 Davide Ferrari
Davide Ferrari Michele Rubini
Michele Rubini Jorge S. Burns
Jorge S. Burns- 1Section of Microbiology and Applied Pathology, University of Ferrara, Ferrara, Italy
- 2Department of Life Science and Biotechnology, University of Ferrara, Ferrara, Italy
- 3Department of Neuroscience and Rehabilitation, University of Ferrara, Ferrara, Italy
- 4Department of Environmental and Prevention Sciences, University of Ferrara, Ferrara, Italy
A long-shared evolutionary history is congruent with the multiple roles played by purinergic signaling in viral infection, replication and host responses that can assist or hinder viral functions. An overview of the involvement of purinergic signaling among a range of viruses is compared and contrasted with what is currently understood for SARS-CoV-2. In particular, we focus on the inflammatory and antiviral responses of infected cells mediated by purinergic receptor activation. Although there is considerable variation in a patient’s response to SARS-CoV-2 infection, a principle immediate concern in Coronavirus disease (COVID-19) is the possibility of an aberrant inflammatory activation causing diffuse lung oedema and respiratory failure. We discuss the most promising potential interventions modulating purinergic signaling that may attenuate the more serious repercussions of SARS-CoV-2 infection and aspects of their implementation.
Introduction to the Purinome and its Druggability
Of broad function, purine containing nucleotides and nucleosides serve as the precursors of RNA and DNA and also have ubiquitous critical roles as cofactors in the enzymatic reactions that create and sustain all living organisms. These include small G proteins, kinases, dehydrogenases, ATPases, non-conventional purine utilizing enzymes, helicases, synthetases, deaminases, lipases, sulfotransferases, cytosolic tyrosine kinases, carboxylases, motor proteins and purinergic receptors. Adenosine 5’-Triphosphate (ATP) of high abundance in every cell (1-10 mM) represents an archetypical molecule that intracellularly is a fundamental energy currency, providing controlled bursts of about 7 kilocalories per mole with the splitting of its terminal phosphoanhydride bond yielding adenosine 5’-diphosphate (ADP) and inorganic phosphate (Pi). The proposition that extracellularly, the ATP molecule also served as a signaling cotransmitter, was confirmed with the cloning and characterization of specific receptors for purines and pyrimidines (1). The large family of receptors has been subdivided into three subtypes according to molecular, biochemical and pharmacological characteristics; comprising four members of the P1 (adenosine) receptor family, seven members of the P2X receptor family and eight members of the P2Y receptor family. These receptors interact with a vast heterogeneity of purinergic ligands that in turn are regulated in substance and concentration by ectonucleotide-metabolizing enzymes hydrolyzing nucleotide phosphates and the activity of nucleotide channels and transporters. The extracellular purine signaling network constitutes only a small percentage of the more than 3,260 proteins utilizing purine cofactors that constitute the total human purinome (2). However, since inter-cellular communication represents a powerful functional class comprised of significant complex and dynamic molecular cross-talk networks, this has itself been termed “purinome”, comprising the numerous extracellular molecular interaction partners responsible for the biological effects of extracellular purine and pyrimidine ligands (3). This review will refer to the total purinome when considering virus interactions with intracellular host proteins that may dictate viral replication, yet will predominantly focus on the extracellular definition of “purinome” and the associated concept of “receptosome” comprising the web of interactions between not only receptors but other proteins at the plasma membrane that contribute to the very heterogeneous responses to epigenetic factors and modified extracellular environments. The receptosome presents a comprehensive regulatory concept of dynamic interactions that collectively invoke the fine tuning to accommodate rapidly changing microenvironmental contexts (4). The receptosome concept is thus broader than the similar-sounding yet alternatively defined “recepterome” of plasma membrane hosted ligand capture molecules that serve as a useful cohort to efficiently discover molecular targets for therapeutic drug discovery (5).
Capture of purinome molecular participants has been achieved using affinity arrays of γ-phosphate linked ATP-sepharose, orientated towards ligand-relevant conformations, to screen small molecule libraries (6, 7). This has generated information about drug targets and also off-target liabilities, an important consideration given that shared molecular spaces within purine binding pockets are not likely to be very diverse, with high probability that seemingly disparate enzymes may bind the same drug by chance. Several hundred purine-utilizing enzymes are implicated in the Online Mendelian Inheritance in Man (OMIM) database as being associated with inborn errors of metabolism, making the unwanted off-target side-effects more likely. Nonetheless, the advantageous ability to simultaneously screen for potency and selectivity has meant drugs targeting purine-utilizing enzymes have been very successful. Galdanamycin that binds the ATP/ADP binding pocket in the N terminus of the HSP90 protein has been shown to inhibit proliferation of a wide variety of tumor cell lines and possess antifungal activity (8).
The anti-folate agent Methotrexate (MTX), a folic acid analogue that is polyglutamated inside the cell to its active form, can inhibit de novo purine synthesis involved in generating the precursor nucleotides required for RNA and DNA. Besides being a chemotherapy agent and immune system suppressant, MTX has also some antiviral effect, as it can reduce the viral titer of Zika virus, which can be rescued through addition of adenosine to virus-infected cells. This evidence suggests that restriction of de novo synthesis ATP pools could suppress viral replication (9). In addition, this disrupts the S phase of the cell cycle in rapidly dividing immune cells and additional immunomodulatory effects mean that MTX can reduce inflammation and joint damage in some rheumatological diseases. However, acute high dose methotrexate treatment for cancer is associated with lactic acidosis and even when used at low doses for arthritis, there is a risk of liver fibrosis (10). Thus when developing pharmacological approaches that target antiviral purinergic mechanisms there is need to go beyond single molecule concepts and adopt a more holistic approach. New public databases of AI-predicted protein structures, incorporating AlphaFold or RoseTTAFold algorithms (11, 12), to improve computational prediction accuracy of protein conformations, could be invaluable for accelerating the development of purinome directed drugs.
Virus-Cell Purinome Interactions Share Ancient History
Contemporary to early stages of the evolution of life, chemically treated and irradiated powdered meteorites are demonstrable sources of amino acids and nucleosides (13). It is considered that within a primordial context emerged viroids, virus-like infectious agents; fundamentally an encapsulated biological code, acquiring biotic potency once the code could be processed and directed towards self-assembly of a unit particle (14). To what extent viruses are likely to have evolved from viroid-like RNA that preceded cellular life (15) is of debate (16). However, the relatively recent discovery of giant viruses, that can mimic bacteria in size, with >1000 gene genomes including genes more significant for cellular rather than viral metabolism, encoding a large yet incomplete repertoir of protein-synthesis translation factors (17), has lent credence to the concept that early plant-interacting viroids were autonomous units.
Selection pressure for increased autonomy has resulted in self-replicating cells and specialisations coordinated to the level of whole organisms that derive nutrition and energy from environmental interactions that sustain life. Molecules serving roles restricted to replication have been adopted for more complex tasks governing intracellular and ultimately intercellular signaling. Conversely, evolutionary pressures favouring selection for replication with reduced energy requirements, exploited endosymbiotic interactions to such a parasitic extent that today’s viruses lack enough genes to live independently.
Given a replicative code chemistry composed of ribonucleic acid (RNA) or deoxyribonucleic acid (DNA), viruses have a critical requirement for purines and pyrimidines as essential small molecules constituting the bases of synthesized nucleotides. Within a cellular microenvironment, these can be sourced from de novo pathways of synthesis or obtained from more energetically efficient salvage pathways whereby component small molecules, such as hypoxanthine, adenine or guanine for purine synthesis or uracil, cytidine or thymidine for pyrimidine are synthesized from catabolic products of nucleic acid turnover, processes that may be therapeutically exploited (18). Characterization of specific cellular receptors for extracellular nucleotides has revealed a repurposing of purine nucleotides and nucleosides. They can also act as extracellular signaling mediators, as ligands that interact with a broad array of purinoreceptors and ectonucleotidases in different tissues and infiltrating immune cells; thereby governing diverse functions in almost every organ and system of organisms. The long shared history of viruses and the human purinome, provides ancient interplay for evolving intricate and complex interactions pertaining to both viral establishment within host cell metabolism, cell-reactive mechanisms aiming to restore homeostasis and viral countermeasures to host defense mechanisms to maintain selfish genetic presence.
Five steps of viral infection include; (i) Binding to a host cell, (ii) receptor downstream interactions facilitating entry and integration within the host cell genome, (iii) Genetic code processing to favor viral genome replication, (iv) Processing of the viral code to commandeer host cell functions supporting translation and assembly of viral core structures, (v) Release of maturated viral particles into the microenvironment allowing recapitulation of processes (i) to (v) in new host cells. It is not surprising that purinergic signaling plays a significant role in all these viral infection steps and host cell responses (19), the challenge is to characterize precisely how viral interactions can impact upon purinergic signaling to influence pathological processes and whether this can reveal key targets for effective therapeutic intervention.
By reviewing responses to viral infection mediated by purinergic signaling discovered in a diverse array of antecedent viruses, we seek to establish clues and principles that may improve our understanding for better control of the newly emerged SARS-CoV-2 pandemic virus (20) and its COVID-19 disease symptoms.
Extracellular Nucleotides and Nucleosides Act as Signaling Molecules
Undoubtedly, nucleotides (ATP, ADP, Uridine triphosphate (UTP), Uridine diphosphate (UDP), UDP- glucose) and nucleosides (adenosine/ADO) are fundamental for cell energy demands, metabolic regulation and building macromolecules such as nucleic acids (21). However, an increased opportunity to control viral pathologies is presented by focus on the additional purinergic role outside the cell, were they behave as intercellular ligands for signals acting both autocrinally and paracrinally to stimulate a plethora of organ and cell responses.
Besides being released as a consequence of membrane stress or damage, ATP can be transferred into the extracellular space by Pannexin-1, Connexin 43, exosomes and microvesicles (22). Resulting responses range from nervous transmission (23, 24), modulation of the cardiovascular system (25), muscle contraction (26), microvessel permeability (27), immune defense (28) and the fundamental processes of cell proliferation, differentiation and cell death (29–31).
Responses to extracellular nucleotides and nucleosides occur by stimulation of plasma membrane proteins named purinergic P1 and P2 receptors. Extracellular ADO binds and activates P1 receptors that are G-protein-coupled, metabotropic seven-membrane-spanning domain receptors, subdivided into A1, A2A, A2B and A3 receptors (32). The nomenclature for the diverse family of P2 receptors has bifurcated according to biochemical characteristics; ionotropic P2X and metabotropic P2Y forms. ATP, ADP, UTP, UDP, UDP-glucose activate P2 receptors, comprising seven P2X subtypes (P2X1-7) and eight G-protein-coupled P2Y receptor subtypes (P2Y1, 2, 4, 6, 11, 12, 13, 14) (33). The receptors have distinct preferential binding avidities for different extracellular nucleotides and ectonucleotidases, present on the extracellular side of the plasma membrane, that play a fundamental role in hydrolyzing nucleotides, thereby modifying their extracellular concentration and availability for the P1 and P2 receptors (34) (Figure 1).
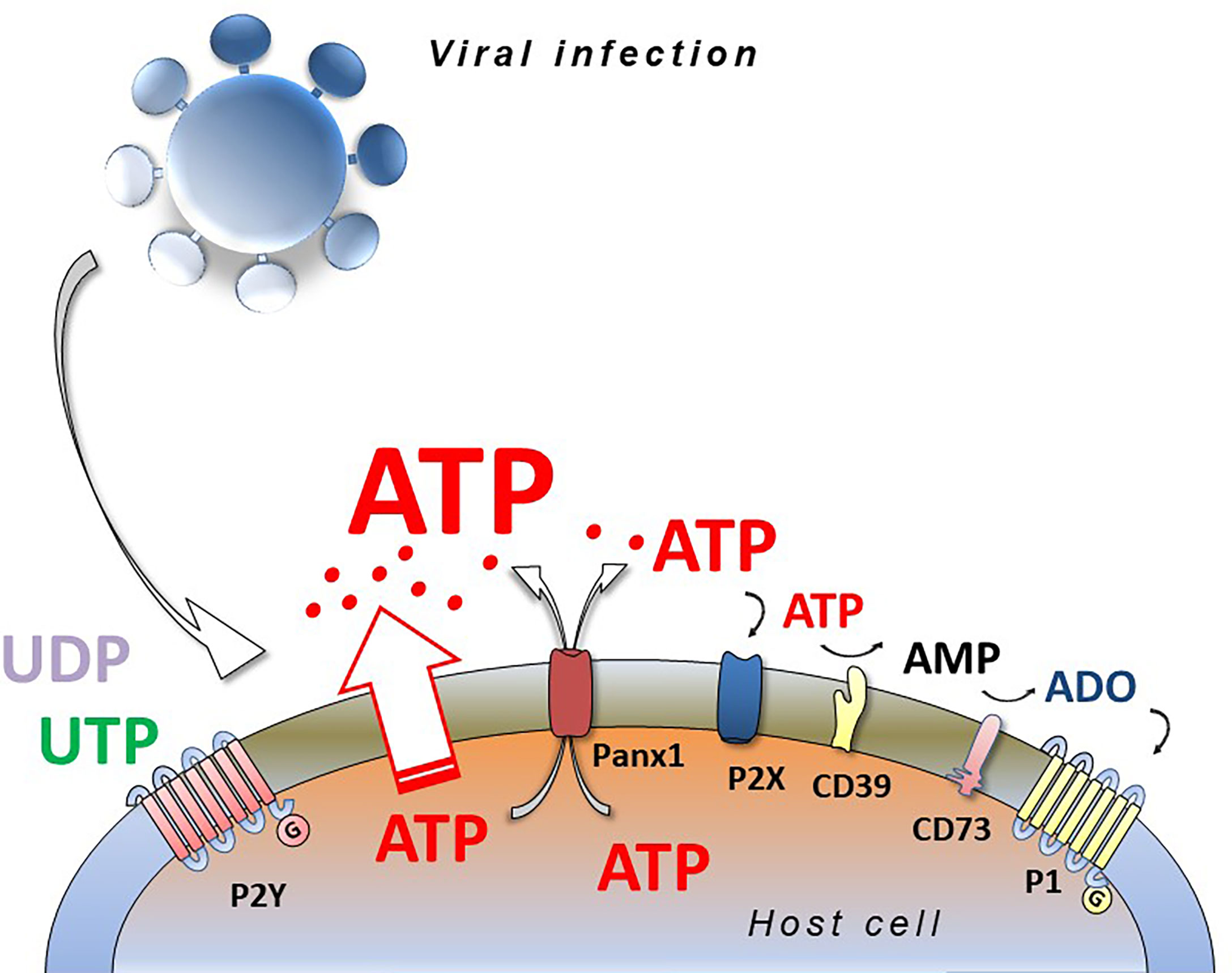
Figure 1 Viral infection and purinergic signalling. Viruses infecting eucariotic cells induce the release of intracellular nucleotides (ATP, ADP, UTP, UDP) by different ways (connexins, pannexins, membrane stress or damage) activating purinergic P2 (P2X and P2Y) and P1 (A1, A2A, A2B, A3) receptors and inducing pro- or anti-viral responses depending on virus species, cell type and purinergic receptors expressed. ATP and ADP are hydrolyzed by CD39 and CD73 ecto-nucleotidases generating adenosine (ADO) that is agonist at P1 receptors.
Release of Intracellular Nucleotides: A Widespread Paradigm for Cell Invading Microorganisms, Including Viruses
Disruption of cellular metabolic processes by “foreign agents” such as microbes and parasites e.g. Plasmodium falciparum infecting human erythrocytes (35), often evokes nucleotide release from infected cells, with extracellular ATP an exemplary contributor to danger-associated molecular patterns (DAMPs) (36). The DAMP response can include neutrophil recruitment (37), macrophage activation (38) and systemic inflammation (39), cellular pathways favoring elimination of different intracellular parasites (28, 40) and this likely explains why several microorganisms express ectonucleotidases that rapidly degrade released ATP to mitigate ATP-mediated cell lysis (41). Cytomegalovirus (CMV) infection induces adenine nucleotide liberation from endothelial cells (42) with disruption of ATP- and KCL-stimulated calcium signaling (43). Even singular viral components were able to provoke this response, although it is to be appreciated that sterile non-infectious trauma can also activate DAMPs and cause a systemic inflammatory response syndrome (39). Liberation of these nucleotides has been detected in numerous examples of virus infected cells including Herpes Simplex Virus 1 (HSV-1), Newcastle disease virus and murine leukemia virus (44). The HIV-1 glycoprotein gp120 induced ATP release from human macrophages and the nucleotide was also found in supernatants of HIV-1 infected cells (19, 45, 46).
Culture media from HIV-infected macrophages also contained large amounts of ADP, AMP and a low concentration of ADO. Notably, however, extracellular ATP binding its receptor P2X7 was the only agonist shown to trigger rapid release of virus containing compartment sequestered HIV-1 virions without simultaneously causing the death of infected macrophages (47). In contrast, extracellular ATP acted in a P2X7 receptor dependent manner to protectively restrict viral replication and facilitated interferon ß (IFN-ß) secretion with subsequent promotion of antiviral immunity (48). These examples highlight how the same purinergic pathway agonist, extracellular ATP, may be regarded as either a friend or foe of viral infectivity according to the viral species.
Different viruses may favor activation of different purinergic mediators, for example vesicular stomatitis virus (VSV) induced the release of UDP (49, 50), whereas infection with respiratory syncytial virus (RSV) caused predominantly UTP release in mice, that subsequently participated in signaling pathways that reduced basal alveolar fluid clearance (AFC) a symptom of lower respiratory tract disease (51). In RSV-induced goblet-cell metaplastic cultures and in bronchoalveolar lavage of influenza H1N1 infected mice, extracellular ATP and ADO accumulation was described (52). Release of these nucleotides and ADO may exacerbate lung inflammation and consequent tissue damage (53). Accordingly, acute injury of the lung tissue due to the viral-induced immune response was greatly reduced in A1 ADO receptor-knockout mice, highlighting a role for these receptors in lung pathogenesis due to influenza virus (54). ATP release was also found in VSV-infected RAW 264.7 and L929 cells (48) and in primary human bronchial epithelial cultures infected by RSV (55).
Purinergic Cell Signaling can Impair Viral Function
The multiple mechanisms underlying purinergic receptor participation in viral biology are attracting the attention of virologists involved in different fields (48, 56, 57). Human anti-viral response is based on transcription of genes encoding interferons, a family of cell host proteins endowed with the ability to potentiate the overall immune response thus heightening body anti-viral defenses (58) (Figure 2).
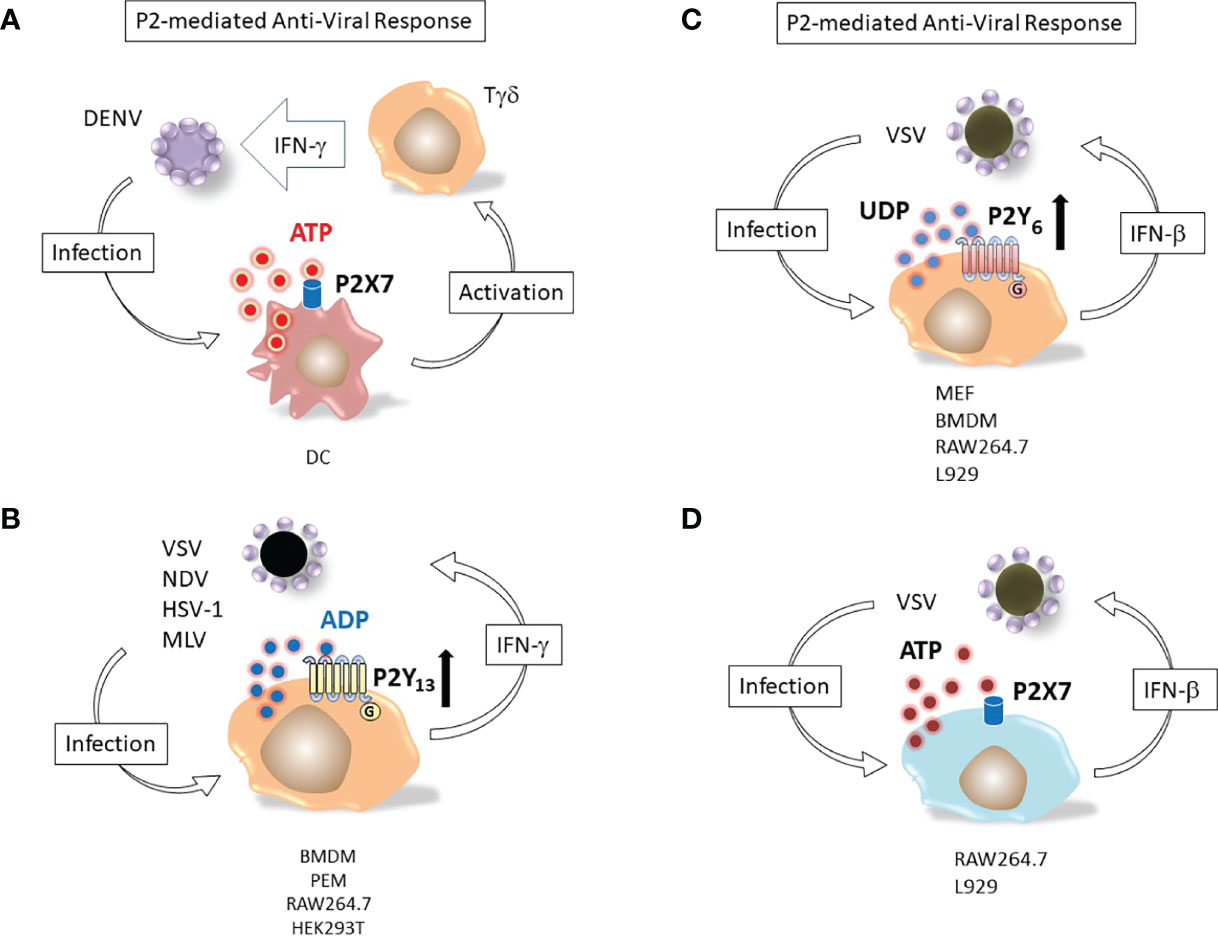
Figure 2 Nucleotide induced defensive responses. (A) ATP and P2X7 dependent immunological response to DENV. Dendritic cells (DC) and Tγδ lymphocytes (Tγδ) major responders against Dengue-2 virus (DENV) infection. Binding of DC P2X7 receptor by extracellular ATP activates Tγδ to produce of interferon-γ (IFN-γ), potentiating the antiviral immune response (59). (B) ADP and P2Y13-dependent antiviral response. Replication of viruses as different as vesicular stomatitis virus (VSV), Newcastle disease virus (NDV), herpes simplex virus 1 (HSV-1) and murine leukemia virus (MLV) is restricted by massive release of ADP from infected cells; (IFN-γ) up-regulates the ADP activated P2Y13 receptor (60). (C) UDP and P2Y6-mediated response against VSV infection. Murine embryonic fibroblasts (MEF), bone marrow-derived macrophages (BMDM), RAW264.7 or L929 cell lines release UDP upon infection with vesicular stomatitis virus (VSV). The nucleotide activates P2Y6 receptor and induces interferon-β (IFN-β) secretion (49). Bone marrow-derived macrophages (BMDM), peritoneal macrophages (PEM). (D) ATP and P2X7-mediated response against VSV infection. RAW264.7 or L929 cell lines release ATP upon infection with vesicular stomatitis virus (VSV). The nucleotide activates P2X7 receptor and induces interferon-β (IFN-β) secretion (48).
P2Y13, a receptor for extracellular ADP, was upregulated in mice treated with interferons (IFNs) and its expression protected mice from different viruses by inhibiting the cAMP/EPAC1 signaling pathway (60). P2Y13 mediated the antiviral response against vesicular stomatitis virus (VSV), Newcastle disease virus (NDV), herpes simplex virus 1 (HSV-1) and murine leukemia virus (MLV) while the other ADP-activated subtypes (P2Y1 and P2Y12) did not (60).
The interplay between nucleotides and IFNs in antiviral responses was also true for extracellular UDP. This uridine nucleotide was found to be released by infected cells and prompted IFN-β secretion by activating the antiviral response mediated by p38/JNK- and ATF-2/c-Jun-signaling pathways (49). VSV-infected cells showed an enhanced expression of the UDP receptor P2Y6. Consistent with the hypothesized protective role of this subtype, MRS2578, a selective P2Y6 antagonist that knocked-out expression of the receptor, increased VSV replication (49). Purinergic stimulation of P2 receptors during virus infection also modulated physiology of infected tissues. Instillation of the P2 antagonists suramin or XAMR-0721 in the airway of mice infected by the human Respiratory Syncytial Virus (hRSV) restored the alveolar fluid clearance capacity of the bronchoalveolar epithelium (61). Degradation of UTP but not ATP had the same effect, suggesting that a uridine activated nucleotide receptor (i.e. of the P2Y subtype) was likely involved in the hRSV-induced deficit of alveolar fluid clearance and that UTP was released into alveolar fluids as a consequence of hRSV infection (61).
A role for the P2X7 receptor subtype in controlling Dengue-2 virus infection in human monocytes has been shown (62). Dengue virus particles infect host cells by receptor-mediated endocytosis, and upon acidification of the endocytic vesicle, the nucelocapsid contents is released into the cytoplasm, where the viral genome is translated into a single polyprotein chain, subsequently subdivided into individual viral proteins by virus and host proteases (63). Pharmacological stimulation of human monocytes with extracellular ATP prior to Dengue-2 (DENV) virus treatment was able to inhibit infection, likely due to activation of P2X7-triggered antiviral responses, since specific inhibition of this subtype expressed in the host cell abrogated this effect (62). Stimulation of monocytes with ATP induced production of reactive oxygen species and nitric oxide, while it reduced secretion of CCL2, CXCL10 and chemokines IL-8 and TNF-α (62). Involvement of P2X7 in the human immune response to DENV infection was also shown. Accordingly, inhibition of the receptor expressed by dendritic cells (DC) significantly reduced the anti-DENV IFN-γ response of γδ T cells (59).
Over-activation of the P2X7 receptor has been linked to severe tissue damage due to an uncontrolled host immune response (64). The presence of viruses or even of adenoviral vectors in co-cultures of macrophages and epithelial cells induced P2X7-dependent inflammasome activation, causing acute respiratory distress syndrome and high mortality in P2X7-expressing mice but not in P2X7 KO mice. According to the well-known pro-inflammatory role of the receptor, the increased survival of P2X7 deficient mice was paralleled by decreased IL-1β, IL-6 levels and reduced neutrophil infiltration (64).Viral infection could induce over-expression of single P2 receptor subtypes. Peripheral blood mononuclear cells (PBMC) isolated from hepatitis C virus (HCV) patients responding to antiviral therapy (PEGylated interferon and Ribavirin) showed an increased expression of the P2X7 and P2X4 mRNA, while cells from non-responders had no up-regulation of messengers of these subtypes (65). Although the role of P2X7 and P2X4 subtypes in HCV infection is still not clear, authors have hypothesized that the latter might have a proviral role as they found increased expression of this subtype as a consequence of over-expression of E1/E2 HCV proteins in huh-7 cells (66). Changes in cell host P2 expression also occurred during Epstein-Barr Virus (EBV) induced cell transformation. In particular, both P2X7 mRNA and protein levels were potently augmented by the presence of EBV, while on the contrary, P2X1, P2X4, P2Y1 and P2Y11 proteins were down-regulated (67).
Purinergic Signaling as a Viral Accomplice
The first scientific evidence that purinergic receptors were involved in the mechanism of virus entry in eukaryotic cells came from a report on hepatitis B virus (HBV) and hepatitis delta virus (HDV), both major causes of acute and chronic hepatitis (68). Generic inhibitors of the P2 receptors such as suramin, were found to block HDV entry into primary human hepatocytes. The same effects were observed for pyridoxal-phosphate-6-azophenyl-2’,4’-disulfonate (PPADS), a P2X antagonist and brilliant blue G (BBG), a selective inhibitor of the P2X7 subtype. It was thus suggested that this last receptor contributed both to viral penetration into hepatocytes and liver inflammation, i.e. hepatitis (68, 69).
Numerous observations concerning involvement of different P2Y and P2X receptors in human immunodeficiency virus 1 (HIV-1) infection of eukaryotic cells have concurred to give a more precise model of the interplay between cell receptors for HIV-1 binding/recognition, ATP-releasing molecules (connexins, pannexins, etc.) and purinergic receptors (19, 45, 46). HIV-1, infects the cell by binding CD4 (primary receptor) with viral glycoproteins gp120 and gp41 plus interactions with the chemokine receptors (secondary receptors or co-receptor) CXCR4 (expressed on lymphocytes) and CCR5 (present in macrophages and peripheral lymphocytes. It has recently been shown that P2 purinergic receptors play a fundamental role in allowing HIV-1 to penetrate eukaryotic cells and replicate, contributing to virion release and disease progression (45, 70–72). Hence, activation of different P2 subtypes (P2Y2, P2X1, P2X7) have been shown to modulate crucial steps of HIV-1 biology (44). Regarding questions about the mechanism adopted by the virus to evoke ATP release, it has emerged that upon virus-host-cell interaction there is opening of pannexin-1 (Panx1) membrane channels with consequent release of ATP and autocrine triggering of the P2Y2 receptor (45) with proline-rich tyrosine kinase 2 (Pyk2) kinase activation and fusion of Env-expressing viral membranes with membranes of the host cell. Accordingly, blocking just one of the pathway constituents (e.g. Panx-1, ATP, P2Y2 or Pyk2) caused a decrease in HIV-1 infection and replication efficiency. Another indication of the importance of extracellular ATP during HIV-1 infection came from the observation that the ATP hydrolyzing enzyme apyrase greatly reduced HIV-1 infection (45).
Another interesting point is that HIV-1 infected both CD4+ lymphocytes and macrophages although it was released through a cytopathic effect only by lymphocytes. In macrophages, the new progeny of HIV-1 virions accumulated into intracellular virus-containing compartments, but little was known about their release. It has recently been shown that ATP can stimulate the release of virions from infected macrophages through stimulation of the P2X7 subtype in the absence cell death (72). Tat HIV-1 exerts direct neurotoxic effects, but much less is known about indirect Tat-induced neurotoxicity and it has recently been reported that HIV Tat-mediated neuronal death can be caused by stimulation from ATP released by astrocytes (73). Hence, Tat upregulated P2X7 expression in astrocytes and its activation evoked intracellular calcium concentration increase, phosphorylation of ERK1/2, with subsequent secretion of CCL2 and ATP from astrocytes (73). Interestingly, P2X7 knockdown in astrocytes reduced Tat-mediated indirect neurotoxicity. Another cell type strongly involved in neuronal damage in HIV-infected individuals is the microglia, at least in primary neuron-glia co-cultures from mouse striatum. Although microglial cells express the P2X7 receptor, the P2X4 subtype has been indicated as a candidate for Tat-dependent neuronal injury (74).
Treg lymphocytes play a fundamental role in limiting the immune response, promoting self-tolerance and preventing chronic inflammation and autoimmune diseases (75). However, their activities towards other lymphocytes prevent them from performing an efficient anti-viral response (76). The ectonucleotidase CD39 plays a role in down-modulating the immune response of Treg lymphocytes. Low clinical outcome in HIV infected patients was associated with a high frequency of CD39+ Treg cells suppressing il-2 gene expression in CD4+ lymphocytes more efficiently than CD39- Treg cells. The CD39/ADO pathway activated A2A receptor expressed by CD4+ lymphocytes thus increasing intracellular cAMP and reducing il-2 gene promoter demethylation, with consequently reduced IL-2 production (77). Treg inhibitory effects were counteracted by down-modulating CD39 expression while they were mimicked by P1 receptor agonists (78). Confirmation of the role played by CD39 and ADO in the pathogenesis of AIDS came from the observation that expansion of CD39+ expressing Treg correlated with lower CD4+ counts in HIV-1 infected patients and more importantly, a CD39 gene polymorphism associated with down-modulation of CD39 expression resulted in slower progression to AIDS (78). Attention should be paid also to the interplay between thromboregulation and antiretroviral therapy, since cardiovascular diseases increased in patients subjected to highly active antiretroviral therapy (HAART) (79). Increased platelet aggregation was found in HAART treated patients compared to a control group. Since nucleotides play a role in blood coagulation and nucleotidases (E-NTPDase, E-5’-nucleotidase) were expressed on the platelet surface, the authors assessed their activity. E-NTPDase mediated ATP hydrolysis was higher in HIV infected individuals compared with uninfected controls and the HIV/HAART patients. Alternatively, ADP hydrolysis by E-NTPDase was higher in HIV patients compared to controls, and lower in HIV/HAART patients compared to controls and HIV infected individuals (80)
SARS-CoV-2 Interactions With the Purinome
Much about the mechanisms of SARS-CoV-2 entry into the target cell determines the host and tissue-specific tropism, pathogenicity and zoonotic transmission (81). Prominently the viral spike protein (S) mediates fusion of the viral envelope and host cell membranes through its binding angiotensin-converting enzyme 2 (ACE2) (82), to date the only bona fide primary receptor for SARS-CoV-2, the highly related SARS-CoV and more distant HCoV-NL63 viruses. Viral entry occurs through the interaction of the Spike (S) protein with the cellular ACE2 receptor (83), facilitated in many cases by the processing of the S protein by proteases, in particular furin, the transmembrane serine protease 2 (TMPRSS2) and endosomal cathepsins. Coronavirus entry cofactors e.g. Neuropilin-1 (NRP-1) and Cluster of Differentiation 147 (CD147) can interact with furin cleaved SARS-CoV-2 spike protein since furin preactivation of the S protein increases the binding affinity to its receptor and co-factors, resulting in a widespread infectious cycle introducing differences in comparison to SARS-CoV despite both viruses targeting ACE2 (84). With reference to other RNA viruses, HIV-1 infection was found to induce the opening of ubiquitously expressed Pannexin-1 (Panx-1) channels on the surface of peripheral blood mononuclear cells, leading to a release of ATP that could stimulate purinergic signaling pathways that enhanced viral binding, accelerating HIV infectious entry and contributed to the compromised synaptic function found in cases of HIV-associated neurocognitive disorder (85). It was proposed SARS-CoV-2 may also induce opening of the large ionic Panx-1 channels, allowing passive outflux of ATP, metabolites and other small anions likely to have a role in exacerbating the inflammatory response (86). Independent research has confirmed this supposition, moreover in contrast to the common cold coronavirus 229E, the SARS-CoV-2 S protein caused more sustained Panx-1 channel opening, resulting in relatively enhanced ATP, PGE2 and IL-1ß release. The S protein induced Panx-1 channel opening required ACE2, endocytosis and furin activity (87). Notably, Tenofovir an AMP nucleotide analogue antiviral used in the treatment of hepatitis B and HIV, inhibited Panx-1 mediated ATP release in a human liver HepG2 cell line, diminishing adenosine levels in the extracellular space (88), but whether this relatively inexpensive and widely-available drug could be repurposed for SARS-CoV-2 infection awaits further in vitro confirmation and subsequent clinical trials (89).
The assumption that insights from successfully treating influenza or HIV-1 with purinome-directed drugs might have direct relevance against SARS-CoV-2 was met with abrupt disappointment. Nucleotide analogues that could be effective antiviral drugs to disrupt the replication of several viruses did not necessarily work against the SARS-CoV-2 coronavirus polymerase (90). This was surprising given the relatively large 30,000 base SARS-CoV-2 genome size (about three times the size of the influenza virus genome), that should have rendered it susceptible to a drug targeting replication machinery, to induce many errors and mutations causing error catastrophe (91). However, unlike other RNA viruses, coronaviruses tend to mutate less because of a correction system (92) that includes a non-structural protein (nsp14-ExoN) encoding a proofreading exonuclease removing mismatched nucleotides during replication and transcription (93, 94). Extending pathological effects, nsp14-ExoN has also been shown to interact with host Inosine-5’-monophosphate dehydrogenase 2 (IMPDH2) protein, known to regulate NFκB activation and signalling leading to induction of cytokines IL-6 and IL-8, both proinflammatory mediators (95). The porcine epidemic diarrhea virus (PEDV) has also been found to express nsp14-ExoN and a crucial mutation lead to high genetic instability during viral replication (96). Small molecular inhibitors of the nsp14 exoribonuclease have become an attractive target for novel SARS-CoV-2 replication inhibitors (97, 98). Notably the Omicron variant had many more mutations than previous SARS-CoV-2 variants with most of the mutations in the spike protein and high conservation in 25 other proteins, including those for replication. The Omicron variant does have a mutation leading to a I42V substitution in the Nsp14 protein, yet this is distant from the active site and needn’t necessarily explain the significant increase in Omicron lineage mutations (99). Whether a greater mutation incidence indicates SARS-CoV-2 Omicron variants are more susceptible to any intracellular purinome-directed drugs has yet to be determined.
SARS-CoV-2 has shown greater similarity with other viruses with regard to a need for host HSP90 protein, a non-conventional class purine utilizing enzyme, as a chaperone for viral proteins, helping prevent proteasomal degradation. Targeting HSP90 could inhibit replication of Kaposi sarcoma-associated herpesvirus (KHSV) (100) hepatitis C virus (HCV) (101) and also the human coronaviruses MERS-CoV and SARS-CoV (102). In a similar manner, ex vivo experiments involving transcriptomic profiling of SARS-CoV-2 infected human cell lines have shown that HSP90 inhibition impaired viral replication and activation of pro-inflammatory genes in primary human airway epithelial cells (103).
Fatty acid synthase (FASN), a multifunctional cytosolic enzyme of 272kDa, uses NADPH to condense acetyl-CoA and malonyl CoA into palmitate. The late stages of HIV-1 replication are modulated by FASN and specific inhibitors e.g. Fasnall could reduce the number of infectious HIV-1 particles, as was also the case for other enveloped viruses (104). Human embryo kidney HEK293T cells engineered to downregulate expression of fatty acid synthesis pathway enzymes FASN or ACACA failed to support SARS-CoV-2 replication (105), consistent with a palmitoylation modification of the SARS-CoV-2 spike protein being essential for viral infectivity (106). In addition to FASN inhibition, targeting fatty acid metabolism with inhibitors to ATP-binding Vacuolar protein sorting 34 (VPS34), a member of the phosphatidylinositol-3-kinase lipid kinase family that controls the canonical autophagy pathway and vesicular trafficking, also inhibited SARS-CoV-2 replication (107). Orlistat a FASN inhibitor used in clinical trials against numerous viruses, may suppress SARS-CoV-2 replication by lowering the lipid synthesis required in replication organelles with prevention of viral palmitoylation (108). This would likely have particular applicability for post-entry inhibition during the early phase of SARS-CoV-2 infection. In patients experiencing a later pathological post-acute phase of SARS-CoV-2 infection, the emphasis of coronavirus disease shifted from viral interactions to host alveolar epithelial and endothelial injury from proinflammatory responses (109), threatening the possibility of acute respiratory distress syndrome characterized by pulmonary edema and respiratory failure with a mortality rate risk of 30-40%. The purinome has important roles to play in inflammation, tissue adaptation to low oxygen conditions and the hypoxia-inducible factors crucial in mediating the crosstalk between hypoxia and inflammation. The mammalian brain is highly oxygen dependent and numerous adaptive responses have evolved to protect brain O2 levels, among them an important homeostatic control whereby C1 noradrenergic neurons, that are powerfully excited by hypoxia, control heart rate and blood pressure. Evidence from rodents points to astrocytes in the pre-Bötzinger Complex, a critical site for generating breathing rhythm, responding to hypoxia with release of ATP that acts via P2Y1 receptors to excite inspiratory neurons and increase ventilation (110). Extracellular ATP is also rapidly broken down by ectonucleotides (e.g. CD39 and CD73) into extracellular adenosine diphosphate (ADP), adenosine monophosphate (AMP) and ultimately extracellular ADO that signals via the 4 types of P1 receptor. Together, P2 and P1 receptor signaling and ectonucleotidase activity shapes the hypoxic ventilatory response. How this network is governed and how alteration of its dynamics influence hypoxia and ATP release from injury of damaged/ruptured cells are precise details that have yet to be determined before one can contemplate ATP/ADO signaling-directed therapy to stimulate ventilation (111).
Notably, ADO could stimulate expression of stress and inflammation related genes in folliculostellate cells including thrombomodulin and endothelial protein C (EPCR) that could play a negative-feedback role in controlling and limiting inflammation (112). Yet lung autopsies of COVID-19 casualties revealed a significantly low endothelial cell expression of the thrombomodulin and EPCR anticoagulants and this was associated with increased immune cell infiltration. Lack of SARS-CoV-2 particles in the lung endothelium implied that the dysfunctional endothelium and severe coagulation abnormalities might reflect pathological host responses in COVID-19 lungs rather than a direct infection with SARS-CoV-2 (113).
An observation consistent with the above interpretation was that like other viruses, SARS-CoV-2 induced a primary stress response in infected cells that evoked cellular senescence (114). This irreversible growth arrest of the cell was accompanied by a senescence-associated secretory phenotype (SASP) that could induce secondary senescence of endothelial cells, with an associated infiltration of macrophages and neutrophils resulting in endothelial damage and lung tissue thrombosis (115). Given involvement of the inflammasome in paracrine senescence (116) and purinergic P2Y14 receptor modulation of stress-induced hematopoietic progenitor cell senescence (117, 118), purinergic pathways may present new pharmacological avenues for controlling senescence-mediated morbidities from SARS-CoV-2 infection.
COVID-19 Interactions With the Purine Signalling Pathway
Coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 infection has clinical features that vary widely between individuals from asymptomatic or mild symptoms to severe disease invoking pneumonia, acute respiratory distress syndrome, multiorgan failure and death. The pathobiology following SARS-CoV-2 infection involves several phases of COVID-19 evolution, typically an upper and then lower respiratory involvement, followed by a systemic inflammatory phase that may lack signs of SARS-CoV-2 presence, yet become excessive and cause cell death and tissue damage. Untargeted metabolomics analyses by liquid chromatography quadrupole time-of-flight mass spectrometry (LC/Q-TOF/MS) demonstrated that in contrast to healthy controls, COVID-19 patients bore significant differences in terms of purine, glutamine, leukotriene D4 (LTD4) and glutathione metabolism with evidence for an association between the pathophysiology of COVID-19 and altered purine metabolism (119). Concepts regarding SARS-CoV-2 co-involvement of the purine signalling pathway have been associated with three main phases from virus transmission to systemic hyperinflammation. In an early infection phase within the first 5 days of contracting the virus mild symptoms (fever, cough, fatigue, myalgia, sore throat, loss of sense of smell and headache) may emerge. In the second pulmonary phase, typically after 5-10 days, there can be respiratory failure (shortness of breath, hypoxemia, tachypnea) if this progresses, the third phase can include a hyperinflammatory reaction, acute respiratory distress syndrome, shock and multiorgan failure (120).
Asymptomatic or presymptomatic incubation periods for people testing positive for SARS-CoV-2 have facilitated pandemic spread. As a possible presenting symptom, about 20% of patients have reported loss of smell or taste impairment, with higher prevalence of such anosmia accompanying upper respiratory tract infections. Other viruses including the common cold virus and many rhinoviruses responsible for upper respiratory tract infections can also evoke post-viral anosmia that persists until damaged cells are repaired. The SARS-CoV-2 induced incidence of anosmia and also loss of taste or ageusia is high, without the virus directly invading the olfactory nerves and taste buds. The cells of these tissues lack the primary ACE2 receptor, nevertheless, the virus can target adjacent tissues e.g. ACE2+ nasal sustentacular cells or cells of the human salivary gland. By binding ACE2, the virus prevents the enzyme from converting angiotensin II to angiotensin, causing high angiotensin II levels to accumulate and stimulate NADPH oxidases (NOXes), leading to high levels of Reactive Oxygen Species (ROS) and a cascade of events leading to lysosomal release of ATP by damaged cells (121). The oxidative damage produced by ROS has numerous consequences, including perturbation of the corticosteroid type I mineralocorticoid (aldosterone) receptor (MR) and the enzyme 11ß-hydroxysteroid dehydrogenase (11ßHSD) a cortisol inactivating enzyme, that converts glucocorticoids (cortisol and corticosterone to cortisone and dehydrocorticosterone respectively) preventing them from binding the MR (122). Without functional 11ßHSD, cortisol could persist to activate the unprotected MR, stimulating release of ATP into the basolateral compartment of cells, that can act on purinergic receptors to open Ca2+ channels. The increased intracellular calcium can evoke contraction of the cell and exocytosis of lysosomes containing ATP from the apical surface of the cell resulting in further increased levels of extracellular ATP (123) (Figure 3). Acting as a neuromodulatory substance, ATP activated both P2Y and P2X receptors to markedly reduce the odour responsiveness of olfactory epithelium (124).
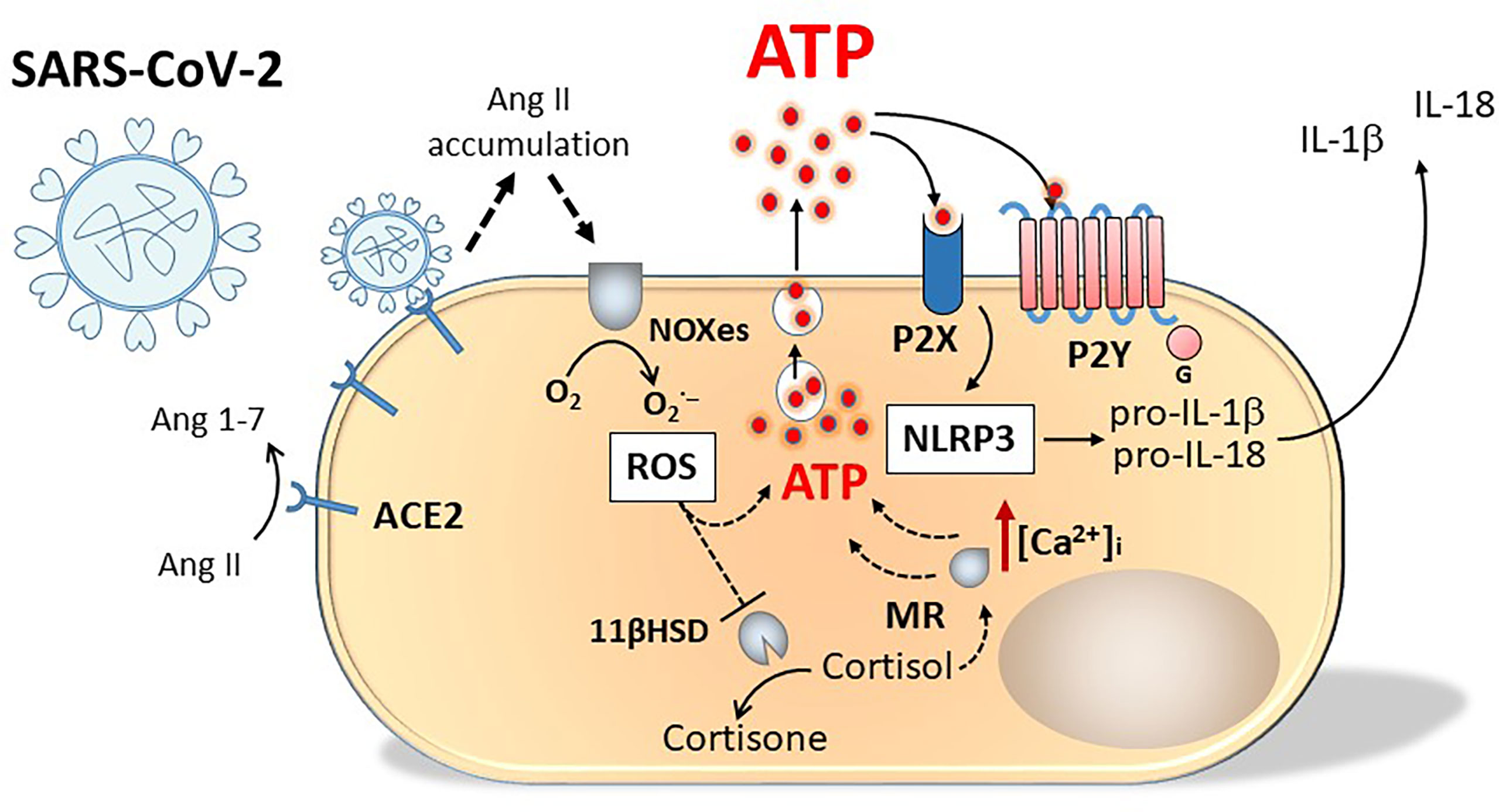
Figure 3 Schematic representation of purinergic signalling involvement in SARS-Cov-2 infection. Engagement of SARS-Cov-2 virus with ACE2 receptors causes angiotensin II accumulation and production of oxygen radicals (ROS) by NADPH oxidases (NOXes). ROS inhibit 11 beta-hydroxysteroid dehydrogenase (11ßHSD), the enzyme transforming cortisol into cortisone. Cortisol accumulation activates unprotected mineralocorticoid (type I) receptor (MR). Both ROS and MR stimulate ATP release from the cell. Activation of P2X and P2Y receptors by ATP increases intracellular Ca2+ concentration [Ca2+]i evoking cell contraction and exocytosis of lysosomes containing ATP from the apical surface of the cell, resulting in a further increase in extracellular ATP concentration and in NALP-3 activation.
The highest levels of ACE2 expression have been found in the nose, with decreasing expression through the lower respiratory tract, a likely explanation for a gradient of SARS-Cov-2 infection that was higher in proximal versus distal pulmonary epithelial cells (125). This agreed with the concept of early infection in the upper respiratory tract (0-5 days) followed by aspiration and infection of the lower lung, whereby there can emerge a more severe pulmonary phase of disease in about 20% of cases according to WHO data. For most cases of non-critically ill classified patients, there was no difference in pulmonary function tests before and after SARS-CoV-2 infection (126). Early use of engineered ACE2 decoys may thus prophylactically mitigate lung disease (127). In contrast, for patients that succumbed to post-acute COVID-19 syndrome (PACS) where breathlessness persists, preliminary indications from novel xenon gas scan Magnetic Resonance Imaging (MRI) methods revealed there can be covert persistent lung damage that significantly impaired gas transfer to the bloodstream (128). Nuclear medicine methods are well suited to studying hyperinflammatory reactions and there are well-established radiopharmaceuticals targeting the purinergic P2X7 receptor (129). Measurement of Immunoglobulin (Ig) signatures based on IgM and IgG3 may provide early indication of a patient’s susceptibility to develop PACS (130).
Regarding acute cases of COVID-19, an induced pluripotent stem cell model of human airway epithelial lineage types indicated that multicilliated cells were primary targets of SARS-Cov-2 infection whereupon they could instigate interferon (IFN) and strong NFκB inflammatory responses, the hallmark of SARS-CoV-2 infection associated with morbidity (131). An initial release of cytokines and recruitment of immune reaction cells, e.g. CD4+ lymphocytes may progress excessively to trigger a so-called “cytokine storm” (132), causing progression from a difficulty in breathing to severe pneumonia, hypoxia, organ failure and risk of death.
Beyond susceptibility to viral infection, a second component for the propensity to develop a qualitatively different critical illness with distinct phenotypes to mild disease, involves activation of the innate immune system by virus infected cells causing macrophage and NK cell migration to the site of infection with release of pro-inflammatory cytokines. After viral replication, cells become lysed, releasing numerous molecules and damage signals including ATP and subsequently large amounts of ADO that in turn can activate purinergic signaling (133). The acute inflammatory response includes blood vessel dilation, increased permeability and leakage of fluid into the alveoli characteristic of pneumonia, that may reflect elevated bradykinin levels (134). This can attract more CD4+ T lymphocytes, compounding further release of pro-inflammatory cytokines, generating a positive feedback hyperinflammatory response. Profiling bronchoalveolar fluid in a comparison between mild or critical COVID-19 patients revealed not only numeric differences in T-cells exhibiting good effector function, but also a discriminatory lower activity of anti-viral type 1 and II IFN signalling pathways in critical COVID-19 cases. Reflecting a responsiveness to IFN signals, macrophages exhibited a pro-homeostatic M2 (anti-inflammatory) phenotype (135) helping to resolve inflammation in mild cases of COVID-19, whereas samples from critical COVID-19 patients had more M1 (proinflammatory) macrophages demonstrating an ATP-purinergic signaling inflammasome and increased levels of ATP in the bronchoalveolar lavage (136).
Extracellular ATP and the purinergic system have a homeostatic role in lung tissue. Alveoli are stabilized during respiration by a reduction in surface tension mediated by a surfactant composed of lipids and specific proteins secreted from the lysosome-related lamellar body organelles of alveolar type II cells. In addition to being important for preventing respiratory distress syndrome, the surfactant contributes to the innate immune system of the lung. The lamellar body membrane expresses a range of P2 purine receptors including the P2X4 receptor (137) with a role in boosting surfactant secretion and fluid clearance during homeostatic responses to ATP controlling local blood flow and surfactant secretion (138). These mechanisms remain also responsive to danger signal rises in ATP levels, triggered by aggravated airflow, changes in mucus hydration and viral infection. Inflammatory pulmonary hypoxia will result in increased extracellular ATP, promptly metabolized into ADP and then AMP by NTPDase that is then hydrolyzed by CD73 to ADO. The effects of ATP, ADP and ADO on tissue remodeling and inflammatory processes can diverge from acute anti-inflammatory and protective effects to chronic pro-inflammatory damage. No specific treatments targeting potentiation of P2X4 receptor have yet been developed, but conceptually they could counteract danger signal rises in ATP by promoting P2X4 receptor mediated Na+ transport into the cell cytoplasm, removing osmotically active Na+ from the alveolar lumen, thereby protecting the alveolus by restoring surface tension.
Intuitively, one might imagine that patients with a pre-existing pulmonary disease may be automatically predisposed to more severe COVID-19 symptoms. However, worldwide data initially reported the phenomenon that cystic fibrosis (CF) patients had a relatively improved Covid-19 survival, even though the defect in cystic fibrosis transmembrane conductance regulator protein (CFTR), impairing chloride and bicarbonate secretion, causes impaired mucociliary clearance and an exaggerated proinflammatory response that could be driven by infection (139). Moreover, the muco-inflammatory environment of CF airways often showed relatively high IL-1ß expression, an inducer of ACE2 expression. This would conceivably allow increased SARS-CoV-2 infection of more distal alveolar epithelial cells. Nonetheless, prior studies for precedent SARS-CoV infection had associated increased ACE2 pathway expression with improved lung function, perhaps by counterbalancing Spike protein-mediated downregulation of ACE2 (140). It has been proposed that the survival benefit demonstrated by homozygous CFTR mutations may involve purinergic pathways, since CF patients may show increased mitochondrial ATP production and elevated systemic levels of ATP (141). However, precise mechanisms were conjectured and more recent multicentric studies have indicated that COVID-19 should not be considered a mild disease in CF patients (142–144).
Cardiovascular disease following SARS-CoV-2 infection may reflect direct or indirect damage as a result of viral infection. The main mechanisms being proposed are inflammatory processes and an exacerbated host immune response involving extensive release of proinflammatory cytokines and chemokines. Myocardial injury increases extracellular ATP and ADO levels that activate different purinergic receptors. Extracellular ATP signals can activate P2X4 on the membrane of mast cells, macrophages and neutrophils that then further promote the inflammatory condition with increases in ATP and further proliferation of immune cells. Activation of T cells by the P2X1 receptor, with signaling of the P2X7 receptors of immune cells and also P2Y1 and P2Y2 in macrophages, can also trigger further release of inflammatory mediators such as IL-1ß, GM-CSF, IL-6, IL-10, INF-γ, γ-chain cytokines, TNF-α and IL-17, thereby amplifying inflammation. Blockade of purinergic receptors and suppression of ATP may be a strategy that helps inhibit purinergic signaling at P2 receptors, with the aim of reducing systemically mediated inflammatory damage to myocardial tissue (145).
Patients progressing to severe COVID-19 pneumonia, can suffer hypoxemia from both inflammation-compromised alveolar function and endothelial dysfunction together with a concerning systemic cytokine storm response ultimately causing micro and macro thromboses in pulmonary vessels and other organs. Colossal platelet activation, following sustained tissue factor (TF) expression in pathologically affected tissues and released procoagulant microvesicles can cause severe coagulopathy (146). The extensive platelet activation in COVID-19 patients was unlikely to reflect a direct effect of the virus, given low SARS-CoV-2 titers in the blood of only a low percentage of infected patients. Rather, numerous host factors as mentioned above, including IL-6 were implicated. IL-6 did not directly activate platelets in vitro, but enhanced the effect of a cytokine storm by potentiating the effect of ADP or thromboxane. Notably, antagonists of platelet P2Y12 receptors, that bound ADP, inhibited thromboxane-dependent pathways of platelet activation to a marked extent, more significantly than aspirin (147). Blockade of the P2Y12 receptor could also partially perturb release of sIL-6R, the specific soluble receptor needed for IL-6 signaling via gp130 (148). Therapeutic approaches addressing platelet alterations during SARS-CoV-2 infection (148) have entered clinical trials with outcomes that may reflect how effectively the IL-6 pathway can be inhibited. The effect of P2Y12 inhibitors on non-critically ill COVID-19 patients did not increase odds of improvement and reduced hospitalization (149), whereas treatment with IL-6 receptor antagonists tocilizumab and sarilumab did improve survival outcomes among critically ill COVID-19 patients (150).
Purinergic Targets for SARS-Cov-2 Induced Pathologies
Natural selection forces favoring evolution of SARS-CoV-2 variants with higher fitness and transmission rates generate variants of the virus with novel properties that may be diversly pathogenic, risking new public health vulnerabilities. With modulation of transmission advantage and immune evasion, five variants of concern (VOCs) have demonstrated these phenotypes to a varying degree, with rapid whole genome sequencing revolutionizing our insights from ongoing surveillance. Beyond the root A lineage defined by the Wuhan/WH04/2020 sequence, the Beta [B.1.351 in Pango nomenclature (151)] and to a lesser extent the Gamma (P.1) variants showed immune evasion in vitro and local geographical spread. More significant VOCs reached a global distribution causing significant waves of population infection with an increase in the effective reproduction number (R naught, the virus’s transmission among a population that has no immunity). A mutation at position 681 within the Spike protein polybasic furin cleavage site, changing to histidine for VOC Alpha (B.1.1.7) and arginine for VOC Delta (B.1.617.2), generated in each case variants that were 50% more transmissible than prior forms. In the latter Delta variant, this alteration caused enhanced fusogenicity and pathogenicity (152). This concerned how upon attaching to the cell surface and binding the ACE2 receptor, SARS-CoV-2 may exploit two different entry pathways to enter the cytosol (1): The early or cell surface pathway involving the virus directly fusing with the plasma membrane via help from the TMPRSS2 surface protease (2); The relatively late virus-triggered endocytic pathway, whereby virus is trafficked to the lysosome/late endosome where proteases such as cathepsin L prime the S protein for membrane fusion. SARS-CoV-2 Alpha, Beta and Delta variants displayed enhanced spike-mediated cell fusion with syncytia formation (153). Interestingly, P2X1-specific antagonistic compounds have implicated a role for this particular purinergic pathway in HIV-1 membrane fusion, although precise mechanisms remain uncertain (154).
Omicron, the fifth World Health Organization (WHO) nominated VOC, has emulated Alpha and Delta to achieve global dominance. The Omicron (B.1.1.529 lineage) first detected in Botswana, South Africa in mid-November 2021, has split into at least three divergent sublineages (BA.1, BA.2 and BA.3) of which BA.1 has spread rapidly around the world (155). The BA.1 Omicron genome encodes an unusual array of mutations, enigmatically evolved outside overt paths of transmission from previous variants, with at least 30 amino acid substitutions in the spike glycoprotein relative to the Wuhan/WH04/2020 sequence. Of these, 15 mutations were found in the receptor-binding domain (RBD) and within its receptor-binding motif (RBM) subdomain that interacts with the human ACE2 receptor, six out of nine mutations enhanced binding affinity. Overall, the Omicron RBD demonstrated over twice the ACE2 binding affinity of the original SARS-CoV-2 virus. However, whilst the Delta variant achieved efficient entry via fusion, Omicron has switched entry route preference, entering predominantly via the second endocytic pathway, reducing dependence on TMPRSS2-mediated activation. This offers a mechanistic explanation for significantly reduced syncytia formation by Omicron infected cells (156–158), yet also emphasizes how a selection pressure to evolve changes in fitness can make SARS-CoV-2 a moving target for treatments that seek to target early stages of virus-host interaction.
Focus on host pathological responses rather than viral infectivity per se is likely to be more useful for implementing therapies that target purinergic signalling involved in potentially more severe chronic aspects of COVID-19 disease (159) in particular post-acute sequelae of COVID-19 (PASC). Quantifiable risk factors for PASC are beginning to be resolved, four that have been determined include type 2 diabetes, RNAemia, Epstein-Barr virus (EBV) viremia and specific autoantibodies (160). The fact that one of the risk factors should be viraemia from EBV suggests that there may be commonalities among responses to virus that impinge upon the pathological potential. EBV reactivated from latency and entering lytic replication could induce ACE2 expression (161) or it may be that both viruses are implicated in compromising mitochondrial health (162). Notably, dissecting the individual roles of the 19 purinergic receptors encompassing the P2Y metabotropic and P2X ionotropic types has revealed close involvement in diabetes and metabolism (163), knockout mice engineered with an adipocyte-specific lack of P2Y6 receptor have a greatly improved glucose homeostasis with improved glucose tolerance and reduced systemic inflammation (164).
A number of different innate immune response cells form a first-line defense against viral infection; macrophages, neutrophils, platelets and mast cells having significant COVID-19 roles in activating the innate immune system that is critical for establishing a subsequent adaptive immune response mediated by numerous T lymphocytes. Principally, these include regulatory T cells (Treg) and T helper cells (Th), CD4-positive cells that release cytokines to “help” the activity of other immune cells. The interleukin twelve (IL-12)-induced Th1 subset generates interferon gamma (IFN-γ) and tumor necrosis factor (TNF-α) whereas the IL-6 driven Th17 cells generate IL-17 and IL-21 (165). Recent studies have shown significant reduction of Tregs in COVID-19 patients, with an imbalance in the Treg/Th17 ratio weakening inflammatory inhibition worsening patient prognosis (166).
The innate immune system is sensitive to pathogen-induced danger-associated molecular patterns (DAMPs) of expression, with responsive pattern-recognition receptors (PRR) including toll-like receptors (TLRs) (167), NOD-like receptors/nucleotide-binding domain and leucine-rich repeat containing receptors (NLRs) including NLR family pyrin domain containing 3 (NLRP3) (168) and retinoic acid inducible gene I (RIG-I)-like receptors (RLR) (169) that can sense viral RNA and collectively serve as components of an inflammasome that detects the pathogen’s presence with activation of an antiviral immune response, including production of type I interferon (170). Like other human respiratory RNA viruses, SARS-CoV-2 proteins can act to suppress innate immune responses (171, 172). The multifunctional nonstructural protein 5 (NSP5) encoded main protease (Mpro), has not only a pivotal role in the replication cycle of all coronaviruses, but acts to evade intracellular innate immunity (173) by targeting RIG-I and mitochondrial antiviral signaling (MAVS) proteins (174). There is evidence that the SARS-CoV-2 envelope (E) protein can have biphasic effects, initially suppressing the host NLRP3 mediated inflammasome activation during the early stages of viral infection helping to protect early viral replication. Yet in the presence of viral RNA derived ssRNA sequences, a non-classical pathway could activate the NLRP3 in human macrophages (175). Although E protein potentiation of this NLRP3 inflammasome activation should serve to advantageously limit viral replication and stimulate an adaptive immune response, it may aberrantly exacerbate inflammasome activation, inducing an autoinflammatory disease and tissue damage (176, 177). Hence, it has been hypothesized that overactivation of NLRP3 inflammasome by SARS-CoV-2 infection would lead to excessive production of master inflammatory cytokines such as IL-1β and IL-18 (178) and could also damage hematopoietic stem/progenitor cells (179).
A number of purinergic receptors are integral to a macrophage surveillance function sensitive to even slight changes in the tissue microenvironment. Of these, P2X7 receptors have a relatively low affinity to ATP, yet respond to high concentrations of extracellular ATP under pathological conditions and then again to still higher concentrations of ATP, when a threshold of repetitive agonist application causes P2X7 receptors to further interact with a range of other membrane channels (e.g. P2X4, transient receptor potential A1 (TRPA1), pannexin-1, ANO6 chloride channel), making membrane permeability increase further (180). In particular, the macrophage P2X7 receptor is coactivated with the lipopolysaccharide-sensitive toll-like receptor 4 (TLR4) to induce the formation of the inflammasome 3 (NLPR3) that then activates pro-interleukin 1ß (pro-IL-1ß) that can degrade caspase-1, leading to IL-1ß release. Concurrently, P2X7 receptor activation induced downstream upregulation of second messengers (e.g. phospholipase A2, p38 mitogen-activated kinase (p38MAPK) can activate reactive oxygen and nitrogen species and rho G proteins). Notably, elevated Rho GTPase and mTOR pathway activities were among features characterizing responses to SARS-CoV-2 that were distinct from those to HIV-1 infections (181). Data from corneal allograft rejection studies demonstrated that blockage of the ligand-gated ion channel P2X7 receptor suppressed Th1/Th17-mediated immune responses and NLRP3 inflammasome activation (182).
In addition, macrophage P2Y family receptors (135) consisting of seven hydrophobic transmembrane domains serve to bind nucleotides or nucleotide sugars at three extracellular loops whilst intracellular regions mediate G protein activation. Under stressful conditions, P2Y receptors also form homodimers and heterotrimers to diversify the biochemical spectrum of activities in response to released nucleotide ligands modulated by ectonucleotidases e.g. CD39 and CD73. COVID-19 patients have been shown to have relatively low CD73 levels on cytotoxic lymphocyte populations including CD8+T, natural killer T (NKT) and natural killer (NK) cells (183) and several such abnormal phenotypes may contribute to an uncoupling between the innate and adaptive immune system in patients experiencing progressive COVID-19 (184).
Neutrophils recruited by activated macrophages are the most abundant immune cells in the circulation. Prompted through TLR activation they release neutrophil extracellular traps (NETs) comprising a mesh of DNA-based cytoplasmic material containing antimicrobial elements such as granule proteins, cathepsins, neutrophil elastase and myeloperoxidase together with cytoskeletal proteins; a process finely regulated by purinergic signaling (185). Viruses e.g. respiratory syncytial virus (RSV) and influenza are known to trigger NET formation and with the requisites of neutrophil ACE2 receptor, TMPRSS2 activity and viral replication, SARS-CoV-2 has been shown to directly stimulate expressing neutrophils to release NETs (186–189) making inhibition of NET formation a potential therapeutic target for COVID-19 treatment (190, 191). At the same time neutrophils are actively involved in triggering the resolution phase of an inflammatory response. Activation of specific adenosine receptors can suppress NET formation via cyclic AMP dependent signaling and the FDA-approved drug Dipyrimadole can potentiate anti-inflammatory adenosine receptor signaling by inhibiting ectonucleoside reuptake and stabilizing intracellular cyclic AMP, with prevention of NET-dependent thrombosis. Additional antiviral and anti-oxidant activities may also contribute to promising initial clinical trials that suggest dipyrimadole may be of benefit for critically ill COVID-19 patients, warranting larger randomized controlled trials (192).
Platelets have important roles to play in the innate immune system and can directly contribute to NET generation when stimulated by viral activation, the interaction of platelets and neutrophils likely to be a relevant factor for thrombogenesis (193). Multiple mechanisms involving a global inflammatory reaction in COVID-19 patients can serve to activate platelets, be it diseased endothelium, increased viscosity or viral infection, considered plausible, despite debate regarding whether the platelet ACE2 expression demonstrated in murine platelets in vitro (194) is consistent for humans in vivo (195). Extrinsic coagulation pathways ultimately lead to the formation of thrombin, a proteinase that initiates platelet activation, aggregation and secretion of proinflammatory and signalling molecules including thromboxane A2. Ordinarily these responses provide controlled hemostasis, however SARS-CoV-2 infection was associated with alterations in platelets (148) resulting in enhanced inflammatory tissue factor (TF) expression and an atypical amount of platelet-leukocyte rather than platelet-platelet aggregates. This was not unique to SARS-CoV-2, since increased platelet-leukocyte interactions have been reported in critically ill patients succumbed to influenza A (H1N1) virus infections (196). Platelet P2Y1 receptor signaling via RhoGTPase was found to be necessary for platelet dependent leukocyte recruitment (197), moreover the study highlighted subtleties and complexities concerning purinergic signaling in platelets according to their contextual role. Platelet functional roles bifurcate between hemostasis (aggregation to stop bleeding) or host defense mechanisms against pathogens (platelet motility and induction of neutrophil chemotaxis). Different orthosteric site molecular docking interactions of P2Y1 receptor agonists, e.g. ADP or the P2Y1 specific agonist MRS2365 could affect the selection of downstream signaling events; MRS2365 induced platelet chemotaxis more effectively than ADP. Modulation of P2Y1 receptor signalling may be distinct according to the context, whether the platelets are functioning for homeostasis or within an inflammatory microenvironment with accordingly biased responses.
In vitro evidence indicated that a P2Y12 inhibitors e.g. clopidogrel or prasugrel could enhance the effect of aspirin to inhibit platelet activation (198) including platelet activation induced by plasma from COVID-19 patients an activity attributed to P2Y12 inhibitors not only blocking ADP, but by also impairing IL-6-mediated platelet activation (199). The different P2Y12 inhibitors showed different clinical efficacy, trial data suggested that prasugrel and ticagrelor were more effective than clopidogrel for cardiac therapy (200). Taking into consideration unique characteristics, there may be preferences for use of a particular P2Y12 inhibitor depending on the circumstances, safety indications, contraindications, cost, efficacy and dose. Routes of administration may assist application of purinergic signalling targeted therapies. For example, when the half-maximal effective concentration of the P2X7 receptor antagonist lidocaine needed to block hyperinflammation was excessive, adverse reactions were minimized by selectively inhibiting P2X7 receptors of the immune cells of the lymphatic system, by inducing clonal expansion of Tregs in local lymph nodes that subsequently migrated to impart systemic anti-inflammatory activity, notably improving outcomes in six critically ill COVID-19 patients (201).
Mast cells (MC), present in lung mucosa have been commonly associated with diseases involving allergic reactions e.g. asthma, yet could also recognize viruses by diverse mechanisms, e.g. TLR3 detection of viral RNA or RIG-I recognition of uncapped viral RNA. Notably, MC also express ACE2 receptors and serine protease tryp2, qualifying a route to host SARS-CoV-2. There is growing evidence that mast cell activation may have an important role in COVID-19 hyperinflammation (202). SARS-CoV-2 activation has been shown to release histamine stored endogenously in MC granules, increasing release of the chemokines IL-8, IL-6. Alveolar macrophages bearing SARS-CoV-2 activated TLR could produce the pleiotropic inflammatory cytokine IL-1, that can stimulated MC IL-6 production and in combination these cytokines contributed to hyperinflammation (203). Purinergic signaling has been strongly implicated in mast cell degranulation. Extracellular ATP activated ionotropic P2X4 receptors and enhanced antigen-triggered degranulation potentiating a high affinity IgE receptor (FcεRI)-mediated tyrosine kinase signaling cascade. Among multiple P2Y receptor subtypes, the P2Y11 receptor was involved in degranulation enhancement via the PI3K/protein kinase B pathway (204). Studies in rhesus macaques, that presented authentic mild to moderate forms of COVID-19 (205), have shown that SARS-CoV-2 triggered mast cell degranulation induced acute alveolar epithelial inflammation and lung injury and that prophylactic administration of MC stabilizers that blocked degranulation could dampen the SARS-CoV-2 response and prevent lung injury (206). Mast cell activation has also been associated with the unclear aetiologically of long COVID or PASC (207) and the repurposing of drugs addressing mast cell activation syndrome may help patients undergoing PASC rehabilitation training (208).
Of concern, lingering neurological symptoms, including impaired information processing, memory and concentration (aka “COVID fog”) are likely to reflect neuroinflammatory mechanisms involving brain-resident macrophages or microglia that can be induced even after relatively mild COVID-19 symptoms (209). Such persistent neurological complications may also follow infection with Ebola, Zika and influenza A viruses, though it remains unclear whether the infectious virus needs to persist in the affected tissue or whether infection alters the tissue to evade an innate immune response, with altered immune processes persisting to invoke impaired function of mitochondria and microglia causing neuroinflammation. Given their strategic role, it is not surprising that viruses should target mitochondria, the key organelles that govern cellular homeostasis, metabolism, innate-immune signalling, aging, apoptosis and other signalling pathways. Either targeted directly by viral proteins or influenced by changes in the cellular microenvironment during viral pathogenesis, the mitochondria constitute a dynamic population of organelles that continuously elongate (by fusion), divide (by fission) and undergo a controlled turnover with elimination by the process of mitophagy (210). Several viruses modulate mitochondrial functions that act as a central hub for the innate immune system. Under excessive stress the mitochondrial inner membrane can become depolarized releasing mitochondrial unique components including mtDNA, cardiolipin, N-formyl-peptides and mitochondrial reactive oxygen species (mROS) that constitute mitochondria-derived DAMPs (mDAMPs). These can bind to and activate the NLRP3 inflammasome that is colocalized with mitochondria via mitochondrial outer membrane proteins; MAVS and mitofusion 2 (MFN2). Together with the involvement of other interconnected organelles, mitochondria activated the cyclic guanosine monophosphate synthetase (cGAS)-stimulator of interferon genes (STING) signaling pathway, leading to the production of host defense type I IFNs and pro-inflammatory cytokines (211). Therefore, beyond producing the ATP that was released and necessary for driving systemic inflammation (39), mitochondria interacted with P2X1, P2X4 and P2Y11 receptors to regulate T cell metabolism (212), migration (213) and activation (214); emphasizing the important role of purinergic signalling pathways in persistent neuroinflammation (215).
Discussion
There is growing appreciation that targeting purinergic receptors may bring benefit to treat SARS-CoV-2 induced processes associated with the pathologies of COVID-19 (216–222). Accumulated clues to the puzzle of SARS-CoV-2 have suggested that when a virus can initiate platelet and neutrophil driven exacerbated macrophage inflammatory circuits, this can spell a pandemic. The molecules of the purinergic signalling pathway receptosome are directly implicated as key mediators for the interactions between cells involved in these principle pathological processes of concern (223–226). Despite available vaccines and emergency use antibodies (EUA), a pharmacological prevention and treatment therapy of COVID-19 is still urgently needed, especially for PACS sequelae. Observations regarding viral infections demonstrated that P2X1 receptor inhibitors e.g. NF279 or NF449 may block HIV-1 productive infection and associated inflammation processes in cultured cells (227), or that absence of the P2X7 receptor in mouse models showed a better outcome to influenza virus infection (228). Such outcomes have yet to be translated into treatment for viral comorbidities in the clinic. Nevertheless, drugs targeting purinergic signaling affected processes are being repurposed with some success against acute COVID-19. Tranilast, an old anti-allergy clinical drug is a direct NLRP3 inhibitor that has entered clinical trials for the treatment of COVID-19 patients (229). Ticagrelor, an orally administered selective reversible P2Y12 receptor antagonist, preventing P2Y12 and ADP mediated platelet activation and aggregation has been approved for prevention of severe cardiovascular events and may be suited for sepsis-induced coagulopathy in COVID-19 (230) especially since in silico analysis suggests ticagrelor also bound the main protease and spike proteins of SARS-CoV-2 (231). Preliminary trials of prehospital antiplatelet therapy in patients with COVID-19 was associated with modest yet significantly lower in-hospital mortality (232). A number of new compounds targeting P2X and P2Y receptors are currently under development. Of these, Cangrelor an anti-platelet nucleotide analogue drug, derived from ATP, capable of acting as a high-potency competitive analogue for most P2Y receptor subtypes (233), also scored highly in computer models seeking potential candidates to inhibit the desirable target Mpro (234). Similarly, Entecavir Hydrate, a guanosine analogue possessing anti-HBV activity was a P2X7 antagonist that could ameliorate platelet activation and thrombus formation (235). It has recently been identified among nucleoside and nucleotide analogues as a potential novel SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) inhibitor (236). It is possible to envisage that agents modulating purinergic signaling may be advantageously capable of a dual role against both SARS-CoV-2 replication and hyperinflammatory COVID-19 symptoms. The complex context-dependent nature of purinergic signaling makes it a difficult yet potentially versatile therapeutic target, therefore, further pre-clinical and clinical studies are needed to confirm preliminary observations that support modulation of SARS-CoV-2 mediated responses by purinergic signaling. Intense global research seeking to limit and effectively control the SARS-CoV-2 pandemic has accelerated new technologies that can greatly improve our understanding of purinergic signaling and the enormous shared scientific progress already demonstrated by an ability to provide a vaccine defense provides cause for optimism concerning ever-improved adjunct pharmacological interventions.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
Local funds of the University of Ferrara.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Burnstock G. Introduction to Purinergic Signaling. Methods Mol Biol (2020) 2041:1–15. doi: 10.1007/978-1-4939-9717-6_1
2. Murray JM, Bussiere DE. Targeting the Purinome. Methods Mol Biol (2009) 575:47–92. doi: 10.1007/978-1-60761-274-2_3
3. Volonté C, D’Ambrosi N. Membrane Compartments and Purinergic Signalling: The Purinome, a Complex Interplay Among Ligands, Degrading Enzymes, Receptors and Transporters. FEBS J (2009) 276:318–29. doi: 10.1111/j.1742-4658.2008.06793.x
4. Volonté C, Amadio S, D’Ambrosi N, Colpi M, Burnstock G. P2 Receptor Web: Complexity and Fine-Tuning. Pharmacol Ther (2006) 112:264–80. doi: 10.1016/j.pharmthera.2005.04.012
5. Strachan RT, Ferrara G, Roth BL. Screening the Receptorome: An Efficient Approach for Drug Discovery and Target Validation. Drug DiscovToday (2006) 11:708–16. doi: 10.1016/j.drudis.2006.06.012
6. Haystead J, Timothy A. The Purinome, a Complex Mix of Drug and Toxicity Targets. Curr Topics Med Chem (2006) 6:1117–27. doi: 10.2174/156802606777812059
7. Felder ER, Badari A, Disingrini T, Mantegani S, Orrenius C, Avanzi N, et al. The Generation of Purinome-Targeted Libraries as a Means to Diversify ATP-Mimetic Chemical Classes for Lead Finding. Mol Divers (2012) 16:27–51. doi: 10.1007/s11030-012-9361-6
8. Yamaki H, Suzuki H, Choi E, Tanaka N. Inhibition of DNA Synthesis in Murine Tumor Cells by Geldanamycin, an Antibiotic of the Benzoquinoid Ansamycin Group. J Antibiot (1982) 35:886–92. doi: 10.7164/antibiotics.35.886
9. Beck S, Zhu Z, Oliveira MF, Smith DM, Rich JN, Bernatchez JA, et al. Mechanism of Action of Methotrexate Against Zika Virus. Viruses (2019) 11:E338. doi: 10.3390/v11040338
10. Ezhilarasan D. Hepatotoxic Potentials of Methotrexate: Understanding the Possible Toxicological Molecular Mechanisms. Toxicology (2021) 458:152840. doi: 10.1016/j.tox.2021.152840
11. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly Accurate Protein Structure Prediction With AlphaFold. Nature (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
12. Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science (2021) 373:871–6. doi: 10.1101/2020.07.22.211482
13. Saladino R, Carota E, Botta G, Kapralov M, Timoshenko GN, Rozanov AY, et al. Meteorite-Catalyzed Syntheses of Nucleosides and of Other Prebiotic Compounds From Formamide Under Proton Irradiation. Proc Natl Acad Sci USA (2015) 112:E2746–55. doi: 10.1073/pnas.1422225112
14. Prosdocimi F, de Farias ST. Life and Living Beings Under the Perspective of Organic Macrocodes. Biosystems (2021) 206:104445. doi: 10.1016/j.biosystems.2021.104445
15. Krupovic M, Dolja VV, Koonin EV. The LUCA and Its Complex Virome. Nat Rev Microbiol (2020) 18:661–70. doi: 10.1038/s41579-020-0408-x
16. Lee BD, Koonin EV. Viroids and Viroid-Like Circular RNAs: Do They Descend From Primordial Replicators. Life (Basel) (2022) 12:103. doi: 10.3390/life12010103
17. Moniruzzaman M, Weinheimer AR, Martinez-Gutierrez CA, Aylward FO. Widespread Endogenization of Giant Viruses Shapes Genomes of Green Algae. Nature (2020) 588:141–5. doi: 10.1038/s41586-020-2924-2
18. Mazzarino RC. Targeting Future Pandemics, a Case for De Novo Purine Synthesis and Basic Research. Front Immunol (2021) 12:694300. doi: 10.3389/fimmu.2021.694300
19. Paoletti A, Raza SQ, Voisin L, Law F, Pipoli da Fonseca J, Caillet M, et al. Multifaceted Roles of Purinergic Receptors in Viral Infection. Microbes Infect (2012) 14:1278–83. doi: 10.1016/j.micinf.2012.05.010
20. Ning S, Yu B, Wang Y, Wang F. SARS-CoV-2: Origin, Evolution, and Targeting Inhibition. Front Cell Infect Microbiol (2021) 11:676451. doi: 10.3389/fcimb.2021.676451
21. Desdín-Micó G, Soto-Heredero G, Mittelbrunn M. Mitochondrial Activity in T Cells. Mitochondrion (2018) 41:51–7. doi: 10.1016/j.mito.2017.10.006
22. Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP Release by Inflammatory Cells. Int J Mol Sci (2018) 19:E1222. doi: 10.3390/ijms19041222
23. Burnstock G. Historical Review: ATP as a Neurotransmitter. Trends Pharmacol Sci (2006) 27:166–76. doi: 10.1016/j.tips.2006.01.005
24. Magni G, Ceruti S. The Role of Adenosine and P2Y Receptors Expressed by Multiple Cell Types in Pain Transmission. Brain Res Bull (2019) 151:132–43. doi: 10.1016/j.brainresbull.2019.02.011
25. Zhou Z, Matsumoto T, Jankowski V, Pernow J, Mustafa SJ, Duncker DJ, et al. Uridine Adenosine Tetraphosphate and Purinergic Signaling in Cardiovascular System: An Update. Pharmacol Res (2019) 141:32–45. doi: 10.1016/j.phrs.2018.12.009
26. Spek A, Li B, Rutz B, Ciotkowska A, Huang R, Liu Y, et al. Purinergic Smooth Muscle Contractions in the Human Prostate: Estimation of Relevance and Characterization of Different Agonists. Naunyn Schmiedebergs Arch Pharmacol (2021) 394:1113–31. doi: 10.1007/s00210-020-02044-4
27. Tanaka N, Nejime N, Kagota S, Kubota Y, Yudo K, Nakamura K, et al. ATP Participates in the Regulation of Microvessel Permeability. J Pharm Pharmacol (2006) 58:481–7. doi: 10.1211/jpp.58.4.0007
28. Coutinho-Silva R, Savio LEB. Purinergic Signalling in Host Innate Immune Defence Against Intracellular Pathogens. Biochem Pharmacol (2021) 187:114405. doi: 10.1016/j.bcp.2021.114405
29. Idzko M, Ferrari D, Eltzschig HK. Nucleotide Signalling During Inflammation. Nature (2014) 509:310–7. doi: 10.1038/nature13085
30. Idzko M, Ferrari D, Riegel AK, Eltzschig HK. Extracellular Nucleotide and Nucleoside Signaling in Vascular and Blood Disease. Blood (2014) 124:1029–37. doi: 10.1182/blood-2013-09-402560
31. Ferrari D, Gambari R, Idzko M, Müller T, Albanesi C, Pastore S, et al. Purinergic Signaling in Scarring. FASEB J (2016) 30:3–12. doi: 10.1096/fj.15-274563
32. Jacobson KA, Costanzi S, Kim SK, Roh E, Joshi BV, Tchilibon S, et al. Action of Nucleosides and Nucleotides at 7 Transmembrane-Spanning Receptors. Nucleosides Nucleotides Nucleic Acids (2006) 25:1425–36. doi: 10.1080/15257770600919027
33. Burnstock G, Knight GE. Cellular Distribution and Functions of P2 Receptor Subtypes in Different Systems. Int Rev Cytol (2004) 240:31–304. doi: 10.1016/S0074-7696(04)40002-3
34. Yegutkin GG. Enzymes Involved in Metabolism of Extracellular Nucleotides and Nucleosides: Functional Implications and Measurement of Activities. Crit Rev Biochem Mol Biol (2014) 49:473–97. doi: 10.3109/10409238.2014.953627
35. Akkaya C, Shumilina E, Bobballa D, Brand VB, Mahmud H, Lang F, et al. The Plasmodium Falciparum-Induced Anion Channel of Human Erythrocytes Is an ATP-Release Pathway. Pflugers Arch (2009) 457:1035–47. doi: 10.1007/s00424-008-0572-8
36. Di Virgilio F, Sarti AC, Coutinho-Silva R. Purinergic Signaling, DAMPs, and Inflammation. Am J Physiol Cell Physiol (2020) 318:C832–5. doi: 10.1152/ajpcell.00053.2020
37. Pittman K, Kubes P. Damage-Associated Molecular Patterns Control Neutrophil Recruitment. J Innate Immun (2013) 5:315–23. doi: 10.1159/000347132
38. Wang J, Takemura N, Saitoh T. Macrophage Response Driven by Extracellular ATP. Biol Pharm Bull (2021) 44:599–604. doi: 10.1248/bpb.b20-00831
39. Cauwels A, Rogge E, Vandendriessche B, Shiva S, Brouckaert P. Extracellular ATP Drives Systemic Inflammation, Tissue Damage and Mortality. Cell Death Dis (2014) 5:e1102. doi: 10.1038/cddis.2014.70
40. Savio LEB, Coutinho-Silva R. Immunomodulatory Effects of P2X7 Receptor in Intracellular Parasite Infections. Curr Opin Pharmacol (2019) 47:53–8. doi: 10.1016/j.coph.2019.02.005
41. Kolli BK, Kostal J, Zaborina O, Chakrabarty AM, Chang KP. Leishmania-Released Nucleoside Diphosphate Kinase Prevents ATP-Mediated Cytolysis of Macrophages. Mol Biochem Parasitol (2008) 158:163–75. doi: 10.1016/j.molbiopara.2007.12.010
42. Kas-Deelen AM, Bakker WW, Olinga P, Visser J, de Maar EF, van Son WJ, et al. Cytomegalovirus Infection Increases the Expression and Activity of Ecto-ATPase (CD39) and Ecto-5’nucleotidase (CD73) on Endothelial Cells. FEBS Lett (2001) 491:21–5. doi: 10.1016/s0014-5793(01)02085-3
43. Sison SL, O’Brien BS, Johnson AJ, Seminary ER, Terhune SS, Ebert AD. Human Cytomegalovirus Disruption of Calcium Signaling in Neural Progenitor Cells and Organoids. J Virol (2019) 93:e00954–19. doi: 10.1128/JVI.00954-19
44. Alves VS, Leite-Aguiar R, Silva JPD, Coutinho-Silva R, Savio LEB. Purinergic Signaling in Infectious Diseases of the Central Nervous System. Brain Behav Immun (2020) 89:480–90. doi: 10.1016/j.bbi.2020.07.026
45. Séror C, Melki MT, Subra F, Raza SQ, Bras M, Saïdi H, et al. Extracellular ATP Acts on P2Y2 Purinergic Receptors to Facilitate HIV-1 Infection. J Exp Med (2011) 208:1823–34. doi: 10.1084/jem.20101805
46. Hazleton JE, Berman JW, Eugenin EA. Purinergic Receptors Are Required for HIV-1 Infection of Primary Human Macrophages. J Immunol (2012) 188:4488–95. doi: 10.4049/jimmunol.1102482
47. Graziano F, Vicenzi E, Poli G. The ATP/P2X7 Axis in Human Immunodeficiency Virus Infection of Macrophages. Curr Opin Pharmacol (2019) 47:46–52. doi: 10.1016/j.coph.2019.02.006
48. Zhang C, He H, Wang L, Zhang N, Huang H, Xiong Q, et al. Virus-Triggered ATP Release Limits Viral Replication Through Facilitating IFN-β Production in a P2X7-Dependent Manner. J Immunol (2017) 199:1372–81. doi: 10.4049/jimmunol.1700187
49. Li R, Tan B, Yan Y, Ma X, Zhang N, Zhang Z, et al. Extracellular UDP and P2Y6 Function as a Danger Signal to Protect Mice From Vesicular Stomatitis Virus Infection Through an Increase in IFN-β Production. J Immunol (2014) 193:4515–26. doi: 10.4049/jimmunol.1301930
50. Jean Beltran PM, Cristea IM. The Life Cycle and Pathogenesis of Human Cytomegalovirus Infection: Lessons From Proteomics. Expert Rev Proteomics (2014) 11:697–711. doi: 10.1586/14789450.2014.971116
51. Davis IC, Sullender WM, Hickman-Davis JM, Lindsey JR, Matalon S. Nucleotide-Mediated Inhibition of Alveolar Fluid Clearance in BALB/c Mice After Respiratory Syncytial Virus Infection. Am J Physiol Lung Cell Mol Physiol (2004) 286:L112–20. doi: 10.1152/ajplung.00218.2003
52. Aeffner F, Woods PS, Davis IC. Activation of A1-Adenosine Receptors Promotes Leukocyte Recruitment to the Lung and Attenuates Acute Lung Injury in Mice Infected With Influenza A/WSN/33 (H1N1) Virus. J Virol (2014) 88:10214–27. doi: 10.1128/JVI.01068-14
53. Koeppen M, Di Virgilio F, Clambey ET, Eltzschig HK. Purinergic Regulation of Airway Inflammation. Subcell Biochem (2011) 55:159–93. doi: 10.1007/978-94-007-1217-1_7
54. Aeffner F, Woods PS, Davis IC. Ecto-5’-Nucleotidase CD73 Modulates the Innate Immune Response to Influenza Infection But Is Not Required for Development of Influenza-Induced Acute Lung Injury. Am J Physiol Lung Cell Mol Physiol (2015) 309:L1313–22. doi: 10.1152/ajplung.00130.2015
55. Okada SF, Zhang L, Kreda SM, Abdullah LH, Davis CW, Pickles RJ, et al. Coupled Nucleotide and Mucin Hypersecretion From Goblet-Cell Metaplastic Human Airway Epithelium. Am J Respir Cell Mol Biol (2011) 45:253–60. doi: 10.1165/rcmb.2010-0253OC
56. Zandberg M, van Son WJ, Harmsen MC, Bakker WW. Infection of Human Endothelium In Vitro by Cytomegalovirus Causes Enhanced Expression of Purinergic Receptors: A Potential Virus Escape Mechanism. Transplantation (2007) 84:1343–7. doi: 10.1097/01.tp.0000287598.25493.a5
57. Pacheco PA, Faria RX, Ferreira LG, Paixão IC. Putative Roles of Purinergic Signaling in Human Immunodeficiency Virus-1 Infection. Biol Direct (2014) 9:21. doi: 10.1186/1745-6150-9-21
58. Levy DE, Marié IJ, Durbin JE. Induction and Function of Type I and III Interferon in Response to Viral Infection. Curr Opin Virol (2011) 1:476–86. doi: 10.1016/j.coviro.2011.11.001
59. Tsai CY, Liong KH, Gunalan MG, Li N, Lim DS, Fisher DA, et al. Type I IFNs and IL-18 Regulate the Antiviral Response of Primary Human γδ T Cells Against Dendritic Cells Infected With Dengue Virus. J Immunol (2015) 194:3890–900. doi: 10.4049/jimmunol.1303343
60. Zhang X, Qin J, Zou J, Lv Z, Tan B, Shi J, et al. Extracellular ADP Facilitates Monocyte Recruitment in Bacterial Infection via ERK Signaling. Cell Mol Immunol (2018) 15:58–73. doi: 10.1038/cmi.2016.56
61. Matthay MA, Wiener-Kronish JP. Intact Epithelial Barrier Function Is Critical for the Resolution of Alveolar Edema in Humans. Am Rev Respir Dis (1990) 142:1250–7. doi: 10.1164/ajrccm/142.6_Pt_1.1250
62. Corrêa G, de A Lindenberg C, Fernandes-Santos C, Gandini M, Petitinga Paiva F, Coutinho-Silva R, et al. The Purinergic Receptor P2X7 Role in Control of Dengue Virus-2 Infection and Cytokine/Chemokine Production in Infected Human Monocytes. Immunobiology (2016) 221:794–802. doi: 10.1016/j.imbio.2016.02.003
63. Ranjit S, Kissoon N. Dengue Hemorrhagic Fever and Shock Syndromes. Pediatr Crit Care Med (2011) 12:90–100. doi: 10.1097/PCC.0b013e3181e911a7
64. Lee BH, Hwang DM, Palaniyar N, Grinstein S, Philpott DJ, Hu J. Activation of P2X(7) Receptor by ATP Plays an Important Role in Regulating Inflammatory Responses During Acute Viral Infection. PloS One (2012) 7:e35812. doi: 10.1371/journal.pone.0035812
65. Manzoor S, Akhtar U, Naseem S, Khalid M, Mazhar M, Parvaiz F, et al. Ionotropic Purinergic Receptors P2X4 and P2X7: Proviral or Antiviral? An Insight Into P2X Receptor Signaling and Hepatitis C Virus Infection. Viral Immunol (2016) 29:401–8. doi: 10.1089/vim.2016.0008
66. Manzoor S, Idrees M, Ashraf J, Mehmood A, Butt S, Fatima K, et al. Identification of Ionotrophic Purinergic Receptors in Huh-7 Cells and Their Response Towards Structural Proteins of HCV Genotype 3a. Virol J (2011) 8:431. doi: 10.1186/1743-422X-8-431
67. Lee DH, Park KS, Kong ID, Kim JW, Han BG. Expression of P2 Receptors in Human B Cells and Epstein-Barr Virus-Transformed Lymphoblastoid Cell Lines. BMC Immunol (2006) 7:22. doi: 10.1186/1471-2172-7-22
68. Taylor JM, Han Z. Purinergic Receptor Functionality Is Necessary for Infection of Human Hepatocytes by Hepatitis Delta Virus and Hepatitis B Virus. PloS One (2010) 5:e15784. doi: 10.1371/journal.pone.0015784
69. Chen S, Shenk T, Nogalski MT. P2Y2 Purinergic Receptor Modulates Virus Yield, Calcium Homeostasis, and Cell Motility in Human Cytomegalovirus-Infected Cells. Proc Natl Acad Sci USA (2019) 116:18971–82. doi: 10.1073/pnas.1907562116
70. Swartz TH, Esposito AM, Durham ND, Hartmann BM, Chen BK. P2X-Selective Purinergic Antagonists Are Strong Inhibitors of HIV-1 Fusion During Both Cell-to-Cell and Cell-Free Infection. J Virol (2014) 88:11504–15. doi: 10.1128/JVI.01158-14
71. Passos DF, Schetinger MR, Leal DB. Purinergic Signaling and Human Immunodeficiency Virus/Acquired Immune Deficiency Syndrome: From Viral Entry to Therapy. World J Virol (2015) 4:285–94. doi: 10.5501/wjv.v4.i3.285
72. Graziano F, Desdouits M, Garzetti L, Podini P, Alfano M, Rubartelli A, et al. Extracellular ATP Induces the Rapid Release of HIV-1 From Virus Containing Compartments of Human Macrophages. Proc Natl Acad Sci USA (2015) 112:E3265–73. doi: 10.1073/pnas.1500656112
73. Tewari M, Monika, Varghse RK, Menon M, Seth P. Astrocytes Mediate HIV-1 Tat-Induced Neuronal Damage via Ligand-Gated Ion Channel P2X7R. J Neurochem (2015) 132:464–76. doi: 10.1111/jnc.12953
74. Schachter J, Delgado KV, Barreto-de-Souza V, Bou-Habib DC, Persechini PM, Meyer-Fernandes JR. Inhibition of Ecto-ATPase Activities Impairs HIV-1 Infection of Macrophages. Immunobiology (2015) 220:589–96. doi: 10.1016/j.imbio.2014.12.004
75. Schachter J, Valadao AL, Aguiar RS, Barreto-de-Souza V, Rossi AD, Arantes PR, et al. 2´,3´-Dialdehyde of ATP, ADP, and Adenosine Inhibit HIV-1 Reverse Transcriptase and HIV-1 Replication. Curr HIV Res (2014) 12:347–58. doi: 10.2174/1570162x12666140826105824
76. Barat C, Gilbert C, Imbeault M, Tremblay MJ. Extracellular ATP Reduces HIV-1 Transfer From Immature Dendritic Cells to CD4+ T Lymphocytes. Retrovirology (2008) 5:30. doi: 10.1186/1742-4690-5-30
77. Jenabian MA, Seddiki N, Yatim A, Carriere M, Hulin A, Younas M, et al. Regulatory T Cells Negatively Affect IL-2 Production of Effector T Cells Through CD39/adenosine Pathway in HIV Infection. PloS Pathog (2013) 9:e1003319. doi: 10.1371/journal.ppat.1003319
78. Nikolova M, Carriere M, Jenabian MA, Limou S, Younas M, Kök A, et al. CD39/adenosine Pathway Is Involved in AIDS Progression. PloS Pathog (2011) 7:e1002110. doi: 10.1371/journal.ppat.1002110
79. Palella FJ, Phair JP. Cardiovascular Disease in HIV Infection. Curr Opin HIV AIDS (2011) 6:266–71. doi: 10.1097/COH.0b013e328347876c
80. Rezer JF, Souza VC, Thorstenberg ML, Ruchel JB, Bertoldo TM, Zanini D, et al. Effect of Antiretroviral Therapy in Thromboregulation Through the Hydrolysis of Adenine Nucleotides in Platelets of HIV Patients. BioMed Pharmacother (2016) 79:321–8. doi: 10.1016/j.biopha.2016.02.008
81. Cheng YR, Li X, Zhao X, Lin H. Cell Entry of Animal Coronaviruses. Viruses (2021) 13:1977. doi: 10.3390/v13101977
82. Starr TN, Zepeda SK, Walls AC, Greaney AJ, Alkhovsky S, Veesler D, et al. ACE2 Binding Is an Ancestral and Evolvable Trait of Sarbecoviruses. Nature (2022) 603:913–98. doi: 10.1038/s41586-022-04464-z
83. Oz M, Lorke DE. Multifunctional Angiotensin Converting Enzyme 2, the SARS-CoV-2 Entry Receptor, and Critical Appraisal of Its Role in Acute Lung Injury. BioMed Pharmacother (2021) 136:111193. doi: 10.1016/j.biopha.2020.111193
84. Evans JP, Liu SL. Role of Host Factors in SARS-CoV-2 Entry. J Biol Chem (2021) 297:100847. doi: 10.1016/j.jbc.2021.100847
85. Gorska AM, Donoso M, Valdebenito S, Prideaux B, Queen S, Scemes E, et al. Human Immunodeficiency Virus-1/Simian Immunodeficiency Virus Infection Induces Opening of Pannexin-1 Channels Resulting in Neuronal Synaptic Compromise: A Novel Therapeutic Opportunity to Prevent NeuroHIV. J Neurochem (2021) 158:500–21. doi: 10.1111/jnc.15374
86. Swayne LA, Johnstone SR, Ng CS, Sanchez-Arias JC, Good ME, Penuela S, et al. Consideration of Pannexin 1 Channels in COVID-19 Pathology and Treatment. Am J Physiology-Lung Cell Mol Physiol (2020) 319:L121–5. doi: 10.1152/ajplung.00146.2020
87. Luu R, Valdebenito S, Scemes E, Cibelli A, Spray DC, Rovegno M, et al. Pannexin-1 Channel Opening Is Critical for COVID-19 Pathogenesis. Iscience (2021) 24:103478. doi: 10.1016/j.isci.2021.103478
88. Feig JL, Mediero A, Corciulo C, Liu H, Zhang J, Perez-Aso M, et al. The Antiviral Drug Tenofovir, an Inhibitor of Pannexin-1-Mediated ATP Release, Prevents Liver and Skin Fibrosis by Downregulating Adenosine Levels in the Liver and Skin. PloS One (2017) 12:e0188135. doi: 10.1371/journal.pone.0188135
89. Zanella I, Zizioli D, Castelli F, Quiros-Roldan E. Tenofovir, Another Inexpensive, Well-Known and Widely Available Old Drug Repurposed for SARS-COV-2 Infection. Pharmaceuticals (Basel) (2021) 14:454. doi: 10.3390/ph14050454
90. Seifert M, Bera SC, van Nies P, Kirchdoerfer RN, Shannon A, Le TT, et al. Inhibition of SARS-CoV-2 Polymerase by Nucleotide Analogs From a Single-Molecule Perspective. Elife (2021) 10:e70968. doi: 10.7554/eLife.70968
91. Tejero H, Montero F, Nuño JC. Theories of Lethal Mutagenesis: From Error Catastrophe to Lethal Defection. Curr Top Microbiol Immunol (2016) 392:161–79. doi: 10.1007/82_2015_463
92. Yan L, Yang Y, Li M, Zhang Y, Zheng L, Ge J, et al. Coupling of N7-Methyltransferase and 3’-5’ Exoribonuclease With SARS-CoV-2 Polymerase Reveals Mechanisms for Capping and Proofreading. Cell (2021) 184:3474–3485.e11. doi: 10.1016/j.cell.2021.05.033
93. Liu C, Shi W, Becker ST, Schatz DG, Liu B, Yang Y. Structural Basis of Mismatch Recognition by a SARS-CoV-2 Proofreading Enzyme. Science (2021) 373:1142–6. doi: 10.1126/science.abi9310
94. Moeller NH, Shi K, Demir Ö, Banerjee S, Yin L, Belica C, et al. Structure and Dynamics of SARS-CoV-2 Proofreading Exoribonuclease ExoN. Proc Natl Acad Sci USA (2022) 119:e2106379119. doi: 10.1073/pnas.2106379119
95. Li T, Kenney AD, Liu H, Fiches GN, Zhou D, Biswas A, et al. SARS-CoV-2 Nsp14 Activates NF-κb Signaling and Induces IL-8 Upregulation. bioRxiv (2021) 2021. doi: 10.1101/2021.05.26.445787
96. Niu X, Kong F, Hou YJ, Wang Q. Crucial Mutation in the Exoribonuclease Domain of Nsp14 of PEDV Leads to High Genetic Instability During Viral Replication. Cell Biosci (2021) 11:106. doi: 10.1186/s13578-021-00598-1
97. Tahir M. Coronavirus Genomic Nsp14-ExoN, Structure, Role, Mechanism, and Potential Application as a Drug Target. J Med Virol (2021) 93:4258–64. doi: 10.1002/jmv.27009
98. Canal B, McClure AW, Curran JF, Wu M, Ulferts R, Weissmann F, et al. Identifying SARS-CoV-2 Antiviral Compounds by Screening for Small Molecule Inhibitors of Nsp14/Nsp10 Exoribonuclease. Biochem J (2021) 478:2445–64. doi: 10.1042/BCJ20210198
99. Wiegand T, McVey A, Nemudraia A, Nemudryi A, Wiedenheft B. The Rise and Fall of SARS-CoV-2 Variants and the Mutational Profile of Omicron. (2021). doi: 10.1101/2021.12.16.473096
100. Wen KW, Damania B. Hsp90 and Hsp40/Erdj3 are Required for the Expression and Anti-Apoptotic Function of KSHV K1. Oncogene (2010) 29:3532–44. doi: 10.1038/onc.2010.124
101. Lillsunde KE, Tomašič T, Schult P, Lohmann V, Kikelj D, Tammela P. Inhibition of Hepatitis C Replication by Targeting the Molecular Chaperone Hsp90: Synthesis and Biological Evaluation of 4,5,6,7-Tetrahydrobenzo[1,2-D]Thiazole Derivatives. ChemMedChem (2019) 14:334–42. doi: 10.1002/cmdc.201800724
102. Li C, Chu H, Liu X, Chiu MC, Zhao X, Wang D, et al. Human Coronavirus Dependency on Host Heat Shock Protein 90 Reveals an Antiviral Target. Emerg Microbes Infect (2020) 9:2663–72. doi: 10.1080/22221751.2020.1850183
103. Wyler E, Mösbauer K, Franke V, Diag A, Gottula LT, Arsiè R, et al. Transcriptomic Profiling of SARS-CoV-2 Infected Human Cell Lines Identifies HSP90 as Target for COVID-19 Therapy. iScience (2021) 24:102151. doi: 10.1016/j.isci.2021.102151
104. Kulkarni MM, Ratcliff AN, Bhat M, Alwarawrah Y, Hughes P, Arcos J, et al. Cellular Fatty Acid Synthase Is Required for Late Stages of HIV-1 Replication. Retrovirology (2017) 14:45. doi: 10.1186/s12977-017-0368-z
105. Chu J, Xing C, Du Y, Duan T, Liu S, Zhang P, et al. Pharmacological Inhibition of Fatty Acid Synthesis Blocks SARS-CoV-2 Replication. Nat Metab (2021) 3:1466–75. doi: 10.1038/s42255-021-00479-4
106. Wu Z, Zhang Z, Wang X, Zhang J, Ren C, Li Y, et al. Palmitoylation of SARS-CoV-2 S Protein Is Essential for Viral Infectivity. Signal Transduct Target Ther (2021) 6:231. doi: 10.1038/s41392-021-00651-y
107. Williams CG, Jureka AS, Silvas JA, Nicolini AM, Chvatal SA, Carlson-Stevermer J, et al. Inhibitors of VPS34 and Fatty-Acid Metabolism Suppress SARS-CoV-2 Replication. Cell Rep (2021) 36:109479. doi: 10.1016/j.celrep.2021.109479
108. Tanner JE, Alfieri C. The Fatty Acid Lipid Metabolism Nexus in COVID-19. Viruses (2021) 13:90. doi: 10.3390/v13010090
109. El Jamal SM, Pujadas E, Ramos I, Bryce C, Grimes ZM, Amanat F, et al. Tissue-Based SARS-CoV-2 Detection in Fatal COVID-19 Infections: Sustained Direct Viral-Induced Damage Is Not Necessary to Drive Disease Progression. Hum Pathol (2021) 114:110–9. doi: 10.1016/j.humpath.2021.04.012
110. Sheikhbahaei S, Turovsky EA, Hosford PS, Hadjihambi A, Theparambil SM, Liu B, et al. Astrocytes Modulate Brainstem Respiratory Rhythm-Generating Circuits and Determine Exercise Capacity. Nat Commun (2018) 9:370. doi: 10.1038/s41467-017-02723-6
111. Reklow RJ, Alvares TS, Zhang Y, Miranda Tapia AP, Biancardi V, Katzell AK, et al. The Purinome and the Prebötzinger Complex - A Ménage of Unexplored Mechanisms That May Modulate/Shape the Hypoxic Ventilatory Response. Front Cell Neurosci (2019) 13:365. doi: 10.3389/fncel.2019.00365
112. Rees DA, Giles P, Lewis MD, Ham J. Adenosine Regulates Thrombomodulin and Endothelial Protein C Receptor Expression in Folliculostellate Cells of the Pituitary Gland. Purinergic Signal (2010) 6:19–29. doi: 10.1007/s11302-009-9172-0
113. Won T, Wood MK, Hughes DM, Talor MV, Ma Z, Schneider J, et al. Endothelial Thrombomodulin Downregulation Caused by Hypoxia Contributes to Severe Infiltration and Coagulopathy in COVID-19 Patient Lungs. EBioMedicine (2022) 75:103812. doi: 10.1016/j.ebiom.2022.103812
114. Lee S, Yu Y, Trimpert J, Benthani F, Mairhofer M, Richter-Pechanska P, et al. Virus-Induced Senescence is a Driver and Therapeutic Target in COVID-19. Nature (2021) 599:283–9. doi: 10.1038/s41586-021-03995-1
115. D’Agnillo F, Walters KA, Xiao Y, Sheng ZM, Scherler K, Park J, et al. Lung Epithelial and Endothelial Damage, Loss of Tissue Repair, Inhibition of Fibrinolysis, and Cellular Senescence in Fatal COVID-19. Sci Transl Med (2021) 13:eabj7790. doi: 10.1126/scitranslmed.abj7790
116. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat Cell Biol (2013) 15:978–90. doi: 10.1038/ncb2784
117. Cho J, Yusuf R, Kook S, Attar E, Lee D, Park B, et al. Purinergic P2Y14 Receptor Modulates Stress-Induced Hematopoietic Stem/Progenitor Cell Senescence. J Clin Invest (2014) 124:3159–71. doi: 10.1172/JCI61636
118. Yang L, Hu M, Lu Y, Han S, Wang J. Inflammasomes and the Maintenance of Hematopoietic Homeostasis: New Perspectives and Opportunities. Molecules (2021) 26:E309. doi: 10.3390/molecules26020309
119. Doğan HO, Şenol O, Bolat S, Yıldız ŞN, Büyüktuna SA, Sarıismailoğlu R, et al. Understanding the Pathophysiological Changes via Untargeted Metabolomics in COVID-19 Patients. J Med Virol (2021) 93:2340–9. doi: 10.1002/jmv.26716
120. Romagnoli S, Peris A, De Gaudio AR, Geppetti P. SARS-CoV-2 and COVID-19: From the Bench to the Bedside. Physiol Rev (2020) 100:1455–66. doi: 10.1152/physrev.00020.2020
121. Edwards C, Klekot O, Halugan L, Korchev Y. Follow Your Nose: A Key Clue to Understanding and Treating COVID-19. Front Endocrinol (Lausanne) (2021) 12:747744. doi: 10.3389/fendo.2021.747744
122. Kern RC, Pitovski DZ. Localization of 11/β-Hydroxysteroid Dehydrogenase: Specific Protector of the Mineralocorticoid Receptor in Mammalian Olfactory Mucosa. Acta Oto-Laryngologica (1997) 117:738–43. doi: 10.3109/00016489709113470
123. Edwards C. New Horizons: Does Mineralocorticoid Receptor Activation by Cortisol Cause ATP Release and COVID-19 Complications. J Clin Endocrinol Metab (2021) 106:622–35. doi: 10.1210/clinem/dgaa874
124. Hegg CC, Greenwood D, Huang W, Han P, Lucero MT. Activation of Purinergic Receptor Subtypes Modulates Odor Sensitivity. J Neurosci (2003) 23:8291–301. doi: 10.1523/JNEUROSCI.23-23-08291.2003
125. Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH, et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell (2020) 182:429–46.e14. doi: 10.1016/j.cell.2020.05.042
126. Lewis KL, Helgeson SA, Tatari MM, Mallea JM, Baig HZ, Patel NM. COVID-19 and the Effects on Pulmonary Function Following Infection: A Retrospective Analysis. EClinicalMedicine (2021) 39:101079. doi: 10.1016/j.eclinm.2021.101079
127. Zhang L, Dutta S, Xiong S, Chan M, Chan KK, Fan TM, et al. Engineered ACE2 Decoy Mitigates Lung Injury and Death Induced by SARS-CoV-2 Variants. Nat Chem Biol (2022) 18:342–351. doi: 10.1038/s41589-021-00965-6
128. Grist JT, Chen M, Collier GJ, Raman B, Abueid G, McIntyre A, et al. Hyperpolarized 129xe MRI Abnormalities in Dyspneic Patients 3 Months After COVID-19 Pneumonia: Preliminary Results. Radiology (2021) 301:E353–60. doi: 10.1148/radiol.2021210033
129. Juengling FD, Maldonado A, Wuest F, Schindler TH. The Role of Nuclear Medicine for COVID-19: Time to Act Now. J Nucl Med (2020) 61:781–2. doi: 10.2967/jnumed.120.246611
130. Cervia C, Zurbuchen Y, Taeschler P, Ballouz T, Menges D, Hasler S, et al. Immunoglobulin Signature Predicts Risk of Post-Acute COVID-19 Syndrome. Nat Commun (2022) 13:446. doi: 10.1038/s41467-021-27797-1
131. Wang R, Hume AJ, Beermann ML, Simone-Roach C, Lindstrom-Vautrin J, Le Suer J, et al. Human Airway Lineages Derived From Pluripotent Stem Cells Reveal the Epithelial Responses to SARS-CoV-2 Infection. Am J Physiol Lung Cell Mol Physiol (2022) 322:L462–L478. doi: 10.1152/ajplung.00397.2021
132. Kim JS, Lee JY, Yang JW, Lee KH, Effenberger M, Szpirt W, et al. Immunopathogenesis and Treatment of Cytokine Storm in COVID-19. Theranostics (2021) 11:316–29. doi: 10.7150/thno.49713
133. Linden J, Koch-Nolte F, Dahl G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu Rev Immunol (2019) 37:325–47. doi: 10.1146/annurev-immunol-051116-052406
134. Garvin MR, Alvarez C, Miller JI, Prates ET, Walker AM, Amos BK, et al. A Mechanistic Model and Therapeutic Interventions for COVID-19 Involving a RAS-Mediated Bradykinin Storm. Elife (2020) 9:e59177. doi: 10.7554/eLife.59177
135. Klaver D, Thurnher M. Control of Macrophage Inflammation by P2Y Purinergic Receptors. Cells (2021) 10:1098. doi: 10.3390/cells10051098
136. Wauters E, Van Mol P, Garg AD, Jansen S, Van Herck Y, Vanderbeke L, et al. Discriminating Mild From Critical COVID-19 by Innate and Adaptive Immune Single-Cell Profiling of Bronchoalveolar Lavages. Cell Res (2021) 31:272–90. doi: 10.1038/s41422-020-00455-9
137. Olotu C, Kiefmann M, Ronneburg C, Lehmensiek F, Cuvenhaus A, Meidl V, et al. Analysis of Purine Receptor Expression and Functionality in Alveolar Epithelial Cells. Purinergic Signal (2020) 16:213–29. doi: 10.1007/s11302-020-09696-0
138. Dietl P, Frick M. Channels and Transporters of the Pulmonary Lamellar Body in Health and Disease. Cells (2021) 11:45. doi: 10.3390/cells11010045
139. Peckham D, McDermott MF, Savic S, Mehta A. COVID-19 Meets Cystic Fibrosis: For Better or Worse? Genes Immun (2020) 21:260–2. doi: 10.1038/s41435-020-0103-y
140. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A Crucial Role of Angiotensin Converting Enzyme 2 (ACE2) in SARS Coronavirus-Induced Lung Injury. Nat Med (2005) 11:875–9. doi: 10.1038/nm1267
141. Abraham EH, Guidotti G, Rapaport E, Bower D, Brown J, Griffin RJ, et al. Cystic Fibrosis Improves COVID-19 Survival and Provides Clues for Treatment of SARS-CoV-2. Purinergic Signal (2021) 17:399–410. doi: 10.1007/s11302-021-09771-0
142. Hadi YB, Lakhani DA, Naqvi SF, Fatima NU, Sarwari AR. Outcomes of SARS-CoV-2 Infection in Patients With Cystic Fibrosis: A Multicenter Retrospective Research Network Study. Respir Med (2021) 188:106606. doi: 10.1016/j.rmed.2021.106606
143. Colombo C, Alicandro G, Daccó V, Gagliano V, Morlacchi LC, Casciaro R, et al. SARS-CoV-2 Infection in Cystic Fibrosis: A Multicentre Prospective Study With a Control Group, Italy, February-July 2020. PloS One (2021) 16:e0251527. doi: 10.1371/journal.pone.0251527
144. Jung A, Orenti A, Dunlevy F, Aleksejeva E, Bakkeheim E, Bobrovnichy V, et al. Factors for Severe Outcomes Following SARS-CoV-2 Infection in People With Cystic Fibrosis in Europe. ERJ Open Res (2021) 7:00411–2021. doi: 10.1183/23120541.00411-2021
145. Dos Anjos F, Simões JLB, Assmann CE, Carvalho FB, Bagatini MD. Potential Therapeutic Role of Purinergic Receptors in Cardiovascular Disease Mediated by SARS-CoV-2. J Immunol Res (2020) 2020:8632048. doi: 10.1155/2020/8632048
146. Iba T, Levy JH. The Roles of Platelets in COVID-19-Associated Coagulopathy and Vaccine-Induced Immune Thrombotic Thrombocytopenia. Trends Cardiovasc Med (2022) 32:1–9. doi: 10.1016/j.tcm.2021.08.012
147. Armstrong PC, Leadbeater PD, Chan MV, Kirkby NS, Jakubowski JA, Mitchell JA, et al. In the Presence of Strong P2Y12 Receptor Blockade, Aspirin Provides Little Additional Inhibition of Platelet Aggregation. J Thromb Haemost (2011) 9:552–61. doi: 10.1111/j.1538-7836.2010.04160.x
148. Brambilla M, Canzano P, Becchetti A, Tremoli E, Camera M. Alterations in Platelets During SARS-CoV-2 Infection. Platelets (2021) 27:1–9. doi: 10.1161/ATVBAHA.121.316188
149. Berger JS, Kornblith LZ, Gong MN, Reynolds HR, Cushman M, Cheng Y, et al. Effect of P2Y12 Inhibitors on Survival Free of Organ Support Among Non-Critically Ill Hospitalized Patients With COVID-19: A Randomized Clinical Trial. JAMA (2022) 327:227–36. doi: 10.1001/jama.2021.23605
150. REMAP-CAP I, Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients With Covid-19. N Engl J Med (2021) 384:1491–502. doi: 10.1056/NEJMoa2100433
151. Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat Microbiol (2020) 5:1403–7. doi: 10.1038/s41564-020-0770-5
152. Saito A, Irie T, Suzuki R, Maemura T, Nasser H, Uriu K, et al. Enhanced Fusogenicity and Pathogenicity of SARS-CoV-2 Delta P681R Mutation. Nature (2021) 602:300–6. doi: 10.1038/s41586-021-04266-9
153. Rajah MM, Hubert M, Bishop E, Saunders N, Robinot R, Grzelak L, et al. SARS-CoV-2 Alpha, Beta, and Delta Variants Display Enhanced Spike-Mediated Syncytia Formation. EMBO J (2021) 40:e108944. doi: 10.15252/embj.2021108944
154. Freeman TL, Swartz TH. Purinergic Receptors: Elucidating the Role of These Immune Mediators in HIV-1 Fusion. Viruses (2020) 12:E290. doi: 10.3390/v12030290
155. Viana R, Moyo S, Amoako DG, Tegally H, Scheepers C, Althaus CL, et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature (2022) 603:79–686. doi: 10.1038/s41586-022-04411-y
156. Shuai H, Chan JF, Hu B, Chai Y, Yuen TT, Yin F, et al. Attenuated Replication and Pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature (2022) 603:693–699. doi: 10.1038/s41586-022-04442-5
157. Zhao H, Lu L, Peng Z, Chen LL, Meng X, Zhang C, et al. SARS-CoV-2 Omicron Variant Shows Less Efficient Replication and Fusion Activity When Compared With Delta Variant in TMPRSS2-Expressed Cells. Emerg Microbes Infect (2022) 11:277–83. doi: 10.1080/22221751.2021.2023329
158. Willett BJ, Grove J, MacLean OA, Wilkie C, Logan N, Lorenzo GD, et al. The Hyper-Transmissible SARS-CoV-2 Omicron Variant Exhibits Significant Antigenic Change, Vaccine Escape and a Switch in Cell Entry Mechanism. (2022). doi: 10.1101/2022.01.03.21268111
159. Sriram K, Insel PA. Inflammation and Thrombosis in COVID-19 Pathophysiology: Proteinase-Activated and Purinergic Receptors as Drivers and Candidate Therapeutic Targets. Physiol Rev (2021) 101:545–67. doi: 10.1152/physrev.00035.2020
160. Su Y, Yuan D, Chen DG, Ng RH, Wang K, Choi J, et al. Multiple Early Factors Anticipate Post-Acute COVID-19 Sequelae. Cell (2022) 185:881–95.e20. doi: 10.1016/j.cell.2022.01.014
161. Verma D, Church TM, Swaminathan S. Epstein-Barr Virus Lytic Replication Induces ACE2 Expression and Enhances SARS-CoV-2 Pseudotyped Virus Entry in Epithelial Cells. J Virol (2021) 95:e0019221. doi: 10.1128/JVI.00192-21
162. Nunn AVW, Guy GW, Botchway SW, Bell JD. SARS-CoV-2 and EBV; the Cost of a Second Mitochondrial “Whammy”. Immun Ageing (2021) 18:40. doi: 10.1186/s12979-021-00252-x
163. Jain S, Jacobson KA. Purinergic Signaling in Diabetes and Metabolism. Biochem Pharmacol (2021) 187:114393. doi: 10.1016/j.bcp.2020.114393
164. Jain S, Pydi SP, Toti KS, Robaye B, Idzko M, Gavrilova O, et al. Lack of Adipocyte Purinergic P2Y6 Receptor Greatly Improves Whole Body Glucose Homeostasis. Proc Natl Acad Sci USA (2020) 117:30763–74. doi: 10.1073/pnas.2006578117
165. Wang H, Wang Z, Cao W, Wu Q, Yuan Y, Zhang X. Regulatory T Cells in COVID-19. Aging Dis (2021) 12:1545–53. doi: 10.14336/AD.2021.0709
166. Caldrer S, Mazzi C, Bernardi M, Prato M, Ronzoni N, Rodari P, et al. Regulatory T Cells as Predictors of Clinical Course in Hospitalised COVID-19 Patients. Front Immunol (2021) 12:789735. doi: 10.3389/fimmu.2021.789735
167. Khalifa AE, Ghoneim AI. Potential Value of Pharmacological Agents Acting on Toll-Like Receptor (TLR) 7 and/or TLR8 in COVID-19. Curr Res Pharmacol Drug Discov (2021) 2:100068. doi: 10.1016/j.crphar.2021.100068
168. Zangiabadi S, Abdul-Sater AA. Regulation of the NLRP3 Inflammasome by Posttranslational Modifications. J Immunol (2022) 208:286–92. doi: 10.4049/jimmunol.2100734
169. Kouwaki T, Nishimura T, Wang G, Oshiumi H. RIG-I-Like Receptor-Mediated Recognition of Viral Genomic RNA of Severe Acute Respiratory Syndrome Coronavirus-2 and Viral Escape From the Host Innate Immune Responses. Front Immunol (2021) 12:700926. doi: 10.3389/fimmu.2021.700926
170. Spel L, Martinon F. Detection of Viruses by Inflammasomes. Curr Opin Virol (2021) 46:59–64. doi: 10.1016/j.coviro.2020.10.001
171. Bouayad A. Innate Immune Evasion by SARS-CoV-2: Comparison With SARS-CoV. Rev Med Virol (2020) 30:1–9. doi: 10.1002/rmv.2135
172. Anaeigoudari A, Mollaei HR, Arababadi MK, Nosratabadi R. Severe Acute Respiratory Syndrome Coronavirus 2: The Role of the Main Components of the Innate Immune System. Inflammation (2021) 44:2151–69. doi: 10.1007/s10753-021-01519-7
173. Wu Y, Ma L, Zhuang Z, Cai S, Zhao Z, Zhou L, et al. Main Protease of SARS-CoV-2 Serves as a Bifunctional Molecule in Restricting Type I Interferon Antiviral Signaling. Signal Transduct Target Ther (2020) 5:221. doi: 10.1038/s41392-020-00332-2
174. Liu Y, Qin C, Rao Y, Ngo C, Feng JJ, Zhao J, et al. SARS-CoV-2 Nsp5 Demonstrates Two Distinct Mechanisms Targeting RIG-I and MAVS To Evade the Innate Immune Response. MBio (2021) 12:e02335–21. doi: 10.1128/mBio.02335-21
175. Campbell GR, To RK, Hanna J, Spector SA. SARS-CoV-2, SARS-CoV-1, and HIV-1 Derived ssRNA Sequences Activate the NLRP3 Inflammasome in Human Macrophages Through a Non-Classical Pathway. iScience (2021) 24:102295. doi: 10.1016/j.isci.2021.102295
176. Yalcinkaya M, Liu W, Islam MN, Kotini AG, Gusarova GA, Fidler TP, et al. Modulation of the NLRP3 Inflammasome by Sars-CoV-2 Envelope Protein. Sci Rep (2021) 11:1–12. doi: 10.1016/j.isci.2021.102295
177. Kim NE, Song YJ. Coordinated Regulation of Interferon and Inflammasome Signaling Pathways by SARS-CoV-2 Proteins. J Microbiol (2022) 60:300–7. doi: 10.1007/s12275-022-1502-8
178. Ratajczak MZ, Kucia M. SARS-CoV-2 Infection and Overactivation of Nlrp3 Inflammasome as a Trigger of Cytokine “Storm” and Risk Factor for Damage of Hematopoietic Stem Cells. Leukemia (2020) 34:1726–9. doi: 10.1038/s41375-020-0887-9
179. Kucia M, Ratajczak J, Bujko K, Adamiak M, Ciechanowicz A, Chumak V, et al. An Evidence That SARS-Cov-2/COVID-19 Spike Protein (SP) Damages Hematopoietic Stem/Progenitor Cells in the Mechanism of Pyroptosis in Nlrp3 Inflammasome-Dependent Manner. Leukemia (2021) 35:3026–9. doi: 10.1038/s41375-021-01332-z
180. Ren W, Rubini P, Tang Y, Engel T, Illes P. Inherent P2X7 Receptors Regulate Macrophage Functions During Inflammatory Diseases. Int J Mol Sci (2021) 23:232. doi: 10.3390/ijms23010232
181. Pan T, Cao G, Tang E, Zhao Y, Penaloza-MacMaster P, Fang Y, et al. A Single-Cell Atlas Reveals Shared and Distinct Immune Responses and Metabolism During SARS-CoV-2 and HIV-1 Infections. bioRxiv (2022) 2022:01. doi: 10.1101/2022.01.10.475725
182. Fan X, Zhang J, Dai Y, Shan K, Xu J. Blockage of P2X7R Suppresses Th1/Th17-Mediated Immune Responses and Corneal Allograft Rejection via Inhibiting NLRP3 Inflammasome Activation. Exp Eye Res (2021) 212:108792. doi: 10.1016/j.exer.2021.108792
183. Ahmadi P, Hartjen P, Kohsar M, Kummer S, Schmiedel S, Bockmann JH, et al. Defining the CD39/CD73 Axis in SARS-CoV-2 Infection: The CD73- Phenotype Identifies Polyfunctional Cytotoxic Lymphocytes. Cells (2020) 9:E1750. doi: 10.3390/cells9081750
184. Unterman A, Sumida TS, Nouri N, Yan X, Zhao AY, Gasque V, et al. Single-Cell Multi-Omics Reveals Dyssynchrony of the Innate and Adaptive Immune System in Progressive COVID-19. Nat Commun (2022) 13:440. doi: 10.1038/s41467-021-27716-4
185. Wang X, Chen D. Purinergic Regulation of Neutrophil Function. Front Immunol (2018) 9:399. doi: 10.3389/fimmu.2018.00399
186. Shaath H, Vishnubalaji R, Elkord E, Alajez NM. Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe COVID-19. Cells (2020) 9:E2374. doi: 10.3390/cells9112374
187. Reusch N, De Domenico E, Bonaguro L, Schulte-Schrepping J, Baßler K, Schultze JL, et al. Neutrophils in COVID-19. Front Immunol (2021) 12:652470. doi: 10.3389/fimmu.2021.652470
188. Cavalcante-Silva LHA, Carvalho DCM, Lima ÉA, Galvão JGFM, da Silva JSF, Sales-Neto JM, et al. Neutrophils and COVID-19: The Road So Far. Int Immunopharmacol (2021) 90:107233. doi: 10.1016/j.intimp.2020.107233
189. Keane C, Coalter M, Martin-Loeches I. Immune System Disequilibrium-Neutrophils, Their Extracellular Traps, and COVID-19-Induced Sepsis. Front Med (Lausanne) (2021) 8:711397. doi: 10.3389/fmed.2021.711397
190. Ackermann M, Anders HJ, Bilyy R, Bowlin GL, Daniel C, De Lorenzo R, et al. Patients With COVID-19: In the Dark-NETs of Neutrophils. Cell Death Differ (2021). doi: 10.1038/s41418-021-00805-z
191. Blanch-Ruiz MA, Ortega-Luna R, Gómez-García G, Martínez-Cuesta MÁ, Álvarez Á. Role of Neutrophil Extracellular Traps in COVID-19 Progression: An Insight for Effective Treatment. Biomedicines (2021) 10:31–47. doi: 10.3390/biomedicines10010031
192. Aliter KF, Al-Horani RA. Potential Therapeutic Benefits of Dipyridamole in COVID-19 Patients. Curr Pharm Des (2021) 27:866–75. doi: 10.2174/1381612826666201001125604
193. Carestia A, Kaufman T, Schattner M. Platelets: New Bricks in the Building of Neutrophil Extracellular Traps. Front Immunol (2016) 7:271. doi: 10.3389/fimmu.2016.00271
194. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 Binds Platelet ACE2 to Enhance Thrombosis in COVID-19. J Hematol Oncol (2020) 13:120. doi: 10.1186/s13045-020-00954-7
195. Campbell RA, Boilard E, Rondina MT. Is There a Role for the ACE2 Receptor in SARS-CoV-2 Interactions With Platelets. J Thromb Haemost (2021) 19:46–50. doi: 10.1111/jth.15156
196. Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, Harris ES, et al. In Vivo Platelet Activation in Critically Ill Patients With Primary 2009 Influenza a(H1N1). Chest (2012) 141:1490–5. doi: 10.1378/chest.11-2860
197. Amison RT, Jamshidi S, Rahman KM, Page CP, Pitchford SC. Diverse Signalling of the Platelet P2Y1 Receptor Leads to a Dichotomy in Platelet Function. Eur J Pharmacol (2018) 827:58–70. doi: 10.1016/j.ejphar.2018.03.014
198. Schrottmaier WC, Kral JB, Badrnya S, Assinger A. Aspirin and P2Y12 Inhibitors in Platelet-Mediated Activation of Neutrophils and Monocytes. Thromb Haemost (2015) 114:478–89. doi: 10.1160/TH14-11-0943
199. Canzano P, Brambilla M, Porro B, Cosentino N, Tortorici E, Vicini S, et al. Platelet and Endothelial Activation as Potential Mechanisms Behind the Thrombotic Complications of COVID-19 Patients. JACC Basic Transl Sci (2021) 6:202–18. doi: 10.1016/j.jacbts.2020.12.009
201. Hasan D, Shono A, van Kalken CK, van der Spek PJ, Krenning EP, Kotani T. A Novel Definition and Treatment of Hyperinflammation in COVID-19 Based on Purinergic Signalling. Purinergic Signal (2021) 18:13–59. doi: 10.1007/s11302-021-09814-6
202. Afrin LB, Weinstock LB, Molderings GJ. Covid-19 Hyperinflammation and Post-Covid-19 Illness May Be Rooted in Mast Cell Activation Syndrome. Int J Infect Dis (2020) 100:327–32. doi: 10.1016/j.ijid.2020.09.016
203. Conti P, Caraffa A, Tetè G, Gallenga CE, Ross R, Kritas SK, et al. Mast Cells Activated by SARS-CoV-2 Release Histamine Which Increases IL-1 Levels Causing Cytokine Storm and Inflammatory Reaction in COVID-19. J Biol Regul Homeost Agents (2020) 34:1629–32. doi: 10.23812/20-2EDIT
204. Nishi H, Niyonsaba F, Pelleg A, Schulman ES. Enhancement of Mast Cell Degranulation Mediated by Purinergic Receptors’ Activation and PI3K Type δ. J Immunol (2021) 207:1001–8. doi: 10.4049/jimmunol.2001002
205. Salguero FJ, White AD, Slack GS, Fotheringham SA, Bewley KR, Gooch KE, et al. Comparison of Rhesus and Cynomolgus Macaques as an Infection Model for COVID-19. Nat Commun (2021) 12:1260. doi: 10.1038/s41467-021-21389-9
206. Wu ML, Liu FL, Sun J, Li X, He XY, Zheng HY, et al. SARS-CoV-2-Triggered Mast Cell Rapid Degranulation Induces Alveolar Epithelial Inflammation and Lung Injury. Signal Transduct Target Ther (2021) 6:428. doi: 10.1038/s41392-021-00849-0
207. Wechsler JB, Butuci M, Wong A, Kamboj AP, Youngblood BA. Mast Cell Activation Is Associated With Post-Acute COVID-19 Syndrome. Allergy (2021) 77:1288–91. doi: 10.1111/all.15188
208. Yong SJ. Long COVID or Post-COVID-19 Syndrome: Putative Pathophysiology, Risk Factors, and Treatments. Infect Dis (Lond) (2021) 53:1–18. doi: 10.1080/23744235.2021.1924397
209. Fernández-Castañeda A, Lu P, Geraghty AC, Song E, Lee MH, Wood J, et al. Mild Respiratory SARS-CoV-2 Infection Can Cause Multi-Lineage Cellular Dysregulation and Myelin Loss in the Brain. bioRxiv (2022) 2022. doi: 10.1101/2022.01.07.475453
210. Khan M, Syed GH, Kim SJ, Siddiqui A. Mitochondrial Dynamics and Viral Infections: A Close Nexus. Biochim Biophys Acta (2015) 1853:2822–33. doi: 10.1016/j.bbamcr.2014.12.040
211. Yasukawa K, Koshiba T. Mitochondrial Reactive Zones in Antiviral Innate Immunity. Biochim Biophys Acta Gen Subj (2021) 1865:129839. doi: 10.1016/j.bbagen.2020.129839
212. Ledderose C, Junger WG. Mitochondria Synergize With P2 Receptors to Regulate Human T Cell Function. Front Immunol (2020) 11:549889. doi: 10.3389/fimmu.2020.549889
213. Ledderose C, Bromberger S, Slubowski CJ, Sueyoshi K, Aytan D, Shen Y, et al. The Purinergic Receptor P2Y11 Choreographs the Polarization, Mitochondrial Metabolism, and Migration of T Lymphocytes. Sci Signal (2020) 13:eaba3300. doi: 10.1126/scisignal.aba3300
214. Ledderose C, Bromberger S, Slubowski CJ, Sueyoshi K, Junger WG. Frontline Science: P2Y11 Receptors Support T Cell Activation by Directing Mitochondrial Trafficking to the Immune Synapse. J Leukoc Biol (2021) 109:497–508. doi: 10.1002/JLB.2HI0520-191R
215. Stefano GB, Büttiker P, Weissenberger S, Martin A, Ptacek R, Kream RM. Editorial: The Pathogenesis of Long-Term Neuropsychiatric COVID-19 and the Role of Microglia, Mitochondria, and Persistent Neuroinflammation: A Hypothesis. Med Sci Monit (2021) 27:e933015. doi: 10.12659/MSM.933015
216. Abouelkhair MA. Targeting Adenosinergic Pathway and Adenosine A2A Receptor Signaling for the Treatment of COVID-19: A Hypothesis. Med Hypotheses (2020) 144:110012. doi: 10.1016/j.mehy.2020.110012
217. Franciosi MLM, Lima MDM, Schetinger MRC, Cardoso AM. Possible Role of Purinergic Signaling in COVID-19. Mol Cell Biochem (2021) 476:2891–8. doi: 10.1007/s11010-021-04130-4
218. Zarei M, Sahebi Vaighan N, Ziai SA. Purinergic Receptor Ligands: The Cytokine Storm Attenuators, Potential Therapeutic Agents for the Treatment of COVID-19. Immunopharmacol Immunotoxicol (2021) 43:633–43. doi: 10.1080/08923973.2021.1988102
219. Leão Batista Simões J, Fornari Basso H, Cristine Kosvoski G, Gavioli J, Marafon F, Elias Assmann C, et al. Targeting Purinergic Receptors to Suppress the Cytokine Storm Induced by SARS-CoV-2 Infection in Pulmonary Tissue. Int Immunopharmacol (2021) 100:108150. doi: 10.1016/j.intimp.2021.108150
220. Simões JLB, Bagatini MD. Purinergic Signaling of ATP in COVID-19 Associated Guillain-Barré Syndrome. J Neuroimmune Pharmacol (2021) 16:48–58. doi: 10.1007/s11481-020-09980-1
221. Simões JLB, de Araújo JB, Bagatini MD. Anti-Inflammatory Therapy by Cholinergic and Purinergic Modulation in Multiple Sclerosis Associated With SARS-CoV-2 Infection. Mol Neurobiol (2021) 58:5090–111. doi: 10.1007/s12035-021-02464-0
222. Schultz IC, Bertoni APS, Wink MR. Purinergic Signaling Elements Are Correlated With Coagulation Players in Peripheral Blood and Leukocyte Samples From COVID-19 Patients. J Mol Med (Berl) (2022) 100:569–84. doi: 10.1007/s00109-021-02175-y
223. Vargas-Martínez EM, Gómez-Coronado KS, Espinosa-Luna R, Valdez-Morales EE, Barrios-García T, Barajas-Espinosa A, et al. Functional Expression of P2X1, P2X4 and P2X7 Purinergic Receptors in Human Monocyte-Derived Macrophages. Eur J Pharmacol (2020) 888:173460. doi: 10.1016/j.ejphar.2020.173460
224. Kvietys PR, Fakhoury HMA, Kadan S, Yaqinuddin A, Al-Mutairy E, Al-Kattan K. COVID-19: Lung-Centric Immunothrombosis. Front Cell Infect Microbiol (2021) 11:679878. doi: 10.3389/fcimb.2021.679878
225. Lintzmaier Petiz L, Glaser T, Scharfstein J, Ratajczak MZ, Ulrich H. P2Y14 Receptor as a Target for Neutrophilia Attenuation in Severe COVID-19 Cases: From Hematopoietic Stem Cell Recruitment and Chemotaxis to Thrombo-Inflammation. Stem Cell Rev Rep (2021) 17:241–52. doi: 10.1007/s12015-021-10129-7
226. Caillon A, Trimaille A, Favre J, Jesel L, Morel O, Kauffenstein G. Role of Neutrophils, Platelets, and Extracellular Vesicles and Their Interactions in COVID-19-Associated Thrombopathy. J Thromb Haemost (2022) 20:17–31. doi: 10.1111/jth.15566
227. Soare AY, Malik HS, Durham ND, Freeman TL, Alvarez R, Patel F, et al. P2X1 Selective Antagonists Block HIV-1 Infection Through Inhibition of Envelope Conformation-Dependent Fusion. J Virol (2020) 94:e01622–19. doi: 10.1128/JVI.01622-19
228. Leyva-Grado VH, Ermler ME, Schotsaert M, Gonzalez MG, Gillespie V, Lim JK, et al. Contribution of the Purinergic Receptor P2X7 to Development of Lung Immunopathology During Influenza Virus Infection. mBio (2017) 8:e00229–17. doi: 10.1128/mBio.00229-17
229. Saeedi-Boroujeni A, Mahmoudian-Sani MR, Nashibi R, Houshmandfar S, Tahmaseby Gandomkari S, Khodadadi A. Tranilast: A Potential Anti-Inflammatory and NLRP3 Inflammasome Inhibitor Drug for COVID-19. Immunopharmacol Immunotoxicol (2021) 43:1–12. doi: 10.1080/08923973.2021.1925293
230. Omarjee L, Meilhac O, Perrot F, Janin A, Mahe G. Can Ticagrelor be Used to Prevent Sepsis-Induced Coagulopathy in COVID-19. Clin Immunol (2020) 216:108468. doi: 10.1016/j.clim.2020.108468
231. Abosheasha MA, El-Gowily AH, Elfiky AA. Potential Antiviral Properties of Antiplatelet Agents Against SARS-CoV-2 Infection: An in Silico Perspective. J Thromb Thromb (2021) 53:273–81. doi: 10.1007/s11239-021-02558-5
232. Chow JH, Yin Y, Yamane DP, Davison D, Keneally RJ, Hawkins K, et al. Association of Prehospital Antiplatelet Therapy With Survival in Patients Hospitalized With COVID-19: A Propensity Score-Matched Analysis. J Thromb Haemost (2021) 19:2814–24. doi: 10.1111/jth.15517
233. Müller CE, Namasivayam V. Recommended Tool Compounds and Drugs for Blocking P2X and P2Y Receptors. Purinergic Signal (2021) 17:633–48. doi: 10.1007/s11302-021-09813-7
234. Paul D, Basu D, Ghosh Dastidar S. Multi-Conformation Representation of Mpro Identifies Promising Candidates for Drug Repurposing Against COVID-19. J Mol Model (2021) 27:128. doi: 10.1007/s00894-021-04732-1
235. Ming Y, Xin G, Ji B, Ji C, Wei Z, Zhang B, et al. Entecavir as a P2X7R Antagonist Ameliorates Platelet Activation and Thrombus Formation. J Pharmacol Sci (2020) 144:43–51. doi: 10.1016/j.jphs.2020.07.002
Keywords: extracellular ATP, extracellular adenosine, P1 receptors, P2 receptors, virus, SARS-CoV-2
Citation: Ferrari D, Rubini M and Burns JS (2022) The Potential of Purinergic Signaling to Thwart Viruses Including SARS-CoV-2. Front. Immunol. 13:904419. doi: 10.3389/fimmu.2022.904419
Received: 25 March 2022; Accepted: 05 May 2022;
Published: 17 June 2022.
Edited by:
Robson Coutinho-Silva, Federal University of Rio de Janeiro, BrazilReviewed by:
Luiz Eduardo Baggio Savio, Federal University of Rio de Janeiro, BrazilMariusz Z. Ratajczak, University of Louisville Physicians, United States
Copyright © 2022 Ferrari, Rubini and Burns. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Davide Ferrari, ZGF2aWRlLmZlcnJhcmlAdW5pZmUuaXQ=