 Chen Sun1†
Chen Sun1† Yunze Han1†Ruoyu Zhang1†
Yunze Han1†Ruoyu Zhang1† Simon Liu2Jing Wang1Yuqing Zhang1
Simon Liu2Jing Wang1Yuqing Zhang1 Xuemei Chen3
Xuemei Chen3 Chao Jiang4
Chao Jiang4 Junmin Wang3*
Junmin Wang3* Xiaochong Fan1*
Xiaochong Fan1* Jian Wang1,3*
Jian Wang1,3*- 1Department of Pain Medicine, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Medical Genomics Unit, National Human Genome Research Institute, Bethesda, MD, United States
- 3Department of Human Anatomy, School of Basic Medical Sciences, Zhengzhou University, Zhengzhou, China
- 4Department of Neurology, Fifth Affiliated Hospital of Zhengzhou University, Zhengzhou, China
COVID-19 caused by SARS-CoV-2 can cause various systemic diseases such as acute pneumonia with cytokine storm. Constituted of necroptosis, pyroptosis, and ferroptosis, regulated necrosis constitutes the cell death patterns under the low apoptosis condition commonly observed in COVID-19. Regulated necrosis is involved in the release of cytokines like TNF-α, IL-1 β, and IL-6 and cell contents such as alarmins, PAMPs, and DAMPs, leading to more severe inflammation. Uncontrolled regulated necrosis may explain the poor prognosis and cytokine storm observed in COVID-19. In this review, the pathophysiology and mechanism of regulated necrosis with the double-edged sword effect in COVID-19 are thoroughly discussed in detail. Furthermore, this review also focuses on the biomarkers and potential therapeutic targets of the regulated necrosis pathway in COVID-19, providing practical guidance to judge the severity, prognosis, and clinical treatment of COVID-19 and guiding the development of clinical anti-SARS-CoV-2 drugs.
Highlights
1. Regulated necrosis plays a critical role in the pathologic process of COVID-19.
2. The mechanisms and “double-edged sword” effects of regulated necrosis are discussed.
3. The potential therapeutic targets can guide the development of anti-SARS-CoV-2 drugs.
1 Introduction
An unprecedented worldwide spread of coronavirus-2 of the severe acute respiratory syndrome (SARS-CoV-2), the cause of coronavirus disease 2019 (COVID-19), has imposed tough challenges on the health and medical infrastructure around the world (1). As of 1 May 2022, 510 million people had been diagnosed with COVID-19, including 6 million deaths worldwide (2). The initial clinical manifestations are mainly nonspecific respiratory syndromes (3–5) followed by complex complications, including multiple organ failure, septicemia, and cytokine storm (6–8). SARS-CoV-2 infects the host with the cell receptor angiotensin-converting enzyme-2 (ACE2) through respiratory droplets and causes various pathophysiological changes and syndromes. Cell metabolism is interpreted as the trigger for cell death through multiple pathways that coexist in COVID-19 (9). More components and increased cell damage lead to systemic inflammatory response syndrome (SIRS) and a SIRS-like immune response (10). With the aggravation of the now dysfunctional inflammatory system, inflammatory monocytes and neutrophils increase, and lymphocytes decrease markedly (6, 11, 12). Cytokine storms and a high burden of systemic inflammation accelerate subclinical disorders and lead to complications through inflammatory cell death (12). In this process, regulated necrosis plays a vital role in the pathophysiology of COVID-19 (13); exploring its connection to COVID-19 can provide a crucial theoretical basis for our treatment of COVID-19.
Necrosis is an old concept that has recently gained new attention (14). Traditional necrosis, characterized by organelle disintegration, oncosis, degeneration of proteins and enzymes in vivo, and plasma membrane rupture (15), evokes inflammatory responses by releasing damage-associated molecular patterns (DAMP) that trigger an immune response known as necroinflammation (16). During inflammation, necrotic cells and neutrophils exude lysosomal enzymes, promoting further necrosis and local parenchymal lysis of multiple cells simultaneously (17).
More recently, regulated necrosis has emerged as a revolutionary concept (18). Unlike traditional necrosis, which is understood to be a disordered, passive, gene-independent, and pathological process, regulated necrosis is genetically programmed. It biochemically represents various signaling pathways, such as kinase-mediated necroptosis, gasdermin-mediated necrosis downstream of inflammasomes, and an iron-catalyzed mechanism (14, 19, 20). Regulated necrosis comes in different forms, such as necroptosis, pyroptosis, and ferroptosis, all of which play an essential role in host defense and maintaining tissue homeostasis (18). Furthermore, regulated necrosis also contributes to the pathophysiology of various inflammatory, infectious, tumor, and degenerative diseases (21, 22). Like apoptosis, regulated necrosis can be controlled by specific molecular modulations on therapeutic targets [e.g., RPM1-induced protein kinase (RIPK1), iron] (14, 18, 23–25). Specific molecules require regulatory pathways that involve the Fas-associated protein with a new death domain, caspase-8, caspase-1, RIPK3, mixed lineage kinase domain-like pseudokinase (MLKL), nuclear factor-κB (NF-κB), etc. (26).
β-coronavirus proteins or infection with the complete virus can lead to coronavirus-induced cell death, necroptosis, and pyroptosis (27). Infection with SARS-CoV-2 activates caspase-8 (a master regulator of pyroptosis and necroptosis) and RIPK3 to initiate inflammatory cytokines within lung epithelial cells (28). Necroptosis depends on forming a molecular complex called the necrosome, which incorporates the phosphorylation of RIPK1, RIPK3, and the recruitment of mixed lineage kinase domain-like pseudokinase (MLKL), and pro–caspase-8 (29). Serum levels of RIPK3 have been demonstrated to be upregulated in patients with COVID-19, suggesting the necroptosis-driven response to host defense of SARS-CoV-2 (16). Pyroptosis depends on inflammasomes, caspase-1/11, gasdermin-D (GSDMD), and the release of interleukin-1β (IL-1 β) and IL-18 (30). Ferroptosis triggered by iron overload or glutathione peroxidase 4 (GPX4) inactivation leads to lipid peroxidation and the release of cell contents (31).
Studies on the molecular mechanism of regulated necrosis have improved our understanding of the role regulated necrosis plays in COVID-19. Although the essential functions of regulated necrosis in the immune system are fully described, their roles in COVID-19 are still quite complex and elusive. For example, necroptosis is vital in maintaining T-cell homeostasis (32). Furthermore, excess activated T cells can be removed after clonal expansion, while deregulation can result in immunodeficiency or autoimmunity (32). This review discusses the potential roles and specific consequences of regulated necrosis, including necroptosis, ferroptosis, and pyroptosis, in the pathophysiological process of COVID-19. Furthermore, the mechanism of regulated necrosis and potential therapeutic targets are comprehensively reviewed. The relevance of regulated necrosis in anti-COVID-19 therapy makes it possible to inhibit further infection of COVID-19, in turn giving the development of anti-SARS-CoV-2 drugs a high clinical value.
2 Regulated necrosis in COVID-19: A double-edged sword?
Previous studies have shown that regulated necrosis is a double-edged sword in cancer development and progression. It has pro- and antitumor effects that drive oncogenesis and defend against the emergence of cancer, respectively (33). Regarding the SARS-CoV-2 infection process, regulated necrosis results from viral replication in permissive cells, which dominates in the early stage of infection. Therefore, it can inhibit virus invasion by killing cells and activating immune defense function. However, later in the disease, regulated necrosis has extremely pro-inflammatory effects. Pro-inflammatory cytokines (tumor necrosis factor (TNF-α), IFN-γ, IL-1, or IL-6) are the main functional activity of the immune system (18, 20). This process leads to the recruitment of immune cells, the generation of immune complexes, and relevant injury (34). Additionally, the release of processed inflammatory cytokines and the amplification of inflammatory reactions together induce necroptosis and pyroptosis in macrophages, which aggravate lymphopenia through the direct killing of lymphocytes and limit the appropriate immune response (28).
Regulated necrosis can play a protective role in limiting SARS-CoV-2 infection. It seems that SARS-CoV-2 infection manipulates the necrosis process by regulating NF-κ B-dependent cell survival and mitochondria among various pathways to inhibit regulated necrosis and allow SARS-CoV-2 to replicate and spread. Additionally, the anti-SARS-CoV-2 role of regulated necrosis may be mediated by killing cells invaded by SARS-CoV-2, activating innate and adaptive immunity, and promoting apoptosis. In infected cells, the initiation of regulated necrosis, such as necroptosis, ferroptosis, and pyroptosis, rapidly eliminates infected cells to limit the viral spread and avoid harmful host pathogenesis (35). Similarly, regulated necrosis, a third mechanism by which TNF contributes to innate immune control of pathogens, strengthens antiviral responses in regulating virus-induced inflammation and other inflammatory processes (22).
Furthermore, necroptosis releases damage-associated molecular patterns (DAMP) to induce robust cross-priming of CD8+ T cells (36). Again, the close interaction between necroptosis and apoptosis also removes infectious cells that terminate virus infection. It seems that apoptosis at the initial stage of the disease wreaks havoc on viral replication (37). Complex IIa induced by deubiquitination of RIPK1 activates caspase-8 and further facilitates apoptosis (13). However, when caspase-8 is blocked, complex IIb is formed and starts necroptosis (13). The two pathways can counteract the upregulation of the cellular FLICE inhibitory protein, an antiapoptotic protein, which defers cell death and benefits SARS-CoV-2 replication (37, 38). Consequently, inhibiting regulated necrosis might be an emerging viral immune evasion strategy.
However, due to the replication and liberation of SARS-CoV-2, regulated necrosis releases a large number of pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP), including inflammatory cytokines and cell contents such as SARS-CoV-2 particles, chemokines, lactate dehydrogenase, adenosine triphosphate (ATP), and reactive oxygen species (ROS). These mediators are beneficial in causing an immune and inflammatory response of the surrounding cells (39, 40). At the same time, adjacent epithelial and endothelial cells are induced to release pro-inflammatory mediators during necrosis (41). In addition, neutrophil infiltration, macrophage activation, Th17, and the high mobility group box-1 protein (HMGB1) promote the inflammatory cascade reaction, which eventually leads to a cytokine storm in host cells and cytokine release syndrome (CRS) in the human body (42–44).
Cytokine storm and inflammatory immune reactions, mainly due to uncontrolled necrosis, occupy a crucial position in COVID-19 complications (45). Due to vascular leakage, inflammatory cells such as T cells, monocytes, and macrophages are recruited from the blood to the lungs (46). Inflammatory mediators such as IL-2, IL-6, and tumor necrosis factor (TNF) are also infiltrated by inflammatory cells into the lungs due to increased production, destroying lung structure (47). In addition, aerobic granular sludge and SARS-CoV-2 RNA spread in the bloodstream, produce immune complexes, and deposit in target organs such as the kidneys, causing a severe inflammatory cascade (48). Furthermore, the cytokine storm reaches other organs through the vascular system (44), leading to multiple organ dysfunction syndromes (MODS) and sequential cell necrosis. For example, pyroptosis and necroptosis are effective mechanisms for the secretion of IL-1β and IL-18, ATP, HMGB1, S100 proteins, and IL-1α, leading to acute phase reactions, inflammatory tissue damage, fever, cytokine release syndrome, neurotoxicity, and potential systemic organ failure in sepsis and other diseases (49).
In addition to regulated necrosis, traditional necrosis can be caused directly by vital pathogenic factors or developed from reversible damage in COVID-19 (13). COVID-19 causes necrotic damage to the lungs, and the resulting cytokine storm can also spread to multiple organs through the circulatory system, resulting in cell necrosis of various organs and leading to MODS (50). Kidney injury and subsequent clinical complications such as hematuria and proteinuria are present in approximately 40% of COVID-19 patients (51). Nephrocortical necrosis is caused by an ACE2 pathway, acute tubular necrosis, and hypercoagulation (51). Neurocyte necrosis is present in patients with acute demyelinating encephalomyelitis (52), and dermatic necrosis in COVID-19 is characterized by infectious exanthemas (41). Data in China showed that one in five patients with COVID-19 has myocardial necrosis (53), with an inflammation response and cytokine storm as possible mechanisms (54). Furthermore, trophoblast necrosis appears in the placentas in pregnant women (55). SARS-CoV-2-induced vasculitis can induce fibrinoid necrosis of small vessel walls (56).
Based on an increasing understanding of the complexity of regulating host cell necrosis in COVID-19 and the connection with the intrinsic immune defense mechanism of SARS-CoV-2 infection (innate immunity of cells), we speculate that regulated necrosis plays a double-edged sword role in COVID-19 and seems to depend on many factors, including downstream effector molecules (various cytokines and chemokines), the specific cell death pathway involved, and the immune response at different clinical stages of COVID-19 progression. However, the driving factors of regulated necrosis still need more studies to fully explain this mechanism.
2.1 Necroptosis in COVID-19
Necroptosis is an essential programmed cell death model with a necrotic morphology mediating by RIPK1, RIPK3, and MLKL (57, 58). Previous research has shown that necroptosis could be a double-edged sword during viral infection (59). Necroptosis can lead to cell suicide to prevent viral replication completion and further block disease progression (60). Furthermore, necroptosis stimulates wild antiviral immune responses and accelerates adequate viral clearance from infected organs when apoptosis is inhibited (61). However, with cell rupture, intracellular viruses successfully evade cell death and spread throughout the body (62). Furthermore, uncontrolled lytic cell death can cause tissue injury and severe diseases, including acute respiratory distress syndrome (ARDS), neurodegenerative diseases, and inflammatory diseases (62).
Investigating whether SARS-CoV-2 can cause necroptosis begins with the human lung cancer cell line Calu-3 (28). After Calu-3 cells were infected with SARS-CoV-2, MLKL phosphorylation (pMLKL) was up-regulated in infected cells, and antibody staining showed that pMLKL was expressed in the cell membrane. Scientists inactivated infected cells with ultraviolet light, which prevented viral replication and phosphorylation of MLKL, suggesting that necroptosis is virus-dependent (28). Excessive immune response and cytokine production were discovered at the SARS-CoV-2 infection site (10). Necroptosis creates an abundant inflammatory environment by releasing DAMPs to recruit immune cells and chemokines or cytokines to prevent virus infection (63). Critical cytokines discovered in the SARS-CoV-2 condition include TNF-α, IL-1β, and IL-6, all common cytokines released in necroptosis (64–66). However, these robust inflammatory responses lead to complications in COVID-19 patients, such as ARDS, vascular injury, and neurological sequelae leading to physiological deterioration and death (67–69). The discussion elaborates on the double-edged sword effects of necroptosis in COVD-19. The mechanism of necroptosis in COVID-19 is described in Figure 1. However, relevant research in this field is still relatively scarce, and detailed mechanisms need to be explored.
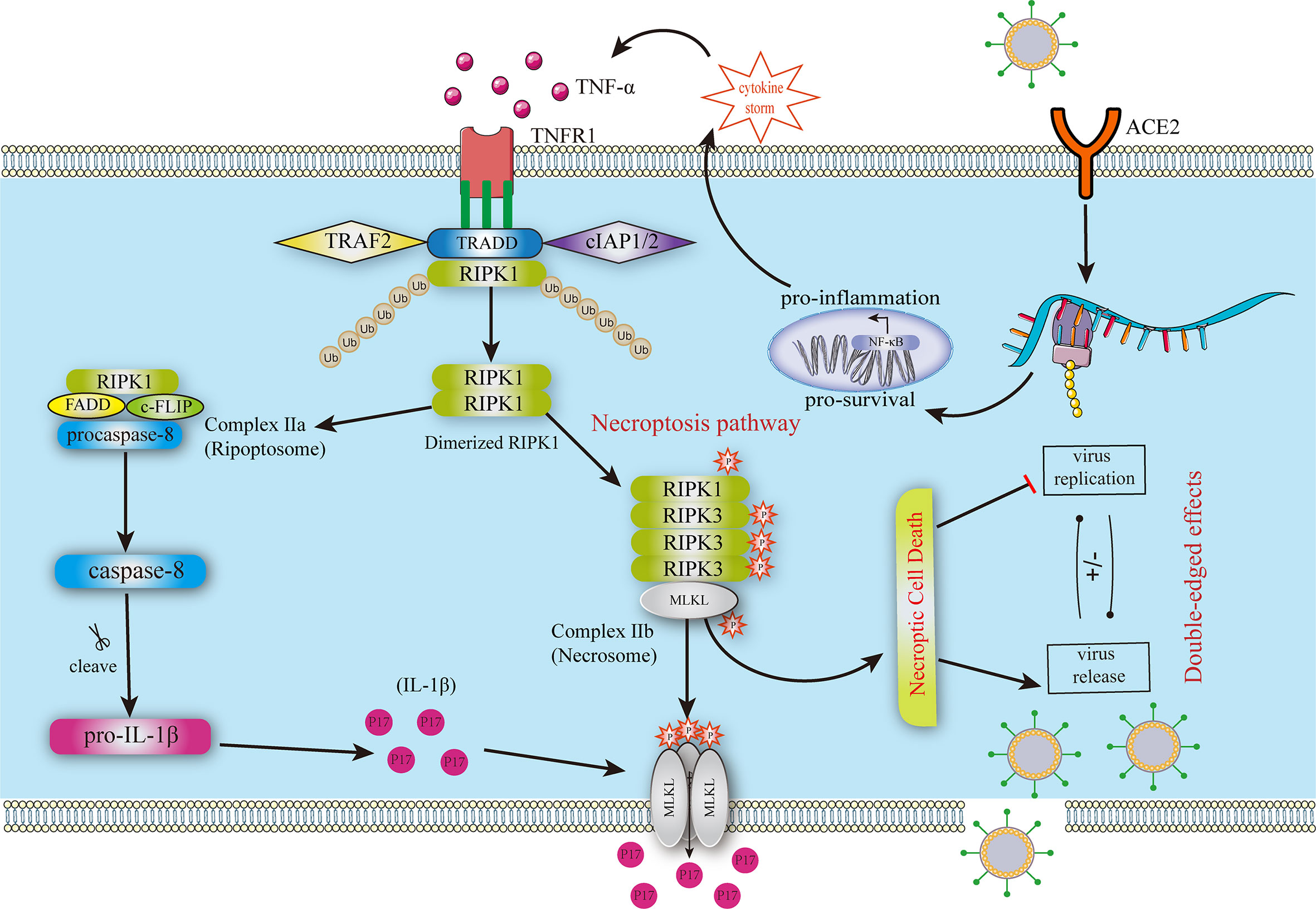
Figure 1 Mechanism of necroptosis in COVID-19. The SRAS-CoV-2 infection leads to severe cytokine storms contributing to necroptosis in uninfected cells. TNF-α binds to TNFR1, forming a stable complex that deubiquitinates and includes complexes II A and II B Complex II b contains phosphorylated RIPK1, RIPK3, and MLKL, triggering the necroptosis pathway. MLKL is oligomerized to form pores in the membrane, leading to cytokine leakage. The other two pathways lead to caspase-8 production, including the RIPK1-independent and the RIPK1-dependent pathways. Then, caspase-8 promotes the production and leakage of IL-1β. FADD, FAS‐associated death domain; MLKL, pseudokinase similar to the mixed lineage kinase domain-like pseudokinase; RIPK1, receptor-interacting protein kinase 1; RIPK3, receptor-interacting protein kinase 3; TNF-α, Tumor Necrosis Factor-alpha; TNFR1, tumor necrosis factor receptor 1; TRADD, TNFR-associated death domain.
As one of the critical cell death pathways in SARS-CoV-2 infection, necroptosis can be orchestrated to alleviate the damage the cytokine storm causes. Therefore, some drugs that can specifically inhibit the necrotic progression of COVID-19 will hopefully contribute to the treatment of COVID-19. Necrostatin-1 (Nec-1) is a specific RIPK1 inhibitor that suppresses the necroptosis signaling pathway. Recently, Nec-1 has been found to prevent COVID-19 complications potentially (23). Furthermore, Nec-1 can alleviate the release of DAMP and pro-inflammatory cytokines, inhibit the inflammatory NF-κB pathway, and reduce ROS damage.
Furthermore, after Nec-1 administration, T cell exhaustion in patients with COVID-19 can be alleviated by modulating host defense (23). However, considering the role of viral promotion in necroptosis, here is the question: Will Nec-1 promote viral infection by inhibiting necroptosis? Relevant studies are needed to explore the connection. Primidone is another inhibitor to block RIPK1 activation and TNF-α-induced necroptosis (70). Necrosulfonamide is an effective inhibitor to decrease pMLKL (59). Additionally, nocodazole, cytochalasin B, and brefeldin A can overtly inhibit pMLKL accumulation in the membrane to prevent rupture of the plasma membrane (71). RIPK3 is a potential target with a predominant increase in SARS-CoV-2 infection (72), but there have been no necroptosis studies relevant for RIPK3 inhibitors. This may be attributed to the fact that RIPK3 also mediates other inflammatory pathways; the removal of RIPK3 does not directly show the performance of necroptosis (73).
2.2 Pyroptosis in COVID-19
Pyroptosis, an essential component of the human antiviral innate immune system and one of the critical pathways of programmed cell death after SARS-CoV-2 infection, depends on recognising pattern recognition receptors and the activation and assembly of the inflammasome. Inflammasome activation triggers the influx of sodium ions and water-mediated by gasdermin D, resulting in pyroptosis and the maturation and release of cytokines. PAMPs and DAMPs, such as SARS-CoV-2 RNA, activate cytoplasmic pattern recognition receptors (74), among which the NACHT, LRR, and PYD domains contain protein 3 (NLRP3), an essential receptor that causes pyroptosis.
Pyroptosis may play a dual role in antiviral and viral promotion in the COVID-19 process: pyroptosis is critical for the induction of effective antiviral immune responses and disease resolution to limit pathogen infection and kill viruses-invading cells. A protective role of inflammasome signaling and IL-1β release has been demonstrated against multiple pathogens, particularly in the acute phase of the disease (75). Studies in patients with mild asymptomatic COVID-19 suggest that the activation of inflammasomes and pyroptosis may benefit the host due to the initiation of the protective response against SARS-CoV-2. However, its prolonged late-stage activation and uncontrolled pyroptosis may be the basis for immunopathology with the rapid release of a substantial number of PAMPs (SARS-CoV-2 particles, cell contents, etc.), DAMPs (ATP, ROS, etc.), and excessive production of pro-inflammatory cytokines (IL-1α/β, IL-6, TNF, etc.) and chemokines into the inflammatory microenvironment to recruit more immune cells (39). The activation of macrophages and HMGB1, as well as neutrophil infiltration, can promote the inflammatory chain reaction of surrounding cells, causing an excessive immune response and tissue damage that eventually leads to severe COVID-19 and an increased risk of ARDS (39, 42–44, 75). ARDS results from dysregulated hyperinflammation, rather than viral replication or infection that causes lung injury (76).
Additionally, SARS-CoV-2 antigens and RNA are disseminated in the bloodstream after pyroptosis and likely produce immune complexes that deposit in target organs, such as the kidneys, to induce a severe inflammatory cascade (48) and may explain the poor prognosis of severe COVID-19. Besides, many biomarkers in the models of death in COVID-19 have been described in Table 1 to predict immune responses and further evaluate the prognosis. Pyroptosis is also the key cause of overexuberant inflammation and cytokine release observed in severe and fatal cases of COVID-19, damage to the lung endothelium accompanied by immune cell infiltration, and systemic hypercoagulability (88). Targeting the NLRP3 Inflammasome and mitigating aberrant inflammatory responses may play an important role in severe COVID-19 and complications of SARS-CoV-2 infection (48). Mechanisms, potential targets, and double-edged sword effects of the NLRP3 inflammasome and pyroptosis in COVID-19 are described in Figure 2.
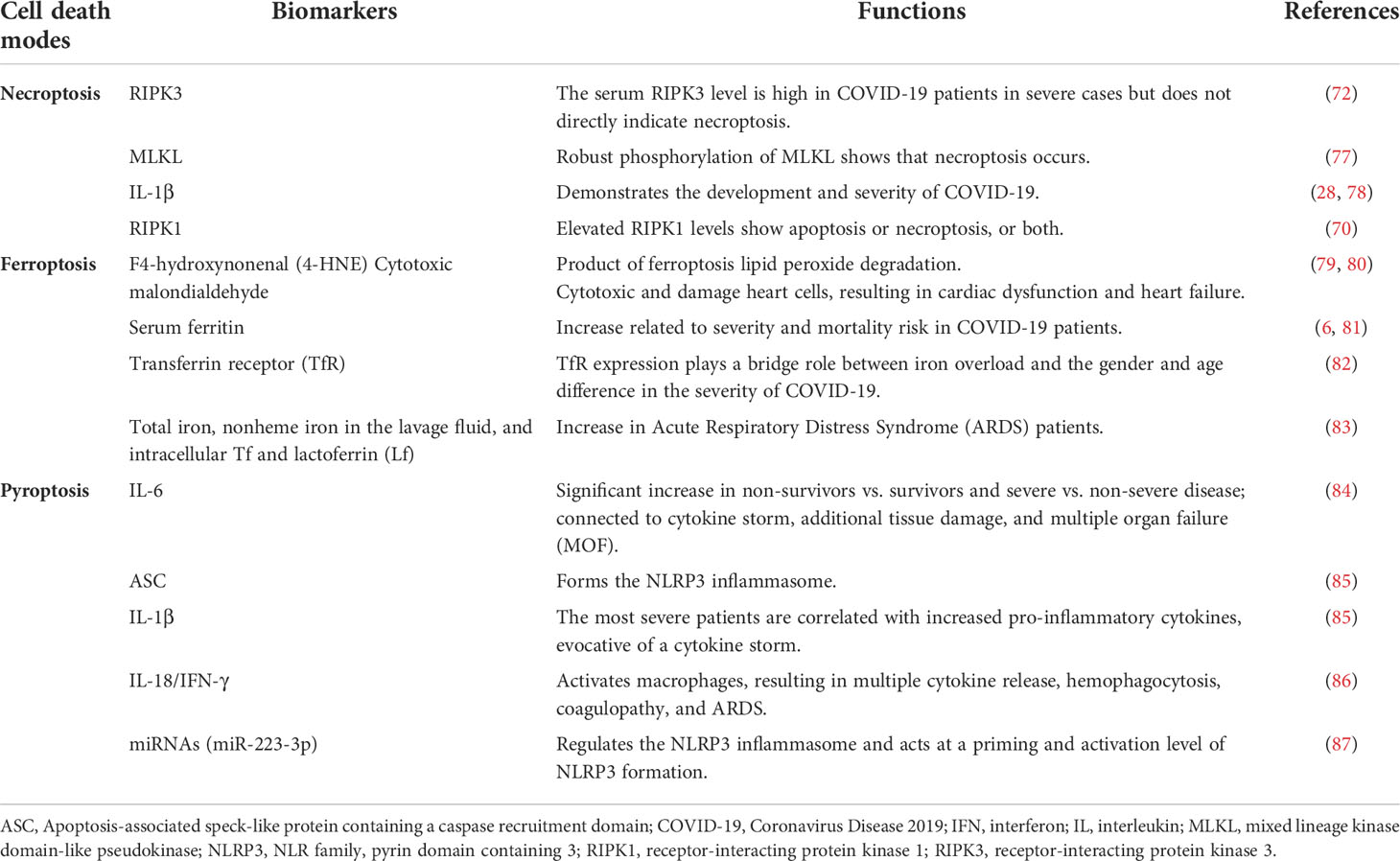
Table 1 Biomarkers in the modes of cell death in COVID-19.
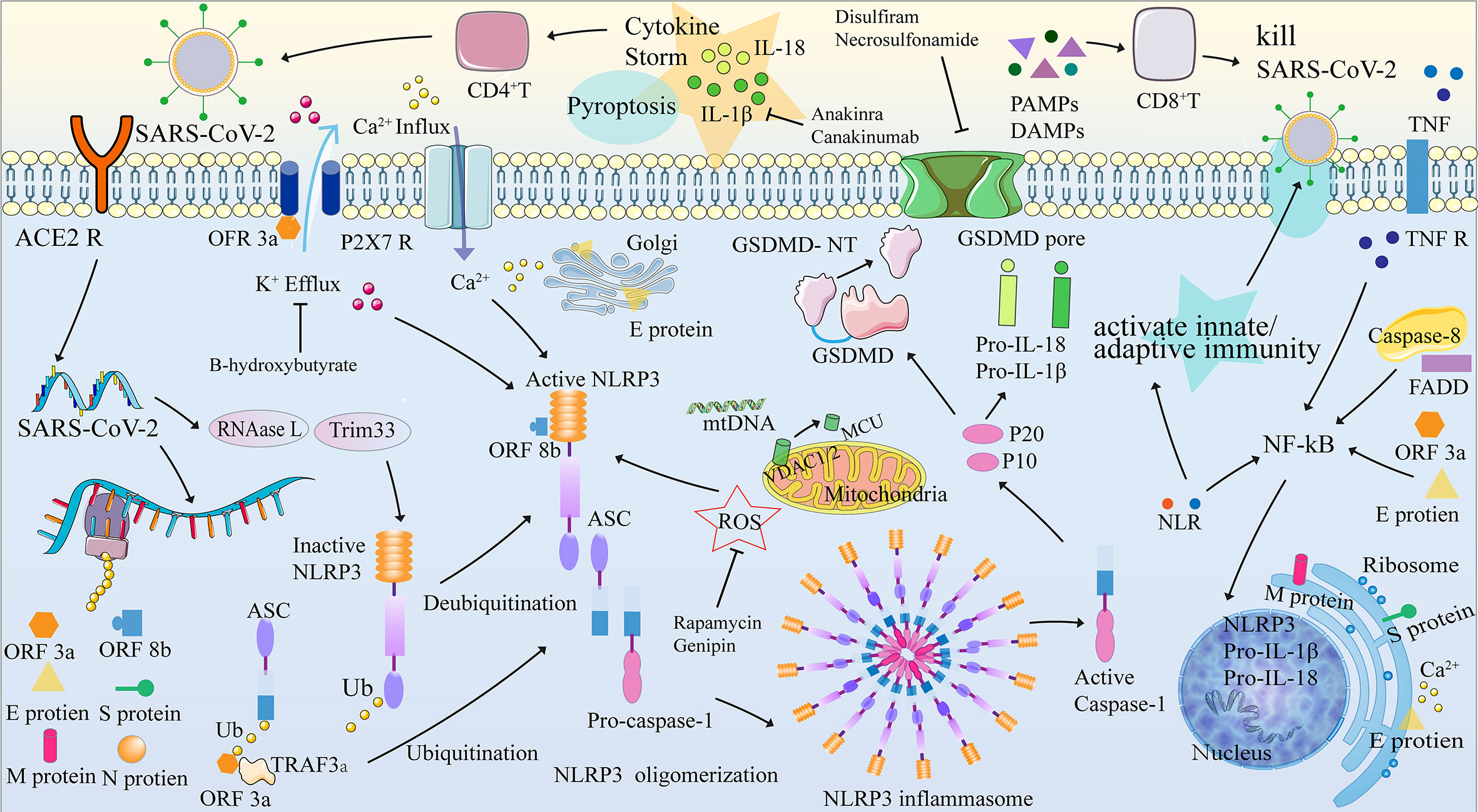
Figure 2 NLRP3 inflammasome, pyroptosis, and their mechanisms in COVID-19. The pyroptosis process can be divided into four stages: (1) activation, (2) formation of the inflammasome, (3) release of IL-1 β and IL-18, and (4) GSDMD-induced pyroptosis. When the SARS-COV-2 protein binds to the ACE2 receptor and is internalized by endocytosis, the SARS-COV-2 RNA is translated and replicated, transcribing the structural proteins ORF3a, ORF8b, and SARS-COV-2 (N, S, M and E proteins). The NLRP3 inflammasome assembly signal, provided by PAMP and DAMP, is involved in the imbalance of intracellular ion concentration (Ca2+ concentration increased and K+ concentration decreased), mitochondrial dysfunction leading to release of ROS, Ca2+ and mtDNA, cardiolipin translocation of cardiolipin from inner to the outer mitochondrial membrane, phagocyte and lysosome rupture that releases cathepsin, etc. Activation of NLRP3 results in recruitment, oligomerization, and binding to ASCs, which then recruit and bind to pro-caspase-1 through their shared domain to drive the assembly of NLRP3-ASC-procaspase-1. The formation of the NLRP3 inflammasome leads to two results: (1) the release of IL-1 β and IL-18 and (2) GSDMD-induced pyroptosis. ACE2R, Angiotensin converting enzyme 2 receptor; ASC, Apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC); ATP, adenosine triphosphate; BTK, Bruton’s tyrosine kinase; Ca/CaMK, Calcium/Calmodulin-dependent protein kinase; CpG, Cytosine Phosphate Guanosine; DAMPs, damage-associated molecular patterns; ER, endoplasmic reticulum; FADD, Fas-associated death domain; Golgi, Golgi apparatus; GSDMD-NT, gasdermin D-N terminal; IL, Interleukin; IRAK, Interleukin-1 receptor-associated kinase; LRR, leucine-rich repeat; MCU, mitochondrial Ca2+ uniporter; MtDNA, mitochondrial DNA; MyD88, myeloid differentiation factor 88; NF-κB, Nuclear factor-κB; NLRP3, NLR family, pyrin domain containing 3; NOD, nucleotide-binding and oligomerization; ORF3a, Open reading frame 3a; ORF8b, Open reading frame 8b; PAMPs pathogen-associated molecular patterns; PRR, pattern recognition receptor; P2X7R, purinergic ligand-gated ion channel 7 receptor; RNAase L, latent endoribonuclease; ROS, reactive oxygen species; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; TRAF3, TNF receptor-associated factor 3; Trim33, Tripartite motif-containing protein 33; TLR 4, Toll-like receptor 4; JAKUb, Ubiquitin; VDAC1/2, voltage-dependent anion-selective channel ½.
A deep understanding of the molecular mechanisms and role of pyroptosis will allow selective interference of the deleterious actions of pyroptosis in pathological contexts and promote the beneficial effects for therapeutic purposes. Many potential therapeutic agents and their targets have been studied to manipulate the COVID-19-related immune response and pyroptosis, summarized in Table 2. As the mechanisms of pro-infective activity by pyroptosis are well understood, inhibiting the pyroptosis process can limit intracellular replication of SARS-CoV-2 and inhibit extensive cytokine storm and tissue inflammation induced by the NLRP inflammasome by causing pyroptosis, which is expected to be used in COVID-19 treatment (88). Disulfiram and necrosulfonamide, effective inhibitors of pyroptosis and GSDMD pore formation (88, 103), can limit intracellular virus replication and inhibit extensive cytokine storm and tissue inflammation by causing apoptosis, which is also expected to be used in COVID-19 treatment (104). A phase 2 randomized (2:1), double-blind placebo-controlled trial of disulfiram evaluates the effect of disulfiram on the severity of COVID-19 symptoms, the viral load of SARS-CoV-2, and biomarkers of inflammation and pyroptosis over 31 days. (ClinicalTrials.gov Identifier: NCT04485130). Furthermore, rapamycin and genipin can significantly inhibit the expression level of the NLRP3 inflammasome-related protein (93) or alleviate the inflammatory response by activating the antioxidant system (24). Recently, a single-center double-blind placebo-controlled randomized clinical trial assessed the clinical effectiveness of rapamycin in minimizing or decreasing the severity of (acute lung injury/ARDS) in participants infected with mild to moderate COVID-19 (ClinicalTrials.gov Identifier: NCT04482712). B-hydroxybutyrate can inhibit K+ outflow and caspase-1 activation (92), which may greatly benefit patients with COVID-19. A randomized placebo-controlled double-blind cross-over acute intervention study showed that beta-hydroxybutyrate had acute beneficial hemodynamic effects in patients with heart failure and healthy controls (ClinicalTrials.gov Identifier: NCT04573764).
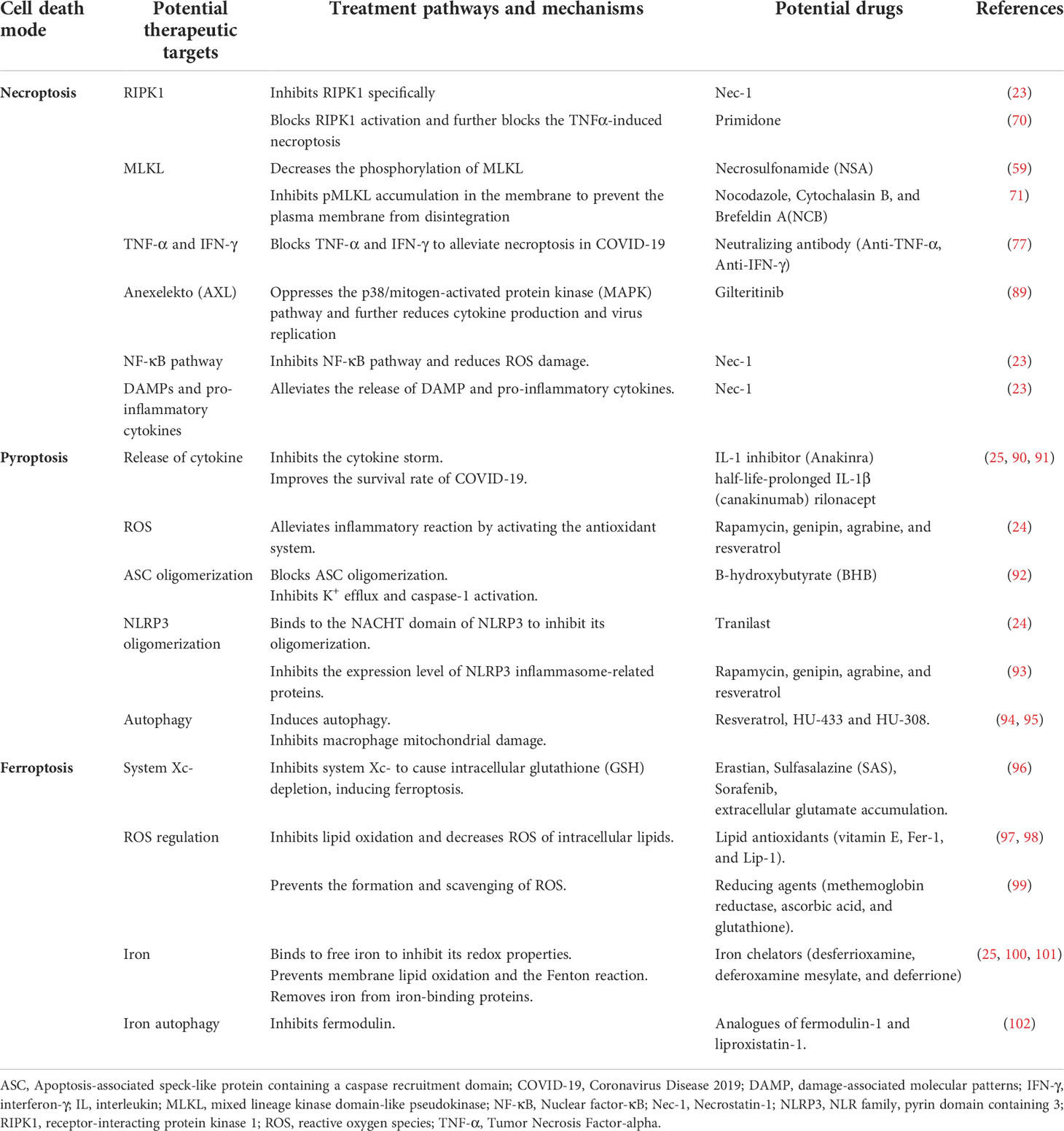
Table 2 Treatment targets and mechanisms in the modes of cell death in COVID-19.
Although the appropriate phage for patients with COVID-19 to accept pharmacological interventions is worth exploring, much remains obscure regarding potential agonists to provide a host for pyroptosis Downstream cytokine antagonists may have advantages in treating COVID-19, given that the overexpression of IL-1β, specifically in the lungs, is sufficient to recapitulate many of the ARDS phenotypes (90). Anakinra, an inhibitor of IL-1, and canakinumab, a half-life-prolonged IL-1β, may have potential benefits in the treatment of COVID-19, for which prospective randomized controlled trials are currently underway (90). RNA sequencing of the early recovery stage of COVID-19 showed that classical CD14++ and CD14++ IL-1β+;monocytes are of greater abundance with high expression of inflammatory genes, indicating that IL-1β may be a potential target for the COVID-19 intervention (105). Furthermore, existing retrospective cohort trials have shown that high doses of anakinra, with or without dexamethasone, can improve the survival rate of COVID-19 (25, 91). However, a prospective study did not show clinical results and required further investigation (106).
2.3 Ferroptosis in COVID-19
Due to intracellular iron overload caused by many factors such as hepcidin, transferrin receptor (TfR), free iron release, and inhibition of GPX4 (107–111), ferroptosis is an iron-dependent programmed cell death separate from pyroptosis. It is characterized by an increase in membrane lipid peroxide levels and is accompanied by a decrease in glutathione and GPX4 expression levels (112–115). Therefore, the imbalance of intracellular iron homeostasis caused by SARS-CoV-2, which leads to fatal ferroptosis, is closely related to the mechanism of COVID-19 and the pathophysiological process of various complications. The means of ferroptosis in COVID-19 are described in Figure 3.
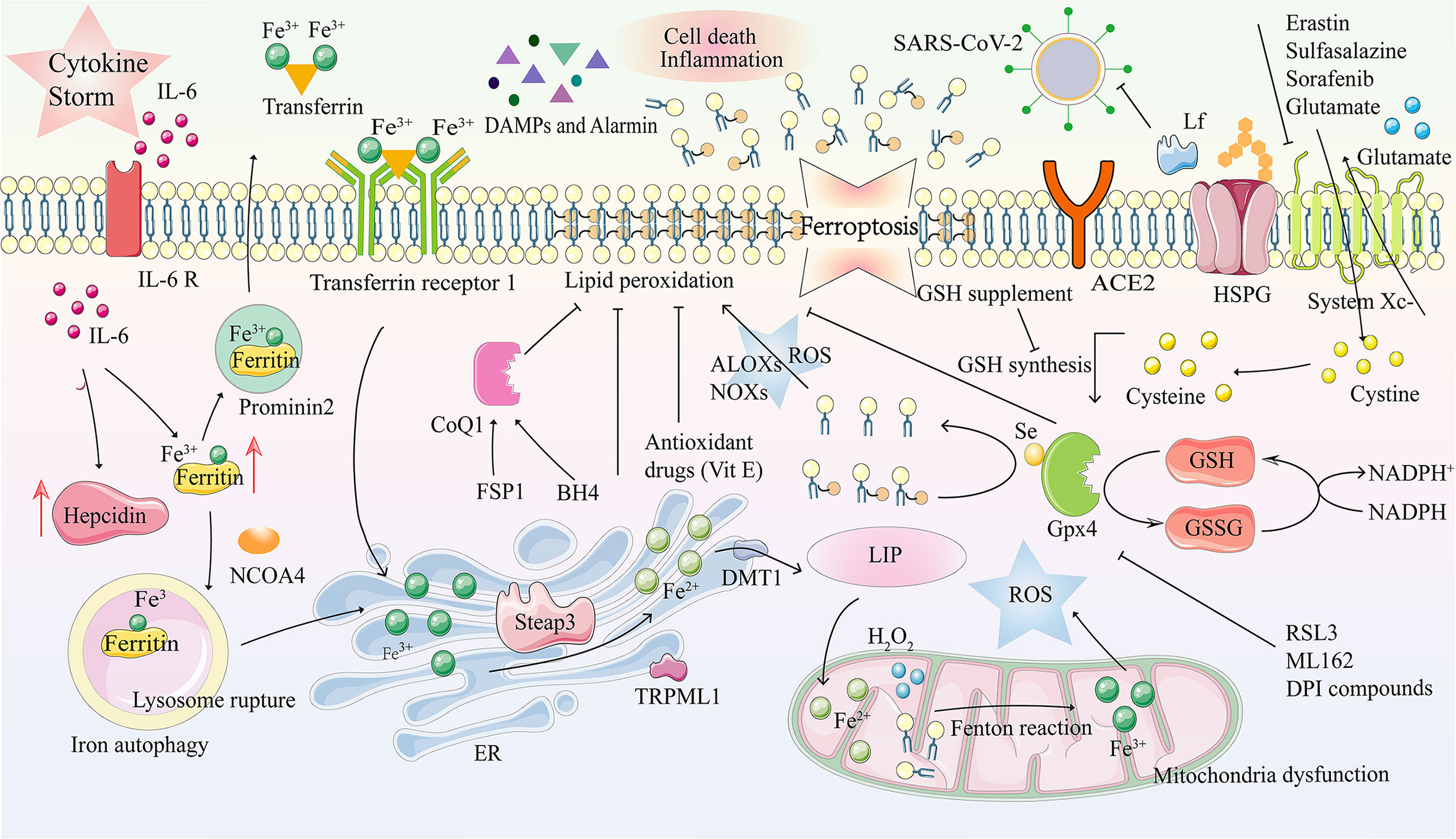
Figure 3 Ferroptosis and its mechanism in COVID-19. Ferroptosis is a kind of programmed cell death characterized by an imbalance in intracellular iron metabolism or a distortion of the glutathione peroxidation pathway. The transferrin receptor recognizes excess transferrin carrying Fe3+ and enters cells through endocytosis after SARS-COV-2 infection. Metal reductase Steap3 reduces Fe ions from trivalent to divalent, while iron channels DMT1 and TRPML1 in the endosome membrane transport Fe2+ to the cytoplasm, accompanied by iron accumulation. In the case of intracellular iron overload, chemical substances in the mitochondrial electron transfer chain react with H2O2, Fe 2+, and lipids, together inducing the Fenton reaction, which produces large amounts of ROS. Due to GPX4 depletion and iron overload in LIP, lipid, nucleic acid, and protein peroxidation results in cell membrane damage due to oxidative stress and ferroptosis. DAMPs and Alarmin (HMGB1, IL-33, TNF) are released, eventually aggravating cell death and inflammation. Tf and prominin 2 can effectively excrete iron from cells and inhibit ferroptosis. ACE2R, Angiotensin-converting enzyme 2 receptor; ALOXs, arachidonate lipoxygenases; BH4, tetrahydrobiopterin; CoQ10, coenzyme Q10; DAMP, damage-associated molecular patterns; DFO, deferoxamine; DMT1, divalent metal transporter 1; DPI, diphenyleneiodonium; ER, endoplasmic reticulum; FSP1, ferroptosis suppressor protein 1; GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, oxidized GSH; HMGB1, high mobility group box-1 protein; HSPGs, heparan sulfate proteoglycans; H2O2, hydrogen peroxide; IL, interleukin; Lf, lactoferrin; LIP, labile iron pool; LOXs, lysyl oxidases; NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, nicotinamide adenine dinucleotide phosphate; NCOA4, nuclear receptor coactivator 4; NOXs, NADPH oxidases; PLOOH, phospholipid hydroperoxide; PUFAs, polyunsaturated fatty acids; ROS, reactive oxygen species; RSL3, 1S,3R-RSL3; Se, selenocysteine; Steap3, six-transmembrane epithelial antigen of prostate 3; TFR1, transferrin receptors 1; TNF, tumor necrosis factor; TRPML1, transient receptor potential mucolipin 1; VitE, vitamin E.
More and more evidence has shown that excessive inflammatory reactions and complications caused by COVID-19, such as acute respiratory distress, heart and kidney injury, and dysfunction of the blood and immune system, are closely related to oxidative stress and ferroptosis (116, 117). Elevated serum ferritin levels caused by COVID-19-related hyperinflammation are likely to trigger further tissue damage (116). Ferroptosis of immune cells during infection is considered immunogenic and pro-inflammatory, beneficial for disease progression, and causes cell death, which may explain the clinical characteristics of MODS in COVID-19 (118). Cell death can induce the release of DAMPs, PAMPs, and alarmins recognized by immune receptors and, finally, aggravate ferroptosis and inflammation (118). One of the main processes in the pathogenesis of COVID-19 infected with SARS-CoV-2 may be the imbalance of intracellular iron homeostasis and fatal ferroptosis (119). Four possible factors mainly induce this process: massive release of the iron homeostasis regulator Hepcidin, excessive iron influx dependent on the transferrin receptor (TfR), which mediates cellular iron uptake through endocytosis of iron-loaded transferrin during SARS-CoV-2 replication, attack of hemoglobin to release free iron into the circulation, and inhibition of GPX4 by SARS-CoV-2 (107–109).
ROS, reactive nitrogen species, and reactive sulfur species (116, 120, 121) produced during pathological conditions can cause oxidative damage and ischemia-reperfusion injury to different organs, such as the lungs, liver, kidney, heart, intestine, and brain (122–124). To a great extent, ferroptosis is associated with hyperinsulinemia, hypercoagulable state, hemorrhagic stroke injury, shock, and MODS (123, 125, 126). Iron overload is also cardiotoxic; the Haber-Weiss and Fenton reactions produce harmful hydroxyl radicals that increase ROS levels in the heart and oxidative stress. Malondialdehyde and F4-hydroxynonenal are cytotoxic and damage heart cells, leading to cardiac dysfunction and heart failure. Furthermore, iron overload may play a role in the hypercoagulable state of patients with severe COVID-19. The destruction of hemoglobin increases the amount of free iron in the blood, leading to iron-induced oxidative stress, thrombocytosis, and changes in red blood cell viscosity. Then fibrinogen is transformed into fibrin clots, leading to pathological thrombosis. Furthermore, hemoglobin loses its ability to bind to oxygen and deliver to major organs, leading to multiple organ failures (127).
Previous studies have shown that iron overload plays a vital role in the pathogenesis of multiple system diseases caused by SARS-CoV-2 infections (31). Reducing iron levels in infected cells can effectively inhibit virus growth and disease progression caused by SARS-CoV-2 (128). Lipophilic antioxidants, including vitamin E, ferrostatin-1, and liproxstatin-1, can alleviate ferroptosis by inhibiting lipid autoxidation, which may have potential value in treating COVID-19 (97, 98). Iron chelators, such as deferoxamine, deferoxamine mesylate, and deferiprone, can bind to free iron to inhibit its redox properties, prevent the Fenton reaction, down-regulate hepcidin, remove iron from iron-binding proteins, etc. (100, 101, 128). Treatment of COVID-19 with reducing agents, including methemoglobin reductase, ascorbic acid, and glutathione to prevent formation and scavenging, may be of great value (99). A new generation of ferroptosis inhibitors, such as improved ferrostatin-1 and liproxstatin-1 analogs, may also be potential drug candidates for COVID-19 (102). The process and therapeutic targets of ferroptosis in COVID-19 are described in Figure 3.
3 Conclusions
In conclusion, this review elaborates on the biochemical and molecular mechanisms of regulated necrosis in COVID-19 and introduces the latest research progress. Furthermore, we discuss the two sides of regulated necrosis to provide a new view on the treatment of COVID-19. After analyzing the difference between traditional and regulated necrosis, we showed the overall effects of necrosis on the pathophysiology of COVID-19. However, to better understand the two sides of regulated necrosis, future research should investigate the following problems: (i) the driving factors of activation in regulated necrosis in COVID-19; (ii) the possible mechanism of how inflammatory molecular interaction and the influence of the inflammatory environment caused by SARS-CoV-2 affect the regulation of regulated necrosis; and (iii) the effects of regulated necrosis on the regulation of immune cells, antiviral immunity, and the efficiency of targeted therapy. In clinical use, diagnostic criteria, disease severity classification system, combined antiviral treatment, secondary infection, and cytokine measurement should be considered. The disease severity classification system is vital to prompt the initiation of immunomodulatory therapy, which may be beneficial only for severe cases of COVID-19. Still, aggressive anti-inflammatory therapy can prevent progression in mild or moderate patients. The timing of treatment, the variability in the course of the disease, and the proper patient stratification will be essential for identifying the ‘sweet spot’ (appropriate time and stage of inflammasome-inhibiting interventions). In short, understanding the double-edged sword of regulated necrosis with SARS-CoV-2-manipulated molecular details and applying drugs to target regulated necrosis or its downstream pathway alone or in combination with other immunotherapy methods will have great potential as a new treatment method for COVD-19. With further studies in progress, promising results will emerge in the future.
Author contributions
CS, YH, and RZ: writing the initial draft preparation, table and figure production in equal contribution, with the help of JinW, YZ, and XC; JMW and JiaW: conceptualization, review, and critical revision. All authors: literature search, review, commentary, and final approval of the manuscript.
Funding
JMW was supported by the Zhengzhou University Education and Teaching Reform Research and Practice Project (2021ZZUJGLX219). This research was also supported in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sreepadmanabh M, Sahu AK, Chande A. COVID-19: Advances in diagnostic tools, treatment strategies, and vaccine development. J Biosci (2020) 45(1):148. doi: 10.1007/s12038-020-00114-6
2. World Health Organization. Coronavirus (COVID-19) dashboard data table. Available at: https://covid19.who.int/.
3. Carvalho T, Krammer F, Iwasaki A. The first 12 months of COVID-19: a timeline of immunological insights. Nat Rev Immunol (2021) 21(4):245–56. doi: 10.1038/s41577-021-00522-1
4. Mallah SI, Ghorab OK, Al-Salmi S, Abdellatif OS, Tharmaratnam T, Iskandar MA, et al. COVID-19: breaking down a global health crisis. Ann Clin Microbiol Antimicrob (2021) 20(1):35. doi: 10.1186/s12941-021-00438-7
5. Aleksova A, Gagno G, Sinagra G, Beltrami AP, Janjusevic M, Ippolito G, et al. Effects of SARS-CoV-2 on cardiovascular system: The dual role of angiotensin-converting enzyme 2 (ACE2) as the virus receptor and homeostasis regulator-review. Int J Mol Sci (2021) 22(9). doi: 10.3390/ijms22094526
6. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
7. Iwasaki M, Saito J, Zhao H, Sakamoto A, Hirota K, Ma D. Inflammation triggered by SARS-CoV-2 and ACE2 augment drives multiple organ failure of severe COVID-19: Molecular mechanisms and implications. Inflammation (2021) 44(1):13–34. doi: 10.1007/s10753-020-01337-3
8. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med (2017) 377(6):562–72. doi: 10.1056/NEJMra1608077
9. Harrison AG, Lin T, Wang P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol (2020) 41(12):1100–15. doi: 10.1016/j.it.2020.10.004
10. Alshammary AF, Al-Sulaiman AM. The journey of SARS-CoV-2 in human hosts: a review of immune responses, immunosuppression, and their consequences. Virulence (2021) 12(1):1771–94. doi: 10.1080/21505594.2021.1929800
11. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature (2020) 584(7821):463–9. doi: 10.1038/s41586-020-2588-y
12. Nishiga M, Wang DW, Han Y, Lewis DB, Wu JC. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol (2020) 17(9):543–58. doi: 10.1038/s41569-020-0413-9
13. D'Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int (2019) 43(6):582–92. doi: 10.1002/cbin.11137
14. Xu T, Ding W, Tariq MA, Wang Y, Wan Q, Li M, et al. Molecular mechanism and therapy application of necrosis during myocardial injury. J Cell Mol Med (2018) 22(5):2547–57. doi: 10.1111/jcmm.13575
15. Schwartz DA, Morotti D. Placental pathology of COVID-19 with and without fetal and neonatal infection: Trophoblast necrosis and chronic histiocytic intervillositis as risk factors for transplacental transmission of SARS-CoV-2. Viruses (2020) 12(11). doi: 10.3390/v12111308
16. Tonnus W, Belavgeni A, Beuschlein F, Eisenhofer G, Fassnacht M, Kroiss M, et al. The role of regulated necrosis in endocrine diseases. Nat Rev Endocrinol (2021) 17(8):497–510. doi: 10.1038/s41574-021-00499-w
17. Yuan X, Nie W, He Z, Yang J, Shao B, Ma X, et al. Carbon black nanoparticles induce cell necrosis through lysosomal membrane permeabilization and cause subsequent inflammatory response. Theranostics (2020) 10(10):4589–605. doi: 10.7150/thno.34065
18. Martin-Sanchez D, Poveda J, Fontecha-Barriuso M, Ruiz-Andres O, Sanchez-Niño MD, Ruiz-Ortega M, et al. Targeting of regulated necrosis in kidney disease. Nefrologia (Engl Ed) (2018) 38(2):125–35. doi: 10.1016/j.nefroe.2018.02.004
19. Tonnus W, Meyer C, Paliege A, Belavgeni A, von Mässenhausen A, Bornstein SR, et al. The pathological features of regulated necrosis. J Pathol (2019) 247(5):697–707. doi: 10.1002/path.5248
20. Giżycka A, Chorostowska-Wynimko J. [Programmed necrosis and necroptosis - molecular mechanisms]. Postepy Hig Med Dosw (Online) (2015) 69:1353–63. doi: 10.5604/17322693.1186337
21. Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: Mechanisms and relevance to disease. Annu Rev Pathol (2017) 12:103–30. doi: 10.1146/annurev-pathol-052016-100247
22. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell (2009) 137(6):1112–23. doi: 10.1016/j.cell.2009.05.037
23. Cao L, Mu W. Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol Res (2021) 163:105297. doi: 10.1016/j.phrs.2020.105297
24. Zhao T, Zhang Y, Mu S, Park JP, Bu H, Leng X, et al. Protective effects of genipin on ethanol-induced acute gastric injury in mice by inhibiting NLRP3 inflammasome activation. Eur J Pharmacol (2020) 867:172800. doi: 10.1016/j.ejphar.2019.172800
25. Bozzi G, Mangioni D, Minoia F, Aliberti S, Grasselli G, Barbetta L, et al. Anakinra combined with methylprednisolone in patients with severe COVID-19 pneumonia and hyperinflammation: An observational cohort study. J Allergy Clin Immunol (2021) 147(2):561–566.e4. doi: 10.1016/j.jaci.2020.11.006
26. Kondylis V, Kumari S, Vlantis K, Pasparakis M. The interplay of IKK, NF-κB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol Rev (2017) 277(1):113–27. doi: 10.1111/imr.12550
27. Zheng M, Williams EP, Malireddi RKS, Karki R, Banoth B, Burton A, et al. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J Biol Chem (2020) 295(41):14040–52. doi: 10.1074/jbc.RA120.015036
28. Li S, Zhang Y, Guan Z, Li H, Ye M, Chen X, et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct Target Ther (2020) 5(1):235. doi: 10.1038/s41392-020-00334-0
29. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol (2017) 18(2):127–36. doi: 10.1038/nrm.2016.149
30. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: A new frontier in cancer. BioMed Pharmacother (2020) 121:109595. doi: 10.1016/j.biopha.2019.109595
31. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
32. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity (2013) 38(2):209–23. doi: 10.1016/j.immuni.2013.02.003
33. Qin X, Ma D, Tan YX, Wang HY, Cai Z. The role of necroptosis in cancer: A double-edged sword? Biochim Biophys Acta Rev Cancer (2019) 1871(2):259–66. doi: 10.1016/j.bbcan.2019.01.006
34. Felsenstein S, Herbert JA, McNamara PS, Hedrich CM. COVID-19: Immunology and treatment options. Clin Immunol (2020) 215:108448. doi: 10.1016/j.clim.2020.108448
35. Nguyen LN, Kanneganti TD. PANoptosis in viral infection: The missing puzzle piece in the cell death field. J Mol Biol (2021) p:167249. doi: 10.1016/j.jmb.2021.167249
36. Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis Sousa C, et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science (2015) 350(6258):328–34. doi: 10.1126/science.aad0395
37. Ivanisenko NV, Seyrek K, Kolchanov NA, Ivanisenko VA, Lavrik IN. The role of death domain proteins in host response upon SARS-CoV-2 infection: modulation of programmed cell death and translational applications. Cell Death Discovery (2020) 6:101. doi: 10.1038/s41420-020-00331-w
38. He MX, He YW. A role for c-FLIP(L) in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ (2013) 20(2):188–97. doi: 10.1038/cdd.2012.148
39. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517(7534):311–20. doi: 10.1038/nature14191
40. Toyokuni S, Yanatori I, Kong Y, Zheng H, Motooka Y, Jiang L. Ferroptosis at the crossroads of infection, aging and cancer. Cancer Sci (2020) 111(8):2665–71. doi: 10.1111/cas.14496
41. Novak N, Peng W, Naegeli MC, Galvan C, Kolm-Djamei I, Brüggen C, et al. SARS-CoV-2, COVID-19, skin and immunology - what do we know so far? Allergy (2021) 76(3):698–713. doi: 10.1111/all.14498
42. Cao X. COVID-19: immunopathology and its implications for therapy. Nat Rev Immunol (2020) 20(5):269–70. doi: 10.1038/s41577-020-0308-3
43. Potey PM, Rossi AG, Lucas CD, Dorward DA. Neutrophils in the initiation and resolution of acute pulmonary inflammation: understanding biological function and therapeutic potential. J Pathol (2019) 247(5):672–85. doi: 10.1002/path.5221
44. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med (2020) 8(4):420–2. doi: 10.1016/S2213-2600(20)30076-X
45. Magadum A, Kishore R. Cardiovascular manifestations of COVID-19 infection. Cells (2020) 9(11). doi: 10.3390/cells9112508
46. Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. COVID-19 infection: the perspectives on immune responses. Cell Death Differ (2020) 27(5):1451–4. doi: 10.1038/s41418-020-0530-3
47. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat Rev Nephrol (2021) 17(1):46–64. doi: 10.1038/s41581-020-00357-4
48. Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J Immunol (2020) 205(2):307–12. doi: 10.4049/jimmunol.2000513
49. Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity (2019) 50(6):1352–64. doi: 10.1016/j.immuni.2019.05.020
50. Devaux CA, Rolain JM, Raoult D. ACE2 receptor polymorphism: Susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. J Microbiol Immunol Infect (2020) 53(3):425–35. doi: 10.1016/j.jmii.2020.04.015
51. Ahmadian E, Hosseiniyan Khatibi SM, Razi Soofiyani S, Abediazar S, Shoja MM, Ardalan M, et al. Covid-19 and kidney injury: Pathophysiology and molecular mechanisms. Rev Med Virol (2021) 31(3):e2176. doi: 10.1002/rmv.2176
52. Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, et al. The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain (2020) 143(10):3104–20. doi: 10.1093/brain/awaa240
53. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol (2020) 5(7):811–8. doi: 10.1001/jamacardio.2020.1017
54. Wilson MG, Hull JH, Rogers J, Pollock N, Dodd M, Haines J, et al. Cardiorespiratory considerations for return-to-play in elite athletes after COVID-19 infection: a practical guide for sport and exercise medicine physicians. Br J Sports Med (2020) 54(19):1157–61. doi: 10.1136/bjsports-2020-102710
55. Schwartz DA, Baldewijns M, Benachi A, Bugatti M, Collins RRJ, De Luca D, et al. Chronic histiocytic intervillositis with trophoblast necrosis is a risk factor associated with placental infection from coronavirus disease 2019 (COVID-19) and intrauterine maternal-fetal severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) transmission in live-born and stillborn infants. Arch Pathol Lab Med (2021) 145(5):517–28. doi: 10.5858/arpa.2020-0771-SA
56. Iba T, Connors JM, Levy JH. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflammation Res (2020) 69(12):1181–9. doi: 10.1007/s00011-020-01401-6
57. Shan B, Pan H, Najafov A, Yuan J. Necroptosis in development and diseases. Genes Dev (2018) 32(5-6):327–40. doi: 10.1101/gad.312561.118
58. Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol (2018) 36:489–517. doi: 10.1146/annurev-immunol-042617-053010
59. Meessen-Pinard M, Le Coupanec A, Desforges M, Talbot PJ. Pivotal role of receptor-interacting protein kinase 1 and mixed lineage kinase domain-like in neuronal cell death induced by the human neuroinvasive coronavirus OC43. J Virol (2017) 91(1). doi: 10.1128/JVI.01513-16
60. Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ (2017) 24(7):1184–95. doi: 10.1038/cdd.2017.65
61. Shubina M, Tummers B, Boyd DF, Zhang T, Yin C, Gautam A, et al. Necroptosis restricts influenza a virus as a stand-alone cell death mechanism. J Exp Med (2020) 217(11). doi: 10.1084/jem.20191259
62. Garcia LR, Tenev T, Newman R, Haich RO, Liccardi G, John SW, et al. Ubiquitylation of MLKL at lysine 219 positively regulates necroptosis-induced tissue injury and pathogen clearance. Nat Commun (2021) 12(1):3364. doi: 10.1038/s41467-021-23474-5
63. Paolini A, Borella R, De Biasi S, Neroni A, Mattioli M, Lo Tartaro D, et al. Cell death in coronavirus infections: Uncovering its role during COVID-19. Cells (2021) 10(7). doi: 10.3390/cells10071585
64. Deepa SS, Unnikrishnan A, Matyi S, Hadad N, Richardson A. Necroptosis increases with age and is reduced by dietary restriction. Aging Cell (2018) 17(4):e12770. doi: 10.1111/acel.12770
65. Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents (2020) 55(5):105954. doi: 10.1016/j.ijantimicag.2020.105954
66. Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents (2020) 34(2):327–31.
67. Ferren M, Favède V, Decimo D, Iampietro M, Lieberman NAP, Weickert JL, et al. Hamster organotypic modeling of SARS-CoV-2 lung and brainstem infection. Nat Commun (2021) 12(1):5809. doi: 10.1038/s41467-021-26096-z
68. Price DR, Benedetti E, Hoffman KL, Gomez-Escobar L, Alvarez-Mulett S, Capili A, et al. Angiopoietin 2 is associated with vascular necroptosis induction in coronavirus disease 2019 acute respiratory distress syndrome. Am J Pathol (2022) 192(7):1001–1015. doi: 10.1016/j.ajpath.2022.04.002
69. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol (2020) 11:1446. doi: 10.3389/fimmu.2020.01446
70. Riebeling T, Jamal K, Wilson R, Kolbrink B, von Samson-Himmelstjerna FA, Moerke C, et al. Primidone blocks RIPK1-driven cell death and inflammation. Cell Death Differ (2021) 28(5):1610–26. doi: 10.1038/s41418-020-00690-y
71. Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun (2020) 11(1):3151. doi: 10.1038/s41467-020-16887-1
72. Nakamura H, Kinjo T, Arakaki W, Miyagi K, Tateyama M, Fujita J. Serum levels of receptor-interacting protein kinase-3 in patients with COVID-19. Crit Care (2020) 24(1):484. doi: 10.1186/s13054-020-03209-6
73. Faust H, Mangalmurti NS. Collateral damage: necroptosis in the development of lung injury. Am J Physiol Lung Cell Mol Physiol (2020) 318(2):L215–l225. doi: 10.1152/ajplung.00065.2019
74. Carty M, Guy C, Bowie AG. Detection of viral infections by innate immunity. Biochem Pharmacol (2021) 183:114316. doi: 10.1016/j.bcp.2020.114316
75. Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor fedratinib. J Microbiol Immunol Infect (2020) 53(3):368–70. doi: 10.1016/j.jmii.2020.03.005
76. Freeman TL, Swartz TH. Targeting the NLRP3 inflammasome in severe COVID-19. Front Immunol (2020) 11:1518. doi: 10.3389/fimmu.2020.01518
77. Karki R, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell (2021) 184(1):149–168.e17. doi: 10.1016/j.cell.2020.11.025
78. Fagone P, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Transcriptional landscape of SARS-CoV-2 infection dismantles pathogenic pathways activated by the virus, proposes unique sex-specific differences and predicts tailored therapeutic strategies. Autoimmun Rev (2020) 19(7):102571. doi: 10.1016/j.autrev.2020.102571
79. Aziza SA, Azab Mel S, El-Shall SK. Ameliorating role of rutin on oxidative stress induced by iron overload in hepatic tissue of rats. Pak J Biol Sci (2014) 17(8):964–77. doi: 10.3923/pjbs.2014.964.977
80. Sripetchwandee J, Pipatpiboon N, Chattipakorn N, Chattipakorn S. Combined therapy of iron chelator and antioxidant completely restores brain dysfunction induced by iron toxicity. PloS One (2014) 9(1):e85115. doi: 10.1371/journal.pone.0085115
81. Phua J, Weng L, Ling L, Egi M, Lim CM, Divatia JV, et al. Intensive care management of coronavirus disease 2019 (COVID-19): challenges and recommendations. Lancet Respir Med (2020) 8(5):506–17. doi: 10.1016/S2213-2600(20)30161-2
82. McLaughlin KM, Bechtel M, Bojkova D, Münch C, Ciesek S, Wass MN, et al. COVID-19-Related coagulopathy-is transferrin a missing link? Diagnostics (Basel) 2020 10(8):539. doi: 10.3390/diagnostics10080539
83. Kim J, Wessling-Resnick M. The role of iron metabolism in lung inflammation and injury. J Allergy Ther (2012) 3(Suppl 4). doi: 10.4172/2155-6121.S4-004
84. Henry BM, de Oliveira MHS, Benoit S, Plebani M, Lippi G. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): a meta-analysis. Clin Chem Lab Med (2020) 58(7):1021–8. doi: 10.1515/cclm-2020-0369
85. O'Brien WT, Pham L, Symons GF, Monif M, Shultz SR, McDonald SJ, et al. The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J Neuroinflamm (2020) 17(1):104. doi: 10.1186/s12974-020-01778-5
86. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol (2019) 10:119. doi: 10.3389/fimmu.2019.00119
87. Boxberger N, Hecker M, Zettl UK. Dysregulation of inflammasome priming and activation by MicroRNAs in human immune-mediated diseases. J Immunol (2019) 202(8):2177–87. doi: 10.4049/jimmunol.1801416
88. Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, et al. Caspase-1 initiates apoptosis in the absence of gasdermin d. Nat Commun (2019) 10(1):2091. doi: 10.1038/s41467-019-09753-2
89. Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell (2020) 182(3):685–712.e19. doi: 10.1016/j.cell.2020.06.034
90. Hartog N, Faber W, Frisch A, Bauss J, Bupp CP, Rajasekaran S, et al. SARS-CoV-2 infection: molecular mechanisms of severe outcomes to suggest therapeutics. Expert Rev Proteomics (2021) 18(2):105–18. doi: 10.1080/14789450.2021.1908894
91. Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol (2020) 2(6):e325–31. doi: 10.1016/S2665-9913(20)30127-2
92. Yamanashi T, Iwata M, Kamiya N, Tsunetomi K, Kajitani N, Wada N, et al. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci Rep (2017) 7(1):7677. doi: 10.1038/s41598-017-08055-1
93. Ko JH, Yoon SO, Lee HJ, Oh JY. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFκB pathways in autophagy- and p62-dependent manners. Oncotarget (2017) 8(25):40817–31. doi: 10.18632/oncotarget.17256
94. Jessop F, Hamilton RF, Rhoderick JF, Shaw PK, Holian A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol Appl Pharmacol (2016) 309:101–10. doi: 10.1016/j.taap.2016.08.029
95. Liu Y, Zhang W, Wu X, Gong J. Foxo3a-dependent bim transcription protects mice from a high fat diet via inhibition of activation of the NLRP3 inflammasome by facilitating autophagy flux in kupffer cells. Oncotarget (2017) 8(21):34258–67. doi: 10.18632/oncotarget.15946
96. Xu X, Lin D, Tu S, Gao S, Shao A, Sheng J. Is ferroptosis a future direction in exploring cryptococcal meningitis? Front Immunol (2021) 12:598601. doi: 10.3389/fimmu.2021.598601
97. Shah R, Margison K, Pratt DA. The potency of diarylamine radical-trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chem Biol (2017) 12(10):2538–45. doi: 10.1021/acschembio.7b00730
98. Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PloS Biol (2018) 16(5):e2006203. doi: 10.1371/journal.pbio.2006203
99. Muhoberac BB. What can cellular redox, iron, and reactive oxygen species suggest about the mechanisms and potential therapy of COVID-19? Front Cell Infect Microbiol (2020) 10:569709. doi: 10.3389/fcimb.2020.569709
100. Wang B, Timilsena YP, Blanch E, Adhikari B. Lactoferrin: Structure, function, denaturation and digestion. Crit Rev Food Sci Nutr (2019) 59(4):580–96. doi: 10.1080/10408398.2017.1381583
101. Cavezzi A, Troiani E, Corrao S. COVID-19: hemoglobin, iron, and hypoxia beyond inflammation. a narrative review. Clin Pract (2020) 10(2):1271. doi: 10.4081/cp.2020.1271
102. Yang M, Lai CL. SARS-CoV-2 infection: can ferroptosis be a potential treatment target for multiple organ involvement? Cell Death Discovery (2020) 6:130. doi: 10.1038/s41420-020-00369-w
103. Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin d inhibits inflammatory cell death and sepsis. Sci Immunol (2018) 3(26). doi: 10.1126/sciimmunol.aat2738
104. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-Approved disulfiram inhibits pyroptosis by blocking gasdermin d pore formation. Nat Immunol (2020) 21(7):736–45. doi: 10.1038/s41590-020-0669-6
105. Wen W, Su W, Tang H, Le W, Zhang X, Zheng Y, et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discovery (2020) 6(1):31. doi: 10.1038/s41421-020-0168-9
106. Kooistra EJ, Waalders NJB, Grondman I, Janssen NAF, de Nooijer AH, Netea MG, et al. Anakinra treatment in critically ill COVID-19 patients: a prospective cohort study. Crit Care (2020) 24(1):688. doi: 10.1186/s13054-020-03364-w
107. Daher R, Manceau H, Karim Z. Iron metabolism and the role of the iron-regulating hormone hepcidin in health and disease. Presse Med (2017) 46(12 Pt 2):e272–8. doi: 10.1016/j.lpm.2017.10.006
108. Bessman NJ, Mathieu JRR, Renassia C, Zhou L, Fung TC, Fernandez KC, et al. Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science (2020) 368(6487):186–9. doi: 10.1126/science.aau6481
109. Ehsani S. COVID-19 and iron dysregulation: distant sequence similarity between hepcidin and the novel coronavirus spike glycoprotein. Biol Direct (2020) 15(1):19. doi: 10.1186/s13062-020-00275-2
110. Wu H, Wu T, Li M, Wang J. Efficacy of the lipid-soluble iron chelator 2,2'-dipyridyl against hemorrhagic brain injury. Neurobiol Dis (2012) 45(1):388–94. doi: 10.1016/j.nbd.2011.08.028
111. Li Q, Wan J, Lan X, Han X, Wang Z, Wang J. Neuroprotection of brain-permeable iron chelator VK-28 against intracerebral hemorrhage in mice. J Cereb Blood Flow Metab (2017) 37(9):3110–23. doi: 10.1177/0271678X17709186
112. Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, et al. A major role for ferroptosis in mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med (2019) 216(3):556–70. doi: 10.1084/jem.20181776
113. Weiland A, Wang Y, Wu W, Lan X, Han X, Li Q, et al. Ferroptosis and its role in diverse brain diseases. Mol Neurobiol (2019) 56(7):4880–93. doi: 10.1007/s12035-018-1403-3
114. Wan J, Ren H, Wang J, toxicity I. Lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc Neurol (2019) 4(2):93–5. doi: 10.1136/svn-2018-000205
115. Qin D, Wang J, Le A, Wang TJ, Chen X, Wang J. Traumatic brain injury: Ultrastructural features in neuronal ferroptosis, glial cell activation and polarization, and blood-brain barrier breakdown. Cells (2021) 10(5). doi: 10.3390/cells10051009
116. Edeas M, Saleh J, Peyssonnaux C. Iron: Innocent bystander or vicious culprit in COVID-19 pathogenesis? Int J Infect Dis (2020) 97:303–5. doi: 10.1016/j.ijid.2020.05.110
117. Wang Y, Huang J, Sun Y, Stubbs D, He J, Li W, et al. SARS-CoV-2 suppresses mRNA expression of selenoproteins associated with ferroptosis, endoplasmic reticulum stress and DNA synthesis. Food Chem Toxicol (2021) 153:112286. doi: 10.1016/j.fct.2021.112286
118. Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol (2014) 14(11):759–67. doi: 10.1038/nri3743
119. Habib HM, Ibrahim S, Zaim A, Ibrahim WH. The role of iron in the pathogenesis of COVID-19 and possible treatment with lactoferrin and other iron chelators. BioMed Pharmacother (2021) 136:111228. doi: 10.1016/j.biopha.2021.111228
120. Kell DB, Pretorius E. Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics (2014) 6(4):748–73. doi: 10.1039/C3MT00347G
121. Cortese-Krott MM, Koning A, Kuhnle GGC, Nagy P, Bianco CL, Pasch A, et al. The reactive species interactome: Evolutionary emergence, biological significance, and opportunities for redox metabolomics and personalized medicine. Antioxid Redox Signal (2017) 27(10):684–712. doi: 10.1089/ars.2017.7083
122. Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. The emerging role of ferroptosis in inflammation. BioMed Pharmacother (2020) 127:110108. doi: 10.1016/j.biopha.2020.110108
123. Jacobs W, Lammens M, Kerckhofs A, Voets E, Van San E, Van Coillie S, et al. Fatal lymphocytic cardiac damage in coronavirus disease 2019 (COVID-19): autopsy reveals a ferroptosis signature. ESC Heart Fail (2020) 7(6):3772–81. doi: 10.1002/ehf2.12958
124. Zhang R, Sun C, Chen X, Han Y, Zang W, Jiang C, et al. COVID-19-Related brain injury: The potential role of ferroptosis. J Inflammation Res (2022) 15:2181–98. doi: 10.2147/JIR.S353467
125. Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight (2017) 2(7):e90777. doi: 10.1172/jci.insight.90777
126. Li Q, Weiland A, Chen X, Lan X, Han X, Durham F, et al. Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage: Coexistence of ferroptosis, autophagy, and necrosis. Front Neurol (2018) 9:581. doi: 10.3389/fneur.2018.00581
127. Gill D, Brewer CF, Monori G, Trégouët DA, Franceschini N, Giambartolomei C, et al. Effects of genetically determined iron status on risk of venous thromboembolism and carotid atherosclerotic disease: A mendelian randomization study. J Am Heart Assoc (2019) 8(15):e012994. doi: 10.1161/JAHA.119.012994
Keywords: COVID-19, necrosis, mechanisms, biomarkers, clinical treatment
Citation: Sun C, Han Y, Zhang R, Liu S, Wang J, Zhang Y, Chen X, Jiang C, Wang J, Fan X and Wang J (2022) Regulated necrosis in COVID-19: A double-edged sword. Front. Immunol. 13:917141. doi: 10.3389/fimmu.2022.917141
Received: 10 April 2022; Accepted: 01 August 2022;
Published: 25 August 2022.
Edited by:
Mainul Haque, National Defence University of Malaysia, MalaysiaReviewed by:
Sreya Ghosh, Harvard Medical School, United StatesRonald B. Corley, Boston University, United States
Copyright © 2022 Sun, Han, Zhang, Liu, Wang, Zhang, Chen, Jiang, Wang, Fan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Wang, amlhbndhbmcyMDIwQG91dGxvb2suY29t; Junmin Wang, d2FuZ2p1bm1pbkB6enUuZWR1LmNu; Xiaochong Fan, ZmNjZnhjQHp6dS5lZHUuY24=
†These authors share first authorship