 Negar Sadeghi Hassanabadi
Negar Sadeghi Hassanabadi Bieke Broux
Bieke Broux Sonja Marinović
Sonja Marinović Dagmar Gotthardt
Dagmar Gotthardt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 June 2022
Sec. NK and Innate Lymphoid Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.909275
This article is part of the Research Topic Drivers of Innate Lymphoid Cell Plasticity View all 9 articles
Multiple sclerosis (MS) is a highly debilitating autoimmune disease affecting millions of individuals worldwide. Although classically viewed as T-cell mediated disease, the role of innate lymphoid cells (ILC) such as natural killer (NK) cells and ILC 1-3s has become a focal point as several findings implicate them in the disease pathology. The role of ILCs in MS is still not completely understood as controversial findings have been reported assigning them either a protective or disease-accelerating role. Recent findings in experimental autoimmune encephalomyelitis (EAE) suggest that ILCs infiltrate the central nervous system (CNS), mediate inflammation, and have a disease exacerbating role by influencing the recruitment of autoreactive T-cells. Elucidating the detailed role of ILCs and altered signaling pathways in MS is essential for a more complete picture of the disease pathology and novel therapeutic targets. We here review the current knowledge about ILCs in the development and progression of MS and preclinical models of MS and discuss their potential for therapeutic applications.
Multiple sclerosis (MS) is a chronic autoimmune, inflammatory neurological disease of the central nervous system (CNS) that most commonly affects young individuals between the age of 20 and 40 (1). The number of people worldwide living with MS has increased from 2.3 million in 2013 to 2.8 million in 2020 becoming the primary cause of non-traumatic disability in young adults (1). Although disease-modifying therapies (DMTs) have alleviated symptoms and reduced the subsequent disability coming from an MS diagnosis, the disease progression still cannot be stopped (2). This is due to a complex disease etiology and pathogenesis, highlighting the unmet need to identify key players and altered drivers in MS. For decades, MS has been viewed as an immune-mediated disease primarily induced by the infiltration of classical T-cells (2). Subsequently, studies on MS and experimental autoimmune encephalomyelitis (EAE) have mostly focused on CD4+ T-cells, and DMTs that have been developed over the years mainly target these cells. The traditional view has been altered to include the involvement of innate immune cells that have long been neglected as disease mediators. Most studies investigating the role of ILCs in MS and preclinical models of MS have been focused on NK cells as ILC1-3 have been identified and classified later. Nonetheless, recent studies suggest that also other ILC family members play a crucial role in the initial activation of the autoimmune response as well as the disease progression, making them a potential target for future therapies (3–19).
MS can be divided into 3 subtypes: relapse remitting MS (RRMS), primary progressive MS (PPMS), and secondary progressive MS (SPMS) (20). The most common type is RRMS, which is characterized by recurring episodes of neurological dysfunction, followed by a clinical recovery. Many RRMS patients develop SPMS within 10 to 15 years after the RRMS diagnosis, whereby inflammatory lesions are no longer the main characteristic. Instead, progressive neurological decline occurs, followed by brain atrophy (2). Only 10 to 15% of the patients have PPMS, whereby disability progression is present from the beginning of the disease and relapses do not occur (20). The disease symptoms are heterogeneous and depending on where the lesions are in the CNS, sensory disturbances, bladder dysfunction, cognitive deficits, limb weakness, ataxia, and fatigue can occur (21). The disease pathology of MS is characterized by confluent demyelinated areas in both the white and gray matter of the brain and spinal cord. These lesions indicate loss of myelin and myelin-producing oligodendrocytes, resulting in disrupted conduction of electrical impulses (2). The pathology is due to an autoimmune response directed against myelin, whereby immune cells such as CD4+ T-cells and B-cells infiltrate the brain parenchyma and cause local tissue damage. In addition, ectopic lymphoid follicles (ELFs), which resemble germinal center-like structures can be found in the meninges of 40% of the SPMS patients. ELFs were also observed in RRMS and PPMS patients but lack features of more developed follicles such as follicular dendritic cells (FDCs), and distinct T and B cell zones (22, 23). ELFs can cause local antigen-specific responses within tissues and thereby support cortical degeneration and clinical disease progression (22). Although the causes of MS are still unknown, next to a genetic predisposition, many different environmental factors such as vitamin D deficiency, obesity, smoking, and infection with the Epstein Barr virus (EBV) have been described to play a role in developing MS (2). Emerging evidence from a longitudinal study shows that an EBV infection increases the risk of getting MS 32-fold, suggesting that EBV is the leading cause of MS (24).
ILCs are a branch of the innate immune system and an important source of innate effector cytokines (25, 26). In the last years, their resemblance to T-cells has been described, which lead to the recognition of ILCs as the innate counterparts of T-cells whereby each subset resembles a specific T-cell population in particular. There are different nomenclatures regarding ILCs, but in this review, we use the nomenclature proposed by Vivier et al. (27). ILCs can be divided into five different subsets: lymphoid tissue inducers (LTis); cytotoxic NK cells, which enter the circulation and migrate through tissues; and ILC1, 2, and 3 which are tissue-resident non-cytotoxic cells that exert their effects locally (25, 28). Interestingly, recent studies have identified ILC1 and ILC3 subsets that also have a cytotoxic nature (25, 28) suggesting that some key features are yet to be discovered in terms of subsets, functions as well as plasticity. LTis, NK cells, and ILC1, 2, and 3 differ in their transcription factor profiles as well as functions. NK cells resemble CD8+ cytotoxic T-cells whereas ILC1-3s, mirror CD4+ T helper (Th)1, Th2, and Th17 in terms of function (25, 26) which is visualized in Figure 1.
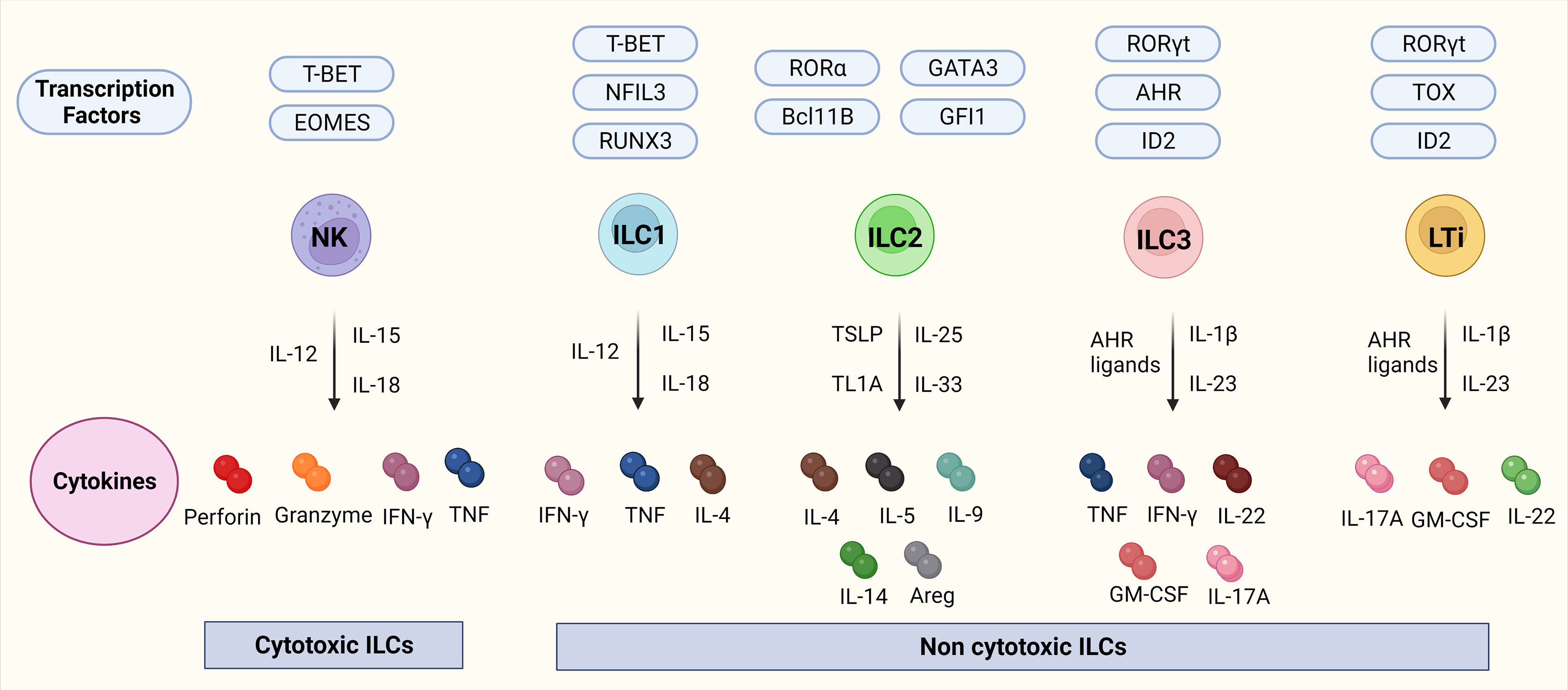
Figure 1 ILCs, their transcription factors, and cytokines. Figure 1 depicts the ILC family, their transcription factors, and cytokines in mice. NK cells are transcription factor T-bet and Eomes dependent cytotoxic ILCs that release cytokines IFN-γ, and TNF together with cytotoxic molecules such as perforin, and granzyme. ILC1-3 and LTis are non-cytotoxic ILCs. ILC1s are dependent on the transcription factors Tbet, NFIL3, and RUNX2 and release IFN-γ, TNF, and IL-4. ILC2s are dependent on the transcription factors RORα, GATA3, Bcl11B, and GFI and release IL-4, IL-5, IL-9, IL-14, and transcription factor Areg. ILC3s are dependent on the transcription factors RORγt, AHR, and ID2 and release TNF, IFN-γ, IL-22, GM-CSF, and IL-17A. LTis are dependent on the transcription factors RORγt, TOX, and ID2 and release IL-17A, GM-CSF, and IL-22. Abbreviations of transcription factors: NFIL3, nuclear factor IL-3 induced; ID2, inhibitor of DNA binding 2; TOX, thymocyte selection associated high mobility group box protein; GATA3, GATA binding protein 3; T-BET, T-box transcription factor; EOMES, Eomesodermin; RUNX3, runt-related transcription factor 3; RORα, RAR-related orphan recepto;, Bcl11b, B cell lymphoma/leukemia 11B; RORγt, RAR- related orphan receptor γt; and AhR, Aryl hydrocarbon receptor. Abbreviations of cytokines: IFN-γ, Interferon-gamma; TNF, Tumor necrosis factor-alpha; IL, Interleukin; GM-CSF, Granulocyte-macrophage colony-stimulating-factor; Areg, amphiregulin.
NK cells are dependent on the transcription factors, T-bet (Tbx21) and Eomesodermin (Eomes) and are cytotoxic cells that control tumors and viral infections by producing IFN-γ, TNF, perforin, and granzymes (25). In addition, NK cells exert regulatory effects by influencing adaptive immune responses (27). In mice, three NK cell populations have been characterized, based on their CD11b and CD27 expression (29). The double-positive and the most mature subset CD11b+CD27+ have cytolytic potential and secrete IFN-γ being the most potent killer cell population while CD11b+ CD27- cells have lower proliferative capacity indicating replicative senescent cells (29, 30). The NK cells subsets not only differ in functions but as well in their tissue distribution; the CD11b- CD27+ NK cells are mostly found in the bone marrow and lymph nodes, whereas the CD11b+ CD27- NK cells are predominantly found in the blood, spleen, lung, and liver. The double-positive NK cells can be found in all tissues and are evenly distributed (29). In humans, NK cell maturation can be distinguished based on CD56 and CD16 expression – functional homolog to the CD27/CD11b subsets in the mouse. Human NK cells are mainly divided into immature CD56brightCD16- and mature CD56dimCD16+ and subsets (31). In the peripheral blood and spleen, around 90% of the NK cells are CD56dimCD16+. These cytotoxic cells express perforin and in vitro, it was shown that they can produce IFN-γ upon interaction with tumor cells (32). In the lymph nodes and tonsils, most NK cells are CD56brightCD16- and lack perforin but produce cytokines such as IFN-γ in response to stimulation with interleukin (IL)-12, 15, and 18 (31, 33). Although there are differences between human and mouse NK cells such as the absence of the NKp30 and NKp44 activating receptors and the CD56 marker in mice, many fundamental principles such as the biology and function of NK cells can be studied in mice and applied to humans (34).
ILC1s defend against intracellular bacteria and parasites and play a role against tumor and virus-infected cells by producing cytokines (25). ILC1s, although having similar features as NK cells such as the production of IFN-γ, can be distinguished from conventional NK cells as they are only dependent on the transcription factor T-bet but not Eomes. Their similarity to NK cells causes a hurdle to correctly identify them, as specific ILC1 markers are lost upon cell activation and are tissue-dependent. As well, many ILC1 markers are shared with NK cells and ILC3s such as NK1.1, NKp44, and NKp46 (26). ILC1s can be further divided into CD127- and CD127+ cells whereby CD127- cells can produce IFN-γ and TNF upon stimulation by IL-12, IL-15, and IL-18, and CD127+ cells produce IFN-γ in response to IL-12 and IL-18 in both mouse and humans (25).
ILC2s are important for the defense against parasites and are involved in asthma and allergic diseases (24). They are dependent on the GATA3 transcription factor and when activated via cytokine receptors, produce type 2 cytokines such as IL-4, IL-5, IL-9, and IL13, and the transcription factor Areg in both mice and humans (24). ILC2 can be identified by ST2, a component of the IL-33 receptor (25).
ILC3s are dependent on the transcription factors RORγt, AHR, and ID2. They are significant producers of IL-17A, IL-22 as well as GM-CSF and TNF and promote antibacterial immunity, chronic inflammation, and tissue repair as well they can regulate adaptive Th17 cell response (25). ILC3s are heterogeneous and in mice, there are two subsets, CCR6-ILC3s whereas in humans all express CCR6 and CD117 but two subsets can be distinguished based on NKp44 expression (25).
LTis are CCR6+ transcription factor RORγt - dependent cells and during embryonic development are crucial for the formation of secondary lymph nodes and Peyer’s patches (PP) (35). In mice, there are two different subsets, which can be differentiated based on their CD4 expression. They produce similar cytokines as ILC3 whereby the only difference is that they produce IL-17A instead of TNF (35).
The experimental autoimmune encephalomyelitis (EAE) model is the most commonly used mouse model to study the immune response in MS. EAE is a CD4+ T cell-driven disease, directed against myelin antigens (36). Since it was first introduced, over 60 years ago, it has been extensively used to understand the autoimmune contribution to the pathogenesis of MS (37). EAE is induced by active immunization with myelin proteins or peptides derived from the myelin sheath, whereby the most common models are myelin oligodendrocyte glycoprotein (MOG33-55) immunized C57BL/6 mice or PLP139-151 SJL mice. The mice present symptoms such as loss of appetite, ascending paralysis, and significant inflammatory infiltration which can be found predominantly in the spinal cord but also the brain and which worsens with time (36). The MOG33-55 immunized C57BL/6 mice and PLP139-151 immunized SJL mice mimic different disease stages. The SJL mice show similarities to a relapse–remitting disease course, which is the most common form of MS. Furthermore, it also mimics a sex-dysmorphism that can be found in humans (38). The MOG33-55 immunized C57BL/6 mice are more useful to mimic the chronic stages of MS (37). Although differences between the pathophysiology of the EAE animal model and human MS exist, EAE is a very powerful tool for understanding the autoimmune and inflammatory parts of the disease. In addition, it is essential for the development of therapies and is used in pre-clinical trials to test the efficacy of drug candidates (37). Indeed, the use of EAE in pre-clinical trials has been essential for MS drugs such as Interferon beta IFN-β, Glatiramer acetate (GA), and Natalizumab (39).
In the last decade, the role of NK cells in MS has been investigated and both the beneficial and harmful role of NK cells has been demonstrated (40, 41). The fact that NK cells could play a role in MS, arises from findings of studies in MS patients, the EAE model, and DMTs influencing NK cell numbers as well as function and migration (3, 4, 40) (Table 1).

Table 1 Overview of ILCs in preclinical models of MS and in MS patients.
A study done in 1997, showed that C57BL/6 mice deprived of NK cells, using an anti-NK1.1 depletion antibody before EAE induction with MOG35-55, resulted in a serious form of EAE with frequent relapses which occurred earlier than in EAE induced control mice (40). To test that the seen effects were not caused by the depletion of NK-T cells, which are also targeted by an anti-NK1.1 antibody, they used a β2-microglobulin-/- β2m-/- mice in which NK-T-cells are absent. The β2m-/- mice depleted from NK cells developed a chronic non-remitting form of EAE with high clinical scores, providing evidence that NK cells can play a regulatory role in a manner independent of CD8+ T cells or NK1.1+ T cells. In addition, in the same study enhanced disease progression was observed upon NK cell depletion in Rag2-/- mice lacking T-, NK-T, and B-cells after disease induction by adoptive transfer of MOG-specific T-cells (40). This study clearly indicates a disease protective role of NK cells (40). Similar results were found in NK-depleted SJL/J mice (5), whereby the depletion of NK cells by an anti-NK1.1 monoclonal antibody, one day before and 14 days after the immunization with PLP136–150 lead to an enhanced form of EAE (5). The authors describe that the disease protection of NK cells was mediated by a cytotoxic effect on autoantigen-specific encephalitogenic T-cells, which are known to play a vital role in the autoimmune attack in MS (5). In contrast to this, another study demonstrated an NK cell depletion with either anti-NK1.1, anti-asialo GM1, and anti-Ly49 in C57BL/6 before EAE induction with MOG35-55 results in a decrease in clinical pathology and relapses (41). The authors also report higher survival, thereby showing a disease-accelerating role of NK cells in EAE (41). It must be noted that an NK1.1 antibody depletes not only NK cells but also ILC1s and partially ILC3s and does not allow for discrimination between the role of these cell types whereas the used anti-asialo GM1 and anti-Ly49H preferentially deplete NK cells. The authors also state without providing the data that depletion of NK cells after the immunization did not alter the clinical symptoms in the mice which supports the notion that NK cells have only a regulatory role early in the disease development. Another study supported the disease-accelerating role of NK cells which was based on the fact that IL-18 can promote the production of IFN-γ by NK and Th1 cells. In the study, IL-18-/- mice were completely protected from EAE after MOG immunization, while IL-18 administration restored the disease partially in the presence of NK cells but not when NK1.1+ cells were depleted before immunization (6). Likewise as mentioned above the study also notes that NK depletion after primary immunization has no effects on the EAE course further providing evidence that NK cells have a role in the initiation of the disease but not in the progression.
Human studies have implicated NK cells directly in the process of demyelination and thus support the notion of a disease-accelerating role. In a study, using human brain-derived oligodendrocytes (OLs) from surgery, it was found that CD3+CD19+CD14+ depleted mononuclear cell preparations (MNCs) were cytotoxic toward OLs (7). This effect was visible when using NK cell-enriched MNCs and OLs from either the same donor (autologous donors) or healthy volunteers and OLs from epilepsy patients (heterogenous donors). NK cell-mediated cytotoxicity towards OLs was further increased when NK cells were activated with IL-2. The same group showed in another study that OLs from patients with MS lesions express MICA/B ligands for the activating NK cell receptor NKG2D that were not detected in healthy control samples. Blocking NKG2D on NK cells significantly inhibited the killing of OLs indicating an NKG2D-mediated killing of OLs by NK cells (8). Using cortical CNS biopsies of patients diagnosed with inflammatory demyelination a study showed the presence of brain perivascular granzyme B+ NK cells. The authors further showed in a Th/+ (BCR transgenic for MOG) mouse model that NK cells strongly aggravate the extent of perivascular cortical demyelination, providing evidence for the relevance of NK cells in perivascular cortical demyelination and contribution to the neurodegeneration of MS (9). In addition, autoreactive 2D2 T-cells were either transferred into RAG1−/− (lacking mature T– and B–cells) or into RAG1−/− γc−/− (no mature T– and B–cells, no NK cells) mice and the latter ones showed significantly less perivascular cortical demyelination. However, the extravasation of NK cells into the cortical parenchyma required activated T cells. These data suggest that NK cells contribute to the demyelination process in the presence of pathogenic antibodies by performing ADCC (10).
Recently, EBV infection has been proven to be a leading cause of MS (24). As NK cells protect against viral infections, they play a vital role in the early defense against an EBV infection (11). Findings suggest that during infectious mononucleosis (IM) caused by a primary EBV infection, NK cell numbers were significantly elevated both at the diagnosis and during the first month of an IM diagnosis (12, 13). Furthermore, significant changes in cell phenotype and function of NK cells were also detected.
Induction of EAE in transcription factor T-bet-deficient mice showed that myelin-reactive pathogenic CD4+ Th-17 cells invaded the CNS and caused major lesions throughout the CNS tissue in immuno-competent hosts but were completely absent from the CNS parenchyma in T-bet-deficient hosts (4). Furthermore, disease induction by an adoptive transfer of autoreactive CD4+ Th17 cells into NK1.1 depleted mice resulted in resistance to EAE. In addition, mice with NKp46-lineage-specific deletion of the transcription factor T-bet significantly decreased the incidence of inflammation using the adoptive transfer model (4). Interestingly, the study also used NKp46 lineage-specific deletion of transcription factor Eomes in mice as a tool to study the absence of conventional NK cells only and found that these NK cell-deficient mice developed a similarly severe paralysis after EAE induction compared to NK cell-sufficient mice. This indicates, in contrast to prior studies, that ILC1s but not NK cells play a role in the immunopathogenesis of EAE. The authors concluded that the pathogenic function of the transcription factor T-bet is rather dependent on ILC1 and the NKp46+ subset of ILC3s. A study investigating phenotypic and functional plasticity of murine ILCs found that the meningeal subsets of NKp46+ ILC1s and ILC3s are also characterized by a specific gene signature that was not found in splenic or meningeal NK cells and might contribute to the disease pathology (14). Another study found an increase of ILC1s in the meninges of EAE-diseased in comparison to healthy wild-type C57BL/6 mice (19). Others also found that in contrast to NK cells, ILC1s are enriched in the choroid plexus as well as in the brain parenchyma and meninges after EAE induction (15). However, while ILC1s maintain stable IFN-γ and TNF levels, NK cells show increased production of these cytokines during EAE progression. These findings at least indicate again that besides NK cells, CNS-ILC1s could be involved in the control of neuroinflammation in the brain (15).
ILC2s are the predominant ILC population in human as well as mouse brains and have been implicated in the development of several diseases such as allergy, asthma, dermatitis, and fibrosis (16). Studies also suggest the involvement of ILC2s in the disease pathology of MS (15, 16, 40). Like ILC1s, ILC2s have also been found in the meninges of wildtype C57BL/6 mice (19). A study from 2020, using an HSV-IL-2 model of CNS demyelination where mice were infected with a recombinant HSV-1 expressing murine interleukin-2 showed that NK cells did not play a role in demyelination in this model (16). However, ILC2-/- mice infected with the HSV-IL-2 virus were protected from demyelination whereas ILC1-/- and ILC3-/- mice showed demyelination to the same extent as wild type mice. Additionally, adoptive transfer of bone marrow-derived ILC2 from wild-type mice into ILC2-/- infected HSV-IL-2 virus restored demyelination visible in the brain, spinal cord, and optic nerve implying that CNS demyelination is dependent on ILC2s but not ILC1s and ILC3s (16). As previously mentioned, studies of EAE in SJL mice serve as a useful model of sex-dimorphism in MS (38). A recent study suggested that the female bias for disease development comes from a lack of male-specific IL-33 expression in mast cells, likely influenced directly by testosterone (17). Interestingly, instead of IL-33, immunized wild-type females express IL-1β and TNF that exert a variety of pathogenic effects and disease promotion. IL-33 plays an important role in the activation of ILC2s, a population that is essential for skewing the immune environment towards a protective Th2-dominated response of wild-type males rather than the harmful Th17-dominated response seen in females (17). The protective role of ILC2s was further corroborated in SJL-cKit mutant mice (SJL-KitW/Wv) where it was shown that c-kit mutations induce severe EAE in males, but not females, due to the deficit of c-kit+ ILC2s and mast cells (18).
A study investigating the contribution of ILCs to autoimmune neuroinflammation via their immediate responsiveness to IL-23 signaling found that ILC3s are not only present at mucosal surfaces but indeed can be found in the CNS during steady-state and inflammation (19). Especially IL-17A-producing ILC3s have been found in the CNS but the depletion of Thy1+ ILCs did not alter the progression of EAE (19). The same study that identified ILC1s and ILC2s in meninges of wildtype C57BL/6 mice identified an ILC3 subset (CD45+Lin-IL-7Ra+RORγt+), which also resides in the meninges (42). To assess the role of this ILC3 subtype, the authors induced EAE with MOG35-55 in the C57BL/6 mice and found an increased number of IFN-γ, GM-CSF, and IL-17 producing ILC3s in the meninges of EAE induced mice. It is therefore assumed that this subset plays an essential role in MS by promoting inflammation and thereby creating an environment that promotes the survival and reactivation of encephalitogenic T-cells (42).
LTi cells are elevated in the CNS and circulation of MS patients, providing evidence for a possible pathogenic role of LTis (42–45). A study published in 2016, used Cerebrospinal fluid (CSF) samples from MS patients and healthy controls to compare the levels of different ILCs (43). They found that CSF samples from MS patients compared to healthy controls have an increased frequency of CD56- RORγt+ LTis while the frequency of CD56+ RORγt ILC3s was unaltered. LTis are important producers of IL-22 and during an MS relapse, the balance of cytokines is shifted towards a pro-inflammatory profile whereby IL-22 is assumed to promote blood-brain barrier (BBB) disruption and CNS inflammation (42). The same study that found an increased number of ILC3s in the meninges of EAE induced mice, showed that also LTis are significantly increased in the meninges and the CNS at the peak of EAE (19). They also showed that Rorc-/- mice are protected against EAE, which might be due to the lack of LTis or their subsequent inability to mobilize to the meninges providing further support for the involvement of LTis in the development of ELFs (42). That LTi subtypes also reside in the meninges, is an important finding as LTis are also speculated to be involved in the development of ELFs in MS patients, and ELFs are commonly formed in the meningeal tissues in humans and mice. It is assumed that the LTi subset residing in the meninges has a direct role in orchestrating the formation of ELFs, which are hallmarks of chronic autoimmune inflammatory diseases such as MS.
Together, these findings provide evidence that ILC subtypes reside in the meninges and can infiltrate the CNS. Next to this, these findings show that ILCs release cytokines that regulate the inflammatory response of T-cells, as well as cytokines that are pro-inflammatory and exacerbate the immune response in MS, which can be seen in Figure 2. The specific role of ILCs is still unclear as contradictory results have been found.
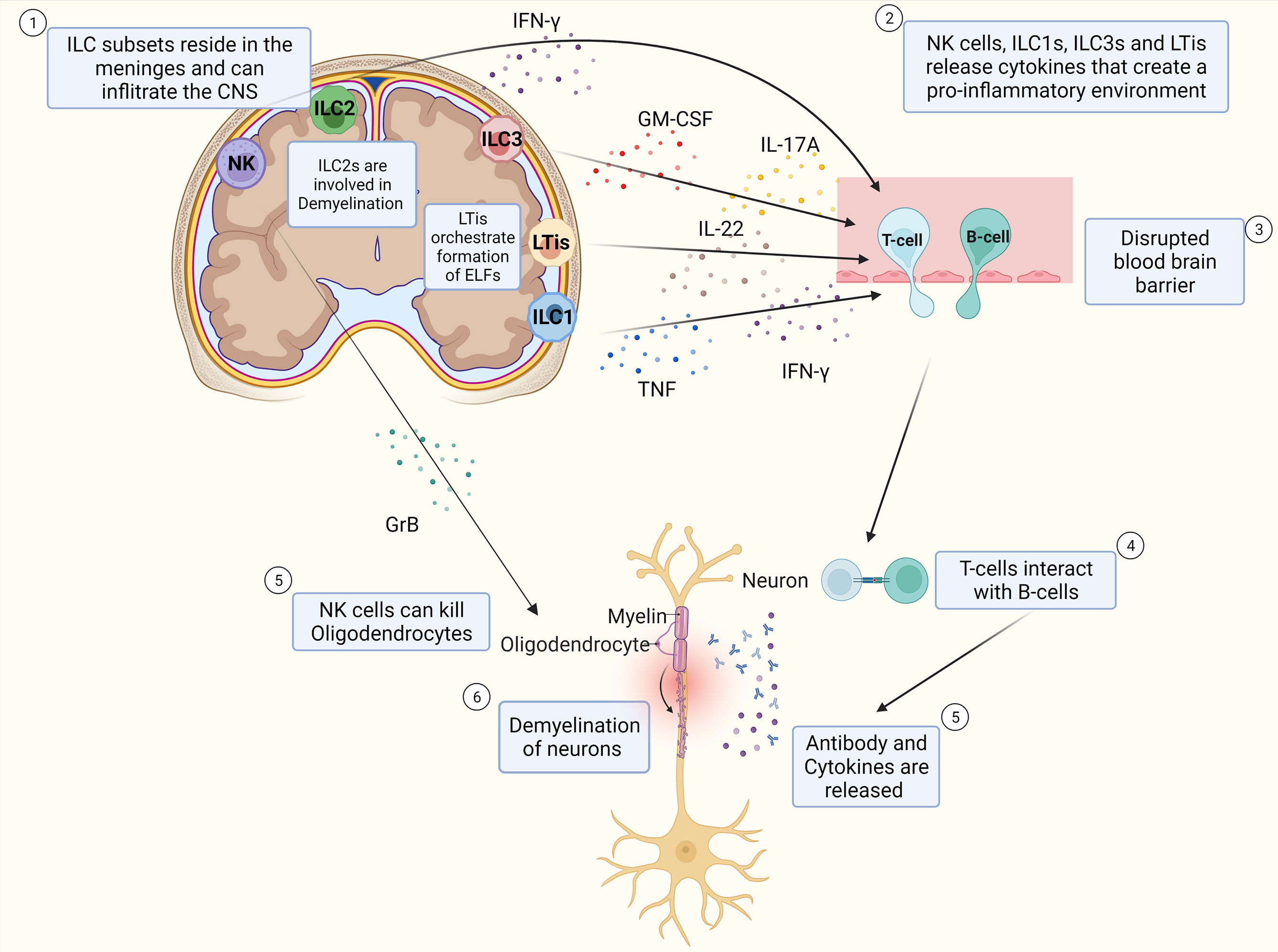
Figure 2 Schematic Overview of potential pathophysiology of MS and role of ILCs. T-cells and B-cells infiltrate the CNS through a leaky blood-brain barrier whereby ILC subtypes that reside in the meninges release pro-inflammatory cytokines that influence the extent of CD4+ T-cell infiltration into the CNS. T-cells interact with B-cells and release cytokines and antibodies that cause inflammation which damages the myelin sheath of neurons and thereby induce demyelination. NK cells can also kill oligodendrocytes directly thereby potentially contributing to the extent of demyelination of the neurons. ILC2s are assumed to play a role in demyelination. LTis are assumed to play a role in the development of ELFs.
Next to IFN-β and Glatiramer acetate other immunomodulatory therapies for MS have been approved in the last years, such as the anti-inflammatory Dimethyl fumarate (DMF), the highly potent migration inhibitors such as Natalizumab and Fingolimod (FTY720), and the IL-2 receptor modulating Daclizumab (10). Importantly, all aforementioned drugs also influence NK cells next to their original target. Some drugs, such as Daclizumab and Fingolimod, show an additional effect on other members of the ILC family (Table 2).
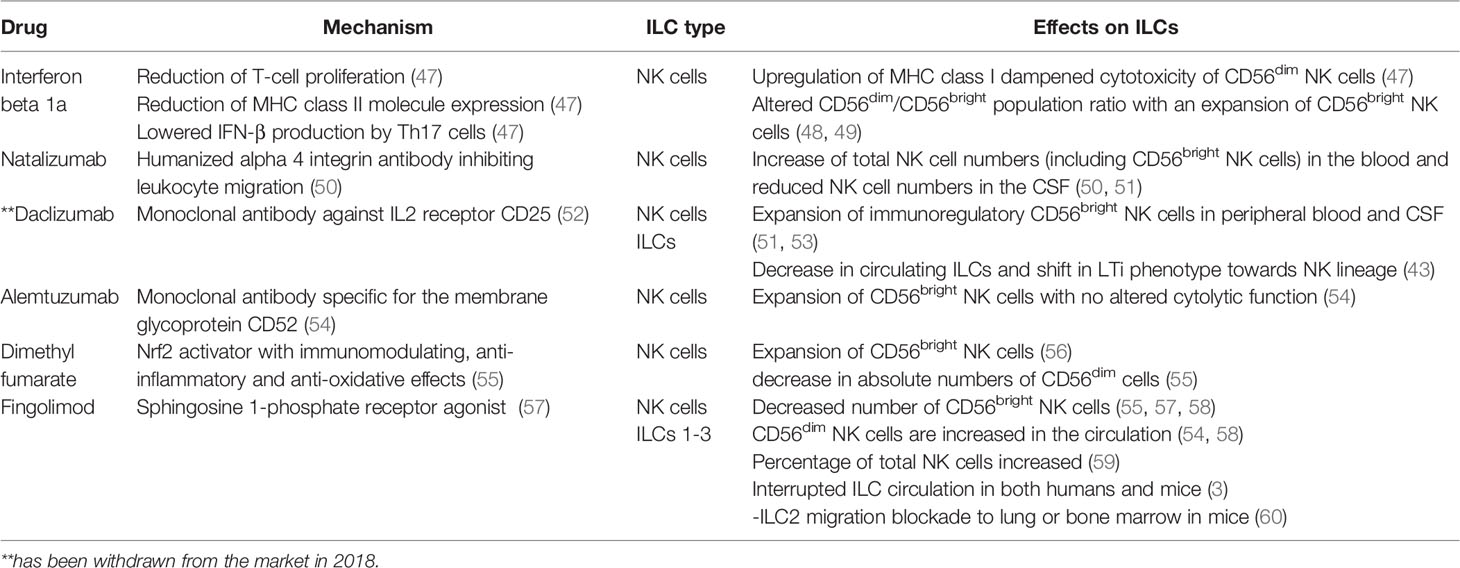
Table 2 Overview of DMTs for MS influencing ILCs.
IFN-β reduces T-cell proliferation, leads to a reduction of MHC class II molecule expression, and lowers IL-17A, TNF, and IFN-γ production in Th17/Th1 cells (47). It also upregulates MHC class I, which is the main inhibitory ligand for CD56dim NK cells thereby dampening cytotoxicity of the CD56dim NK cell population (61). In parallel, IFN-β alters the CD56bright and dim population ratio in the peripheral blood with an expansion of CD56bright NK cells after treatment (48). Whether these changes in the NK cell immune phenotype are relevant for therapeutic success is not understood (49, 61). In patients treated with Natalizumab- a humanized alpha 4 integrin (CD49d) antibody inhibiting leukocyte migration, an increase of total NK cells and CD56bright NK cells in blood comes concomitantly with reduced NK cell numbers in the CSF suggesting that transmigration of NK cells into the CNS is CD49d dependent (50, 51, 53). Daclizumab - a monoclonal antibody blocking the IL2 receptor α (CD25) is assumed to reduce early T-cell activation by the expansion of CD56bright NK cells (52, 62). Daclizumab both boosts NK cell cytolytic function in a DC-dependent manner and renders antigen-activated T cells more sensitive toward NK-mediated lysis, thus restoring defective NK cell-mediated control of T cell activity in MS (51, 52). Next to this, in MS patients treated with Daclizumab, a decrease in circulating ILCs can be found as well as a shift in the phenotype of ILCs, from LTis toward an NK cell lineage (44). However, Daclizumab has been withdrawn from the market in 2018 due to cases of severe inflammatory brain disease with fatal outcomes (63).
Alemtuzumab, a humanized monoclonal antibody that is specific for the membrane glycoprotein CD52, is highly effective in long-lasting suppression of the disease activity in RRMS patients (54). A study investigating the phenotype and effector function of innate immune cells in RRMS patients shows that the decrease of CD4+ T-cells was accompanied by an increase in ILCs which mainly was due to an expansion of the CD56bright NK cells although it has to be mentioned that the cytolytic function of NK cells was not altered 6 months after alemtuzumab treatment (54).
DMF is an Nrf2 activator with immunomodulating, as well as anti-inflammatory and anti-oxidative effects (55). Studies show that MS patients treated with DMF have reduced percentages of pro-inflammatory CD4+ and CD8+ T cells and an expansion of CD56bright NK cells and a modest increase in absolute numbers of CD56dim cells (55, 56). FYT20, a sphingosine 1-phosphate receptor agonist used as an oral compound for the treatment of MS shows efficacy in reducing inflammation in the CNS of MS patients (57). NK cells express two receptors affected by this treatment- S1PR1 and S1PR5 consequently affecting their egress from the lymph nodes (3). Data on the effect of long-term FTY20 treatment on NK cells are however contradictory (57, 59, 64). In a clinical phase IV trial in 17 RRMS patients, the longitude impact of FYT20 treatment on NK cells was assessed (58). An increased frequency of circulating CD56dim mature NK cells was found while the frequency of CD56bright and CD127+ ILCs decreased over time (58). This is in line with two other studies reporting a significant decrease of CD56bright NK cells in the peripheral blood of FYT20 treated patients (55, 57). Other studies found that in FYT20 treated MS patients the percentage of NK cells increased when compared to treatment-naïve patients (56) or that NK cells were not affected by FYT20 (59).
A study investigating whether ILC subsets such as ILC1-3, like NK cells, also express S1PR1, which is targeted by FYT20 found that ILCs 1-3 indeed express S1PR1 in both humans and mice (65). A comparison between treatment-free MS patients and patients receiving FYT20 further showed that the absolute number of total ILC from all ILC subsets was reduced in the peripheral blood. Similar results were also found in mice (20). In mice, injections with FYT20 resulted also in a blocked ILC2 migration from intestines to lung or bone marrow during an inflammatory state (65) indicating that FYT20 treatment affects several immune subsets.
NK cells and other ILCs have long been neglected as players in MS but accumulating evidence shows that ILCs indeed play a role in the disease pathology. While on the one hand ILCs have been described to prevent autoimmunity, several studies implicated them in a disease-promoting role in EAE by influencing T-cells and releasing cytokines that enhance the pro-inflammatory response.
In addition, in vitro, and in vivo mouse studies suggest that NK cells are also able to directly target oligodendrocytes- adding a layer of complexity as they might not only be involved in the neuroinflammation aspect but also directly in the neurodegenerative portion of MS.
Different depletion methods of NK cells and additional different experimental conditions such as gender and age of mice, peptides used for EAE induction, or housing conditions (SPF status, microbiota) might play a role in the contradictory findings of the aforementioned studies. As well the amount of peptides used and whether a direct EAE induction or passive EAE induction was used, could additionally have influenced the outcome of the studies. Next to this, several studies have been performed before the discovery or strict classification of different ILC subsets making it hard to clearly interpret their role in MS. In addition, a certain plasticity of ILCs has been reported and, likely, ILCs can also adapt to environmental cues during neuroinflammation changing their transcription factor and cytokine profile.
Given that ILCs interact with other immune cells such as T- and B-cells, it would be important to get a deeper understanding of the interaction of the different innate and adaptive immune subsets during the disease initiation and progression. The fact that all types of ILCs are recruited to the meninges and CNS indicates a complex interaction of these cell types. Another question that still needs to be addressed is whether ILC subsets are only involved in the initiation phase of disease by opening the blood-brain barrier and recruiting other immune cells, thereby triggering local T-cell and B-cell responses, or whether they are also crucial for the maintenance and progression of the disease. Deepening our understanding of the role of ILCs in the progression and not the initiation phase would be of importance to design better therapies.
Although there are several treatment options for MS, most of the treatments are perceived as burdening the patients due to the administration route and severe side effects Consequently, there is a high unmet medical need for new therapeutics which are more patient-friendly and possibly delay and minimize the neurodegenerative part of the disease (20). Due to their pharmacological safety, tolerability, and efficacy profiles, peptides present a unique starting point for creating new treatments for a wide range of diseases (58, 60). Thus, it is attractive to further explore the option of peptide therapeutics for MS. Indeed, the circular plant peptide T20J kB1 (T20K) has been shown to silence T-cell proliferation in an IL-2-dependent mechanism, and treating MOG-induced mice with the peptide led to a significantly delayed onset of clinical symptoms in the EAE model (58). This compound is currently tested in clinical trials and it is attractive to speculate that T20K might have an additional impact on innate lymphoid cells as IL-2 can promote activation and cell growth of ILCs.
Next to this, the finding of ILC subset residing in the meninges could be of deep interest since they lie outside of the blood brain barrier and are more targetable. It is therefore essential to further investigate how ILCs accumulate in the meninges. It is not clear yet whether meningeal ILCs are homeostatic gatekeepers and have the potential to locally proliferate in a disease-promoting environment or whether circulating ILCs are recruited to the CNS during the disease.
In conlusion, increasing our understanding of the complex interplay of different immune cells including ILCs during the disease initiation and progression will be crucial to find alternative treatment options.
NH, DG, SM, and BB discussed the literature and wrote the manuscript. NH generated all figures. All authors contributed to the article and approved the submitted version.
NH and DG are supported by a grant from the Austrian Science Fund (FWF): ZK-81B. SM is supported by the JESH program of the Austrian Academy of Sciences. BB is supported by the Fonds voor Wetenschappelijk Onderzoek (FWO), the Belgian Charcot Foundation, Stichting MS Research, Multiple Sclerosis International Federation (MSIF) and MoveS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We want to thank our laboratory members and the entire Peptaides/ZK-81B community as well as K. Klein for numerous discussions and to apologize to all investigators whose research was not cited. BioRender.com was used for all Figures.
1. Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, et al. Rising Prevalence of Multiple Sclerosis Worldwide: Insights From the Atlas of Ms, Third Edition. Mult Scler (2020) 26(14):1816–21. doi: 10.1177/1352458520970841
2. Dendrou CA, Fugger L, Friese MA. Immunopathology of Multiple Sclerosis. Nat Rev Immunol (2015) 15(9):545–58. doi: 10.1038/nri3871
3. Eken A, Yetkin MF, Vural A, Okus FZ, Erdem S, Azizoglu ZB, et al. Fingolimod Alters Tissue Distribution and Cytokine Production of Human and Murine Innate Lymphoid Cells. Front Immunol (2019) 10:217. doi: 10.3389/fimmu.2019.00217
4. Kwong B, Rua R, Gao Y, Flickinger J Jr., Wang Y, Kruhlak MJ, et al. T-Bet-Dependent Nkp46(+) Innate Lymphoid Cells Regulate the Onset of Th17-Induced Neuroinflammation. Nat Immunol (2017) 18(10):1117–27. doi: 10.1038/ni.3816
5. Xu W, Fazekas G, Hara H, Tabira T. Mechanism of Natural Killer (Nk) Cell Regulatory Role in Experimental Autoimmune Encephalomyelitis. J Neuroimmunol (2005) 163(1-2):24–30. doi: 10.1016/j.jneuroim.2005.02.011
6. Shi FD, Takeda K, Akira S, Sarvetnick N, Ljunggren HG. Il-18 Directs Autoreactive T Cells and Promotes Autodestruction in the Central Nervous System Via Induction of Ifn-Gamma by Nk Cells. J Immunol (2000) 165(6):3099–104. doi: 10.4049/jimmunol.165.6.3099
7. Morse RH, Seguin R, McCrea EL, Antel JP. Nk Cell-Mediated Lysis of Autologous Human Oligodendrocytes. J Neuroimmunol (2001) 116(1):107–15. doi: 10.1016/s0165-5728(01)00289-2
8. Saikali P, Antel JP, Newcombe J, Chen Z, Freedman M, Blain M, et al. Nkg2d-Mediated Cytotoxicity Toward Oligodendrocytes Suggests a Mechanism for Tissue Injury in Multiple Sclerosis. J Neurosci (2007) 27(5):1220–8. doi: 10.1523/JNEUROSCI.4402-06.2007
9. Lagumersindez-Denis N, Wrzos C, Mack M, Winkler A, van der Meer F, Reinert MC, et al. Differential Contribution of Immune Effector Mechanisms to Cortical Demyelination in Multiple Sclerosis. Acta Neuropathol (2017) 134(1):15–34. doi: 10.1007/s00401-017-1706-x
10. Gross CC, Schulte-Mecklenbeck A, Wiendl H, Marcenaro E, Kerlero de Rosbo N, Uccelli A, et al. Regulatory Functions of Natural Killer Cells in Multiple Sclerosis. Front Immunol (2016) 7:606. doi: 10.3389/fimmu.2016.00606
11. Azzi T, Lunemann A, Murer A, Ueda S, Beziat V, Malmberg KJ, et al. Role for Early-Differentiated Natural Killer Cells in Infectious Mononucleosis. Blood (2014) 124(16):2533–43. doi: 10.1182/blood-2014-01-553024
12. Hendricks DW, Balfour HH Jr., Dunmire SK, Schmeling DO, Hogquist KA, Lanier LL. Cutting Edge: Nkg2c(Hi)Cd57+ Nk Cells Respond Specifically to Acute Infection With Cytomegalovirus and Not Epstein-Barr Virus. J Immunol (2014) 192(10):4492–6. doi: 10.4049/jimmunol.1303211
13. Williams H, McAulay K, Macsween KF, Gallacher NJ, Higgins CD, Harrison N, et al. The Immune Response to Primary Ebv Infection: A Role for Natural Killer Cells. Br J Haematol (2005) 129(2):266–74. doi: 10.1111/j.1365-2141.2005.05452.x
14. Verrier T, Satoh-Takayama N, Serafini N, Marie S, Di Santo JP, Vosshenrich CA. Phenotypic and Functional Plasticity of Murine Intestinal Nkp46+ Group 3 Innate Lymphoid Cells. J Immunol (2016) 196(11):4731–8. doi: 10.4049/jimmunol.1502673
15. Romero-Suarez S, Del Rio Serrato A, Bueno RJ, Brunotte-Strecker D, Stehle C, Figueiredo CA, et al. The Central Nervous System Contains Ilc1s That Differ From Nk Cells in the Response to Inflammation. Front Immunol (2019) 10:2337. doi: 10.3389/fimmu.2019.02337
16. Hirose S, Jahani PS, Wang S, Jaggi U, Tormanen K, Yu J, et al. Type 2 Innate Lymphoid Cells Induce Cns Demyelination in an Hsv-Il-2 Mouse Model of Multiple Sclerosis. iScience (2020) 23(10):101549. doi: 10.1016/j.isci.2020.101549
17. Russi AE, Walker-Caulfield ME, Ebel ME, Brown MA. Cutting Edge: C-Kit Signaling Differentially Regulates Type 2 Innate Lymphoid Cell Accumulation and Susceptibility to Central Nervous System Demyelination in Male and Female Sjl Mice. J Immunol (2015) 194(12):5609–13. doi: 10.4049/jimmunol.1500068
18. Stier MT, Zhang J, Goleniewska K, Cephus JY, Rusznak M, Wu L, et al. Il-33 Promotes the Egress of Group 2 Innate Lymphoid Cells From the Bone Marrow. J Exp Med (2018) 215(1):263–81. doi: 10.1084/jem.20170449
19. Mair F, Becher B. Thy1+ Sca1+ Innate Lymphoid Cells Infiltrate the Cns During Autoimmune Inflammation, But Do Not Contribute to Disease Development. Eur J Immunol (2014) 44(1):37–45. doi: 10.1002/eji.201343653
20. Yong HYF, Yong VW. Mechanism-Based Criteria to Improve Therapeutic Outcomes in Progressive Multiple Sclerosis. Nat Rev Neurol (2022) 18(1):40–55. doi: 10.1038/s41582-021-00581-x
21. Huang WJ, Chen WW, Zhang X. Multiple Sclerosis: Pathology, Diagnosis and Treatments. Exp Ther Med (2017) 13(6):3163–6. doi: 10.3892/etm.2017.4410
22. Zhan J, Kipp M, Han W, Kaddatz H. Ectopic Lymphoid Follicles in Progressive Multiple Sclerosis: From Patients to Animal Models. Immunology (2021) 164(3):450–66. doi: 10.1111/imm.13395
23. Negron A, Stuve O, Forsthuber TG. Ectopic Lymphoid Follicles in Multiple Sclerosis: Centers for Disease Control? Front Neurol (2020) 11:607766. doi: 10.3389/fneur.2020.607766
24. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated With Multiple Sclerosis. Science (2022) 375(6578):296–301. doi: 10.1126/science.abj8222
25. Artis D, Spits H. The Biology of Innate Lymphoid Cells. Nature (2015) 517(7534):293–301. doi: 10.1038/nature14189
26. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years on. Cell (2018) 174(5):1054–66. doi: 10.1016/j.cell.2018.07.017
27. Lunemann A, Lunemann JD, Munz C. Regulatory Nk-Cell Functions in Inflammation and Autoimmunity. Mol Med (2009) 15(9-10):352–8. doi: 10.2119/molmed.2009.00035
28. Krabbendam L, Bernink JH, Spits H. Innate Lymphoid Cells: From Helper to Killer. Curr Opin Immunol (2021) 68:28–33. doi: 10.1016/j.coi.2020.08.007
29. Hayakawa Y, Smyth MJ. Cd27 Dissects Mature Nk Cells Into Two Subsets With Distinct Responsiveness and Migratory Capacity. J Immunol (2006) 176(3):1517–24. doi: 10.4049/jimmunol.176.3.1517
30. Kim S, Iizuka K, Kang HS, Dokun A, French AR, Greco S, et al. In Vivo Developmental Stages in Murine Natural Killer Cell Maturation. Nat Immunol (2002) 3(6):523–8. doi: 10.1038/ni796
31. Cooper MA, Fehniger TA, Caligiuri MA. The Biology of Human Natural Killer-Cell Subsets. Trends Immunol (2001) 22(11):633–40. doi: 10.1016/s1471-4906(01)02060-9
32. Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human Nk Cell Education by Inhibitory Receptors for Mhc Class I. Immunity (2006) 25(2):331–42. doi: 10.1016/j.immuni.2006.06.013
33. Ferlazzo G, Munz C. Nk Cell Compartments and Their Activation by Dendritic Cells. J Immunol (2004) 172(3):1333–9. doi: 10.4049/jimmunol.172.3.1333
34. Sungur CM, Murphy WJ. Utilization of Mouse Models to Decipher Natural Killer Cell Biology and Potential Clinical Applications. Hematol Am Soc Hematol Educ Program (2013) 2013:227–33. doi: 10.1182/asheducation-2013.1.227
35. Bar-Ephraim YE, Mebius RE. Innate Lymphoid Cells in Secondary Lymphoid Organs. Immunol Rev (2016) 271(1):185–99. doi: 10.1111/imr.12407
36. Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental Autoimmune Encephalomyelitis (Eae) as a Model for Multiple Sclerosis (Ms). Br J Pharmacol (2011) 164(4):1079–106. doi: 10.1111/j.1476-5381.2011.01302.x
37. Robinson AP, Harp CT, Noronha A, Miller SD. The Experimental Autoimmune Encephalomyelitis (Eae) Model of Ms: Utility for Understanding Disease Pathophysiology and Treatment. Handb Clin Neurol (2014) 122:173–89. doi: 10.1016/B978-0-444-52001-2.00008-X
38. Papenfuss TL, Rogers CJ, Gienapp I, Yurrita M, McClain M, Damico N, et al. Sex Differences in Experimental Autoimmune Encephalomyelitis in Multiple Murine Strains. J Neuroimmunol (2004) 150(1-2):59–69. doi: 10.1016/j.jneuroim.2004.01.018
39. Steinman L, Zamvil SS. Virtues and Pitfalls of Eae for the Development of Therapies for Multiple Sclerosis. Trends Immunol (2005) 26(11):565–71. doi: 10.1016/j.it.2005.08.014
40. Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T. Regulation of Experimental Autoimmune Encephalomyelitis by Natural Killer (Nk) Cells. J Exp Med (1997) 186(10):1677–87. doi: 10.1084/jem.186.10.1677
41. Winkler-Pickett R, Young HA, Cherry JM, Diehl J, Wine J, Back T, et al. In Vivo Regulation of Experimental Autoimmune Encephalomyelitis by Nk Cells: Alteration of Primary Adaptive Responses. J Immunol (2008) 180(7):4495–506. doi: 10.4049/jimmunol.180.7.4495
42. Hatfield JK, Brown MA. Group 3 Innate Lymphoid Cells Accumulate and Exhibit Disease-Induced Activation in the Meninges in Eae. Cell Immunol (2015) 297(2):69–79. doi: 10.1016/j.cellimm.2015.06.006
43. Degn M, Modvig S, Dyring-Andersen B, Bonefeld CM, Frederiksen JL, Geisler C, et al. Increased Prevalence of Lymphoid Tissue Inducer Cells in the Cerebrospinal Fluid of Patients With Early Multiple Sclerosis. Mult Scler (2016) 22(8):1013–20. doi: 10.1177/1352458515609795
44. Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, et al. Inhibition of Lti Cell Development by Cd25 Blockade Is Associated With Decreased Intrathecal Inflammation in Multiple Sclerosis. Sci Transl Med (2012) 4(145):145ra06. doi: 10.1126/scitranslmed.3004140
45. Serafini B, Rosicarelli B, Veroni C, Zhou L, Reali C, Aloisi F. Rorgammat Expression and Lymphoid Neogenesis in the Brain of Patients With Secondary Progressive Multiple Sclerosis. J Neuropathol Exp Neurol (2016) 75(9):877–88. doi: 10.1093/jnen/nlw063
46. Gross CC, Schulte-Mecklenbeck A, Hanning U, Posevitz-Fejfar A, Korsukewitz C, Schwab N, et al. Distinct Pattern of Lesion Distribution in Multiple Sclerosis Is Associated With Different Circulating T-Helper and Helper-Like Innate Lymphoid Cell Subsets. Mult Scler (2017) 23(7):1025–30. doi: 10.1177/1352458516662726
47. Filipi M, Jack S. Interferons in the Treatment of Multiple Sclerosis: A Clinical Efficacy, Safety, and Tolerability Update. Int J MS Care (2020) 22(4):165–72. doi: 10.7224/1537-2073.2018-063
48. Martinez-Rodriguez JE, Lopez-Botet M, Munteis E, Rio J, Roquer J, Montalban X, et al. Natural Killer Cell Phenotype and Clinical Response to Interferon-Beta Therapy in Multiple Sclerosis. Clin Immunol (2011) 141(3):348–56. doi: 10.1016/j.clim.2011.09.006
49. Saraste M, Irjala H, Airas L. Expansion of Cd56bright Natural Killer Cells in the Peripheral Blood of Multiple Sclerosis Patients Treated With Interferon-Beta. Neurol Sci (2007) 28(3):121–6. doi: 10.1007/s10072-007-0803-3
50. Putzki N, Baranwal MK, Tettenborn B, Limmroth V, Kreuzfelder E. Effects of Natalizumab on Circulating B Cells, T Regulatory Cells and Natural Killer Cells. Eur Neurol (2010) 63(5):311–7. doi: 10.1159/000302687
51. Gross CC, Schulte-Mecklenbeck A, Runzi A, Kuhlmann T, Posevitz-Fejfar A, Schwab N, et al. Impaired Nk-Mediated Regulation of T-Cell Activity in Multiple Sclerosis Is Reconstituted by Il-2 Receptor Modulation. Proc Natl Acad Sci U.S.A. (2016) 113(21):E2973–82. doi: 10.1073/pnas.1524924113
52. Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, et al. Regulatory Cd56(Bright) Natural Killer Cells Mediate Immunomodulatory Effects of Il-2ralpha-Targeted Therapy (Daclizumab) in Multiple Sclerosis. Proc Natl Acad Sci U S A (2006) 103(15):5941–6. doi: 10.1073/pnas.0601335103
53. Skarica M, Eckstein C, Whartenby KA, Calabresi PA. Novel Mechanisms of Immune Modulation of Natalizumab in Multiple Sclerosis Patients. J Neuroimmunol (2011) 235(1-2):70–6. doi: 10.1016/j.jneuroim.2011.02.010
54. Gross CC, Ahmetspahic D, Ruck T, Schulte-Mecklenbeck A, Schwarte K, Jorgens S, et al. Alemtuzumab Treatment Alters Circulating Innate Immune Cells in Multiple Sclerosis. Neurol Neuroimmunol Neuroinflamm (2016) 3(6):e289. doi: 10.1212/NXI.0000000000000289
55. Montes Diaz G, Fraussen J, Van Wijmeersch B, Hupperts R, Somers V. Dimethyl Fumarate Induces a Persistent Change in the Composition of the Innate and Adaptive Immune System in Multiple Sclerosis Patients. Sci Rep (2018) 8(1):8194. doi: 10.1038/s41598-018-26519-w
56. Marastoni D, Buriani A, Pisani AI, Crescenzo F, Zuco C, Fortinguerra S, et al. Increased Nk Cell Count in Multiple Sclerosis Patients Treated With Dimethyl Fumarate: A 2-Year Longitudinal Study. Front Immunol (2019) 10:1666. doi: 10.3389/fimmu.2019.01666
57. Acar NP, Tuncer A, Ozkazanc D, Ozbay FG, Karaosmanoglu B, Goksen S, et al. An Immunological and Transcriptomics Approach on Differential Modulation of Nk Cells in Multiple Sclerosis Patients Under Interferon-Beta1 and Fingolimod Therapy. J Neuroimmunol (2020) 347:577353. doi: 10.1016/j.jneuroim.2020.577353
58. Thell K, Hellinger R, Sahin E, Michenthaler P, Gold-Binder M, Haider T, et al. Oral Activity of a Nature-Derived Cyclic Peptide for the Treatment of Multiple Sclerosis. Proc Natl Acad Sci U.S.A. (2016) 113(15):3960–5. doi: 10.1073/pnas.1519960113
59. Kowarik MC, Pellkofer HL, Cepok S, Korn T, Kumpfel T, Buck D, et al. Differential Effects of Fingolimod (Fty720) on Immune Cells in the Csf and Blood of Patients With Ms. Neurology (2011) 76(14):1214–21. doi: 10.1212/WNL.0b013e3182143564
60. Fosgerau K, Hoffmann T. Peptide Therapeutics: Current Status and Future Directions. Drug Discovery Today (2015) 20(1):122–8. doi: 10.1016/j.drudis.2014.10.003
61. Yong VW. Differential Mechanisms of Action of Interferon-Beta and Glatiramer Aetate in Ms. Neurology (2002) 59(6):802–8. doi: 10.1212/wnl.59.6.802
62. Bielekova B, Richert N, Herman ML, Ohayon J, Waldmann TA, McFarland H, et al. Intrathecal Effects of Daclizumab Treatment of Multiple Sclerosis. Neurology (2011) 77(21):1877–86. doi: 10.1212/WNL.0b013e318239f7ef
63. Rommer PS, Berger K, Ellenberger D, Fneish F, Simbrich A, Stahmann A, et al. Management of Ms Patients Treated With Daclizumab - a Case Series of 267 Patients. Front Neurol (2020) 11:996. doi: 10.3389/fneur.2020.00996
64. Mehling M, Burgener AV, Brinkmann V, Bantug GR, Dimeloe S, Hoenger G, et al. Tissue Distribution Dynamics of Human Nk Cells Inferred From Peripheral Blood Depletion Kinetics After Sphingosine-1-Phosphate Receptor Blockade. Scand J Immunol (2015) 82(5):460–6. doi: 10.1111/sji.12347
Keywords: multiple sclerosis, innate lymphoid cells (ILCs), natural killer cells, experimental autoimmune encephalomyelitis (EAE), autoimmune disease, disease-modifying therapies (DMTs)
Citation: Sadeghi Hassanabadi N, Broux B, Marinović S and Gotthardt D (2022) Innate Lymphoid Cells - Neglected Players in Multiple Sclerosis. Front. Immunol. 13:909275. doi: 10.3389/fimmu.2022.909275
Received: 31 March 2022; Accepted: 20 May 2022;
Published: 17 June 2022.
Edited by:
Marina Cella, Washington University in St. Louis, United StatesReviewed by:
Laura Piccio, Washington University in St. Louis, United StatesCopyright © 2022 Sadeghi Hassanabadi, Broux, Marinović and Gotthardt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dagmar Gotthardt, ZGFnbWFyLmdvdHRoYXJkdEB2ZXRtZWR1bmkuYWMuYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.