 Diana Coman
Diana Coman Isabelle Coales1†
Isabelle Coales1† Luke B. Roberts
Luke B. Roberts Joana F. Neves
Joana F. Neves
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 23 June 2022
Sec. NK and Innate Lymphoid Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.903688
This article is part of the Research Topic Innate Lymphoid Cells: Characterization and Classification View all 16 articles
Inflammatory bowel disease (IBD) is an idiopathic condition characterized by chronic relapsing inflammation in the intestine. While the precise etiology of IBD remains unknown, genetics, the gut microbiome, environmental factors, and the immune system have all been shown to contribute to the disease pathophysiology. In recent years, attention has shifted towards the role that innate lymphoid cells (ILCs) may play in the dysregulation of intestinal immunity observed in IBD. ILCs are a group of heterogenous immune cells which can be found at mucosal barriers. They act as critical mediators of the regulation of intestinal homeostasis and the orchestration of its inflammatory response. Despite helper-like type 1 ILCs (ILC1s) constituting a particularly rare ILC population in the intestine, recent work has suggested that an accumulation of intestinal ILC1s in individuals with IBD may act to exacerbate its pathology. In this review, we summarize existing knowledge on helper-like ILC1 plasticity and their classification in murine and human settings. Moreover, we discuss what is currently understood about the roles that ILC1s may play in the progression of IBD pathogenesis.
Inflammatory bowel disease (IBD) primarily describes two idiopathic conditions characterized by chronic relapsing inflammation in the intestine: ulcerative colitis (UC) and Crohn’s disease (CD). Despite many similarities between the two, UC and CD are differentiated based upon their histopathological features, the primary affected anatomical location, and their associated symptoms. Within the gastrointestinal tract, UC is exclusively colorectal, with mucosal inflammation found continuously extending from the rectum throughout the length of the colon (1). In contrast, individuals with CD exhibit discontinuous pockets of transmural inflammation which can be found scattered along the length of the gastrointestinal tract (2). In comparison to the relatively superficial legions of UC, CD is destructive, with around half of all patients exhibiting complications such as strictures or fistulae, within 10 years of being diagnosed (2). Yet while the precise etiology of IBD remains unknown, its progression has been shown to be influenced by complex interactions between genetic predispositions (3), perturbations to the gut microbiome and its associated metabolites (4–8), and the dysregulation of innate and adaptive immune responses (2, 9). Recently, attention has shifted towards the role that innate lymphoid cells (ILCs) may play in the pathophysiology of IBD. ILCs are a highly heterogenous family of lymphocytes which are considered the innate counterpart of adaptive T cells. Broadly, they have been split into 5 subtypes based on their development, transcription factor expression, and effector functions; these comprise natural killer (NK) cells, lymphoid tissue inducer, and the helper-like type 1, 2 and 3 ILCs (ILC1, ILC2, and ILC3, respectively) (10).
As extensively reviewed elsewhere, the abundance and phenotype of ILCs were shown to vary substantially across tissues (11). However, in contrast to other mucosal sites, such as the salivary gland and liver, ILC1s constitute an extremely rare ILC population in the intestine. Despite this, recent work suggests that they may play a critical role in the pathology of IBD (12–16). However, due to their substantial phenotypic overlap with NK cells, the identification and characterization of intestinal ILC1 remains under debate. Here we will discuss the complexities surrounding the differentiation of ILC1s from NK cells, with a focus on those found in the intestine, and highlight the potential roles of ILC1s in the exacerbation of IBD pathogenesis and its associated complications.
In mice, both ILC1s and NK cells can be identified by their shared co-expression of the activating receptors NK1.1 and NKp46 in the absence of other cell lineage (Lin-)-defining markers, including CD3, CD14, CD19, and T cell receptor proteins (17) (Table 1). Additionally, they share the expression of T-bet, a canonical master transcriptional regulator of type 1 immunity which includes the production of IFN-γ in lymphocytes (19). Within the murine intestine, four distinct populations of Lin- NK1.1+ NKp46+ cells have been described, which were differentiated based on their anatomical location, transcription factor expression, cell surface marker profile, and effector functions (10). These are comprised of conventional NK (cNK) cells, lamina propria (LP) ILC1s, intra-epithelial ILC1s (ieILC1), and a population of ILC1-like ex-ILC3s.
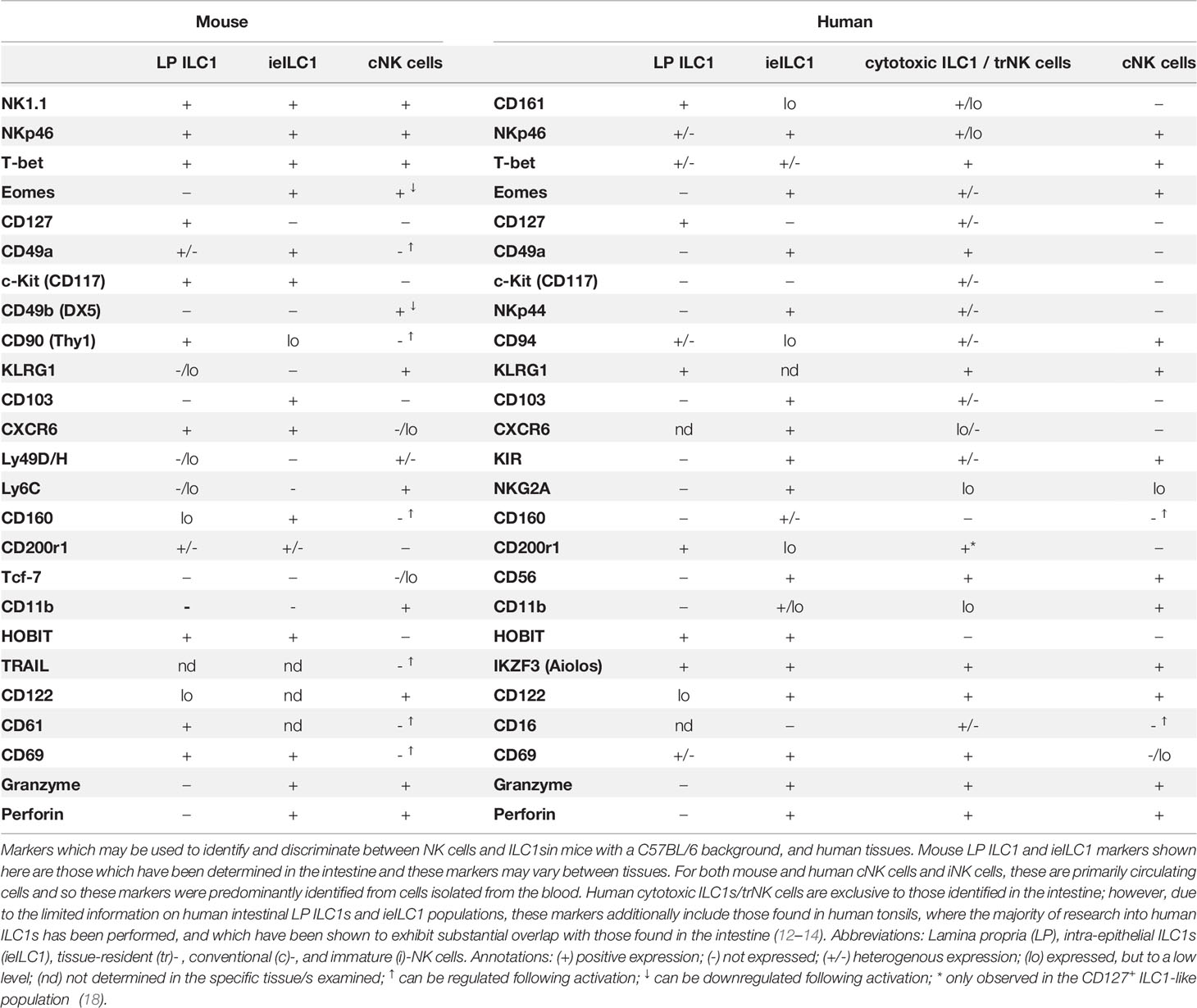
Table 1 NK cell and ILC1 Markers.
Predominantly found circulating in the blood or residing in the bone marrow and lymphoid organs, cNK cells are critical for the recognition and elimination of virus-infected and transformed cells, mediated via their secretion of cytotoxic effector molecules, such as perforin and granzyme B, and of proinflammatory cytokines, principally that of IFN-γ (10). In contrast, ILC1s are primarily a tissue-resident population of cells (20, 21). Within the LP, ILC1s harbor little to no cytotoxic capacity but exhibit significantly greater IFN-γ secretion than cNK cells, coupled to the production of TNFα upon their activation (12, 22, 23). Moreover, they lack the characteristic expression of Eomes found in cNK cells, instead expressing the IL-7 receptor subunit CD127, a marker of all helper-like ILC lineages (12, 22, 24–26). In the intestine, homeostatic cNK cells and LP ILC1s have further been shown to exhibit a dichotomous expression of cell surface markers including CD49a, CD49b, CXCR6, and CD200r1 and the inhibitory Ly49 family of receptors (21, 27, 28). Consequentially, under steady-state conditions, murine ILC1s and cNK cells form distinct clusters following global analyses of both their chromatin and transcriptional landscapes (21, 27, 29). However, none of those cited above acts alone as a population-defining marker. The advent of single-cell RNA sequencing technologies has highlighted the complex tissue-specific expression patterns of ILC1s and revealed that, even within tissues, ILC1 populations exhibit substantial transcriptional, functional, and developmental heterogeneity (11, 21, 27, 28, 30). Consequentially, many markers previously proposed to define ILC1s, including CD49a, TRAIL, Ly6C, CD103, CD200r1, and CD127 (10, 11), have been found inconsistently expressed within ILC1 populations and/or have been found expressed in immature NK cells and their progenitors (17, 21). This is exemplified by Eomes. While only cNK cells depend on the expression of Eomes for their maturation (31), a lack of Eomes is insufficient as a marker for ILC1s, as it is not expressed on immature NK cells (26). To complicate matters further, activated cNK cells have been shown to downregulate Eomes expression and upregulate ILC1 markers (32–34), resulting in increasingly blurred boundaries between the cell types under inflammatory conditions—for example, NK cells have recently been shown to be able to transition towards an ILC1-like phenotype (Eomes- CD49a+ Ly6C+) as a consequence of IL-12 s following infection with Toxoplasma gondii (32), or in response to TGF-β signaling in the tumor microenvironment (35), in a manner which may be mediated via STAT4 signaling (36). Consequentially, no universal cell surface marker that is able to conclusively distinguish ILC1s from NK cells has been identified.
As with the ability of cNK cells to differentiate to ILC1s, there is substantial and convincing evidence demonstrating a high level of plasticity between the helper-like ILCs in mice (37), particularly among ILC3s and ILC1s (23, 27, 28, 38, 44). In homeostasis, ILC3s form the major ILC population in the intestine, characterized via a high expression of the transcription factor RORγt and secretion of the cytokine IL-22 (10). However, in response to an infection or IL-12 signaling, ILC3s can downregulate RORγt while upregulating the expression of T-bet (38–42, 44), of which ILC1s are completely dependent upon for their differentiation (10) and post-developmental maintenance (43). Consequentially, the IL-22-expressing population of intestinal ILC3s is depleted, replaced by an expansion of IFN-γ-producing cells exhibiting a phenotype indistinguishable from ILC1s. Notably, using RORγt fate mapping analysis, an accumulation of these ILC1-like ex-ILC3s (Lin- Eomes- NK1.1- IFNγ+ T-bet+) was shown to be the principal driver of Campylobacter jejuni-induced intestinal pathology (44).
Finally, the remaining Lin- NK1.1+ NKp46+ population described in mice are CD103+ ILC1s, resembling the well-characterized ieILC1s in the human intestine (27).
Two distinct types of ILC1s have been described in the human intestine (Table 1). Intestinal LP Lin- c-Kit- NKp44- NKp46+ CD127+ CD161+ ILC1s exhibit T-bet expression and produce IFNγ in response to inflammation, resemblant of cNK cells (12). However, human LP ILC1s are Eomes- and do not express perforin or granzyme B or other markers typically seen in cNK cells in humans with cNK cells, including the expression of KIR3DL1, CD56, and/or IL1R1 (12, 13). These intestinal LP ILC1s are resemblant of ILC1s identified within other human mucosal tissues, such as the tonsils (45) where the majority of research into human ILCs have been performed. The second ILC1 subtype is a cytotoxic population found within the intra-epithelial lymphocyte compartment, characterized by their high expression of CD103 relative to other local ILC populations (13, 14). Contrasting those found in the LP, human CD103+ ILC1s are NKp46+ CD56+ Eomes+ CD127- CD161low, thus exhibiting a transcriptional and effector function signature reflecting that of both cNK cells and LP ILC1s (13, 14, 46).
Importantly, while the presence of NK1.1+ NKp46+ CD127+ ILC1s within the intestinal LP is consistently observed in mice, a question remains as to the existence of their NKp46+ CD127+ CD161+ LP ILC1 counterparts within the non-inflamed human intestine (30, 47, 48). Critically, the level of T-bet expression is heterogeneous in both ILC1 and NK cell populations in human tissues (46, 49), and a lack of T-bet in a putative ILC1 population (Lin- c-Kit- NKp44- CD45+ CD127+) has led some to question the very existence of ILC1s themselves (47). In contrast, the presence of cytotoxic populations of ILC1s, comparable to ieILC1s (CD103+/- T-bet+ Eomes+ CD56+ NKp46+ CD49a+ CD127-), is more consistently observed in various human tissues (13, 14, 47, 50, 51). However, these constitute highly heterogenous populations of cells which have been proposed to resemble cNK cells more closely than other helper-like ILC lineages (46, 47, 49). Accordingly, they are often instead classed as tissue-resident NK (trNK) cells.
While the subpopulations of LP ILC1, ieILC1, cytotoxic/trNK cells, and cNK cells exhibit highly overlapping phenotypes, a direct transition between these populations in the intestine has yet to be demonstrated. However, as described in mice, ILC3 to ILC1 phenotypic plasticity may also be present in human intestinal tissues (13, 15, 47, 52, 53)—for example, CD14+ dendritic cells from CD resection specimens have been found to promote the differentiation of ILC3s into ILC1s in vitro (13). Moreover, human tonsillar ILC3s have shown that they are able to convert to ILC1-like ex-ILC3s in a manner which may be driven, in part, by TGF-β and IL-23 acting coordinately to regulate the expression of the transcription factors Aiolos, T-bet, and RORγt (15, 53). These transitionary cells were also identified in the ileum of both individuals with CD and non-disease controls (15), suggesting that it is not a tissue-specific phenomenon. In the absence of fate mapping within human tissues, this plasticity acts to further complicate the identification of bona fide human ILC1s.
The consequence of their overlapping transcriptional phenotypes and the plasticity between populations result in a lack of consistently used nomenclature to define and differentiate between ILC1 and NK cells, particularly within human intestinal tissues. Until markers which conclusively discriminate between the two are identified, this remains a major challenge to the field.
LP ILC1s act in the first line of defense to external pathogens, rapidly inducing cytokine secretion upon their activation and promoting the recruitment of other immune cells to the site of inflammation (10) (Figure 1). Most studies addressing the potential roles of LP ILC1s in murine intestinal inflammation are primarily focused on their secretion of pro-inflammatory cytokines, namely, IFN-γ and TNFα (14, 18, 33, 38, 39, 44, 54). While these have been shown to aid the clearance of infections, such as mouse cytomegalovirus (33), Toxoplasma gondii (23), and Clostridium difficile (54), the excess production of IFN-γ and TNFα is highly disruptive to intestinal homeostasis, inhibiting proliferation and inducing apoptosis of local epithelial cells (55, 56). Consequentially, signaling via these pro-inflammatory cytokines has been shown to exacerbate IBD-associated pathology in response to infection with Helicobacter typhlonius and in mouse models of dextran sulfate sodium (DSS)-induced colitis (12, 25, 43, 55, 57–59)—for example, in response to DSS treatment, IFN-γ disrupts the vascular barrier integrity by altering the adherens junction protein VE-cadherin. As a result, this drives an increase in vascular permeability and an accumulation of immune cells at the site of inflammation, enabling the migration of intestinal bacteria into the blood and other organs (55). Concordantly, the depletion of LP ILC1s and/or their associated cytokines have been shown to significantly attenuate the symptoms of colitis in vivo (43, 57, 59).
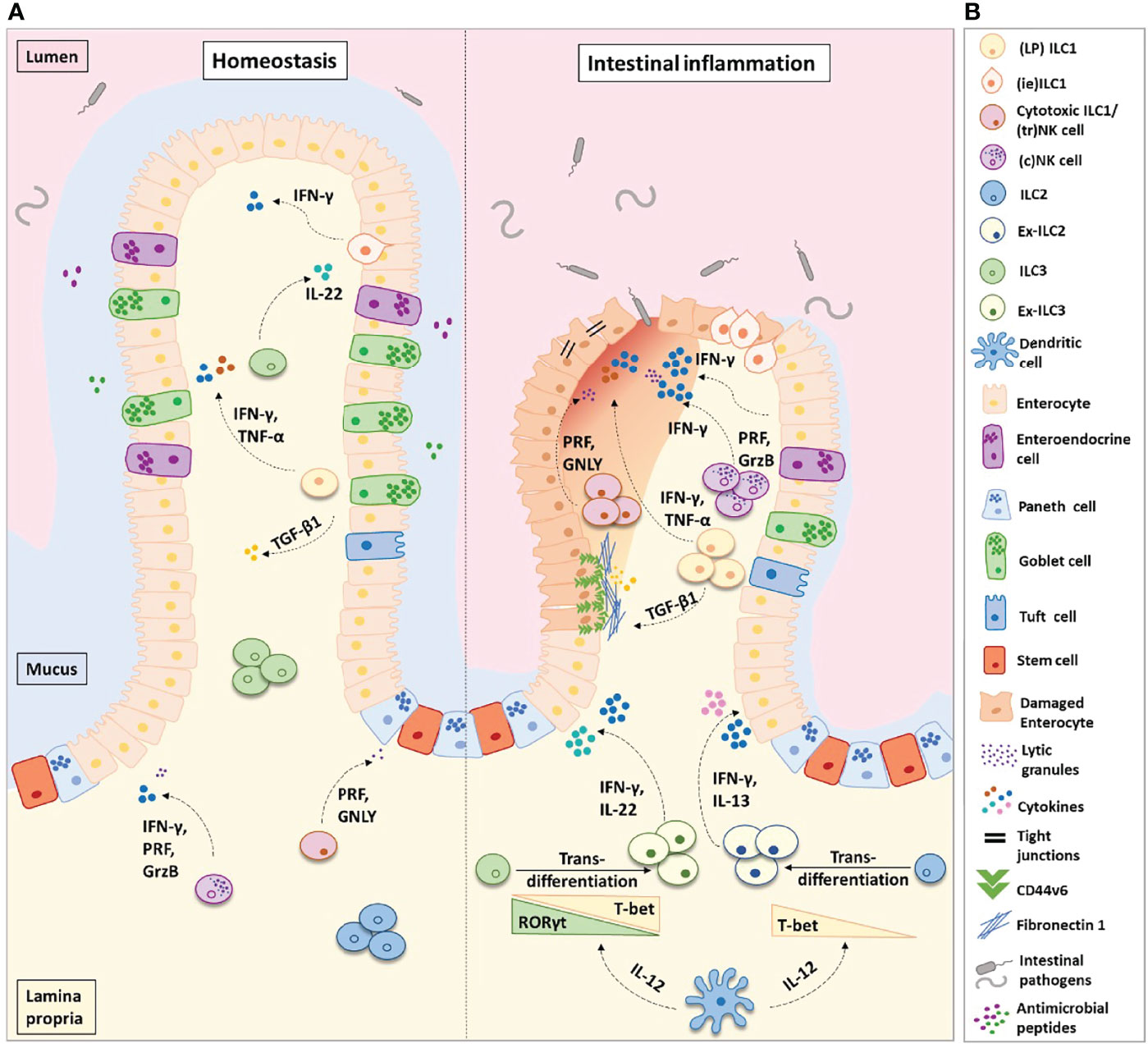
Figure 1 Helper-like type 1 ILCs (ILC1s) in homeostasis and intestinal inflammation. (A) ILC1s comprise of lamina propria (LP) ILC1s, intra-epithelial (ie) ILC1s, and cytotoxic ILC1s/tissue-resident (tr) natural killer cells. In the healthy intestinal microenvironment, ILC1s are the least frequent ILC population. In inflammatory bowel disease (IBD), the frequency of ILC1s is increased at the expense of ILC3s and ILC2s. ILC3s and ILC2s transdifferentiate into ILC1s aided by the secretion of IL-12 from dendritic cells. Ex-ILC3s can secrete both IFN-γ and IL-22 upon stimulation, while ex-ILC2s can produce both IFN-γ and IL-13. The increase in ILC1 frequency leads to an increase in pro-inflammatory cytokine secretion and augmented levels of ILC1-derived TGF-β1. TGF-β1 has been shown to increase CD44v6+ expression in epithelial and mesenchymal cells. In homeostasis, this may aid wound healing; however, in IBD this is associated with enhanced fibronectin-1 deposition and extracellular matrix degradation, consequently promoting tissue scarring. Abundant levels of IFN-γ disrupt the epithelial tight junctions, leading to increased epithelial permeability. This allows commensal, pathogenic, and opportunistic bacterial species (such as Bacteroides fragilis, Escherichia coli, Mycobacterium avium subsp. paratuberculosis, and C. difficile) to penetrate the lamina propria, thus perpetuating the inflammation. (B) Figure key illustrating the different cell types and molecules participating in intestinal inflammation. IFN-γ, interferon-gamma; IL-22, interleukin-22; IL-17, interleukin-17; IL-13, interleukin-13; IL-12, interleukin-12; TGF-β1, transforming growth factor-beta 1; TNF-α, tumor necrosis factor-alpha; PRF, perforin; GrzB, granzyme-B; GNLY, granulysin.
As observed in mice, VE-cadherin-mediated disruption of the intestinal vascular barrier by IFN-γ has additionally been described in human IBD patients, suggesting a direct causative role of IFN-γ in the pathogenicity of the disease (55). Moreover, an accumulation of ILC1s in the inflamed intestine has repeatedly been identified in patients with IBD (12–14, 18, 52, 60). This was first observed by Bernink et al., who demonstrated an accumulation of IFNG-expressing c-Kit- NKp44- CD127+ ILC1s in the intestinal LP of Crohn’s disease patients and following DSS-induced colitis in mice with a humanized immune system (12). Subsequently, these results have been confirmed in larger patient groups (15, 60) and have been shown not to be restricted to LP ILC1s but to include an increase in the proportions of cytotoxic ieILC1s/trNK cells and ILC1-like cells (CD127+ CD94+) in individuals with CD (14, 18). However, it is not currently clear whether an increase in the frequencies of ILC1s is a universal feature of IBD. Recent investigations into ILC1s in the mucosal tissue of patients newly diagnosed with CD and UC found that while LP ILC1 proportions increased at the sites of active inflammation in CD, ILC2s instead accumulated in patients with UC (48). This finding supports the controversial Th1/Th2 paradigm, in which CD is a Th1-mediated disease and UC a Th2-mediated disease (61). In contrast, using cytometry by time-of-flight analysis, the presence of NK cells and ILC1s (Lin- CD161+ T-bet+ CD56lo-) has been demonstrated in the mucosa of UC patients, albeit to a lesser extent than was present in individuals with CD (62).
In patients with active IBD, perturbations in the ILC compartment may not be restricted to the intestine, with a small increase in the frequency of c-Kit- NKp44- CD127+ CD161+ ILC1s identified in their peripheral blood (63). However, this finding is not universal with earlier reports demonstrating no alterations in circulating ILC subtypes between IBD patients and/or healthy controls (48, 62). Recently, in the absence of changes in the total frequencies of helper-like ILCs, an expansion of those exhibiting putative markers of activation was observed, including HLR-DR+ ILC1s and both HLR-DR+ and FMALF1+ ILC2s (64). This suggested that existing peripheral ILC populations may be switching from a homeostatic to an activated phenotype. While a question remains as to what extent this may impact IBD pathogenesis (65), it is notable that such a switch was additionally observed in the population of ILC precursors (ILCp); an increase in NKp44+ and CD56+ ILCp was detected in CD patients, coupled to a decrease in their corresponding naïve CD45RA+ ILCp (64). However, while circulating human ILCp has been shown to contribute to the pool of ILCs in tissues (66), the extent to which perturbations in peripheral ILCp in IBD patents may impact the ILC compartment within the intestinal mucosae remains to be determined—for example, recent research has shown that CD45RA+ ILCp exhibits tissue-specific differentiation potential (67). Moreover, the authors demonstrated an accumulation of a CD62L- subpopulation of CD45RA+ ILCp in the inflamed mucosa of patients with both UC and CD. The expansion of CD26L- ILCp was found to be inversely proportional to the frequency of NKp44+ ILC3s, suggesting that disruptions to ILCp compartments may act to promote the disequilibrium of ILCs observed in IBD (67).
The lack of long-term treatment options for IBD brings into question the principal role of pro-inflammatory cytokines as key drivers of the disease pathology—for example, the current gold-standard treatment is the use of TNF-α blockers (e.g., infliximab, adalimumab, certolizumab pegol, and golimumab), yet while this has dramatically improved the IBD outcomes for both CD and UC patients (68), only 1 in 3 initially responded to the drug, and in 30–50% of these patients, the efficacy of treatment decreases over time (69). Furthermore, despite IFN-γ signaling being shown to drive colitis in mice, a lack of efficacy was observed in treatments aimed at suppressing signaling by this pro-inflammatory cytokine in humans (70, 71). However, recent work within our laboratory has demonstrated an alternative signaling method by which ILC1s may act to exacerbate IBD-associated pathology. Using both human and murine intestinal organoids co-cultured with ILC1s, it was shown that LP ILC1-derived TGF-β can drive the degradation of the extracellular matrix, enhance fibronectin deposition, and induce the non-specific expansion of intestinal epithelial cells (16). While in homeostasis these pathways may drive epithelial regeneration and wound healing, under conditions of chronic inflammation, they have the potential to be pathological (15)—for example, TGF-β has previously been characterized as an upstream regulator of fibrosis in CD (72, 73). Notably, despite the organoids used here being genetically, epigenetically, and environmentally identical, pathological matrix remodeling was only observed in the hyper-proliferative ILC1s isolated from the inflamed tissue of CD patients (16). This concurred with previous research in 2019 by Cella et al., who observed a significant increase in a population of proliferative ILC1s in the ileum of individuals with CD (15). These data together highlight a novel non-canonical function of ILC1 in the exacerbation of IBD, in which their augmented expansion may be coupled to enhanced TGF-β signaling, consequentially driving the inflamed tissues toward fibrotic scar tissue formation and/or promoting the growth of epithelial tumors during chronic inflammation, two life-threatening sequalae of IBD (74).
Interestingly, Cella et al. additionally identified a transitionary population of ILC3–ILC1 cells in both CD patients and non-disease controls. As with the conversion of NK cells to ILC1s in the TGF-β-rich tumor microenvironment (35), this transition was demonstrated to be mediated via TGF-β signaling in both mice and humans (15, 42). Considering that the accumulation of ILC1s in IBD is coupled to a decrease in the frequency of ILC3s (12, 48, 60), a question remains as to what extent this is reflective of an expansion of the existing ILC1 population or an increased conversion of other ILC subtypes to an “ILC1-like” phenotype or both. Moreover, TGF-β has also been shown to impair NK cell activity (75), and the presence of dysregulated NK cells has been observed in the blood of IBD patients (76). Thus, while these data do not show a direct relationship to intestinal inflammation, they highlight the need for further investigations into the pleiotropic role of TGF-β signaling in the dysregulated immune compartment found within IBD.
ILC1s represent a minor fraction of the innate cell population in the intestinal LP of healthy individuals. When present in small numbers, ILC1-derived TGF-β may contribute to the maintenance of epithelial regeneration (16). However, the expansion of their population at sites of active intestinal inflammation may indicate their potential role as inducer and/or driver of IBD pathogenicity, which is linked to the development of IBD sequelae such as fibrosis and cancer (16). Critically, there are still many key questions that need to be addressed regarding their presence in inflamed tissues. Do ILC1s accumulate in the intestinal lamina propria of CD patients as a cause or consequence of inflammation? Does the pro-inflammatory microenvironment lead to the augmented plasticity of ILC2s and ILC3s, driving their transition towards ILC1s? Or does it promote ILC1 survival and expansion via the secretion of the cytokines and growth factors that they depend on? To what extent may perturbed blood ILC compartments impact or reflect on those observed within the intestine? Does the inflamed microenvironment observed in CD imprint a highly proliferative state on ILC1s? Are the effects on ILC1s a result of “trained” immunity as recently reported for murine ILC3 (77)?
The development of new experimental techniques, such as the co-culture of human organoids with ILCs (78, 79) and single-cell multiomics, provides new avenues to address these questions. Such studies will lead to a better understanding of the role of ILC1 in IBD. Importantly, they will contribute to the development of new therapeutic approaches to revert the pathogenic accumulation of ILC1s in inflamed tissues.
JN and DC conceptualized the manuscript’s theme. DC, IC, LR, and JN designed and reviewed the manuscript. DC and IC researched and wrote the manuscript. DC designed the figure. IC constructed the table. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
DC acknowledges a Ph.D. studentship from the National Institute of Health and Care Research (NIHR) Biomedical Research Centre (BRC) based at Guy’s and St Thomas’ (GSTT) NHS Foundation Trust and King’s College London (KCL), and LR is also supported by NIHR BRC based at GSTT and KCL. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care. We apologize to authors whose papers we could not include due to space and scope limitations.
1. Kobayashi T, Siegmund B, le Berre C, Wei SC, Ferrante M, Shen B, et al. Ulcerative Colitis. Nat Rev Dis Primers (2020) 6(1):1–20. doi: 10.1038/s41572-020-0205-x
2. Roda G, Chien Ng S, Kotze PG, Argollo M, Panaccione R, Spinelli A, et al. Crohn’s Disease. Nat Rev Dis Primers (2020) 6(1):1–19. doi: 10.1038/s41572-020-0156-2
3. Graham DB, Xavier RJ. Pathway Paradigms Revealed From the Genetics of Inflammatory Bowel Disease. Nature (2020) 578(7796):527–39. doi: 10.1038/s41586-020-2025-2
4. Ungaro F, Massimino L, Furfaro F, Rimoldi V, Peyrin-Biroulet L, D’Alessio S, et al. Metagenomic Analysis of Intestinal Mucosa Revealed a Specific Eukaryotic Gut Virome Signature in Early-Diagnosed Inflammatory Bowel Disease. Gut Microbes (2019) 10:149–58. doi: 10.1080/19490976.2018.1511664
5. Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases. Nature (2019) 569(7758):655–62. doi: 10.1038/s41586-019-1237-9
6. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host–microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature (2012) 491(7422):119–24. doi: 10.1038/nature11582
7. Caruso R, Lo BC, Núñez G. Host–microbiota Interactions in Inflammatory Bowel Disease. Nat Rev Immunol (2020) 20(7):411–26. doi: 10.1038/s41577-019-0268-7
8. Ma Y, Zhang Y, Xiang J, Xiang S, Zhao Y, Xiao M, et al. Metagenome Analysis of Intestinal Bacteria in Healthy People, Patients With Inflammatory Bowel Disease and Colorectal Cancer. Front Cell Infect Microbiol (2021) 11. doi: 10.3389/FCIMB.2021.599734
9. Zhang M, Sun K, Wu Y, Yang Y, Tso P, Wu Z, et al. Interactions Between Intestinal Microbiota and Host Immune Response in Inflammatory Bowel Disease. Front Immunol (2017) 8:942. doi: 10.3389/fimmu.2017.00942
10. Vivier E, Artis D, Colonna M, Diefenbach A, di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years on. Cell (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
11. Meininger I, Carrasco A, Rao A, Soini T, Kokkinou E, Mjösberg J, et al. Tissue-Specific Features of Innate Lymphoid Cells. Trends Immunol (2020) 41:902–17. doi: 10.1016/j.it.2020.08.009
12. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human Type 1 Innate Lymphoid Cells Accumulate in Inflamed Mucosal Tissues. Nat Immunol (2013) 14(3):221–9. doi: 10.1038/ni.2534
13. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 Control Plasticity of CD127+ Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity (2015) 43:146–60. doi: 10.1016/j.immuni.2015.06.019
14. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial Type 1 Innate Lymphoid Cells are a Unique Subset of Il-12- and Il-15-Responsive Ifn-γ-Producing Cells. Immunity (2013) 38:769–81. doi: 10.1016/j.immuni.2013.02.010
15. Cella M, Gamini R, Sécca C, Collins PL, Zhao S, Peng V, et al. Subsets of ILC3–ILC1-Like Cells Generate a Diversity Spectrum of Innate Lymphoid Cells in Human Mucosal Tissues. Nat Immunol (2019) 20(8):980–91. doi: 10.1038/s41590-019-0425-y
16. Jowett GM, Norman MDA, Yu TTL, Rosell Arévalo P, Hoogland D, Lust ST, et al. ILC1 Drive Intestinal Epithelial and Matrix Remodelling. Nat Mater (2021) 20(2):250–9. doi: 10.1038/s41563-020-0783-8
17. Riggan L, Freud AG, O’Sullivan TE. True Detective: Unraveling Group 1 Innate Lymphocyte Heterogeneity. Trends Immunol (2019) 40:909–21. doi: 10.1016/j.it.2019.08.005
18. Krabbendam L, Heesters BA, Kradolfer CMA, Haverkate NJE, Becker MAJ, Buskens CJ, et al. CD127+ CD94+ Innate Lymphoid Cells Expressing Granulysin and Perforin Are Expanded in Patients With Crohn’s Disease. Nat Commun (2021) 12(1):1–11. doi: 10.1038/s41467-021-26187-x
19. Mohamed R, Lord GM. T-Bet as a Key Regulator of Mucosal Immunity. Immunology (2016) 147:367–76. doi: 10.1111/imm.12575
20. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue Residency of Innate Lymphoid Cells in Lymphoid and Nonlymphoid Organs. Science (2015) 350:981–5. doi: 10.1126/science.aac9593
21. McFarland AP, Yalin A, Wang SY, Cortez VS, Landsberger T, Sudan R, et al. Multi-Tissue Single-Cell Analysis Deconstructs the Complex Programs of Mouse Natural Killer and Type 1 Innate Lymphoid Cells in Tissues and Circulation. Immunity (2021) 54:1320–37.e4. doi: 10.1016/j.immuni.2021.03.024
22. Vosshenrich CAJ, García-Ojeda ME, Samson-Villéger SI, Pasqualetto V, Enault L, Goff le OR, et al. A Thymic Pathway of Mouse Natural Killer Cell Development Characterized by Expression of GATA-3 and CD127. Nat Immunol (2006) 7(11):1217–24. doi: 10.1038/ni1395
23. Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of Type 1 ILCs From a Common Progenitor to All Helper-Like Innate Lymphoid Cell Lineages. Cell (2014) 157:340–56. doi: 10.1016/j.cell.2014.03.030
24. Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, et al. Involvement of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Surveillance of Tumor Metastasis by Liver Natural Killer Cells. Nat Med (2001) 7(1):94–100. doi: 10.1038/83416
25. Cortez VS, Fuchs A, Cella M, Gilfillan S, Colonna M. Cutting Edge: Salivary Gland NK Cells Develop Independently of Nfil3 in Steady-State. J Immunol (2014) 192:4487–91. doi: 10.4049/jimmunol.1303469
26. Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, et al. Tissue-Resident Natural Killer (NK) Cells are Cell Lineages Distinct From Thymic and Conventional Splenic NK Cells. Elife (2014) 3:e01659. doi: 10.7554/eLife.01659
27. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional Programs Define Molecular Characteristics of Innate Lymphoid Cell Classes and Subsets. Nat Immunol (2015) 16(3):306–17. doi: 10.1038/ni.3094
28. Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell (2016) 166:1231–46.e13. doi: 10.1016/j.cell.2016.07.043
29. Shih HY, Sciumè G, Mikami Y, Guo L, Sun HW, Brooks SR, et al. Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell (2016) 165:1120–33. doi: 10.1016/j.cell.2016.04.029
30. Mazzurana L, Czarnewski P, Jonsson V, Wigge L, Ringnér M, Williams TC, et al. Tissue-Specific Transcriptional Imprinting and Heterogeneity in Human Innate Lymphoid Cells Revealed by Full-Length Single-Cell RNA-Sequencing. Cell Res (2021) 31(5):554–68. doi: 10.1038/s41422-020-00445-x
31. Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The Transcription Factors T-Bet and Eomes Control Key Checkpoints of Natural Killer Cell Maturation. Immunity (2012) 36:55–67. doi: 10.1016/j.immuni.2011.11.016
32. Park E, Patel S, Wang Q, Andhey P, Zaitsev K, Porter S, et al. Toxoplasma Gondii Infection Drives Conversion of NK Cells Into ILC1-Like Cells. Elife (2019) 8. doi: 10.7554/eLife.47605
33. Weizman O, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli-Esposti MA, et al, et al. ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell (2017) 171:795–808.e12. doi: 10.1016/j.cell.2017.09.052
34. Bezman NA, Kim CC, Sun JC, Min-Oo G, Hendricks DW, Kamimura Y, et al. Molecular Definition of the Identity and Activation of Natural Killer Cells. Nat Immunol (2012) 13. doi: 10.1038/ni.2395
35. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor Immunoevasion by the Conversion of Effector NK Cells Into Type 1 Innate Lymphoid Cells. Nat Immunol (2017) 18(9):1004–15. doi: 10.1038/ni.3800
36. Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, et al. SMAD4 Impedes the Conversion of NK Cells Into ILC1-Like Cells by Curtailing non-Canonical TGF-β Signaling. Nat Immunol (2017) 18(9):995–1003. doi: 10.1038/ni.3809
37. Bal SM, Golebski K, Spits H. Plasticity of Innate Lymphoid Cell Subsets. Nat Rev Immunol (2020) 20(9):552–65. doi: 10.1038/s41577-020-0282-9
38. Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, et al. Regulated Expression of Nuclear Receptor Rorγt Confers Distinct Functional Fates to NK Cell Receptor-Expressing Rorγt+ Innate Lymphocytes. Immunity (2010) 33:736–51. doi: 10.1016/j.immuni.2010.10.017
39. Klose CSN, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, D’Hargues Y, et al. A T-Bet Gradient Controls the Fate and Function of CCR6–Rorγt+ Innate Lymphoid Cells. Nature (2013) 494(7436):261–5. doi: 10.1038/nature11813
40. Rankin LC, Groom JR, Chopin M, Herold MJ, Walker JA, Mielke LA, et al. The Transcription Factor T-Bet is Essential for the Development of NKp46+ Innate Lymphocytes via the Notch Pathway. Nat Immunol (2013) 14:389–95. doi: 10.1038/ni.2545
41. Tizian C, Lahmann A, Hölsken O, Cosovanu C, Kofoed-Branzk M, Heinrich F, et al. C-Maf Restrains T-Bet-Driven Programming of CCR6-Negative Group 3 Innate Lymphoid Cells. Elife (2020) 9:e52549. doi: 10.7554/eLife.52549
42. Viant C, Rankin LC, Girard-Madoux MJH, Seillet C, Shi W, Smyth MJ, et al. Transforming Growth Factor-β and Notch Ligands Act as Opposing Environmental Cues in Regulating the Plasticity of Type 3 Innate Lymphoid Cells. Sci Signaling (2016) 9:426. doi: 10.1126/scisignal.aaf2176
43. Schroeder JH, Roberts LB, Meissl K, Lo JW, Hromadová D, Hayes K, et al. Sustained Post-Developmental T-Bet Expression Is Critical for the Maintenance of Type One Innate Lymphoid Cells In Vivo. Front Immunol (2021) 12:4446. doi: 10.3389/fimmu.2021.760198
44. Muraoka WT, Korchagina AA, Xia Q, Shein SA, Jing X, Lai Z, et al. Campylobacter Infection Promotes Ifnγ-Dependent Intestinal Pathology via ILC3 to ILC1 Conversion. Mucosal Immunol (2021) 14:703–16. doi: 10.1038/s41385-020-00353-8
45. Björklund AK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, et al. The Heterogeneity of Human CD127+ Innate Lymphoid Cells Revealed by Single-Cell RNA Sequencing. Nat Immunol (2016) 17(4):451–60. doi: 10.1038/ni.3368
46. Sagebiel AF, Steinert F, Lunemann S, Körner C, Schreurs RRCE, Altfeld M, et al. Tissue-Resident Eomes+ NK Cells are the Major Innate Lymphoid Cell Population in Human Infant Intestine. Nat Commun (2019) 10(1):1–13. doi: 10.1038/s41467-018-08267-7
47. Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo SL, Loh CY, et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity (2017) 46:148–61. doi: 10.1016/j.immuni.2016.11.005
48. Forkel M, VanTol S, Höög C, Michaëlsson J, Almer S, Mjösberg J, et al. Distinct Alterations in the Composition of Mucosal Innate Lymphoid Cells in Newly Diagnosed and Established Crohn’s Disease and Ulcerative Colitis. J Crohn’s Colitis (2019) 13:67–78. doi: 10.1093/ecco-jcc/jjy119
49. Yudanin NA, Schmitz F, Flamar AL, Thome JJC, Tait Wojno E, Moeller JB, et al. Spatial and Temporal Mapping of Human Innate Lymphoid Cells Reveals Elements of Tissue Specificity. Immunity (2019) 50:505–19.e4. doi: 10.1016/j.immuni.2019.01.012
50. Koues OI, Collins PL, Cella M, Robinette ML, Porter SI, Pyfrom SC, et al. Distinct Gene Regulatory Pathways for Human Innate Versus Adaptive Lymphoid Cells. Cell (2016) 165:1134–46. doi: 10.1016/j.cell.2016.04.014
51. Collins PL, Cella M, Porter SI, Li S, Gurewitz GL, Hong HS, et al. Gene Regulatory Programs Conferring Phenotypic Identities to Human NK Cells. Cell (2019) 176:348–60.e12. doi: 10.1016/j.cell.2018.11.045
52. Cella M, Otero K, Colonna M. Expansion of Human NK-22 Cells With IL-7, IL-2, and IL-1β Reveals Intrinsic Functional Plasticity. Proc Natl Acad Sci USA (2010) 107:10961–6. doi: 10.1073/pnas.1005641107
53. Mazzurana L, Forkel M, Rao A, van Acker A, Kokkinou E, Ichiya T, et al. Suppression of Aiolos and Ikaros Expression by Lenalidomide Reduces Human ILC3–ILC1/NK Cell Transdifferentiation. Eur J Immunol (2019) 49:1344–55. doi: 10.1002/eji.201848075
54. Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Susac B, et al. Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection Against Acute Clostridium Difficile Infection. Cell Host Microbe (2015) 18:27–37. doi: 10.1016/j.chom.2015.06.011
55. Langer V, Vivi E, Regensburger D, Winkler TH, Waldner MJ, Rath T, et al. IFN-γ Drives Inflammatory Bowel Disease Pathogenesis Through VE-Cadherin-Directed Vascular Barrier Disruption. J Clin Invest (2019) 129:4691–707. doi: 10.1172/JCI124884
56. Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, et al. Interferon-Gamma Regulates Intestinal Epithelial Homeostasis Through Converging Beta-Catenin Signaling Pathways. Immunity (2010) 32:392–402. doi: 10.1016/j.immuni.2010.03.001
57. Ito R, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, et al. Interferon-Gamma Is Causatively Involved in Experimental Inflammatory Bowel Disease in Mice. Clin Exp Immunol (2006) 146:330–8. doi: 10.1111/j.1365-2249.2006.03214.x
58. Powell N, Walker AW, Stolarczyk E, Canavan JB, Gökmen MR, Marks E, et al. The Transcription Factor T-Bet Regulates Intestinal Inflammation Mediated by Interleukin-7 Receptor+ Innate Lymphoid Cells. Immunity (2012) 37:674–84. doi: 10.1016/j.immuni.2012.09.008
59. Xiao YT, Yan WH, Cao Y, Yan JK, Cai W. Neutralization of IL-6 and TNF-α Ameliorates Intestinal Permeability in DSS-Induced Colitis. Cytokine (2016) 83:189–92. doi: 10.1016/j.cyto.2016.04.012
60. Li J, Doty AL, Tang Y, Berrie D, Iqbal A, Tan SA, et al. Enrichment of IL-17a + IFN-γ + and IL-22 + IFN-γ + T Cell Subsets is Associated With Reduction of NKp44 + ILC3s in the Terminal Ileum of Crohn’s Disease Patients. Clin Exp Immunol (2017) 190:143–53. doi: 10.1111/cei.12996
61. Li J, Ueno A, Gasia MF, Luider J, Wang T, Hirota C, et al. Profiles of Lamina Propria T Helper Cell Subsets Discriminate Between Ulcerative Colitis and Crohn’s Disease. Inflamma Bowel Dis (2016) 22:1779–92. doi: 10.1097/MIB.0000000000000811
62. Mitsialis V, Wall S, Liu P, Ordovas-Montanes J, Parmet T, Vukovic M, et al. Single-Cell Analyses of Colon and Blood Reveal Distinct Immune Cell Signatures of Ulcerative Colitis and Crohn’s Disease. Gastroenterology (2020) 159:591–608.e10. doi: 10.1053/J.GASTRO.2020.04.074
63. Creyns B, Jacobs I, Verstockt B, Cremer J, Ballet V, Vandecasteele R, et al. Biological Therapy in Inflammatory Bowel Disease Patients Partly Restores Intestinal Innate Lymphoid Cell Subtype Equilibrium. Front Immunol (2020) 11:1847. doi: 10.3389/fimmu.2020.01847
64. Mazzurana L, Bonfiglio F, Forkel M, D’Amato M, Halfvarson J, Mjösberg J, et al. Crohn’s Disease Is Associated With Activation of Circulating Innate Lymphoid Cells. Inflamma Bowel Dis (2021) 27:1128–38. doi: 10.1093/ibd/izaa316
65. Farber DL. Tissues, Not Blood, are Where Immune Cells Function. Nature (2021) 593(7860):506–9. doi: 10.1038/d41586-021-01396-y
66. Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, et al. Systemic Human ILC Precursors Provide a Substrate for Tissue ILC Differentiation. Cell (2017) 168:1086–100.e10. doi: 10.1016/j.cell.2017.02.021
67. Kokkinou E, Pandey RV, Mazzurana L, Gutierrez-Perez I, Tibbitt CA, Weigel W, et al. CD45RA+CD62L– ILCs in Human Tissues Represent a Quiescent Local Reservoir for the Generation of Differentiated ILCs. Sci Immunol (2022) 7:8301. doi: 10.1126/sciimmunol.abj8301
68. Vulliemoz M, Brand S, Juillerat P, Mottet C, Ben-Horin S, Michetti P, et al. TNF-Alpha Blockers in Inflammatory Bowel Diseases: Practical Recommendations and a User’s Guide: An Update. Digestion (2020) 101 Suppl 1:16–26. doi: 10.1159/000506898
69. Bank S, Andersen PS, Burisch J, Pedersen N, Roug S, Galsgaard J, et al. Genetically Determined High Activity of IL-12 and IL-18 in Ulcerative Colitis and TLR5 in Crohns Disease Were Associated With Non-Response to Anti-TNF Therapy. Pharmacogenom J (2018) 18:87–97. doi: 10.1038/tpj.2016.84
70. Hommes DW, Mikhajlova TL, Stoinov S, Štimac D, Vucelic B, Lonovics J, et al. Fontolizumab, a Humanised Anti-Interferon Gamma Antibody, Demonstrates Safety and Clinical Activity in Patients With Moderate to Severe Crohn’s Disease. Gut (2006) 55:1131–7. doi: 10.1136/gut.2005.079392
71. Reinisch W, de Villiers W, Bene L, Simon L, Rácz I, Katz S, et al. Fontolizumab in Moderate to Severe Crohn’s Disease: A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Multiple-Dose Study. Inflammation Bowel Dis (2010) 16:233–42. doi: 10.1002/ibd.21038
72. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: The Master Regulator of Fibrosis. Nat Rev Nephrol (2016) 12(6):325–38. doi: 10.1038/nrneph.2016.48
73. Györfi AH, Matei AE, Distler JHW. Targeting TGF-β Signaling for the Treatment of Fibrosis. Matrix Biol (2018) 68–69:8–27. doi: 10.1016/j.matbio.2017.12.016
74. Shimshoni E, Yablecovitch D, Baram L, Dotan I, Sagi I. ECM Remodelling in IBD: Innocent Bystander or Partner in Crime? The Emerging Role of Extracellular Molecular Events in Sustaining Intestinal Inflammation. Gut (2015) 64:367–72. doi: 10.1136/gutjnl-2014-308048
75. Viel S, Marçais A, Guimaraes FSF, Loftus R, Rabilloud J, Grau M, Degouve S, et al, et al. TGF-β Inhibits the Activation and Functions of NK Cells by Repressing the mTOR Pathway. Sci Signaling (2016) 9:415. doi: 10.1126/scisignal.aad1884
76. Zaiatz Bittencourt V, Jones F, Tosetto M, Doherty GA, Ryan EJ. Dysregulation of Metabolic Pathways in Circulating Natural Killer Cells Isolated From Inflammatory Bowel Disease Patients. J Crohn’s Colitis (2021) 15:1316–25. doi: 10.1093/ecco-jcc/jjab014
77. Serafini N, Jarade A, Surace L, Goncalves P, Sismeiro O, Varet H, et al. Trained ILC3 Responses Promote Intestinal Defense. Science (2022) 375:859–63. doi: 10.1126/science.aaz8777
78. Read E, Jowett GM, Coman D, Neves JF. Co-Culture of Murine Small Intestine Epithelial Organoids With Innate Lymphoid Cells. JoVE (Journal Visualized Experiments) (2022) 181:e63554. doi: 10.3791/63554
Keywords: innate lymphocyte cells, intestine, inflammatory bowel disease, inflammation, natural killer cell, type 1 innate lymphoid cells
Citation: Coman D, Coales I, Roberts LB and Neves JF (2022) Helper-Like Type-1 Innate Lymphoid Cells in Inflammatory Bowel Disease. Front. Immunol. 13:903688. doi: 10.3389/fimmu.2022.903688
Received: 24 March 2022; Accepted: 17 May 2022;
Published: 23 June 2022.
Edited by:
Carolina Jancic, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Sabrina B. Bennstein, Heinrich Heine University of Düsseldorf, GermanyCopyright © 2022 Coman, Coales, Roberts and Neves. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joana F. Neves, am9hbmEucGVyZWlyYV9kYXNfbmV2ZXNAa2NsLmFjLnVr
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.