
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol., 24 June 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.917601
This article is part of the Research TopicAdvances in primary Immunodeficiencies (Inborn Errors of Immunity) in Central-Eastern Europe: Volume IIView all 20 articles
 Eduardo de la Fuente-Munoz1†
Eduardo de la Fuente-Munoz1† Ana Van Den Rym2,3,4†
Ana Van Den Rym2,3,4† Blanca García-Solis2,3,4
Blanca García-Solis2,3,4 Juliana Ochoa Grullón1
Juliana Ochoa Grullón1 Kissy Guevara-Hoyer1
Kissy Guevara-Hoyer1 Miguel Fernández-Arquero1,4Lucía Galán Dávila5
Miguel Fernández-Arquero1,4Lucía Galán Dávila5 Jorge Matías-Guiú5
Jorge Matías-Guiú5 Silvia Sánchez-Ramón1,4‡
Silvia Sánchez-Ramón1,4‡ Rebeca Pérez de Diego2,3,4*‡
Rebeca Pérez de Diego2,3,4*‡Gain-of-function (GOF) mutations in STIM1 are responsible for tubular aggregate myopathy and Stormorken syndrome (TAM/STRMK), a clinically overlapping multisystemic disease characterised by muscle weakness, miosis, thrombocytopaenia, hyposplenism, ichthyosis, dyslexia, and short stature. Several mutations have been reported as responsible for the disease. Herein, we describe a patient with TAM/STRMK due to a novel L303P STIM1 mutation, who not only presented clinical manifestations characteristic of TAM/STRMK but also manifested immunological involvement with respiratory infections since childhood, with chronic cough and chronic bronchiectasis. Despite the seemingly normal main immunological parameters, immune cells revealed GOF in calcium signalling compared with healthy donors. The calcium flux dysregulation in the immune cells could be responsible for our patient’s immune involvement. The patient’s mother carried the mutation but did not exhibit TAM/STRMK, manifesting an incomplete penetrance of the mutation. More cases and evidence are necessary to clarify the dual role of STIM1 in immune system dysregulation and myopathy.
Calcium (Ca2+) signalling, in which Ca2+ acts as a second messenger, controls numerous cellular functions, such as proliferation, apoptosis, exocytosis, differentiation, neurotransmission, hormone secretion, blood coagulation, and muscle contraction. Store-operated Ca2+ entry (SOCE) is a ubiquitous mechanism for Ca2+ entry into eukaryotic cells mediated by STIM1 and ORAI1 proteins. Human disorders have been related to SOCE, including autosomal recessive STIM1 and ORAI1 loss-of-function (LOF) mutations, resulting in insufficient SOCE and subsequently altering Ca2+ release-activated calcium (CRAC) channels (1). The alteration of CRAC channels generates a severe combined immunodeficiency, which includes recurrent and chronic infections, autoimmunity, muscular hypotonia, ectodermal dysplasia, anhidrosis, and mydriasis. Most STIM1 and ORAI1 LOF mutations do not express protein; however, mutations have been described that disrupt the STIM1 function and interfere with the STIM1-ORAI1 interaction (such as R426C and R429C mutations) or generate an obstructed ORAI1 channel, such as the R91W mutation (2). In contrast, gain-of-function (GOF) STIM1 and ORAI1 mutations are autosomal dominant forms that induce overactivation due to excessive Ca2+ entry through SOCE. These patients experience tubular aggregate myopathy and Stormorken syndrome (TAM/STRMK), with progressive muscle weakness, myalgia, miosis, ichthyosis, short stature, hyposplenism, thrombocytopaenia (3), and dyslexia (2). All GOF mutations share missense mutations that affect highly conserved amino acids in the EF-hand Ca2+-binding motif (H72Q, N80T, G81D, D84G, D84E, S88G, L92V, L96V, Y98C, K104N, F108I, F108L, H109N, H109R, H109Y, I115F), in the sterile alpha motif domain (V138I), in the luminal coiled-coil domains of STIM1 (4) (CC1: R304W and R304Q, and CC2: K365N), between the S/P and K domains (S630F and H632fs*), in the K domain (R749H), and in the ORAI1 transmembrane domains forming the channel pore or concentric rings surrounding the pore (G97C, G98S, V107M, L138F, T184M and P245L) (2, 5–8). Missense mutations in the muscle-specific sarcoplasmic reticulum Ca2+ buffer calsequestrin-1 (CASQ1) have been also reported in patients with late-onset muscle weakness and myalgia, forming the mild end of the TAM/STRMK spectrum (9, 10).
We examined a 52-year-old European man from Spain with non-consanguineous parents. Written informed consent was obtained from the patient for the publication of any potentially identifiable data included in this article. The patient had a clear case of TAM/STRMK, with marked myopathy, defective dental enamel, numerous dental caries and root canals, brittle nails, no dystrophy, congenital pes cavus of the right foot, congenital hammer toes (fourth and fifth digits), arthrosis, generalised myalgia, muscle atrophy with myoclonus, and incapacitating fatigue after physical exercise.
The patient also experienced photosensitivity, with erythema and desquamation after sun exposure (doubtful association with drugs); however, there was no report of oral aphthous ulcers, cold sores, arthritis, or Raynaud’s disease. The patient presented skin rashes that worsened in the summer, as well as solar urticaria and dermographism (erythema). He reported that each episode was accompanied by digestive symptoms, with a tendency to diarrhoea and with clinical worsening of his myopathy.
The patient experienced myalgia in the mornings, which decreased with exercise, as well as muscle stiffness, muscle fatigue, myalgia in the peripheral forearms and legs, and muscle contractures that had progressed in the past year. The patient had experienced muscle mass loss even while performing exercise (walking 1 h daily, Pilates), as well as intense post-exercise myalgia. He has always been in good physical shape; however, after 1 hour of exercise, the patient presented fatigue with extreme exhaustion. He woke at night because of muscle pain, which decreased with short walks. The patient experienced myoclonus in the arms with exercise, as well as pain in both wrists, with functional disability and spontaneous resolution.
The patient’s skeletal muscle has preserved architecture but without increased endomysial connective tissue or adipose infiltration. A skeletal muscle biopsy showed discrete variability in muscle fibre size, with the presence of fibres with multiple internalised nuclei (approximately 8%). We observed no necrotic or regenerative fibres and no structural disorders (vacuoles or inclusions). Through ATPase techniques, we determined that the fibre type distribution was normal. With oxidative techniques, we observed a few COX-negative fibres with sorbitol dehydrogenase overexpression (approximately 3%), as well as NADH-TR alterations. We observed no inflammatory infiltrates and no alterations using the histochemical technique for phosphorylase and myoadenylate deaminase. Although the patient’s phosphofructokinase level was not assessable, he presented high creatine kinase levels (Table 1) and pseudomyotonia, which was observed on an electromyogram.
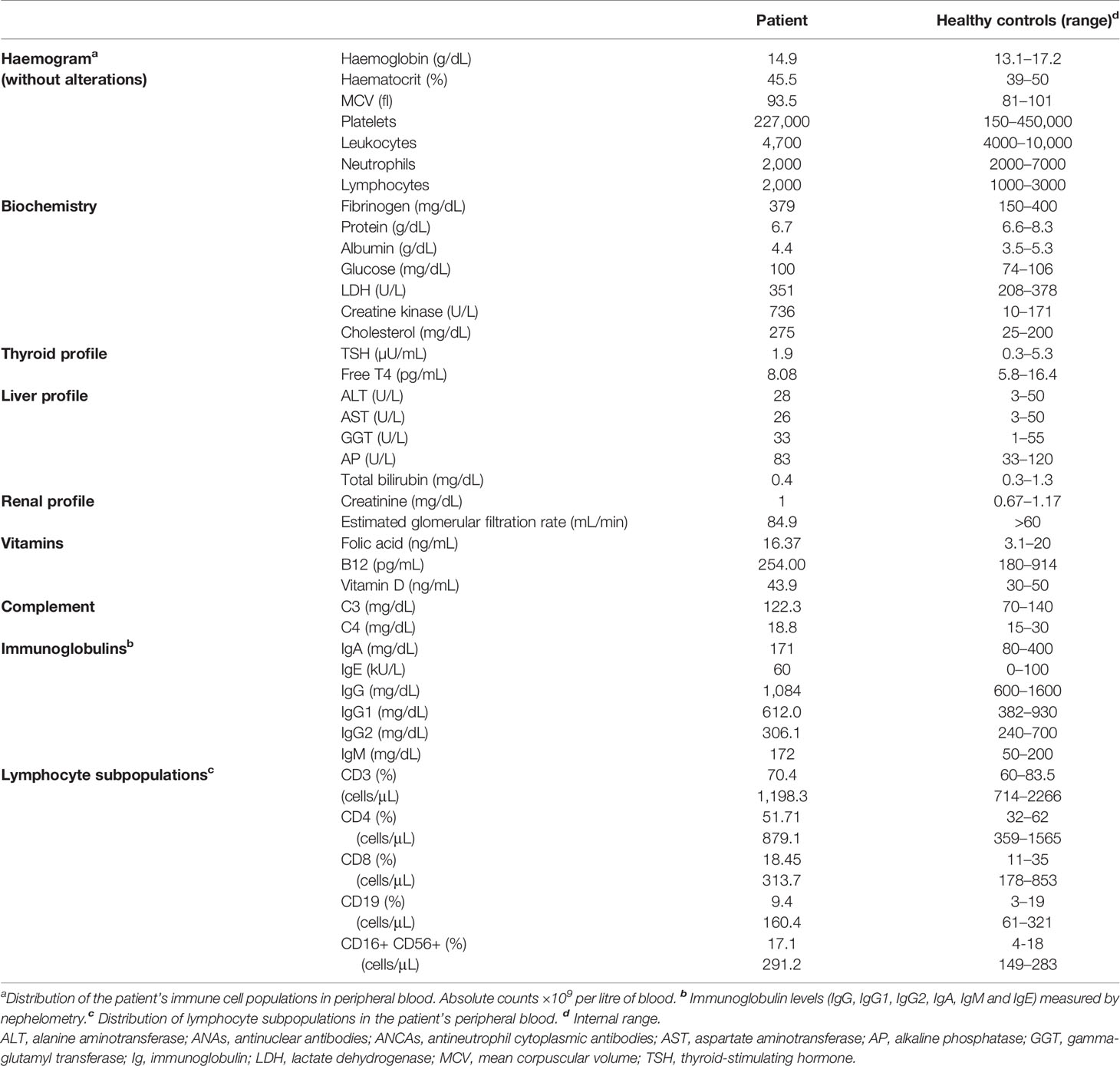
Table 1 Analytical studies.
Beyond the TAM/STRMK disease pattern, the patient presented immunological involvement, with respiratory infections since childhood, chronic cough, and chronic bronchiectasis. At the immunological level, a lymphocyte subpopulation study showed normal T, B, and natural killer cell counts (Table 1). The patient had normal activity of complement factors C3, C4, and CH50, as well as normal immunoglobulin levels (Table 1).
The patient presented adequate specific antibody response against protein and polysaccharide immunisation. The results of a study of lymphocyte proliferation to mitogens with CD3-CD28 and phytohaemagglutinin were within normal limits. The antibody studies were negative for antinuclear antibodies, antineutrophil cytoplasmic antibodies, celiac antibodies, and Helicobacter pylori antibodies.
The acid phosphatase test, and immunohistochemistry for human leukocyte antigen showed no abnormalities. The immunohistochemistry did show preserved membrane proteins (dystrophins 1 and 2, alpha and gamma sarcoglycans, caveolin, and merosin).
The patient showed a good response to the salmonella and tetanus toxoid vaccine. Functional respiratory tests showed reduced baseline spirometry: 11/19 forced vital capacity (FVC) of 4130 mL (77%); forced expiratory volume in 1 second (FEV1) of 3620 mL (91%); and a FEV1/FVC ratio of 88%.
The patient also presented various allergies (pollen, olive trees, reeds, banana, and grasses) and dyslipidaemia, which is under treatment. His allergic symptoms have worsened at the dermatological level, with particular involvement of the chest, back, and legs.
The patient takes eslicarbazepine (800 mg; 0-0-0.5), ezetimibe (10 mg; 0-0-1), chondroitin sulphate (400 mg; 2 doses every 24 h), bilastine (20 mg; 0-1-0), rupatadine (10 mg; 0-0-0-1), and budesonide/formoterol (1-0-1).
The experimental protocol was approved by the ethics committee of Clinico San Carlos University Hospital (Madrid, Spain) and La Paz University Hospital (Madrid, Spain), and written informed consent was obtained from the family for participation in this study.
The patient underwent a next-generation sequencing gene panel for the diagnosis of primary immunodeficiencies. We found a variant of the STIM1 gene on chromosome 11, a heterozygous missense mutation (T/C) affecting the nucleotide position g.4095848 (GRCh37.p13) of exon 7 of the gene encoding STIM1 in the genomic DNA extracted from the leukocytes (g.4095848T>C). This mutation affects leucine at position 303, generating a missense mutation by a proline (c.1477T>C, p.L303P, transcript ID ENST00000300737.4), a previously unreported variant, which we validated by Sanger sequencing of genomic DNA from peripheral leukocytes (Figure 1A). His mother, who has cardiomyopathy, has the same STIM1 mutation (L303P) (data not shown). No more data could be obtained from the mother, the father could not be tested, and the patient has no offspring. No other mutations were found in the STIM1 coding region. Alignment of the human STIM1 protein sequence with sequences from the 7 animal species in which STIM1 has been sequenced showed L303 to be highly conserved throughout evolution (Figure 1B). Protein structure modelling showed how the nonpolar lateral chain of leucine 303 is oriented inside the interaction of the 2 alpha chains of CC1-IH STIM1 (4). The L303P mutation modifies the position of the lateral chain, affecting the proper conformation of the CC1-IH region of STIM1 (Figure 1C) (4). A mutation significance cut-off study (http://pec630.rockefeller.edu:8080/MSC/) predicted this variant as likely to be damaging (Figure 1D). These data suggest that a heterozygous germline missense STIM1 mutation (g.4095848T>C; p.L303P) might be responsible for the novel autosomal dominant form of the patient’s GOF STIM1 mutation.
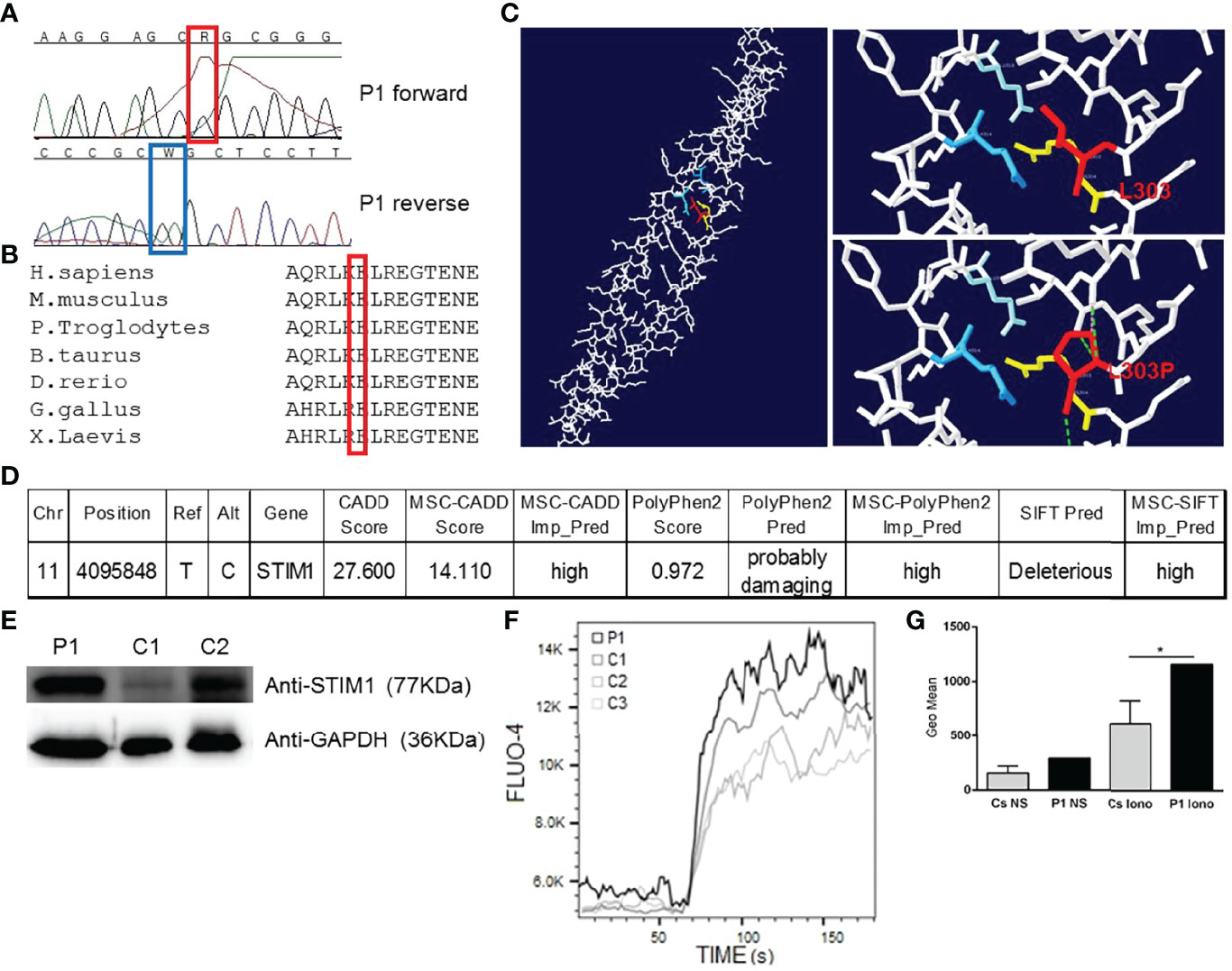
Figure 1 Heterozygous STIM1 mutation in a patient with myopathy. (A) The sequence of the PCR products of genomic DNA from the patient’s leukocytes is shown. g.4095848T>C (c.1477T>C, p.L303P). This mutation has not been previously reported. (B) Multiple alignment of the sequences from humans and 6 other species, showing that L303 is a conserved amino acid in 7 analysed species. (C) The left panel shows the structure of human CC1-IH STIM1 (4). The top right panel shows the interactions between 2 alpha helices of the CC1-IH region of STIM1. Blue is Q314 and light blue is E318 of one alpha helix, and L303 and R304 are the red and yellow residues, respectively, of the other alpha helix (4). The bottom right panel shows the L303P mutation. The figure was produced using Swiss-PdbViewer. (D) Mutation significance cut-off (http://pec630.rockefeller.edu:8080/MSC/) of STIM1 L303P mutation. (E) Immunoblot analysis of STIM1 protein from the patient’s (P1) peripheral blood mononuclear cells (PBMCs) and from 2 healthy donors (C1 and C2). We employed GAPDH as a loading control. The panels illustrate the results from a single experiment, representative of 3. (F) Calcium flux analysis in the PBMCs of P1 and 3 healthy donors (C1, C2, and C3) in response to ionomycin. The panels illustrate the results from a single experiment, representative of 3. (G) Calcium flux geometric mean (Geo Mean) is represented for C1, C2, and C3. ± SD and P1 in non-stimulated PBMCs (NS) or ionomycin-stimulated PBMCs (Iono). p < 0.05 (*).
Numerous studies have described the role of STIM1 mutations in the cells implicated in TAM/STRMK, such as myoblasts and myotubes (5). Given that the patient showed immunological involvement, we wanted to test the protein expression and calcium signalling in immune cells. We then assessed STIM1 protein levels in the patient’s peripheral blood mononuclear cells (PBMCs), detecting normal STIM1 protein levels compared with healthy donors (Figure 1D). Given that STIM1 GOF mutations induce excessive Ca2+ entry in muscle cells, thereby causing myopathy (10), we wanted to test whether the L303P STIM1 mutation affects calcium homeostasis in PBMCs. We stimulated PBMCs with the calcium ionophore ionomycin, measuring calcium mobilisation by changes in fluorescence of the Fluo4 dye loaded into the cells. Higher calcium flux in response to ionomycin was detected in the patient compared with the 3 healthy donors (Figures 1E–G), confirming that the L303P mutation generates a GOF in PBMCs that affects calcium homeostasis. A study of lymphocyte proliferation response to mitogens with CD3-CD28 and phytohaemagglutinin showed normal results (data not shown).
The present case describes a patient with an autosomal dominant GOF STIM1 mutation responsible for TAM/STRMK. The L303P mutation in the STIM1 gene showed an incomplete penetrance in the mother, who carried the mutation without TAM/STRMK symptoms. STIM1 mutations can be divided into those with GOF that manifest TAM/STRMK (2) and those with LOF that have an immune effect responsible for severe combined immunodeficiency, involving recurrent and chronic infections, muscular hypotonia, autoimmunity, ectodermal dysplasia, anhidrosis, and mydriasis (2). However, our patient has a GOF STIM1 mutation responsible for TAM/STRMK and has had respiratory infections since childhood, including chronic cough and chronic bronchiectasis, despite the main immunological features in terms of immune response and levels of immune cells and other immune parameters being within normal limits. The analysis of immune cells (PBMCs) of the Ca2+ signalling revealed a GOF with higher levels of calcium flux compared with healthy donors (Figures 1E, F), revealing calcium flux misregulation in the immune cells that could be responsible for the patient’s immune involvement. Worth to mention that the very close residue R304, the only mutated residue described in luminal coiled-coil CC1 domain, in mice harboring GOF R304W mutation presenting TAM/STRMK and abnormal immune cell counts as well as skin abnormalities (11) as shown by our patient.
Despite the study’s limitations resulting from the lack of access to information and studies of the patient’s relatives (incomplete penetrance of the mutation in the mother being the only datum), this is an important clinical case that warrants careful examination to clarify the dual role of STIM1 in immune system dysregulation and myopathy.
It has been widely reported that STIM1 GOF mutations are responsible for TAM/STRMK and that STIM1 LOF mutations cause severe combined immunodeficiency. However, it is plausible that a STIM1 GOF mutation in a patient with TAM/STRMK can also cause immune involvement, given that STIM1 is a protein involved in the immune system and that a misfunction (either by an excess or lack of calcium signalling) would affect the proper functioning of the immune system. Patients with TAM/STRMK should be followed up by clinical immunology departments to find more candidates with whom to study this preliminary finding in more depth. We cannot rule-out the possibility that this immunological involvement would be due to a digenic or polygenic condition by other genes involved in the immune system; however, excess calcium signalling in immune cells highlights a misfunction in the immune system. More cases need to be reported to accumulate sufficient evidence.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Study approval: The experimental protocol was approved by the ethics committee of Clinico San Carlos University Hospital (Madrid, Spain) and La Paz University Hospital (Madrid, Spain), and written informed consent was obtained from the family for participation in this study. Written informed consent was obtained from the patient for the publication of any potentially identifiable data included in this article.
EF: Physicians in charge of the patient’s care. Clinical report and analytical studies. AR: Calcium signalling and Sanger sequencing. BG-S: Protein expression, Sanger sequencing and protein modelling. JO, KG-H, and MF-A: Clinical study, analytical parameters, manuscript editing. LG: Physician in charge of the patient’s study and care. JM-G: Physician in charge of the patient’s study and care. SS-R: Physician in charge of the patient’s study and care, has read and revised the manuscript. RP: Laboratory head, experiment design, manuscript drafting and editing, Corresponding author. All authors contributed to the article and approved the submitted version.
Support was provided by FIS grant Ref. PI17/00543, BG-S is supported by PEJD2019-PRE/BMD-16556 Predoctoral Fellowships CAM and ESID Bridge Fellowship. AR was provided support by FIS grant Ref. PI17/00543.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the patient for participating in this study and the Clinical Immunology Department, San Carlos Clinical Hospital for their helpful advice.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.917601/full#supplementary-material
1. Feske S. Immunodeficiency Due to Defects in Store-Operated Calcium Entry. Ann N Y Acad Sci (2011) 1238:74–90. doi: 10.1111/j.1749-6632.2011.06240.x
2. Silva-Rojas R, Laporte J, Böhm J. STIM1/ORAI1 Loss-Of-Function and Gain-Of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases. Front Physiol (2020) 11:604941. doi: 10.3389/fphys.2020.604941
3. Markello T, Chen D, Kwan JY, Horkayne-Szakaly I, Morrison A, Simakova O, et al. York Platelet Syndrome Is a CRAC Channelopathy Due to Gain-of-Function Mutations in STIM1. Mol Genet Metab (2015) 114(3):474–82. doi: 10.1016/j.ymgme.2014.12.307
4. Cui B, Yang X, Li S, Lin Z, Wang Z, Dong C, et al. The Inhibitory Helix Controls the Intramolecular Conformational Switching of the C-Terminus of STIM1. PLoS One (2013) 8:e74735. doi: 10.1371/journal.pone.0074735
5. Conte E, Pannunzio A, Imbrici P, Camerino GM, Maggi L, Mora M, et al. Gain-Of-Function STIM1 L96V Mutation Causes Myogenesis Alteration in Muscle Cells From a Patient Affected by Tubular Aggregate Myopathy. Front Cell Dev Biol (2021) 9:635063. doi: 10.3389/fcell.2021.635063
6. Morin G, Bruechle NO, Singh AR, Knopp C, Jedraszak G, Elbracht M, et al. Gain-Of-Function Mutation in STIM1 (P.R304W) Is Associated With Stormorken Syndrome. Hum Mutat (2014) 35(10):1221–32. doi: 10.1002/humu.22621
7. Ticci C, Cassandrini D, Rubegni A, Riva B, Vattemi G, Matà S, et al. Expanding the Clinical and Genetic Spectrum of Pathogenic Variants in STIM1. Muscle Nerve (2021) 64:567–75. doi: 10.1002/mus.27391
8. Jiang L-J, Zhao X, Dou Z-Y, Su Q-X, Rong Z-H. Stormorken Syndrome Caused by a Novel STIM1 Mutation: A Case Report. Front Neurol (2021) 12:522513. doi: 10.3389/fneur.2021.522513
9. Michelucci A, García-Castañeda M, Boncompagni S, Dirksen RT. Role of STIM1/ORAI1-Mediated Store-Operated Ca2+ Entry in Skeletal Muscle Physiology and Disease. Cell Calcium (2018) 76:101–15. doi: 10.1016/j.ceca.2018.10.004
10. Böhm J, Laporte J. Gain-Of-Function Mutations in STIM1 and ORAI1 Causing Tubular Aggregate Myopathy and Stormorken Syndrome. Cell Calcium (2018) 76:1–9. doi: 10.1016/j.ceca.2018.07.008
Keywords: STIM1, infection, gain of function, myopathy, calcium signalling
Citation: de la Fuente-Munoz E, Van Den Rym A, García-Solis B, Ochoa Grullón J, Guevara-Hoyer K, Fernández-Arquero M, Galán Dávila L, Matías-Guiú J, Sánchez-Ramón S and Pérez de Diego R (2022) Case Report: Novel STIM1 Gain-of-Function Mutation in a Patient With TAM/STRMK and Immunological Involvement. Front. Immunol. 13:917601. doi: 10.3389/fimmu.2022.917601
Received: 11 April 2022; Accepted: 27 May 2022;
Published: 24 June 2022.
Edited by:
László Dr. Maródi, The Rockefeller University, United StatesReviewed by:
Nourhen Agrebi, Sidra Medicine, QatarCopyright © 2022 de la Fuente-Munoz, Van Den Rym, García-Solis, Ochoa Grullón, Guevara-Hoyer, Fernández-Arquero, Galán Dávila, Matías-Guiú, Sánchez-Ramón and Pérez de Diego. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebeca Pérez de Diego, cGVyZXpkZWRpZWdvckBnbWFpbC5jb20=
†These authors have contributed equally to this work
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.