
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 July 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.907733
This article is part of the Research Topic Insights in Autoimmune and Autoinflammatory Disorders: 2021 View all 14 articles
 Jianan Zhao1,2,3
Jianan Zhao1,2,3 Kai Wei1,2,3
Kai Wei1,2,3 Ping Jiang1,2,3
Ping Jiang1,2,3 Cen Chang1,2,3
Cen Chang1,2,3 Lingxia Xu1,2,3
Lingxia Xu1,2,3 Linshuai Xu1,2,3
Linshuai Xu1,2,3 Yiming Shi1,2,3
Yiming Shi1,2,3 Shicheng Guo4,5*
Shicheng Guo4,5* Dongyi He1,2,3,6*
Dongyi He1,2,3,6*Rheumatoid arthritis (RA) is a chronic inflammatory disease that leads to joint damage and even disability. Although there are various clinical therapies for RA, some patients still have poor or no response. Thus, the development of new drug targets remains a high priority. In this review, we discuss the role of G-protein-coupled receptors (GPCRs), including chemokine receptors, melanocortin receptors, lipid metabolism-related receptors, adenosine receptors, and other inflammation-related receptors, on mechanisms of RA, such as inflammation, lipid metabolism, angiogenesis, and bone destruction. Additionally, we summarize the latest clinical trials on GPCR targeting to provide a theoretical basis and guidance for the development of innovative GPCR-based clinical drugs for RA.
Rheumatoid arthritis (RA) is an autoimmune disease characterized by synovial inflammation, joint destruction, and other clinical symptoms, including joint swelling, pain, morning stiffness, and weakness (1). The global prevalence of RA is approximately 0.5% to 1% (2). The risk factors for RA include genetic and metabolic aspects, genetic-environmental interactions, and microbial communities, all of which are involved in the pathogenesis of RA (3). For example, oxidative stress in multiple immune cells is thought to be an important factor in the chronic inflammatory destruction of RA, which in turn leads to joint destruction and extra-articular damage, including atherosclerosis, subcutaneous nodules and leg ulcers, systemic vasculitis, pulmonary fibrosis, scleritis and outer scleral inflammation, valvular heart disease and conduction abnormalities, and spinal cervical spondylosis (4). Currently, treatment options for RA include disease-modifying antirheumatic drugs (DMARDs), nonsteroidal anti-inflammatory drugs (NSAIDs), and biologics. Among them, analgesics and NSAIDs can reduce pain and stiffness, but NSAIDs have limited effectiveness and often have gastrointestinal and cardiac toxicity (5). DMARDs are the primary treatment and have been tried in combination, but some DMARDs have multiple adverse effects such as nausea, hepatotoxicity, hematometabolic disorders, and interstitial lung disease (5). Biological agents, including anti-tumor necrosis factor (TNF)-α antibodies, are also effective, but there are still adverse events, such as infusion and injection site infections, and differences in efficacy (5). With the advent of these new therapies, treatment of patients with RA has improved. However, due to heterogeneous factors and complex pathological mechanisms in RA, some patients have a poor clinical response, and the targeted development of new therapies is still a priority.
G-protein-coupled receptors (GPCRs), also known as seven-transmembrane domain receptors, respond to signals such as hormones, neurotransmitters, odors, and light, and transmit signals to cells for physiological functions (6). GPCRs can be classified into glutamate, rhodopsin, adhesion, frizzled/taste2, and secretin families (7). The classical signaling process of GPCR has been well described and consists of three parts: receptor, G protein, and effector. G protein is a heterotrimer composed of α, β, and γ subunits (see Figure 1) (8). Briefly, in response to stimuli, the receptor’s structure begins to change to enhance binding to G proteins; the guanosine diphosphate (GDP) in the Gα subunit of resting G proteins is released; and is converted to guanosine triphosphate (GTP). The Gβγ dimer dissociates, which in turn activates downstream effectors to continue signaling, often accompanied by an increase in cyclic adenosine monophosphate (cAMP) and activation of protein kinase C (PKC). The specific downstream transmission signal depends on the α subunit species, including primarily Gαs, Gαi/o, Gαq/11, and Gα12/13 (6). GPCRs and their signaling pathways are involved in multiple human physiological and pathological processes. Drugs targeting GPCRs account for approximately 27% of the global drug therapy market (6). Multiple multifamily GPCRs in are linked to immune mechanisms in RA, including inflammatory responses. Therefore, in this review, we have investigated the mechanisms of GPCRs in RA. Based on our findings, GPCR-targeted drug development has an excellent potential and economic translational value for clinical therapy development in RA.
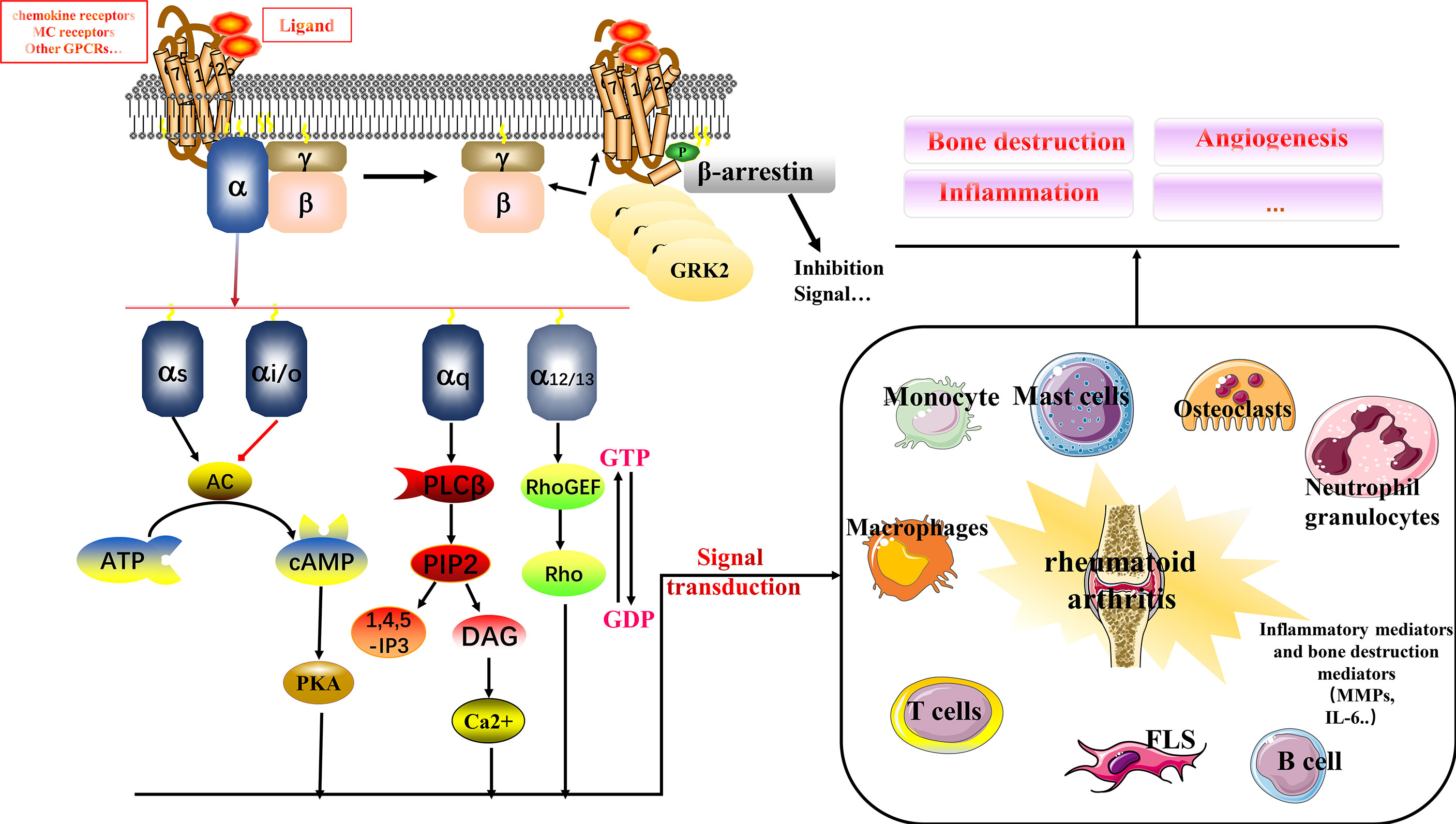
Figure 1 Relationship between multiple GPCRs and RA. GPCRs signal through the αβγ subunit of heterotrimeric G proteins. In response to ligand signals, GTP binds to the Gα subunit, and Gβγ dissociates to form a dimer for downstream signaling, binding to biological mediators. GRK2 is a kinase that phosphorylates activated GPCRs, making them a high-affinity substrate for the binding of the uncoupling protein arrestin. Arrestin binding abolishes (“arrests”) G protein-mediated signaling. The binding of GRK2 to Gβγ recruits cytosolic GRK2 to the plasma membrane, where the activated receptor is located. This is its primary function. It can also inhibit Gβγ-mediated signaling by sequestering Gβγ, which is a secondary effect. GPCRs interact with immune cells in RA to influence multiple mechanisms including bone destruction, inflammation, and angiogenesis.
Chemokine receptors are a class of GPCRs that regulate immunity. This class includes C-C motif chemokine receptor(CCR) 1-10, C-X-C motif chemokine receptors (CXCR) 1-7, X-C motif chemokine receptor 1 (XCR1), and C-X3-C motif chemokine receptor 1 (CX3CR1) (9). Chemokine receptors appear to be mostly Gαi-coupled, with some chemokine receptors also coupled to other G proteins, as verified by manual transfection in some contexts lacking Gαi/o proteins. Therefore, in the absence of specific information, chemokine receptors are by default considered Gαi/o-coupled (10). The overall chemokine and chemokine receptor cellular expression patterns in RA have been studied. For example, surface molecular assays of many peripheral blood B cells from RA patients revealed that CCR5, CCR6, CCR7, CXCR3, CXCR4, and CXCR5 play an essential role in B cell synovial migration, proliferation, and cytokine production (11). CCR1, CXCR4, and CCR5 are abundantly expressed in the RA synovium (12). Fibroblast-like synoviocytes (FLSs) in RA express CCR2, CCR5, CXCR3, and CXCR4. They have a pro-migration, proliferation, and matrix metalloproteinase production effect on FLSs under the stimulation of different ligands (13). Monocytes in synovial fluid mainly express CCR1, CCR2, CCR3, and CCR5, whereas peripheral blood expresses CCR1-5 at different levels of expression (14). Macrophages in rats with AIA mainly express CCR1, CCR2, and CCR5, which may maintain an inflammatory environment. CXCR4 is upregulated in endothelial cells and may be mainly associated with cell migration, angiogenesis, and inflammation (15). CCR1 and CXCR4 expression is upregulated in osteoblastic monocytes in RA (16). The roles of chemokines and chemokine receptors in RA have been previously reviewed (17–19). Therefore, we briefly summarize and update this information in the following sections (see Tables 1 and 2).
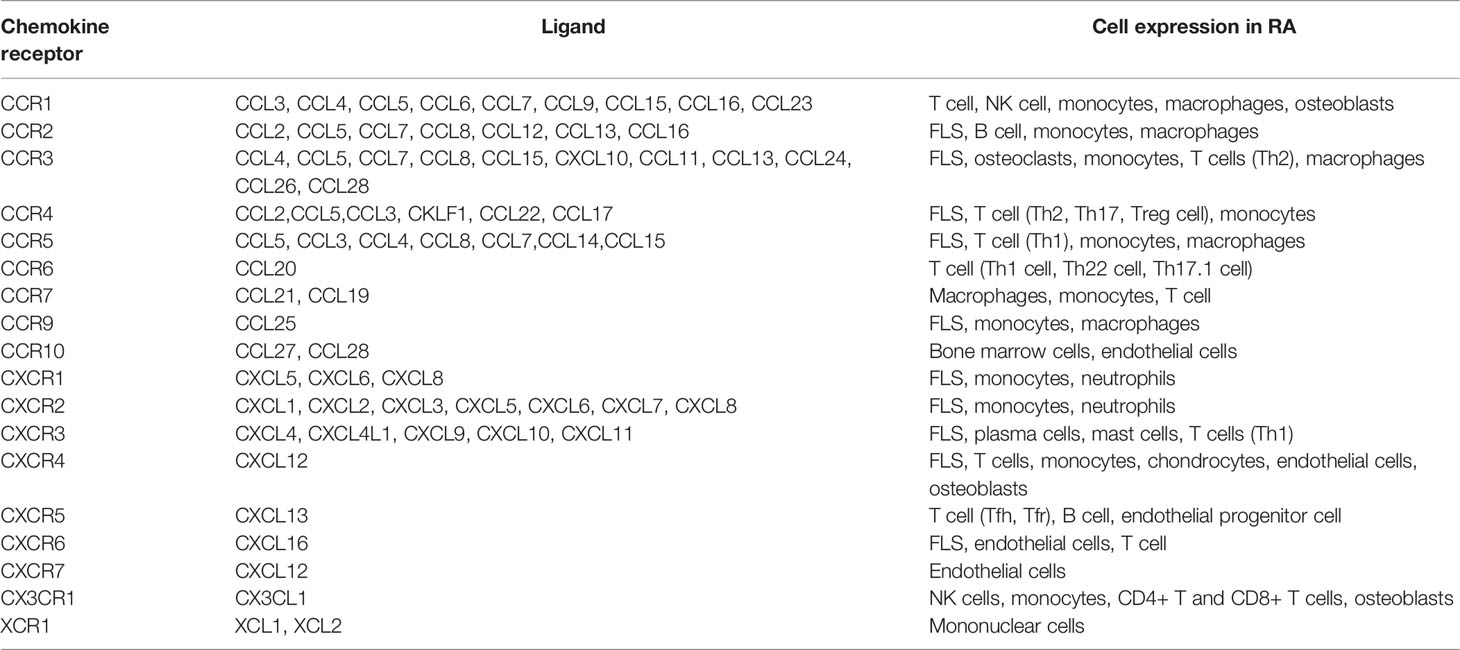
Table 1 Chemokine receptors and their ligands.
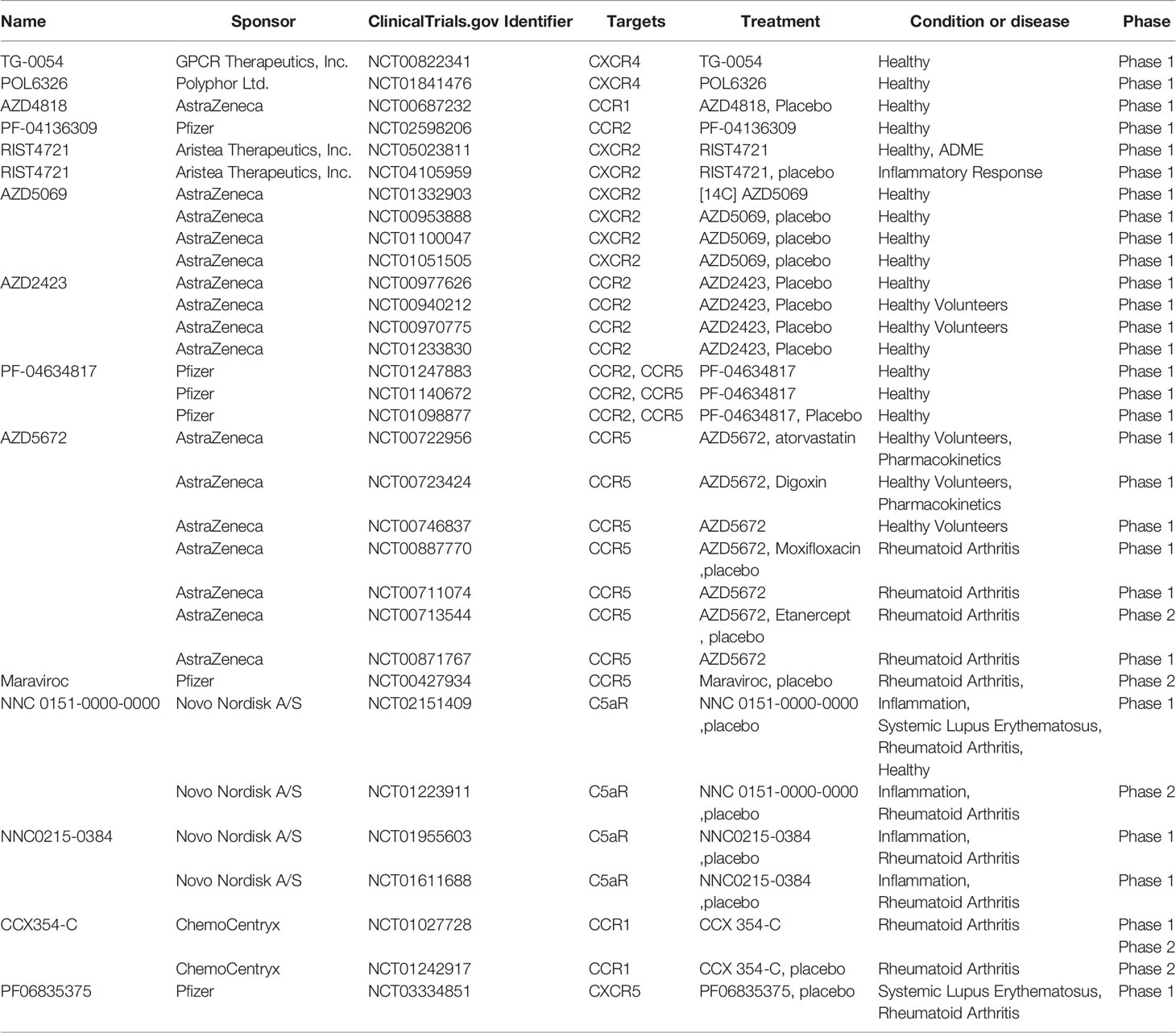
Table 2 Clinical trials of GPCR related to RA.
CCR1 is coupled with Gαi/o (20). CCL3 (MIP-1α), CCL4, CCL5 (RANTES), CCL6 (MIP-related protein-1), CCL7, CCL8, CCL9 (MIP-1γ/MIP-related protein-2), CCL15 (MIP-1δ/hemofiltrate CC chemokine-2/leukotactin-1), CCL16, and CCL23/CKβ8/myeloid progenitor inhibitory factor-1) have been described as CCR1 ligands (21, 22). The number of peripheral blood CCR1+ T/NK lymphocytes in patients with RA is negatively correlated with IL-10, whereas the number of CCR2+ B cells is positively correlated with IL-6 (23). The expression of CCR1 was positively correlated with serum antibodies against anti-cyclic citrullinated peptide (anti-CCP) in patients with RA (24). Several preclinical experimental studies have shown that CCR1 inhibition improves arthritic symptoms. For example, CCR1 inhibition reduces inflammation, joint damage, and cellular infiltration in the collagen-induced arthritis (CIA) mouse model. The specific mechanism may involve reduced recruitment of inflammatory cells; however, it is noteworthy that CCR1 inhibition spontaneously increases TNF-α levels in certain settings (25). The favorable animal results for CCR1 further support the clinical development of CCR1 antagonists. A phase IIa, double-blind, placebo-controlled, randomized, proof-of-concept study investigated the efficacy of a CCR1 antagonist (MLN3897) in RA patients; MLN3897 was well tolerated, but the results were not favorable, and there was no significant difference in its efficacy (26). Clinical blockade of CCR1 is likely to be effective and may always require maintenance of high levels of receptor occupancy (27). The CCR1 antagonist (CCX354-C) has shown initial clinical effectiveness. In a randomized placebo-controlled clinical trial in patients with RA, a CCR1 antagonist (CCX354-C) demonstrated good safety, tolerability, and clinical activity. CCX354 showed good tolerability and a linear dose-exposure profile in healthy subjects (28, 29).
The ligands of CCR2 include CCL2, CCL5, CCL7, CCL8, CCL12, CCL13, and CCL16 (22, 30, 31). In RA, the human isoforms of CCR2 include CCR2a and CCR2b, which are coupled to Gαi/o in most cases and Gαq in some cases and trigger the canonical activation of phospholipase Cβ isoenzymes downstream (32, 33). The mRNA expression of CCR2 was significantly higher in the range of 2.6< the disease activity score-28 (DAS28) <5.1 than DAS28>5.1 and control. CCL2 was negatively correlated with DAS28 index. Thus, CCR2 may contribute to early and rapid progression of inflammation (24). For example, the binding of CCR2 and CCL2 can help monocytes migrate to sites of RA inflammation and differentiate into M1 proinflammatory macrophages, possibly linking RA inflammation to insulin resistance (34). However, a double-blind, randomized, placebo-controlled clinical trial investigating the efficacy of a CCR2-blocking antibody (MLN1202) in patients with RA found that CCR2 blockade was not sufficient to improve multiple symptoms of RA (35).
CCR5 couples with Gαi/o and Gαq, and its ligands include CCL5, CCL3, CCL4, CCL8, CCL7,CCL14, and CCL15 (22, 31, 36, 37). CCR5 expression in rheumatoid factor (RF)-negative patients with RA is markedly higher than that in RF-positive patients with RA, which may be a molecular basis for the differences in RF expression in patients with RA (24). Investigators have found that the number of CCR5 molecules expressed on the cell surface correlates with the intensity of tumor necrosis factor α (TNFα) -induced T cell migration to the joint in RA. Anti-CCR5 antibodies can block this migration effect, thus possibly linking it with joint inflammation (38). Similar to the failed clinical trial of CCR2, clinical trials of CCR5 antagonists suggest that silencing CCR5 alone is not a truly effective target for RA (39–41). In conclusion, the lack of clinical efficacy of CCR2 and CCR5 blockade may be because their role in the migration of monocytes to the synovial membrane in patients with RA is not critical.
CCR3 may be coupled with Gαi/o (42). The ligands of CCR3 are CCL4, CCL5 (RANTES), CCL7 (MCP-3), CCL8 (MCP-2), CCL15 (HCC-1), CXCL10 (interferon (IFN)-γ inducible protein-10), CCL11 (eotaxin), CCL13, CCL24 (eotaxin-2), and CCL26 (eotaxin-3) (17, 22, 31). The role of CCR3 in RA may include the induction of cell migration and promotion of bone destruction and act as a predictor of the efficacy of certain clinical therapies. For example, flow cytometry analysis of synovial cells from patients with RA revealed that most CCR3+ cells were FLSs and that CCL11 expression was upregulated in the plasma and synovial fluid (43). IL-1β enhances the release of CCL11 from FLSs; CCL11 induces upregulation of CCR3 and matrix metalloproteinase (MMP)-9 mRNA expression in FLSs (43) and can induce the migration of FLSs and monocytes (44). CCL11 is expressed in osteoblasts and its expression is enhanced in response to inflammatory stimuli. Osteoclasts expressing CCR3 interact with exogenous CCL11 to stimulate cell migration and bone resorption (45). CXCL10, with an increased expression in synovial cells of patients with RA in response to TNF-α stimulation, interacts with CCR3 in T-cells and mediates an increase in receptor activation of nuclear factor kappa-B ligand (RANKL) expression via the Gαi subunit. As a result, osteoclast genesis and bone destruction are enhanced (46).Reduced CCR3 expression in serum CD4+ lymphocytes and reduced number of synovial CCR3+ monocytes in RA patients treated with steroids and anti-TNFα (47). In contrast, CD4 T cells and CD14 monocytes expressing CCR3 and CD8 T cells expressing CCR5 were increased in the peripheral blood of RA patients treated with anti-TNFα antibodies. This suggests the restoration of peripheral cell-mediated immune function, thereby blocking aggregation in the joints and inhibiting inflammation (48). The role of other CCR3 ligands in RA has been well studied and summarized (17–19). For example, CCL5 is mainly involved in the migration of leukocytes, and the use of anti-CCL5 antibodies can reduce the pathological manifestations of arthritis in the CIA mouse model (49). CCL24 is a major chemokine for inflammatory cells, and adjuvant arthritic mice treated with anti-CCL24 antibody showed significantly improved arthritic symptoms and inflammatory responses (50). Clinical inhibitors of CCR3 are in development, but currently do not target RA directly; most inhibitors target other diseases to test their effects, such as Parkinson’s disease, macular degeneration, and diabetic retinopathy.
The ligands of CCR4 include chemokine-like factor 1 (CKLF1), CCL2, CCL3, CCL5, CCL17, and CCL22 (22, 31, 51, 52). CKLF1 is significantly positively correlated with C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) (51). The expression of FLS in CCR4 may promote cell migration and proliferation (53). CCR4 expression in pro-inflammatory T-cell populations may promote inflammation by facilitating cell migration process. For example, a significant increase in circulating CCR4+ CXCR3-helper T cells (Th2 and Th17 cells) has been observed in clinically untreated patients with early RA (54). In addition, the expression of Treg cells of CCR4 may be a regulatory negative feedback mechanism of inflammation. For example, CCR4 and CCR6 expression was upregulated in peripheral blood Treg cells from patients with active RA and it was positively correlated with DAS28, suggesting that they could migrate to joints (55). CCR4+ T cells exert anti-inflammatory effects in patients with juvenile rheumatoid arthritis (JRA) by producing anti-inflammatory cytokines (IL-4) (56). However, this is not sufficient for the suppression of inflammation, so there are many proinflammatory mechanisms, such as CCL22, which is increased in both synovial fluid and serum of patients with RA, and may bind Treg cells CCR4 suppresses the number of Treg cells through the signal transducer and activator of transcription 5 (STAT5) signaling pathway (57).
CCL20 is the ligand of CCR6 (31). CCR6 expression may be primarily associated with T-cell subsets in RA. The proportion of CCR6+ memory T helper cell populations in anti-cyclic citrullinated peptide antibody (ACPA)+ versus ACPA- patients with RA is significantly different. This suggests that other cell subpopulations may be involved in various mechanisms of RA (58). The CCR6+ memory T helper cell population contains Th1/Th22 and Th17.1 cells, which activate FLSs in an IFN-γ-independent manner (59). In response to TNF-α, interleukin (IL)-6, and IL-1β stimulation, Th22 cells expressing CCR4, CCR6, and CCR10 migrate to the synovial tissues of patients with active RA and produce IL-22 to stimulate osteoclast differentiation for bone destruction (60). T cells and synovial cells in RA synovium produce CCL20, which binds to CCR6. High expression of RAR-related orphan nuclear receptor (ROR) γt promotes CCR6 expression, enhancing Th17 cell migration into the joint and promoting inflammation (61, 62).
The ligands of CXCR3 are CXCL4, CXCL4L1, CXCL9, CXCL10, and CXCL11 (22, 31). CXCL10-induced cell migration was found to require CXCR3, EGFR, and Gβγ subunits downstream of CXCR3, but not Gαi/o (63). CXCR3 expression in plasma cells, mast cells, and T-cell subsets is associated with RA. Plasma cells from patients with RA express CXCR3 by interacting with sub-synovial FLS-expressing Mig/CXCL9 recruited to the subsynovial lamina (64). In addition, mast cells in RA synovial tissue abundantly express CXCR3; this may maintain the synovial inflammatory environment by binding CXCL9 and CXCL10 and producing mediators, including histamine, proteases, arachidonic acid metabolites, and cytokines (65). Patients with RA appear to express CCR5 and CXCR3 preferentially on Th1 cells, and Th2 cells preferentially express CCR3, CCR4, and CCR8 (66, 67). The combination of MTX and anti-TNFα antibodies decreased CXCR3 and IL-12R expression (considered a Th1 cell markers) and upregulated CCR4 and IL-4R expression (considered a Th2 cell markers) in peripheral blood CD4 cells in patients with RA, with a high percentage of apoptotic cells (68). Paradoxically, peripheral blood CD4 and CD8 CXCR3+ T lymphocytes were increased in RA patients treated with infliximab and etanercept. CXCR3+ CD4 T lymphocytes negatively correlated with DAS28 (69). This seems to validate the findings of Nanki et al. that chemokine receptor expression did not differ significantly across T-cell subpopulations (70). Further experiments are needed, but restoring the suppression of inflammation by regulating the balance between Th1/2 cells is a feasible strategy to improve RA. Several preclinical trials have demonstrated that targeted inhibition of CXCR3 is beneficial for the treatment of RA. Specifically, it reduced the recruitment of Th1 cells to sites of inflammation (71) and restored the balance between Th17 and Treg cells (72).
CCR7 may be coupled with Gαi/o and Gαq (73). CCR7 ligands, including CCL21 and CCL19, are associated with the homing and localization of dendritic cells and T cells (74). CCR7 is a surface marker of macrophages in the RA synovial fluid. Its upregulation in cells may promote inflammation and is positively correlated with DAS28 and inflammatory factor levels, and negatively correlated with anti-inflammatory factor levels in patients with RA. For example, CCR7+CD95+CD4+ peripheral lymphocytes were significantly elevated in patients with active RA and were positively correlated with IL-6 (75). Lipopolysaccharides (LPS) and IFN-γ promote CCR7 expression. IL-4 inhibits CCR7 expression. CCL21 promotes Th17 differentiation, osteoclast formation, angiogenesis, and proinflammatory macrophage differentiation (76, 77). In addition, miR-155 expression was higher in ACPA+ patients than in ACPA − patients and was correlated with DAS28. miR-155 can promote CCR7 expression and downregulate CCR2 expression in RA monocytes (78). The progression of arthritis in CIA mice can be inhibited using an anti-CCR7 antibody (74). The development of clinical inhibitors of CCR7 for non-Hodgkin’s lymphoma and chronic lymphocytic leukemia is currently underway.
The association of CCR9 and CCR10 with RA is less direct, with CCR9 associated primarily with cell migration of FLS, proinflammatory differentiation of macrophages, and related inflammatory and bone destruction processes. CCR10 is primarily associated with angiogenesis. CCL25 is the ligand of CCR9. In response to TNF-α, CCR9 elevated expression in peripheral blood monocytes and macrophages of RA and in combination with CCL25 induces cell migration (79) and differentiation of monocytes into M1 macrophages via the P38 and extracellular signal-regulated kinase (ERK) pathways (80). FLSs and macrophages release CCL25 in RA synovial fluid in response to stimulation by IL-1β and IL-6. CCL25 binds to CCR9 on FLSs and macrophages to promote osteoclast formation and vascular opacification (80). Additionally, it stimulates IL-6 and MMP-3 production in FLSs and IL-6 and TNF-α production in peripheral blood mononuclear cells. The antagonism of CCR9 suppressed arthritic symptoms in the CIA mouse model (81).
The ligands of CCR10 are CCL27 and CCL28 (82). CCR10 and ligand CCL28 expression is elevated in the synovial tissue and synovial fluid of patients with RA, mainly in the bone marrow and endothelial cells; inhibition of CCL28 or CCR10 reduces endothelial cell migration and angiogenesis (83). Crohn’s disease is a focus in the development of clinical inhibitors for CCR9 and a gap exists in clinical inhibitors for CCR10. Therefore, further clinical trials of both for RA are still necessary.
Both CXCR1 and CXCR2 are coupled to Gαi/o proteins (22, 84). The ligands of CXCR1 include CXCL5, CXCL6, and CXCL8. The ligands of CXCR2 include CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8, and interact with GRK6 to negatively regulate receptor sensitization and transport, thereby affecting cell signaling and angiogenesis (22, 84). CXCR1 and CXCR2 are involved in the migration of neutrophils, FLS, T-cells, and monocytes in RA, influencing the subsequent inflammation and bone destruction processes and are expressed in their cell populations (85, 86). For example, CXCR1 and CXCR2 ligands, such as CXCL1, CXCL5, and LTB4, are highly expressed in the joints and bind to CXCR1 and CXCR2. Here, they promote joint migration of neutrophils in AIA mice (87). Targeted inhibition of neutrophil migration by the CXCR1 and CXCR2 antagonists SCH563705 reduces disease activity scores, joint inflammation, and bone and joint destruction in CIA mice (88). In addition, CXCL5 [epithelial neutrophil-activating peptide 78 (ENA-78)] of citrulline is significantly elevated in the synovial fluid of RA patients. It correlates with CRP and ESR, inducing inflammatory cell focus and inflammation through CXCR1-and CXCR2-induced chemotaxis of monocytes (89). Reduced CXCR1 and CCR2 expression in blood T cells after treating patients with RA with the anti-TNF-α antibody infliximab may inhibit their migration to sites of inflammation (90).
CXCR4 may be coupled to Gαi/o and Gαq (73). The ligand of CXCR4 is mainly CXCL12 [stromal cell-derived factor 1 (SDF-1)]. High expression of CXCL12 and CXCR4 in the serum and synovial fluid of patients with RA is positively correlated with ESR, CRP, RF, and DAS28 scores (91). T-cell expression of CXCR4 may promote cell migration and inflammatory processes. CXCR4 is positively correlated with memory-activated CD4+ T cells and follicular helper T (Tfh) cells (92).Pablos et al. found that FLSs specifically express CXCL12, which aggregates and fixes in the heparan sulfate molecules of vascular endothelial cells, promoting angiogenesis and inflammatory cell infiltration. CXCL12 activates CXCR4 on the surface of T cells. Simultaneously, very late activation antigen 4 (VLA-4) interacts with vascular cell adhesion molecule 1 (VCAM-1) to promote the recruitment of T cells and their retention in the joints to promote inflammation (93, 94). The expression of CXCR4 in B cells may be indicative of disease activity; for example, considerable infiltration of P-gp+CXCR4+ CD19+ B cells can be observed in patients with RA, which may correlate with disease activity, drug resistance, and progressive joint destruction (95).
In addition, monocyte expression of CXCR4 may promote the process of cell migration and differentiation into proinflammatory macrophages, which can affect subsequent inflammatory and bone destruction processes. The hypoxic microenvironment induces SDF-1 expression in RA FLSs, leading to the accumulation of CXCR4-expressing monocytes in the synovial membrane and their differentiation into macrophages and secretion of proinflammatory factors (IL-1β, IL-6, and TNFα) and MMP. This process mediates inflammation and osteoarthritic destruction (96). The specific mechanism may involve SDF-1α enhancing the binding of c-Jun to AP-1 on the IL-6 promoter and enhancing AP-1 transcriptional activity to regulate IL-6 expression (97). Similarly, SDF-1α activates ERK/c-Fos/c-Jun via CXCR4 to mediate AP-1 activation of MMP13 to promote cartilage destruction (98). SDF-1a also stimulates CXCR4+ chondrocytes to release MMP3, which destroys bone joints (99).In addition, the endogenously released reduced form of HMGB forms a heterotrimeric complex with CXCL12; this complex interacts with CXCR4 to promote cell migration and inflammation via the toll-like receptor (TLR) 2/4 (100).
Therefore, the inhibition of CXCR4 is beneficial for improving RA. The herbal compound QLY can reduce joint lesions and paw swelling in adjuvant arthritis rats by a mechanism involving the inhibition of the CXCL12/CXCR4/nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) pathway (101). In addition, targeted CXCR4 inhibitors are now well developed, and described in many excellent reviews (102–105). Many small-molecule compounds have significant CXCR4 inhibitory effects. We have focused on the progress of CXCR4 inhibitor development in clinical trials and have summarized and updated them here. The CXCR4 antagonist AMD3100 has been approved by the FDA for use as a hematopoietic stem cell mobilizer. All information was obtained from www.clinicaltrials.gov (See Table 1).
CXCL13 is the ligand of CXCR5. Changes in CXCR5 expression in different T-cell subsets may be associated with RA. The circulating Tfr/Tfh cell ratio in RA is negatively correlated with serum CRP, ESR, RF, anti-CCP, IgG, and the DAS28 index (106). Tfh cells can activate B cells and produce specific antibodies, and the number of Tfh cells in peripheral blood is positively correlated with anti-CCP antibody levels (107). In contrast, in RA remission, circulating Tfr cell subsets increase and are negatively correlated with RF, anti-cyclic citrullinated peptide, and DAS28 (108). A decrease in CD4+CXCR5+Foxp3+ Tfr cells/CD4+CXCR5+ Tfh cells in the peripheral blood of patients with RA may contribute to multiple humoral immune mechanisms (109). In vitro co-culture of RA FLSs and peripheral blood mononuclear cells (PBMCs) revealed that increased production of TNF-α, IL-1β, IL-6, and reactive oxygen species (ROS) could promote CD4 + CXCR5 + ICOS + Tfh differentiation (110). CXCR5 expression in other cells (endothelial progenitor cells and regulatory B cells) has also been linked to RA. CXCL13 expression is elevated in the synovial fluid of CIA mice and patients with RA. CXCL13 interacts with CXCR5 in endothelial progenitor cells and promotes homing and angiogenesis through the PLC, MEK, and AP-1 signaling pathways (111). CXCR5 expression on the surface of regulatory B cells (Bregs) is downregulated. The response to CXCL13 is not sufficient for preferential migration to the synovial fluid to produce sufficient anti-inflammatory factor IL-10, which may be one of the mechanisms of RA inflammation (112). CXCR5-deficient mice with impaired germinal response centers are resistant to collagen-induced arthritis (113). The application of clinical inhibitors of CXCR5 in RA is ongoing, and promising results are expected to be published.
The ligand of CXCR6 is CXCL16. CXCR6 is expressed in FLS, endothelial cells, and T-cells in RA. CXCR6 and CXCL16 levels are elevated in both RA FLSs and may stimulate FLS proliferation (114). CXCL16 also stimulates RANKL expression in RA FLSs through Janus kinase (JAK)2/STAT3 and P38/mitogen-activated protein kinase (MAPK) signaling (115). CXCR6 is also expressed in endothelial cells and may be involved in endothelial cell recruitment and angiogenesis in the RA joints by binding to CXCL16 (116). CXCR6 and CXCL16 can promote inflammation by affecting T cell differentiation and homing to joints. CXCR6 knockout CIA mice exhibit multiple arthritic symptoms (117, 118).
The ligand of CXCR7 is CXCL12 and was found to be internalized and signaled downstream in a chemokine-dependent manner only through recruitment of the β-arrestin protein (119, 120). CXCR7 binds to chemokines with a high affinity, but ligand binding does not lead to G protein-mediated intracellular calcium mobilization or chemotaxis (121). CXCR7 is expressed on endothelial cells in the synovium of RA patients and can be significantly upregulated in response to IL-1β and CXCL12 stimulation. It promotes angiogenesis, and is mainly used as an alternative receptor for CXCR4. CXCR7 inhibitors significantly reduced arthritic symptoms and vascularity in CIA mice (122).
CX3CR1 is mainly expressed in NK cells, monocytes, and CD4+ and CD8+ T cells. Its ligand CX3CL1/FKN (fractalkine) is primarily involved in cell adhesion (123). CX3CR1/CX3CL1 are also associated with the production of inflammatory mediators by macrophages, T cells, and FLSs in RA (124). CX3CR1+HLA-DRhiCD11c+CD80-CD86+ cells, an osteoclast subpopulation, are present in the synovium of patients with RA. In mice, the corresponding subpopulation CX3CR1hiLy6CintF4/80+I-A+/I-E+ cells can be inhibited by inhibiting FOXM1 (125). Patients with active RA treated with infliximab and etanercept had reduced CX3CR1 expression in peripheral blood PBMC and T cells and serum CX3CL1 levels (126, 127).
The ligands of XCR1 are XCL1 and XCL2 (22). XCR1 expression is upregulated in mononuclear cells (MNCs) of synovial fluid and venous blood samples from patients with RA, suggesting that XCR1 may be involved in the mechanism of RA (128). The connection between CX3CR1 or XCR1 and RA has not been well studied, and the specific mechanisms require further study.
The structure and function of MC and its receptors have been well-reviewed (129). MCR mainly included MC1-5R. MC1-5R is coupled with Gαs to stimulate downstream cAMP/PKA signaling. In addition, M3CR promotes downstream ERK1/2 signaling to promote cell proliferation by coupling with Gαi/o. MC4R also promotes downstream protein kinase C by coupling with Gαq and inhibits apoptosis by coupling with Gαi to promote downstream ERK1/2 signaling. MC5R can also couple Gαi/o to promote downstream PI3K/C-Raf/MEK1/2/ERK1/2 signaling (130). Activation of MCR may have an ameliorative effect on RA. MC3R knockdown with serum transfer-induced arthritis exacerbated significant bone erosion, high expression of RANKL in the joint, increased time of NF-kB activation, and upregulation of proinflammatory genes [IL-1β, IL-6, and nitric oxide synthase 2 (NOS2)]. Conversely, overexpression of MC3R and MC3R agonist D [Trp8]-γ-MSH significantly reduced the degree of arthritis, suggesting that MC3R may be an important target in preventing bone destruction and inflammation progression in arthritis (131). Endogenous MCR agonists include ACTH and MSH (α, β, and γ), with different affinities for the five MCRs, where MC2R was activated only in response to ACTH (130).
Alpha-melanocyte-stimulating hormone (α-MSH) is a 13-amino acid peptide (132). α-MSH can inhibit inflammation by interacting with the MCR of multiple cells in RA via multiple mechanisms (132). Elevation of α-MSH may be a measure of suppression of inflammation in joint tissues. Catania et al. observed significantly elevated synovial fluid α-MSH, interleukin 1 receptor antagonist (IL-1ra), and soluble tumor necrosis factor receptor (sTNFr) levels in patients with RA (133). In vitro, α-MSH inhibits LPS-induced protein hydrolase release from macrophages, oxidative burst response, production of reactive oxygen and reactive nitrogen species, adhesion molecule expression, NF-kB activation, and downregulation of cell surface CD14 expression to inhibit inflammation (134). The inhibitory effect of NF-kB may be mediated by an increase in the cAMP-mediated activity (135). Similarly, Bhardwaj et al. found that the C-terminal tripeptide of α-MSH induces the production of the anti-inflammatory factor IL-10 in monocytes (136). α-MSH stimulation of T cells in vitro promotes phenotypic conversion of T cells to CD25+CD4+ regulatory T cells. It induces TGF-β production to suppress IFN-γ production by other effector T cells; eventually, inflammation is suppressed via a mechanism involving α-MSH binding to MC5R on T cells (137). Thus, synthetic MCR agonists may also inhibit inflammation in patients with RA. For example, Montero-Melendez et al. found that the pro-senescence effect of FLSs via agonist-induced MC1R expression suppressed the inflammatory response in RA (138). The MC1R agonist BMS interferes with FLSs cell cycle, anti-inflammatory, and arthroprotective features These include cell proliferation, cycle arrest, lysosomal amplification, expression of the cellular senescence marker p16 INK4, downregulation of cell cycle promoters and anti-apoptotic signals, downregulation of proinflammatory factors (CCL2, IL6, and IL8), increased expression of MMPs, and downregulation of matrix-degrading enzymes (ADAMTS1 and ADAMTS2) (138). Montero-Melendez et al. also found that MC Pan Agonist AP214 reduced disease scores and paw edema (which primarily affects IL-1β release) in a K/BxN serum transfer arthritis model, exerting an anti-inflammatory effect (139).
α-MSH is also linked to RA and is osteoprotective. α-MSH acts directly on the bone, increases bone turnover, and reduces bone volume, possibly through the synergistic action of other cells such as adipocytes or islet B cells (132). Human chondrocytes express a variety of MCRs such as MC2R and MC5R. In vitro, a-MSH stimulation can regulate cAMP, proinflammatory factors, and MMP in chondrocytes, which may play a role in inflammation, development, and cartilage degeneration (140). Similarly, Kaneva et al. found that α-MSH and D[Trp8]-γ-MSH inhibited TNF-α-induced proinflammatory factor release (IL-1, IL-6, and IL-8) from C-20/A4 chondrocytes, increased anti-inflammatory factor IL-10 release, decreased the expression of MMP1, MMP3, and MMP13, and inhibited the apoptosis of key molecules caspase3/7 and cell death. MC3R/4R antagonists inhibited the effects of D[Trp8]-γ-MSH, suggesting that MC1R and MC3R may be the primary MCRs for these mechanisms (141). Zaidi et al. found that ATCH induced vascular endothelial growth factor (VEGF) secretion via MC2R and stimulated osteoblast maturation and survival. This reduced experimental osteonecrosis induced by methylprednisolone acetate, which may be a mechanism to inhibit bone destruction in RA (142).
In addition to the intensively studied MCRs and chemokine receptors described above, there are also some GPCRs that are associated with RA For example, GPR120 regulates lipid metabolism and GPCR (CD97) is related to adhesion. Further experiments are needed to investigate their specific roles in depth.
C5aR is Gαi/o-coupled (143). C5aR is mainly expressed in neutrophils and macrophages. Elevated C5aR and C5a levels have been found in the blood and synovial fluid of RA patients (144). Patient mast cells also express C5aR and release histamine to participate in the inflammatory response (145). In the presence of multiple cytokines, neutrophils express chemokine receptors in the joint to release LTB4 and IL-1β to promote their recruitment. Simultaneously, immune complexes in RA can activate C5a production by neutrophils and bind C5aR to further amplify the inflammatory response (146). In addition, the plasma kallikrein-kinin system (KKS) is present in RA. KKS activation activates prekallikrein (pKal) and factor XII (FXII) cleavage of high-MW kininogen (HK) to release bradykinin. Kal also activates monocytes to promote proinflammatory cytokines, upregulate C5aR and FcRIII expression, and release C5a. Inhibition of KKS alleviates symptoms in arthritic mice (147). Macrophage infiltration in zymosan-induced arthritis (ZIA) mice; increased expression of C5aR and C3aR in joints; and elevated levels of C5a and soluble receptor activator of nuclear factor kappa B ligand (sRANKL) in synovial fluid are possible mechanisms of inflammation (148). Targeted inhibition of C5aR remains challenging in terms of clinical translation, although preclinical data have shown that anti-C5aR antibodies can improve symptoms in experimental arthritic mice (149). However, a double-blind placebo-controlled study found no clinical improvement in patients with RA receiving a C5aR inhibitor (150).
Adenosine is an anti-inflammatory mediator that acts mainly through four receptors (A1, A2A, A2B, and A3) to inhibit proinflammatory factors (IL-6 and TNF-α) and to promote the synthesis of anti-inflammatory factors (IL-10) (151). Its role in RA has been extensively reviewed and is mainly related to the mechanism of action of methotrexate, phosphoinositide 3-kinases (PI3Ks)/protein kinase B (PKB), and NF-kB signaling pathways. The detailed mechanism of action has previously been explained (152–156).
GPR120 is Gαi/o- or Gαq-coupled, and its ligands are LCFAs, unsaturated fatty acids, omega-3 fatty acids, and omega-6 fatty acids (157). Abnormalities in lipid metabolism were observed in CIA mice. For example, the antioxidant enzymes LDH and lipoproteins are positively correlated with the lipid fractions in the plasma and joint tissue. Lipid content is negatively correlated with lipid levels, and cytokine content is correlated with lipid fractions and the saturated fatty acid/unsaturated fatty acid ratio (158). Therefore, in addition to the effects of multiple immune mechanisms on RA, lipid metabolism may also regulate RA. GPCRs have been shown to regulate lipid metabolism, which may have potential therapeutic effects on RA. Some endogenous n-6 polyunsaturated fatty acids (PUFA) can cause inflammation and pain by promoting the synthesis of leukotrienes, prostaglandins, IL-6, TNF-α, and ROS. Simultaneously, exogenous linolenic acid, an n-3 PUFA that activates FFA4 (GPR120) and has potent anti-inflammatory effects, has been shown to improve inflammation and disease severity in RA (159, 160). Omega-3 fatty acids activate GRP120, which decreases transforming growth factor-β activated kinase 1 (TAK1) and inhibits the downstream IKKβ/I-κB pathway and terminal NF-κB. Thereby it suppresses T cell activation and inflammatory responses, and improves symptoms in arthritic mice (161). GPR120 inhibits CD40L-induced activation of dendritic cells and proinflammatory responses, and GPR120 agonists have similar effects (162).
GPR43 is Gαi/o- or Gαq-coupled, and its ligands are acetate, propionate, and butyrate (157). GRP43/FFA2R is expressed on RA FLSs and upregulated in response to TNFα stimulation, and its inhibition of GPR43 can significantly inhibit a variety of biological mediators and signaling pathways in RA, including IL-6, IL-8, high mobility group protein 1 (HMG-1), monocyte chemoattractant protein 1 (MCP-1), intercellular adhesion molecule 1 (ICAM-1), and vascular cellular adhesion molecule 1 (VCAM-1), production of ROS and 4-hydroxynoneal, MMP-3, and MMP-13, and activation of the NF-κB inflammatory signaling pathway (163).
GPR91 is Gαi/o- or Gαq-coupled, and its ligands is succinate (157). GPR91 is mainly expressed in macrophages and dendritic cells. GPR91 in dendritic cells triggers intracellular calcium flow by binding succinate; induces cell migration; and interacts with TLR to release proinflammatory factors. Succinate also enhances T helper cell activation (164). Succinate is abundant in the RA synovial fluid. Macrophage sensing of endogenous LPS activates TLR, releases succinate, and upregulates GPR91 sensing of succinate to activate glycolytic pathways and promote IL-1β production. GPR91 knockout mice have reduced macrophage activation and IL-1β release in response to antigen-induced arthritis (165). Intracellular succinate promotes VEGF production by activating GPR91 and inducing angiogenesis via HIF-1α (166).
CD97 is primarily coupled to the Gαi/o protein (167). CD97 plays multiple roles in RA through its N-terminal epidermal growth factor structural domain combined with the N-terminal short consensus repeat structural domain of CD55 (168). For example, CD97 is expressed in granulocytes and monocytes, and is upregulated in T and B cells combined with CD55 activation to promote T cell activation (169). Neutralizing anti-CD97 antibody attenuates multiple arthritic manifestations in a collagen-induced murine joint model (170).
CasR is Gαi/o- or Gαq-coupled, and its ligands are Ca2+, L-amino acids, and oligopeptides (157). NLRP3 inflammasome and associated IL-1β release are closely associated with inflammatory progression in RA (171, 172). Monocytes show increased CasR expression (173, 174). Monocytes and macrophages can phagocytose colloidal calmodulin particles to prevent extracellular calcification in vivo. In this process, increased extracellular Ca2+ concentrations can activate CasR, and thus activate the NLRP3 inflammasome in monocytes to promote IL-1β release for inflammation (175).
TGR5 is Gαs-coupled and its ligands are lithocholic acid, deoxycholic acid, chenodeoxycholic acid, and cholic acid (157). Bile acids mainly activate TGR5. TGR5 mRNA expression is reduced in PBMCs from RA patients and correlates with CRP and DAS28 levels. Lithocholic acid inhibits NF-kB activity and inflammation by binding TGR5 in PBMC, while it reduces proinflammatory factors in CIA mice, such as TNF-α, IL-6, IL-8, and IL-1β (176).
FRP1 and FPRL1s are Gαi/o- or Gαq-coupled, and their ligands are N-formyl-methionine and N-formyl-metoligopeptides (157). Acute-phase serum amyloid A (A-SAA) and FPRL1 are expressed in RA FLSs, macrophages, and endothelial cells. A-SAA is associated with RA disease activity and is regulated by proinflammatory factors, which may be involved in the stromal degradation of RA through FPRL1-induced secretion of MMP-1 and MMP-3 from FLSs (177). In addition, A-SAA can promote FLS proliferation and angiogenesis by binding to FPRL1 (178). Similarly, annexin-1 may also induce MMP-1 secretion from FLS via FPRL1 (179).
GRK2 is a key enzyme involved in desensitization of many G proteins (180). Reduced T-cell expression of GRK2 promotes increased responses in CCL3, CCL4, and CCL5 (181). GRK2 in endothelial cells may regulate inflammatory responses by regulating Weibel-Palade body formation-mediated cytokinesis and histamine-stimulated aggregation of leukocytes to endothelial cells (180).
The structure and signaling pathways of GPCRs have been investigated and explained in various diseases; however, their specific mechanisms and roles in RA remain to be elucidated. In this review, we discussed multiple GPCR receptors to clarify some of their roles and mechanisms. Nevertheless, many questions remain. First, chemokine receptors are currently the most intensively studied GPCRs in RA. There are two main aspects of RA drug strategies developed for chemokines and chemokine receptors: on one hand, corresponding ligands that can be selected to inhibit chemokine receptors and on the other, direct inhibition of chemokine receptors. Both options are currently investigated in several preclinical and clinical studies. However, the exact clinical efficacy still needs to be further observed in well-designed clinical trials. Unfortunately, some good preclinical trial results do not translate into an excellent clinical protocol. Some clinical trials on chemokines have faced difficulties, possibly owing to the widespread expression of chemokine receptors in a variety of cells, implying that a portion of chemokine receptors may be necessary for certain normal physiological processes, and therefore, potentially effective chemokine receptor inhibitors may specifically target receptor expression in certain pathogenic cell subpopulations. Second, the expression of GPCRs at different disease stages of RA may have diverse functional roles, such as CCR4. Clarifying the role of receptors at different stages is vital for drug development. Finally, the application of novel variable conformation modulators and biased agonists of GPCRs may be an essential tool for the future development of GPCR drugs for RA. So far, inhibition of single GPCR has failed to bring the desired outcomes. The combined inhibition of multiple GPCRs showed better efficacy. In addition, better use of modern techniques, such as synovial biopsy and arthroscopic surgery with the aid of computer imaging, may allow for better evaluation of the overall disease situation, and combined analysis using multi-omics and multiple molecular biology techniques may improve the current situation. Although the translation of preclinical GPCR results to clinical results for RA is still challenging, in-depth studies present a direction with a great potential.
JZ is responsible for the collection, collation, and writing of the original manuscript. KW, CC, PJ, LXX, LSX, and YS are accountable for the collection. SG and DH are responsible for the concept development, revision, and manuscript review. All authors reviewed and accepted the final version.
This work was funded by the National Natural Science Funds of China (82074234 and 82071756), National Key Research and Development Project (2018YFC1705200 and 2018YFC1705203), Shanghai Chinese Medicine Development Office, National Administration of Traditional Chinese Medicine, Regional Chinese Medicine (Specialist) Diagnosis and Treatment Center Construction Project-Rheumatology, State Administration of Traditional Chinese Medicine, National TCM Evidence-Based Medicine Research and Construction Project, Basic TCM Evidence-Based Capacity Development Program, Shanghai Municipal Health Commission, and East China Region-based Chinese and Western Medicine Joint Disease Specialist Alliance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid ArthritIs. Nat Rev DIs Primers (2018) 4:18002. doi: 10.1038/nrdp.2018.2
2. Silman AJ, Pearson JE. Epidemiology and Genetics of Rheumatoid ArthritIs. ArthritIs Res (2002) 4 Suppl 3(Suppl 3):S265–72. doi: 10.1186/ar578
3. Zhao J, Guo S, Schrodi SJ, He D. Molecular and Cellular Heterogeneity in Rheumatoid ArthritIs: MechanIsms and Clinical Implications. Front Immunol (2021) 12:790122. doi: 10.3389/fimmu.2021.790122
4. da Fonseca LJS, Nunes-Souza V, Goulart MOF, Rabelo LA. Oxidative Stress in Rheumatoid ArthritIs: What the Future Might Hold Regarding Novel Biomarkers and Add-On Therapies. Oxid Med Cell Longev (2019) 2019:7536805. doi: 10.1155/2019/7536805
5. Scott DL, Wolfe F, Huizinga TW. Rheumatoid ArthritIs. Lancet (2010) 376(9746):1094–108. doi: 10.1016/s0140-6736(10)60826-4
6. Calebiro D, Koszegi Z, LanoIselée Y, Miljus T, O'Brien S. G Protein-Coupled Receptor-G Protein Interactions: A Single-Molecule Perspective. Physiol Rev (2021) 101(3):857–906. doi: 10.1152/physrev.00021.2020
7. Lagerström MC, Schiöth HB. Structural Diversity of G Protein-Coupled Receptors and Significance for Drug DIscovery. Nat Rev Drug Discov (2008) 7(4):339–57. doi: 10.1038/nrd2518
8. Gilman AG. G Proteins: Transducers of Receptor-Generated Signals. Annu Rev Biochem (1987) 56:615–49. doi: 10.1146/annurev.bi.56.070187.003151
9. Charo IF, Ransohoff RM. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N Engl J Med (2006) 354(6):610–21. doi: 10.1056/NEJMra052723
10. Thelen M, Stein JV. How Chemokines Invite Leukocytes to Dance. Nat Immunol (2008) 9(9):953–9. doi: 10.1038/ni.f.207
11. Nanki T, Takada K, Komano Y, Morio T, Kanegane H, Nakajima A, et al. Chemokine Receptor Expression and Functional Effects of Chemokines on B Cells: Implication in the PathogenesIs of Rheumatoid ArthritIs. Arthritis Res Ther (2009) 11(5):R149. doi: 10.1186/ar2823
12. Haringman JJ, Smeets TJ, Reinders-Blankert P, Tak PP. Chemokine and Chemokine Receptor Expression in Paired Peripheral Blood Mononuclear Cells and Synovial TIssue of Patients With Rheumatoid Arthritis, OsteoarthritIs, and Reactive ArthritIs. Ann Rheum DIs (2006) 65(3):294–300. doi: 10.1136/ard.2005.037176
13. García-Vicuña R, Gómez-Gaviro MV, Domínguez-LuIs MJ, Pec MK, González-Alvaro I, Alvaro-Gracia JM, et al. CC and CXC Chemokine Receptors Mediate Migration, Proliferation, and Matrix Metalloproteinase Production by Fibroblast-Like Synoviocytes From Rheumatoid Arthritis Patients. ArthritIs Rheumatol (2004) 50(12):3866–77. doi: 10.1002/art.20615
14. Katschke KJ Jr., Rottman JB, Ruth JH, Qin S, Wu L, LaRosa G, et al. Differential Expression of Chemokine Receptors on Peripheral Blood, Synovial Fluid, and Synovial TIssue Monocytes/Macrophages in Rheumatoid ArthritIs. ArthritIs Rheumatol (2001) 44(5):1022–32. doi: 10.1002/1529-0131(200105)44:5<1022::Aid-anr181>3.0.Co;2-n
15. Haas CS, Martinez RJ, Attia N, Haines GK 3rd, Campbell PL, Koch AE. Chemokine Receptor Expression in Rat Adjuvant-Induced ArthritIs. ArthritIs Rheumatol (2005) 52(12):3718–30. doi: 10.1002/art.21476
16. Šućur A, Jajić Z, Ikić Matijašević M, Stipić Marković A, Flegar D, Lukač N, et al. Combined Manual and Automated ImmunophenotypIsation Identified DIsease-Specific Peripheral Blood Immune Subpopulations in Rheumatoid ArthritIs, Ankylosing Spondylitis and Psoriatic Arthritis. Clin Exp Rheumatol (2020) 38(5):903–16.
17. Szekanecz Z, Vegvari A, Szabo Z, Koch AE. Chemokines and Chemokine Receptors in ArthritIs. Front Biosci (Schol Ed) (2010) 2(1):153–67. doi: 10.2741/s53
18. Koch AE. Chemokines and Their Receptors in Rheumatoid ArthritIs: Future Targets? ArthritIs Rheum (2005) 52(3):710–21. doi: 10.1002/art.20932
19. Elemam NM, Hannawi S, Maghazachi AA. Role of Chemokines and Chemokine Receptors in Rheumatoid ArthritIs. Immunotargets Ther (2020) 9:43–56. doi: 10.2147/itt.S243636
20. Gilliland C, Salanga C, Kawamura T, Trejo J, Handel T. The Chemokine Receptor CCR1 Is Constitutively Active, Which Leads to G Protein-Independent, β-Arrestin-Mediated Internalization. J Biol Chem (2013) 288(45):32194–210. doi: 10.1074/jbc.M113.503797
21. Berahovich RD, Miao Z, Wang Y, Premack B, Howard MC, Schall TJ. Proteolytic Activation of Alternative CCR1 Ligands in Inflammation. J Immunol (2005) 174(11):7341–51. doi: 10.4049/jimmunol.174.11.7341
22. Solari R, Pease JE. Targeting Chemokine Receptors in DIsease–A Case Study of CCR4. Eur J Pharmacol (2015) 763(Pt B):169–77. doi: 10.1016/j.ejphar.2015.05.018
23. Kholodnyuk I, KadIsa A, SvirskIs S, Gravelsina S, Studers P, Spaka I, et al. Proportion of the CD19-Positive and CD19-Negative Lymphocytes and Monocytes Within the Peripheral Blood Mononuclear Cell Set Is CharacterIstic for Rheumatoid ArthritIs. Med (Kaunas) (2019) 55(10):630. doi: 10.3390/medicina55100630
24. Zhebrun DA, Totolyan AA, Maslyanskii AL, Titov AG, Patrukhin AP, KostAreva AA, et al. SynthesIs of Some CC Chemokines and Their Receptors in the Synovium in Rheumatoid ArthritIs. Bull Exp Biol Med (2014) 158(2):192–6. doi: 10.1007/s10517-014-2720-9
25. Amat M, Benjamim CF, Williams LM, Prats N, Terricabras E, Beleta J, et al. Pharmacological Blockade of CCR1 Ameliorates Murine ArthritIs and Alters Cytokine Networks In Vivo. Br J Pharmacol (2006) 149(6):666–75. doi: 10.1038/sj.bjp.0706912
26. Vergunst CE, Gerlag DM, von Moltke L, Karol M, Wyant T, Chi X, et al. MLN3897 Plus Methotrexate in Patients With Rheumatoid ArthritIs: Safety, Efficacy, Pharmacokinetics, and Pharmacodynamics of an Oral CCR1 AntagonIst in a Phase IIa, Double-Blind, Placebo-Controlled, Randomized, Proof-of-Concept Study. ArthritIs Rheumatol (2009) 60(12):3572–81. doi: 10.1002/art.24978
27. Lebre MC, Vergunst CE, Choi IY, Aarrass S, Oliveira AS, Wyant T, et al. Why CCR2 and CCR5 Blockade Failed and Why CCR1 Blockade Might Still be Effective in the Treatment of Rheumatoid ArthritIs. PloS One (2011) 6(7):e21772. doi: 10.1371/journal.pone.0021772
28. Tak PP, Balanescu A, Tseluyko V, Bojin S, Drescher E, Dairaghi D, et al. Chemokine Receptor CCR1 AntagonIst CCX354-C Treatment for Rheumatoid ArthritIs: CARAT-2, a RandomIsed, Placebo Controlled Clinical Trial. Ann Rheum DIs (2013) 72(3):337–44. doi: 10.1136/annrheumdIs-2011-201605
29. Dairaghi DJ, Zhang P, Wang Y, Seitz LC, Johnson DA, Miao S, et al. Pharmacokinetic and Pharmacodynamic Evaluation of the Novel CCR1 AntagonIst CCX354 in Healthy Human Subjects: Implications for Selection of Clinical Dose. Clin Pharmacol Ther (2011) 89(5):726–34. doi: 10.1038/clpt.2011.33
30. Li M, Chen L, Gao Y, Li M, Wang X, Qiang L, et al. Recent Advances Targeting C-C Chemokine Receptor Type 2 for Liver Diseases in Monocyte/Macrophage. Liver Int Off J Int Assoc Study Liver (2020) 40(12):2928–36. doi: 10.1111/liv.14687
31. O'Hayre M, Salanga CL, Handel TM, Allen SJ. Chemokines and Cancer: Migration, Intracellular Signalling and Intercellular Communication in the Microenvironment. Biochem J (2008) 409(3):635–49. doi: 10.1042/bj20071493
32. Markx D, Schuhholz J, Abadier M, Beier S, Lang M, Moepps B. Arginine 313 of the Putative 8th Helix Mediates Gα Coupling of Human CC Chemokine Receptors CCR2a and CCR2b. Cell Signal (2019) 53:170–83. doi: 10.1016/j.cellsig.2018.10.007
33. Vatter P, Schuhholz J, Koenig C, Pfreimer M, Moepps B. Ligand-Dependent Serum Response Factor Activation by the Human CC Chemokine Receptors CCR2a and CCR2b Is Mediated by G Proteins of the Gq Family. J Leuk Biol (2016) 99(6):979–91. doi: 10.1189/jlb.2MA0815-386R
34. Diaz-Rubio GI, Corona-Meraz FI, Madrigal-Ruiz PM, Robles-De Anda JA, Gómez-Bañuelos E, Castro-Albarran J, et al. CCR2/CCL2 and CMKLR1/RvE1 Chemokines System Levels Are Associated With Insulin ResIstance in Rheumatoid ArthritIs. PloS One (2021) 16(1):e0246054. doi: 10.1371/journal.pone.0246054
35. Vergunst CE, Gerlag DM, Lopatinskaya L, KlAreskog L, Smith MD, van den Bosch F, et al. Modulation of CCR2 in Rheumatoid ArthritIs: A Double-Blind, Randomized, Placebo-Controlled Clinical Trial. ArthritIs Rheumatol (2008) 58(7):1931–9. doi: 10.1002/art.23591
36. Horioka M, Ceraudo E, Lorenzen E, Sakmar T, Huber T. Purinergic Receptors Crosstalk With CCR5 to Amplify Ca Signaling. Cell Mol Neurobiol (2021) 41(5):1085–101. doi: 10.1007/s10571-020-01002-1
37. Qi B, Fang Q, Liu S, Hou W, Li J, Huang Y, et al. Advances of CCR5 AntagonIsts: From Small Molecules to Macromolecules. Eur J Med Chem (2020) 208:112819. doi: 10.1016/j.ejmech.2020.112819
38. Desmetz C, Lin YL, Mettling C, Portalès P, Noël D, Clot J, et al. Cell Surface CCR5 Density Determines the Intensity of T Cell Migration Towards Rheumatoid ArthritIs Synoviocytes. Clin Immunol (2007) 123(2):148–54. doi: 10.1016/j.clim.2007.01.004
39. FleIshaker DL, Garcia Meijide JA, Petrov A, Kohen MD, Wang X, Menon S, et al. Maraviroc, a Chemokine Receptor-5 AntagonIst, Fails to Demonstrate Efficacy in the Treatment of Patients With Rheumatoid ArthritIs in a Randomized, Double-Blind Placebo-Controlled Trial. ArthritIs Res Ther (2012) 14(1):R11. doi: 10.1186/ar3685
40. van Kuijk AW, Vergunst CE, Gerlag DM, Bresnihan B, Gomez-Reino JJ, Rouzier R, et al. CCR5 Blockade in Rheumatoid ArthritIs: A RandomIsed, Double-Blind, Placebo-Controlled Clinical Trial. Ann Rheum DIs (2010) 69(11):2013–6. doi: 10.1136/ard.2010.131235
41. Gerlag DM, HollIs S, Layton M, Vencovský J, Szekanecz Z, Braddock M, et al. Preclinical and Clinical Investigation of a CCR5 AntagonIst, AZD5672, in Patients With Rheumatoid ArthritIs Receiving Methotrexate. ArthritIs Rheumatol (2010) 62(11):3154–60. doi: 10.1002/art.27652
42. van Aalst E, Wylie BJ. Cholesterol Is a Dose-Dependent Positive Allosteric Modulator of CCR3 Ligand Affinity and G Protein Coupling. Front Mol Biosci (2021) 8:724603. doi: 10.3389/fmolb.2021.724603
43. Liu X, Zhang H, Chang X, Shen J, Zheng W, Xu Y, et al. Upregulated Expression of CCR3 in Rheumatoid ArthritIs and CCR3-Dependent Activation of Fibroblast-Like Synoviocytes. Cell Biol Toxicol (2017) 33(1):15–26. doi: 10.1007/s10565-016-9356-7
44. Wakabayashi K, Isozaki T, Tsubokura Y, Fukuse S, Kasama T. Eotaxin-1/CCL11 Is Involved in Cell Migration in Rheumatoid ArthritIs. Sci Rep (2021) 11(1):7937. doi: 10.1038/s41598-021-87199-7
45. Kindstedt E, Holm CK, Sulniute R, Martinez-Carrasco I, Lundmark R, Lundberg P. CCL11, a Novel Mediator of Inflammatory Bone Resorption. Sci Rep (2017) 7(1):5334. doi: 10.1038/s41598-017-05654-w
46. Lee EY, Seo M, Juhnn YS, Kim JY, Hong YJ, Lee YJ, et al. Potential Role and MechanIsm of IFN-Gamma Inducible Protein-10 on Receptor Activator of Nuclear Factor Kappa-B Ligand (RANKL) Expression in Rheumatoid ArthritIs. ArthritIs Res Ther (2011) 13(3):R104. doi: 10.1186/ar3385
47. Aloush V, George J, Elkayam O, Wigler I, Oren S, Entin-Meer M, et al. Decreased Levels of CCR3 in CD4+ Lymphocytes of Rheumatoid ArthritIs Patients. Clin Exp Rheumatol (2010) 28(4):462–7.
48. NIssinen R, LeirIsalo-Repo M, Peltomaa R, Palosuo T, Vaarala O. Cytokine and Chemokine Receptor Profile of Peripheral Blood Mononuclear Cells During Treatment With Infliximab in Patients With Active Rheumatoid ArthritIs. Ann Rheum DIs (2004) 63(6):681–7. doi: 10.1136/ard.2003.008599
49. Barnes DA, Tse J, Kaufhold M, Owen M, Hesselgesser J, Strieter R, et al. Polyclonal Antibody Directed Against Human RANTES Ameliorates DIsease in the LewIs Rat Adjuvant-Induced ArthritIs Model. J Clin Invest (1998) 101(12):2910–9. doi: 10.1172/jci2172
50. Ablin JN, Entin-Meer M, Aloush V, Oren S, Elkayam O, George J, et al. Protective Effect of Eotaxin-2 Inhibition in Adjuvant-Induced ArthritIs. Clin Exp Immunol (2010) 161(2):276–83. doi: 10.1111/j.1365-2249.2010.04172.x
51. Tao K, Tang X, Wang B, Li RJ, Zhang BQ, Lin JH, et al. DIstinct Expression of Chemokine-Like Factor 1 in Synovium of OsteoarthritIs, Rheumatoid ArthritIs and Ankylosing SpondylitIs. J Huazhong Univ Sci Technolog Med Sci (2016) 36(1):70–6. doi: 10.1007/s11596-016-1544-4
52. Takei J, Suzuki H, Asano T, Tanaka T, Kaneko M, Kato Y. Development of a Novel Anti-Mouse CCR4 Monoclonal Antibody (CMab-1) by N-Terminal Peptide Immunization. Monoclonal Antibodies ImmunodiagnosIs Immunother (2022) 41(2):87–93. doi: 10.1089/mab.2021.0064
53. Xu W, Li S, Chang X. E2F2 Stimulates CCR4 Expression and Activates Synovial Fibroblast-Like Cells in Rheumatoid ArthritIs. Cent Eur J Immunol (2021) 46(1):27–37. doi: 10.5114/ceji.2021.105243
54. Pandya JM, Lundell AC, Hallström M, Andersson K, Nordström I, Rudin A. Circulating T Helper and T Regulatory Subsets in Untreated Early Rheumatoid ArthritIs and Healthy Control Subjects. J Leukoc Biol (2016) 100(4):823–33. doi: 10.1189/jlb.5A0116-025R
55. Li N, Wei W, Yin F, Chen M, Ma TR, Wu Q, et al. The Abnormal Expression of CCR4 and CCR6 on Tregs in Rheumatoid ArthritIs. Int J Clin Exp Med (2015) 8(9):15043–53.
56. Thompson SD, Luyrink LK, Graham TB, Tsoras M, Ryan M, Passo MH, et al. Chemokine Receptor CCR4 on CD4+ T Cells in Juvenile Rheumatoid ArthritIs Synovial Fluid Defines a Subset of Cells With Increased IL-4:IFN-Gamma mRNA Ratios. J Immunol (2001) 166(11):6899–906. doi: 10.4049/jimmunol.166.11.6899
57. Wang L, Wang L, Hao P, Cao Q, Zhang Z. Anti-CCL22 Increases Regulatory T Cells in CD4(+) T Cells of Rheumatoid ArthritIs Patients via STAT5 Pathway. Exp Ther Med (2020) 19(3):2127–32. doi: 10.3892/etm.2019.8404
58. PaulIssen SM, van Hamburg JP, Davelaar N, Vroman H, Hazes JM, de Jong PH, et al. CCR6(+) Th Cell Populations DIstinguIsh ACPA Positive From ACPA Negative Rheumatoid ArthritIs. ArthritIs Res Ther (2015) 17:344. doi: 10.1186/s13075-015-0800-5
59. Dankers W, den Braanker H, PaulIssen SMJ, van Hamburg JP, Davelaar N, Colin EM, et al. The Heterogeneous Human Memory CCR6+ T Helper-17 Populations Differ in T-Bet and Cytokine Expression But All Activate Synovial Fibroblasts in an Ifnγ-Independent Manner. ArthritIs Res Ther (2021) 23(1):157. doi: 10.1186/s13075-021-02532-9
60. Miyazaki Y, Nakayamada S, Kubo S, Nakano K, Iwata S, Miyagawa I, et al. Th22 Cells Promote Osteoclast Differentiation via Production of IL-22 in Rheumatoid ArthritIs. Front Immunol (2018) 9:2901. doi: 10.3389/fimmu.2018.02901
61. Kaneko S, Kondo Y, Yokosawa M, Furuyama K, Segawa S, Tsuboi H, et al. The Rorγt-CCR6-CCL20 AxIs Augments Th17 Cells Invasion Into the Synovia of Rheumatoid ArthritIs Patients. Mod Rheumatol (2018) 28(5):814–25. doi: 10.1080/14397595.2017.1416923
62. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential Recruitment of CCR6-Expressing Th17 Cells to Inflamed Joints via CCL20 in Rheumatoid ArthritIs and Its Animal Model. J Exp Med (2007) 204(12):2803–12. doi: 10.1084/jem.20071397
63. Tsutsumi E, Stricklin J, Peterson E, Schroeder J, Kim S. Cxcl10 Chemokine Induces Migration of ING4-Deficient Breast Cancer Cells via a Novel Cross Talk MechanIsm Between the Cxcr3 and Egfr Receptors. Mol Cell Biol (2022) 42(2):e0038221. doi: 10.1128/mcb.00382-21
64. Tsubaki T, Takegawa S, Hanamoto H, Arita N, Kamogawa J, Yamamoto H, et al. Accumulation of Plasma Cells Expressing CXCR3 in the Synovial Sublining Regions of Early Rheumatoid ArthritIs in Association With Production of Mig/CXCL9 by Synovial Fibroblasts. Clin Exp Immunol (2005) 141(2):363–71. doi: 10.1111/j.1365-2249.2005.02850.x
65. Ruschpler P, Lorenz P, Eichler W, Koczan D, Hänel C, Scholz R, et al. High CXCR3 Expression in Synovial Mast Cells Associated With CXCL9 and CXCL10 Expression in Inflammatory Synovial TIssues of Patients With Rheumatoid ArthritIs. ArthritIs Res Ther (2003) 5(5):R241–52. doi: 10.1186/ar783
66. Motoki Y, Tani K, Shimizu T, Tamiya H, Hase K, Ohmoto Y, et al. The Expression of Chemokine Receptor CXCR3: Relevance to DIsease Activity of Rheumatoid ArthritIs. Mod Rheumatol (2003) 13(2):114–20. doi: 10.3109/s10165-002-0209-2
67. Campbell JD, HayGlass KT. T Cell Chemokine Receptor Expression in Human Th1- and Th2-Associated DIseases. Arch Immunol Ther Exp (Warsz) (2000) 48(6):451–6.
68. Herman S, Zurgil N, Machlav S, Shinberg A, Langevitz P, Ehrenfeld M, et al. DIstinct Effects of Anti-Tumor NecrosIs Factor Combined Therapy on TH1/TH2 Balance in Rheumatoid ArthritIs Patients. Clin Vaccine Immunol (2011) 18(7):1077–82. doi: 10.1128/cvi.00061-11
69. Aeberli D, Seitz M, Jüni P, Villiger PM. Increase of Peripheral CXCR3 Positive T Lymphocytes Upon Treatment of RA Patients With TNF-Alpha Inhibitors. Rheumatol (Oxford) (2005) 44(2):172–5. doi: 10.1093/rheumatology/keh437
70. Nanki T, Lipsky PE. Lack of Correlation Between Chemokine Receptor and T(h)1/T(h)2 Cytokine Expression by Individual Memory T Cells. Int Immunol (2000) 12(12):1659–67. doi: 10.1093/intimm/12.12.1659
71. Xie JH, Nomura N, Lu M, Chen SL, Koch GE, Weng Y, et al. Antibody-Mediated Blockade of the CXCR3 Chemokine Receptor Results in DiminIshed Recruitment of T Helper 1 Cells Into Sites of Inflammation. J Leukoc Biol (2003) 73(6):771–80. doi: 10.1189/jlb.1102573
72. Bakheet SA, Ansari MA, Nadeem A, Attia SM, Alhoshani AR, Gul G, et al. CXCR3 AntagonIst AMG487 Suppresses Rheumatoid ArthritIs PathogenesIs and Progression by Shifting the Th17/Treg Cell Balance. Cell Signal (2019) 64:109395. doi: 10.1016/j.cellsig.2019.109395
73. Shi G, Partida-Sánchez S, MIsra RS, Tighe M, Borchers MT, Lee JJ, et al. Identification of an Alternative G{alpha}q-Dependent Chemokine Receptor Signal Transduction Pathway in Dendritic Cells and Granulocytes. J Exp Med (2007) 204(11):2705–18. doi: 10.1084/jem.20071267
74. MoschovakIs GL, Bubke A, Friedrichsen M, RIstenpart J, Back JW, Falk CS, et al. The Chemokine Receptor CCR7 Is a PromIsing Target for Rheumatoid ArthritIs Therapy. Cell Mol Immunol (2019) 16(10):791–9. doi: 10.1038/s41423-018-0056-5
75. Aldahlawi AM, Elshal MF, Ashgan FT, Bahlas S. Chemokine Receptors Expression on Peripheral CD4-Lymphocytes in Rheumatoid ArthritIs: Coexpression of CCR7 and CD95 Is Associated With DIsease Activity. Saudi J Biol Sci (2015) 22(4):453–8. doi: 10.1016/j.sjbs.2015.02.011
76. Van Raemdonck K, Umar S, Palasiewicz K, Volkov S, Volin MV, Arami S, et al. CCL21/CCR7 Signaling in Macrophages Promotes Joint Inflammation and Th17-Mediated Osteoclast Formation in Rheumatoid ArthritIs. Cell Mol Life Sci (2020) 77(7):1387–99. doi: 10.1007/s00018-019-03235-w
77. Van Raemdonck K, Umar S, Palasiewicz K, Meyer A, Volin MV, Chang HJ, et al. Metabolic Reprogramming of Macrophages Instigates CCL21-Induced ArthritIs. Immunol Cell Biol (2022) 100(2):127–35. doi: 10.1111/imcb.12512
78. Elmesmari A, Fraser AR, Wood C, GilchrIst D, Vaughan D, Stewart L, et al. MicroRNA-155 Regulates Monocyte Chemokine and Chemokine Receptor Expression in Rheumatoid ArthritIs. Rheumatol (Oxford) (2016) 55(11):2056–65. doi: 10.1093/rheumatology/kew272
79. Schmutz C, Cartwright A, Williams H, Haworth O, Williams JH, Filer A, et al. Monocytes/macrophages Express Chemokine Receptor CCR9 in Rheumatoid ArthritIs and CCL25 Stimulates Their Differentiation. ArthritIs Res Ther (2010) 12(4):R161. doi: 10.1186/ar3120
80. Umar S, Palasiewicz K, Van Raemdonck K, Volin MV, Romay B, Ahmad I, et al. CCL25 and CCR9 Is a Unique Pathway That Potentiates Pannus Formation by Remodeling RA Macrophages Into Mature Osteoclasts. Eur J Immunol (2021) 51(4):903–14. doi: 10.1002/eji.202048681
81. Yokoyama W, Kohsaka H, Kaneko K, Walters M, Takayasu A, Fukuda S, et al. Abrogation of CC Chemokine Receptor 9 Ameliorates Collagen-Induced ArthritIs of Mice. ArthritIs Res Ther (2014) 16(5):445. doi: 10.1186/s13075-014-0445-9
82. Hessner F, Dlugos C, Chehab T, Schaefer C, Homey B, Gerke V, et al. CC Chemokine Receptor 10 Cell Surface Presentation in Melanocytes Is Regulated by the Novel Interaction Partner S100A10. Sci Rep (2016) 6:22649. doi: 10.1038/srep22649
83. Chen Z, Kim SJ, Essani AB, Volin MV, Vila OM, Swedler W, et al. CharacterIsing the Expression and Function of CCL28 and Its Corresponding Receptor, CCR10, in RA PathogenesIs. Ann Rheum DIs (2015) 74(10):1898–906. doi: 10.1136/annrheumdIs-2013-204530
84. Raghuwanshi S, Su Y, Singh V, Haynes K, Richmond A, Richardson R. The Chemokine Receptors CXCR1 and CXCR2 Couple to DIstinct G Protein-Coupled Receptor Kinases to Mediate and Regulate Leukocyte Functions. J Immunol (Baltimore Md 1950) (2012) 189(6):2824–32. doi: 10.4049/jimmunol.1201114
85. Elhaj Mahmoud D, Kaabachi W, Sassi N, Mokhtar A, Ben Ammar L, Rekik S, et al. Expression of Extracellular Matrix Components and Cytokine Receptors in Human Fibrocytes During Rheumatoid ArthritIs. Connect TIssue Res (2021) 62(6):720–31. doi: 10.1080/03008207.2021.1873962
86. Podolin PL, Bolognese BJ, Foley JJ, Schmidt DB, Buckley PT, Widdowson KL, et al. A Potent and Selective Nonpeptide AntagonIst of CXCR2 InhibIts Acute and Chronic Models of ArthritIs in the Rabbit. J Immunol (2002) 169(11):6435–44. doi: 10.4049/jimmunol.169.11.6435
87. Grespan R, Fukada SY, Lemos HP, Vieira SM, Napimoga MH, Teixeira MM, et al. CXCR2-Specific Chemokines Mediate Leukotriene B4-Dependent Recruitment of Neutrophils to Inflamed Joints in Mice With Antigen-Induced ArthritIs. ArthritIs Rheumatol (2008) 58(7):2030–40. doi: 10.1002/art.23597
88. Min SH, Wang Y, Gonsiorek W, Anilkumar G, Kozlowski J, Lundell D, et al. Pharmacological Targeting Reveals DIstinct Roles for CXCR2/CXCR1 and CCR2 in a Mouse Model of ArthritIs. Biochem Biophys Res Commun (2010) 391(1):1080–6. doi: 10.1016/j.bbrc.2009.12.025
89. Yoshida K, Korchynskyi O, Tak PP, Isozaki T, Ruth JH, Campbell PL, et al. Citrullination of Epithelial Neutrophil-Activating Peptide 78/CXCL5 Results in Conversion From a Non-Monocyte-Recruiting Chemokine to a Monocyte-Recruiting Chemokine. ArthritIs Rheumatol (2014) 66(10):2716–27. doi: 10.1002/art.38750
90. Eriksson C, Rantapää-DahlqvIst S, SundqvIst KG. Changes in Chemokines and Their Receptors in Blood During Treatment With the TNF Inhibitor Infliximab in Patients With Rheumatoid ArthritIs. Scand J Rheumatol (2013) 42(4):260–5. doi: 10.3109/03009742.2012.754937
91. Peng L, Zhu N, Mao J, Huang L, Yang Y, Zhou Z, et al. Expression Levels of CXCR4 and CXCL12 in Patients With Rheumatoid ArthritIs and Its Correlation With DIsease Activity. Exp Ther Med (2020) 20(3):1925–34. doi: 10.3892/etm.2020.8950
92. Zhou S, Lu H, Xiong M. Identifying Immune Cell Infiltration and Effective Diagnostic Biomarkers in Rheumatoid ArthritIs by Bioinformatics AnalysIs. Front Immunol (2021) 12:726747. doi: 10.3389/fimmu.2021.726747
93. Pablos JL, Santiago B, Galindo M, Torres C, Brehmer MT, Blanco FJ, et al. Synoviocyte-Derived CXCL12 Is DIsplayed on Endothelium and Induces AngiogenesIs in Rheumatoid ArthritIs. J Immunol (2003) 170(4):2147–52. doi: 10.4049/jimmunol.170.4.2147
94. Bryant J, Ahern DJ, Brennan FM. CXCR4 and Vascular Cell Adhesion Molecule 1 Are Key Chemokine/Adhesion Receptors in the Migration of Cytokine-Activated T Cells. ArthritIs Rheumatol (2012) 64(7):2137–46. doi: 10.1002/art.34394
95. Tsujimura S, Adachi T, Saito K, Kawabe A, Tanaka Y. Relevance of P-Glycoprotein on CXCR4(+) B Cells to Organ Manifestation in Highly Active Rheumatoid ArthritIs. Mod Rheumatol (2018) 28(2):276–86. doi: 10.1080/14397595.2017.1341458
96. Yang R, Yao Y, Wang P. Hypoxia-Induced the Upregulation of Stromal Cell-Derived Factor 1 in Fibroblast-Like Synoviocytes Contributes to Migration of Monocytes Into Synovium TIssue in Rheumatoid ArthritIs. Cell Biosci (2018) 8:11. doi: 10.1186/s13578-018-0210-x
97. Chen HT, Tsou HK, Hsu CJ, Tsai CH, Kao CH, Fong YC, et al. Stromal Cell-Derived Factor-1/CXCR4 Promotes IL-6 Production in Human Synovial Fibroblasts. J Cell Biochem (2011) 112(4):1219–27. doi: 10.1002/jcb.23043
98. Chiu YC, Yang RS, Hsieh KH, Fong YC, Way TD, Lee TS, et al. Stromal Cell-Derived Factor-1 Induces Matrix Metalloprotease-13 Expression in Human Chondrocytes. Mol Pharmacol (2007) 72(3):695–703. doi: 10.1124/mol.107.036541
99. Kanbe K, TakagIshi K, Chen Q. Stimulation of Matrix Metalloprotease 3 Release From Human Chondrocytes by the Interaction of Stromal Cell-Derived Factor 1 and CXC Chemokine Receptor 4. ArthritIs Rheumatol (2002) 46(1):130–7. doi: 10.1002/1529-0131(200201)46:1<130::aid-art10020>3.0.co;2-d
100. Cecchinato V, D'Agostino G, Raeli L, Nerviani A, Schiraldi M, Danelon G, et al. Redox-Mediated MechanIsms Fuel Monocyte Responses to CXCL12/HMGB1 in Active Rheumatoid ArthritIs. Front Immunol (2018) 9:2118. doi: 10.3389/fimmu.2018.02118
101. Si M, Ma Z, Zhang J, Li X, Li R, Wang C, et al. Qingluoyin Granules Protect Against Adjuvant-Induced ArthritIs in Rats via Downregulating the CXCL12/CXCR4-NF-κb Signalling Pathway. Pharm Biol (2021) 59(1):1441–51. doi: 10.1080/13880209.2021.1991386
102. Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Small Molecule Inhibitors of CXCR4. Theranostics (2013) 3(1):47–75. doi: 10.7150/thno.5376
103. Khan A, Greenman J, Archibald SJ. Small Molecule CXCR4 Chemokine Receptor AntagonIsts: Developing Drug Candidates. Curr Med Chem (2007) 14(21):2257–77. doi: 10.2174/092986707781696618
104. Choi WT, Duggineni S, Xu Y, Huang Z, An J. Drug DIscovery Research Targeting the CXC Chemokine Receptor 4 (CXCR4). J Med Chem (2012) 55(3):977–94. doi: 10.1021/jm200568c
105. Tamamura H, Tsutsumi H, Nomura W, Fujii N. Exploratory Studies on Development of the Chemokine Receptor CXCR4 AntagonIsts Toward Downsizing. Perspect Medicin Chem (2008) 2:1–9. doi: 10.4137/pmc.s422
106. Wang X, Yang C, Xu F, Qi L, Wang J, Yang P. Imbalance of Circulating Tfr/Tfh Ratio in Patients With Rheumatoid ArthritIs. Clin Exp Med (2019) 19(1):55–64. doi: 10.1007/s10238-018-0530-5
107. Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C, et al. Increased Frequency of Circulating Follicular Helper T Cells in Patients With Rheumatoid ArthritIs. Clin Dev Immunol (2012) 2012:827480. doi: 10.1155/2012/827480
108. Liu C, Wang D, Lu S, Xu Q, Zhao L, Zhao J, et al. Increased Circulating Follicular Treg Cells Are Associated With Lower Levels of Autoantibodies in Patients With Rheumatoid ArthritIs in Stable RemIssion. ArthritIs Rheumatol (2018) 70(5):711–21. doi: 10.1002/art.40430
109. Cao G, Wang P, Cui Z, Yue X, Chi S, Ma A, et al. An Imbalance Between Blood CD4(+)CXCR5(+)Foxp3(+) Tfr Cells and CD4(+)CXCR5(+)Tfh Cells May Contribute to the ImmunopathogenesIs of Rheumatoid ArthritIs. Mol Immunol (2020) 125:1–8. doi: 10.1016/j.molimm.2020.06.003
110. Tang Y, Wang B, Sun X, Li H, Ouyang X, Wei J, et al. Rheumatoid ArthritIs Fibroblast-Like Synoviocytes Co-Cultured With PBMC Increased Peripheral CD4(+) CXCR5(+) ICOS(+) T Cell Numbers. Clin Exp Immunol (2017) 190(3):384–93. doi: 10.1111/cei.13025
111. Tsai CH, Chen CJ, Gong CL, Liu SC, Chen PC, Huang CC, et al. CXCL13/CXCR5 AxIs Facilitates Endothelial Progenitor Cell Homing and AngiogenesIs During Rheumatoid ArthritIs Progression. Cell Death DIs (2021) 12(9):846. doi: 10.1038/s41419-021-04136-2
112. Rempenault C, Mielle J, Schreiber K, Corbeau P, Macia L, Combe B, et al. CXCR5/CXCL13 Pathway, a Key Driver for Migration of Regulatory B10 Cells, Is Defective in Patients With Rheumatoid ArthritIs. Rheumatol (Oxford) (2021) 61(3):2185–96. doi: 10.1093/rheumatology/keab639
113. MoschovakIs GL, Bubke A, Friedrichsen M, Falk CS, Feederle R, Förster R. T Cell Specific Cxcr5 Deficiency Prevents Rheumatoid ArthritIs. Sci Rep (2017) 7(1):8933. doi: 10.1038/s41598-017-08935-6
114. Zhang X, Zhao JX, Sun L, Liu XY. Expression of CXCL16/CXCR6 in Fibroblast-Like Synoviocytes in Rheumatoid ArthritIs and Its Role in Synoviocyte Proliferation. Beijing Da Xue Xue Bao Yi Xue Ban (2017) 49(4):663–8.
115. Li CH, Xu LL, Zhao JX, Sun L, Yao ZQ, Deng XL, et al. CXCL16 Upregulates RANKL Expression in Rheumatoid ArthritIs Synovial Fibroblasts Through the JAK2/STAT3 and P38/MAPK Signaling Pathway. Inflammation Res (2016) 65(3):193–202. doi: 10.1007/s00011-015-0905-y
116. Isozaki T, Arbab AS, Haas CS, Amin MA, Arendt MD, Koch AE, et al. Evidence That CXCL16 Is a Potent Mediator of AngiogenesIs and Is Involved in Endothelial Progenitor Cell ChemotaxIs: Studies in Mice With K/BxN Serum-Induced ArthritIs. ArthritIs Rheumatol (2013) 65(7):1736–46. doi: 10.1002/art.37981
117. Slauenwhite D, Gebremeskel S, Doucette CD, Hoskin DW, Johnston B. Regulation of Cytokine Polarization and T Cell Recruitment to Inflamed Paws in Mouse Collagen-Induced ArthritIs by the Chemokine Receptor CXCR6. ArthritIs Rheumatol (2014) 66(11):3001–12. doi: 10.1002/art.38816
118. van der Voort R, van Lieshout AW, Toonen LW, Slöetjes AW, van den Berg WB, Figdor CG, et al. Elevated CXCL16 Expression by Synovial Macrophages RecruIts Memory T Cells Into Rheumatoid Joints. ArthritIs Rheumatol (2005) 52(5):1381–91. doi: 10.1002/art.21004
119. Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, et al. Beta-Arrestin- But Not G Protein-Mediated Signaling by the "Decoy" Receptor CXCR7. Proc Natl Acad Sci U S A (2010) 107(2):628–32. doi: 10.1073/pnas.0912852107
120. Luker KE, Gupta M, Luker GD. Imaging Chemokine Receptor Dimerization With Firefly Luciferase Complementation. FASEB J (2009) 23(3):823–34. doi: 10.1096/fj.08-116749
121. Ulvmar MH, Hub E, Rot A. Atypical Chemokine Receptors. Exp Cell Res (2011) 317(5):556–68. doi: 10.1016/j.yexcr.2011.01.012
122. Watanabe K, Penfold ME, Matsuda A, Ohyanagi N, Kaneko K, Miyabe Y, et al. Pathogenic Role of CXCR7 in Rheumatoid ArthritIs. ArthritIs Rheumatol (2010) 62(11):3211–20. doi: 10.1002/art.27650
123. Umehara H, Tanaka M, Sawaki T, Jin ZX, Huang CR, Dong L, et al. Fractalkine in Rheumatoid ArthritIs and Allied Conditions. Mod Rheumatol (2006) 16(3):124–30. doi: 10.1007/s10165-006-0471-9
124. Nanki T, Imai T, Kawai S. Fractalkine/CX3CL1 in Rheumatoid ArthritIs. Mod Rheumatol (2017) 27(3):392–7. doi: 10.1080/14397595.2016.1213481
125. Hasegawa T, Kikuta J, Sudo T, Matsuura Y, Matsui T, Simmons S, et al. Identification of a Novel ArthritIs-Associated Osteoclast Precursor Macrophage Regulated by Foxm1. Nat Immunol (2019) 20(12):1631–43. doi: 10.1038/s41590-019-0526-7
126. Sato M, Ohtsuka K, Takahashi R, Wakabayashi K, Odai T, Isozaki T, et al. Involvement of CX3CL1/CX3CR1 AxIs in Etanercept Therapy for Patients With Active Rheumatoid ArthritIs. Open Access Rheumatol (2011) 3:1–7. doi: 10.2147/oarrr.S16210
127. Odai T, Matsunawa M, Takahashi R, Wakabayashi K, Isozaki T, Yajima N, et al. Correlation of CX3CL1 and CX3CR1 Levels With Response to Infliximab Therapy in Patients With Rheumatoid ArthritIs. J Rheumatol (2009) 36(6):1158–65. doi: 10.3899/jrheum.081074
128. Wang CR, Liu MF, Huang YH, Chen HC. Up-Regulation of XCR1 Expression in Rheumatoid Joints. Rheumatol (Oxford) (2004) 43(5):569–73. doi: 10.1093/rheumatology/keh147
129. Montero-Melendez T. ACTH: The Forgotten Therapy. Semin Immunol (2015) 27(3):216–26. doi: 10.1016/j.smim.2015.02.003
130. Rodrigues AR, Almeida H, Gouveia AM. Intracellular Signaling MechanIsms of the Melanocortin Receptors: Current State of the Art. Cell Mol Life Sci (2015) 72(7):1331–45. doi: 10.1007/s00018-014-1800-3
131. Patel HB, Bombardieri M, Sampaio AL, D'AcquIsto F, Gray M, Grieco P, et al. Anti-Inflammatory and AntiosteoclastogenesIs Properties of Endogenous Melanocortin Receptor Type 3 in Experimental ArthritIs. FASEB J (2010) 24(12):4835–43. doi: 10.1096/fj.10-167759
132. CornIsh J, Callon KE, Mountjoy KG, Bava U, Lin JM, Myers DE, et al. Alpha -Melanocyte-Stimulating Hormone Is a Novel Regulator of Bone. Am J Physiol Endocrinol Metab (2003) 284(6):E1181–90. doi: 10.1152/ajpendo.00412.2002
133. Catania A, Gerloni V, Procaccia S, Airaghi L, Manfredi MG, Lomater C, et al. The Anticytokine Neuropeptide Alpha-Melanocyte-Stimulating Hormone in Synovial Fluid of Patients With Rheumatic DIseases: ComparIsons With Other Anticytokine Molecules. Neuroimmunomodulation (1994) 1(5):321–8. doi: 10.1159/000097183
134. Sarkar A, Sreenivasan Y, Manna SK. Alpha-Melanocyte-Stimulating Hormone InhibIts Lipopolysaccharide-Induced Biological Responses by Downregulating CD14 From Macrophages. FEBS Lett (2003) 553(3):286–94. doi: 10.1016/s0014-5793(03)01029-9
135. Manna SK, Aggarwal BB. Alpha-Melanocyte-Stimulating Hormone InhibIts the Nuclear Transcription Factor NF-Kappa B Activation Induced by Various Inflammatory Agents. J Immunol (1998) 161(6):2873–80.
136. Bhardwaj RS, Schwarz A, Becher E, Mahnke K, Aragane Y, Schwarz T, et al. Pro-Opiomelanocortin-Derived Peptides Induce IL-10 Production in Human Monocytes. J Immunol (1996) 156(7):2517–21.
137. Taylor A, Namba K. In Vitro Induction of CD25+ CD4+ Regulatory T Cells by the Neuropeptide Alpha-Melanocyte Stimulating Hormone (Alpha-MSH). Immunol Cell Biol (2001) 79(4):358–67. doi: 10.1046/j.1440-1711.2001.01022.x
138. Montero-Melendez T, Nagano A, Chelala C, Filer A, Buckley CD, Perretti M. Therapeutic Senescence via GPCR Activation in Synovial Fibroblasts Facilitates Resolution of ArthritIs. Nat Commun (2020) 11(1):745. doi: 10.1038/s41467-020-14421-x
139. Montero-Melendez T, Patel HB, Seed M, Nielsen S, Jonassen TE, Perretti M. The Melanocortin AgonIst AP214 Exerts Anti-Inflammatory and Proresolving Properties. Am J Pathol (2011) 179(1):259–69. doi: 10.1016/j.ajpath.2011.03.042
140. Grässel S, Opolka A, Anders S, Straub RH, Grifka J, Luger TA, et al. The Melanocortin System in Articular Chondrocytes: Melanocortin Receptors, Pro-Opiomelanocortin, Precursor Proteases, and a Regulatory Effect of Alpha-Melanocyte-Stimulating Hormone on Proinflammatory Cytokines and Extracellular Matrix Components. ArthritIs Rheumatol (2009) 60(10):3017–27. doi: 10.1002/art.24846
141. Kaneva MK, Kerrigan MJ, Grieco P, Curley GP, Locke IC, Getting SJ. Chondroprotective and Anti-Inflammatory Role of Melanocortin Peptides in TNF-α Activated Human C-20/A4 Chondrocytes. Br J Pharmacol (2012) 167(1):67–79. doi: 10.1111/j.1476-5381.2012.01968.x
142. Zaidi M, Sun L, Robinson LJ, Tourkova IL, Liu L, Wang Y, et al. ACTH Protects Against Glucocorticoid-Induced OsteonecrosIs of Bone. Proc Natl Acad Sci U S A (2010) 107(19):8782–7. doi: 10.1073/pnas.0912176107
143. Sayah S, Ischenko A, Zhakhov A, Bonnard A, Fontaine M. Expression of Cytokines by Human Astrocytomas Following Stimulation by C3a and C5a Anaphylatoxins: Specific Increase in Interleukin-6 mRNA Expression. J Neurochem (1999) 72(6):2426–36. doi: 10.1046/j.1471-4159.1999.0722426.x
144. Hornum L, Hansen AJ, Tornehave D, Fjording MS, Colmenero P, Wätjen IF, et al. C5a and C5aR Are Elevated in Joints of Rheumatoid and Psoriatic ArthritIs Patients, and C5aR Blockade Attenuates Leukocyte Migration to Synovial Fluid. PloS One (2017) 12(12):e0189017. doi: 10.1371/journal.pone.0189017
145. Kiener HP, Baghestanian M, Dominkus M, Walchshofer S, Ghannadan M, Willheim M, et al. Expression of the C5a Receptor (CD88) on Synovial Mast Cells in Patients With Rheumatoid ArthritIs. ArthritIs Rheumatol (1998) 41(2):233–45. doi: 10.1002/1529-0131(199802)41:2<233::Aid-art7>3.0.Co;2-v
146. Sadik CD, Kim ND, Iwakura Y, Luster AD. Neutrophils Orchestrate Their Own Recruitment in Murine ArthritIs Through C5aR and Fcγr Signaling. Proc Natl Acad Sci U S A (2012) 109(46):E3177–85. doi: 10.1073/pnas.1213797109
147. Yang A, Zhou J, Wang B, Dai J, Colman RW, Song W, et al. A Critical Role for Plasma Kallikrein in the PathogenesIs of Autoantibody-Induced ArthritIs. FASEB J (2017) 31(12):5419–31. doi: 10.1096/fj.201700018R
148. Belenska-Todorova L, Gyurkovska V, Ivanovska N. How Complement Activation Influences the Development of Chronic SynovitIs in a Mouse Model of Rheumatoid ArthritIs. Scand J Rheumatol (2016) 45(1):13–22. doi: 10.3109/03009742.2015.1036114
149. Andersson C, Wenander CS, Usher PA, Hebsgaard JB, Sondergaard BC, Rønø B, et al. Rapid-Onset Clinical and MechanIstic Effects of Anti-C5aR Treatment in the Mouse Collagen-Induced ArthritIs Model. Clin Exp Immunol (2014) 177(1):219–33. doi: 10.1111/cei.12338
150. Vergunst CE, Gerlag DM, Dinant H, Schulz L, Vinkenoog M, Smeets TJ, et al. Blocking the Receptor for C5a in Patients With Rheumatoid ArthritIs Does Not Reduce Synovial Inflammation. Rheumatol (Oxford) (2007) 46(12):1773–8. doi: 10.1093/rheumatology/kem222
151. Sohn R, Junker M, Meurer A, Zaucke F, Straub RH, Jenei-Lanzl Z. Anti-Inflammatory Effects of Endogenously Released Adenosine in Synovial Cells of OsteoarthritIs and Rheumatoid ArthritIs Patients. Int J Mol Sci (2021) 22(16):8956. doi: 10.3390/ijms22168956
152. Pal Y, Bandyopadhyay N, Pal RS, Ahmed S, Bandopadhyay S. Perspective and Potential of A2A and A3 Adenosine Receptors as Therapeutic Targets for the Treatment of Rheumatoid ArthritIs. Curr Pharm Des (2019) 25(26):2859–74. doi: 10.2174/1381612825666190710111658
153. Cronstein BN, Sitkovsky M. Adenosine and Adenosine Receptors in the PathogenesIs and Treatment of Rheumatic DIseases. Nat Rev Rheumatol (2017) 13(1):41–51. doi: 10.1038/nrrheum.2016.178
154. Shakya AK, Naik RR, Almasri IM, Kaur A. Role and Function of Adenosine and Its Receptors in Inflammation, Neuroinflammation, IBS, Autoimmune Inflammatory DIsorders, Rheumatoid ArthritIs and PsoriasIs. Curr Pharm Des (2019) 25(26):2875–91. doi: 10.2174/1381612825666190716145206
155. Varani K, Padovan M, Govoni M, Vincenzi F, Trotta F, Borea PA. The Role of Adenosine Receptors in Rheumatoid ArthritIs. Autoimmun Rev (2010) 10(2):61–4. doi: 10.1016/j.autrev.2010.07.019
156. Magni G, Ceruti S. Adenosine Signaling in Autoimmune DIsorders. Pharm (Basel) (2020) 13(9):260. doi: 10.3390/ph13090260
157. Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-Mediated Signaling of Metabolites. Cell Metab (2017) 25(4):777–96. doi: 10.1016/j.cmet.2017.03.008
158. Srivastava NK, Sharma S, Sinha N, Mandal SK, Sharma D. Abnormal Lipid MetabolIsm in a Rat Model of ArthritIs: One Possible Pathway. Mol Cell Biochem (2018) 448(1-2):107–24. doi: 10.1007/s11010-018-3318-8
159. Berbert AA, Kondo CR, Almendra CL, Matsuo T, Dichi I. Supplementation of FIsh Oil and Olive Oil in Patients With Rheumatoid ArthritIs. Nutrition (2005) 21(2):131–6. doi: 10.1016/j.nut.2004.03.023
160. Veselinovic M, Vasiljevic D, Vucic V, Arsic A, Petrovic S, Tomic-Lucic A, et al. Clinical BenefIts of N-3 PUFA and ɤ-Linolenic Acid in Patients With Rheumatoid ArthritIs. Nutrients (2017) 9(4):325. doi: 10.3390/nu9040325
161. Ouyang L, Dan Y, Hua W, Shao Z, Duan D. Therapeutic Effect of Omega-3 Fatty Acids on T Cell-Mediated Autoimmune DIseases. Microbiol Immunol (2020) 64(8):563–9. doi: 10.1111/1348-0421.12800
162. Guo Y, Walsh AM, Fearon U, Smith MD, Wechalekar MD, Yin X, et al. CD40L-Dependent Pathway Is Active at Various Stages of Rheumatoid ArthritIs DIsease Progression. J Immunol (2017) 198(11):4490–501. doi: 10.4049/jimmunol.1601988
163. Zhang C, Chang J, Wu W, Deng Y, Zhou P, Jiang W, et al. Activation of GPR43 Suppresses TNF-α-Induced Inflammatory Response in Human Fibroblast-Like Synoviocytes. Arch Biochem Biophys (2020) 684:108297. doi: 10.1016/j.abb.2020.108297
164. Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, et al. Triggering the Succinate Receptor GPR91 on Dendritic Cells Enhances Immunity. Nat Immunol (2008) 9(11):1261–9. doi: 10.1038/ni.1657
165. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 Senses Extracellular Succinate Released From Inflammatory Macrophages and Exacerbates Rheumatoid ArthritIs. J Exp Med (2016) 213(9):1655–62. doi: 10.1084/jem.20160061
166. Li Y, Liu Y, Wang C, Xia WR, Zheng JY, Yang J, et al. Succinate Induces Synovial AngiogenesIs in Rheumatoid ArthritIs Through Metabolic Remodeling and HIF-1α/VEGF AxIs. Free Radic Biol Med (2018) 126:1–14. doi: 10.1016/j.freeradbiomed.2018.07.009
167. Park S, Lee K, Kang S, Chung H, Bae Y, Okajima F, et al. Lysophosphatidylethanolamine Utilizes LPA(1) and CD97 in MDA-MB-231 Breast Cancer Cells. Cell Signal (2013) 25(11):2147–54. doi: 10.1016/j.cellsig.2013.07.001
168. Niu M, Xu S, Yang J, Yao D, Li N, Yan J, et al. Structural BasIs for CD97 Recognition of the Decay-Accelerating Factor CD55 Suggests Mechanosensitive Activation of Adhesion GPCRs. J Biol Chem (2021) 296:100776. doi: 10.1016/j.jbc.2021.100776
169. Capasso M, Durrant LG, Stacey M, Gordon S, Ramage J, Spendlove I. Costimulation via CD55 on Human CD4+ T Cells Mediated by CD97. J Immunol (2006) 177(2):1070–7. doi: 10.4049/jimmunol.177.2.1070
170. de Groot DM, Vogel G, Dulos J, Teeuwen L, Stebbins K, Hamann J, et al. Therapeutic Antibody Targeting of CD97 in Experimental ArthritIs: The Role of Antigen Expression, Shedding, and Internalization on the Pharmacokinetics of Anti-CD97 Monoclonal Antibody 1B2. J Immunol (2009) 183(6):4127–34. doi: 10.4049/jimmunol.0901253
171. Zhao J, Jiang P, Guo S, Schrodi SJ, He D. ApoptosIs, Autophagy, NETosIs, NecroptosIs, and PyroptosIs Mediated Programmed Cell Death as Targets for Innovative Therapy in Rheumatoid ArthritIs. Front Immunol (2021) 12:809806. doi: 10.3389/fimmu.2021.809806
172. Zhao J, Hu Y, Peng J. Targeting Programmed Cell Death in Metabolic Dysfunction-Associated Fatty Liver DIsease (MAFLD): A PromIsing New Therapy. Cell Mol Biol Lett (2021) 26(1):17. doi: 10.1186/s11658-021-00254-z
173. Séjourné A, Boudot C, ObjoIs T, Fardellone P, Brazier M, Six I, et al. Expression of the Calcium-Sensing Receptor in Monocytes From Synovial Fluid Is Increased in OsteoarthritIs. Joint Bone Spine (2017) 84(2):175–81. doi: 10.1016/j.jbspin.2016.03.012
174. Paccou J, Boudot C, Renard C, Liabeuf S, Kamel S, Fardellone P, et al. Total Calcium-Sensing Receptor Expression in Circulating Monocytes Is Increased in Rheumatoid ArthritIs Patients With Severe Coronary Artery Calcification. ArthritIs Res Ther (2014) 16(5):412. doi: 10.1186/s13075-014-0412-5
175. Jäger E, Murthy S, Schmidt C, Hahn M, Strobel S, Peters A, et al. Calcium-Sensing Receptor-Mediated NLRP3 Inflammasome Response to Calciprotein Particles Drives Inflammation in Rheumatoid ArthritIs. Nat Commun (2020) 11(1):4243. doi: 10.1038/s41467-020-17749-6
176. Li ZY, Zhou JJ, Luo CL, Zhang LM. Activation of TGR5 Alleviates Inflammation in Rheumatoid ArthritIs Peripheral Blood Mononuclear Cells and in Mice With Collagen II−induced ArthritIs. Mol Med Rep (2019) 20(5):4540–50. doi: 10.3892/mmr.2019.10711
177. O'Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. Local Expression of the Serum Amyloid A and Formyl Peptide Receptor-Like 1 Genes in Synovial TIssue Is Associated With Matrix Metalloproteinase Production in Patients With Inflammatory ArthritIs. ArthritIs Rheumatol (2004) 50(6):1788–99. doi: 10.1002/art.20301
178. Lee MS, Yoo SA, Cho CS, Suh PG, Kim WU, Ryu SH. Serum Amyloid A Binding to Formyl Peptide Receptor-Like 1 Induces Synovial Hyperplasia and AngiogenesIs. J Immunol (2006) 177(8):5585–94. doi: 10.4049/jimmunol.177.8.5585
179. Tagoe CE, Marjanovic N, Park JY, Chan ES, Abeles AM, Attur M, et al. Annexin-1 Mediates TNF-Alpha-Stimulated Matrix Metalloproteinase Secretion From Rheumatoid ArthritIs Synovial Fibroblasts. J Immunol (2008) 181(4):2813–20. doi: 10.4049/jimmunol.181.4.2813
180. Stevenson NL, Martin-Martin B, Freeman J, KrIston-Vizi J, Ketteler R, Cutler DF. G Protein-Coupled Receptor Kinase 2 Moderates Recruitment of THP-1 Cells to the Endothelium by Limiting HIstamine-Invoked Weibel-Palade Body ExocytosIs. J Thromb Haemost (2014) 12(2):261–72.
Keywords: G-protein-coupled receptors, rheumatoid arthritis, inflammation, bone destruction, lipid metabolism, angiogenesis
Citation: Zhao J, Wei K, Jiang P, Chang C, Xu L, Xu L, Shi Y, Guo S and He D (2022) G-Protein-Coupled Receptors in Rheumatoid Arthritis: Recent Insights into Mechanisms and Functional Roles. Front. Immunol. 13:907733. doi: 10.3389/fimmu.2022.907733
Received: 30 March 2022; Accepted: 20 June 2022;
Published: 08 July 2022.
Edited by:
Betty Diamond, Feinstein Institute for Medical Research, United StatesReviewed by:
Eugenia Gurevich, Vanderbilt University, United StatesCopyright © 2022 Zhao, Wei, Jiang, Chang, Xu, Xu, Shi, Guo and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shicheng Guo, U2hpY2hlbmcuR3VvQHdpc2MuZWR1; Dongyi He, ZG9uZ3lpaGVAbWVkbWFpbC5jb20uY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.