 Arielle Raugh
Arielle Raugh Denise Allard
Denise Allard Maria Bettini
Maria Bettini- 1Department of Pathology, Microbiology and Immunology, University of Utah, Salt Lake City, UT, United States
- 2Translational Biology and Molecular Medicine Graduate Program, Baylor College of Medicine, Houston, TX, United States
The importance of regulatory T cells (Tregs) in preventing autoimmunity has been well established; however, the precise alterations in Treg function in autoimmune individuals and how underlying genetic associations impact the development and function of Tregs is still not well understood. Polygenetic susceptibly is a key driving factor in the development of autoimmunity, and many of the pathways implicated in genetic association studies point to a potential alteration or defect in regulatory T cell function. In this review transcriptomic control of Treg development and function is highlighted with a focus on how these pathways are altered during autoimmunity. In combination, observations from autoimmune mouse models and human patients now provide insights into epigenetic control of Treg function and stability. How tissue microenvironment influences Treg function, lineage stability, and functional plasticity is also explored. In conclusion, the current efficacy and future direction of Treg-based therapies for Type 1 Diabetes and other autoimmune diseases is discussed. In total, this review examines Treg function with focuses on genetic, epigenetic, and environmental mechanisms and how Treg functions are altered within the context of autoimmunity.
Introduction
At the crossroads of autoimmunity and health are regulatory T cells (Tregs) - a crucial immune cell involved in tolerance towards self and suppression of auto-antigen specific T cells. Tregs were first identified as a subpopulation of CD4 T cells that expressed the high affinity IL-2 receptor chain CD25 (1). However, it took several more years to identify the lineage specific transcription factor, Forkhead Box Protein 3 (FOXP3), that is a core regulator of suppressive Treg function (2–4), and acts as both a positive and negative regulator of gene expression (2–4). For example, FOXP3 directly upregulates CD25 expression, but suppresses IL-2 production (5). With the knowledge of how to identify Tregs and a basic understanding of their function, the field was propelled towards key findings regarding their developmental source, suppressive mechanisms, and therapeutic potential (6–8).
While the transcription factor FOXP3 was initially considered the “master regulator” of CD4 Treg development and function (7, 8) we now understand that a more complex system is at work. Rather than a single element, the Treg suppressive program is regulated by a combination of transcription factors, genetic and epigenetic elements, as well as tissue-microenvironment cues. Due to the complexity that underlies the Treg suppressive phenotype, it has become apparent that loss of Treg lineage commitment can occur through either loss of FOXP3 or through a number of alternative genetic and/or transcriptional dysregulations. However, the precise alterations that occur in autoimmune individuals that affect Treg-mediated tolerance, and how underlying genetic variations impact the development and function of Tregs during autoimmunity are only partially elucidated. Polygenetic susceptibly is a key driving factor of many autoimmune diseases. However, while genome wide association studies (GWAS) alone were suggestive, they were not sufficient to formally link Treg dysfunction to disease. Integration of GWAS studies with functional and other omics-data now implicate alterations or defects in regulatory T cell function in autoimmune pathogenesis (9–11).
In this review we consider the function and regulation of FOXP3 both during homeostasis and autoimmunity, as well as how FOXP3 and mutations in key Treg genes influence Treg function and stability. In addition, we examine epigenetic modifications that regulate FOXP3 activity and how inflammation in the surrounding tissue environment impacts Tregs. Finally, we feature how Treg based therapies for autoimmunity have changed since their inception as well as factors that need to be improved in order to make these therapies efficacious as treatments for autoimmunity.
Mutations in the FOXP3 gene
Immune dysregulation, polyendocrinopathy, enteropathy X-linked (IPEX) syndrome is a rare disorder that often results from mutations within the FOXP3 gene (2, 12, 13). However, in a cohort of 173 patients with IPEX syndrome symptoms, only 50.9% had direct mutations in FOXP3, underscoring the fragility of Treg function and its sensitivity to modulation of alternative pathways (14). Of the 85 patients that had no discernable FOXP3 mutation, 25% had mutations in key Treg genes such as LRBA, STAT1, STAT3, CTLA4, IL2RA, STAT5B, and DOCK8 which are responsible for various aspects of Treg differentiation and function. This suggests that although FOXP3 is critical for Treg mediated tolerance, other factors also participate in maintaining a functional Treg population (Figure 1A). For example, mice lacking the inhibitory molecule CTLA4 develop severe lymphoproliferative disease reminiscent of Foxp3 mutant mice (37, 38). Furthermore, another study of 15 IPEX patients bearing FOXP3 mutations revealed that Treg signature genes were still expressed, although with variable expression levels, indicating that Tregs can still maintain partial lineage characteristics after loss of FOXP3 expression (28). A transcriptomic disease signature was observed across both Tregs and conventional CD4 cells and was likely induced by global immune dysregulation. To put it differently, transcriptomic changes occur as a result of both cell-intrinsic and cell-extrinsic mechanisms, where Tregs first have dysregulated core genes involved in Treg stability and suppressive function (i.e. Il2ra, Tnfrsf4, Tnfrsf9, Tnfrsf18, Capg, Ikzf2, and Ctla4), which in turn alter the tissue environment, ultimately leading to enhanced broad transcriptomic changes affecting all T cells (28). In the absence of cell-extrinsic inflammatory signals in heterozygous mothers of IPEX patients, patient FOXP3 mutations impacted only a narrow set of genes directly under FOXP3 control. In combination, these observations point to limited direct impacts of FOXP3 mutations and an increased role for activation of inflammatory feedback loops leading to cumulative dysregulation of both regulatory and effector T cells. This further underscores the importance of Tregs’ ability to integrate information from their environment and alter their subsequent functions.
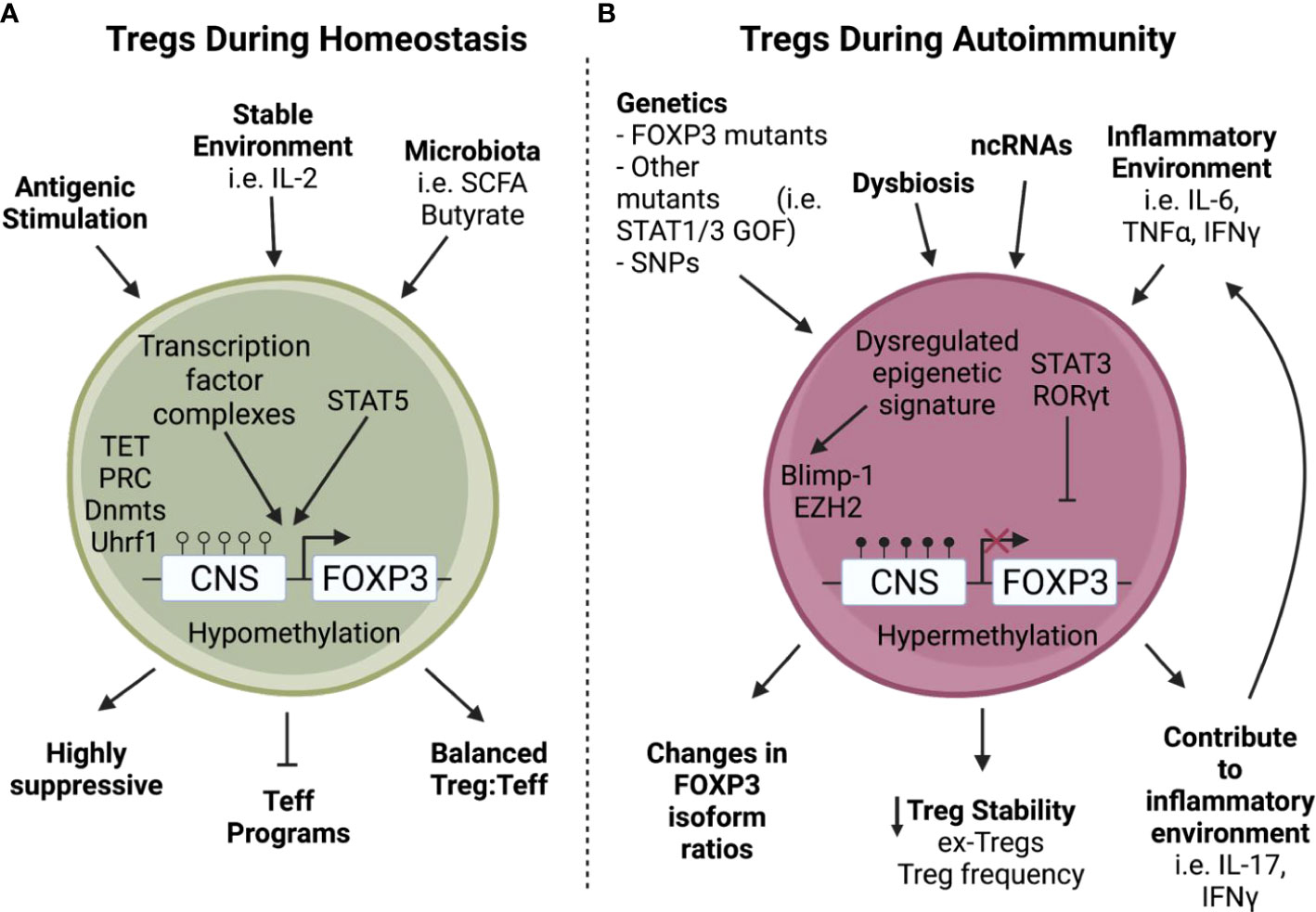
Figure 1 Tregs encounter increased stressors during autoimmunity. A) During homeostasis, Tregs are stimulated through TCR activation and proliferate using IL-2 from the surrounding environment (1, 5), allowing downstream transcription factor complexes to bind to a hypomethylated Foxp3 and enact critical Treg functions such as suppressive capabilities and repression of Teff programming (15–18). B) In contrast, Tregs found during autoimmunity often have intrinsic defects in addition to environmental stressors (2, 12–14, 19–23). For example, non-coding RNAs can be dysregulated during autoimmunity (24–26) leading to a dysregulated epigenetic signature with increased methylation of the Foxp3 Treg Specific Demethylated Region (TSDR) which can cause a loss of Foxp3 (27). In addition, a tissue environment rich in inflammatory cytokines can convert Tregs into Th17-like cells leading to the creation of ex-Tregs and decreased Treg stability (32–34). Furthermore, these stressors encountered during autoimmunity can also lead to perturbations in FOXP3 isoform ratios (35, 36) and expression of inflammatory cytokines (32, 33) which in turn leads to decreased Treg stability.
FOXP3 has four structural domains that are used to interact with diverse binding partners to exert transcriptional regulation. The examination of FOXP3 mutations in IPEX patients and in mouse models has provided important insights into the function of the specific domains within FOXP3. Mutations in FOXP3 identified in IPEX patients have been localized to all four structural domains of the transcription factor, although to some level they are concentrated in the DNA-binding FKH domain (29). For example, identification of a patient with a mutation within the dimerization motif in the FKH domain of FOXP3 showed that FOXP3’s domain swap interface is crucial for restricting Th2 immune responses in Tregs. When the domain swap interface is mutated, FOXP3 interacts with Th2 specific loci inducing expression of Th2 signature cytokines that are normally repressed in Tregs (29).
While most mutations within FOXP3 result in systemic immune dysregulation and global autoimmune manifestations, partial disruption of interactions between FOXP3 and its binding partners can have understated effects on FOXP3-driven gene activity. Foxp3-GFP reporter mice that express GFP fused to Foxp3 at its N-terminus provided a system to observe how subtle changes can have disease-specific impacts (30, 31). The Foxp3-GFP reporter mouse shows no abnormal Treg function on the C57BL/6 genetic background; however, when backcrossed to the NOD autoimmune-susceptible strain it resulted in rapidly accelerated autoimmune diabetes development (30, 31). Foxp3-GFP showed reduced interaction with several binding partners involved in Foxp3 gene regulation, suggesting Foxp3 instability and loss of Treg function under increased inflammatory stress (30). Interestingly, the GFP-modified Foxp3 was protective in a model of arthritis due to disruption in HIF1α binding and increasing Foxp3 interactions with Interferon Regulatory Factor 4 (IRF4) leading to improved Treg control of Th2 and Th17 responses (31). Perturbations in Treg function were observed in autoimmune prone, but not autoimmune resistant mice suggesting that the genetic or inflammatory environment has a direct influence on the ultimate functionality of Tregs. The loss of Treg stability under inflammatory conditions has been a concern in situations of chronic autoimmunity and has been directly observed in mouse models of autoimmune diabetes, multiple sclerosis, and rheumatoid arthritis (32–34). Loss of Foxp3 expression in these situations resulted in the formation of ‘ex-Tregs’ that acquired an effector pro-inflammatory phenotype (Figure 1B). However, such ex-Tregs have not been directly observed in human autoimmune conditions, and if they exist are more likely to be localized directly in inflammatory tissues.
Polygenetic susceptibilities and Treg function
Autoimmune manifestations that result from direct mutations of the FOXP3 gene and related Treg functional genes, such as CTLA4, can be traced to loss of Treg numbers and/or function (38–40). However, it has been more challenging to infer the target immune cell population in polygenetic autoimmune susceptibilities. HLA alleles associated with autoimmunity contribute the largest risk for development of autoimmunity, including type 1 diabetes (9). For some HLA alleles, such as DQ8 and DQ2, loss of self-tolerance is thought to be prompted by the structure of the peptide binding grooves, which lead to increased selection or peripheral activation of autoimmune T cells (41). Many other T1D associated SNPs are located in close proximity to immune genes, such as CTLA4, and components of the IL-2 and TCR signaling pathways among others. The cytokine IL-2 binds to CD25 (IL2RA) and signals through STAT5 to regulate FOXP3 expression in Tregs (Figure 1A) (42). Complete IL2RA deficiency can lead to severe autoimmunity with IPEX like symptoms (43), and IL2RA variants have been associated with reduced Treg numbers, suboptimal Treg function, and an increased risk for development of T1D (44). Since HLA alleles, CTLA4, and IL2RA among others are implicated in both T effector (Teff) and Treg function, the ultimate impact on either population is difficult to determine. Nevertheless, several T1D related SNPs have been connected to Treg function (45–47) and the Treg to T effector cell ratio (48). Additionally, evidence suggests that Tregs from T1D patients may not be as suppressive and may have a more inflammatory phenotype (49). Therefore, there is a growing consensus that Treg function is altered in T1D, and Treg dysregulation might be in part due to genetics.
In many other autoimmune and inflammatory disorders it is not so clear whether there is an underlying defect in regulatory T cells. Many polymorphic variants are shared between several autoimmune diseases, including PTPN22 (TCR signaling), TKY2 (cytokine signaling), and TNFAIP3 (TNF signaling) among others (50, 51). These variants point to genes besides FOXP3 that could influence T cell and Treg function during autoimmunity. For example, in the context of rheumatoid arthritis there is still an ongoing debate regarding Treg dysfunction. There are a number of conflicting observations on whether Treg frequency decreases or remains stable (52–55), whether there are changes in Treg suppressive capability, or the relative expression of Treg associated regulatory molecules, such as CTLA-4 (52, 56, 57). The markers used to define Tregs as well as disease severity should be carefully considered in these studies, and could potentially explain some of the discrepancies in observations. Nevertheless, the lack of clear loss in Treg number or function in RA supports the idea that Treg dysfunction is disease specific.
The majority of disease-associated genetic variants defined by GWAS studies are found in non-coding areas of the genome, which presents a challenge in determining the ultimate relationship between SNPs, gene expression, and downstream effects on cellular function. Importantly, many disease associated SNPs are mapped to regulated chromatin regions and enhancers, i.e. epigenetically regulated transcription factor binding sites (58–60). Several mechanisms for non-coding regions’ impact on immune genes have been described. These range from direct disruption of transcription factor binding at SNPs located within enhancer regions (61) to distal effects mediated by genomic misfolding and interconnection of enhancers in 3D chromatin organization (62). Recent studies have coupled epigenetic profile analyses of isolated T cell populations to determine the effects of particular SNPs on chromatin accessibility in the context of T cell populations. Interestingly, the chromatin accessibility at these loci is preferentially associated with naive and activated Tregs, rather than conventional T cells (10, 63, 64). These observations imply that genetic susceptibility disproportionally effects Treg function compared to effector T cells in the context of autoimmunity. Based on cumulative genetic studies we can infer that genetic polymorphisms have connections to FOPX3+ Treg function and predisposition to autoimmunity (9, 44–48). Therefore, it is critical to examine the transcriptional regulation of the Treg lineage and the factors that impinge on Treg stability.
Genetic regulation of the FOXP3 locus
Genetic control and regulation of FOXP3 plays a major role in Treg development and function during both homeostasis and disease. While several FOXP3 isoforms have been identified in humans, there are two distinct isoforms that are necessary for optimal Treg function; the full length FOXP3 isoform and the alternatively spliced FOXP3 isoform which lacks exon 2 (FOXP3Δ2) (65). The full length FOXP3 isoform has recently been identified as a critical component of regulating FOXP3 activity and maintaining Treg stability (66). FOXP3Δ2 on the other hand, has been shown to be upregulated during Treg activation, and is linked to transcription of the transmembrane protein, Glycoprotein A Repetitions Predominant (GARP), which tethers TGFβ to the cell membrane and potentiates cell-contact dependent TGFβ function (67, 68). While both isoforms are necessary for optimal Treg function (65), regulation of FOXP3 isoform ratios appears to alter the disease course in some autoimmune diseases (Figure 1B) (35, 36).
Regulation of the FOXP3 locus is multifaceted and involves several key enhancer regions that recruit a number of regulators that control Treg development and stabilize the Treg lineage (Figure 1A). The FOXP3 locus has four enhancer regions known as conserved non-coding sequences (CNS; CNS0, CNS1, CNS2, and CNS3) that work in tandem to drive FOXP3 transcription and downstream gene expression necessary for Treg stability (15–17, 69, 70). These enhancer regions are embedded throughout upstream-promoter and intronic regions of FOXP3 (71, 72) and alter FOXP3 transcription and activity by controlling methylation status, chromatin accessibility, and act as docking sites for unique sets of binding partner complexes (15–17, 73, 74). For example, the transcription factor SATB1 binds CNS0 (18) which along with the transcription factor HIVEP2 co-regulates pathways involved in Treg immunosuppression (75). SATB1 is an important transcription factor in regulating T cell differentiation (76); however, it is repressed by FOXP3 in Tregs to balance Treg proliferation and function. Loss of SATB1 increases Treg frequency but diminishes Treg suppressive function (77, 78). In Tregs, SATB1 is epigenetically regulated through histone trimethylation and acetylation changes, as well as by microRNAs such as mir-155, mir-21a, mir-7, mir-34a, and mir-18a (79). During development, IL-2 signaling directs the pioneer factor SATB1 to bind nucleosome dense regions in Tregs leading to chromatin remodeling and accessibility of critical Treg signature genes (77). This is aided by the transcription factor Foxp1 which enhances IL-2 signaling and Foxp3 expression (78), making IL-2 signaling a critical step in differentiating Tregs from CD25+Foxp3- Treg precursors in the thymus (15).
CNS1 is primarily associated with peripheral induction of Tregs and is bound by several transcription factors including AP-1, NFAT, Foxo1, Hhex, Batf3, and importantly Smad3 induced by TGFβ signaling. Batf3 represses FOXP3 expression and downregulates the differentiation of naïve CD4 T cells into Tregs (80). In addition, Hhex (Hematopoietically expressed homeobox) is a transcription factor that binds to CNS1/CNS2 and represses FOXP3 expression; particularly under inflammatory conditions (81). CNS2 is a critical response element during thymic Treg development, and is bound by Ets-1, CREB, Stat5, NFAT, c-Rel, Runx, Foxp3, and AP-1. Importantly, CNS2 contains the Regulatory T cell Specific Demethylated Region (TSDR) (82), which maintains FOXP3 expression in Tregs and allows FOXP3 to positively regulate its own transcription even in the absence of TCR signaling (18). Lastly, CNS3 is another region important for the development of thymic Tregs and can bind Foxo and c-Rel (17, 83–85). These transcription factor binding complexes can alter FOXP3 activity, downstream targets of FOXP3, and additional pathways involved in Treg function (75). In addition, CNS regions CNS0 and CNS3, were recently determined to be sites that help initiate Treg development when bound by transcription factor complexes that allow chromatin remodeling and drive FOXP3 transcription (16). Beyond FOXP3 enhancer regions, transcription of Treg signature genes is also regulated by cooperation of Foxp3 and one of the five transcription factors Eos, IRF4, GATA-1, Lef1, and Satb1. These cofactors, referred to as the “quintet”, enhance Foxp3 activity by ‘locking in’ and stabilizing Foxp3 to its binding sites (18).
Furthermore, demethylation status of the FOXP3 TSDR was determined to be key for maintaining FOXP3 expression and stabilizing Treg identity. However, while demethylation of the TSDR is enough to stabilize FOXP3 expression in Tregs, it is not enough to confer suppressive function (86). This suggests that Treg suppressive function is not solely linked to FOXP3 expression, and that additional transcription factors are required. As an example, the transcription factor Helios is expressed in approximately 70% of Tregs and helps to maintain Treg stability by controlling certain aspects of Treg function, differentiation, and survival (87). However, mice lacking Helios are still able to convert naïve T cells into functional Tregs; indicating a level of redundancy in transcriptional regulation of Treg function (88).
Heterogeneity within the Treg population
The FOXP3+ Treg population exhibits phenotypic and functional complexity driven by tissue and context specific transcription factors. Similar to conventional T cells (Tconv), the majority of lymphoid derived thymic Tregs maintain a non-activated phenotype, characterized by expression of CD62L, CCR7 and TCF1 (a transcription factor associated with stemness) (89). However, Tregs can also be derived from naïve CD4 T cells in the periphery through TGFβ signaling (90, 91) (Figure 2). TGFβ signal can be provided in the form of latent TGFβ on the cell surface of tTregs, which leads to induction of additional Foxp3+ T cells (pTregs), in a process that is described as “infectious tolerance” (Figure 2) (98). Upon differentiation from naïve T cells, in vivo induced pTregs repress CD4 effector T cell programming, stabilize expression of FOXP3, and maintain a fully demethylated TSDR, similar to tTregs (92–94). In addition, recent work suggests that type 1 interferons can stabilize expression of STAT3, STAT5, and FOXP3 in peripheral CD4 T cells allowing their differentiation into pTregs (Figure 2) (99). However, Type 1 interferons have been shown to have opposing effects on Tregs depending on the timing of exposure. In the short-term, Type 1 interferons lead to decreased Treg frequency and function; however, in the long run they can stabilize expression of FOXP3 and promote Treg expansion (100). Nevertheless, since no definitive markers of pTregs have been identified, the functional importance of pTregs during autoimmunity is still heavily debated (101–103).
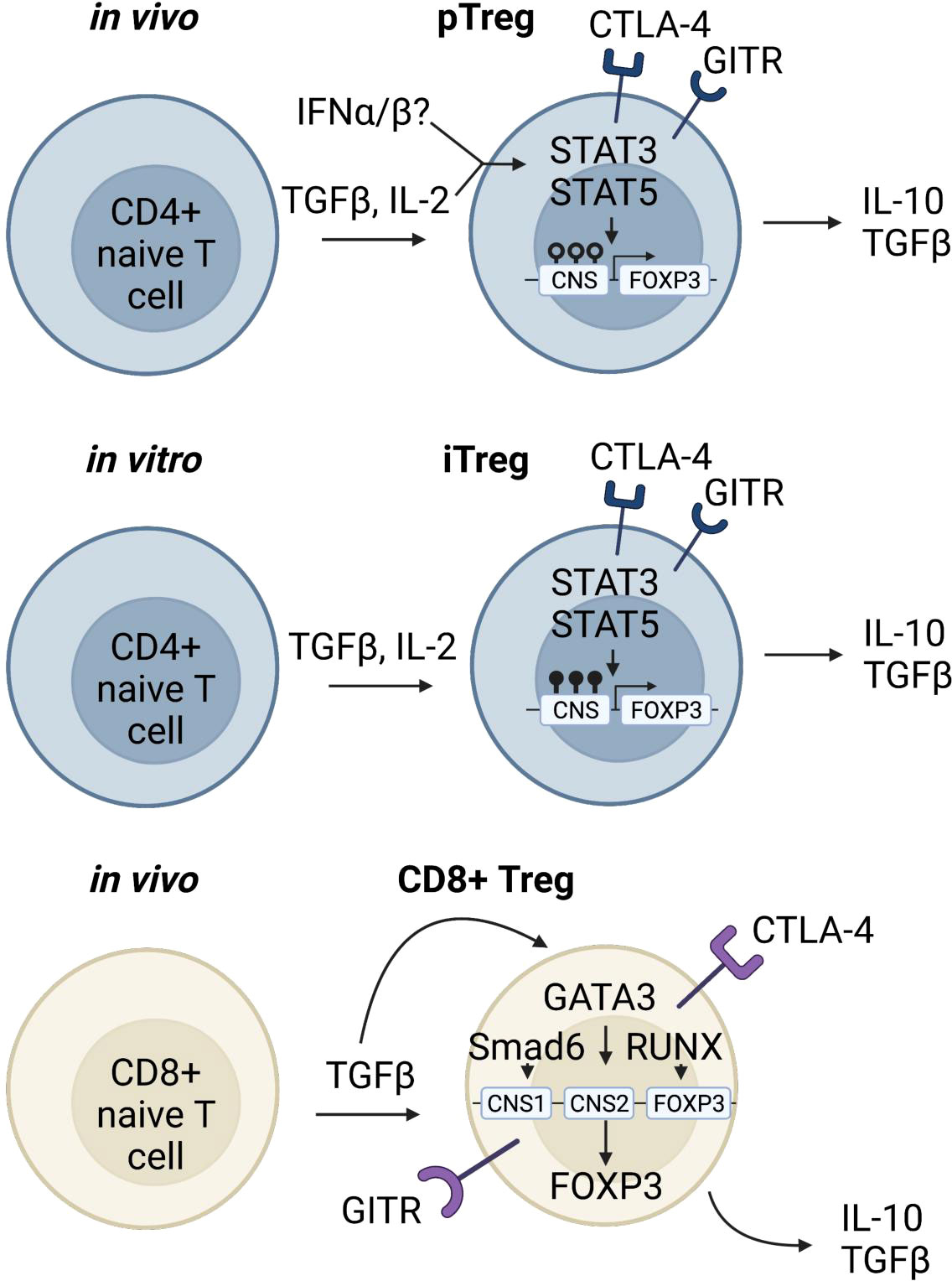
Figure 2 Induced regulatory T cells. In the periphery, T lymphocytes can encounter stimuli that turn on downstream signaling leading to genetic reprogramming and a regulatory phenotype (90, 91). pTregs which have the capacity to be immunosuppressive and traffic to inflamed tissue sites are differentiated from CD4 naïve T cells under inflammatory conditions (92–94). A rare and unique subpopulation of Tregs is the CD8+Foxp3+ Treg. In the periphery, when naïve CD8 Tconv cells encounter TGFβ, pSmad3 binds to CNS1 of Foxp3, and along with transcription factors Runx3 and Gata3 promote expression of Foxp3 (95–97). CD8+Foxp3+ Tregs express similar markers as CD4 Tregs and have immunosuppressive functions.
The widely accepted approach to induce Treg differentiation in vitro relies on a combination of TCR ligation in the context of TGFβ and high concentrations of IL-2 (Figure 2) (104). While studies show that iTregs have suppressive function both in vitro and in vivo, their long-term stability is more controversial (93, 105). Stability is measured by quantification of methylation at the TSDR region, and TCR and IL-2 stimulation can promote demethylation of TSDR in iTregs, thus stabilizing the lineage (106, 107). However, iTregs that have a hypermethylated TSDR can still be functional (108, 109).
As Tregs migrate from lymphoid organs to peripheral tissues they accumulate a common tissue-resident signature and are further differentiated into unique phenotypes dependent on tissue-specific signals. These tissue-resident Tregs (tissue Tregs) have the potential to be derived from both tTregs and pTregs, with the change from a lymphoid-resident phenotype to a tissue-resident phenotype, a process that is mediated by a combination of transcriptional regulators (Figure 3) (111, 112). In the spleen and lymph nodes, the transcription factor BATF drives the stepwise progression of tissue Treg precursors into tissue Tregs by increasing chromatin accessibility of tissue specific Treg genes (113). Repression of BATF impairs tissue Treg function and contributes to induction of autoimmunity (120). In addition, tissue Tregs often exhibit specialized functions associated with upregulation of tissue specific transcription factors, such as PPARγ in visceral fat tissue and Eos in the skin (89, 110). Although, more recently PPARγ has been linked to skin and liver Tregs as well (121, 122). Interestingly, upregulation of IL-33R (ST2) and its downstream target cytokine, amphiregulin, is a trait shared among many Tregs that are transitioning towards tissue phenotype; indicative of an acquired ability to participate in tissue repair in response to inflammation or injury (Figure 3) (89, 123–125). The growth factor amphiregulin is expressed by tissue Tregs in response to alarmin cytokines released by injured tissue cells, including IL-33 (114–116). The ramifications of this discovery show that Tregs upregulate receptors necessary to sense the tissue microenvironment in order to rapidly respond to environmental changes.
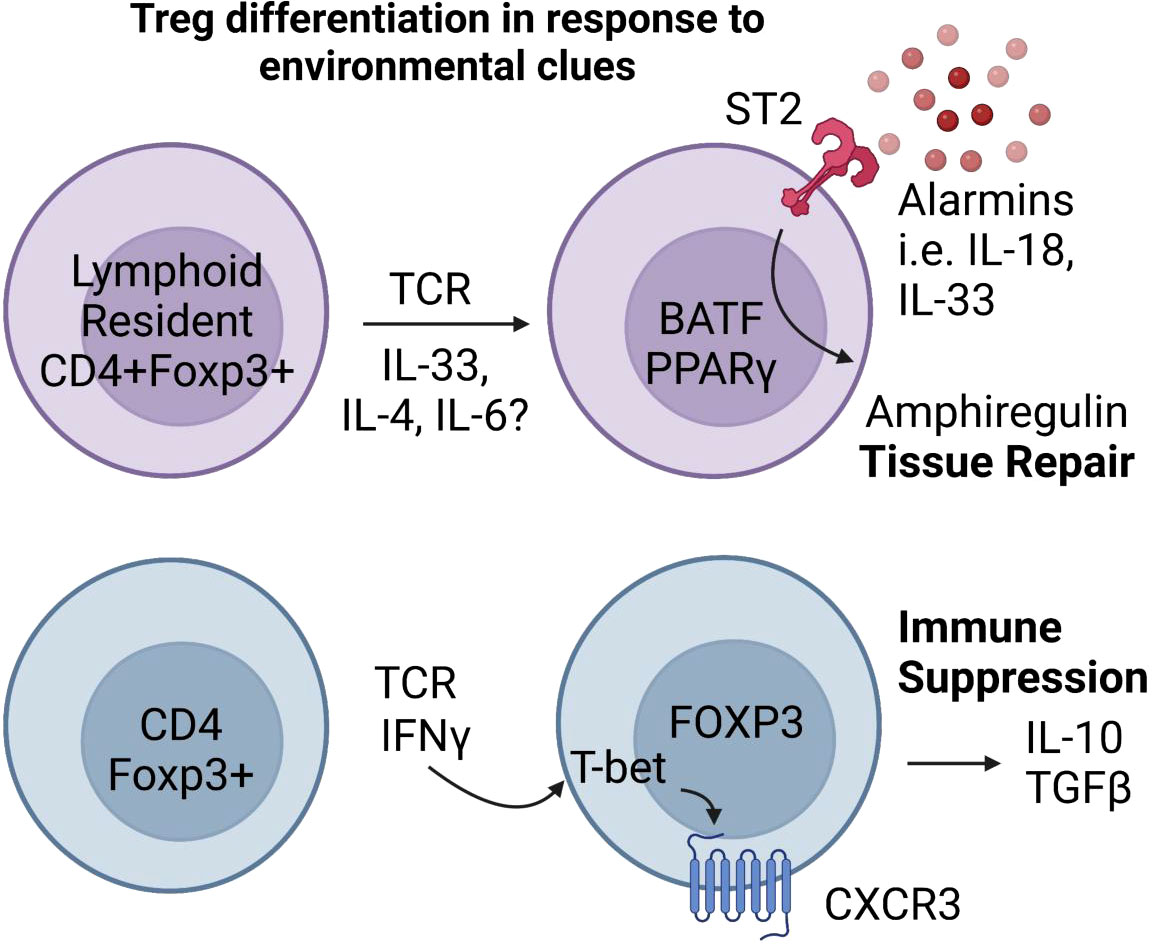
Figure 3 Inflammatory and tissue specific signals shape Treg responses. Tissue Tregs are poised to respond to inflammatory tissue environments. In the presence of alarmin cytokines, tissue Tregs expressing the transcriptional regulators Batf and PPARγ (89, 110–113) secrete the wound repair factor Amphiregulin (114–116). Inflammatory cytokines that normally drive T-helper lineage specific factors like T-bet can similarly induce T-helper transcription factor expression in Tregs. IFNγ and TCR stimulation induce T-bet expression in Foxp3+ Tregs, which provides them with increased ability to suppress Th1 effector T cells (117–119).
In addition, CD4 T-helper lineage defining transcription factors can shape Treg responses during inflammation. A prime example of this is T-bet, which in addition to being the major Th1 lineage-defining transcription factor, provides Tregs with increased ability to suppress Th1 effectors (117). T-bet is upregulated in Tregs in response to IFNγ and TCR ligation and is directly responsible for the upregulation of chemokine receptor CXCR3, allowing Tregs to traffic to sites of inflammation (Figure 3) (118, 119).
Furthermore, CD8+FOXP3+ regulatory T cells constitute a smaller proportion of the Treg compartment but are still functional contributors to the regulatory arm of the immune system. They are transcriptionally similar to CD4+FOXP3+ T cells; and although they seem to be less potent than CD4+FOXP3+ Tregs they have been shown to be effective in models of GVHD and lupus (126, 127). In contrast to CD4+FOXP3+ T cells, CD8 Treg suppressor programs are controlled by the transcription factors RUNX3 and GATA3 (Figure 2). In naïve CD8 T cells, GATA3 binds to the CNS1 region of FOXP3 to inhibit FOXP3 expression, however in CD8 Tregs, GATA3 binds to the CNS2 region to maintain FOXP3 expression (95). Furthermore, RUNX3 binds to the promoter region of FOXP3 to initiate transcription, and under conditions with high levels of TGFβ, Smad3 is phosphorylated and binds to CNS1 inducing FOXP3 expression (Figure 2). nCD8+CD25+ Tregs are also somewhat functionally similar to CD4+FOXP3+ Tregs as they express suppressive markers such as GITR and CTLA4 (96), as well as cytokines such as IL-10 and TGFβ (Figure 2) (95, 97).
Epigenetic regulation of Treg lineage
Epigenetic regulation of gene expression can have major consequences for cells. During development, thymocytes that are fated to become Tregs undergo a series of epigenetic modifications to CNS regions of FOXP3 to activate transcription of FOXP3 and downstream Treg signature genes. At the same time, T cell effector cellular differentiation programs are repressed (Figure 1A) (128, 129). Stepwise histone tail acetylation at the FOXP3 promoter initiates chromatin remodeling and FOXP3 transcription (130). FOXP3 histone tail acetylation allows ten-eleven translocation (TET)-mediated DNA demethylation to occur in the CNS2 region of the FOXP3 locus and maintains FOXP3 transcription by increasing chromatin accessibility; thus, removing the need for further histone acetylation (130). Positive regulation of Treg lineage is also accomplished by repression of alternative T-helper lineage programs. Polycomb-repressive complexes (PRC) are multi-protein enzymes that transcriptionally silence genes through histone H2A ubiquitylation and H3K27 methylation (131). PRCs silence Th17 related genes and enhance the Wnt signaling pathway to favor Treg development and stability (132, 133).
Once chromatin remodeling and access to core Treg genes is achieved, additional epigenetic changes occur that maintain stable chromatin accessibility. For example, the protein ubiquitin like with PHD and ring finger domains 1 (Uhrf1) is an epigenetic regulator that recruits DNA methyltransferases (Dnmt) such as Dnmt1, Dnmt3a, and Dnmt3b to stabilize methylation patterns (134–137) during Treg development (Figure 1A), as well as following TCR engagement in the periphery (138). Similarly, ablation of Dnmt1 in Tregs severely impairs their function through global changes in methylation (139).
However, many of these normal epigenetic modifications fail to function and/or maintain Treg stability during autoimmunity, as Tregs derived from autoimmune patients often have epigenetic and transcriptomic changes. For example, effector Tregs derived from juvenile idiopathic arthritis patients present with consistent changes that include methylation changes in enhancer regions, as well as upregulation of functional and core Treg genes (140). The upstream regulatory elements that are dysregulated can be numerous due to the complexity of epigenetic mechanisms that control Treg lineage. For instance, in a model of multiple sclerosis, methylation of CNS2 normally repressed by Dnmt3a and controlled by Blimp1 is disrupted and leads to loss of Treg identity (141). There are indications that similar disruptions occur in human autoimmunity. The chromatin-modifying enzyme Ezh2 maintains Treg identity after activation, and its reduction is observed in RA patients (Figure 1B) (142, 143). Moreover, tissue antigens themselves can produce variable epigenetic responses in antigen-specific Tregs. For example, Tregs expanded in vitro using APCs expressing insulin B:9-23 peptide were found to have transcriptomic and epigenetic signatures representative of highly suppressive Tregs compared to Tregs expanded using whole insulin peptide (144). This provides evidence for the importance of T cell receptor signaling and antigen specificity in the development of optimally functional and stable Tregs.
Understanding the epigenetic changes that Tregs undergo during chronic inflammation is important for gaining new targeting strategies for autoimmune therapies. Tregs function differently during homeostasis and acute infection compared to chronic inflammatory conditions, implying context and inflammation specific Treg functional programs potentially regulated at the epigenetic level (19–21). In addition, Treg frequency and core signature gene expression mainly associated with DNA accessibility, transcription, translation, signal transduction, and cytokine receptors are prone to changing throughout the span of autoimmune disease pathology (145).
Microbiota have also been shown to influence Treg function and stability. Interestingly, some microbial-derived signals directly engage with Treg epigenetic elements (146). While still a new field of study, there is increasing evidence that short chain fatty acids (SCFA), such as butyrate, can be produced by commensal bacteria and positively regulate Treg differentiation (147). This appears to be CNS1 dependent, and is mediated by enhanced acetylation at the FOXP3 locus (148, 149). However, it is still unclear whether SCFAs are the key signal for pTreg induction in the mesenteric lymph nodes (150). Importantly, gut dysbiosis is a feature of several autoimmune diseases such as IBD, SLE, RA, Graves’ Disease and T1D, and it might contribute to disbalance of immune homeostasis (Figure 1B) (151–156).Thus, it is relevant to ask if inflammation or other microenvironmental cues at tissue sites can play a direct role in changing Treg function through epigenetic and/or transcriptomic changes.
Shifting the Treg/Teff equilibrium
A major question that remains regarding Tregs in autoimmunity is how they inevitably fail throughout the course of disease. One hypothesis is a decrease in the ratio of Tregs : Teffs, which can be seen in several autoimmune diseases (157–160). The shifts seen in this equilibrium could be the result of direct mutations in FOXP3 such as in IPEX syndrome, other polymorphisms that affect Treg function or stability, or could occur due to the influence of the surrounding tissue environment, since normal cellular mechanisms of differentiation and function that work to maintain the Treg : Teff balance are often dysregulated during autoimmunity (Figure 1).
For example, a major pathway that diverts CD4 T cells away from Treg differentiation and towards a Th17 program is the IL-6/STAT3 pathway commonly associated with inflammation. Dysregulation of the IL-6/STAT3 pathway seen in patients with gain of function mutations in STAT3 is correlated with increased susceptibility to T1D; most likely related to the Treg : Th17 imbalance seen in these patients (22). Inflammatory environments high in IL-6 have been shown to increase the Th17 transcription factor RORγt in both tTregs and pTregs (23), and lead to the creation of ex-Tregs that are capable of secreting inflammatory cytokines (32, 33). These ex-Treg cells lose FOXP3 expression and convert into pathogenic Th17 cells capable of producing IFNγ and destabilizing Tregs in the surrounding environment (33, 161–163). Formation of ex-Tregs promotes a shift in the Treg : Teff ratio skewed towards destructive Teff cells. Treg-derived IFNγ can also act as a negative feedback regulator of Treg stability and lead to further loss of suppressive function, indicating an important role for the environment in continually shaping and sometimes destabilizing Treg responses (164). Additionally, antigen exposure and/or scarcity can impact the balance between Th17 and Treg differentiation (165). Recent evidence suggests that T cells can trogocytose MHCII molecules from APCs displaying specific antigens, and subsequently display the MHCII to other antigen-specific T cells. When differentiation is favored towards Tregs there is a high APC:T cell ratio, however, when the reverse occurs (high T cell:APC ratio) differentiation is skewed towards Th17 cells (166).
These inflammatory pathways implicated in Treg lineage destabilization can be effectively targeted for therapeutic purposes. Small molecule targeted inhibition of IL-6 or STAT3 promotes Treg development and leads to the establishment of homeostasis between Treg and Th17 cells in a model of multiple sclerosis (MS) (167, 168). MS patients often exhibit dysregulated cytokine levels - including an increase in IL-6 in their cerebral spinal fluid, which could be targeted with the goal of shifting the balance between anti- and pro-Treg micro-environment cues (169). However, blocking the IL-6R in early onset T1D patients with a mAb did not prevent or delay beta cell loss (170), illuminating the limitations of therapies that target a single inflammatory pathway.
Non-coding RNAs during autoimmunity
Evidence shows dysregulated microRNA (miRNA) and long non-coding RNA (lncRNA) expression is also associated with many autoimmune diseases (24, 25). miRNAs are small non-coding RNAs that regulate proteins largely by binding to the 3’ UTR of mRNA and preventing translation, or by targeting the mRNA for degradation. Similarly, lncRNAs modulate chromatin architecture and mRNA stability (171). Both miRNA and lncRNA can impact Treg genetic regulation by altering expression of epigenetic regulators, directly targeting FOXP3, and by altering the signaling pathways that allow Tregs to respond to the surrounding microenvironment. Through these mechanisms, miRNAs influence Treg frequency and modify Treg functional capabilities.
Further, miRNAs can play an important role in regulating Treg epigenetics. For example, miR-142-3p, which is upregulated during T1D in humans and mice, can bind to lysine demethylase 6A (KDM6A) and demethylate H3K27me3 in Tregs leading to increased autophagy, decreased apoptosis, and increased Treg function (172). While two different Treg-specific miR-142 deficient mouse models showed impaired Treg function, whether or not Treg frequency is altered in these mice remains unclear since the two studies showed conflicting results (173, 174). Furthermore, miR-142-3p function in Tregs may operate through multiple pathways as miRNA142-3p also destabilizes Tregs by interacting with TET2 to alter Treg methylation in both humans and mice (27).
In addition to modifying Treg epigenetic signatures, non-coding RNAs can target FOXP3 and other Treg signature genes. In humans, several miRNAs including, mi-R206, miR-133a, miR-133b, and miR-31 have been identified that directly target the 3’ UTR of FOXP3 mRNA leading to FOXP3 translational downregulation (175, 176). miR-31 is among the better studied miRNAs that target FOXP3 and has been implicated in numerous autoimmune diseases. In murine models of autoimmunity, mi-R31 is upregulated upon TCR stimulation, but is inhibited by TGFβ/NF-κB signaling (177). miR-31 functions by directly targeting FOXP3, and also acts indirectly by promoting HIF1α and downregulating Nrp1 and retinoic acid-inducible protein 3 (Gprc5a) (178). miR-31 also inhibits carcinoembryonic antigen related cell adhesion molecule 1 (CEACAM1)-S, which represses Treg development in a model of murine liver autoimmunity but promotes Treg development in peripheral blood mononuclear cells (PBMCs) isolated from systemic lupus erythematosus (SLE) patients (179). The ultimate effect of miR-31 on Treg development and frequency depends on the balance between its inhibitory and enhancer functions. However, the factors that determine this require further investigation.
In addition, miRNAs can also influence FOXP3 by targeting pathways that regulate its expression. miR-21, which is among the best studied miRNAs that regulate Tregs in this manner, is dysregulated in several autoimmune disorders in both humans and mice. miR-21 acts indirectly to positively regulate Foxp3 expression (180); however, in autoimmunity, reduced miR-21 expression is correlated with increased STAT3 and reduced STAT5 and Foxp3 expression (26, 181–183). miR-21 directly targets STAT3 resulting in its downregulation and subsequently reduces effector molecules IL-17 and IL-22 (182, 184). Maresin 1 (MaR1) and the EGF/c-Jun pathway have both been shown to induce miR-21, restore Treg : Teff ratios through FOXP3 induction, and reduce autoimmunity (183, 185).
While some studies show that transfection of naïve human CD4 T cells with miR-21 is sufficient to induce Treg development by increasing Foxp3, TGFβ, and IL-10, another study found that miR-21 promotes RORγt and suppresses Foxp3 and IL-10 (180, 186, 187). Indeed, Treg specific depletion of miR-21 in mice induced the expression of both IL-17 and IL-10 indicating that miR-21 may play a role in opposing pathways (184). In line with these opposing observations, increased miR-21 expression inhibited FOXP3+ Tregs in human gastric cancer (188) whereas it induced FOXP3 in human and mouse autoimmunity (182, 183). Interestingly, LPS stimulation of PBMCs from RA patients down-regulated miR-21; however, PBMCs from healthy controls responded to LPS in the opposite fashion by up-regulating miR-21 (175). The opposite regulation and effects of miR-21 in autoimmune patients compared to healthy controls and in cancer settings suggests that a complex network of factors determines whether miR-21 promotes or inhibits Treg stability and function.
Furthermore, lncRNAs can also modulate Treg epigenetics. For example, FOXP3 long intergenic noncoding RNA, Flicr, reduces chromatin accessibility to the CNS3/Accessible Region 5 in mature Tregs and represses FOXP3 expression in both humans and mice. Knockout of Flicr on the NOD mouse background results in stabilized Foxp3 expression with a reduction in diabetes incidence (25). Additionally, in both humans and mice the lncRNA lnc-Smad3 interacts with the histone deacetylase HDAC1 to silence SMAD3 transcription. Upon TGFβ stimulation SMAD3 inhibits lnc-Smad3, thus allowing for greater SMAD3 transcription (189).
lncRNAs are also integral in regulating key Treg transcription factors. For example, Homeobox D gene cluster antisense growth-associated long noncoding RNA (HAGLR) is another lncRNA involved in autoimmunity. In human Tregs, HAGLR suppresses RUNX3 expression resulting in reduced Treg frequency (190). Additionally, lncRNA DQ786243 induces FOXP3 expression in human Tregs and promotes Treg suppressive function (191).
Noncoding RNAs are also important participants in regulating and responding to environmental cues. In inflammatory environments rich in IL-6 and TNFα, NF-κb upregulates the expression of miR-34a in humans and mice (192), which attenuates FOXP3 expression and can result in a shift of the Treg : Teff ratio. miR-124, which is dysregulated in numerous autoimmune diseases (24, 26), inhibits IL-6/STAT3 signaling and promotes Treg development (193). Similarly, miR-146a normally targets STAT5b to enhance Treg function and differentiation, but loss of miR-146a during inflammatory conditions leads to reduced FOXP3 expression and reduced Treg frequency. IL-2 represses Flicr thus removing Flicr’s inhibition of FOXP3 expression, while TGFβ inhibits the Foxp3-repressive noncoding RNAs miR-31 and lnc-Smad3 (194). The anti-inflammatory molecules MaR1 and EGF promote miR-21 (183, 185). The field of noncoding RNAs and their role in Treg development and function is growing, but additional studies are still required to reveal the full extent they may have in autoimmunity.
Treg-based therapies
With the central role for regulatory T cells in autoimmune diseases, it is unsurprising that investigation is underway as to how Tregs can be used therapeutically (195). One example is the use of Tregs as a treatment for T1D. The current standard of care for T1D patients is exogenous replacement of insulin. When managed well, the administration of synthetic insulin results in more stable blood glucose levels but does not entirely negate the risk of comorbidities (196). Thus, having an immunomodulatory therapy that prevents, attenuates, or reverses the course of pancreatic islet destruction is crucial.
Altering Treg to Teff ratio using immunomodulation
Due to potential imbalance in the Treg : Teff homeostasis seen during T1D, much attention has been focused on changing the ratio either by depleting effector T cells or expanding the Treg population. One of the earliest immunomodulatory therapies attempted in T1D patients was the use of anti-CD3 antibodies (197–199). Even a single dose of anti-CD3 lessened T1D progression and allowed reduction or complete withdrawal from exogenous insulin replacement therapy in some patients (197). Following initial positive observations in early diagnosed patients, anti-CD3 mAb therapy was used in a clinical trial of relatives of T1D patients who had at least two diabetes related auto-antibodies and confirmed dysglycemia prior to the start of the trial (200). A subgroup of participants in the treatment arm of the trial displayed delayed onset of T1D compared to controls, showing that modulation of T cell function after loss of tolerance but prior to overt disease can influence disease outcomes. Anti-CD3 antibodies appear to function by altering the ratio of Tregs : Teffs, as Teffs are susceptible to depletion by anti-CD3, whereas Tregs are more resistant (201). Additionally, following anti-CD3 mAb therapy a temporary increase in PD1+FOXP3+ Tregs was seen that paralleled a rise in anergic/exhausted CD4 and CD8 Teff cells (202). While early versions of anti-CD3 mAbs resulted in significant side effects that limited their use, genetic engineering and proteolytical removal of Fc domains alleviated many of the side effects (203, 204). The recent successes obtained with the anti-CD3 mAb therapy in T1D allow us to conclude that (1) immunotherapeutic interventions can be successful in T1D, (2) timing of immunotherapy is important, but success can be achieved even after anti-beta cell responses are detected, and (3) shifting the balance between inflammatory and regulatory pathways might be sufficient to acquire long-term tolerance. Although anti-CD3 mAb therapy is highly promising, it is not effective for ~25% of T1D patients and its positive effects can be temporary, which necessitates further investigation of the mechanisms underlying persistence of autoimmune T cells and their resistance to anti-CD3 therapy in certain individuals (197, 198).
Direct expansion of Tregs
Another avenue to address Treg frequency is by isolating and expanding endogenous Tregs from T1D patients directly in vitro followed by adoptive transfer back into the patient (Figure 4) (196). One way to expand Tregs utilizes the IL-2 pathway. For example, several studies have used low-dose IL-2 as a way to expand Tregs in vivo and increase their suppressive function (223, 224). Careful dosing of IL-2 in this approach is critical since high-dose IL-2 also expands effector T cells and other immune cell populations. Recent studies have addressed this dosing issue and improved upon this approach by modifying the IL-2 cytokine so that it selectively binds to Tregs (225–227). Targeting the IL-2 pathway is logical, as the decrease in Tregs seen during NOD diabetes progression is thought to be due to dysregulated IL-2 production within the pancreatic islets leading to loss in Treg function and survival (158), and IL-2R dysfunction is implicated in development of T1D (9). However, combining IL-2 therapy with autologous polyclonal expanded Treg infusion can have the potential to induce more harm than good. When IL-2 and Tregs are concomitantly administered to T1D recipients, IL-2 induces the proliferation not only of Tregs, but also of potentially cytotoxic cells, highlighting the need for Treg specific IL-2 (228). Although, low-dose IL-2 was well tolerated and specifically expanded Tregs in individuals of other autoimmune diseases (229).
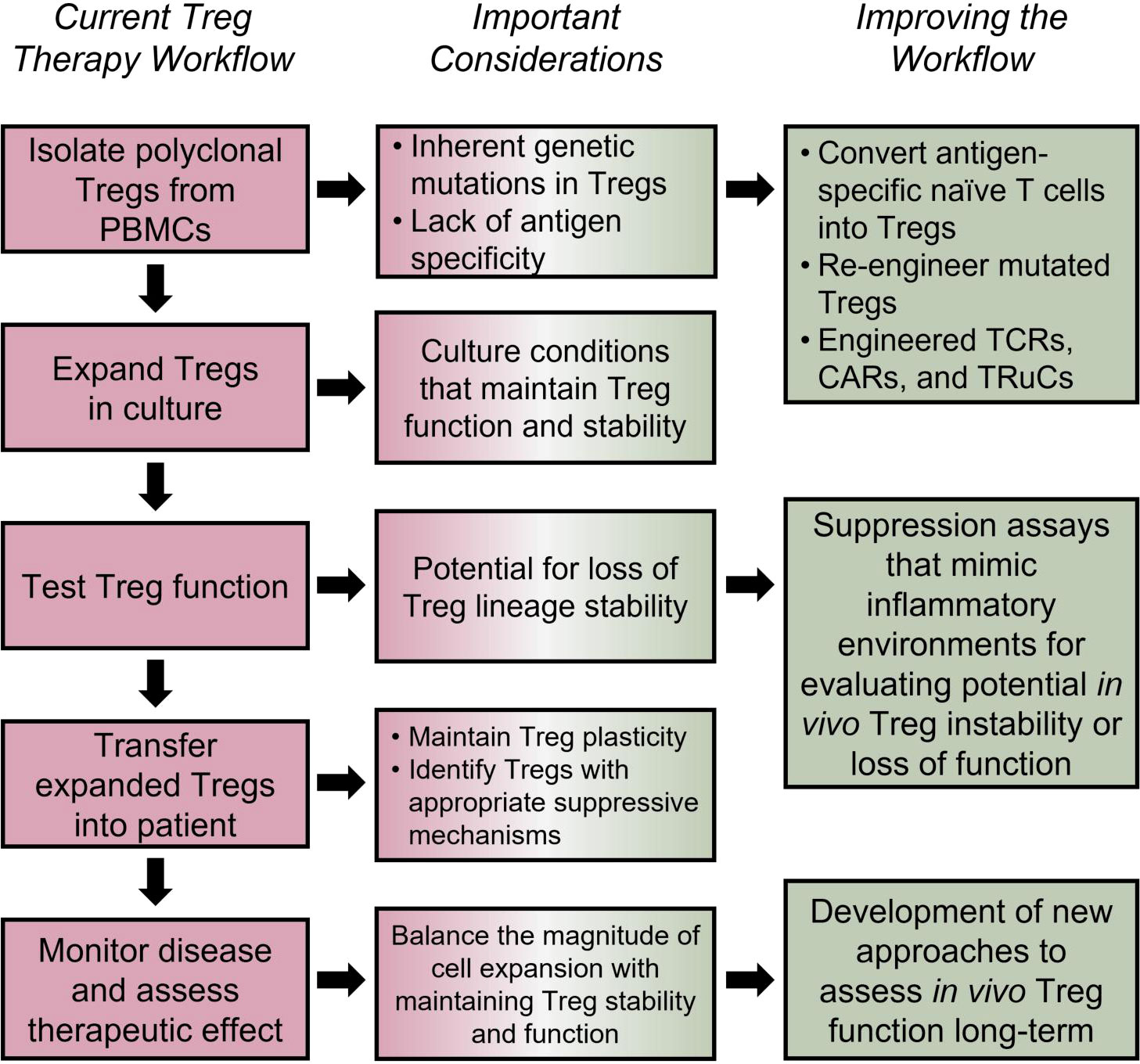
Figure 4 Tailoring Treg therapies for improved efficacy. Using human autologous Tregs is a promising approach for treatment of autoimmune and inflammatory disorders (196); however, the efficacy of such approaches depends on several factors. Loss of Treg suppressive capacity, stability, or stemness could be a side effect of in vitro expansion protocols (205–207). The potential inherent defects in Tregs, lack of antigen specificity (208–211), TSDR methylation status post expansion (206), and long-term functionality must also be considered. Potential solutions include engineering antigen specific TCRs (212), TRuCs (213–215), and CARs (216–219), utilizing Cas9/CRISPR technology for targeted demethylation of the TSDR (220), and using cytokine cocktails to optimize Treg expansion and functionality long-term (221, 222). Furthermore, clinical studies should be focused on accurately assessing the long-term in vivo Treg lineage stability, survival and disease-specific Treg suppressive mechanisms.
An additional caveat to Treg therapy is how Tregs may change during the manufacturing process, i.e., expansion of Tregs ex vivo. While ex vivo-expanded Tregs maintain suppressive capacity (205), they can also upregulate inflammatory effector T cell-associated cytokines, such as IFNγ, which can lead to loss of Treg stability (Figure 4) (207). Genome wide DNA methylation sequencing on Tregs undergoing in vitro expansion show increased methylation in enhancer and promoter regions of genes associated with T cell activation and function, as well as hypomethylation of genes associated with T cell exhaustion. These results are donor independent and are consistent throughout manufacturing runs (206), raising the question of whether Tregs expanded under current in vitro protocols are poised for long term function in vivo, regardless of their transcriptomic landscape or suppressive capacities at the end of expansion. Findings such as this could elucidate why current Treg therapies often fail to suppress disease long term. Fortunately, recent experiments have shown that the Cas9/CRISPR system can be used for targeted TET-mediated demethylation of the Treg TSDR (220); potentially providing a solution for Treg manufacturing complications. Indeed, simply using a chemical inducer of TSDR demethylation was shown to decrease NOD diabetes disease (230).
Another standard approach for in vitro expansion of T cells, including Tregs, is based on anti-CD3/CD28 crosslinking that leads to engagement of TCR and co-stimulatory pathways. However, strong and continuous TCR stimulation might result in loss of Treg stability or lead to Treg exhaustion. As an alternative to using anti-CD3/CD28, a combination of cytokines and CD28 superagonist antibodies (CD28SA) can induce robust Treg expansion while maintaining superior Treg stability (221, 222). Collectively, these findings suggest that a more tailored approach is necessary to create Treg-based treatments, and that increased Treg frequency, while helpful, needs to be accompanied by a high suppressive capacity in order to fully curtail disease.
Antigen specificity in Treg therapy
Another important consideration for effective Treg therapy is their tissue antigen specificity, which was shown to be necessary for optimal Treg function in mouse models of T1D (209, 211). Indeed, islet auto-antigen specific, but not polyclonal Tregs transferred into NOD mice are capable of engrafting and expanding following anti-CD3 Ab treatment (208). This may be due to antigen specific Tregs’ ability to traffic to the site of autoimmune inflammation more efficiently than polyclonal Tregs. For example, a clinical trial that recently concluded in MS patients saw that ex vivo expanded polyclonal CD4+CD25highCD127-FoxP3+ Tregs injected intrathecally, but not intravenously, had the ability to reduce disease severity, suggesting that inflammatory signals alone are not sufficient for recruitment of Tregs to the autoimmune tissue (210). Various approaches have been in development to increase antigen specific Tregs. One approach involved expansion of antigen specific Tregs in vitro using CD8+ splenic dendritic cells presenting islet antigens. Islet-antigen specific Tregs generated using this method had the ability to suppress diabetogenic T cells (231). Antigen specific Tregs can also be induced directly in vivo, as was observed in a recent clinical trial that utilized the in vivo delivery of beta cell peptide antigens (232). One potential problem that exists with this approach, however, is that some patients have inherent defects in their Treg populations, and thus it may be difficult to increase the number of functional Tregs. To address a potential lack of Treg precursors, one approach is to insert an enhancer before the FOXP3 coding region in bulk CD4 T cells (233). This approach overcomes epigenetic repression of the FOXP3 gene and can be used on antigen-specific CD4 conventional T cells (Figure 4). In addition, these edited Tregs express Treg signature genes and have a similar suppressive potential as naturally derived tTregs (234).
Understanding and identifying various subpopulations of Tregs is an important step to improving Treg-based therapies for autoimmune diseases, as the ability to isolate highly functional Tregs would be beneficial in enriching potentially more efficacious Tregs. As an example, TIGIT+ human Tregs positively correlate with stable FOXP3 expression (demethylated TSDR) while CD226+ Tregs are associated with effector cytokine expression and increased TSDR methylation (235). Furthermore, additional Treg subpopulations have been identified, that may increase our understanding of Treg biology and function (236).
Another approach to conferring antigen specificity to Tregs is with engineered TCRs, TCR-fusion constructs (TRuCs), or chimeric antigen receptors (CARs) (Figure 4). As the name suggests, engineered TCR Tregs are Tregs transfected with an antigen-specific TCR, however this approach may not create TCRs with a high enough affinity to be effective in resolving autoimmunity (212). Alternatively, CD4+FOXP3+ T cells can be transduced with a high affinity CAR specific for an autoimmune antigen (216, 217). Current results suggest that CAR Tregs specific for autoimmune antigens can traffic to the correct tissue site and maintain suppressive function (218, 219). TRuCs on the other hand, are tissue-protein specific antibody fragments fused to TCR, allowing for antigen recognition to be combined with natural TCR signaling (214). This approach may be superior to CAR Tregs when there is low density of the antigen available at the tissue site (213, 215).
Discussion
Understanding the genetic elements that lead to loss of regulatory T cell function in autoimmunity requires a foundational understanding of Treg function in a homeostatic environment. Control of Treg lineage and stability often revolve around the transcription factor FOXP3, although FOXP3 activity only accounts for a part of all Treg signature gene expression. Recent evidence has shown that FOXP3 expression and activity is tightly controlled through many different cis- and trans-regulatory factors including enhancer regions, transcription factor complexes, and epigenetic modifications. In turn, these regulatory factors can be influenced by the surrounding tissue environment, allowing for tight control of tolerance in healthy individuals. Thus, ultimate Treg function is a matter of both nature and nurture.
Genetic mutations leading to IPEX syndrome and polygenetic autoimmune susceptibilities revealed through GWAS analyses (9–11) converge on several pathways crucial to Treg stability and function and imply their dysregulation during autoimmunity. The dysregulation can be caused by mutations in FOXP3 itself, mutations in Treg functional genes, or SNPs that affect regulatory elements such as enhancer regions or genes critical for proper Treg function. In addition, transcription factor complexes that associate with CNS regions of FOXP3, are another component that give Tregs a ‘manual’ for how they should function in maintaining immune tolerance. However, this so-called manual often becomes distorted or destroyed during pathological autoimmunity, which might be attributed to chronic inflammation present in the tissue environment.
We know that Tregs are poised to interact with their environment and to make functional changes in response to seemingly minute alterations; especially compared to their effector T cell counterparts. The ability for a lymphoid resident Treg to undergo transcriptional reprogramming in order to become a tissue Treg is only one example of such functional changes. Additional evidence can be found in the sensitivity Tregs have to IL-2 in their surrounding environment, and the ability of Tregs to utilize unique metabolites (237–239). The idea that Tregs are influenced by their environment is not novel; however, there is growing appreciation that the environment or so-called ‘nurture’ can impose permanent changes in Treg nature.
GWAS and other -omics studies point to Treg defects as a partial contribution to autoimmune susceptibility. However, the ultimate trigger that destabilizes the immune system and leads to autoimmunity is hard to define. Do Tregs become dysfunctional due to the tissue environment created during inflammation or autoimmune attack, or are they dysfunctional prior to the initial triggering event? Perhaps, Tregs in autoimmune patients may be poised for dysregulation, but are only partially impaired and progressively lose function in response to specific environmental changes. Perturbations in the environment might provoke a series of downstream events related to epigenetic and transcriptomic changes of Tregs; ultimately leading to a loss of function and self-tolerance.
Author contributions
AR, DA, and MB conceptualized and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
The work was supported by National Institutes of Health, R01 AI125301 to MB. Figures were generated using Biorender.com. The authors would like to thank Matt Bettini, Yi Jing, Viva Rase, and Nouf Aljobaily for their critical reading and insightful feedback during the preparation of this review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol Baltim Md 1950 (1995) 155:1151–64.
2. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova J-L, Buist N, et al. X-Linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27:18–20. doi: 10.1038/83707
3. Khattri R, Kasprowicz D, Cox T, Mortrud M, Appleby MW, Brunkow ME, et al. The amount of scurfin protein determines peripheral T cell number and responsiveness. J Immunol (2001) 167:6312–20. doi: 10.4049/jimmunol.167.11.6312
4. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko S-A, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27:68–73. doi: 10.1038/83784
5. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell (2006) 126:375–87. doi: 10.1016/j.cell.2006.05.042
6. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi: 10.1126/science.1079490
7. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi: 10.1038/ni904
8. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor Foxp3. Immunity (2005) 22:329–41. doi: 10.1016/j.immuni.2005.01.016
9. Todd JA. Etiology of type 1 diabetes. Immunity (2010) 32:457–67. doi: 10.1016/j.immuni.2010.04.001
10. Ohkura N, Yasumizu Y, Kitagawa Y, Tanaka A, Nakamura Y, Motooka D, et al. Regulatory T cell-specific epigenomic region variants are a key determinant of susceptibility to common autoimmune diseases. Immunity (2020) 52:1119–1132.e4. doi: 10.1016/j.immuni.2020.04.006
11. Kim S-S, Hudgins AD, Yang J, Zhu Y, Tu Z, Rosenfeld MG, et al. A comprehensive integrated post-GWAS analysis of type 1 diabetes reveals enhancer-based immune dysregulation. PloS One (2021) 16:e0257265. doi: 10.1371/journal.pone.0257265
12. Powell BR, Buist NRM, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr (1982) 100:731–7. doi: 10.1016/s0022-3476(82)80573-8
13. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi: 10.1038/83713
14. Gambineri E, Mannurita SC, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front Immunol (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
15. Dikiy S, Li J, Bai L, Jiang M, Janke L, Zong X, et al. A distal Foxp3 enhancer enables interleukin-2 dependent thymic treg cell lineage commitment for robust immune tolerance. Immunity (2021) 54:931–946.e11. doi: 10.1016/j.immuni.2021.03.020
16. Kawakami R, Kitagawa Y, Chen KY, Arai M, Ohara D, Nakamura Y, et al. Distinct Foxp3 enhancer elements coordinate development, maintenance, and function of regulatory T cells. Immunity (2021) 54:947–961.e8. doi: 10.1016/j.immuni.2021.04.005
17. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463:808–12. doi: 10.1038/nature08750
18. Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, et al. A multiply redundant genetic switch “in” the transcriptional signature of regulatory T cells. Nat Immunol (2012) 13:972–80. doi: 10.1038/ni.2420
19. Joosten SA, Ottenhoff THM. Human CD4 and CD8 regulatory T cells in infectious diseases and vaccination. Hum Immunol (2008) 69:760–70. doi: 10.1016/j.humimm.2008.07.017
20. Pearson SS, Mason LG, Klarquist J, Burton JR, Tester IA, Wang CC, et al. Functional suppression by FoxP3+CD4+CD25high regulatory T cells during acute hepatitis c virus infection. J Infect Dis (2008) 197:46–57. doi: 10.1086/523651
21. Xu D, Fu J, Jin L, Zhang H, Zhou C, Zou Z, et al. Circulating and liver resident CD4+CD25+ regulatory T cells actively influence the antiviral immune response and disease progression in patients with hepatitis b. J Immunol (2006) 177:739–47. doi: 10.4049/jimmunol.177.1.739
22. Fabbri M, Frixou M, Degano M, Fousteri G. Type 1 diabetes in STAT protein family mutations: Regulating the Th17/Treg equilibrium and beyond. Diabetes (2019) 68:258–65. doi: 10.2337/db18-0627
23. Yang J, Zou M, Pezoldt J, Zhou X, Huehn J. Thymus-derived Foxp3+ regulatory T cells upregulate RORγt expression under inflammatory conditions. J Mol Med (2018) 96:1387–94. doi: 10.1007/s00109-018-1706-x
24. Jin F, Hu H, Xu M, Zhan S, Wang Y, Zhang H, et al. Serum microRNA profiles serve as novel biomarkers for autoimmune diseases. Front Immunol (2018) 9:2381. doi: 10.3389/fimmu.2018.02381
25. Zemmour D, Pratama A, Loughhead SM, Mathis D, Benoist C. Flicr, a long noncoding RNA, modulates Foxp3 expression and autoimmunity. Proc Natl Acad Sci USA (2017) 114:E3472–80. doi: 10.1073/pnas.1700946114
26. Mohammadnia-Afrouzi M, Hosseini AZ, Khalili A, Abediankenari S, Amari A, Aghili B, et al. Altered microRNA expression and immunosuppressive cytokine production by regulatory T cells of ulcerative colitis patients. Immunol Invest (2016) 45:63–74. doi: 10.3109/08820139.2015.1103749
27. Scherm MG, Serr I, Zahm AM, Schug J, Bellusci S, Manfredini R, et al. miRNA142-3p targets Tet2 and impairs treg differentiation and stability in models of type 1 diabetes. Nat Commun (2019) 10:5697. doi: 10.1038/s41467-019-13587-3
28. Zemmour D, Charbonnier L-M, Leon J, Six E, Keles S, Delville M, et al. Single-cell analysis of FOXP3 deficiencies in humans and mice unmasks intrinsic and extrinsic CD4+ T cell perturbations. Nat Immunol (2021) 22:607–19. doi: 10.1038/s41590-021-00910-8
29. Gool FV, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, et al. A mutation in the transcription factor Foxp3 drives T helper 2 effector function in regulatory T cells. Immunity (2019) 50:362–377.e6. doi: 10.1016/j.immuni.2018.12.016
30. Bettini ML, Pan F, Bettini M, Finkelstein D, Rehg JE, Floess S, et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity (2012) 36:717–30. doi: 10.1016/j.immuni.2012.03.020
31. Darce J, Rudra D, Li L, Nishio J, Cipolletta D, Rudensky AY, et al. An n-terminal mutation of the Foxp3 transcription factor alleviates arthritis but exacerbates diabetes. Immunity (2012) 36:731–41. doi: 10.1016/j.immuni.2012.04.007
32. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells. Vivo Nat Immunol (2009) 10:1000–7. doi: 10.1038/ni.1774
33. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2013) 20:62–8. doi: 10.1038/nm.3432
34. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-Driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity (2013) 39:949–62. doi: 10.1016/j.immuni.2013.10.016
35. Sambucci M, Gargano F, Rosa VD, Bardi MD, Picozza M, Placido R, et al. FoxP3 isoforms and PD-1 expression by T regulatory cells in multiple sclerosis. Sci Rep-uk (2018) 8:3674. doi: 10.1038/s41598-018-21861-5
36. Serena G, Yan S, Camhi S, Patel S, Lima RS, Sapone A, et al. Proinflammatory cytokine interferon-γ and microbiome-derived metabolites dictate epigenetic switch between forkhead box protein 3 isoforms in coeliac disease. Clin Exp Immunol (2017) 187:490–506. doi: 10.1111/cei.12911
37. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
38. Jain N, Nguyen H, Chambers C, Kang J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci (2010) 107:1524–8. doi: 10.1073/pnas.0910341107
39. Lin T-W, Hu Y-C, Yang Y-H, Chien Y-H, Lee N-C, Yu H-H, et al. CTLA-4 gene mutation and multiple sclerosis: A case report and literature review. J Microbiol Immunol Infect (2021) 55:545–8. doi: 10.1016/j.jmii.2021.10.009
40. Verma N, Burns SO, Walker LSK, Sansom DM. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin Amp Exp Immunol (2017) 190:1–7. doi: 10.1111/cei.12997
41. Bettini ML, Bettini M. Understanding autoimmune diabetes through the prism of the tri-molecular complex. Front Endocrinol (2017) 8:351. doi: 10.3389/fendo.2017.00351
42. Murawski MR, Litherland SA, Clare-Salzler MJ, Davoodi-Semiromi A. Upregulation of Foxp3 expression in mouse and human treg is IL-2/STAT5 dependent. Ann Ny Acad Sci (2006) 1079:198–204. doi: 10.1196/annals.1375.031
43. Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Mannurita SC, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol Orlando Fla (2013) 146:248–61. doi: 10.1016/j.clim.2013.01.004
44. Garg G, Tyler JR, Yang JHM, Cutler AJ, Downes K, Pekalski M, et al. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol (2012) 188:4644–53. doi: 10.4049/jimmunol.1100272
45. Chen Y, Chen S, Gu Y, Feng Y, Shi Y, Fu Q, et al. CTLA-4 +49 G/A, a functional T1D risk SNP, affects CTLA-4 level in treg subsets and IA-2A positivity, but not beta-cell function. Sci Rep-uk (2018) 8:10074. doi: 10.1038/s41598-018-28423-9
46. Long SA, Cerosaletti K, Wan JY, Ho J-C, Tatum M, Wei S, et al. An autoimmune-associated variant in PTPN2 reveals an impairment of IL-2R signaling in CD4+ T cells. Genes Immun (2011) 12:116–25. doi: 10.1038/gene.2010.54
47. Marwaha AK, Panagiotopoulos C, Biggs CM, Staiger S, Bel KLD, Hirschfeld AF, et al. Pre-diagnostic genotyping identifies T1D subjects with impaired treg IL-2 signaling and an elevated proportion of FOXP3+IL-17+ cells. Genes Immun (2017) 18:15–21. doi: 10.1038/gene.2016.44
48. Valta M, Gazali AM, Viisanen T, Ihantola E, Ekman I, Toppari J, et al. Type 1 diabetes linked PTPN22 gene polymorphism is associated with the frequency of circulating regulatory T cells. Eur J Immunol (2020) 50:581–8. doi: 10.1002/eji.201948378
49. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TIM. Defective suppressor function in CD4+CD25+ T-cells from patients with type 1 diabetes. Diabetes (2004) 54:92–9. doi: 10.2337/diabetes.54.1.92
50. Consortium T 1 DG, Onengut-Gumuscu S, Chen W-M, Burren O, Cooper NJ, Quinlan AR, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet (2015) 47:381–6. doi: 10.1038/ng.3245
51. Westra H-J, Martínez-Bonet M, Onengut-Gumuscu S, Lee A, Luo Y, Teslovich N, et al. Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat Genet (2018) 50:1366–74. doi: 10.1038/s41588-018-0216-7
52. Walter GJ, Fleskens V, Frederiksen KS, Rajasekhar M, Menon B, Gerwien JG, et al. Phenotypic, functional, and gene expression profiling of peripheral CD45RA+ and CD45RO+ CD4+CD25+CD127low treg cells in patients with chronic rheumatoid arthritis. Arthritis Rheumatol Hoboken N J (2016) 68:103–16. doi: 10.1002/art.39408
53. Sempere-Ortells JM, Pérez-García V, Marín-Alberca G, Peris-Pertusa A, Benito JM, Marco FM, et al. Quantification and phenotype of regulatory T cells in rheumatoid arthritis according to disease activity score-28. Autoimmunity (2009) 42:636–45. doi: 10.3109/08916930903061491
54. Avdeeva A, Rubtsov Y, Dyikanov D, Popkova T, Nasonov E. Regulatory T cells in patients with early untreated rheumatoid arthritis: Phenotypic changes in the course of methotrexate treatment. Biochimie (2020) 174:9–17. doi: 10.1016/j.biochi.2020.03.014
55. Wang Y, Su R, Li B, Guo Q, Hu F, Yu X, et al. Reduction of peripheral regulatory T cells in active rheumatoid arthritis patients with coronary artery disease. BMC Immunol (2021) 22:76. doi: 10.1186/s12865-021-00466-0
56. Shevyrev D, Tereshchenko V, Kozlov V, Sizikov A, Chumasova O, Koksharova V. T-Regulatory cells from patients with rheumatoid arthritis retain suppressor functions in vitro. Exp Ther Med (2021) 21:209. doi: 10.3892/etm.2021.9641
57. Fessler J, Raicht A, Husic R, Ficjan A, Schwarz C, Duftner C, et al. Novel senescent regulatory T-cell subset with impaired suppressive function in rheumatoid arthritis. Front Immunol (2017) 8:300. doi: 10.3389/fimmu.2017.00300
58. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science (2012) 337:1190–5. doi: 10.1126/science.1222794
59. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell (2013) 155:934–47. doi: 10.1016/j.cell.2013.09.053
60. Vahedi G, Kanno Y, Furumoto Y, Jiang K, Parker SCJ, Erdos MR, et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature (2015) 520:558–62. doi: 10.1038/nature14154
61. Gao P, Uzun Y, He B, Salamati SE, Coffey JKM, Tsalikian E, et al. Risk variants disrupting enhancers of TH1 and TREG cells in type 1 diabetes. Proc Natl Acad Sci USA (2019) 116:7581–90. doi: 10.1073/pnas.1815336116
62. Fasolino M, Goldman N, Wang W, Cattau B, Zhou Y, Petrovic J, et al. Genetic variation in type 1 diabetes reconfigures the 3D chromatin organization of T cells and alters gene expression. Immunity (2020) 52:257–274.e11. doi: 10.1016/j.immuni.2020.01.003
63. Zhang Z, Liu L, Shen Y, Meng Z, Chen M, Lu Z, et al. Characterization of chromatin accessibility in psoriasis. Front Med-prc (2021), 16:1–13. doi: 10.1007/s11684-021-0872-3
64. Arvey A, van der Veeken J, Plitas G, Rich SS, Concannon P, Rudensky AY. Genetic and epigenetic variation in the lineage specification of regulatory T cells. Elife (2015) 4:e07571. doi: 10.7554/elife.07571
65. Sato Y, Liu J, Lee E, Perriman R, Roncarolo MG, Bacchetta R. Co-Expression of FOXP3FL and FOXP3Δ2 isoforms is required for optimal treg-like cell phenotypes and suppressive function. Front Immunol (2021) 12:752394. doi: 10.3389/fimmu.2021.752394
66. Seitz C, Joly A-L, Fang F, Frith K, Gray P, Andersson J. The FOXP3 full-length isoform controls the lineage-stability of CD4+FOXP3+ regulatory T cells. Clin Immunol (2022) 237:108957. doi: 10.1016/j.clim.2022.108957
67. Frith K, Joly A-L, Ma CS, Tangye SG, Lohse Z, Seitz C, et al. The FOXP3Δ2 isoform supports regulatory T cell development and protects against severe IPEX. J Allergy Clin Immun (2019) 144:317–320.e8. doi: 10.1016/j.jaci.2019.03.003
68. Joly A-L, Seitz C, Liu S, Kuznetsov NV, Gertow K, Westerberg LS, et al. Alternative splicing of FOXP3 controls regulatory T cell effector functions and is associated with human atherosclerotic plaque stability. Circ Res (2018) 122:1385–94. doi: 10.1161/circresaha.117.312340
69. Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda-Martínez JA, et al. Control of Foxp3 stability through modulation of TET activity. J Exp Med (2016) 213:377–97. doi: 10.1084/jem.20151438
70. Zong X, Hao X, Xu B, Crawford JC, Wright S, Li J, et al. Foxp3 enhancers synergize to maximize regulatory T cell suppressive capacity. J Exp Med (2021) 218:e20202415. doi: 10.1084/jem.20202415
71. Iizuka-Koga M, Nakatsukasa H, Ito M, Akanuma T, Lu Q, Yoshimura A. Induction and maintenance of regulatory T cells by transcription factors and epigenetic modifications. J Autoimmun (2017) 83:113–21. doi: 10.1016/j.jaut.2017.07.002
72. Lee W, Lee GR. Transcriptional regulation and development of regulatory T cells. Exp Mol Med (2018) 50:e456. doi: 10.1038/emm.2017.313
73. Feng Y, van der Veeken J, Shugay M, Putintseva EV, Osmanbeyoglu HU, Dikiy S, et al. A mechanism for expansion of regulatory T-cell repertoire and its role in self-tolerance. Nature (2015) 528:132–6. doi: 10.1038/nature16141
74. Kwon H-K, Chen H-M, Mathis D, Benoist C. Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nat Immunol (2017) 18:1238–48. doi: 10.1038/ni.3835
75. Schumann K, Raju SS, Lauber M, Kolb S, Shifrut E, Cortez JT, et al. Functional CRISPR dissection of gene networks controlling human regulatory T cell identity. Nat Immunol (2020) 21:1456–66. doi: 10.1038/s41590-020-0784-4
76. Alvarez JD, Yasui DH, Niida H, Joh T, Loh DY, Kohwi-Shigematsu T. The MAR-binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T-cell development. Gene Dev (2000) 14:521–35. doi: 10.1101/gad.14.5.521
77. Chorro L, Suzuki M, Chin SS, Williams TM, Snapp EL, Odagiu L, et al. Interleukin 2 modulates thymic-derived regulatory T cell epigenetic landscape. Nat Commun (2018) 9:5368. doi: 10.1038/s41467-018-07806-6
78. Konopacki C, Pritykin Y, Rubtsov Y, Leslie CS, Rudensky AY. Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat Immunol (2019) 20:232–42. doi: 10.1038/s41590-018-0291-z
79. Beyer M, Thabet Y, Müller R-U, Sadlon T, Classen S, Lahl K, et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat Immunol (2011) 12:898–907. doi: 10.1038/ni.2084
80. Lee W, Kim HS, Hwang SS, Lee GR. The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Exp Mol Med (2017) 49:e393–3. doi: 10.1038/emm.2017.157
81. Jang SW, Hwang SS, Kim HS, Kim MK, Lee WH, Hwang SU, et al. Homeobox protein hhex negatively regulates treg cells by inhibiting Foxp3 expression and function. P Natl Acad Sci USA (2019) 116:25790–9. doi: 10.1073/pnas.1907224116
82. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PloS Biol (2007) 5:e38. doi: 10.1371/journal.pbio.0050038
83. Long M, Park S-G, Strickland I, Hayden MS, Ghosh S. Nuclear factor-κB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity (2009) 31:921–31. doi: 10.1016/j.immuni.2009.09.022
84. Harada Y, Harada Y, Elly C, Ying G, Paik J-H, DePinho RA, et al. Transcription factors Foxo3a and Foxo1 couple the E3 ligase cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med (2010) 207:1381–91. doi: 10.1084/jem.20100004
85. Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, et al. Novel Foxo1-dependent transcriptional programs control treg cell function. Nature (2012) 491:554–9. doi: 10.1038/nature11581
86. Kressler C, Gasparoni G, Nordström K, Hamo D, Salhab A, Dimitropoulos C, et al. Targeted de-methylation of the FOXP3-TSDR is sufficient to induce physiological FOXP3 expression but not a functional treg phenotype. Front Immunol (2020) 11:609891. doi: 10.3389/fimmu.2020.609891
87. Sebastian M, Lopez-Ocasio M, Metidji A, Rieder SA, Shevach EM, Thornton AM. Helios Controls a limited subset of regulatory T cell functions. J Immunol (2016) 196:144–55. doi: 10.4049/jimmunol.1501704
88. Skadow M, Penna VR, Galant-Swafford J, Shevach EM, Thornton AM. Helios Deficiency predisposes the differentiation of CD4 + Foxp3 – T cells into peripherally derived regulatory T cells. J Immunol (2019) 203:370–8. doi: 10.4049/jimmunol.1900388
89. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity (2019) 50:493–504.e7. doi: 10.1016/j.immuni.2019.01.001
90. Goldstein JD, Pérol L, Zaragoza B, Baeyens A, Marodon G, Piaggio E. Role of cytokines in thymus- versus peripherally derived-regulatory T cell differentiation and function. Front Immunol (2013) 4:155. doi: 10.3389/fimmu.2013.00155
91. Chen W, Jin W, Hardegen N, Lei K, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25– naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi: 10.1084/jem.20030152
92. Jones A, Opejin A, Henderson JG, Gross C, Jain R, Epstein JA, et al. Peripherally induced tolerance depends on peripheral regulatory T cells that require hopx to inhibit intrinsic IL-2 expression. J Immunol (2015) 195:1489–97. doi: 10.4049/jimmunol.1500174
93. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev (2014) 259:88–102. doi: 10.1111/imr.12160
94. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA Methylation controls Foxp3 gene expression. Eur J Immunol (2008) 38:1654–63. doi: 10.1002/eji.200838105
95. Chakraborty S, Panda AK, Bose S, Roy D, Kajal K, Guha D, et al. Transcriptional regulation of FOXP3 requires integrated activation of both promoter and CNS regions in tumor-induced CD8+ treg cells. Sci Rep-uk (2017) 7:1628. doi: 10.1038/s41598-017-01788-z
96. Cosmi L, Liotta F, Lazzeri E, Francalanci M, Angeli R, Mazzinghi B, et al. Human CD8+CD25+ thymocytes share phenotypic and functional features with CD4+CD25+ regulatory thymocytes. Blood (2003) 102:4107–14. doi: 10.1182/blood-2003-04-1320
97. Zhang S, Ke X, Zeng S, Wu M, Lou J, Wu L, et al. Analysis of CD8+ treg cells in patients with ovarian cancer: a possible mechanism for immune impairment. Cell Mol Immunol (2015) 12:580–91. doi: 10.1038/cmi.2015.57
98. Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O’Shea JJ, et al. CD4+FoxP3+ regulatory T cells confer infectious tolerance in a TGF-β–dependent manner. J Exp Med (2008) 205:1975–81. doi: 10.1084/jem.20080308
99. Vitale S, Russo V, Dettori B, Palombi C, Baev D, Proietti E, et al. Type I interferons induce peripheral T regulatory cell differentiation under tolerogenic conditions. Int Immunol (2020) 33:59–77. doi: 10.1093/intimm/dxaa058
100. Piconese S, Pacella I, Timperi E, Barnaba V. Divergent effects of type-I interferons on regulatory T cells. Cytokine Growth F R (2015) 26:133–41. doi: 10.1016/j.cytogfr.2014.10.012
101. Schlenner SM, Weigmann B, Ruan Q, Chen Y, von Boehmer H. Smad3 binding to the foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J Exp Med (2012) 209:1529–35. doi: 10.1084/jem.20112646
102. Schuster C, Jonas F, Zhao F, Kissler S. Peripherally-induced regulatory T cells contribute to the control of autoimmune diabetes in the NOD mouse model. Eur J Immunol (2018) 48:1211–6. doi: 10.1002/eji.201847498
103. Holohan DR, Gool FV, Bluestone JA. Thymically-derived Foxp3+ regulatory T cells are the primary regulators of type 1 diabetes in the non-obese diabetic mouse model. PloS One (2019) 14:e0217728. doi: 10.1371/journal.pone.0217728
104. Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting edge: IL-2 is essential for TGF-β-Mediated induction of Foxp3+ T regulatory cells. J Immunol (2007) 178:4022–6. doi: 10.4049/jimmunol.178.7.4022
105. Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, et al. Plasticity of Foxp3+ T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity (2012) 36:262–75. doi: 10.1016/j.immuni.2011.12.012
106. Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, et al. In vivo expansion of T reg cells with IL-2–mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med (2009) 206:751–60. doi: 10.1084/jem.20082824
107. Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-β–induced Foxp3+ T cells In vivo. J Immunol (2011) 186:6329–37. doi: 10.4049/jimmunol.1100061
108. Alvarez-Salazar EK, Cortés-Hernández A, Arteaga-Cruz S, Alberú-Gómez J, Soldevila G. Large-Scale generation of human allospecific induced tregs with functional stability for use in immunotherapy in transplantation. Front Immunol (2020) 11:375. doi: 10.3389/fimmu.2020.00375
109. Huter EN, Punkosdy GA, Glass DD, Cheng LI, Ward JM, Shevach EM. TGF-β-induced Foxp3 + regulatory T cells rescue scurfy mice. Eur J Immunol (2008) 38:1814–21. doi: 10.1002/eji.200838346
110. Zeng Q, Sun X, Xiao L, Xie Z, Bettini M, Deng T. A unique population: Adipose-resident regulatory T cells. Front Immunol (2018) 9:2075. doi: 10.3389/fimmu.2018.02075
111. Lui PP, Cho I, Ali N. Tissue regulatory T cells. Immunology (2020) 161:4–17. doi: 10.1111/imm.13208
112. Shao Q, Gu J, Zhou J, Wang Q, Li X, Deng Z, et al. Tissue tregs and maintenance of tissue homeostasis. Front Cell Dev Biol (2021) 9:717903. doi: 10.3389/fcell.2021.717903
113. Delacher M, Imbusch CD, Hotz-Wagenblatt A, Mallm J-P, Bauer K, Simon M, et al. Precursors for nonlymphoid-tissue treg cells reside in secondary lymphoid organs and are programmed by the transcription factor BATF. Immunity (2020) 52:295–312.e11. doi: 10.1016/j.immuni.2019.12.002
114. Schiaffino S, Pereira MG, Ciciliot S, Rovere-Querini P. Regulatory T cells and skeletal muscle regeneration. FEBS J (2017) 284:517–24. doi: 10.1111/febs.13827
115. Zaiss DM, Minutti CM, Knipper JA. Immune- and non-immune-mediated roles of regulatory T-cells during wound healing. Immunology (2019) 157:190–7. doi: 10.1111/imm.13057
116. Sanchez-Solares J, Sanchez L, Pablo-Torres C, Diaz-Fernandez C, Sørensen P, Barber D, et al. Celiac disease causes epithelial disruption and regulatory T cell recruitment in the oral mucosa. Front Immunol (2021) 12:623805. doi: 10.3389/fimmu.2021.623805
117. Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature (2017) 546:421–5. doi: 10.1038/nature22360
118. Tan TG, Mathis D, Benoist C. Singular role for T-BET+CXCR3+ regulatory T cells in protection from autoimmune diabetes. Proc Natl Acad Sci (2016) 113:14103–8. doi: 10.1073/pnas.1616710113
119. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol (2009) 10:595–602. doi: 10.1038/ni.1731
120. Hayatsu N, Miyao T, Tachibana M, Murakami R, Kimura A, Kato T, et al. Analyses of a mutant Foxp3 allele reveal BATF as a critical transcription factor in the differentiation and accumulation of tissue regulatory T cells. Immunity (2017) 47:268–283.e9. doi: 10.1016/j.immuni.2017.07.008
121. Li C, DiSpirito JR, Zemmour D, Spallanzani RG, Kuswanto W, Benoist C, et al. TCR transgenic mice reveal stepwise, multi-site acquisition of the distinctive fat-treg phenotype. Cell (2018) 174:285–299.e12. doi: 10.1016/j.cell.2018.05.004
122. Li C, Muñoz-Rojas AR, Wang G, Mann AO, Benoist C, Mathis D. PPARγ marks splenic precursors of multiple nonlymphoid-tissue treg compartments. Proc Natl Acad Sci (2021) 118. doi: 10.1073/pnas.2025197118
123. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
124. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
125. Sakai R, Ito M, Komai K, Iizuka-Koga M, Matsuo K, Nakayama T, et al. Kidney GATA3+ regulatory T cells play roles in the convalescence stage after antibody-mediated renal injury. Cell Mol Immunol (2021) 18:1249–61. doi: 10.1038/s41423-020-00547-x
126. Agle K, Vincent BG, Piper C, Belle L, Zhou V, Shlomchik W, et al. Bim regulates the survival and suppressive capability of CD8+ FOXP3+ regulatory T cells during murine GVHD. Blood (2018) 132:435–47. doi: 10.1182/blood-2017-09-807156
127. Singh RP, Cava AL, Wong M, Ebling F, Hahn BH. CD8+ T cell-mediated suppression of autoimmunity in a murine lupus model of peptide-induced immune tolerance depends on Foxp3 expression. J Immunol (2007) 178:7649–57. doi: 10.4049/jimmunol.178.12.7649
128. Gonzalez MM, Bamidele AO, Svingen PA, Sagstetter MR, Smyrk TC, Gaballa JM, et al. BMI1 maintains the treg epigenomic landscape to prevent inflammatory bowel disease. J Clin Invest (2021) 131:e140755. doi: 10.1172/jci140755
129. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity (2008) 29:44–56. doi: 10.1016/j.immuni.2008.05.007
130. Li J, Xu B, He M, Zong X, Cunningham T, Sha C, et al. Control of Foxp3 induction and maintenance by sequential histone acetylation and DNA demethylation. Cell Rep (2021) 37:110124–4. doi: 10.1016/j.celrep.2021.110124
131. Piunti A, Shilatifard A. The roles of polycomb repressive complexes in mammalian development and cancer. Nat Rev Mol Cell Bio (2021) 22:326–45. doi: 10.1038/s41580-021-00341-1
132. Escobar TM, Kanellopoulou C, Kugler DG, Kilaru G, Nguyen CK, Nagarajan V, et al. miR-155 activates cytokine gene expression in Th17 cells by regulating the DNA-binding protein Jarid2 to relieve polycomb-mediated repression. Immunity (2014) 40:865–79. doi: 10.1016/j.immuni.2014.03.014
133. Zhu F, Li H, Liu Y, Tan C, Liu X, Fan H, et al. miR-155 antagomir protect against DSS-induced colitis in mice through regulating Th17/Treg cell balance by Jarid2/Wnt/β-catenin. BioMed Pharmacother (2020) 126:109909. doi: 10.1016/j.biopha.2020.109909
134. Bostick M, Kim JK, Estéve P-O, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science (2007) 317:1760–4. doi: 10.1126/science.1147939
135. Nishiyama A, Yamaguchi L, Sharif J, Johmura Y, Kawamura T, Nakanishi K, et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature (2013) 502:249–53. doi: 10.1038/nature12488
136. Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature (2007) 450:908–12. doi: 10.1038/nature06397
137. Maenohara S, Unoki M, Toh H, Ohishi H, Sharif J, Koseki H, et al. Role of UHRF1 in de novo DNA methylation in oocytes and maintenance methylation in preimplantation embryos. PloS Genet (2017) 13:e1007042. doi: 10.1371/journal.pgen.1007042
138. Helmin KA, Morales-Nebreda L, Acosta MAT, Anekalla KR, Chen S-Y, Abdala-Valencia H, et al. Maintenance DNA methylation is essential for regulatory T cell development and stability of suppressive function. J Clin Invest (2020) 130:6571–87. doi: 10.1172/jci137712
139. Sun X, Cui Y, Feng H, Liu H, Liu X. TGF-β signaling controls Foxp3 methylation and T reg cell differentiation by modulating Uhrf1 activity. J Exp Med (2019) 216:2819–37. doi: 10.1084/jem.20190550
140. Mijnheer G, Lutter L, Mokry M, van der Wal M, Scholman R, Fleskens V, et al. Conserved human effector treg cell transcriptomic and epigenetic signature in arthritic joint inflammation. Nat Commun (2021) 12:2710. doi: 10.1038/s41467-021-22975-7
141. Garg G, Muschaweckh A, Moreno H, Vasanthakumar A, Floess S, Lepennetier G, et al. Blimp1 prevents methylation of Foxp3 and loss of regulatory T cell identity at sites of inflammation. Cell Rep (2019) 26:1854–1868.e5. doi: 10.1016/j.celrep.2019.01.070
142. DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity (2015) 42:227–38. doi: 10.1016/j.immuni.2015.01.007
143. Xiao X, Li Y, Jiang X, Ji X, Lu X, Yang B, et al. EZH2 deficiency attenuates treg differentiation in rheumatoid arthritis. J Autoimmun (2020) 108:102404. doi: 10.1016/j.jaut.2020.102404
144. Iwaszkiewicz-Grzes D, Piotrowska M, Gliwinski M, Urban-Wójciuk Z, Trzonkowski P. Antigenic challenge influences epigenetic changes in antigen-specific T regulatory cells. Front Immunol (2021) 12:642678. doi: 10.3389/fimmu.2021.642678
145. Pesenacker AM, Chen V, Gillies J, Speake C, Marwaha AK, Sun AC, et al. Treg gene signatures predict and measure type 1 diabetes trajectory. JCI Insight (2019) 4, 739–47. doi: 10.1172/jci.insight.123879
146. Takahashi K. Influence of bacteria on epigenetic gene control. Cell Mol Life Sci (2014) 71:1045–54. doi: 10.1007/s00018-013-1487-x
147. Luo A, Leach ST, Barres R, Hesson LB, Grimm MC, Simar D. The microbiota and epigenetic regulation of T helper 17/Regulatory T cells: In search of a balanced immune system. Front Immunol (2017) 8:417. doi: 10.3389/fimmu.2017.00417
148. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi: 10.1038/nature12726
149. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi: 10.1038/nature12721
150. Wiechers C, Zou M, Galvez E, Beckstette M, Ebel M, Strowig T, et al. The microbiota is dispensable for the early stages of peripheral regulatory T cell induction within mesenteric lymph nodes. Cell Mol Immunol (2021) 18:1211–21. doi: 10.1038/s41423-021-00647-2
151. Edwards C, Costenbader K. Epigenetics and the microbiome: developing areas in the understanding of the aetiology of lupus. Lupus (2014) 23:505–6. doi: 10.1177/0961203314531636
152. Conrad MA, Wu GD, Kelsen JR. (2017). The gut microbiota and inflammatory bowel disease. In: Mamula P, Grossman A, Baldassano R, Kelsen J, Markowitz J. (eds) Pediatric inflammatory bowel disease. Springer, Cham. doi: 10.1007/978-3-319-49215-5_4
153. Jiang W, Yu X, Kosik RO, Song Y, Qiao T, Tong J, et al. Gut microbiota may play a significant role in the pathogenesis of graves’ disease. Thyroid (2021) 31:810–20. doi: 10.1089/thy.2020.0193
154. Su X, Yin X, Liu Y, Yan X, Zhang S, Wang X, et al. Gut dysbiosis contributes to the imbalance of treg and Th17 cells in graves’ disease patients by propionic acid. J Clin Endocrinol Metab (2020) 105:3526–47. doi: 10.1210/clinem/dgaa511
155. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med (2015) 21:895–905. doi: 10.1038/nm.3914
156. Kugelberg E. Diet can protect against type 1 diabetes. Nat Rev Immunol (2017) 17:279–9. doi: 10.1038/nri.2017.40
157. Baraut J, Grigore EI, Jean-Louis F, Khelifa SH, Durand C, Verrecchia F, et al. Peripheral blood regulatory T cells in patients with diffuse systemic sclerosis (SSc) before and after autologous hematopoietic SCT: a pilot study. Bone Marrow Transpl (2014) 49:349–54. doi: 10.1038/bmt.2013.202
158. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity (2008) 28:687–97. doi: 10.1016/j.immuni.2008.03.016
159. Tang M, Cheng L, Li F, Wu B, Chen P, Zhan Y, et al. Transcription factor IRF4 dysfunction affects the immunosuppressive function of treg cells in patients with primary immune thrombocytopenia. BioMed Res Int (2019) 2019:1050285. doi: 10.1155/2019/1050285
160. Erikçi AA, Karagöz B, Bilgi O. Regulatory T cells in patients with idiopathic thrombocytopenic purpura. Turk J Hematol (2016) 33:153–5. doi: 10.4274/tjh.2015.0335
161. Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ treg cells that phenotypically mirror effector Th cells. Blood (2012) 119:4430–40. doi: 10.1182/blood-2011-11-392324
162. Koenecke C, Lee C-W, Thamm K, Föhse L, Schafferus M, Mittrücker H-W, et al. IFN-γ production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-Host disease. J Immunol (2012) 189:2890–6. doi: 10.4049/jimmunol.1200413
163. Pandiyan P, Zhu J. Origin and functions of pro-inflammatory cytokine producing Foxp3+ regulatory T cells. Cytokine (2015) 76:13–24. doi: 10.1016/j.cyto.2015.07.005
164. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, et al. Interferon-γ drives treg fragility to promote anti-tumor immunity. Cell (2017) 169:1130–1141.e11. doi: 10.1016/j.cell.2017.05.005
165. Su LF, del Alcazar D, Stelekati E, Wherry EJ, Davis MM. Antigen exposure shapes the ratio between antigen-specific tregs and conventional T cells in human peripheral blood. Proc Natl Acad Sci (2016) 113:E6192–8. doi: 10.1073/pnas.1611723113
166. Boccasavia VL, Bovolenta ER, Villanueva A, Borroto A, Oeste CL, van Santen HM, et al. Antigen presentation between T cells drives Th17 polarization under conditions of limiting antigen. Cell Rep (2021) 34:108861. doi: 10.1016/j.celrep.2021.108861
167. Aqel SI, Kraus EE, Jena N, Kumari V, Granitto MC, Mao L, et al. Novel small molecule IL-6 inhibitor suppresses autoreactive Th17 development and promotes treg development. Clin Exp Immunol (2019) 196:215–25. doi: 10.1111/cei.13258
168. Aqel SI, Yang X, Kraus EE, Song J, Farinas MF, Zhao EY, et al. A STAT3 inhibitor ameliorates CNS autoimmunity by restoring Teff:Treg balance. JCI Insight (2021) 6:e142376. doi: 10.1172/jci.insight.142376
169. Maimone D, Gregory S, Arnason BGW, Reder AT. Cytokine levels in the cerebrospinal fluid and serum of patients with multiple sclerosis. J Neuroimmunol (1991) 32:67–74. doi: 10.1016/0165-5728(91)90073-g
170. Greenbaum CJ, Serti E, Lambert K, Weiner LJ, Kanaparthi S, Lord S, et al. IL-6 receptor blockade does not slow β cell loss in new-onset type 1 diabetes. JCI Insight (2021) 6:e150074. doi: 10.1172/jci.insight.150074
171. Yao R-W, Wang Y, Chen L-L. Cellular functions of long noncoding RNAs. Nat Cell Biol (2019) 21:542–51. doi: 10.1038/s41556-019-0311-8
172. Gao J, Gu J, Pan X, Gan X, Ju Z, Zhang S, et al. Blockade of miR-142-3p promotes anti-apoptotic and suppressive function by inducing KDM6A-mediated H3K27me3 demethylation in induced regulatory T cells. Cell Death Dis (2019) 10:332. doi: 10.1038/s41419-019-1565-6
173. Anandagoda N, Willis JCD, Hertweck A, Roberts LB, Jackson I, Gökmen MR, et al. microRNA-142–mediated repression of phosphodiesterase 3B critically regulates peripheral immune tolerance. J Clin Invest (2019) 129:1257–71. doi: 10.1172/jci124725
174. Wang W-L, Ouyang C, Graham NM, Zhang Y, Cassady K, Reyes EY, et al. microRNA-142 guards against autoimmunity by controlling treg cell homeostasis and function. PloS Biol (2022) 20:e3001552. doi: 10.1371/journal.pbio.3001552
175. Li J, Zhang H, Liu M, Xiang Y, Li H, Huang F, et al. miR-133a-3p/FOXP3 axis regulates cell proliferation and autophagy in gastric cancer. J Cell Biochem (2020) 121:3392–405. doi: 10.1002/jcb.29613
176. Ye X, Lu Q, Yang A, Rao J, Xie W, He C, et al. MiR-206 regulates the Th17/Treg ratio during osteoarthritis. Mol Med (2021) 27:64. doi: 10.1186/s10020-021-00315-1
177. Zhang L, Ke F, Liu Z, Bai J, Liu J, Yan S, et al. MicroRNA-31 negatively regulates peripherally derived regulatory T-cell generation by repressing retinoic acid-inducible protein 3. Nat Commun (2015) 6:7639. doi: 10.1038/ncomms8639
178. Wu Y, Mealer C, Schutt SD, Wilson CL, Bastian D, Sofi MHH, et al. MicroRNA-31 regulates T-cell metabolism via HIF1α and promotes chronic GVHD pathogenesis in mice. Blood Adv (2022). doi: 10.1182/bloodadvances.2021005103
179. Liu Y, Li C, Yang Y, Li T, Xu Y, Zhang W, et al. The TGF-β/miR-31/CEACAM1-S axis inhibits CD4+CD25+ treg differentiation in systemic lupus erythematosus. Immunol Cell Biol (2021) 99:697–710. doi: 10.1111/imcb.12449
180. Rouas R, Fayyad-Kazan H, Zein NE, Lewalle P, Rothé F, Simion A, et al. Human natural treg microRNA signature: Role of microRNA-31 and microRNA-21 in FOXP3 expression. Eur J Immunol (2009) 39:1608–18. doi: 10.1002/eji.200838509
181. Dong L, Wang X, Tan J, Li H, Qian W, Chen J, et al. Decreased expression of microRNA-21 correlates with the imbalance of Th17 and treg cells in patients with rheumatoid arthritis. J Cell Mol Med (2014) 18:2213–24. doi: 10.1111/jcmm.12353
182. Huo J, Liu T, Li F, Song X, Hou X. MicroRNA-21-5p protects melanocytes via targeting STAT3 and modulating Treg/Teff balance to alleviate vitiligo. Mol Med Rep (2021) 23:51. doi: 10.3892/mmr.2020.11689
183. Zhu H, Lan L, Zhang Y, Chen Q, Zeng Y, Luo X, et al. Epidermal growth factor stimulates exosomal microRNA-21 derived from mesenchymal stem cells to ameliorate aGVHD by modulating regulatory T cells. FASEB J (2020) 34:7372–86. doi: 10.1096/fj.201900847rrrr
184. Sun J, Liu R, He X, Bian J, Zhao W, Shi W, et al. MicroRNA-21 regulates diametrically opposed biological functions of regulatory T cells. Front Immunol (2021) 12:766757. doi: 10.3389/fimmu.2021.766757
185. Jin S, Chen H, Li Y, Zhong H, Sun W, Wang J, et al. Maresin 1 improves the Treg/Th17 imbalance in rheumatoid arthritis through miR-21. Ann Rheum Dis (2018) 77:1644. doi: 10.1136/annrheumdis-2018-213511
186. Ji Q, Liu J, Dong Y, Wang L, Dong K, Setiz B, et al. Exosomes derived from thymic stromal lymphopoietin-treated dendritic cells regulate T helper 17/regulatory T cell differentiation via miR-21/Smad7 axis. Exp Cell Res (2021) 398:112393. doi: 10.1016/j.yexcr.2020.112393
187. Namdari H, Ghayedi M, Hadjati J, Rezaei F, Kalantar K, Rahimzadeh P, et al. Effect of MicroRNA-21 transfection on In-vitro differentiation of human naive CD4+ T cells to regulatory T cells. Iranian J Allergy Asthma Immunol (2017) 16:235–44.
188. Zheng X, Dong L, Wang K, Zou H, Zhao S, Wang Y, et al. MiR-21 participates in the PD-1/PD-L1 pathway-mediated imbalance of Th17/Treg cells in patients after gastric cancer resection. Ann Surg Oncol (2019) 26:884–93. doi: 10.1245/s10434-018-07117-6
189. Xia M, Liu J, Liu S, Chen K, Lin H, Jiang M, et al. Ash1l and lnc-Smad3 coordinate Smad3 locus accessibility to modulate iTreg polarization and T cell autoimmunity. Nat Commun (2017) 8:15818. doi: 10.1038/ncomms15818
190. Yan W, Wang L, Chen Z, Gu C, Chen C, Liu X, et al. Knockdown of lncRNA HAGLR promotes treg cell differentiation through increasing the RUNX3 level in dermatomyositis. J Mol Histol (2022) 53, 1–9. doi: 10.1007/s10735-021-10051-9
191. Wang J, Zhai X, Guo J, Li Y, Yang Y, Wang L, et al. Long non-coding RNA DQ786243 modulates the induction and function of CD4+ treg cells through Foxp3-miR-146a-NF-κB axis: Implications for alleviating oral lichen planus. Int Immunopharmacol (2019) 75:105761. doi: 10.1016/j.intimp.2019.105761
192. Xie M, Wang J, Gong W, Xu H, Pan X, Chen Y, et al. NF-κB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J Autoimmun (2019) 102:96–113. doi: 10.1016/j.jaut.2019.04.018
193. Zhou L, Wang J, Li J, Li T, Chen Y, June RR, et al. 1,25-dihydroxyvitamin D3 ameliorates collagen-induced arthritis via suppression of Th17 cells through miR-124 mediated inhibition of IL-6 signaling. Front Immunol (2019) 10:178. doi: 10.3389/fimmu.2019.00178
194. Gao X, Liu L, Min X, Jia S, Zhao M. Non-coding RNAs in CD4+ T cells: New insights into the pathogenesis of systemic lupus erythematosus. Front Immunol (2020) 11:568. doi: 10.3389/fimmu.2020.00568
195. Esensten JH, Muller YD, Bluestone JA, Tang Q. Regulatory T-cell therapy for autoimmune and autoinflammatory diseases: The next frontier. J Allergy Clin Immun (2018) 142:1710–8. doi: 10.1016/j.jaci.2018.10.015
196. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
197. Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, et al. A single course of anti-CD3 monoclonal antibody hOKT3γ1(Ala-ala) results in improvement in c-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes (2005) 54:1763–9. doi: 10.2337/diabetes.54.6.1763
198. Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med (2002) 346:1692–8. doi: 10.1056/nejmoa012864
199. Herold KC, Gitelman SE, Ehlers MR, Gottlieb PA, Greenbaum CJ, Hagopian W, et al. Teplizumab (Anti-CD3 mAb) treatment preserves c-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial. Diabetes (2013) 62:3766–74. doi: 10.2337/db13-0345
200. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. New Engl J Med (2019) 381:603–13. doi: 10.1056/nejmoa1902226
201. Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol (2011) 187:2015–22. doi: 10.4049/jimmunol.1100713
202. Long SA, Thorpe J, Herold KC, Ehlers M, Sanda S, Lim N, et al. Remodeling T cell compartments during anti-CD3 immunotherapy of type 1 diabetes. Cell Immunol (2017) 319:3–9. doi: 10.1016/j.cellimm.2017.07.007
203. Gaglia J, Kissler S. Anti-CD3 antibody for the prevention of type 1 diabetes: A story of perseverance. Biochemistry-us (2019) 58:4107–11. doi: 10.1021/acs.biochem.9b00707
204. Ke Q, Kroger CJ, Clark M, Tisch RM. Evolving antibody therapies for the treatment of type 1 diabetes. Front Immunol (2021) 11:624568. doi: 10.3389/fimmu.2020.624568
205. Balcerek J, Shy BR, Putnam AL, Masiello LM, Lares A, Dekovic F, et al. Polyclonal regulatory T cell manufacturing under cGMP: A decade of experience. Front Immunol (2021) 12:744763. doi: 10.3389/fimmu.2021.744763
206. Ou K, Hamo D, Schulze A, Roemhild A, Kaiser D, Gasparoni G, et al. Strong expansion of human regulatory T cells for adoptive cell therapy results in epigenetic changes which may impact their survival and function. Front Cell Dev Biol (2021) 9:751590. doi: 10.3389/fcell.2021.751590
207. Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, et al. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes (2009) 58:652–62. doi: 10.2337/db08-1168
208. Cabello-Kindelan C, Mackey S, Sands A, Rodriguez J, Vazquez C, Pugliese A, et al. Immunomodulation followed by antigen-specific treg infusion controls islet autoimmunity. Diabetes (2019) 69:215–27. doi: 10.2337/db19-0061
209. Masteller EL, Warner MR, Tang Q, Tarbell KV, McDevitt H, Bluestone JA. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol (2005) 175:3053–9. doi: 10.4049/jimmunol.175.5.3053
210. Chwojnicki K, Iwaszkiewicz-Grześ D, Jankowska A, Zieliński M, Łowiec P, Gliwiński M, et al. Administration of CD4+CD25highCD127–FoxP3+ regulatory T cells for relapsing-remitting multiple sclerosis: A phase 1 study. Biodrugs (2021) 35:47–60. doi: 10.1007/s40259-020-00462-7
211. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro–expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199:1455–65. doi: 10.1084/jem.20040139
212. Liu Y, Yan X, Zhang F, Zhang X, Tang F, Han Z, et al. TCR-T immunotherapy: The challenges and solutions. Front Oncol (2022) 11:794183. doi: 10.3389/fonc.2021.794183
213. Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol (2020) 20:158–72. doi: 10.1038/s41577-019-0232-6
214. Baeuerle PA, Ding J, Patel E, Thorausch N, Horton H, Gierut J, et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat Commun (2019) 10:2087. doi: 10.1038/s41467-019-10097-0
215. Rana J, Perry DJ, Kumar SRP, Muñoz-Melero M, Saboungi R, Brusko TM, et al. CAR- and TRuC-redirected regulatory T cells differ in capacity to control adaptive immunity to FVIII. Mol Ther (2021) 29:2660–76. doi: 10.1016/j.ymthe.2021.04.034
216. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J Autoimmun (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
217. Boardman D, Maher J, Lechler R, Smyth L, Lombardi G. Antigen-specificity using chimeric antigen receptors: the future of regulatory T-cell therapy? Biochem Soc T (2016) 44:342–8. doi: 10.1042/bst20150247
218. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflamm (2012) 9:112–2. doi: 10.1186/1742-2094-9-112
219. Pohl ADP, Schmidt A, Zhang A-H, Maldonado T, Königs C, Scott DW. Engineered regulatory T cells expressing myelin-specific chimeric antigen receptors suppress EAE progression. Cell Immunol (2020) 358:104222. doi: 10.1016/j.cellimm.2020.104222
220. Wilk C, Effenberg L, Abberger H, Steenpass L, Hansen W, Zeschnigk M, et al. CRISPR/Cas9-mediated demethylation of FOXP3-TSDR toward treg-characteristic programming of jurkat T cells. Cell Immunol (2022) 371:104471. doi: 10.1016/j.cellimm.2021.104471
221. Skartsis N, Peng Y, Ferreira LMR, Nguyen V, Ronin E, Muller YD, et al. IL-6 and TNFα drive extensive proliferation of human tregs without compromising their lineage stability or function. Front Immunol (2021) 12:783282. doi: 10.3389/fimmu.2021.783282
222. He X, Smeets RL, van Rijssen E, Boots AMH, Joosten I, Koenen HJPM. Single CD28 stimulation induces stable and polyclonal expansion of human regulatory T cells. Sci Rep-uk (2017) 7:43003. doi: 10.1038/srep43003
223. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol (2013) 1:295–305. doi: 10.1016/s2213-8587(13)70113-x
224. Rosenzwajg M, Salet R, Lorenzon R, Tchitchek N, Roux A, Bernard C, et al. Low-dose IL-2 in children with recently diagnosed type 1 diabetes: a phase I/II randomised, double-blind, placebo-controlled, dose-finding study. Diabetologia (2020) 63:1808–21. doi: 10.1007/s00125-020-05200-w
225. Spangler JB, Trotta E, Tomala J, Peck A, Young TA, Savvides CS, et al. Engineering a single-agent Cytokine/Antibody fusion that selectively expands regulatory T cells for autoimmune disease therapy. J Immunol (2018) 201:2094–106. doi: 10.4049/jimmunol.1800578
226. Peterson LB, Bell CJM, Howlett SK, Pekalski ML, Brady K, Hinton H, et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun (2018) 95:1–14. doi: 10.1016/j.jaut.2018.10.017
227. Khoryati L, Pham MN, Sherve M, Kumari S, Cook K, Pearson J, et al. An IL-2 mutein engineered to promote expansion of regulatory T cells arrests ongoing autoimmunity in mice. Sci Immunol (2020) 5. doi: 10.1126/sciimmunol.aba5264
228. Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, et al. The effect of low-dose IL-2 and treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight (2021) 6:e147474. doi: 10.1172/jci.insight.147474
229. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, Soufi KE, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Dis (2019) 78:209. doi: 10.1136/annrheumdis-2018-214229
230. Zheng Q, Xu Y, Liu Y, Zhang B, Li X, Guo F, et al. Induction of Foxp3 demethylation increases regulatory CD4+CD25+ T cells and prevents the occurrence of diabetes in mice. J Mol Med Berlin Ger (2009) 87:1191–205. doi: 10.1007/s00109-009-0530-8
231. Fisson S, Djelti F, Trenado A, Billiard F, Liblau R, Klatzmann D, et al. Therapeutic potential of self-antigen-specific CD4+CD25+ regulatory T cells selected in vitro from a polyclonal repertoire. Eur J Immunol (2006) 36:817–27. doi: 10.1002/eji.200535445
232. Liu Y-F, Powrie J, Arif S, Yang JHM, Williams E, Khatri L, et al. Immune and metabolic effects of antigen-specific immunotherapy using multiple β-cell peptides in type 1 diabetes. Diabetes (2022) 71:722–32. doi: 10.2337/db21-0728
233. Amini L, Greig J, Schmueck-Henneresse M, Volk H-D, Bézie S, Reinke P, et al. Super-treg: Toward a new era of adoptive treg therapy enabled by genetic modifications. Front Immunol (2021) 11:611638. doi: 10.3389/fimmu.2020.611638
234. Honaker Y, Hubbard N, Xiang Y, Fisher L, Hagin D, Sommer K, et al. Gene editing to induce FOXP3 expression in human CD4+ T cells leads to a stable regulatory phenotype and function. Sci Transl Med (2020) 12. doi: 10.1126/scitranslmed.aay6422
235. Fuhrman CA, Yeh W-I, Seay HR, Lakshmi PS, Chopra G, Zhang L, et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol (2015) 195:145–55. doi: 10.4049/jimmunol.1402381
236. Mason GM, Lowe K, Melchiotti R, Ellis R, de Rinaldis E, Peakman M, et al. Phenotypic complexity of the human regulatory T cell compartment revealed by mass cytometry. J Immunol (2015) 195:2030–7. doi: 10.4049/jimmunol.1500703
237. Wong HS, Park K, Gola A, Baptista AP, Miller CH, Deep D, et al. A local regulatory T cell feedback circuit maintains immune homeostasis by pruning self-activated T cells. Cell (2021) 184:3981–3997.e22. doi: 10.1016/j.cell.2021.05.028
238. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25:1282–1293.e7. doi: 10.1016/j.cmet.2016.12.018
Keywords: Treg - regulatory T cell, T cell, autoimmunity, type 1 diabetes, genetic, FOXP3
Citation: Raugh A, Allard D and Bettini M (2022) Nature vs. nurture: FOXP3, genetics, and tissue environment shape Treg function. Front. Immunol. 13:911151. doi: 10.3389/fimmu.2022.911151
Received: 02 April 2022; Accepted: 11 July 2022;
Published: 12 August 2022.
Edited by:
David Serreze, Jackson Laboratory, United StatesReviewed by:
Georgia Fousteri, San Raffaele Hospital (IRCCS), ItalyTatjana Nikolic, Leiden University, Netherlands
Copyright © 2022 Raugh, Allard and Bettini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Bettini, TWFyaWEuQmV0dGluaUBwYXRoLnV0YWguZWR1