 Jesse Santos
Jesse Santos Daniel R. Calabrese
Daniel R. Calabrese John R. Greenland
John R. Greenland- 1Department of Medicine University of California, San Francisco, San Francisco, CA, United States
- 2Medical Service, Veterans Affairs Health Care System, San Francisco, CA, United States
Lung transplant remains a key therapeutic option for patients with end stage lung disease but short- and long-term survival lag other solid organ transplants. Early ischemia-reperfusion injury in the form of primary graft dysfunction (PGD) and acute cellular rejection are risk factors for chronic lung allograft dysfunction (CLAD), a syndrome of airway and parenchymal fibrosis that is the major barrier to long term survival. An increasing body of research suggests lymphocytic airway inflammation plays a significant role in these important clinical syndromes. Cytotoxic T cells are observed in airway rejection, and transcriptional analysis of airways reveal common cytotoxic gene patterns across solid organ transplant rejection. Natural killer (NK) cells have also been implicated in the early allograft damage response to PGD, acute rejection, cytomegalovirus, and CLAD. This review will examine the roles of lymphocytic airway inflammation across the lifespan of the allograft, including: 1) The contribution of innate lymphocytes to PGD and the impact of PGD on the adaptive immune response. 2) Acute cellular rejection pathologies and the limitations in identifying airway inflammation by transbronchial biopsy. 3) Potentiators of airway inflammation and heterologous immunity, such as respiratory infections, aspiration, and the airway microbiome. 4) Airway contributions to CLAD pathogenesis, including epithelial to mesenchymal transition (EMT), club cell loss, and the evolution from constrictive bronchiolitis to parenchymal fibrosis. 5) Protective mechanisms of fibrosis involving regulatory T cells. In summary, this review will examine our current understanding of the complex interplay between the transplanted airway epithelium, lymphocytic airway infiltration, and rejection pathologies.
Introduction
Since the first successful series of heart-lung and lung transplants in the 1980s, obliterative bronchiolitis has been recognized as the predominant pathologic finding of chronic lung allograft rejection. Both proliferative bronchiolitis, characterized by transluminal fibroproliferative tissue or Masson bodies, and constrictive bronchiolitis, characterized by concentric subepithelial fibrosis, were observed in these early allografts, typically surrounded by lymphocytes (1). Chronic lung allograft dysfunction (CLAD) is the syndrome of lung function decline in transplant recipients that is the major barrier to long term survival following lung transplant and includes both obstructive and restrictive phenotypes (2, 3). The obstructive phenotype is termed Bronchiolitis Obliterans Syndrome (BOS), because the predominant decline in one-second forced expiratory volume (FEV1) is presumed to be secondary to obliterative bronchiolitis pathology (3). Restrictive allograft syndrome (RAS) is pathologically associated with pleuro-parenchymal fibroelastosis (2). As was demonstrated in this original autopsy series, the pathologic hallmarks of BOS and RAS were frequently observed together (4, 5). Although, autopsy and explant studies typically reflect advanced or end stage lung disease which may limit conclusions drawn regarding disease processes marked by significant evolution. Further, advanced lung disease has significant tissue heterogeneity, rendering temporal conclusions involving focal or diffuse pathology challenging.
A similar syndrome of bronchiolitis obliterans is seen following allogeneic, but not autologous, stem cell transplant, suggesting that bronchiolitis obliterans results from an immune-mediated process. Indeed, increasing numbers of donor-recipient major histocompatibility complex (MHC) mismatches have been associated with risk of CLAD (6, 7). Even a minor histocompatibility antigen mismatch encoded by a single amino acid can drive obliterative airway disease, a murine analog of bronchiolitis obliterans, via CD8+ T cell-mediated alloimmune responses (8). In the absence of MHC mismatch between the lung and immune system, obliterative bronchiolitis is associated with some unusual exposures. Identified as a result of environmental exposures among popcorn factory workers, the butter flavoring butane-2,3-dione (diacetyl) covalently binds arginine residues in the small airways, forming haptens that trigger lymphocytic inflammation as a precursor to obliterative bronchiolitis (9–11). Together, these findings implicate lymphocytic immune responses in the airways as central to CLAD pathogenesis, as this review will explicate.
Innate and Adaptive Lymphocytes in the Lung
Among transplanted solid organs, lung and intestine allografts have continual exposure to microbes and non-infectious environmental stimuli, necessitating mucosal-associated lymphoid tissue. Accordingly, lung allografts are predisposed to lymphocytic inflammation. The lung is notable for a diverse resident lymphocytic cell population at rest, and is a site for lymphocyte trafficking from peripheral reservoirs during acute injury (12). As such, across the various lung transplant inflammatory syndromes, lymphocytes can play a variety of roles. Where possible, this review attempts to distinguish disease processes where it is known that lymphocytes directly mediate injuries from those where there may be non-specific recruitment.
Innate lymphoid cells (ILCs) provide a first line of immunologic defense and are distinct from adaptive immune cells, discussed further below (Table 1). ILC activation is dependent upon integration of signals from cytokine stimulation, activating and inhibitory receptors, and physiological cues from their microenvironment (13, 14). There are three major ILC subsets: ILC1s defend against viruses and some bacteria primarily through cytokines like IFN-γ (interferon-gamma) and TNF-α (tumor necrosis factor-alpha). ILC2s classically respond to parasites and play important roles in allergic responses with cytokines like IL-4, IL-5, and IL-13 (18); and ILC3s play important antibacterial roles though IL-1β, IL-22, and IL-17. ILC1s and natural killer (NK) cells have overlapping roles and functions. Functionally, ILC1s are largely tissue-resident, while NK cells more commonly circulate and have greater cytotoxic function (12, 19). NK cells are a major source of IFN-γ in the lung and comprise up to 10% of the resident lymphocyte populations. NK cells mediate infectious and sterile lung diseases and have been implicated in both allograft injury and tolerance through a variety of mechanisms (12). NK cell function is determined by the integration of multiple activating and inhibiting signals from a variety of somatically-encoded receptors (20). As such, their role in directly mediating versus trafficking to sites of injury depends upon tissue contexts. For example, the role of NK cells during influenza infection is contested as some studies show NK depletion in experimental models leads to worse outcomes; whereas, other studies show no differences in experimental lung injury (21–23).
Table 1 Overview of airway lymphocyte types.
T cells develop in the thymus, where T cell receptor (TCR) genes rearrange to generate a diverse array of receptors that are subsequently selected for low-level binding to self-antigens. Recognition of near-self antigens makes T cells adept at recognizing virally infected cells, but also explains how auto- and alloimmune responses develop. In fact, 5-15% of circulating T cells will typically react to donor alloantigen, depending on HLA (human leukocyte antigen) mismatching and recipient immune status (24, 25). T cells are further subdivided based on function and cellular markers into 3 major groups: CD4+ T cells, CD8+ T cells, and γδ T cells. Helper CD4+ T cells primarily secrete cytokines to drive immune response and provide co-stimulation to drive cytotoxic CD8+ T cell and B cell humoral responses (26, 27). The types of cytokines produced by helper T cells lends to their subcategorization into Th1, Th2, Th17, and T regulatory subsets. There is some debate in the literature over the relative contributions of helper T subtypes, but there is evidence supporting a role for all four (15, 28, 29). Th1, Th2, and Th17 phenotypes are analogous to ILC1, ILC2, and ILC3 subclasses and are mediated by similar transcription factors, Tbet, GATA3, and RORγT, respectively (30–32). Like NK cells, CD8+ T cells have cytotoxic properties and secrete perforin and granzymes to lyse virally infected or malignant cells. Within this construct of innate and adaptive lymphoid cells lies a multitude of pathways to mediate injury, either non-specifically or in a targeted fashion. Following transplantation, donor antigens can be presented on either donor or host antigen presenting cells, resulting in direct or indirect antigen presentation, respectively (33).
B cells and plasma cells comprise the final major category of lymphoid cells and are responsible for producing antibodies. As with T cell maturation, B cell diversity is determined by somatic recombination, although B cells undergo a subsequent optimization step, called somatic hypermutation to heighten antigen specificity. B cells are activated by APCs (antigen presenting cell) and CD4+ T cells and contribute to acute and chronic allograft dysfunction through the process of antibody mediated rejection (AMR) (16, 17). As such B cells and plasma cells mediate allograft injury by directing effector cells to tissue deemed “non-self.”
While most lymphocyte populations amplify the cascade of responses that promote inflammation, there are a collection of T cell and B cell subsets which work to dampen this process. Regulatory immune cells help to limit the amount of collateral damage from the innate and adaptive immune systems. Regulatory T cells (Tregs) impair the expansion of conventional T lymphocytes, dampen T cell function, secrete immunosuppressive cytokines, adsorb proinflammatory cytokines, potentiate tolerogenic APCs, and create an environment to facilitate expansion of other Tregs (34). Preclinical studies in mouse models of solid organ transplant, have shown long-term graft acceptance by augmentation of Treg populations in transplant recipients (35, 36). Regulatory B cells (Bregs) are proposed to play a key role in homeostasis after lung transplant (37–39). Breg features may contribute to tolerance, making it possible to reduce immunosuppression (40). Finally, while NK cells do not have a specific regulatory subset, their actions may be curtailed via inhibitory surface receptor signaling. NK cells may also perform regulatory functions such as targeting pro-inflammatory cells, in certain contexts (41).
Lymphocytic Inflammation in the Context of Primary Graft Dysfunction (PGD)
Primary graft dysfunction (PGD) is a syndrome of acute lung dysfunction in the early transplant period. Clinically, it is defined as multi-lobar chest X-ray opacifications and a decreased ratio of arterial oxygen to inspired oxygen (PaO2/FiO2) within the first 72 hours post-transplant. PGD is graded from absent (grade 0) to severe (grade 3). Severe PGD accounts for 30% of mortality in the first 30 days after transplant, 50% of the mortality within the first year of transplant and has been associated with lower baseline lung function and risk of CLAD (42, 43). PGD is the clinical manifestation of the pathologic process of ischemia-reperfusion injury (IRI) (44). Accordingly, PGD risk is dependent on the severity of ischemic injury, including warm and cold ischemic time. Allograft ischemia is further potentiated by chronic hypoperfusion, as bronchial arteries are not typically re-anastomosed during transplant. Advancements in surgical technique and allograft handling have reduced rates and severity of ischemia through limited use of cardiopulmonary bypass, limiting intra-operative blood transfusions, and limiting fraction of inspired oxygen intraoperatively (45–49). PGD risk is also driven by non-ischemic mediators of graft injury, including recipient BMI, donor tobacco use, and operative transfusions as stated above (47). Such factors may contribute to PGD by potentiating inflammation.
IRI is primarily mediated by the innate immune system but can be further amplified through adaptive immune responses (Figure 1). Experimental and clinical data suggest a biphasic nature to this inflammatory process. The early phase of IRI is marked by oxidative stress, epithelial and endothelial dysfunction leading to further injury. Airway epithelial cells release chemokines and damage-associated molecular patterns (DAMPs) (50, 51), while endothelial cells upregulate adhesion markers (50, 52). Within murine models, oxidative stress measured via isoprostanes, was increased after IRI and could be mitigated by administration of azithromycin (53), These signals recruit and activate innate immune cells, including neutrophils and macrophages, and drive antigen presentation (54, 55). Accordingly, macrophage depletion is associated with reduced lung injury in murine models of PGD (56, 57). IL-17 and DAMPs promote neutrophil migration to the interstitial space. Neutrophils can amplify IRI through neutrophil extracellular traps (58, 59). CD1d-restricted NKT cells (natural killer T cell) have been shown to secrete IFN-γ; and help recruitment of neutrophils to the site of injury, suggesting innate immune cells may play an important role as a major source of IFN-γ in the lung (60).
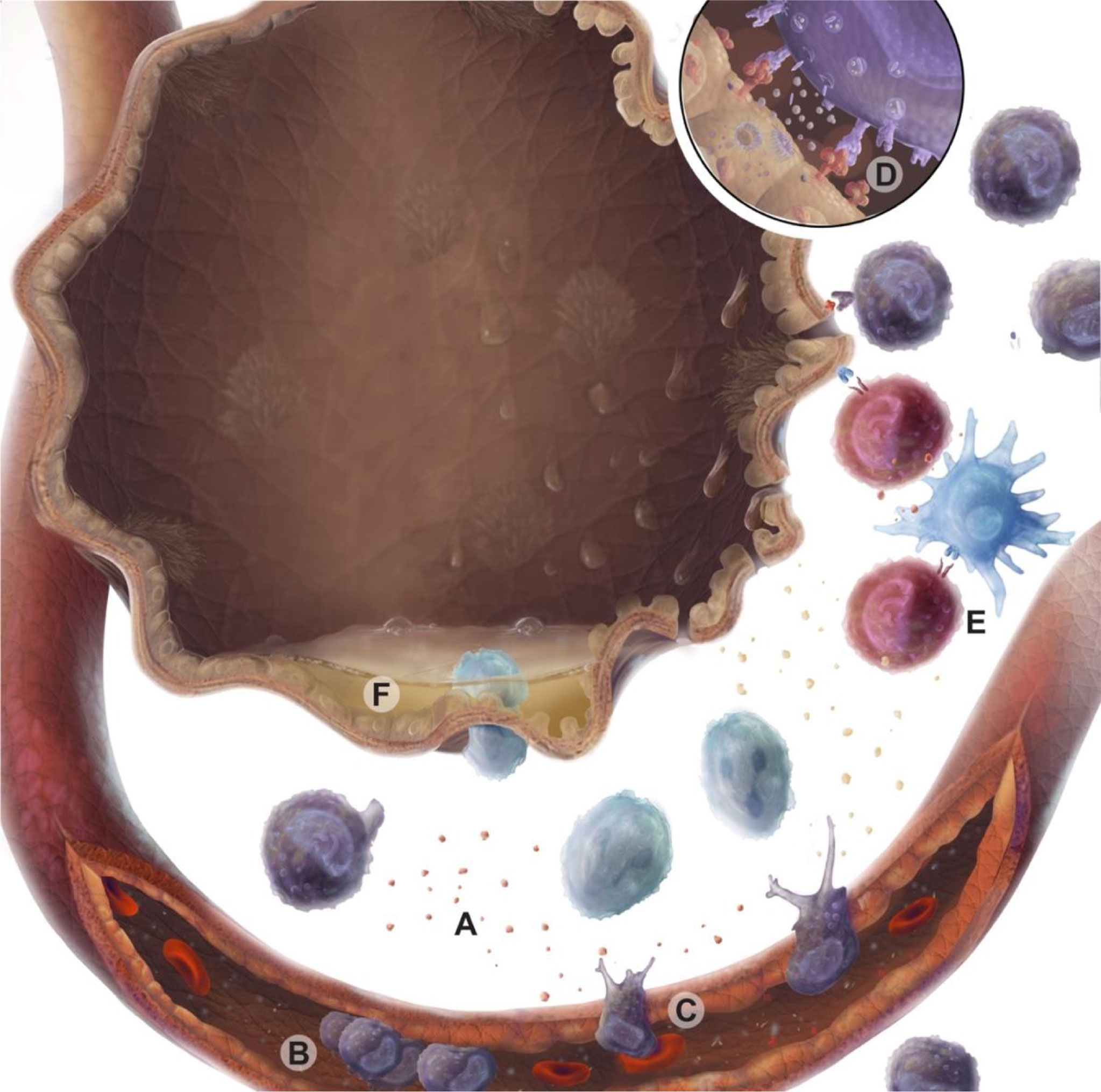
Figure 1 Immune cell responses during ischemia reperfusion injury (IRI). Warm ischemia, cold ischemia, and subsequent reperfusion with oxygenated blood lead to oxidative and mitochondrial cell stress, which are associated with epithelial injury. These injured epithelial cells produce damage molecular patterns (DAMPs) and chemokines (A) that recruit and activate immune cells via the vascular endothelium. Activated endothelium tether passing leukocytes from the circulation via selectins and integrins, causing immune cells to roll and adhere (B) prior to transmigration across a chemotactic gradient (C). Lymphocyte activation is driven through MHC binding to T cell receptors or NK cell receptor ligand interactions. These activated lymphocytes may secrete cytotoxic perforin and granzyme molecules (D). Professional antigen presenting cells can also present alloantigen to T cells amplifying graft-specific responses in response to injury (E). Epithelial cell loss of tight junctions and breakdown results in barrier dysfunction and interstitial edema (F).
Innate and adaptive lymphocytes play a key role in bridging early and late IRI. In both mouse models of IRI and in human lung transplant recipients following PGD, NK cells are observed in and around airways (61). By contrast, in lung allograft biopsies taken peripherally (excluding airways) before implantation and immediately after reperfusion NK cell populations are decreased (62). This would suggest the airways as central sites of NK-cell mediated IRI. The NKG2D receptor on NK cells recognizes stress molecules that are absent or lowly expressed at baseline but rapidly increased in response to a variety of injurious stimuli (63). In mouse models of IRI, NKG2D receptor stress ligands were shown to be increased on pulmonary endothelial and epithelial cells (61). Further, blockade of the NKG2D receptor or genetic deletion of the receptor on NK cells alone, was enough to abrogate pulmonary injury in these mouse models. Although, it should be repeated that NK cells predominantly influence the early phase of IRI, with other cell populations becoming more important as the initial wave of injury subsides. Consequently, renal models of IRI also show a similar role for NK cells in mediating renal tubule epithelial cell injury. This suggests that NK cells, from the moment of reperfusion, may be critical in translating epithelial cell stress during IRI to allograft damage.
These early reperfusion responses of NK cells may potentiate long-term outcomes by killing graft APCs. NK cell activity against APCs occurs in the setting of licensing mismatch (61). During NK cell development, NK cells express inhibitory receptors to host MHCI molecules to avoid self-cytotoxicity. During transplant with a mismatch in donor and recipient MHCI, this inhibitory signal is absent which releases NK cells for activation. For example, NK cells from an HLA-Bw4 positive recipient are licensed to Bw4 antigen and will kill APC lacking Bw4 antigens. This phenomenon plays a critical role in allogenic stem cell transplant, where graft versus host NK activity can prevent leukemia relapse (64). In a mouse skin transplant model of NK licensing mismatch, graft-derived APCs were largely destroyed by donor NK cells and skin allograft survival was improved via reduced antigen presentation to recipient lymphocytes (41, 65). A similar phenomenon has been observed in mouse lung transplant models, where NK cells could improve tolerance of an orthotopic lung allograft in a perforin dependent manner and in association with dendritic cell depletion (66). While this is predominantly animal model evidence there is some data pointing to HLA Bw4 mismatching that potentiates NK cell host-versus-graft activity has been linked to improved outcomes in two cohorts of lung transplant recipients (41).
Conventional lymphocytes are also implicated in driving the lung injury of IRI (67). IRI may potentiate HLA- or neo-antigen presentation and subsequent alloimmune responses (68, 69). While there is not a prominent influx of CD4+ T cells into the allograft during experimental IRI, depletion of CD4+ T cells attenuates injury. This suggests that CD4+ T cells have other roles than direct injury, such as recruitment of effector cells (70). Although, this also points towards CD4+ T cells being complimentary to other underlying disease processes. Within severe combined immunodeficient (SCID) mice a documented lack of lymphocytes caused decreased neutrophil invasion into ischemic lungs (71). A deeper look into this process shows that lymphocyte attraction of neutrophils occurs as early as during warm ischemia time (72, 73). Finally, IRI may also amplify anti-donor anti-MHC and anti-autoantigen antibody production (61).
Acute Cellular Rejection Pathologies and the Significance of Airway Inflammation
Acute lung allograft rejection is mediated via two primary pathologies: acute cellular rejection and antibody mediated rejection. Some degree of acute cellular rejection (ACR) occurs in up to 30% of all lung transplants within the first post-operative year (74). ACR is predominantly a T cell mediated process. Recipient-derived effector memory T cells infiltrate the allograft traversing vascular endothelium, proliferate and migrate to the airways, where they can persist as resident memory cells (75). The diagnosis of ACR is currently confirmed with transbronchial biopsies and quantified based on standardized histopathologic patterns (76, 77). Risk factors for ACR include the degree of human leukocyte antigen mismatching and genetically determined differences within the innate and adaptive immunologic responses of the recipient (78–80). A-grade rejection refers to a mononuclear perivascular infiltrate. B-grade rejection refers to lymphocytic bronchitis or small airway inflammation. Lymphocytic bronchiolitis after transplant is linked to worse CLAD-free survival (81). C-grade rejection refers to obliterative bronchiolitis on transbronchial biopsy. However, this finding is neither sensitive nor specific for CLAD. D-grade rejection denotes accelerated graft atherosclerosis, which is not typically seen on transbronchial biopsies. Finally, E-grade rejection is not a part of standard ISHLT criteria but refers to lymphocytic inflammation on endobronchial (large airway) biopsies (82).
While B-grade rejection is generally assessed on transbronchial biopsies, similar criteria can be used to grade airway inflammation on large airway endobronchial biopsies. In a single center study, diagnosis of E-grade rejection within the first year after transplant was associated with a subsequent 1.8-fold increased risk of CLAD or death. Interestingly, gene expression profiling of A-, B-, and E-grade rejection pathologies identified signatures of allograft rejection that are shared across solid organ transplant, suggesting that these histopathologic findings may share a common pathobiology (82).
Much of the effect seen in E-grade rejection was attributable to high-grade lymphocytic bronchitis (83). The presence of lymphocytic inflammation on transbronchial or endobronchial biopsies has been termed Lymphocytic Airway Disease (LAD). In a separate study, LAD was associated with a 1.6-fold increased risk of CLAD or death. Interestingly, this association was limited to the cohort not taking azithromycin for CLAD prophylaxis (84). The use of azithromycin has been suggested to improved lung function after development of BOS as well as improve overall survival, when used as rescue therapy (85–87). There is evidence in animal models that azithromycin may be linked to reduced production of IL-17 from Th17 cells (88). At our center, we observed a decreased incidence of lymphocytic bronchitis since the introduction of azithromycin for CLAD prophylaxis (89). However, data are mixed regarding the effectiveness of azithromycin on improving CLAD-free survival or overall survival when used prophylactically (90–92). Additionally, the mechanism whereby azithromycin reduces airway inflammation remains unclear. However, there is some evidence supporting multiple pathways via; the reduction in free radicals, suppression of vascular endothelial growth factor’s (VEGF) effects on angiogenesis, and the reduction of gastroesophageal reflux owning to azithromycin’s gut motility effects (53, 93, 94).
Young age is also associated with a higher rate of acute rejection within the first year after transplantation, perhaps owning to a stronger immune response or exposure to a diverse antigens as recipients age lends to less immunogenic responses (95). ACR in the pulmonary allograft is a serious complication that is both an acute cause of graft-dysfunction and inflammation-related morbidity, but also a major risk factor for the development of CLAD (96). Acute rejection contributes to some low risk of mortality, particularly in the first year after lung transplantation, representing approximately 3.3% of all deaths within the first 30 days (95).
Antibody-mediated rejection is rarer in the context of lung transplantation and occurs when de novo or pre-formed antibodies against donor antigens trigger cell injury via two primary pathways. In the classic pathway, complement-binding antibodies activate the complement cascade resulting in membrane attack complex formation and direct target cell death. However, injury may also occur when antibodies bound to target are non-specifically recognized by cells carrying Fc-receptors leading to a process termed antibody-dependent cell mediated cytotoxicity (ADCC). Irrespective of mechanism, the increased frequency of de novo donor-specific antibodies (DSA) is associated with increased risk of CLAD (97, 98). While antibodies against donor antigens are common and associated with CLAD, definitive acute AMR occurs in fewer than 5% of all lung transplant recipients (99, 100). The development of DSA depends on T follicular helper cell interactions with B cells, including CD28-dependent co-stimulation (101). Thus, DSA may be a marker for alloimmune activation as much as biological mediator. Neutrophils, macrophages, and NK cells have been implicated in ADCC. Though, NK cells are thought to be the primary effector cell in human ADCC as their Fc receptor, CD16, is activating-only. In contrast, CD32 and CD64 lead to a mix of activating and inhibiting signals. In support of this mechanism, CD16 polymorphisms that enhance ADCC are associated with increased CLAD risk (102, 103). Thus, the roles of lymphocytes and airway inflammation in AMR require further investigation.
There are two pathways of allorecognition implicated within ACR, the direct and indirect pathways. In the direct pathway, donor APCs migrate to secondary lymphoid tissue and present alloantigen directly to recipient T cells. In the indirect pathway, recipient APCs present alloantigen derived from dying donor APCs to T cells, either in the secondary lymphoid organs or in the allograft itself (104). ACR is suspected to reflect the direct pathway (105), and ACR is associated with increased in donor-specific CD8+, conventional CD4+, and regulatory T cell responses in the peripheral blood (24). Within other solid organ transplant models, recipients one year post-transplantation may demonstrate hypo-responsiveness to alloantigen via the direct pathway (105–107). This type of partial tolerance to donor MHC is inconsistently observed following lung transplantation and may depend on conventional or regulatory T cell immune senescence (108). Conversely, one year post-transplantation, recipients show hyper-responsiveness towards alloantigen via the indirect pathway, where “primed” T cells have been identified on bronchoalveolar lavage (BAL) (106, 107). Thus, repeated rejection could lead to CLAD via either pathway.
ACR has important limitations as a predictor of CLAD development. While both A- and B-grade rejection have been linked to CLAD (81), the association between A1-grade ACR and CLAD risk is inconsistent (83, 109, 110). This perhaps points to a common theme among several studies that although in the acute setting lymphocytic inflammation is a major contributor of injury long term outcomes likely are underpinned by a multitude of inflammatory mediators and effects.
ACR is typically heterogenous and sometimes a symptomatically silent process. There is poor interobserver reliability for ACR grading across sites, with a Cohen’s kappa value of 0.18 to 0.48 for A-grade rejection and 0.04 to 0.47 for B-grade (111, 112). Inadequate tissue sampling is an issue for both grades, but insufficient airway tissue for confident assessment of B-grade rejection has been reported in up to two-thirds of transbronchial biopsies (113). During incipient CLAD, with active decline in FEV1, there are no reliable histopathologic correlates on transbronchial biopsy (113). ACR diagnosis can depend upon institutional surveillance and biopsy protocols. Multiple studies have identified gene expression or BAL cell counts or cytology as better predictors of CLAD than ACR itself (114–117). For example, a gene signature of lymphocytic bronchitis assessed in small airway cytologic brushings identified cases of FEV1 decline that would go on to death or retransplant in the next two years, even when transbronchial biopsies showed no evidence of rejection (113). These inconsistencies suggest that these sampling and interpretation issues may be under appreciated on transbronchial biopsy and have led to an underappreciation of the importance of airway inflammation leading to CLAD. Gene expression-based diagnostics using BAL or airway brushes would sample a larger proportion of small airway tissue, may facilitate detection of airway inflammation, and could guide potential therapies to reduce CLAD progression (29, 115).
Potentiators of Lymphocytic Airway Inflammation and Heterologous Immunity
Airway inflammation may be challenging to quantify but can yield insights into alloimmune responses and the risk for progression to CLAD. However, there are multiple drivers of airway inflammation outside of alloimmune responses that are relevant to long term lung transplant outcomes including air pollution, infections, and aspiration of gastric acid (Figure 2) (118).
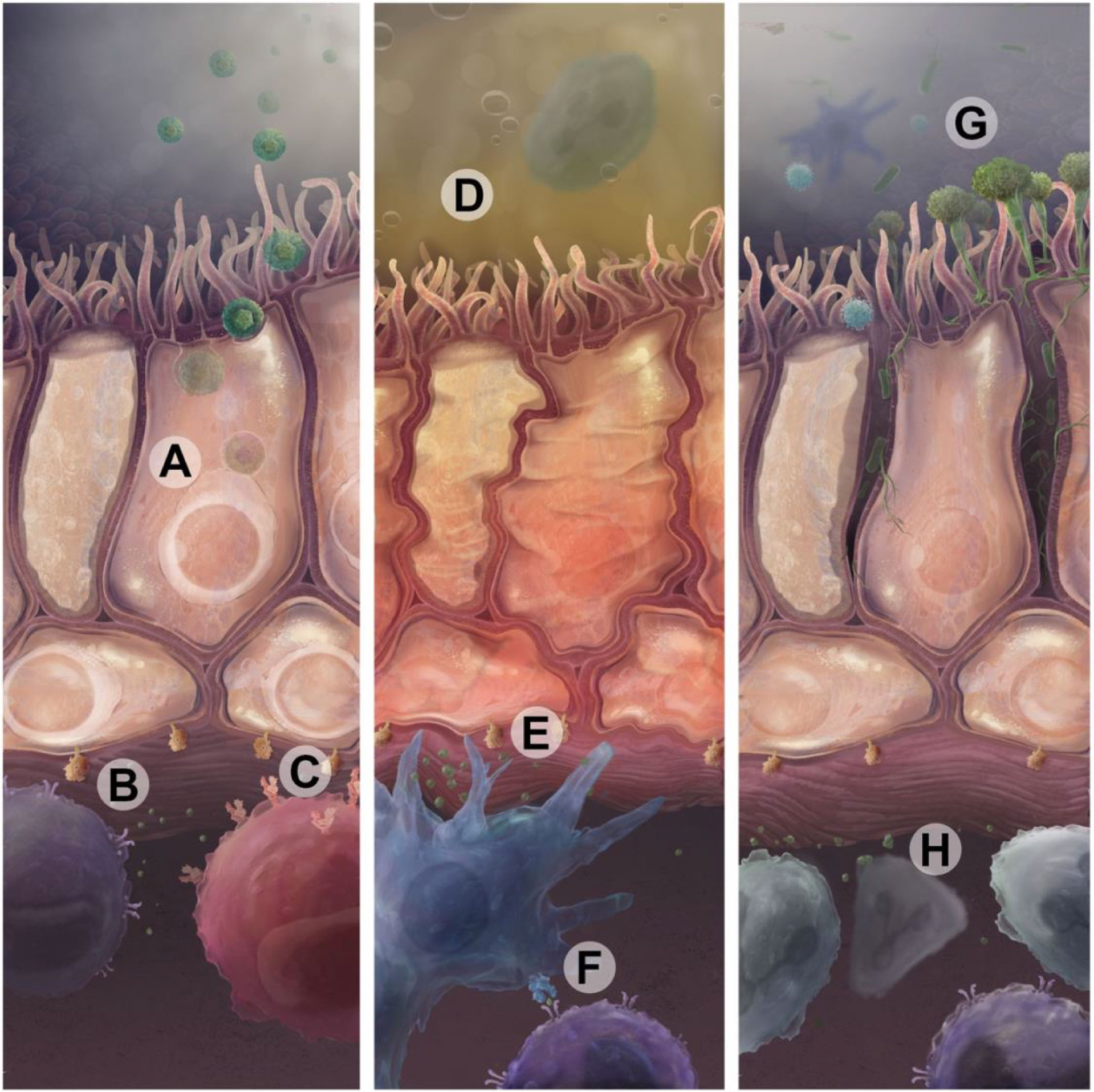
Figure 2 Infectious and non-infectious insults drive immune activation that can lead to CLAD. (A) CMV or other respiratory viral infections in epithelial cells augment antigen presentation through upregulation of donor-derived MHC and β2-microglobuilin, shown in (B). These MHC complexes present viral antigens and participate in direct presentation of donor antigens to T cell receptors. CMV antigens are also presented on HLA-E to activating NKG2C receptors on NK cells (C). Sterile injury, such as through exposure to gastric acid reflux (D) or air pollution, can cause direct airway cell injury which also leads to upregulation of antigen presentation and proinflammatory cytokines (E). Recipient antigen presenting cells then present alloantigens through the indirect pathway using recipient MCH or through the semi-direct pathway using acquired donor MHC molecules (F). This can drive lymphocytic immune responses specific to donor antigens or unmasked self-antigens. (G) Bacterial and fungal infections can serve as an acute or persistent source of pathogen-associated molecular patterns that drive immune responses in lymphoid and myeloid immune cells (H) via Toll-like receptors, Dectin-1, or other pathways.
Lung transplant recipient exposure to air pollution, as quantified by the concentration of particulate matter less than 10 micrometers in diameter (PM10), is associated with increased risk of airway inflammation on biopsy and in BAL in the 2–3 days following exposure (84). In a study including 13 centers in Europe, PM10 and proximity to roads were associated with worse CLAD-free survival (119). Interestingly, azithromycin appeared to mitigate this effect.
Infections may stimulate alloimmune responses and precipitate CLAD development directly and through increased ACR (120). Bacterial infections like Pseudomonas, as well as infections from fungi like Aspergillus may affect CLAD risk through impacts on inflammation, airway epithelial cells, and other constituents of the respiratory microbiome (121, 122). Lung transplant recipients are at particular risk for community-acquired respiratory virus (CARV) infections: respiratory syncytial virus (RSV), coronavirus, rhinovirus, influenza, and parainfluenza viruses (123). Several studies independently demonstrate that community respiratory virus infections convey an increased risk of CLAD development. When stratified between upper and lower viral respiratory tract infections there is an increased risk, almost 3-fold, for lower respiratory tract viral infections (124). Additionally, there appears to be a temporal component to the development of CLAD and onset of respiratory viral infection (RVI), where a recent infection confers a larger risk of CLAD development (125). CARV infection within the first year of transplant confers a risk to CLAD development several years thereafter (126). Early treatment of RSV infection decreased the incidence of new or progressive CLAD (127).
CARV infection may drive airway inflammation and subsequent CLAD through multiple mechanisms. In a rat model of lung transplantation, parainfluenza virus infection potentiated lymphocytic inflammation and obliterative airway disease in allogeneic lungs relative to syngeneic or uninfected lungs (128). Viruses are potent inducers of interferons and interferon-associated chemokines can recruit cytotoxic lymphocytes to airways. Specifically, in CARV-infected lung transplant recipients, higher concentrations of chemokine C-X-C motif ligand 10 (CXCL10) and C-C motif chemokine ligand 11 (CCL11) predicted FEV1 decline over the next 6 months (129). CARV infection can impair regulatory T cells and expose cryptic antigens leading to de novo anti-ColV and k-alpha1 tubulin antibodies that are associated with CLAD (130). Viral infections can also lead to the release of exosomes containing self-antigens that can trigger responses to self-antigens and CLAD pathology (131). Viral infections can potentiate donor-specific immune responses through heterologous immunity. For example, CD8+ T cells specific for Human cytomegalovirus (CMV) or Epstein-Barr virus (EBV) have been shown to cross react with donor alloantigen (132). NK cells can also mediate recall immune responses to CMV through the NKG2C receptor, and elevations in NKG2C+ NK cells in the BAL is a risk factor for CLAD (63).
CMV infection, within immunocompetent hosts, establishes immunity which controls infection even if the virus is reactivated (133). However, there is evidence to suggest CMV infection may cause life-threatening complications in organ transplant recipients and has been associated with more frequent acute and chronic rejection (134–136). CMV-reactive T cells can cause tissue damage by several mechanisms: (i) direct cytotoxic effect on CMV infected (allograft) cells, (ii) indirect bystander activation and proinflammatory milieu formation, and (iii) heterologous (cross-reactive) allorecognition (137). The cross-reactivity of CMV-reactive effector T cells to HLA class I antigens is widely accepted and have been isolated from the peripheral blood of kidney transplant recipients (132, 138, 139).
Chronic exposure to gastric acid secondary gastroesophageal reflux disease (GERD) has also been shown to be associated with the development of CLAD (140). Gastric acid may directly trigger lymphocytic airway inflammation. For example, chronic exposure to gastric fluid in rodent lung transplant models is associated with ACR, peribronchial T cell infiltration, T cell-dependent cytokine release in BAL, and increased frequencies of obliterative bronchiolitis lesions (141, 142).Conversely, anti-reflux surgery is associated with decreased BAL lymphocytes and neutrophils (143). For these reasons, many centers will perform anti-reflux surgery for lung transplant recipients with uncontrolled GERD and risk of CLAD progression (143–145).
Airway Inflammation in the Pathogenesis of CLAD
CLAD pathology may reflect a final common pathway of injury responses leading to airway remodeling and fibrosis. For example, neutrophils in BAL fluid are identified as a reversible CLAD risk factor. A syndrome >15% BAL neutrophils and ≥10% decreased in FEV1 that reverses with azithromycin treatment is termed azithromycin-responsive allograft dysfunction (ARAD), previously known as neutrophilic reversible allograft dysfunction (NRAD) (146). ARAD is closely linked with lymphocytic airway inflammation and may reflect a paradoxical IL-17-dependent production of IL-8 in airway epithelial cells exposed to tacrolimus that is reversed by azithromycin (87, 147). Nonetheless, while azithromycin prophylaxis can potently reduce airway inflammation, it has been inconsistently associated with CLAD prevention (91, 148). That lung transplant recipients continue to develop CLAD despite azithromycin prophylaxis suggests multiple pathways to CLAD.
Airway inflammation can induce and activate myofibroblasts. These cells deposit the extracellular proteins like collagen and fibronectin that constitute airway fibrosis (149). Myofibroblasts may derive from airway epithelial cells via epithelial to mesenchymal transition (EMT) as well as from pericytes via pericyte-mesenchymal transition (PMT) (150–152). Pathologic EMT can be triggered by lymphocyte activation and secretion of transforming growth factor-beta (TGF-β). Mouse models with knockout of TGF-β show protection from fibrosis and EMT (153–155). Growth factors such as VEGF and TGF-β also mediate interactions between the lung endothelium and pericytes and have been independently studied as drivers of fibrosis (156, 157). Myofibroblasts can also differentiate from donor-derived resident mesenchymal stem cells in response to Th2 lymphocytic inflammation (158).
Club cell dysfunction can also drive CLAD pathology. Club cells are non-ciliated epithelial cells typically found within bronchioles that promote injury repair and secrete anti-inflammatory proteins (159–162). Club cell depletion leads to CLAD-like pathology that can be prevented with CD8+ T cell depletion (163). Club cells can proliferate rapidly and differentiate into airway epithelial cell populations. Such proliferation puts stress on cell replication machinery, including telomeres, the nucleoprotein caps on chromosomes. Telomere dysfunction in the allograft has been associated with CLAD risk and induction of club cell telomere dysfunction in mice drives both lymphocytic airway inflammation and CLAD-like pathology (164, 165).
Conclusions
CLAD is primarily a disease of airway or parenchymal fibrosis resulting from alloimmune responses and lymphocytic airway inflammation is likely to be a major driver of CLAD pathology. However, lymphocytic airway inflammation can be challenging to detect using standard of care histopathologic analysis on transbronchial biopsies. Transcriptional analysis of airway brushings biopsies, or BAL fluid may allow more reliably detection of pathogenic airway inflammation (29, 113, 121). Airway lymphocytes include ILCs, T cells, B cells, and NK cells, which have distinct roles in PGD, ACR, AMR, and CLAD. Together with acute peri-vascular rejection, antibody-mediated responses, ischemia-reperfusion injury, graft infections, and gastroesophageal reflux disease, airway inflammation appears to drive an inflammatory milieu leading to airway-centric fibrosis (6, 24, 81, 83, 116, 166–169).
At the same time there are some limitations to the current data linking airway lymphocytes to rejection pathology. The observation of lymphocytes coincident with graft pathology does not imply lymphocytes are causal. These lymphocytes could be a consequence of injury, or actively counteracting pathology, such as with regulatory T and B cells (24). While there are some causal data from rodent lung transplant models, the models have limitations and may not always match human immunobiology (170). Additionally, lymphocytes are only a component of the immune cells contributing to lung injury, as neutrophils, monocytes, and other cells also play key roles.
Targeting immune suppression to airway lymphocytes is a promising strategy to prevent or delay CLAD. For example, a trial of inhaled cyclosporin showed encouraging results, even though it was terminated early for business reasons (171). The JAK-1 inhibitor itacitinib has shown promise as inhibitor of lymphocytic mucosal inflammation and is under investigation to address inflammation in the context of early CLAD (172). The use of azithromycin as prophylaxis for CLAD or as a rescue from BOS has been implemented by several institutions, as detailed previously with varying degrees of success (85–87, 90, 92). Also, an adenosine A2A receptor antagonist is under investigation to reduce invariant NKT cell mediated inflammation in PGD (173). Other strategies to dampen airway inflammation, such as regulatory T cell adoptive therapy and/or pretransplant allograft modification during ex vivo lung perfusion, have shown preclinical promise as adjuncts to traditional immune suppression (174). A fair portion of our understanding of allograft injury comes from in vitro, ex vivo, and animal models which are extremely important in studying the biology that informs our clinical pursuits. However, it is vital to continue to test these theories within robust and safe clinical trials.
Author Contributions
All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Nicholas Bezio of Beizo Studios (nickbezio.com) created the illustrations for Figures 1 and 2.
Glossary

References
1. Yousem SA, Burke CM, Billingham ME. Pathologic Pulmonary Alterations in Long-Term Human Heart-Lung Transplantation. Hum Pathol (1985) 16(9):911–23. doi: 10.1016/S0046-8177(85)80130-1
2. Glanville AR, Verleden GM, Todd JL, Benden C, Calabrese F, Gottlieb J, et al. Chronic Lung Allograft Dysfunction: Definition and Update of Restrictive Allograft Syndrome-A Consensus Report From the Pulmonary Council of the ISHLT. J Heart Lung Transplant (2019) 38(5):483–92. doi: 10.1016/j.healun.2019.03.008
3. Verleden GM, Glanville AR, Lease ED, Fisher AJ, Calabrese F, Corris PA, et al. Chronic Lung Allograft Dysfunction: Definition, Diagnostic Criteria, and Approaches to Treatment-A Consensus Report From the Pulmonary Council of the ISHLT. J Heart Lung Transplant (2019) 38(5):493–503. doi: 10.1016/j.healun.2019.03.009
4. Burke CM, Theodore J, Dawkins KD, Yousem SA, Blank N, Billingham ME, et al. Post-Transplant Obliterative Bronchiolitis and Other Late Lung Sequelae in Human Heart-Lung Transplantation. Chest (1984) 86(6):824–9. doi: 10.1378/chest.86.6.824
5. Tazelaar HD, Yousem SA. The Pathology of Combined Heart-Lung Transplantation: An Autopsy Study. Hum Pathol (1988) 19(12):1403–16. doi: 10.1016/S0046-8177(88)80233-8
6. Belperio JA, Weigt SS, Fishbein MC, Lynch JP 3rd. Chronic Lung Allograft Rejection: Mechanisms and Therapy. Proc Am Thorac Soc (2009) 6(1):108–21. doi: 10.1513/pats.200807-073GO
7. Ditschkowski M, Elmaagacli AH, Koldehoff M, Gromke T, Trenschel R, Beelen DW. Bronchiolitis Obliterans After Allogeneic Hematopoietic SCT: Further Insight–New Perspectives? Bone Marrow Transplant (2013) 48(9):1224–9. doi: 10.1038/bmt.2013.17
8. Higuchi T, Maruyama T, Jaramillo A, Mohanakumar T. Induction of Obliterative Airway Disease in Murine Tracheal Allografts by CD8+ CTLs Recognizing a Single Minor Histocompatibility Antigen. J Immunol (2005) 174(4):1871–8. doi: 10.4049/jimmunol.174.4.1871
9. Wallace KB, Veith GD. Safe Exposure Level for Diacetyl. Int J Occup Environ Health (2014) 20(1):4–5.doi: 10.1179/1077352513Z.000000000100
10. Greenland JR, Jones K, Singer JP. Bronchiolitis. In: Broaddus VC, et al, editors. Murray & Nadel’s Textbook of Respiratory Medicine. Philadelphia, PA: Philadelphia:Elsevier (2021). 994–1004.
11. Mathews JM, Watson SL, Snyder RW, Burgess JP, Morgan DL. Reaction of the Butter Flavorant Diacetyl (2,3-Butanedione) With N-Alpha-Acetylarginine: A Model for Epitope Formation With Pulmonary Proteins in the Etiology of Obliterative Bronchiolitis. J Agric Food Chem (2010) 58(24):12761–8. doi: 10.1021/jf103251w
12. Calabrese DR, Lanier LL, Greenland JR. Natural Killer Cells in Lung Transplantation. Thorax (2019) 74(4):397–404. doi: 10.1136/thoraxjnl-2018-212345
13. Vivier E, Artis D, Colonna M, Diefenbach A, Santo Di JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years on. Cell (2018) 174(5):1054–66. doi: 10.1016/j.cell.2018.07.017
14. Artis D, Spits H. The Biology of Innate Lymphoid Cells. Nature (2015) 517(7534):293–301. doi: 10.1038/nature14189
15. Bos S, Filby AJ, Vos R, Fisher AJ. Effector Immune Cells in Chronic Lung Allograft Dysfunction: A Systematic Review. Immunology (2022). doi: 10.1111/imm.13458
16. Matsuda Y, Sarwal MM. Unraveling the Role of Allo-Antibodies and Transplant Injury. Front Immunol (2016) 7:432. doi: 10.3389/fimmu.2016.00432
17. Brynjolfsson SF, Berg Persson L, Ekerhult Olsen T, Rimkute I, Wick MJ, Martensson IL, et al. Long-Lived Plasma Cells in Mice and Men. Front Immunol (2018) 9:2673. doi: 10.3389/fimmu.2018.02673
18. Uddin S, Amour A, Lewis DJ, Edwards CD, Williamson MG, Hall S, et al. PI3Kdelta Inhibition Prevents IL33, ILC2s and Inflammatory Eosinophils in Persistent Airway Inflammation. BMC Immunol (2021) 22(1):78–78.
19. Seillet C, Brossay L, Vivier E. Natural Killers or ILC1s? That is the Question. Curr Opin Immunol (2021) 68:48–53. doi: 10.1016/j.coi.2020.08.009
20. Barrow AD, Martin CJ, Colonna M. The Natural Cytotoxicity Receptors in Health and Disease. Front Immunol (2019) 10:909. doi: 10.3389/fimmu.2019.00909
21. Abdul-Careem MF, Mian MF, Yue G, Gillgrass A, Chenoweth MJ, Barra NG, et al. Critical Role of Natural Killer Cells in Lung Immunopathology During Influenza Infection in Mice. J Infect Dis (2012) 206(2):167–77. doi: 10.1093/infdis/jis340
22. Carlin LE, Hemann EA, Zacharias ZR, Heusel JW, Legge KL. Natural Killer Cell Recruitment to the Lung During Influenza A Virus Infection Is Dependent on CXCR3, CCR5, and Virus Exposure Dose. Front Immunol (2018) 9(781). doi: 10.3389/fimmu.2018.00781
23. Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, et al. Lethal Influenza Infection in the Absence of the Natural Killer Cell Receptor Gene Ncr1. Nat Immunol (2006) 7(5):517–23. doi: 10.1038/ni1322
24. Greenland JR, Wong CM, Ahuja R, Wang AS, Uchida C, Golden JA, et al. Donor-Reactive Regulatory T Cell Frequency Increases During Acute Cellular Rejection of Lung Allografts. Transplantation (2016) 100(10):2090–8. doi: 10.1097/TP.0000000000001191
25. Wang P, Leung J, Lam A, Lee S, Calabrese DR, Hays SR, et al. Lung Transplant Recipients With Idiopathic Pulmonary Fibrosis Have Impaired Alloreactive Immune Responses. J Heart Lung Transplant (2021). doi: 10.1016/j.healun.2021.01.1888
26. Yang W, Chen X, Hu H. CD4(+) T-Cell Differentiation In Vitro. Methods Mol Biol (2020) 2111:91–9. doi: 10.1007/978-1-0716-0266-9_8
27. Harper SJ, Ali JM, Wlodek E, Negus MC, Harper IG, Chhabra M, et al. CD8 T-Cell Recognition of Acquired Alloantigen Promotes Acute Allograft Rejection. Proc Natl Acad Sci U.S.A. (2015) 112(41):12788–93. doi: 10.1073/pnas.1513533112
28. Wu Q, Gupta PK, Suzuki H, Wagner SR, Zhang C, W Cummings O, et al. CD4 T Cells But Not Th17 Cells Are Required for Mouse Lung Transplant Obliterative Bronchiolitis. Am J Transplant (2015) 15(7):1793–804. doi: 10.1111/ajt.13215
29. Iasella CJ, Hoji A, Popescu I, Wei J, Snyder ME, Zhang Y, et al. Type-1 Immunity and Endogenous Immune Regulators Predominate in the Airway Transcriptome During Chronic Lung Allograft Dysfunction. Am J Transplant (2021) 21(6):2145–60. doi: 10.1111/ajt.16360
30. Berger A. Th1 and Th2 Responses: What Are They? BMJ (2000) 321(7258):424. doi: 10.1136/bmj.321.7258.424
31. Guo B. IL-10 Modulates Th17 Pathogenicity During Autoimmune Diseases. J Clin Cell Immunol (2016) 7(2). doi: 10.4172/2155-9899.1000400
32. d'Alessandro M, Bergantini L, Cameli P, Fanetti M, Alderighi L, Armati M, et al. Immunologic Responses to Antifibrotic Treatment in IPF Patients. Int Immunopharmacol (2021) 95:107525. doi: 10.1016/j.intimp.2021.107525
33. Siu JHY, Surendrakumar V, Richards JA, Pettigrew GJ. T Cell Allorecognition Pathways in Solid Organ Transplantation. Front Immunol (2018) 9:2548. doi: 10.3389/fimmu.2018.02548
34. Tang Q, Vincenti F. Transplant Trials With Tregs: Perils and Promises. J Clin Invest (2017) 127(7):2505–12. doi: 10.1172/JCI90598
35. Bharat A, Kuo E, Saini D, Steward N, Hachem R, Trulock EP, et al. Respiratory Virus-Induced Dysregulation of T-Regulatory Cells Leads to Chronic Rejection. Ann Thorac Surg (2010) 90(5):1637–44. doi: 10.1016/j.athoracsur.2010.06.048
36. Piloni D, Morosini M, Magni S, Balderacchi A, Scudeller L, Cova E, et al. Analysis of Long Term CD4+CD25highCD127- T-Reg Cells Kinetics in Peripheral Blood of Lung Transplant Recipients. BMC Pulm Med (2017) 17(1):102. doi: 10.1186/s12890-017-0446-y
37. Qin J, Zhou J, Fan C, Zhao N, Liu Y, Wang S, et al. Increased Circulating Th17 But Decreased CD4(+)Foxp3(+) Treg and CD19(+)CD1d(hi)CD5(+) Breg Subsets in New-Onset Graves' Disease. BioMed Res Int (2017) 2017:8431838. doi: 10.1155/2017/8431838
38. Wang WW, Yuan XL, Chen H, Xie GH, Ma YH, Zheng YX, et al. CD19+CD24hiCD38hiBregs Involved in Downregulate Helper T Cells and Upregulate Regulatory T Cells in Gastric Cancer. Oncotarget (2015) 6(32):33486–99. doi: 10.18632/oncotarget.5588
39. Rosser EC, Mauri C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity (2015) 42(4):607–12. doi: 10.1016/j.immuni.2015.04.005
40. Peng B, Ming Y, Yang C. Regulatory B Cells: The Cutting Edge of Immune Tolerance in Kidney Transplantation. Cell Death Dis (2018) 9(2):109. doi: 10.1038/s41419-017-0152-y
41. Greenland JR, Sun H, Calabrese D, Chong T, Singer JP, Kukreja J, et al. HLA Mismatching Favoring Host-Versus-Graft NK Cell Activity Via KIR3DL1 Is Associated With Improved Outcomes Following Lung Transplantation. Am J Transplant (2017) 17(8):2192–9. doi: 10.1111/ajt.14295
42. Li D, Weinkauf J, Kapasi A, Hirji A, Varughese R, Lien D, et al. Baseline Lung Allograft Dysfunction in Primary Graft Dysfunction Survivors After Lung Transplantation. Respir Med (2021) 188:106617. doi: 10.1016/j.rmed.2021.106617
43. Keller M, Sun J, Mutebi C, Shah P, Levine D, Aryal S, et al. Donor-Derived Cell-Free DNA as a Composite Marker of Acute Lung Allograft Dysfunction in Clinical Care. J Heart Lung Transplant (2021) 41(4):p. 458–66. doi: 10.1016/j.healun.2021.12.009
44. Christie JD, Bavaria JE, Palevsky HI, Litzky L, Blumenthal NP, Kaiser LR, et al. Primary Graft Failure Following Lung Transplantation. Chest (1998) 114(1):51–60. doi: 10.1378/chest.114.1.51
45. Kuntz CL, Hadjiliadis D, Ahya VN, Kotloff RM, Pochettino A, Lewis J, et al. Risk Factors for Early Primary Graft Dysfunction After Lung Transplantation: A Registry Study. Clin Transplant (2009) 23(6):819–30. doi: 10.1111/j.1399-0012.2008.00951.x
46. Daud SA, Yusen RD, Meyers BF, Chakinala MM, Walter MJ, Aloush AA, et al. Impact of Immediate Primary Lung Allograft Dysfunction on Bronchiolitis Obliterans Syndrome. Am J Respir Crit Care Med (2007) 175(5):507–13. doi: 10.1164/rccm.200608-1079OC
47. Diamond JM, Lee JC, Kawut SM, Shah RJ, Localio AR, Bellamy SL, et al. Clinical Risk Factors for Primary Graft Dysfunction After Lung Transplantation. Am J Respir Crit Care Med (2013) 187(5):527–34. doi: 10.1164/rccm.201210-1865OC
48. Shah RJ, Diamond JM, Cantu E, Lee JC, Lederer DJ, Lama VN, et al. Latent Class Analysis Identifies Distinct Phenotypes of Primary Graft Dysfunction After Lung Transplantation. Chest (2013) 144(2):616–22. doi: 10.1378/chest.12-1480
49. Shaver CM, Wickersham N, McNeil JB, Nagata H, Miller A, Landstreet SR, et al. Cell-Free Hemoglobin Promotes Primary Graft Dysfunction Through Oxidative Lung Endothelial Injury. JCI Insight (2018) 3(2):e98546. doi: 10.1172/jci.insight.98546
50. Shah RJ, Diamond JM, Lederer DJ, Arcasoy SM, Cantu EM, Demissie EJ, et al. Plasma Monocyte Chemotactic Protein-1 Levels at 24 Hours are a Biomarker of Primary Graft Dysfunction After Lung Transplantation. Transl Res (2012) 160(6):435–42. doi: 10.1016/j.trsl.2012.08.003
51. Hoffman SA, Wang L, Shah CV, Ahya VN, Pochettino A, Olthoff K, et al. Plasma Cytokines and Chemokines in Primary Graft Dysfunction Post-Lung Transplantation. Am J Transplant (2009) 9(2):389–96. doi: 10.1111/j.1600-6143.2008.02497.x
52. Liu G, Place A.T, Chen Z, Brovkovych VM, Vogel SM, Muller WA, et al. ICAM-1-Activated Src and eNOS Signaling Increase Endothelial Cell Surface PECAM-1 Adhesivity and Neutrophil Transmigration. Blood (2012) 120(9):1942–52. doi: 10.1182/blood-2011-12-397430
53. Geudens N, Timmermans L, Vanhooren H, Vanaudenaerde B.M, Vos R, Wauwer De Van C, et al. Azithromycin Reduces Airway Inflammation in a Murine Model of Lung Ischaemia Reperfusion Injury. Transpl Int (2008) 21(7):688–95. doi: 10.1111/j.1432-2277.2008.00670.x
54. El-Sawy T, Fahmy NM, Fairchild RL. Chemokines: Directing Leukocyte Infiltration Into Allografts. Curr Opin Immunol (2002) 14(5):562–8. doi: 10.1016/S0952-7915(02)00382-5
55. Hancock WW, Gao W, Faia K.L, Csizmadia V. Chemokines and Their Receptors in Allograft Rejection. Curr Opin Immunol (2000) 12(5):511–6. doi: 10.1016/S0952-7915(00)00130-8
56. Dhaliwal K, Scholefield E, Ferenbach D, Gibbons M, Duffin R, Dorward D.A, et al. Monocytes Control Second-Phase Neutrophil Emigration in Established Lipopolysaccharide-Induced Murine Lung Injury. Am J Respir Crit Care Med (2012) 186(6):514–24. doi: 10.1164/rccm.201112-2132OC
57. Tsushima Y, Jang JH, Yamada Y, Schwendener R, Suzuki K, Weder W, et al. The Depletion of Donor Macrophages Reduces Ischaemia-Reperfusion Injury After Mouse Lung Transplantation. Eur J Cardiothorac Surg (2014) 45(4):703–9. doi: 10.1093/ejcts/ezt489
58. Shah D, Romero F, Stafstrom W, Duong M, Summer R. Extracellular ATP Mediates the Late Phase of Neutrophil Recruitment to the Lung in Murine Models of Acute Lung Injury. Am J Physiol Lung Cell Mol Physiol (2014) 306(2):L152–61. doi: 10.1152/ajplung.00229.2013
59. Sayah DM, Mallavia B, Liu F, Ortiz-Munoz G, Caudrillier A, DerHovanessian A, et al. Neutrophil Extracellular Traps are Pathogenic in Primary Graft Dysfunction After Lung Transplantation. Am J Respir Crit Care Med (2015) 191(4):455–63. doi: 10.1164/rccm.201406-1086OC
60. Li L, Huang L, Sung SS, Lobo PI, Brown MG, Gregg RK, et al. NKT Cell Activation Mediates Neutrophil IFN-Gamma Production and Renal Ischemia-Reperfusion Injury. J Immunol (2007) 178(9):5899–911. doi: 10.4049/jimmunol.178.9.5899
61. Calabrese DR, Aminian E, Mallavia B, Liu F, Cleary SJ, Aguilar OA, et al. Natural Killer Cells Activated Through NKG2D Mediate Lung Ischemia-Reperfusion Injury. J Clin Invest (2021) 131(3):e137047. doi: 10.1172/JCI137047
62. Monticelli LA, Diamond JM, Saenz SA, Wojno Tait ED, Porteous MK, Cantu E, et al. Lung Innate Lymphoid Cell Composition Is Altered in Primary Graft Dysfunction. Am J Respir Crit Care Med (2020) 201(1):63–72. doi: 10.1164/rccm.201906-1113OC
63. Calabrese DR, Chong T, Wang A, Singer JP, Gottschall M, Hays SR, et al. NKG2C Natural Killer Cells in Bronchoalveolar Lavage Are Associated With Cytomegalovirus Viremia and Poor Outcomes in Lung Allograft Recipients. Transplantation (2019) 103(3):493–501. doi: 10.1097/TP.0000000000002450
64. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science (2002) 295(5562):2097–100. doi: 10.1126/science.1068440
65. Yu G, Xu X, Vu MD, Kilpatrick ED, Li XC. NK Cells Promote Transplant Tolerance by Killing Donor Antigen-Presenting Cells. J Exp Med (2006) 203(8):1851–8. doi: 10.1084/jem.20060603
66. Jungraithmayr W, Codarri L, Bouchaud G, Krieg C, Boyman O, Gyulveszi G, et al. Cytokine Complex-Expanded Natural Killer Cells Improve Allogeneic Lung Transplant Function via Depletion of Donor Dendritic Cells. Am J Respir Crit Care Med (2013) 187(12):1349–59. doi: 10.1164/rccm.201209-1749OC
67. Watanabe T, Martinu T, Chruscinski A, Boonstra K, Joe B, Horie M, et al. A B Cell-Dependent Pathway Drives Chronic Lung Allograft Rejection After Ischemia-Reperfusion Injury in Mice. Am J Transplant (2019) 19(12):3377–89. doi: 10.1111/ajt.15550
68. Aiello S, Podesta MA, Rodriguez-Ordonez PY, Pezzuto F, Azzollini N, Solini S, et al. Transplantation-Induced Ischemia-Reperfusion Injury Modulates Antigen Presentation by Donor Renal CD11c(+)F4/80(+) Macrophages Through IL-1r8 Regulation. J Am Soc Nephrol (2020) 31(3):517–31. doi: 10.1681/ASN.2019080778
69. Yoshida S, Haque A, Mizobuchi T, Iwata T, Chiyo M, Webb TJ, et al. Anti-Type V Collagen Lymphocytes That Express IL-17 and IL-23 Induce Rejection Pathology in Fresh and Well-Healed Lung Transplants. Am J Transplant (2006) 6(4):724–35. doi: 10.1111/j.1600-6143.2006.01236.x
70. Yang Z, Sharma AK, Linden J, Kron IL, Laubach VE. CD4+ T Lymphocytes Mediate Acute Pulmonary Ischemia-Reperfusion Injury. J Thorac Cardiovasc Surg (2009) 137(3):695–702. doi: 10.1016/j.jtcvs.2008.10.044
71. Geudens N, Vanaudenaerde BM, Neyrinck AP, Wauwer De Van C, Vos R, Verleden GM, et al. The Importance of Lymphocytes in Lung Ischemia-Reperfusion Injury. Transplant Proc (2007) 39(8):2659–62. doi: 10.1016/j.transproceed.2007.08.001
72. Godfrey DI, Hammond KJ, Poulton LD, Smyth MJ, Baxter AG. NKT Cells: Facts, Functions and Fallacies. Immunol Today (2000) 21(11):573–83. doi: 10.1016/S0167-5699(00)01735-7
73. Kronenberg M. Toward an Understanding of NKT Cell Biology: Progress and Paradoxes. Annu Rev Immunol (2005) 23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742
74. Yusen RD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Goldfarb SB, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-Second Official Adult Lung and Heart-Lung Transplantation Report–2015; Focus Theme: Early Graft Failure. J Heart Lung Transplant (2015) 34(10):1264–77. doi: 10.1016/j.healun.2015.08.014
75. Snyder ME, Moghbeli K, Bondonese A, Craig A, Popescu I, Fan L, et al. Modulation of Tissue Resident Memory T Cells by Glucocorticoids After Acute Cellular Rejection in Lung Transplantation. J Exp Med (2022) 219(4):e20212059. doi: 10.1084/jem.20212059
76. Roden AC, Aisner DL, Allen TC, Aubry MC, Barrios RJ, Beasley MB, et al. Diagnosis of Acute Cellular Rejection and Antibody-Mediated Rejection on Lung Transplant Biopsies: A Perspective From Members of the Pulmonary Pathology Society. Arch Pathol Lab Med (2017) 141(3):437–44. doi: 10.5858/arpa.2016-0459-SA
77. Stewart S, Fishbein MC, Snell GI, Berry GJ, Boehler A, Burke MM, et al. Revision of the 1996 Working Formulation for the Standardization of Nomenclature in the Diagnosis of Lung Rejection. J Heart Lung Transplant (2007) 26(12):1229–42. doi: 10.1016/j.healun.2007.10.017
78. Quantz MA, Bennett LE, Meyer DM, Novick RJ. Does Human Leukocyte Antigen Matching Influence the Outcome of Lung Transplantation? An Analysis of 3,549 Lung Transplantations. J Heart Lung Transplant (2000) 19(5):473–9. doi: 10.1016/S1053-2498(00)00081-4
79. Girnita DM, Webber SA, Zeevi A. Clinical Impact of Cytokine and Growth Factor Genetic Polymorphisms in Thoracic Organ Transplantation. Clin Lab Med (2008) 28(3):423–40. doi: 10.1016/j.cll.2008.08.002
80. Colobran R, Casamitjana N, Roman A, Faner R, Pedrosa E, Arostegui JI, et al. Copy Number Variation in the CCL4L Gene is Associated With Susceptibility to Acute Rejection in Lung Transplantation. Genes Immun (2009) 10(3):254–9. doi: 10.1038/gene.2008.96
81. Glanville AR, Aboyoun CL, Havryk A, Plit M, Rainer S, Malouf MA. Severity of Lymphocytic Bronchiolitis Predicts Long-Term Outcome After Lung Transplantation. Am J Respir Crit Care Med (2008) 177(9):1033–40. doi: 10.1164/rccm.200706-951OC
82. Greenland JR, Wang P, Brotman JJ, Ahuja R, Chong TA, Kleinhenz ME, et al. Gene Signatures Common to Allograft Rejection are Associated With Lymphocytic Bronchitis. Clin Transplant (2019) 33(5):e13515. doi: 10.1111/ctr.13515
83. Greenland JR, Jones KD, Hays SR, Golden JA, Urisman A, Jewell NP, et al. Association of Large-Airway Lymphocytic Bronchitis With Bronchiolitis Obliterans Syndrome. Am J Respir Crit Care Med (2013) 187(4):417–23. doi: 10.1164/rccm.201206-1025OC
84. Verleden SE, Scheers H, Nawrot TS, Vos R, Fierens F, Geenens R, et al. Lymphocytic Bronchiolitis After Lung Transplantation Is Associated With Daily Changes in Air Pollution. Am J Transplant (2012) 12(7):1831–8. doi: 10.1111/j.1600-6143.2012.04134.x
85. Jain R, Hachem RR, Morrell MR, Trulock EP, Chakinala MM, Yusen RD, et al. Azithromycin Is Associated With Increased Survival in Lung Transplant Recipients With Bronchiolitis Obliterans Syndrome. J Heart Lung Transplant (2010) 29(5):531–7. doi: 10.1016/j.healun.2009.12.003
86. Corris PA, Ryan VA, Small T, Lordan J, Fisher AJ, Meachery G, et al. A Randomised Controlled Trial of Azithromycin Therapy in Bronchiolitis Obliterans Syndrome (BOS) Post Lung Transplantation. Thorax (2015) 70(5):442–50. doi: 10.1136/thoraxjnl-2014-205998
87. Vos R, Verleden SE, Ruttens D, Vandermeulen E, Bellon H, Neyrinck A, et al. Azithromycin and the Treatment of Lymphocytic Airway Inflammation After Lung Transplantation. Am J Transplant (2014) 14(12):2736–48. doi: 10.1111/ajt.12942
88. Borkner L, Misiak A, Wilk MM, Mills KHG. Azithromycin Clears Bordetella Pertussis Infection in Mice But Also Modulates Innate and Adaptive Immune Responses and T Cell Memory. Front Immunol (2018) 9:1764. doi: 10.3389/fimmu.2018.01764
89. Santos J, Hays S, Golden JA, Calabrese D, Kolaitis NA, Kleinenz M, Rupal S, et al. Decreased Lymphocytic Bronchitis Severity in the Era of Azithromycin Prophylaxis. J Heart Lung Transplantation. (2022):S310–S311. doi: 10.1016/j.healun.2022.01.769
90. Ruttens D, Verleden SE, Vandermeulen E, Bellon H, Vanaudenaerde BM, Somers J, et al. Prophylactic Azithromycin Therapy After Lung Transplantation: Post Hoc Analysis of a Randomized Controlled Trial. Am J Transplant (2016) 16(1):254–61. doi: 10.1111/ajt.13417
91. Vos R, Vanaudenaerde BM, Verleden SE, Vleeschauwer De SI, Willems-Widyastuti A, Raemdonck Van DE, et al. A Randomised Controlled Trial of Azithromycin to Prevent Chronic Rejection After Lung Transplantation. Eur Respir J (2011) 37(1):164–72. doi: 10.1183/09031936.00068310
92. Li D, Duan Q, Weinkauf J, Kapasi A, Varughese R, Hirji A, et al. Azithromycin Prophylaxis After Lung Transplantation Is Associated With Improved Overall Survival. J Heart Lung Transplant (2020) 39(12):1426–34. doi: 10.1016/j.healun.2020.09.006
93. Willems-Widyastuti A, Vanaudenaerde BM, Vos R, Dilisen E, Verleden SE, Vleeschauwer De SI, et al. Azithromycin Attenuates Fibroblast Growth Factors Induced Vascular Endothelial Growth Factor via P38(MAPK) Signaling in Human Airway Smooth Muscle Cells. Cell Biochem Biophys (2013) 67(2):331–9. doi: 10.1007/s12013-011-9331-0
94. Mertens V, Blondeau K, Pauwels A, Farre R, Vanaudenaerde B, Vos R, et al. Azithromycin Reduces Gastroesophageal Reflux and Aspiration in Lung Transplant Recipients. Dig Dis Sci (2009) 54(5):972–9. doi: 10.1007/s10620-009-0725-4
95. Yusen RD, Edwards LB, Dipchand AI, Goldfarb SB, Kucheryavaya AY, Levvey BJ, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-Third Adult Lung and Heart-Lung Transplant Report-2016; Focus Theme: Primary Diagnostic Indications for Transplant. J Heart Lung Transplant (2016) 35(10):1170–84. doi: 10.1016/j.healun.2016.09.001
96. Benzimra M, Calligaro GL, Glanville AR. Acute Rejection. J Thorac Dis (2017) 9(12):5440–57. doi: 10.21037/jtd.2017.11.83
97. Halverson LP, Hachem RR. Antibody-Mediated Rejection and Lung Transplantation. Semin Respir Crit Care Med (2021) 42(3):428–35. doi: 10.1055/s-0041-1728796
98. Cleary SJ, Kwaan N, Tian JJ, Calabrese DR, Mallavia B, Magnen M, et al. Complement Activation on Endothelium Initiates Antibody-Mediated Acute Lung Injury. J Clin Invest (2020) 130(11):5909–23. doi: 10.1172/JCI138136
99. Kulkarni HS, Bemiss BC, Hachem RR. Antibody-Mediated Rejection in Lung Transplantation. Curr Transplant Rep (2015) 2(4):316–23. doi: 10.1007/s40472-015-0074-5
100. Wijewickreme AN, Kitts DD. Modulation of Metal-Induced Genotoxicity by Maillard Reaction Products Isolated From Coffee. Food Chem Toxicol (1998) 36(7):543–53. doi: 10.1016/S0278-6915(98)00007-6
101. Parsons RF, Larsen CP, Pearson TC, Badell IR. Belatacept and CD28 Costimulation Blockade: Preventing and Reducing Alloantibodies Over the Long Term. Curr Transplant Rep (2019) 6(4):277–84. doi: 10.1007/s40472-019-00260-3
102. Sun H, Greenland JR, Kopchaliiska D, Gae DD, Singer JP, Golden JA, et al. OR24 Recipient FCGR3A-158V Homozygous Genotype Is Associated With an Increased Risk of Chronic Lung Allograft Dysfunction. Hum Immunol (2016) 77:19–20. doi: 10.1016/j.humimm.2016.07.036
103. Paul P, Pedini P, Lyonnet L, Cristofaro Di J, Loundou A, Pelardy M, et al. FCGR3A and FCGR2A Genotypes Differentially Impact Allograft Rejection and Patients' Survival After Lung Transplant. Front Immunol (2019) 10:1208. doi: 10.3389/fimmu.2019.01208
104. Ingulli E. Mechanism of Cellular Rejection in Transplantation. Pediatr Nephrol (2010) 25(1):61–74. doi: 10.1007/s00467-008-1020-x
105. Stanford RE, Ahmed S, Hodson M, Banner NR, Rose ML. A Role for Indirect Allorecognition in Lung Transplant Recipients With Obliterative Bronchiolitis. Am J Transplant (2003) 3(6):736–42. doi: 10.1034/j.1600-6143.2003.00142.x
106. Hornick PI, Mason PD, Baker RJ, Hernandez-Fuentes M, Frasca L, Lombardi G, et al. Significant Frequencies of T Cells With Indirect Anti-Donor Specificity in Heart Graft Recipients With Chronic Rejection. Circulation (2000) 101(20):2405–10. doi: 10.1161/01.CIR.101.20.2405
107. Baker RJ, Hernandez-Fuentes MP, Brookes PA, Chaudhry AN, Cook HT, Lechler RI. Loss of Direct and Maintenance of Indirect Alloresponses in Renal Allograft Recipients: Implications for the Pathogenesis of Chronic Allograft Nephropathy. J Immunol (2001) 167(12):7199–206. doi: 10.4049/jimmunol.167.12.7199
108. Wang P, Leung J, Lam A, Lee J, Calabrese DR, Hays SR, et al. Lung Transplant Recipients With Idiopathic Pulmonary Fibrosis and Telomere Dysfunction Have Impaired Donor-Specific Immune Responses. J Heart Lung Transplant (2021) 40(4):S60–1. doi: 10.1016/j.healun.2021.01.1888
109. Levy L, Huszti E, Tikkanen J, Ghany R, Klement W, Ahmed M, et al. The Impact of First Untreated Subclinical Minimal Acute Rejection on Risk for Chronic Lung Allograft Dysfunction or Death After Lung Transplantation. Am J Transplant (2020) 20(1):p.241–249. doi: 10.1111/ajt.15561
110. Khalifah AP, Hachem RR, Chakinala MM, Yusen RD, Aloush A, Patterson GA, et al. Minimal Acute Rejection After Lung Transplantation: A Risk for Bronchiolitis Obliterans Syndrome. Am J Transplant (2005) 5(8):2022–30. doi: 10.1111/j.1600-6143.2005.00953.x
111. Arcasoy SM, Berry G, Marboe CC, Tazelaar HD, Zamora MR, Wolters HJ, et al. Pathologic Interpretation of Transbronchial Biopsy for Acute Rejection of Lung Allograft Is Highly Variable. Am J Transplant (2011) 11(2):320–8. doi: 10.1111/j.1600-6143.2010.03382.x
112. Bhorade SM, Husain AN, Liao C, Li LC, Ahya VN, Baz MA, et al. Interobserver Variability in Grading Transbronchial Lung Biopsy Specimens After Lung Transplantation. Chest (2013) 143(6):1717–24. doi: 10.1378/chest.12-2107
113. Dugger DT, Fung M, Hays SR, Singer JP, Kleinhenz ME, Leard LE, et al. Chronic Lung Allograft Dysfunction Small Airways Reveal a Lymphocytic Inflammation Gene Signature. Am J Transplant (2021) 21(1):362–71. doi: 10.1111/ajt.16293
114. Pomerance A, Madden B, Burke MM, Yacoub MH. Transbronchial Biopsy in Heart and Lung Transplantation: Clinicopathologic Correlations. J Heart Lung Transplant (1995) 14(4):761–73.
115. Weigt SS, Wang X, Palchevskiy V, Li X, Patel N, Ross DJ, et al. Usefulness of Gene Expression Profiling of Bronchoalveolar Lavage Cells in Acute Lung Allograft Rejection. J Heart Lung Transplant (2019) 38(8):845–55. doi: 10.1016/j.healun.2019.05.001
116. Greenland JR, Jewell NP, Gottschall M, Trivedi NN, Kukreja J, Hays SR, et al. Bronchoalveolar Lavage Cell Immunophenotyping Facilitates Diagnosis of Lung Allograft Rejection. Am J Transplant (2014) 14(4):831–40. doi: 10.1111/ajt.12630
117. Greenland NY, Deiter F, Calabrese DR, Hays SR, Kukreja J, Leard LE, et al. Inflammation on Bronchoalveolar Lavage Cytology Is Associated With Decreased Chronic Lung Allograft Dysfunction-Free Survival. Clin Transplant (2022) p:e14639. doi: 10.1111/ctr.14639
118. Sato M. Bronchiolitis Obliterans Syndrome and Restrictive Allograft Syndrome After Lung Transplantation: Why Are There Two Distinct Forms of Chronic Lung Allograft Dysfunction? Ann Transl Med (2020) 8(6):418. doi: 10.21037/atm.2020.02.159
119. Ruttens D, Verleden SE, Bijnens EM, Winckelmans E, Gottlieb J, Warnecke G, et al. An Association of Particulate Air Pollution and Traffic Exposure With Mortality After Lung Transplantation in Europe. Eur Respir J (2017) 49(1):254–61. doi: 10.1183/13993003.00484-2016
120. Vandervest KM, Zamora MR. Respiratory Viral Infections Post-Lung Transplantation. Curr Respir Care Rep (2012) 1(3):162–7. doi: 10.1007/s13665-012-0017-x
121. Dugger DT, Fung M, Zlock L, Caldera S, Sharp L, Hays SR, et al. Cystic Fibrosis Lung Transplant Recipients Have Suppressed Airway Interferon Responses During Pseudomonas Infection. Cell Rep Med (2020) 1(4):100055. doi: 10.1016/j.xcrm.2020.100055
122. Calabrese DR, Wang P, Chong T, Hoover J, Singer JP, Torgerson D, et al. Dectin-1 Genetic Deficiency Predicts Chronic Lung Allograft Dysfunction and Death. JCI Insight (2019) 4(22):e133083. doi: 10.1172/jci.insight.133083
123. Hoover J, Mintz MA, Deiter F, Aminian E, Chen J, Hays SR, et al. Rapid Molecular Detection of Airway Pathogens in Lung Transplant Recipients. Transpl Infect Dis (2021) 23(4):e13579. doi: 10.1111/tid.13579
124. Khalifah AP, Hachem R.R, Chakinala MM, Schechtman KB, Patterson GA, Schuster DP, et al. Respiratory Viral Infections Are a Distinct Risk for Bronchiolitis Obliterans Syndrome and Death. Am J Respir Crit Care Med (2004) 170(2):181–7. doi: 10.1164/rccm.200310-1359OC
125. Fisher CE, Preiksaitis CM, Lease ED, Edelman J, Kirby KA, Leisenring WM, et al. Symptomatic Respiratory Virus Infection and Chronic Lung Allograft Dysfunction. Clin Infect Dis (2016) 62(3):313–9. doi: 10.1093/cid/civ871
126. Magnusson J, Westin J, Andersson LM, Brittain-Long R, Riise GC. The Impact of Viral Respiratory Tract Infections on Long-Term Morbidity and Mortality Following Lung Transplantation: A Retrospective Cohort Study Using a Multiplex PCR Panel. Transplantation (2013) 95(2):383–8. doi: 10.1097/TP.0b013e318271d7f0
127. Gottlieb J, Zamora MR, Hodges T, Musk AW, Sommerwerk U, Dilling D, et al. ALN-RSV01 for Prevention of Bronchiolitis Obliterans Syndrome After Respiratory Syncytial Virus Infection in Lung Transplant Recipients. J Heart Lung Transplant (2016) 35(2):213–21. doi: 10.1016/j.healun.2015.08.012
128. Winter JB, Gouw AS, Groen M, Wildevuur C, Prop J. Respiratory Viral Infections Aggravate Airway Damage Caused by Chronic Rejection in Rat Lung Allografts. Transplantation (1994) 57(3):418–22. doi: 10.1097/00007890-199402150-00018
129. Weigt SS, Derhovanessian A, Liao E, Hu S, Gregson AL, Kubak BM, et al. CXCR3 Chemokine Ligands During Respiratory Viral Infections Predict Lung Allograft Dysfunction. Am J Transplant (2012) 12(2):477–84. doi: 10.1111/j.1600-6143.2011.03859.x
130. Chiu S, Fernandez R, Subramanian V, Sun H, DeCamp MM, Kreisel D, et al. Lung Injury Combined With Loss of Regulatory T Cells Leads to De Novo Lung-Restricted Autoimmunity. J Immunol (2016) 197(1):51–7. doi: 10.4049/jimmunol.1502539
131. Gunasekaran M, Bansal S, Ravichandran R, Sharma M, Perincheri S, Rodriguez F, et al. Respiratory Viral Infection in Lung Transplantation Induces Exosomes That Trigger Chronic Rejection. J Heart Lung Transplant (2020) 39(4):379–88. doi: 10.1016/j.healun.2019.12.009
132. Heutinck KM, Yong SL, Tonneijck L, Heuvel den van H, Weerd der van NC, Pant der van KA, et al. Virus-Specific CD8(+) T Cells Cross-Reactive to Donor-Alloantigen Are Transiently Present in the Circulation of Kidney Transplant Recipients Infected With CMV and/or EBV. Am J Transplant (2016) 16(5):1480–91. doi: 10.1111/ajt.13618
133. Rafailidis PI, Mourtzoukou EG, Varbobitis IC, Falagas ME. Severe Cytomegalovirus Infection in Apparently Immunocompetent Patients: A Systematic Review. Virol J (2008) 5:47. doi: 10.1186/1743-422X-5-47
134. Klenerman P, Oxenius A. T Cell Responses to Cytomegalovirus. Nat Rev Immunol (2016) 16(6):367–77. doi: 10.1038/nri.2016.38
135. Reinke P, Fietze E, Ode-Hakim S, Prosch S, Lippert J, Ewert R, Volk HD, et al. Late-Acute Renal Allograft Rejection and Symptomless Cytomegalovirus Infection. Lancet (1994) 344(8939-8940):1737–8. doi: 10.1016/S0140-6736(94)92887-8
136. Cainelli F, Vento S. Infections and Solid Organ Transplant Rejection: A Cause-and-Effect Relationship? Lancet Infect Dis (2002) 2(9):539–49. doi: 10.1016/S1473-3099(02)00370-5
137. Sharma S, Thomas PG. The Two Faces of Heterologous Immunity: Protection or Immunopathology. J Leukoc Biol (2014) 95(3):405–16. doi: 10.1189/jlb.0713386
138. Gamadia LE, Remmerswaal EB, Surachno S, Lardy NM, Dillen Wertheim-van PM, Lier van RA, et al. Cross-Reactivity of Cytomegalovirus-Specific CD8+ T Cells to Allo-Major Histocompatibility Complex Class I Molecules. Transplantation (2004) 77(12):1879–85. doi: 10.1097/01.TP.0000131158.81346.64
139. Stranavova L, Pelak O, Svaton M, Hruba P, Fronkova E, Slavcev A, et al. Heterologous Cytomegalovirus and Allo-Reactivity by Shared T Cell Receptor Repertoire in Kidney Transplantation. Front Immunol (2019) 10:2549. doi: 10.3389/fimmu.2019.02549
140. Hathorn KE, Chan WW, Lo WK. Role of Gastroesophageal Reflux Disease in Lung Transplantation. World J Transplant (2017) 7(2):103–16. doi: 10.5500/wjt.v7.i2.103
141. Li B, Hartwig MG, Appel JZ, Bush EL, Balsara KR, Holzknecht ZE, et al. Chronic Aspiration of Gastric Fluid Induces the Development of Obliterative Bronchiolitis in Rat Lung Transplants. Am J Transplant (2008) 8(8):1614–21. doi: 10.1111/j.1600-6143.2008.02298.x
142. Hartwig MG, Appel JZ, Li B, Hsieh CC, Yoon YH, Lin SS, et al. Chronic Aspiration of Gastric Fluid Accelerates Pulmonary Allograft Dysfunction in a Rat Model of Lung Transplantation. J Thorac Cardiovasc Surg (2006) 131(1):209–17. doi: 10.1016/j.jtcvs.2005.06.054
143. Fisichella PM, Davis CS, Lowery E, Pittman M, Gagermeier J, Love RB, et al. Pulmonary Immune Changes Early After Laparoscopic Antireflux Surgery in Lung Transplant Patients With Gastroesophageal Reflux Disease. J Surg Res (2012) 177(2):e65–73. doi: 10.1016/j.jss.2012.03.066
144. Robertson AG, Krishnan A, Ward C, Pearson JP, Small T, Corris PA, et al. Anti-Reflux Surgery in Lung Transplant Recipients: Outcomes and Effects on Quality of Life. Eur Respir J (2012) 39(3):691–7. doi: 10.1183/09031936.00061811
145. Abbassi-Ghadi N, Kumar S, Cheung B, McDermott A, Knaggs A, Zacharakis E, et al. Anti-Reflux Surgery for Lung Transplant Recipients in the Presence of Impedance-Detected Duodenogastroesophageal Reflux and Bronchiolitis Obliterans Syndrome: A Study of Efficacy and Safety. J Heart Lung Transplant (2013) 32(6):588–95. doi: 10.1016/j.healun.2013.02.009
146. Verleden SE, Vandermeulen E, Ruttens D, Vos R, Vaneylen A, Dupont LJ, et al. Neutrophilic Reversible Allograft Dysfunction (NRAD) and Restrictive Allograft Syndrome (RAS). Semin Respir Crit Care Med (2013) 34(3):352–60. doi: 10.1055/s-0033-1348463
147. Vanaudenaerde BM, Wuyts WA, Geudens N, Dupont LJ, Schoofs K, Smeets S, et al. Macrolides Inhibit IL17-Induced IL8 and 8-Isoprostane Release From Human Airway Smooth Muscle Cells. Am J Transplant (2007) 7(1):76–82. doi: 10.1111/j.1600-6143.2006.01586.x
148. Van Herck A, Frick AE, Schaevers V, Vranckx A, Verbeken EK, Vanaudenaerde BM, et al. Azithromycin and Early Allograft Function After Lung Transplantation: A Randomized, Controlled Trial. J Heart Lung Transplant (2019) 38(3):252–9. doi: 10.1016/j.healun.2018.12.006
149. El Agha E, Kramann R, Schneider RK, Li X, Seeger W, Humphreys BD, et al. Mesenchymal Stem Cells in Fibrotic Disease. Cell Stem Cell (2017) 21(2):166–77. doi: 10.1016/j.stem.2017.07.011
150. Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-Mesenchymal Transition in Lung Development and Disease: Does it Exist and is it Important? Thorax (2014) 69(8):760–5. doi: 10.1136/thoraxjnl-2013-204608
151. Barnes JL, Glass Ii WF. Renal Interstitial Fibrosis: A Critical Evaluation of the Origin of Myofibroblasts. Contrib Nephrol (2011) 169:73–93. doi: 10.1159/000313946
152. Barron L, Gharib SA, Duffield JS. Lung Pericytes and Resident Fibroblasts: Busy Multitaskers. Am J Pathol (2016) 186(10):2519–31. doi: 10.1016/j.ajpath.2016.07.004
153. Qiu J, Wang Y, Guo W, Xu L, Mou Y, Cui L, et al. Role of TGF-Beta1-Mediated Epithelial-Mesenchymal Transition in the Pathogenesis of Tympanosclerosis. Exp Ther Med (2021) 21(1):6. doi: 10.3892/etm.2020.9438
154. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, Bois RM, et al. Induction of Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells by Transforming Growth Factor-Beta1: Potential Role in Idiopathic Pulmonary Fibrosis. Am J Pathol (2005) 166(5):1321–32. doi: 10.1016/S0002-9440(10)62351-6
155. Hao Y, Baker D, Ten Dijke P. TGF-Beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int J Mol Sci (2019) 20(11):2767. doi: 10.3390/ijms20112767
156. Ballermann BJ, Obeidat M. Tipping the Balance From Angiogenesis to Fibrosis in CKD. Kidney Int Suppl (2011) 4(1):45–52. doi: 10.1038/kisup.2014.9
157. Shammout B, Johnson JR. Pericytes in Chronic Lung Disease. Adv Exp Med Biol (2019) 164(3): 299–317. doi: 10.1007/978-3-030-16908-4_14
158. Walker N, Badri L, Wettlaufer S, Flint A, Sajjan U, Krebsbach PH, et al. Resident Tissue-Specific Mesenchymal Progenitor Cells Contribute to Fibrogenesis in Human Lung Allografts. Am J Pathol (2011) 178(6):2461–9. doi: 10.1016/j.ajpath.2011.01.058
159. Boers JE, Ambergen AW, Thunnissen FB. Number and Proliferation of Clara Cells in Normal Human Airway Epithelium. Am J Respir Crit Care Med (1999) 159(5 Pt 1):1585–91. doi: 10.1164/ajrccm.159.5.9806044
160. Hiemstra PS, Bourdin A. Club Cells, CC10 and Self-Control at the Epithelial Surface. Eur Respir J (2014) 44(4):831–2. doi: 10.1183/09031936.00089214
161. Barnes PJ. Club Cells, Their Secretory Protein, and COPD. Chest (2015) 147(6):1447–8. doi: 10.1378/chest.14-3171
162. Wong AP, Keating A, Waddell TK. Airway Regeneration: The Role of the Clara Cell Secretory Protein and the Cells That Express it. Cytotherapy (2009) 11(6):676–87. doi: 10.3109/14653240903313974
163. Liu Z, Liao F, Scozzi D, Furuya Y, Pugh KN, Hachem R, et al. An Obligatory Role for Club Cells in Preventing Obliterative Bronchiolitis in Lung Transplants. JCI Insight (2019):e124732 5. doi: 10.1172/jci.insight.124732
164. Faust HE, Golden JA, Rajalingam R, Wang AS, Green G, Hays SR, et al. Short Lung Transplant Donor Telomere Length Is Associated With Decreased CLAD-Free Survival. Thorax (2017) 72(11):1052–4. doi: 10.1136/thoraxjnl-2016-209897
165. Naikawadi RP, Green G, Jones KD, Achtar-Zadeh N, Mieleszko JE, Arnould I, et al. Airway Epithelial Telomere Dysfunction Drives Remodeling Similar to Chronic Lung Allograft Dysfunction. Am J Respir Cell Mol Biol (2020) 63(4):490–501. doi: 10.1165/rcmb.2019-0374OC
166. Husain AN, Siddiqui MT, Holmes EW, Chandrasekhar AJ, McCabe M, Radvany R, et al. Analysis of Risk Factors for the Development of Bronchiolitis Obliterans Syndrome. Am J Respir Crit Care Med (1999) 159(3):829–33. doi: 10.1164/ajrccm.159.3.9607099
167. Jungraithmayr W, Ji L, Yang L, Weder W, and Korom S, Hersberger M. Increased T-Bet to GATA-3 Ratio During Acute Allograft Rejection in the Rat Lung. Transplant Proc (2009) 41(10):4316–20. doi: 10.1016/j.transproceed.2009.08.057
168. Jaramillo A, Smith CR, Maruyama T, Zhang L, Patterson GA, Mohanakumar T. Anti-HLA Class I Antibody Binding to Airway Epithelial Cells Induces Production of Fibrogenic Growth Factors and Apoptotic Cell Death: A Possible Mechanism for Bronchiolitis Obliterans Syndrome. Hum Immunol (2003) 64(5):521–9. doi: 10.1016/S0198-8859(03)00038-7
169. Estenne M, Hertz MI. Bronchiolitis Obliterans After Human Lung Transplantation. Am J Respir Crit Care Med (2002) 166(4):440–4. doi: 10.1164/rccm.200201-003PP
170. Lama VN, Belperio JA, Christie JD, El-Chemaly S, Fishbein MC, Gelman AE, et al. Models of Lung Transplant Research: A Consensus Statement From the National Heart, Lung, and Blood Institute Workshop. JCI Insight (2017) 2(9):e93121. doi: 10.1172/jci.insight.93121
171. Neurohr C, Kneidinger N, Ghiani A, Monforte V, Knoop C, Jaksch P, et al. A Randomized Controlled Trial of Liposomal Cyclosporine A for Inhalation in the Prevention of Bronchiolitis Obliterans Syndrome Following Lung Transplantation. Am J Transplant (2022) 22(1):222–9. doi: 10.1111/ajt.16858
172. Covington M, He X, Scuron M, Li J, Collins R, Juvekar A, et al. Preclinical Characterization of Itacitinib (INCB039110), A Novel Selective Inhibitor of JAK1, for the Treatment of Inflammatory Diseases. Eur J Pharmacol (2020) 885:173505. doi: 10.1016/j.ejphar.2020.173505
173. Lau CL, Beller J.P, Boys J.A, Zhao Y, Phillips J, Cosner M, et al. Adenosine A2A Receptor Agonist (Regadenoson) in Human Lung Transplantation. J Heart Lung Transplant (2020) 39(6):563–70. doi: 10.1016/j.healun.2020.02.003
Keywords: lung allograft, lung allograft immunity, lung allograft inflammation, NK cell, T cell, inflammation, lymphocyte
Citation: Santos J, Calabrese DR and Greenland JR (2022) Lymphocytic Airway Inflammation in Lung Allografts. Front. Immunol. 13:908693. doi: 10.3389/fimmu.2022.908693
Received: 30 March 2022; Accepted: 16 June 2022;
Published: 12 July 2022.
Edited by:
Jiangnan Xu, Capital Medical University, ChinaReviewed by:
Robin Vos, KU Leuven and UZ Leuven, BelgiumPhil Halloran, University of Alberta, Canada
Stefan Brocke, University of Connecticut Health Center, United States
Copyright © 2022 Santos, Calabrese and Greenland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Calabrese, ZGFuaWVsLmNhbGFicmVzZUB1Y3NmLmVkdQ==; John R. Greenland, Sm9obi5HcmVlbmxhbmRAdWNzZi5lZHU=