 Weizheng Liang
Weizheng Liang Yanxu Qi
Yanxu Qi Hongyang Yi
Hongyang Yi Chenyu Mao
Chenyu Mao Qingxue Meng1
Qingxue Meng1 Hao Wang
Hao Wang Chunfu Zheng
Chunfu Zheng- 1Central Laboratory, The First Affiliated Hospital of Hebei North University, Zhangjiakou, China
- 2Department of Immunology, School of Basic Medical Sciences, Fujian Medical University, Fuzhou, China
- 3Department of Oral and Maxillofacial Surgery, School and Hospital of Stomatology, Cheeloo College of Medicine, Shandong University & Shandong Key Laboratory of Oral Tissue Regeneration & Shandong Engineering Laboratory for Dental Materials and Oral Tissue Regeneration, Jinan, China
- 4National Clinical Research Centre for Infectious Diseases, The Third People's Hospital of Shenzhen and The Second Affiliated Hospital of Southern University of Science and Technology, Shenzhen, China
- 5School of Engineering and Applied Science, University of Pennsylvania, Philadelphia, PA, United States
- 6Shenzhen Key Laboratory, Shenzhen University General Hospital, Shenzhen, China
- 7Department of Obstetrics and Gynecology, Shenzhen University General Hospital, Shenzhen, China
- 8Department of Microbiology, Immunology and Infectious Diseases, University of Calgary, Calgary, AB, Canada
Macrophages are a population of immune cells functioning in antigen presentation and inflammatory response. Research has demonstrated that macrophages belong to a cell lineage with strong plasticity and heterogeneity and can be polarized into different phenotypes under different microenvironments or stimuli. Many macrophages can be recruited by various cytokines secreted by adipose tissue. The recruited macrophages further secrete various inflammatory factors to act on adipocytes, and the interaction between the two leads to chronic inflammation. Previous studies have indicated that adipose tissue macrophages (ATMs) are closely related to metabolic diseases like obesity and diabetes. Here, we will not only conclude the current progress of factors affecting the polarization of adipose tissue macrophages but also elucidate the relationship between ATMs and human diseases. Furthermore, we will highlight its potential in preventing and treating metabolic diseases as immunotherapy targets.
Introduction
Obesity is caused by the excessive accumulation of lipids in adipose tissues. In recent years, obesity has become the causing factor of many chronic diseases, including type 2 diabetes mellitus (T2DM), hypertension, cardiovascular and cerebrovascular diseases, and breast cancer, thus posing a burden on not only patients’ health and finance but also social, medical system (1–4). Apart from storing nutrients, adipose tissue is also an important immune organ containing many immune cells, among which macrophages function in maintaining immune levels. “Obesity is metabolic inflammation” was first proposed by Spiegelman in 1993 (5). It was not until 2003 that researchers discovered macrophage markers in the adipose tissue of obese animals, finding that the higher the macrophage content, the higher the obesity level of the animal (6). The traditional theory holds that macrophages in peripheral tissues are derived from monocytes in the blood (7). Visceral adipose tissue (VAT), a type of white adipose tissue (WAT), is the primary location of inflammatory response in obesity. Although many immune cells participate in the inflammatory response, adipose tissue macrophages (ATM) are considered the most important and characteristic immune cells (8). The proportion of macrophages in total cells in normal adipose tissue is only 10%, but it can reach 50% in obese people (6). Based on the difference in function and activation markers, macrophages are divided into pro-inflammatory M1 and anti-inflammatory M2, with M1 macrophages contributing mostly to the increase in obesity (8–10).
T2DM poses a serious threat to human health, with 80% of its patients caused by overweight or obese. Insulin resistance (Figure 1), a common pathological feature of obesity, occurs when organs are insensitive to insulin stimulation, leading to high blood sugar levels, thus causing diabetes (11–13). Obesity and age-related factors are major risk factors for insulin resistance (14). Obesity stimulates NF-κB, JNK, and other signaling pathways to promote the expression of inflammatory factors, thus influencing the insulin signaling pathway and causing insulin resistance (15). Here, we will summarize the role of ATMs in human diseases and mainly focus on obesity and T2DM, thus providing new insight into the treatment of these diseases as therapeutic targets.
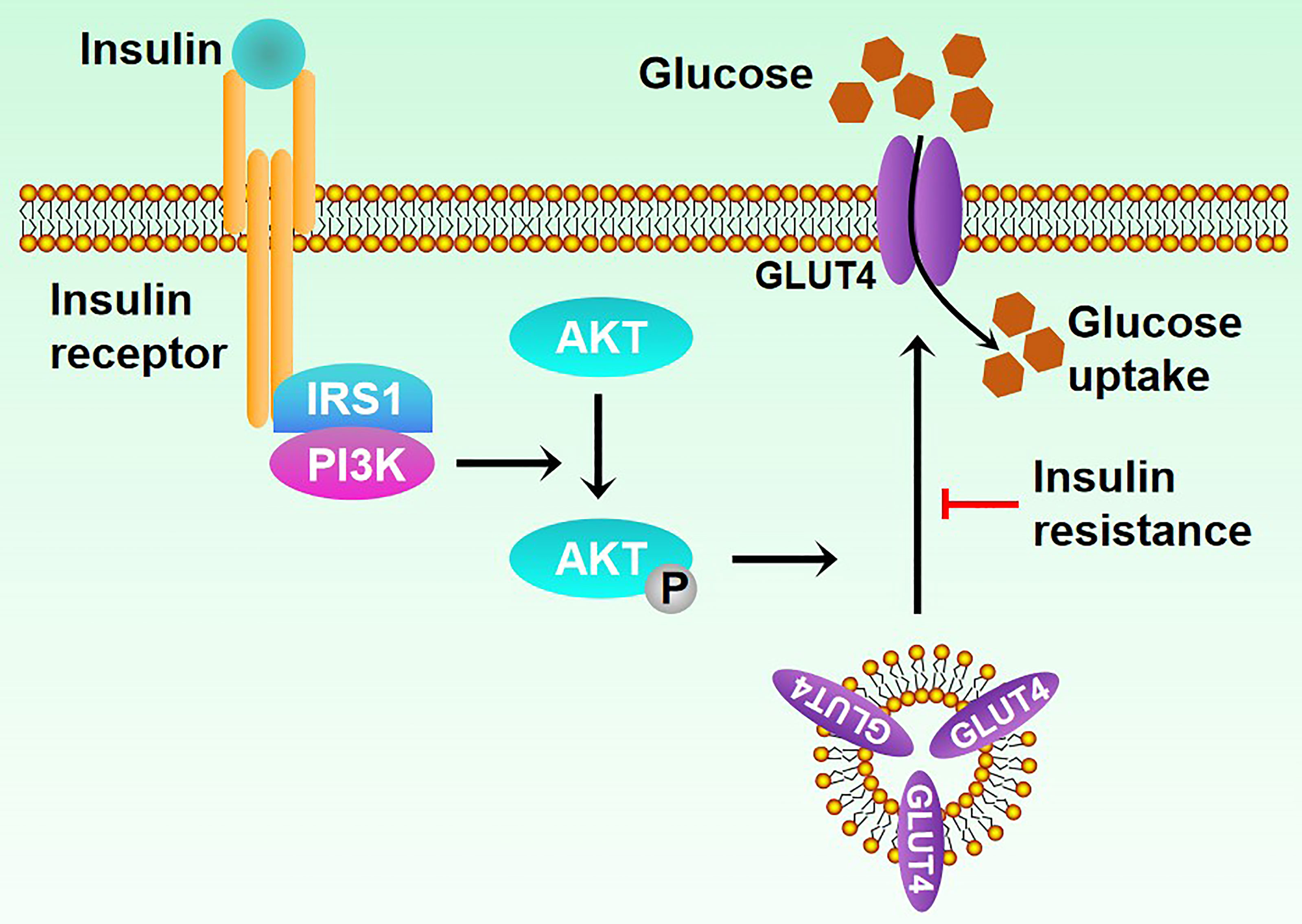
Figure 1 Mechanisms of insulin resistance.
Overview of ATMs
Macrophages are pivotal in the body’s immune system, and they are distributed in various tissues and organs throughout the body, including adipose tissue. Hematopoietic progenitor cells (HPCs) in the bone marrow can differentiate into monocytes upon being stimulated by various cytokines, which will transfer to VAT through the bloodstream to form ATM, thus producing corresponding inflammatory mediators and promoting HPC differentiation (16). Previous studies have shown that ATM mostly appears during embryonic development and will polarize into different phenotypes based on environment, like body weight (17, 18). When an individual is obese, macrophages are often polarized to a pro-inflammatory type, the M1 type (19, 20). With the induction of lipopolysaccharide and saturated fatty acid, M1 macrophages can activate and secrete tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), interleukin-12 (IL-12), interleukin-1β (IL-1β) and other pro-inflammatory factors, leading to inflammation and insulin resistance (Table 1) (21). ATM manifests as an anti-inflammatory type when the individual is thin, namely the M2 type. Both M1 and M2 types have CD11b molecules on the surface. In addition, the M1 type expresses CD11c molecules, and the M2 type expresses CD206, CD301, and macrophage galactose type C-type lectin 1 specifically (29). Different from the M1 type, ATM undergoes the M2 type polarization with the induction of IL-4 and IL-13 and secretes anti-inflammatory mediators such as IL-10 and IL-1 receptor antagonists to play an anti-inflammatory role and maintain insulin sensitivity (21, 22).

Table 1 Properties of adipose tissue macrophages.
Mechanisms of ATMs Polarization and Recruitment
M1 Recruitment and Polarization
M1 macrophages are activated by helper T lymphocyte Th1 cytokines such as interferon, TNF, and LPS (lipopolysaccharide). The pathogenesis of obesity is closely related to the recruitment of ATMs polarized to the pro-inflammatory M1 phenotype (23). The proportion of CD11c-positive monocytes in obese patients was higher than that of normal people, which would decrease after a low-fat diet (30). Therefore, identifying the factors that can polarize ATMs to M1 and recruit macrophages to peripheral tissues in the process of obesity is of great significance for the prevention and treatment of obesity. Accumulating studies have indicated that various signaling pathways contribute to the recruitment and polarization of M1 ATMs during the progression of obesity.
MAPK (Mitogen-activated protein kinase) is a family of serine-threonine protein kinases that can be activated by different extracellular stimuli and cell adhesion, including four subfamilies: ERK, P38, JNK, and ERK5, and it is significant in the pathophysiological process of obesity (31). An early study observed the overexpression of the genes involved in p38 and JNK signaling pathways in adipose tissue of obese people (32). An animal study in mice showed that the increase of M1 ATMs proportion is achieved by increasing mRNA transcription and protein expression levels of JNK (33). In a classical study, the researchers constructed a JNK KO mouse, then fed JNK knockout mice and WT mice with a normal diet and a high-fat diet, and found that high-fat feeding increased ATMs in WT mice had few effects on KO mice (34). Moreover, the increase of macrophages in WT mice was attributed to a significant increase of M1 macrophages, while the numbers of M1 and M2 macrophages in KO mice did not show significant changes. Furthermore, the expression of M1-related genes was down-regulated, and the expression of M2-related genes was up-regulated in KO mice. These data together suggest that the activation of the MAPK signaling pathway may be related to the polarization of ATMs towards the M1 type.
Toll-like receptors are a class of innate immune receptors that are widely expressed on the surface of monocytes, macrophages, and lymphocytes, among which TLR-4 contributes to the LPS response (35). A previous study found that the transcription level of TLR4 mRNA in obese patients was remarkably higher than that in normal people, suggesting that the activation of the TLR-4 receptor may be related to the infiltration of ATMs in the process of obesity (36). Results from different labs confirmed that TLR-4 receptor deficiency reduces inflammation in adipose tissue, and TLR-4 has a positive role in the polarization of ATMs towards M1 (37, 38).
The transcription factor NF-κB is the main regulator of immune homeostasis and inflammation, discovered 30 years ago (39). Studies have demonstrated that activation of NF-κB signaling could facilitate the M1 polarization of macrophages in 3T3-L1 cell lines (40, 41). Other studies have shown that inhibiting NF-κB signaling can promote the release of IL-10 and other anti-inflammatory factors from ATMs (42). In addition, Cao et al. also observed this phenomenon in the mouse model (43). These studies strongly demonstrate that NF-κB can mediate the polarization of ATMs towards M1.
In addition to the above signaling pathways, other factors can also lead to the polarization of ATMs towards M1, including lysosomes and the AMPK signaling pathway (44–47), indicating that the polarization of ATMs towards M1 is a complex process with the coordination of multiple pathways, which needs further investigation.
M2 Recruitment and Polarization
Th2 cytokines can activate anti-inflammatory M2 macrophage in three ways: M2a subtypes activated by IL-4 and IL-13; M2b subtype activated by immune complexes combined with IL-1β or bacterial lipopolysaccharide; M2c subtype induced by IL-10, TGFβ or glucocorticoids. During the process of inflammation resolution, M1 phenotype macrophages are polarized towards the M2 phenotype and are accompanied by the recruitment of M2 phenotype macrophages. Nuclear receptor transcription factors are significant in macrophage polarization, such as PPAR family members.
PPARγ is highly expressed in anti-inflammation macrophages and is important (48, 49). Previous research has found that activation of PPARγ can promote the conversion of M1 type macrophages to M2 type macrophages, improve insulin resistance caused by obesity, and reduce the expression of inflammatory factors (50, 51). After specific activation of PPARγ signaling in mice, it was found that the number of M1 macrophages in ATMs decreased along with the expression of M1-related genes, and the number of M2 macrophages increased, along with the expression of M2-related genes (52, 53). Furthermore, the ex vivo therapy model also demonstrated that activation of PPARγ signaling could induce the polarization of macrophages toward M2 macrophages and induce the recruitment of M2 macrophages (54). The above studies demonstrate that PPARγ is involved in ATM polarization towards M2 and M2 macrophage recruitment.
Previous studies also prove that adiponectin can promote the M2 polarization of macrophages (55). After adiponectin knockout in mice, the expression of M1-related genes was up-regulated, and the expression of M2-related genes was down-regulated. In addition, recombinant adiponectin can up-regulate the expression of M2-related genes as well (56). These results suggest that adiponectin can facilitate the polarization of adipose tissue macrophage towards M2.
IL-4 secreted by immune cells in adipose tissue can also mediate M2 polarization (24). Overexpression or knockout of IL-4 was shown to up-regulate or down-regulate the expression of M2-related genes, respectively (25). These studies demonstrate that IL-4 can mediate adipose tissue macrophage polarization toward M2. Some subsequent studies also found that cytokines such as IL-10, IL-13, and IL-33 can also mediate the polarization of macrophages towards the M2 phenotype (26–28).
ATMs and Obesity
In the obese state, the adipose tissue is under low-intensity inflammation, and the infiltration of ATMs in it is significantly increased to a percentage of 41% compared to a normal state, accompanied by M1 polarization (6). The histological method shows that many M1 type ATMs gather around the dying adipocytes, and crown-like structures (CLSs) appear, associated with obesity-related insulin resistance (57). Further studies have shown that Mincle (macrophage-inducible C-type lectin) in ATMs is involved in the formation of CLSs, and its expression level is positively related to adipose tissue interstitial fibrosis, thus promoting liver fibrosis, progression of hepatic steatosis, and insulin resistance (58–60).
During obesity, ATM is stimulated by inflammatory factors such as IFN-γ, leukotriene B4 (LTB4), and monocyte chemoattractant protein-1 (MCP-1) released by fatty tissue, followed by M1 polarization (8, 61). Previous studies also found that the expression of IL-6, monocyte MCP-1, resistin, lipase (Adip-sin), leptin, and other factors in obese adipose tissue is up-regulated, which increases the expression of vascular endothelial cell adhesion molecules, thus recruiting monocytes in the blood, and promoting the infiltration of ATMs (Figure 2). Further studies confirmed that MCP-1 recruits ATMs through CCR2, while LTB4 recruits ATMs through its receptor BLT1 (62–64). The M1 type ATM secretes inflammatory factors such as IL-6, TNF-α, IL-1β, MCP-1, and PAI-1 (plasminogen activator inhibitor-1), which further increase ATM levels and maintain the M1 phenotype, thus forming a vicious circle. Studies have shown that the occurrence of various obesity-related chronic diseases, such as type 2 diabetes and atherosclerosis, are inseparable from inflammatory factors such as IL-6 and TNF-α (65, 66). In addition, in a previous study, Shimizu et al. verified that neuronal guidance molecules are also involved in the recruitment of ATMs, such as Sema3E, which can promote adipose tissue inflammation through its receptor PlexinD1 (67). Other molecules such as osteocalcin are also involved in the recruitment of ATMs and the progression of adipose tissue inflammation and may be targeted for intervention in metabolic-related diseases such as obesity (68). There is also a positive feedback loop between ATMs derived from blood monocytes and myeloid progenitors in bone marrow tissue. The NLRP3 inflammasome of ATMs is activated to stimulate myeloid progenitor cells to differentiate into monocytes and neutrophils by secreting IL-1β, and intervening in this circuit can reduce adipose tissue inflammation (16). Besides, Zhuang et al. found that miR-223 can inhibit the polarization of ATM to M1 type and ultimately inhibit the inflammatory response of adipose tissue while knocking out the miR-223 gene can aggravate the inflammatory response and increase the proportion of M1 type in ATM (69). Other studies have shown that adipose tissue inflammatory response is closely related to the β1 subunit of AMPK, and the results suggest that these molecules and enzymes may provide new entry points for future obesity treatment (70). Another study showed that IL-6 could induce the IL-4 receptor expression of ATM. ATM was significantly polarized towards the M1 type in mice that did not express the IL-6R α chain, suggesting that IL-6 may affect ATM polarization, reducing inflammation in adipose tissue (71).
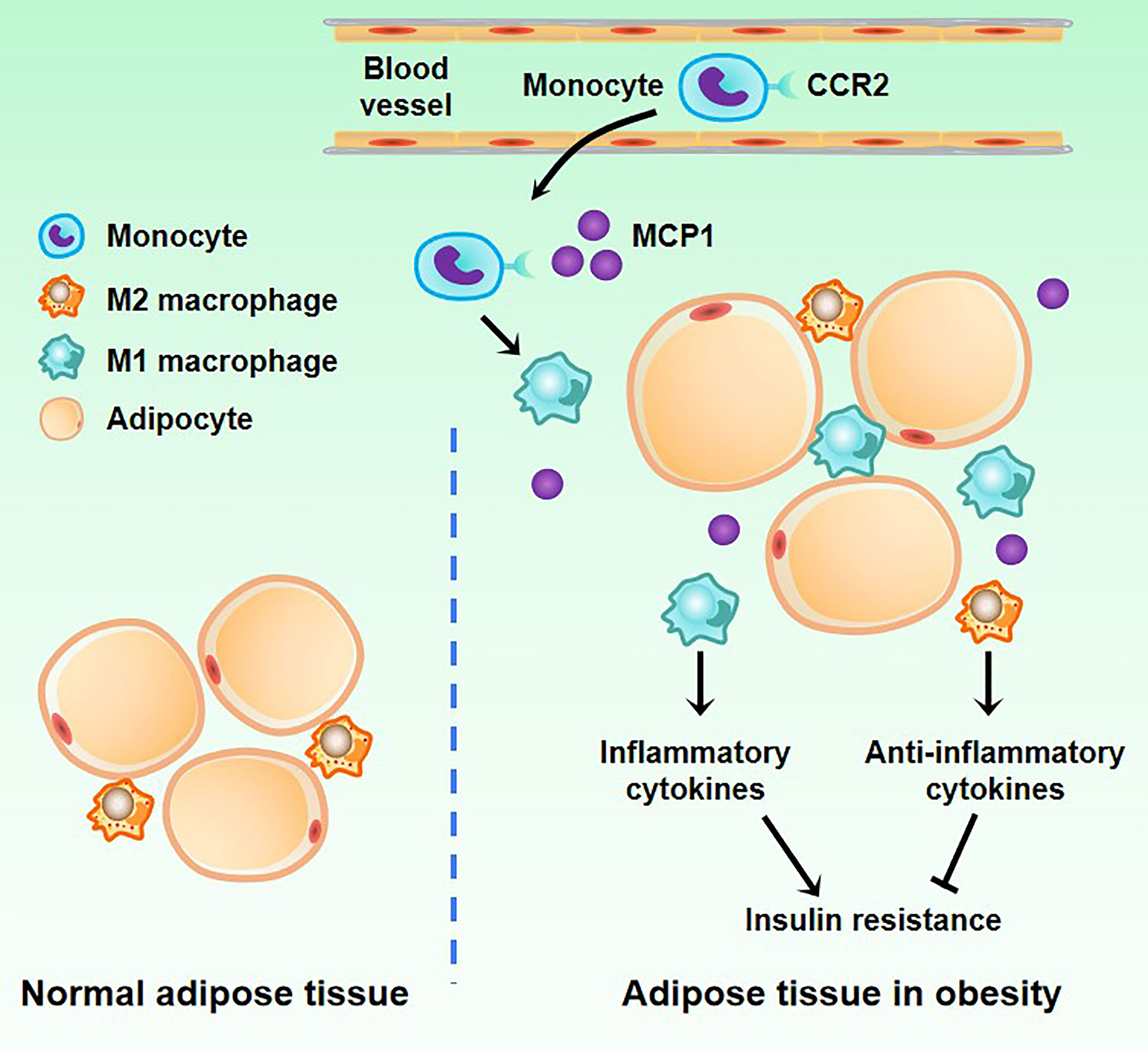
Figure 2 Changes of macrophages in adipose tissue in obesity. There are a small number of M2 macrophages in normal adipose tissue. When obesity occurs, blood monocytes accumulate in adipose tissue. Under the induction of the MCP1 factor secreted by adipose tissue, monocytes differentiate into M1 macrophages.
Growing evidence has indicated that macrophages have a greater impact on the remodeling process of adipose tissue. First, the adipose tissue of obese animals has a higher number of macrophages, which are an important component of adipose tissue. In addition, M1 macrophages can produce some inflammatory mediators and reactive oxygen that have a certain impact on the structure and function of adipocytes. These substances will affect the normal metabolism of adipocytes and increase the release of free fatty acids (FFA), leading to increased lipotoxicity and reduction in the synthesis and secretion of adiponectin (72). Compared with normal mice, adipocyte death was significantly increased in mice with higher fat content, and a similar situation occurred in obese people, indicating that an important pathological manifestation of obesity is adipocyte death (73). A study of adipose tissue of obese patients showed that after adipocyte apoptosis, ATM surrounded it with a coronal structure, forming huge multinucleated cells, but this phenomenon was not observed in the adipose tissue of non-obese people (20). Therefore, the infiltration and activation of ATM during obesity is a powerful mechanism of adipose tissue remodeling.
In addition to the abnormal recruitment and polarization of ATMs in the adipose tissue of obese animals, their emigration is also abnormal, which is mediated by signaling molecules such as chemokines and neural guidance molecules. One previous study reported that Netrin-1 was up-regulated in ATMs of obese patients and mouse models, thus inhibiting the migration of ATMs through its receptor Unc5b (74).
M1 type ATMs are considered pro-inflammatory phenotypes in adipose tissue, and M2 type ATMs are considered anti-inflammatory phenotypes, but ATMs cannot be mechanically recognized in practice. A growing number of studies have shown that ATMs have multiple origins, with their functions spanning pure pro- or anti-inflammatory effects, and they are highly plastic and can achieve phenotypic transformation under specific circumstances, which can be therapeutic targets in the future (75, 76).
ATMs and IR, T2MD
More and more studies have demonstrated that ATMs are important in IR and T2MD. Next, we will clarify the relationship between ATMs and IR and T2MD (Figure 3).
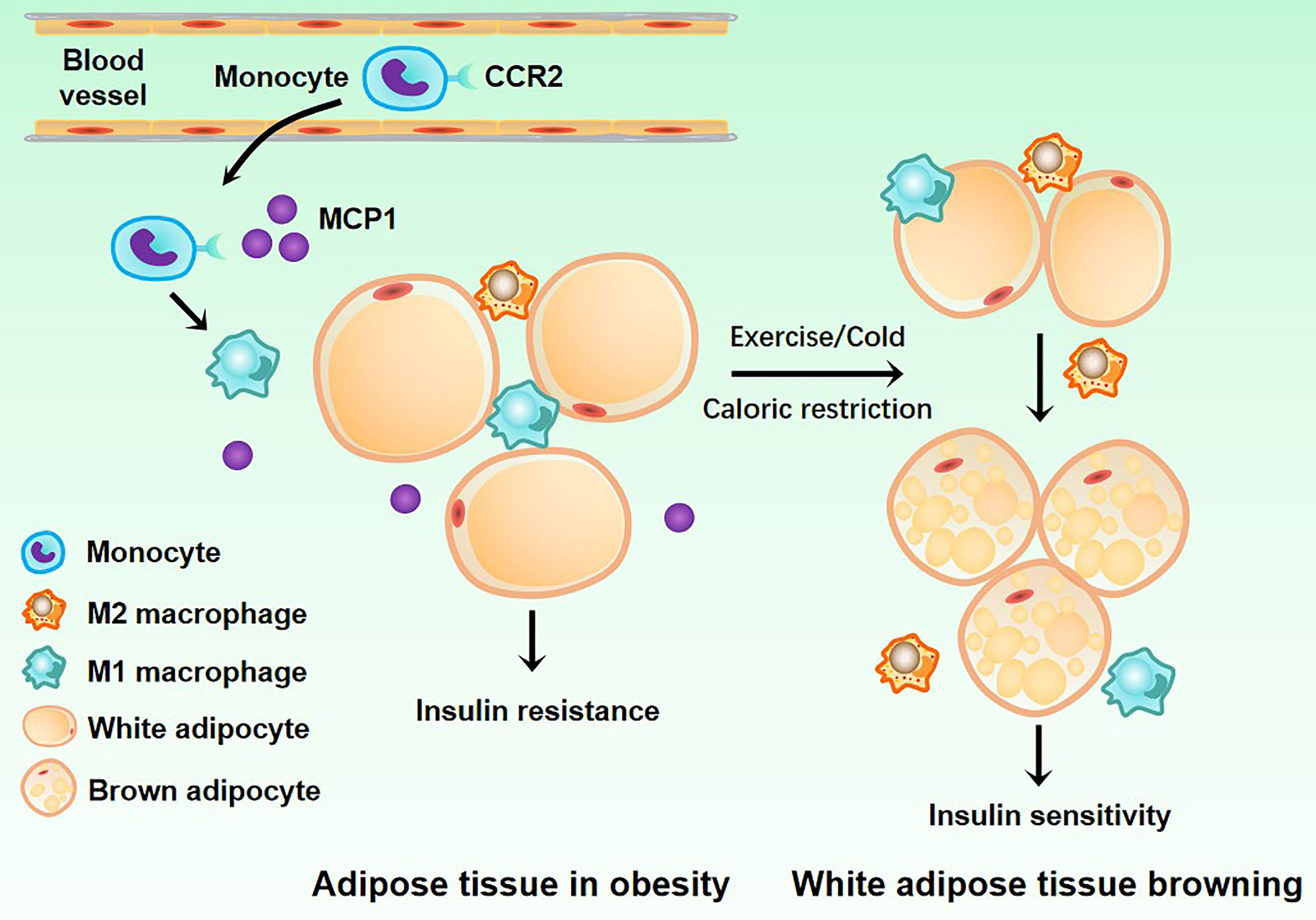
Figure 3 Related mechanisms of adipose tissue macrophages and type2 diabetes mellitus. White adipose tissue acquires insulin resistance under the action of M1 macrophages. When the body is exercising or dieting, M2 macrophages can induce the transformation of white adipose tissue into brown adipose tissue, allowing it to regain insulin sensitivity.
In a previous study, scientists demonstrated that M1 macrophages could aggravate insulin resistance, and CD11c+ cell depletion led to decreased adipose tissue inflammation and rapid normalization of insulin sensitivity (77). In another study, scientists observed that CD11c+ ATM ablation could reduce adipose tissue inflammatory gene expression and improve insulin resistance in the Ccr2 KO mice model (78). It can be seen that CD11c+ ATM infiltration of adipose tissue is one of the reasons for insulin resistance, where increasing FFAs may aggravate insulin resistance. Fetuin-A is a glycoprotein secreted by the liver, and its plasma concentration increases in obesity (79). With the mediation of Fetuin-A, FFAs can indirectly activate TLR4 of CD11c+ ATM so that nuclear factor downstream of TLR4 inhibits the phosphorylation of protein kinase β/nuclear factor-B (IKKβ/NF-κB) and c-Jun N-terminal kinase-activator protein 1 (JNK/AP-1) inflammatory signaling pathway, enhancing inflammatory gene expression and secreting more inflammatory factors like TNF-α, IL-6, and MCP-1. Some studies have found that FFAs also activate TLR2 of ATM to participate in insulin resistance (80). Physiologically, insulin mediates the tyrosine phosphorylation of the insulin receptor substrate (IRS) through the insulin receptor, thus enhancing the downstream PI3K/Akt signaling pathway, promoting glucose uptake, and exerting the hypoglycemic effect. However, activated IKKβ and JNK can cause insulin resistance through IRS serine phosphorylation and blockage of IRS tyrosine phosphorylation and the downstream PI3K/Akt pathway (81). In addition, inflammatory factors secreted by macrophages, such as TNF-α, can further activate inflammatory pathways such as IKKβ/NF-kB, JNK/AP-1, and mTOR signaling pathway, forming a vicious circle (82).
Saturated fatty acids are also involved in insulin resistance. The researchers found that knockout of the CGI-58 (comparative gene identification-58) gene in obese mouse macrophages resulted in mitochondrial dysfunction and reactive oxygen species-mediated oxidative stress in ATM, resulting in the activation of NLRP3 inflammasome and downstream caspase-1, leading to the exacerbation of insulin resistance and hyperglycemia (83). NLRP3 inflammasome is a protein complex in the cell cytoplasm, a member of the NLRs family, and its expression is increased in the adipose tissue of obese diabetic patients (84). Activated NLRP3 inflammasome and downstream caspase-1 do not affect the ratio of M1/M2 in adipose tissue but promote the secretion of IL-1β and IL-18, leading to insulin resistance (84).
Unlike the M1 type, M2 type ATMs secrete the anti-inflammatory factor IL-10, thus inhibiting inflammation and enhancing insulin resistance (85). Therefore, activating factors of M2 macrophages also indirectly affect insulin sensitivity. PPARγ is a fatty acid sensor widely expressed in M2 ATMs, which mediates the activation of monocytes’ polarization towards M2 macrophages (86, 87). With the PPARγ gene knocked out in obese mice, the expression levels of related genes in M2 type macrophages in adipose tissue decreased by 70% to 80%, while the expression levels of inflammatory genes in M1 type macrophages increased, accompanied by insulin resistance and exacerbated hyperglycemia, suggesting that PPARγ is vital in maintaining M2 macrophage phenotype and recovering insulin sensitivity (86). KLF4, as another M2-related cytokine, can synergize with IL-4 to activate STAT6 and inhibit the NF-kB signaling pathway, thus activating M2 type polarization and inhibiting M1 type polarization (88). Knockout of KLF4 in macrophages of obese mice would decrease the proportion of M2 type ATMs and worsen insulin resistance and hyperglycemia (88). In addition, compared with normal people, the expression level of KLF4 in subcutaneous adipose tissue of obese patients decreased by 50%, which may be one of the reasons for the increased M1/M2 ratio in adipose tissue.
In addition, ATM can secrete an exosome (Exos) containing microRNA (miRNA). Intravenous injection of ATM-secreted Exos (ATM−Exos) from obese mice into normal mice for 2 weeks resulted in impaired glucose tolerance and IS, suggesting the occurrence of T2DM. In contrast, when ATM-Exos from normal mice were injected into obese mice, their glucose tolerance and IS were significantly improved, and the overexpression of miR-155 in obese mice ATM-Exos inhibited the expression of its downstream IS-promoting target gene PPARγ, thereby impairing insulin signaling, leading to IR (89). Another study found that miR-29a was overexpressed in ATM-Exos of obese mice and transferred to adipocytes, cardiomyocytes, and hepatocytes, causing IR (90). ATM−Exos can be paracrine to insulin target cells, impacting intracellular insulin and glucose homeostasis. However, there are hundreds of miRNAs in ATM−Exos, and none of them affects IS alone, which may be that multiple miRNAs work together to affect adipose tissue metabolism. The above studies have shown that ATMs secrete exosomes carrying miRNAs, which can be transported to insulin target cells through paracrine or endocrine mechanisms, significantly enhancing the action of intracellular insulin, improving insulin sensitivity and overall glucose homeostasis.
Obesity is closely related to T2MD observed in clinical practice (91). Recent studies suggest that ATMs and the inflammatory response play a bridge role in this process (63, 69). The c-Jun N-terminal kinase (JNK) signaling pathway is significant in obesity-related metabolic responses. In a high-fat diet-induced obesity mouse model, although macrophage-specific JNK knockout did not affect the bodyweight of mice, it reduced ATMs infiltration and improved insulin sensitivity, and JNK knockout could inhibit the polarization of ATMs towards M1 (34). These suggest that ATMs-related inflammatory responses, rather than obesity itself, contribute to the development of obesity-related T2MD.
Further studies have shown that ATMs are involved in obesity-related T2MD by secreting cytokines such as upd3. Studies in Drosophila have shown that depletion of macrophages or macrophage-specific knockout of upd3 can inhibit the activation of the JAK-STAT signaling pathway, thus increasing insulin sensitivity without affecting body weight (92). Mincle in ATMs plays a role in the formation of CLSs and participates in obesity-related insulin resistance (58). Furthermore, the inflammatory cytokines secreted by ATMs may be causative factors leading to insulin resistance and T2MD. In addition, PAI-1 blood levels were significantly increased in obese individuals, and further studies confirmed that it was derived from ATMs stimulated by free fatty acids (93). It is worth noting that breaking the link between ATMs and NK cells, CD8+T cells, and myeloid progenitor cells can inhibit ATMs-mediated inflammatory response and ultimately reduce insulin resistance, which may bring light to the treatment of T2MD (16, 94, 95).
ATMs and Clinical Therapy
Metformin is still the first-line T2MD drug especially caused by obesity. Since macrophages are involved in insulin resistance, they are likely to be ideal targets for treating metabolic diseases. The strategy is to regulate the inflammation-related signaling pathways in macrophages, thus inhibiting their polarization toward M1 and reducing macrophages’ production of inflammatory factors. Some small interfering RNAs and small molecule drugs block the activity of M1 macrophages by inhibiting NF-κB, JNK, and other signaling pathways in macrophages, reducing their infiltration in adipose tissue, thereby improving the body’s sensitivity to insulin (96, 97). Nevertheless, the models used in most studies are mice, which did not enter clinical trials.
However, most clinical research reduces the level of inflammatory factors secreted by macrophages through inflammatory factor inhibitors to treat insulin resistance. TNF-α is the first pro-inflammatory cytokine involved in insulin resistance, but limited data can show that TNF-α is involved in glucose regulation in humans. Early research suggested that short-term administration of a single TNF-α antagonist could not modulate blood glucose homeostasis (98, 99). However, 50 patients with obesity-related metabolic diseases were treated with TNF-α inhibitor etanercept for 6 months, which could significantly improve fasting blood glucose and increase adiponectin content in blood (100). The mechanism by which TNF-α inhibitors improve blood sugar still needs further investigation.
The interaction of CCR2 with its ligand MCP-1 affects monocyte migration into tissues and regulates monocyte-to-macrophage differentiation, producing pro-inflammatory cytokines and amplifying adipose tissue inflammation (20). Accumulating studies in mice have demonstrated that CCR2 selective inhibitors or CCR2/5 inhibitors can significantly improve type 2 diabetes (101–104). Combined with CCR2 inhibitors, metformin can treat diabetes by lowering blood sugar and inhibiting inflammation. A clinical trial involving 332 diabetic nephropathy patients showed that based on standard treatment, taking the CCR2 selective inhibitor, CCX140-B, could further reduce urinary protein and protect the kidneys. Compared with the placebo group, fasting blood glucose levels were significantly lower in the inhibitor group compared to the placebo group, although there was little change in HbA1C level (102). TRIM29 inhibits the secretion of IL6 and CCL2/5 in alveolar macrophages (105). CCL2/CCR2 is not the only pathway affecting the recruitment and differentiation of macrophages. The chemokine regulatory network is very complex, with CCR1-CCL3/4/5, CX3CR1-CX3CL1, and CXCR3-CXCL10 involved in macrophage differentiation. Therefore, utilizing CCR2 inhibitors to regulate macrophages to improve insulin resistance requires the support of more clinical trial data.
Concluding Remarks and Perspectives
The infiltration of pro-inflammatory macrophages in adipose tissue increases in obesity, and many inflammatory factors are secreted, resulting in adipose tissue inflammation. Inflammatory responses inhibit adipocyte insulin signaling, leading to insulin resistance. An adipose tissue macrophage is a key factor in obesity-induced insulin resistance by regulating a series of insulin-related and inflammatory factor-related signaling pathways through paracrine interactions between adipocytes and macrophages. In recent years, adipose macrophages have become a research hotspot based on their important role in insulin resistance. The in-depth study of macrophages has added new insights to the pathogenesis of metabolic diseases. In different microenvironments or under different stimuli, macrophages can show different activation modes and polarize into subtypes with different functions. Each subtype is involved in obesity, insulin resistance, T2MD, and other diseases such as atherosclerosis and severe acute pancreatitis (SAP). Therefore, the polarization direction of macrophages can be induced by regulating various factors affecting the polarization of macrophages, thereby stabilizing the balance between M1/M2 types of macrophages in vivo, which will make macrophages a potential new target for the treatment of metabolic diseases and bring a boon to human health (Figure 4).
Figure 4 Adipose tissue macrophages can be used as a potential therapeutic target for treating obesity and diabetes.
Author Contributions
CZ contributed directly to this review. WL wrote the preliminary version of the manuscript. YQ and WL polished the manuscript’s language and prepared figures. All authors were involved in the manuscript preparation, including figure modification, paper discussion, manuscript writing, and editing. All authors have read and approved the final manuscript.
Funding
This work was supported by the Natural Science Foundation of Shandong Province (No. ZR2020MH190, ZR2021MH086), Medical Science and Technology Development Plans of Shandong province (No. 202008021019), and Project of Department of education of Guangdong province (No. 2021KQNCX074).
Conflict of Interest
The authors declare that the research was conducted without any commercial or financial relationships construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Koliaki C, Liatis S, Kokkinos A. Obesity and Cardiovascular Disease: Revisiting an Old Relationship. Metabolism (2019) 92:98–107. doi: 10.1016/j.metabol.2018.10.011
2. Fernández-Sánchez A, Madrigal-Santillán E, Bautista M, Esquivel-Soto J, Morales-González A, Esquivel-Chirino C, et al. Inflammation, Oxidative Stress, and Obesity. Int J Mol Sci (2011) 12(5):3117–32. doi: 10.3390/ijms12053117
3. Powell-Wiley TM, Poirier P, Burke LE, Després JP, Gordon-Larsen P, Lavie CJ, et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation (2021) 143(21):e984–e1010. doi: 10.1161/CIR.0000000000000973
4. Khan S, Luck H, Winer S, Winer DA. Emerging Concepts in Intestinal Immune Control of Obesity-Related Metabolic Disease. Nat Commun (2021) 12(1):2598. doi: 10.1038/s41467-021-22727-7
5. Spiegelman BM, Hotamisligil GS. Through Thick and Thin: Wasting, Obesity, and TNF Alpha. Cell (1993) 73(4):625–7. doi: 10.1016/0092-8674(93)90243-J
6. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW, et al. Obesity is Associated With Macrophage Accumulation in Adipose Tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/JCI200319246
7. Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, et al. Local Proliferation of Macrophages Contributes to Obesity-Associated Adipose Tissue Inflammation. Cell Metab (2014) 19(1):162–71. doi: 10.1016/j.cmet.2013.11.017
8. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
9. Lumeng CN, Bodzin JL, Saltiel AR. Obesity Induces a Phenotypic Switch in Adipose Tissue Macrophage Polarization. J Clin Invest (2007) 117(1):175–84. doi: 10.1172/JCI29881
10. Hill AA, Reid Bolus W, Hasty AH. A Decade of Progress in Adipose Tissue Macrophage Biology. Immunol Rev (2014) 262(1):134–52. doi: 10.1111/imr.12216
11. Meshkani R, Vakili S. Tissue Resident Macrophages: Key Players in the Pathogenesis of Type 2 Diabetes and its Complications. Clin Chim Acta (2016) 462:77–89. doi: 10.1016/j.cca.2016.08.015
12. Castoldi A, Naffah de Souza C, Câmara NO, Moraes-Vieira PM. The Macrophage Switch in Obesity Development. Front Immunol (2015) 6:637. doi: 10.3389/fimmu.2015.00637
13. Veit M, van Asten R, Olie A, Prinz P. The Role of Dietary Sugars, Overweight, and Obesity in Type 2 Diabetes Mellitus: A Narrative Review. Eur J Clin Nutr (2022). doi: 10.1038/s41430-022-01114-5
14. Ye J. Mechanism of Insulin Resistance in Obesity: A Role of ATP. Front Med (2021) 15(3):372–82. doi: 10.1007/s11684-021-0862-5
15. Ray I, Mahata SK, De RK. Obesity: An Immunometabolic Perspective. Front Endocrinol (Lausanne) (2016) 7:157. doi: 10.3389/fendo.2016.00157
16. Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, et al. Adipose Tissue Macrophages Promote Myelopoiesis and Monocytosis in Obesity. Cell Metab (2014) 19(5):821–35. doi: 10.1016/j.cmet.2014.03.029
17. Epelman S, Lavine KJ, Randolph GJ. Origin and Functions of Tissue Macrophages. Immunity (2014) 41(1):21–35. doi: 10.1016/j.immuni.2014.06.013
18. Wang N, Liang H, Zen K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front Immunol (2014) 5:614. doi: 10.3389/fimmu.2014.00614
19. Chylikova J, Dvorackova J, Tauber Z, Kamarad V. M1/M2 Macrophage Polarization in Human Obese Adipose Tissue. BioMed Pap Med Fac Univ Palacky Olomouc Czech Repub (2018) 162(2):79–82. doi: 10.5507/bp.2018.015
20. Engin AB. Adipocyte-Macrophage Cross-Talk in Obesity. Adv Exp Med Biol (2017) 960:327–43. doi: 10.1007/978-3-319-48382-5_14
21. Thomas D, Apovian C. Macrophage Functions in Lean and Obese Adipose Tissue. Metabolism (2017) 72:120–43. doi: 10.1016/j.metabol.2017.04.005
22. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep (2016) 17(3):684–96. doi: 10.1016/j.celrep.2016.09.008
23. Makki K, Froguel P, Wolowczuk I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflammation (2013) 2013:139239. doi: 10.1155/2013/139239
24. Oeser K, Schwartz C, Voehringer D. Conditional IL-4/IL-13-Deficient Mice Reveal a Critical Role of Innate Immune Cells for Protective Immunity Against Gastrointestinal Helminths. Mucosal Immunol (2015) 8(3):672–82. doi: 10.1038/mi.2014.101
25. Xie L, Fu Q, Ortega TM, Zhou L, Rasmussen D, O'Keefe J, et al. Overexpression of IL-10 in C2D Macrophages Promotes a Macrophage Phenotypic Switch in Adipose Tissue Environments. PLoS One (2014) 9(1):e86541. doi: 10.1371/journal.pone.0086541
26. Yoshida S, Kobayashi Y, Nakama T, Zhou Y, Ishikawa K, Arita R, et al. Increased Expression of M-CSF and IL-13 in Vitreous of Patients With Proliferative Diabetic Retinopathy: Implications for M2 Macrophage-Involving Fibrovascular Membrane Formation. Br J Ophthalmol (2015) 99(5):629–34. doi: 10.1136/bjophthalmol-2014-305860
27. Zdrenghea MT, Makrinioti H, Muresan A, Johnston SL, Stanciu LA. The Role of Macrophage IL-10/Innate IFN Interplay During Virus-Induced Asthma. Rev Med Virol (2015) 25(1):33–49. doi: 10.1002/rmv.1817
28. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 Promotes ST2-Dependent Lung Fibrosis by the Induction of Alternatively Activated Macrophages and Innate Lymphoid Cells in Mice. J Allergy Clin Immunol (2014) 134(6):1422–32.e11. doi: 10.1016/j.jaci.2014.05.011
29. Chmelar J, Chung KJ, Chavakis T. The Role of Innate Immune Cells in Obese Adipose Tissue Inflammation and Development of Insulin Resistance. Thromb Haemost (2013) 109(3):399–406. doi: 10.1160/TH12-09-0703
30. Wu H, Perrard XD, Wang Q, Perrard JL, Polsani VR, Jones PH. CD11c Expression in Adipose Tissue and Blood and its Role in Diet-Induced Obesity. Arterioscler Thromb Vasc Biol (2010) 30(2):186–92. doi: 10.1161/ATVBAHA.109.198044
31. Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK Pathway Across Different Malignancies: A New Perspective. Cancer (2014) 120(22):3446–56. doi: 10.1002/cncr.28864
32. Blüher M, Bashan N, Shai I, Harman-Boehm I, Tarnovscki T, Avinaoch E, et al. Activated Ask1-MKK4-P38mapk/JNK Stress Signaling Pathway in Human Omental Fat Tissue may Link Macrophage Infiltration to Whole-Body Insulin Sensitivity. J Clin Endocrinol Metab (2009) 94(7):2507–15. doi: 10.1210/jc.2009-0002
33. Nakamitsu PZ, Compri CM, de Fraia Pinto L, Gotardo ÉM, de Oliveira CC, Ribeiro ML, et al. Thalidomide Controls Adipose Tissue Inflammation Associated With High-Fat Diet-Induced Obesity in Mice. Endocr Metab Immune Disord Drug Targets (2015) 15(2):151–8. doi: 10.2174/1871530314666141128115225
34. Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, et al. JNK Expression by Macrophages Promotes Obesity-Induced Insulin Resistance and Inflammation. Science (2013) 339(6116):218–22. doi: 10.1126/science.1227568
35. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
36. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valentí V, et al. Increased Tenascin C and Toll-Like Receptor 4 Levels in Visceral Adipose Tissue as a Link Between Inflammation and Extracellular Matrix Remodeling in Obesity. J Clin Endocrinol Metab (2012) 97(10):E1880–9. doi: 10.1210/jc.2012-1670
37. Poggi M, Bastelica D, Gual P, Iglesias MA, Gremeaux T, Knauf C, et al. C3H/HeJ Mice Carrying a Toll-Like Receptor 4 Mutation are Protected Against the Development of Insulin Resistance in White Adipose Tissue in Response to a High-Fat Diet. Diabetologia (2007) 50(6):1267–76. doi: 10.1007/s00125-007-0654-8
38. Ni W, Zhang Q, Liu G, Wang F, Yuan H, Guo Y, et al. Escherichia Coli Maltose-Binding Protein Activates Mouse Peritoneal Macrophages and Induces M1 Polarization via TLR2/4 In Vivo and In Vitro. Int Immunopharmacol (2014) 21(1):171–80. doi: 10.1016/j.intimp.2014.04.025
39. Barnabei L, Laplantine E, Mbongo W, Rieux-Laucat F, Weil R. NF-κb: At the Borders of Autoimmunity and Inflammation. Front Immunol (2021) 12:716469. doi: 10.3389/fimmu.2021.716469
40. Park KS. Aucubin, a Naturally Occurring Iridoid Glycoside Inhibits TNF-α-Induced Inflammatory Responses Through Suppression of NF-κb Activation in 3T3-L1 Adipocytes. Cytokine (2013) 62(3):407–12. doi: 10.1016/j.cyto.2013.04.005
41. Hsu CL, Lin YJ, Ho CT, Yen GC. The Inhibitory Effect of Pterostilbene on Inflammatory Responses During the Interaction of 3T3-L1 Adipocytes and RAW 264.7 Macrophages. J Agric Food Chem (2013) 61(3):602–10. doi: 10.1021/jf304487v
42. Turner JJ, Foxwell KM, Kanji R, Brenner C, Wood S, Foxwell BM, et al. Investigation of Nuclear Factor-κb Inhibitors and Interleukin-10 as Regulators of Inflammatory Signalling in Human Adipocytes. Clin Exp Immunol (2010) 162(3):487–93. doi: 10.1111/j.1365-2249.2010.04260.x
43. Cao C, Li L, Chen W, Zhu Y, Qi Y, Wang X, et al. Deficiency of Ikkϵ Inhibits Inflammation and Induces Cardiac Protection in High-Fat Diet-Induced Obesity in Mice. Int J Mol Med (2014) 34(1):244–52. doi: 10.3892/ijmm.2014.1746
44. Linden MA, Pincu Y, Martin SA, Woods JA, Baynard T. Moderate Exercise Training Provides Modest Protection Against Adipose Tissue Inflammatory Gene Expression in Response to High-Fat Feeding. Physiol Rep (2014) 2(7). doi: 10.14814/phy2.12071
45. Gabriel TL, Tol MJ, Ottenhof R, van Roomen C, Aten J, Claessen N, et al. Lysosomal Stress in Obese Adipose Tissue Macrophages Contributes to MITF-Dependent Gpnmb Induction. Diabetes (2014) 63(10):3310–23. doi: 10.2337/db13-1720
46. Shin E, Shin S, Kong H, Lee S, Do SG, Jo TH, et al. Dietary Aloe Reduces Adipogenesis via the Activation of AMPK and Suppresses Obesity-Related Inflammation in Obese Mice. Immune Netw (2011) 11(2):107–13. doi: 10.4110/in.2011.11.2.107
47. Weng SY, Schuppan D. AMPK Regulates Macrophage Polarization in Adipose Tissue Inflammation and NASH. J Hepatol (2013) 58(3):619–21. doi: 10.1016/j.jhep.2012.09.031
48. Kidani Y, Bensinger SJ. Liver X Receptor and Peroxisome Proliferator-Activated Receptor as Integrators of Lipid Homeostasis and Immunity. Immunol Rev (2012) 249(1):72–83. doi: 10.1111/j.1600-065X.2012.01153.x
49. Matsushita Y, Ogawa D, Wada J, Yamamoto N, Shikata K, Sato C, et al. Activation of Peroxisome Proliferator-Activated Receptor Delta Inhibits Streptozotocin-Induced Diabetic Nephropathy Through Anti-Inflammatory Mechanisms in Mice. Diabetes (2011) 60(3):960–8. doi: 10.2337/db10-1361
50. Hulsmans M, Geeraert B, Arnould T, Tsatsanis C, Holvoet P. PPAR Agonist-Induced Reduction of Mcp1 in Atherosclerotic Plaques of Obese, Insulin-Resistant Mice Depends on Adiponectin-Induced Irak3 Expression. PLoS One (2013) 8(4):e62253. doi: 10.1371/journal.pone.0062253
51. Ohno H, Shinoda K, Spiegelman BM, Kajimura S. Pparγ Agonists Induce a White-to-Brown Fat Conversion Through Stabilization of PRDM16 Protein. Cell Metab (2012) 15(3):395–404. doi: 10.1016/j.cmet.2012.01.019
52. Penna-de-Carvalho A, Graus-Nunes F, Rabelo-Andrade J, Mandarim-de-Lacerda CA, Souza-Mello V. Enhanced Pan-Peroxisome Proliferator-Activated Receptor Gene and Protein Expression in Adipose Tissue of Diet-Induced Obese Mice Treated With Telmisartan. Exp Physiol (2014) 99(12):1663–78. doi: 10.1113/expphysiol.2014.081596
53. Fujisaka S, Usui I, Kanatani Y, Ikutani M, Takasaki I, Tsuneyama K, et al. Telmisartan Improves Insulin Resistance and Modulates Adipose Tissue Macrophage Polarization in High-Fat-Fed Mice. Endocrinology (2011) 152(5):1789–99. doi: 10.1210/en.2010-1312
54. Bullers SJ, Baker SC, Ingham E, Southgate J. The Human Tissue-Biomaterial Interface: A Role for Pparγ-Dependent Glucocorticoid Receptor Activation in Regulating the CD163+ M2 Macrophage Phenotype. Tissue Eng Part A (2014) 20(17-18):2390–401. doi: 10.1089/ten.tea.2013.0628
55. Lovren F, Pan Y, Quan A, Szmitko PE, Singh KK, Shukla PC, et al. Adiponectin Primes Human Monocytes Into Alternative Anti-Inflammatory M2 Macrophages. Am J Physiol Heart Circ Physiol (2010) 299(3):H656–63. doi: 10.1152/ajpheart.00115.2010
56. Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin Promotes Macrophage Polarization Toward an Anti-Inflammatory Phenotype. J Biol Chem (2010) 285(9):6153–60. doi: 10.1074/jbc.M109.088708
57. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic Switching of Adipose Tissue Macrophages With Obesity is Generated by Spatiotemporal Differences in Macrophage Subtypes. Diabetes (2008) 57(12):3239–46. doi: 10.2337/db08-0872
58. Tanaka M, Ikeda K, Suganami T, Komiya C, Ochi K, Shirakawa I, et al. Macrophage-Inducible C-Type Lectin Underlies Obesity-Induced Adipose Tissue Fibrosis. Nat Commun (2014) 5:4982. doi: 10.1038/ncomms5982
59. Tanaka M. Molecular Mechanism of Obesity-Induced Adipose Tissue Inflammation; the Role of Mincle in Adipose Tissue Fibrosis and Ectopic Lipid Accumulation. Endocr J (2020) 67(2):107–11. doi: 10.1507/endocrj.EJ19-0417
60. Ichioka M, Suganami T, Tsuda N, Shirakawa I, Hirata Y, Satoh-Asahara N, et al. Increased Expression of Macrophage-Inducible C-Type Lectin in Adipose Tissue of Obese Mice and Humans. Diabetes (2011) 60(3):819–26. doi: 10.2337/db10-0864
61. Maurizi G, Della Guardia L, Maurizi A, Poloni A. Adipocytes Properties and Crosstalk With Immune System in Obesity-Related Inflammation. J Cell Physiol (2018) 233(1):88–97. doi: 10.1002/jcp.25855
62. Gutierrez DA, Kennedy A, Orr JS, Anderson EK, Webb CD, Gerrald WK, et al. Aberrant Accumulation of Undifferentiated Myeloid Cells in the Adipose Tissue of CCR2-Deficient Mice Delays Improvements in Insulin Sensitivity. Diabetes (2011) 60(11):2820–9. doi: 10.2337/db11-0314
63. Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, et al. LTB4 Promotes Insulin Resistance in Obese Mice by Acting on Macrophages, Hepatocytes and Myocytes. Nat Med (2015) 21(3):239–47. doi: 10.1038/nm.3800
64. Spite M, Hellmann J, Tang Y, Mathis SP, Kosuri M, Bhatnagar A, et al. Deficiency of the Leukotriene B4 Receptor, BLT-1, Protects Against Systemic Insulin Resistance in Diet-Induced Obesity. J Immunol (2011) 187(4):1942–9. doi: 10.4049/jimmunol.1100196
65. Arai S, Miyazaki T. Impacts of the Apoptosis Inhibitor of Macrophage (AIM) on Obesity-Associated Inflammatory Diseases. Semin Immunopathol (2014) 36(1):3–12. doi: 10.1007/s00281-013-0405-5
66. Komohara Y, Fujiwara Y, Ohnishi K, Shiraishi D, Takeya M. Contribution of Macrophage Polarization to Metabolic Diseases. J Atheroscler Thromb (2016) 23(1):10–7. doi: 10.5551/jat.32359
67. Shimizu I, Yoshida Y, Moriya J, Nojima A, Uemura A, Kobayashi Y, et al. Semaphorin3E-Induced Inflammation Contributes to Insulin Resistance in Dietary Obesity. Cell Metab (2013) 18(4):491–504. doi: 10.1016/j.cmet.2013.09.001
68. Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, et al. Osteopontin Mediates Obesity-Induced Adipose Tissue Macrophage Infiltration and Insulin Resistance in Mice. J Clin Invest (2007) 117(10):2877–88. doi: 10.1172/JCI31986
69. Zhuang G, Meng C, Guo X, Cheruku PS, Shi L, Xu H, et al. A Novel Regulator of Macrophage Activation: miR-223 in Obesity-Associated Adipose Tissue Inflammation. Circulation (2012) 125(23):2892–903. doi: 10.1161/CIRCULATIONAHA.111.087817
70. Galic S, Fullerton MD, Schertzer JD, Sikkema S, Marcinko K, Walkley CR, et al. Hematopoietic AMPK β1 Reduces Mouse Adipose Tissue Macrophage Inflammation and Insulin Resistance in Obesity. J Clin Invest (2011) 121(12):4903–15. doi: 10.1172/JCI58577
71. Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. Signaling by IL-6 Promotes Alternative Activation of Macrophages to Limit Endotoxemia and Obesity-Associated Resistance to Insulin. Nat Immunol (2014) 15(5):423–30. doi: 10.1038/ni.2865
72. Martinez-Santibañez G, Lumeng CN. Macrophages and the Regulation of Adipose Tissue Remodeling. Annu Rev Nutr (2014) 34:57–76. doi: 10.1146/annurev-nutr-071812-161113
73. Kuroda M, Sakaue H. Adipocyte Death and Chronic Inflammation in Obesity. J Med Invest (2017) 64(3.4):193–6. doi: 10.2152/jmi.64.193
74. Ramkhelawon B, Hennessy EJ, Ménager M, Ray TD, Sheedy FJ, Hutchison S, et al. Netrin-1 Promotes Adipose Tissue Macrophage Retention and Insulin Resistance in Obesity. Nat Med (2014) 20(4):377–84. doi: 10.1038/nm.3467
75. Russo L, Lumeng CN. Properties and Functions of Adipose Tissue Macrophages in Obesity. Immunology (2018) 155(4):407–17. doi: 10.1111/imm.13002
76. Herrada AA, Olate-Briones A, Rojas A, Liu C, Escobedo N, Piesche M, et al. Adipose Tissue Macrophages as a Therapeutic Target in Obesity-Associated Diseases. Obes Rev (2021) 22(6):e13200. doi: 10.1111/obr.13200
77. Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-Positive Cells Normalizes Insulin Sensitivity in Obese Insulin Resistant Animals. Cell Metab (2008) 8(4):301–9. doi: 10.1016/j.cmet.2008.08.015
78. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 Modulates Inflammatory and Metabolic Effects of High-Fat Feeding. J Clin Invest (2006) 116(1):115–24. doi: 10.1172/JCI24335
79. Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A Acts as an Endogenous Ligand of TLR4 to Promote Lipid-Induced Insulin Resistance. Nat Med (2012) 18(8):1279–85. doi: 10.1038/nm.2851
80. Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, et al. A Subpopulation of Macrophages Infiltrates Hypertrophic Adipose Tissue and is Activated by Free Fatty Acids via Toll-Like Receptors 2 and 4 and JNK-Dependent Pathways. J Biol Chem (2007) 282(48):35279–92. doi: 10.1074/jbc.M706762200
81. Solinas G, Karin M. JNK1 and IKKbeta: Molecular Links Between Obesity and Metabolic Dysfunction. FASEB J (2010) 24(8):2596–611. doi: 10.1096/fj.09-151340
82. Gao Z, Zuberi A, Quon MJ, Dong Z, Ye J. Aspirin Inhibits Serine Phosphorylation of Insulin Receptor Substrate 1 in Tumor Necrosis Factor-Treated Cells Through Targeting Multiple Serine Kinases. J Biol Chem (2003) 278(27):24944–50. doi: 10.1074/jbc.M300423200
83. Miao H, Ou J, Ma Y, Guo F, Yang Z, Wiggins M, et al. Macrophage CGI-58 Deficiency Activates ROS-Inflammasome Pathway to Promote Insulin Resistance in Mice. Cell Rep (2014) 7(1):223–35. doi: 10.1016/j.celrep.2014.02.047
84. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat Med (2011) 17(2):179–88. doi: 10.1038/nm.2279
85. Toita R, Kawano T, Murata M, Kang JH. Anti-Obesity and Anti-Inflammatory Effects of Macrophage-Targeted Interleukin-10-Conjugated Liposomes in Obese Mice. Biomaterials (2016) 110:81–8. doi: 10.1016/j.biomaterials.2016.09.018
86. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-Specific PPARgamma Controls Alternative Activation and Improves Insulin Resistance. Nature (2007) 447(7148):1116–20. doi: 10.1038/nature05894
87. Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. PPARgamma Activation Primes Human Monocytes Into Alternative M2 Macrophages With Anti-Inflammatory Properties. Cell Metab (2007) 6(2):137–43. doi: 10.1016/j.cmet.2007.06.010
88. Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, et al. Krüppel-Like Factor 4 Regulates Macrophage Polarization. J Clin Invest (2011) 121(7):2736–49. doi: 10.1172/JCI45444
89. Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB, et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell (2017) 171(2):372–384.e12. doi: 10.1016/j.cell.2017.08.035
90. Liu T, Sun YC, Cheng P, Shao HG. Adipose Tissue Macrophage-Derived Exosomal miR-29a Regulates Obesity-Associated Insulin Resistance. Biochem Biophys Res Commun (2019) 515(2):352–8. doi: 10.1016/j.bbrc.2019.05.113
91. Kell J. The Addition of Gemtuzumab Ozogamicin to Chemotherapy in Adult Patients With Acute Myeloid Leukemia. Expert Rev Anticancer Ther (2016) 16(4):377–82. doi: 10.1586/14737140.2016.1162099
92. Woodcock KJ, Kierdorf K, Pouchelon CA, Vivancos V, Dionne MS, Geissmann F, et al. Macrophage-Derived Upd3 Cytokine Causes Impaired Glucose Homeostasis and Reduced Lifespan in Drosophila Fed a Lipid-Rich Diet. Immunity (2015) 42(1):133–44. doi: 10.1016/j.immuni.2014.12.023
93. Kishore P, Li W, Tonelli J, Lee DE, Koppaka S, Zhang K, et al. Adipocyte-Derived Factors Potentiate Nutrient-Induced Production of Plasminogen Activator Inhibitor-1 by Macrophages. Sci Transl Med (2010) 2(20):20ra15. doi: 10.1126/scitranslmed.3000292
94. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ Effector T Cells Contribute to Macrophage Recruitment and Adipose Tissue Inflammation in Obesity. Nat Med (2009) 15(8):914–20. doi: 10.1038/nm.1964
95. McNelis JC, Olefsky JM. Macrophages, Immunity, and Metabolic Disease. Immunity (2014) 41(1):36–48. doi: 10.1016/j.immuni.2014.05.010
96. Ye L, Liang S, Guo C, Yu X, Zhao J, Zhang H, et al. Inhibition of M1 Macrophage Activation in Adipose Tissue by Berberine Improves Insulin Resistance. Life Sci (2016) 166:82–91. doi: 10.1016/j.lfs.2016.09.025
97. Zhang C, Qian D, Zhao H, Lv N, Yu P, Sun Z, et al. MiR17 Improves Insulin Sensitivity Through Inhibiting Expression of ASK1 and Anti-Inflammation of Macrophages. BioMed Pharmacother (2018) 100:448–54. doi: 10.1016/j.biopha.2018.02.012
98. Paquot N, Castillo MJ, Lefèbvre PJ, Scheen AJ. No Increased Insulin Sensitivity After a Single Intravenous Administration of a Recombinant Human Tumor Necrosis Factor Receptor: Fc Fusion Protein in Obese Insulin-Resistant Patients. J Clin Endocrinol Metab (2000) 85(3):1316–9. doi: 10.1210/jcem.85.3.6417
99. Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R. Effects of an Engineered Human Anti-TNF-Alpha Antibody (CDP571) on Insulin Sensitivity and Glycemic Control in Patients With NIDDM. Diabetes (1996) 45(7):881–5. doi: 10.2337/diab.45.7.881
100. Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, et al. TNF-Alpha Antagonism With Etanercept Decreases Glucose and Increases the Proportion of High Molecular Weight Adiponectin in Obese Subjects With Features of the Metabolic Syndrome. J Clin Endocrinol Metab (2011) 96(1):E146–50. doi: 10.1210/jc.2010-1170
101. Sullivan T, Miao Z, Dairaghi DJ, Krasinski A, Wang Y, Zhao BN, et al. CCR2 Antagonist CCX140-B Provides Renal and Glycemic Benefits in Diabetic Transgenic Human CCR2 Knockin Mice. Am J Physiol Renal Physiol (2013) 305(9):F1288–97. doi: 10.1152/ajprenal.00316.2013
102. de Zeeuw D, Bekker P, Henkel E, Hasslacher C, Gouni-Berthold I, Mehling H, et al. The Effect of CCR2 Inhibitor CCX140-B on Residual Albuminuria in Patients With Type 2 Diabetes and Nephropathy: A Randomised Trial. Lancet Diabetes Endocrinol (2015) 3(9):687–96. doi: 10.1016/S2213-8587(15)00261-2
103. Kang YS, Lee MH, Song HK, Ko GJ, Kwon OS, Lim TK, et al. CCR2 Antagonism Improves Insulin Resistance, Lipid Metabolism, and Diabetic Nephropathy in Type 2 Diabetic Mice. Kidney Int (2010) 78(9):883–94. doi: 10.1038/ki.2010.263
104. Sullivan TJ, Miao Z, Zhao BN, Ertl LS, Wang Y, Krasinski A, et al. Experimental Evidence for the Use of CCR2 Antagonists in the Treatment of Type 2 Diabetes. Metabolism (2013) 62(11):1623–32. doi: 10.1016/j.metabol.2013.06.008
Keywords: Adipose tissue macrophages, inflammation, obesity, diabetes, insulin resistance (IR), insulin sensitivity (IS)
Citation: Liang W, Qi Y, Yi H, Mao C, Meng Q, Wang H and Zheng C (2022) The Roles of Adipose Tissue Macrophages in Human Disease. Front. Immunol. 13:908749. doi: 10.3389/fimmu.2022.908749
Received: 31 March 2022; Accepted: 12 May 2022;
Published: 09 June 2022.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Yue Wan, University of California, San Francisco, United StatesMingliang Zhang, Shanghai Jiao Tong University, China
Copyright © 2022 Liang, Qi, Yi, Mao, Meng, Wang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunfu Zheng, emhlbmcuYWxhbkBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work