltration, ameliorate intestinal barrier dysfunction, and prolong survival (70). B and T lymphocyte attenuator (BTLA), a receptor that is structurally similar to PD-1, is expressed on T lymphocytes, B lymphocytes, monocytes, macrophages, and DCs (73, 74). A previous study showed that treatment with the anti-BTLA monoclonal [6A6] antibody (25μg/g body weight) can abolish HS followed by sepsis-induced reduction of cytokines and chemokines (TNF-α, IL-12, IL-10, KC, MIP-2, MCP-1) and decreased recruitment of neutrophils, macrophages, and DCs to the peritoneal cavity, which in turn relieve organ injury and reduce mortality (75).
Clinical studies have shown patients with HS with a poor prognosis within 24 h after admission have higher type 2 cytokines in serum, such as IL-5 (76). IL-33-stimulated ILC2, the resident innate lymphocytes that potently regulate host immunity in the lung, are the primary source of type 2 cytokines response to HS injury (34, 77). A recent finding has shown that anti-IL-5 antibody, IL-33, or ILC2 deletion, significantly increased IL-5 expression in neutrophils and decreased lung injury scores at 6 h in the HS-induced mice injury model (77). DAMPs can activate systemic inflammation and organ injury in HS through binding to TLR2 on immune cells (78). Similarly, anti-TLR2 monoclonal antibody or TLR2-/- mice exhibited significantly less liver damage, and lower NF-κB and inflammatory cell infiltrate in HS at 20 h (78). Consistently, the phenotype of TLR2-/- mice shows reduced intestinal injury accompanied by reduced complement (CD55, Factor H, and C3) and inflammatory (IL-12, IL-6, and TNF-α) factors, compared to wild-type mice (79).
Extracellular CIRP, an 18-kDa RNA chaperone protein, acts as an endogenous proinflammatory mediator, binds to TLR4, and leads to mitochondrial DNA fragmentation that triggers innate immunity and inflammatory responses in patients with HS (17, 80). Continuity studies demonstrated that the purified recombinant murine CIRP (rmCIRP) induces cytokine release in macrophages and deficiency or blockade of CIRP using antisera leads to attenuated TNF-α and IL-6 release, neutrophil accumulation, and lethality in HS injury (17, 80). Interestingly, wound-associated TNF-α enhancement and neutrophil infiltration is also attenuated in CIRP-/- mice compared to WT mice (81). Mitochondrial DNA binds to the stimulator of interferon genes (STING) as a ligand, activating ype I interferon and proinflammatory cytokines-producing signals (82, 83). Kehong Chen et al. reported that the HS-induced increase in IL-6 and IFN-β levels in the serum and the high mRNAs expression of TNF-α, IL-6, and IL-1β in the lung were significantly counteracted by STING knockout, which suggests that the absence of STING significantly reduces inflammation and lung injury after HS (84).
Many studies have focused on the role of PD-1 and its ligand, PD-L1 (B7H1) in the cellular immunotherapy (85–87). The population of PD-1+ blood leukocytes in patients is positively correlated with interleukin levels in the serum, which suggests that PD-1 is a key indicator in the assessment of HS-induced immune dysfunction (87). Indeed, in terms of immune regulation, animals deficient in PD-1 or PD-L1 expression exhibited an attenuation in the neutrophil influx in HS injury, while PD-L1 knockout produced a marked suppression in the secretion of TNF-α, IL-6, and MCP-1, which were consistently elevated induced by HS in the WT mice group (88).
Clinical and preclinical studies have observed that nuclear factor-erythroid 2 p45-related factor-2 (Nrf2), a major mediator in innate immunity and inflammation, is significantly increased in the leukocytes collected from patients with HS (89–92). Haige Zhao et al. reported that HS-induced secretion of HMGB1, L-6, IL-1β, and TNF-αwere higher at 2 h in Nrf2 knockout mice (92). Likewise, Nrf2-KO offers no benefit over the hepatoprotection of remote ischemic conditioning in reductions in HS-induced TNF-α and IL-6 (93).
CD226, a costimulatory adhesion molecule expressed on both immune and endothelial cells, can regulate immune metabolic activity and function (94, 95). Recent studies have illustrated that CD226 deficiency in vascular endothelial cells can alleviate HS-induced intestinal damage and the inflammatory response (96). Emerging evidence shows that microRNAs play essential roles in pathophysiological responses by regulating inflammation and immunity (97, 98). Moreover, data suggest that miR-18b-5p knockdown notably reduced the levels of SOD1, iNOS, and IL-6 in macrophages, decreased the M1/M2 ratio of macrophages, and reduced the Th1/Th2 ratio of CD4+ T cells in splenic tissues after HS injury (99).
Significant advances have been made in the identification of immune therapies for HS injury, including antibodies (anti-IL-6, anti-TLR2, anti-IL-5, anti-IL-BTLA, and anti-MAdCAM-1), RNAi-based deficiency (PD-1, CD226, and miR-18b-5p), and gene knockout (TLR2, IL33, CIRP, or STING). In summary, these antibody therapies and genetic approaches for HS are associated with a potent innate immune response that not only regulates the levels of inflammatory factors, the lymphocyte influx, and neutrophil infiltration, but also reduces complement, the ratio of M1/M2 macrophages, the Th1/Th2 ratio in CD4+ T cells, and increases recruitment of DCs (Figure 3 and Table 2).
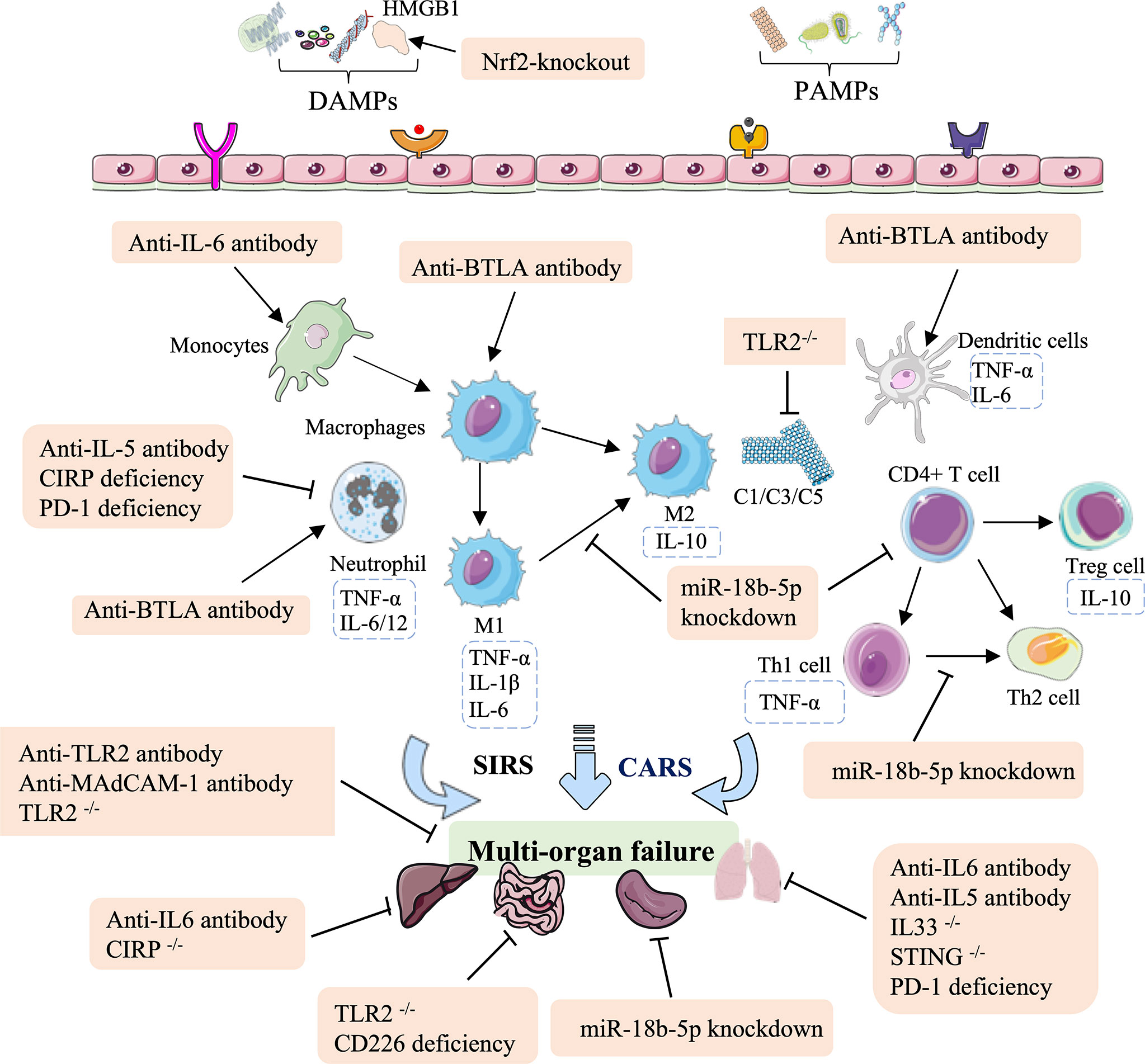
Figure 3 Overview of the immune therapeutic strategies of antibodies, and genetic approaches in hemorrhagic shock. The antibody of anti-IL-6, anti-TLR2, anti-IL-5, anti-IL-BTLA, and anti-MAdCAM-1; genetic approaches (RNAi-based deficiency such as PD-1, CD226, and miR-18b-5p); and gene knockout such as TLR2, IL33, CIRP, and STING can relieve lung, intestinal, splenic, liver, and multi-organ failure. These effects are mainly mediated by regulating the population of monocytes and macrophages, mediating neutrophil infiltration, reducing complement, the M1/M2 ratio in macrophages and the Th1/Th2 ratio in CD4+ T cells, and increasing recruitment of DCs.

Table 2 Summary of the applications of antibody therapy and genetic approaches in the hemorrhagic shock model.
Small molecule inhibitor or agonist therapy in HS
As a master alarm system and a major fluid defense system of innate immunity after HS injury, the complement cascade can be rapidly activated by DAMPs or PAMPs, and lead to elevated plasma levels of complement activation products such as complement factor 1 (C1), complement factor 3 (C3) and complement factor 5 (C5) (100). As reviewed earlier, such exuberant complement activation evokes systemic inflammation, which is associated with increased susceptibility to infections and HS-induced MOF (8, 101). Early studies have shown that C3 deficiency attenuates HS-related hepatic injury and SIRS (102). The therapeutic inhibition of C3 by C3 inhibitor compstatin-40 (Cp40) is capable of improving immune, coagulation, and organ (kidney and intestine) functions by decreasing IL-6, MIF, IL-1RA, MIP-1, MCP-1, and IFN-γ (103). Another C3 inhibitor-soluble form of CR1 (sCR1) was confirmed to significantly mitigate the over-expression of NO, ET-1, TNF-α, and reactive oxygen species in serum to relieve vascular hyperreactivity in HS rats (104). Recombinant human C1-esterase inhibitor (rhC1-INH) has been found to particularly reduce tissue damage (kidney, gut, and lung), tissue complement activation, and cytokine release in an HS-induced porcine injury model (105). Additionally, the alternative complement activation in response to HS injury can induce macrophage infiltration and IL-12 secretion in the intestine (106). Multiple studies have indicated that treatment with complement inhibitors (C5 receptor antagonist or cobra venom factor) can significantly attenuate HS-induced intestinal injury (107–109). Furthermore, the mucosal damage, macrophage infiltration, and intestinal inflammation induced by HS injury were decreased by reducing leukotriene B4, IL-12p40, and TNF-α in the absence of IL-12p70 or treatment with complement receptor 2-targeted factor H (CR2-fH), a targeted inhibitor of the alternative complement pathway (106).
In addition to inhibitors targeting complement, many inhibitors targeting key proteins of innate immunity decrease HS-induced organ dysfunction. Recent studies have indicated that early intravenous treatment of tranexamic acid, a serine protease inhibitor, can protect the intestinal barrier by inhibiting neutrophil extracellular trap formation in the development of HS (110). CIRP acts as a DAMP to activate innate immunity and increases complications caused by HS (17). CIRP-derived oligopeptide-23 (C23) is homologous to the human CIRP protein (Ser110-Glu125) that binds to the CIRP receptor with high affinity and inhibits the secretion of TNF-α (111). Fangming Zhang et al. reported that the mRNA levels of IL-1β, TNF-α, and IL-6 in the lungs were reduced by adjuvant treatment with C23 (8 mg/kg) in HS-induced lung injury (111). Cyclosporine A (CsA) acts as a calcineurin inhibitor that participate in the innate immune response to pathogens in an inflammation storm (112). Some studies have shown that CsA could increase the survival time of HS rats by inhibiting proinflammatory cytokine production (IL-6) and reducing liver injury (113, 114).
Emerging evidence suggests that treatment with HDAC inhibitors (HDACIs) can attenuate MOF and improve early survival in animal models of HS by restoring “acetylation homeostasis” of histones and inducing transcriptional activation (115, 116). Transcriptomic studies in peripheral blood mononuclear cells (PBMC) and brain tissue suggested that valproic acid (VPA, one of HDACIs) can reduce HS-induced neurologic injury by downregulating genes associated with cell death and inflammation (IL-6, TLR4, JAK2, NLRP1, TNFα, IL-1α, IL-1B, NF-κB) (117, 118). In addition, treatment with VPA (150 mg/kg) significantly decreased brain lesion size and improved neurologic recovery by activating nuclear factor- k B (NF-κB) and degrading of cytosolic IκB in Yorkshire swine models of HS (119). Elizabeth A. Sailhamer et al. demonstrated that suberoylanilide hydroxamic acid (SAHA), one of HDACIs, can normalizes inflammatory cytokines (TNFα and IL-1β) levels by acetylating the transcription factor NF-κB following HS in the rats (120).
FTY720, an immunomodulator targeting receptors of sphingosine 1-phosphate (S1P), which can disrupt lymphocyte trafficking, prevent lymphocytes from accumulating in secondary lymphoid organs, and decrease lymphocytes in the blood circulation (121, 122). Jason S. Hawksworth et al. reported that FTY720 (0.3 mg/kg) could sequestrate the central lymphocytes, resulting in attenuation in innate cellular and signal activation following HS in a swine liver and lung injury model (123). FTY720 (1 mg/kg) has also been shown to reduce HS-induced MOD syndromes, red cell injury, and neutrophil priming in a rat model (124). The direct administration by a receptor agonist can block the binding of TLR ligands with their receptor, interfere with intracellular signaling molecules, and prevent signal amplification, which is a promising approach for treating HS-induced immune dysregulation. Xu Ding et al. reported that macrophage-activating lipopeptide-2 (MALP-2), as an agonist of TLR, given at the earliest can reduce pulmonary damage and polymorphonuclear neutrophil infiltration in an HS mouse model (125).
Latest studies have shown that some activators, such as sulforaphane, an Nrf2 pathway agonist, can modulate immunity against HS damage (126). Weiqiang Liang et al. demonstrated that sulforaphane, a potential immune modulator, could protect the liver from HS-induced inflammation storm by decreasing the secretion of TNF-α, MCP-1, KC/CXCL1, IL-6, and IL-10 and abolishing neutrophil infiltration in kupffer cells (126). Moreover, in a mouse HS model, sulforaphane administration reduced lung and liver injury via down-regulating pro-inflammatory cytokines, such as TNF-α, COX-2, iNOS, and IL-1β (127, 128).
As discussed above, small molecule inhibitors, especially complement-related target inhibitors, inhibitors of serine protease, CIRP, sphingosine-1-phosphate, toll-like receptors, mPTP, and agonists of the Nrf2 pathway, can reduce HS-induced liver, kidney, intestinal, renal, and lung and vascular hyperreactivity injury in monkey, swine, and rodent animal models by modulating innate immune responses (Figure 4, Table 3).
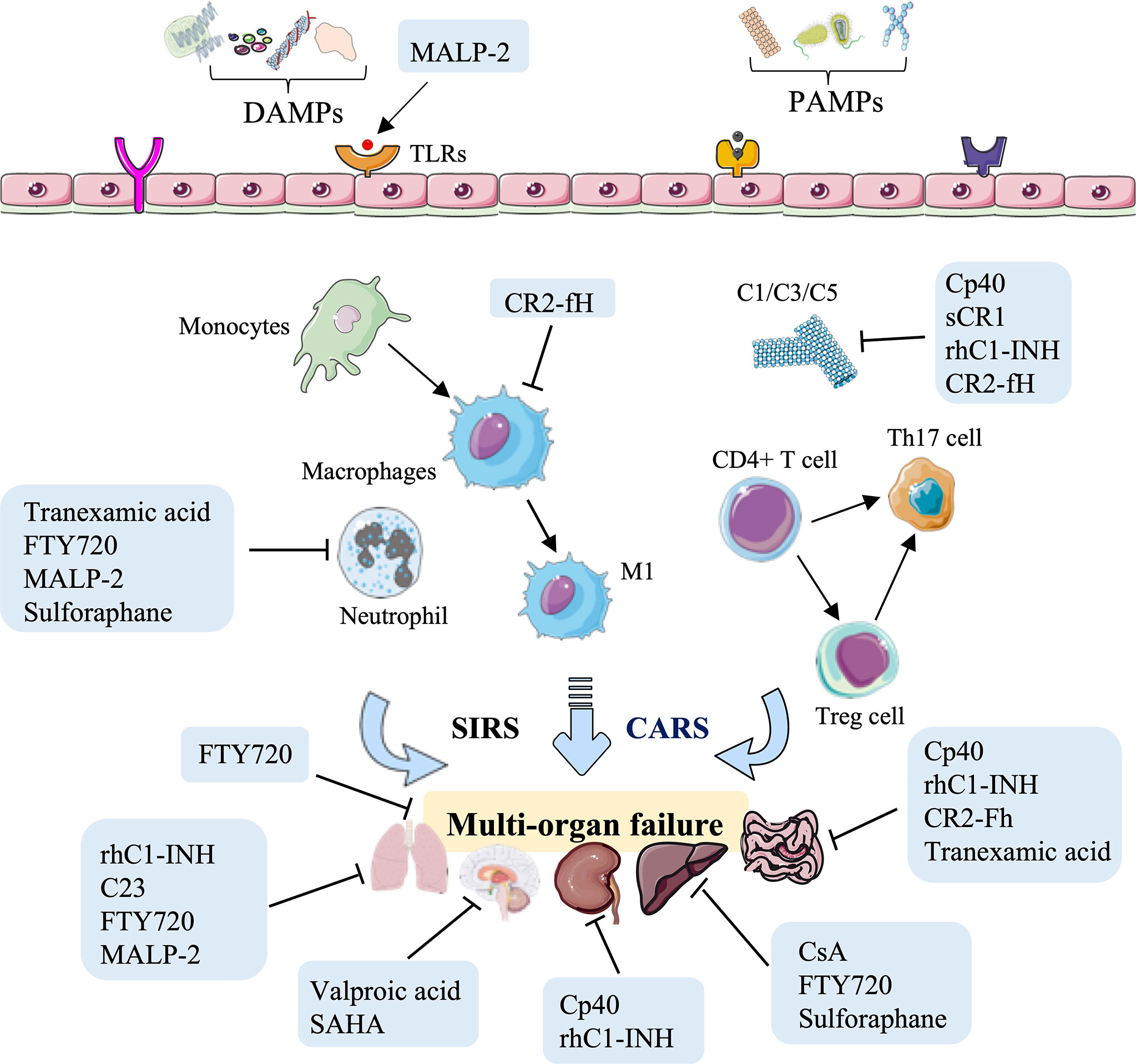
Figure 4 Overview of the immune therapeutic strategies of small molecule inhibitors or agonists in hemorrhagic shock. Small molecule inhibitors or agonists, especially complement-related target inhibitors, such as compstatin-40 (Cp40), soluble form of CR1 (sCR1), recombinant human C1-esterase inhibitor (rhC1-INH), complement receptor 2-targeted factor H (CR2-fH), tranexamic acid, CIRP-derived oligopeptide-23 (C23), FTY720, macrophage-activating lipopeptide-2 (MALP-2), cyclosporine A (CsA), and sulforaphane can reduce HS-induced liver, kidney, intestinal, lung, and multi-organ failure injury by reducing complement activation, macrophage infiltration, neutrophil priming, and systemic inflammation.
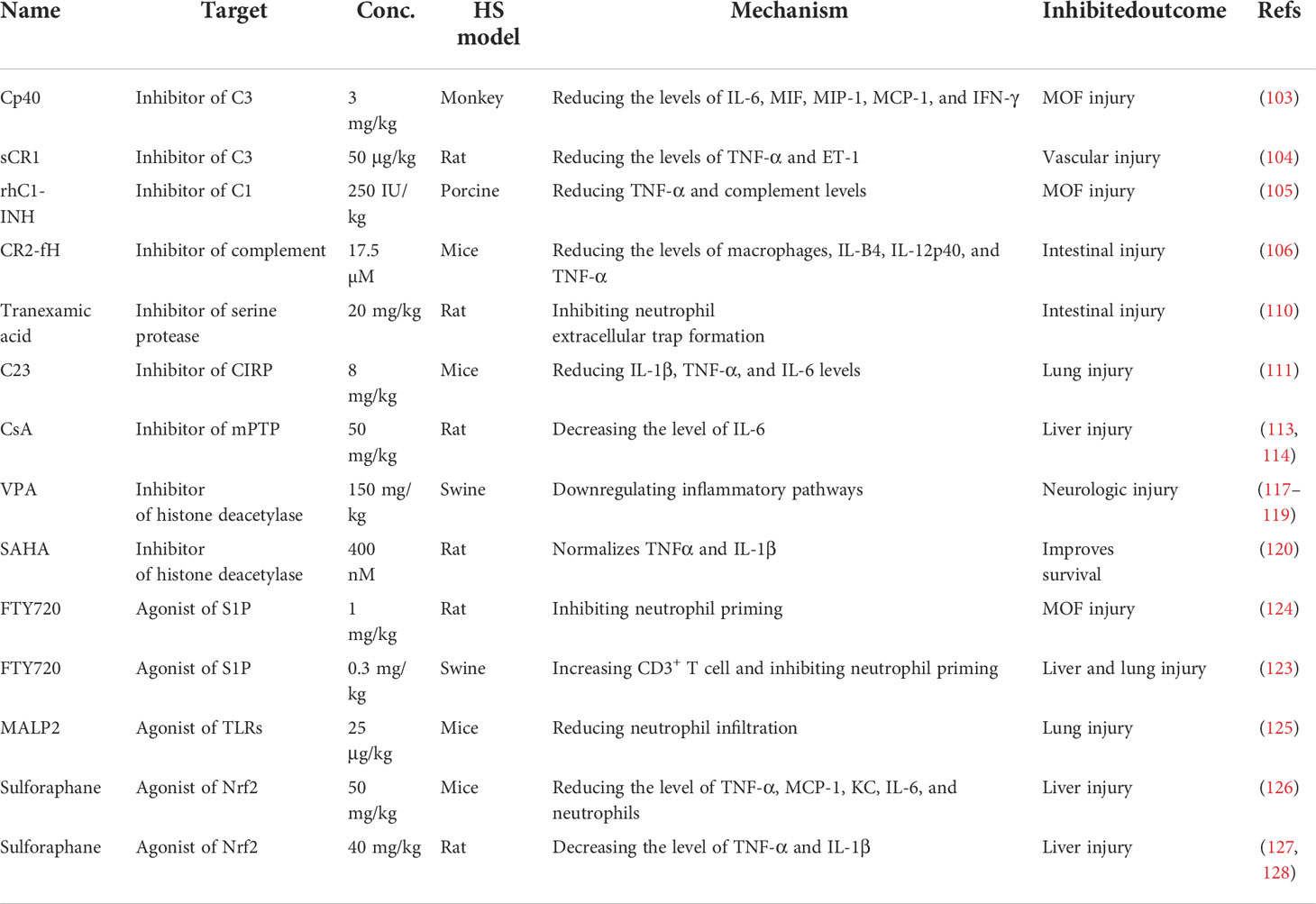
Table 3 Summary of the applications of small molecule inhibitor or agonist therapy in a hemorrhagic shock model.
Natural medicine therapy for HS
Increasing research has confirmed the role of resveratrol, a natural polyphenol widely found in plants and fruits, in improving survival and prolonging lifespan following HS by improving immune function and reducing inflammation (129, 130). Phosphorylation and acetylation on the p65 subunit of NF-κB regulate the inflammatory cascade (131). In the HS-induced rat injury model, the ratio of the phosphorylated p65 subunit of NF-κB to the unphosphorylated form demonstrated a noticeable decline following resveratrol treatment (18). Resveratrol also counteracts the increase in gene expression and plasma secretion levels of IL-2, IL-6, IL-10, TNF-α, and MIP-1α at 2 h following HS in heart tissue (18). It has been reported that the release of proinflammatory cytokines caused by HS, participates in the development of kidney injury (132). Ophiopogonin A, an effective active component extracted from ophiopogonis radix, can dose-dependently downregulate the levels of iNOS, TNF-α, IL-1β and IL-6, and decrease HS-induced renal injury (133).
Intestinal DCs play essential roles in regulating the function of the intestinal immune barrier and intestinal bacterial translocation (134). Experimental evidence suggests that treatment with allicin, a thiosulfonate extract from freshly minced garlic, can block intraintestinal bacterial translocation and reduce the permeability of the intestinal barrier by assisting the immunologic barrier function of the ML node and facilitating the maturation of DCs (135, 136). Ursolic acid, a natural pentacyclic triterpenoid carboxylic acid isolated from uncaria rhynchophylla, reduces immune-mediated lung inflammation, assists human DCs via TLRs, and accelerates the production of IFNγ by CD4+ T cells (137, 138). Additionally, ursolic acid suppresses superoxide production in activated neutrophils and restrains HS-induced hepatic and lung injuries in rats (139). The development of complications from HS is accompanied by the activation of neutrophils (77, 140). In the rat HS model, the neutrophilic oxidative stress and lung injury were restrained after administration of osthol, a natural coumarin found in traditional medicinal plants (141).
In conclusion, natural medicines, such as polyphenol, saponins, thiosulfonate, carboxylic acid, and coumarin represent an essential therapy against HS-induced injury in terms of regulating immune processes, including subsiding inflammatory cytokine release, assisting the immunologic barrier function of the ML node, facilitating DC maturation, reducing superoxide production in activated neutrophils, and attenuating neutrophil-dominated inflammation, to improve cardiac function, block intraintestinal bacterial translocation, and relieve hepatic, lung, and kidney tissue injury (Figure 5, Table 4).
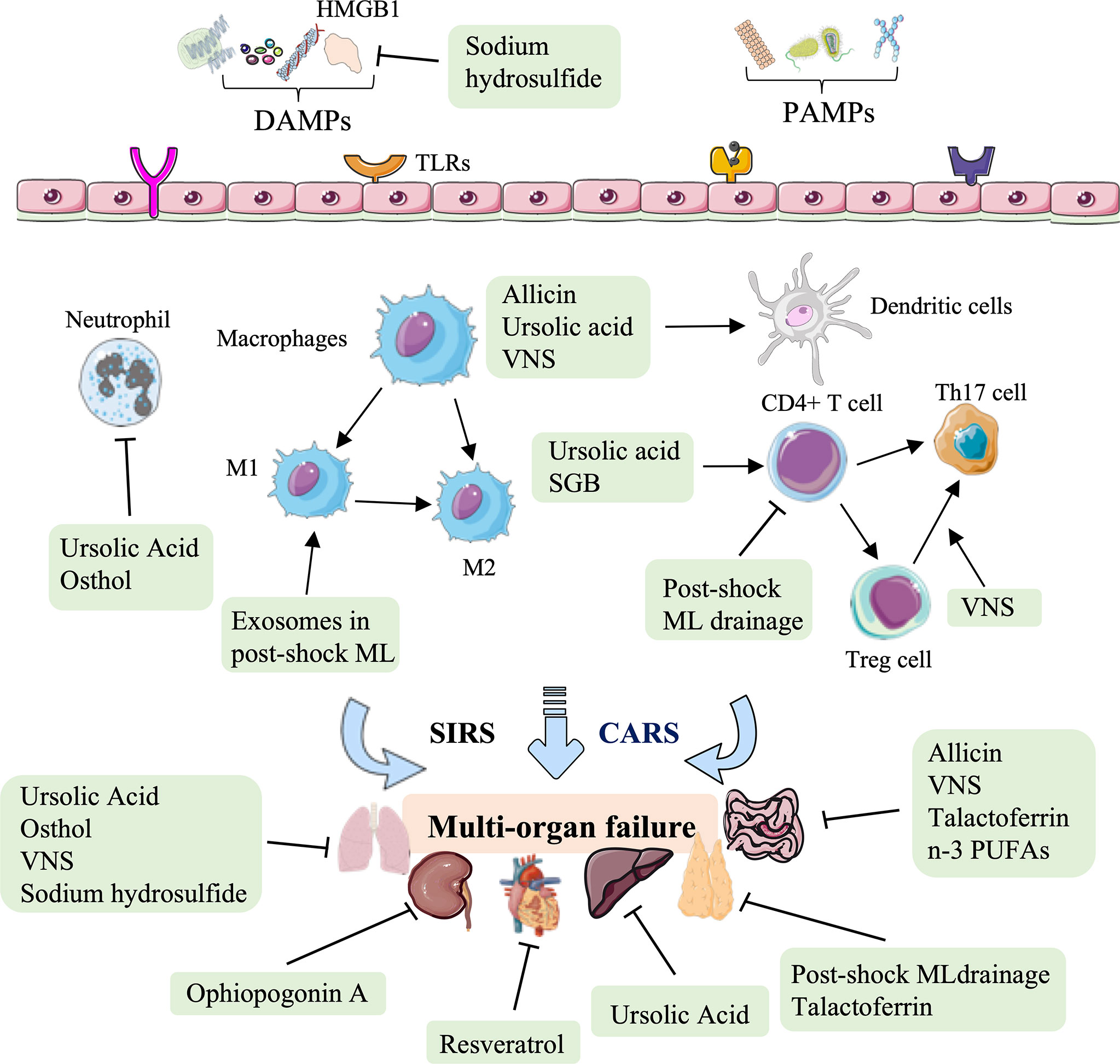
Figure 5 Overview of the natural medicine, vagus nerve stimulation, and other therapy approaches in hemorrhagic shock. Natural medicines, such as resveratrol, ophiopogonin A, allicin, ursolic acid, and osthol, act on several immune biological processes against HS-induced cardiac, liver, lung, and kidney injury, mainly rely on the inhibition of inflammatory cytokine release, modulating DC maturation and neutrophil-dominated inflammation. Other therapeutic approaches to HS, such as post-shock ML drainage, vagus nerve stimulation (VNS), stellate ganglion block (SGB), talactoferrin, n-3 polyunsaturated fatty acids (PUFAs), and hydrogen sulfide, restore the critical organs (thymus, lung, and gut) mainly by regulating the secretion of inflammatory and chemokines, and regulating the proportion of CD4 + T cell, Treg cells, Th17 cells, M1-type macrophages, and DCs.
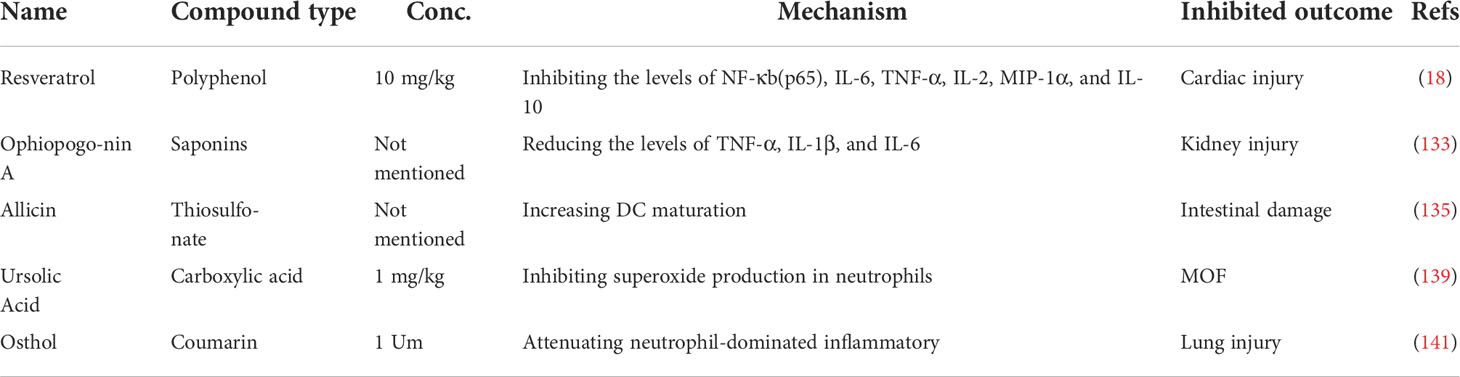
Table 4 Overview of the applications of natural medicine therapy in a hemorrhagic shock model.
Other therapies in HS
Other therapies, such as physical therapy (post-shock ML drainage, VNS, stellate ganglion block), glycoprotein, fatty acids, and inorganic compounds can also be used to modulate immunity against HS injury (Figure 5, Table 5). Previous studies have shown that the diversion of the ML or lymphatic duct ligation can reduce vascular permeability, subside systemic neutrophil priming, and decrease lung injury in HS models (143). It has been recently shown that post-shock ML drainage can decrease the levels of the T lymphocyte subgroup, including the population of CD3+ cells, CD3+ cells, CD4+ cells, and CD4+CD25+ cells, and reduce IFN-γ and IL-4 secretion in the HS rat model at 3 h after resuscitation, which suggests that post-shock ML drainage can markedly improve hyperimmunity occurred at early stages (142). Conversely, exosomes isolated from post-shock ML significantly increase lung injury by recruitment of inflammatory cells to the alveolar space and lung parenchyma, inducing mRNA expression of NF-κB, iNOS, TNF-α, and CINC-1 during HS (150).

Table 5 Summary of the applications of physical therapy, glycoprotein, fatty acids, and inorganic compounds in the hemorrhagic shock model.
The continuous migration of CD103+ DCs from the intestine to the ML nodes is considered to induce Treg cell maturation and promote tolerance to intestinal inflammation (151, 152). The balance of Treg and Th17 cells determines the intestinal tolerance to inflammation and immune response (153). Previous studies have demonstrated that VNS prevents HS-induced impairment in intestinal blood flow, alters the DC profile, and prevents incompleteness of the gut barrier in the ML (154–158). Additionally, Koji Morishita et al. reported that VNS could promote tolerance to HS-induced inflammation by increasing the CD103+ DC population in the ML and facilitating the ratio of Treg cells to Th17 cells (143). VNS has also been shown to increase the level of IL-10 and decrease HS-induced lung and gut barrier injury, with a marked decrease in the mRNA level of TNF-α, IL-6, NF-κB, and MPO (144, 145). Previous studies have shown that stellate ganglion block (SGB), a standard method of blocking sympathetic nerves, can reduce intestinal barrier dysfunction and prolong the survival time in the HS rat model (159). The latest research shows that SGB administration significantly normalized the population of CD4+ T cells and the level of IL-2, IL-4, and TNFα-induced protein 8 like 2 (TIPE2) in the development of HS (146).
Lactoferrin, as a pleiotropic glycoprotein, was proven to limit HS-induced gut injury and inhibit the biological activity of ML by enhancing the gastrointestinal barrier and assisting mucosal immunity (147). Talactoferrin, a unique recombinant form of human lactoferrin and an oral DC cell-mediated immunotherapy, has demonstrated safety and preliminary efficacy in clinical trials (160–162). It has been recently shown that talactoferrin (1000 mg/kg/day) pretreatment 5 d before being subjected to HS injury has gut-protective effects by reducing the respiratory burst activity of lymph (147). Intestinal mucosa innate immunity is involved in removing pathogenic bacteria and alleviating intestinal injury (163). Feng Tian et al. reported that n-3 polyunsaturated fatty acids (PUFAs) could restore the function of the intestinal barrier by improving the innate immunity of the intestinal mucosa, increasing the expression of lysozyme, mucin 2, and IL-4, and stabilizing the intestinal microbiota in mice after HS (148). An early study revealed that hydrogen sulfide could increase survival in rodent models of lethal hemorrhage (164). Moreover, Dunquan Xu et al. demonstrated that sodium hydrosulfide administration could protect lungs against HS injury by suppressing the levels of IL-6, TNF-α, and HMGB1 in rat bronchoalveolar lavage fluid (149).
In summary, increasing therapeutic approaches to HS have been verified, focusing on various mechanisms involving innate immunity. These therapeutic approaches restore the thymus, lung, and gut. In addition to regulating the secretion of inflammatory cytokines and chemokines (IFN-γ, IL-4, NF-κB, iNOS, TNF-α, IL-10, IL-6, IL-2, and CINC-1), these novel therapeutic options for treating HS to restore critical organ function mainly rely on regulating the population of innate immune cells, involving T lymphocyte subgroup, CD4 + T cell, Treg cells, monocytes, Th17 cells and DC cells (Figure 5, Table 5).
Clinical opportunities of innate immune modulation in HS
Although some attempts at targeting innate immunity against HS injury and subsequent organ damage in preclinical models have been successful, only a few clinical trials have evaluated the treatment strategies with immune-related indicators as primary or second outcome measures in patients with HS.
Dexmedetomidine (Dex; α2 adrenergic receptor agonist) targeted activation of α2 receptors can produce sedative, analgesic, antisympathetic, and hemodynamic effects (165). New clinical evidence indicates that early intervention with Dex can effectively prevent postoperative renal insufficiency or renal failure and improve microcirculation in patients with HS requiring surgery, mainly by inhibiting the release of oxygen free radicals, IL-6, and IL-8 (166). Another promising therapeutic compound is ulinastatin, a glycoprotein derived from human urine; combined with thymosin α1, ulinastatin improves the survival rate of patients with bacterial infection by significantly increasing the CD4+CD8+ population and restoring the balance between proinflammatory mediators (TNFα, IL-1β, IL-6, and IL-8) and anti-inflammatory cytokines (IL-4 and IL-10) (167). Park et al. reported that ulinastatin administration (300,000 IU) neutralizes the serum polymorphonuclear leukocyte elastase (PMNE) levels and decreases the secretion of TNF-α and IL-6 in trauma patients with HS at 48 h after administration (168). Pre-clinical studies have certificated the benefit of estrogen in reducing MOF injury and mortality in HS (169, 170). More importantly, the levels of Treg cell, monocytes, and inflammatory factors were significantly balanced in patients with HS after receiving estrogen treatment (171). Conversely, the results of one clinical study on the effect of remote ischemic conditioning on trauma patients with HS using immune regulation (neutrophil activity and plasma inflammatory factor expression) as the primary outcome measures were disappointing, limited by prolonged emergency transport time and delayed application of therapy (ClinicalTrials.gov Identifier: NCT02071290). One therapeutic method for HS injury currently under clinical investigation is hypertonic resuscitation, with the primary outcome measures being neutrophil activation, coagulation parameters, and monocyte activation (ClinicalTrials.gov Identifier: NCT00750997).
Most of the clinical studies of HS therapy have focused on resuscitation fluid and modulators (nitroglycerine, polydatin, vasopressin, estrogen, and the combination of norepinephrine with octreotide) using hospital admission rate, urinary output, blood pressure, heart rate, Glasgow coma scale value, microcirculatory flow index, perfusion index, mean arterial pressure, survival, or organ dysfunction as the primary outcome measures (ClinicalTrials.gov Identifier: NCT01780129, NCT03891849, NCT03235921, NCT01433276, NCT00379522, NCT00973102) (172–175). There remains an immense need to validate these promising strategies targeting innate immunity against HS injury in non-human primate models, organoid models, and clinical patients.
Conclusion and prospects
Increasing experimental and clinical evidence has contributed to a profound understanding of the pathophysiology of HS injuries in recent years. Furthermore, regulation of innate immunity is recognized as an attractive pharmacological target offering encouraging future directions for the R&D of novel therapeutics. In this regard, numerous strategies, including MSCs, exosomes, genetic approaches, antibody therapy, small molecule inhibitors, and natural medicine, have been successfully employed for protection from HS damage and MOF in rodents, porcine, and non-human primate models. The immunomodulatory mechanisms of therapeutic approaches in HS injury discussed above are not only reflected in the regulation of inflammatory cytokines and chemokines but also in the balance of complement, DCs, macrophages polarization, T lymphocyte differentiation, and neutrophil infiltration.
The systematic and comprehensive research focus on HS injury and innate immunity regulation has led to many advances; however, gaps in the translation from basic research to clinic capability remain. As immune activation and immunosuppression are inseparable and sequential during the development of MOF in HS, the time point of starting and stopping therapeutic interventions is crucial. Furthermore, reliable monitoring of the remaining immunomodulatory functions of the intervening strategies within the HS-induced immunity response cascade are equally important. Ongoing studies should accelerate the progression of the most promising strategy targeting innate immunity to clinical trials in HS injury. Overall, the modulation of the innate immune response by specific intervening strategies might provide the key to closing the cascading damage resulting from the vicious danger response after HS injury.
Author contributions
QH, DZ, and XL conceived and designed the review. QH wrote the first draft of the manuscript. DZ and XL wrote sections of the manuscript. SG and YY conceived and drafted the figures. YW and JL guided part of the manuscript. JC and CG critically revised the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82104432 and U19A2013), and the Science and Technology Development Plan Project of Jilin Province (Grant No. 202002053JC and 20200201419JC).
Conflict of interest
Author SG is employed by JX Pharmaceutical New Drug Development Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Halmin M, Chiesa F, Vasan SK, Wikman A, Norda R, Rostgaard K, et al. Epidemiology of massive transfusion: A binational study from Sweden and Denmark. Crit Care Med (2016) 443:468–77. doi: 10.1097/CCM.0000000000001410
3. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. Lancet (London England) (2012) 3809859:2095–128. doi: 10.1016/S0140-67361261728-0
4. Jiang SY, Zhao YY, Zhao XG. Potential role of therapeutic hypothermia in the salvage of traumatic hemorrhagic shock. Crit Care (2013) 173:318. doi: 10.1186/cc12559
5. Dutton RP. Haemostatic resuscitation. Br J Anaesthesia (2012) 109 Suppl 1:i39–46. doi: 10.1093/bja/aes389
6. Sperry JL, Guyette FX, Brown JB, Yazer MH, Triulzi DJ, Early-Young BJ, et al. Prehospital plasma during air medical transport in trauma patients at risk for hemorrhagic shock. New Engl J Med (2018) 3794:315–26. doi: 10.1056/NEJMoa1802345
7. Mitra B, Gabbe BJ, Kaukonen KM, Olaussen A, Cooper DJ, Cameron PA. Long-term outcomes of patients receiving a massive transfusion after trauma. Shock (2014) 424:307–12. doi: 10.1097/SHK.0000000000000219
8. Huber-Lang M, Gebhard F, Schmidt C, Palmer A, Denk S, Wiegner R. Complement therapeutic strategies in trauma, hemorrhagic shock and systemic inflammation - closing pandora's box? Semin Immunol (2016) 283:278–84. doi: 10.1016/j.smim.2016.04.005
9. Valade G, Libert N, Martinaud C, Vicaut E, Banzet S, Peltzer J. Therapeutic potential of mesenchymal stromal cell-derived extracellular vesicles in the prevention of organ injuries induced by traumatic hemorrhagic shock. Front Immunol (2021) 12:749659. doi: 10.3389/fimmu.2021.749659
10. von Burg N, Chappaz S, Baerenwaldt A, Horvath E, Bose Dasgupta S, Ashok D, et al. Activated group 3 innate lymphoid cells promote T-Cell-Mediated immune responses. Proc Natl Acad Sci USA. (2014) 11135:12835–40. doi: 10.1073/pnas.1406908111
11. Bortolotti P, Faure E, Kipnis E. Inflammasomes in tissue damages and immune disorders after trauma. Front Immunol (2018) 9:1900. doi: 10.3389/fimmu.2018.01900
12. Manson J, Hoffman R, Chen S, Ramadan MH, Billiar TR. Innate-like lymphocytes are immediate participants in the hyper-acute immune response to trauma and hemorrhagic shock. Front Immunol (2019) 10:1501. doi: 10.3389/fimmu.2019.01501
13. Wilson N, Wall J, Naganathar V, Brohi K, De'Ath H. Mechanisms involved in secondary cardiac dysfunction in animal models of trauma and hemorrhagic shock. Shock (Augusta Ga) (2017) 484:401–10. doi: 10.1097/shk.0000000000000882
14. Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial n-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol (2015) 3087:H768–77. doi: 10.1152/ajpheart.00779.2014
15. Itagaki K, Kaczmarek E, Lee YT, Tang IT, Isal B, Adibnia Y, et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PloS One (2015) 103:e0120549. doi: 10.1371/journal.pone.0120549
16. Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated levels of plasma mitochondrial DNA damps are linked to clinical outcome in severely injured human subjects. Ann Surg (2013) 2584:591–6. doi: 10.1097/SLA.0b013e3182a4ea46
17. Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W, et al. Cold-inducible rna-binding protein (Cirp) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med (2013) 1911:1489–95. doi: 10.1038/nm.3368
18. Lu S, Aguilar A, Subramani K, Poulose N, Ayub A, Raju R. Alteration of cytokine profile following hemorrhagic shock. Cytokine (2016) 81:35–8. doi: 10.1016/j.cyto.2016.01.022
19. Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol (2018) 194:327–41. doi: 10.1038/s41590-018-0064-8
20. Ma KC, Schenck EJ, Pabon MA, Choi AMK. The role of danger signals in the pathogenesis and perpetuation of critical illness. Am J Respir Crit Care Med (2018) 1973:300–9. doi: 10.1164/rccm.201612-2460PP
21. Pantalone D, Bergamini C, Martellucci J, Alemanno G, Bruscino A, Maltinti G, et al. The role of damps in burns and hemorrhagic shock immune response: Pathophysiology and clinical issues. Review Int J Mol Sci (2021) 2213:7020. doi: 10.3390/ijms22137020
22. Xiang M, Yuan Y, Fan L, Li Y, Li A, Yin L, et al. Role of macrophages in mobilization of hematopoietic progenitor cells from bone marrow after hemorrhagic shock. Shock (2012) 375:518–23. doi: 10.1097/SHK.0b013e318249b81d
23. Mehta AK, Kadel S, Townsend MG, Oliwa M, Guerriero JL. Macrophage biology and mechanisms of immune suppression in breast cancer. Front Immunol (2021) 12:643771. doi: 10.3389/fimmu.2021.643771
24. Gordon S, Martinez FO. Alternative activation of macrophages: Mechanism and functions. Immunity (2010) 325:593–604. doi: 10.1016/j.immuni.2010.05.007
25. Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukoc Biol (2013) 936:875–81. doi: 10.1189/jlb.1012512
26. Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P, et al. Aging and cancer: The role of macrophages and neutrophils. Ageing Res Rev (2017) 36:105–16. doi: 10.1016/j.arr.2017.03.008
27. Keeley T, Costanzo-Garvey DL, Cook LM. Unmasking the many faces of tumor-associated neutrophils and macrophages: Considerations for targeting innate immune cells in cancer. Trends Cancer (2019) 512:789–98. doi: 10.1016/j.trecan.2019.10.013
28. Scapini P, Cassatella MA. Social networking of human neutrophils within the immune system. Blood (2014) 1245:710–9. doi: 10.1182/blood-2014-03-453217
29. Zettel K, Korff S, Zamora R, Morelli AE, Darwiche S, Loughran PA, et al. Toll-like receptor 4 on both myeloid cells and dendritic cells is required for systemic inflammation and organ damage after hemorrhagic shock with tissue trauma in mice. Front Immunol (2017) 8:1672. doi: 10.3389/fimmu.2017.01672
30. Namas RA, Mi Q, Namas R, Almahmoud K, Zaaqoq AM, Abdul-Malak O, et al. Insights into the role of chemokines, damage-associated molecular patterns, and lymphocyte-derived mediators from computational models of trauma-induced inflammation. Antioxid Redox Signal (2015) 2317:1370–87. doi: 10.1089/ars.2015.6398
31. Wang P, Jiang LN, Wang C, Li Y, Yin M, Du HB, et al. Estradiol-induced inhibition of endoplasmic reticulum stress normalizes splenic Cd4 + T lymphocytes following hemorrhagic shock. Sci Rep (2021) 111:7508. doi: 10.1038/s41598-021-87159-1
32. Lohani N, Rajeswari MR. Dichotomous life of DNA binding high mobility group Box1 protein in human health and disease. Curr Protein Pept Sci (2016) 178:762–75. doi: 10.2174/1389203717666160226145217
33. Liu GQ, Zuo XH, Jiang LN, Zhang YP, Zhang LM, Zhao ZG, et al. Inhibitory effect of post-hemorrhagic shock mesenteric lymph drainage on the Hmgb1 and rage in mouse kidney. Renal failure (2016) 381:131–6. doi: 10.3109/0886022X.2015.1105026
34. Zhang K, Jin Y, Lai D, Wang J, Wang Y, Wu X, et al. Rage-induced Ilc2 expansion in acute lung injury due to haemorrhagic shock. Thorax (2020) 753:209–19. doi: 10.1136/thoraxjnl-2019-213613
35. Zhang H, Jiang Z, Zhang L. Dual effect of T helper cell 17 (Th17) and regulatory T cell (Treg) in liver pathological process: From occurrence to end stage of disease. Int Immunopharmacol (2019) 69:50–9. doi: 10.1016/j.intimp.2019.01.005
36. Wang X, Jiang Z, Xing M, Fu J, Su Y, Sun L, et al. Interleukin-17 mediates triptolide-induced liver injury in mice. Food Chem Toxicol an Int J published Br Ind Biol Res Assoc (2014) 71:33–41. doi: 10.1016/j.fct.2014.06.004
37. Gupta DL, Bhoi S, Mohan T, Galwnkar S, Rao DN. Coexistence of Th1/Th2 and Th17/Treg imbalances in patients with post traumatic sepsis. Cytokine (2016) 88:214–21. doi: 10.1016/j.cyto.2016.09.010
38. Yao F, Lu YQ, Jiang JK, Gu LH, Mou HZ. Immune recovery after fluid resuscitation in rats with severe hemorrhagic shock. J Zhejiang Univ Sci B (2017) 185:402–9. doi: 10.1631/jzus.B1600370
39. Zhang J, Zhang Y, Xu T, Pan SJ, Nie G, Miao XY, et al. Severe traumatic hemorrhagic shock induces compromised immune barrier function of the mesenteric lymph node leading to an increase in intestinal bacterial translocation. Am J Transl Res (2017) 95:2363–73. doi: 10.1002/ptr.5847
40. Sauaia A, Moore EE, Johnson JL, Chin TL, Banerjee A, Sperry JL, et al. Temporal trends of postinjury multiple-organ failure: Still resource intensive, morbid, and lethal. J Trauma Acute Care Surg (2014) 763:582–92. doi: 10.1097/TA.0000000000000147
41. Veith NT, Histing T, Menger MD, Pohlemann T, Tschernig T. Helping Prometheus: Liver protection in acute hemorrhagic shock. Ann Trans Med (2017) 510:206. doi: 10.21037/atm.2017.03.109
42. De Luca M, Aiuti A, Cossu G, Parmar M, Pellegrini G, Robey PG. Advances in stem cell research and therapeutic development. Nat Cell Biol (2019) 217:801–11. doi: 10.1038/s41556-019-0344-z
43. Johnson TC, Siegel D. Directing stem cell fate: The synthetic natural product connection. Chem Rev (2017) 11718:12052–86. doi: 10.1021/acs.chemrev.7b00015
44. Gore AV, Bible LE, Song K, Livingston DH, Mohr AM, Sifri ZC. Mesenchymal stem cells increase T-regulatory cells and improve healing following trauma and hemorrhagic shock. J Trauma Acute Care Surg (2015) 791:48–52. doi: 10.1097/TA.0000000000000681
45. Gore A, Bible L, Livingston D, Mohr A, Sifri Z. Mesenchymal stem cells enhance lung recovery after injury, shock, and chronic stress. Surgery (2016) 1595:1430–5. doi: 10.1016/j.surg.2015.12.006
46. Pati S, Gerber MH, Menge TD, Wataha KA, Zhao Y, Baumgartner JA, et al. Bone marrow derived mesenchymal stem cells inhibit inflammation and preserve vascular endothelial integrity in the lungs after hemorrhagic shock. PloS One (2011) 69:e25171. doi: 10.1371/journal.pone.0025171
47. Potter DR, Miyazawa BY, Gibb SL, Deng X, Togaratti PP, Croze RH, et al. Mesenchymal stem cell-derived extracellular vesicles attenuate pulmonary vascular permeability and lung injury induced by hemorrhagic shock and trauma. J Trauma Acute Care Surg (2018) 842:245–56. doi: 10.1097/TA.0000000000001744
48. Bambakidis T, Dekker SE, Williams AM, Biesterveld BE, Bhatti UF, Liu B, et al. Early treatment with a single dose of mesenchymal stem cell derived extracellular vesicles modulates the brain transcriptome to create neuroprotective changes in a porcine model of traumatic brain injury and hemorrhagic shock. Shock (2022) 572:281–90. doi: 10.1097/SHK.0000000000001889
49. Zhang Y, Zhang X, Zhang H, Song P, Pan W, Xu P, et al. Viamesenchymal stem cells derived extracellular vesicles alleviate traumatic hemorrhagic shock induced hepatic injury il-10/Ptpn22-Mediated M2 kupffer cell polarization. Front Immunol (2021) 12:811164. doi: 10.3389/fimmu.2021.811164
50. Dias VL, Braga KAO, Nepomuceno NA, Ruiz LM, Perez JDR, Correia AT, et al. Soluble factors of mesenchimal stem cells (Fs-msc) as a potential tool to reduce inflammation in donor's lungs after hypovolemic shock. Jornal brasileiro pneumologia publicacao oficial da Sociedade Bras Pneumologia e Tisilogia (2021) 474:e20200452. doi: 10.36416/1806-3756/e20200452
51. Williams AM, Wu Z, Bhatti UF, Biesterveld BE, Kemp MT, Wakam GK, et al. Early single-dose exosome treatment improves neurologic outcomes in a 7-day swine model of traumatic brain injury and hemorrhagic shock. J Trauma Acute Care Surg (2020) 892:388–96. doi: 10.1097/TA.0000000000002698
52. Williams AM, Higgins GA, Bhatti UF, Biesterveld BE, Dekker SE, Kathawate RG, et al. Early treatment with exosomes following traumatic brain injury and hemorrhagic shock in a swine model promotes transcriptional changes associated with neuroprotection. J Trauma Acute Care Surg (2020) 893:536–43. doi: 10.1097/TA.0000000000002815
53. Aussel C, Baudry N, Grosbot M, Caron C, Vicaut E, Banzet S, et al. Il-1beta primed mesenchymal stromal cells moderate hemorrhagic shock-induced organ injuries. Stem Cell Res Ther (2021) 121:438. doi: 10.1186/s13287-021-02505-4
54. Ashmwe M, Penzenstadler C, Bahrami A, Klotz A, Jafarmadar M, Banerjee A, et al. Secretome conveys the protective effects of ascs: Therapeutic potential following hemorrhagic shock? Shock (2018) 504:442–8. doi: 10.1097/SHK.0000000000001047
55. Cohen J, Danise M, Machan J, Zhao R, Lefort C. Murine myeloid progenitors attenuate immune dysfunction induced by hemorrhagic shock. Stem Cell Rep (2021) 162:324–36. doi: 10.1016/j.stemcr.2020.12.014
56. Spees JL, Lee RH, Gregory CA. Mechanisms of mesenchymal Stem/Stromal cell function. Stem Cell Res Ther (2016) 71:125. doi: 10.1186/s13287-016-0363-7
57. Ohkura N, Sakaguchi S. Transcriptional and epigenetic basis of treg cell development and function: Its genetic anomalies or variations in autoimmune diseases. Cell Res (2020) 306:465–74. doi: 10.1038/s41422-020-0324-7
58. Minshawi F, Lanvermann S, McKenzie E, Jeffery R, Couper K, Papoutsopoulou S, et al. The generation of an engineered interleukin-10 protein with improved stability and biological function. Front Immunol (2020) 11:1794. doi: 10.3389/fimmu.2020.01794
59. Kobbe P, Lichte P, Schreiber H, Reiss LK, Uhlig S, Pape HC, et al. Inhalative il-10 attenuates pulmonary inflammation following hemorrhagic shock without major alterations of the systemic inflammatory response. Mediators Inflammation (2012) 2012:512974. doi: 10.1155/2012/512974
60. Kupani M, Pandey RK, Mehrotra S. Neutrophils and visceral leishmaniasis: Impact on innate immune response and cross-talks with macrophages and dendritic cells. J Cell Physiol (2021) 2364:2255–67. doi: 10.1002/jcp.30029
61. Kraus RF, Gruber MA. Neutrophils-from bone marrow to first-line defense of the innate immune system. Front Immunol (2021) 12:767175. doi: 10.3389/fimmu.2021.767175
62. Briggs GD, Lemmert K, Lott NJ, de Malmanche T, Balogh ZJ. Biomarkers to guide the timing of surgery: Neutrophil and monocyte l-selectin predict postoperative sepsis in orthopaedic trauma patients. J Clin Med (2021) 1010:2207. doi: 10.3390/jcm10102207
63. Schietroma M, Pessia B, Mattei A, Romano L, Giuliani A, Carlei F. Temperature-Neutrophils-Multiple organ failure grading for complicated intra-abdominal infections. Surg infections (2020) 211:69–74. doi: 10.1089/sur.2019.092
64. Ionescu L, Byrne RN, van Haaften T, Vadivel A, Alphonse RS, Rey-Parra GJ, et al. Stem cell conditioned medium improves acute lung injury in mice: In vivo evidence for stem cell paracrine action. Am J Physiol Lung Cell Mol Physiol (2012) 30311:L967–77. doi: 10.1152/ajplung.00144.2011
65. Carrero R, Cerrada I, Lledo E, Dopazo J, Garcia-Garcia F, Rubio MP, et al. Il1beta induces mesenchymal stem cells migration and leucocyte chemotaxis through nf-kappab. Stem Cell Rev Rep (2012) 83:905–16. doi: 10.1007/s12015-012-9364-9
66. Fan H, Zhao G, Liu L, Liu F, Gong W, Liu X, et al. Pre-treatment with il-1beta enhances the efficacy of msc transplantation in dss-induced colitis. Cell Mol Immunol (2012) 96:473–81. doi: 10.1038/cmi.2012.40
67. Zhang Y, Zhang J, Korff S, Ayoob F, Vodovotz Y, Billiar TR. Delayed neutralization of interleukin 6 reduces organ injury, selectively suppresses inflammatory mediator, and partially normalizes immune dysfunction following trauma and hemorrhagic shock. Shock (2014) 423:218–27. doi: 10.1097/SHK.0000000000000211
68. Halbgebauer R, Karasu E, Braun CK, Palmer A, Braumuller S, Schultze A, et al. Thirty-Eight-Negative kinase 1 is a mediator of acute kidney injury in experimental and clinical traumatic hemorrhagic shock. Front Immunol (2020) 11:2081. doi: 10.3389/fimmu.2020.02081
69. Barayan D, Abdullahi A, Vinaik R, Knuth CM, Auger C, Jeschke MG. Interleukin-6 blockade, a potential adjunct therapy for post-burn hypermetabolism. FASEB J (2021) 355:e21596. doi: 10.1096/fj.202100388R
70. Zhang H, Besner G, Feng J. Antibody blockade of mucosal addressin cell adhesion molecule-1 attenuates proinflammatory activity of mesenteric lymph after hemorrhagic shock and resuscitation. Surgery (2016) 1595:1449–60. doi: 10.1016/j.surg.2015.12.013
71. August KJ, Chiang KY, Qayed M, Dulson A, Worthington-White D, Cole CR, et al. Relative defects in mucosal immunity predict acute graft-Versus-Host disease. Biol Blood marrow Transplant J Am Soc Blood Marrow Transplant (2014) 207:1056–9. doi: 10.1016/j.bbmt.2014.03.012
72. Habtezion A, Nguyen LP, Hadeiba H, Butcher EC. Leukocyte trafficking to the small intestine and colon. Gastroenterology (2016) 1502:340–54. doi: 10.1053/j.gastro.2015.10.046
73. Mintz MA, Felce JH, Chou MY, Mayya V, Xu Y, Shui JW, et al. The hvem-btla axis restrains T cell help to germinal center b cells and functions as a cell-extrinsic suppressor in lymphomagenesis. Immunity (2019) 512:310–23.e7. doi: 10.1016/j.immuni.2019.05.022
74. Jones A, Bourque J, Kuehm L, Opejin A, Teague RM, Gross C, et al. Immunomodulatory functions of btla and hvem govern induction of extrathymic regulatory T cells and tolerance by dendritic cells. Immunity (2016) 455:1066–77. doi: 10.1016/j.immuni.2016.10.008
75. Cheng T, Bai J, Chung CS, Chen Y, Biron BM, Ayala A. Enhanced innate inflammation induced by anti-btla antibody in dual insult model of hemorrhagic Shock/Sepsis. Shock (2016) 451:40–9. doi: 10.1097/SHK.0000000000000479
76. Namas RA, Vodovotz Y, Almahmoud K, Abdul-Malak O, Zaaqoq A, Namas R, et al. Temporal patterns of circulating inflammation biomarker networks differentiate susceptibility to nosocomial infection following blunt trauma in humans. Ann Surg (2016) 2631:191–8. doi: 10.1097/SLA.0000000000001001
77. Xu J, Guardado J, Hoffman R, Xu H, Namas R, Vodovotz Y, et al. Il33-mediated Ilc2 activation and neutrophil Il5 production in the lung response after severe trauma: A reverse translation study from a human cohort to a mouse trauma model. PloS Med (2017) 147:e1002365. doi: 10.1371/journal.pmed.1002365
78. Korff S, Loughran P, Cai C, Fan J, Elson G, Shang L, et al. Tlr2 on bone marrow and non-bone marrow derived cells regulates inflammation and organ injury in cooperation with Tlr4 during resuscitated hemorrhagic shock. Shock (2016) 465:519–26. doi: 10.1097/SHK.0000000000000650
79. Goering J, Pope MR, Fleming SD. Tlr2 regulates complement-mediated inflammation induced by blood loss during hemorrhage. Shock (2016) 451:33–9. doi: 10.1097/SHK.0000000000000477
80. Aziz M, Brenner M, Wang P. Extracellular cirp (Ecirp) and inflammation. J Leukoc Biol (2019) 1061:133–46. doi: 10.1002/JLB.3MIR1118-443R
81. Idrovo JP, Jacob A, Yang WL, Wang Z, Yen HT, Nicastro J, et al. A deficiency in cold-inducible rna-binding protein accelerates the inflammation phase and improves wound healing. Int J Mol Med (2016) 372:423–8. doi: 10.3892/ijmm.2016.2451
82. Ishikawa H, Ma Z, Barber GN. Sting regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 4617265:788–92. doi: 10.1038/nature08476
83. Ishikawa H, Barber GN. Sting is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 4557213:674–8. doi: 10.1038/nature07317
84. Chen K, Cagliani J, Aziz M, Tan C, Brenner M, Wang P. Extracellular cirp activates sting to exacerbate hemorrhagic shock. JCI Insight (2021) 614:e143715. doi: 10.1172/jci.insight.143715
85. Huang X, Chen Y, Chung CS, Yuan Z, Monaghan SF, Wang F, et al. Identification of B7-H1 as a novel mediator of the innate Immune/Proinflammatory response as well as a possible myeloid cell prognostic biomarker in sepsis. J Immunol (Baltimore Md 1950) (2014) 1923:1091–9. doi: 10.4049/jimmunol.1302252
86. Tang L, Bai J, Chung CS, Lomas-Neira J, Chen Y, Huang X, et al. Active players in resolution of Shock/Sepsis induced indirect lung injury: Immunomodulatory effects of tregs and pd-1. J Leukoc Biol (2014) 965:809–20. doi: 10.1189/jlb.4MA1213-647RR
87. Monaghan SF, Thakkar RK, Tran ML, Huang X, Cioffi WG, Ayala A, et al. Programmed death 1 expression as a marker for immune and physiological dysfunction in the critically ill surgical patient. Shock (2012) 382:117–22. doi: 10.1097/SHK.0b013e31825de6a3
88. Lomas-Neira J, Monaghan S, Huang X, Fallon E, Chung C, Ayala A. Novel role for pd-1:Pd-L1 as mediator of pulmonary vascular endothelial cell functions in pathogenesis of indirect Ards in mice. Front Immunol (2018) 9:3030. doi: 10.3389/fimmu.2018.03030
89. Battino M, Giampieri F, Pistollato F, Sureda A, de Oliveira MR, Pittala V, et al. Nrf2 as regulator of innate immunity: A molecular Swiss army knife! Biotechnol Adv (2018) 362:358–70. doi: 10.1016/j.biotechadv.2017.12.012
90. Borcherding DC, Siefert ME, Lin S, Brewington J, Sadek H, Clancy JP, et al. Clinically-approved cftr modulators rescue Nrf2 dysfunction in cystic fibrosis airway epithelia. J Clin Invest (2019) 1298:3448–63. doi: 10.1172/JCI96273
91. Zhou YQ, Mei W, Tian XB, Tian YK, Liu DQ, Ye DW. The therapeutic potential of Nrf2 inducers in chronic pain: Evidence from preclinical studies. Pharmacol Ther (2021) 225:107846. doi: 10.1016/j.pharmthera.2021.107846
92. Zhao H, Hao S, Xu H, Ma L, Zhang Z, Ni Y, et al. Protective role of nuclear factor erythroid 2-related factor 2 in the hemorrhagic shock-induced inflammatory response. Int J Mol Med (2016) 374:1014–22. doi: 10.3892/ijmm.2016.2507
93. Leung CH, Caldarone CA, Guan R, Wen XY, Ailenberg M, Kapus A, et al. Nuclear factor (Erythroid-derived 2)-like 2 regulates the hepatoprotective effects of remote ischemic conditioning in hemorrhagic shock. Antioxid Redox Signal (2019) 3014:1760–73. doi: 10.1089/ars.2018.7541
94. Long Y, Wang C, Xia C, Li X, Fan C, Zhao X, et al. Recovery of Cd226-Tigit(+)Foxp3(+) and Cd226-Tigit-Foxp3(+) regulatory T cells contributes to clinical remission from active stage in ulcerative colitis patients. Immunol Lett (2020) 218:30–9. doi: 10.1016/j.imlet.2019.12.007
95. Lozano E, Joller N, Cao Y, Kuchroo VK, Hafler DA. The Cd226/Cd155 interaction regulates the proinflammatory (Th1/Th17)/Anti-inflammatory (Th2) balance in humans. J Immunol (Baltimore Md 1950) (2013) 1917:3673–80. doi: 10.4049/jimmunol.1300945
96. Zhou S, Xie J, Yu C, Feng Z, Cheng K, Ma J, et al. Cd226 deficiency promotes glutaminolysis and alleviates mitochondria damage in vascular endothelial cells under hemorrhagic shock. FASEB J Off Publ Fed Am Societies Exp Biol (2021) 3511:e21998. doi: 10.1096/fj.202101134R
97. Neudecker V, Yuan X, Bowser JL, Eltzschig HK. Micrornas in mucosal inflammation. J Mol Med (2017) 959:935–49. doi: 10.1007/s00109-017-1568-7
98. Wu XQ, Dai Y, Yang Y, Huang C, Meng XM, Wu BM, et al. Emerging role of micrornas in regulating macrophage activation and polarization in immune response and inflammation. Immunology (2016) 1483:237–48. doi: 10.1111/imm.12608
99. Sheng X, Yang Y, Liu J, Yu J, Guo Q, Guan W, et al. Down-regulation of mir-18b-5p protects against splenic hemorrhagic shock by directly targeting hif-1α/Inos pathway. Immunobiology (2022) 2272:152188. doi: 10.1016/j.imbio.2022.152188
100. Messerer DAC, Halbgebauer R, Nilsson B, Pavenstadt H, Radermacher P, Huber-Lang M. Immunopathophysiology of trauma-related acute kidney injury. Nat Rev Nephrol (2021) 172:91–111. doi: 10.1038/s41581-020-00344-9
101. Karasu E, Nilsson B, Köhl J, Lambris J, Huber-Lang M. Targeting complement pathways in polytrauma- and sepsis-induced multiple-organ dysfunction. Front Immunol (2019) 10:543. doi: 10.3389/fimmu.2019.00543
102. Cai C, Gill R, Eum HA, Cao Z, Loughran PA, Darwiche S, et al. Complement factor 3 deficiency attenuates hemorrhagic shock-related hepatic injury and systemic inflammatory response syndrome. Am J Physiol Regulatory Integr Comp Physiol (2010) 2995:R1175–82. doi: 10.1152/ajpregu.00282.2010
103. van Griensven M, Ricklin D, Denk S, Halbgebauer R, Braun C, Schultze A, et al. Protective effects of the complement inhibitor compstatin Cp40 in hemorrhagic shock. Shock (Augusta Ga) (2019) 511:78–87. doi: 10.1097/shk.0000000000001127
104. Chen D, Song MQ, Liu YJ, Xue YK, Cheng P, Zheng H, et al. Inhibition of complement C3 might rescue vascular hyporeactivity in a conscious hemorrhagic shock rat model. Microvascular Res (2016) 105:23–9. doi: 10.1016/j.mvr.2015.12.006
105. Dalle Lucca J, Li Y, Simovic M, Pusateri A, Falabella M, Dubick M, et al. Effects of C1 inhibitor on tissue damage in a porcine model of controlled hemorrhage. Shock (Augusta Ga) (2012) 381:82–91. doi: 10.1097/SHK.0b013e31825a3522
106. Hylton DJ, Hoffman SM, Van Rooijen N, Tomlinson S, Fleming SD. Macrophage-produced il-12p70 mediates hemorrhage-induced damage in a complement-dependent manner. Shock (2011) 352:134–40. doi: 10.1097/SHK.0b013e3181ed8ec9
107. Hylton DJ, Phillips LM, Hoffman SM, Fleming SD. Hemorrhage-induced intestinal damage is complement-independent in helicobacter hepaticus-infected mice. Shock (2010) 345:467–74. doi: 10.1097/SHK.0b013e3181dc077e
108. Fleming SD, Phillips LM, Lambris JD, Tsokos GC. Complement component C5a mediates hemorrhage-induced intestinal damage. J Surg Res (2008) 1502:196–203. doi: 10.1016/j.jss.2008.02.010
109. Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in Ischemia/Reperfusion injury. J Immunol (Baltimore Md 1950) (2008) 18111:8068–76. doi: 10.4049/jimmunol.181.11.8068
110. Chu C, Yang C, Wang X, Xie T, Sun S, Liu B, et al. Early intravenous administration of tranexamic acid ameliorates intestinal barrier injury induced by neutrophil extracellular traps in a rat model of Trauma/Hemorrhagic shock. Surgery (2020) 1672:340–51. doi: 10.1016/j.surg.2019.10.009
111. Zhang F, Yang W, Brenner M, Wang P. Attenuation of hemorrhage-associated lung injury by adjuvant treatment with C23, an oligopeptide derived from cold-inducible rna-binding protein. J Trauma acute Care Surg (2017) 834:690–7. doi: 10.1097/ta.0000000000001566
112. Fenizia C, Galbiati S, Vanetti C, Vago R, Clerici M, Tacchetti C, et al. Cyclosporine a inhibits viral infection and release as well as cytokine production in lung cells by three sars-Cov-2 variants. Microbiol Spectr (2022) 101:e0150421. doi: 10.1128/spectrum.01504-21
113. Kim K, Shin J, Lee J, Jo Y, Kim M, Lee K, et al. The effect of cyclosporine a in hemorrhagic shock model of rats. J Trauma acute Care Surg (2015) 782:370–7. doi: 10.1097/ta.0000000000000511
114. Gui Q, Jiang Z, Zhang L. Insights into the modulatory role of cyclosporine a and its research advances in acute inflammation. Int Immunopharmacol (2021) 93:107420. doi: 10.1016/j.intimp.2021.107420
115. Biesterveld B, Wakam G, Kemp M, Williams A, Shamshad A, O'Connell R, et al. Histone deacetylase 6 inhibition improves survival in a swine model of lethal hemorrhage, polytrauma, and bacteremia. J Trauma acute Care Surg (2020) 895:932–9. doi: 10.1097/ta.0000000000002677
116. Nikolian V, Dennahy I, Weykamp M, Williams A, Bhatti U, Eidy H, et al. Isoform 6-selective histone deacetylase inhibition reduces lesion size and brain swelling following traumatic brain injury and hemorrhagic shock. J Trauma acute Care Surg (2019) 862:232–9. doi: 10.1097/ta.0000000000002119
117. Georgoff P, Nikolian V, Higgins G, Chtraklin K, Eidy H, Ghandour M, et al. Valproic acid induces prosurvival transcriptomic changes in swine subjected to traumatic injury and hemorrhagic shock. J Trauma Acute Care Surg (2018) 844:642–9. doi: 10.1097/ta.0000000000001763
118. Bambakidis T, Dekker S, Sillesen M, Liu B, Johnson C, Jin G, et al. Resuscitation with valproic acid alters inflammatory genes in a porcine model of combined traumatic brain injury and hemorrhagic shock. J Neurotrauma (2016) 3316:1514–21. doi: 10.1089/neu.2015.4163
119. Chang P, Williams A, Bhatti U, Biesterveld B, Liu B, Nikolian V, et al. Valproic acid and neural apoptosis, inflammation, and degeneration 30 days after traumatic brain injury, hemorrhagic shock, and polytrauma in a swine model. J Am Coll Surgeons (2019) 2283:265–75. doi: 10.1016/j.jamcollsurg.2018.12.026
120. Sailhamer E, Li Y, Smith E, Shuja F, Shults C, Liu B, et al. Acetylation: A novel method for modulation of the immune response following Trauma/Hemorrhage and inflammatory second hit in animals and humans. Surgery (2008) 1442:204–16. doi: 10.1016/j.surg.2008.03.034
121. Mazzola MA, Raheja R, Murugaiyan G, Rajabi H, Kumar D, Pertel T, et al. Identification of a novel mechanism of action of fingolimod (Fty720) on human effector T cell function through tcf-1 upregulation. J Neuroinflamm (2015) 12:245. doi: 10.1186/s12974-015-0460-z
122. Luessi F, Kraus S, Trinschek B, Lerch S, Ploen R, Paterka M, et al. Fty720 (Fingolimod) treatment tips the balance towards less immunogenic antigen-presenting cells in patients with multiple sclerosis. Multiple Sclerosis (Houndmills Basingstoke England) (2015) 2114:1811–22. doi: 10.1177/1352458515574895
123. Hawksworth J, Graybill J, Brown T, Wallace S, Davis T, Tadaki D, et al. Lymphocyte modulation with Fty720 improves hemorrhagic shock survival in swine. PloS One (2012) 74:e34224. doi: 10.1371/journal.pone.0034224
124. Bonitz J, Son J, Chandler B, Tomaio J, Qin Y, Prescott L, et al. A sphingosine-1 phosphate agonist (Fty720) limits Trauma/Hemorrhagic shock-induced multiple organ dysfunction syndrome. Shock (Augusta Ga) (2014) 425:448–55. doi: 10.1097/shk.0000000000000227
125. Xu D, Horst K, Wang W, Luo P, Shi Y, Tschernig T, et al. The influence of macrophage-activating lipopeptide 2 (Malp-2) on local and systemic inflammatory response in a murine two-hit model of hemorrhagic shock and subsequent sepsis. Inflammation (2021) 442:481–92. doi: 10.1007/s10753-020-01329-3
126. Liang W, Greven J, Qin K, Fragoulis A, Horst K, Blasius F, et al. Sulforaphane exerts beneficial immunomodulatory effects on liver tissue Via a Nrf2 pathway-related mechanism in a murine model of hemorrhagic shock and resuscitation. Front Immunol (2022) 13:822895. doi: 10.3389/fimmu.2022.822895
127. Liang W, Greven J, Fragoulis A, Horst K, Blasius F, Wruck C, et al. Sulforaphane-dependent up-regulation of Nrf2 activity alleviates both systemic inflammatory response and lung injury after hemorrhagic Shock/Resuscitation in mice. Shock (2022) 572:221–9. doi: 10.1097/SHK.0000000000001859
128. Guan Z, Zhou L, Zhang Y, Zhang Y, Chen H, Shao F. Sulforaphane ameliorates the liver injury of traumatic hemorrhagic shock rats. J Surg Res (2021) 267:293–301. doi: 10.1016/j.jss.2021.05.004
129. Kulkarni SS, Canto C. The molecular targets of resveratrol. Biochim Biophys Acta (2015) 18526:1114–23. doi: 10.1016/j.bbadis.2014.10.005
130. Ayub A, Poulose N, Raju R. Resveratrol improves survival and prolongs life following hemorrhagic shock. Mol Med (Cambridge Mass) (2015) 21:305–12. doi: 10.2119/molmed.2015.00013
131. Pradere JP, Hernandez C, Koppe C, Friedman RA, Luedde T, Schwabe RF. Negative regulation of nf-kappab P65 activity by serine 536 phosphorylation. Sci Signaling (2016) 9442:ra85. doi: 10.1126/scisignal.aab2820
132. Rabb H, Griffin MD, McKay DB, Swaminathan S, Pickkers P, Rosner MH, et al. Inflammation in aki: Current understanding, key questions, and knowledge gaps. J Am Soc Nephrol (2016) 272:371–9. doi: 10.1681/ASN.2015030261
133. Sheng X, Yang Y, Liu J, Yu J, Guo Q, Guan W, et al. Ophiopogonin a alleviates hemorrhagic shock-induced renal injury Via induction of Nrf2 expression. Front Physiol (2020) 11:619740. doi: 10.3389/fphys.2020.619740
134. Luciani C, Hager FT, Cerovic V, Lelouard H. Dendritic cell functions in the inductive and effector sites of intestinal immunity. Mucosal Immunol (2022) 151:40–50. doi: 10.1038/s41385-021-00448-w
135. Zhang Y, Zhang J, Xu T, Wu W, Huang F, Yu W, et al. Allicin ameliorates intraintestinal bacterial translocation after Trauma/Hemorrhagic shock in rats: The role of mesenteric lymph node dendritic cell. Surgery (2017) 1612:546–55. doi: 10.1016/j.surg.2016.08.029
136. Xu S, Liao Y, Wang Q, Liu L, Yang W. Current studies and potential future research directions on biological effects and related mechanisms of allicin. Crit Rev Food Sci Nutr (2022) 2049691:1–27. doi: 10.1080/10408398.2022.2049691
137. Slate JR, Chriswell BO, Briggs RE, McGill JL. The effects of ursolic acid treatment on immunopathogenesis following mannheimia haemolytica infections. Front Veterinary Sci (2021) 8:782872. doi: 10.3389/fvets.2021.782872
138. Jung TY, Pham TN, Umeyama A, Shoji N, Hashimoto T, Lee JJ, et al. Ursolic acid isolated from uncaria rhynchophylla activates human dendritic cells via Tlr2 and/or Tlr4 and induces the production of ifn-gamma by Cd4+ naive T cells. Eur J Pharmacol (2010) 6432-3:297–303. doi: 10.1016/j.ejphar.2010.06.030
139. Hwang TL, Shen HI, Liu FC, Tsai HI, Wu YC, Chang FR, et al. Ursolic acid inhibits superoxide production in activated neutrophils and attenuates trauma-hemorrhage shock-induced organ injury in rats. PloS One (2014) 910:e111365. doi: 10.1371/journal.pone.0111365
140. Xiang M, Yin L, Li Y, Xiao G, Vodovotz Y, Billiar TR, et al. Hemorrhagic shock activates lung endothelial reduced nicotinamide adenine dinucleotide phosphate (Nadph) oxidase Via neutrophil nadph oxidase. Am J Respir Cell Mol Biol (2011) 443:333–40. doi: 10.1165/rcmb.2009-0408OC
141. Tsai YF, Yu HP, Chung PJ, Leu YL, Kuo LM, Chen CY, et al. Osthol attenuates neutrophilic oxidative stress and hemorrhagic shock-induced lung injury Via inhibition of phosphodiesterase 4. Free Radical Biol Med (2015) 89:387–400. doi: 10.1016/j.freeradbiomed.2015.08.008
142. Liu H, Zhao Z, Xing L, Zhang L, Niu C. Post-shock mesenteric lymph drainage ameliorates cellular immune function in rats following hemorrhagic shock. Inflammation (2015) 382:584–94. doi: 10.1007/s10753-014-9965-3
143. Morishita K, Coimbra R, Langness S, Eliceiri BP, Costantini TW. Neuroenteric axis modulates the balance of regulatory T cells and T-helper 17 cells in the mesenteric lymph node following Trauma/Hemorrhagic shock. Am J Physiol Gastrointestinal Liver Physiol (2015) 3093:G202–8. doi: 10.1152/ajpgi.00097.2015
144. Wu J, Yin Y, Qin M, Li K, Liu F, Zhou X, et al. Vagus nerve stimulation protects enterocyte glycocalyx after hemorrhagic shock Via the cholinergic anti-inflammatory pathway. Shock (2021) 565:832–9. doi: 10.1097/SHK.0000000000001791
145. Reys LG, Ortiz-Pomales YT, Lopez N, Cheadle G, de Oliveira PG, Eliceiri B, et al. Uncovering the neuroenteric-pulmonary axis: Vagal nerve stimulation prevents acute lung injury following hemorrhagic shock. Life Sci (2013) 9213:783–92. doi: 10.1016/j.lfs.2013.02.009
146. Li Y, Du H, Jiang L, Wang C, Yin M, Zhang L, et al. Stellate ganglion block improves the proliferation and function of splenic Cd4 + T cells through inhibition of posthemorrhagic shock mesenteric lymph-mediated autophagy. Inflammation (2021) 44:254353. doi: 10.1007/s10753-021-01523-x
147. Son J, Chandler B, Feketova E, Qin Y, Quackenbush E, Deitch E. Oral pretreatment with recombinant human lactoferrin limits trauma-hemorrhagic shock-induced gut injury and the biological activity of mesenteric lymph. J Surg Res (2014) 1871:270–7. doi: 10.1016/j.jss.2013.10.026
148. Tian F, Gao X, Zhang L, Wang X, Wan X, Jiang T, et al. Effects of n-3 pufas on intestinal mucosa innate immunity and intestinal microbiota in mice after hemorrhagic shock resuscitation. Nutrients (2016) 810:609. doi: 10.3390/nu8100609
149. Xu DQ, Gao C, Niu W, Li Y, Wang YX, Gao CJ, et al. Sodium hydrosulfide alleviates lung inflammation and cell apoptosis following resuscitated hemorrhagic shock in rats. Acta Pharmacologica Sin (2013) 3412:1515–25. doi: 10.1038/aps.2013.96
150. Kojima M, Gimenes-Junior J, Chan T, Eliceiri B, Baird A, Costantini T, et al. Viaexosomes in postshock mesenteric lymph are key mediators of acute lung injury triggering the macrophage activation toll-like receptor 4. FASEB J Off Publ Fed Am Societies Exp Biol (2018) 321:97–110. doi: 10.1096/fj.201700488R
151. Cerovic V, Houston SA, Scott CL, Aumeunier A, Yrlid U, Mowat AM, et al. Intestinal Cd103- dendritic cells migrate in lymph and prime effector T cells. Mucosal Immunol (2013) 61:104–13. doi: 10.1038/mi.2012.53
152. Farache J, Zigmond E, Shakhar G, Jung S. Contributions of dendritic cells and macrophages to intestinal homeostasis and immune defense. Immunol Cell Biol (2013) 913:232–9. doi: 10.1038/icb.2012.79
153. Dai H, Sun T, Liu Z, Zhang J, Zhou M. The imbalance between regulatory and il-17-Secreting Cd4(+)T cells in multiple-trauma rat. Injury (2013) 4411:1521–7. doi: 10.1016/j.injury.2013.03.015
154. Yagi M, Morishita K, Ueno A, Nakamura H, Akabori H, Senda A, et al. Electrical stimulation of the vagus nerve improves intestinal blood flow after trauma and hemorrhagic shock. Surgery (2020) 1673:638–45. doi: 10.1016/j.surg.2019.09.024
155. Morishita K, Costantini TW, Ueno A, Bansal V, Eliceiri B, Coimbra R. A pharmacologic approach to vagal nerve stimulation prevents mesenteric lymph toxicity after hemorrhagic shock. J Trauma Acute Care Surg (2015) 781:52–8. doi: 10.1097/TA.0000000000000489
156. Morishita K, Costantini TW, Eliceiri B, Bansal V, Coimbra R. Vagal nerve stimulation modulates the dendritic cell profile in posthemorrhagic shock mesenteric lymph. J Trauma Acute Care Surg (2014) 763:610–7. doi: 10.1097/TA.0000000000000137
157. Luyer MD, de Haan JJ, Lubbers T, Greve JW, Buurman WA. Parasympathetic stimulation Via the vagus nerve prevents systemic organ dysfunction by abrogating gut injury and lymph toxicity in trauma and hemorrhagic shock. Shock (2013) 395:460–1. doi: 10.1097/SHK.0b013e31828def5a
158. Levy G, Fishman JE, Xu DZ, Dong W, Palange D, Vida G, et al. Vagal nerve stimulation modulates gut injury and lung permeability in trauma-hemorrhagic shock. J Trauma Acute Care Surg (2012) 732:338–42. doi: 10.1097/TA.0b013e31825debd3
159. Zhang J, Lin XR, Zhang YP, Zhang LM, Du HB, Jiang LN, et al. Blockade of stellate ganglion remediates hemorrhagic shock-induced intestinal barrier dysfunction. J Surg Res (2019) 244:69–76. doi: 10.1016/j.jss.2019.06.007
160. Ramalingam S, Crawford J, Chang A, Manegold C, Perez-Soler R, Douillard JY, et al. Talactoferrin Alfa versus placebo in patients with refractory advanced non-Small-Cell lung cancer (Fortis-m trial). Ann Oncol Off J Eur Soc Med Oncol (2013) 2411:2875–80. doi: 10.1093/annonc/mdt371
161. Riess JW, Bhattacharya N, Blenman KR, Neal JW, Hwang G, Pultar P, et al. Immune correlates of talactoferrin Alfa in biopsied tumor of Relapsed/Refractory metastatic non-small cell lung cancer patients. Immunopharmacol Immunotoxicol (2014) 362:182–6. doi: 10.3109/08923973.2013.864671
162. Madan RA, Tsang KY, Bilusic M, Vergati M, Poole DJ, Jochems C, et al. Effect of talactoferrin Alfa on the immune system in adults with non-small cell lung cancer. Oncologist (2013) 187:821–2. doi: 10.1634/theoncologist.2013-0199
163. Perez-Lopez A, Behnsen J, Nuccio SP, Raffatellu M. Mucosal immunity to pathogenic intestinal bacteria. Nat Rev Immunol (2016) 163:135–48. doi: 10.1038/nri.2015.17
164. Ganster F, Burban M, de la Bourdonnaye M, Fizanne L, Douay O, Loufrani L, et al. Effects of hydrogen sulfide on hemodynamics, inflammatory response and oxidative stress during resuscitated hemorrhagic shock in rats. Crit Care (2010) 145:R165. doi: 10.1186/cc9257
165. Mahmoud M, Mason KP. Dexmedetomidine: Review, update, and future considerations of paediatric perioperative and periprocedural applications and limitations. Br J Anaesthesia (2015) 1152:171–82. doi: 10.1093/bja/aev226
166. Jia Z, Chen X, Sun P, Liu M, Li X. The protective mechanism of dexmedetomidine on renal in hemorrhagic shock. Comput Math Methods Med (2022) 2022:6394544. doi: 10.1155/2022/6394544
167. Zhang Y, Chen H, Li YM, Zheng SS, Chen YG, Li LJ, et al. Thymosin Alpha1- and ulinastatin-based immunomodulatory strategy for sepsis arising from intra-abdominal infection due to carbapenem-resistant bacteria. J Infect Dis (2008) 1985:723–30. doi: 10.1086/590500
168. Park KH, Lee KH, Kim H, Hwang SO. The anti-inflammatory effects of ulinastatin in trauma patients with hemorrhagic shock. J Korean Med Sci (2010) 251:128–34. doi: 10.3346/jkms.2010.25.1.128
169. Abdou H, Morrison JJ, Edwards J, Patel N, Lang E, Richmond MJ, et al. An estrogen (17alpha-ethinyl estradiol-3-Sulfate) reduces mortality in a swine model of multiple injuries and hemorrhagic shock. J Trauma Acute Care Surg (2022) 921:57–64. doi: 10.1097/TA.0000000000003434
170. Buleon M, Cuny M, Grellier J, Charles PY, Belliere J, Casemayou A, et al. A single dose of estrogen during hemorrhagic shock protects against kidney injury whereas estrogen restoration in ovariectomized mice is ineffective. Sci Rep (2020) 101:17240. doi: 10.1038/s41598-020-73974-5
171. Gupta D, Tiwari S, Sinha T, Soni K, Galwnkar S, Kumar S, et al. Estrogen as a safe therapeutic adjunct in reducing the inflammatory storm in trauma hemorrhagic shock patients. Shock (Augusta Ga) (2021) 56:514–21. doi: 10.1097/shk.0000000000001779
172. Guyette FX, Sperry JL, Peitzman AB, Billiar TR, Daley BJ, Miller RS, et al. Prehospital blood product and crystalloid resuscitation in the severely injured patient: A secondary analysis of the prehospital air medical plasma trial. Ann Surg (2021) 2732:358–64. doi: 10.1097/SLA.0000000000003324
173. Moore HB, Moore EE, Chapman MP, McVaney K, Bryskiewicz G, Blechar R, et al. Plasma-first resuscitation to treat haemorrhagic shock during emergency ground transportation in an urban area: A randomised trial. Lancet (London England) (2018) 39210144:283–91. doi: 10.1016/S0140-67361831553-8
174. Bulger EM, May S, Kerby JD, Emerson S, Stiell IG, Schreiber MA, et al. Out-of-Hospital hypertonic resuscitation after traumatic hypovolemic shock: A randomized, placebo controlled trial. Ann Surg (2011) 2533:431–41. doi: 10.1097/SLA.0b013e3181fcdb22
Keywords: Innate immunity, hemorrhagic shock, immunotherapy, multiple organ failure, mesenchymal stem cell, antibody therapy, small molecule inhibitor
Citation: Huang Q, Gao S, Yao Y, Wang Y, Li J, Chen J, guo C, Zhao D and Li X (2022) Innate immunity and immunotherapy for hemorrhagic shock. Front. Immunol. 13:918380. doi: 10.3389/fimmu.2022.918380
Received: 12 April 2022; Accepted: 04 August 2022;
Published: 25 August 2022.
Edited by:
Yongqing Li, School of Medicine, University of Michigan, United StatesReviewed by:
Morgan Salmon, Michigan Medicine, University of Michigan, United StatesLu Wang, Renmin Hospital of Wuhan University, China
Wei Chong, The First Affiliated Hospital of China Medical University, China
Copyright © 2022 Huang, Gao, Yao, Wang, Li, Chen, guo, Zhao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daqing Zhao, emhhb2RhcWluZzE5NjNAMTYzLmNvbQ==; Xiangyan Li, eGlhbmd5YW5fbGkxOTgxQDE2My5jb20=