 Hemant Joshi
Hemant Joshi Sharon Celeste Morley
Sharon Celeste Morley
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 29 June 2022
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.916137
This article is part of the Research Topic The Cytoskeleton in T Cell Migration and Activation View all 12 articles
Rapid re-organization of the actin cytoskeleton supports T-cell trafficking towards immune sites and interaction with antigen presenting cells (APCs). F-actin rearrangement enables T-cell trafficking by stabilizing adhesion to vascular endothelial cells and promoting transendothelial migration. T-cell/APC immune synapse (IS) maturation also relies upon f-actin-anchored LFA-1:ICAM-1 ligation. Therefore, efficient T-cell responses require tight regulation of f-actin dynamics. In this review, we summarize how the actin-bundling protein L-plastin (LPL) regulates T-cell activation and migration. LPL enhances f-actin polymerization and also directly binds to the β2 chain of the integrin LFA-1 to support intercellular adhesion and IS formation in human and murine T cells. LPL- deficient T cells migrate slowly in response to chemo-attractants such as CXCL12, CCL19, and poorly polarize towards ICAM-1. Loss of LPL impairs thymic egress and intranodal motility. LPL is also required for T-cell IS maturation with APCs, and therefore for efficient cytokine production and proliferation. LPL-/- mice are less susceptible to T-cell mediated pathologies, such as allograft rejection and experimental autoimmune encephalomyelitis (EAE). LPL activity is regulated by its N-terminal “headpiece”, which contains serine and threonine phosphorylation and calcium- and calmodulin-binding sites. LPL phosphorylation is required for lamellipodia formation during adhesion and migration, and also for LFA-1 clustering during IS formation. However, the precise molecular interactions by which LPL supports T-cell functional responses remain unclear. Future studies elucidating LPL-mediated regulation of T-cell migration and/or activation may illuminate pathways for therapeutic targeting in T-cell-mediated diseases.
Activated T cells drive adaptive immune responses. Effective T cell responses require rapid migration towards immune sites, followed by engagement with antigen presenting cells (APCs) (1, 2). By supporting migration and adhesion, cytoskeletal rearrangements regulate the quality and magnitude of the T cell response. The cytoskeleton comprises filamentous proteins including microtubules, actin filaments, and intermediate filaments (3). Receptor engagement, internal organization of the cytoplasm, and stabilization against applied forces trigger cytoskeleton rearrangement (4). For instance, chemokine receptor engagement promotes T-cell motility, shear flow activates T cell adhesion, and T cell receptor (TCR) engagement to peptide-major histocompatibility complex (pMHCs) on APCs triggers immune synapse (IS) formation (5–7).
Actin filaments are the most dynamic cytoskeletal proteins that respond to and direct T-cell biological processes. Actin filaments (f-actin) are formed by the rapid assembly of globular-actin (g-actin) monomers. Bundling, or cross-linking filaments, further remodels f-actin (8, 9). F-actin polymerization is ATP-dependent; ATP binding to monomeric g-actin enhances its affinity for f-actin association (polymerization), while hydrolysis of bound ATP to ADP triggers dissociation (depolymerization). An array of actin-binding proteins regulate actin turnover through g-actin sequestration, nucleation, ATP hydrolysis, nucleotide exchange, f-actin severing, capping, and bundling (10, 11). This review focus on how efficient T cell activation and migration rely on the f-actin bundling protein, leukocyte plastin (L-plastin; LPL).
LPL, also known as lcp1 or plastin-2, bundles f-actin (8, 12, 13). LPL belongs to the plastin family of actin-binding proteins, comprising three isotypes: LPL, T-plastin and I –plastin. Each isoform exhibits distinct tissue expression. Only LPL is expressed in immune cells. Consistent with its expression in hematopoietic cells, LPL specifically binds to β-actin, and does not interact with skeletal muscle α-actin or smooth muscle γ-actin (8). Physiologic LPL expression is restricted to replicating hematopoietic cells, but LPL is also ectopically overexpressed in multiple transformed cancer cells (14). LPL is a 66 kDa protein, consisting of an N-terminal regulatory “headpiece” and 2 actin-binding domains (ABDs); ABD1 and ABD2 (Figure 1A). LPL is distinguished from I- and T-plastin by its N-terminal regulatory sequence, as it is the only isoform containing two serine phosphorylation sites at Ser5 and Ser7. Each ABD contains tandem calponin homology (CH) domains (11, 15). How ABD1 and ABD2 of LPL coordinate to engage two actin filaments has not been fully described, because full-length LPL crystallization is unavailable. However, analysis of the available ABD2 structure along with available T-plastin and the Arabidopsis thalinana fimbrin (LPL orthologue) protein structures (16) suggests that binding of LPL-ABD2 to f-actin induces “closure” of the actin monomeric ATP binding site. The “closed” conformation inhibits hydrolysis to ADP, reducing depolymerization and stabilizing the actin filament (17). To stabilize bundles, LPL may adopt a twisted conformation, supporting the binding of ABD2 to one filament and activating ABD1 to bind another actin filament, generating cross-linked f-actin (17). Thus, LPL binding stabilizes polymerized actin, as well as altering the twist and tilt of each filament, while LPL bundling arranges filaments into parallel arrays (Figure 1B) (16, 18). LPL actin-binding is separable from its function in actin-bundling.
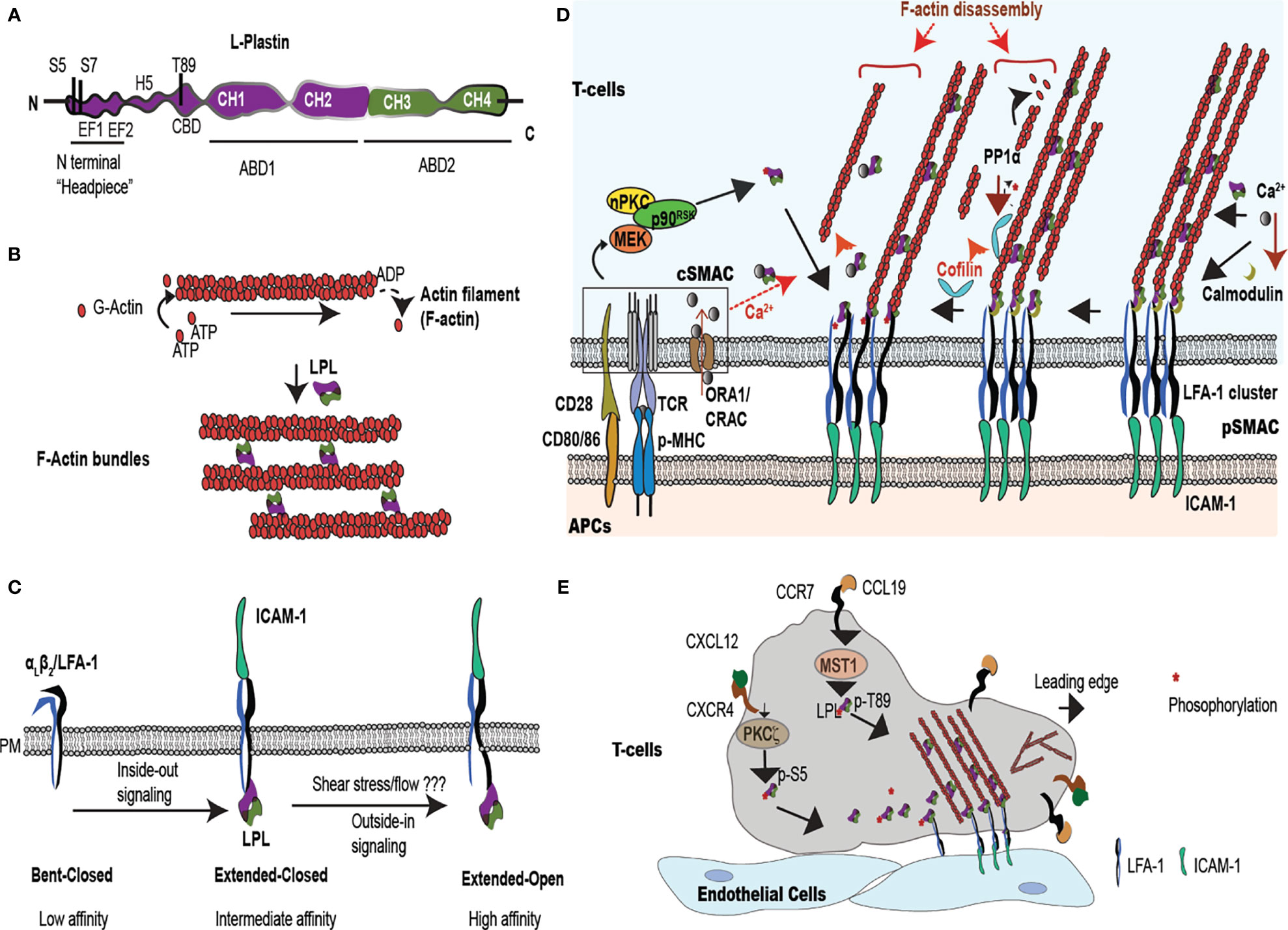
Figure 1 LPL is an actin-bundling protein that supports T cell synapse maturation and migration. (A) LPL comprises a regulatory ‘N-terminal headpiece’ and two actin binding domains (ABDs). The regulatory headpiece includes three known phosphorylation sites (Ser-5, Ser-7, Thr89), two calcium binding EF domains, a regulatory H5 “switch helix,” and a calmodulin binding domain (CBD). (B) LPL cross-links two actin filaments to generates f-actin bundles. (C) LPL interacts with the cytoplasmic tail of the β2 subunit of the integrin LFA-1. In T cells, inside-out signaling induces a conformational change from low to intermediate affinity, whereas shear flow or ICAM-1 engagement triggers the high-affinity, extended-open conformation. (D) Multiple regulatory pathways converge upon LPL in the pSMAC, where LFA-1 mediates tight approximation to ICAM-expressing APCs. The IS, the interface at the site of T-cell:APC contact site, generates a cSMAC, pSMAC and a dSMAC (dSMAC not shown). In the IS, TCR ligation to peptide-loaded MHC (pMHC) and costimulatory CD28:CD80/86 interactions occur in the central super-molecular activation complex (cSMAC), which activates a kinase cascade [nPKC-MEK- ribosomal protein S6 kinase p90 (p90RSK)]. This kinase cascade results in Ser-5 phosphorylation of LPL. LPL phosphorylation accelerates LFA-1 clustering in the peripheral SMAC (pSMAC). Phosphorylated LPL is largely associated with high affinity LFA-1, and may stabilize the IS. LPL can also bind to LFA-1 independently of phosphorylation. LPL-induced LFA-1 clusters further accumulate at IS by cofilin-mediated f-actin remodeling. Cofilin is activated by PP1α phosphatase. Simultaneously, LPL also recruits f-actin bundles to connect the IS to actin cytoskeleton. Ca2+ influx at cSMAC also regulates LPL. Ca2+ binding reduces the LPL bundling efficiency, perhaps allowing f-actin clearing at IS. However, lower Ca2+ in the pSMAC permits calmodulin binding to LPL, stabilizing actin bundles and maintainting LFA-1 clustering. (E) Intravascular T cells traffic to peripheral immunological sites via the binding of the integrin LFA-1 to the adhesion molecule, ICAM-1, expressed on vascular endothelial cells. In response to chemoattractants, such as CXCL12 and CCL19, T cells polarize the respective chemokine receptors CXCR4 and CCR7 to the leading edge and activate kinases such Mst1 and PKCζ. Mst1, PKCζ, and other kinases phosphorylate the regulatory headpiece of LPL. Phosphorylated LPL initiates LFA-1 clustering, sustaining ICAM-1 binding. Branched f-actin forms in the lamellipod, while the ‘uropod’ propels cells in a forward direction. The symbol "*" indicates the phosphate group during phosphorylation/dephosphorylation reaction.
The actin-binding and actin-bundling activities are regulated by the N-terminal headpiece of LPL. The headpiece harbors at least three phosphorylation sites (Ser-5, Ser-7 and Thr-89), two calcium-binding EF-hand loops, and a consensus sequence for calmodulin binding (19–21). Under some conditions, phosphorylation of Ser-5 and Ser-7 enhances actin-bundling activity, and localizes LPL to sites of actin assembly (13, 22, 23). For example, IL-2 stimulation, CD3/CD28 or CD3/CD2 receptor engagement, and CXCL12 treatment all induce Ser-5 phosphorylation (24, 25). However, recent biochemical evidence suggests that single Ser-5 phosphorylation is irrelevant to LPL bundling activity (26), but enhances binding to f-actin structures in non-immune cells and under in vitro conditions (13). In T cells, LPL-mediated actin-bundling activity was first noted in a calcium-regulated manner (8). Calcium chelation by the high-affinity EF hands reduces the actin-bundling, but not the actin-binding, activity of LPL (9) (26). Jurkat T cells show increased actin-bundling activity in low intracellular Ca2+ concentration of 10-7 M, while incubation in higher concentrations, such as 10-6 M Ca2+, destabilized f-actin bundles (8). Immune cells maintain 10-8-10-7 M Ca2+ in resting phase while elevating to 10-6 upon activation, inducing f-actin rearrangement (e.g. intracellular Ca2+ is estimated to 50 nM in resting cells and > 1 mM in activated T cells) (27, 28). The physiological range of Ca2+ found in T cells correlates with the concentrations of Ca2+ that modulate LPL bundling activity. The spatiotemporal regulation of Ca2+ flux and LPL activity at the T cell immunological synapse (IS) is discussed in detail later.
In addition to bundling actin, LPL also directly binds to the cytoplasmic tail of the β1 or β2 subunits of integrins. The regulation of integrin affinity and/or avidity by LPL is poorly understood. In brief, integrins are transmembrane heterodimers that bind extracellular matrix proteins or receptors expressed on other cells and mediate cellular adhesion. The dominant integrin mediating T cell adhesion, LFA-1 (αLβ2; CD11a/CD18), exists in three affinity states: a low-affinity, “bent-closed” state, an intermediate affinity “extended-closed” state, and the high-affinity “extended-open” state (Figure 1C) (29). Conversion from the low- to intermediate-affinity state is induced by inside-out signaling; limited evidence suggests that LPL may be dispensable for this conversion (30, 31). However, studies in neutrophils indicated that binding of the β2 cytoplasmic tail by the isolated N-terminal peptide sequence of LPL converted the integrin to a high-affinity state (Figure 1C) (32). LPL was also required for integrin-mediated signaling to the oxidative burst in PMNs, though dispensable for integrin-mediated cell spreading (33), indicating that LPL participates in integrin-mediated signaling to downstream events beyond actin remodeling. LPL directly binds the β2 subunit of the integrin LFA-1 to connect with the actin cytoskeleton during migration and activation (34). Finally, LPL was shown to preferentially bind the “clasped” (or closed) cytoplasmic tail of CR3 (Mac-1; αmβ2; CD11b/CD18). Phosphomimetics of Ser5/Ser7 dual phosphorylation reduced the binding of LPL to β2, and binding of LPL to β2 reduced CR3-mediated adhesion in the cell line RAW (35). The participation of LPL in T-cell activation and motility therefore extends beyond actin-bundling and into integrin signaling. LPL enables the actin cytoskeletal rearrangements and integrin-mediated adhesion that are core drivers of T-cell migration and T-cell adhesion to APCs and/or target cells. We review the increased knowledge about LPL in T-cell activation and motility, the heart of most adaptive immune responses.
T cells mount adaptive immune responses by activating other immune cells through cytokine production and/or direct contact, and by direct killing of target cells. TCR engagement by APCs initiates T-cell activation through interaction at a specialized intercellular contact site, the IS. The IS is stabilized by cytoskeleton machinery via lamellipodia formation that enhances the contact area between APCs and T cells, increasing TCR signaling (5, 6). The T cell-APC contact site comprises three supramolecular activation clusters (SMAC) that organize signaling proteins into the central, peripheral, and distal SMACs (cSMAC, pSMAC, and dSMAC) (36). The four different actin formations that characterize the IS are extensively reviewed in (36). Notably, the f-actin network is “cleared,” or hypodense, in the cSMAC, where the TCR/CD3 signaling complexes accumulate (37). The pSMAC forms a ring around the centrally cleared area, and is marked by LFA-1 accumulation and generation of actomyosin arcs. These actomysoin arcs are force generating, enabling close approximation of the T cell to its target APC (Figure 1D). Larger molecules, such as CD43, localize to the outer dSMAC, where f-actin is organized in a branching network (36, 38, 39). Throughout the pSMAC and dSMAC are small actin foci, areas of integrin engagement that resemble macrophage podosomes. IS formation and T cell activation are absolutely dependent upon active actin rearrangements that create these varied actin structures (13, 40, 41).
Efficient IS formation also requires LPL. Murine T cells isolated from genetically-deficient LPL mice (LPL-/- mice) exhibited impaired IS formation (42), with reduced T-cell:APC contact area. Decreased T-cell activation in LPL-/- mice correlated with enhanced tolerance to T cell-mediated diseases such as skin allograft rejection and experimental autoimmune encephalomyelitis (EAE) (42). Furthermore, LPL supported Tfh cell-B cell interactions and was required for germinal center formation and subsequent T-dependent antibody production (43). In human T cells, LPL directly binds the β2 subunit (CD18) of LFA-1 to link the actin cytoskeleton to the IS (34, 44). Reduction of LPL (<10% normal expression through siRNA knockdown), impaired LFA-1 concentration to the pSMAC and generated smaller T-cell:APC contact zone, confirming a crucial need for LPL in IS formation (34).
The requirement for LPL Ser-5 phosphorylation during T-cell activation is unclear. CD3/CD2-mediated Ser-5 phosphorylation was first reported in 1994 (45). Subsequent extensive work using knock-down of LPL in human T cells, followed by re-expression of phosphomimetic or non-phosphorylatable LPL, has suggested a significant regulatory role for Ser-5. In human T cells, Ser-5 phosphorylation enabled translocation of the activation markers CD69 and CD25 to the cell surface after TCR/CD3, CD2 or CD28 engagement, although LPL localized to the IS independently of Ser-5 phosphorylation (24). In the IS, LPL maximizes accumulation of LFA-1 at pSMAC by directly interacting with LFA-1 (34). LPL localization to the pSMAC was stabilized by calmodulin binding and by ABD-mediated actin-bundling activity. LPL bound to LFA-1 equivalently in naive or CD3/CD28-activated T cells, independently of the EF-hand loops and Ser-5 phosphorylation (34). However, Ser-5 phosphorylation of LPL (LFA-1 bound) increased upon CD3/CD28 stimulation and at the IS generated after superantigen [S. aureus enterotoxin B (SEB)]-mediated cross-linking of Raji B cells and T cells (31).
Mechanistically, in CD3/CD28-costimulated T cells, activation of the nPKC-MEK- ribosomal protein S6 kinase p90 (p90RSK) pathway phosphorylated LPL (Ser-5) to initiate LFA-1 clustering at the IS (31). Association of phosphorylated LPL with high affinity LFA-1 may sustain IS formation (31). Further, LPL-induced LFA-1 clustering was enhanced by another actin-binding protein, cofilin. Cofilin is an f-actin severing protein that promotes f-actin remodeling by generating new filament ends for addition or removal of g-actin monomers (46). Cofilin binds the ADP-bound actin monomer within an existing filament, increasing the likelihood of f-actin depolymerization, accelerating f-actin turn-over, untangling filaments, and facilitating re-arrangement (47). Cofilin enhances LFA-1 accumulation at the IS by accelerating f-actin remodeling (31). Cofilin is activated by PP1α-mediated dephosphorylation, while LPL is dephosphorylated by the serine/threonine phosphatase PP2A (31). LFA-1 clustering and accumulation at the IS enhances the interaction of LFA-1 with its ligand ICAM-1 (expressed by APCs), and amplifies T-cell activation (Figure 1D). Pharmacological inhibition of nPKC (Gö6983), MEK (U0126), or p90RSK (BI-D1870) reduced LPL phosphorylation and cofilin activation, which correlated with IS destabilization, diminished T cell-APC contact, and impaired T-cell activation (31). Similarly, blockade of LPL using nanobodies directed against the EF-hands or the ABDs impaired the binding of LPL to LFA-1, and subsequently inhibited IS formation and downstream T-cell proliferation (48). IS stabilization by LPL : LFA-1 binding is targeted by bacterial infections (e.g. Bordetella pertussis and Bacillus anthracis), in which cAMP production at the IS suppressed T-cell activation (49). Treatment with the glucocorticoid dexamethasone inhibited LPL Ser-5 phosphorylation after CD3/CD2 and CD3/CD28-mediated T cell activation, and dexamethasone also impaired IS formation (50). Thus, a major function of LPL during T cell activation is maintenance of the IS. LPL-mediated IS stabilization is further suggested by the observation that enhanced phosphorylation of LPL amplified LFA-1 clustering and inhibited serial killing by cytotoxic T cells (CTL). Amplified LFA-1 clustering prevented detachment from target cells, thereby prolonging IS maintenance, and thus reduced interactions with new target cells. The pro-oxidative drug WF-10 increased LPL phosphorylation, rigidified the cytolytic IS, and suppressed CTL activity (51). Thus, LPL phosphorylation activates naïve/effector T cells, while interrupting cytolytic activity, making it an important intrinsic modulator during critical immune reactions (44, 51).
Intriguingly, a murine model in which the Ser5 of LPL was converted to the non-phosphorylatable alanine, and thus ablated the Ser5 phosphorylation site, did not exhibit obvious defects in TCR-mediated activation (52). TCR-mediated activation was assessed by proliferation and upregulation of CD25 and CD69. Possible explanations for a lack of effect on T cell activation by ablation of Ser5 phosphorylation include the differences in experimental systems. These explanations will be discussed in detail later.
T cells constantly migrate to scan APCs for foreign antigens, to activate immune responses, and to directly kill target cells. This rapid migration (average velocity of 8-15 μm/min) depends on the actin cytoskeleton (27). During chemotaxis, chemokine engagement induces lamellipodia formation and chemokine receptor concentration at the leading edges of cells. Contractile elements at the rear edges, called “uropods,” provide propulsive forces (53). LPL actin-bundling activity is essential for efficient T lymphocyte motility (34, 54, 55).
LPL was first identified as a critical component of T cell chemotaxis during CCL20-induced transwell migration (55). A more detailed analysis of the role of LPL in T cell migration was explored using LPL-/- mice (54). LPL-/- mice showed higher thymic retention of CD4+ (3-fold increase) and CD8+ (2-fold increase) single positive thymocytes, which resulted from impaired thymic egress. LPL-deficient mature T cells also exhibited impaired intranodal velocity and motility. During CCL19-mediated transwell migration, LPL-deficient T cells showed reduced polarization of CCR7 to the leading edge and reduced uropod formation (54). LPL phosphorylation at Thr-89 by Mst1 kinase (Ste20 kinase/STK4) supported T cell lamellipodia formation and thus T cell migration (21). Mst1 kinase had been previously shown to enable T-cell lamellipodia formation, polarization and migration (13, 56). Identification of a Mst1 consensus sequence in the regulatory headpiece of LPL provided a downstream molecular target by which Mst1 regulated T cell motility. Notably, expression of LPL-T89A, which ablated the Mst1 phosphorylation site (Thr-89), but not Ser-5, failed to restore CCL19-induced CCR7 polarization and transwell migration in LPL-deficient T cells, whereas expression of wild-type LPL did restore the chemokine-induced events. Furthermore, reconstitution of T cells in LPL-/- mice with lentivirally-expressed LPL-T89A showed greater accumulations of CD4+ and CD8+ single positive thymocytes than did LPL-/- mice, consistent with a requirement for Thr-89 phosphorylation during thymic egress (21).
Investigation in human T cells further confirmed a crucial role for LPL in migration (25). Reduction of LPL by 75-95% by siRNA knock-down caused impaired polarization of CXCR4 receptors in CXCL12 (SDF-1α)-stimulated T cells (25). In human T cells (CD3+), CXCL12 activated atypical PKC-ζ to phosphorylate Ser-5 of LPL, triggering lamellipodial localization during polarization (25). However, T cell migration was not impaired in S5A mice, suggesting that Ser-5 phosphorylation of LPL correlates with, but may not be required for, T-cell migration (52). Chemokine signals induce a conformational change in LFA-1, through inside-out signaling, which converts LFA-1 to a moderate-affinity state. Moderate-affinity LFA-1 promotes cell adhesion to vascular endothelial cells via ICAM-1 binding (Figure 1E). LFA-1:ICAM-1 ligation reorganizes f-actin to initiate T-cell polarization, crawling and migration (57, 58). When LPL expression is reduced, unstimulated and effector (CD3/CD28 stimulated) T cells migrate more slowly and with higher migratory persistence on surfaces coated with immobilized CXCL12 and ICAM-1 (25). Thus, LPL-supported f-actin cytoskeletal polarization is essential for efficient T-cell migration (59).
A comprehensive model that reconciles apparently disparate observations regarding the regulation of LPL during T-cell migration and activation remains elusive. With multiple regulatory sites, and separable functions of actin-binding, actin-bundling, and integrin-association, too much remains unknown to formulate a complete description of the mechanism(s) by which LPL enables migration and IS formation. To provide a framework for future exploration, we suggest that the primary role of LPL is to stabilize multimolecular complexes that arise at sites of integrin-mediated adhesion, and thereby enhance or promote mechanotransduction at those sites. Mechanotransduction is the translation of external mechanical forces exerted upon the cell into intracellular biochemical signaling (60). For example, IS formation is strengthened by mechanosensation generated by interactions with mature APCs that actively increase cortical cytoskeletal stiffness, thereby boosting T-cell activation (61). Similarly, enhanced adhesion of T cells to stiffer culture surfaces (elastic modulus 25 kilopascal (kPa) to 100 kPa) triggers higher IL-2 production upon CD3 activation (62). Finally, T cells migrate faster on stiffer substrates, demonstrating that mechanotransduction also regulates T cell motility (63). A proposal that LPL supports formation of integrin-mediated adhesion sites and mechanotransduction through these sites provides a single mechanism common to both migration and IS formation (64), and explains how LPL deficiency could disrupt both these processes (Figure 2). As discussed below, this framework draws on studies of LPL in hematopoietic cell types including macrophages, B cells, and neutrophils, in addition to T cells.
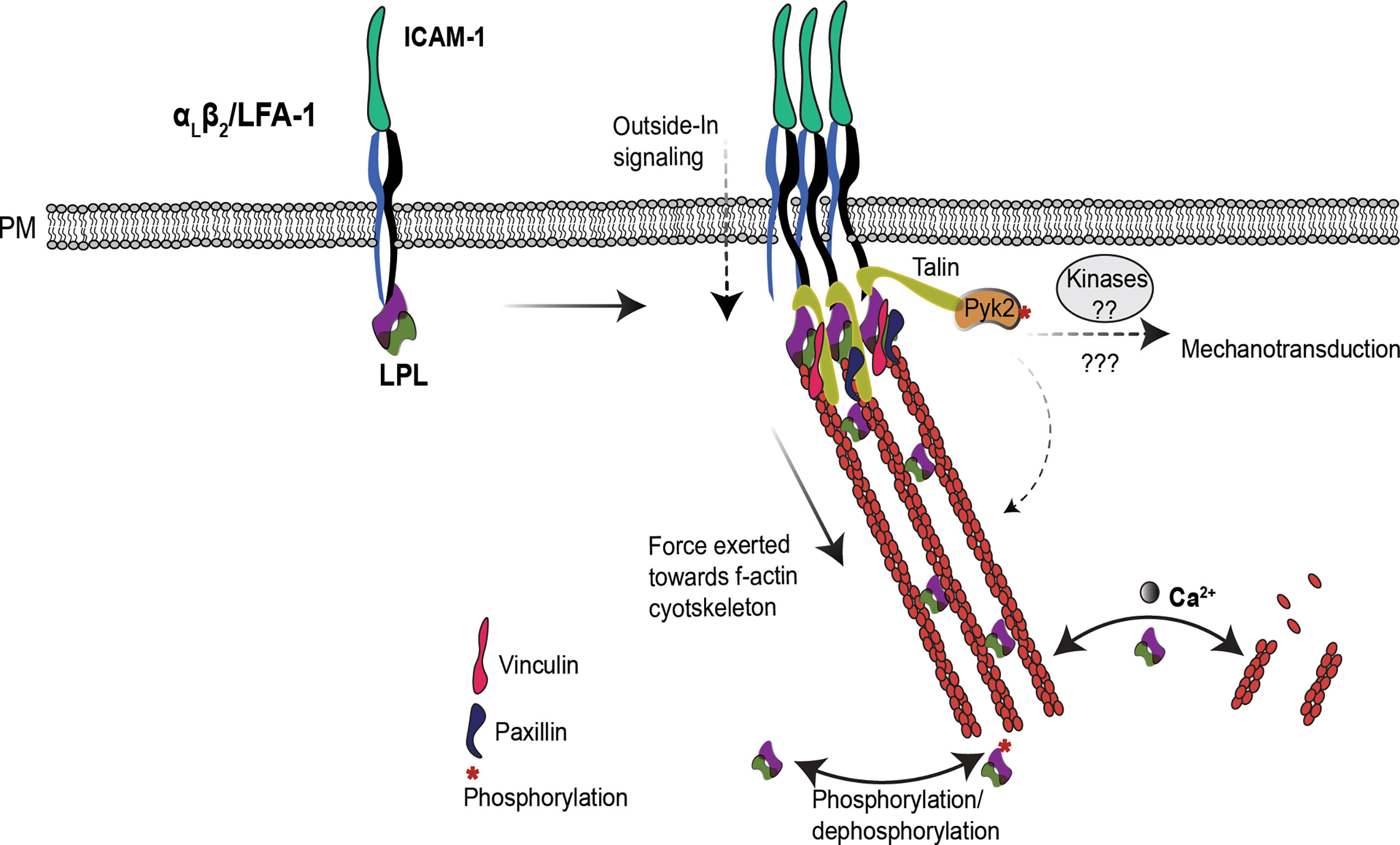
Figure 2 LPL may regulate T cell integrins induced mechanotransduction. In T cells, LPL binds to LFA-1. Under certain conditions, e.g. shear flow or ICAM-1 engagement, LFA-1 shifts to the extended-open (high-affinity) conformation. LPL binding to LFA-1 recruits the mechanosensing protein Talin, and also connects it to the f-actin cytoskeleton. Talin recruits additional cytoskeleton-associated proteins, such as vinculin, paxillin and the tyrosine kinase Pyk2. Collectively, this cascade generates mechanotransduction through Pyk2 and other kinases, and triggers f-actin remodeling required for efficient T cell activation and migration. The separable activities of LPL–LFA-1 binding, f-actin binding, and f-actin-bundling—are coordinately regulated to support mechanotransduction. How the multiple regulatory sites on LPL (phosphorylation, Ca2+, and calmodulin-binding) converge to modulate the varied activities of LPL are yet to be illuminated.
LPL has been shown to regulate mechanotransduction in macrophages (65). WT macrophages incubated upon softer substrates produced more IL-1β in response to NLRP3 inflammasome activation. However, LPL-deficient macrophages produced the same, reduced amount of IL-1 β following NLRP3 activation, and were unresponsive to varying substrate stiffnesses (65). Mechanotransudction in LPL-deficient macrophages was disrupted due to the mislocalization of the kinase Pyk2. In macrophages, β2 integrin activation phosphorylates Pyk2 to induce podosome signaling (66). Macrophage podosomes are multimolecular signaling complexes generated by integrin-mediated adhesion and anchored by f-actin. Podosomes support macrophage adhesion, signaling and motility. Podosome stabilization requires LPL; thus, LPL also regulates macrophage motility (67). The f-actin foci described as one of the four actin structures present in the T-cell IS have been likened to macrophage podosomes (36). LPL is also required for CXCL12-induced migration and Pyk2 activation (Tyr-402 phosphorylation) in B cells (68). In T cells, Pyk2 regulates LFA-1:ICAM-1 signaling. Pyk2-/- T cells weakly adhere to ICAM-1 and migrate slowly upon CXCL12 stimulation (69), and thus are similar in phenotype to LPL-deficient T cells. While not yet experimentally determined in T cells, it is reasonable to propose that LPL serves to link sites of integrin-mediated adhesion to downstream Pyk2 activation and signaling in all hemaopoietic cells. However, the molecular mechanism by which LPL and Pyk2 interact is unclear, and further investigation in understanding of synergistic functioning of LPL and Pyk2 via f-actin cytoskeleton will help in understanding their role in T cell function and mechanotransduction.
In T cells, LPL participates in the “inside-out” cascade for LFA-1 clustering to establish adhesion and IS formation (34). LFA-1 engagement also initiates an “outside -in” signal via linking to f-actin (60) to fine-tune T-cell migration, differentiation, and effector functions (70, 71). LPL helps recruit a mechanosensing protein, Talin, to LFA-1 clusters, connecting f-actin to the IS (34). Talin further recruits various cytoskeleton proteins, such as vinculin and paxillin and f-actin, by direct binding, and propagates mechanical signals (72, 73). Since the β2-binding site also lies in ABDs (30), it has not yet been shown if a single molecule of LPL can bind both β2 integrin and f-actin, or if binding of one precludes binding of the other. Direct cross-linking of f-actin to the cytoplasmic tail of integrins by LPL would be one possible mechanism by which LPL could sustain integrin-mediated adhesion sites.
During T cell migration, LFA-1 binds to ICAM-1 on endothelial cells to enable transmigration (74). F-actin cytoskeleton retrograde flow aligns ICAM-1 bound integrins at the leading edge to reinforce cellular adhesions (75). While LPL appears to regulate the binding of integrins, such as LFA-1 and VLA-4, to their respective ligands, ICAM-1 and VCAM-1, during chemotaxis on immobilized CXCL12, LPL is dispensable under shear flow conditions (25). This differential requirement for LPL during static chemotaxis and under shear flow could be due to differential f-actin reorganization and/or recruitment of additional actin binding partners, such as Cofilin (31). LFA-1 changes from an inactive, clasped conformation to an active, open conformation to bind ICAM-1 (58, 76, 77). Perhaps the binding of LPL to the intracellular β2 integrin is conformation specific, and varies with T-cell migration under stasis versus shear flow. A recent study in human myeloid (PBL-985) cells showed preferential binding of LPL to the clasped αMβ2 (CD11β/CD18, also called CR3 or Mac-1) conformation to maintain the inactive state under flow (35). By analogy, LPL may likewise stabilize the inactive LFA-1 conformation when T cells are under flow, while under static conditions LPL promotes LFA-1 clustering to initiate adherence. However, conformation-specific binding of LPL to LFA-1 (CD11α/CD18) in T cells is unresolved and future studies will address this important open question.
Further studies are required to determine how signals from the multiple regulatory sites on the LPL N-terminal headpiece are integrated to modify the integrin-binding, actin-binding, and actin-bundling activities of LPL during T-cell activation and migration. Current studies have analyzed single regulatory sites without clearly defining how modulation at other sites impact LPL functioning. For instance, LPL Ser-5 phosphorylation is essential for activation and IS formation in human T cells, while Thr-89 phosphorylation supports T cell migration in murine cells (21). However, no studies have examined the combined effects of Ser5/Thr89 phosphorylation, or if phosphorylation at Ser5 prevents Thr89 phosphorylation. Furthermore, no studies have examined the effect of Ca2+ binding on phosphorylated vs non-phosphorylated LPL (at any site). Thr-89 phosphorylation is not yet studied in human T cells, and may not function similarly as it does in mice. Regulation of LPL could vary between human and murine T cells during activation and migration because of different (and as yet undefined) pathways that phosphorylate Ser-5 and Thr-89, or because of different inter-molecular interactions. In human T cells, overexpression of non-phosphorylatable Ser-5 (S5A) LPL abrogated CD3-induced surface translocation of activation markers CD25 and CD69 (24), which was not observed in LPL-S5A reconstituted LPL deficient murine T cells (42). Similarly, our study in LPL-S5A mice showed no defect in CD25 and CD69 upregulation in CD3/CD28 activated T cells (52). However, the exact molecular cascade for LPL-dependent CD69 and CD25 upregulation is not known. Possibly, a dominant negative effect of the S5A mutant in overexpressing human T cells generated a different phenotype than endogenous expression of S5A-LPL-only expressing murine T- cells. Further detailed molecular studies are required to resolve the disparities of LPL regulation in human and mouse T cells.
In addition to phosphorylation, LPL activity is regulated by calcium influx in T cells. The LPL N-terminal headpiece possesses two EF-hand calcium-binding sites and a binding site for the calcium-binding protein calmodulin (15, 78). Calcium binding (increased intracellular calcium concentration above 10-7 M) at EF-hands induces a conformational change that inhibits LPL’s bundling activity (8, 9). Recent NMR solution structures of the LPL EF-hand domains revealed a calcium sensor ‘switch-helix’ motif (H5) in-between EF-hand motifs and ABD1 (Figure 1A) (79). In the absence of Ca2+, the H5 motif remains flexible and unstructured, which stabilizes the orientation of LPL-ABDs when bound to f-actin to promote efficient f-actin bundling. Whereas, when Ca2+ increases, the H5 motif changes conformation to a more rigid α-helix, which induces the release of the H5 motif from f-actin-ABD pocket and permits binding of H5 to EF hands. This helix switch in the H5 motif generates a conformation unfavorable to binding of LPL-ABDs to f-actin, thus reducing f-actin bundling (79). The coordinate spatial and temporal dynamics of Ca2+ flux and LPL activity during IS formation have not been fully defined. The Ca2+ channels ORA1 and CRAC are localized to the cSMAC, suggesting that Ca2+ flux is tightly spatially regulated, with increased flux in the cSMAC. Perhaps the higher levels of Ca2+ in the cSMAC inhibits LPL bundling activity, contributing to the decreased f-actin polymerization and f-actin density (actin “clearing”) in the cSMAC (37). Lower levels of Ca2+ in the pSMAC may correlate with increased LPL-mediated actin-bundling and formation of actomyosin arcs. Calmodulin binding to LPL occurs in the absence of calcium. Calmodulin binding stabilizes LPL binding to LFA-1 clusters in the pSMAC to enhance T-cell/APC contact at IS (34) (Figure 1D). Additional studies are required to determine the spatiotemporal regulation of LPL phosphorylation, Ca2+ binding, and calmodulin binding during IS formation and maintenance.
The requirement for LPL in T-cell activation reveals new intrinsic regulators for therapeutic targeting to ameliorate T-cell-driven pathologies. For instance, reducing intracellular levels of LPL by genetic knock-down (48) or nanobody-mediated LPL inactivation (48) impair T cell activation and IS formation. Similarly, pharmacological inhibition of LPL directly using dexamethasone (50) or LPL kinase nPKC using Gö6983 (31) can be used to inhibit T-cell immune responses. Conversely, enhancing LPL phosphorylation using WF-10 could be employed to suppress T cell mediated cytotoxicity during allograft transplantation (51). In addition to activation, inhibiting cell migration by targeting kinases [e.g. Mst1 (21) and PKCζ (25)] could be explored to maximize T-cell retention in tumors microenvironment. The urgent need for therapies for autoimmune and oncologic processes compels further elucidation of the key T-cell migration and activation pathways dependent upon LPL.
HJ and SM both have equally contributed to idea conceptualization and manuscript writing. All authors contributed to the article and approved the submitted version.
This work was supported by National Institutes of Health grants R01- AI104732, R21EB030171, R01 AI118719, HL148033, R01AI139540, and R56 AI104732, National Science Foundation grants CBET-1900277 and CMMI1548571, American Lung Association grant ETRA 736343, Washington University in Saint Louis Children’s Discovery Institute grants CDI-CORE-2019-813 and CDI-CORE-2015-505, Foundation for Barnes-Jewish Hospital grants 3770 and 4642, American Association of Immunologists AAI Careers in immunology fellowship. The content of this study is the authors’ sole responsibility and does not necessarily represent official NIH views.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Negulescu PA, Krasieva TB, Khan A, Kerschbaum HH, Cahalan MD. Polarity of T Cell Shape, Motility, and Sensitivity to Antigen. Immunity (1996) 4(5):421–30. doi: 10.1016/s1074-7613(00)80409-4
2. Iezzi G, Karjalainen K, Lanzavecchia A. The Duration of Antigenic Stimulation Determines the Fate of Naive and Effector T Cells. Immunity (1998) 8(1):89–95. doi: 10.1016/s1074-7613(00)80461-6
3. Thauland TJ, Hu KH, Bruce MA, Butt MJ. Cytoskeletal Adaptivity Regulates T Cell Receptor Signaling. Sci Signal (2017) 10(469):eaah3737. doi: 10.1126/scisignal.aah3737
4. Hu KH, Butte MJ. T Cell Activation Requires Force Generation. J Cell Biol (2016) 213(5):535–42. doi: 10.1083/jcb.201511053
5. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The Immunological Synapse: A Molecular Machine Controlling T Cell Activation. Science (1999) 285(5425):221–7. doi: 10.1126/science.285.5425.221
6. Krummel MF, Davis MM. Dynamics of the Immunological Synapse: Finding, Establishing and Solidifying a Connection. Curr Opin Immunol (2002) 14(1):66–74. doi: 10.1016/S0952-7915(01)00299-0
7. Dustin ML. Cell Adhesion Molecules and Actin Cytoskeleton at Immune Synapses and Kinapses. Curr Opin Cell Biol (2007) 19(5):529–33. doi: 10.1016/j.ceb.2007.08.003
8. Namba Y, Ito M, Zu Y, Shigesada K, Maruyama K. Human T Cell L-Plastin Bundles Actin Filaments in a Calcium-Dependent Manner. J Biochem (1992) 112(4):503–7. doi: 10.1093/oxfordjournals.jbchem.a123929
9. Miyakawa T, Shinomiya H, Yumoto F, Miyauchi Y, Tanaka H, Ojima T, et al. Different Ca²⁺-Sensitivities Between the EF-Hands of T- and L-Plastins. Biochem Biophys Res Commun (2012) 429(3-4):137–41. doi: 10.1016/j.bbrc.2012.10.126
10. Al Tanoury Z, Schaffner-Reckinger E, Halavatyi A, Hoffmann C, Moes M, Hadzic E, et al. Quantitative Kinetic Study of the Actin-Bundling Protein L-Plastin and of its Impact on Actin Turn-Over. PLoS One (2010) 5(2):e9210. doi: 10.1371/journal.pone.0009210
11. Morley SC. The Actin-Bundling Protein L-Plastin: A Critical Regulator of Immune Cell Function. Int J Cell Biol 2012 (2012) p:935173. doi: 10.1155/2012/935173
12. Lin CS, Aebersold R, Kent H S, Varma B M, Leavitt J. Molecular Cloning and Characterization of Plastin, a Human Leukocyte Protein Expressed in Transformed Human Fibroblasts. Mol Cell Biol (1988) 8(11):4659–68. doi: 10.1128/mcb.8.11.4659-4668.1988
13. Janji B, Giganti A, De Corte V, Catillon M, Bruyneel E, Lentz D, et al. Phosphorylation on Ser5 Increases the F-Actin-Binding Activity of L-Plastin and Promotes its Targeting to Sites of Actin Assembly in Cells. J Cell Sci (2006) 119(Pt 9):1947–60. doi: 10.1242/jcs.02874
14. Samstag Y, Klemke M. Ectopic Expression of L-Plastin in Human Tumor Cells: Diagnostic and Therapeutic Implications. Adv Enzyme Regul (2007) 47:118–26. doi: 10.1016/j.advenzreg.2006.12.008
15. Pacaud M, Derancourt J. Purification and Further Characterization of Macrophage 70-kDa Protein, a Calcium-Regulated, Actin-Binding Protein Identical to L-Plastin. Biochemistry (1993) 32(13):3448–55. doi: 10.1021/bi00064a031
16. Volkmann N, DeRosier D, Matsudaira P, Hanein D. An Atomic Model of Actin Filaments Cross-Linked by Fimbrin and Its Implications for Bundle Assembly and Function. J Cell Biol (2001) 153(5):947–56. doi: 10.1083/jcb.153.5.947
17. Galkin VE, Orlova A, Cherepanova O, Lebart MC, Egelman EH. High-Resolution Cryo-EM Structure of the F-Actin-Fimbrin/Plastin ABD2 Complex. Proc Natl Acad Sci USA (2008) 105(5):1494–8. doi: 10.1073/pnas.0708667105
18. Evans JG, Correia I, Krasavina O, Watson N, Matsudaira P. Macrophage Podosomes Assemble at the Leading Lamella by Growth and Fragmentation. J Cell Biol (2003) 161(4):697–705. doi: 10.1083/jcb.200212037
19. de Arruda MV. Fimbrin is a Homologue of the Cytoplasmic Phosphoprotein Plastin and has Domains Homologous With Calmodulin and Actin Gelation Proteins. J Cell Biol (1990) 111(3):1069–79. doi: 10.1083/jcb.111.3.1069
20. Shinomiya H, Hagi A, Fukuzumi M, Mizobuchi M, Hirata H, Utsumi S. Complete Primary Structure and Phosphorylation Site of the 65-kDa Macrophage Protein Phosphorylated by Stimulation With Bacterial Lipopolysaccharide. J Immunol (1995) 154(7):3471–8.
21. Xu X, Wang X, Todd EM, Jaeger ER, Vella JL, Mooren OL, et al. Mst1 Kinase Regulates the Actin-Bundling Protein L-Plastin To Promote T Cell Migration. J Immunol (2016) 197(5):1683–91. doi: 10.4049/jimmunol.1600874
22. Chellaiah MA, Ma T, Majumdar S. L-Plastin Phosphorylation Regulates the Early Phase of Sealing Ring Formation by Actin Bundling Process in Mouse Osteoclasts. Exp Cell Res (2018) 372(1):73–82. doi: 10.1016/j.yexcr.2018.09.014
23. Lin CS, Lau A, Lue TF. Analysis and Mapping of Plastin Phosphorylation. DNA Cell Biol (1998) 17(12):1041–6. doi: 10.1089/dna.1998.17.1041
24. Wabnitz GH, Köcher T, Lohneis P, Stober C, Konstandin MH, Funk B, et al. Costimulation Induced Phosphorylation of L-Plastin Facilitates Surface Transport of the T Cell Activation Molecules CD69 and CD25. Eur J Immunol (2007) 37(3):649–62. doi: 10.1002/eji.200636320
25. Freeley M, O'Dowd F, Paul T, Kashanin D, Davies A, Kelleher D. L-Plastin Regulates Polarization and Migration in Chemokine-Stimulated Human T Lymphocytes. J Immunol (2012) 188(12):6357–70. doi: 10.4049/jimmunol.1103242
26. Schwebach CL, Agrawal R, Lindert S, Kudryashova E, Kudryashov DS. The Roles of Actin-Binding Domains 1 and 2 in the Calcium-Dependent Regulation of Actin Filament Bundling by Human Plastins. J Mol Biol (2017) 429(16):2490–508. doi: 10.1016/j.jmb.2017.06.021
27. Krummel MF, Cahalan MD. The Immunological Synapse: A Dynamic Platform for Local Signaling. J Clin Immunol (2010) 30(3):364–72. doi: 10.1007/s10875-010-9393-6
28. Donnadieu E, Bismuth G, Trautmann A. Calcium Fluxes in T Lymphocytes. J Biol Chem (1992) 267(36):25864–72. doi: 10.1016/S0021-9258(18)35689-8
29. Walling BL, Kim M. LFA-1 in T Cell Migration and Differentiation. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00952
30. Le Goff E, Vallentin A, Harmand PO, Aldrian-Herrada G, Rebière B, Roy C, et al. Characterization of L-Plastin Interaction With Beta Integrin and Its Regulation by Micro-Calpain. Cytoskeleton (Hoboken) (2010) 67(5):286–96. doi: 10.1002/cm.20442
31. Wabnitz GH, Honus S, Habicht J, Orlik C, Kirchgessner H, Samstag Y, et al. LFA-1 Cluster Formation in T-Cells Depends on L-Plastin Phosphorylation Regulated by P90(RSK) and PP2A. Cell Mol Life Sci (2021) 78(7):3543–64. doi: 10.1007/s00018-020-03744-z
32. Jones SL, Wang J, Turck CW, Brown EJ. A Role for the Actin-Bundling Protein L-Plastin in the Regulation of Leukocyte Integrin Function. Proc Natl Acad Sci USA (1998) 95(16):9331–6. doi: 10.1073/pnas.95.16.9331
33. Chen H, Mocsai A, Zhang H, Ding RX, Morisaki JH, Wh M, et al. Role for Plastin in Host Defense Distinguishes Integrin Signaling From Cell Adhesion and Spreading. Immunity (2003) 19(1):95–104. doi: 10.1016/s1074-7613(03)00172-9
34. Wabnitz GH, Lohneis P, Kirchgessner H, Jahraus B, Gottwald S, Konstandin M, et al. Sustained LFA-1 Cluster Formation in the Immune Synapse Requires the Combined Activities of L-Plastin and Calmodulin. Eur J Immunol (2010) 40(9):2437–49. doi: 10.1002/eji.201040345
35. Tseng HY, Samarelli AV, Kammerer P, Scholze S, Ziegler T, Immler R, et al. LCP1 Preferentially Binds Clasped Alphambeta2 Integrin and Attenuates Leukocyte Adhesion Under Flow. J Cell Sci (2018) 131(22):jcs218214. doi: 10.1242/jcs.218214
36. Hammer JA, Wang JC, Saeed M, Pedrosa AT. Origin, Organization, Dynamics, and Function of Actin and Actomyosin Networks at the T Cell Immunological Synapse. Annu Rev Immunol (2019) 37:201–24. doi: 10.1146/annurev-immunol-042718-041341
37. Hartzell CA, Jankowska KI, Burkhardt JK, Lewi RS. Calcium Influx Through CRAC Channels Controls Actin Organization and Dynamics at the Immune Synapse. Elife (2016) 5:e14850. doi: 10.7554/eLife.14850
38. Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-Dimensional Segregation of Supramolecular Activation Clusters in T Cells. Nature (1998) 395(6697):82–6. doi: 10.1038/25764
39. Freiberg BA, Kupfer H, Maslanik W, Delli J, Kappler J, Zaller DM, et al. Staging and Resetting T Cell Activation in SMACs. Nat Immunol (2002) 3(10):911–7. doi: 10.1038/ni836
40. Poenie M, Kuhn J, Combs J. Real-Time Visualization of the Cytoskeleton and Effector Functions in T Cells. Curr Opin Immunol (2004) 16(4):428–38. doi: 10.1016/j.coi.2004.05.016
41. Campi G, Varma R, Dustin ML. Actin and Agonist MHC-Peptide Complex-Dependent T Cell Receptor Microclusters as Scaffolds for Signaling. J Exp Med (2005) 202(8):1031–6. doi: 10.1084/jem.20051182
42. Wang C, Morley SC, Donermeyer D, Peng I, Lee WP, Devoss J, et al. Actin-Bundling Protein L-Plastin Regulates T Cell Activation. J Immunol (2010) 185(12):7487–97. doi: 10.4049/jimmunol.1001424
43. Todd EM, Deady LE, Morley SC. Intrinsic T- and B-Cell Defects Impair T-Cell-Dependent Antibody Responses in Mice Lacking the Actin-Bundling Protein L-Plastin. Eur J Immunol (2013) 43(7):1735–44. doi: 10.1002/eji.201242780
44. Wabnitz G, Balta E, Samstag Y. L-Plastin Regulates the Stability of the Immune Synapse of Naive and Effector T-Cells. Adv Biol Regul (2017) 63:107–14. doi: 10.1016/j.jbior.2016.09.009
45. Henning SW, Meuer SC, Samstag Y. Serine Phosphorylation of a 67-kDa Protein in Human T Lymphocytes Represents an Accessory Receptor-Mediated Signaling Event. J Immunol (1994) 152(10):4808–15.
46. Tanaka K, Takeda S, Mitsuoka K, Oda T, Kimura-Sakiyama C, Maéda Y, et al. Structural Basis for Cofilin Binding and Actin Filament Disassembly. Nat Commun (2018) 9(1):1860. doi: 10.1038/s41467-018-04290-w
47. McCall PM, MacKintosh FC, Kovar DR, Gardel ML. Cofilin Drives Rapid Turnover and Fluidization of Entangled F-Actin. Proc Natl Acad Sci USA (2019) 116(26):12629–37. doi: 10.1073/pnas.1818808116
48. De Clercq S, Zwaenepoel O, Martens E, Vandekerckhove J, Guillabert A, Gettemans J. Nanobody-Induced Perturbation of LFA-1/L-Plastin Phosphorylation Impairs MTOC Docking, Immune Synapse Formation and T Cell Activation. Cell Mol Life Sci (2013) 70(5):909–22. doi: 10.1007/s00018-012-1169-0
49. Arumugham VB, Ulivieri C, Onnis A, Finetti F, Tonello F, Ladant D, et al. Compartmentalized Cyclic AMP Production by the Bordetella Pertussis and Bacillus Anthracis Adenylate Cyclase Toxins Differentially Affects the Immune Synapse in T Lymphocytes. Front Immunol (2018) 9:919. doi: 10.3389/fimmu.2018.00919
50. Wabnitz GH, Michalke F, Stober C, Kirchgessner H, Jahraus B, van denBoomen D, et al. L-Plastin Phosphorylation: A Novel Target for the Immunosuppressive Drug Dexamethasone in Primary Human T Cells. Eur J Immunol (2011) 41(11):3157–69. doi: 10.1002/eji.201041366
51. Wabnitz GH, Balta E, Schindler S, Kirchgessner H, Jahraus B, Meuer S, et al. The Pro-Oxidative Drug WF-10 Inhibits Serial Killing by Primary Human Cytotoxic T-Cells. Cell Death Discovery (2016) 2:16057. doi: 10.1038/cddiscovery.2016.57
52. Anaya EP, Lin X, Todd EM, Szasz TP, Morley SC. Novel Mouse Model Reveals That Serine Phosphorylation of L-Plastin Is Essential for Effective Splenic Clearance of Pneumococcus. J Immunol (2021) 206(9):2135–45. doi: 10.4049/jimmunol.2000899
53. Nieto M, Frade JM, Sancho D, Mellado M, Martinez AC, Sánchez-Madrid F, et al. Polarization of Chemokine Receptors to the Leading Edge During Lymphocyte Chemotaxis. J Exp Med (1997) 186(1):153–8. doi: 10.1084/jem.186.1.153
54. Morley SC, Wang C, Lo WL, Lio CW, Zinselmeyer BH, Miller MJ, et al. The Actin-Bundling Protein L-Plastin Dissociates CCR7 Proximal Signaling From CCR7-Induced Motility. J Immunol (2010) 184(7):3628–38. doi: 10.4049/jimmunol.0903851
55. Lin SL, Lin CW, Chien CL, Han ES, Chen SH, Kao YJ, et al. Temporal Proteomics Profiling of Lipid Rafts In CCR6-Activated T Cells Reveals the Integration of Actin Cytoskeleton Dynamics. J Proteome Res (2010) 9(1):283–97. doi: 10.1021/pr9006156
56. Katagiri K, Imamura M, Kinashi T. Spatiotemporal Regulation of the Kinase Mst1 by Binding Protein RAPL Is Critical for Lymphocyte Polarity and Adhesion. Nat Immunol (2006) 7(9):919–28. doi: 10.1038/ni1374
57. Hogg N, Patzak I, Willenbrock F. The Insider's Guide to Leukocyte Integrin Signalling and Function. Nat Rev Immunol (2011) 11(6):416–26. doi: 10.1038/nri2986
58. Alon R, Shulman Z. Chemokine Triggered Integrin Activation and Actin Remodeling Events Guiding Lymphocyte Migration Across Vascular Barriers. Exp Cell Res (2011) 317(5):632–41. doi: 10.1016/j.yexcr.2010.12.007
59. Morley SC. The Actin-Bundling Protein L-Plastin Supports T-Cell Motility and Activation. Immunol Rev (2013) 256(1):48–62. doi: 10.1111/imr.12102
60. Joshi H, Morley SC. Cells Under Stress: The Mechanical Environment Shapes Inflammasome Responses to Danger Signals. J Leukoc Biol (2019) 106(1):119–25. doi: 10.1002/JLB.3MIR1118-417R
61. Blumenthal D, Chandra V, Avery L, Burkhardt JK. Mouse T Cell Priming is Enhanced by Maturation-Dependent Stiffening of the Dendritic Cell Cortex. Elife (2020) 9:e55995. doi: 10.7554/eLife.55995
62. Judokusumo E, Tabdanov E, Kumari S, Dustin ML, Kam LC. Mechanosensing in T Lymphocyte Activation. Biophys J (2012) 102(2):L5–7. doi: 10.1016/j.bpj.2011.12.011
63. Saitakis M, Dogniaux S, Goudot C, Bufi N, Asnacios S, Maurin M, et al. Different TCR-Induced T Lymphocyte Responses are Potentiated by Stiffness With Variable Sensitivity. Elife (2017) 6:e23190. doi: 10.7554/eLife.23190
64. Rossy J, Laufer JM, Legler DF. Role of Mechanotransduction and Tension in T Cell Function. Front Immunol (2018) 9:2638. doi: 10.3389/fimmu.2018.02638
65. Joshi H, Almgren-Bell A, Anaya EP, Todd EM, Van Dyken SJ, Seth A, et al. L-Plastin Enhances NLRP3 Inflammasome Assembly and Bleomycin-Induced Lung Fibrosis. Cell Rep (2022) 38(11):110507. doi: 10.1016/j.celrep.2022.110507
66. Rodan GA, Duong LT. PYK2 Is an Adhesion Kinase in Macrophages, Localized in Podosomes and Activated by B2-Integrin Ligation. Cell Motil Cytoskeleton (2000) 47:15. doi: 10.1002/1097-0169(200011)47:3<174::AID-CM2>3.0.CO;2-N
67. Zhou JY, Szasz TP, Stewart-Hutchinson PJ, Sivapalan J, Todd EM, Deady LE, et al. L-Plastin Promotes Podosome Longevity and Supports Macrophage Motility. Mol Immunol (2016) 78:79–88. doi: 10.1016/j.molimm.2016.08.012
68. Todd EM, Deady LE, Morley SC. The Actin-Bundling Protein L-Plastin is Essential for Marginal Zone B Cell Development. J Immunol (2011) 187(6):3015–25. doi: 10.4049/jimmunol.1101033
69. Beinke S, Phee H, Clingan JM, Schlessinger J, Matloubian M, Weiss A. Proline-Rich Tyrosine Kinase-2 is Critical for CD8 T-Cell Short-Lived Effector Fate. Proc Natl Acad Sci USA (2010) 107(37):16234–9. doi: 10.1073/pnas.1011556107
70. Buxboim A, Ivanovska IL, Discher DE. Matrix Elasticity, Cytoskeletal Forces and Physics of the Nucleus: How Deeply do Cells 'Feel' Outside and in? J Cell Sci (2010) 123(Pt 3):297–308. doi: 10.1242/jcs.041186
71. Gérard A, Cope AP, Kemper C, Alon R, Köchl R. LFA-1 in T Cell Priming, Differentiation, and Effector Functions. Trends Immunol (2021) 42(8):706–22. doi: 10.1016/j.it.2021.06.004
72. Jankowska KI, Williamson EK, Roy NH, Blumenthal D, Chandra V, Baumgart T, et al. Integrins Modulate T Cell Receptor Signaling by Constraining Actin Flow at the Immunological Synapse. Front Immunol (2018) 9:25. doi: 10.3389/fimmu.2018.00025
73. Owen LM, Bax NA, Weis WI, Dunn AR. The C-Terminal Actin-Binding Domain of Talin Forms an Asymmetric Catch Bond With F-Actin. Proc Natl Acad Sci USA (2022) 119(10):e2109329119. doi: 10.1073/pnas.2109329119
74. Friedl P, Bröcker EB. T Cell Migration in Three-Dimensional Extracellular Matrix: Guidance by Polarity and Sensations. Dev Immunol (2000) 7(2-4):249–66. doi: 10.1155/2000/56473
75. Swaminathan V, Kalappurakkal JM, Mehta SB, Nordenfelt P, Moore TI, Koga N, et al. Actin Retrograde Flow Actively Aligns and Orients Ligand-Engaged Integrins in Focal Adhesions. Proc Natl Acad Sci USA (2017) 114(40):10648–53. doi: 10.1073/pnas.1701136114
76. Fischer RS, Myers KA, Gardel ML, Waterman CM. Stiffness-Controlled Three-Dimensional Extracellular Matrices for High-Resolution Imaging of Cell Behavior. Nat Protoc (2012) 7(11):2056–66. doi: 10.1038/nprot.2012.127
77. Alon R, Feigelson SW. Chemokine-Triggered Leukocyte Arrest: Force-Regulated Bi-Directional Integrin Activation in Quantal Adhesive Contacts. Curr Opin Cell Biol (2012) 24(5):670–6. doi: 10.1016/j.ceb.2012.06.001
78. Lin CS, Park T, Chen ZP, Leavitt J. Human Plastin Genes. Comparative Gene Structure, Chromosome Location, and Differential Expression in Normal and Neoplastic Cells. J Biol Chem (1993) 268(4):2781–92. doi: 10.1016/S0021-9258(18)53842-4
Keywords: T cells, L-plastin, immune synapse formation, immune cell adhesion and migration, mechanotransduction, LFA-1 (CD11A/CD18; ITGAL/ITGB2), F-actin assembly, cytoskeleton
Citation: Joshi H and Morley SC (2022) Efficient T Cell Migration and Activation Require L-Plastin. Front. Immunol. 13:916137. doi: 10.3389/fimmu.2022.916137
Received: 08 April 2022; Accepted: 02 June 2022;
Published: 29 June 2022.
Edited by:
Manish Butte, University of California, Los Angeles, United StatesReviewed by:
Jeremie Rossy, Biotechnology Institute Thurgau, SwitzerlandCopyright © 2022 Joshi and Morley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sharon Celeste Morley, bW9ybGV5c0B3dXN0bC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.