 Sonia Aristin Revilla
Sonia Aristin Revilla Onno Kranenburg
Onno Kranenburg Paul J. Coffer
Paul J. Coffer- 1Center Molecular Medicine, University Medical Center Utrecht, Utrecht, Netherlands
- 2Regenerative Medicine Center, University Medical Center Utrecht, Utrecht, Netherlands
- 3Laboratory Translational Oncology, University Medical Center Utrecht, Utrecht, Netherlands
Colorectal cancer (CRC) is a heterogeneous disease with one of the highest rates of incidence and mortality among cancers worldwide. Understanding the CRC tumor microenvironment (TME) is essential to improve diagnosis and treatment. Within the CRC TME, tumor-infiltrating lymphocytes (TILs) consist of a heterogeneous mixture of adaptive immune cells composed of mainly anti-tumor effector T cells (CD4+ and CD8+ subpopulations), and suppressive regulatory CD4+ T (Treg) cells. The balance between these two populations is critical in anti-tumor immunity. In general, while tumor antigen-specific T cell responses are observed, tumor clearance frequently does not occur. Treg cells are considered to play an important role in tumor immune escape by hampering effective anti-tumor immune responses. Therefore, CRC-tumors with increased numbers of Treg cells have been associated with promoting tumor development, immunotherapy failure, and a poorer prognosis. Enrichment of Treg cells in CRC can have multiple causes including their differentiation, recruitment, and preferential transcriptional and metabolic adaptation to the TME. Targeting tumor-associated Treg cell may be an effective addition to current immunotherapy approaches. Strategies for depleting Treg cells, such as low-dose cyclophosphamide treatment, or targeting one or more checkpoint receptors such as CTLA-4 with PD-1 with monoclonal antibodies, have been explored. These have resulted in activation of anti-tumor immune responses in CRC-patients. Overall, it seems likely that CRC-associated Treg cells play an important role in determining the success of such therapeutic approaches. Here, we review our understanding of the role of Treg cells in CRC, the possible mechanisms that support their homeostasis in the tumor microenvironment, and current approaches for manipulating Treg cells function in cancer.
Introduction
Colorectal cancer (CRC) is the third most diagnosed cancer worldwide, and the second main cause of cancer-related death (1). The main causes of high mortality are late diagnosis and high relapse after treatment, with 70% of patients only being diagnosed when the tumor has metastasized to distant organs such as liver (20%-70%) and lung (10%-20%) (2, 3). CRC patients are a heterogeneous group, where differences at the molecular and genetic level influence clinical outcomes and response to therapy (4, 5). Seventy percent of CRC is of mostly sporadic origin, while 20-30% of cases are familial. From these, approximately one-third are caused by highly penetrant inherited mutations that have been well characterized, so-called hereditary CRC. The etiology of the remaining familial cases is not completely understood but is often associated with lower-penetrance susceptibility genes and specific polymorphisms regulated by environmental, or other genetic inherited factors, that when co-inherited increase cancer risk (6–8). Additionally, intestinal inflammatory responses related to inflammatory bowel diseases (IBDs) can result in the development of CRC (9). Prolonged and severe inflammation often correlates with increased cancer risk (10). Colorectal polyps or adenomas arise from the epithelial cells lining the colon and are considered precursors to tumor development. During the initiation of CRC, epithelial cells acquire genetic and epigenetic modifications increasing the risk of malignant transformation and conferring a selective advantage. These can increase in size and the degree of dysplasia eventually leading to the development of dysplastic adenomas that subsequently evolve to adenoma-carcinoma (4, 5). The immune response can recognize and target tumor-specific and tumor-associated neo-antigens thereby arresting CRC development. Here, CD8+ cytotoxic T lymphocytes (CTLs) play an essential role, with the cooperation of CD4+ helper T (Th) cells. However, CRC tumors can manipulate the tumor microenvironment (TME), promoting cancer outgrowth through regulation of immune infiltration, and generating an immunosuppressive and inflammatory environment. In this way, the TME plays a key role in modulating the plasticity of tumor cells and providing immune escape mechanisms (11–14).
The CRC Tumor Microenvironment and Tumor-Infiltrating Lymphocytes
The TME includes malignant tumor cells together with immune cells, stromal cells, endothelial cells, extracellular matrix (ECM), cytokines, chemokines, and other soluble mediators (14–17). Cancer development in general, and CRC progression in particular, are linked with the complex role that the immune response plays in the early stages of tumor development (15, 18–20). The immune system has a multi-faceted role in CRC development (15) and has a substantial impact on patient outcome (18). Tumor-infiltrating immune cells (TIICs) include adaptive immune cells (T cells and B cells), natural killer (NK) cells, macrophages, and additional innate cells such as myeloid-derived suppressor cells (MDSCs) (14, 21, 22). TIICs directly participate in the generation and maintenance of an actively suppressive anti-tumor immune response supporting CRC progression (23). MDSCs secrete cytokines and express immunosuppressive molecules at their surface that can inhibit T cells, B cells, and NK cells while promoting Treg cells and tumor-associated macrophages (TAMs) (24, 25). The most enriched population of TIICs are tumor-infiltrating lymphocytes (TILs) that promote inflammation and are often used to evaluate disease prognosis (26, 27). TILs are a heterogeneous mixture of lymphocytes composed mainly of anti-tumor effector T cells (CD4+ and CD8+ subpopulations) and immunosuppressive Treg cells (27, 28). The balance between these two populations is critical in anti-tumor immunity. In general, while tumor antigen-specific T cell responses are observed, tumor clearance frequently does not occur, highlighting the important role of immunosuppressive Treg cells (29, 30). Since tumors can directly shape TIICs populations and function, this interplay provides the capacity to escape immunosurveillance (15, 18). TILs are also associated with the molecular classification of CRC tumors, and with patient prognosis and immunotherapy responses (21, 23, 31). CRC-tumors composed of mainly anti-tumor T cells, particularly type 1 helper T cells (Th1) and cytotoxic CD8+ T cells, correlate with favorable prognosis and survival (32, 33). In contrast, increased accumulation of Treg cells is generally associated with CRC progression and metastasis, immunotherapy failure, and a poorer prognosis, although this correlation is not definitive (34–40). Understanding the dynamics between CRC tumors and immune cells in the TME is essential to improve both diagnosis and treatment.
FOXP3+ Regulatory T Cells
FOXP3+ regulatory T cells, hereafter termed Treg cells, are essential in the maintenance of immunological homeostasis and self-tolerance (41–43). They can have broad suppressive activity, secreting immunomodulatory cytokines and cytolytic molecules allowing them to regulate immune responses (44). Treg cells have been well-studied due to their capacity to regulate the function of a wide variety immune cells including lymphocytes, dendritic cells (DCs), and macrophages (42). The depletion or reduction of Treg cells is associated with beneficial anti-tumor immune responses and eradication of microbes during chronic infections (45, 46). The concept of Treg cells as suppressor T cells was first proposed in the 1970s (47–49) and was later further defined as a subpopulation of approximately 10% of peripheral CD4+ T-cells expressing the IL-2 receptor alpha-chain (CD25) (50). However, the use of CD25 to identify Treg cells is problematic since it is a general marker of activated T-cells (51, 52). Subsequently, in a breakthrough for the field, the forkhead box P3 transcription factor (Foxp3) was found to be an additional marker of Treg cell ontogeny (53–57). Subsequent studies in both mice and humans identified FOXP3 is a master regulator of both Treg cell development and function (58, 59). Naïve CD4+ T cells can transiently upregulate CD25 and FOXP3 expression upon activation (60) and human Treg cells are usually characterized by the expression of FOXP3, CD25, and the low expression of the IL-7 receptor alpha chain, CD127 (61).
In the thymus, tTreg cell differentiation is driven by strong T cell receptor (TCR)-mediated recognition of self-antigens (62, 63). In secondary lymphoid organs, peripheral Treg cells (pTreg) can differentiate from naïve or effector CD4+ T cells through TCR stimulation in the presence of TGF-β and IL-2, inducing FOXP3 expression (63–65). The pTreg cell TCR repertoire recognizes foreign-antigens, as opposed to tTreg that are biased towards self-recognition (66, 67). Furthermore, the expression of transcription factor Helios and the cell-surface glycoprotein neuropilin-1 (Nrp1) can be used to distinguish tTreg that highly express both factors from pTreg that poorly express them. However, pTreg can also upregulate Helios and Nrp1 under inflammatory conditions, or in response to certain activation signals such as IL-2 stimulation (68–70).
FOXP3-expressing CD4+ T cells can be further subdivided into two main subsets: central Treg cells (cTreg cells) and effector Treg cells (eTreg cells). However, in humans, activated Teff cells can transiently express FOXP3 and secrete pro-inflammatory cytokines (IL-17, IL-2, and IFNγ), however they are considered to be non-suppressive (71, 72). cTreg cells are recent thymic emigrants that have not yet been activated and exhibit a naïve phenotype (CD45RA+ FOXP3lo) with low suppressive activity. They are enriched in lymphoid tissues where they express lymphoid-tissue homing molecules including CD62Lhigh, CD44low and CCR7, and are dependent on IL-2 for maintaining their quiescent state (73). In secondary lymphoid organs, cTreg cells undergo further differentiation upon TCR engagement to become eTreg cells (CD45RA- FOXP3hi). These cells proliferate, further and develop into highly suppressive cells, upregulating activation markers, immunosuppressive cytokines, chemokines, and their receptors. They subsequently migrate to non-lymphoid tissues, downregulate lymphoid-tissue homing molecules (CD62Llow, CD44high) and acquire a tissue-specific transcriptional signature associated with their role in each location (63, 74, 75). eTreg cell suppressive function correlates with co-expression of FOXP3 and lineage-specific transcription factors common the Teff cells population (Table 1).
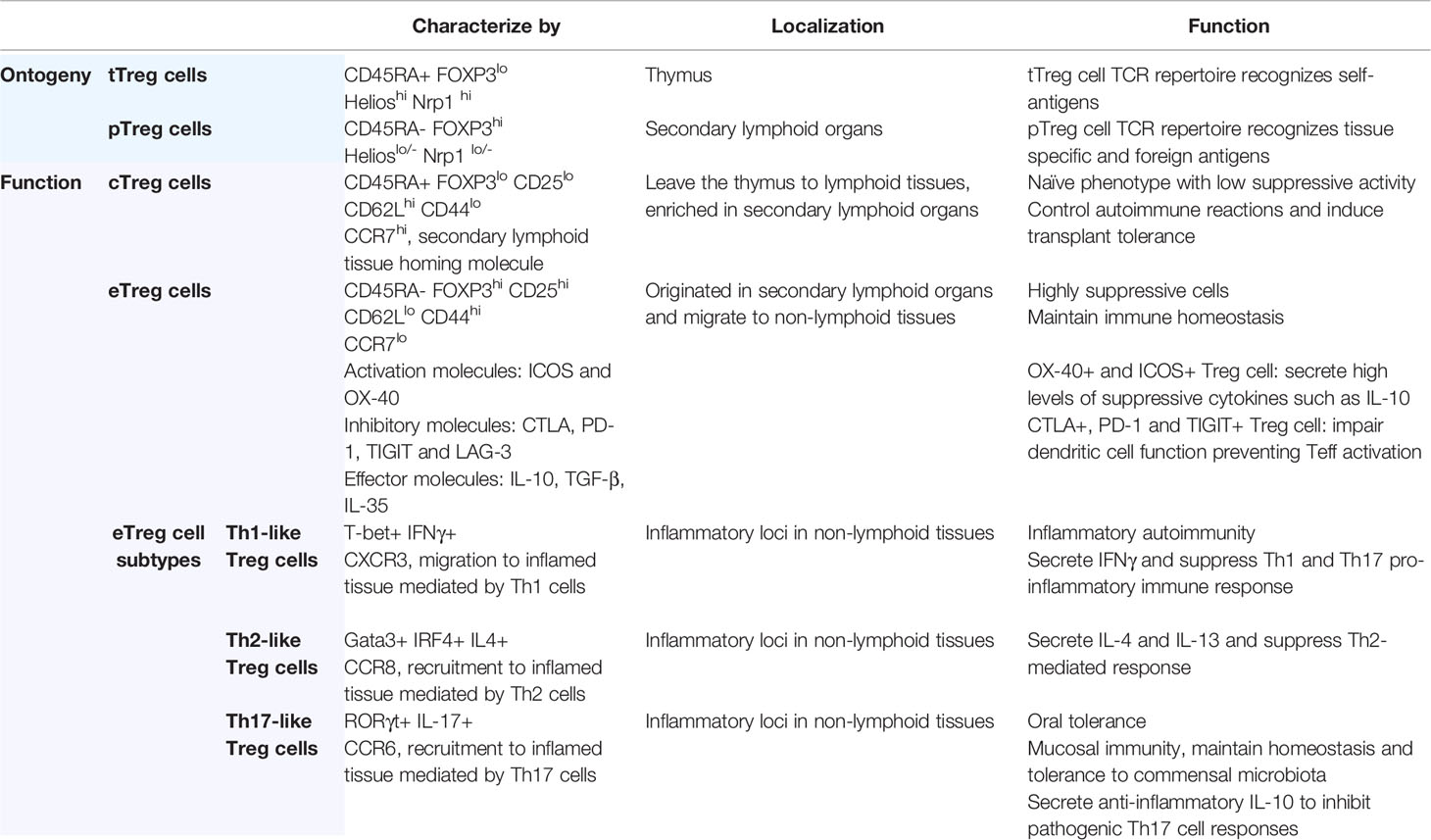
Table 1 Treg cells subtypes.
Histone post-translational modifications and DNA methylation patterns are intimately associated with the differentiation and function of Treg cells (76–78). FOXP3 transcription is epigenetically controlled through regulation of its promoter and several intronic enhancers, termed conserved non-coding DNA sequences (CNS) 0–3 (77, 79). The demethylation status of CNS1 and CNS2 is regulated by TGF-β and IL-2 and associated with the stable expression of Foxp3 (80–82). TET1/2 methylcytosine dioxygenases demethylate CNS1/2 thereby stabilizing Foxp3 expression and other Treg cell signature genes including Cd25, Nrp1, and Il1rl1 (82). Mice with CNS1‐deficiencies can generate tTreg cells but not pTreg cells, leading to a deficiency of colonic Treg cells, development of aggressive inflammation in the mucosa, and spontaneous colitis (77, 83, 84). For tTreg cell stability, Foxp3 expression alone is insufficient and additionally requires genome-wide CpG hypomethylation driven by TCR stimulation (85).
The mechanisms by which Treg cells modulate immune suppression have been the subject of many studies (43, 44, 86). In general, Treg cells exert their immunosuppressive function through three main mechanisms. Firstly, Treg cells repress the immune response of effector immune cells by secreting immunomodulatory cytokines and cytotoxic molecules. Secondly, Treg cells express immune checkpoint receptors by which they interact and suppress Teff cells or antigen-presenting cells. Furthermore, Treg cells interfere with the metabolism of effector cells thereby affecting their function.
Tissue-Resident Intestinal Treg Cells
Over the last decade, it has become clear that Treg cells undergo tissue-specific adaptation in non-lymphoid tissues and acquire context-dependent tissue-specific gene signatures (75, 87, 88). As described above, cTreg cells express lymphoid tissue homing molecules involved in trafficking to secondary lymphoid organs. In these secondary organs, tissue-specific self-antigens or microbial antigens recognized by cTreg cells lead to their differentiation to eTreg cells, upregulating activation markers (CD44hi), effector molecules (CTLA4, GZMB, KLRG1), chemokines and their receptors (CCR4), and immunosuppressive cytokines (IL-10) (75, 89). Upon TCR stimulation, the upregulation of homing receptors, including chemokine receptors and adhesion molecules, further directs the migration and localization of the eTreg cells to non-lymphoid peripheral sites in response to specific stimuli (89).
Tissue-resident eTreg cells are found in almost all peripheral tissues where they adapt to environmental cues and exhibit a tissue-specific transcriptional adaptation associated with their function (88, 90). They are specialized in controlling peripheral immune homeostasis by acquiring a combination of homing receptors, transcription factors, immune-regulatory mechanisms, and a specific TCR repertoire (74). eTreg cells not only dampen immune responses but also promote the regeneration and repair of injured tissue or stimulate stem cell differentiation (91, 92). In the intestinal tract, the majority of the immunoregulatory processes occur in the mucosal lamina propria (LP). Among the total CD4+ T-cell population in the LP, 10-15% in the small intestine and 25-35% in the large intestine are Treg cells. To maintain intestinal immune homeostasis, Treg cells control immune responses against innocuous food and microbial antigens (75, 93). Colonic pTreg cells are mainly directed towards microbial antigens, as highlighted in studies using germ-free mice, where a reduced number of colonic Treg cells was observed compared to specific pathogen-free (SPF) mice (94). Some colonic eTreg cells originate from tTreg cells and are characterized by expression of the transcription factors Gata3, Helios, and Nrp1 (95). However, most eTreg cells result from the conversion of CD4+Foxp3− T cells to pTreg cells expressing the nuclear hormone receptor RORγt, with low Helios or Nrp1 expression (96). Control of Foxp3 expression in pTregs generated in the digestive-system lymphoid-tissue has been attributed to regulation of CNS1 (77). CNS1 contains binding sites for several transcription factors including NFAT, Smad, and retinoic acid receptor (RAR) and retinoid X receptor (RXR) whose binding promotes Foxp3 expression. TCR stimulation activates NFAT, while TGF-β signaling activates Smad3 whose association with CNS1 contributes to histone acetylation and enhancer activation. This synergistic binding of NFAT and Smad to CNS1 is essential for Foxp3 expression (83).
Transcriptional and functional analysis of both murine and human colonic Treg cells has defined two specialized subsets based on the expression of transcription factors and cell surface markers. One population, the FOXP3+ GATA3+ Helios+ Nrp1+ Treg cells are similar to so-called Th2-like eTreg cells. GATA3 is a canonical Th2 transcription factor with a fundamental role in controlling Treg cell fate and accumulation in tissues during inflammation. It is expressed in Treg cells homing to barrier sites such as skin and intestine, constituting one-third of the colonic Treg cells (97). Most of the GATA3+ Treg cells also express IL-1RL1 (also known as ST2), whose expression is GATA3-dependent. ST2 binds to tissue-danger signal IL-33, which is elevated in the colon after inflammation-driven tissue damage. The IL-33-ST2 pathway is crucial for Treg cell homing and accumulation in the intestine, along with high expression of the intestinal-homing receptors CCR9 and α4β7. In a positive feedback loop, IL-33 induces the recruitment of GATA3 to the il1r1 enhancer and foxp3 promoter, resulting in increased FOXP3 and ST2 expression establishing the eTreg cell transcriptional program. GATA3+ ST2+ Treg cells also secrete amphiregulin, a tissue-remodeling factor that mediates tissue-regeneration, and IL-10, a cytokine that supports intestinal stem cell renewal, restraining their proliferation and aberrant differentiation (98–100). In this way, Treg cell accumulation limits tissue damage by a rapid adaptation to tissue inflammation.
A second, larger proportion of eTreg cells express FOXP3+ RORγt+ Helios- Nrp1- and are classed as Th17-like Treg cells. RORγt Treg cells constitute 40-60% of total colonic Treg cells (96, 101) and are normally directed against microbiota, constraining intestinal immune responses (94, 102). Recently, RORγt expression in colonic Treg cells has been associated with maintenance of Foxp3 expression during colitis (103). Deletion of RORγt was found to lead to an upregulation of T-bet and IFN-γ, loss of Foxp3 expression, and severe intestinal inflammation in mice. T-bet deletion in RORγt knockout animals was found to restore Foxp3 expression and immunosuppressive function during inflammation. These results suggest that RORγt, by suppressing T-bet expression, promotes Foxp3 expression and thereby Treg cell function (103). Moreover, RORγt expression correlates with upregulation of immunosuppressive receptors including PD-1, ICOS, and CTLA4, the nucleotidases CD39 and CD73, and secretion of high amounts of anti-inflammatory cytokines including IL-10 and TGF-β. This transcriptional reprogramming enhances regulatory function to restrain inflammatory responses critical for controlling chronic inflammation (96, 101, 104).
Taken together, these findings illustrate a range of Treg cell phenotypes and functions in the intestinal mucosa, with remarkable flexibility to maintain tissue homeostasis. While transcriptional analysis of mice colonic Treg cells identified Gata3+Helios+ and RORγt+ Helios− subpopulations, in humans this is less clear. GATA3+ Treg cells have been described in blood, however, they have not yet been identified in human intestine (97). RORγt+ Treg cells are present in comparable levels in biopsies from healthy colonic LP and patients with Crohn’s disease (96). Furthermore, RORγt+ Treg cells are elevated both in blood and tissues of IBD patients, and in the blood and tumors of CRC patients during different stages of the disease (105, 106).
Tumor-Infiltrating Treg Cells and CRC
Since the observation of a population of immunosuppressive CD4+ T-cells in sarcoma tumor-bearing mice (107), a steadily increasing number of studies has demonstrated an important role for Treg cells regulating anti-tumor immunity (108–110). Treg cells can account for more than 50% of all CD4+ T cells in the TME of solid tumors including gastric (111), lung (112), breast (113), ovarian (114), cervical (115), melanomas (116) and hepatocellular (112) cancer. As already mentioned, high numbers of TI-Treg cells is often associated with poor prognosis and low survival rates (117, 118). In CRC, increased numbers of FOXP3+ T cells correlate with both, improved (30, 71, 117, 119–121) or worsened prognosis and overall survival (122). This discrepancy may be explained by the difficulties in defining the identity of suppressive FOXP3+ Treg cells at the tumor site. In line with this, analysis of TILs in human CRC identified two heterogeneous subpopulations of FOXP3+ T-cells associated with patient outcome (123). Better prognosis was associated with increased infiltration of non-suppressive FOXP3lo non-Treg cells (CD45RA- FOXP3lo). These were found to generate a strong pro-inflammatory environment through the secretion of inflammatory cytokines such as TGF-β, IL-12, and TNF-α. In contrast, increased numbers of suppressive CD45RA- FOXP3hi eTreg cells correlate with poorer outcomes and lower disease-free survival (123).
The percentage of Treg cells, as a proportion of CD4+ T cells, infiltrating the TME in CRC is significantly higher compared to healthy colon (124). While some studies support the concept that TI-Treg cells may originate from tTreg cells that migrate and/or expand in the TME, others support in situ generation of pTreg cells (28, 125–127). TI-Treg cells expressing Helios have also been identified suggesting their thymic origin in human CRC (128). Studies analyzing CD4+ TCR repertoires in murine solid tumor models have demonstrated largely distinct TCR repertoires in Foxp3+ and Foxp3− CD4+ T cells. This suggests that increased numbers of TI-Treg cells is due to the in situ proliferation of Treg cell clones in tumors, and not conversion of CD4+ T cells to Treg cells (129, 130). Consistent with these findings, additional studies of TCR repertoires support that TI-Treg cells have little overlap with conventional CD4+ T-cells in murine models of prostate cancer (131) or human breast cancer (128). In CRC patients, Teff and Treg cells have been shown to develop distinct TCR repertoires against tumor-associated antigens, leading to specific tumor-associated antigen responses. Treg cells have high specificity for a limited repertoire of tumor antigens and exert their immunosuppressive function against Teff cells to damp their responses. Since the repertoire of antigens that Teff and Treg cells can detect is different, this provides the potential for therapeutic approaches that identify peptides that can stimulate anti-tumor T-cell but not Treg cell responses (132). A recent study characterizing the TCR repertoires of TI-Treg cells in human metastatic gastrointestinal melanoma, and ovarian cancers showed a significant overlap with circulating Treg cells but not with conventional CD4+ T cells found in either tumor or blood (133).
TI-Treg cells generally exhibit CpG hypomethylation patterns required for the induction of Foxp3 expression and generation of stable, functional Treg cells (78, 85, 134). In colonic RORγt+ Treg cells, the CNS2 region, also referred to as the major Treg-specific demethylated region (TSDR), is significantly demethylated (101, 135). In CRC tumors, TSDR demethylation has been associated with STAT5 and TET2, and the expression of both is upregulated in CRC tumor-infiltrating CD4+ T cells. Here, STAT5 and TET2 coordinately bind the FOXP3-TSDR, promoting DNA hypomethylation and FOXP3 expression (136). Analysis of the FOXP3-TSDR demethylation rates (TSDR-DMRs) in Treg cells from CRC tumors and adjacent healthy colon samples demonstrated higher TSDR-DMRs demethylation in TI-Treg cells (137). In another study utilizing 130 paired samples of CRC tumor tissue and adjacent healthy colonic mucosa samples, significantly higher TSDR-DMRs and increased FOXP3 mRNA and protein expression were also observed in TI-Treg cells (138). These observations provide an explanation for increased FOXP3 expression in CRC tumor-infiltrating Treg cells compared to healthy colon tissue.
The CRC TME can also promote the conversion of CD4+ T-cells to TI-Treg cells through a variety of ways, for example increasing the availability of TGF-β (Figure 1C) (139). Other mechanisms associated with the differentiation of CD4+ T cells to Treg cells include the increased expression of indoleamine 2,3-dioxygenase (IDO) in mouse and human CRC TME. IDO converts tryptophan to kynurenine, which can bind the aromatic hydrocarbon receptor (AhR) in CD4+ T cells thereby promoting Foxp3 expression (140, 141). Treg cells localize to the CRC TME through their expression of specific chemokine receptors. These include: CC chemokine receptor 4 (CCR4), attracted to inflammatory loci by the ligands CCL22 or CCL17 produced by CRC tumor cells or macrophages (142); CCR5, highly expressed in TI-Treg cells and associated with Treg cell recruitment towards the ligands CCL3, and CCL4 expressed by CRC tumor cells (34, 143); CCR6, involved in migration to the TME in response to CCL20 produced by tumor-associated macrophages (144, 145); CCR8, exclusively expressed by TI-Treg cells (146) and CCL1, expressed by tumor-infiltrating myeloid cells (128). Taken together, multiple mechanisms are involved in the increased numbers of Treg cells in the context of CRC. This can involve accumulation in the TME by chemotaxis and clonal expansion by recognition of tumor-associated antigens.
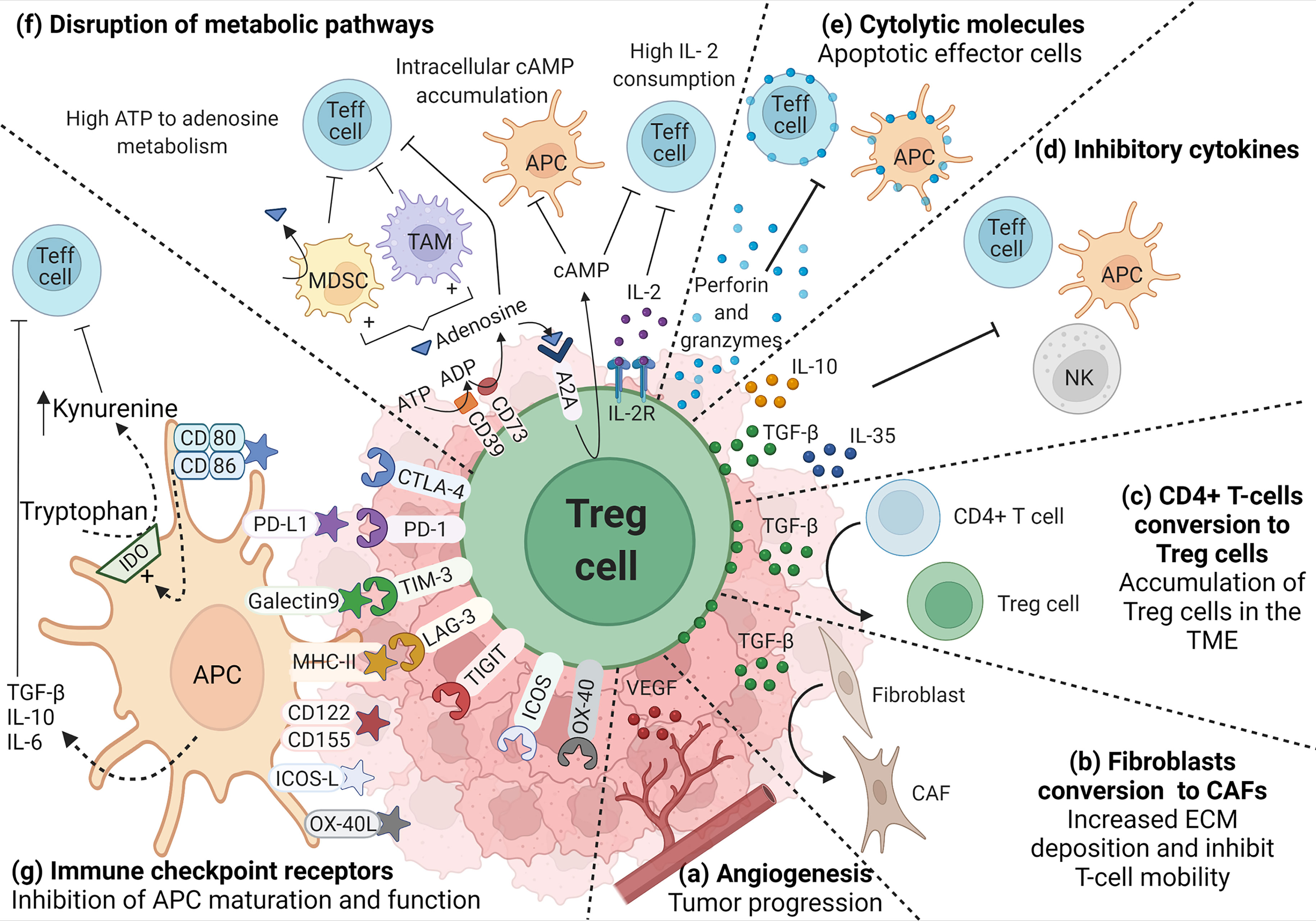
Figure 1 Role of TI-Treg cells in the TME. TI-Treg cells produce (A) VEGF to promote dysregulated angiogenesis associated with tumor progression and TGF-β that, (B) promote the conversion of fibroblast to cancer-associated fibroblasts (CAFs), and (C) the conversion of CD4+ T-cells to Treg cells promoting their accumulation in the TME. TI-Treg cells also regulate anti-tumor immune responses by producing (D) inhibitory cytokines, such as IL-10, TGF-β and IL-35, inhibiting Teff cells, NKs and APCs, the last two are also inhibited through membrane-bound TGF-β, and (E) cytotoxic molecules such as granzymes and perforin that can directly kill Teff cells and APCs. TI-Treg cells also (F) disrupt Teff cell intracellular metabolism impairing their function by depleting IL-2 in the TME. They express CD39 and CD73 ectonucleotidases that covert ATP and ADP into adenosine, which can engage adenosine receptor A2A on the surface of Teff cells, increasing intracellular cAMP and disrupting their metabolism and function. cAMP also binds to APCs and macrophages inducing tolerogenic myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) that further impact Teff cells. Furthermore, adenosine can also bind A2A in Treg cells promoting the intracellular accumulation of cAMP that can be transferred through gap junctions to Teff cells interfering with their metabolism. Among different molecules that participate in the suppression process, Treg cells highly express (G) immune checkpoint receptors that bind their corresponding ligand in APCs and differently regulate their function (increasing the release of inhibitory cytokines or the upregulation of IDO) promoting an immunosuppressive TME.
TI-Treg cells are more proliferative and have a more activated immunosuppressive phenotype when compared to Treg cells from healthy tissue or peripheral blood (147). Helios, associated with Treg cell thymic origin, is highly expressed in CRC Foxp3hi TI-Treg cells which also co-express activation markers such as ICOS and OX-40, and inhibitory molecules such as PD-1, CTLA-4, TIM-3 (Figure 1G), and CD39 that mediate their highly suppressive phenotype (Figure 1F) (148). CD39 is an ectonucleotidase that catalyzes the generation of adenosine from ATP, negatively regulating immune responses (149). A recent study comparing CRC tumors and healthy colon tissues identified three clusters of Treg cells differentially expressing CD39. The subgroups with the highest and intermediate expression of CD39 were found in the tumor, while the subgroup with the lowest expression was found in healthy colon tissue. Higher expression of CD39 correlated with poorer outcome (150).
In the context of CRC, the most significantly enriched TI-Treg cell population has a Th17-like profile. IL-17+ TI-Treg cells, originated from memory CCR6+ T cells, cTreg cells, or the transdifferentiation of Th17-to-Treg cells, accumulate in colitis inflamed tissue and are associated with CRC progression (151–153). These cells maintain transcriptional and epigenetic signatures of both Th17 cells (Folr4, GARP, Itgb8, Pglyrp1, Il1rl1 and Itgae) and Treg cells (Foxp3, Tigit and Icos) (135, 153). IL-17+ TI-Treg cells also acquire RORγt expression contributing to local inflammation by producing IL-17, IL-2, IL-6, TNF and IFN-γ, in combination with the capacity to inhibit T cell immunity, thereby driving initiation of tumorigenesis (105, 106, 153–156). RORγt expression is increased and promoted by Wnt signaling. In healthy colonic Treg cells, TCF-1 and FOXP3 co-bind to the enhancer regions of pro-inflammatory genes such as Rorc (RORγt), Il17a (IL-17) and Ifng (IFNγ), repressing their expression. However, in CRC RORγt+ TI-Treg cells, sustained Wnt and/or TCR signaling leads to nuclear translocation of β-catenin where it binds to TCF-1 facilitating the transcription of pro-inflammatory genes. Increased numbers of pathogenic β-catenin(hi) RORγt+ TI-Treg cells correlates with the progression from IBD to CRC in humans and in murine colitis-associated dysplasia models (105, 157). CRC-associated Treg cells have decreased TCF-1 expression and increased Th17 expression signatures compared to normal tissues. Reduced TCF-1 activity in Treg cells increases their immunosuppressive function against T cell proliferation, but at the same time promotes their pro-inflammatory activity, promoting tumor growth in polyp-prone mice (158). Conversely, the specific ablation of RORγt in Foxp3+ Treg cells of polyp-prone mice improves anti-tumor responses and reduces polyposis (106, 159).
Epigenetic modifications at the promoter regions of TI-Treg cell genes promote immunosuppressive function and support tumor immune evasion. Expression levels of immune checkpoint proteins such as PD-1, CTLA-4, TIM-3 and TIGIT, and checkpoint ligands including PD-L1 and galectin-9 are higher in peripheral blood or tumor tissue for CRC patients, compared with blood or colon tissue from healthy donors (160, 161). Increased DNA hypomethylation at the promoters of CTLA-4 and TIGIT in TI-Treg cells, compared to healthy colon tissue, correlates with increased expression of TETs. Moreover, the distribution of the repressive histone modifications H3K27me3 in CTLA-4 and TIM-3, and H3K9me3 in PD-1, TIM-3 and TIGIT, is lower in TI-Treg cells correlating with higher expression of these immune checkpoint receptors (160). RORγt+ Treg cells retain a suppressive phenotype through hypomethylation of Treg-specific signature genes including Foxp3, Ctla-4, Gitr, Eos, and Helios (135, 151, 152, 154). Their immunosuppressive activity in the TME is thought to occur through the release of factors such as TGF-β (Figure 1D), which is upregulated in around 80% of the RORγt+ TI-Treg cells. A smaller proportion also expresses IL-10 (Figure 1D), and they can also release IL-6, IL-4, IFN-γ, and TNF-α (106, 154). RORγt+ TI-Treg cells can also co-express the BLIMP1 transcription factor identified as the primary regulator of IL-10 expression in the colon, suppressing inflammation-driven CRC (162). BLIMP1+ TI-Treg cells have been associated with longer disease-free survival in CRC patients (163). In summary, RORγt has an important role in the balance between suppressive Treg cells and pathogenic RORγt+ Treg cells that can contribute to an inflammatory and immunosuppressive environment that can promote both CRC tumor development and immune evasion.
The majority of TI-Treg cells express co-inhibitory receptors such as LAG-3 and TIM-3 that can modulate T cell responses (Figure 1G) (164). LAG-3+ Treg cells have increased IL-10 and TGF-β production and are enriched both in the TME and peripheral blood of CRC patients (165). TIM-3+ Treg cells have been associated with multiple cancers including CRC and have a role in regulating immune responses by driving T cell inhibition or exhaustion. They express CD39/CD73 (Figure 1F), IL-10, and TGF-β (Figure 1D) and upregulate the expression of checkpoint receptors (Figure 1G), such as CTLA-4, PD-1, and LAG-3 (166–170). CRC tumors implanted in TIM-3 deficient mice have a reduced infiltration of Treg cells, reduced CD8+ Teff cell exhaustion, and slower tumor growth (170). LAG-3+TIM-3+ TI-Treg cells are also enriched in CRC tumors compared to healthy colon, with increased TGF-β and IL-10 release, and upregulation of CTLA-4 expression compared to LAG-3−TIM-3− TI-Treg cells. In addition, LAG-3+TIM-3+ TI-Treg cells can reduce macrophage expression of MHC class II, CD80/CD86, and TNF-α and increase IL-10 secretion thereby supporting immunosuppression (171).
Additional CRC-related Treg cell subsets have been described, although less is known about their functional relevance. TI-Treg cells producing IL-35 have been identified in CRC patients (Figure 1D); however, their origin, phenotype, and function remain to be defined. IL-35 has immunosuppressive activity both in vitro and in vivo, inhibiting Teff cell proliferation and inducing conversion to IL-35-producing Treg cells (172–174). Studies with CRC patients and mouse models found increased TI-Treg cell numbers correlated with high levels of IL-35 expression in serum and TME (173–176). Latency-associated peptide (LAP) non-covalently binds to TGF-β and forms a latent TGF-β complex inhibiting the interaction with its cognate receptors on immune cells (177). LAP+ TI-Treg are functionally more immunosuppressive than LAP- TI-Tregs, and their numbers increase with CRC progression and the metastatic stage (178, 179). A study of 42 CRC patients concluded that LAP+ TI-Treg cells were increased in the peripheral blood and tumor tissue of patients compared to healthy colon controls. These Treg cells upregulated effector molecules including tumor necrosis factor receptor II, granzyme B, perforin, Ki67, and CCR5 that further support their immunosuppressive function. LAP+ TI-Treg cells can direct cell-mediated cytotoxicity through the expression of perforin and granzyme B, thereby also suppressing Teff cells (Figure 1E) (178).
Taken together, Treg cell plasticity and heterogeneity provide a broad range of mechanisms to temper anti-tumor immune responses and promote immune evasion. It is therefore important to accurately characterize Treg cell populations to improve the diagnosis and outcome of CRC patients and more specifically target TI-Treg cells.
The TME Promotes Treg Cell Metabolic Reprogramming
The reprograming of energy metabolism is one of the hallmarks of cancer whereby tumors can influence the immunosuppressive TME. In CRC, this already takes place at the colon adenoma stage (180) where CRC initiation, proliferation, invasion, and metastasis are closely associated with the metabolic crosstalk between tumor cells, the TME, and the microbiota (181). CRC tumor cells reprogram their metabolism in order to fulfill their excessive energy and nutrient needs, relying on aerobic glycolysis, glutaminolysis, and fatty acid synthesis. As a result of the rapid proliferation of tumor cells and the immaturity of tumor vasculature, the TME is characterized by hypoxia, acidity, and nutrient deficiency including glucose, glutamine, and tryptophan, with enrichment of lactate and kynurenine (182, 183). These metabolic changes hamper effective Teff cell activation and proliferation, which themselves rely on aerobic glycolysis. In contrast, this harsh metabolic environment promotes the recruitment and differentiation of Treg cells (184). FOXP3 is a central regulator of TI-Treg cell metabolic adaptation, driving a distinct metabolic profile compared to Teff cells. TI-Treg cells are less dependent on glycolysis, increasing FA-oxidation (FAO) and oxidative phosphorylation (OXOPHOS) to support their differentiation and function (185, 186). In CRC, as in other solid tumors, TI-Treg cells utilize these alternative metabolic pathways to produce energy, proliferate and perform their immunosuppressive functions (187–189).
Accumulation of lactate (Figure 2A), the end-product of tumor cell glycolysis, affects the TME through multiple mechanisms. Treg cells can metabolically adapt to increased lactate in the TME, utilizing it as a carbon source for intracellular metabolism. TI-Treg cells upregulate pathways related to the metabolism of lactate such as lactate dehydrogenase (LDH), and the lactate transporter MCT1 (SLC16A1) (190, 191). FOXP3 directly alters T cell metabolism to maintain Treg cell suppressive function in lactate-rich environments (185). By binding to the MYC promoter, FOXP3 can suppress c-myc expression, a transcription factor regulating glycolytic gene expression, thereby suppressing glycolysis. FOXP3 also influences LDH activity, promoting the conversion of lactate to pyruvate and increasing oxidative phosphorylation (OXPHOS), and the NAD : NADH ratio. These adaptations permit Treg cells to differentiate and work effectively under conditions of low-glucose and high lactate as found in the TME (185).
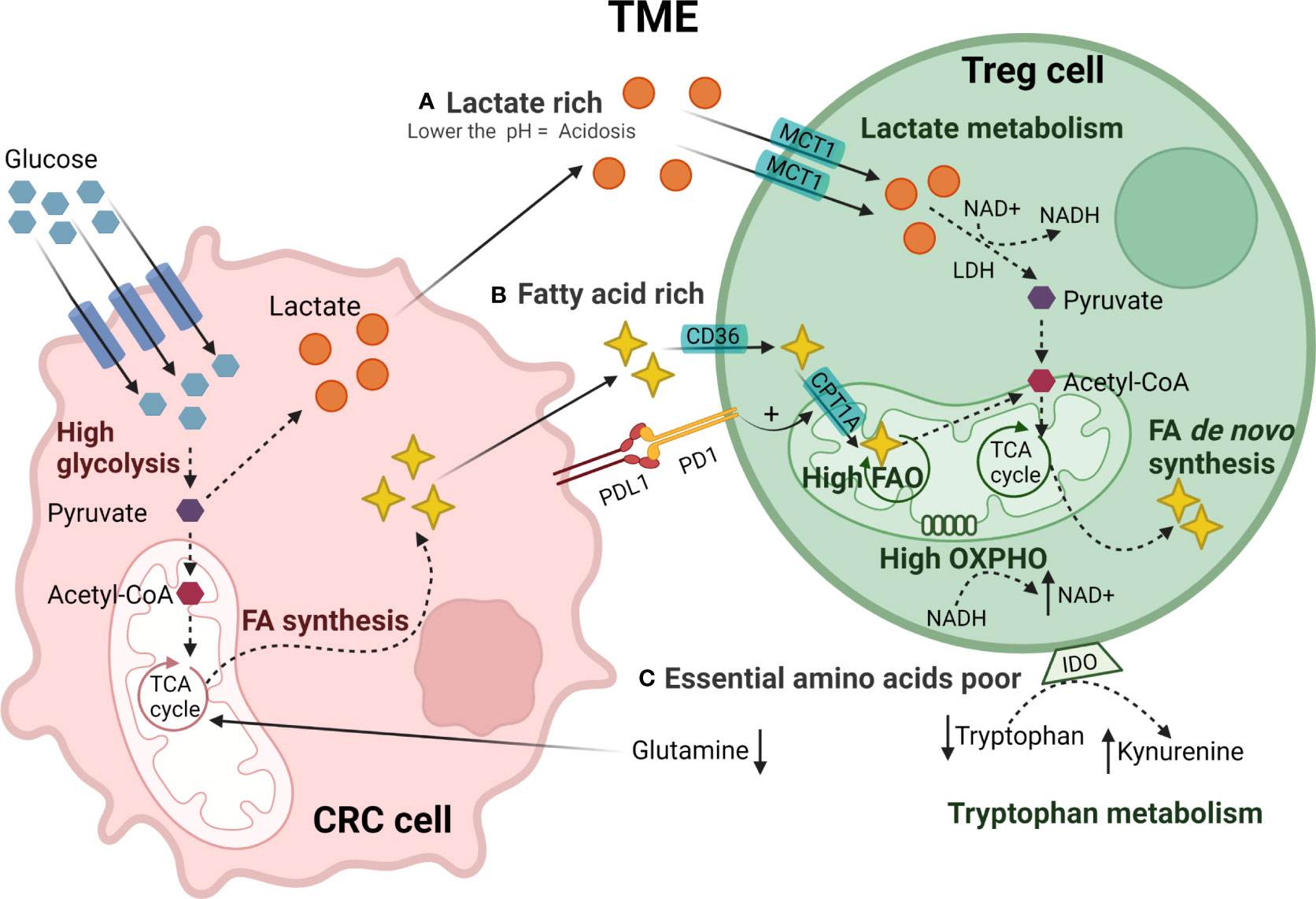
Figure 2 Metabolic reprogramming of TI-Treg cells in CRC TME. CRC tumor cells have a high glucose uptake generating pyruvate through glycolysis. Pyruvate is converted to acetyl-CoA or lactate that is secreted creating (A) a lactate-rich TME. TI-Treg cells adapt to these metabolic stresses present in the TME. TI-Treg cells can take up lactate through the MCT1 transporter that is subsequently converted to pyruvate by LDH and further to acetyl-CoA. Moreover, CRC tumor cells fuel the tri-carboxylic acid (TCA) cycle in the mitochondria for FA synthesis creating a (B) fatty acid-rich TME. TI-Treg cells can take up FA from the TME through the fatty acid transporter CD36. The CPTR1A transporter drives FA uptake into the mitochondria where it is oxidized by fatty acid oxidation (FAO) to acetyl-CoA. Acetyl-CoA fuels the TCA cycle in the mitochondria for de novo fatty acid synthesis that can be accumulated intracellularly or exported to the TME. The TME has (C) low availability of essential amino acids, particularly glutamine, that is consumed by tumor cells, and tryptophan, that is catabolized to kynurenine by IDO highly expressed by Treg cells.
Besides lactate, TI-Treg cells also rely on fatty acids (Figure 2B), utilizing lipid metabolism to meet both their energy demands and immunosuppressive function through FAO-driven OXPHOS (185, 186). Fatty acids stimulate the AMPK pathway, inhibiting mTOR signaling and promoting mitochondrial FAO (186). TI-Treg cells also upregulate SREBP transcription factors in order to adapt to the TME. SREBPs regulate the expression of genes required for de novo synthesis of lipids by fatty acid synthase (FAS), promoting the functional maturation of TI-Treg cells. SREBP-deficient Treg cells show decreased expression of PD-1 and have been associated with an effective anti-tumor immune response and reduced tumor growth (192). PD-1 signaling in TI-Treg cells can also promote FAO by upregulating palmitoyltransferase-1A (CPT1A), an importer of fatty acids to the mitochondria (193). A study in CRC, among other solid tumors, showed that high levels of fatty acids in the TME are an essential energy source for TI-Treg cells (188). TI-Treg cells also increase intracellular lipid accumulation through increases in fatty acid synthesis (188). In a CRC murine model, TI-Treg cells were found to upregulate CD36, an FA-importer, increasing FA uptake and FAO (194). This metabolic flexibility allows TI-Treg cells to thrive and supports their suppressive activity in the TME under conditions of high concentrations of tumor-derived lipids (195).
Under homeostatic conditions, amino acids regulate the activation of mTORC1 which is important for the functional fitness of eTreg cells (196, 197). However, the TME generally has low availability of many amino acids (Figure 2C), particularly glutamine. Reduced levels of glutamine, with the subsequent reduction of glutamine catabolism, stimulates the conversion of CD4+ T cells to Treg cells during Th1 polarization in vitro (198, 199). In addition, the TME often has high levels of amino acid-degrading enzymes such as IDO. IDO is expressed by both tumor cells and Treg cells and, as already mentioned, catabolizes tryptophan to kynurenine, binding to AhR in Teff cells and promoting the Treg cell differentiation (200–205). High IDO expression by CRC cells and tryptophan depletion in the TME has been associated with tumor immune evasion and increased Treg infiltration in CRC (206).
Other metabolites abundant in the TME include retinoic acid (RA) and adenosine. RA can act as a cofactor to induce Foxp3 expression and promote the conversion of CD4+ T cells into Treg cells while inhibiting the generation of Th17 cells. In the intestine, CD103+ DCs can generate RA from vitamin A and drive Treg cells differentiation in the presence of TGF-β (207, 208). A colitis-associated CRC murine model treated with RA showed an increased frequency of TI-Treg cells in the tumor and reduced inflammation (209). Adenosine levels are increased in response to hypoxia and chronic inflammation, both characteristics of CRC. Treg cells can produce adenosine by CD39/CD73 action but can also respond to it by engaging the adenosine receptors (A2A AR) (Figure 1F). A2A engagement promotes the intracellular accumulation of cAMP and, subsequently, the activation of the cAMP response element-binding protein (CREB) that drives the expression of anti-inflammatory cytokines, including IL-10 and TGF-β (210, 211).
Despite the metabolic challenges present in the TME such as low glucose and amino acid availability, high lactate and lipid concentration, acidity, and hypoxia, TI-Treg cells show a remarkable ability to engage unique metabolic reprogramming, compared to other Teff cells, to support their bioenergetic and functional needs. Understanding the TI-Treg cell metabolic reprogramming potentially provides novel therapeutic approaches for targeting in the control CRC pathogenesis.
Therapeutic Approaches to Target Treg Cells in CRC
Modulating TI-Treg cell function offers an important strategy to improve therapeutic responses for a wide variety of tumors. A variety of approaches are currently under development including Treg cell depletion, suppressing their activity, impeding their recruitment, and preventing their differentiation within the TME. The challenge presented is to specifically target TI-Treg cells without affecting other Treg populations, thereby diminishing the risk of developing unwanted autoimmune responses. In the past decade, the efficacy of checkpoint inhibitors to block immune checkpoint receptors or their ligands has greatly benefited the survival of patients with solid tumors (212–216). As discussed earlier, CRC TI-Treg cells express immune checkpoint receptors including PD-1, CTLA-4, TIM-3, and the NTPDase CD39 that regulate their immunosuppressive phenotype and support tumor cells in escaping immune surveillance (Figure 1G) (148, 217). A variety of humanized monoclonal antibodies developed against these receptors have been evaluated in CRC patients in order to alter TI-Treg cells numbers and functionality (Table 2).
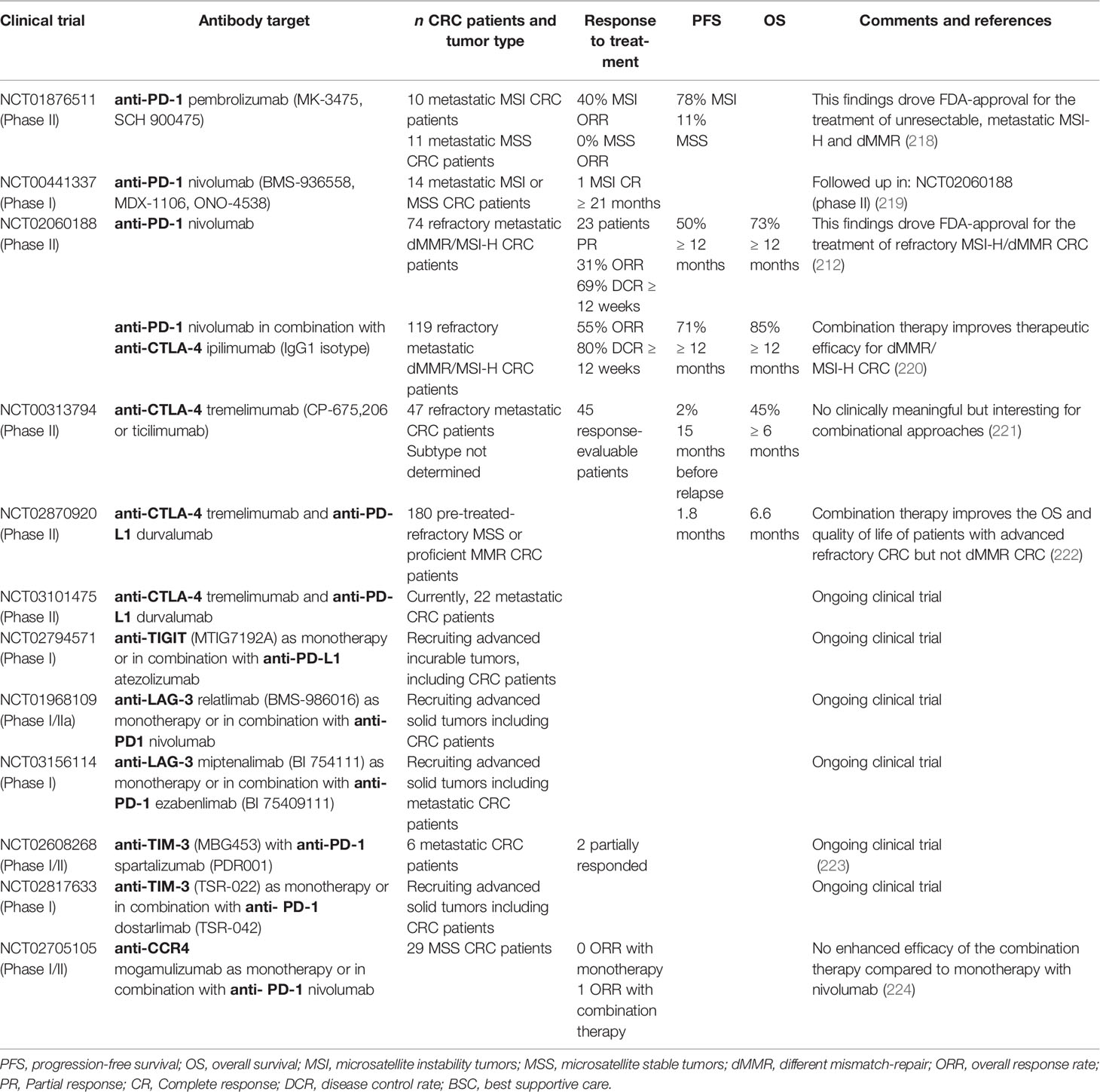
Table 2 CRC clinical trials potentially targeting Treg cell populations.
PD-1 is highly expressed in TI-Treg cells and plays a role in their homeostasis and function. PD-1 signaling also promotes FAO to support their bioenergetics needs in the TME (193). PD-1-expressing Treg cells are considered a critical element in tumor immune evasion and progression of CRC (225). PD-1, expressed on tumor cells and Treg cells, can bind its ligand PD-L1 on Teff cells activating inhibitory signals that interfere with the TCR signal transduction thereby blocking anti-tumor immune responses (226, 227). Humanized monoclonal anti-PD-1 antibodies (mAbs) block interaction with PD-L1 and can thereby benefit the anti-tumor immune response (217, 228). Recently, it has been demonstrated that the balance of PD-1 expressing CD8+ Teff cells and eTreg cells in the TME is relevant in predicting the efficacy of anti-PD-1 mAbs (229), although how PD-1 signaling promotes Treg cell suppressive function is unclear. A study in CRC patients showed that anti-PD-1 mAb therapy has a better outcome for patients with high numbers of PD-1+ CD8+ T cells compared to non-responders that have higher numbers of eTreg cells. In line with these previous results, a CRC murine model demonstrated the capacity of anti-PD-1 mAbs to restore the effector function of PD-1+ CD8+ T cells and promote tumor regression (230). Clinical trials have investigated the possibility of using humanized anti-PD-1 monoclonal antibodies for the treatment of CRC patients (Table 2). Promising clinical results with the first mAb against PD-1, pembrolizumab (MK-3475, SCH 900475) and, later, nivolumab (BMS-936558, MDX-1106, ONO-4538) in CRC patients lead the U.S. Food and Drug Administration (FDA) to approve them for the treatment of refractory metastatic CRC that still progressed after prior treatment with chemotherapeutics (212, 231). The diverse outcomes observed in CRC patients treated with different anti-PD-1 mAbs can be explained according to the subtype of CRC tumor. Unfortunately, few studies have reported responses regarding the targeting of TI-Treg cells by these antibodies in CRC patients. However, based on current knowledge it is likely that anti-PD-1 mAbs target Treg cells and in this way support activation of an anti-tumor immune response.
Treg cells express high levels of cytotoxic T lymphocyte antigen 4 (CTLA-4) in a FOXP3-dependent manner. CTLA-4 binds and inhibits B7 molecules (CD80 and CD86) on the surface of APCs such as DCs with higher affinity than their CD28 co-stimulatory signal. This results in the induction of immunological tolerance (230). Murine CRC models have shown that anti-CTLA4 mAbs induce a potent immune response, rejection of the tumor, and significantly prolonged survival (232). Humanized anti-CTLA-4 mAbs such as ipilimumab (IgG1 isotype) and tremelimumab (IgG2 isotype) have been tested in multiple clinical trials with solid tumors (233–235). The anti-tumor mechanism of these anti-CTLA-4 mAb was first attributed to preventing interaction of CTLA-4 with its ligand B7 allowing APCs to present antigens and activate anti-tumor T cells response. Recently, it has been shown that anti-CTLA-4 mAbs also deplete TI-Treg cells, but not pTreg cells in secondary lymphoid organs. This selective Treg cell depletion depends on their ability to activate Fc receptors on tumor-associated macrophages or NK cells that can subsequently phagocytose or kill Treg cells (234, 236–241). The use of anti-CTLA-4 (IgG2a isotype) in two murine subcutaneous CRC tumor models demonstrated successful reduction of TI-Treg cells together with the expansion of CD8+ Teff cells promoting anti-tumor activity (238). The use of anti-CTLA-4 mAb in clinical trials for patients with metastatic CRC, overall, has shown improved therapeutic efficacy in combination with anti-PD-1 mAbs compared to anti-CTLA-4 mAb monotherapy (Table 2) (212, 220, 222). Currently, the dual combination of checkpoint inhibitors is being evaluated in various phase II clinical trials for metastatic CRC patients, for example, durvalumab, an anti-PD-L1 mAb, and tremelimumab, an anti-CTLA-4 mAb (NCT03101475). There have been no specific reports on anti-CTLA-4 mAb targeting TI-Treg cells in these CRC patients, but again it is likely that TI-Treg cell function and numbers are impacted. Anti-CTLA4 mAbs have also been shown to have a T-cell intrinsic mechanism of action that enhances the proliferation of Teff and Treg cells in response to self-antigens as shown in human and mouse models (130, 242, 243).
Novel immune checkpoint targets such as TIGIT, LAG-3 and TIM-3 are currently under pre-clinical investigation and are being evaluated for their safety profiles in phase I trials (244, 245). In CRC, these receptors are highly expressed on TI-Treg cells compared to healthy colon tissue (150). TIGIT is a co-inhibitory molecule, a member of the CD28 family, expressed preferentially in T cells and NK cells. TIGIT competes with the co-stimulatory receptor CD226 in T-cells to bind CD155 on APCs which become tolerogenic, cannot activate T-cells, and release IL-10 inhibiting of Teff cell anti-tumor responses (246–248). A phase I clinical trial (NCT02794571; Genentech) utilizing humanized anti-TIGIT mAb (MTIG7192A) as monotherapy or in combination with anti-PD-L1 mAb (atezolizumab) is ongoing in advanced or metastatic tumors including CRC (Table 2). LAG-3 is expressed on activated CD4+ and CD8+ T cells (249), Tregs (250), a subpopulation of natural killer (NK) cells (251), B cells (252), and plasmacytoid dendritic cells (pDCs) (253). The LAG-3 co-inhibitory receptor can bind stable peptide-MHC-II complexes impairing DC function, maturation, and proliferation. LAG-3 can also induce the production of IDO, impairing Teff cell and DC proliferation, but promoting eTreg cell differentiation (165, 230, 254, 255). In CRC patients, LAG-3+ Treg cells have been identified in both TILs and in peripheral blood (165). Currently, there are two ongoing clinical trials that aim to investigate the efficacy of anti-LAG-3 mAb alone or in combination with anti-PD-L1 mAb in patients with advanced solid tumors including CRC (Table 2). The rationale behind this combinational treatment is to synergistically restore T cell activation and enhance antitumor immunity.
TIM-3 is another immune checkpoint molecule expressed on innate immune cells such as DCs and NK cells (256). pTreg cells do not normally express TIM-3, however, TIM-3+ TI-Treg cells have been identified in the TME where they exert an inhibitory role against Teff cell responses mainly by driving their exhaustion (166–168, 257). This suggests a therapeutic advantage over other checkpoint receptors, such as CTLA-4 and PD-1, due to expression predominantly on TI-Treg cells. For CRC patients, the combination therapy anti-TIM-3 mAb and anti-PD-1 mAb has already been tested showing partial response (Table 2) (223). An ongoing Phase 1 clinical trial using a humanized anti-TIM-3 mAb (TSR-022) aims to evaluate first the dosage and, then, the anti-tumor capacity of the antibody in advanced solid tumors (NCT02817633). For CRC patients, anti-TIM-3 was administrated as monotherapy or in combination with an anti-PD-1 mAb, dostarlimab (TSR-042) (Table 2).
As already discussed, TI-Treg cells express high levels of CD39/CD73 ectonucleotidases that convert ATP to adenosine, a potent suppressor of tumor immunity (Figure 1f) (258). CD39+ TI-Treg cells have been found to play an important role during CRC tumor growth (150) and inhibition of CD39 enzymatic activity has been evaluated in a CRC hepatic-metastatic murine model. Here, CD39+ Treg cells modulate NK reactivity against the tumor, and targeting CD39 was found to both inhibit Treg cell activity in vitro and reduce tumor growth in vivo (259). Adoptive reconstitution of either wild-type or CD73-deficient Treg cells has been performed in Treg cell-depleted mice implanted with CRC-tumors. Only mice reconstituted with wild-type Treg cells effectively support tumor growth, suggesting a possible therapeutic benefit of targeting CD73-Treg cells (260).
CCR8 expression can discriminate between Treg cells infiltrating CRC and other solid tumors, from those found in secondary lymphoid organs (261). In solid tumors, CCR8 expressing cells migrate in response to CCL1 ligand secreted by cancer-associated fibroblasts (CAFs), M2-polarized tumor-associated macrophages and Treg cells (262–264). Murine tumor models including colorectal adenocarcinoma have been used to evaluate the efficacy of targeting CCR8+ Treg cells in the TME. Administration of monoclonal antibodies targeting either the receptor or ligand reduces the number of TI-Treg cells without affecting the pTreg cells, reinforcing the anti-tumor immune response (265). CCR4+ TI-Treg cells also contribute to the CRCTME, and CCR4 expression is increased in TI-Treg cells compared to those of healthy colon tissue (266). Mogamulizumab is an anti-CCR4 mAb that has been used in clinical trials for the treatment of solid tumors with the aim of depleting TI-Treg cells. It has been evaluated as monotherapy (NCT01929486) or in combination with other targeted therapies such as anti-PD-1 mAb (nivolumab) (NCT02705105). Dose, safety, and efficacy for combination therapy has been assessed in 114 patients with locally advanced or metastatic solid tumors, including 29 MSS-CRC patients. The result showed no enhanced efficacy of the combination therapy compared to monotherapy with nivolumab. In the case of the CRC patients, only one out of the 29 showed ORR, similar to previously reported results of single-nivolumab treatment where none responded (224). Further evaluation of chemotaxis receptor-targeting therapeutics may provide more selective therapeutic strategies avoiding targeting pTreg cells.
Other broader strategies effective in Treg cell depletion include drugs such as cyclophosphamide (CY). The administration of low-dose CY, a chemotherapeutic agent used to treat cancer, seems to predominantly affect Treg cells over other effector cells (Teff cells or NKs), as has been demonstrated in a variety of human cancers and animal models. Low-dose CY can lead to a reduction in Treg cell numbers, upregulation of pro-inflammatory cytokines, and boosts the innate antitumor immune response (267–270). In CRC murine and rat models, the efficacy of low-dose CY, inhibiting Treg cells, in combination treatment with mycobacterium bovis Bacillus Calmette–Guérin (BCG) has been evaluated. BCG has TLR-agonist activity and increases the capacity of DCs to mediate an efficient anti-tumor immune response. The combination therapy resulted in tumor regression with Treg cell depletion in the blood and lymphoid organs but also decreased the number of TI-Treg cells (271). The synergistic combination of IL-12 gene therapy in mice with subcutaneous CRC, pre-treated with low-dose CY, was found to induce a potent anti-tumor effect. Combined treatment reduced the number of MDSCs and increased the anti-tumor DC responses and the number of IFN-γ-secreting CD4+ T cells (272, 273). Depletion of Treg cells by low-dose CY also results in significant anti-tumor responses in advanced chemotherapy-resistant solid cancers (268). In a randomized clinical trial with inoperable metastatic CRC patients, low-dose CY treatment resulted in delayed tumor progression associated with an increase in IFNγ+ anti-tumor T-cell responses, and reduction of Treg cells, B cells, and NK cells (274, 275).
Refinement of combinational therapies targeting Treg cells while promoting anti-tumor immune responses is clearly a promising approach to treat CRC more effectively. However, it remains critical to consider CRC subtypes, and to specifically target TI-Treg cells, rather than systemically depleting all Treg or other Teff cells. These are issues that still require proper evaluation when considering new therapeutic strategies.
Future Directions
CRC FOXP3+ TI-Treg cells are a heterogeneous immune population with specific gene signatures and distinct functional properties. The phenotype and function of Treg cells are both modulated and exploited by the TME to promote immune evasion, tumor progression, and resulting in poor prognosis. Knowledge concerning Treg cell adaptation to the TME is invaluable in designing therapeutic strategies to target TI-Treg cells. However, it remains hard to specifically target Treg cells in the context of the TME. This is compounded by the difficulty of studying Treg biology in vivo or developing valid models for evaluating immune cell-TME interactions in vitro. Immunotherapy approaches can stimulate anti-tumor immune responses of effector cells or inhibit immunosuppressive mechanisms. Refinement of combinational therapies targeting Treg cells together with promoting Teff cell anti-tumor immune responses provides promising strategies to treat CRC more effectively. However, Treg cell-directed immunotherapy for CRC patients has several limitations. To start with, the different CRC molecular subtypes likely have a critical role in determining the success of certain therapeutic approaches. Due to CRC heterogeneity, treatment stratification to the tumor subtype will likely be required. Moreover, a better understanding of the (sub)phenotypes and functional diversity of CRC TI-Treg cells is still needed. This is essential to avoid compromising immune hemostasis and thereby developing unwanted side effects. Utilizing bivalent antibody approaches may help relieve the problem of specifically targeting only TI-Treg cells, as long as appropriate tumor markers are also available (276). Importantly, the expression of therapeutic targets on both TI-Treg cells and Teff cells does not necessarily prevent their application. Differential expression levels and dynamic expression profiles can still provide preferential Treg cell depletion in the context of the TME. Besides antibody-based immunotherapy approaches, the application of small molecule inhibitors remains an avenue of active exploration to disable TI-Treg cells. For example, FOXP3 protein stability is maintained by both active acetylation and deubiquitination, and inhibiting these processes effectively disables Treg cell function (277, 278). As already discussed, Treg cells can thrive in the altered metabolic environment of the TME. Increasing our understanding of the metabolic interactions between infiltrating T cells and tumor cells may help to expose an Achille’s heel that can be used to inhibit TI-Treg cell function more specifically. Combining such approaches with checkpoint inhibitor therapy may help drive the differentiation of anti-tumor effector T cells, at the expense of Treg cells, which are then free to reduce tumor progression. Similarly, understanding the contribution of epigenetic mechanisms to the control of Treg cell suppressive capacity is also relevant. Particularly as several small-molecule epigenetic modifiers are FDA approved and utilized in the clinic (279). Hypomethylating agents, inhibitors of histone deacetylases, and bromodomain inhibitors are all relevant in this context. For example, both the bromodomain inhibitor JQ1, and the HDAC6 inhibitor ricolinostat were found to attenuate Treg cell suppressive function, facilitate immune-mediated tumor growth arrest, and lead to prolonged survival of mice with lung adenocarcinomas (280).
Taken together, these studies further highlight the potential of not only targeting Treg cell markers but also Treg cell biology, in developing novel approaches to effectively re-activate anti-tumor immunity in CRC.
Author Contributions
SAR conceptualized, wrote, edited and approved the manuscript. PJC conceptualized, edited, and approved the manuscript. OK contributed to the revision and final approval of the manuscript.
Funding
This work was supported by a Worldwide Cancer Research grant (Reference: 19-0371).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank all members of the Coffer and Kranenberg labs for helpful discussions. All figures were created using BioRender.com.
References
1. World Health Organization (WHO). International Agency for Research on Cancer. World. In: GLOBOCAN 2020. WHO Website (2021). https://gco.iarc.fr/today/data/factsheets/populations/900-world-fact-sheets.pdf\.
2. Yamashita K, Watanabe M. Clinical Significance of Tumor Markers and an Emerging Perspective on Colorectal Cancer. Cancer Sci (2009) 100(2):195–9. doi: 10.1111/j.1349-7006.2008.01022.x
3. Dubois RN. Role of Inflammation and Inflammatory Mediators in Colorectal Cancer. Trans Am Clin Climatol Assoc (2014) 125:358–73.
4. Yamauchi M, Lochhead P, Morikawa T, Huttenhower C, Chan AT, Giovannucci E, et al. Colorectal Cancer: A Tale of Two Sides or a Continuum? Gut (2012) 61(6):794–7. doi: 10.1136/gutjnl-2012-302014
5. Markowitz SD, Bertagnolli MM. Molecular Basis of Colorectal Cancer. N Engl J Med (2009) 361(25):2449–60. doi: 10.1056/nejmra0804588
6. Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol (2017) 3(4):464–71. doi: 10.1001/jamaoncol.2016.5194
7. Stigliano V, Sanchez-Mete L, Martayan A, Anti M. Early-Onset Colorectal Cancer: A Sporadic or Inherited Disease? World J Gastroenterol (2014) 20(35):12420–30. doi: 10.3748/wjg.v20.i35.12420
8. Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and Familial Colon Cancer. Gastroenterology (2010) 138(6):2044–58. doi: 10.1053/j.gastro.2010.01.054
9. Grivennikov SI. Inflammation and Colorectal Cancer: Colitis-Associated Neoplasia. Semin Immunopathol (2013) 35(2):229–44. doi: 10.1007/S00281-012-0352-6
10. Sturlan S, Oberhuber G, Beinhauer BG, Tichy B, Kappel S, Wang J, et al. Interleukin-10-Deficient Mice and Inflammatory Bowel Disease Associated Cancer Development. Carcinogenesis (2001) 22(4):665–71. doi: 10.1093/carcin/22.4.665
11. Teng MWL, Swann JB, Koebel CM, Schreiber RD, Smyth MJ. Immune-Mediated Dormancy: An Equilibrium With Cancer. J Leukoc Biol (2008) 84(4):988–93. doi: 10.1189/JLB.1107774
12. Hanahan D, Coussens LM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell (2012) 21(3):309–22. doi: 10.1016/j.ccr.2012.02.022
13. Quail DF, Joyce JA. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
14. Joyce JA, Pollard JW. Microenvironmental Regulation of Metastasis. Nat Rev Cancer (2009) 9(4):239–52. doi: 10.1038/nrc2618
15. Markman JL, Shiao SL. Impact of the Immune System and Immunotherapy in Colorectal Cancer. J Gastrointest Oncol (2015) 6(2):208–23. doi: 10.3978/j.issn.2078-6891.2014.077
16. Liotta LA, Kohn EC. The Microenvironment of the Tumour - Host Interface. Nature (2001) 411(6835):375–9. doi: 10.1038/35077241
17. Li M, Cha DJ, Lai Y, Villaruz AE, Sturdevant DE, Otto M. The Antimicrobial Peptide-Sensing System Aps of Staphylococcus Aureus. Mol Microbiol (2007) 66(5):1136–47. doi: 10.1111/J.1365-2958.2007.05986.X
18. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Sci (80-) (2006) 313(5795):1960–4. doi: 10.1126/science.1129139
19. Gatenbee CD, Baker AM, Schenck RO, Strobl M, West J, Neves MP, et al. Immunosuppressive Niche Engineering at the Onset of Human Colorectal Cancer. Nat Commun (2022) 13(1):1798. doi: 10.1038/s41467-022-29027-8
20. Becker WR, Nevins SA, Chen DC, Chiu R, Horning A, Laquindanum R, et al. Single-Cell Analyses Reveal a Continuum of Cell State and Composition Changes in the Malignant Transformation of Polyps to Colorectal Cancer. bioRxiv (2021) 2021:3. doi: 10.1101/2021.03.24.436532
21. Galon J, Pagès F, Marincola FM, Angell HK, Thurin M, Lugli A, et al. Cancer Classification Using the Immunoscore: A Worldwide Task Force. J Transl Med (2012) 10(1):1–10. doi: 10.1186/1479-5876-10-205/FIGURES/2
22. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity (2013) 39(4):782–95. doi: 10.1016/j.immuni.2013.10.003
23. Guo L, Wang C, Qiu X, Pu X, Chang P. Colorectal Cancer Immune Infiltrates: Significance in Patient Prognosis and Immunotherapeutic Efficacy. Front Immunol (2020) 11:1052. doi: 10.3389/fimmu.2020.01052
24. Yang Y, Li C, Liu T, Dai X, Bazhin AV. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front Immunol (2020) 11:1371. doi: 10.3389/fimmu.2020.01371
25. Liu S, Ren J, ten Dijke P. Targeting Tgfβ Signal Transduction for Cancer Therapy. Signal Transduct Target Ther (2021) 6(8):1–20. doi: 10.1038/s41392-020-00436-9
26. Chen J, Pitmon E, Wang K. Microbiome, Inflammation and Colorectal Cancer. Semin Immunol (2017) 32:43–53. doi: 10.1016/J.SMIM.2017.09.006
27. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ Tumor-Infiltrating Lymphocytes and a High CD8+/regulatory T Cell Ratio are Associated With Favorable Prognosis in Ovarian Cancer. Proc Natl Acad Sci U S A (2005) 102(51):18538–43. doi: 10.1073/PNAS.0509182102
28. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat Med (2004) 10(9):942–9. doi: 10.1038/nm1093
29. Yoon HH, Orrock JM, Foster NR, Sargent DJ, Smyrk TC, Sinicrope FA. Prognostic Impact of FoxP3+ Regulatory T Cells in Relation to CD8+ T Lymphocyte Density in Human Colon Carcinomas. PloS One (2012) 7(8):e42274. doi: 10.1371/journal.pone.0042274
30. DeLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The Prognostic Value of FoxP3+ Tumor-Infiltrating Lymphocytes in Cancer: A Critical Review of the Literature. Clin Cancer Res (2012) 18(11):3022–9. doi: 10.1158/1078-0432.CCR-11-3216
31. Phan V, Disis ML. Tumor Stromal Barriers to the Success of Adoptive T Cell Therapy. Cancer Immunol Immunother (2008) 57(2):281–3. doi: 10.1007/s00262-007-0356-6
32. Pagès F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, Bindea G, et al. In Situ Cytotoxic and Memory T Cells Predict Outcome in Patients With Early-Stage Colorectal Cancer. J Clin Oncol (2009) 27(35):5944–51. doi: 10.1200/JCO.2008.19.6147
33. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I Interferons in Anticancer Immunity. Nat Rev Immunol (2015) 15(7):405–14. doi: 10.1038/nri3845
34. Chang LY, Lin YC, Mahalingam J, Huang CT, Chen TW, Kang CW, et al. Tumor-Derived Chemokine CCL5 Enhances TGF-β-Mediated Killing of CD8 + T Cells in Colon Cancer by T-Regulatory Cells. Cancer Res (2012) 72(5):1092–102. doi: 10.1158/0008-5472.CAN-11-2493
35. Cui G, Shi Y, Cui J, Tang F, Florholmen J. Immune Microenvironmental Shift Along Human Colorectal Adenoma-Carcinoma Sequence: Is it Relevant to Tumor Development, Biomarkers and Biotherapeutic Targets? Scand J Gastroenterol (2012) 47(4):367–77. doi: 10.3109/00365521.2011.648950
36. Chaput N, Louafi S, Bardier A, Charlotte F, Vaillant JC, FMénégaux, et al. Identification of CD8 +CD25 +Foxp 3+ Suppressive T Cells in Colorectal Cancer Tissue. Gut (2009) 58(4):520–9. doi: 10.1136/gut.2008.158824
37. Clarke SL, Betts GJ, Plant A, Wright KL, El-Shanawany TM, Harrop R, et al. CD4+CD25+FOXP3+ Regulatory T Cells Suppress Anti-Tumor Immune Responses in Patients With Colorectal Cancer. PloS One (2006) 1(1):e129. doi: 10.1371/journal.pone.0000129
38. Katz SC, Bamboat ZM, Maker AV, Shia J, Pillarisetty VG, Yopp AC, et al. Regulatory T Cell Infiltration Predicts Outcome Following Resection of Colorectal Cancer Liver Metastases. Ann Surg Oncol (2013) 20(3):946–55. doi: 10.1245/s10434-012-2668-9
39. Nagorsen D, Voigt S, Berg E, Stein H, Thiel E, Loddenkemper C. Tumor-Infiltrating Macrophages and Dendritic Cells in Human Colorectal Cancer: Relation to Local Regulatory T Cells, Systemic T-Cell Response Against Tumor-Associated Antigens and Survival. J Transl Med (2007) 5(1):1–8. doi: 10.1186/1479-5876-5-62/FIGURES/2
40. Brudvik KW, Henjum K, Aandahl EM, Bjørnbeth BA, Taskén K. Regulatory T-Cell-Mediated Inhibition of Antitumor Immune Responses is Associated With Clinical Outcome in Patients With Liver Metastasis From Colorectal Cancer. Cancer Immunol Immunother (2012) 61(7):1045–53. doi: 10.1007/s00262-011-1174-4
41. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell. (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
42. Okeke EB, Uzonna JE. The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front Immunol (2019) 10:680(APR). doi: 10.3389/fimmu.2019.00680
43. Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol (2020) 10:3100. doi: 10.3389/fimmu.2019.03100
44. Vignali DAA, Collison LW, Workman CJ. How Regulatory T Cells Work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
45. Belkaid Y, Rouse BT. Natural Regulatory T Cells in Infectious Disease. Nat Immunol (2005) 6(4):353–60. doi: 10.1038/ni1181
46. Wang HY, Wang RF. Regulatory T Cells and Cancer. Curr Opin Immunol (2007) 19(2):217–23. doi: 10.1016/j.coi.2007.02.004
47. Gershon RK, Kondo K. Cell Interactions in the Induction of Tolerance: The Role of Thymic Lymphocytes. Immunology (1970) 18(5):723–37.
48. Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-Specific Autoimmune Diseases Induced in Mice by Elimination of T Cell Subset. I. Evidence for the Active Participation of T Cells in Natural Self-Tolerance; Deficit of a T Cell Subset as a Possible Cause of Autoimmune Disease. J Exp Med (1985) 161(1):72–87. doi: 10.1084/JEM.161.1.72
49. Möller G. Do Suppressor T Cells Exist? Scand J Immunol (1988) 27(3):247–50. doi: 10.1111/J.1365-3083.1988.TB02344.X
50. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155(3):1151–64.
51. Ashwell JD, Robb RJ, Malek TR. Proliferation of T Lymphocytes in Response to Interleukin 2 Varies With Their State of Activation. J Immunol (1986) 137(8):2572–8.
52. Lenardo MJ. Lnterleukin-2 Programs Mouse αβ T Lymphocytes for Apoptosis. Nature. (1991) 353(6347):858–61. doi: 10.1038/353858a0
53. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a New Forkhead/Winged-Helix Protein, Scurfin, Results in the Fatal Lymphoproliferative Disorder of the Scurfy Mouse. Nat Genet (2001) 27(1):68–73. doi: 10.1038/83784
54. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. J Immunol (2017) 198(3):986–92. doi: 10.1038/ni904
55. Hori S, Nomura T, Sakaguchi S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. J Immunol (2017) 198(3):981–5. doi: 10.1126/science.1079490
56. Khattri R, Cox T, Yasayko SA, Ramsdell F. An Essential Role for Scurfin in CD4+CD25+T Regulatory Cells. J Immunol (2017) 198(3):993–8. doi: 10.1038/ni909
57. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T Cell Lineage Specification by the Forkhead Transcription Factor Foxp3. Immunity (2005) 22(3):329–41. doi: 10.1016/j.immuni.2005.01.016
58. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
59. Roncador G, Brown PJ, Maestre L, Hue S, Martínez-Torrecuadrada JL, Ling KL, et al. Analysis of FOXP3 Protein Expression in Human CD4+CD25+ Regulatory T Cells at the Single-Cell Level. Eur J Immunol (2005) 35(6):1681–91. doi: 10.1002/eji.200526189
60. Gavin MA, Torgerson TR, Houston E, DeRoos P, Ho WY, Stray-Pedersen A, et al. Single-Cell Analysis of Normal and FOXP3-Mutant Human T Cells: FOXP3 Expression Without Regulatory T Cell Development. Proc Natl Acad Sci (2006) 103(17):6659–64. doi: 10.1073/PNAS.0509484103
61. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3 + Regulatory T Cells in the Human Immune System. Nat Rev Immunol (2010) 10(7):490–500. doi: 10.1038/nri2785
62. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of Regulatory T Cell Development by Satb1-Dependent Super-Enhancer Establishment. Nat Immunol (2017) 18(2):173–83. doi: 10.1038/ni.3646
63. Togashi Y, Nishikawa H. Regulatory T Cells: Molecular and Cellular Basis for Immunoregulation. Curr Top Microbiol Immunol (2017) 410:3–27. doi: 10.1007/82_2017_58
64. Chen WJ, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of Peripheral CD4+CD25– Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-β Induction of Transcription Factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi: 10.1084/JEM.20030152
65. Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 Is Essential for TGF-β to Convert Naive CD4 + CD25 – Cells to CD25 + Foxp3 + Regulatory T Cells and for Expansion of These Cells. J Immunol (2007) 178(4):2018–27. doi: 10.4049/jimmunol.178.4.2018
66. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral Education of the Immune System by Colonic Commensal Microbiota. Nature (2011) 478(7368):250–4. doi: 10.1038/nature10434
67. Yadav M, Stephan S, Bluestone JA. Peripherally Induced Tregs-Role in Immune Homeostasis and Autoimmunity. Front Immunol (2013) 4:232. doi: 10.3389/fimmu.2013.00232
68. Elkord E. Helios Should Not be Cited as a Marker of Human Thymus-Derived Tregs. Commentary: Helios+ and Helios- Cells Coexist Within the Natural FOXP3+ T Regulatory Cell Subset in Humans. Front Immunol (2016) 7:276. doi: 10.3389/fimmu.2016.00276
69. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 Distinguishes Natural and Inducible Regulatory T Cells Among Regulatory T Cell Subsets In Vivo. J Exp Med (2012) 209(10):1713–22. doi: 10.1084/JEM.20120822
70. Weiss JM, Bilate AM, Gobert M, Ding Y, de Lafaille MAC, Parkhurst CN, et al. Neuropilin 1 is Expressed on Thymus-Derived Natural Regulatory T Cells, But Not Mucosa-Generated Induced Foxp3+ T Reg Cells. J Exp Med (2012) 209(10):1723–42. doi: 10.1084/JEM.20120914
71. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing the FoxP3 Transcription Factor. Immunity. (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
72. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 Expression in Naive Human CD4+FOXP3 - T Cells by T-Cell Receptor Stimulation is Transforming Growth Factor-β-Dependent But Does Not Confer a Regulatory Phenotype. Blood. (2007) 110(8):2983–90. doi: 10.1182/blood-2007-06-094656
73. Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 Provides Localized Access to IL-2 and Defines Homeostatically Distinct Regulatory T Cell Subsets. J Exp Med (2014) 211(1):121–36. doi: 10.1084/jem.20131142
74. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity. (2019) 50(2):493–504.e7. doi: 10.1016/j.immuni.2019.01.001
75. Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
76. Kim HP, Leonard WJ. CREB/ATF-Dependent T Cell Receptor-Induced FoxP3 Gene Expression: A Role for DNA Methylation. J Exp Med (2007) 204(7):1543–51. doi: 10.1084/jem.20070109
77. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of Conserved non-Coding DNA Elements in the Foxp3 Gene in Regulatory T-Cell Fate. Nature. (2010) 463(7282):808–12. doi: 10.1038/nature08750
78. Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, et al. Lineage-Specific DNA Methylation in T Cells Correlates With Histone Methylation and Enhancer Activity. Genome Res (2009) 19(7):1165–74. doi: 10.1101/gr.091470.109
79. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the Inheritance of Regulatory T Cell Identity by a Cis Element in the Foxp3 Locus. Cell. (2014) 158(4):749–63. doi: 10.1016/j.cell.2014.07.031
80. Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda-Martínez JA, et al. Control of Foxp3 Stability Through Modulation of TET Activity. J Exp Med (2016) 213(3):377–97. doi: 10.1084/JEM.20151438
81. Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, et al. Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 Demethylation to Drive Regulatory T Cell Differentiation and Maintain Immune Homeostasis. Immunity. (2015) 43(2):251–63. doi: 10.1016/j.immuni.2015.07.017
82. Yue X, Lio CWJ, Samaniego-Castruita D, Li X, Rao A. Loss of TET2 and TET3 in Regulatory T Cells Unleashes Effector Function. Nat Commun (2019) 10(1):1–14. doi: 10.1038/s41467-019-09541-y
83. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT Cooperate to Induce Foxp3 Expression Through its Enhancer. Nat Immunol (2008) 9(2):194–202. doi: 10.1038/ni1549
84. Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, et al. Extrathymically Generated Regulatory T Cells Control Mucosal T H 2 Inflammation. Nature. (2012) 482(7385):395–9. doi: 10.1038/nature10772
85. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T Cell Receptor Stimulation-Induced Epigenetic Changes and Foxp3 Expression Are Independent and Complementary Events Required for Treg Cell Development. Immunity. (2012) 37(5):785–99. doi: 10.1016/j.immuni.2012.09.010
86. Zhao H, Liao X, Kang Y. Tregs: Where We are and What Comes Next? Front Immunol (2017) 8:1578. doi: 10.3389/fimmu.2017.01578
87. Bollrath J, Powrie FM. Controlling the Frontier: Regulatory T-Cells and Intestinal Homeostasis. Semin Immunol (2013) 25(5):352–7. doi: 10.1016/J.SMIM.2013.09.002
88. Liston A, Gray DHD. Homeostatic Control of Regulatory T Cell Diversity. Nat Rev Immunol (2014) 14(3):154–65. doi: 10.1038/nri3605
89. Huehn J, Hamann A. Homing to Suppress: Address Codes for Treg Migration. Trends Immunol (2005) 26(12):632–6. doi: 10.1016/J.IT.2005.10.001
90. Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, Träger U, et al. Genome-Wide DNA-Methylation Landscape Defines Specialization of Regulatory T Cells in Tissues. Nat Immunol (2017) 18(10):1160–72. doi: 10.1038/ni.3799
91. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A Special Population of Regulatory T Cells Potentiates Muscle Repair. Cell (2013) 155(6):1282–95. doi: 10.1016/j.cell.2013.10.054
92. Kalekar LA, Cohen JN, Prevel N, Sandoval PM, Mathur AN, Moreau JM, et al. Regulatory T Cells in Skin are Uniquely Poised to Suppress Profibrotic Immune Responses. Sci Immunol (2019) 4(39):eaaw2910. doi: 10.1126/SCIIMMUNOL.AAW2910
93. Tanoue T, Atarashi K, Honda K. Development and Maintenance of Intestinal Regulatory T Cells. Nat Rev Immunol (2016) 16(5):295–309. doi: 10.1038/nri.2016.36
94. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science. (2011) 331(6015):337–41. doi: 10.1126/science.1198469
95. Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL, et al. Thymus-Derived Regulatory T Cells Contribute to Tolerance to Commensal Microbiota. Nature. (2013) 497(7448):258–62. doi: 10.1038/nature12079
96. Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, et al. Individual Intestinal Symbionts Induce a Distinct Population of Rorγ+ Regulatory T Cells. Science. (2015) 349(6251):993–7. doi: 10.1126/science.aaa9420
97. Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, et al. GATA3 Controls Foxp3+ Regulatory T Cell Fate During Inflammation in Mice. J Clin Invest. (2011) 121(11):4503–15. doi: 10.1172/JCI57456
98. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The Alarmin IL-33 Promotes Regulatory T-Cell Function in the Intestine. Nature. (2014) 513(7519):564–8. doi: 10.1038/nature13577
99. Siede J, Fröhlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 Receptor-Expressing Regulatory T Cells are Highly Activated, Th2 Biased and Suppress CD4 T Cell Proliferation Through IL-10 and Tgfβ Release. PloS One (2016) 11(8):e0161507. doi: 10.1371/journal.pone.0161507
100. Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, et al. T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell. (2018) 175(5):1307–20.e22. doi: 10.1016/j.cell.2018.10.008
101. Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, et al. The Microbiota Regulates Type 2 Immunity Through Rorγt+ T Cells. Science. (2015) 349(6251):989–93. doi: 10.1126/science.aac4263
102. Geuking MB, Cahenzli J, Lawson MAE, Ng DCK, Slack E, Hapfelmeier S, et al. Intestinal Bacterial Colonization Induces Mutualistic Regulatory T Cell Responses. Immunity (2011) 34(5):794–806. doi: 10.1016/j.immuni.2011.03.021
103. Bhaumik S, Mickael ME, Moran M, Spell M, Basu R. Rorγt Promotes Foxp3 Expression by Antagonizing the Effector Program in Colonic Regulatory T Cells. J Immunol (2021) 207(8):2027–38. doi: 10.4049/jimmunol.2100175
104. Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, De Vries JE, et al. A CD4+ T-Cell Subset Inhibits Antigen-Specific T-Cell Responses and Prevents Colitis. Nature (1997) 389(6652):737–42. doi: 10.1038/39614
105. Quandt J, Arnovitz S, Haghi L, Woehlk J, Mohsin A, Okoreeh M, et al. Wnt–β-Catenin Activation Epigenetically Reprograms Treg Cells in Inflammatory Bowel Disease and Dysplastic Progression. Nat Immunol (2021) 22(4):471–84. doi: 10.1038/s41590-021-00889-2
106. Blatner NR, Mulcahy MF, Dennis KL, Scholtens D, Bentrem DJ, Phillips JD, et al. Expression of Rorγt Marks a Pathogenic Regulatory T Cell Subset in Human Colon Cancer. Sci Transl Med (2012) 4(164):164ra159. doi: 10.1126/scitranslmed.3004566
107. Fujimoto S, Greene M, Sehon AH. Immunosuppressor T Cells in Tumor Bearing Hosts. Immunol Invest (1975) 4(3):201–17. doi: 10.3109/08820137409055774
108. Berendt MJ, North RJ. T-Cell-Mediated Suppression of Anti-Tumor Immunity. An Explanation for Progressive Growth of an Immunogenic Tumor. J Exp Med (1980) 151(1):69–80. doi: 10.1084/jem.151.1.69
109. Awwad M, North RJ. Immunologically Mediated Regression of a Murine Lymphoma After Treatment With Anti-L3T4 Antibody. A Consequence of Removing L3T4+ Suppressor T Cells From a Host Generating Predominantly Lyt-2+ T Cell-Mediated Immunity. J Exp Med (1988) 168(6):2193–206. doi: 10.1084/JEM.168.6.2193
110. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor Rejection by In Vivo Administration of Anti-CD25 (Interleukin-2 Receptor α) Monoclonal Antibody. Cancer Res (1999) 59(13):3128–33.
111. Kiyozumi Y, Iwatsuki M, Yamashita K, Koga Y, Yoshida N, Baba H. Update on Targeted Therapy and Immune Therapy for Gastric Cancer. J Cancer Metastasis Treat (2018) 4:31. doi: 10.20517/2394-4722.2017.77
112. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell (2017) 169(7):1342–1356.e16. doi: 10.1016/j.cell.2017.05.035
113. Loyher PL, Rochefort J, Baudesson De Chanville C, Hamon P, Lescaille G, Bertolus C, et al. CCR2 Influences T Regulatory Cell Migration to Tumors and Serves as a Biomarker of Cyclophosphamide Sensitivity. Cancer Res (2016) 76(22):6483–94. doi: 10.1158/0008-5472.CAN-16-0984
114. You Y, Li Y, Li M, Lei M, Wu M, Qu Y, et al. Ovarian Cancer Stem Cells Promote Tumour Immune Privilege and Invasion via CCL5 and Regulatory T Cells. Clin Exp Immunol (2018) 191(1):60–73. doi: 10.1111/cei.13044
115. Tang Y, Xu X, Guo S, Zhang C, Tang Y, Tian Y, et al. An Increased Abundance of Tumor-Infiltrating Regulatory T Cells is Correlated With the Progression and Prognosis of Pancreatic Ductal Adenocarcinoma. PloS One (2014) 9(3):e91551. doi: 10.1371/journal.pone.0091551
116. Vence L, Palucka AK, Fay JW, Ito T, Liu YJ, Banchereau J, et al. Circulating Tumor Antigen-Specific Regulatory T Cells in Patients With Metastatic Melanoma. Proc Natl Acad Sci U S A (2007) 104(52):20884–9. doi: 10.1073/pnas.0710557105
117. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci Rep (2015) 5:15179. doi: 10.1038/srep15179
118. Nishikawa H, Sakaguchi S. Regulatory T Cells in Cancer Immunotherapy. Curr Opin Immunol (2014) 27(1):1–7. doi: 10.1016/j.coi.2013.12.005
119. Sinicrope FA, Rego RL, Ansell SM, Knutson KL, Foster NR, Sargent DJ. Intraepithelial Effector (CD3+)/Regulatory (FoxP3+) T-Cell Ratio Predicts a Clinical Outcome of Human Colon Carcinoma. Gastroenterology (2009) 137(4):1270–9. doi: 10.1053/j.gastro.2009.06.053
120. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, et al. Tumor-Infiltrating FOXP3+ T Regulatory Cells Show Strong Prognostic Significance in Colorectal Cancer. J Clin Oncol (2009) 27(2):186–92. doi: 10.1200/JCO.2008.18.7229
121. Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U, et al. High Frequency of Tumor-Infiltrating FOXP3+ Regulatory T Cells Predicts Improved Survival in Mismatch Repair-Proficient Colorectal Cancer Patients. Wiley Online Libr (2010) 126(11):2635–43. doi: 10.1002/ijc.24989
122. Ladoire S, Martin F, Ghiringhelli F. Prognostic Role of FOXP3+ Regulatory T Cells Infiltrating Human Carcinomas: The Paradox of Colorectal Cancer. Cancer Immunol Immunother. (2011) 60(7):909–18. doi: 10.1007/s00262-011-1046-y
123. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3 + CD4 + T Cell Subpopulations Distinctly Control the Prognosis of Colorectal Cancers. Nat Med (2016) 22(6):679–84. doi: 10.1038/nm.4086
124. Loddenkemper C, Schernus M, Noutsias M, Stein H, Thiel E, Nagorsen D. In Situ Analysis of FOXP3+ Regulatory T Cells in Human Colorectal Cancer. J Transl Med (2006) 4(1):1–8. doi: 10.1186/1479-5876-4-52/FIGURES/1
125. Valzasina B, Piconese S, Guiducci C, Colombo MP. Tumor-Induced Expansion of Regulatory T Cells by Conversion of CD4+CD25- Lymphocytes is Thymus and Proliferation Independent. Cancer Res (2006) 66(8):4488–95. doi: 10.1158/0008-5472.CAN-05-4217
126. Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, et al. Tumor Evasion of the Immune System by Converting CD4 + CD25 – T Cells Into CD4 + CD25 + T Regulatory Cells: Role of Tumor-Derived TGF-β. J Immunol (2007) 178(5):2883–92. doi: 10.4049/jimmunol.178.5.2883
127. Hindley JP, Ferreira C, Jones E, Lauder SN, Ladell K, Wynn KK, et al. Analysis of the T-Cell Receptor Repertoires of Tumor-Infiltrating Conventional and Regulatory T Cells Reveals No Evidence for Conversion in Carcinogen-Induced Tumors. Cancer Res (2011) 71(3):736–46. doi: 10.1158/0008-5472.CAN-10-1797
128. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity (2016) 45(5):1122–34. doi: 10.1016/j.immuni.2016.10.032
129. Sainz-Perez A, Lim A, Lemercier B, Leclerc C. The T-Cell Receptor Repertoire of Tumor-Infiltrating Regulatory T Lymphocytes is Skewed Toward Public Sequences. Cancer Res (2012) 72(14):3557–69. doi: 10.1158/0008-5472.CAN-12-0277
130. Marangoni F, Zhakyp A, Corsini M, Geels SN, Carrizosa E, Thelen M, et al. Expansion of Tumor-Associated Treg Cells Upon Disruption of a CTLA-4-Dependent Feedback Loop. Cell (2021) 184(15):3998–4015.e19. doi: 10.1016/j.cell.2021.05.027
131. Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, et al. Aire-Dependent Thymic Development of Tumor-Associated Regulatory T Cells. Science. (2013) 339(6124):1219–24. doi: 10.1126/science.1233913
132. Bonertz A, Weitz J, Pietsch DHK, Rahbari NN, Schlude C, Ge Y, et al. Antigen-Specific Tregs Control T Cell Responses Against a Limited Repertoire of Tumor Antigens in Patients With Colorectal Carcinoma. J Clin Invest. (2009) 119(11):3311–21. doi: 10.1172/JCI39608
133. Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanović S, Robbins PF, et al. Tumor-Infiltrating Human CD4+ Regulatory T Cells Display a Distinct TCR Repertoire and Exhibit Tumor and Neoantigen Reactivity. Sci Immunol (2019) 4:eaao4310. doi: 10.1126/SCIIMMUNOL.AAO4310
134. Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, Mccune RA, et al. Amount and Distribution of 5-Methylcytosine in Human DNA From Different Types of Tissues or Cells. Nucleic Acids Res (1982) 10(8):2709–21. doi: 10.1093/nar/10.8.2709
135. Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, et al. Foxp3(+) T Cells Expressing Rorγt Represent a Stable Regulatory T-Cell Effector Lineage With Enhanced Suppressive Capacity During Intestinal Inflammation. Mucosal Immunol (2016) 9(2):444–57. doi: 10.1038/MI.2015.74
136. Ma H, Gao W, Sun X, Wang W. STAT5 and TET2 Cooperate to Regulate FOXP3-TSDR Demethylation in CD4+ T Cells of Patients With Colorectal Cancer. J Immunol Res (2018) 2018:6985031. doi: 10.1155/2018/6985031
137. Wieczorek G, Asemissen A, Model F, Turbachova I, Floess S, Liebenberg V, et al. Quantitative DNA Methylation Analysis of FOXP3 as a New Method for Counting Regulatory T Cells in Peripheral Blood and Solid Tissue. Cancer Res (2009) 69(2):599–608. doi: 10.1158/0008-5472.CAN-08-2361
138. Zhuo C, Li Z, Xu Y, Wang Y, Li Q, Peng J, et al. Higher FOXP3-TSDR Demethylation Rates in Adjacent Normal Tissues in Patients With Colon Cancer Were Associated With Worse Survival. Mol Cancer (2014) 13:153. doi: 10.1186/1476-4598-13-153
139. Pickup M, Novitskiy S, Moses HL. The Roles of Tgfβ in the Tumour Microenvironment. Nat Rev Cancer (2013) 13(11):788–99. doi: 10.1038/nrc3603
140. Tang S, Wang J, Yu S. IDO in Colorectal Tumorigenesis: Involvement of Immune Tolerance and Significance in Prevention and Therapy. Cmgh (2021) 12(4):1503–4. doi: 10.1016/j.jcmgh.2021.06.021
141. Takamatsu M, Hirata A, Ohtaki H, Hoshi M, Ando T, Ito H, et al. Inhibition of Indoleamine 2,3-Dioxygenase 1 Expression Alters Immune Response in Colon Tumor Microenvironment in Mice. Cancer Sci (2015) 106(8):1008–15. doi: 10.1111/cas.12705
142. Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and Functional Characterization of Human CD4+CD25+ T Cells With Regulatory Properties Isolated From Peripheral Blood. J Exp Med (2001) 193(11):1285–94. doi: 10.1084/jem.193.11.1285
143. Ward ST, Li KK, Hepburn E, Weston CJ, Curbishley SM, Reynolds GM, et al. The Effects of CCR5 Inhibition on Regulatory T-Cell Recruitment to Colorectal Cancer. Br J Cancer (2015) 112(2):319–28. doi: 10.1038/bjc.2014.572
144. Waniczek D, Lorenc Z, Śnietura M, Wesecki M, Kopec A, Muc-Wierzgoń M. Tumor-Associated Macrophages and Regulatory T Cells Infiltration and the Clinical Outcome in Colorectal Cancer. Arch Immunol Ther Exp (Warsz) (2017) 65(5):445–54. doi: 10.1007/s00005-017-0463-9
145. Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, et al. Tumor-Associated Macrophages Recruit CCR6+ Regulatory T Cells and Promote the Development of Colorectal Cancer via Enhancing CCL20 Production in Mice. PloS One (2011) 6(4):e19495. doi: 10.1371/journal.pone.0019495
146. Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase Ia Study of FoxP3+ CD4 Treg Depletion by Infusion of a Humanized Anti-CCR4 Antibody, KW-0761, in Cancer Patients. Clin Cancer Res (2015) 21(19):4327–36. doi: 10.1158/1078-0432.CCR-15-0357
147. Lee GR. Phenotypic and Functional Properties of Tumor-Infiltrating Regulatory T Cells. Mediators Inflammation (2017) 2017:9. doi: 10.1155/2017/5458178
148. Khaja ASS, Toor SM, Salhat HE, Ali BR, Elkord E. Intratumoral FoxP3+Helios+ Regulatory T Cells Upregulating Immunosuppressive Molecules are Expanded in Human Colorectal Cancer. Front Immunol (2017) 8:619. doi: 10.3389/fimmu.2017.00619
149. Stagg J, Smyth MJ. Extracellular Adenosine Triphosphate and Adenosine in Cancer. Oncogene (2010) 29(39):5346–58. doi: 10.1038/onc.2010.292
150. Szeponik L, Ahlmanner F, Sundström P, Rodin W, Gustavsson B, Bexe Lindskog E, et al. Intratumoral Regulatory T Cells From Colon Cancer Patients Comprise Several Activated Effector Populations. BMC Immunol (2021) 22(1):1–12. doi: 10.1186/s12865-021-00449-1
151. Ma C, Dong X. Colorectal Cancer-Derived Foxp3(+) IL-17(+) T Cells Suppress Tumour-Specific CD8+ T Cells. Scand J Immunol (2011) 74(1):47–51. doi: 10.1111/J.1365-3083.2011.02539.X
152. Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, et al. IL-17 + Regulatory T Cells in the Microenvironments of Chronic Inflammation and Cancer. J Immunol (2011) 186(7):4388–95. doi: 10.4049/jimmunol.1003251
153. Downs-Canner S, Berkey S, Delgoffe GM, Edwards RP, Curiel T, Odunsi K, et al. Suppressive IL-17a+ Foxp3+ and Ex-Th17 IL-17aneg Foxp3+ Treg Cells are a Source of Tumour-Associated Treg Cells. Nat Commun (2017) 8:14649. doi: 10.1038/ncomms14649
154. Gounaris E, Blatner NR, Dennis K, Magnusson F, Gurish MF, Strom TB, et al. T-Regulatory Cells Shift From a Protective Anti-Inflammatory to a Cancer-Promoting Proinflammatory Phenotype in Polyposis. Cancer Res (2009) 69(13):5490–7. doi: 10.1158/0008-5472.CAN-09-0304
155. Blatner NR, Bonertz A-D, Beckhove P, Cheon EC, Krantz S, Strouch M, et al. In Colorectal Cancer Mast Cells Contribute to Systemic Regulatory T-Cell Dysfunction. Natl Acad Sci (2010) 107(27):12405. doi: 10.1073/pnas.0913683107
156. Keerthivasan S, Aghajani K, Dose M, Molinero L, Khan MW, Venkateswaran V, et al. β-Catenin Promotes Colitis and Colon Cancer Through Imprinting of Proinflammatory Properties in T Cells. Sci Transl Med (2014) 6(225):225ra28. doi: 10.1126/SCITRANSLMED.3007607
157. Canè S, Bronte V. Wnt–β-Catenin as an Epigenetic Switcher in Colonic Treg Cells. Nat Immunol (2021) 22(4):400–1. doi: 10.1038/s41590-021-00904-6
158. Osman A, Yan B, Li Y, Pavelko KD, Quandt J, Saadalla A, et al. TCF-1 Controls T Reg Cell Functions That Regulate Inflammation, CD8 + T Cell Cytotoxicity and Severity of Colon Cancer. Nat Immunol (2021) 22(9):1152–62. doi: 10.1038/S41590-021-00987-1
159. Rizzo A, Di GM, Stolfi C, Franze E, Fehling HJ, Carsetti R, et al. RORGT-Expressing Tregs Drive the Growth of Colitis-Associated Colorectal Cancer by Controlling IL6 in Dendritic Cells. Cancer Immunol Res (2018) 6(9):1082–92. doi: 10.1158/2326-6066.CIR-17-0698
160. Sasidharan Nair V, Toor SM, Taha RZ, Shaath H, Elkord E. DNA Methylation and Repressive Histones in the Promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and Galectin-9 Genes in Human Colorectal Cancer. Clin Epigenetics (2018) 10(1):1–9. doi: 10.1186/s13148-018-0539-3
161. Elashi AA, Sasidharan Nair V, Taha RZ, Shaath H, Elkord E. DNA Methylation of Immune Checkpoints in the Peripheral Blood of Breast and Colorectal Cancer Patients. Oncoimmunology (2019) 8(2):e1542918. doi: 10.1080/2162402X.2018.1542918
162. Norton SE, Ward-Hartstonge KA, McCall JL, Leman JKH, Taylor ES, Munro F, et al. High-Dimensional Mass Cytometric Analysis Reveals an Increase in Effector Regulatory T Cells as a Distinguishing Feature of Colorectal Tumors. J Immunol (2019) 202(6):1871–84. doi: 10.4049/jimmunol.1801368
163. Ward-Hartstonge KA, McCall JL, McCulloch TR, Kamps AK, Girardin A, Cretney E, et al. Inclusion of BLIMP-1+ Effector Regulatory T Cells Improves the Immunoscore in a Cohort of New Zealand Colorectal Cancer Patients: A Pilot Study. Cancer Immunol Immunother (2017) 66(4):515–22. doi: 10.1007/s00262-016-1951-1
164. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors With Specialized Functions in Immune Regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
165. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 Expression Defines a Subset of CD4 + CD25 High Foxp3 + Regulatory T Cells That Are Expanded at Tumor Sites. J Immunol (2010) 184(11):6545–51. doi: 10.4049/jimmunol.0903879
166. Yan J, Zhang Y, Zhang JP, Liang J, Li L, Zheng L. Tim-3 Expression Defines Regulatory T Cells in Human Tumors. PloS One (2013) 8(3):e58006. doi: 10.1371/journal.pone.0058006
167. Das M, Zhu C, Kuchroo VK. Tim-3 and its Role in Regulating Anti-Tumor Immunity. Immunol Rev (2017) 276(1):97–111. doi: 10.1111/imr.12520
168. Sakuishi K, Ngiow SF, Sullivan JM, Teng MWL, Kuchroo VK, Smyth MJ, et al. TIM3+FOXP3+ Regulatory T Cells are Tissue-Specific Promoters of T-Cell Dysfunction in Cancer. Oncoimmunology. (2013) 2(4):e23849. doi: 10.4161/onci.23849
169. Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced Suppressor Function of TIM-3+FoxP3+ Regulatory T Cells. Eur J Immunol (2014) 44(9):2703–11. doi: 10.1002/eji.201344392
170. Banerjee H, Nieves-Rosado H, Kulkarni A, Murter B, McGrath KV, Chandran UR, et al. Expression of Tim-3 Drives Phenotypic and Functional Changes in Treg Cells in Secondary Lymphoid Organs and the Tumor Microenvironment. Cell Rep (2021) 36(11):109699. doi: 10.1016/j.celrep.2021.109699
171. Ma Q, Liu J, Wu G, Teng M, Wang S, Cui M, et al. Co-Expression of LAG3 and TIM3 Identifies a Potent Treg Population That Suppresses Macrophage Functions in Colorectal Cancer Patients. Clin Exp Pharmacol Physiol (2018) 45(10):1002–9. doi: 10.1111/1440-1681.12992
172. Sawant DV, Hamilton K, Vignali DAA. Interleukin-35: Expanding Its Job Profile. J Interf Cytokine Res (2015) 35(7):499–512. doi: 10.1089/jir.2015.0015
173. Ma Y, Chen L, Xie G, Zhou Y, Yue C, Yuan X, et al. Elevated Level of Interleukin-35 in Colorectal Cancer Induces Conversion of T Cells Into Itr35 by Activating STAT1/STAT3. Oncotarget. (2016) 7(45):73003–15. doi: 10.18632/oncotarget.12193
174. Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, et al. IL-35-Mediated Induction of a Potent Regulatory T Cell Population. Nat Immunol (2010) 11(12):1093–101. doi: 10.1038/ni.1952
175. Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, et al. The Composition and Signaling of the IL-35 Receptor are Unconventional. Nat Immunol (2012) 13(3):290–9. doi: 10.1038/ni.2227
176. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The Inhibitory Cytokine IL-35 Contributes to Regulatory T-Cell Function. Nature. (2007) 450(7169):566–9. doi: 10.1038/nature06306
177. Oida T, Zhang X, Goto M, Hachimura S, Totsuka M, Kaminogawa S, et al. CD4 + CD25 – T Cells That Express Latency-Associated Peptide on the Surface Suppress CD4 + CD45RB High -Induced Colitis by a TGF-β-Dependent Mechanism. J Immunol (2003) 170(5):2516–22. doi: 10.4049/jimmunol.170.5.2516
178. Mahalingam J, Lin CY, Chiang JM, Su PJ, Chu YY, Lai HY, et al. CD4+ T Cells Expressing Latency-Associated Peptide and Foxp3 Are an Activated Subgroup of Regulatory T Cells Enriched in Patients With Colorectal Cancer. PloS One (2014) 9(9):e108554. doi: 10.1371/JOURNAL.PONE.0108554
179. Zhong W, Jiang ZY, Zhang L, Huang JH, Wang SJ, Liao C, et al. Role of LAP+CD4+ T Cells in the Tumor Microenvironment of Colorectal Cancer. World J Gastroenterol (2017) 23(3):455. doi: 10.3748/WJG.V23.I3.455
180. Satoh K, Saitoh K, Kojima Y, Kushida Y, Designed Research, TS. Global Metabolic Reprogramming of Colorectal Cancer Occurs at Adenoma Stage and is Induced by MYC. Natl Acad Sci (2017) 114(37):E7697–706. doi: 10.1073/pnas.1710366114
181. La Vecchia S, Sebastián C. Metabolic Pathways Regulating Colorectal Cancer Initiation and Progression. Semin Cell Dev Biol (2020) 98:63–70. doi: 10.1016/j.semcdb.2019.05.018
182. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
183. Bensard CL, Wisidagama DR, Olson KA, Berg JA, Krah NM, Schell JC, et al. Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell Metab (2020) 31(2):284–300.e7. doi: 10.1016/j.cmet.2019.11.002
184. Kisielow P. How Does the Immune System Learn to Distinguish Between Good and Evil? The First Definitive Studies of T Cell Central Tolerance and Positive Selection. Immunogenetics (2019) 71(8–9):513–8. doi: 10.1007/s00251-019-01127-8
185. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25(6):1282–1293.e7. doi: 10.1016/j.cmet.2016.12.018
186. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4 + T Cell Subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
187. Ding C, Shan Z, Li M, Chen H, Li X, Jin Z. Characterization of the Fatty Acid Metabolism in Colorectal Cancer to Guide Clinical Therapy. Mol Ther - Oncolytics. (2021) 20:532–44. doi: 10.1016/J.OMTO.2021.02.010
188. Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty Acid Metabolism Complements Glycolysis in Th Selective Regulatory T Cell Expansion During Tumor Growth. Proc Natl Acad Sci U S A. (2018) 115(28):E6546–55. doi: 10.1073/pnas.1720113115
189. Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab (2020) 31(2):422–437.e5. doi: 10.1016/j.cmet.2019.11.021
190. Watson MLJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic Support of Tumour-Infiltrating Regulatory T Cells by Lactic Acid. Nature. (2021) 591(7851):645–51. doi: 10.1038/s41586-020-03045-2
191. Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y, et al. CTLA-4 Blockade Drives Loss of Treg Stability in Glycolysis-Low Tumours. Nature. (2021) 591(7851):652–8. doi: 10.1038/s41586-021-03326-4
192. Lim SA, Wei J, Nguyen TLM, Shi H, Su W, Palacios G, et al. Lipid Signalling Enforces Functional Specialization of Treg Cells in Tumours. Nature. (2021) 591(7849):306–11. doi: 10.1038/s41586-021-03235-6
193. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
194. Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-Mediated Metabolic Adaptation Supports Regulatory T Cell Survival and Function in Tumors. Nat Immunol (2020) 21(3):298–308. doi: 10.1038/s41590-019-0589-5
195. Howie D, Cobbold SP, Adams E, Ten BA, AS N, Zhang W, et al. Foxp3 Drives Oxidative Phosphorylation and Protection From Lipotoxicity. JCI Insight (2017) 2(3):e89160. doi: 10.1172/jci.insight.89160
196. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. MTORC1 Couples Immune Signals and Metabolic Programming to Establish T Reg-Cell Function. Nature. (2013) 499(7459):485–90. doi: 10.1038/nature12297
197. Shi H, Chapman NM, Wen J, Guy C, Long L, Dhungana Y, et al. Amino Acids License Kinase Mtorc1 Activity and Treg Cell Function via Small G Proteins Rag and Rheb. Immunity. (2019) 51(6):1012–1027.e7. doi: 10.1016/j.immuni.2019.10.001
198. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-Dependent α-Ketoglutarate Production Regulates the Balance Between T Helper 1 Cell and Regulatory T Cell Generation. Sci Signal (2015) 8(396):ra97. doi: 10.1126/SCISIGNAL.AAB2610
199. Araujo L, Khim P, Mkhikian H, Mortales CL, Demetriou M. Glycolysis and Glutaminolysis Cooperatively Control T Cell Function by Limiting Metabolite Supply to N-Glycosylation. Elife. (2017) 6:e21330. doi: 10.7554/eLife.21330
200. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J Immunol (2006) 176(11):6752–61. doi: 10.4049/jimmunol.176.11.6752
201. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity. (2005) 22(5):633–42. doi: 10.1016/j.immuni.2005.03.013
202. Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. The Indoleamine 2,3-Dioxygenase Pathway Is Essential for Human Plasmacytoid Dendritic Cell-Induced Adaptive T Regulatory Cell Generation. J Immunol (2008) 181(8):5396–404. doi: 10.4049/jimmunol.181.8.5396
203. Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid Dendritic Cells From Mouse Tumor-Draining Lymph Nodes Directly Activate Mature Tregs via Indoleamine 2,3-Dioxygenase. J Clin Invest. (2007) 117(9):2570–82. doi: 10.1172/JCI31911
204. Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO Activates Regulatory T Cells and Blocks Their Conversion Into Th17-Like T Cells. J Immunol (2009) 183(4):2475–83. doi: 10.4049/jimmunol.0900986
205. Yu J, Sun J, Wang SE, Li H, Cao S, Cong Y, et al. Upregulated Expression of Indoleamine 2, 3-Dioxygenase in Primary Breast Cancer Correlates With Increase of Infiltrated Regulatory T Cells in Situ and Lymph Node Metastasis. Clin Dev Immunol (2011) 2011:469135. doi: 10.1155/2011/469135
206. Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C, et al. Prognostic Value of Indoleamine 2,3-Dioxygenase Expression in Colorectal Cancer: Effect on Tumor-Infiltrating T Cells. Clin Cancer Res (2006) 12(4):1144–51. doi: 10.1158/1078-0432.CCR-05-1966
207. Coombes JL, Siddiqui KRR, Arancibia-Cárcamo CV, Hall J, Sun CM, Belkaid Y, et al. A Functionally Specialized Population of Mucosal CD103+ DCs Induces Foxp3+ Regulatory T Cells via a TGF-β -and Retinoic Acid-Dependent Mechanism. J Exp Med (2007) 204(8):1757–64. doi: 10.1084/jem.20070590
208. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small Intestine Lamina Propria Dendritic Cells Promote De Novo Generation of Foxp3 T Reg Cells via Retinoic Acid. J Exp Med (2007) 204(8):1775–85. doi: 10.1084/jem.20070602
209. Bhattacharya N, Yuan R, Prestwood TR, Penny HL, DiMaio MA, Reticker-Flynn NE, et al. Normalizing Microbiota-Induced Retinoic Acid Deficiency Stimulates Protective CD8+ T Cell-Mediated Immunity in Colorectal Cancer. Immunity. (2016) 45(3):641–55. doi: 10.1016/j.immuni.2016.08.008
210. Naganuma M, Wiznerowicz EB, Lappas CM, Linden J, Worthington MT, Ernst PB. Cutting Edge: Critical Role for A 2a Adenosine Receptors in the T Cell-Mediated Regulation of Colitis. J Immunol (2006) 177(5):2765–9. doi: 10.4049/jimmunol.177.5.2765
211. Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A Receptor Signaling Promotes Peripheral Tolerance by Inducing T-Cell Anergy and the Generation of Adaptive Regulatory T Cells. Blood. (2008) 111(1):251–9. doi: 10.1182/blood-2007-03-081646
212. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in Patients With Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol (2017) 18(9):1182–91. doi: 10.1016/S1470-2045(17)30422-9
213. Khan S, Burt DJ, Ralph C, Thistlethwaite FC, Hawkins RE, Elkord E. Tremelimumab (Anti-CTLA4) Mediates Immune Responses Mainly by Direct Activation of T Effector Cells Rather Than by Affecting T Regulatory Cells. Clin Immunol (2011) 138(1):85–96. doi: 10.1016/j.clim.2010.09.011
214. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in Patients With Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet. (2017) 389(10088):2492–502. doi: 10.1016/S0140-6736(17)31046-2
215. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science. (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
216. Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
217. Jiao Q, Ren Y, Ariston Gabrie AN, Wang Q, Wang Y, Du L, et al. Advances of Immune Checkpoints in Colorectal Cancer Treatment. BioMed Pharmacother. (2020) 123:109745. doi: 10.1016/j.biopha.2019.109745
218. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors With Mismatch-Repair Deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMOA1500596
219. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I Study of Single-Agent Anti-Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J Clin Oncol (2010) 28(19):3167–75. doi: 10.1200/JCO.2009.26.7609
220. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol (2018) 36(8):773–9. doi: 10.1200/JCO.2017.76.9901
221. Chung KY, Gore I, Fong L, Venook A, Beck SB, Dorazio P, et al. Phase II Study of the Anti-Cytotoxic T-Lymphocyte-Associated Antigen 4 Monoclonal Antibody, Tremelimumab, in Patients With Refractory Metastatic Colorectal Cancer. J Clin Oncol (2010) 28(21):3485–90. doi: 10.1200/JCO.2010.28.3994
222. Chen EX, Jonker DJ, Loree JM, Kennecke HF, Berry SR, Couture F, et al. Effect of Combined Immune Checkpoint Inhibition vs Best Supportive Care Alone in Patients With Advanced Colorectal Cancer: The Canadian Cancer Trials Group Co. 26 Study. JAMA Oncol (2020) 6(6):831–8. doi: 10.1001/jamaoncol.2020.0910
223. Curigliano G, Gelderblom H, Mach N, Doi T, Tai WMD, Forde P, et al. Abstract CT183: Phase (Ph) I/II Study of MBG453± Spartalizumab (PDR001) in Patients (Pts) With Advanced Malignancies. Cancer Res (2019) 79(13_Supplement):CT183–3. doi: 10.1158/1538-7445.am2019-ct183
224. Hong DS, Rixe O, Chiu VK, Forde PM, Dragovich T, Lou Y, et al. Mogamulizumab in Combination With Nivolumab in a Phase I/II Study of Patients With Locally Advanced or Metastatic Solid Tumors. Clin Cancer Res (2022) 28(3):479–88. doi: 10.1158/1078-0432.CCR-21-2781
225. Fujimoto H, Saito Y, Ohuchida K, Kawakami E, Fujiki S, Watanabe T, et al. Deregulated Mucosal Immune Surveillance Through Gut-Associated Regulatory T Cells and PD-1 + T Cells in Human Colorectal Cancer. J Immunol (2018) 200(9):3291–303. doi: 10.4049/jimmunol.1701222
226. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J Exp Med (2000) 192(7):1027–34. doi: 10.1084/JEM.192.7.1027
227. Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 Pathway. Sci Adv (2020) 6(38):eabd2712. doi: 10.1126/sciadv.abd2712
228. Na Z, Yeo SP, Bharath SR, Bowler MW, Balikçi E, Wang CI, et al. Structural Basis for Blocking PD-1-Mediated Immune Suppression by Therapeutic Antibody Pembrolizumab. Cell Res (2017) 27(1):147–50. doi: 10.1038/cr.2016.77
229. Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 Expression Balance Between Effector and Regulatory T Cells Predicts the Clinical Efficacy of PD-1 Blockade Therapies. Nat Immunol (2020) 21(11):1346–58. doi: 10.1038/s41590-020-0769-3
230. Walker LSK, Sansom DM. The Emerging Role of CTLA4 as a Cell-Extrinsic Regulator of T Cell Responses. Nat Rev Immunol (2011) 11(12):852–63. doi: 10.1038/nri3108
231. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin Cancer Res (2019) 25(13):3753–8. doi: 10.1158/1078-0432.CCR-18-4070
232. Lute KD, May KF, Lu P, Zhang H, Kocak E, Mosinger B, et al. Human CTLA4 Knock-in Mice Unravel the Quantitative Link Between Tumor Immunity and Autoimmunity Induced by Anti-CTLA-4 Antibodies. Blood. (2005) 106(9):3127–33. doi: 10.1182/blood-2005-06-2298
233. Ascierto PA, Marincola FM, Ribas A. Anti-CTLA4 Monoclonal Antibodies: The Past and the Future in Clinical Application. J Transl Med (2011) 9(1):196. doi: 10.1186/1479-5876-9-196
234. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-Dependent Depletion of Tumor-Infiltrating Regulatory T Cells Co-Defines the Efficacy of Anti-CTLA-4 Therapy Against Melanoma. J Exp Med (2013) 210(9):1695–710. doi: 10.1084/jem.20130579
235. Hoos A, Ibrahim R, Korman A, Abdallah K, Berman D, Shahabi V, et al. Development of Ipilimumab: Contribution to a New Paradigm for Cancer Immunotherapy. Semin Oncol (2010) 37(5):533–46. doi: 10.1053/j.seminoncol.2010.09.015
236. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 Control Over Foxp3+ Regulatory T Cell Function. Science. (2008) 322(5899):271–5. doi: 10.1126/science.1160062
237. Tang F, Du X, Liu M, Zheng P, Liu Y. Anti-CTLA-4 Antibodies in Cancer Immunotherapy: Selective Depletion of Intratumoral Regulatory T Cells or Checkpoint Blockade? Cell Biosci (2018) 8:30. doi: 10.1186/s13578-018-0229-z
238. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity Through Reduction of Intratumoral Regulatory T Cells. Cancer Immunol Res (2013) 1(1):32–42. doi: 10.1158/2326-6066.CIR-13-0013
239. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating Fc γ Receptors Contribute to the Antitumor Activities of Immunoregulatory Receptor-Targeting Antibodies. J Exp Med (2013) 210(9):1685–93. doi: 10.1084/jem.20130573
240. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science. (2011) 332(6029):600–3. doi: 10.1126/science.1202947
241. Vargas FA, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell (2018) 33(4):649–663.e4. doi: 10.1016/j.ccell.2018.02.010
242. Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 Blockade and GM-CSF Combination Immunotherapy Alters the Intratumor Balance of Effector and Regulatory T Cells. J Clin Invest. (2006) 116(7):1935–45. doi: 10.1172/JCI27745
243. Kavanagh B, O’Brien S, Lee D, Hou Y, Weinberg V, Rini B, et al. CTLA4 Blockade Expands FoxP3+ Regulatory and Activated Effector CD4 + T Cells in a Dose-Dependent Fashion. Blood. (2008) 112(4):1175–83. doi: 10.1182/blood-2007-11-125435
244. Zhou E, Huang Q, Wang J, Fang C, Yang L, Zhu M, et al. Up-Regulation of Tim-3 is Associated With Poor Prognosis of Patients With Colon Cancer. Int J Clin Exp Pathol (2015) 8(7):8018–27.
245. Yu X, Huang X, Chen X, Liu J, Wu C, Pu Q, et al. Characterization of a Novel Anti-Human Lymphocyte Activation Gene 3 (LAG-3) Antibody for Cancer Immunotherapy. MAbs. (2019) 11(6):1139–48. doi: 10.1080/19420862.2019.1629239
246. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
247. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a Member of the CD28 Family and an Important Modulator of T-Cell Function. Eur J Immunol (2011) 41(4):902–15. doi: 10.1002/eji.201041136
248. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8+ T Cell Effector Function. Cancer Cell (2014) 26(6):923–37. doi: 10.1016/j.ccell.2014.10.018
249. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a Novel Lymphocyte Activation Gene Closely Related to CD4. J Exp Med (1990) 171(5):1393–405. doi: 10.1084/JEM.171.5.1393
250. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in Regulatory T Cells. Immunity. (2004) 21(4):503–13. doi: 10.1016/j.immuni.2004.08.010
251. Huard B, Tournier M, Triebel F. LAG-3 Does Not Define a Specific Mode of Natural Killing in Human. Immunol Lett (1998) 61(2–3):109–12. doi: 10.1016/S0165-2478(97)00170-3
252. Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of Lymphocyte Activation Gene 3 (LAG-3) on B Cells is Induced by T Cells. Eur J Immunol (2005) 35(7):2081–8. doi: 10.1002/eji.200526090
253. Andreae S, Piras F, Burdin N, Triebel F. Maturation and Activation of Dendritic Cells Induced by Lymphocyte Activation Gene-3 (Cd223). J Immunol (2002) 168(8):3874–80. doi: 10.4049/jimmunol.168.8.3874
254. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of Tryptophan Catabolism by Regulatory T Cells. Nat Immunol (2003) 4(12):1206–12. doi: 10.1038/ni1003
255. Maruhashi T, Okazaki I, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 Inhibits the Activation of CD4 + T Cells That Recognize Stable pMHCII Through its Conformation-Dependent Recognition of pMHCII. . Nat Immunol (2018) 19(12):1415–26. doi: 10.1038/s41590-018-0217-9
256. Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The TIM Gene Family Regulates Autoimmune and Allergic Diseases. Trends Mol Med (2005) 11(8):362–9. doi: 10.1016/J.MOLMED.2005.06.008
257. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 Expression Characterizes Regulatory T Cells in Tumor Tissues and is Associated With Lung Cancer Progression. PloS One (2012) 7(2):e30676. doi: 10.1371/journal.pone.0030676
258. Scurr M, Ladell K, Besneux M, Christian A, Hockey T, Smart K, et al. Highly Prevalent Colorectal Cancer-Infiltrating LAP+ Foxp3– T Cells Exhibit More Potent Immunosuppressive Activity Than Foxp3+ Regulatory T Cells. Mucosal Immunol (2014) 7(2):428–39. doi: 10.1038/MI.2013.62
259. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Müller CE, et al. CD39/ENTPD1 Expression by CD4+Foxp3+ Regulatory T Cells Promotes Hepatic Metastatic Tumor Growth in Mice. Gastroenterology. (2010) 139(3):1030–40. doi: 10.1053/j.gastro.2010.05.007
260. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MWL, Darcy PK, et al. CD73-Deficient Mice Have Increased Antitumor Immunity and are Resistant to Experimental Metastasis. Cancer Res (2011) 71(8):2892–900. doi: 10.1158/0008-5472.CAN-10-4246
261. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity. (2016) 45(5):1135–47. doi: 10.1016/j.immuni.2016.10.021
262. Li Z, Chan K, Qi Y, Lu L, Ning F, Wu M, et al. Participation of Ccl1 in Snail-Positive Fibroblasts in Colorectal Cancer Contribute to 5-Fluorouracil/Paclitaxel Chemoresistance. Cancer Res Treat (2018) 50(3):894–907. doi: 10.4143/crt.2017.356
263. Sun D, Luo T, Dong P, Zhang N, Chen J, Zhang S, et al. M2-Polarized Tumor-Associated Macrophages Promote Epithelial-Mesenchymal Transition via Activation of the AKT3/PRAS40 Signaling Pathway in Intrahepatic Cholangiocarcinoma. J Cell Biochem (2020) 121(4):2828–38. doi: 10.1002/jcb.29514
264. Hoelzinger DB, Smith SE, Mirza N, Dominguez AL, Manrique SZ, Lustgarten J. Blockade of CCL1 Inhibits T Regulatory Cell Suppressive Function Enhancing Tumor Immunity Without Affecting T Effector Responses. J Immunol (2010) 184(12):6833–42. doi: 10.4049/jimmunol.0904084
265. Villarreal DO, L’Huillier A, Armington S, Mottershead C, Filippova EV, Coder BD, et al. Targeting CCR8 Induces Protective Antitumor Immunity and Enhances Vaccine-Induced Responses in Colon Cancer. Cancer Res (2018) 78(18):5340–8. doi: 10.1158/0008-5472.CAN-18-1119
266. Halim L, Romano M, McGregor R, Correa I, Pavlidis P, Grageda N, et al. An Atlas of Human Regulatory T Helper-Like Cells Reveals Features of Th2-Like Tregs That Support a Tumorigenic Environment. Cell Rep (2017) 20(3):757–70. doi: 10.1016/j.celrep.2017.06.079
267. Lutsiak MEC, Semnani RT, De Pascalis R, Kashmiri SVS, Schlom J, Sabzevari H. Inhibition of CD4+25+ T Regulatory Cell Function Implicated in Enhanced Immune Response by Low-Dose Cyclophosphamide. Blood. (2005) 105(7):2862–8. doi: 10.1182/blood-2004-06-2410
268. Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic Cyclophosphamide Regimen Selectively Depletes CD4 +CD25+ Regulatory T Cells and Restores T and NK Effector Functions in End Stage Cancer Patients. Cancer Immunol Immunother. (2007) 56(5):641–8. doi: 10.1007/s00262-006-0225-8
269. Terme M, Chaput N, Combadiere B, Ma A, Ohteki T, Zitvogel L. Regulatory T Cells Control Dendritic Cell/NK Cell Cross-Talk in Lymph Nodes at the Steady State by Inhibiting CD4+ Self-Reactive T Cells. J Immunol (2008) 180(7):4679–86. doi: 10.4049/JIMMUNOL.180.7.4679
270. Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory Effects of Cyclophosphamide and Implementations for Vaccine Design. Semin Immunopathol (2011) 33(4):369–83. doi: 10.1007/S00281-011-0245-0
271. Roux S, Apetoh L, Chalmin F, Ladoire S, Mignot G, Puig PE, et al. CD4+CD25+ Tregs Control the TRAIL-Dependent Cytotoxicity of Tumor-Infiltrating DCs in Rodent Models of Colon Cancer. J Clin Invest. (2008) 118(11):3751–61. doi: 10.1172/JCI35890
272. Malvicini M, Ingolotti M, Piccioni F, Garcia M, Bayo J, Atorrasagasti C, et al. Reversal of Gastrointestinal Carcinoma-Induced Immunosuppression and Induction of Antitumoural Immunity by a Combination of Cyclophosphamide and Gene Transfer of IL-12. Mol Oncol (2011) 5(3):242–55. doi: 10.1016/J.MOLONC.2011.03.007
273. Malvicini M, Alaniz L, Bayo J, García M, Piccioni F, Fiore E, et al. Single Low-Dose Cyclophosphamide Combined With Interleukin-12 Gene Therapy is Superior to a Metronomic Schedule in Inducing Immunity Against Colorectal Carcinoma in Mice. Oncoimmunology. (2012) 1(7):1038–47. doi: 10.4161/onci.20684
274. Scurr M, Pembroke T, Bloom A, Roberts D, Thomson A, Smart K, et al. Low-Dose Cyclophosphamide Induces Antitumor T-Cell Responses, Which Associate With Survival in Metastatic Colorectal Cancer. Clin Cancer Res (2017) 23(22):6771–80. doi: 10.1158/1078-0432.CCR-17-0895
275. Scurr M, Pembroke T, Bloom A, Roberts D, Thomson A, Smart K, et al. Effect of Modified Vaccinia Ankara–5T4 and Low-Dose Cyclophosphamide on Antitumor Immunity in Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA Oncol (2017) 3(10):e172579. doi: 10.1001/jamaoncol.2017.2579
276. Suurs FV, Lub-de Hooge MN, de Vries EGE, de Groot DJA. A Review of Bispecific Antibodies and Antibody Constructs in Oncology and Clinical Challenges. Pharmacol Ther (2019) 201:103–19. doi: 10.1016/j.pharmthera.2019.04.006
277. Barbi J, Pardoll DM, Pan F. Ubiquitin-Dependent Regulation of Foxp3 and Treg Function. Immunol Rev (2015) 266(1):27–45. doi: 10.1111/imr.12312
278. Wang L, Beier UH, Akimova T, Dahiya S, Han R, Samanta A, et al. Histone/protein Deacetylase Inhibitor Therapy for Enhancement of Foxp3+ T-Regulatory Cell Function Posttransplantation. Am J Transplant. (2018) 18(7):1596–603. doi: 10.1111/ajt.14749
279. Xiao W, Zhou Q, Wen X, Wang R, Liu R, Wang T, et al. Small-Molecule Inhibitors Overcome Epigenetic Reprogramming for Cancer Therapy. Front Pharmacol (2021) 12:702360. doi: 10.3389/fphar.2021.702360
Keywords: colorectal cancer, CRC, T regulatory cells, Treg, tumor microenvironment, immunotherapy, immunometabolism, tumor infiltrating cells
Citation: Aristin Revilla S, Kranenburg O and Coffer PJ (2022) Colorectal Cancer-Infiltrating Regulatory T Cells: Functional Heterogeneity, Metabolic Adaptation, and Therapeutic Targeting. Front. Immunol. 13:903564. doi: 10.3389/fimmu.2022.903564
Received: 24 March 2022; Accepted: 06 June 2022;
Published: 08 July 2022.
Edited by:
Paula D. Bos, Virginia Commonwealth University, United StatesReviewed by:
Theresa L. Whiteside, University of Pittsburgh, United StatesKhashayarsha Khazaie, Mayo Clinic Arizona, United States
Copyright © 2022 Aristin Revilla, Kranenburg and Coffer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul J. Coffer, cGNvZmZlckB1bWN1dHJlY2h0Lm5s