 Michael Conner
Michael Conner Jeremy D. Waight
Jeremy D. Waight- Oncology R&D, GlaxoSmithKline, Collegeville, PA, United States
In recent years, a set of immune receptors that interact with members of the nectin/nectin-like (necl) family has garnered significant attention as possible points of manipulation in cancer. Central to this axis, CD226, TIGIT, and CD96 represent ligand (CD155)-competitive co-stimulatory/inhibitory receptors, analogous to the CTLA-4/B7/CD28 tripartite. The identification of PVRIG (CD112R) and CD112 has introduced complexity and enabled additional nodes of therapeutic intervention. By virtue of the clinical progression of TIGIT antagonists and emergence of novel CD96- and PVRIG-based approaches, our overall understanding of the ‘CD226 axis’ in cancer immunotherapy is starting to take shape. However, several questions remain regarding the unique characteristics of, and mechanistic interplay between, each receptor-ligand pair. This review provides an overview of the CD226 axis in the context of cancer, with a focus on the status of immunotherapeutic strategies (TIGIT, CD96, and PVRIG) and their underlying biology (i.e., cis/trans interactions). We also integrate our emerging knowledge of the immune populations involved, key considerations for Fc gamma (γ) receptor biology in therapeutic activity, and a snapshot of the rapidly evolving clinical landscape.
Introduction
With the widespread clinical application of the ‘first generation’ of immune checkpoint inhibitors (ICI), namely antibody-mediated blockade of cytotoxic T lymphocyte-associated protein-4 (CTLA-4) and programmed cell death protein/ligand-1 (PD-[L]1), immunotherapy has become a mainstay approach for the treatment of cancer (1). However, despite a wealth of evidence supporting the use of anti-CTLA-4 and anti-PD-1/L1, many patients fail to derive meaningful benefit – highlighting the need for alternative and complementary immunotherapeutic interventions (2). In this regard, engagement of novel pathways, cell types, and combinations may provide therapeutic options for patients wherein the pre-existing host and tumor microenvironment factors do not favor current immunotherapeutic agents or where adaptive resistance has occurred (3).
For more than a decade, the CD226 axis have been characterized in the context of natural killer (NK) and T cell biology (4). At the core of this family, T cell immunoreceptor with Ig and ITIM domains (TIGIT) and CD96 (TACTILE) effectively compete with CD226 (DNAX Accessory Molecule-1 [DNAM-1] for binding to the necl protein CD155 (poliovirus receptor [PVR). This regulatory network is reminiscent of the interplay between cytotoxic T-lymphocyte-associated protein-4 (CTLA-4)/CD28 and B7 (CD80/CD86), where a common ligand (CD155) is shared between costimulatory (CD226) and co-inhibitory receptors (CD96 and TIGIT) (Figure 1) (5). While a parallel with the CTLA-4 axis could provide some mechanistic insight to the CD226 axis, there are some important differences in cellular expression, potential for direct inhibitory receptor signaling, and the potential impact of soluble ligand (6, 7). Adding to this complexity, the recently described PVRIG has been shown to compete with CD226 binding to CD112, another ligand in this axis (Figure 1) (8–10).
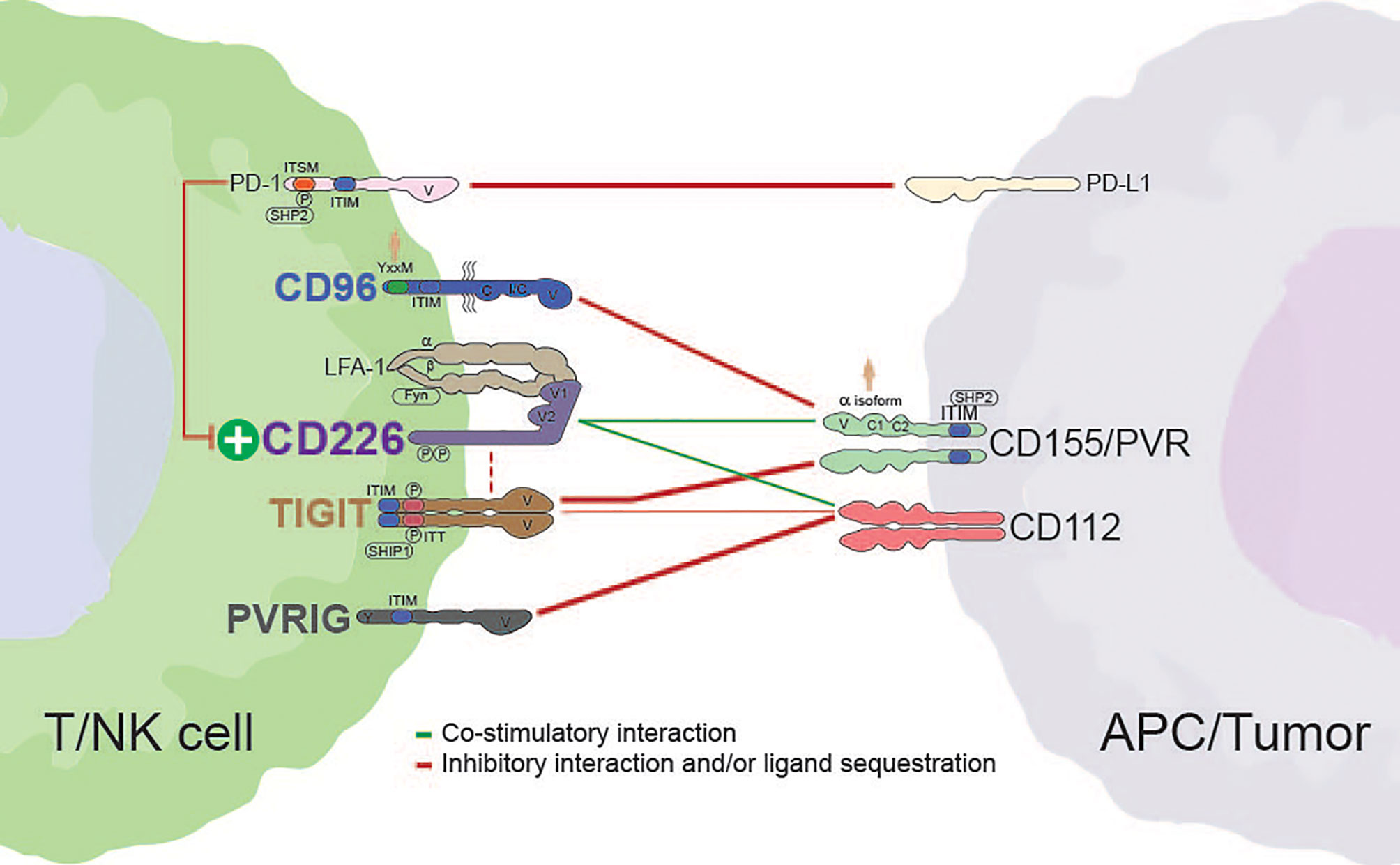
Figure 1 Interactions between members of the CD226 axis. Dashed red lines indicate the potential for cis interactions/inhibition. The weight of each line is representative of the relative strength of interaction. Alternative interactions not shown (e.g., CD96 with CD111; TIGIT with CD113 or PVRL4). Human silhouettes signify that a motif or isoform is not present in rodents. APC, antigen presenting cell; V, variable-like domain; C, constant-like domain; I/C, I/C-like folding pattern present in isoform 2 of human CD96; P, tyrosine phosphorylation site; SHP2, Src homology 2-containing phosphotyrosine phosphatase; ITIM, immunoreceptor tyrosine-based inhibition motif; SHIP1, Src homology 2-containing-inositol-phosphatase-1.
The contiguous nature of the CD226 axis begs several questions. For example, are the family members redundant such that concurrent antagonism of multiple receptors is necessary to reveal their full functional potential, or are individual receptors dominant under distinct contexts? A thorough understanding of the dynamics of each ligand-receptor pair will be critical for the mechanistic deconvolution of a seemingly redundant family. These relationships may also inform the best approaches for successful therapeutic intervention (i.e., best indications to target individual or multiple receptors, mono- or bi-specific strategies, etc.). To help address this, we explore the structural characteristics, reported interactions, and expression patterns for each immune receptor in the CD226 axis (Table 1). We also discuss the potential for cell-intrinsic activity and present the available evidence supporting combinations with antibodies targeting the CD226 axis. In addition, given the importance of Fc-Fc gamma (γ) receptor co-engagement to CTLA-4 antibody function, and inherent similarities with the CTLA-4/B7/CD28 family, we briefly discuss the potential role of FcγRs in promoting the functional activity of antibodies targeting the immune receptors in the CD226 axis (14–16). Finally, we provide a snapshot view of the current therapeutic landscape for the CD226 axis, surveying the available clinical data for each target and highlighting current indications, safety considerations, and combination strategies for each target.
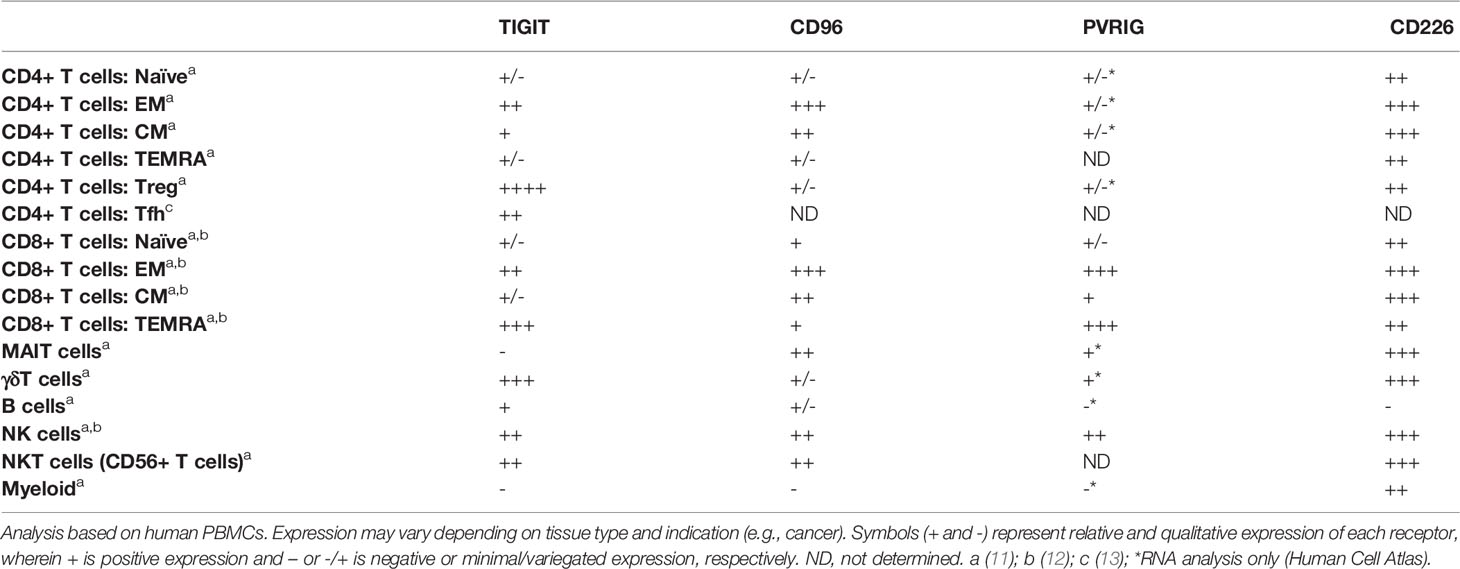
Table 1 Expression of CD226 axis members on human immune populations.
The Core of the Axis: CD226
Discovery, Structure, and Interactions
By virtue of its role in cytotoxic T cell maturation and platelet activation, CD226 was initially identified as T lineage-specific antigen (TLiSA1) and platelet and T cell antigen 1 (PTA1) shortly thereafter (17, 18). CD226, or DNAX accessory molecule 1 (DNAM-1), has since been thoroughly characterized as a T and NK cell co-stimulatory receptor responsible for orchestrating the signaling of shared ligands CD155 and CD112 (8, 19–22). Analogous to CD28 in the B7/CTLA-4 axis, CD226 has a reduced affinity for shared ligands CD155 and CD112 relative to the inhibitory receptors TIGIT, CD96, and PVRIG, thus creating a layer of immune regulation via competitive inhibition (9, 19, 23–26). Exemplifying its important role in immune homeostasis, CD226 genetic polymorphisms are associated with various immune pathologies (27–29). Similar correlations are lacking for TIGIT, CD96, and PVRIG, highlighting the central nature of CD226 in controlling immune activity within the family.
The extracellular region of CD226 forms a unique structure whereby its two IgV domains (domain [D]1 and D2) are linked in a side-by-side arrangement (Figure 2). As a result, while interactions are primarily mediated by a conserved ‘lock-and-key’ motif in D1, the second extracellular domain (D2) can also contribute to ligand binding (25, 26). The intracellular region of CD226 harbors a conserved tyrosine (Y)/asparagine (N) motif (D/EIYV/MNY), which engages with multiple proteins, including growth factor receptor bound protein 2 (Grb2) (30). Site-directed mutagenesis of Y319 abrogates CD226-induced cellular cytotoxicity (30) (Table 2). This residue (Y319) has also been associated with regulation of CD226 expression via Casitas B-lineage lymphoma proto-oncogene-b (Cbl-b)-dependent ubiquitination/degradation following CD155 engagement (37). Additionally, although it appears to be contextual, co-localization with lymphocyte function-associated antigen 1 (LFA-1) during immune synapse formation has also been described (Figure 1) (30, 32).
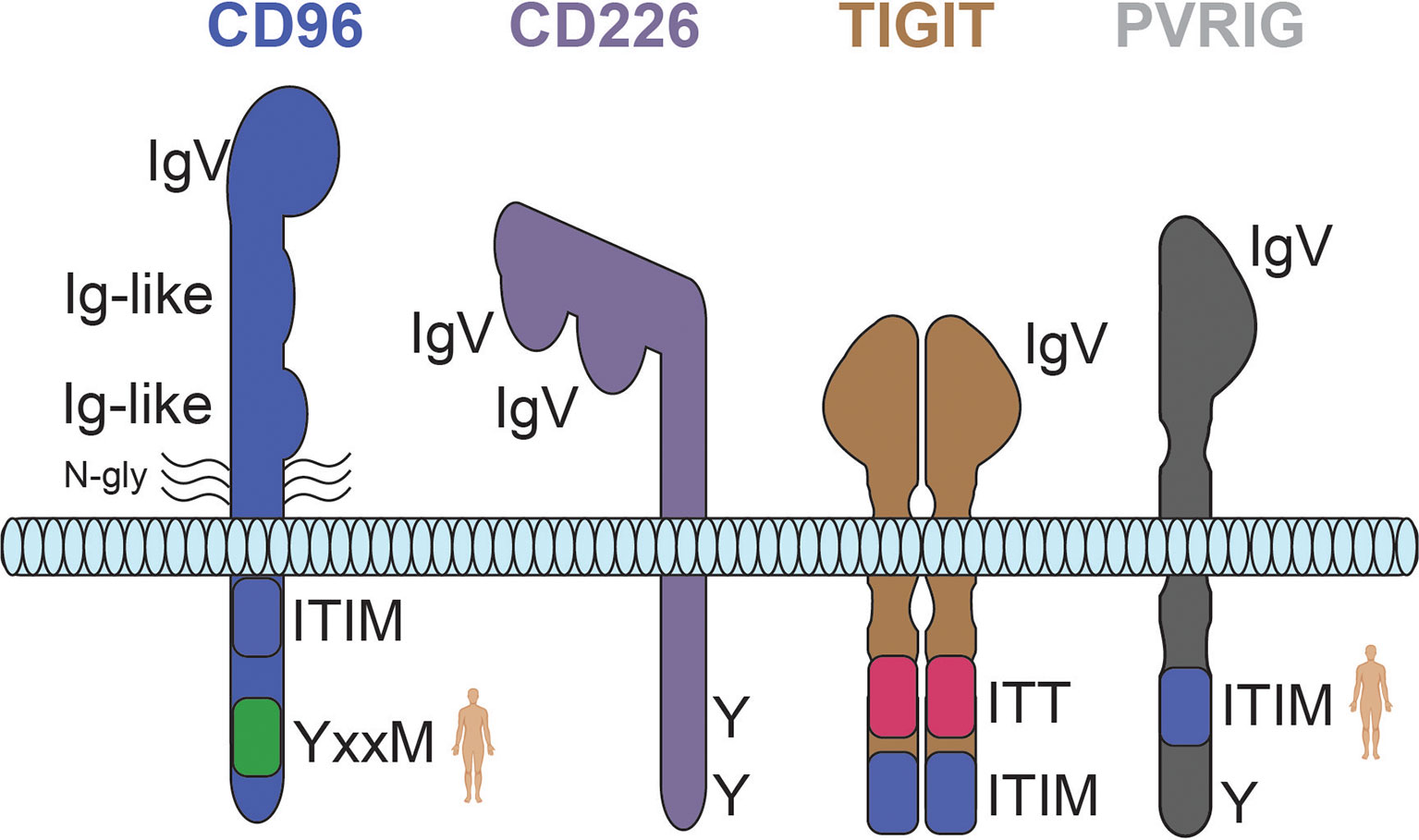
Figure 2 Predicted structures for the CD226 axis receptors CD96, CD226, TIGIT, and PVRIG. The weight of each line is representative of the relative strength of interaction. Human silhouettes signify that a motif is not present in rodents. N-gly, n-linked glycosylation; Y, tyrosine residue; ITT, immunoglobulin tail tyrosine motif; ITIM, immunoreceptor tyrosine-based inhibition motif.
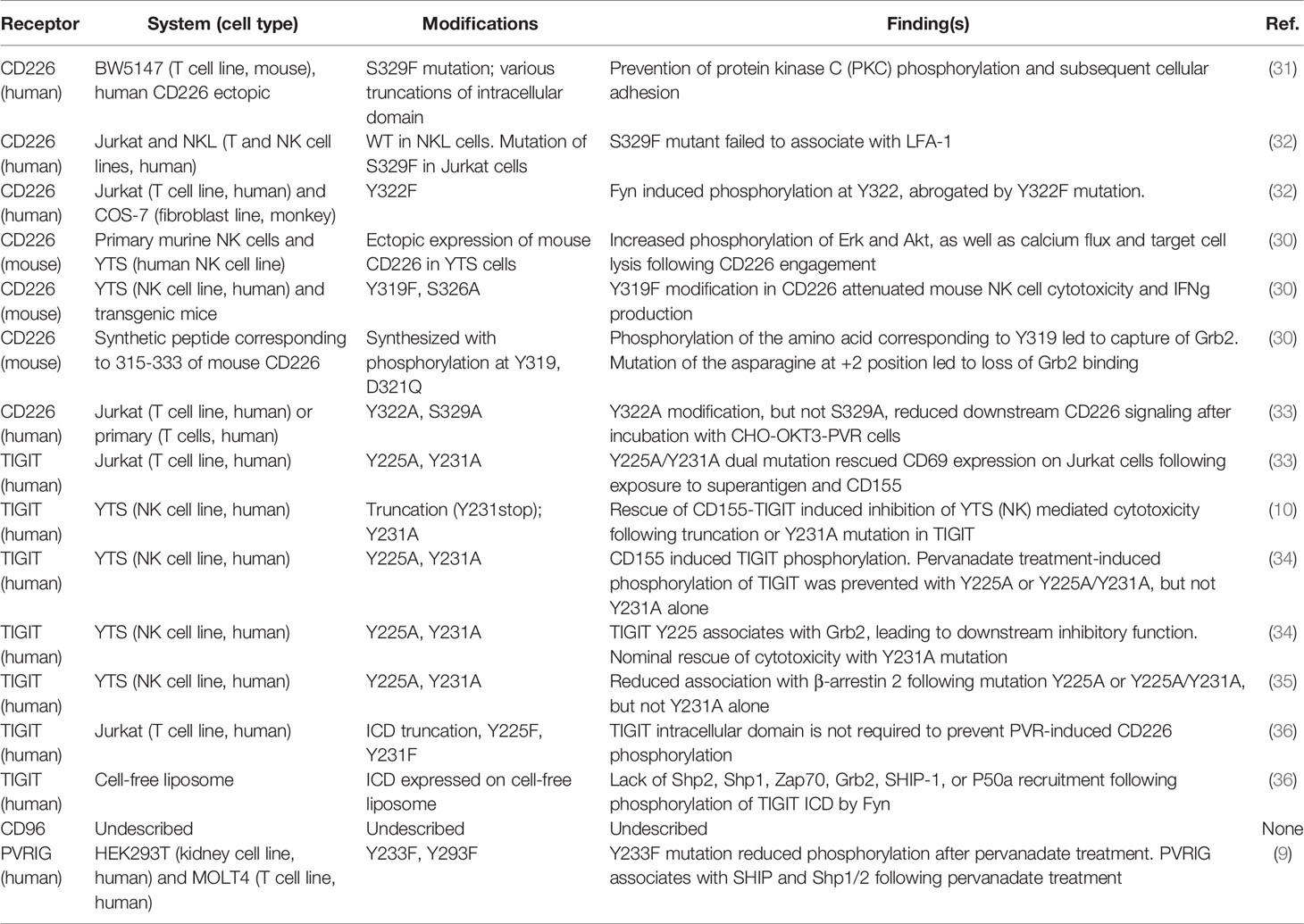
Table 2 CD226 axis receptor ICD mutational studies and associated functional effects.
Expression and Regulation
CD226 is broadly expressed on innate and adaptive immune populations and, like the associated inhibitory receptors within the axis, can be induced on peripheral human T cells following T cell receptor (TCR) activation (Table 1) (11, 38). Notably, the expression of CD226 on activated human T and intratumoral NK cells tracks closely to that of CD96 (11, 39). While co-expression could suggest some level of reactive regulation within the axis, it remains unclear if it is simply correlative or if it is biologically meaningful.
Another mechanism to temper immune activation could be the direct regulation of CD226 expression (i.e., by ligand-induced internalization/endocytosis or cleavage), as evidenced by its modulation in various disease settings including chronic viral infection and cancer (37, 40, 41). For example, in non-small cell lung cancer (NSCLC), CD226 expression is reduced on tumor-infiltrating NK cells relative to cells from normal adjacent tissues (NAT) and peripheral blood (42, 43). Moreover, CD226 has been shown to be sensitive to changes elicited by various therapeutics, such as chemotherapy (44).
While ligand-based interactions have primarily been implicated in driving the loss of cell surface CD226, recent work by Sun et al. also suggests that this effect could be mediated by soluble factors like transforming growth factor beta (TGF-β1) (37, 39, 45, 46). It is unclear if this is a direct or indirect effect, however, TGF-β-dependent modulation of CD226 has the potential to skew axis signaling in the tumor microenvironment (TME), enhancing the potential for immune evasion. Additionally, increased soluble CD226, and related loss of cell-surface expression, has been observed in the sera of cancer patients, suggesting some level of protease-dependent biology (47). Regardless of mechanism, the reduction of CD226 on immune cells in tumor-bearing hosts and prevalence of other axis members, raises several questions. Most notably, how much CD226 expression is necessary to drive functional responses following blockade of inhibitory receptors in the axis, and is this something that needs to be monitored to predict responses? As clinical efforts progress, it will be of interest to interrogate the relevance of CD226 dynamics in therapeutic responses.
Functional and Therapeutic Implications
The importance of CD226 in shaping the overall immune response has been thoroughly described in the context of autoimmunity, cancer, and viral infections (48). CD226 orchestrates the net activity of innate (NK cells) and adaptive (T cells) immunity via interplay with CD155 and CD112 (8, 49). For example, high expression of cell-surface CD155 coupled with low human leukocyte antigen (HLA) expression increases the susceptibility of immature DCs to CD226-mediated killing by NK cells. This process bridges both innate and adaptive immunity by removing Th2-polarizing iDCs, thus skewing T helper cell polarization (50–52). CD226 has been shown to play a critical role in promoting broad T cell expansion, CD8+ T cell antitumor activity, and “adaptive” NK cell responses (37, 53–55).
A significant amount of information regarding the contribution of CD226 to immune responses has been gleaned from genetic- and biologics-based approaches in mice. In contrast to delayed tumor progression in TIGIT-/- and CD96-/-, CD226-deficient mice exhibit increased susceptibility to MCA-induced fibrosarcomas as well as metastatic lung colonization (e.g., LLC lung and RM-1 prostate tumors) (19, 20, 56–58). Moreover, genetic ablation or antibody-mediated blockade of CD226 has been shown to abrogate the antitumor activity observed in CD96-deficient mice and in mice treated with anti-TIGIT, anti-PD-(L)1, and/or GITR (19, 20, 59, 60). Tumors propagated in CD226-deficient mice also exhibit increased expression of CD155 and CD112, further highlighting the dynamics within the CD226 axis (54, 57).
Given its critical immunostimulatory role, agonist approaches for CD226 seem attractive as a means to generate antitumor responses. However, CD226 is also expressed on platelets and has been associated with their adhesion/activation, potentially complicating the desired pharmacology profile (18, 61). Thus far, only one agent for CD226, LY3435151 (anti-CD226 agonist antibody, Eli Lilly), has progressed to clinical testing (NCT04099277, Table 3). Nevertheless, shortly after initiation, the phase 1 study for LY3435151 was terminated. Although one can speculate as to the reason(s) for study termination, given the critical position of CD226 in CD155 and CD112 axes, it will be of great interest to understand the limitations surrounding CD226 as a target for cancer immunotherapy.
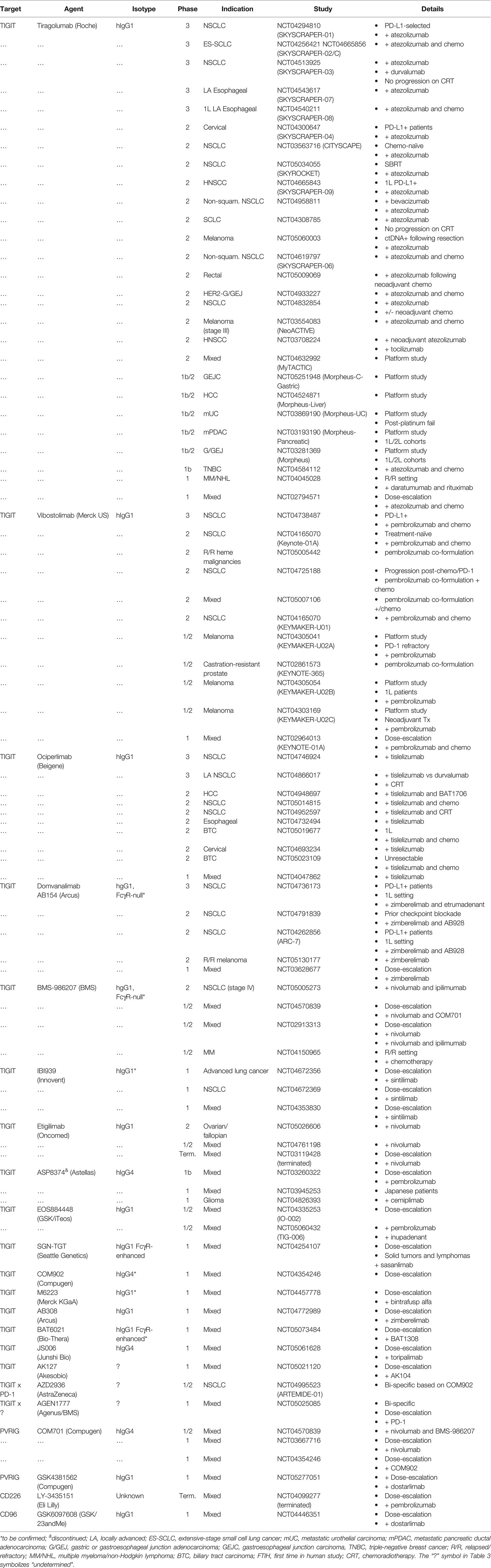
Table 3 Ongoing or discontinued (terminated) clinical trials evaluating CD226 axis-related antibody-based therapies in cancer patients.
Key Ligands in the Axis: CD155 and CD112
Discovery, Structure, and Interactions
Identified several decades before an association with relevant immune receptors, CD155 (poliovirus receptor [PVR], necl-5) and CD112 (PVRL2, nectin-2) represent structurally similar immunoglobulin superfamily (IgSF) adhesion glycoproteins (62–64). There are several isoforms of CD155 (α, β, γ, δ) and CD112 (short, CD112α; long, CD112δ), each with slightly different structural characteristics and tissue distribution (8, 65, 66). While the α and δ isoforms of CD155 code for cell-surface expressed CD155, the β and γ isoforms lack transmembrane (TM) domains, resulting in a soluble/secreted proteins that are not present in rodents (7, 65, 67). The extracellular region of CD155 and CD112 is comprised of an N-terminal variable (V) domain and constant (C1-C2) domains (24, 68–70). Like CD226, the V domain contains conserved ‘lock’ (AX6G) and ‘key’ (T[F/Y]P) motifs that are critical for mediating homo- and heterophilic interactions (24, 25, 68). Appropriately, CD155 and CD112 are capable of trans interactions, which can facilitate sustained cell-to-cell contact and immunoregulation (71, 72). Contrary to many nectin proteins, CD155 fails to exhibit strong homophilic interactions (71, 73). Similar evidence suggests that CD112 functions as a monomer, but also exhibits proclivity for homodimerization (74).
Outside of their conserved binding motifs, CD155 and CD112 exhibit nuances in binding orientation depending on the partner. For example, both CD155 and CD112 have been suggested to form heterotetrameric structures with TIGIT, where TIGIT dimers are sandwiched between CD155 or CD112 monomers; while other CD155 (e.g., CD226 and CD96) and CD112 complexes (e.g., CD226) are thought to be more conventional monomeric interactions (Figure 1) (25, 68–70). Differential structural attributes of TIGIT, CD96, and CD226 provide a partial explanation for their relative affinity to cell surface CD155 (26, 75). However, this is likely more complex given the variegated glycosylation patterns of the axis proteins. As mentioned previously, this affinity gradient, which favors inhibitory receptor binding (i.e., TIGIT and CD96), is reminiscent of the archetype CTLA-4 axis, whereby CTLA-4 effectively outcompetes costimulatory receptor CD28 from binding to B7 (CD80/86) (5). In a similar fashion, the inhibitory receptor PVRIG demonstrates a greater affinity for CD112 relative to CD226 (9).
Expression and Regulation
Despite detectable levels on multiple cell types (e.g., myeloid and epithelial cells) under physiological conditions, CD155 and CD112 are often elevated in various solid and hematological malignancies, correlating with a worse overall survival (12, 76–83). These correlations are not ubiquitous, however, as lack of expression in hepatocellular carcinoma (HCC) has been shown to be prognostically unfavorable (84, 85). Indeed, akin to PD-L1, CD155 expression has also been shown to be predictive of response to ICI (e.g., anti-PD-1 and anti-PD-1/CTLA-4) (86). This underscores the need for a clear etiological understanding and cancer-immune interplay in each indication. Moreover, treatment status must be considered, as CD155 and CD112 can be induced downstream of a range of cellular insults or stimuli known to activate the DNA damage response (DDR) pathway (87–92).
In addition to relatively broad expression by human tumor tissue and stromal populations, CD155 and CD112 are expressed on myeloid cells, such as monocytes and various subsets of dendritic cells (DCs) (50, 93). Given the reported expression of CD155 on follicular DCs, a key component of B cell follicles/germinal centers (GC), the involvement of the CD226 axis in tertiary lymphoid structure (TLS) biology may warrant further exploration (93, 94). This is particularly attractive in light of recent correlations between TLS generation/presence and response to cancer immunotherapy (95–97). One area where CD155 and CD112 appear to diverge is in lymphocyte expression, whereby CD155 can be induced on highly activated T cells (87, 98, 99). While the functional consequences of CD155 on activated T cells remains to be determined, CD155 has been associated with thymic selection via lymphocyte retention, underscoring its adhesion properties and potential impact on T cells (100). The expression of CD155 on T cells may also introduce complexity into the mechanistic interpretation and be derivative of certain experimental models. For example, CD155 induction on T cells may complicate our understanding of how anti-CD96 antibodies mediate functional activity even under conditions of T cell isolation (i.e., agonist activity or blockade of CD96:CD155 T cell-to-T cell interactions).
As the name suggests, CD155 or poliovirus receptor (PVR) serves as a point of cellular entry for poliovirus and, similar to the described role for soluble intercellular adhesion molecule-1 (ICAM-1) in response to rhinovirus infection, soluble CD155 has been proposed to be a partial serum-based sink for poliovirus (65, 101). Interestingly, soluble CD155 and CD112, have also been described in the serum of cancer patients and are often correlated with disease stage (7, 66, 102, 103). However, the specific contribution of soluble CD155 and CD112 to disease progression remains to be determined. Despite the longstanding awareness of CD155 and CD112 expression in cancer, an understanding of their contribution to the regulation of immune function and migration was lacking until a connection with the CD226 was established (8–10, 70, 104, 105). Functional characteristics and therapeutic implications for each interaction will be discussed in the following sections.
TIGIT
Discovery, Interactions and Structure
Several years after the description of CD226 and its association with CD155, T cell immunoglobulin and ITIM domain (TIGIT; V-set and transmembrane domain-containing 3 [Vstm3]; V-set and Ig domain-containing 9 [Vsig9]; Washington University Cell Adhesion Molecule [WUCAM]) emerged as an important member of the CD226 axis (10, 23, 93). The discovery of TIGIT was aided by searching for predicted structural similarities with cell-surface immune receptors, like PD-1. After its initial identification as a receptor for CD155, an additional ligand (CD112) and a role for TIGIT in NK cell modulation was described (10).
The extracellular region of TIGIT is comprised of a single IgV, and, consistent with other receptor-ligand pairs in the axis, trans interactions are facilitated by a lock-and-key motif in the N-terminal domain (68) (Figure 2). The majority of TIGIT function is tied to CD155, with binding to CD112 representing a lower affinity interaction (~30-fold via surface plasmon resonance [SPR]) (106). TIGIT has also been shown to interact with CD113 (PVRL3) and nectin-4 (PVRL4); however, clear functional implications remain to be seen (23, 107). Primarily facilitated by isoleucine (Ile) 42 (human; an analogous residue exists in mice) and the associated parallel interface, TIGIT exhibits a propensity for dimerization on the surface of cells (68). Accordingly, cis multimeric interactions with CD226 have also been reported, providing an additional means of CD226 regulation (36, 60).
The intracellular domain of TIGIT harbors an immunoreceptor tyrosine tail (ITT)-like phosphorylation motif and a conserved ITIM (LSYRSL) (106). While the role of each motif has been evaluated in mice and man, the relative contribution of ITT/ITIM to cell intrinsic TIGIT activity appears to be more contextual in humans (Table 2). Using a modified cytotoxicity system (YTS and 721.221 cells), Stanietsky et al. demonstrated that rescue of murine CD155-mediated NK cell suppression required mutation of both ITT and ITIM tyrosine residues (108). The same authors demonstrated that site-directed mutagenesis of only the ITIM (Y221) in human cells was able to abrogate CD155- and CD112-induced suppression of NK cell cytolytic activity (10). Further, a separate set of studies ascribed the ITT as the critical moiety for TIGIT-mediated inhibition of human NK cell IFNγ production, granule polarization, cellular cytotoxicity (34, 35). Recently, Banta and colleagues demonstrated that the intracellular domain of TIGIT was largely dispensable for impairment of CD226 phosphorylation (36). Rather, as previously mentioned, cis multimeric interactions with CD226 and/or ligand competition were found to drive TIGIT inhibition of CD226 co-stimulation. While this work brings into question the relative contribution of intrinsic signaling to TIGIT function (Table 2), it will be necessary to understand if it is selective to the experimental system, specific to TIGIT/CD226, or a more ubiquitous phenomenon.
Apart from ligand sequestration, TIGIT has been credited with additional mechanisms of action, which may be extrinsic or intrinsic depending on cell type. For instance, Yu et al. utilized human T cell and monocyte-derived DC co-cultures to demonstrate that unabated TIGIT : CD155 ligation alters antigen presenting cell (APC) cytokine profiles, indirectly impairing T cell responses (23). While multiple studies have revealed similar cell-extrinsic function of TIGIT, T and NK cell-intrinsic TIGIT signaling has also been described in both mice and man (22, 58, 108–112). For example, Joller et al. utilized an anti-mouse agonistic TIGIT antibody, in concert with TIGIT-/- mice, to characterize intrinsic TIGIT signaling in CD4+ T cells (111). The authors found that components of the T cell receptor complex (e.g., TCRα and CD3ε) were directly modulated following TIGIT engagement, adding another layer of immunoregulation to the mechanistic story of TIGIT.
Expression and Regulation
In the peripheral compartment, TIGIT is broadly represented on T and NK cell subsets, with noteworthy representation on regulatory T (Treg) cells, NKT cells, T follicular helper (Tfh) cells, and γδ T cells (Table 1) (11, 13). Apart from prominent expression of TIGIT on Treg cells, expression patterns for CD226 axis members ostensibly diverge when it comes to memory T cell populations (12). TIGIT and PVRIG are elevated on human terminally differentiated CD45RA+ TEMRA cells, whereas CD96 expression is mainly restricted to central memory (CM)/effector memory (EM) T cells (113, 114). It is intriguing to speculate about the functional effects that blockade of each immune receptor may have on peripheral or tumor/tissue-resident memory T cells. However, additional characterization, particularly on tumor-infiltrating immune populations, is required to determine if these expression patterns are indicative of any meaningful functional differentiation.
Consistent with its relatively high level of expression, TIGIT has been ascribed a role in the homeostasis and function of Treg cells. Indeed, TIGIT+ Treg cells have been shown to be highly immunosuppressive relative to their TIGIT- counterparts, inhibiting T helper (Th)1 and Th17 responses via induction of fibrinogen-like 2 (Fgl2) (59, 113, 115). This observation may be particularly relevant in cancer due to the elevated expression of TIGIT on intratumoral Treg cells (58, 116). As such, cellular depletion represents a plausible therapeutic mechanism for Fc-enabled TIGIT antibodies in cancer patients (discussed later).
In addition to readily detectable baseline expression, TIGIT is also upregulated on T and NK cells following activation. As exhausted T cells (TEX) are largely a product of tonic TCR stimulation, TIGIT has become a mainstay in several T cell ‘exhaustion signatures’ and has been implicated as a potential node for functional reversion of TEX (117, 118). However, because T cell exhaustion is a complicated process that is encumbered by progressive stages of epigenetic modification, TIGIT blockade alone is likely insufficient for meaningful functional rescue of fully exhausted T cells (119). Perhaps one way to determine a functional role for TIGIT in this process is to longitudinally characterize the impact of TIGIT inhibition on the arc of T cell activation to terminal exhaustion. Adapting these observations to the TME may inform whether TIGIT blockade could effectively prevent exhaustion or simply accelerate it. Alternatively, one could consider TIGIT blockade in concert with epigenetic modulators as a potential strategy to unmask and concomitantly enhance the activity of TEX (120). Regardless of potential functional implications, robust expression on TEX is consistent with progressive TIGIT induction following immune activation.
TIGIT expression is detectable in a wide range of human cancers and is often tightly correlated with T cell transcripts (CD4 and CD8A) (11). While its expression profile is largely consistent between the periphery and TME, TIGIT is upregulated on various TIL populations and is often accompanied by the expression of other activation-induce immune receptors, such as CD244 (2B4), TIM-3, and PD-1 (121, 122). Similar to the expression of TIGIT on TEMRA cells in the periphery, terminally differentiated intratumoral CD8+ T cells also express elevated levels of TIGIT. One could hypothesize that these cells are derivative of TIGITlow stem-like T cells residing within defined TME niches or in the periphery. Alternately, these could be newly infiltrating cytotoxic T cells that progressively acquire TIGIT during in situ differentiation or repeated TCR activation (123, 124).
Functional and Therapeutic Implications
No baseline developmental or immunological defects have been described in TIGIT (VSTM3)-deficient mice (C57BL/6 background) (125). However, the severity of induced autoimmune manifestations, such as myelin oligodendrocyte glycoprotein (MOG)-dependent encephalomyelitis (EAE) or graft-vs-host disease (GVHD) is increased relative to wildtype mice. In the context of cancer, TIGIT deficiency, or prophylactic antibody blockade, yields a modest level of protection against primary tumor growth in mice (19, 126–128). By contrast, TIGIT monotherapy is often ineffective in mice with established lesions (60). Therefore, combination with other ICI, such as anti-PD-(L)1, has been utilized to achieve more pronounced antitumor responses in a range of tumor models (60, 129, 130). In addition, the antitumor activity of murine anti-TIGIT antibodies has been tied to Fc-Fcγ receptor co-engagement (15, 131). This dependency is noteworthy given the myriad of anti-TIGIT isotypes currently under clinical evaluation (discussed later).
While the focus has largely been on solid tumors, a role for TIGIT in hematological malignancies has also been described. TIGIT is most notably upregulated on CD8+ T cells in multiple myeloma (MM), in both mice and man (128, 132, 133). Akin to what has been suggested in solid tumors, repeat antigen exposure and associated progressive exhaustion have been implicated in the upregulation of TIGIT in this setting and is of particular interest in the context of relapsed/refractory (R/R) disease. Accordingly, inhibition of TIGIT (genetic- or antibody-based) results in T cell-dependent antitumor responses in several syngeneic models of MM and is currently being evaluated in clinical studies (e.g., NCT04045028 and NCT04150965) (128). TIGIT has also been implicated in other heme cancers, such as follicular lymphoma (FL) and acute myeloid leukemia (AML) (134–136).
CD96
Discovery, Interactions and Structure
CD96 (T-cell activation, increased late expression [TACTILE]) was first identified as an orphan receptor on AML and T-cell acute lymphoblastic leukemia (T-ALL) cell lines (137). A functional role for CD96 wasn’t identified until half a decade later when the interaction between CD96 on NK cells and CD155 on tumor cells was described (105). Interestingly, this finding was influenced by observations involving NK cell expressed CD226 (8): The authors noted that, while the human NK cell line NK92 bound to the extracellular domain of CD155, the cells lacked detectable expression of CD226, thereby implicating a similarly structured receptor, CD96. This finding introduced a possible functional role for CD96 on immune cells and, with CD226, provided a framework for a novel immunoregulatory axis.
CD96 is comprised of three Ig-like extracellular domains and a flexible membrane-proximal stalk region containing multiple O-linked glycosylation sites (138) (Figure 2). Only the N-terminal domain (D1) contains a lock-and-key motif critical for binding CD155. In addition to CD155 binding, CD96 has also recently been shown to interact with human CD111; however, a functional role for this interaction remains to be elucidated (70, 75). While much of the biophysical data generated suggest that CD96 functions as a monomer, the possibility of CD96 oligomerization (i.e., cis interactions) needs to be evaluated in more complex systems to better understand how CD96 behaves on cells. As an example, the anti-mouse CD96 clone 8B10 (D2 binder) exhibits partial tumor control in an experimental model of metastases, yet is not entirely dependent on the presence of CD155 (139). This leads one to question if cis interference has a contextual biological role for CD96, if there are other important trans interactions, or whether this is the result of technical limitations.
Three isoforms of CD96 have been identified in humans, with two membrane-tethered isoforms (a longer variant [1] and a shorter variant [2]) and a less studied soluble isoform (variant 3) (75, 138, 140). Due to a truncated exon 4, the V2 isoform lacks a stretch of amino acids (~18) in the second Ig domain, resulting in an abbreviated loop structure. While less is known about the soluble form (V3), the V2 isoform is reported to be the most widely expressed and exhibits the highest affinity for CD155. The first domain of CD96 is reported to contain the epitope(s) required for CD155 binding while the second domain supports the magnitude/strength of binding (138). The intracellular domain of CD96 contains multiple tyrosine residues, with a prototypical inhibitory motif (ITIM, IXYXXI) that is conserved across species (138). The presence of an ITIM, coupled with the capacity for direct cross-competition with CD226/CD155 binding, suggests that CD96 functions as an inhibitory receptor. However, the categorization of CD96 as an inhibitory receptor has been mired due to (i) limited knowledge of CD96 signaling, (ii) conflicting functional data (particularly with NK cells), and (iii) the existence of a YXXM motif (YHEM) in primate CD96 (Figure 1) (105). Recently, Chiang et al. ascribed costimulatory properties to both mouse and human CD96 (141). While certainly not the only description of CD96 as a costimulatory receptor, caution should be taken regarding the interpretation of data in certain experimental systems. Because CD155 can be induced on TCR-activated T cells and the primordial involvement of nectin/necl proteins in cell adhesion, it is important to understand if the functional effects elicited by anti-CD96 are simply the result of blocking CD155-CD96 trans interactions (87, 98, 99). In addition, while several costimulatory receptors (e.g., inducible T-cell co-stimulator [ICOS]) harbor a YXXM motif, defining anti-CD96 functional directionality based on its presence may be shortsighted, as CD96 lacks a YxxM in mice and inhibitory receptors like CTLA-4 also contain similar sequences without a clearly defined functional role (142, 143).
Expression and Regulation
CD96 is expressed at baseline by several T cell populations (αβ and γδ), NK/NKT cells, and select B cell subsets in both mice and man (Table 1). The expression of CD96 on primary human immune cells is most evident on CD56+ NK cells and CD8+ T cells, with prominent representation on central and effector memory T cells (11, 113). Interestingly, human bone marrow-resident lymphoid tissue (lt) NK cells, which exhibit an overlapping transcriptional profile with tissue-resident memory CD8+ T cells, express both CD96 and TIGIT (144). Consistent with its alias (T-cell activation, increased late expression or ‘TACTILE’), CD96 expression also increases following TCR- or cytokine-based activation (e.g., interleukin [IL]-18 for T cells and TGF-β for NK cells) (39, 145, 146). In a recent study, Lepletier et al. described a near-homogenous level of CD96 and CD226 co-expression on TCR-activated peripheral human CD8+ T cells (11).
CD96 is also highly represented on rodent and human T cells and NK cells within the TME. A marked correlation between CD96/TIGIT messenger(m) RNA levels and CD3E/CD8a/CD4, with similar observations at the protein level (i.e., T cells), can be seen across multiple tumor types (11). Relative to T cells, the correlation between CD96 and NK cell-related genes is more infrequent despite being strong for specific indications such as HCC, head and neck squamous cell carcinoma (HNSCC), stomach adenocarcinoma, and melanoma (11, 147). Relative expression of CD96 and TIGIT on HCC-derived NK cells was shown to be dependent on tissue sub-localization, with TIGIT evenly represented across NK cells in the normal liver and intra/peritumoral space and CD96 more restricted to intratumoral NK cells (39, 148). CD96 expression in HCC is inversely correlated with several functional markers of NK cells, including T-bet (TBX21), perforin (PRF1), and granzyme B (GZMB) (39). Interestingly, TGF-β1 has been implicated in the induction of CD96 and associated CD226 downregulation in HCC, establishing a connection between two immunosuppressive pathways (39). More recently, CD96 was found to be co-expressed with PD-1 on TCF1+ exhausted precursor T cells in cervical tumors, a characteristic that the authors tied to therapeutic insensitivity (149). CD96 expression has also been noted in various hematological malignancies, such as T-ALL, myelodysplastic syndrome (MDS), and leukemic stem cells (LSCs) in AML (137, 150, 151). However, the biological relevance of CD96 in heme malignancies and its potential as a therapeutic target remains to be determined.
Functional and Therapeutic Implications
Similar to TIGIT, CD96-deficient mice (C57BL/6 background) do not exhibit overt baseline immunological defects (19, 152). However, challenge of CD96-/- mice with lipopolysaccharide (LPS) results in hyperinflammation characterized by enhanced IFNγ production by NK cells. Augmented NK cell function in CD96-/- mice is best exemplified by improved antimetastatic activity in B16F10 melanoma, RM-1 prostate cancer, and EO771 breast cancer models (127). Similar CD8+ T cell-dependent effects have been observed in CD96-defificent mice, albeit in the context of concomitant PD-1 (Pdcd1) knockout (114, 152). Notably, inflammatory responses and related antitumor activity in CD96-/- mice are abrogated following genetic ablation of CD226, underscoring the level of axis interplay (19, 152). Similar improvements in antitumor responses have been observed in mice following therapeutic inhibition of CD96, alone or in combination with other immune checkpoint inhibitors (114, 149). As an example, following neoadjuvant anti-PD-1 and gemcitabine, adjuvant therapy with anti-CD96 resulted in a significant survival benefit in pancreatic ductal adenocarcinoma (PDAC) tumor-bearing mice (153). Improvement in overall survival in this model was not solely driven by attenuation of primary tumor growth, but also by NK cell-dependent control of metastatic spread (153). Combination benefit of CD96 and PD-1 blockade has also been shown in other immunotherapy-insensitive models, such as the TC-1 (HPV+) tumor model (149).
Despite the wealth of information supporting an inhibitory role for CD96 in mice, functional data in humans is largely absent. Therefore, the progression of clinical-stage anti-CD96 antibodies, such as GSK6097608 will add important data to better understand mechanism of CD226 axis dynamics.
PVRIG
Discovery, Interactions and Structure
PVR-related Ig domain-containing (PVRIG) is the most recently discovered CD226 axis member (9). While much less is known about PVRIG, the sequestration of CD112 away from CD226 represents a regulatory mechanism similar to that utilized by TIGIT and CD96 co-inhibitory receptors (Figure 1) (9, 12). Alongside this potential for ligand competition, PVRIG has the capacity for intrinsic inhibitory signaling – which is likely facilitated by an ITIM-like motif found in the intracellular domain of PVRIG (9) (Table 2). Interestingly, this inhibitory domain is lacking in mice, suggesting that PVRIG function in mice is more dependent on ligand competition or is tied to an alternative mechanism that has yet to be described (Figure 2).
Expression and Regulation
Like TIGIT and CD96, PVRIG is expressed on various T and NK cell subsets and is upregulated on T cells following TCR stimulation (9). Elevated PVRIG expression has been described on exhausted T cells in human tumors, with noteworthy expression on TILs from ovarian, kidney, lung, endometrial, and breast cancers (12, 154). Similarly, antigen specific CD8+ T cells from virally infected mice exhibit similar upregulation of PVRIG as cells transition from activation to exhaustion (155). Despite lack of induction on in vitro-activated NK cells, increased PVRIG expression is observed on tumor-infiltrating NK cells (12). To date, little is known about regulation of PVRIG expression on different immune cell subsets.
Functional and Therapeutic Implications
Genetic- or biologics-based inhibition of PVRIG has been shown to impair tumor growth, particularly when combined with anti-PD-L1 (76, 155, 156). Notably, tumor-infiltrating CD8+ T cells and NK cells from PVRIG-deficient mice exhibit increased proinflammatory cytokine production, suggesting that PVRIG is involved in direct or indirect modulation of tumor-infiltrating T/NK cells (155, 156). In humans, in vitro blockade of PVRIG : CD112 binding results in enhanced TCR signaling and, when combined with trastuzumab (anti-HER2) and anti-TIGIT, potentiates ADCC activity and IFNγ production by NK cells (9, 157).
Clinical Landscape
Overview
This current clinical landscape for the CD226 axis is heavily represented by TIGIT antibodies, with an increasing number of agents at various stages of clinical evaluation (158, 159). Two PVRIG antagonist antibodies (COM-701, Compugen; GSK4381562, GlaxoSmithKline [GSK]), a single CD96 antibody (GSK6097608; GSK/23andMe), and a recently discontinued CD226 agonist antibody (LY-3435151, Eli Lilly & Co) round out the rest of the agents under clinical testing (Table 3).
TIGIT
As peculiar as it may sound, given that several agents have progressed into late-stage studies, the clinical landscape for TIGIT is just starting to take shape. Recent positive signs from CITYSCAPE (NCT03563716), a first-line NSCLC (1L) phase 2 clinical trial exploring atezolizumab (anti-PD-L1)/tiragolumab (anti-TIGIT) versus atezolizumab/placebo, were on display at American Society of Clinical Oncology (ASCO) 2020 (160). The most striking observation was the large jump in overall response rate (ORR) in PD-L1high patients (PD-L1 TPS > 50%; N=29 patients in each group) from 24% in the control arm (atezolizumab + placebo) to 66% with the combination. At first glance, this ORR compares favorably to similar ICI combinations (e.g., anti-PD-1/CTLA-4, Checkmate-227/NCT02477826) and is comparable to anti-PD-1 + chemotherapy (Keynote-189/NCT02578680) in a similar NSCLC patient subset (N=202 patients) (161). These data have recently enabled a breakthrough therapy designation by the FDA for 1L PD-L1high NSCLC. However, given the relatively small number of patients in CITYSCAPE, phase 3 studies from Roche and others will be critical for determining the robustness of the combination. Moreover, despite these promising results, both atezolizumab (18% [N=39 patients]) and the combination (16% [N=38 patients]) failed to elicit meaningful responses in PD-L1low patients (TPS 1-49%) relative to historical use of anti-PD-1 or anti-PD-L1 + chemotherapy (e.g., Keynote-407/NCT02775435 and Impower150/NCT02366143) (162, 163). This lack of overt activity across NSCLC patients may not be all that surprising given the nature of TIGIT antibodies (i.e., activated T cell-orientation and combination dependence), however as clinical studies progress, it will be important to establish a mechanistic understanding for the reduced activity of the combination in these patients.
The safety profile of tiragolumab combined with atezolizumab was similar to that of atezolizumab alone. Immune-related adverse events (IRAEs) were more frequent with the combination (69% versus 47%) but these were primarily manageable grade 1 and 2 immune-mediated AEs. There were also a similar number of grade 3+ AEs in the two groups (48% versus 44%) suggesting that this ICI combination will be better tolerated than ICI/chemo combinations (Keynote-189/NCT02578680, pembrolizumab + chemo [67.2%]) (161). With respect to AEs that demonstrated a >5% difference between arms, infusion-related reactions (IRR), pruritus, rash, were more frequent with the combination whereas dyspnea, productive cough, and hypercalcemia were more frequent with atezolizumab monotherapy.
Tiragolumab/atezolizumab combinations have since expanded into a suite of late-stage clinical trials dubbed the SKYSCRAPER trials. These trails are spread across indications, with SKYSCRAPER-01, -02, -03 and -06 in lung cancers, SKYSCRAPER-04 in PD-L1+ cervical cancer, and SKYSCRAPER-07, -08 and -09 in the ENT (esophageal or head and neck) sphere. Other indications including urothelial carcinoma, pancreatic cancer, and esophageal cancer are being explored via MORPHEUS umbrella studies (Table 3).
Other TIGIT molecules, including vibostolimab (MK-7684, Merck US) and ociperlimab (BGB-A1217, Beigene) are close behind with similar studies in NSCLC and a wave of studies in alternative indications (Table 3). Merck is also progressing vibostolimab and pembrolizumab as a co-formulation through phase 2 studies. This effort underscores the marriage between PD-1 or PD-L1 and TIGIT intervention strategies, however, time will tell if concurrent administration is sufficient or if dosing flexibility is required to enable optimal responses. Other agents, including Arcus (domvanalimab) and BMS (BMS-986207) are entering phase 2 and 3 studies. As opposed to the number of Fc-enabled TIGIT antibodies like tiragolumab and vibostolimab, agents like domvanalimab and BMS-986207 have attenuated Fc regions. Adding to this diversity, Fc-enhanced (SGN-TGT and BAT6021) as well as bi-specific (AZD2936 and AGEN1777) TIGIT antibodies have recently entered clinical development (Table 3). Given the non-clinical data suggesting the importance of Fc-FcγR co-engagement for anti-TIGIT function, the divergence in Fc biology between clinical molecules will be something to consider as studies read out.
Outside of solid tumor indications, TIGIT is also being evaluated in heme malignancies, such as MM and non-Hodgkin lymphoma (B-NHL) (Table 3). As discussed previously, TIGIT has been mechanistically linked to T cell exhaustion in MM and has shown promise as a therapeutic target in non-clinical models (128, 164). It will be interesting to see how well these findings translate to the clinical space, and if this is unique to TIGIT or if the therapeutic potential extends to other CD226 axis members.
PVRIG and CD96
As of Q2 2022, COM701 (IgG4) and GSK4381562 (IgG1) are the only two PVRIG molecules under clinical evaluation. Notably, COM701 demonstrated some early signs of activity, with a preliminary single-agent disease control rate (DCR) of 69% (165). Compugen also initiated a phase 1/2 study evaluating the triple combination of COM701, anti-PD-1 (nivolumab), and BMS-96820 (anti-TIGIT) in ovarian, endometrial, and select PVRL2high cancers. Clinical studies for CD96 are even more nascent, with a single molecule (GSK6097608, IgG1) in dose escalation as a monotherapy and in combination with anti-PD-1 (Table 3).
Considerations for the CD226 Axis
Stronger Together? Targeting Multiple CD226 Axis Members
Given the nuances in expression profiles and potential for promiscuity within the CD226 axis, it is intriguing to consider the possibility for compensatory regulation between different axis members, particularly under therapeutic pressure (Figure 1 and Table 1). These characteristics also highlight the potential of therapeutic collaboration in order to prevent inhibitory exigencies and/or increase coverage of various immune subsets and regulatory nodes within the axis (12). Several lines of non-clinical evidence directly or indirectly support co-inhibition of CD226 axis members. For example, antibody-mediated blockade of PVRIG has been shown to both induce rapid receptor internalization and increase TIGIT expression on antigen-specific CD8+ T cells (12). Similar dynamics have been observed with the ligands in the axis (83, 166). Moreover, genetic- or biologics-based co-blockade of TIGIT and CD96 has been shown to improve tumor control while co-blockade of TIGIT and PVRIG has also been shown to promote NK cell function (152, 157).
A PD-(L)1 Partnership
Although difficult to accurately assess due to the potential concomitant impact on TCR and CD28 activity, PD-1 signaling has been described to attenuate CD226 activity via SHP2-mediated dephosphorylation (20, 167) (Figure 1). This suggests that any CD226 signaling mediated by anti-TIGIT-, CD96- or PVRIG-based ligand redirection has the potential to be undercut by PD-(L)1 activity. Therefore, it is logical that CD226 axis therapeutics may need to be considered in the context of PD-(L)1 pathway blockade in order to reveal their full functional potential. Thus far, the available non-clinical data are consistent with this hypothesis. However, it will be interesting to see how the clinical space evolves, and whether or not PD-(L)1 blockade will become a prerequisite for the efficacy of all of the CD226 axis-based strategies.
Potential Utility of Fc-FcγR Co-Engagement
Given the relative breadth of non-clinical studies, it is not surprising that much of the data describing a potential role for Fc biology in the efficacy of antibodies targeting CD226 axis members has been restricted to TIGIT (15, 16, 114). An underlying point of contention has been the potential for antibody-mediated cellular depletion, and whether this is beneficial or detrimental, either due to safety concerns or impairment of efficacy (60, 130, 168–170). TIGIT is highly expressed on both peripheral and tumor infiltrating Treg cells. While selective depletion of Treg cells in the TME could relieve a suppressive barrier and promote antitumor responses, depletion of Treg cells in other tissues could impair peripheral tolerance and result in autoimmune manifestations, as seen in patients treated with mogamulizumab, an afucosylated antibody targeting CCR4-expressingTreg cells (11, 58, 171, 172). One could postulate that TIGIT+ effector/cytotoxic T cells would also be a target for depletion, which may attenuate the desired therapeutic effect. Thus, if these concerns were overwhelming, pursuing an Fc-attenuated TIGIT antibody would appear logical. However, given that TIGIT expression is elevated on terminally exhausted T cells, one could also posit that depleting these cells in the TME would permit the establishment of more functional T cell populations, leading to a net beneficial effect. Multiple factors, including differential cellular thresholds for target opsonization, antibody affinity, presence of effector cells mediating depletion, and FcγR polymorphisms need to be considered (14, 173, 174). Moreover, while safety concerns have been allayed with the clinical progression of Fc-competent antibodies, various non-clinical studies have demonstrated a benefit and, in some cases, a requirement for intact Fc biology to facilitate anti-TIGIT function (15, 60, 114, 169). Although the exact mechanism(s) responsible for improved anti-TIGIT activity remains to be determined, it has been suggested that Fc-FcγR co-engagement drives myeloid activation and/or T cell-APC immune synapse quality (15, 131, 169). Clinical efficacy and safety data for recently developed Fc-enhanced TIGIT antibodies should also provide visibility on potential advantages or disadvantages associated with this format. Ultimately, the clinic will be the proving ground for the seemingly disparate Fc variants under evaluation; that is, to determine if pure antagonism, effector biology, or a mixture of mechanisms are required for optimal patient responses.
Limited Fc-based characterization has been conducted for rodent/human PVRIG or human CD96 antibodies (139). However, it is important to consider that the optimal therapeutic potential for each target must integrate a thorough understanding of Fc biology. Finally, given the breadth of TIGIT antibodies with distinct Fc regions currently under clinical evaluation (Table 3), broader mechanistic insights into anti-TIGIT biology and whether these characteristics extend to other members of the CD226 axis, may begin to surface in the coming years.
Closing Remarks
In an effort to provide a framework for our evolving understanding of the CD226 axis in cancer, we discuss available non-clinical data and give an overview of the current clinical landscape. However, despite an understanding of the differential functional characteristics and expression profiles of each axis member, many questions remain regarding the mechanistic dynamics and contextual roles for each receptor. Some examples include (i) the functional implications of the variegated receptor expression (particularly with memory or stem-like memory T cell populations and regulatory T cells), (ii) the cell-intrinsic impact of individual receptor signaling, (iii) the functional consequence of ligand/receptor dynamics in different tissues (e.g., TME versus peripheral blood), (iv) the trans/cis interactions critical for activity, and (v) the contribution of FcγR biology to the function of antibodies for each receptor. As clinical programs advance and interest expands, some of these questions may begin to be addressed. Overall, it will be intriguing to see how therapeutic strategies for each receptor evolves and what mechanistic learnings precipitate from an increased amount of activity around the targets.
Author Contributions
MC and JW generated the original draft manuscript. JW, KH, SY, and JS reviewed, edited, and added content. All authors contributed to the article and approved the submitted version.
Conflict of Interest
All authors are employees of GlaxoSmithKline.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Iris Roth, Julia Cuende, Rachael Easton, and Gregory Driessens for their thoughtful review.
References
1. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discovery (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
2. Andrews LP, Yano H, Vignali DAA. Inhibitory Receptors and Ligands Beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or Backups. Nat Immunol (2019) 20:1425–34. doi: 10.1038/s41590-019-0512-0
3. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
4. de Andrade LF, Smyth MJ, Martinet L. DNAM-1 Control of Natural Killer Cells Functions Through Nectin and Nectin-Like Proteins. Immunol Cell Biol (2014) 92:237–44. doi: 10.1038/icb.2013.95
5. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell (2017) 170:1120–33. e1117. doi: 10.1016/j.cell.2017.07.024
6. Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers (Basel) (2019) 11(6):877. doi: 10.3390/cancers11060877
7. Okumura G, Iguchi-Manaka A, Murata R, Yamashita-Kanemaru Y, Shibuya A, Shibuya K. Tumor-Derived Soluble CD155 Inhibits DNAM-1-Mediated Antitumor Activity of Natural Killer Cells. J Exp Med (2020) 217(4):e20191290. doi: 10.1084/jem.20191290
8. Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B. Identification of PVR (CD155) and Nectin-2 (CD112) as Cell Surface Ligands for the Human DNAM-1 (CD226) Activating Molecule. J Exp Med (2003) 198:557–67. doi: 10.1084/jem.20030788
9. Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT. Identification of CD112R as a Novel Checkpoint for Human T Cells. J Exp Med (2016) 213:167–76. doi: 10.1084/jem.20150785
10. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A. The Interaction of TIGIT With PVR and PVRL2 Inhibits Human NK Cell Cytotoxicity. Proc Natl Acad Sci U S A (2009) 106:17858–63. doi: 10.1073/pnas.0903474106
11. Lepletier A, Lutzky VP, Mittal D, Stannard K, Watkins TS, Ratnatunga CN. The Immune Checkpoint CD96 Defines a Distinct Lymphocyte Phenotype and is Highly Expressed on Tumor-Infiltrating T Cells. Immunol Cell Biol (2019) 97:152–64. doi: 10.1111/imcb.12205
12. Whelan S, Ophir E, Kotturi MF, Levy O, Ganguly S, Leung L. PVRIG and PVRL2 Are Induced in Cancer and Inhibit CD8(+) T-Cell Function. Cancer Immunol Res (2019) 7:257–68. doi: 10.1158/2326-6066.CIR-18-0442
13. Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T. A Distinct Subpopulation of CD25(-) T-Follicular Regulatory Cells Localizes in the Germinal Centers. Proc Natl Acad Sci USA (2017) 114:E6400–9. doi: 10.1073/pnas.1705551114
14. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell (2018) 33:649–663 e644. doi: 10.1016/j.ccell.2018.02.010
15. Waight JD, Chand D, Dietrich S, Gombos R, Horn T, Gonzalez AM. Selective FcgammaR Co-Engagement on APCs Modulates the Activity of Therapeutic Antibodies Targeting T Cell Antigens. Cancer Cell (2018) 33:1033–1047 e1035. doi: 10.1016/j.ccell.2018.05.005
16. Preillon J, et al. Restoration of T-Cell Effector Function, Depletion of Tregs, and Direct Killing of Tumor Cells: The Multiple Mechanisms of Action of a-TIGIT Antagonist Antibodies. Mol Cancer Ther (2021) 20:121–31. doi: 10.1158/1535-7163.MCT-20-0464
17. Burns GF, Triglia T, Werkmeister JA, Begley CG, Boyd AW. TLiSA1, a Human T Lineage-Specific Activation Antigen Involved in the Differentiation of Cytotoxic T Lymphocytes and Anomalous Killer Cells From Their Precursors. J Exp Med (1985) 161:1063–78. doi: 10.1084/jem.161.5.1063
18. Scott JL, Dunn SM, Jin B, Hillam AJ, Walton S, Berndt MC. Characterization of a Novel Membrane Glycoprotein Involved in Platelet Activation. J Biol Chem (1989) 264:13475–82. doi: 10.1016/S0021-9258(18)80021-7
19. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L. The Receptors CD96 and CD226 Oppose Each Other in the Regulation of Natural Killer Cell Functions. Nat Immunol (2014) 15:431–8. doi: 10.1038/ni.2850
20. Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM. Combination Cancer Immunotherapy Targeting PD-1 and GITR can Rescue CD8(+) T Cell Dysfunction and Maintain Memory Phenotype. Sci Immunol (2018) 3(29):eaat7061. doi: 10.1126/sciimmunol.aat7061
21. Tahara-Hanaoka S, Shibuya K, Onoda Y, Zhang H, Yamazaki S, Miyamoto A. Functional Characterization of DNAM-1 (CD226) Interaction With its Ligands PVR (CD155) and Nectin-2 (PRR-2/Cd112). Int Immunol (2004) 16:533–8. doi: 10.1093/intimm/dxh059
22. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 Axis Regulates Human T Cell Function. J Immunol (2012) 188:3869–75. doi: 10.4049/jimmunol.1103627
23. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10:48–57. doi: 10.1038/ni.1674
24. Liu J, Qian X, Chen Z, Xu X, Gao F, Zhang S. Crystal Structure of Cell Adhesion Molecule Nectin-2/CD112 and its Binding to Immune Receptor DNAM-1/Cd226. J Immunol (2012) 188:5511–20. doi: 10.4049/jimmunol.1200324
25. Wang H, Qi J, Zhang S, Li Y, Tan S, Gao GF. Binding Mode of the Side-by-Side Two-IgV Molecule CD226/DNAM-1 to its Ligand CD155/Necl-5. Proc Natl Acad Sci USA (2019) 116:988–96. doi: 10.1073/pnas.1815716116
26. Deuss FA, Watson GM, Goodall KJ, Leece I., Chatterjee S, Fu Z. Structural Basis for the Recognition of Nectin-Like Protein-5 by the Human-Activating Immune Receptor, DNAM-1. J Biol Chem (2019) 294:12534–46. doi: 10.1074/jbc.RA119.009261
27. Liu G, Hu Y, Jin S, Jiang Q. Genetic Variant Rs763361 Regulates Multiple Sclerosis CD226 Gene Expression. Proc Natl Acad Sci USA (2017) 114:E906–7. doi: 10.1073/pnas.1618520114
28. Maiti AK, Kim-Howard X, Viswanathan P, Guillen L, Qian X, Rojas-Villarraga A. Non-Synonymous Variant (Gly307Ser) in CD226 is Associated With Susceptibility to Multiple Autoimmune Diseases. Rheumatol (Oxford) (2010) 49:1239–44. doi: 10.1093/rheumatology/kep470
29. Hafler JP, Maier LM, Cooper JD, Plagnol V, Hinks A, Simmonds MJ. CD226 Gly307Ser Association With Multiple Autoimmune Diseases. Genes Immun (2009) 10:5–10. doi: 10.1038/gene.2008.82
30. Zhang Z, Wu N, Lu Y, Davidson D, Colonna M, Veillette A. DNAM-1 Controls NK Cell Activation via an ITT-like motif. J Exp Med (2015) 212:2165–82. doi: 10.1084/jem.20150792
31. Shibuya A, Lanier LL, Phillips JH. Protein Kinase C Is Involved in the Regulation of Both Signaling and Adhesion Mediated by DNAX Accessory Molecule-1 Receptor. J Immunol (1998) 161:1671–6.
32. Shibuya K, Lanier LL, Phillips JH, Ochs HD, Shimizu K, Nakayama E. Physical and Functional Association of LFA-1 With DNAM-1 Adhesion Molecule. Immunity (1999) 11:615–23. doi: 10.1016/s1074-7613(00)80136-3
33. Jin HS, Ko M, Choi DS, Kim JH, Lee DH, Kang SH. CD226(hi)CD8(+) T Cells Are a Prerequisite for Anti-TIGIT Immunotherapy. Cancer Immunol Res (2020) 8:912–25. doi: 10.1158/2326-6066.CIR-19-0877
34. Liu S, Zhang H, Li M, Hu D, Li C, Ge B. Recruitment of Grb2 and SHIP1 by the ITT-Like Motif of TIGIT Suppresses Granule Polarization and Cytotoxicity of NK Cells. Cell Death Differ (2013) 20:456–64. doi: 10.1038/cdd.2012.141
35. Li M, Xia P, Du Y, Liu S, Huang G, Chen J. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Receptor/Poliovirus Receptor (PVR) Ligand Engagement Suppresses Interferon-Gamma Production of Natural Killer Cells. via beta-arrestin 2-mediated negative Signaling J Biol Chem (2014) 289:17647–57. doi: 10.1074/jbc.M114.572420
36. Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O'Gorman WE. Mechanistic Convergence of the TIGIT and PD-1 Inhibitory Pathways Necessitates Co-Blockade to Optimize Anti-Tumor CD8(+) T Cell Responses. Immunity (2022) 55:512–526 e519. doi: 10.1016/j.immuni.2022.02.005
37. Braun M, Aguilera AR, Sundarrajan A, Corvino D, Stannard K, Krumeich S. CD155 on Tumor Cells Drives Resistance to Immunotherapy by Inducing the Degradation of the Activating Receptor CD226 in CD8(+) T Cells. Immunity (2020) 53:805–823 e815. doi: 10.1016/j.immuni.2020.09.010
38. Vo AV, Takenaka E, Shibuya A, Shibuya K. Expression of DNAM-1 (CD226) on Inflammatory Monocytes. Mol Immunol (2016) 69:70–6. doi: 10.1016/j.molimm.2015.11.009
39. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology (2019) 70:168–83. doi: 10.1002/hep.30347
40. Cifaldi L, Doria M, Cotugno N, Zicari S, Cancrini C, Palma P. DNAM-1 Activating Receptor and Its Ligands: How Do Viruses Affect the NK Cell-Mediated Immune Surveillance During the Various Phases of Infection? Int J Mol Sci (2019) 20(15):3715. doi: 10.3390/ijms20153715
41. Chauvin JM, Ka M, Pagliano O, Menna C, Ding Q, DeBlasio R. IL15 Stimulation With TIGIT Blockade Reverses CD155-Mediated NK-Cell Dysfunction in Melanoma. Clin Cancer Res (2020) 26:5520–33. doi: 10.1158/1078-0432.CCR-20-0575
42. Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P. Profound Coordinated Alterations of Intratumoral NK Cell Phenotype and Function in Lung Carcinoma. Cancer Res (2011) 71:5412–22. doi: 10.1158/0008-5472.CAN-10-4179
43. Gillard-Bocquet M, Caer C, Cagnard N, Crozet L, Perez M, Fridman WH. Lung Tumor Microenvironment Induces Specific Gene Expression Signature in Intratumoral NK Cells. Front Immunol (2013) 4:19. doi: 10.3389/fimmu.2013.00019
44. Niu C, Jin H, Li M, Zhu S, Zhou L, Jin F. Low-Dose Bortezomib Increases the Expression of NKG2D and DNAM-1 Ligands and Enhances Induced NK and Gammadelta T Cell-Mediated Lysis in Multiple Myeloma. Oncotarget (2017) 8:5954–64. doi: 10.18632/oncotarget.13979
45. Carlsten M, Norell H, Bryceson YT, Poschke I, Schedvins K, Ljunggren HG. Primary Human Tumor Cells Expressing CD155 Impair Tumor Targeting by Down-Regulating DNAM-1 on NK Cells. J Immunol (2009) 183:4921–30. doi: 10.4049/jimmunol.0901226
46. Sanchez-Correa B, Gayoso I., Bergua JM, Casado JG, Morgado S, Solana R, et al. Decreased Expression of DNAM-1 on NK Cells From Acute Myeloid Leukemia Patients. Immunol Cell Biol (2012) 90:109–15. doi: 10.1038/icb.2011.15
47. Xu Z, Zhang T, Zhuang R, Zhang Y, Jia W, Song C. Increased Levels of Soluble CD226 in Sera Accompanied by Decreased Membrane CD226 Expression on Peripheral Blood Mononuclear Cells From Cancer Patients. BMC Immunol (2009) 10:34. doi: 10.1186/1471-2172-10-34
48. Huang Z, Qi G, Miller JS, Zheng SG. CD226: An Emerging Role in Immunologic Diseases. Front Cell Dev Biol (2020) 8:564. doi: 10.3389/fcell.2020.00564
49. Pauken KE, Wherry EJ. TIGIT and CD226: Tipping the Balance Between Costimulatory and Coinhibitory Molecules to Augment the Cancer Immunotherapy Toolkit. Cancer Cell (2014) 26:785–7. doi: 10.1016/j.ccell.2014.11.016
50. Pende D, Castriconi R, Romagnani P, Spaggiari GM, Marcenaro S, Dondero A. Expression of the DNAM-1 Ligands, Nectin-2 (CD112) and Poliovirus Receptor (CD155), On Dendritic Cells: Relevance for Natural Killer-Dendritic Cell Interaction. Blood (2006) 107:2030–6. doi: 10.1182/blood-2005-07-2696
51. Walwyn-Brown K, Guldevall K, Saeed M, Pende D, Onfelt B, MacDonald AS, et al. Human NK Cells Lyse Th2-Polarizing Dendritic Cells via NKp30 and DNAM-1. J Immunol (2018) 201:2028–41. doi: 10.4049/jimmunol.1800475
52. Seth S, Georgoudaki AM, Chambers BJ, Qiu Q., Kremmer E, Maier MK. Heterogeneous Expression of the Adhesion Receptor CD226 on Murine NK and T Cells and its Function in NK-Mediated Killing of Immature Dendritic Cells. J Leukoc Biol (2009) 86:91–101. doi: 10.1189/jlb.1208745
53. Sun JC, Beilke JN, Lanier LL. Adaptive Immune Features of Natural Killer Cells. Nature (2009) 457:557–61. doi: 10.1038/nature07665
54. Gilfillan S, Chan CJ, Cella M, Haynes NM, Rapaport AS, Boles KS. DNAM-1 Promotes Activation of Cytotoxic Lymphocytes by Nonprofessional Antigen-Presenting Cells and Tumors. J Exp Med (2008) 205:2965–73. doi: 10.1084/jem.20081752
55. Nabekura T, Kanaya M, Shibuya A, Fu G, Gascoigne NR, Lanier LL. Costimulatory Molecule DNAM-1 Is Essential for Optimal Differentiation of Memory Natural Killer Cells During Mouse Cytomegalovirus Infection. Immunity (2014) 40:225–34. doi: 10.1016/j.immuni.2013.12.011
56. Du X, de Almeida P, Manieri N, de AlmeidaNagata D, Wu TD, Harden Bowles K. CD226 Regulates Natural Killer Cell Antitumor Responses via phosphorylation-mediated inactivation of transcription factor FOXO1. Proc Natl Acad Sci USA (2018) 115:E11731–40. doi: 10.1073/pnas.1814052115
57. Iguchi-Manaka A, Kai H., Yamashita Y., Shibata K., Tahara-Hanaoka S., Honda S.. Accelerated Tumor Growth in Mice Deficient in DNAM-1 Receptor. J Exp Med (2008) 205:2959–64. doi: 10.1084/jem.20081611
58. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW. TIGIT Predominantly Regulates the Immune Response via regulatory T cells. J Clin Invest (2015) 125:4053–62. doi: 10.1172/JCI81187
59. Weulersse M, Asrir A, Pichler AC, Lemaitre L, Braun M, Carrie N. Eomes-Dependent Loss of the Co-Activating Receptor CD226 Restrains CD8(+) T Cell Anti-Tumor Functions and Limits the Efficacy of Cancer Immunotherapy. Immunity (2020) 53:824–39.e810. doi: 10.1016/j.immuni.2020.09.006
60. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8(+) T Cell Effector Function. Cancer Cell (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
61. Kojima H, Kanada H, Shimizu S, Kasama E, Shibuya K, Nakauchi H. CD226 Mediates Platelet and Megakaryocytic Cell Adhesion to Vascular Endothelial Cells. J Biol Chem (2003) 278:36748–53. doi: 10.1074/jbc.M300702200
62. Mendelsohn CL, Wimmer E, Racaniello VR. Cellular Receptor for Poliovirus: Molecular Cloning, Nucleotide Sequence, and Expression of a New Member of the Immunoglobulin Superfamily. Cell (1989) 56:855–65. doi: 10.1016/0092-8674(89)90690-9
63. Eberle F, Dubreuil P, Mattei MG, Devilard E, Lopez M. The Human PRR2 Gene, Related to the Human Poliovirus Receptor Gene (PVR), is the True Homolog of the Murine MPH Gene. Gene (1995) 159:267–72. doi: 10.1016/0378-1119(95)00180-e
64. Lopez M, Aoubala M, Jordier F, Isnardon D, Gomez S, Dubreuil P. The Human Poliovirus Receptor Related 2 Protein is a New Hematopoietic/Endothelial Homophilic Adhesion Molecule. Blood (1998) 92:4602–11. doi: 10.1182/blood.V92.12.4602
65. Baury B, Masson D, McDermott BM Jr., Jarry A., Blottiere HM, Blanchardie P. Identification of Secreted CD155 Isoforms. Biochem Biophys Res Commun (2003) 309:175–82. doi: 10.1016/s0006-291x(03)01560-2
66. Iguchi-Manaka A, Okumura G, Kojima H, Cho Y, Hirochika R, Bando H. Increased Soluble CD155 in the Serum of Cancer Patients. PloS One (2016) 11:e0152982. doi: 10.1371/journal.pone.0152982
67. Ravens I, Seth S, Forster R, Bernhardt G. Characterization and Identification of Tage4 as the Murine Orthologue of Human Poliovirus Receptor/CD155. Biochem Biophys Res Commun (2003) 312:1364–71. doi: 10.1016/j.bbrc.2003.11.067
68. Stengel KF, Harden-Bowles K, Yu X, Rouge L, Yin J, Comps-Agrar L. Structure of TIGIT Immunoreceptor Bound to Poliovirus Receptor Reveals a Cell-Cell Adhesion and Signaling Mechanism That Requires Cis-Trans Receptor Clustering. Proc Natl Acad Sci USA (2012) 109:5399–404. doi: 10.1073/pnas.1120606109
69. Deuss FA, Gully BS, Rossjohn J, Berry R. Recognition of Nectin-2 by the Natural Killer Cell Receptor T Cell Immunoglobulin and ITIM Domain (TIGIT). J Biol Chem (2017) 292:11413–22. doi: 10.1074/jbc.M117.786483
70. Deuss FA, Watson GM, Fu Z, Rossjohn J, Berry R. Structural Basis for CD96 Immune Receptor Recognition of Nectin-Like Protein-5, Cd155. Structure (2019) 27:219–28.e213. doi: 10.1016/j.str.2018.10.023
71. Ikeda W, Kakunaga S, Itoh S, Shingai T, Takekuni K, Satoh K. Tage4/Nectin-Like Molecule-5 Heterophilically Trans-Interacts With Cell Adhesion Molecule Nectin-3 and Enhances Cell Migration. J Biol Chem (2003) 278:28167–72. doi: 10.1074/jbc.M303586200
72. Devilard E, Xerri L, Dubreuil P, Lopez M, Reymond N. Nectin-3 (CD113) Interacts With Nectin-2 (CD112) to Promote Lymphocyte Transendothelial Migration. PloS One (2013) 8:e77424. doi: 10.1371/journal.pone.0077424
73. Harrison OJ, Vendome J, Brasch J, Jin X, Hong S, Katsamba PS. Nectin Ectodomain Structures Reveal a Canonical Adhesive Interface. Nat Struct Mol Biol (2012) 19:906–15. doi: 10.1038/nsmb.2366
74. Samanta D, Ramagopal UA, Rubinstein R, Vigdorovich V, Nathenson SG, Almo SC. Structure of Nectin-2 Reveals Determinants of Homophilic and Heterophilic Interactions That Control Cell-Cell Adhesion. Proc Natl Acad Sci U S A (2012) 109:14836–40. doi: 10.1073/pnas.1212912109
75. Holmes VM, Maluquer deMotes C, Richards PT, Roldan J, Bhargava AK, Orange JS. Interaction Between Nectin-1 and the Human Natural Killer Cell Receptor CD96. PloS One (2019) 14:e0212443. doi: 10.1371/journal.pone.0212443
76. Stamm H, Klingler F, Grossjohann EM, Muschhammer J, Vettorazzi E, Heuser M. Immune Checkpoints PVR and PVRL2 are Prognostic Markers in AML and Their Blockade Represents a New Therapeutic Option. Oncogene (2018) 37:5269–80. doi: 10.1038/s41388-018-0288-y
77. Huang DW, Huang M, Lin XS, Huang Q. CD155 Expression and its Correlation With Clinicopathologic Characteristics, Angiogenesis, and Prognosis in Human Cholangiocarcinoma. Onco Targets Ther (2017) 10:3817–25. doi: 10.2147/OTT.S141476
78. Nakai R, Maniwa Y, Tanaka Y, Nishio W, Yoshimura M, Okita Y. Overexpression of Necl-5 Correlates With Unfavorable Prognosis in Patients With Lung Adenocarcinoma. Cancer Sci (2010) 101:1326–30. doi: 10.1111/j.1349-7006.2010.01530.x
79. Nishiwada S, Sho M, Yasuda S, Shimada K, Yamato I, Akahori T. Clinical Significance of CD155 Expression in Human Pancreatic Cancer. Anticancer Res (2015) 35:2287–97.
80. Bevelacqua V, Bevelacqua Y, Candido S, Skarmoutsou E, Amoroso A, Guarneri C. Nectin Like-5 Overexpression Correlates With the Malignant Phenotype in Cutaneous Melanoma. Oncotarget (2012) 3:882–92. doi: 10.18632/oncotarget.594
81. Sloan KE, Eustace BK, Stewart JK, Zehetmeier C, Torella C, Simeone M. CD155/PVR Plays a Key Role in Cell Motility During Tumor Cell Invasion and Migration. BMC Cancer (2004) 4:73. doi: 10.1186/1471-2407-4-73
82. Masson D, Jarry A, Baury B, Blanchardie P, Laboisse C, Lustenberger P, et al. Overexpression of the CD155 Gene in Human Colorectal Carcinoma. Gut (2001) 49:236–40. doi: 10.1136/gut.49.2.236
83. Nagumo Y, Iguchi-Manaka A, Yamashita-Kanemaru Y, Abe F, Bernhardt G, Shibuya A, et al. Increased CD112 Expression in Methylcholanthrene-Induced Tumors in CD155-Deficient Mice. PloS One (2014) 9:e112415. doi: 10.1371/journal.pone.0112415
84. Qu P, Huang X, Zhou X, Lu Z, Liu F, Shi Z. Loss of CD155 Expression Predicts Poor Prognosis in Hepatocellular Carcinoma. Histopathology (2015) 66:706–14. doi: 10.1111/his.12584
85. Huang X, Qu P, Chen Y, Zhou X, Wu Y, Liu F. Low Expression of CD112 is Associated With Poor Overall Survival in Patients With Hepatocellular Carcinoma. Hum Pathol (2014) 45:1944–50. doi: 10.1016/j.humpath.2014.06.001
86. Lepletier A, Madore J, O'Donnell JS, Johnston RL, Li XY, McDonald E. Tumor CD155 Expression Is Associated With Resistance to Anti-PD1 Immunotherapy in Metastatic Melanoma. Clin Cancer Res (2020) 26:3671–81. doi: 10.1158/1078-0432.CCR-19-3925
87. Ardolino M, Zingoni A, Cerboni C, Cecere F, Soriani A, Iannitto ML, et al. DNAM-1 Ligand Expression on Ag-Stimulated T Lymphocytes Is Mediated by ROS-Dependent Activation of DNA-Damage Response: Relevance for NK-T Cell Interaction. Blood (2011) 117:4778–86. doi: 10.1182/blood-2010-08-300954
88. Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V. ATM-ATR-Dependent Up-Regulation of DNAM-1 and NKG2D Ligands on Multiple Myeloma Cells by Therapeutic Agents Results in Enhanced NK-Cell Susceptibility and Is Associated With a Senescent Phenotype. Blood (2009) 113:3503–11. doi: 10.1182/blood-2008-08-173914
89. Gao J, Zheng Q, Shao Y, Wang W, Zhao C. CD155 Downregulation Synergizes With Adriamycin to Induce Breast Cancer Cell Apoptosis. Apoptosis (2018) 23:512–20. doi: 10.1007/s10495-018-1473-8
90. Fionda C, Abruzzese MP, Zingoni A, Soriani A, Ricci B, Molfetta R. Nitric Oxide Donors Increase PVR/CD155 DNAM-1 Ligand Expression in Multiple Myeloma Cells: Role of DNA Damage Response Activation. BMC Cancer (2015) 15:17. doi: 10.1186/s12885-015-1023-5
91. Cerboni C, Fionda C, Soriani A, Zingoni A, Doria M, Cippitelli M, et al. The DNA Damage Response: A Common Pathway in the Regulation of NKG2D and DNAM-1 Ligand Expression in Normal, Infected, and Cancer Cells. Front Immunol (2014) 4:508. doi: 10.3389/fimmu.2013.00508
92. Tang ML, Khan MK, Croxford JL, Tan KW, Angeli V, Gasser S. The DNA Damage Response Induces Antigen Presenting Cell-Like Functions in Fibroblasts. Eur J Immunol (2014) 44:1108–18. doi: 10.1002/eji.201343781
93. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG. A Novel Molecular Interaction for the Adhesion of Follicular CD4 T Cells to Follicular DC. Eur J Immunol (2009) 39:695–703. doi: 10.1002/eji.200839116
94. Heesters BA, Myers RC, Carroll MC. Follicular Dendritic Cells: Dynamic Antigen Libraries. Nat Rev Immunol (2014) 14:495–504. doi: 10.1038/nri3689
95. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R. B Cells and Tertiary Lymphoid Structures Promote Immunotherapy Response. Nature (2020) 577:549–55. doi: 10.1038/s41586-019-1922-8
96. Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J. B Cells are Associated With Survival and Immunotherapy Response in Sarcoma. Nature (2020) 577:556–60. doi: 10.1038/s41586-019-1906-8
97. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S. Author Correction: Tertiary Lymphoid Structures Improve Immunotherapy and Survival in Melanoma. Nature (2020) 580:E1. doi: 10.1038/s41586-020-2155-6
98. Maier MK, Seth S, Czeloth N, Qiu Q, Ravens I, Kremmer E. The Adhesion Receptor CD155 Determines the Magnitude of Humoral Immune Responses Against Orally Ingested Antigens. Eur J Immunol (2007) 37:2214–25. doi: 10.1002/eji.200737072
99. Davis ZB, Sowrirajan B, Cogswell A, Ward JP, Planelles V, Barker E. CD155 on HIV-Infected Cells Is Not Modulated by HIV-1 Vpu and Nef But Synergizes With NKG2D Ligands to Trigger NK Cell Lysis of Autologous Primary HIV-Infected Cells. AIDS Res Hum Retroviruses (2017) 33:93–100. doi: 10.1089/AID.2015.0375
100. Qiu Q, Ravens I, Seth S, Rathinasamy A, Maier MK, Davalos-Misslitz A. CD155 is Involved in Negative Selection and is Required to Retain Terminally Maturing CD8 T Cells in Thymus. J Immunol (2010) 184:1681–9. doi: 10.4049/jimmunol.0900062
101. Marlin SD, Staunton DE, Springer TA, Stratowa C., Sommergruber W, Merluzzi VJ. A Soluble Form of Intercellular Adhesion Molecule-1 Inhibits Rhinovirus Infection. Nature (1990) 344:70–2. doi: 10.1038/344070a0
102. Iguchi-Manaka A, Okumura G, Ichioka E, Kiyomatsu H, Ikeda T, Bando H. High Expression of Soluble CD155 in Estrogen Receptor-Negative Breast Cancer. Breast Cancer (2020) 27:92–9. doi: 10.1007/s12282-019-00999-8
103. Karabulut M, Gunaldi M, Alis H, Afsar CU, Karabulut S, Serilmez M. Serum Nectin-2 Levels are Diagnostic and Prognostic in Patients With Colorectal Carcinoma. Clin Transl Oncol (2016) 18:160–71. doi: 10.1007/s12094-015-1348-1
104. Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T.. DNAM-1, A Novel Adhesion Molecule Involved in the Cytolytic Function of T Lymphocytes. Immunity (1996) 4:573–81. doi: 10.1016/s1074-7613(00)70060-4
105. Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting Edge: CD96 (Tactile) Promotes NK Cell-Target Cell Adhesion by Interacting With the Poliovirus Receptor (CD155). J Immunol (2004) 172:3994–8. doi: 10.4049/jimmunol.172.7.3994
106. Samanta D, Guo H, Rubinstein R, Ramagopal UA, Almo SC. Structural, Mutational and Biophysical Studies Reveal a Canonical Mode of Molecular Recognition Between Immune Receptor TIGIT and Nectin-2. Mol Immunol (2017) 81:151–9. doi: 10.1016/j.molimm.2016.12.003
107. Reches A, Ophir Y, Stein N, Kol I., Isaacson B, Charpak Amikam Y. Nectin4 Is a Novel TIGIT Ligand Which Combines Checkpoint Inhibition and Tumor Specificity. J Immunother Cancer (2020) 8(29):e000266. doi: 10.1136/jitc-2019-000266
108. Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R. Mouse TIGIT Inhibits NK-Cell Cytotoxicity Upon Interaction With PVR. Eur J Immunol (2013) 43:2138–50. doi: 10.1002/eji.201243072
109. Chen X, Lu PH, Liu L, Fang ZM, Duan W, Liu ZL. TIGIT Negatively Regulates Inflammation by Altering Macrophage Phenotype. Immunobiology (2016) 221:48–55. doi: 10.1016/j.imbio.2015.08.003
110. Schorer M, Rakebrandt N, Lambert K, Hunziker A, Pallmer K, Oxenius A. TIGIT Limits Immune Pathology During Viral Infections. Nat Commun (2020) 11:1288. doi: 10.1038/s41467-020-15025-1
111. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD. Cutting Edge: TIGIT has T Cell-Intrinsic Inhibitory Functions. J Immunol (2011) 186:1338–42. doi: 10.4049/jimmunol.1003081
112. Lucca LE, Axisa PP, Singer ER, Nolan NM, Dominguez-Villar M, Hafler DA. TIGIT Signaling Restores Suppressor Function of Th1 Tregs. JCI Insight (2019) 4(3):e124427. doi: 10.1172/jci.insight.124427
113. Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J Immunol (2015) 195:145–55. doi: 10.4049/jimmunol.1402381
114. Mittal D, Lepletier A, Madore J, Aguilera AR, Stannard K, Blake SJ. CD96 Is an Immune Checkpoint That Regulates CD8(+) T-Cell Antitumor Function. Cancer Immunol Res (2019) 7:559–71. doi: 10.1158/2326-6066.CIR-18-0637
115. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C. Treg Cells Expressing the Coinhibitory Molecule TIGIT Selectively Inhibit Proinflammatory Th1 and Th17 Cell Responses. Immunity (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
116. Ge Z, Zhou G, Campos Carrascosa L, Gausvik E, Boor PPC, Noordam L. TIGIT and PD1 Co-Blockade Restores Ex Vivo Functions of Human Tumor-Infiltrating CD8(+) T Cells in Hepatocellular Carcinoma. Cell Mol Gastroenterol Hepatol (2021) 12:443–64. doi: 10.1016/j.jcmgh.2021.03.003
117. Chew GM, Fujita T, Webb GM, Burwitz BJ, Wu HL, Reed JS. TIGIT Marks Exhausted T Cells, Correlates With Disease Progression, and Serves as a Target for Immune Restoration in HIV and SIV Infection. PloS Pathog (2016) 12:e1005349. doi: 10.1371/journal.ppat.1005349
118. Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O'Brien S. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity (2018) 48:1029–1045 e1025. doi: 10.1016/j.immuni.2018.04.026
119. Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity (2020) 52:825–41.e828. doi: 10.1016/j.immuni.2020.04.014
120. McGoverne I, Dunn J, Batham J, Tu WJ, Chrisp J, Rao S. Epitherapy and Immune Checkpoint Blockade: Using Epigenetic Reinvigoration of Exhausted and Dysfunctional T Cells to Reimburse Immunotherapy Response. BMC Immunol (2020) 21:22. doi: 10.1186/s12865-020-00353-0
121. Li X, Wang R, Fan P, Yao X, Qin L, Peng Y. A Comprehensive Analysis of Key Immune Checkpoint Receptors on Tumor-Infiltrating T Cells From Multiple Types of Cancer. Front Oncol (2019) 9:1066. doi: 10.3389/fonc.2019.01066
122. Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(-)CD8(+) Tumor-Infiltrating T Cells. Immunity (2019) 50:181–94.e186. doi: 10.1016/j.immuni.2018.11.014
123. Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA. An Intra-Tumoral Niche Maintains and Differentiates Stem-Like CD8 T Cells. Nature (2019) 576:465–70. doi: 10.1038/s41586-019-1836-5
124. Galletti G, De Simone G, Mazza EMC, Puccio S, Mezzanotte C, Bi TM. Two Subsets of Stem-Like CD8(+) Memory T Cell Progenitors With Distinct Fate Commitments in Humans. Nat Immunol (2020) 21(12):1552–62. doi: 10.1038/s41590-020-0791-5
125. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM. Vstm3 is a Member of the CD28 Family and an Important Modulator of T-Cell Function. Eur J Immunol (2011) 41:902–15. doi: 10.1002/eji.201041136
126. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W. Blockade of the Checkpoint Receptor TIGIT Prevents NK Cell Exhaustion and Elicits Potent Anti-Tumor Immunity. Nat Immunol (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
127. Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D. Suppression of Metastases Using a New Lymphocyte Checkpoint Target for Cancer Immunotherapy. Cancer Discovery (2016) 6:446–59. doi: 10.1158/2159-8290.CD-15-0944
128. Guillerey C, Harjunpaa H, Carrie N, Kassem S, Teo T, Miles K. TIGIT Immune Checkpoint Blockade Restores CD8(+) T-Cell Immunity Against Multiple Myeloma. Blood (2018) 132:1689–94. doi: 10.1182/blood-2018-01-825265
129. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS. TIGIT and PD-1 Dual Checkpoint Blockade Enhances Antitumor Immunity and Survival in GBM. Oncoimmunology (2018) 7:e1466769. doi: 10.1080/2162402X.2018.1466769
130. Dixon KO, Schorer M, Nevin J, Etminan Y, Amoozgar Z, Kondo T. Functional Anti-TIGIT Antibodies Regulate Development of Autoimmunity and Antitumor Immunity. J Immunol (2018) 200:3000–7. doi: 10.4049/jimmunol.1700407
131. Han JH, Cai M, Grein J, Perera S, Wang H, Bigler M. Effective Anti-Tumor Response by TIGIT Blockade Associated With FcgammaR Engagement and Myeloid Cell Activation. Front Immunol (2020) 11:573405. doi: 10.3389/fimmu.2020.573405
132. Lozano E, Mena MP, Diaz T, Martin-Antonio B, Leon S, Rodriguez-Lobato LG. Nectin-2 Expression on Malignant Plasma Cells Is Associated With Better Response to TIGIT Blockade in Multiple Myeloma. Clin Cancer Res (2020) 26:4688–98. doi: 10.1158/1078-0432.CCR-19-3673
133. Minnie SA, Kuns RD, Gartlan KH, Zhang P, Wilkinson AN, Samson L. Myeloma Escape After Stem Cell Transplantation is a Consequence of T-Cell Exhaustion and is Prevented by TIGIT Blockade. Blood (2018) 132:1675–88. doi: 10.1182/blood-2018-01-825240
134. Josefsson SE, Beiske K, Blaker YN, Forsund MS, Holte H, Ostenstad B. TIGIT and PD-1 Mark Intratumoral T Cells With Reduced Effector Function in B-Cell Non-Hodgkin Lymphoma. Cancer Immunol Res (2019) 7:355–62. doi: 10.1158/2326-6066.CIR-18-0351
135. Josefsson SE, Huse K, Kolstad A, Beiske K, Pende D, Steen CB. T Cells Expressing Checkpoint Receptor TIGIT Are Enriched in Follicular Lymphoma Tumors and Characterized by Reversible Suppression of T-Cell Receptor Signaling. Clin Cancer Res (2018) 24:870–81. doi: 10.1158/1078-0432.CCR-17-2337
136. Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates With CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin Cancer Res (2016) 22:3057–66. doi: 10.1158/1078-0432.CCR-15-2626
137. Gramatzki M, Ludwig WD, Burger R, Moos P, Rohwer P, Grunert C. Antibodies TC-12 ("Unique") and TH-111 (CD96) Characterize T-Cell Acute Lymphoblastic Leukemia and a Subgroup of Acute Myeloid Leukemia. Exp Hematol (1998) 26:1209–14.
138. Meyer D, Seth S, Albrecht J, Maier MK, du Pasquier L, Ravens I. CD96 Interaction With CD155 via its first Ig-like domain is modulated by alternative splicing or mutations in distal Ig-like domains. J Biol Chem (2009) 284:2235–44. doi: 10.1074/jbc.M807698200
139. Roman Aguilera A, Lutzky VP, Mittal D, Li XY, Stannard K, Takeda K. CD96 Targeted Antibodies Need Not Block CD96-CD155 Interactions to Promote NK Cell Anti-Metastatic Activity. Oncoimmunology (2018) 7:e1424677. doi: 10.1080/2162402X.2018.1424677
140. Gong J, Zhu C, Zhuang R, Song C, Li Q, Xu Z. Establishment of an Enzyme-Linked Immunosorbent Assay System for Determining Soluble CD96 and its Application in the Measurement of Scd96 in Patients With Viral Hepatitis B and Hepatic Cirrhosis. Clin Exp Immunol (2009) 155:207–15. doi: 10.1111/j.1365-2249.2008.03829.x
141. Chiang EY, de Almeida PE, de AlmeidaNagata DE, Bowles KH, Du X, Chitre AS. CD96 Functions as a Co-Stimulatory Receptor to Enhance CD8(+) T Cell Activation and Effector Responses. Eur J Immunol (2020) 50:891–902. doi: 10.1002/eji.201948405
142. Rudd CE, Schneider H. Unifying Concepts in CD28, ICOS and CTLA4 Co-Receptor Signalling. Nat Rev Immunol (2003) 3:544–56. doi: 10.1038/nri1131
143. Kirchgessner H, Dietrich J, Scherer J, Isomaki P, Korinek V, Hilgert I. The Transmembrane Adaptor Protein TRIM Regulates T Cell Receptor (TCR) Expression and TCR-Mediated Signaling via an association with the TCR zeta chain. J Exp Med (2001) 193:1269–84. doi: 10.1084/jem.193.11.1269
144. Melsen JE, Lugthart G, Vervat C, Kielbasa SM, van derZeeuw SAJ, Buermans HPJ. Human Bone Marrow-Resident Natural Killer Cells Have a Unique Transcriptional Profile and Resemble Resident Memory CD8(+) T Cells. Front Immunol (2018) 9:1829. doi: 10.3389/fimmu.2018.01829
145. Eriksson EM, Keh CE, Deeks SG, Martin JN, Hecht FM, Nixon DF. Differential Expression of CD96 Surface Molecule Represents CD8(+) T Cells With Dissimilar Effector Function During HIV-1 Infection. PloS One (2012) 7:e51696. doi: 10.1371/journal.pone.0051696
146. Cluxton CD, Spillane C, O'Toole SA, Sheils O, Gardiner CM, O'Leary JJ. Suppression of Natural Killer Cell NKG2D and CD226 Anti-Tumour Cascades by Platelet Cloaked Cancer Cells: Implications for the Metastatic Cascade. PloS One (2019) 14:e0211538. doi: 10.1371/journal.pone.0211538
147. Wu M, Mei F, Liu W, Jiang J. Comprehensive Characterization of Tumor Infiltrating Natural Killer Cells and Clinical Significance in Hepatocellular Carcinoma Based on Gene Expression Profiles. BioMed Pharmacother (2020) 121:109637. doi: 10.1016/j.biopha.2019.109637
148. Sun H, Liu L, Huang Q, Liu H, Huang M, Wang J.. Accumulation of Tumor-Infiltrating CD49a(+) NK Cells Correlates With Poor Prognosis for Human Hepatocellular Carcinoma. Cancer Immunol Res (2019) 7:1535–46. doi: 10.1158/2326-6066.CIR-18-0757
149. Wang Y, Wang C, Qiu J, Qu X, Peng J, Lu C. Targeting CD96 Overcomes PD-1 Blockade Resistance by Enhancing CD8+ TIL Function in Cervical Cancer. J Immunother Cancer (2022) 10(3):e003667. doi: 10.1136/jitc-2021-003667
150. Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M. CD96 is a Leukemic Stem Cell-Specific Marker in Human Acute Myeloid Leukemia. Proc Natl Acad Sci U.S.A. (2007) 104:11008–13. doi: 10.1073/pnas.0704271104
151. Zhang W, Shao Z, Fu R, Wang H, Li L, Liu H. Expressions of CD96 and CD123 in Bone Marrow Cells of Patients With Myelodysplastic Syndromes. Clin Lab (2015) 61:1429–34. doi: 10.7754/clin.lab.2015.141240
152. Harjunpaa H, Blake SJ, Ahern E, Allen S, Liu J, Yan J. Deficiency of Host CD96 and PD-1 or TIGIT Enhances Tumor Immunity Without Significantly Compromising Immune Homeostasis. Oncoimmunology (2018) 7:e1445949. doi: 10.1080/2162402X.2018.1445949
153. Brooks J, Fleischmann-Mundt B, Woller N, Niemann J, Ribback S, Peters K. Perioperative, Spatiotemporally Coordinated Activation of T and NK Cells Prevents Recurrence of Pancreatic Cancer. Cancer Res (2018) 78:475–88. doi: 10.1158/0008-5472.CAN-17-2415
154. Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P. Proliferating Transitory T Cells With an Effector-Like Transcriptional Signature Emerge From PD-1(+) Stem-Like CD8(+) T Cells During Chronic Infection. Immunity (2019) 51:1043–1058 e1044. doi: 10.1016/j.immuni.2019.11.002
155. Murter B, Pan X, Ophir E, Alteber Z, Azulay M, Sen R. Mouse PVRIG Has CD8(+) T Cell-Specific Coinhibitory Functions and Dampens Antitumor Immunity. Cancer Immunol Res (2019) 7:244–56. doi: 10.1158/2326-6066.CIR-18-0460
156. Li Y, Zhang Y, Cao G, Zheng X, Sun C, Wei H. Blockade of Checkpoint Receptor PVRIG Unleashes Anti-Tumor Immunity of NK Cells in Murine and Human Solid Tumors. J Hematol Oncol (2021) 14:100. doi: 10.1186/s13045-021-01112-3
157. Xu F, Sunderland A, Zhou Y, Schulick RD, Edil BH, Zhu Y. Blockade of CD112R and TIGIT Signaling Sensitizes Human Natural Killer Cell Functions. Cancer Immunol Immunother (2017) 66:1367–75. doi: 10.1007/s00262-017-2031-x
158. Harjunpaa H, Guillerey C. TIGIT as an Emerging Immune Checkpoint. Clin Exp Immunol (2020) 200:108–19. doi: 10.1111/cei.13407
159. Chen X, Song X, Li K, Zhang T. FcgammaR-Binding Is an Important Functional Attribute for Immune Checkpoint Antibodies in Cancer Immunotherapy. Front Immunol (2019) 10:292. doi: 10.3389/fimmu.2019.00292
160. Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM. Primary Analysis of a Randomized, Double-Blind, Phase II Study of the Anti-TIGIT Antibody Tiragolumab (Tira) Plus Atezolizumab (Atezo) Versus Placebo Plus Atezo as First-Line (1L) Treatment in Patients With PD-L1-Selected NSCLC (CITYSCAPE). J Clin Onco (2020) 38:9503–3. doi: 10.1200/JCO.2020.38.15_suppl.9503
161. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F. Pembrolizumab Plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
162. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J. Pembrolizumab Plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med (2018) 379:2040–51. doi: 10.1056/NEJMoa1810865
163. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
164. Li W, Blessin NC, Simon R, Kluth M, Fischer K, Hube-Magg C. Expression of the Immune Checkpoint Receptor TIGIT in Hodgkin's Lymphoma. BMC Cancer (2018) 18:1209. doi: 10.1186/s12885-018-5111-1
165. Sullivan R, Rasco D, Lim E, Sharma M, Shepard D, Patnaik A. Abstract CT031: COM701 Demonstrates Preliminary Antitumor Activity as Monotherapy and in Combination With Nivolumab in Patients With Advanced Solid Tumors. J Cancer Res (2020) 80:CT031–1. doi: 10.1158/1538-7445.AM2020-CT031
166. Li XY, Das I, Lepletier A, Addala V, Bald T, Stannard K. CD155 Loss Enhances Tumor Suppression. via combined Host tumor-intrinsic mechanisms J Clin Invest (2018) 128:2613–25. doi: 10.1172/JCI98769
167. Celis-Gutierrez J, Blattmann P, Zhai Y, Jarmuzynski N, Ruminski K, Gregoire C. Quantitative Interactomics in Primary T Cells Provides a Rationale for Concomitant PD-1 and BTLA Coinhibitor Blockade in Cancer Immunotherapy. Cell Rep (2019) 27:3315–30.e3317. doi: 10.1016/j.celrep.2019.05.041
168. Smith A, Zeng W, Grogan B, Haass J, Blackmarr A, Peterson S, et al. Abstract 4986: TIGIT Directed Human Antibody Modulates T-Regulatory and Effector Cell Function. J Cancer Res (2019) 79:4986–6. doi: 10.1158/1538-7445.AM2019-4986
169. Chand D, Waight JD, Paltrinieri E, Dietrich S, Bushell M, Costa M. Abstract 2390: FcgR Co-Engagement by Anti-TIGIT Monoclonal Antibodies Enhances T Cell Functionality and Antitumor Immune Responses. J Cancer Res (2019) 79:2390–0. doi: 10.1158/1538-7445.AM2019-2390
170. Argast GM, Cancilla B, Cattaruzza F, Yeung P, Scolan E, Harris R. Abstract 5627: Anti-TIGIT Biomarker Study: Inhibition of TIGIT Induces Loss of Tregs From Tumors and Requires Effector Function for Tumor Growth Inhibition. J Cancer Res (2018) 78:5627–7. doi: 10.1158/1538-7445.AM2018-5627
171. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
172. Ishitsuka K, Yurimoto S, Tsuji Y, Iwabuchi M, Takahashi T, Tobinai K. Safety and Effectiveness of Mogamulizumab in Relapsed or Refractory Adult T-Cell Leukemia-Lymphoma. Eur J Haematol (2019) 102:407–15. doi: 10.1111/ejh.13220
173. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA. Activating Fc Gamma Receptors Contribute to the Antitumor Activities of Immunoregulatory Receptor-Targeting Antibodies. J Exp Med (2013) 210:1685–93. doi: 10.1084/jem.20130573
Keywords: cancer immunotherapy, NK cells, T cells, Treg cells, antibodies, fc gamma receptors (FcγR)
Citation: Conner M, Hance KW, Yadavilli S, Smothers J and Waight JD (2022) Emergence of the CD226 Axis in Cancer Immunotherapy. Front. Immunol. 13:914406. doi: 10.3389/fimmu.2022.914406
Received: 06 April 2022; Accepted: 26 May 2022;
Published: 24 June 2022.
Edited by:
Xian Zeng, Fudan University, ChinaReviewed by:
Romain Roncagalli, INSERM U1104 Centre d’immunologie de Marseille-Luminy (CIML), FranceAndrey Shaw, Genentech, Inc., United States
Copyright © 2022 Conner, Hance, Yadavilli, Smothers and Waight. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremy D. Waight, amVyZW15Lngud2FpZ2h0QGdzay5jb20=