 Maximilian Haist
Maximilian Haist Volker Mailänder
Volker Mailänder Matthias Bros
Matthias Bros
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 May 2022
Sec. Vaccines and Molecular Therapeutics
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.912594
This article is part of the Research Topic Current Concepts of Cellular and Biological Drugs to Modulate Regulatory T cell Activity in the Clinic, volume II View all 7 articles
In contrast to conventional anti-tumor agents, nano-carriers allow co-delivery of distinct drugs in a cell type-specific manner. So far, many nanodrug-based immunotherapeutic approaches aim to target and kill tumor cells directly or to address antigen presenting cells (APC) like dendritic cells (DC) in order to elicit tumor antigen-specific T cell responses. Regulatory T cells (Treg) constitute a major obstacle in tumor therapy by inducing a pro-tolerogenic state in APC and inhibiting T cell activation and T effector cell activity. This review aims to summarize nanodrug-based strategies that aim to address and reprogram Treg to overcome their immunomodulatory activity and to revert the exhaustive state of T effector cells. Further, we will also discuss nano-carrier-based approaches to introduce tumor antigen-specific chimeric antigen receptors (CAR) into T cells for CAR-T cell therapy which constitutes a complementary approach to DC-focused vaccination.
So far, the vast majority of nano therapeutics applied for tumor therapy aims to target either tumor cells for direct killing by delivery of cytotoxic drugs (1) or antigen presenting cells (APC) like dendritic cells (DC) to evoke tumor antigen-specific T cell responses by co-delivery of antigen and adjuvant (2). The latter, immunotherapeutic nano vaccination approach is quite attractive since DC, which are considered the most potent APC due to their capacity to induce even primary T cell responses, are specialized in the uptake of exogenous material via several receptors allowing endocytosis and phagocytosis (3). Further, APC are equipped with numerous cell surface as well as intracellular receptors that sense danger signals (4). Nano-vaccines can be considered as minimal pathogens, providing a source of antigen and adjuvant, i.e. APC-stimulating danger signals.
However, this vaccination approach is limited by immunoregulatory immune cells, including myeloid-derived suppressor cells (MDSC) as well as regulatory T cells (Treg), that are induced and expanded by molecular cues of the tumor as well as of accessory cells like tumor-associated macrophages (TAM) and cancer-associated fibroblasts (CAF), forming the tumor microenvironment (TME) (5). MDSC and Treg are able to spread tumor tolerance by imprinting a pro-tolerogenic state in APC, by inhibiting activation of (naïve) T cells, and by deactivating T effector cells (6, 7). Therefore, the development of nanodrugs that address and either deplete or reprogram immunoregulatory cell types have come into focus (8). Whereas MDSC and TAM are equipped with numerous uptake receptors and thereby may readily internalize nanodrugs, especially T cells have proven more difficult in that regard. Therefore, approaches employing nanodrugs to induce T cell activation have so far only scarcly been applied.
This review aims to present current nanodrug-based strategies developed to address and modulate T cells (an overview is given in Figure 1). In most cases, according studies have focused on the activation and expansion of T effectors cells and the generation of chimeric antigen receptor (CAR-)T cells, genetically altered to express a defined tumor antigen-specific T cell receptor. Both approaches focus on expanding the pool of tumor antigen-specific T cells. In contrast, the complementary strategy to reprogram Treg towards T effector cells by nanodrugs has been issued in a limited number of studies only.
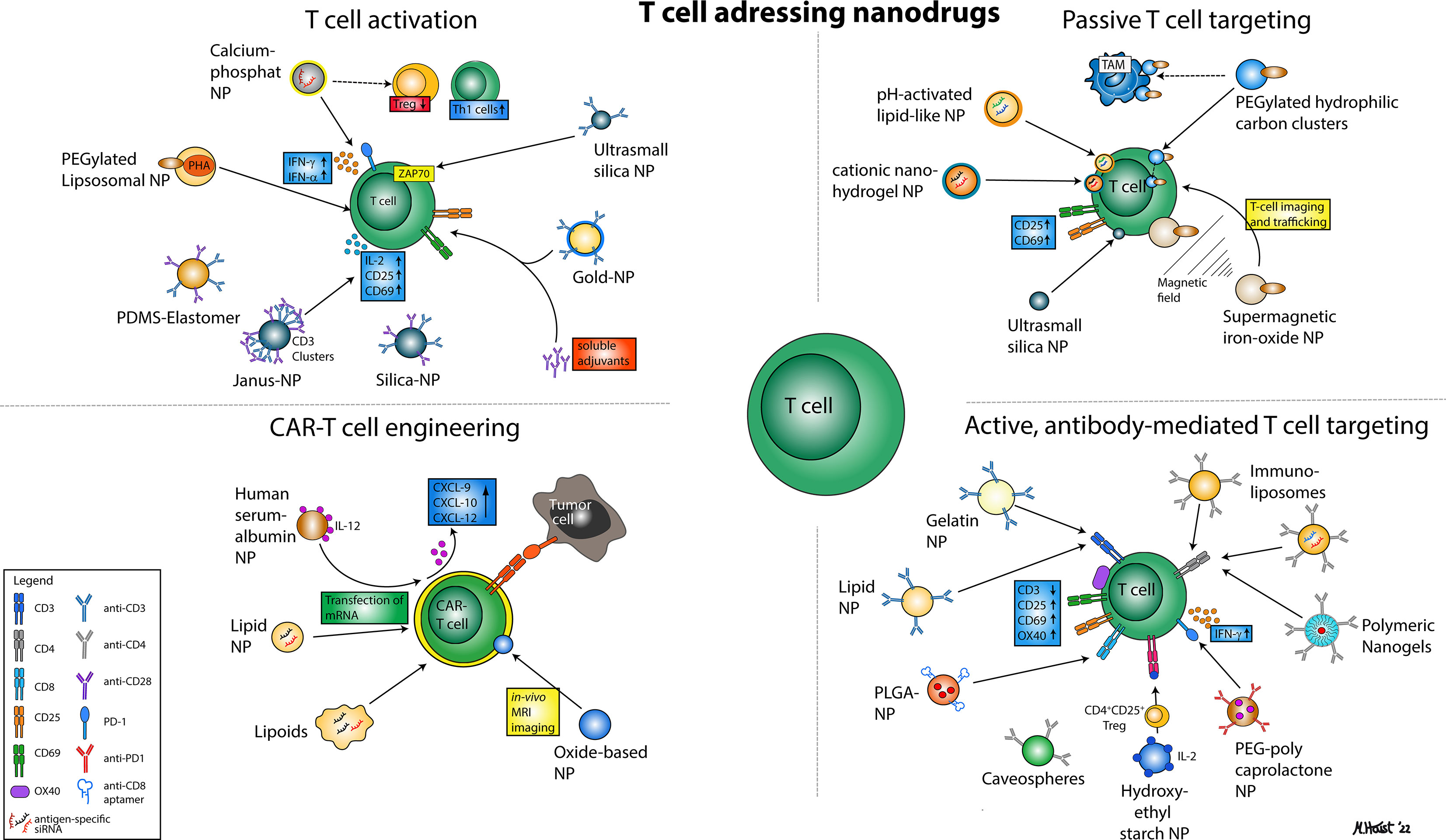
Figure 1 Illustration of various nanoparticular approaches to address T cells for immunotherapy. In general, four major mechanisms can be distinguished that comprise nanodrugs aiming to activate T cells (1), nanodrugs that allow for passive (2) or active, antibody-mediated T cell targeting (3) and nanodrugs that may enhance the efficacy of CAR T cell therapies.
So far, most clinical studies focused on the generation and adoptive transfer of CAR-T cells, while mRNA-based nanodrugs directly targeting T cells have not been translated widely into the clinic. However, a number of clinical trials have applied immune-checkpoint inhibitors (ICI) in combination with nanodrugs in order to indirectly address the T cell compartment and induce a robust anti-tumor T cell response. Most commonly, these indirect approaches act via the induction of immunogenic cell-death (ICD), the depletion of immunosuppressive cell types and DC-mediated T cell activation.
For in vitro activation of T cells in the context of adoptive T cell transfer, several types of nano structures have been developed to improve polyclonal stimulation with immobilized agonistic anti-CD3 and CD28-specific antibody. In many cases the T cell stimulatory antibodies are attached to nano-sized beads, termed artificial (a)APC (9).
Matic and coworkers observed an inverse relation between the density of immobilized anti-CD3 antibody and the extent of primary human CD4+ T cell activation as reflected by CD69 expression and IL-2 levels (10). T cell proliferation required the co-application of anti-CD28 antibody, which was applied in soluble form. Later, beads that were conjugated with agonistic anti-CD3 and -CD28 antibodies were introduced for T cell stimulation, which displayed elevated stimulation properties due to the structural similarity of the stimulatory beads and APC (11). This concept was developed further by using polydimethylsiloxane (PDMS) elastomer as a carrier substrate, which was coated with albumin for biocompatibility (12). According beads (Ø 28-40 nm) induced faster and longer-lasting T cell divisions as compared to antibody-coated `stiff´ beads of comparable size, namely DynaBeads and polystyrene NP. This demonstrates that besides the density of the stimulating agents also physical properties of nano-carriers like stiffness/softness of the underlying material are important to optimize T cell stimulation.
The activation of primary CD4+ T cells was also improved by employing oligo peptides that enhanced cell adhesion (13). To this end, PEG hydrogel was conjugated with fibronectin-derived peptides, known to be engaged by integrin receptors expressed by T cells (14). These functionalized PEG hydrogels were decorated in addition with Au-NP, which in turn were conjugated with agonistic CD3-specific antibody. Agonistic anti-CD28 antibody was applied in soluble form. CD4+ T cell activation (CD69 expression, IL-2 production) and proliferation was improved in the presence of the integrin-binding Arg-Gly-Asp tripeptide (15), and correlated inversely with Au-NP density on PEG hydrogels (minimal distance: 20 nm). In a follow-up study T cell activation and proliferation was further increased by applying a cocktail of phorbol 12-myristate 13-acetate (PMA), ionomycin and Golgi inhibitors (16) that enhanced IL-2 production via activation of protein kinase C and elevation of cytosolic calcium levels (17).
Recently, Hammink and coworkers showed that as compared to direct conjugation of agonistic CD3 and CD28 antibodies to the bead surface the inclusion of semi-flexible polyisocyanopeptide polymer linkers of about 400 nm in length enhanced the activation of primary human CD4+ and CD8+ T cells in terms of proliferation in terms of proliferation, as well as the production of IL-2 and interferon (IFN)-γ production (18).
Whereas in the aforementioned approach as well as in case of standard aAPC both types of agonistic antibody are evenly distributed, Lee and Yu asked for the potency of aAPC based on silica NP (Ø ~500 nm) on which anti-CD3 antibody was clustered in segregated patches, surrounded by anti-CD28 antibody (19). These so-called Janus NP conferred much stronger T cell activation as in case of homogenous antibody distribution.
NP may exert intrinsic T cell stimulatory activity as shown for ultrasmall silica NP (Ø <10 nm) that engaged the T cell receptor and CD3, and thereby induced ζ-chain-associated protein kinase 70 (ZAP70) phosphorylation as well as induction of activation markers (CD25, CD69) (20). However, T cell proliferation and IL-2 production required the co-application of agonistic CD28 antibody or PMA.
The lectin phytohemagglutinin (PHA) stimulates T cells by cross-linking of the T cell receptor (21), but is highly toxic (22). Most recently, Alhallak and coworkers showed that PEGylated liposomes, comprised of DPPC and cholesterol, allowed efficient loading with PHA (23). These liposomes conferred strong T cell activation in vitro. In contrast to soluble PHA, the liposomal PHA formulation exerted no increased toxicity in vivo, and mediated T cell activation as well. Further, in a murine multiple myeloma tumor model, in vivo application of liposomal PHA resulted in a strong decrease in tumor growth, which has been attributed to T cell activation. It was not clarified, however, which additional cell types engaged liposomal PHA and were affected by the cargo.
Some types of nanoparticles (NP) have been demonstrated to possess an intrinsic T cell targeting property, termed passive targeting. However, in a growing number of studies active T cell targeting has been achieved by conjugation of surface receptor binding ligand derivatives or antibodies.
In a proof of concept study we issued the potential of non-functionalized NP for uptake by T cells (24). To this end, we compared binding and uptake of polystyrene particles of different sizes (Ø 63-121 nm) and surface functionalization by human pre-activated CD4+ and CD8+ T cells. In the course of these experiments amino-functionalized polystyrene beads (Ø 65 nm) were most efficient as evaluated by confocal microscopy. NP were apparent in membrane-surrounded vesicles. Of note, we observed a considerable release of NP after longer periods of incubation. NP uptake exerted no detrimental effects on cell viability. Interestingly, NP/T cell interaction was attenuated in the presence of human serum in a dose-dependent manner. Both IFN-γ production (CD4+ and CD8+ T cells) and target cell killing (CD8+ T cells) were not affected at low to intermediate NP concentrations.
The potential of superparamagnetic iron oxide NP (SPION) to label T cells in vitro in the context of adaptive T cell transfer for tumor therapy was assessed in several studies. For example, Liu and coworkers demonstrated that PEG-coated SPION could be used to efficiently address T cells without causing any detrimental effects on their physiological functions (25). SPION-labeled T cells could be tracked in vivo by magnetic resonance imaging (MRI) as shown in a rodent transplantation model. Besides in vivo tracking, SPION-labeled pre-isolated CD3+ T cells that were labeled ex vivo with SPION and systemically injected could be directed in vivo e.g. to tumor sites by applying a magnetic field as shown in several proof of concept studies (26, 27). Polyclonal T cell activation increased SPION (Ø 54 nm) binding, but also resulted in a drop in overall T cell viability.
Huq and coworkers demonstrated specific uptake of poly(ethylene glycol) (PEG) functionalized hydrophilic carbon clusters (HCC) by splenic rat T cells in vivo after systemic application (28), but not by lymph node T cells. Subsequent in vitro spleen cell studies corroborated these findings and showed that both CD4+ and CD8+ T cells internalized PEG-HCC by clathrin-mediated endocytosis both at resting state and at enhanced level after mitogenic stimulation. In accordance with the previously reported scavenger activity of PEG-HCC (29), accordingly treated T cells contained lower levels of oxygen radicals, and displayed lower activation in response to antigen-specific stimulation (28). Interestingly, myeloid cell types displayed considerable binding, but no uptake of PEG-HCC. Accordingly, macrophages were not affected in terms of activation. In contrast to PEG-HCC, ultra-small silica NP Staff(Ø 3.6-5.1 nm) displayed no T cell-specific uptake when assayed with human peripheral blood mononuclear cell (PBMC) preparations, but conferred strong T cell activation (30). In this regard, CD4+ and CD8+ T cells displayed strong upregulation of surface activation markers (CD25, CD69) and IFN-γ release, whereas Th2-associated cytokines were not induced. Of note, T cell activation did not result in T cell proliferation. Somewhat surprisingly, we observed an inverse correlation between the size of silica nanocapsules (SiNC; Ø 50-400 nm) and their cytotoxic effect on human primary CD8+ T cells (31). Therefore, we chose SiNC of larger size for subsequent experiments. We demonstrated that SiNC (Ø 248 nm) were internalized by human primary CD8+ T cells, and successfully transferred siRNA specific for the T cell inhibitory receptor programmed cell death protein (PD-)1 (32). The downregulation of PD-1 was accompanied by upregulated expression of the surface activation markers CD25, CD69 and CD71. Besides SiNC we also assayed other types of NP for their ability to transfect T cells with siRNA, namely lipophilic triphenylphosphonium cation-modified diblock copolymer structures with a terminal 3-guanidinopropyl methacrylamide block for the complexation of siRNA, and cationic nanohydrogel NP (Ø <28 nm) (33). The latter were based on an amphiphilic block copolymer harboring a hydrophilic block, which consisted of triethylenglycol methylether methacrylate and a hydrophobic block of pentafluorophenyl methacrylate as reactive ester. The derived amphiphilic polymers self-assembled into polymeric micelles with a reactive ester core, substituted from hydrophobic to cationic by using bi-functional amine containing cross-linkers. Primary human CD8+ T cells isolated from PBMC and stimulated with agonistic anti-CD3 antibody and IL-2 showed uptake of either siRNA carrier.
Whereas the aforementioned carrier systems have been evaluated to alter T cell activation by RNA interference using siRNA, Tanaka and coworkers optimized lipid NP for mRNA transfection of T cells using the human CD4+ T cell line Jurkat as a model (34). Stepwise improvements resulted in a formulation consisting of SS-cleavable and pH-activated lipid-like material (ssPalmO-Phe-P4C2), previously shown to promote endosomal escape and release of mRNA into the cytoplasm (35), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine, cholesterol and 1-(monomethoxy PEG2000)2,3-dimyristoylglycerol.
Taken together, the aforementioned studies confirmed that nanodrugs may successfully deliver cargo into (activated) T cells. However, the obstacle of competitive uptake of NP by other types of immune cells with more pronounced endocytic activity, thereby interfering with T cell -specific targeting, was scarcely addressed.
In order to specifically address T cells and to enhance NP uptake, a number of approaches evaluated T cell-specific targeting by addressing suitable surface receptors like CD3 and CD4 with according antibodies.
Dinauer and coworkers demonstrated that targeting of the pan-T cell marker CD3 using gelatin NP coated with anti-CD3 antibody yielded strong uptake of that functionalized NP by T cell leukemia cells as well as primary T cells in vitro (36). Most recently, T cell-specific transfection with mRNA in vivo was achieved by using lipid NP containing an ionizable cationic lipid (DLin-MC3-DMA), that was conjugated with a CD3-specific antibody (37). Intravenous application resulted in an accumulation of fluorescent reporter mRNA containing lipid NP in the spleen, and a transfection efficiency of up to 4% of all splenic and 7% of circulating T cells. In accordance with the agonistic property of the CD3 antibody, transfected T cells expressed activation markers (CD25, CD69, OX40), accompanied by downregulation of CD3ε, and displayed elevated plasma levels of T cell-associated cytokines. Further, when injected into tumor-bearing mice undergoing immunotherapy, transfected T cells were apparent in the tumor and tumor-draining lymph nodes.
Several studies aimed to address CD4+ T cells by NP conjugated with CD4-binding antibodies. Ramana and coworkers evaluated the efficiency of this targeting strategy by using anti-CD4 antibody conjugated immunoliposomes consisting of phosphatidyl choline (PC), distearoylphosphatidylethanolamine (DSPE)–PEG), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) and cholesterol (38). These immunoliposomes were loaded with two anti-retroviral drugs (Ø ~71 nm). CD4-targeting immunoliposomes were efficiently internalized by cells of the Jurkat T cell line, and both drugs exerted biological activity. CD4-targeting lipid NP were also employed to deliver CD45-specific siRNA into murine primary CD4+ T cells (39). In vivo administered lipid NP, consisting of cholesterol, distearoylphosphatidylcholine, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-PEG, and Dlin-MC3-DMA (40), with a diameter of ~130 nm, were detectable in various organs only in CD4+ T cells, and caused CD45 downregulation, which was associated with a decrease in CD4 surface levels due to endocytosis-mediated sequestration (41). Recently, Tombácz and coworkers demonstrated efficient mRNA transfection of murine CD4+ cells in vivo after intravenous injection using CD4 antibody-conjugated lipid NP consisting of the proprietary ionizable cationic lipid ALC-0307 (42), PC, cholesterol and PEG-lipid NP (43). To demonstrate efficacy of their lipid NP in vivo, reporter mice were used which were engineered to express a fluorescent protein after Cre recombinase mediated recombination. When injecting these reporter mice with complexes composed of Cre recombinase encoding mRNA and lipid NP high frequencies of CD4+ T cells (spleen: ~ 60 %, lymph nodes: ~ 40 %) were found to express the fluoresecent reporter. Canakci and coworkers used anti-CD4 antibody conjugated polymeric nanogels generated by polymerization of monomeric PEG methyl ether methacrylate (PEGMA) and pyridyl disulfide ethyl methacrylate to deliver the tubulin inhibitor mertansine to T cell lymphoman cell lines (44).
Aptamers are peptides or oligonucleotides which are identified by library screenings to bind a given target molecule at high affinity and are much smaller than according antibodies (45). The aptamer CD8AP17 specifically binds human CD8 (46). Mansouri and coworkers used poly(lactic-co-glycolic acid) (PLGA) based NP that were modified with chitosan to promote endosomal escape (47), conjugated with the aptamer CD8AP17 (Ø 345 nm) to deliver the calcineurin inhibitor tacrolimus (48) specifically to cells of an immortalized CD8+ T cell line (49). Glass and coworkers generated so-called caveospheres, which consist of self-assembling Caveolin-1 fused with Staphylococcal IgG Fc part binding peptide with a diameter of ~40 nm (50). These caveospheres could be functionalized with an anti-CD4 antibody, and showed specific binding to CD4+ cells within a mixed PBMC population. Likewise, functionalization with a C-C chemokine receptor type 5 specific antibody resulted in specific binding and uptake by CD4+ T cells.
As an alternative approach for T cell targeting, we conjugated interleukin (IL-)2 to hydroxyethyl starch (HES-)NP (Ø ~215 nm) and showed that IL-2 retained its biological activity as tested in an IL-2 bioassay (51). Further, human CD4+ T cells polyclonally pre-activated to express the IL-2 receptor CD25 internalized IL-2-coated HES-NP, and showed a higher extent of proliferation, confirming that IL-2 in a dual manner served both as a targeting and costimulatory agent by addressing CD25. Efficient engagement was also observed for CD4+CD25high Treg. CD25-dependent T cell binding was achieved as well in in vivo experiments using immunodeficient Rag2–/–γc–/– mice that had been reconstituted with human CD4+ T cells.
In the following, strategies that aimed to revert the exhausted state of T effector cells and to overcome the immunoregulatory function of Treg are discussed (Figure 2). In a proof-of-concept study focusing on NP-mediated RNA interference in Treg, laser-generated Au-based NP were coupled with a locked nucleic acid, that encompassed a green fluorescent protein (GFP)-specific siRNA, and a cell penetrating peptide to enhance endocytic particle internalization (52). This NP formulation (Ø ~6.8 nm) was internalized at high extent by splenic CD11b+ myeloid cell populations, and within the CD4+ T cell population by double as many CD4+CD25+ Treg (~ 10%) as CD4+CD25- T cells. Incubation of Treg isolated from GFP-expressing reporter mice with GFP-siRNA coupled Au-NP resulted in a strong down-regulation of the GFP reporter on protein level after two days (52).
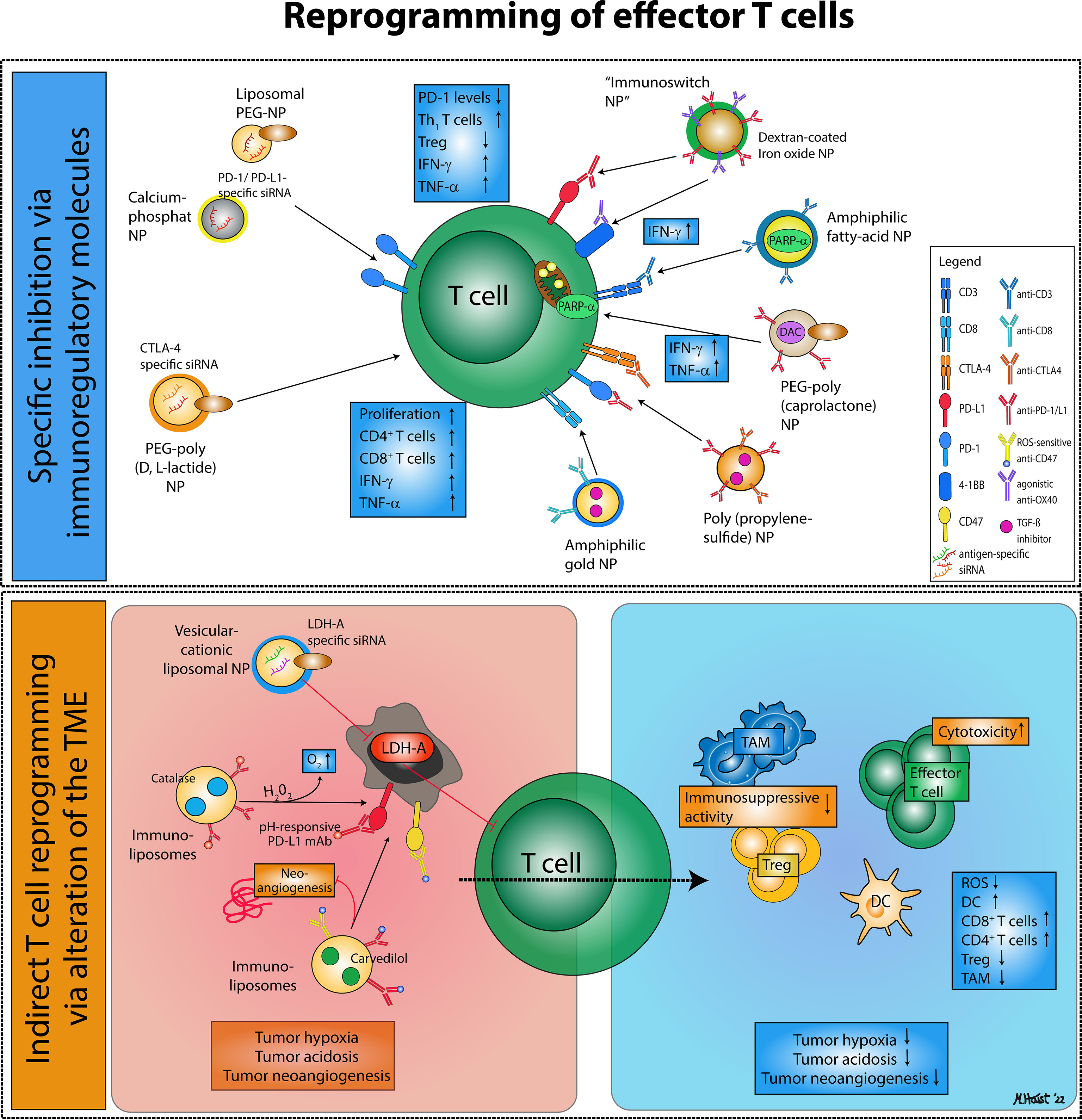
Figure 2 Nanodrug-based approaches for effector T cell reprogramming. Reprogramming of exhausted effector T cells and regulatory T cells via nanodrugs can be distinguished into two major categories: (1) Nanoparticles that specifically inhibit immunoregulatory molecules on effector T cells or regulatory T cells, thereby enhancing effector T cell activity. (2) Nanoparticles targeting immunosuppressive mechanisms characteristic of the tumor microenvironment that favor the accumulation of immunosuppressive cell types and an alleviate an effective anti-tumor immune response.
Another early study aimed to inhibit the expression of the immune checkpoint cytotoxic T lymphocyte-associated protein (CTLA-)4 in T cells by siRNA using NP composed of PEG-poly(D,L-lactide) and the cationic lipid N,N-bis(2-hydroxyethyl)-N-methyl-N-(2-cholesteryoxycarbonyl-aminoethyl) ammonium bromide as a carrier (Ø ~142 nm) (53). Treatment of antigen-specifically stimulated CD4+ T cells with CTLA-4 siRNA-delivering NP enhanced their proliferative activity. Systemic administration of these NP to melanoma-burdened mice resulted in strongly reduced tumor growth, which was accompanied which was accompanied by an overall increased tumor infiltration of CD4+ and CD8+ T effector cells, higher levels of T helper (Th)1/CTL-associated IFN-γ and tumor necrosis factor (TNF)-α in serum, whereas the number of Treg remained unaltered. T cell activation may also be extended by NP-aided inhibition of the inhibitory PD-1 signaling axis as demonstrated by Wu and coworkers by using calcium phosphate NP coated with (dioleoyl-3-trimethylammonium propane) DOTAP, dioleoylphosphatidylcholine and cholesterol, that encapsulated siRNA specific for PD-1 and PD ligand(L)1 (Ø ~30 nm) (54). These NP yielded downregulation of PD-1 expression in primary human tumor-infiltrating leukocytes (TIL) obtained from breast cancer specimen, and enhanced their tumor cell killing activity in vitro, accompanied by elevated IFN-γ and TNF-α contents. In a similar approach, liposomes composed of cholesterol, DOTAP, dioleoylphosphatidylethoanolamine (DOPE), and PEG were complexed with PD-1 specific siRNA (Ø ~87 nm), which engaged splenic CD8+ T cells in vitro, conferring downregulation of PD-1 expression (55). When systemically administered to melanoma-burdened mice, these liposomes enriched at the tumor site and in lymph nodes. By this approach the composition of tumor-infiltrating T cells was altered, displaying a higher frequency of Th1 cells on the expense of Treg and Th2 cells, and accompanied by lowered tumor growth.
Kosmides and co-workers aimed to enhance T cell effector activities by generating dextran-coated iron oxide NP that were conjugated with a PD-L1 blocking and an agonistic 4-1BB antibody (56). This so-called immunoswitch NP (Ø ~80 nm) was intended to target 4-1BB+ and PD-L1+ tumor cells. On the one hand this NP prevented binding of PD-L1 to PD-1 as expressed by T effector cells (57). On the other hand, the agonistic 4-1BB antibody engaged and triggered the costimulatory receptor 4-1BB on T cells (58). Thereby, the NP acted in a dual manner as a bridging and stimulatory agent. Immunoswitch NP enhanced the interaction of pre-activated murine CD8+ T cells with PD-L1 expressing melanoma, resulting in a stronger IFN-γ production and tumor cell killing in vitro. In a murine melanoma model, both intratumoral and intravenous injection of immunoswitch NP conferred strongly attenuated tumor growth, which was accompanied by higher frequencies of tumor-infiltrating IFN-γ+CD8+ T effector cells. In contrast, co-administration of soluble PD-L1 and 4-1BB antibodies was not effective.
Other studies aimed to inhibit the immunoregulatory effects of tumor growth factor (TFG-)β. In this regard, Yang and coworkers generated amphiphilic Au-NP that were loaded with a hydrophobic TGF-β inhibitor (SB525334) into the shell (Ø ~2.4 nm) (59). Additional conjugation with a CD8-specific antibody strongly enhanced the binding of this NP formulation to CD8+ T cells and at the same time attenuated its binding to other types of leukocytes in vitro. When applied systemically in vivo, besides CD8+ T cells also myeloid cell types, including conventional DC type 2, macrophages and granulocytic neutrophils engaged this NP at high-levels as well. Of note, also CD4+ T cells displayed considerable interaction with this NP formulation. Nonetheless, peripheral blood of tumor-burdened mice that were treated with this type of NP contained higher frequencies of CD8+TNF-α+IFN-γ+ T cells. However, tumor growth was not affected. In a similar approach Francis and coworkers generated poly(propylene sulfide) NP which upon conjugation of immune checkpoint PD-1- and CTLA-4- specific blocking antibodies, respectively, preferentially engaged CD4+ and CD8+ T cells in mice (60). In a melanoma model, co-administration of these NP formulations exerted similar inhibition of tumor growth as observed after the application of equimolar amounts of soluble antibodies applied in combination. Moreover, immune checkpoint-targeting NP loaded with a TGF-β inhibitor (SB-431542) conferred an even stronger tumor inhibition and prolonged the life span of melanoma-burdened mice.
Besides specific inhibition of distinct immunoregulatory molecules, another approach aimed to exploit that Treg within the TME depend on enhanced fatty acid oxidation (61). Muriski and coworkers generated fatty acid-conjugated quantum dots, which enabled quantification of fatty acid uptake by T cells (62). This quantum dot-derivative labeled tumor-derived CD4+Foxp3+ Treg at higher extent than any other tumor-infiltrating T cell population in a mouse model of intracranial tumors. In order to metabolically reprogram T cells within the TME, Kim and co-workers encapsulated the peroxisome proliferator-activated receptor (PARP-)α inducer fenofibrate in amphiphilic polygamma glutamic acid-based NP that were conjugated with a CD3-specific F(ab´)2 antibody fragment for pan T cell targeting (Ø ~150 nm) (63). This NP formulation enhanced the mitochondrial oxygen consumption rate of T cells under glucose-deficient conditions via PARP-α induced activation of fatty acid metabolism in vitro. In a murine melanoma model, intratumoral administration of this NP formulation resulted in higher frequencies of tumor-infiltrating T cells, elevated frequencies of IFN-γ and granzyme B expressing CD3+ T cells, and virtually abrogated tumor growth.
Based on the observation that the effector functions of tumor-infiltrating T effector cells are inhibited on genomic level by de novo gene promoter methylation (64), Hu and co-workers tested the efficacy of T cell-focused delivery of the DNA methyltransferase inhibitor 5-Aza-2´-deoxycytidine (DAC) (65). For this, PEG-poly(ε-caprolactone) NP were conjugated with the clinically employed PD-1 blocking antibody nivolumab to target PD-1 expressing T cells and were loaded with DAC. This formulation (Ø ~218 nm) enhanced IFN-γ production and cytotoxicity of PD-1 expressing CD8+ T cells in an in vitro assay, which suggested suitability for in vivo therapy.
In other approaches, enhanced tumor infiltration of T effector cells was not achieved by direct addressing, but by altering TME conditions. To overcome the acidic state of the TME, known to inhibit T effector cells (66), Zhang and coworkers aimed to inhibit lactate dehydrogenase A (LDHA) in the TME (67). For this, vesicular cationic lipid-assisted NP consisting of DOTAP and PEG-PLGA were loaded with Ldha-specific siRNA (Ø ∼95 nm). In a murine melanoma model, intraveneously administered NP accumulated intratumorally, and reduced Ldha expression, which elevated the pH value. Further, NP application decelerated tumor growth, which was further attenuated upon co-administration of a PD-1 blocking antibody. Tumor analysis revealed enhanced tumor infiltration of CD4+ and CD8+ T effector cells and lower numbers of CD4+Foxp3+ Treg. To overcome hypoxia, which constitutes another hallmark of solid tumors, immunoliposomes composed of lecithin, cholesterol and DSPE-PEG were loaded with Catalase to generate oxygen from break down of hydrogen peroxide (68). These NP were conjugated with a PD-L1 specific blocking antibody via a pH-responsive linker to target PD-L1 expressing tumor cells. In a murine melanoma model, immunoliposomes (Ø ∼118 nm) accumulated in the tumor, PD-L1 blocking antibody was released due to the acidic conditions, and the NP was internalized by tumor cells. Furthermore, the intratumoral hypoxic state was normalized, and a higher infiltration of the TME with CD8+ T cells, but not CD4+ T cells was observed. Treatment of tumor-burdened mice inhibited tumor growth at a higher extent as observed in response to the application of soluble PD-L1 blocking antibody, resulting in enhanced overall survival. In a subsequent study, the aforementioned immunoliposome platform was used to encapsulate carvedilol, intended to block neoangiogenesis within the tumor (69), and to conjugate CD47 and PD-L1 blocking antibodies via reactive oxygen species (ROS)-sensitive thioketal linkers (70). Functionalization with CD47-blocking antibody was dedicated to inhibit `don´t-eat-me´ signaling by tumor cells, which may express this receptor at high level (71). In melanoma-burdened mice intravenous administration of these multi-functionalized immunoliposomes (Ø ∼101 nm) attenuated ROS concentrations due to their reaction with the thioketal linkers (70). Moreover, TIL composition was strongly altered, characterized by attenuated levels of Treg and TAM and elevated frequencies of activated DC, M1-like macrophages and CD8+ T cells. Furthermore, blood vessel density was strongly reduced in the course of treatment, and the frequency of apoptotic tumor cells increased.
So far, CAR-T cells are generated ex vivo by transduction of patient-derived T cells with viral vectors encoding for the chimeric TCR, costimulatory moieties and eventually additional stimulatory factors (72). By now, CAR-T therapy has been approved by the FDA for treatment of lymphoma and leukemia (73). In order to overcome especially safety concerns associated with the use of viral entities, in a number of studies T cells have been transfected with mRNA by electroporation (74). However, also mRNA transfection of T cells by electroporation bears limitations with regard to method-associated cytotoxicity and low transfection efficacy. As an alternative especially lipid NP that complex mRNA and contain helper lipids for stabilization have been tested for ex vivo T cell transfection with mRNA (75–78) and plasmid DNA (79). Based on library screening, Billingsley and coworkers identified an ionizable lipid which induced comparable mRNA transfection efficiency at lower cytotoxicity on Jurkat T cells (75), and subsequently optimized this vector by varying the molar ratios of DOPE, cholesterol and lipid-anchored PEG (76). The optimized formulation conferred CAR mRNA transfection of primary T cells at similar efficacy as electroporation and viral transfer, yielding comparable CAR-T-mediated target cell killing. By screening a library encompassing chemically differing lipid-like formulations, termed lipidoids, Zhao and coworkers showed that lipidoids with an imidazole group yielded stronger reporter mRNA transfection efficacies of primary human CD8+ T cells in vitro (77), and was suitable for transfer of CAR-T mRNA into CD8+ T cells as reflected from potent killing activity of lymphoma cells (78). Furthermore, lipoids also mediated efficient in vivo transfection of T cells as assessed by applying Cre recombinase-encoding mRNA into a reporter mouse strain yielding dTomato expression in response to Cre recombinase-mediated recombination (77). Yu and coworkers screened a number of self-assembled NP composed of cyclodextrin-grafted branched PEI and adamantine-grafted polyamidoamine dendrimers for their suitability to mediate plasmid DNA transfection of human Jurkat T cells (79). Based on initial reporter mRNA transfection studies a formulation (Ø ~100 nm) was chosen and successfully evaluated for CAR plasmid DNA transfection and tumor cell killing.
NP have also been used to enable in vivo MRI of CAR-T cells: In two recent studies CAR-T cells were labeled with iron oxide-based spheric NP (Ø ~25 nm) (80) and nanoworms (Ø ~120 nm) (81). Luo and coworkers aimed to increase the stimulatory properties of CAR-T cells by binding interleukin-12 –coated human serum albumin-based NP onto the CAR-T cell surface by means of bioorthogenal chemistry (82). IL-12 was released in response to binding to tumor target cells, which resulted in an enhanced release of T cell-attracting chemokines and thereby increased CAR-T infiltration, yielding a positive feedback loop.
In the past decade immune-oncology research has undergone substantial progress in understanding the regulatory mechanisms of anti-tumor immunity paving the way for novel immunotherapeutic agents such as ICI and CAR-T cells, which demonstrated impressive outcomes in various cancer types. In particular, the advent of ICI, that block PD-1, PD-L1 and CTLA-4, significantly improved treatment for a number of solid tumors, such as metastatic melanoma (83), non-small-cell lung cancer (84), merkel cell carcinoma (85), colon carcinoma (86), head-and-neck squamous cell carcinoma (HNSCC) (87) and others (88, 89). Also, CAR-T cell therapy showed remarkable long-term results in hematological diseases such as B cell malignancies (90–93). Still, a majority of patients may not derive significant benefit from immunotherapy treatment (35, 94), attributed to low immune cell infiltration into solid tumors, tumor antigen loss and the potent immunosuppressive character of the TME (95, 96).
In order to overcome treatment resistance and to enhance clinical responses to immunotherapy, new therapeutic strategies are needed that may synergize with current immunotherapeutic agents. In this context, nanodrugs are showing high potential in cancer immunotherapy as demonstrated in various preclinical trials as outlined above because they (1) allow for the selective targeted delivery of drugs, (2) may enhance immunogenic cell death (ICD), and (3) may reduce off-target toxicity (97). Also co-administration of several agents in a physically coupled form by one delivery platform is feasible with nanocarriers. Based on the promising results of preclinical trials a steadily increasing number of clinical studies are underway that investigate the clinical efficacy of T cell addressing nanodrugs. The number of these trials substantially increased after the approval of ICI and CAR-T cell therapies, which may be attributed to the potent synergistic effects hypothesized for T cell targeting nanodrugs and ICI treatments (98). Therefore, the majority of these studies are investigating on the combination of ICI and T cell addressing nanodrugs, whereas only a small number of trials examine the clinical efficacy of T cell addressing nanodrugs as monotherapy.
In the following, we will summarize and discuss the most important clinical trials on T cell-targeting nanodrugs for cancer immunotherapy. Table 1 gives an overview of landmark clinical trials on T cell-targeting nanodrugs that mainly comprise indirect approaches to address T cells, such as DC-mediated T cell stimulation or immunomodulation of T cells as a bystander effect of an applied agent. Given the great number of clinical trials we will focus on the most advanced studies and further distinguish between nanodrugs directly addressing T cells and those indirectly affecting T cells. Among indirect T cell targeting nanodrugs, the majority of trials uses nano-formulated chemotherapeutics and mRNA-based nano-vaccines in combination with ICI therapies, which will be outlined in the following.
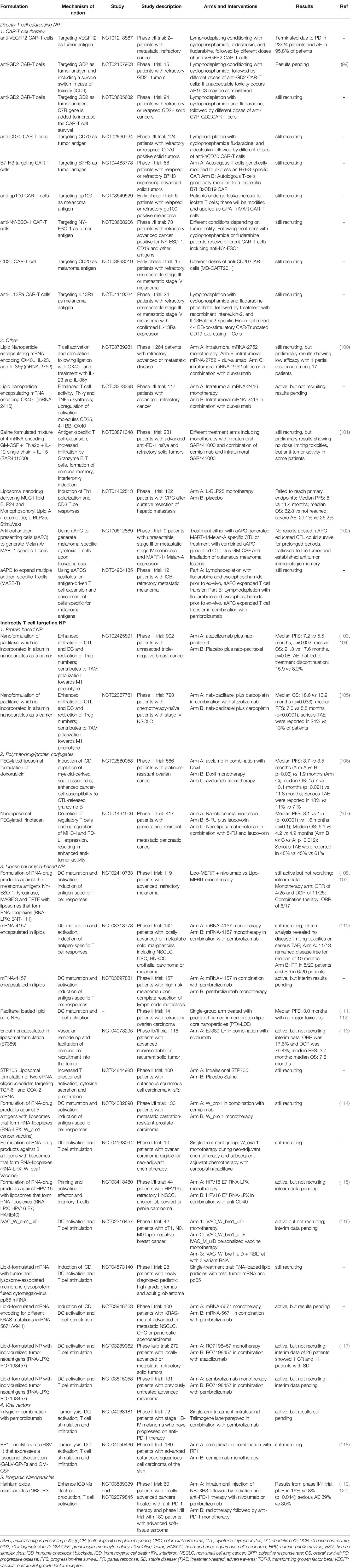
Table 1 Selection of clinical trials in cancer immunotherapy involving T cell targeting nanodrugs and biomaterials.
Nanodrugs modulating T cells encompass a wide spectrum of different anticancer modalities, that act (1) via induction of ICD, (2) DC-mediated T cell activation or (3) depletion/reprogramming of immunosuppressive cell types. In the following we will present three different types of such indirectly T cell modulating nanodrugs that include protein-based NP, polymer-drug conjugates and liposomal mRNA vaccines, as well as viral vectors.
The most common protein-based NP that affect T cells are nano-formulations of chemotherapeutic agents, which are incorporated in albumin-based NP as a carrier. In this regard, Abraxane was the first protein-based NP formulation that was approved by the FDA (121). Abraxane is a nano-formulation of the chemotherapeutic paclitaxel. Apart from direct tumoricidal effects of paclitaxel various immunomodulatory properties have been reported for this agent. In particular, paclitaxel was reported to enhance tumor infiltration of immune cells, such as lymphocytes and DC (111, 122). Also it has been shown that paclitaxel reduced the number of Tregs (123) and contributed to the repolarization of TAM towards an inflammatory M1 macrophage phenotype (124). More importantly, application of Abraxane resulted in higher response rates and substantially decreased side effects as compared to conventional paclitaxel treatment, which led to FDA approval for treatment of metastatic breast cancer, metastatic non-small cell lung cancer (NSCLC) and metastatic pancreatic cancer (125). Most clinical trials investigating the efficacy of Abraxane have been conducted in combination with other immunotherapy agents, in particular with PD-1/PD-L1 blocking antibodies (e.g. atezolizumab). Here, the IMpassion130 trial showed that Abraxane in combination with the PD-L1 inhibitor atezolizumab significantly improved the median progression-free survival (PFS) and overall survival (OS) of patients with advanced triple negative breast cancer (TNBC) as compared to Abraxane plus placebo, without inducing further adverse events (126). The combination of Abraxane and atezolizumab also demonstrated improved clinical efficacy when compared to anti-PD-1/PD-L1 antibodies, such as pembrolizumab or avelumab (127, 128). Therefore, Abraxane plus atezolizumab was the first immunotherapeutic regimen that obtained FDA approval for advanced TNBC, and later Abraxane, atezolizumab and carboplatin obtained also approval for metastatic non-squamous and squamous NSCLC (105, 129). Next to the improved delivery of Abraxane, the reduced toxicities, which avoided premedication with immunosuppressive corticosteroids, are considered particularly important for the clinical efficacy of the combination of Abraxane and PD-(L1) inhibitors and contribute to the immunomodulatory effects of Abraxane on T effector cells (130).
Polymer-protein conjugates that are applied in cancer treatment are mainly PEGylated proteins as PEGylation effectively protects proteins from degradation and improves drug distribution (131). The PEGylated liposomal formulation of doxorubicin, Doxil, was the first nano-medicine approved by the FDA for ovarian cancer (OC), acquired immune deficiency syndrome (AIDS)-related Kaposi´s sarcoma and multiple myeloma (98). Apart from the direct tumoricidal effects of Doxil, it also displayed various immunomodulatory properties, such as the induction of ICD, the depletion of MDSC (132), and enhanced susceptibility of cancer cells to CTL-released granzyme B (133), all contributing to an elevated anti-tumor immune response (134, 135). Doxil enhanced these immunomodulatory properties as opposed to the overall immunosuppressive nature of free doxorubicin, which was also related to decreased off-target toxicities of the former (136). In contrast to doxorubicin, which has numerous toxicities including a cumulative dose-depending cardiotoxicity, its liposomal formulation (Doxil) showed effectively reduced cardiotoxicity and avoided premedication with corticosteroids (137). Most clinical trials evaluating the clinical efficacy of Doxil were conducted in combination with PD-1/PD-L1 targeting ICI. In the most advanced phase III JAVELIN Ovarian 2000 trial, which compared the combination of avelumab and Doxil and either agent alone in platinum-resistant OC patients, it has however been found that the combination of both agents did not significantly improve PFS or OS (106). Notably, the best response was observed for patients with PD-L1 positive tumors, indicating that the clinical benefit of combined ICI and nanodrug treatment may also be achieved by adequate biomarker stratification of patients. Also, Doxil has been tested in combination with various other ICI agents, such as pembrolizumab for breast cancer (138) or platinum-resistant OC (139), and showed similar or improved efficacy as compared to doxorubicin at reduced systemic toxicity. In order to better understand the role of biomarker stratification and ICD to improve overall ICI efficacy, a number of ongoing clinical trials investigate the combination of immunogenic chemotherapy with Doxil in combination with ICI such as atezolizumab, and the combination of ipilimumab and nivolumab in metastatic TNBC (NCT03164993 and NCT03409198) (140, 141).
In addition to the aforementioned trials Doxil has also been tested in combination with cytokines such as IL-12 in AIDS-related Kaposi´s sarcoma, and this regimen yielded rapid and sustained tumor responses (NCT00020449) (142). By contrast, in platinum-resistant OC the combination of IL-18 and Doxil showed no significantly improved response and survival data (143), highlighting the overall observation that the combination of Doxil and immunotherapy did not enhance clinical efficacy for all cancer patients, but a certain subgroup of patients only.
Cancer vaccines may consist of mRNA that encodes for tumor-associated antigens, which after internalization by APC are translated, processed and presented to T cells, thus initiating tumor antigen-specific T cell responses (144). Despite promising results many earlier clinical trials investigating on cancer vaccines have failed due to poor cell entry of mRNA vaccines and enzymatic degradation of the cargo (145). Thus, nanocarriers developed to deliver mRNA vaccines to the site of interest without the risk of degradation are of special interest to cancer immunotherapy and liposomal formulations of mRNA vaccines have proven to be particularly efficient in stimulating DC-mediated T cell activity (98, 146).
One of the best known liposomal formulations of mRNA cancer vaccines has previously been tested in the Lipo-MERIT trial (NCT02410733) and demonstrated durable objective responses in ICI-experienced patients with unresectable stage III or stage IV melanoma (108, 109). Here, the nano vaccine is composed of a mixture of mRNA species encoding the melanoma tumor antigens New York esophageal squamous cell carcinoma (NY-ESO-)1, melanoma-associated antigen (MAGE-)3, tyrosinase and putative tyrosine-protein phosphatase (TPTE), and cationic liposomes, which upon intravenous injection effectively transfected APC such as DC and macrophages, thereby inducing strong tumor antigen-specific CD4+ and CD8+ T cell immunity (109). Due to the promising results observed in the Lipo-MERIT trial, further mRNA-based nano vaccines encoding for tumor-associated antigens (TAA) using a similar liposomal formulation have been initiated for patients with advanced prostate cancer (NCT04382898) (114), advanced HNSCC (NCT04534205), and advanced OC. In accordance, the phase I dose escalation study of mRNA-4157 in combination with pembrolizumab reported safety and efficacy in patients with resected solid tumors, including melanoma, NSCLC, colon carcinoma and bladder carcinoma (NCT03313778). Hence, a phase II study investigating on adjuvant treatment mRNA-4157 in patients with high-risk melanoma has been initiated recently (NCT03897881).
Immunization with the tumor-specific MAGE-3 antigen has been shown to induce robust immune responses for patients with metastatic melanoma (147), albeit in a phase III trial the immunostimulatory lipid AS15 failed to improve PFS as an adjuvant treatment strategy in patients with MAGE-A3 positive NSCLC (148). Also, liposomal DepoVax-0907 (DPX) that delivered seven peptide TAA increased persistent antigen-specific T cell responses in 39% of patients with breast, ovarian or prostate cancer (149). In the following trial the liposomal DPX platform using survivin class I peptides (DPX-Survivac) was applied in combination with immunomodulatory cyclophosphamide in patients with advanced OC and all patients receiving combination therapy presented with an antigen-specific immune response, which has also been attributed to the Treg depleting effects of cyclophosphamide (150). Due to the strong immune response observed in this trial, DPX-Survivac and cyclophosphamide are currently investigated in a phase II trial in combination with pembrolizumab for patients with recurrent diffuse large B cell lymphoma (NCT03349450) (151).
T-cell targeting nanodrugs also comprise viral vectors that encode tumor proteins, cytokines and enzymes aiming to improve anti-cancer efficacy. In this regard, FDA-approved intralesional treatment of patients with unresectable melanoma and cutaneous or lymph node metastasis with Talimogen laherparepvec (T-VEC), an oncolytic herpes-simples type 1 virus encoding for granulocyte-macrophage colony-stimulating factor (GM-CSF), has demonstrated to enhance immune response to tumor antigens released after virus replication (152). While efficacy has been demonstrated mainly on the injected lesion, in some patients an abscopal systemic effect was also evident in distant organ metastasis, which has been attributed to the recruitment and activation of CTL (153). However, the overall response to T-VEC treatment was relatively low with an overall response rate of 26%, although the limited toxicity profile and survival rates prompted follow-up trials for metastatic melanoma and advanced cutaneous squamous cell carcinoma (NCT04050436) (118, 154–156).
As mentioned above, in contrast to indirectly T cell addressing agents, nanodrugs that directly address T cells have not widely progressed into clinical trials so far. The most prominent clinical trials involve CAR-T cell therapies, which have shown high efficacy in hematological malignancies (73), but could not reproduce these effects in many solid tumors as outlined in the following. Additionally, T cell addressing mRNA-based nanodrugs are increasingly moving into clinical trials and will therefore also be addressed in this chapter.
A number of clinical trials that assess the efficacy and safety of CAR-T cell therapy for solid tumors including melanoma are currently underway. As melanoma constitutes a model tumor for immunotherapy, we will in the following focus on CAR-T cell therapies for this deadliest type of skin cancers (see Table 1).
Only two studies are completed by now, and the only published results came from a single study conducted on 24 patients (results available in NCT01218867). Here, the safety and efficacy of anti- vascular endothelial growth factor receptor CAR-T cells subsequent to lymphodepleting conditioning with cyclophosphamide, Aldesleukin, fludarabine and IL-2 was investigated. As opposed to previous pre-clinical studies in B16 melanoma-bearing mice (157), this study was terminated due to lack in efficacy with 95.8% showing progressive disease and high rates of serious adverse events (95.8%), that mainly involved elevated liver enzymes. The other clinical trial that has been completed by now is a phase I trial on the 3rd generation of anti-disialoganglioside (GD)2 CAR-T cells that have been tested in combination with pembrolizumab after prior lymphodepleting conditioning with cyclophosphamide in young adults with GD2 expressing solid tumors, such as sarcoma, neuroblastoma and melanoma (NCT02107963). GD2 is expressed specifically on tumors of neuroectodermal origin such as melanoma and neuroblastoma (158). CAR-T cells specific for GD2 have previously been reported to show potent effector functions without evidence of functional exhaustion, which persisted in patients treated within the phase I trial and could be augmented with additional anti-PD-1 combination therapy (99). In addition, another phase I trial using anti-GD2-C7R CAR-T cells after prior lymphodepleting conditioning with cyclophosphamide and fludarabine is currently underway for patients with GD2-positive tumors such as uveal melanoma (GAIL-N; NCT03635632). Moreover, a more recent phase I clinical trial is investigating the safety and efficacy of anti-B7H3 CAR-T cells and bispecific anti-B7H3xCD19 CAR-T cells in recurrent solid tumors including melanoma (NCT04483778). Further, phase I clinical trials that evaluate the efficacy of various CAR-T cell therapies for metastatic melanoma are summarized in Table 1 and include CAR-T cells specific for the tumor antigens gp100, NY-ESO1, CD20, CD70 and IL-13a2.
Despite the success of CAR-T cells in the treatment of hematological cancers, several challenges still limit the efficacy of this approach in solid tumors, such as antigen selection, antigen loss and heterogeneity, the immunosuppressive TME, the lack of T cell infiltration into the tumor and off-target toxicity. In order to increase the efficacy of CAR-T cell therapy, a combinational approach has been proposed to overcome some of these challenges and indeed combination treatments with ICI or oncolytic viruses have previously been reported to augment overall efficacy in preclinical tumor models (159–161). Despite the potential synergistic effects of ICI and CAR-T cell therapy there are yet no clinical trials that assess the efficacy and safety of this combinational approach for advanced melanoma, but only in other solid tumors (NCT03726515).
Next to intravenously administered mRNA encoding nano-vaccines that act via DC-mediated T cell activation, some clinical trials investigate the efficacy of intratumorally administered mRNA nanodrugs that directly address T cells. Here, the mRNA nanovaccine-2752, which encodes for human TNF superfamily member 4, IL-23 and IL-36γ, administered in combination with the PD-1 inhibitor durvalumab is currently under investigation in a phase I trial in patients suffering from advanced solid tumors such as melanoma, NSCLC, HNSCC and TNBC (NCT03739931). Preliminary data showed less promising results with only 1/17 patients presenting with a partial response upon intratumoral injection of mRNA-2752 (100). Another phase I clinical trial aimed to investigate the safety and efficacy of intratumorally applied SAR441000, delivering mRNA species that encode IL-12, interferon-α, GM-CSF, and IL-15, in combination with the PD-1 blocking antibody cemiplimab for patients with melanoma, breast cancer and other skin cancers (NCT03871348). Here, tumor responses have been observed more frequently, and were accompanied by increased T cell infiltration and antitumor activity even beyond the site of injection (101).
Additionally, Tecemotide, (L-BLP25, StimuVax), a liposomal nanodrug delivering mucin1 glycoprotein and the TLR4 agonist monophosphoryl lipid A (MPL) using the immunostimulatory lipid BLP24 has been tested in several clinical trials. MPL has been reported to induce Th1 polarization and CTL responses (162, 163). Despite these promising preclinical data and an acceptable safety profile in clinical trials, phase II and III trials failed to demonstrate a significant survival benefit for patients with NSCLC (164), but proved beneficial when applied in combination with cyclophosphamide in patients with stage IIIB locoregional disease (162). The subsequent trial investigating Tecemotide versus placebo in patients with colorectal cancer (CRC) after resection of liver metastases (NCT01462513) also failed to reach its primary endpoints as the median PFS (median PFS: 6.1 months vs. 11.4 months; p=0.18) and 3-year OS rates (3-year OS rates: 69.1 vs 79.1%; p=0.21) were non-significantly shorter in Tecemotide-treated patients as compared to the placebo control group (165).
Last, there are a number of clinical trials which have investigated the application of aAPC for tumor therapy. Here, it has been shown that aAPC were able to educate anti-tumor CTL to acquire a central memory and effector memory phenotype, resulting in T cell trafficking to the tumor site, and mediating clinical responses in advanced melanoma patients (NCT00512889) (102). In the ongoing phase I ImmPACT trial it will be investigated whether aAPC alone or in combination with pembrolizumab may expand antigen-specific endogenous T cells that may result in better objective responses and PFS for patients with metastatic melanoma upon T cell infusion and subsequent lymphocyte depleting chemotherapy with cyclophosphamide and fludarabine (NCT04904185).
The advent of ICI treatment has substantially improved the treatment landscape of many solid tumor types (83). However, innate and acquired resistance to these immunotherapies is frequently observed as solid tumors present with resistance mechanisms, such as an immunosuppressive TME and antigen loss, that impair clinical efficacy of currently available immunotherapies. To address these mechanisms of tumor immune evasion, the clinical application of T cell addressing nanodrugs is a promising approach as it may enhance the efficacy of other immunotherapeutics such as ICI, while reducing off-target toxicities.
In particular, it has been shown that local tumor responsiveness to ICI can be improved by codelivery with of adjuvants within the same nanocarrier or a combination of ICI with nano-formulated immune-modulatory agents. For example, coencapsulation of a PD-L1 blocking antibody and an inhibitor of indoleamine 2,3-dioxygenase 1 in a ROS-responsive polypeptide gel enabled the specific release of the cargo within the TME, characterized by at low pH and high ROS levels, subsequently reducing the local ROS level and enhancing anti-melanoma efficacy in vivo (166). Also, the coadministration of stimulator of IFN genes (STING) agonist-loaded poly(beta-amino-ester) NP with PD-1 blocking antibodies yielded strongly improved anti-tumor effects in established B16 melanomas-burdened mice as compared to either monotherapy and using STING agonist applied in its soluble form (167). Similarly, complexation of immunostimulatory CpG-rich DNA oligo (TLR9 ligand) with a nano-carrier carrying tumor-binding peptides enhanced NP accumulation within the tumor and improved responsiveness to coapplied CTLA-4 blocking antibodies (168).
Moreover, it has been found that nano-formulated TAA-specific mRNA vaccines (RNA-LPX) may induce potent anti-tumor T cell responses and enhance the efficacy of PD-(L)1 blocking antibodies and CAR-T cells (169) via selective delivery of RNA-LPX to APC in mice (170) and patients (171). These studies demonstrate the therapeutic potential of neoantigen-specific anti-tumor vaccines both when applied as a monotherapy as well as in combination with ICI. More recently, the same group also showed that the therapeutic benefit of RNA-LPX vaccination was preserved when employing non-mutant TAA that are primarily expressed by melanoma cells instead of neoantigens, and that this therapeutic approach may help to overcome ICI resistance (108). Next to the application of ICI in combination with other nano-formulated immune-modulatory agents, it has also been shown that nano-carriers may enhance the tumor-specific activity of ICI per se due to selective targeting of tumor-specific surface receptors, thereby additionally reducing off-target toxicities. In this regard, Ishihara and coworkers demonstrated that the conjugation of ICI (anti-CTLA4 and anti-PD-L1 mAb) to a matrix-binding peptide enhanced tissue retention of ICI, delayed tumor growth and prolonged survival in a B16-melanoma model while reducing systemic side effects (172). Finally, nanodrugs may also serve to overcome ICI resistance by altering hostile TME conditions. More specifically, it has been shown that TME-targeted nanoparticles can (a) overcome the physico-chemical barriers (ECM) of the TME (173), the acidic (67) and hypoxic (174) metabolic state of the TME, and (b) modulate immunosuppressive cells, such as Tregs, TAM or MDSC that frequently restrain therapy response in cancer immunotherapy (175). For example, Ou and coworkers demonstrated in a B16 melanoma model a synergistic anti-tumor effect of a Treg-specific tLyp1 peptide-conjugated and Imatinib-loaded NP in combination with an anti-CTLA4 antibody, that has been attributed to the downregulation of immunosuppressive Treg and an enhanced activation of CD8+ T cells (176). The tLyp peptide is a cell-penetrating peptide that has a high affinity for the Neuopilin-1 (Nrp1) surface receptor, which is expressed by many cancer types and by the majority of Treg (175). While the aforementioned combinational regimens may help to overcome ICI resistance in general, it will however be necessary to identify the individual resistance mechanisms that are present in each patient as a prerequisite to target these with personalized combination therapies of T cell-addressing nanodrugs and other immunotherapeutics in order to derive the maximal clinical benefit of specific nanodrug-combination regimens. Therefore, the role of immune biomarkers in clinical immune-oncology will also be critical with regard to T cell addressing nanodrugs (98, 177).
In general, the design the nanodrugs will need to take into account the potential of unwanted cellular interactions in vivo, which may not be recognized in standard in vitro assays when testing NP interaction with the intended target cell type only. In this regard, we have recently shown that non-directed conjugation of antibodies to the NP surface for cell type-specific targeting promoted strong liver accumulation of the NP due to binding of the exposed constant Fc part to Fc receptors expressed by liver sinusoidal endothelial cells (LSEC) at high density (178). Besides LSEC, many other immune cell types also express Fc receptors, which in general serve to recognize antibody-opsonized pathogens (179) and immune complexes (180). Such unwanted effects can be minimized e.g. by either Fc-directed antibody conjugation (181) and by antibody fragments devoid of the Fc part (182). Furthermore, NP may adsorb serum factors in vivo which in turn may alter their biodistribution and cellular interaction as shown by us and others (183, 184). The composition of this so-called protein corona depends on the surface characteristics of the nanodrug like charge and hydrophobicity/hydrophilicity. Besides passive adsorption of serum factors, the NP surface may also trigger the innate immune system as shown by us and others for NP coated with lectin like dextran and starch to achieve biocompatibility that triggered lectin-dependent complement activation (103, 184). This resulted in opsonization of the NP with active complement C3, which in turn yielded strong binding to B cells (103) and myeloid cell types (184) via their complement receptors and subsequent internalization. The formation of a protein corona and accordingly uptake by unwanted cell types can be strongly reduced by conjugation with PEG e.g. in a brush-like conformation at high density as recently shown by us (185) or by using materials that scarcely bind serum factors as e.g. polysarcosin (178, 186).
MH, VM, and MB wrote the manuscript. MH prepared the figures and table. All authors contributed to the article and approved the submitted version.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (SFB1066, Q2, Q6N, B11 [VM] and B15N [MB], and a Walter-Benjamin Fellowship (project number: 507666201) [MH]), and the University Medical Center Mainz (intramural grants: MH, MB; Research Center for Immunotherapy [FZI]: VM, MB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Xiang J, Zhao R, Wang B, Sun X, Guo X, Tan S, et al. Advanced Nano-Carriers for Anti-Tumor Drug Loading. Front Oncol (2021) 11:758143. doi: 10.3389/fonc.2021.758143
2. Chen F, Wang Y, Gao J, Saeed M, Li T, Wang W, et al. Nanobiomaterial-Based Vaccination Immunotherapy of Cancer. Biomaterials (2021) 270:120709. doi: 10.1016/j.biomaterials.2021.120709
3. Germic N, Frangez Z, Yousefi S, Simon HU. Regulation of the Innate Immune System by Autophagy: Monocytes, Macrophages, Dendritic Cells and Antigen Presentation. Cell Death Differ (2019) 26(4):715–27. doi: 10.1038/s41418-019-0297-6
4. Dowling JK, Mansell A. Toll-Like Receptors: The Swiss Army Knife of Immunity and Vaccine Development. Clin Transl Immunol (2016) 5(5):e85. doi: 10.1038/cti.2016.22
5. Haist M, Stege H, Grabbe S, Bros M. The Functional Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells Within the Immunosuppressive Tumor Microenvironment. Cancers (Basel) (2021) 13(2):3. doi: 10.3390/cancers13020210
6. Groth C, Weber R, Lasser S, Özbay FG, Kurzay A, Petrova V, et al. Tumor Promoting Capacity of Polymorphonuclear Myeloid-Derived Suppressor Cells and Their Neutralization. Int J Cancer (2021) 149(9):1628–38. doi: 10.1002/ijc.33731
7. Yano H, Andrews LP, Workman CJ, Vignali DAA. Intratumoral Regulatory T Cells: Markers, Subsets and Their Impact on Anti-Tumor Immunity. Immunology (2019) 157(3):232–47. doi: 10.1111/imm.13067
8. Peng SY, Chen L, Deng RH, Li H, Liu XH, Zheng DW, et al. Non-Depleting Reformation of Immunosuppressive Myeloid Cells to Broaden the Application of Anti-PD Therapy. Nanoscale (2021) 13(8):4420–31. doi: 10.1039/D1NR00830G
9. Rhodes KR, Green JJ. Nanoscale Artificial Antigen Presenting Cells for Cancer Immunotherapy. Mol Immunol (2018) 98:13–8. doi: 10.1016/j.molimm.2018.02.016
10. Matic J, Deeg J, Scheffold A, Goldstein I, Spatz JP. Fine Tuning and Efficient T Cell Activation With Stimulatory Acd3 Nanoarrays. Nano Lett (2013) 13(11):5090–7. doi: 10.1021/nl4022623
11. Est-Witte SE, Livingston NK, Omotoso MO, Green JJ, Schneck JP. Nanoparticles for Generating Antigen-Specific T Cells for Immunotherapy. Semin Immunol (2021) 56:101541. doi: 10.1016/j.smim.2021.101541
12. Lambert LH, Goebrecht GK, De Leo SE, O'Connor RS, Nunez-Cruz S, Li TD, et al. Improving T Cell Expansion With a Soft Touch. Nano Lett (2017) 17(2):821–6. doi: 10.1021/acs.nanolett.6b04071
13. Guasch J, Muth CA, Diemer J, Riahinezhad H, Spatz JP. Integrin-Assisted T-Cell Activation on Nanostructured Hydrogels. Nano Lett (2017) 17(10):6110–6. doi: 10.1021/acs.nanolett.7b02636
14. Bednarczyk M, Stege H, Grabbe S, Bros M. β2 Integrins-Multi-Functional Leukocyte Receptors in Health and Disease. Int J Mol Sci (2020) 21(4):8. doi: 10.3390/ijms21041402
15. Wu PH, Opadele AE, Onodera Y, Nam JM. Targeting Integrins in Cancer Nanomedicine: Applications in Cancer Diagnosis and Therapy. Cancers (Basel) (2019) 11(11):4. doi: 10.3390/cancers11111783
16. Guasch J, Hoffmann M, Diemer J, Riahinezhad H, Neubauer S, Kessler H, et al. Combining Adhesive Nanostructured Surfaces and Costimulatory Signals to Increase T Cell Activation. Nano Lett (2018) 18(9):5899–904. doi: 10.1021/acs.nanolett.8b02588
17. Chatila T, Silverman L, Miller R, Geha R. Mechanisms of T Cell Activation by the Calcium Ionophore Ionomycin. J Immunol (1989) 143(4):1283–9.
18. Hammink R, Weiden J, Voerman D, Popelier C, Eggermont LJ, Schluck M, et al. Semiflexible Immunobrushes Induce Enhanced T Cell Activation and Expansion. ACS Appl Mater Interf (2021) 13(14):16007–18. doi: 10.1021/acsami.0c21994
19. Lee K, Yu Y. Janus Nanoparticles for T Cell Activation: Clustering Ligands to Enhance Stimulation. J Mater Chem B (2017) 5(23):4410–5. doi: 10.1039/C7TB00150A
20. Vis B, Hewitt RE, Monie TP, Fairbairn C, Turner SD, Kinrade SD, et al. Ultrasmall Silica Nanoparticles Directly Ligate the T Cell Receptor Complex. Proc Natl Acad Sci USA (2020) 117(1):285–91. doi: 10.1073/pnas.1911360117
21. Ceuppens JL, Baroja ML, Lorre K, Van Damme J, Billiau A. Human T Cell Activation With Phytohemagglutinin. The Function of IL-6 as an Accessory Signal. J Immunol (1988) 141(11):3868–74.
22. He S, Simpson BK, Sun H, Ngadi MO, Ma Y, Huang T. Phaseolus Vulgaris Lectins: A Systematic Review of Characteristics and Health Implications. Crit Rev Food Sci Nutr (2018) 58(1):70–83. doi: 10.1080/10408398.2015.1096234
23. Alhallak K, Sun J, Muz B, Jeske A, O'Neal J, Ritchey JK, et al. Liposomal Phytohemagglutinin: In Vivo T-Cell Activator as a Novel Pan-Cancer Immunotherapy. J Cell Mol Med (2022) 26(3):940–4. doi: 10.1111/jcmm.16885
24. Zupke O, Distler E, Jürchott A, Paiphansiri U, Dass M, Thomas S, et al. Nanoparticles and Antigen-Specific T-Cell Therapeutics: A Comprehensive Study on Uptake and Release. Nanomed (Lond) (2015) 10(7):1063–76. doi: 10.2217/nnm.14.160
25. Liu L, Ye Q, Wu Y, Hsieh WY, Chen CL, Shen HH, et al. Tracking T-Cells In Vivo With a New Nano-Sized MRI Contrast Agent. Nanomedicine (2012) 8(8):1345–54. doi: 10.1016/j.nano.2012.02.017
26. Mühlberger M, Unterweger H, Band J, Lehmann C, Heger L, Dudziak D, et al. Loading of Primary Human T Lymphocytes With Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Does Not Impair Their Activation After Polyclonal Stimulation. Cells (2020) 9(2):2. doi: 10.3390/cells9020342
27. Boosz P, Pfister F, Stein R, Friedrich B, Fester L, Band J, et al. Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Enable a Stable Non-Spilling Loading of T Cells and Their Magnetic Accumulation. Cancers (Basel) (2021) 13(16):2. doi: 10.3390/cancers13164143
28. Huq R, Samuel EL, Sikkema WK, Nilewski LG, Lee T, Tanner MR, et al. Preferential Uptake of Antioxidant Carbon Nanoparticles by T Lymphocytes for Immunomodulation. Sci Rep (2016) 6:33808. doi: 10.1038/srep33808
29. Jalilov AS, Zhang C, Samuel EL, Sikkema WK, Wu G, Berka V, et al. Mechanistic Study of the Conversion of Superoxide to Oxygen and Hydrogen Peroxide in Carbon Nanoparticles. ACS Appl Mater Interf (2016) 8(24):15086–92. doi: 10.1021/acsami.6b03502
30. Vis B, Hewitt RE, Faria N, Bastos C, Chappell H, Pele L, et al. Non-Functionalized Ultrasmall Silica Nanoparticles Directly and Size-Selectively Activate T Cells. ACS Nano (2018) 12(11):10843–54. doi: 10.1021/acsnano.8b03363
31. Thiramanas R, Jiang S, Simon J, Landfester K, Mailänder V. Silica Nanocapsules With Different Sizes and Physicochemical Properties as Suitable Nanocarriers for Uptake in T-Cells. Int J Nanomed (2020) 15:6069–84. doi: 10.2147/IJN.S246322
32. Thiramanas R, Li M, Jiang S, Landfester K, Mailänder V. Cellular Uptake of siRNA-Loaded Nanocarriers to Knockdown PD-L1: Strategies to Improve T-Cell Functions. Cells (2020) 9(9):1. doi: 10.3390/cells9092043
33. Tabujew I, Willig M, Leber N, Freidel C, Negwer I, Koynov K, et al. Overcoming the Barrier of CD8(+)T Cells: Two Types of Nano-Sized Carriers for siRNA Transport. Acta Biomater (2019) 100:338–51. doi: 10.1016/j.actbio.2019.10.006
34. Tanaka H, Miyama R, Sakurai Y, Tamagawa S, Nakai Y, Tange K, et al. Improvement of mRNA Delivery Efficiency to a T Cell Line by Modulating PEG-Lipid Content and Phospholipid Components of Lipid Nanoparticles. Pharmaceutics (2021) 13(12):4. doi: 10.3390/pharmaceutics13122097
35. Tanaka H, Takahashi T, Konishi M, Takata N, Gomi M, Shirane D, et al. Self-Degradable Lipid-Like Materials Based on “Hydrolysis Accelerated by the Intra-Particle Enrichment of Reactant (HyPER)” for Messenger RNA Delivery. Adv Funct Mater (2020) 30(34):1910575. doi: 10.1002/adfm.201910575
36. Dinauer N, Balthasar S, Weber C, Kreuter J, Langer K, von Briesen H. Selective Targeting of Antibody-Conjugated Nanoparticles to Leukemic Cells and Primary T-Lymphocytes. Biomaterials (2005) 26(29):5898–906. doi: 10.1016/j.biomaterials.2005.02.038
37. Kheirolomoom A, Kare AJ, Ingham ES, Paulmurugan R, Robinson ER, Baikoghli M, et al. In Situ T-Cell Transfection by Anti-CD3-Conjugated Lipid Nanoparticles Leads to T-Cell Activation, Migration, and Phenotypic Shift. Biomaterials (2022) 281:121339. doi: 10.1016/j.biomaterials.2021.121339
38. Ramana LN, Sharma S, Sethuraman S, Ranga U, Krishnan UM. Stealth Anti-CD4 Conjugated Immunoliposomes With Dual Antiretroviral Drugs–Modern Trojan Horses to Combat HIV. Eur J Pharm Biopharm (2015) 89:300–11. doi: 10.1016/j.ejpb.2014.11.021
39. Ramishetti S, Kedmi R, Goldsmith M, Leonard F, Sprague AG, Godin B, et al. Systemic Gene Silencing in Primary T Lymphocytes Using Targeted Lipid Nanoparticles. ACS Nano (2015) 9(7):6706–16. doi: 10.1021/acsnano.5b02796
40. Ermilova I, Swenson J. DOPC Versus DOPE as a Helper Lipid for Gene-Therapies: Molecular Dynamics Simulations With DLin-MC3-DMA. Phys Chem Chem Phys (2020) 22(48):28256–68. doi: 10.1039/D0CP05111J
41. Zheng Y, Tang L, Mabardi L, Kumari S, Irvine DJ. Enhancing Adoptive Cell Therapy of Cancer Through Targeted Delivery of Small-Molecule Immunomodulators to Internalizing or Noninternalizing Receptors. ACS Nano (2017) 11(3):3089–100. doi: 10.1021/acsnano.7b00078
42. Maier MA, Jayaraman M, Matsuda S, Liu J, Barros S, Querbes W, et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol Ther (2013) 21(8):1570–8. doi: 10.1038/mt.2013.124
43. Tombácz I, Laczkó D, Shahnawaz H, Muramatsu H, Natesan A, Yadegari A, et al. Highly Efficient CD4+ T Cell Targeting and Genetic Recombination Using Engineered CD4+ Cell-Homing mRNA-LNPs. Mol Ther (2021) 29(11):3293–304. doi: 10.1016/j.ymthe.2021.06.004
44. Canakci M, Singh K, Munkhbat O, Shanthalingam S, Mitra A, Gordon M, et al. Targeting CD4(+) Cells With Anti-CD4 Conjugated Mertansine-Loaded Nanogels. Biomacromolecules (2020) 21(6):2473–81. doi: 10.1021/acs.biomac.0c00442
45. He F, Wen N, Xiao D, Yan J, Xiong H, Cai S, et al. Aptamer-Based Targeted Drug Delivery Systems: Current Potential and Challenges. Curr Med Chem (2020) 27(13):2189–219. doi: 10.2174/0929867325666181008142831
46. Wang CW, Chung WH, Cheng YF, Ying NW, Peck K, Chen YT, et al. A New Nucleic Acid-Based Agent Inhibits Cytotoxic T Lymphocyte-Mediated Immune Disorders. J Allergy Clin Immunol (2013) 132(3):713–22.e11. doi: 10.1016/j.jaci.2013.04.036
47. Luesakul U, Puthong S, Neamati N, Muangsin N. pH-Responsive Selenium Nanoparticles Stabilized by Folate-Chitosan Delivering Doxorubicin for Overcoming Drug-Resistant Cancer Cells. Carbohydr Polym (2018) 181:841–50. doi: 10.1016/j.carbpol.2017.11.068
48. Laskin BL, Jiao J, Baluarte HJ, Amaral S, Furth SL, Akimova T, et al. The Effects of Tacrolimus on T-Cell Proliferation Are Short-Lived: A Pilot Analysis of Immune Function Testing. Transplant Direct (2017) 3(8):e199. doi: 10.1097/TXD.0000000000000715
49. Mansouri A, Abnous K, Alibolandi M, Taghdisi SM, Ramezani M. Targeted Delivery of Tacrolimus to T Cells by pH-Responsive Aptamer-Chitosan- Poly(Lactic-Co-Glycolic Acid) Nanocomplex. J Cell Physiol (2019) 234(10):18262–71. doi: 10.1002/jcp.28458
50. Glass JJ, Yuen D, Rae J, Johnston AP, Parton RG, Kent SJ, et al. Human Immune Cell Targeting of Protein Nanoparticles–Caveospheres. Nanoscale (2016) 8(15):8255–65. doi: 10.1039/C6NR00506C
51. Frick SU, Domogalla MP, Baier G, Wurm FR, Mailänder V, Landfester K, et al. Interleukin-2 Functionalized Nanocapsules for T Cell-Based Immunotherapy. ACS Nano (2016) 10(10):9216–26. doi: 10.1021/acsnano.5b07973
52. Gamrad L, Rehbock C, Westendorf AM, Buer J, Barcikowski S, Hansen W. Efficient Nucleic Acid Delivery to Murine Regulatory T Cells by Gold Nanoparticle Conjugates. Sci Rep (2016) 6:28709. doi: 10.1038/srep28709
53. Li SY, Liu Y, Xu CF, Shen S, Sun R, Du XJ, et al. Restoring Anti-Tumor Functions of T Cells via Nanoparticle-Mediated Immune Checkpoint Modulation. J Control Release (2016) 231:17–28. doi: 10.1016/j.jconrel.2016.01.044
54. Wu Y, Gu W, Li L, Chen C, Xu ZP. Enhancing PD-1 Gene Silence in T Lymphocytes by Comparing the Delivery Performance of Two Inorganic Nanoparticle Platforms. Nanomater (Basel) (2019) 9(2):2. doi: 10.3390/nano9020159
55. Barati M, Mirzavi F, Nikpoor AR, Sankian M, Namdar Ahmadabad H, Soleimani A, et al. Enhanced Antitumor Immune Response in Melanoma Tumor Model by Anti-PD-1 Small Interference RNA Encapsulated in Nanoliposomes. Cancer Gene Ther (2021). doi: 10.1038/s41417-021-00367-9
56. Kosmides AK, Sidhom JW, Fraser A, Bessell CA, Schneck JP. Dual Targeting Nanoparticle Stimulates the Immune System To Inhibit Tumor Growth. ACS Nano (2017) 11(6):5417–29. doi: 10.1021/acsnano.6b08152
57. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor Antigen-Specific CD8 T Cells Infiltrating the Tumor Express High Levels of PD-1 and are Functionally Impaired. Blood (2009) 114(8):1537–44. doi: 10.1182/blood-2008-12-195792
58. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy Targeting 4-1BB: Mechanistic Rationale, Clinical Results, and Future Strategies. Blood (2018) 131(1):49–57. doi: 10.1182/blood-2017-06-741041
59. Yang YS, Moynihan KD, Bekdemir A, Dichwalkar TM, Noh MM, Watson N, et al. Targeting Small Molecule Drugs to T Cells With Antibody-Directed Cell-Penetrating Gold Nanoparticles. Biomater Sci (2018) 7(1):113–24. doi: 10.1039/C8BM01208C
60. Francis DM, Manspeaker MP, Archer PA, Sestito LF, Heiler AJ, Schudel A, et al. Drug-Eluting Immune Checkpoint Blockade Antibody-Nanoparticle Conjugate Enhances Locoregional and Systemic Combination Cancer Immunotherapy Through T Lymphocyte Targeting. Biomaterials (2021) 279:121184. doi: 10.1016/j.biomaterials.2021.121184
61. Cluxton D, Petrasca A, Moran B, Fletcher JM. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front Immunol (2019) 10:115. doi: 10.3389/fimmu.2019.00115
62. Muroski ME, Miska J, Chang AL, Zhang P, Rashidi A, Moore H, et al. Fatty Acid Uptake in T Cell Subsets Using a Quantum Dot Fatty Acid Conjugate. Sci Rep (2017) 7(1):5790. doi: 10.1038/s41598-017-05556-x
63. Kim D, Wu Y, Li Q, Oh YK. Nanoparticle-Mediated Lipid Metabolic Reprogramming of T Cells in Tumor Microenvironments for Immunometabolic Therapy. Nanomicro Lett (2021) 13(1):31. doi: 10.1007/s40820-020-00555-6
64. Bam M, Chintala S, Fetcko K, Williamsen BC, Siraj S, Liu S, et al. Genome Wide DNA Methylation Landscape Reveals Glioblastoma's Influence on Epigenetic Changes in Tumor Infiltrating CD4+ T Cells. Oncotarget (2021) 12(10):967–81. doi: 10.18632/oncotarget.27955
65. Hu N, Li W, Hong Y, Zeng Z, Zhang J, Wu X, et al. A PD1 Targeted Nano-Delivery System Based on Epigenetic Alterations of T Cell Responses in the Treatment of Gastric Cancer. Mol Ther Oncolytics (2022) 24:148–59. doi: 10.1016/j.omto.2021.12.006
66. Wang JX, Choi SYC, Niu X, Kang N, Xue H, Killam J, et al. Lactic Acid and an Acidic Tumor Microenvironment Suppress Anticancer Immunity. Int J Mol Sci (2020) 21(21):2. doi: 10.3390/ijms21218363
67. Zhang YX, Zhao YY, Shen J, Sun X, Liu Y, Liu H, et al. Nanoenabled Modulation of Acidic Tumor Microenvironment Reverses Anergy of Infiltrating T Cells and Potentiates Anti-PD-1 Therapy. Nano Lett (2019) 19(5):2774–83. doi: 10.1021/acs.nanolett.8b04296
68. Hei Y, Teng B, Zeng Z, Zhang S, Li Q, Pan J, et al. Multifunctional Immunoliposomes Combining Catalase and PD-L1 Antibodies Overcome Tumor Hypoxia and Enhance Immunotherapeutic Effects Against Melanoma. Int J Nanomed (2020) 15:1677–91. doi: 10.2147/IJN.S225807
69. Li T, Kang G, Wang T, Huang H. Tumor Angiogenesis and Anti-Angiogenic Gene Therapy for Cancer. Oncol Lett (2018) 16(1):687–702. doi: 10.3892/ol.2018.8733
70. Hei Y, Chen Y, Li Q, Mei Z, Pan J, Zhang S, et al. Multifunctional Immunoliposomes Enhance the Immunotherapeutic Effects of PD-L1 Antibodies Against Melanoma by Reprogramming Immunosuppressive Tumor Microenvironment. Small (2022) 18(9):e2105118. doi: 10.1002/smll.202105118
71. Russ A, Hua AB, Montfort WR, Rahman B, Riaz IB, Khalid MU, et al. Blocking "Don't Eat Me" Signal of CD47-Sirpα in Hematological Malignancies, an in-Depth Review. Blood Rev (2018) 32(6):480–9. doi: 10.1016/j.blre.2018.04.005
72. Labanieh L, Majzner RG, Mackall CL. Programming CAR-T Cells to Kill Cancer. Nat BioMed Eng (2018) 2(6):377–91. doi: 10.1038/s41551-018-0235-9
73. Dana H, Chalbatani GM, Jalali SA, Mirzaei HR, Grupp SA, Suarez ER, et al. CAR-T Cells: Early Successes in Blood Cancer and Challenges in Solid Tumors. Acta Pharm Sin B (2021) 11(5):1129–47. doi: 10.1016/j.apsb.2020.10.020
74. Lukjanov V, Koutná I, Šimara P. CAR T-Cell Production Using Nonviral Approaches. J Immunol Res (2021) 2021:6644685. doi: 10.1155/2021/6644685
75. Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett (2020) 20(3):1578–89. doi: 10.1021/acs.nanolett.9b04246
76. Billingsley MM, Hamilton AG, Mai D, Patel SK, Swingle KL, Sheppard NC, et al. Orthogonal Design of Experiments for Optimization of Lipid Nanoparticles for mRNA Engineering of CAR T Cells. Nano Lett (2022) 22(1):533–42. doi: 10.1021/acs.nanolett.1c02503
77. Zhao X, Chen J, Qiu M, Li Y, Glass Z, Xu Q. Imidazole-Based Synthetic Lipidoids for In Vivo mRNA Delivery Into Primary T Lymphocytes. Angew Chem Int Ed Engl (2020) 59(45):20083–9. doi: 10.1002/anie.202008082
78. Ye Z, Chen J, Zhao X, Li Y, Harmon J, Huang C, et al. In Vitro Engineering Chimeric Antigen Receptor Macrophages and T Cells by Lipid Nanoparticle-Mediated mRNA Delivery. ACS Biomater Sci Eng (2022) 8(2):722–33. doi: 10.1021/acsbiomaterials.1c01532
79. Yu Q, Zhang M, Chen Y, Chen X, Shi S, Sun K, et al. Self-Assembled Nanoparticles Prepared From Low-Molecular-Weight PEI and Low-Generation PAMAM for EGFRvIII-Chimeric Antigen Receptor Gene Loading and T-Cell Transient Modification. Int J Nanomed (2020) 15:483–95. doi: 10.2147/IJN.S229858
80. Kiru L, Zlitni A, Tousley AM, Dalton GN, Wu W, Lafortune F, et al. In Vivo Imaging of Nanoparticle-Labeled CAR T Cells. Proc Natl Acad Sci USA (2022) 119(6):1. doi: 10.1073/pnas.2102363119
81. Zhang W, Gaikwad H, Groman EV, Purev E, Simberg D, Wang G. Highly Aminated Iron Oxide Nanoworms for Simultaneous Manufacturing and Labeling of Chimeric Antigen Receptor T Cells. J Magn Magn Mater (2022) 541:1. doi: 10.1016/j.jmmm.2021.168480
82. Luo Y, Chen Z, Sun M, Li B, Pan F, Ma A, et al. IL-12 Nanochaperone-Engineered CAR T Cell for Robust Tumor-Immunotherapy. Biomaterials (2022) 281:121341. doi: 10.1016/j.biomaterials.2021.121341
83. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
84. Antonia SJ, López-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab Alone and Nivolumab Plus Ipilimumab in Recurrent Small-Cell Lung Cancer (CheckMate 032): A Multicentre, Open-Label, Phase 1/2 Trial. Lancet Oncol (2016) 17(7):883–95. doi: 10.1016/S1470-2045(16)30098-5
85. Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Avelumab in Patients With Chemotherapy-Refractory Metastatic Merkel Cell Carcinoma: A Multicentre, Single-Group, Open-Label, Phase 2 Trial. Lancet Oncol (2016) 17(10):1374–85. doi: 10.1016/S1470-2045(16)30364-3
86. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in Patients With Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol (2017) 18(9):1182–91. doi: 10.1016/S1470-2045(17)30422-9
87. Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. New Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
88. Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, et al. Nivolumab in Metastatic Urothelial Carcinoma After Platinum Therapy (CheckMate 275): A Multicentre, Single-Arm, Phase 2 Trial. Lancet Oncol (2017) 18(3):312–22. doi: 10.1016/S1470-2045(17)30065-7
89. Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J Clin Oncol (2015) 33(13):1430–7. doi: 10.1200/JCO.2014.59.0703
90. Majzner RG, Mackall CL. Clinical Lessons Learned From the First Leg of the CAR T Cell Journey. Nat Med (2019) 25(9):1341–55. doi: 10.1038/s41591-019-0564-6
91. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
92. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene Maraleucel for Patients With Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. Lancet (2020) 396(10254):839–52. doi: 10.1016/S0140-6736(20)31366-0
93. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med (2019) 380(1):45–56. doi: 10.1056/NEJMoa1804980
94. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of Resistance to Immune Checkpoint Inhibitors. Br J Cancer (2018) 118(1):9–16. doi: 10.1038/bjc.2017.434
95. Fesnak AD, June CH, Levine BL. Engineered T Cells: The Promise and Challenges of Cancer Immunotherapy. Nat Rev Cancer (2016) 16(9):566–81. doi: 10.1038/nrc.2016.97
96. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat Rev Cancer (2012) 12(4):298–306. doi: 10.1038/nrc3245
97. Debele TA, Yeh CF, Su WP. Cancer Immunotherapy and Application of Nanoparticles in Cancers Immunotherapy as the Delivery of Immunotherapeutic Agents and as the Immunomodulators. Cancers (Basel) (2020) 12(12):3773. doi: 10.3390/cancers12123773
98. Shi Y. Clinical Translation of Nanomedicine and Biomaterials for Cancer Immunotherapy: Progress and Perspectives. Adv Ther (2020) 3(9):1900215. doi: 10.1002/adtp.201900215
99. Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-Specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter But can be Protected From Activation-Induced Cell Death by PD-1 Blockade. Mol Ther (2016) 24(6):1135–49. doi: 10.1038/mt.2016.63
100. Patel MR, Bauer TM, Jimeno A, Wang D, LoRusso P, Do KT, et al. A Phase I Study of mRNA-2752, a Lipid Nanoparticle Encapsulating mRNAs Encoding Human OX40L, IL-23, and IL-36γ, for Intratumoral (iTu) Injection Alone and in Combination With Durvalumab. J Clin Oncol (2020) 38(15_suppl):3092. doi: 10.1200/JCO.2020.38.15_suppl.3092
101. Bechter O, Utikal J, Baurain J-F, Massard C, Sahin U, Derhovanessian E, et al. 391 A First-in-Human Study of Intratumoral SAR441000, an mRNA Mixture Encoding IL-12sc, Interferon Alpha2b, GM-CSF and IL-15sushi as Monotherapy and in Combination With Cemiplimab in Advanced Solid Tumors. J ImmunoTher Cancer (2020) 8(Suppl 3):A237–A8. doi: 10.1136/jitc-2020-SITC2020.0391
102. Butler MO, Friedlander P, Milstein MI, Mooney MM, Metzler G, Murray AP, et al. Establishment of Antitumor Memory in Humans Using In Vitro-Educated CD8+ T Cells. Sci Transl Med (2011) 3(80):80ra34. doi: 10.1126/scitranslmed.3002207
103. Shen L, Tenzer S, Storck W, Hobernik D, Raker VK, Fischer K, et al. Protein corona-mediated targeting of nanocarriers to B cells allows redirection of allergic immune responses. J Allergy Clin Immunol (2018) 142(5):1558–70. doi: 10.1016/j.jaci.2017.08.049
104. Kang C, Syed YY. Atezolizumab (in Combination With Nab-Paclitaxel): A Review in Advanced Triple-Negative Breast Cancer. Drugs (2020) 80(6):601–7. doi: 10.1007/s40265-020-01295-y
105. West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, et al. Atezolizumab in Combination With Carboplatin Plus Nab-Paclitaxel Chemotherapy Compared With Chemotherapy Alone as First-Line Treatment for Metastatic non-Squamous non-Small-Cell Lung Cancer (IMpower130): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol (2019) 20(7):924–37. doi: 10.1016/S1470-2045(19)30167-6
106. Pujade-Lauraine E, Fujiwara K, Dychter SS, Devgan G, Monk BJ. Avelumab (Anti-PD-L1) in Platinum-Resistant/Refractory Ovarian Cancer: JAVELIN Ovarian 200 Phase III Study Design. Future Oncol (2018) 14(21):2103–13. doi: 10.2217/fon-2018-0070
107. Wang-Gillam A, Li CP, Bodoky G, Dean A, Shan YS, Jameson G, et al. Nanoliposomal Irinotecan With Fluorouracil and Folinic Acid in Metastatic Pancreatic Cancer After Previous Gemcitabine-Based Therapy (NAPOLI-1): A Global, Randomised, Open-Label, Phase 3 Trial. Lancet (2016) 387(10018):545–57. doi: 10.1016/s0140-6736(15)00986-1
108. Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, et al. An RNA Vaccine Drives Immunity in Checkpoint-Inhibitor-Treated Melanoma. Nature (2020) 585(7823):107–12. doi: 10.1038/s41586-020-2537-9
109. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy. Nature (2016) 534(7607):396–401. doi: 10.1038/nature18300
110. Burris Iii HA, Patel MR, Cho DC, Clarke JM, Gutierrez M, Zaks TZ, et al. A Phase 1, Open-Label, Multicenter Study to Assess the Safety, Tolerability, and Immunogenicity of mRNA-4157 Alone in Subjects With Resected Solid Tumors and in Combination With Pembrolizumab in Subjects With Unresectable Solid Tumors (Keynote-603). J Global Oncol (2019) 5(suppl):93. doi: 10.1200/JGO.2019.5.suppl.93
111. Pfannenstiel LW, Lam SS, Emens LA, Jaffee EM, Armstrong TD. Paclitaxel Enhances Early Dendritic Cell Maturation and Function Through TLR4 Signaling in Mice. Cell Immunol (2010) 263(1):79–87. doi: 10.1016/j.cellimm.2010.03.001
112. Graziani SR, Vital CG, Morikawa AT, Van Eyll BM, Fernandes Junior HJ, Kalil Filho R, et al. Phase II Study of Paclitaxel Associated With Lipid Core Nanoparticles (LDE) as Third-Line Treatment of Patients With Epithelial Ovarian Carcinoma. Med Oncol (2017) 34(9):151. doi: 10.1007/s12032-017-1009-z
113. Yamaguchi K, Iwasa S, Hirao M, Oshima T, Harada K, Sato Y, et al. Phase 1 Study of the Liposomal Formulation of Eribulin (E7389-LF): Results From the Advanced Gastric Cancer Expansion Cohort. J Clin Oncol (2021) 39(15):4025. doi: 10.1200/JCO.2021.39.15_suppl.4025
114. Linch M, Papai Z, Takacs I, Imedio ER, Kühnle M-C, Derhovanessian E, et al. 421 A First-in-Human (FIH) Phase I/IIa Clinical Trial Assessing a Ribonucleic Acid Lipoplex (RNA-LPX) Encoding Shared Tumor Antigens for Immunotherapy of Prostate Cancer; Preliminary Analysis of PRO-MERIT. J ImmunoTher Cancer (2021) 9(Suppl 2):A451–A. doi: 10.1136/jitc-2021-SITC2021.421
115. Grunwitz C, Salomon N, Vascotto F, Selmi A, Bukur T, Diken M, et al. HPV16 RNA-LPX Vaccine Mediates Complete Regression of Aggressively Growing HPV-Positive Mouse Tumors and Establishes Protective T Cell Memory. Oncoimmunology (2019) 8(9):e1629259. doi: 10.1080/2162402X.2019.1629259
116. Schmidt M, Bolte S, Frenzel K, Heesen L, Derhovanessian E, Bukur V, et al. Abstract OT2-06-01: Highly Innovative Personalized RNA-Immunotherapy for Patients With Triple Negative Breast Cancer. Cancer Res (2019) 79(4_Supplement):OT2-06-1-OT2–1. doi: 10.1158/1538-7445.SABCS18-OT2-06-01
117. Braiteh F, LoRusso P, Balmanoukian A, Klempner S, Camidge DR, Hellmann M, et al. Abstract CT169: A Phase Ia Study to Evaluate RO7198457, an Individualized Neoantigen Specific Immunotherapy (Inest), in Patients With Locally Advanced or Metastatic Solid Tumors. Cancer Res (2020) 80(16_Supplement):CT169–CT. doi: 10.1158/1538-7445.AM2020-CT169
118. Haydon A, Alamgeer M, Brungs D, Collichio F, Khushalani N, Colevas D, et al. 547 CERPASS: A Randomized, Controlled, Open-Label, Phase 2 Study of Cemiplimab ± RP1 in Patients With Advanced Cutaneous Squamous Cell Carcinoma. J ImmunoTher Cancer (2021) 9(Suppl 2):A577–A. doi: 10.1136/jitc-2021-SITC2021.547
119. Shen C, Frakes J, Weiss J, Caudell JJ, Hackman TG, Akulian JA, et al. Phase I Study of NBTXR3 Activated by Radiotherapy in Patients With Advanced Cancers Treated With an Anti-PD-1 Therapy. J Clin Oncol (2020) 38(15):TPS3173–TPS. doi: 10.1200/JCO.2020.38.15_suppl.TPS3173
120. Bonvalot S, Rutkowski PL, Thariat J, Carrere S, Ducassou A, Sunyach MP, et al. NBTXR3, a First-in-Class Radioenhancer Hafnium Oxide Nanoparticle, Plus Radiotherapy Versus Radiotherapy Alone in Patients With Locally Advanced Soft-Tissue Sarcoma (Act.In.Sarc): A Multicentre, Phase 2-3, Randomised, Controlled Trial. Lancet Oncol (2019) 20(8):1148–59. doi: 10.1016/S1470-2045(19)30326-2
121. Shan X, Gong X, Li J, Wen J, Li Y, Zhang Z. Current Approaches of Nanomedicines in the Market and Various Stage of Clinical Translation. Acta Pharm Sin B (2022). doi: 10.1016/j.apsb.2022.02.025
122. Demaria S, Volm MD, Shapiro RL, Yee HT, Oratz R, Formenti SC, et al. Development of Tumor-Infiltrating Lymphocytes in Breast Cancer After Neoadjuvant Paclitaxel Chemotherapy1. Clin Cancer Res (2001) 7(10):3025–30.
123. Vicari AP, Luu R, Zhang N, Patel S, Makinen SR, Hanson DC, et al. Paclitaxel Reduces Regulatory T Cell Numbers and Inhibitory Function and Enhances the Anti-Tumor Effects of the TLR9 Agonist PF-3512676 in the Mouse. Cancer Immunol Immunother (2009) 58(4):615–28. doi: 10.1007/s00262-008-0586-2
124. Ran S. The Role of TLR4 in Chemotherapy-Driven Metastasis. Cancer Res (2015) 75(12):2405–10. doi: 10.1158/0008-5472.CAN-14-3525
125. Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, et al. Phase III Trial of Nanoparticle Albumin-Bound Paclitaxel Compared With Polyethylated Castor Oil-Based Paclitaxel in Women With Breast Cancer. J Clin Oncol (2005) 23(31):7794–803. doi: 10.1200/JCO.2005.04.937
126. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med (2018) 379(22):2108–21. doi: 10.1056/NEJMoa1809615
127. Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau HT, Forero-Torres A, et al. Avelumab, an Anti-PD-L1 Antibody, in Patients With Locally Advanced or Metastatic Breast Cancer: A Phase 1b JAVELIN Solid Tumor Study. Breast Cancer Res Treat (2018) 167(3):671–86. doi: 10.1007/s10549-017-4537-5
128. Emens LA, Cruz C, Eder JP, Braiteh F, Chung C, Tolaney SM, et al. Long-Term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol (2019) 5(1):74–82. doi: 10.1001/jamaoncol.2018.4224
129. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, et al. Pembrolizumab Plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med (2018) 379(21):2040–51. doi: 10.1056/NEJMoa1810865
130. Aigner J, Marme F, Smetanay K, Schuetz F, Jaeger D, Schneeweiss A. Nab-Paclitaxel Monotherapy as a Treatment of Patients With Metastatic Breast Cancer in Routine Clinical Practice. Anticancer Res (2013) 33(8):3407–13.
131. Zhang Z, Zhang Y, Song S, Yin L, Sun D, Gu J. Recent Advances in the Bioanalytical Methods of Polyethylene Glycols and PEGylated Pharmaceuticals. J Separation Sci (2020) 43(9-10):1978–97. doi: 10.1002/jssc.201901340
132. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, et al. Doxorubicin Eliminates Myeloid-Derived Suppressor Cells and Enhances the Efficacy of Adoptive T-Cell Transfer in Breast Cancer. Cancer Res (2014) 74(1):104–18. doi: 10.1158/0008-5472.CAN-13-1545
133. Ramakrishnan R, Assudani D, Nagaraj S, Hunter T, Cho HI, Antonia S, et al. Chemotherapy Enhances Tumor Cell Susceptibility to CTL-Mediated Killing During Cancer Immunotherapy in Mice. J Clin Invest (2010) 120(4):1111–24. doi: 10.1172/JCI40269
134. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat Med (2007) 13(1):54–61. doi: 10.1038/nm1523
135. Rios-Doria J, Durham N, Wetzel L, Rothstein R, Chesebrough J, Holoweckyj N, et al. Doxil Synergizes With Cancer Immunotherapies to Enhance Antitumor Responses in Syngeneic Mouse Models. Neoplasia (2015) 17(8):661–70. doi: 10.1016/j.neo.2015.08.004
136. Shi Y, Lammers T. Combining Nanomedicine and Immunotherapy. Acc Chem Res (2019) 52(6):1543–54. doi: 10.1021/acs.accounts.9b00148
137. Marina NM, Cochrane D, Harney E, Zomorodi K, Blaney S, Winick N, et al. Dose Escalation and Pharmacokinetics of Pegylated Liposomal Doxorubicin (Doxil) in Children With Solid Tumors: A Pediatric Oncology Group Study1. Clin Cancer Res (2002) 8(2):413–8.
138. O'Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, et al. Reduced Cardiotoxicity and Comparable Efficacy in a Phase III Trial of Pegylated Liposomal Doxorubicin HCl (CAELYX/Doxil) Versus Conventional Doxorubicin for First-Line Treatment of Metastatic Breast Cancer. Ann Oncol (2004) 15(3):440–9. doi: 10.1093/annonc/mdh097
139. Lee EK, Xiong N, Cheng SC, Barry WT, Penson RT, Konstantinopoulos PA, et al. Combined Pembrolizumab and Pegylated Liposomal Doxorubicin in Platinum Resistant Ovarian Cancer: A Phase 2 Clinical Trial. Gynecol Oncol (2020) 159(1):72–8. doi: 10.1016/j.ygyno.2020.07.028
140. Kyte JA, Rossevold A, Falk RS, Naume B. ALICE: A Randomized Placebo-Controlled Phase II Study Evaluating Atezolizumab Combined With Immunogenic Chemotherapy in Patients With Metastatic Triple-Negative Breast Cancer. J Transl Med (2020) 18(1):252. doi: 10.1186/s12967-020-02424-7
141. Kyte JA, Andresen NK, Russnes HG, Fretland SO, Falk RS, Lingjaerde OC, et al. ICON: A Randomized Phase IIb Study Evaluating Immunogenic Chemotherapy Combined With Ipilimumab and Nivolumab in Patients With Metastatic Hormone Receptor Positive Breast Cancer. J Transl Med (2020) 18(1):269. doi: 10.1186/s12967-020-02421-w
142. Little RF, Aleman K, Kumar P, Wyvill KM, Pluda JM, Read-Connole E, et al. Phase 2 Study of Pegylated Liposomal Doxorubicin in Combination With Interleukin-12 for AIDS-Related Kaposi Sarcoma. Blood (2007) 110(13):4165–71. doi: 10.1182/blood-2007-06-097568
143. Simpkins F, Flores AM, Chu C, Lucci JA, Berek JS, Coukos G, et al. Dose Escalation Trial to Assess the Safety and Biological Activity of Recombinant Human Interleukin-18 (SB-485232) in Combination With Pegylated Liposomal Doxorubicin in Platinum-Resistant Recurrent Ovarian Cancer. J Clin Oncol (2012) 30(15):5065. doi: 10.1200/jco.2012.30.15_suppl.5065
144. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA Vaccines - a New Era in Vaccinology. Nat Rev Drug Discovery (2018) 17(4):261–79. doi: 10.1038/nrd.2017.243
145. Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, et al. Therapeutic Vaccines for Cancer: An Overview of Clinical Trials. Nat Rev Clin Oncol (2014) 11(9):509–24. doi: 10.1038/nrclinonc.2014.111
146. Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol Ther (2019) 27(4):710–28. doi: 10.1016/j.ymthe.2019.02.012
147. Kruit W, Suciu S, Dreno B, Mortier L, Robert C, Chiarion-Sileni V, et al. Selection of Immunostimulant AS15 for Active Immunization With MAGE-A3 Protein: Results of a Randomized Phase II Study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J Clin Oncol (2013) 31(19):2413–20. doi: 10.1200/JCO.2012.43.7111
148. Vansteenkiste J, Cho B, Vanakesa T, De Pas T, Zielinski M, Kim M, et al. MAGRIT, a Double-Blind, Randomized, Placebo-Controlled Phase III Study to Assess the Efficacy of the recMAGE-A3+ AS15 Cancer Immunotherapeutic as Adjuvant Therapy in Patients With Resected MAGE-A3-Positive non-Small Cell Lung Cancer (NSCLC). Ann Oncol (2014) 25:iv409. doi: 10.1093/annonc/mdu347.1
149. Berinstein NL, Karkada M, Morse MA, Nemunaitis JJ, Chatta G, Kaufman H, et al. First-In-Man Application of a Novel Therapeutic Cancer Vaccine Formulation With the Capacity to Induce Multi-Functional T Cell Responses in Ovarian, Breast and Prostate Cancer Patients. J Transl Med (2012) 10:156. doi: 10.1186/1479-5876-10-156
150. Berinstein NL, Karkada M, Oza AM, Odunsi K, Villella JA, Nemunaitis JJ, et al. Survivin-Targeted Immunotherapy Drives Robust Polyfunctional T Cell Generation and Differentiation in Advanced Ovarian Cancer Patients. OncoImmunology (2015) 4(8):e1026529. doi: 10.1080/2162402X.2015.1026529
151. Berinstein NL, Bence-Bruckler I, Laneuville P, Stewart DA, Forward NA, Smyth L, et al. Combination of DPX-Survivac, Low Dose Cyclophosphamide, and Pembrolizumab in Recurrent/Refractory DLBCL: The Spirel Study. Blood (2019) 134(Supplement_1):3236. doi: 10.1182/blood-2019-125963
152. Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, et al. Phase II Clinical Trial of a Granulocyte-Macrophage Colony-Stimulating Factor-Encoding, Second-Generation Oncolytic Herpesvirus in Patients With Unresectable Metastatic Melanoma. J Clin Oncol (2009) 27(34):5763–71. doi: 10.1200/JCO.2009.24.3675
153. Ferrucci PF, Pala L, Conforti F, Cocorocchio E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers (Basel) (2021) 13(6):1383. doi: 10.3390/cancers13061383
154. Andtbacka RH, Ross MI, Agarwala SS, Taylor MH, Vetto JT, Neves RI, et al. Final Results of a Phase II Multicenter Trial of HF10, a Replication-Competent HSV-1 Oncolytic Virus, and Ipilimumab Combination Treatment in Patients With Stage IIIB-IV Unresectable or Metastatic Melanoma. J Clin Oncol (2017) 35(15_suppl):9510. doi: 10.1200/JCO.2017.35.15_suppl.9510
155. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell (2017) 170(6):1109–19.e10. doi: 10.1016/j.cell.2017.08.027
156. Malvehy J, Samoylenko I, Schadendorf D, Gutzmer R, Grob JJ, Sacco JJ, et al. Talimogene Laherparepvec Upregulates Immune-Cell Populations in non-Injected Lesions: Findings From a Phase II, Multicenter, Open-Label Study in Patients With Stage IIIB-IVM1c Melanoma. J Immunother Cancer (2021) 9(3):1. doi: 10.1136/jitc-2020-001621
157. Chinnasamy D, Tran E, Yu Z, Morgan RA, Restifo NP, Rosenberg SA. Simultaneous Targeting of Tumor Antigens and the Tumor Vasculature Using T Lymphocyte Transfer Synergize to Induce Regression of Established Tumors in Mice. Cancer Res (2013) 73(11):3371–80. doi: 10.1158/0008-5472.CAN-12-3913
158. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front Oncol (2020) 10:1000. doi: 10.3389/fonc.2020.01000
159. Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, Gottschalk S, et al. Armed Oncolytic Adenovirus–Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res (2017) 77(8):2040–51. doi: 10.1158/0008-5472.CAN-16-1577
160. Chapuis AG, Roberts IM, Thompson JA, Margolin KA, Bhatia S, Lee SM, et al. T-Cell Therapy Using Interleukin-21-Primed Cytotoxic T-Cell Lymphocytes Combined With Cytotoxic T-Cell Lymphocyte Antigen-4 Blockade Results in Long-Term Cell Persistence and Durable Tumor Regression. J Clin Oncol (2016) 34(31):3787–95. doi: 10.1200/JCO.2015.65.5142
161. Soltantoyeh T, Akbari B, Karimi A, Mahmoodi Chalbatani G, Ghahri-Saremi N, Hadjati J, et al. Chimeric Antigen Receptor (CAR) T Cell Therapy for Metastatic Melanoma: Challenges and Road Ahead. Cells (2021) 10(6):1450. doi: 10.3390/cells10061450
162. Butts C, Murray N, Maksymiuk A, Goss G, Marshall E, Soulières D, et al. Randomized Phase IIB Trial of BLP25 Liposome Vaccine in Stage IIIB and IV non-Small-Cell Lung Cancer. J Clin Oncol (2005) 23(27):6674–81. doi: 10.1200/JCO.2005.13.011
163. ten Brinke A, Karsten ML, Dieker MC, Zwaginga JJ, van Ham SM. The Clinical Grade Maturation Cocktail Monophosphoryl Lipid A Plus Ifnγ Generates Monocyte-Derived Dendritic Cells With the Capacity to Migrate and Induce Th1 Polarization. Vaccine (2007) 25(41):7145–52. doi: 10.1016/j.vaccine.2007.07.031
164. Butts C, Socinski MA, Mitchell PL, Thatcher N, Havel L, Krzakowski M, et al. Tecemotide (L-BLP25) Versus Placebo After Chemoradiotherapy for Stage III non-Small-Cell Lung Cancer (START): A Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol (2014) 15(1):59–68. doi: 10.1016/S1470-2045(13)70510-2
165. Schimanski CC, Kasper S, Hegewisch-Becker S, Schröder J, Overkamp F, Kullmann F, et al. Adjuvant MUC Vaccination With Tecemotide After Resection of Colorectal Liver Metastases: A Randomized, Double-Blind, Placebo-Controlled, Multicenter AIO Phase II Trial (LICC). Oncoimmunology (2020) 9(1):1806680. doi: 10.1080/2162402X.2020.1806680
166. Yu S, Wang C, Yu J, Wang J, Lu Y, Zhang Y, et al. Injectable Bioresponsive Gel Depot for Enhanced Immune Checkpoint Blockade. Adv Mater (2018) 30(28):e1801527. doi: 10.1002/adma.201801527
167. Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, Kim YJ. Biodegradable STING Agonist Nanoparticles for Enhanced Cancer Immunotherapy. Nanomedicine (2018) 14(2):237–46. doi: 10.1016/j.nano.2017.10.013
168. Buss CG, Bhatia SN. Nanoparticle Delivery of Immunostimulatory Oligonucleotides Enhances Response to Checkpoint Inhibitor Therapeutics. Proc Natl Acad Sci U S A (2020) 117(24):13428–36. doi: 10.1073/pnas.2001569117
169. Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA Vaccine Drives Expansion and Efficacy of Claudin-CAR-T Cells Against Solid Tumors. Science (2020) 367(6476):446–53. doi: 10.1126/science.aay5967
170. Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, et al. Mutant MHC Class II Epitopes Drive Therapeutic Immune Responses to Cancer. Nature (2015) 520(7549):692–6. doi: 10.1038/nature14426
171. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity Against Cancer. Nature (2017) 547(7662):222–6. doi: 10.1038/nature23003
172. Ishihara J, Fukunaga K, Ishihara A, Larsson HM, Potin L, Hosseinchi P, et al. Matrix-Binding Checkpoint Immunotherapies Enhance Antitumor Efficacy and Reduce Adverse Events. Sci Transl Med (2017) 9(415):1. doi: 10.1126/scitranslmed.aan0401
173. Zinger A, Koren L, Adir O, Poley M, Alyan M, Yaari Z, et al. Collagenase Nanoparticles Enhance the Penetration of Drugs Into Pancreatic Tumors. ACS Nano (2019) 13(10):11008–21. doi: 10.1021/acsnano.9b02395
174. Le QV, Suh J, Oh YK. Nanomaterial-Based Modulation of Tumor Microenvironments for Enhancing Chemo/Immunotherapy. AAPS J (2019) 21(4):64. doi: 10.1208/s12248-019-0333-y
175. Bockamp E, Rosigkeit S, Siegl D, Schuppan D. Nano-Enhanced Cancer Immunotherapy: Immunology Encounters Nanotechnology. Cells (2020) 9(9):9–:10. doi: 10.3390/cells9092102
176. Ou W, Thapa RK, Jiang L, Soe ZC, Gautam M, Chang JH, et al. Regulatory T Cell-Targeted Hybrid Nanoparticles Combined With Immuno-Checkpoint Blockage for Cancer Immunotherapy. J Control Release (2018) 281:84–96. doi: 10.1016/j.jconrel.2018.05.018
177. Schumacher Ton N, Kesmir C, van Buuren Marit M. Biomarkers in Cancer Immunotherapy. Cancer Cell (2015) 27(1):12–4. doi: 10.1016/j.ccell.2014.12.004
178. Kappel C, Seidl C, Medina-Montano C, Schinnerer M, Alberg I, Leps C, et al. Density of Conjugated Antibody Determines the Extent of Fc Receptor Dependent Capture of Nanoparticles by Liver Sinusoidal Endothelial Cells. ACS Nano (2021) 15(9):15191–209. doi: 10.1021/acsnano.1c05713
179. Heineke MH, van Egmond M. Immunoglobulin A: Magic Bullet or Trojan Horse? Eur J Clin Invest (2017) 47(2):184–92. doi: 10.1111/eci.12716
180. Wen YM, Shi Y. Immune Complex Vaccination. Curr Top Microbiol Immunol (2019) 423:95–118. doi: 10.1007/82_2019_153
181. Sadiki A, Vaidya SR, Abdollahi M, Bhardwaj G, Dolan ME, Turna H, et al. Site-Specific Conjugation of Native Antibody. Antib Ther (2020) 3(4):271–84. doi: 10.1093/abt/tbaa027
182. Lin X, O'Reilly Beringhs A, Lu X. Applications of Nanoparticle-Antibody Conjugates in Immunoassays and Tumor Imaging. AAPS J (2021) 23(2):43. doi: 10.1208/s12248-021-00561-5
183. Bros M, Nuhn L, Simon J, Moll L, Mailänder V, Landfester K, et al. The Protein Corona as a Confounding Variable of Nanoparticle-Mediated Targeted Vaccine Delivery. Front Immunol (2018) 9:1760. doi: 10.3389/fimmu.2018.01760
184. Bednarczyk M, Medina-Montano C, Fittler FJ, Stege H, Roskamp M, Kuske M, et al. Complement-Opsonized Nano-Carriers Are Bound by Dendritic Cells (DC) via Complement Receptor (CR)3, and by B Cell Subpopulations via CR-1/2, and Affect the Activation of DC and B-1 Cells. Int J Mol Sci (2021) 22(6):1. doi: 10.3390/ijms22062869
185. Li M, Jiang S, Simon J, Paßlick D, Frey ML, Wagner M, et al. Brush Conformation of Polyethylene Glycol Determines the Stealth Effect of Nanocarriers in the Low Protein Adsorption Regime. Nano Lett (2021) 21(4):1591–8. doi: 10.1021/acs.nanolett.0c03756
Keywords: nanodrug, nanoparticle, T cell, regulatory T cell, immunotherapy, CAR-T cell, mRNA, melanoma
Citation: Haist M, Mailänder V and Bros M (2022) Nanodrugs Targeting T Cells in Tumor Therapy. Front. Immunol. 13:912594. doi: 10.3389/fimmu.2022.912594
Received: 05 April 2022; Accepted: 27 April 2022;
Published: 25 May 2022.
Edited by:
Robert Zeiser, University of Freiburg, GermanyReviewed by:
Rebecca Kesselring, University of Freiburg Medical Center, GermanyCopyright © 2022 Haist, Mailänder and Bros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias Bros, bWJyb3NAdW5pLW1haW56LmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.